 Xiuzhi Jia
Xiuzhi Jia Chunyuan Zhao
Chunyuan Zhao Wei Zhao
Wei Zhao- Department of Pathogenic Biology, School of Basic Medical Science, Cheeloo College of Medicine, Shandong University, Jinan, China
The major histocompatibility complex (MHC) class I (MHC-I) region contains a multitude of genes relevant to immune response. Multiple E3 ubiquitin ligase genes, including tripartite motif 10 (TRIM10), TRIM15, TRIM26, TRIM27, TRIM31, TRIM38, TRIM39, TRIM40, and RING finger protein 39 (RNF39), are organized in a tight cluster, and an additional two TRIM genes (namely TRIM38 and TRIM27) telomeric of the cluster within the MHC-I region. The E3 ubiquitin ligases encoded by these genes possess important roles in controlling the intensity of innate immune responses. In this review, we discuss the E3 ubiquitin ligases encoded within the MHC-I region, highlight their regulatory roles in innate immunity, and outline their potential functions in infection, inflammatory and autoimmune diseases.
Introduction
Innate immunity is the first line of defense against invading pathogens and cancers. A variety of germline-encoded pattern recognition receptors (PRRs) recognize conserved structures present in pathogenic microorganisms (termed as pathogen-associated molecular patterns) and danger signals (termed as damage-associated molecular patterns), in turn initiating innate immune responses. The different types of PRRs include Toll-like receptors (TLRs), retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), and cytosolic DNA sensors [e.g., cyclic GMP-AMP synthase (cGAS)]. These PRRs transduce activation signals by recruiting cellular adaptors including myeloid differentiation factor 88, Toll/IL-1 receptor (TIR) domain-containing adapter inducing interferon (IFN)-β (TRIF), mitochondrial antiviral signaling protein (MAVS), and stimulator of interferon genes (STING). These activation signals then activate the transcription factors nuclear factor kappa-B (NF-κB) and interferon regulatory factor 3 (IRF3), leading to the expression of proinflammatory cytokines and type I IFNs (1–3).
