 Abbey Lightfoot
Abbey Lightfoot Helen M. McGettrick
Helen M. McGettrick Asif J. Iqbal
Asif J. Iqbal- 1Institute of Cardiovascular Sciences, University of Birmingham, Birmingham, United Kingdom
- 2Institute of Inflammation and Ageing, University of Birmingham, Birmingham, United Kingdom
Leukocyte recruitment to the site of injury is a crucial event in the regulation of an inflammatory response. Tight regulation of interactions between the endothelium and circulating leukocytes is necessary to ensure a protective response to injury does not result in inflammatory disease. Rising interest in the broad immunoregulatory roles displayed by members of the glycan-binding galectin family suggests that these proteins could be an attractive target for therapeutic intervention, since their expression is significantly altered in disease. The focus of this review is to summarize current knowledge on the role of galectins in leukocyte trafficking during inflammation and the clinical approaches being taken to target these interactions for treatment of inflammatory disease.
Introduction
Glycans are one of the four major components that constitute cells, accompanied by nucleic acids, proteins and lipids. Recognition of specific glycan motifs by glycan-binding proteins (lectins) is crucial for facilitating highly sophisticated cross-communication between leukocytes in the bloodstream and endothelial cells (ECs) lining the blood vessels (1). Several key lectins (E-, L- and P-selectins and Cluster of differentiation [CD] 44) have already been identified as mediators of leukocyte recruitment and trafficking at the site of injury in the initial stages of the immune response (1). β-galactoside-binding galectins have emerged as an interesting family of glycan-binding proteins involved in the initiation and resolution stages of the inflammatory response (2, 3). Our understanding of the roles of endothelial-expressed galectins in the leukocyte trafficking cascade is relatively poor when compared to other known glycan-binding proteins involved in this process. As such, this review will provide an up-to-date overview on the role of endothelial derived galectins in leukocyte trafficking; the factors that regulate their expression and function; and discusses the therapeutic potential of targeting these interactions to treat immune-mediated chronic inflammatory disease (IMID).
Upon injury or infection, tissue resident immune cells release pro-inflammatory factors such as reactive oxygen species and proteases that partially degrade glycoprotein components of the glycocalyx; an EC-expressed matrix that serves to shield the vascular walls from direct exposure to blood components and flow (4, 5). Glycosaminoglycans (GAGs) in the reduced layer of glycocalyx bind and immobilize chemokines, accompanied by the activation of ECs and upregulation of surface expressed adhesion molecules (6). Hemodynamic forces facilitate the outward movement of leukocytes towards the venular endothelium in a process known as margination, enabling interactions between leukocytes and newly exposed molecules displayed on the endothelium (6). Tight regulatory mechanisms are required to ensure leukocyte recruitment occurs only when appropriate, as such ECs are able to detect and respond to environmental changes such as the hemodynamic forces of blood flow (7, 8). Continuous pulsatile and laminar flow activates mechanosensitive channels and suppresses nuclear-factor kappa-B (NF-κB) via the MEK5/ERK5 (mitogen-activated protein kinase 5-extracellular signal-regulated kinase 5) pathway. Consequently, this prevents initiation of the inflammatory response through the downregulation of adhesion molecules and thus reduced leukocyte recruitment to the endothelium (9).
Adhesion molecules displayed on the surface of activated ECs, particularly selectins (E- and P-selectins), reduce the velocity of leukocytes rolling along the endothelium through binding to heavily glycosylated counter-receptors [PSGL-1, P-selectin glycoprotein ligand-1 (CD162)] involved in the capture and subsequent rolling events of the leukocyte adhesion cascade (Figure 1A) (10). This interaction induces conformational changes in integrins [MAC-1, Macrophage antigen-1 (CD11b/CD18); VLA-4, very late antigen-4 (CD49d/CD29); and LFA-1, lymphocyte function-associated antigen-1 (CD11a/CD18)] and facilitates firm adhesion of leukocytes to the endothelium by binding EC-expressed immunoglobulin superfamily adhesion molecules [ICAM-1, Intercellular Adhesion Molecule 1 (CD54) and VCAM-1, Vascular cell adhesion protein 1 (CD106)] (11). Additionally, the interaction between integrins and adhesion molecules facilitates the intraluminal crawling of leukocytes across ECs (12). Migration of leukocytes through the endothelium occurs either at endothelial junctions between ECs (paracellular) or through the body of the EC itself (transcellular). This migration of leukocytes into the surrounding tissue is facilitated by leukocyte-integrin binding to junctional adhesion molecules (JAMs) and PECAM-1 [CD31] expressed on the EC surface (Figure 1A) (12).
