 Manikuntala Kundu
Manikuntala Kundu Joyoti Basu
Joyoti Basu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 24 June 2021
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.687962
This article is part of the Research TopicStrategies Played by Immune Cells and Mycobacteria in the Battle between Antimicrobial Activity and Bacterial SurvivalView all 13 articles
Non-coding RNAs have emerged as critical regulators of the immune response to infection. MicroRNAs (miRNAs) are small non-coding RNAs which regulate host defense mechanisms against viruses, bacteria and fungi. They are involved in the delicate interplay between Mycobacterium tuberculosis, the causative agent of tuberculosis (TB), and its host, which dictates the course of infection. Differential expression of miRNAs upon infection with M. tuberculosis, regulates host signaling pathways linked to inflammation, autophagy, apoptosis and polarization of macrophages. Experimental evidence suggests that virulent M. tuberculosis often utilize host miRNAs to promote pathogenicity by restricting host-mediated antibacterial signaling pathways. At the same time, host- induced miRNAs augment antibacterial processes such as autophagy, to limit bacterial proliferation. Targeting miRNAs is an emerging option for host-directed therapies. Recent studies have explored the role of long non-coding RNA (lncRNAs) in the regulation of the host response to mycobacterial infection. Among other functions, lncRNAs interact with chromatin remodelers to regulate gene expression and also function as miRNA sponges. In this review we attempt to summarize recent literature on how miRNAs and lncRNAs are differentially expressed during the course of M. tuberculosis infection, and how they influence the outcome of infection. We also discuss the potential use of non-coding RNAs as biomarkers of active and latent tuberculosis. Comprehensive understanding of the role of these non-coding RNAs is the first step towards developing RNA-based therapeutics and diagnostic tools for the treatment of TB.
Tuberculosis (TB) is a global health problem. About one third of the world’s population is infected with Mycobacterium tuberculosis, the causative agent of TB. Of these, about 5 to 10% of infected individuals develop active disease. Around 10 million new TB infections and 1.4 million deaths were reported in 2019 (1). In latent TB (LTBI), M. tuberculosis remains walled off within a granuloma in the lung for long periods of time in a dormant state with no symptoms of disease and without a marked immune response. The bacilli then reactivate under favorable conditions, most notably when the host is immunocompromised as in the case of HIV infection. A reduction in the global burden of TB, requires a means of diagnosing as well as treating latent TB. The problems of TB are further exacerbated by the growing increase in multidrug-resistant (MDR), extensively drug-resistant (XDR) and totally drug resistant TB. With this background, it is evident that there is an increasing need to develop newer approaches towards therapy of TB. Host-directed therapies (HDTs) provide the option of manipulating the host immune response to thwart disease, without the development of drug-resistant bacilli. Effective HDTs require a detailed knowledge of the immune response to infection.
MicroRNAs (miRNAs) are non-coding RNAs which are conserved across species and phyla and are typically 20 to 22 nucleotides in length (2). In recent years it has become evident that miRNAs regulate the interactions between hosts and pathogens (3–5). They play important roles in regulating the immune response to bacterial pathogens such as Listeria monocytogenes, Helicobacter pylori, Pseudomonas aeruginosa, Salmonella typhimurium and M. tuberculosis (5). miRNAs regulate the response of innate immune cells such as macrophages, to infection. Macrophages are the sentinels of the host immune defense system. One of the first steps of bacterial infection is the sensing of the pathogen associated molecular patterns (PAMPs) by host pattern recognition receptors (PRRs) expressed on macrophages. This triggers a cascade of signaling events which culminate in reprogramming of the host transcriptome so that the bacterium is effectively thwarted. The bacterium responds by remodeling its own transcriptome to adjust metabolism and to express virulence genes. Bacteria also hijack the innate immune pathways of the host for their own benefit (6). In the case of M. tuberculosis infection, the host-pathogen interaction impacts processes such as apoptosis, autophagy, cytokine production, macrophage polarization and MHC class II expression. Recent studies have shown that many of these processes are regulated by miRNAs (7–9). Long noncoding RNAs (lncRNAs) are transcripts that are longer than 200 nucleotides, but do not code for proteins (10–12). LncRNAs interact with proteins such as chromatin remodelers and are regulators of innate immunity (13, 14). Differential regulation of lncRNAs modulates the response of immune cells to mycobacterial infection (15, 16).
In addition to the potential use of miRNA or lncRNA-targeted drugs in HDT, several studies have explored the potential of miRNAs (17) and lncRNAs (18) as biomarkers for active TB as well as LTBI, thus underscoring the translational potential of miRNAs as markers of disease. Here, we will focus on regulation of the macrophage immune response to M. tuberculosis infection by miRNAs and lncRNAs, and touch upon their potential as biomarkers of disease. We will restrict our discussion primarily to how these ncRNAs rewire the innate immune response to regulate inflammation, apoptosis, macrophage polarization and autophagy, which determine the outcome of M. tuberculosis infection. The immune responses to other pathogenic mycobacteria, have not been discussed in detail in this review.
miRBase, an online repository of miRNAs, lists 4885 mature miRNAs in 271 species (release 22, March 2018). miRNA genes are transcribed by RNA polymerase II, as 5’-capped and 3’-polyadenylated precursors of 200 to 300 nucleotides (19). They are processed by nuclear RNase Drosha and the RNA binding protein DiGeorge syndrome critical region 8 (DGCR8) into 60-70 nucleotide premiRNAs with hairpin structures and exported to the cytoplasm with the help of Exportin 5 and RAN GTPase. In the cytoplasm, the pre-miRNA is cleaved by the RNase III Dicer in combination with the HIV TAR RNA‐binding protein (TRBP) or Protein Activator of PKR (PACT) RNA-binding proteins into 16-24 bp double stranded RNA. The guide RNA strand then associates with a protein of the Argonaute (AGO) family while the passenger strand is degraded. The AGO-associated miRNA strand is a part of the RNA-induced silencing complex (RISC). It interacts with mRNA via base pairing. The AGO proteins along with other partners recruit other players such as the deadenylase complexes PAN2-PAN3 and CCR4-NOT to repress translation and promote degradation of target mRNAs (20). miRNA biogenesis is regulated at the post-translational level through modifications such as phosphorylation and sumoylation (19).
MiRNAs regulate innate immune responses by targeting inflammatory pathways. PRRs recognize PAMPS to trigger inflammatory signaling. The role of Toll-like receptors (TLRs) has been extensively studied in the context of M. tuberculosis infection (21). The cytosolic sensors cyclic GMP-AMP synthase (cGAS), interferon-activable protein 204 (IFI204), and absent in melanoma 2 (AIM2) recognize M. tuberculosis DNA, while retinoic acid-inducible gene-I (RIG-I), melanoma differentiation‐associated protein 5 (MDA5), and protein kinase R (PKR) detect RNA (22). Nucleotide‐binding oligomerization domain (NOD) 2 recognizes muramyl dipeptide and regulates inflammatory cytokine production during M. tuberculosis infection (23).
The best studied pathway inhibited by miRNAs involves signaling through TLRs. TLRs are expressed on the plasma membrane [TLRS 1, 2, 5, 6; and 11 (expressed in mice but not in humans)] or on endosomes [TLRs 3, 7-8, 9; and 13 (expressed in mice but not in humans)] (24). Each TLR (or a combination of TLRs) senses PAMPs such as tricayllipopeptides (TLR1/2), diacyllipopeptides (TLR2/TLR6), lipopolysaccharide (LPS) (TLR4), flagellin (TLR5), dsRNA (TLR3), ssRNA (TLRs 7 and 8) or CpG DNA and hemozoin (TLR9). Once a ligand binds to the extracellular domain of a TLR, the intracellular Toll-IL-1-resistance (TIR) domains dimerize and recruit adaptor proteins such as myeloid differentiating factor 88 (MyD88) and TIR domain-containing adaptor inducing interferon-β (TRIF), followed by the sequential recruitment and activation of IL-1R-associated kinase (IRAK)-4, IRAK-1 and IRAK-2. Subsequently, there is engagement of downstream adaptor molecules such as TNF receptor-associated factors (TRAFs), which then undergo K63-linked ubiquitination to activate the IκB kinase (IKK) complex (25). The IKK complex phosphorylates IκB-α, triggering its ubiquitination and proteasomal degradation, release of nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) p65, its translocation to the nucleus, and regulation of gene transcription, including those associated with inflammation (26). TLR3- or TLR4-mediated TRIF-dependent signaling leads to activation of the non-canonical IKKs Tank-binding protein kinase 1 (TBK1) and IKKε, and the phosphorylation and nuclear translocation of IFN regulatory factor (IRF) 3. miRNAs limit inflammation by targeting intermediates in this pathway.
RIG-I receptors (RLRs) are RNA sensors localized in the cytosol (27–29). There are three members, RIG-I, MDA5 and laboratory of genetics and physiology 2 (LGP2). RIG-I and MDA5 have two amino-terminal caspase activation and recruitment domains (CARDs), which mediate downstream signal transduction. RIG-I and MDA5 are essential for antiviral defense and type I interferon induction (30, 31). Interaction of RIG-I with mitochondrial anti-viral signaling protein (MAVS) is followed by activation of TBK1 and IKKϵ, which in turn activate IRF3 and IRF7 (32).
Phagocytosis and maturation of the phagosome are key steps in the clearance of bacterial pathogens by macrophages. Pathogenic mycobacteria evade maturation of the phagosome where they reside at least in part by blocking actin assembly. Early during infection of macrophages with M. tuberculosis there is an increase in miR-142-3p which directly targets N-Wiskott-Aldrich syndrome protein (N-WASp) to inhibit actin assembly (33). This demonstrates the miRNA-mediated regulation of phagocytosis during M. tuberculosis infection. MiR-146a/b, miR-155 and miR-21, form a trinity of miRNAs which regulate multiple steps of the TLR and RLR pathways (34–38) to regulate inflammation. These miRNAs are regulated upon activation of TLRs. miR-21 upregulation during Mycobacterium leprae infection is a bacterial strategy to escape the vitamin D-dependent induction of antimicrobial peptides (39). Downregulation of let-7 family members is also characteristic of the response of macrophages to bacterial infection (40, 41). Phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase (SHIP1) is a negative regulator of NF-κB signaling. miR-155 targets SHIP1 in M. tuberculosis-infected macrophages (42), leading to the activation of the serine/threonine kinase Akt, likely facilitating the survival of M. tuberculosis in macrophages. miR-155 also targets BTB and CNC homology 1 (Bach1), a transcriptional repressor of heme oxygenase-1 (HO-1) and inhibits expression of interleukin-6 (Il6) to emerge as a regulator of the macrophage response to M. tuberculosis. miR-155 plays a dual role during M. tuberculosis infection in vivo. It enhances survival of macrophages as well as M. tuberculosis-specific T cells, providing on the one hand a niche for bacterial replication and on the other hand enabling an effective immune response (43). In support of this, miR-155-/- mice control infection at the initial stages, but fail to do so after the onset of adaptive immunity. Overall, the knockout of miR-155 exacerbates infection. This has also been borne out by the studies of Iwai et al. (44) using miR-155-/- mice challenged with M. tuberculosis. miRNA-mediated repression of the NF-κB pathway prevents exacerbated immune responses to infection. M. tuberculosis-induced miR-21-5p attenuates the secretion of IL-1β, IL-6 and TNF-α in RAW264.7 and THP-1 macrophages (45), whereas miR-27a directly targets IRAK4 to attenuate the production of IFN-γ, IL-β, IL-6, and TNF-α (46). There have been few studies on the regulation of miRNAs during infection of alveolar macrophages. In M. bovis BCG-infected alveolar macrophages, miR-124 negatively regulates inflammatory responses by directly targeting multiple components of the TLR pathways including MyD88, TRAF6 and TNF-α (47). miR-203, miR-30a and miR-149 target MyD88 in infected murine or human macrophages to downregulate proinflammatory cytokines as well as nitric oxide (NO) production (48–50). miR-20b inhibits M. tuberculosis-induced inflammation by directly targeting the NLRP3/casapase1/IL1β pathway (51). Further, miRNA-mediated NF-κB repression promotes the survival of M. tuberculosis in macrophages. The downregulation of let-7f during infection increases the expression of the A20 deubiquitinase. A20 targets the K63-linked ubiquitination of TRAF6 (52, 53), thereby negatively regulating the NF-κB pathway, and inhibiting inflammatory cytokine and nitric oxide production thereby facilitating the survival of M. tuberculosis in macrophages (40). MiRNA-125a targets TRAF6 during M. tuberculosis infection, to repress NF-κB (54). The TLR4/miRNA-32-5p/FSTL1 (follistin like protein 1) axis attenuates IL-1β, IL-6 and TNF-α in THP-1 and U937 cells after M. tuberculosis infection (55). M. tuberculosis-induced miR-1178 targets TLR4 and attenuates release of IFN-γ, IL-6, IL-1β, and TNF-α in human macrophages (56). miR-378d targets Rab10. It is downregulated in M. tuberculosis-infected THP-1 and RAW264.7 macrophages leading to enhanced production of IL-6, IL-1β and TNF-α (57). miR-206 targets tissue inhibitor of metalloproteinase 3 (TIMP3) (58). leading to increased production of MMP9 as well as inflammatory cytokines during M. tuberculosis infection. miR-99b expression is highly upregulated in infected macrophages as well as in dendritic cells. It targets TNF-α and TNFRSF-4 receptor genes to attenuate IL-12, IL-1β, IL-6 and TNF-α (59). miR-125b targets TNF-α, whereas miR-155 enhances TNF-α production by increasing TNF mRNA half-life and limiting expression of SHIP1, a negative regulator of the PI3K/Akt pathway. Rajaram et al. (60) have shown that human macrophages infected with M. tuberculosis induce high miR-125b expression and low miR-155 expression with correspondingly low TNF-α production. miR‐140 is upregulated in human peripheral blood mononuclear cells (PBMCs) from patients with TB. It dampens the production of IL-1β, IL-6 and TNF-α in THP-1 macrophages infected with M. tuberculosis (61). In M. bovis BCG-infected macrophages, miR-142-3p targets IRAK1 (62) and miR-146a targets IRAK-1 and TRAF-6 (63) to dampen the production of proinflammatory cytokines TNF-α, IL-6, IL-1β and the chemokine MCP-1. miR-223 targets the chemoattractants CXCL2, CCL3, and IL-6 in myeloid cells to regulate recruitment of myeloid cells to the lungs and as a result, neutrophil-driven inflammation (64). MiR-223 is upregulated in the blood and lung parenchyma of tuberculosis patients. From the above, it is evident that miRNAs play a central role in the attenuation of inflammatory responses during M. tuberculosis infection. Table 1 summarizes the role of miRNAs in regulating inflammatory responses in M. tuberculosis-infected macrophages.