Optimal activation of innate immunity is crucial for the elimination of invading pathogens and mutant cells, as well as for the maintenance of immune homeostasis. A magnitude of sophisticated strategies have been developed by our body to manipulate the intensity of innate immune response, including epigenetic regulation and post-translational modifications (PTMs) of key immune signaling adaptors (4). Ubiquitination is an important PTM that is dynamically controlled by multiple E3 ubiquitin ligases and deubiquitinases, and it has been implicated in innate immune response (5). Recently, a series of E3 ubiquitin ligases encoded within the MHC-I region (Figure 1) have been reported as important regulators of innate immunity. In this review, we will introduce the MHC-I region genes encoded E3 ubiquitin ligases, highlight their regulatory roles in innate immunity and potential functions in infection, inflammatory and autoimmune diseases.
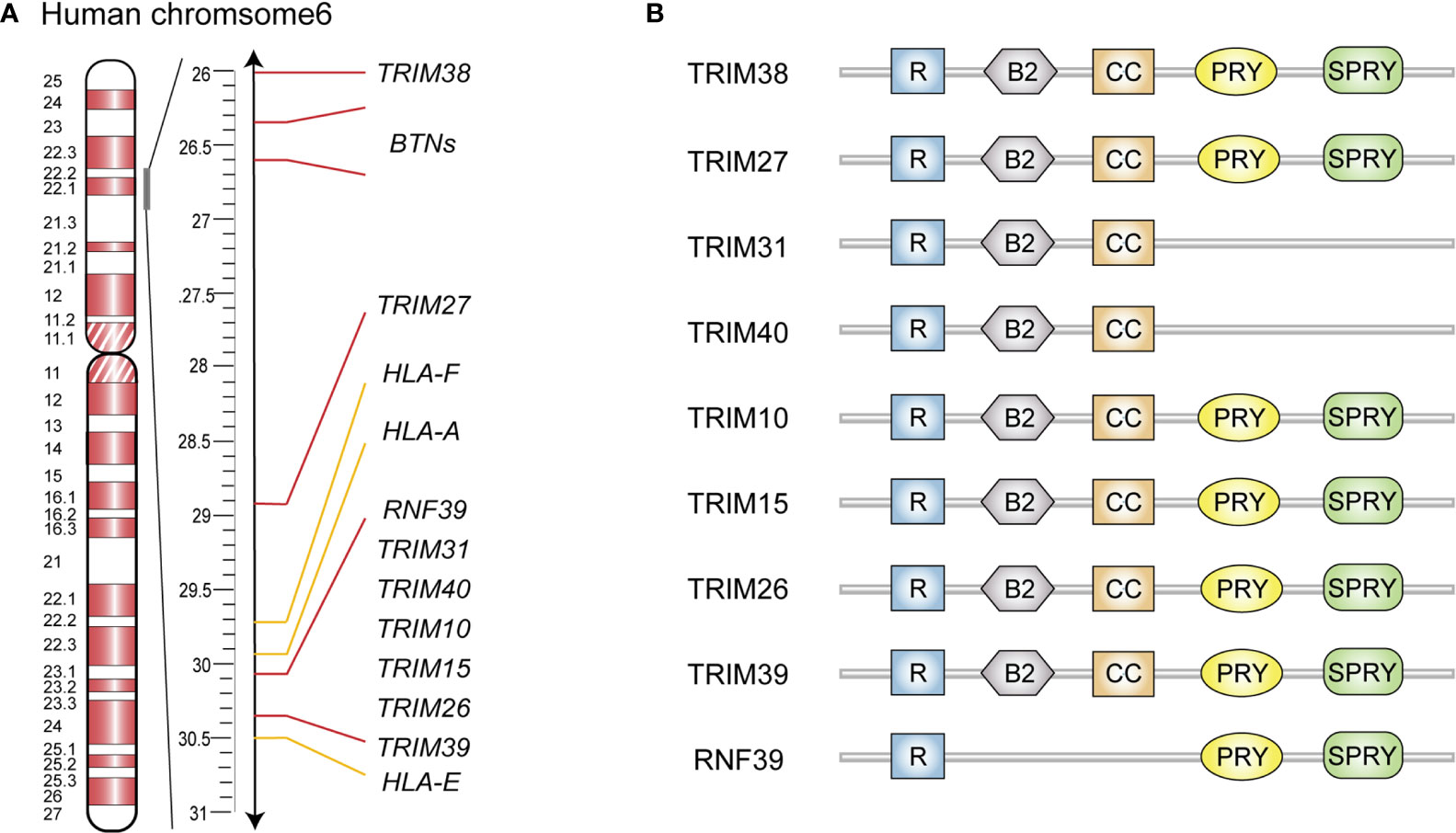
Figure 1 Schematic representation of gene clusters and structure domain of MHC-I region encoded E3 ubiquitin ligases. (A) Gene cluster of human MHC-I region. (B) Schematic diagram of structure domain of MHC-I region encoded E3 ubiquitin ligases. R, RING finger; B, B-box; CC, coiled-coil.
Cluster of E3 Ubiquitin Ligases Genes in the MHC-I Region
MHC-I region contains a large number of immune-related genes, which are often polymorphic and closely linked as a result of their genomic proximity (6). In addition, many of these genes are associated with infections and autoimmune diseases, such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) (6). The human MHC-I genomic region locates on chromosome 6p21.33-6p22.2, known as human leukocyte antigen (HLA) class I region which contains HLA gene loci and several non-HLA gene clusters (7). Several E3 ubiquitin ligase genes in this region are organized in a tight cluster from HLA-E to HLA-A (Figure 1A), and they comprise six TRIM family members (including TRIM10, TRIM15, TRIM26, TRIM31, TRIM39 and TRIM40) as well as RNF39 (8). Two additional TRIM genes (namely TRIM27 and TRIM38) are found telomeric from this cluster and near to the butyrophilin genes (8). In the mouse genome, Trim10, Trim15, Trim26, Trim31, Trim39, Trim40 and Rnf39 are located on chromosome 17 in the B1 region within the histocompatibility(H)2-I genetic group, while Trim27 and Trim38, are located on chromosome 13 in the A3.1 region.