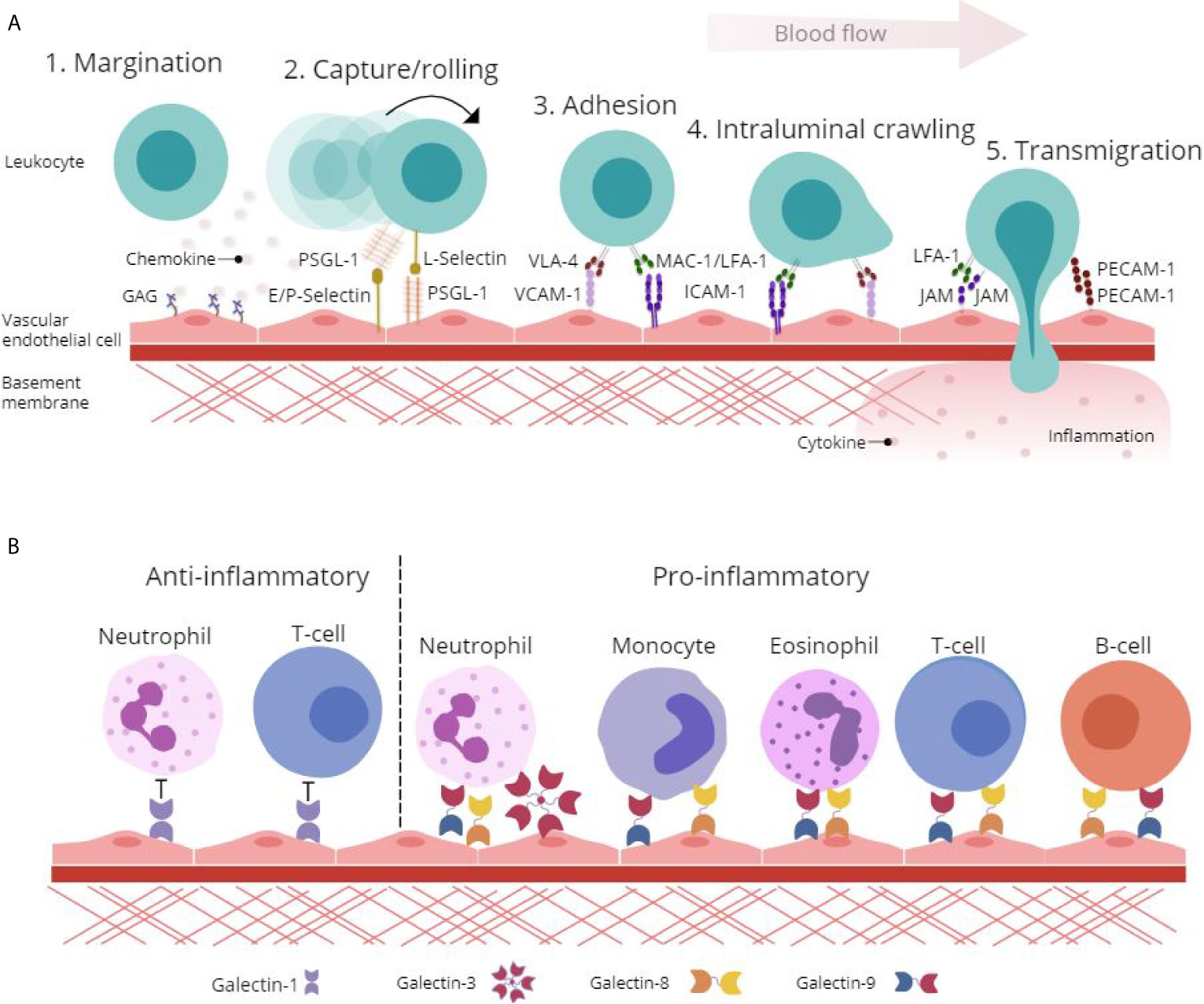
Figure 1 Glycan-binding proteins regulate endothelial-leukocyte interactions in inflammation. (A) Protein-protein and protein-glycan interactions are crucial regulators of leukocyte-endothelial interactions involved in the leukocyte adhesion cascade. Firstly, glycosaminoglycan (GAG)-presented chemokines promote leukocyte margination to the vessel wall. Capture and rolling of leukocytes along the endothelium is facilitated by interactions between selectins and their ligands such as P-selectin glycoprotein ligand (PSGL)-1. This interaction leads to cell activation and conformational changes in leukocyte integrins including Very Late Antigen (VLA)-4, Macrophage (Mac)-1 antigen and lymphocyte function-associated antigen (LFA)-1 that then interact with adhesion molecules such as Vascular Cell Adhesion Molecule (VCAM)-1 and Intracellular Cell Adhesion Molecule (ICAM)-1 to facilitate adhesion and intraluminal crawling. Finally, transmigration through the endothelial cell (EC) layer is mediated by homophilic interactions between platelet EC adhesion molecule (PECAM)-1 and junctional adhesion molecule (JAM). (B) β-galactoside binding galectins facilitate and inhibit leukocyte trafficking to the endothelium to promote pro- and anti-inflammatory responses. Galectin-1 inhibits leukocyte-endothelial interactions to promote an anti-inflammatory phenotype, whilst galectin-3, -8 and -9 facilitate pro-inflammatory leukocyte trafficking.
Whilst many aspects of the leukocyte trafficking cascade have been studied in detail, a lot still remains unknown. Specifically, the galectin family of glycan-binding proteins have recently come to light as functionally important immunoregulatory proteins with involvement in leukocyte activation, apoptosis and in mediating leukocyte adhesion and migration (Figure 1B) (13, 14). Enhancing our understanding of the regulators of galectin expression and their roles in mediating leukocyte trafficking could uncover novel mechanisms of inflammation and highlight distinctive opportunities for the treatment of IMIDs.
Galectin Structure and Signaling
Galectins recognize β-galactoside-containing glycan side chains on proteins and lipids via their unique carbohydrate recognition domains (CRDs) (15). At present, 15 members of galectins have been identified in mammals, of which 12 are associated with genes found in humans (https://www.ncbi.nlm.nih.gov/genbank/) Galectins-1, -3, -8 and -9 are amongst the most studied members since many of their functions have been closely linked to inflammation and disease (2). Importantly, murine and human galectins share approximately ~79% protein sequence homology (16), allowing their function to be studied in pre-clinical murine models. Galectins can be classified into three distinct groups; prototype, tandem repeat, and chimeric as determined by their structural properties (Figure 2). Oligomerization of galectin monomers is typically required for the formation of functional galectin-glycan lattices on the surface of cells (17). Additionally, and despite the absence of a classical secretory signal, galectins are released into the extracellular compartment and mediates extracellular matrix (ECM) assembly and remodeling through binding to ECM components (laminin, fibronectin and vitronectin) (18).