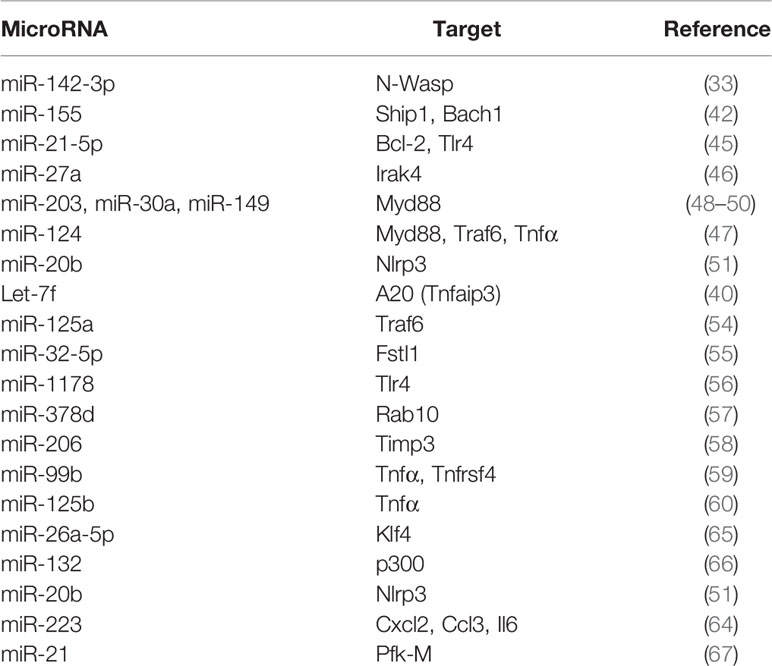
Table 1 MicroRNAs that target signaling in myeloid cells during M. tuberculosis infection to regulate phagocytosis, cytokines, chemokines, macrophage polarization and glycolysis.
Depending on the stimulus, macrophages can be directed towards distinct phenotypes in a process termed macrophage polarization. In the simplest scenario, macrophage polarization results in M1 (classically activated macrophages, proinflammatory state) or M2 (alternatively activated macrophages, anti-inflammatory state) phenotypes, a molecular event crucial for inflammation. M1 macrophages produce proinflammatory cytokines TNF-α, IL-1β, IL-12, IL-6 and IL-23 along with reactive oxygen species (ROS) and nitric oxide. They recruit other immune cells including neutrophils through the production of chemokines such as CXCL8, CCL2, CXCL11, CXCL9 and CXCL10. M2 macrophages produce anti-inflammatory cytokines such as IL-10 and TGF-β, as well as arginase which represses nitric oxide production. M2 macrophages express the mannose receptor (MR) which signals production of cytokines such as CCL17, CCL18, CCL22 and CCL24 that recruit Th2 lymphocytes, eosinophils, basophils and T regulatory (Treg) cells (68). M1 polarized macrophages efficiently clear M. tuberculosis, whereas M2 polarization enhances survival of the bacterium in macrophages. Virulent strains of M. tuberculosis drive M2 polarization (69). Macrophage polarization is influenced by miRNAs. For example, M. tuberculosis infection of murine macrophages, decreases miR-26a-5p, consequently derepressing the transcription factor Kruppel-like factor 4 (KLF4), favoring M2 macrophage polarization in murine macrophages (65). However, in human macrophages miR-26a and miR-132 have been reported to be upregulated (66). These target p300 to attenuate IFNγ responses. In contrast, miR-20b directly targets nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing (NLRP) 3, and its downregulation during M. tuberculosis infection favors M1 polarization through activation of the NLRP3/IL-1β/caspase-1 axis (51). It is evident that inflammatory responses in the macrophage can be regulated by targeting miRNAs which interfere with pathways linked to macrophage polarization.
The phenotype of the macrophage is intimately linked to its metabolism (70). The relationship between metabolic pathways and macrophage polarization, is more complex than previously thought, However, in a broad sense, M1 macrophages rely on glycolysis (71). They present two breaks in the tricarboxylic acid (TCA) cycle which lead to accumulation of itaconate and succinate, which directly impact macrophage functions (72). Succinate stabilizes hypoxia inducible factor 1-α (HIF1-α). This activates the transcription of glycolytic genes and enhances glycolysis. M2 macrophages, on the other hand, are more dependent on oxidative phosphorylation (OXPHOS), their TCA cycle being intact. Evidence for the prevalence of glycolysis during M. tuberculosis infection was obtained by RNA-sequencing of the infected mouse lung tissue at 12, 18 and 30 days (73). It showed the upregulation of glycolytic genes including hexokinases (Hk2 and 3), phosphofructokinase (Pfk) family 1 and 2, glyceraldehye-3-phosphate dehydrogenase (Gapdh), phosphoglycerate kinase (Pgk1), enolase (Eno1), and lactate dehydrogenase A (Ldha); glucose transporters (Glut1, 3 and 6); a transporter for lactate (Mct4); an H+-ATPase involved in cytosolic pH homeostasis; and Hif-1α (regulatory unit of HIF-1, a transcriptional activator of several glycolytic genes which regulates inflammatory processes under normoxia (74). Treating M. tuberculosis-infected mice with 2-deoxyglucose depleted the highly glycolytic subset of infiltrating macrophages and increased the burden of M. tuberculosis. The importance of glycolysis in host defense against M. tuberculosis has been documented in several studies (75–78). Hackett et al. (67) have shown that persistent M. tuberculosis infection of macrophages is associated with miR-21-dependent negative regulation of host glycolysis. Dampening of glycolysis is mediated through targeting of phosphofructokinase muscle (PFK-M) isoform which in turn facilitates bacterial growth by limiting pro-inflammatory mediators such as IL-1β. M. tuberculosis-infected MiR-21−/− bone marrow derived macrophages (BMDMs) showed increased levels of pro-inflammatory mediators including nitric oxide synthase 2 (Nos2) mRNA, arginase 1 (Arg1), and ROS with concomitant containment of M. tuberculosis. Increased containment of bacteria in MiR-21−/− BMDMs is lost when expression of PFK-M is silenced by specific siRNA. In summary, metabolic reprogramming as a consequence of infection, can be influenced by targeting miRNA-regulated pathways. In this context, it is important to evaluate how much information obtained from experiments with mice, can be extrapolated to human infection.
Autophagy is regarded as a homeostatic process by which the eukaryotic cell delivers cytosolic cargo such as misfolded proteins or damaged organelles to the lysosomes for degradation. It can be classified into macroautophagy, chaperone-mediated autophagy and microautophagy. For this review, the term autophagy will refer to macroautophagy. Autophagy impacts diseases linked to inflammation such as infections, cancer, metabolic disorders, autoimmunity, neurodegeneration, cardiovascular and liver diseases (79). In brief, autophagy requires the capture of cargo destined for the lysosomes in double-membrane organelles termed autophagosomes. This occurs through the induction of a signal followed by membrane targeting, vesicle expansion and autophagosome formation. A range of signals such as starvation triggers formation of an isolation membrane usually derived from the endoplasmic reticulum (80). The Unc-51-like kinase 1 (ULK1) is released from the mammalian target of rapamycin complex 1 (mTORC1). Phosphorylated AMP-activated protein kinase (AMPK) phosphorylates ULK1 which in turn forms a complex with the 200 kDa focal adhesion kinase family-interacting protein (FIP200) and autophagy related gene (ATG) 13 to induce autophagy (81). The ULK1 complex recruits Beclin-1 (BECN1) and phosphoinositide 3-kinase regulatory subunit 4 (PIK3R4) in the phagophore membrane nucleation step. The engagement of PtdIns3-kinase class III (PtdIns3KC3) results in the production of PtdIns3P at the phagophore, recruitment of PtdIns3P binding proteins such as WD-repeat protein interacting with phosphoinositides (WIPI) 1 and WIPI2, finally leading to phagophore assembly. The tumor suppressor protein, UV-radiation resistance associated gene (UVRAG) interacts with Beclin-1 and promotes Beclin-1–PI3KC3-mediated autophagy by functioning as an adaptor (82, 83). Autophagosome elongation involves two ubiquitin-like conjugation systems, ATG12-ATG5 which is activated by ATG7 and ATG10 (E1 like and E2 like enzymes respectively) and light chain 3 (LC3 or MAP1LC3B)-phosphatidylethanolamine (PE) which is activated by ATG7 and ATG3 (E2 like enzyme) (84). ATG12-ATG5 complex directs LC3 to the target membrane where it is cleaved by the cysteine protease ATG4B, followed by conjugation of PE to the exposed glycine residue (Figure 1). Eventually, the autophagosomes are directed to lysosomes in a process requiring small GTPases such as Rab7 and soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins (SNAREs). In a variation of autophagy termed xenophagy, intracellular bacteria and viruses are captured with the help of autophagy adaptors for targeting to lysosomes (85, 86). Adaptors such as sequestosome 1 (SQSTM1/p62), nuclear dot protein 52 (NDP52), optineurin (OPTN), and neighbor of BRCA1 gene 1 (NBR1) form a bridge between the cargo and LC3 (87). For a more detailed understanding of autophagy and its role in the immune response to bacteria and viruses, the reader may refer to several excellent reviews (85, 86, 88–90).
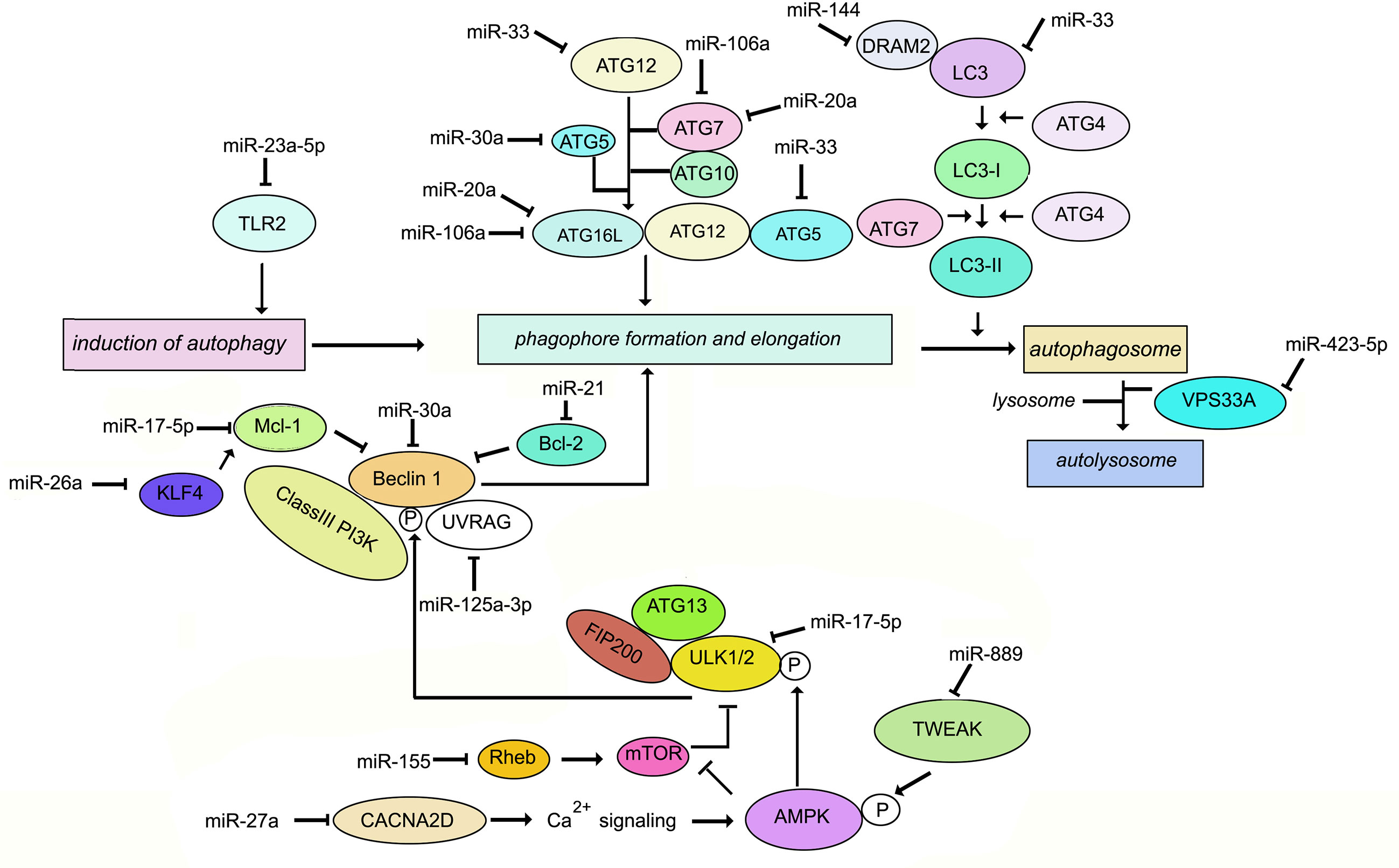
Figure 1 Schematic representation of the key molecules of autophagy targeted by miRNAs during M. tuberculosis infection.