Structure of MHC-I Region-Encoded E3 Ubiquitin Ligases
Most MHC-I region encoded E3 ubiquitin ligases are members of the TRIM protein family and possess RING finger, B-box 2, and coiled-coil (CC) domain (Figure 1B) (9, 10). The N-terminal RING finger domain confers the E3 ubiquitin ligase activity, which is essential for TRIMs to exert their antiviral effects and regulate innate immune signaling pathways (11). Specifically, the RING domain functions by recognizing E2 ubiquitin-conjugating enzymes via zinc finger motifs, subsequently transferring the ubiquitins or ubiquitin-like proteins to their substrates (11). The second signature sequence of MHC-I region encoded TRIM proteins is the B-box 2 domain, which also exhibits zinc-finger motifs similar to RING domain. Currently, the unified function of B-box 2 remains unclear, but there is evidence that it can potentiate the ability of TRIM5α to mediate human immunodeficiency virus 1 (HIV-1) restriction; lead to higher-order self-assembly of TRIM5α; and offer an E2 binding site resembling RING, which endows E3 ligase activity in some TRIMs lacking a RING domain (12–14). Following the B-box 2 domain is the CC domain, a typical hyper-secondary structure that can assemble with other CC structures to mediate homo- or hetero- oligomeric interactions among TRIM proteins (15). This oligomerization promotes the generation of high-molecular-mass complexes that are compartmentalized either in distinct cellular compartments such as nuclear bodies (PML/TRIM19) or microtubules (MID1/TRIM18) (16). In addition, structural analyses of several TRIM CC dimers have indicated that they are formed by antiparallel dimeric architecture, which places the RING and B-box domains on opposite sides of the CC domain. This architecture permits the dimerization of RING domains in some cases, endowing E3 ligase activity to some TRIM proteins (15). Furthermore, TRIM10, TRIM15, TRIM26, TRIM27, TRIM38, TRIM39 and RNF39 share a C-terminal PRY-SPRY domain (Figure 1B) that binds with high specificity to a diverse set of substrates, including peptides and proteins, and even RNA molecules (17, 18).
Emerging Roles of MHC-I Region Encoded E3 Ubiquitin Ligases in Innate Immunity
Increasing evidences indicate that the MHC-I region encoded E3 ubiquitin ligase genes possess moderate levels of polymorphism, play regulatory roles in innate immune responses (Figure 2), and their expressions are associated with a variety of autoimmune diseases (8).