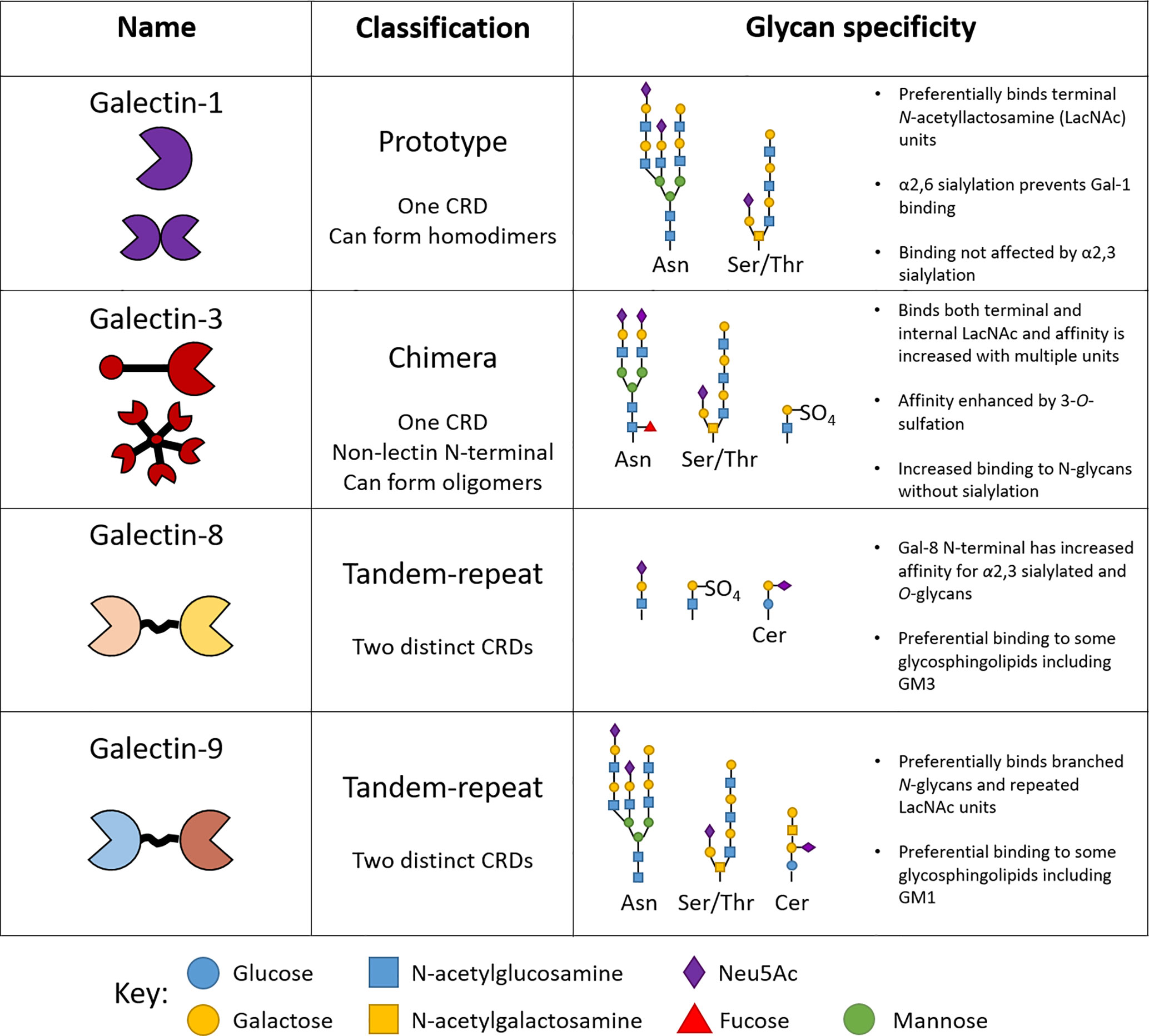
Figure 2 Structure and specificity of human endothelial-expressed β-galactoside binding galectins. The classification of galectins into prototype, tandem-repeat and chimera types is based on the number of carbohydrate recognition domains (CRDs) they contain. Each galectin type reserves high specificity for certain glycan motifs.
Key conserved amino acids in the CRDs across different galectins are responsible for the specific binding to β-galactoside-containing glycans. Despite these similarities, each galectin type reserves specificity for particular branched glycan’s and glycan motifs attached to proteins and lipids alike (Figure 2) (19). For example, the presence of α2,6-linked sialic acid prevents binding of galectin-1 to glycan chains, whilst the same motif enhances the affinity of galectin-8 for its ligand, determining important interactions regulating inflammation (Figure 2) (17). The structural determinants of galectin-glycan recognition have recently been reviewed in (20). The effects of glycan modification on galectin–glycan interactions has been reviewed elsewhere and are summarized in Figure 2 (17, 21). Whether or not structural differences contribute to distinct roles for individual galectin members is yet to be fully elucidated and since galectins are expressed by many cell types it is important to consider both the exogenous and endogenous function of galectins in the context of specific cell types and their physiological interactions.
Regulation of Galectin Expression in Inflammation
To play a role in leukocyte trafficking, galectins must be present at the site of inflammation where endothelial-leukocyte interactions occur. Expression of galectin-1, -3, -8 and -9 mRNA and protein have been detected in in vitro cultured human macro and microvascular ECs, with expression largely intracellular (22). The release of soluble galectin-1 and -8 from human macro- or microvascular ECs respectively, has also been reported (23, 24). Immunohistochemistry (IHC) on sections of human placenta, liver, kidney and colon tissue revealed that low levels of galectin-1 were consistently detected, whilst galectin-3, -8 and -9 expression was detected at variable levels in some but not all of the tissue sections (22). Interestingly, the cellular localization of endothelial galectin expression is highly variable between the different vascular tissue beds (22, 25). Specialized high endothelial venules (HEV) present in sections of human lymph node and tonsil tissue showed higher levels of galectin-9 expression than ECs in other vascular beds (26). Conversely, galectin-3 was not detected and galectin-1 expression was relatively low in HEV from healthy human lymph node sections (25, 27). The variable expression of galectins reported in lymphoid tissues could be indicative of distinct immunoregulatory roles between the galectin types, since the HEV reside within close proximity to leukocytes within the tissue.