Autophagy has long been recognized as a defense against M. tuberculosis (91–93). M. tuberculosis employs a range of factors to evade autophagy. For example, “enhanced intracellular survival” (eis) gene inhibits host autophagy (94). MiRNAs play a role in the regulation of autophagy during M. tuberculosis infection (95). Differentially regulated miR-23a-5p induction by M. tuberculosis inhibits autophagy in murine RAW264.7 macrophages by targeting TLR2 (96). Induction of Ca2+ acts as a major regulator of autophagy. Upon stimulation, entry of Ca2+ into the cytoplasm activates Ca2+/calmodulin-dependent serine/threonine kinase leading to activation of AMPK and phosphorylation of ULK1. Therefore downregulation of Ca2+ signaling inhibits autophagy promoting the intracellular survival of M. tuberculosis in macrophages. miR-27a targets the Ca2+ transporter voltage-dependent calcium channel subunit alfa2delta (CACNA2D) to limit Ca2+ entry into the cytoplasm and downregulate Ca2+ signaling (97). Mycobacterial infection induces TNF-like weak inducer of apoptosis (TWEAK) in the early phase of infection. TWEAK induces autophagy and promotes mycobacterial autophagosome maturation through activation of AMPK. M. tuberculosis induces MiR-889 which inhibits autophagy via post-transcriptional suppression of TWEAK expression to maintain mycobacterial survival in granulomas (98). The Bcl-2 family member Mcl-1 sequesters Beclin-1 to inhibit autophagy (99). M. tuberculosis inhibits miR17-5p and upregulates its targets Mcl-1 and STAT3, which is a transcriptional activator of Mcl-1 (100), thereby inhibiting autophagy during M. tuberculosis infection. miR-125a-3p regulates autophagy by targeting UVRAG (101). M. tuberculosis induces miR106-a in THP1 macrophages. miR-106-a could inhibit autophagy by targeting ULK1, ATG7, and ATG16L1 (102). Infection of macrophages with M. bovis BCG results in enhanced expression of miR-20a, which inhibits autophagy by targeting ATG7 and ATG16L1 (103). A negative correlation has been demonstrated between miR-30a expression and levels of Beclin-1 as well as ATG5 in M. tuberculosis-infected THP-1 cells (104). Ouimet et al. (8) have reported that miR-33 inhibits autophagy flux through repression of ATG5, ATG12 and LC3B in M. tuberculosis-infected macrophages. M. tuberculosis infection upregulates miR-144 which targets DNA damage-regulated autophagy modulator (DRAM2). DRAM2 releases Beclin-1 from the BECN1 inhibitory complex by directly interacting with Beclin1 (105). Vacuolar protein sorting 33A(VPS33A) is a direct target of miR-423-5p which is upregulated in the serum of TB patients (106). It is therefore plausible that upregulation of miR-423-5p suppresses autophagosome-lysosome fusion during active tuberculosis. Upregulation of miR-26a has been demonstrated to modulate autophagy and M. tuberculosis survival in human and murine macrophages by targeting Mcl-1 (65). In summary, during M. tuberculosis infection, multiple miRNAs target intermediates which are involved in autophagy. There are fewer reports of miRNA-mediated induction of autophagy during M. tuberculosis infection. One report is the induction of miR-155 in M. tuberculosis-infected macrophages, which leads to repression of the negative regulator of autophagy, RHEB, ultimately inducing autophagy and compromised bacterial survival (107). On the contrary, M. bovis BCG reportedly inhibits IFNγ-induced autophagy by induction of miR-155 and miR-31 which target the protein phosphatase 2A (PP2A) regulatory subunit, PPP2R5A (108). These differences could be due to miR-155 functioning differently in resting macrophages vs. IFNγ-activated macrophages, or due to differences in cell types infected. The role of miRNAs in the regulation of autophagy during M. tuberculosis infection, is summarized in Figure 1. Exploiting miRNA-directed therapeutics to augment autophagy, is a plausible option for the management of TB.
There are principally two major pathways leading to apoptosis that are characterized by the activation of caspases 3 and 7 (109, 110). The extrinsic apoptotic pathway involves death receptors, such as Fas and TNFR1, which are membrane-bound. Upon binding of their ligands FasL and TNFR1, they oligomerize and initiate a cascade of signaling events involving assembly of a death-inducing signaling complex (DISC) on the cytoplasmic side of the membrane. The Fas/FasL pair recruits the adaptor protein Fas-associated protein with death domain (FADD), while the TNFR1/TNF pair, recruits tumor necrosis factor receptor type 1-associated death domain protein (TRADD). This is followed by formation of the death inducing complex (DISC) which causes the self-cleavage of procaspase 8 into its active form. Caspase 8 then activates downstream caspases -3, -6 and -7. Intrinsic apoptosis occurs when the cell experiences intracellular stress such as DNA damage or oxidative stress, leading to damage of the mitochondrial outer membrane, and mitochondrial outer membrane permeability (MOMP). Cytochrome c is released into the cytosol, where it associates with the adaptor protein apoptosis protease activating factor 1 (Apaf-1) to trigger its oligomerization and formation of the apoptosome complex. The apoptosome recruits and activates caspase 9, finally leading to the activation of caspase 3. The release of cytochrome c is regulated by the Bcl-2 family of proteins that are either pro- or anti-apoptotic (111). Pro-apoptotic members such as Bax and Bak bind directly to the mitochondrial outer membrane forming pores. Anti-apoptotic members such as Mcl-1 and Bcl-2 bind and sequester the proapoptotic Bcl-2 proteins. The BH3 only proteins such as Bim, p53 upregulated modulator of apoptosis (PUMA), Bid and Bad, bind to the pro-survival Bcl-2 family proteins to sequester them (112) (Figure 2). Unlike apoptosis, necrotic cells undergo swelling of organelles and rupturing of cytoplasmic content into the extracellular space. The necrotic debris and damage-associated molecular pattern molecules (DAMPs) trigger inflammation and tissue damage (113). M. tuberculosis initiates apoptosis or necrosis in the infected host. Apoptosis protects the host and kills the bacteria through efferocytosis of the apoptotic vesicles (114). Apoptosis of infected macrophages is required for dendritic cells to acquire mycobacterial antigens for cross priming. Necrosis, on the other hand, benefits the pathogen. It releases the intracellular bacteria into the extracellular milieu to promote dissemination, and triggers inflammation. Virulent strains of M. tuberculosis induce necrosis (115, 116) and evade apoptosis (117). The differential regulation of apoptosis and necrosis by virulent as opposed to avirulent M. tuberculosis or members of the M. tuberculosis complex, depends at least in part, on the differential expression of miRNAs. For example, M. bovis BCG induces IFNγ by inhibiting miR-29 (118), whereas M. tuberculosis H37Rv upregulates miR-99b expression in murine dendritic cells and macrophages to decrease their targets TNF and TNFRSF45 (TNFR1) (59). Cytokine expression in the cells is altered to affect activation of the host immune response and the survival of intracellular mycobacteria. miR-20a-5p targets JNK2 to repress Bim expression and apoptosis in macrophages. Downregulation of miR-20a-5p in THP-1 macrophages triggers apoptosis and facilitates clearance of M. tuberculosis (119). miR-20b-5p on the other hand, targets Mcl-1 (120), and miR-21-5p targets Bcl-2 (45) to activate apoptosis in M. tuberculosis-infected macrophages. Cyclophilin D (CyPD) associates with p53 to induce apoptosis (121). Overexpression of miRNA-1281 which targets CyPD, protects human macrophages from M. tuberculosis-induced programmed necrosis and apoptosis (122). The nuclear body protein, Sp110 regulates miR-125a, miR-146a, miR-155, miR-21a and miR-99b expression in M. tuberculosis-infected RAW264.7 (123). It upregulates Bcl2 modifying factor (Bmf) by inhibiting miR-125a, thereby enhancing apoptosis. In primary human macrophages infected with M. tuberculosis, miR-579 induction downregulates its targets sirtuin 1 (SIRT1) and phosphoinositide-dependent protein kinase 1 (PDK1) and leads to macrophage apoptosis (124). miR-325-3p directly targets ligand of numb-protein X 1 (LNX1) (125), an E3 ubiquitin ligase of NIMA related kinase 6 (NEK6) which promotes NEK6 proteasomal degradation. The accumulation of NEK6 activates signal transducer and activator of transcription 3 (STAT3) signaling and inhibits apoptosis. On the other hand, Wu et al. (126) have shown that miR-21 is increased in alveolar macrophages of BCG-vaccinated mice, and miR-21 enhances apoptosis of bone marrow derived dendritic cells (BMDCs) from BCG-vaccinated mice most likely by targeting Bcl-2. 6 kDa early secretory antigenic target (ESAT-6)-driven miR-155 targets suppressor of cytokine signaling 1 (SOCS1) to augment macrophage apoptosis by elevating caspase 3 activity (127). TLR-2/MyD88/NF-κB dependent induction of miR-27b suppresses NF-κB (128). Further, miR-27b targets Bcl-2-associated athanogene 2 (Bag2) to regulate apoptosis. miR-27b increases p53-dependent apoptosis to lower bacterial burden. Induction of let-7b-5p in M. tuberculosis infected THP-1 macrophages leads to reduction in its target Fas, and inhibition of apoptosis (29). Activated Akt phosphorylates and inhibits the transcriptional activity of forkhead box O3 (FOXO3) (129). Upon Akt inactivation, dephosphorylated FOXO3 translocates to the nucleus where it activates transcription of multiple pro‐apoptotic genes harboring the forkhead response elements (FRE) in their promoter regions (130). miR-223 targets FOXO3 (131). The upregulation of miR-223 during infection, therefore, likely plays a role in the attenuation of macrophage apoptosis. A list of miRNAs regulating apoptosis during M. tuberculosis infection is given in Table 2, and their roles are schematically presented in Figure 2. Manipulation of apoptosis by administration of miRNA agonists or antagonists deserves further exploration as a means of containing M. tuberculosis in vivo.
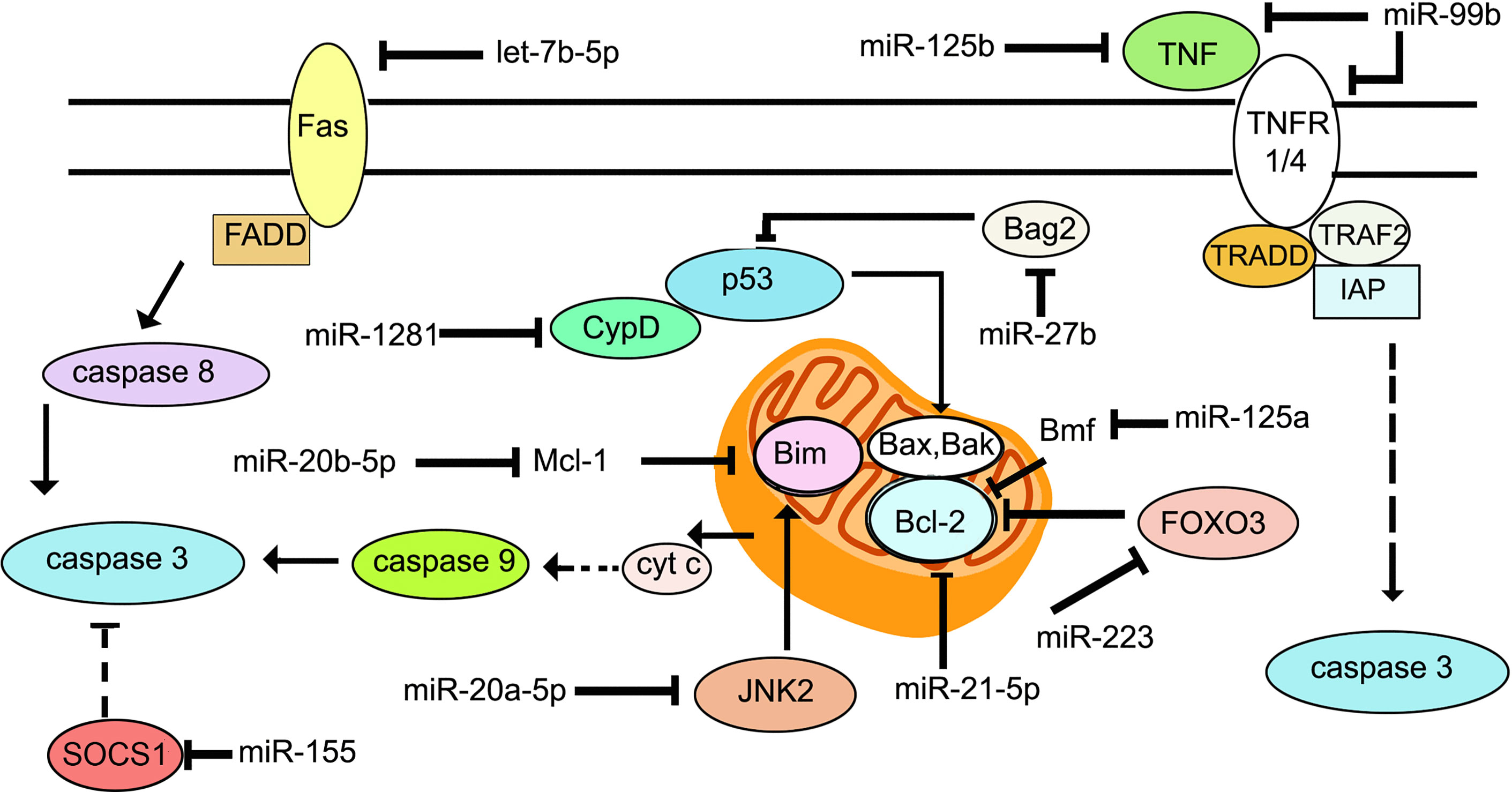
Figure 2 Schematic representation of the key molecules of apoptosis targeted by miRNAs during M. tuberculosis infection.
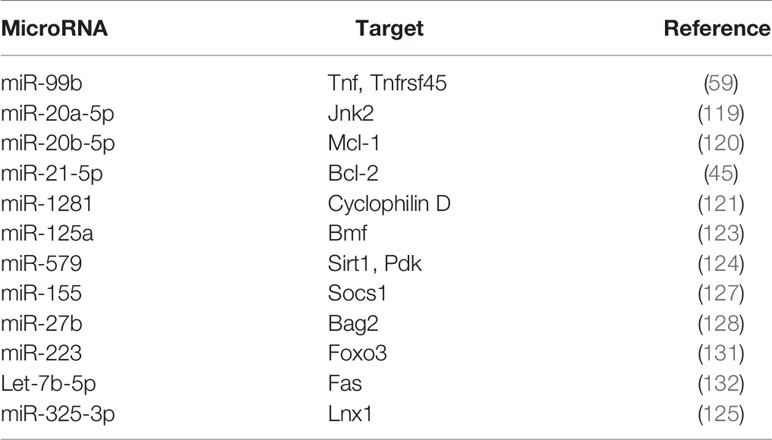
Table 2 MicroRNAs involved in the regulation of apoptosis during M. tuberculosis infection.
The majority of lncRNAs are transcribed by RNA polymerase II (RNAPII) (133), although there are some that are transcribed by RNA polymerase III (RNAPIII) including the 7SL RNA genes (134). LncRNAs undergo transcriptional editing such as splicing and polyadenylation before finally adopting a stable structure. They are expressed in a manner that is dependent on cell and tissue types, the stage of development, and association with disease (10, 135). They show poor evolutionary conservation (136), although a small number of lncRNAs are well conserved (137). LncRNAs fall into different categories. Antisense lncRNAs are transcribed across the exons of protein coding genes, but from the opposite strand. The transcripts for long intergenic non-coding RNAs (lincRNAs) are located between protein coding genes. Enhancer RNA (eRNA) transcripts are bidirectionally expressed at active enhancer regions of the genome. These are mainly cis-acting and control interactions between promoters and enhancers to regulate gene expression. Intronic lncRNAs are transcribed from the introns of annotated protein coding genes. circRNAs interact with and interfere with the functions of miRNAs.