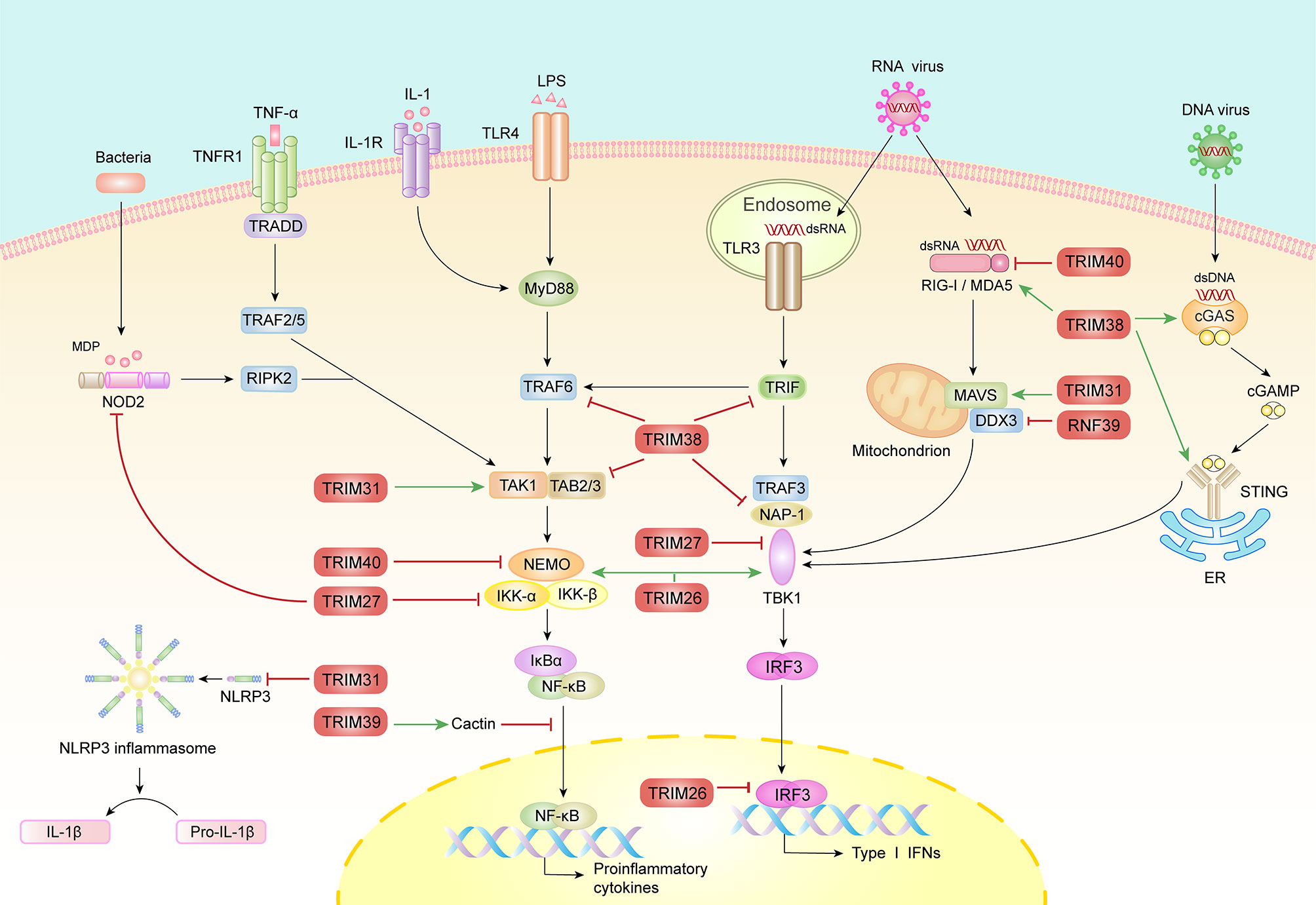
Figure 2 The roles of MHC-I region encoded E3 ubiquitin ligases in innate immunity. Multiple PRRs including TLRs, RLRs, NLRs and DNA sensors detect the invasion of pathogen and trigger downstream complex signaling pathways that culminate in the activation of transcription factor, IRF3 and NF-κB, resulting in the induction of type I IFNs and proinflammatory factors. Conversely, inflammatory cytokines, such as TNFα and IL-1β further active NF-κB signaling pathway. During these processes, E3 ubiquitin ligases catalyze diverse molecules including polyubiquitin, SUMO and Nedd8 to adaptors to ensure optimal activation or timely turned-off of signals. This figure has displayed the positive (green arrows) and negative (red lines) function of the MHC-I region encoded E3 ubiquitin ligases in PRR-mediated innate immune signaling pathways.
TRIM38
TRIM38 may be involved in the development of various autoimmune diseases and generally in the innate immune response. In one study, the presence of autoantibodies to TRIM38 significantly correlated with disease severity in patients with primary Sjögren’s syndrome (SS, a disease in which circulating autoantibodies react with multiple cellular proteins to cause glandular dysfunction) (19). Similarly, in patients with dermatomyositis (DM, an autoimmune connective tissue disease characterized by erythema in the eyes and hands, and weakness in the proximal muscles), skin and muscle biopsy analyses showed that TRIM38 gene expression was upregulated (20). In fact, TRIM38 is an intriguing regulator of innate immunity (21). As a negative regulator, TRIM38 mediates lysine 48 (K48)-linked polyubiquitination of TNF receptor associated factor 6, TRIF and NF-κB-activating kinase-associated protein 1 to promote their proteasomal degradation, resulting in the inhibition of TLR and RLR pathways (22–24). In addition, TRIM38 facilitates the lysosome-dependent degradation of TAK1-binding protein 2 (TAB2) in TNF- and IL-1β-triggered signaling, independent of its E3 ubiquitin ligase activity; however, the specific mechanism remains to be explored (25). Notably, the interaction between TRIM38 and TAB2/3 is weakened in RA, resulting in an excess expression of TAB2/3 and proinflammatory cytokines, indicating the essential roles of TRIM38 in modulating autoimmune disease severity (26).