An additional consideration when studying ECs is that they possess a complex system of mechanosensitive proteins that respond to hemodynamic forces (28). In response to physiological blood flow, ECs maintain an atheroprotective environment by suppressing gene expression of inflammatory, pro-apoptotic and proliferative pathways via mechanotransduction pathways (29). The expression of integrins and adhesion molecules, including PECAM, are modulated via this mechanism. Perturbations in shear stress can disturb EC mechanosensitive protein signaling, contributing to the pathophysiological angiogenesis in tumor vasculature where low shear stress is experienced, and driving inflammation in atherosclerosis-prone vascular niches and flow-obstructing pathologies where shear stress is disturbed (30, 31). Deep sequencing of the transcriptome of human umbilical vein EC (HUVEC) and human aortic EC (HAEC) in response to patterns of shear stress has identified changes in expression of galectins not only in response to specific flow patterns, but also across EC type (32). Collectively, these findings suggest that expression of individual galectins is highly dependent on the tissue microenvironment and that a more in-depth comparison of the patterns of galectin expression across different vascular beds could be insightful for understanding galectin regulation and function in this context.
As is the case with many immunoregulatory proteins, galectin expression is modulated with EC activation in response to inflammatory mediators, pathogen exposure, and injury (22, 33–35). A concentration-dependent increase in expression of galectin-9 mRNA and protein was observed in vitro following treatment of primary HUVEC with double-stranded RNA viral mimetic, polyinosinic-polycytidylic acid [poly(I:C)], a Toll-like receptor (TLR) 3 ligand (33, 34). Similarly, through the TLR4 pathway, lipopolysaccharide (LPS) stimulation up-regulated surface expression of galectin-8 in human microsvascular EC (HMVEC), suggesting a specific role for galectins in viral and bacterial related infections (24). More basic chemical mediators involved in acute and chronic immune responses have also been shown to differentially regulate endothelial galectin expression. Surface expression of galectin-3 on HUVEC is increased in response to treatment with IL-1β (36). Interestingly, stimulation of HUVEC with IFN-β/γ did not increase expression of galectin-3 (or galectin-1), though a significant increase in galectin-9 surface expression was observed (26). More complex cocktails of soluble disease mediators, such as those released in the conditioned culture medium from colon carcinoma cell lines and in mixtures of oxidized low-density lipoproteins, a known driver of atherosclerosis and cardiovascular disease, stimulated galectin-1 translocation to the cell surface in HUVEC and HAEC (22, 25). The link between increased galectin expression and inflammation is well established, and elevated levels of galectin-3 in the serum and tissue of IMID patients is now recognized as a biomarker for detecting early stages of autoimmune and chronic inflammatory disease (37). Similarly, elevated serum levels of galectin-3 are detected in various cancers and even more so in metastatic disease, correlating with increased levels of metastasis-promoting cytokines released from vascular endothelial cells (38). The galectin expression profile within the tumor vasculature itself highlights a role for endothelial galectins in disease pathogenesis, where soluble factors secreted from tumor cells have been shown to induce galectin-1 expression and translocation to the EC surface to promote tumor angiogenesis and inhibit T-cell migration across the endothelium (22, 23, 39). The link between galectins and tumor angiogenesis, metastasis, and immune suppression could make for an attractive immunotherapy target in combination with currently available cancer therapeutics. Blockade of vascular endothelial growth factor (VEGF) is the current standard of targeted anti-angiogenic therapy and offers variable treatment efficacy dependent on the cancer type (40). Perhaps galectin-1 could be a more appropriate and effective target, even more so if used in combination with targeted therapy such as CAR-T cells to increase tumor infiltration. As has been confirmed with in vitro studies, galectin expression in leukocytes is also modulated in response to stimulation with various inflammatory mediators and could be a major influence on the differential expression observed in inflamed tissue and patient serum samples (41). As such, soluble galectins released from immune cells can contribute to the differential levels of galectins observed in inflamed tissue and circulation, potentially counteracting endothelial cell-bound and expressed galectin functions. Distinguishing the specific effects of endothelial galectins on leukocyte trafficking and pathogenesis from the roles of alternative sources of galectins in the inflamed microenvironment is necessary to fully explore the potential for therapeutic intervention.
Leukocyte Trafficking Regulation by Endothelial Galectins
The interaction between glycans and glycan-binding proteins is crucial at all stages of leukocyte trafficking (1) and remains an active area of research. A large proportion of the literature investigating galectins in inflammation report pro- and anti-inflammatory functions of exogenous galectins. In the absence of inflammatory stimuli, galectin-1 has been reported to promote neutrophil migration in vitro (42). Conversely, in the presence of acute inflammatory mediators IL-8 and TNFα, neutrophil chemotaxis was inhibited by galectin-1 (43, 44). Similarly, the transmigration of modified T-lymphocyte cell lines across stimulated HUVEC was significantly inhibited by galectin-1 compared to unstimulated HUVEC (23). Local injection of galectin-3 into the dorsal air pouch was shown to increase monocyte infiltration (45). More recently, galectin-3 was shown to directly dimerize with the chemokine CXCL-12 via an interface containing the GAG‐binding motif, inducing anti‐chemokine effects and inhibiting CXCL-12‐induced migration of monocytic THP‐1 cells and neutrophils in vitro (46). This latter finding could indicate an endothelial-specific galectin function whereby soluble galectin-3 released from ECs binds to the cell surface to mediate the interaction between leukocytes and CXCL-12. This concept is supported by the study from Yamamoto et al., which reported enhanced binding of T-cells, B-cells, neutrophils, eosinophils and monocytes to HUVEC following pre-incubation with increasing concentrations of both galectin-8 and -9 (47). Interestingly enhanced binding was not observed with galectin-1 or -3, although others have shown that oligomerization of galectin-3 at the EC surface was observed by fluorescence resonance energy transfer (FRET) and facilitated leukocyte clustering at the tricellular corners of HUVEC monolayers where leukocytes preferentially transmigrate (48). Galectin-9 was originally identified as a potent chemoattractant and activator of eosinophils (49). It has since also been shown to bind to protein disulfide isomerases on T-helper 2 cells (Th2), influencing their migration through recombinant galectin-9 coated matrigel by increasing the reduction of disulfide bonds on integrins (50). In the absence of inflammatory stimuli, galectin-9 has been shown to significantly induce monocyte chemotaxis in vitro compared to control (51). This finding was also supported in vivo, with increased monocyte and macrophage infiltrate in isolated knee tissue from mice receiving galectin-9 via intra-articular knee injection (51). The earlier reports strongly suggest a positive role for galectin-9 in driving Th2-type immune responses that could be contributing to the development of diseases such as allergic asthma. However, the substantial increase in galectin-9 expression in ECs following stimulation with Poly : IC and IFNγ, and the increased monocyte and macrophage infiltrate observed in response to galectin-9 injection, suggests that galectin-9 mediated immune responses may be broader than Th2-type only. Together, these findings highlight a potential role for galectins as both pro- and anti-inflammatory molecules affecting leukocyte trafficking dependent on the mediators and cell types in the local microenvironment. It is also worth noting that the glycosylation patterns of leukocyte surface molecules change upon cell activation, and as such, are worth considering when studying the interactions between glycans and glycan-binding proteins to elucidate context specific roles and functions. With only a limited amount of research on the secretion of galectins from ECs, we cannot predict that the results observed in response to exogenous galectins would translate to such function in vivo. Thus, further investigation is required to understand the regulators of endothelial galectin secretion and the impact of this on leukocyte migration in vivo.