LncRNAs perform diverse functions among which is the regulation of immunity and host-pathogen interactions (138–140). They interact with other molecules through base pairing or secondary structures (141). Nuclear lncRNAs interact with chromatin remodelers to regulate the expression of neighboring or distal genes. Cytoplasmic lncRNAs interfere with stability and translation of mRNA and signaling pathways (142). They also interact with a range of other molecules. In summary, they can act as protein scaffolds, regulators of transcription, antisense RNA or miRNA sponges (143–147). LncRNAs are now acknowledged as major players in maintaining homeostasis and in disease (148, 149). Transcriptomic data have shown the differential expression of hundreds of lncRNAs during PRR stimulation (150–153). Several of these lncRNAs are trans‐acting regulators of protein‐coding genes such as TNF and HNRNPL related immunoregulatory long non-coding RNAc(THRIL) which regulates TNF‐α expression by forming a complex with the ribonucleoprotein (RNP) hnRNPL that acts at the TNF‐α promoter (154), or the lincRNA‐COX2 which regulates inflammatory gene expression in LPS‐stimulated bone marrow‐derived dendritic cells by interacting with hnRNP‐A/B and hnRNP‐A2/B1 (151) or the lincRNA-EPS which associates with chromatin and interacts with hnRNPL to repress immune-responsive gene-1 (IRG) expression (150).
LncRNA expression profiles have been documented by transcriptome analysis from pulmonary TB (PTB) patients vs. healthy individuals (155). PTB patients showed differential expression of 449 lncRNAs. The expression profiles of lncRNAs were investigated by transcriptome sequencing. Lnc-HNRNPU-1:7 and lnc-FAM76B-4:1 were the most upregulated and downregulated lncRNAs respectively, in PTB patients compared to healthy individuals. In separate studies (156), a comprehensive analysis has shown that compared to healthy volunteers, 1,429 and 2,040 lncRNAs are deregulated in the PBMCs from MDR-TB and drug-sensitive TB patients, respectively. However, these results have not been analyzed with respect to their relevance and implications in the context of infection. During TB infection, the expression of the lnc-TGS1–1 target miR-143, is increased due to elimination of the sponge effect of lnc-TGS1-1. This likely suppresses downstream innate immune signaling (157). Along similar lines, removal of the inhibitory effect of lnc-AC145676.2.1–6 results in upregulation of miR-29a and interference with the TLR pathway (158).
LncRNA nuclear paraspeckle assembly transcript 1 (NEAT1) is expressed at higher levels in PBMCs from patients with tuberculosis than in healthy individuals, and in infected THP1 cells compared to uninfected ones (159). Knockdown of NEAT1 increases bacterial CFUs in infected THP1 cells, suggesting that NEAT1 is required for mounting a bactericidal response against M. tuberculosis. Comprehensive expression profiles of murine RAW264.7 macrophages infected with M. tuberculosis showed differential regulation of 1,487 lncRNAs (791 up and 696 down) (160). LncRNA profiles have also been generated for human macrophages infected with M. tuberculosis (161). In a study involving 473 healthy individuals and 467 TB patients, lnc-AC145676.2.1-6 and lnc-TGS1-1 expression levels were lower in TB patients. LincRNA cyclooxygenase 2 (Cox2) is activated by the TLR signaling pathway in macrophages and mediates both activation and repression of genes (151). LincRNACox2 is increased in macrophages infected with M. tuberculosis (15). Knockdown of lincRNA Cox2 decreased NF-κB and STAT3 while increasing apoptosis in infected macrophages. LncRNA HOX Transcript Antisense RNA (HOTAIR) targets the H3K27 methylase complex polycomb repressive complex 2 (PRC2) to distinct loci to regulate H3K27 methylation at these loci (162). Subuddhi et al. (163) have shown that lncRNA HOTAIR is downregulated in M. tuberculosis H37Rv-infected THP-1 macrophages, but upregulated in M. tuberculosis H37Ra-infected macrophages, leading to differential association of H3K27 methylation marks at the dual specificity phosphatase 4 (DUSP4) and SATB homeobox 1 (SATB1) loci. Downregulation of HOTAIR, facilitated increased transcription of DUSP4 and SATB1, both of which favored the survival of M. tuberculosis H37Rv in macrophages. Li et al. (164) showed a significant reduction in PC-esterase domain containing 1B antisense RNA1 (PCED1B-AS1) expression in monocytes from patients with active TB compared with that in monocytes from healthy individuals. Suppression of PCED1B-AS1 in infected macrophages significantly attenuated TNF-α-induced apoptosis with decreased cleaved caspase-3 and a concomitant increase in Bcl-2. Further, PCED1B-AS1 serves a miR-155 sponge. Its knockdown leads to upregulation of miR-155 and inhibition of its targets RHEB and FOXO3, culminating in enhanced autophagy (107, 165). Sun et al. (166) have reported that LncRNA maternally expressed 3 (MEG3) is activated by vitamin D and plays a regulatory role in carcinogenesis (167). It is highly expressed in pulmonary tuberculosis (PTB) (166). Lnc-MEG3 binds miR-145-5p (168). The relationship between Lnc-MEG3, miR-145-5p and apoptosis has been strengthened by infecting RAW264.7 with M. bovis BCG under conditions of overexpression or knock down of MEG3 (166). The authors have shown that inhibition of Lnc-MEG3 is associated with increased miR-145-5p and decreased apoptosis. Lnc-MEG3 also induces autophagy in macrophages infected with M. bovis BCG (16).
LncRNAs regulate the response of T and B cells to M. tuberculosis by regulating chromatin modification states. During M. tuberculosis infection, CD244, a T cell-inhibitory molecule, mediates inhibition of IFN-γ and TNF-α expression by inducing lncRNA-CD244 which interacts with the H3K27 methylase enhancer of zeste homolog 2 (EZH2), and mediates modification of a more repressive chromatin state at the infg and tnfa loci (169). Knock down of lncRNA-CD244 significantly enhances IFN-γ and TNF-α expression and improves protective immunity of CD8+ T cells.
Differentially expressed lncRNAs have been identified in CD4+ T cells in latent TB (170). Fu et al. (171) have reported that 844 lncRNAs were differentially expressed in B cells from individuals with TB. Concomitant with the dysregulation of several lncRNAs, adjacent protein-coding genes were also deregulated. For example, SOCS3 and its neighboring lncRNA XLOC_012582 were highly expressed in B cells from TB patients. The relevance of this observation in the context of TB, awaits further investigation. LncRNAs that regulate host signaling pathways during M. tuberculosis infection, are shown in Figure 3.
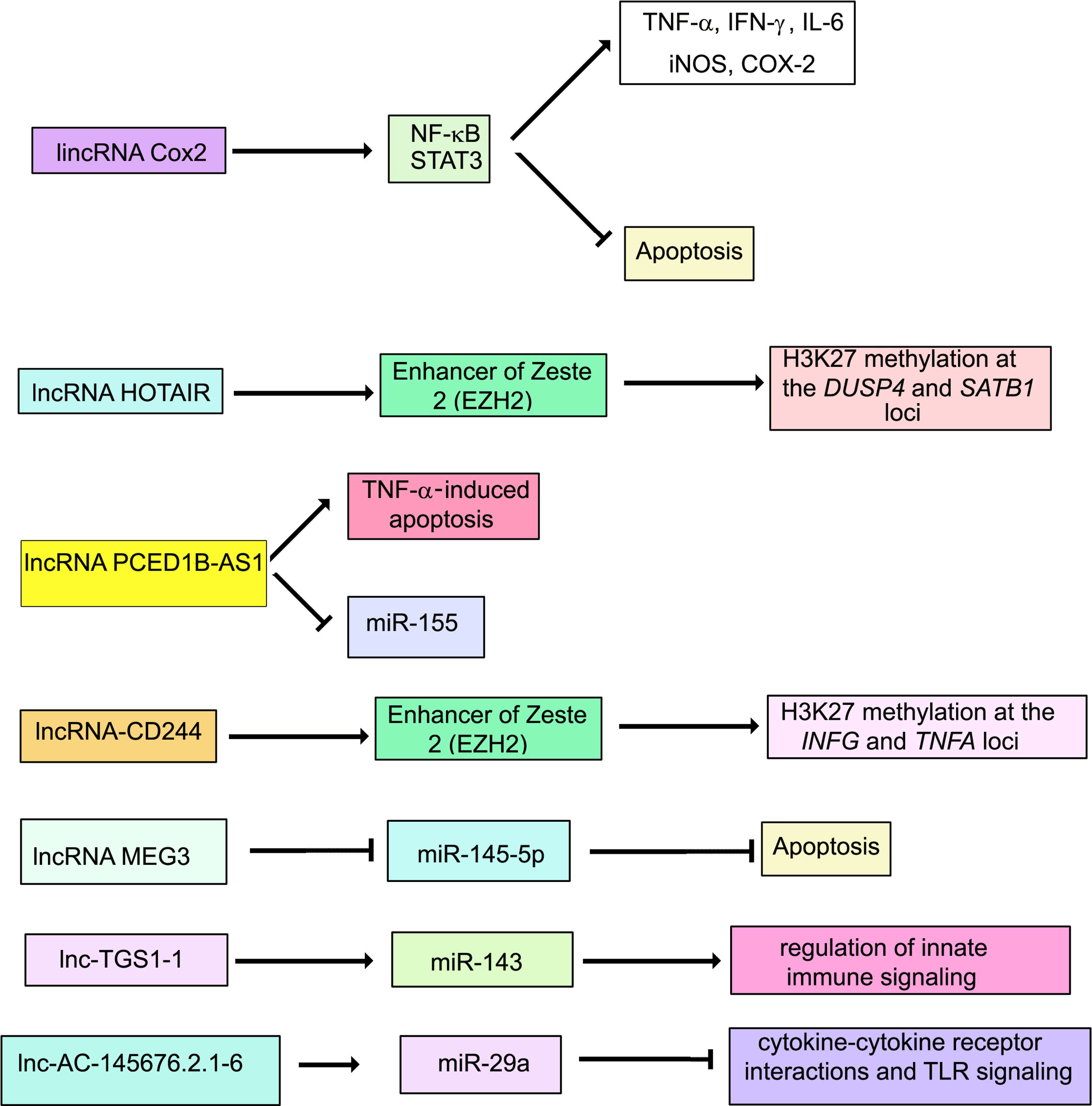
Figure 3 Schematic representation of the role of lncRNAs during M. tuberculosis infection.
The potential use of miRNAs in the diagnosis of tuberculosis has been reviewed by Sabir et al. (172). In particular, the role of exosomal miRNAs as biomarkers of TB is of interest (173). MiRNAs miR-20a, miR-20b, miR-26a, miR-106a, miR-191, miR-486 are differentially expressed in exosomes from TB compared to healthy individuals (174). The overexpressed miRNAs showed reduction in expression after two months of anti-tuberculosis therapy. In a separate study, miR-484, miR-425, and miR-96-3-p were reported to be enriched in exosomes from patients with active TB (175). Some studies have specifically explored the potential of exosomal miRNAs as biomarkers of LTBI. Small RNA sequencing of serum exosomes from LTBI and TB patients showed that let-7e-5p, let-7d-5p, miR-450a-5p and miR-140-5p were specifically expressed in LTBI, whereas miR-1246, miR-2110, miR-370-3p, miR-28-3p and miR-193-5p, were specifically associated with active TB (176). In a separate study, miR-122-5p expression was observed to be significantly higher in the exosomes of LTBI than in active TB (177). On the other hand, let-7i-5p, miR-148a-3p, miR-21-5p and miR-423-5p showed higher expression in active TB than in LTBI. Independent miRNA profiling from PBMCs or serum suggest that a group of differentially regulated miRNAs distinguish active TB from latent TB (178, 179). miR-889 expression was increased in patients with latent TB compared to uninfected individuals (98). Latorre et al. (180) have identified whole blood-derived miRNA signatures that enable distinguishing active TB from latent TB. A systems biology approach has been adopted by Lin et al. (181) to generate a unique miRNA-gene regulatory network for LTBI by analyzing numerous microarray data sets. The miRNAs in this network were identified with the Hippo signaling pathway (miR-212-3p, miR-29a-5p, miR-29b-3p), ECM–receptor interaction (miR-432-5p, miR-148b-5p, miR-29b-3p, miR-532-5p), and the PI3K-Akt signaling pathway (miR-29b-3p). The reciprocal relationship between MAPK1 and miR-212-3p was validated. The authors suggest that the interaction of miR-212-3p with MAPK1 could regulate the PI3K-Akt signaling pathway to affect the transmission of M. tuberculosis.
In a similar vein, attempts have been made to use lncRNA expression as a means of TB diagnosis. Fang et al. (182) have analyzed the GEO dataset (GSE94907) and identified the differentially regulated lncRNAs NONHSAT101518.2, NONHSAT067134.2, NONHSAT148822.1 and NONHSAT078957.2 which are downregulated in the plasma of active TB patient plasma compared with the healthy individuals, as potential biomarkers. In a separate study, a total of 163 upregulated lncRNAs and 348 downregulated lncRNAs were identified in the plasma of PTB patients (18). Four differentially expressed lncRNAs, NR_038221, NR_003142, ENST00000570366 and ENST00000422183 were confirmed by qRT-PCR. The potential target genes of NR_003142 in immune pathways were TLR6, nucleotide binding oligomerization domain containing 2 (NOD2), HLA-DQB and IL6ST suggesting that it could influence TB pathology. Hu et al. (183) have shown that three lncRNAs, ENST00000497872, n333737, and n335265 are differentially expressed in blood samples of active TB patients compared to healthy individuals and suggested that these may serve as potential biomarkers for clinically diagnosed PTB patients. Of these, lncRNA ENST00000497872 is located close to the immune related gene immunoglobulin heavy constant alfa 1 (IGHA1) and n333737 is located close to the immune related gene T cell receptor alfa variable 1-2 (TRAV1-2). Lnc-TGS1–1 and lnc-AC145676.2.1–6 are downregulated in TB patients (184). In separate studies, 41 lncRNAs have been reported to be deregulated in a comparison between healthy subjects, active TB and LTBI (170). These studies suggest that targeting miRNAs and lncRNAs for development of HDTs, and as biomarkers of TB, deserve detailed investigation.
This review outlines the role of miRNAs and lncRNAs in regulating functions in myeloid cells that are critical in determining the outcome of M. tuberculosis infection such as inflammation, macrophage polarization, metabolism, autophagy and apoptosis. In spite of the extensive literature presently available, there are several roadblocks and contradictions along the path towards understanding the role of miRNAs in human tuberculosis, arising out of differences between animal infections and human disease and variations in methodology (such as strains used, multiplicity of infection, and time of infection) between different studies. One such example is the role of miR-21 in infection. Mir-21, on the one hand, inhibits glycolysis and the production of bactericidal effectors such as ROS and NO (67), providing a permissive milieu for the growth of M. tuberculosis. On the other hand, Zhao et al. (45) report that miR-21 targets Bcl-2, suggesting that it accelerates apoptosis. This would deprive M. tuberculosis of its niche within the host. As of now, there are no in vivo studies to resolve such contradictions on the actual role of miR-21 in infection.