Besides its E3 ubiquitin ligase activity, TRIM38 has also been identified as an E3 small ubiquitin-like modifier (SUMO) ligase and mediates the SUMOylation of RIG-I and MDA5 (27). First, TRIM38 catalyzes the SUMOylation of RIG-I and MDA5, subsequently inhibiting K48-linked ubiquitination and degradation through steric hindrance both in rest and infection states (27). Second, this SUMOylation of RIG-I and MDA5 at K96/K889 and K43/K865 mediated by TRIM38 facilitates PP1-mediated dephosphorylation and K63-linked polyubiquitination of RIG-I/MDA5, leading to RLRs activation (27). Third, the SUMOylation of RIG-I/MDA5 impaired and the K48-linked ubiquitination increased in the late phase of viral infection, restricting both the intensity and duration of RLRs activation (27). This may also explain why in a different study, Enterovirus 71, another RNA virus, escaped immune surveillance through promoting degradation of TRIM38 (28). In DNA-sensing signaling pathways, TRIM38 also regulates the activation and expression of both cGAS and STING by mediating their SUMOylation by different mechanisms (29). Thus, TRIM38 may be the key in establishing an efficient antiviral state in the early phase of viral infection, while also terminating the activation of RLRs and cGAS/STING in the late phase. These results indicate that TRIM38 is a multifunctional molecule in innate immunity, and even affects the same pathway through different mechanisms (such as RLRs).
TRIM27
TRIM27, also known as RET finger protein, plays important regulatory roles in innate immune responses; its dysregulation may cause several inflammatory diseases. TRIM27 binds to NOD2 via its PRY-SPRY domain, subsequently promoting K48-linked polyubiquitination and degradation of NOD2 and leading to the inhibition of NF-κB signal (30). An immunohistological inspection indicated that when compared to healthy tissue, TRIM27 expression appeared higher in tissue derived from patients with Crohn disease (CD), implying that TRIM27 has a role in CD, which is a known NOD2-related inflammatory disease (30). Additionally, in a model of hepatic ischemia-reperfusion injury (IRI, which initiates from oxidative stress and inflammation caused by insufficient blood supply and subsequent reperfusion owing to trauma, resection or transplantation of the liver), TRIM27 alleviated liver damage and inflammation by suppressing the recruitment of TAK1 via TAB2/3 degradation (31). Moreover, TRIM27 interacts with multiple IKKs, including IKKα, IKKβ, IKKϵ and TBK1, to attenuate both NF-κB and IRF triggered IFN-β expression (32). Furthermore, TRIM27 promotes IL-6-induced STAT3 activation by mediating ubiquitination of protein inhibitor of activated STAT3, thereby aggravating psoriasis (a chronic inflammatory disease that predominantly affects the skin and joints), colitis, and colitis-associated cancer (33, 34). In addition, it has been reported that TRIM27 is a host restriction factor that suppresses the survival of Mycobacterium tuberculosis in macrophages by upregulating the NF-κB and JNK/p38 pathways (35). Another study showed that TRIM27 could serve as a potential biomarker for discriminating active tuberculosis from latent tuberculosis infection and healthy people (36).
Recently, TRIM27 was reported to promote K48-linked ubiquitination at Lys251/372 and protein degradation of TBK1 during Vesicular stomatitis virus (VSV) infection (37). Furthermore, TRIM27 facilitates hepatitis C virus (HCV) replication through inhibiting IRF3 and NF-κB pathway (38). However, TRIM27 possesses contrary roles in Herpes simplex virus type 1 (HSV-1), HIV-1 and N-tropic murine leukemia virus (N-MLV)-infected cells; the contrary effects of TRIM27 may be related to varying effects of TRIM27 upon the lifecycles of different viruses (39, 40). While TRIM27 may regulate different innate immune pathways to control a type I IFN response during RNA virus and DNA virus infection, its effect during retrovirus infection may depend on direct interaction with viral components. Moreover, considering its vital role in regulating host antiviral immune responses, TRIM27 turn out to be the evasion target of HSV-1. The ICP0 protein of HSV-1, which has its own RING domain and E3 ubiquitin ligase activity, has been shown to interact with TRIM27 to promote its polyubiquitination and degradation (41). These contradictory results suggest complex roles of TRIM27 in innate immunity.