Galectin knockout mice and knock-down studies have been invaluable for studying the endogenous functions of galectins in a more physiological context. Galectin-1 appears to exhibit anti-inflammatory functions since extravasation to inflamed cremaster tissue was enhanced in galectin-1 knockout mice compared to wildtype (44). This mirrored in vitro results showing neutrophil recruitment and rolling along TNF-α stimulated HUVEC was enhanced following galectin-1 knockdown in ECs (44). Similarly, endogenous galectin-1 appeared to inhibit T-lymphocyte capture, rolling and adhesion to stimulated HUVEC under physiological flow (52). Interestingly in an acute model of edema, we found that the absence of galectin-1 did not exacerbate the inflammatory response and recruitment of leukocytes as predicted. This in part was due to redundancy amongst galectins, as we demonstrated galectin-9 levels were significantly elevated in these mice and could therefore potentially compensate for the absence of galectin-1 and promote resolution (53). Galectin-3 on the other hand, has been shown to promote leukocyte recruitment in vivo, as impaired slow rolling and emigration of leukocytes to IL-1β stimulated cremasteric postcapillary venules of galectin-3 knockout mouse was observed by intravital microscopy (54). Interestingly, these galectin-3 null mouse ECs showed reduced surface expression of ICAM-1 and E-selectin following treatment with IL-1β and vehicle compared to wildtype, suggesting endothelial function might be impaired in the absence of endogenous galectin-3 (54). There is limited research on the roles of galectin-8 and -9 on leukocyte trafficking, especially in vivo. The correlation between the results from in vivo studies using galectin knockout mouse models and in vitro studies using galectin-knockdown ECs support the endogenous galectin function on leukocyte trafficking and inflammation. Despite these convincing reports, an endothelial-specific galectin knockout mouse model is necessary to explore and understand mechanistic pathways and regulators of galectin function in the endothelium. Several endothelial-specific Cre and Cre/ERT2 mouse models have successfully been used in the vascular biology field and should be exploited to study endothelial galectin function.
Therapeutic Potential of Targeting Endothelial Galectins
The therapeutic potential of targeting galectins for the treatment of inflammatory and autoimmune diseases is currently being explored and showing promising signs of success in early clinical trials (55). Whilst the current and most developed approach is focused on inhibiting galectin-3 with complex carbohydrate mimetics, the potential benefits of administering galectin-1 to mimic its anti-inflammatory effects are also being considered as treatment options in acute myocardial infarction, ischemic stroke and autoimmune diseases (56, 57). Whilst both approaches offer a seemingly promising route to treating chronic inflammation, the systemic approach to treatment might lead to non-specific effects and reduced treatment efficacy. The use of targeted delivery to affected tissues and cells might optimize the potential benefits of drug delivery mimicking or modulating the effects of galectins in vivo. Whilst more remains to be understood about the regulation and contribution of endothelial galectins to leukocyte trafficking in acute and chronic inflammation, the interaction between EC-galectins and leukocytes observed in vivo and in vitro suggests specific pro- and anti- inflammatory, or even pro-resolution roles that could be manipulated for patient benefit.
Conclusions and Perspectives
Over the last two decades, there has been a steady rise in the number of publications detailing the involvement of galectins in widespread physiological functions, including inflammation, immune responses, apoptosis, autophagy and angiogenesis. Despite a clear link between galectins and disease there still remains ambiguities around the mechanisms by which galectins contribute to pathology, particularly in a cell-specific context since galectin expression and function is diverse and complex. Thus, the challenge remains to gain mechanistic insight, particularly with regards to endothelial galectins in leukocyte trafficking and inflammation to uncover the most appropriate routes for clinical intervention in treatment of IMIDs.
The conditions under which cells and their surface expressed molecules are glycosylated is complex and differs depending on cell state of differentiation, activation and disease (58). As such, attention to the physiological context under which the function of sugar-binding proteins are studied is crucial. A recent study from the Huang group examined the glycosylation-dependent interactions of galectins with intracellular and surface expressed ligands through proximal labeling of cells with galectin-fusion proteins (59). The application of this technique to ECs exposed to inflammatory mediators and/or culture under fluid shear stress could be extremely valuable for understanding the mechanisms and regulators of galectin-glycan interactions in context.
Critically, in vitro culture of ECs still remains problematic. The distinct expressional changes between human EC types in response to different shear stress patterns emphasizes the importance of choosing the right models, with consideration for cell type and contextual interactions, to study endothelial-specific responses in vitro (32). Whilst many research groups have attempted to mimic physiological flow conditions using expensive and/or specialist equipment, the problem still remains that the complex physiological EC environment cannot be mimicked ex vivo. One such example of this is that cultured ECs display deficiencies in the glycocalyx, casting doubts on the reliability of using ECs in vitro as a method to study vascular function and role in pathology (60). With major developments in organ on chip technology, we may have greater success in elucidating the role and mechanisms of endothelial galectins in a tissue specific context (61). Finally, the imminent prospect of galectin-targeted therapeutics is encouraging, and whilst interest in galectins remains high and spread across multidisciplinary subjects our understanding of galectin function will only develop further in the coming years.