Methods for rapid diagnosis of TB are urgently required. miRNAs have been explored for the diagnosis of a range of diseases such as cancer. Several studies have tested the potential of miRNAs as diagnostic markers for TB. Among the miRNAs that have been discussed here, at least four have been consistently associated with TB namely miR-144-3p, miR-144-5p, miR-146a and miR-155. miR-144-3p levels are higher in sputum and serum from patients with active TB compared to healthy uninfected individuals (185). miR-144-3p targets ATG4a to inhibit autophagy during M. bovis BCG infection. miR-144-5p levels are increased in PBMCs from active TB patients compared to healthy individuals (186). miR-144-5p targets DRAM2 to regulate autophagy. Alveolar macrophages from smear-positive patients show lower levels of miR-146a than those from smear-negative patients or healthy uninfected individuals (187). miR-146a targets IRAK1 and TRAF6. miR-155 levels are reportedly reduced in serum from patients with active TB (188), and increased after TB therapy. miR-155 has been reported to regulate inflammatory cytokines, autophagy and apoptosis, during infection of macrophages. miR-196b and miR-376c have been proposed as serum markers of active TB and LTBI (179). The differential expression of miRNAs in body fluids such as blood and sputum, distinguishes TB patients from healthy individuals, or LTBI; treated from untreated patients; and those infected with hypervirulent or drug-resistant strains (189). Serum exosomes have also been evaluated for the presence of miRNA biomarkers capable of distinguishing latent TB, active TB and healthy individuals. A recent study has suggested the differential expression of miR-625-3p in urine samples, as a marker for TB diagnosis (190). This highlights the pivotal role of miRNAs in TB. However, as of now miRNA signatures characteristic of tuberculosis disease states, remain to be defined (191). In particular, detailed investigation needs to be done in terms of identifying biomarkers for latent TB. There are obvious pitfalls in comparing miRNA levels during in vitro and in vivo infection across different studies, arising out of difficulties in pinning down different stages of the disease, choice of platform for miRNA identification, choice of strain of M. tuberculosis, age and sex of individuals, methods of RNA extraction, and sample size, to mention a few. However, there is substantial literature that underscores the need to explore development of miRNA delivery vehicles and miRNA-based therapeutics as possible strategies for shortening the duration of therapy (9). Some studies in animal models have shown promise. Lou et al. (51) have demonstrated that intravenous administration of miR-20b mimics could attenuate the inflammatory response in a mouse model of infection by inhibiting activation of the inflammosome. In a M. marinum model, transfection of microglia with miR-124-3p promoted apoptosis through targeting of STAT3 to decrease mycobacterial proliferation (192). It is therefore important to devise efficient delivery methods for miRNAs that would protect them from circulating RNAse and target them to the site of infection. The available technologies for miRNA delivery include nanoparticles as vehicles, use of viral vectors, exosome-like vesicles and lipid-based delivery systems. A silica nanoparticle-based targeted delivery of miR-34a, to neuroblastoma tumors in a murine orthotopic xenograft model, has been demonstrated (193). It must be mentioned that nanoparticle based delivery platforms are associated with high cost of production, non-specific distribution and cytotoxicity. Administration of miR-26a in a mouse model of hepatocellular carcinoma using adeno-associated virus (AAV) inhibited cancer cell proliferation (194). However, viral delivery methods may elicit unwanted inflammatory responses. There are several reports that suggest that miRNA could probably be delivered to the lung for the treatment of TB. Mice treated with pre-miR-133b containing lipoplexes through the tail vein, showed 52-fold higher expression of miR-33b in the lungs compared to untreated mice (195). miRNA targeting drugs are in clinical trials for the treatment of hepatitis C virus infection via targeting miR-122, polycystic kidney disease via targeting miR-17, cutaneous T cell lymphoma via targeting miR-155 and keloids via targeting miR-19 (196). miRNA based therapeutic options need to be pursued in earnest for the management of TB.
The association of lncRNAs with tuberculosis, and their functions have been less extensively explored than the role of miRNAs. Much more work needs to carried out, before we understand their link to disease outcomes.
JB and MK reviewed the literature and wrote the manuscript. All authors contributed to the article and approved the submitted version.
JB is supported by funding from the J.C. Bose Fellowship (SB/S2/JCB-049/2016). MK is supported by the Council for Scientific and Industrial Research, Emeritus Scientist Scheme (21(1088)/19/EMR-II).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledge the contributions of all the scientists in this area and apologize for failing to cite any work due to constraints of space.
2. Gurtan AM, Sharp PA. The Role of miRNAs in Regulating Gene Expression Networks. J Mol Biol (2013) 425:3582–600. doi: 10.1016/j.jmb.2013.03.007
3. Aguilar C, Mano M, Eulalio A. MicroRNAs at the Host-Bacteria Interface: Host Defense or Bacterial Offense. Trends Microbiol (2019) 27:206–18. doi: 10.1016/j.tim.2018.10.011
4. Skalsky RL, Cullen BR. Viruses, MicroRNAs, and Host Interactions. Annu Rev Microbiol (2010) 64:123–41. doi: 10.1146/annurev.micro.112408.134243
5. Staedel C, Darfeuille F. MicroRNAs and Bacterial Infection. Cell Microbiol (2013) 15:1496–507. doi: 10.1111/cmi.12159
6. Duval M, Cossart C, Lebretonde A. Mammalian MicroRNAs and Long Noncoding RNAs in the Host-Bacterial Pathogen Crosstalk. Semin Cell Dev Biol (2017) 65:11–9. doi: 10.1016/j.semcdb.2016.06.016
7. Das K, Garnica O, Dhandayuthapani S. Modulation of Host miRNAs by Intracellular Bacterial Pathogens. Front Cell Infect Microbiol (2016) 6:79. doi: 10.3389/fcimb.2016.00079
8. Ouimet M, Koster S, Sakowski E, Ramkelawon B, van Solingen C, Oldebeken S, et al. Mycobacterium Tuberculosis Induces mir-33 Locus to Reprogram Autophagy and Host Lipid Metabolism. Nat Immunol (2016) 6:677–86. doi: 10.1038/ni.3434
9. Singh AK, Ghosh M, Kumar V, Aggarwal S, Patil SA. Interplay Between miRNAs and Mycobacterium Tuberculosis: Diagnostic and Therapeutic Implications. Drug Discov Today (2021) 26:1245–55. doi: 10.1016/j/drudis.2021.01.021
10. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE V7 Catalog of Human Long Noncoding RNAs: Analysis of Their Gene Structure, Evolution, and Expression. Genome Res (2012) 22:1775–89. doi: 10.1101/gr.132159.111
11. Häfner SJ, Talvard TG, Lund AH. Long Noncoding RNAs in Normal and Pathological Pluripotency. Semin Cell Dev . Biol (2017) 65:1–10. doi: 10.1016/j.semcdb.2016.07.011
12. Rinn JL, Chang HY. Genome Regulation by Long Noncoding RNAs. Annu Rev Biochem (2012) 81:145–66. doi: 10.1146/annurev-biochem-051410-092902
13. Fathizadeh H, Hayat SMG, Dao S, Ganbarov K, Tanomand A, Asgharzadeh M, et al. Long Non-Coding RNA Molecules in Tuberculosis. Int J Biol Macromol (2020) 156:340–6. doi: 10.1016/j.ijbiomac.2020.04.030
14. Hadjicharalambous MR, Lindsay MA. Long Non-Coding RNAs and the Innate Immune Response. Non-Coding RNA (2019) 5:34. doi: 10.3390/ncrna5020034
15. Li D, Gao C, Zhao L, Zhang Y. Inflammatory Response Is Modulated by LincRNAcox2 Via the NF−κB Pathway in Macrophages Infected by Mycobacterium Tuberculosis. Mol Med Rep (2020) 21:2513–21. doi: 10.3892/mmr.2020.11053
16. Pawar K, Hanisch C, Vera SEP, Einspanier R, Sharbati S. Down Regulated lncRNA MEG3 Eliminates Mycobacteria in Macrophages Via Autophagy. Sci Rep (2016) 6:19416. doi: 10.1038/srep19416
17. Wang C, Yang S, Liu CM, Jiang TT, Chen ZL, Tu HH, et al. Screening and Identification of Four Serum miRNAs as Novel Potential Biomarkers for Cured Pulmonary Tuberculosis. Tuberculosis (2018) 10826:–34. doi: 10.1016/j.tube.2017.08.010
18. Chen Z-L, Wei L-L, Shi L-Y, Li M, Jiang T-T, Chen J, et al. Screening and Identification of lncRNAs as Potential Biomarkers for Pulmonary Tuberculosis. Sci Rep (2017b) 7:16751. doi: 10.1038/s41598-017-17146-y
19. Treiber T, Treiber N, Meister G. Regulation of MicroRNA Biogenesis and Its Crosstalk With Other Cellular Pathways. Nat Rev Mol Cell Biol (2019) 20:5–20. doi: 10.1038/s41580-018-0059-1
20. Huntzinger E, Izaurralde E. Gene Silencing by MicroRNAs: Contributions of Translational Repression and mRNA Decay. Nat Rev Genet (2011) 12:99–110. doi: 10.1038/nrg2936
21. Basu J, Shin D-M, Jo E-K. Mycobacterial Signaling Through Toll-Like Receptors. Front Cell Infect Microbiol (2012) 2:145. doi: 10.3389/fcimb.2012.00145
22. Chai Q, Wang L, Liu CH, Baoxue G. New Insights Into the Evasion of Host Innate Immunity by Mycobacterium Tuberculosis. Cell Mol Immunol (2020) 17:901–13. doi: 10.1038/s41423-020-0502-z
23. Brooks MN, Rajaram MVS, Azad AK, Amer AO, Valdivia-Arenas MA, Park J-H, et al. Nod2 Controls the Nature of the Inflammatory Response and Subsequent Fate of Mycobacterium Tuberculosis and M. Bovis BCG in Human Macrophages. Cell Microbiol (2011) 13:402–18. doi: 10.1111/j.1462-5822.2010.01544.x
24. Kawasaki T, Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
25. Chen ZJ. Ubiquitin Signalling in the NF-κB Pathway. Nat Cell Biol (2005) 7:758–65. doi: 10.1038/ncb0805-758
26. Hayden MS, Ghosh S. Nf-κb, the First Quarter-Century: Remarkable Progress and Outstanding Questions. Genes Dev (2012) 26:203–34. doi: 10.1101/gad.183434.111
27. Goubau D, Deddouche S, Reis e Sousa C. Cytosolic Sensing of Viruses. Immunity (2013) 38:855–69. doi: 10.1016/j.immuni.2013.05.007
28. Hartmann G. Nucleic Acid Immunity. Adv Immunol (2017) 133:121–69. doi: 10.1016/bs.ai.2016.11.001
29. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA Helicase RIG-I Has An Essential Function in Double-Stranded RNA-Induced Innate Antiviral Responses. Nat Immunol (2004) 5:730–7. doi: 10.1038/ni1087
30. Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, et al. Essential Role of Mda-5 in Type I Ifn Responses to Polyriboinosinic:Polyribocytidylic Acid and Encephalomyocarditis PicoRNAvirus. Proc Natl Acad Sci USA (2006) 103:8459–64. doi: 10.1073/pnas.0603082103
31. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential Roles of MDA5 and RIG-I Helicases in the Recognition of RNA Viruses. Nature (2006) 441:101–5. doi: 10.1038/nature04734
32. Rehwinkel J, Gack MU. Rig-I-Like Receptors: Their Regulation and Roles in RNA Sensing. Nat Rev Immunol (2020) 20:537–51. doi: 10.1038/s41577-020-0288-3
33. Bettencourt P, Marion S, Pires D, Santos LF, Lastrucci C, Carmo N, et al. Actin-Binding Protein Regulation by MicroRNAs as a Novel Microbial Strategy to Modulate Phagocytosis by Host Cells: The Case of N-Wasp and Mir-142-3p. Front Cell Infect Microbiol (2013) 3:19. doi: 10.3389/fcimb.2013.00019
34. Hou J, Wang P, Lin L, Liu X, Ma F, An H, et al. MicroRNA-146a Feedback Inhibits RIG-I-Dependent Type I IFN Production in Macrophages by Targeting Traf6, IRAK1, and IRAK2. J Immunol (2009) 183:2150–8. doi: 10.4049/jimmunol.0900707
35. O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 Is Induced During the Macrophage Inflammatory Response. Prof Natl Acad Sci USA (2007) 104:1604–9. doi: 10.1073/pnas.0610731104
36. Nejad C, Stunden HJ, Gantie MP. A Guide to miRNAs in Inflammation and Innate Immune Responses. FEBS Lett (2018) 285:3695–716. doi: 10.1111/febs.14482
37. Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O’Leary JJ, Ruan Q, et al. Negative Regulation of TLR4 Via Targeting of the Proinflammatory Tumor Suppressor PDCD4 by the MicroRNA Mir-21. Nat Immunol (2010) 11:141–7. doi: 10.1038/ni.1828
38. Taganov KD, Boldin MP, Chang KJ, Baltimore D. Nf-κB-Dependent Induction of MicroRNA Mir-146, An Inhibitor Target to Signaling Proteins of Innate Immune Responses. Proc Natl Acad Sci USA (2006) 103:12481–6. doi: 10.1073/pnas.0605298103
39. Liu PT, Wheelwright M, Teles R, Komisopoulos E, Edfeldt K, Ferguson B, et al. MicroRNA-21 Targets the Vitamin D-Dependent Antimicrobial Pathway in Leprosy. Nat Med (2012) 18:267–73. doi: 10.1038/nm.2584
40. Kumar M, Sahu SK, Kumar R, Subuddhi A, Maji RK, Jana K, et al. MicroRNA Let-7 Modulates the Immune Response to Mycobacterium Tuberculosis Infection Via Control of A20, an Inhibitor of the NF-κB Pathway. Cell Host Microbe (2015) 17:345–56. doi: 10.1016/j.chom.2015.01.007
41. Schulte LN, Eulalio A, Mollenkopf H-J, Reinhardt R, Vogel J. Analysis of the Host MicroRNA Response to Salmonella Uncovers the Control of Major Cytokines by the Let-7 Family. EMBO J (2011) 30:1977–89. doi: 10.1038/emboj.2011.94
42. Kumar R, Halder P, Sahu S, Kumar M, Kumari M, Jana K, et al. Identification of a Novel Role of ESAT-6-Dependent Mir-155 Induction During Infection of Macrophages With Mycobacterium Tuberculosis. Cell Microbiol (2012) 14:1620–31. doi: 10.1111/j.1462-5822.2012.01827.x
43. Rothchild AC, Sissons JR, Shafiani S, Plaisier C, Min D, Mai D, et al. Mir-155–Regulated Molecular Network Orchestrates Cell Fate in the Innate and Adaptive Immune Response to Mycobacterium Tuberculosis. Proc Natl Acad Sci USA (2016) 113:E6172–81. doi: 10.1073/pnas.1608255113
44. Iwai H, Funatogawa K, Matsumura K, Kato-Miyazawa M, Kirikae F, Kiga K, et al. MicroRNA-155 Knockout Mice Are Susceptible to Mycobacterium Tuberculosis Infection. Tuberculosis (2015) 95:246–50. doi: 10.1016/j.tube.2015.03.006
45. Zhao Z, Hao J, Li X, Chen Y, Qi X. Mir-21-5p Regulates Mycobacterial Survival and Inflammatory Responses by Targeting Bcl-2 and TLR4 in Mycobacterium Tuberculosis-Infected Macrophages. FEBS Lett (2019) 593:1326–35. doi: 10.1002/1873-3468.13438
46. Wang J, Jia Z, Wei B, Zhou Y, Niu C, Bai S, et al. MicroRNA-27a Restrains the Immune Response to Mycobacterium Tuberculosis Infection by Targeting IRAK4, a Promoter of the NF-κB Pathway. Int J Clin Exp Pathol (2017) 10:9894–901.