TRIM31
Single nucleotide polymorphisms (SNPs) of TRIM31, also called hemochromatosis candidate gene I, are associated with a variety of inflammatory diseases including inflammatory bowel disease (IBD, a chronic nonspecific inflammatory disease characterized by recurrent inflammation of the intestinal mucosa, comprising two main distinctive entities, ulcerative colitis and CD) and irritant contact dermatitis (ICD, a kind of skin inflammatory reaction after contact with exogenous irritants) (42, 43). Additionally, TRIM31 expression correlates with reduced risk of nasopharyngeal carcinoma associated with Epstein–Barr virus infection (44). These evidences suggest the potential roles of TRIM31 in antiviral immune responses and autoimmune disorders. A confirmatory study identified that TRIM31 suppressed NLR family pyrin domain containing 3 (NLRP3) inflammasome activation by promoting K48-linked ubiquitination and proteasomal degradation of NLRP3 (45). The study also showed that Trim31 deficiency aggravates alum-induced peritonitis and attenuates the severity of DSS-induced colitis (45). Furthermore, TRIM31 reduces the risk of other NLRP3 inflammasome-associated diseases such as apical periodontitis (AP, an acute suppurative inflammation caused by endodontic microbial infections) and age-related macular degeneration (AMD, a major cause of blindness in the elderly in developed countries, induced by dysfunction of retinal pigment epithelial cells, which constitute the immune defense barrier of the macula) (46, 47). In addition, TRIM31 is involved in the development of sepsis and colorectal cancer through regulating the NF-κB signaling pathway (48, 49).
In addition to its role in inflammation, TRIM31 has been shown to modify multiple MAVS sites with K63-linked polyubiquitin, leading to its aggregation and thus the enhancement of IFN-I expression upon RNA virus infection (50). The PB1-F2 protein of avian influenza A (H7N9) virus, scaffold protein FAF1 and Rho family small guanosine triphosphatase Rac1 limit the interaction between MAVS and TRIM31, resulting in the inhibition of MAVS ubiquitination, aggregation, and activation (51–53). TRIM31 also triggers ubiquitination and degradation of the hepatitis B virus (HBV) component HBx and therefore plays a potential role in IFN-resistant HBV infection (54). Furthermore, while TRIM31 can inhibit HIV-1 entry, downregulation of endogenously expressed TRIM 31 inhibits both HIV-1 and MLV release, suggesting that TRIM31 plays different roles at early and late stages of the retroviral lifecycle (40). Overall, these findings provide the possibility of TRIM31 as a potential antiviral drug target.
TRIM40
Genome-wide association studies show strong association between genetic variants of TRIM40 and common diseases. For example, one of TRIM40 SNPs, rs757262, can balance the risk of developing different autoimmune diseases (55). Accordingly, several functional studies indicate that TRIM40 is a regulator of innate immunity. TRIM40 physically combines with Nedd8 to promote the neddylation of IKKγ, thereby preventing gastrointestinal neoplasia caused by chronic inflammation; reduces inflammation and liver injury in septic mice via attenuating the activation of TLR4 pathway; and suppresses RLRs pathway by promoting both K27- and K48-linked polyubiquitination of RIG-I and MDA5, thus enhancing their proteasomal degradation (56–58). As RIG-I and MDA5 are associated with autoimmune diseases including Aicardi-Goutières syndrome (AGS, a severe autoimmune encephalopathy caused by aberrant activation of the IFN-I axis), Singleton–Merten syndrome(SMS, a type I interferonopathy characterized by aortic calcifications, psoriasis, glaucoma and skeletal abnormalities) and type 1 diabetes (T1D), TRIM40 may attenuate the pathogenesis of autoimmune diseases by regulating the activation of RIG-I and MDA5 (59–61).
TRIM26
TRIM26, also called RNF95 or ZNF173, exhibits polymorphisms associated with several autoimmune disorders including T1D and multiple sclerosis (MS, an autoimmune disease characterized by inflammatory demyelination of the central nervous system) and with nasopharyngeal carcinoma caused by viral disease (62–64). In antiviral immunity, TRIM26 mediates the K48-linked polyubiquitination and protein degradation of nuclear IRF3, attenuating the antiviral response (65). However, another study reported that TRIM26 actually enhanced innate immunity against RNA viruses, by recruiting NEMO to facilitate the interaction between TBK1 and MAVS (66). Furthermore, a genome-wide CRISPR-Cas9 screening identified TRIM26 as a critical HCV host factor, where it mediates K27-linked ubiquitination of HCV-encoded NS5B protein, enhances the interaction between NS5B-NS5A, and ultimately promotes HCV genome replication (67). Thus, as a key E3 ubiquitin ligase, TRIM26 plays multiple roles through catalyzing the conjugation of multiple ubiquitin chains to variety of substrates.