Author Contributions
AL, HM, and AI wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
AL is supported by Wellcome Trust Mechanisms of Inflammatory Disease (MIDAS) PhD studentship [222392/Z/21/Z]. HM is supported by the MCR [MR/T028025/1]. AI is supported by Birmingham Fellowship and AMS Springboard Award [SBF003\1156].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Krautter F, Iqbal AJ. Glycans and Glycan-Binding Proteins as Regulators and Potential Targets in Leukocyte Recruitment. Front Cell Dev Biol (2021) 9:624082. doi: 10.3389/fcell.2021.624082
2. Liu F-T, Rabinovich GA. Galectins: Regulators of Acute and Chronic Inflammation. Ann N Y Acad Sci (2010) 1183(1):158–82. doi: 10.1111/j.1749-6632.2009.05131.x
3. Law HL, Wright RD, Iqbal AJ, Norling LV, Cooper D. A Pro-resolving Role for Galectin-1 in Acute Inflammation. Front Pharmacol (2020) 11:274–. doi: 10.3389/fphar.2020.00274
4. Sieve I, Münster-Kühnel AK, Hilfiker-Kleiner D. Regulation and Function of Endothelial Glycocalyx Layer in Vascular Diseases. Vasc Pharmacol (2018) 100:26–33. doi: 10.1016/j.vph.2017.09.002
5. Reitsma S, Slaaf DW, Vink H, van Zandvoort MAMJ, oude Egbrink MGA. The Endothelial Glycocalyx: Composition, Functions, and Visualization. Pflugers Arch (2007) 454(3):345–59. doi: 10.1007/s00424-007-0212-8
6. Marki A, Esko JD, Pries AR, Ley K. Role of the Endothelial Surface Layer in Neutrophil Recruitment. J Leukoc Biol (2015) 98(4):503–15. doi: 10.1189/jlb.3MR0115-011R
7. Matharu NM, Rainger GE, Vohra R, Nash GB. Effects of Disturbed Flow on Endothelial Cell Function: Pathogenic Implications of Modified Leukocyte Recruitment. Biorheology (2006) 43(1):31–44.
8. Nash GB, Buckley CD, Ed Rainger G. The Local Physicochemical Environment Conditions the Proinflammatory Response of Endothelial Cells and Thus Modulates Leukocyte Recruitment. FEBS Lett (2004) 569(1-3):13–7. doi: 10.1016/j.febslet.2004.05.040
9. Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, et al. Integration of Flow-Dependent Endothelial Phenotypes by Kruppel-like Factor 2. J Clin Invest (2006) 116(1):49–58. doi: 10.1172/JCI24787
10. Nourshargh S, Alon R. Leukocyte Migration Into Inflamed Tissues. Immunity (2014) 41(5):694–707. doi: 10.1016/j.immuni.2014.10.008
11. Herter J, Zarbock A. Integrin Regulation During Leukocyte Recruitment. J Immunol (2013) 190(9):4451–7. doi: 10.4049/jimmunol.1203179
12. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the Site of Inflammation: The Leukocyte Adhesion Cascade Updated. Nat Rev Immunol (2007) 7(9):678–89. doi: 10.1038/nri2156
13. Rabinovich GA, Liu F-T, Hirashima M, Anderson A. An Emerging Role for Galectins in Tuning the Immune Response: Lessons From Experimental Models of Inflammatory Disease, Autoimmunity and Cancer. Scand J Immunol (2007) 66(2-3):143–58. doi: 10.1111/j.1365-3083.2007.01986.x
14. Cooper D, Iqbal AJ, Gittens BR, Cervone C, Perretti M. The Effect of Galectins on Leukocyte Trafficking in Inflammation: Sweet or Sour? Ann N Y Acad Sci (2012) 1253:181–92. doi: 10.1111/j.1749-6632.2011.06291.x
15. Nabi IR, Shankar J, Dennis JW. The Galectin Lattice at a Glance. J Cell Sci (2015) 128(13):2213–9. doi: 10.1242/jcs.151159
16. Consortium TU. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res (2020) 49(D1):D480–D9. doi: 10.1093/nar/gkaa1100
17. Rabinovich GA, Toscano MA. Turning ‘Sweet’ on Immunity: Galectin-Glycan Interactions in Immune Tolerance and Inflammation. Nat Rev Immunol (2009) 9(5):338–52. doi: 10.1038/nri2536
18. Camby I, Le Mercier M, Lefranc F, Kiss R. Galectin-1: A Small Protein With Major Functions. Glycobiology (2006) 16(11):137R–57R. doi: 10.1093/glycob/cwl025
19. Hirabayashi J, Hashidate T, Arata Y, Nishi N, Nakamura T, Hirashima M, et al. Oligosaccharide Specificity of Galectins: A Search by Frontal Affinity Chromatography. Biochim Biophys Acta (2002) 1572(2-3):232–54. doi: 10.1016/S0304-4165(02)00311-2
20. Modenutti CP, Capurro JIB, Di Lella S, Martí MA. The Structural Biology of Galectin-Ligand Recognition: Current Advances in Modeling Tools, Protein Engineering, and Inhibitor Design. Front Chem (2019) 7:823. doi: 10.3389/fchem.2019.00823
21. Nielsen MI, Stegmayr J, Grant OC, Yang Z, Nilsson UJ, Boos I, et al. Galectin Binding to Cells and Glycoproteins With Genetically Modified Glycosylation Reveals Galectin–Glycan Specificities in a Natural Context. J Biol Chem (2018) 293(52):20249–62. doi: 10.1074/jbc.RA118.004636
22. Thijssen VL, Hulsmans S, Griffioen AW. The Galectin Profile of the Endothelium: Altered Expression and Localization in Activated and Tumor Endothelial Cells. Am J Pathol (2008) 172(2):545–53. doi: 10.2353/ajpath.2008.070938