47. Maa C, Lia Y, Lia M, Deng G, Wu X, Zeng J, et al. MicroRNA-124 Negatively Regulates TLR Signaling in Alveolar Macrophages in Response to Mycobacterial Infection. Mol Immunol (2014) 62:150–8. doi: 10.1016/j.molimm.2014.06.014
48. Wei J, Huang Z, Zhang W, Jia Z, Zhao Z, Zhang Y, et al. Myd88 as a Target of MicroRNA-203 in Regulation of Lipopolysaccharide or Bacille Calmette-Guerin Induced Inflammatory Response of Macrophage Raw264.7 Cells. Mol Immunol (2013) 55:303–9. doi: 10.1016/j.molimm.2013.03.004
49. Wu Y, Sun Q, Dai L. Immune Regulation of Mir-30 on the Mycobacterium Tuberculosis-Induced TLR/Myd88 Signaling Pathway in THP-1 Cells. Exp Ther Med (2017) 14:3299–303. doi: 10.3892/etm.2017.4872
50. Xu G, Zhang Z, Xing Y, Wei J, Ge Z, Liu X, et al. MicroRNA-149 Negatively Regulates TLR-Triggered Inflammatory Response in Macrophages by Targeting Myd88. J Cell Biochem (2014) 115:919–27. doi: 10.1002/jcb.24734
51. Lou J, Wang Y, Zhang Z, Qiu W. Mir-20b Inhibits Mycobacterium Tuberculosis Induced Inflammation in the Lung of Mice Through Targeting Nlrp3. Exp Cell Res (2017) 358:120–8. doi: 10.1016/j.yexcr.2017.06.007
52. Boone DL, Turer EE, Lee EG, Ahmad C, Wheeler MT, Tsui C, et al. The Ubiquitin-Modifying Enzyme A20 Is Required for Termination of Toll-Like Receptor Response. Nat Immunol (2004) 5:1052–60. doi: 10.1038/ni1110
53. Liu YC, Penninger J, Karin M. Immunity by Ubiquitylation: A Reversible Process of Modification. Nat Rev Immunol (2005) 5:941–52. doi: 10.1038/nri1731
54. Niu W, Sun B, Li M, Cui J, Huang J, Zhang L. TLR-4/MicroRNA-125a/NF-κB Signaling Modulates the Immune Response to Mycobacterium Tuberculosis Infection. Cell Cycle (2018) 17:1931–45. doi: 10.1080/15384101.2018.1509636
55. Zhang Z-M, Zhang A-R, Xu M, Lou J, Qiu W-Q. TLR-4/miRNA-32-5p/FSTL1 Signaling Regulates Mycobacterial Survival and Inflammatory Responses in Mycobacterium Tuberculosis-Infected Macrophages. Exp Cell Res (2017) 352:313–21. doi: 10.1016/j.yexcr.2017.02.025
56. Shi G, Mao G, Xe K, Wu D, Wang W. Mir-1178 Regulates Mycobacterial Survival and Inflammatory Responses in Mycobacterium Tuberculosis-Infected Macrophages Partly Via Tlr4. J. Cell Biochem (2018) 119:7449–57. doi: 10.1002/jcb.27054
57. Zhu Y, Xiao Y, Kong D, Liu H, Chen X, Chen Y, et al. Down-Regulation of Mir-378d Increased Rab10 Expression to Help Clearance of Mycobacterium Tuberculosis in Macrophages. Front Cell Infect Microbiol (2020) 17:108. doi: 10.3389/fcimb.2020.00108
58. Fu X, Zeng L, Liu Z, Ke X, Lei L, Li G. MicroRNA-206 Regulates the Secretion of Inflammatory Cytokines and MMP9 Expression by Targeting TIMP3 in Mycobacterium Tuberculosis–Infected THP-1 Human Macrophages. Biochim Biophys Acta (2016) 477:167–73. doi: 10.1016/j.bbrc.2016.06.038
59. Singh Y, Kaul V, Mehra A, Chatterjee S, Tousif S, Dwivedi VP, et al. Mycobacterium Tuberculosis Controls MicroRNA-99b (Mir-99b) Expression in Infected Murine Dendritic Cells to Modulate Host Immunity. J Biol Chem (2013) 288:5056–61. doi: 10.1074/jbc.C112.439778
60. Rajaram MVS, Ni B, Morris JD, Brooks MN, Carlson TK, Bakthavachalu B, et al. Mycobacterium Tuberculosis Lipomannan Blocks TNF Biosynthesis by Regulating Macrophage MAPK-Activated Protein Kinase 2 (MK2) and MicroRNA miR-125b. Proc Natl Acad Sci USA (2011) 108:17408–13. doi: 10.1073/pnas.1112660108
61. Li X, Huang S, Yu T, Liang G, Liu H, Pu D, et al. Mir-140 Modulates the Inflammatory Responses of Mycobacterium Tuberculosis-Infected Macrophages by Targeting Traf6. J Cell Mol Med (2019) 23:5642–53. doi: 10.1111/jcmm.14472
62. Xu G, Zhang Z, Wei J, Zhang Y, Zhang Y, Guo L, et al. Micror-142-3p Down-Regulates IRAK-1 in Response to Mycobacterium Bovis BCG Infection in Macrophages. Tuberculosis (2013) 93:606–11. doi: 10.1016/j.tube.2013.08.006
63. Li S, Yue Y, Xu W, Xiong S. MicroRNA-146a Represses Mycobacteria-Induced Inflammatory Response and Facilitates Bacterial Replication Via Targeting IRAK-1 and TRAF-6. PloS One (2013) 8:e81438. doi: 10.1371/journal.pone.0081438
64. Dorhoi A, Iannaccone M, Farinacci M, Faé KC, Schreiber J, Moura-Alves P, et al. MicroRNA-223 Controls Susceptibility to Tuberculosis by Regulating Lung Neutrophil Recruitment. J Clin Invest (2013) 123:4386–848. doi: 10.1172/JCI67604
65. Sahu SK, Kumar M, Chakraborty S, Banerjee SK, Kumar R, Gupta P, et al. MicroRNA 26a (miR-26a)/KLF4 and CREB-C/Ebpβ Regulate Innate Immune Signaling, the Polarization of Macrophages and the Trafficking of Mycobacterium Tuberculosis to Lysosomes During Infection. PloS Pathog (2017) 13:e1006410. doi: 10.1371/journal.ppat.1006410
66. Ni B, Rajaram MVS, Lafuse WP, Landes MB, Schlesinger LS. Mycobacterium Tuberculosis Decreases Human Macrophage IFN-γ Responsiveness Through miR-132 and miR-26a. J Immunol (2014) 193:4537–47. doi: 10.4049/jimmunol.1400124
67. Hackett EE, Charles-Messance H, O’Leary SM, Gleeson LE, Munoz-Wolf N, Case S, et al. Mycobacterium Tuberculosis Limits Host Glycolysis and IL-1b by Restriction of PFK-M Via MicroRNA-21. Cell Rep (2020) 30:124–36. doi: 10.1016/j.celrep.2019.12.015
68. Thiriot JD, Martinez-Martinez YB, Endsley JJ, Torres AG. Hacking the Host: Exploitation of Macrophage Polarization by Intracellular Bacterial Pathogens. Pathog Dis (2020) 78:ftaa009. doi: 10.1093/femspd/ftaa009
69. Lim Y-J, Yi MH, Choi J-A, Lee J, Han J-Y, Jo S-H, et al. Roles of Endoplasmic Reticulum Stress-Mediated Apoptosis in M1-Polarized Macrophages During Mycobacterial Infections. Sci Rep (2016) 6:37211. doi: 10.1038/srep37211
70. Van den Bossche J, O’Neill LA, Menon D. Macrophage Immunometabolism: Where Are We Going? Trends Immunol (2017) 38:395–406. doi: 10.1016/j.it.2017.03.001
71. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The Metabolic Signature of Macrophage Responses. Front Immunol (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
72. O’Neill LA, Pearce EJ. Immunometabolism Governs Dendritic Cell and Macrophage Function. J Exp Med (2016) 213:15–23. doi: 10.1084/jem.20151570
73. Shi L, Salamon H, Eugenin EA, Pine R, Cooper A, Gennaro ML. Infection With Mycobacterium Tuberculosis Induces the Warburg Effect in Mouse Lungs. Sci Rep (2015) 5:18176. doi: 10.1038/srep18176
74. Imtiyaz HZ, Simon MC. Hypoxia-Inducible Factors as Essential Regulators of Inflammation. Curr Top Microbiol Immunol (2010) 345:105–20. doi: 10.1007/82_2010_74
75. Gleeson LE, Sheedy FJ, McDermott P, Triglia D, O’Leary SM, O’Sullivan MP, et al. Mycobacterium Tuberculosis Induces Aerobic Glycolysis in Human Alveolar Macrophages That Is Required for Control of Intracellular Bacillary Replication. J Immunol (2016) 196:2444–9. doi: 10.4049/jimmunol.1501612
76. Gleeson LE, O’Leary SM, Ryan D, McLaughlin AM, Sheedy FJ, Keane J. Cigarette Smoking Impairs the Bioenergetic Immune Response to Mycobacterium Tuberculosis Infection. Am J Respir Cell Mol Biol (2018) 59:572–9. doi: 10.1165/rcmb.2018-0162OC
77. Lachmandas E, Beigier-Bompadre M, Cheng SC, Kumar V, van Laarhoven A, Wang X, et al. Rewiring Cellular Metabolism Via the AKT/Mtor Pathway Contributes to Host Defence Against Mycobacterium Tuberculosis in Human and Murine Cells. Eur J Immunol (2016a) 46:2574–86. doi: 10.1002/eji.201546259
78. Lachmandas E, Boutens L, Ratter JM, Hijmans A, Hooiveld GJ, Joosten LAB, et al. Microbial Stimulation of Different Toll-Like Receptor Signalling Pathways Induces Diverse Metabolic Programmes in Human Monocytes. Nat Microbiol (2016b) 2:16246. doi: 10.1038/nmicrobiol.2016.246
79. Deretic V. Autophagy in Inflammation, Infection, and Immunometabolism. Immunity (2021) 54:437–53. doi: 10.1016/j.immuni.2021.01.018
80. Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A Subdomain of the Endoplasmic Reticulum Forms a Cradle for Autophagosome Formation. Nat Cell Biol (2009) 11:1433–7. doi: 10.1038/ncb1991
81. Mizushima N. The Role of the Atg1/ULK1 Complex in Autophagy Regulation. Curr Opin Cell Biol (2010) 22:132–9. doi: 10.1016/j.ceb.2009.12.004
82. Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh B-H, et al. Autophagic and Tumour Suppressor Activity of a Novel Beclin1-Binding Protein Uvrag. Nat Cell Biol (2006) 8:688–99. doi: 10.1038/ncb1426
83. Liang C, Lee J-S, Inn K-S, Gack MU, Li Q, Roberts EA, et al. Beclin1-Binding UVRAG Targets the Class C Vps Complex to Coordinate Autophagosome Maturation and Endocytic Trafficking. Nat Cell Biol (2008) 10:776–87. doi: 10.1038/ncb1740
84. Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A Protein Conjugation System Essential for Autophagy. Nature (1998) 395:395–8. doi: 10.1038/26506
85. Levine B. Eating Oneself and Uninvited Guests: Autophagy-Related Pathways in Cellular Defense. Cell (2005) 120:159–62. doi: 10.1016/S0092-8674(05)00043-7
86. Levine B, Mizushima N, Virgin HW. Autophagy in Immunity and Inflammation. Nature (2011) 469:323–35. doi: 10.1038/nature09782
87. Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and Selective Autophagy. Cell Death Differ (2013) 20:21–30. doi: 10.1038/cdd.2012.72
88. Deretic V. Autophagy in Tuberculosis. Cold Spring Harb. Perspect Med (2014) 4:a018481. doi: 10.1101/cshperspect.a018481
89. Jo EK, Yuk J-M, Shin DM, Sasakawa C. Roles of Autophagy in Elimination of Intracellular Bacterial Pathogens. Front Immunol (2013) 4:97. doi: 10.3389/fimmu.2013.00097
90. Siqueira MDS, Ribeiro RM, Travassos LH. Autophagy and Its Interaction With Intracellular Bacterial Pathogens. Front Immunol (2018) 9:935. doi: 10.3389/fimmu.2018.00935
91. Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. Autophagy Protects Against Active Tuberculosis by Suppressing Bacterial Burden and Inflammation. Proc Natl Acad Sci USA (2012) 109:E3168–76. doi: 10.1073/pnas.1210500109
92. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy Is a Defense Mechanism Inhibiting BCG and Mycobacterium Tuberculosis Survival in Infected Macrophages. Cell (2004) 119:753–66. doi: 10.1016/j.cell.2004.11.038
93. Watson RO, Manzanillo PS, Cox JS. Extracellular M. Tuberculosis DNA Targets Bacteria for Autophagy by Activating the Host DNA-Sensing Pathway. Cell (2012) 150:803–15. doi: 10.1016/j.cell.2012.06.040
94. Shin DM, Jeon BY, Lee HM, Jin HS, Yuk JM, Song CH, et al. Mycobacterium Tuberculosis Eis Regulates Autophagy, Inflammation, and Cell Death Through Redox-Dependent Signaling. PloS Pathog (2010) 6:e1001230. doi: 10.1371/journal.ppat.1001230
95. Kim JK, Kim TS, Basu J, Jo E-K. MicroRNA in Innate Immunity and Autophagy During Mycobacterial Infection. Cell Microbiol (2017) 19:e12687. doi: 10.1111/cmi.12687
96. Gu X, Gao Y, Mu DG, Fu EQ. Mir-23a-5p Modulates Mycobacterial Survival and Autophagy During Mycobacterium Tuberculosis Infection Through Tlr2/Myd88/Nf-κb Pathway by Targeting Tlr2. Exp Cell Res (2017) 354:71–7. doi: 10.1016/j.yexcr.2017.03.039
97. Liu F, Chen J, Wang P, Li H, Zhou Y, Liu H, et al. MicroRNA-27a Controls the Intracellular Survival of Mycobacterium Tuberculosis by Regulating Calcium Associated Autophagy. Nat Commun (2018) 9:4295. doi: 10.1038/s41467-018-06836-4