RNF39
Emerging evidences indicate that RNF39 is a potential immune regulator. Genetic variants of RNF39 are associated with a variety of viral diseases and autoimmune diseases, such as the progression of HIV-1 and Behcet’s disease (BD, a chronic systemic vasculitis resulting in ulcerative in the oral cavity and on the genitals, as well as inflammatory damage of the eyes) (68). The DNA methylation state of RNF39 impacts autoimmune disorders, including MS, SLE and allergic rhinitis (AR, a delayed hypersensitivity nasal mucosa reaction to environmental allergens, caused by IgE mediated release of autacoids) and confers poor responsiveness to HBV vaccination (69–72). Furthermore, RNF39 genetic variants are related to HIV-1 plasma viral loads, CD4+ T cell count, and the clinical course of HIV-1 infection (73–75). Besides its role in DNA viruses and retrovirus infection, RNF39 has also been identified as a feedback suppressor of RNA virus-induced signaling and antiviral immunity. RNF39 mediates K48-linked polyubiquitination and proteasomal degradation of DDX3X, a scaffold vital for the formation of the MAVS-TRAF3 complex, to subsequently inhibit the RLR pathway (76). Rnf39 deficiency enhances RLR activation and inhibits RNA viral replication (76). Further studies will identify more immune targets of RNF39, which will help to explain the regulatory roles of RNF39 in antiviral and autoimmune responses.
Other MHC-I Region Encoded E3 Ubiquitin Ligases
Several other MHC-I region-encoded E3 ubiquitin ligases have also been reported to regulate viral infection and innate immunity. SNPs in TRIM15 show a correlation with lupus nephritis (LN, an autoimmune disease characterized by the hypersecretion of autoantibodies and deposition of immune complexes in the kidneys); this correlation varies significantly according to ethnicity (77). Furthermore, human TRIM15 interferes with the release of HIV-1 and MLV from cells (40). TRIM15 targets adaptors upstream of MAVS to potentiate RIG-I-mediated IFN-β production and suppress viral infection (78). TRIM39 stabilizes Cactin to attenuate TLR- and TNF-α-mediated RelA/p65 translocation, inhibiting the expression of IL-6 and IL-8 (79). SNP analyses indicate that the genetic variants of TRIM39 are also associated with inflammatory diseases including psoriasis, and autoimmune diseases including BD and cutaneous lupus erythematosus (CLE, an autoimmune connective tissue disease with cutaneous lesion) (68, 80, 81). Another study found a differentially methylated site in the promoter region of TRIM39-RPP2 that was associated with IBD patients (82). Lastly, TRIM39 also regulates the type I IFN response to exert its antiviral functions (83). TRIM10 contributes to the restriction of HIV-1 entry and shows high correlation with the risk of developing MS, RA, LN, ankylosing spondylitis (AS, a chronic autoimmune-mediated arthritis that predominantly affects the axial skeleton and peripheral joints), autoimmune thyroid disease (AITD, various conditions caused by autoantibodies attacking the thyroid, including Hashimoto’s thyroiditis and Graves’ disease) and T1D, although its exact functions in these conditions remain unknown (40, 77, 84). In summary, the detailed mechanisms of action of these lesser-studied MHC-I region-encoded E3 ubiquitin ligases (including TRIM10, TRIM15 and TRIM39) need to be further investigated.
Conclusion and Perspective
Many MHC-I region-encoded E3 ubiquitin ligases are highly polymorphic, participate in the regulation of inflammation and antiviral innate immunity, and play a key role in the interaction between virus and host. At present, the known mechanisms of action include ubiquitination and SUMOylation; however, more mechanisms will be elucidated with further study. Concordantly, these MHC-I region genes are related to a variety of inflammatory, viral and autoimmune diseases. Nonetheless, their exact roles in the development of these diseases remain largely unclarified. In addition, same E3 ubiquitin ligases may play contrary roles in different inflammatory diseases. So, how does the organism regulate the action of the same MHC-I region-encoded E3 ubiquitin ligase (such as TRIM26 and TRIM38) to exert multiple or even opposing activities in different pathways? Further studies with varied cell types and disease models using gene-deficient mice are required to answer these questions. Given the important role of MHC-I region-encoded E3 ubiquitin ligases in antiviral innate immunity, it is tempting to speculate that viruses have evolved immune escape mechanisms by manipulating TRIM proteins. In addition, as some viruses are known to participate in oncogenesis or cancer progression, whether these E3 ubiquitin ligases could regulate the development of virus-related cancer deserves further investigation.