23. He J, Baum LG. Endothelial Cell Expression of Galectin-1 Induced by Prostate Cancer Cells Inhibits T-cell Transendothelial Migration. Lab Invest (2006) 86(6):578–90. doi: 10.1038/labinvest.3700420
24. Cattaneo V, Tribulatti MV, Carabelli J, Carestia A, Schattner M, Campetella O. Galectin-8 Elicits Pro-Inflammatory Activities in the Endothelium. Glycobiology (2014) 24(10):966–73. doi: 10.1093/glycob/cwu060
25. Baum LG, Seilhamer JJ, Pang M, Levine WB, Beynon D, Berliner JA. Synthesis of an Endogeneous Lectin, Galectin-1, by Human Endothelial Cells is Up-Regulated by Endothelial Cell Activation. Glycoconj J (1995) 12(1):63–8. doi: 10.1007/BF00731870
26. Chakraborty A, Staudinger C, King SL, Erickson FC, Lau LS, Bernasconi A, et al. Galectin-9 Bridges Human B Cells to Vascular Endothelium While Programming Regulatory Pathways. J Autoimmun (2021) 117:102575. doi: 10.1016/j.jaut.2020.102575
27. Stillman BN, Hsu DK, Pang M, Brewer CF, Johnson P, Liu F-T, et al. Galectin-3 and Galectin-1 Bind Distinct Cell Surface Glycoprotein Receptors to Induce T Cell Death. J Immunol (2006) 176(2):778–89. doi: 10.4049/jimmunol.176.2.778
28. Wragg JW, Durant S, McGettrick HM, Sample KM, Egginton S, Bicknell R. Shear Stress Regulated Gene Expression and Angiogenesis in Vascular Endothelium. Microcirculation (2014) 21(4):290–300. doi: 10.1111/micc.12119
29. Zhang J, Friedman MH. Adaptive Response of Vascular Endothelial Cells to an Acute Increase in Shear Stress Magnitude. Am J Physiol Heart Circ Physiol (2012) 302(4):H983–H91. doi: 10.1152/ajpheart.00168.2011
30. Cunningham KS, Gotlieb AI. The Role of Shear Stress in the Pathogenesis of Atherosclerosis. Lab Invest (2005) 85(1):9–23. doi: 10.1038/labinvest.3700215
31. Mura M, Swain RK, Zhuang X, Vorschmitt H, Reynolds G, Durant S, et al. Identification and Angiogenic Role of the Novel Tumor Endothelial Marker CLEC14A. Oncogene (2012) 31(3):293–305. doi: 10.1038/onc.2011.233
32. Maurya MR, Gupta S, Li JY-S, Ajami NE, Chen ZB, Shyy JY-J, et al. Longitudinal Shear Stress Response in Human Endothelial Cells to Atheroprone and Atheroprotective Conditions. Proc Natl Acad Sci (2021) 118(4):e2023236118. doi: 10.1073/pnas.2023236118
33. Ishikawa A, Imaizumi T, Yoshida H, Nishi N, Nakamura T, Hirashima M, et al. Double-Stranded RNA Enhances the Expression of Galectin-9 in Vascular Endothelial Cells. Immunol Cell Biol (2004) 82(4):410–4. doi: 10.1111/j.0818-9641.2004.01248.x
34. Imaizumi T, Kumagai M, Sasaki N, Kurotaki H, Mori F, Seki M, et al. Interferon-γ Stimulates the Expression of Galectin-9 in Cultured Human Endothelial Cells. J Leukoc Biol (2002) 72(3):486–91. doi: 10.1189/jlb.72.3.486
35. Ayona D, Fournier PE, Henrissat B, Desnues B. Utilization of Galectins by Pathogens for Infection. Front Immunol (2020) 11:1877. doi: 10.3389/fimmu.2020.01877
36. Rao SP, Wang Z, Zuberi RI, Sikora L, Bahaie NS, Zuraw BL, et al. Galectin-3 Functions as an Adhesion Molecule to Support Eosinophil Rolling and Adhesion Under Conditions of Flow. J Immunol (2007) 179(11):7800–7. doi: 10.4049/jimmunol.179.11.7800
37. Hara A, Niwa M, Noguchi K, Kanayama T, Niwa A, Matsuo M, et al. Galectin-3 as a Next-Generation Biomarker for Detecting Early Stage of Various Diseases. Biomolecules (2020) 10(3):389. doi: 10.3390/biom10030389
38. Chen C, Duckworth CA, Zhao Q, Pritchard DM, Rhodes JM, Yu L-G. Increased Circulation of Galectin-3 in Cancer Induces Secretion of Metastasis-Promoting Cytokines From Blood Vascular Endothelium. Clin Cancer Res (2013) 19(7):1693–704. doi: 10.1158/1078-0432.CCR-12-2940
39. Thijssen VL, Barkan B, Shoji H, Aries IM, Mathieu V, Deltour L, et al. Tumor Cells Secrete Galectin-1 to Enhance Endothelial Cell Activity. Cancer Res (2010) 70(15):6216–24. doi: 10.1158/0008-5472.CAN-09-4150
40. Vasudev NS, Reynolds AR. Anti-Angiogenic Therapy for Cancer: Current Progress, Unresolved Questions and Future Directions. Angiogenesis (2014) 17(3):471–94. doi: 10.1007/s10456-014-9420-y
41. Krautter F, Recio C, Hussain MT, Lezama DR, Maione F, Chimen M, et al. Characterisation of Endogenous Galectin-1 and -9 Expression in Monocyte and Macrophage Subsets Under Resting and Inflammatory Conditions. Biomed Pharmacother (2020) 130:110595. doi: 10.1016/j.biopha.2020.110595
42. Auvynet C, Moreno S, Melchy E, Coronado-Martínez I, Montiel JL, Aguilar-Delfin I, et al. Galectin-1 Promotes Human Neutrophil Migration. Glycobiology (2012) 23(1):32–42. doi: 10.1093/glycob/cws128