98. Chen DY, Chen Y-M, Lin CF, Lo CM, Liu HJ, Liao TL. MicroRNA-889 Inhibits Autophagy to Maintain Mycobacterial Survival in Patients With Latent Tuberculosis Infection by Targeting Tweak. mBio (2020) 11:e03045–19. doi: 10.1128/mBio.03045-19
99. Germain M, Slack R. Mcl-1 Regulates the Balance Between Autophagy and Apoptosis. Autophagy (2011) 7:549–51. doi: 10.4161/auto.7.5.15098
100. Kumar R, Sahu S, Kumar M, Jana K, Gupta P, Gupta U, et al. MicroRNA 17-5p Regulates Autophagy in Mycobacterium Tuberculosis-Infected Macrophages by Targeting Mcl-1 and STAT3. Cell Microbiol (2016) 18:679–91. doi: 10.1111/cmi.12540
101. Kim J, Yuk JY, Kim S, Kim TS, Jin HS, Young CS, et al. MicroRNA-125a Inhibits Autophagy Activation and Antimicrobial Responses During Mycobacterial Infection. J Immunol (2015) 11:5355–65. doi: 10.4049/jimmunol.1402557
102. Liu K, Hong D, Zhang F, Li X, He M, Han X. MicroRNA-106a Inhibits Autophagy Process and Antimicrobial Responses by Targeting Ulk1, ATG7, and ATG16L1 During Mycobacterial Infection. Front Immunol (2021) 11:610021. doi: 10.3389/fimmu.2020.610021
103. Guo L, Zhao J, Qu Y, Yin R, Gao Q, Ding S, et al. MicroRNA-20a Inhibits Autophagic Process by Targeting ATG7 and ATG16L1 and Favors Mycobacterial Survival in Macrophage Cells. Front Cell Infect Microbiol (2016) 6:134. doi: 10.3389/fcimb.2016.00134
104. Chen Z, Wang T, Liu Z, Zhang G, Wang J, Feng S, et al. Inhibition of Autophagy by miR-30A Induced by Mycobacterium Tuberculosis as a Possible Mechanism of Immune Escape in Human Macrophages. Japan J Infect Dis (2015) 68:420–4. doi: 10.7883/yoken.JJID.2014.466
105. Kim J, Lee HM, Park KS, Shin DM, Kim TS, Kim YS, et al. MIR144* Inhibits Antimicrobial Responses Against Mycobacterium Tuberculosis in Human Monocytes and Macrophages by Targeting the Autophagy Protein Dram2. Autophagy (2017) 13:423–41. doi: 10.1080/15548627.2016.1241922
106. Tu H, Yang S, Jiang T, Wei L, Shi L, Liu C, et al. Elevated Pulmonary Tuberculosis Biomarker miR-423-5p Plays Critical Role in the Occurrence of Active TB by Inhibiting Autophagosome-Lysosome Fusion. Emerg Microb Infect (2019) 8:448–60. doi: 10.1080/22221751.2019.1590129
107. Wang J, Yang K, Zhou L, Wu M, Wu Y, Zhu M, et al. MicroRNA-155 Promotes Autophagy to Eliminate Intracellular Mycobacteria by Targeting Rheb. PloS Pathog (2013) 9:e1003697. doi: 10.1371/journal.ppat.1003697
108. Holla S, Kurowska-Stolarska M, Bayry J, Balaji KN. Selective Inhibition of IFNG-Induced Autophagy by Mir155- and Mir31- Responsive WNT5A and SHH Signalling. Autophagy (2014) 2:311–30. doi: 10.4161/auto.27225
109. Creagh EM, Conroy H, Martin SJ. Caspase Activation Pathways in Apoptosis and Immunity. Immunol Rev (2003) 193:10–21. doi: 10.1034/j.1600-065X.2003.00048.x
111. Youle RJ, Strasser A. The Bcl-2 Protein Family: Opposing Activities That Mediate Cell Death. Nat Rev Mol Cell Biol (2008) 9:47–59. doi: 10.1038/nrm2308
112. Giam M, Huang DCS, Bouillet P. Bh3-Only Proteins and Their Roles in Programmed Cell Death. Oncogene (2008) 27:S128–36. doi: 10.1038/onc.2009.50
113. Lamkanfi M, Dixit VM. Manipulation of Host Cell Death Pathways During Microbial Infections. Cell Host Microbe (2010) 8:44–54. doi: 10.1016/j.chom.2010.06.007
114. Martin CJ, Booty MG, Rosebrock TR, Nunes-Alves C, Desjardins DM, Keren I, et al. Efferocytosis Is an Innate Antibacterial Mechanism. Cell Host Microbe (2012) 12:289–300. doi: 10.1016/j.chom.2012.06.010
115. Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, et al. Mycobacterium Tuberculosis Evades Macrophage Defenses by Inhibiting Plasma Membrane Repair. Nat Immunol (2009) 10:899–906. doi: 10.1038/ni.1758
116. Park JS, Tamayo MH, Gonzalez-Juarrero M, Orme IM, Ordway DJ. Virulent Clinical Isolates of Mycobacterium Tuberculosis Grow Rapidly and Induce Cellular Necrosis But Minimal Apoptosis in Murine Macrophages. J Leukocyte Biol (2006) 79:80–6. doi: 10.1189/jlb.0505250
117. Keane J, Remold HG, Kornfeld H. VirulentMycobacterium Tuberculosis Strains Evade Apoptosis of Infected Alveolar Macrophages. J Immunol (2000) 164:2016–20. doi: 10.4049/jimmunol.164.4.2016
118. Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M, et al. The MicroRNA Mir-29 Controls Innate and Adaptive Immune Responses to Intracellular Bacterial Infection by Targeting Interferon-γ. Nat Immunol (2011) 12:861–9. doi: 10.1038/ni.2073
119. Zhang G, Liu X, Wang W, Cai Y, Li S, Chin Q, et al. Downregulation of miR-20a-5p Triggers Cell Apoptosis to Facilitate Mycobacterial Clearance Through Targeting JNK2 in Human Macrophages. Cell Cycle (2016) 15:2527–38. doi: 10.1080/15384101.2016.1215386
120. Zhang D, Yi Z, Fu Y. Downregulation of miR-20b-5p Facilitates Mycobacterium Tuberculosis Survival in RAW 264.7 Macrophages Via Attenuating the Cell Apoptosis by Mcl-1 Upregulation. Cell Biochem (2019) 120:5889–96. doi: 10.1002/jcb.27874
121. Qin LS, Jia PF, Zhang ZQ, Zhang SM. Ros-P53-Cyclophilin-D Signaling Mediates Salinomycin-Induced Glioma Cell Necrosis. J Exp Clin Cancer Res (2015) 34:57. doi: 10.1186/s13046-015-0174-1
122. Sun Q, Shen X, Wang P, Ma J, Sha W. Targeting Cyclophilin-D by miR-1281 Protects Human Macrophages From Mycobacterium Tuberculosis-Induced Programmed Necrosis and Apoptosis. Aging (2019) 11:12661–73. doi: 10.18632/aging.102593
123. Wu Y, Guo Z, Yao K, Miao Y, Liang S, Liu F, et al. The Transcriptional Foundations of Sp110-Mediated Macrophage (Raw264.7) Resistance to Mycobacterium Tuberculosis H37ra. Sci Rep (2016) 6:22041. doi: 10.1038/srep22041
124. Ma J, Chen X-L, Sun Q. MicroRNA-579 Upregulation Mediates Death of Human Macrophages With Mycobacterium Tuberculosis Infection. Biochem. Biophys. Res Commun (2019) 518:219–26. doi: 10.1016/j.bbrc.2019.08.035
125. Fu B, Xue W, Zhang H, Zhang R, Feldman K, Zhao Q, et al. MicroRNA-325-3p Facilitates Immune Escape of Mycobacterium Tuberculosis Through Targeting LNX1 Via NEK6 Accumulation to Promote Anti-Apoptotic STAT3 Signaling. mBIO (2020) 11:e00557–20. doi: 10.1128/mBio.00557-20
126. Wu Z, Lu H, Sheng J, Li L. Inductive MicroRNA-21 Impairs Anti-Mycobacterial Responses by Targeting IL-12 and Bcl-2. FEBS Lett (2012) 586:2459–67. doi: 10.1016/j.febslet.2012.06.004
127. Yang S, Li F, Jia S, Zhang K, Jiang W, Shang Y, et al. Early Secreted Antigen ESAT-6 of Mycobacterium Tuberculosis Promotes Apoptosis of Macrophages Via Targeting the MicroRNA155-SOCS1 Interaction. Cell Physiol Biochem (2015) 35:1276–88. doi: 10.1159/000373950
128. Liang S, Song Z, Wu Y, Gao Y, Gao M, Liu F, et al. MicroRNA-27b Modulates Inflammatory Response and Apoptosis During Mycobacterium Tuberculosis Infection. J Immunol (2018) 200:3506–18. doi: 10.4049/jimmunol.1701448
129. Tzivion G, Dobson M, Ramakrishnan G. Foxo Transcription Factors; Regulation by AKT and 14-3-3 Proteins. Biochim Biophys Acta (2011) 1813:1938– 1945. doi: 10.1016/j.bbamcr.2011.06.002
130. Gilley J, Coffer PJ, Ham J. Foxo Transcription Factors Directly Activate Bim Gene Expression and Promote Apoptosis in Sympathetic Neurons. J Cell Biol (2003) 162:613–22. doi: 10.1083/jcb.200303026
131. Xi X, Zhang C, Han W, Zhao H, Zhang H, Jiao J. MicroRNA-223 Is Upregulated in Active Tuberculosis Patients and Inhibits Apoptosis of Macrophages by Targeting Foxo3. Genet Test Mol Biomark (2015) 19:650–6. doi: 10.1089/gtmb.2015.0090
132. Tripathi A, Srivastava V, Singh BN. Hsa-Let-7b-5p Facilitates Mycobacterium Tuberculosis Survival in THP-1 Human Macrophages by Fas Downregulation. FEMS Microbiol Lett (2018) 365:1–7. doi: 10.1093/femsle/fny040
133. Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin Signature Reveals Over a Thousand Highly Conserved Large Non-Coding RNAs in Mammals. Nature (2009) 458:223–7. doi: 10.1038/nature07672
134. Englert M, Felis M, Junker V, Beier H. Novel Upstream and Intragenic Control Elements for the RNA Polymerase III-Dependent Transcription of Human 7sl RNA Genes. Biochimie (2004) 86:867–74. doi: 10.1016/j.biochi.2004.10.012
135. Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, et al. Integrative Annotation of Human Large Intergenic Noncoding RNAs Reveals Global Properties and Specific Subclasses. Genes Dev (2011) 25:1915–27. doi: 10.1101/gad.17446611
136. Necsulea A, Necsulea A, Kaessmann H, Kaessmann H. Evolutionary Dynamics of Coding and Non-Coding Transcriptomes. Nat Rev Genet (2014) 15:734–48. doi: 10.1038/nrg3802
137. Johnsson P, Lipovich L, Grandér D, Morris KV. Evolutionary Conservation of Long Non-Coding RNAs; Sequence, Structure, Function. Biochim Biophys Acta (2014) 1840:1063–71. doi: 10.1016/j.bbagen.2013.10.035
138. Agliano F, Rathinam VA, Medvedev AE, Vanaja SK, Vella AT. Long Noncoding RNAs in Host–Pathogen Interactions. Trends Immunol (2019) 40:492–510. doi: 10.1016/j.it.2019.04.001
139. Chen YG, Satpathy AT, Chang HY. Gene Regulation in the Immune System by Long Noncoding RNAs. Nat Immunol (2017a) 18:962–72. doi: 10.1038/ni.3771
140. Zhang Q, Chao T-C, Patil VS, Qin Y, Tiwari SK, Chiou J, et al. The Long Noncoding RNA ROCKI Regulates Inflammatory Gene Expression. EMBO J (2019) 38:e100041. doi: 10.15252/embj.2018100041
141. Boon RA, Jaé N, Holdt L, Dimmeler S. Long Noncoding RNAs: From Clinical Genetics to Therapeutic Targets? J Am Coll Cardiol (2016) 67:1214–26. doi: 10.1016/j.jacc.2015.12.051
142. Statello L, Guo C-J, Chen L-L, Huarte M. Gene Regulation by Long Non-Coding RNAs and Its Biological Functions. Nat Rev Mol Cell Biol (2021) 22:96–118. doi: 10.1038/s41580-020-00315-9
143. Ebert MS, Sharp PA. Emerging Roles for Natural MicroRNA Sponges. Curr Biol (2010) 20:R858–861. doi: 10.1016/j.cub.2010.08.052
144. Liu P, Yang H, Zhang J, Peng X, Lu Z, Tong W, et al. The LncRNA MALAT1 Acts as a Competing Endogenous RNA to Regulate Kras Expression by Sponging Mir-217 in Pancreatic Ductal Adenocarcinoma. Sci Rep (2017) 7:5186. doi: 10.1038/s41598-017-05274-4
145. Wang SH, Zhang WJ, Wu XC, Weng MZ, Zhang MD, Cai Q, et al. The LncRNA Malat1 Functions as a Competing Endogenous RNA to Regulate Mcl-1 Expression by Sponging miR-363-3p in Gallbladder Cancer. J Cell Mol Med (2016) 20:2299–308. doi: 10.1111/jcmm.12920
146. Xiao H, Tang K, Liu P, Chen K, Hu J, Zeng J, et al. LncRNA MALAT1 Functions as a Competing Endogenous RNA to Regulate Zeb2 Expression by Sponging miR-200s in Clear Cell Kidney Carcinoma. Oncotarget (2015) 6:38005–15. doi: 10.18632/oncotarget.5357
147. Xi Y, Jiang T, Wang W, Yu J, Wang Y, Wu X, et al. Long Non-Coding HCG18 Promotes Intervertebral Disc Degeneration by Sponging miR-146a-5p and Regulating Traf6 Expression. Sci Rep (2017) 7:13234. doi: 10.1038/s41598-017-13364-6
148. Esteller M. Non-Coding RNAs in Human Disease. Nat Rev Genet (2011) 12:861–74. doi: 10.1038/nrg3074
149. Yan X, Hu Z, Feng Y, Hu X, Yuan J, Zhao SD, et al. Comprehensive Genomic Characterization of Long Non-Coding RNAs Across Human Cancers. Cancer Cell (2015) 28:529–40. doi: 10.1016/j.ccell.2015.09.006
150. Atianand MK, Hu W, Satpathy AT, Shen Y, Ricci EP, Alvarez-Dominguez JR, et al. A Long Noncoding RNA LincRNA-EPS Acts as a Transcriptional Brake to Restrain Inflammation. Cell (2016) 165:1672–85. doi: 10.1016/j.cell.2016.05.075
151. Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, et al. A Long Noncoding RNA Mediates Both Activation and Repression of Immune Response Genes. Science (2013) 341:789–92. doi: 10.1126/science.1240925
152. Rapicavoli NA, Qu K, Zhang J, Mikhail M, Laberge RM, Chang HY. A Mammalian Pseudogene LncRNA at the Interface of Inflammation and Anti-Inflammatory Therapeutics. Elife (2013) 2:e00762. doi: 10.7554/eLife.00762
153. Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S, et al. The STAT3-Binding Long Noncoding RNA lnc-DC Controls Human Dendritic Cell Differentiation. Science (2014) 344:310–3. doi: 10.1126/science.1251456
154. Li Z, Chao TC, Chang KY, Lin N, Patil VS, Shimizu C, et al. The Long Noncoding RNA Thril Regulates Tnfα Expression Through Its Interaction With Hnrnpl. Proc Natl Acad Sci USA (2014) 111:1002–7. doi: 10.1073/pnas.1313768111
155. Zhang X, Liang Z, Zhang Y, Min DD, Wang ZJ, Hu X. Comprehensive Analysis of Long Non-Coding RNAs Expression Pattern in the Pathogenesis of Pulmonary Tuberculosis. Genomics (2020) 112:1970–7. doi: 10.1016/j.ygeno.2019.11.009
156. Yan H, Xu R, Zhang X, Wang Q, Pang J, Zhang X, et al. Identifying Differentially Expressed Long Non−Coding RNAs in PBMC in Response to the Infection of Multidrug-Resistant Tuberculosis. Infect Drug Resist (2018) 11:945–59. doi: 10.2147/IDR.S154255
157. Zhou J, Chaudhry H, Zhong Y, Ali MM, Perkins LA, Owens WB, et al. Dysregulation in MicroRNA Expression in Peripheral Blood Mononuclear Cells of Sepsis Patients Is Associated With Immunopathology. Cytokine (2015) 71:89–100. doi: 10.1016/j.cyto.2014.09.003
158. Siddle KJ, Deschamps M, Tailleux L, Nedelec Y, Pothlichet J, Lugo-Villarino G, et al. A Genomic Portrait of the Genetic Architecture and Regulatory Impact of MicroRNA Expression in Response to Infection. Genome Res (2014) 24:85–859. doi: 10.1101/gr.161471.113
159. Huang S, Huang Z, Luo Q, Qing C. The Expression of LncRNA NEAT1 in Human Tuberculosis and Its Antituberculosis Effect. Biomed Res Intl (2018) 2018:9529072. doi: 10.1155/2018/9529072
160. Huang Z, Liu J, Li L, Guo Y, Luo Q, Li J. Long Non-Coding RNA Expression Profiling of Macrophage Line RAW264.7 Infected by Mycobacterium Tuberculosis. Biotech Histochem (2020) 95:403–10. doi: 10.1080/10520295.2019.1707874
161. Yang X, Yang J, Wang J, Wen Q, Wang H, He J, et al. Microarray Analysis of Long Noncoding RNA and mRNA Expression Profiles in Human Macrophages Infected With Mycobacterium Tuberculosis. Sci Rep (2016) 6:38963. doi: 10.1038/srep38963
162. Margueron R, Reinberg D. The Polycomb Complex PRC2 and Its Mark in Life. Nature (2011) 469:343–9. doi: 10.1038/nature09784
163. Subuddhi A, Kumar M, Majumder D, Sarkar A, Ghosh Z, Madavan V, et al. Unraveling the Role of H3K4 Trimethylation and LncRNA HOTAIR in SATB1 and DUSP4-Dependent Survival of Virulent Mycobacterium Tuberculosis in Macrophages. Tuberculosis (2020) 120:101897. doi: 10.1016/j.tube.2019.101897
164. Li M, Cui J, Niu W, Huang J, Feng T, Sun B, et al. Long Non-Coding PCED1B-AS1 Regulates Macrophage Apoptosis and Autophagy by Sponging Mir-155 in Active Tuberculosis. Biochem Biophys Res Commun (2019) 509:803–9. doi: 10.1016/j.bbrc.2019.01.005
165. Huang J, Jiao J, Xu W, Zhao H, Zhang C, Shi Y, et al. Mir-155 Is Upregulated in Patients With Active Tuberculosis and Inhibits Apoptosis of Monocytes by Targeting Foxo3. Mol Med Rep (2015) 12:7102–8. doi: 10.3892/mmr.2015.4250
166. Sun W, Lou H, Cao J, Wang P, Sha W, Sun Q. LncRNA MEG3 Controls Mycobacterium Tuberculosis Infection Via Controlled Mir-145-5p Expression and Modulation of Macrophage Proliferation. Microbial Pathogenesis (2020) 149:104550. doi: 10.1016/j.micpath.2020.104550
167. Zuo S, Wu L, Wang Y, Yuan X. Long Non-Coding RNA Meg3 Activated by Vitamin D Suppresses Glycolysis in Colorectal Cancer Via Promoting C-Myc Degradation. Front Oncol (2020) 10:274. doi: 10.3389/fonc.2020.00274
168. Liu D, Liu Y, Zheng X, Liu N. C-MYC-Induced Long Noncoding RNA Meg3 Aggravates Kidney Ischemia–Reperfusion Injury Through Activating Mitophagy by Upregulation of RTKN to Trigger the Wnt/β-Catenin Pathway. Cell Death Dis (2021) 12:191. doi: 10.1038/s41419-021-03466-5
169. Wang Y, Zhong H, Xie X, Chen CY, Huang D, Shen L, et al. Long Noncoding RNA Derived From CD244 Signaling Epigenetically Controls CD8+ T-Cell Immune Responses in Tuberculosis Infection. Proc Natl Acad Sci USA (2015) 112:E3883–92. doi: 10.1073/pnas.1501662112
170. Yi Z, Li J, Gao K, Fu Y. Identification of Differentially Expressed Long Non-Coding RNAs in CD4+ T Cells Response to Latent Tuberculosis Infection. J Infection (2014) 69:558–68. doi: 10.1016/j.jinf.2014.06.016
171. Fu Y, Xu X, Xue J, Duan W, Yi Z. Deregulated LncRNAs in B Cells From Patients With Active Tuberculosis. PloS One (2017) 12:e0170712. doi: 10.1371/journal.pone.0170712
172. Sabir N, Hussain T, Shah SZA, Peramo A, Zhao D, Zhou X. miRNAs in Tuberculosis: New Avenues for Diagnosis and Host-Directed Therapy. Front Microbiol (2018) 9:602. doi: 10.3389/fmicb.2018.00602
173. Mirzaei R, Babakhani S, Ajorloo P, Ahmadi RH, Hosseini-Fard SR, Keyvani H, et al. The Emerging Role of Exosomal miRNAs as a Diagnostic and Therapeutic Biomarker in Mycobacterium Tuberculosis Infection. Mol Med (2021) 27:34. doi: 10.1186/s10020-021-00296-1
174. Hu X, Liao S, Bai H, Wu L, Wang M, Wu Q, et al. Integrating Exosomal MicroRNAs and Electronic Health Data Improved Tuberculosis Diagnosis. EBioMedicine (2019) 40:564–73. doi: 10.1016/j.ebiom.2019.01.023
175. Alipoor SD, Tabarsi P, Varahram M, Movassaghi M, Dizaji MK, Folkerts G. Serum Exosomal miRNAs Are Associated With Active Pulmonary Tuberculosis. Dis Markers (2019) 2019:1907426. doi: 10.1155/2019/1907426
176. Lyu L, Zhang X, Li C, Yang T, Wang J, Pan L, et al. Small RNA Profiles of Serum Exosomes Derived From Individuals With Latent and Active Tuberculosis. Front Microbiol (2019) 10:1174. doi: 10.3389/fmicb.2019.01174
177. Hashimoto S, Zhao H, Hayakawa M, Nakajima K, Taguchi Y-H, Murakami Y. Developing a Diagnostic Method for Latent Tuberculosis Infection Using Circulating MiRNA. Trans Med Commun (2020) 5:25. doi: 10.1186/s41231-020-00078-7
178. Wang C, Yang S, Sun G, Tang X, Lu S, Neyrolles O, et al. Comparative MiRNA Expression Profiles in Individuals With Latent and Active Tuberculosis. PloS One (2011) 6:e25832. doi: 10.1371/journal.pone.0025832
179. Zhang H, Sun Z, Wei W, Liu Z, Fleming J, Zhang S, et al. Identification of Serum MicroRNA Biomarkers for Tuberculosis Using RNA-Seq. PloS One (2014) 9:e88909. doi: 10.1371/journal.pone.0088909
180. Latorre I, Leidinger P, Backes C, Domínguez J, de Souza-Galvão ML, Maldonado J, et al. A Novel Whole-Blood MiRNA Signature for a Rapid Diagnosis of Pulmonary Tuberculosis. Eur Respir J (2015) 45:1173–6. doi: 10.1183/09031936.00221514
181. Lin Y, Zhang Y, Yue H, Tian R, Wang G, Li F. Identification of Unique Key Genes and miRNAs in Latent Tuberculosis Infection by Network Analysis. Mol Immunol (2019) 112:103–14. doi: 10.1016/j.molimm.2019.04.032
182. Fang Y, Zhao J, Wang X, Wang X, Wang L, Liu L, et al. Identification of Differentially Expressed LncRNAs as Potential Plasma Biomarkers for Active Tuberculosis. Tuberculosis (2021) 128:102065. doi: 10.1016/j.tube.2021.102065
183. Hu X, Liao S, Bai H, Gupta S, Zhou Y, Zhou J, et al. LncRNA and Predictive Model to Improve the Diagnosis of Clinically Diagnosed Pulmonary Tuberculosis. J Clin Microbiol (2020) 58:e01973–19. doi: 10.1128/JCM.01973-19
184. Bai H, Wu Q, Hu X, Wu T, Song J, Liu T, et al. Clinical Significance of lnc-AC145676.2.1-6 and lnc-TGS1-1 and Their Variants in Western Chinese Tuberculosis Patients. Int J Infect Dis (2019) 84:8–14. doi: 10.1016/j.ijid.2019.04.018
185. Lv Y, Guo S, Li XG, Chi JY, Qu YQ, Zhong HL. Sputum and Serum MicroRNA-144 Levels in Patients With Tuberculosis Before and After Treatment. Int J Infect Dis (2016) 43:68–73. doi: 10.1016/j.ijid.2015.12.014
186. Spinelli SV, Diaz A, D’Attilio L, Marchesini MM, Bogue C, Bay ML, et al. Altered MicroRNA Expression Levels in Mononuclear Cells of Patients With Pulmonary and Pleural Tuberculosis and Their Relation With Components of the Immune Response. Mol Immunol (2013) 53:265–9. doi: 10.1016/j.molimm.2012.08.008
187. Liu Z, Zhou G, Deng X, Yu Q, Hu Y, Sun H, et al. Analysis of MiRNA Expression Profiling in Human Macrophages Responding to Mycobacterium Infection: Induction of the Immune Regulator Mir-146a. J Infect (2014) 68:553–6. doi: 10.1016/j.jinf.2013.12.017
188. Wagh V, Urhekar A, Modi D. Levels of MicroRNA Mir-16 and Mir-155 Are Altered in Serum of Patients With Tuberculosis and Associate With Responses to Therapy. Tuberculosis (2017) 102:24–30. doi: 10.1016/j.tube.2016.10.007
189. Fu Y, Yi Z, Wu X, Li J, Xu F. Circulating MicroRNAs in Patients With Active Pulmonary Tuberculosis. J Clin Microbiol (2011) 49:4246– 4251. doi: 10.1128/JCM.05459-11
190. Wang J, Zhu X, Xiong X, Ge P, Liu H, Ren N, et al. Identification of Potential Urine Proteins and MicroRNA Biomarkers for the Diagnosis of Pulmonary Tuberculosis Patients. Emerg Microbes Infect (2018) 7:63. doi: 10.1038/s41426-018-0066-5
191. Ruiz-Tagle C, Naves R, Balcells ME. Unraveling the Role of MicroRNAs in Mycobacterium Tuberculosis Infection and Disease: Advances and Pitfalls. Infect Immun (2020) 88:e00649–19. doi: 10.1128/IAI.00649-19
192. Zhou YL, Zhang L, Zhou Z, Liu W, Lu Y, He S, et al. Antibody Modified Nanoparticle-Mediated Delivery of Mir-124 Regulates Apoptosis Via Repression the Stat3 Signal in Mycobacterial-Infected Microglia. J Biomed Nanotechnol (2018) 14:2185–97. doi: 10.1166/jbn.2018.2650
193. Tivnan A, Orr WS, Gubala V, Nooney R, Williams DE, McDonagh C, et al. Inhibition of Neuroblastoma Tumor Growth by Targeted Delivery of MicroRNA-34a Using Anti-Disialoganglioside GD2 Coated Nanoparticles. PloS One (2012) 7:e38129. doi: 10.1371/journal.pone.0038129
194. Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, et al. Therapeutic MicroRNA Delivery Suppresses Tumorigenesis in a Murine Liver Cancer Model. Cell (2009) 137:1005–17. doi: 10.1016/j.cell.2009.04.021
195. Wu Y, Crawford M, Yu B, Mao Y, Nana-Sinkam SP, Lee LJ. MicroRNA Delivery by Cationic Lipoplexes for Lung Cancer Therapy. Mol Pharm (2011) 8:1381–9. doi: 10.1021/mp2002076
Keywords: Mycobacterium tuberculosis, microRNAs, long non-coding RNAs, immune response, autophagy, apoptosis, inflammation
Citation: Kundu M and Basu J (2021) The Role of microRNAs and Long Non-Coding RNAs in the Regulation of the Immune Response to Mycobacterium tuberculosis Infection. Front. Immunol. 12:687962. doi: 10.3389/fimmu.2021.687962
Received: 30 March 2021; Accepted: 09 June 2021;
Published: 24 June 2021.
Edited by:
Rosane M. B. Teles, University of California, Los Angeles, United StatesReviewed by:
Shamila D. Alipoor, National Institute for Genetic Engineering and Biotechnology, IranCopyright © 2021 Kundu and Basu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manikuntala Kundu, bWFuaWt1bnRhbGEua3VuZHVAZ21haWwuY29t; Joyoti Basu, am95b3RpYmFzdUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.