43. La M, Cao TV, Cerchiaro G, Chilton K, Hirabayashi J, Kasai K-I, et al. A Novel Biological Activity for Galectin-1: Inhibition of Leukocyte-Endothelial Cell Interactions in Experimental Inflammation. Am J Pathol (2003) 163(4):1505–15. doi: 10.1016/S0002-9440(10)63507-9
44. Cooper D, Norling LV, Perretti M. Novel Insights Into the Inhibitory Effects of Galectin-1 on Neutrophil Recruitment Under Flow. J Leukoc Biol (2008) 83(6):1459–66. doi: 10.1189/jlb.1207831
45. Sano H, Hsu DK, Yu L, Apgar JR, Kuwabara I, Yamanaka T, et al. Human Galectin-3 is a Novel Chemoattractant for Monocytes and Macrophages. J Immunol (2000) 165(4):2156–64. doi: 10.4049/jimmunol.165.4.2156
46. Eckardt V, Miller MC, Blanchet X, Duan R, Leberzammer J, Duchene J, et al. Chemokines and Galectins Form Heterodimers to Modulate Inflammation. EMBO Rep (2020) 21(4):e47852. doi: 10.15252/embr.201947852
47. Yamamoto H, Nishi N, Shoji H, Itoh A, Lu L-H, Hirashima M, et al. Induction of Cell Adhesion by Galectin-8 and its Target Molecules in Jurkat T-Cells. J Biochem (2007) 143(3):311–24. doi: 10.1093/jb/mvm223
48. Nieminen J, Kuno A, Hirabayashi J, Sato S. Visualization of Galectin-3 Oligomerization on the Surface of Neutrophils and Endothelial Cells Using Fluorescence Resonance Energy Transfer *. J Biol Chem (2007) 282(2):1374–83. doi: 10.1074/jbc.M604506200
49. Matsumoto R, Matsumoto H, Seki M, Hata M, Asano Y, Kanegasaki S, et al. Human Ecalectin, a Variant of Human Galectin-9, Is a Novel Eosinophil Chemoattractant Produced by T Lymphocytes *. J Biol Chem (1998) 273(27):16976–84. doi: 10.1074/jbc.273.27.16976
50. Bi S, Hong PW, Lee B, Baum LG. Galectin-9 Binding to Cell Surface Protein Disulfide Isomerase Regulates the Redox Environment to Enhance T-Cell Migration and HIV Entry. Proc Natl Acad Sci (2011) 108(26):10650–5. doi: 10.1073/pnas.1017954108
51. O’Brien MJ, Shu Q, Stinson WA, Tsou P-S, Ruth JH, Isozaki T, et al. A Unique Role for Galectin-9 in Angiogenesis and Inflammatory Arthritis. Arthritis Res Ther (2018) 20(1):31–. doi: 10.1186/s13075-018-1519-x
52. Norling LV, Sampaio AL, Cooper D, Perretti M. Inhibitory Control of Endothelial Galectin-1 on In Vitro and In Vivo Lymphocyte Trafficking. FASEB J (2008) 22(3):682–90. doi: 10.1096/fj.07-9268com
53. Iqbal AJ, Sampaio AL, Maione F, Greco KV, Niki T, Hirashima M, et al. Endogenous Galectin-1 and Acute Inflammation: Emerging Notion of a Galectin-9 Pro-Resolving Effect. Am J Pathol (2011) 178(3):1201–9. doi: 10.1016/j.ajpath.2010.11.073
54. Gittens BR, Bodkin JV, Nourshargh S, Perretti M, Cooper D. Galectin-3: A Positive Regulator of Leukocyte Recruitment in the Inflamed Microcirculation. J Immunol (Baltimore Md 1950) (2017) 198(11):4458–69. doi: 10.4049/jimmunol.1600709
55. Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients With Nonalcoholic Steatohepatitis With Cirrhosis and Portal Hypertension. Gastroenterology (2020) 158(5):1334–45.e5. doi: 10.1053/j.gastro.2019.11.296
56. Seropian IM, González GE, Maller SM, Berrocal DH, Abbate A, Rabinovich GA. Galectin-1 as an Emerging Mediator of Cardiovascular Inflammation: Mechanisms and Therapeutic Opportunities. Mediators Inflamm (2018) 2018:8696543. doi: 10.1155/2018/8696543
57. Sundblad V, Morosi LG, Geffner JR, Rabinovich GA. Galectin-1: A Jack-of-All-Trades in the Resolution of Acute and Chronic Inflammation. J Immunol (2017) 199(11):3721–30. doi: 10.4049/jimmunol.1701172
58. Wolfert MA, Boons G-J. Adaptive Immune Activation: Glycosylation Does Matter. Nat Chem Biol (2013) 9(12):776–84. doi: 10.1038/nchembio.1403
59. Joeh E, O’Leary T, Li W, Hawkins R, Hung JR, Parker CG, et al. Mapping Glycan-Mediated Galectin-3 Interactions by Live Cell Proximity Labeling. Proc Natl Acad Sci U S A (2020) 177(44):27329–38. doi: 10.1073/pnas.2009206117
60. Potter DR, Damiano ER. The Hydrodynamically Relevant Endothelial Cell Glycocalyx Observed In Vivo Is Absent In Vitro. Circ Res (2008) 102(7):770–6. doi: 10.1161/CIRCRESAHA.107.160226
Keywords: galectins, leukocyte trafficking, glycan-binding protein, endothelial cell, vascular biology
Citation: Lightfoot A, McGettrick HM and Iqbal AJ (2021) Vascular Endothelial Galectins in Leukocyte Trafficking. Front. Immunol. 12:687711. doi: 10.3389/fimmu.2021.687711
Received: 29 March 2021; Accepted: 12 May 2021;
Published: 01 June 2021.
Edited by:
Joao P.B. Viola, National Cancer Institute (INCA), BrazilReviewed by:
Roger Chammas, University of São Paulo, BrazilVinicius Cotta-de-Almeida, Oswaldo Cruz Foundation (Fiocruz), Brazil
Copyright © 2021 Lightfoot, McGettrick and Iqbal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Asif J. Iqbal, YS5qLmlxYmFsQGJoYW0uYWMudWs=