In this review, we have focused on the roles of MHC-I region-encoded E3 ubiquitin ligases in controlling the intensity of innate immune responses; however, whether these MHC-I region-encoded E3 ubiquitin ligases could modulate adaptive immunity requires further investigation. Elucidating their specific functions and molecular details in immune regulation will help us to further understand the vital roles of MHC-I region genes in immunity and provide promising diagnostic and therapeutic targets for diseases characterized by aberrant activation of innate immunity.
Author Contributions
XJ drafted the manuscript and figures, CZ and WZ edited the manuscript, WZ supervised and edited the figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81622030, 31870866 and 81901609). WZ is a Newton Advanced Fellow awarded by the Academy of Medical Sciences.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Fitzgerald KA, Kagan JC. Toll-Like Receptors and the Control of Immunity. Cell (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
2. Rehwinkel J, Gack MU. Rig-I-like Receptors: Their Regulation and Roles in RNA Sensing. Nat Rev Immunol (2020) 20:537–51. doi: 10.1038/s41577-020-0288-3
3. Zhang X, Bai XC, Chen ZJ. Structures and Mechanisms in the Cgas-STING Innate Immunity Pathway. Immunity (2020) 53:43–53. doi: 10.1016/j.immuni.2020.05.013
4. Zhang Q, Cao X. Epigenetic Remodeling in Innate Immunity and Inflammation. Annu Rev Immunol (2021) 39:279–311. doi: 10.1146/annurev-immunol-093019-123619
5. Liu J, Qian C, Cao X. Post-Translational Modification Control of Innate Immunity. Immunity (2016) 45:15–30. doi: 10.1016/j.immuni.2016.06.020
6. Al Naqbi H, Mawart A, Alshamsi J, Al Safar H, Tay GK. Major Histocompatibility Complex (MHC) Associations With Diseases in Ethnic Groups of the Arabian Peninsula. Immunogenetics (2021) 73:131–52. doi: 10.1007/s00251-021-01204-x
7. Ruddy DA, Kronmal GS, Lee VK, Mintier GA, Quintana L, Domingo R Jr, et al. A 1.1-Mb Transcript Map of the Hereditary Hemochromatosis Locus. Genome Res (1997) 7:441–56. doi: 10.1101/gr.7.5.441
8. Meyer M, Gaudieri S, Rhodes DA, Trowsdale J. Cluster of TRIM Genes in the Human MHC Class I Region Sharing the B30.2 Domain. Tissue Antigens (2003) 61:63–71. doi: 10.1034/j.1399-0039.2003.610105.x
9. Nisole S, Stoye JP, Saïb A. TRIM Family Proteins: Retroviral Restriction and Antiviral Defence. Nat Rev Microbiol (2005) 3:799–808. doi: 10.1038/nrmicro1248
10. van Gent M, Sparrer K, Gack MU. Trim Proteins and Their Roles in Antiviral Host Defenses. Annu Rev Virol (2018) 5:385–405. doi: 10.1146/annurev-virology-092917-043323
11. Joazeiro CA, Weissman AM. RING Finger Proteins: Mediators of Ubiquitin Ligase Activity. Cell (2000) 102:549–52. doi: 10.1016/s0092-8674(00)00077-5
12. Diaz-Griffero F, Qin XR, Hayashi F, Kigawa T, Finzi A, Sarnak Z, et al. A B-box 2 Surface Patch Important for TRIM5alpha Self-Association, Capsid Binding Avidity, and Retrovirus Restriction. J Virol (2009) 83:10737–51. doi: 10.1128/JVI.01307-09
13. Wagner JM, Roganowicz MD, Skorupka K, Alam SL, Christensen D, Doss G, et al. Mechanism of B-box 2 Domain-Mediated Higher-Order Assembly of the Retroviral Restriction Factor TRIM5α. Elife (2016) 5:e16309. doi: 10.7554/eLife.16309
14. Bell JL, Malyukova A, Holien JK, Koach J, Parker MW, Kavallaris M, et al. TRIM16 Acts as an E3 Ubiquitin Ligase and can Heterodimerize With Other TRIM Family Members. PloS One (2012) 7:e37470. doi: 10.1371/journal.pone.0037470
15. Esposito D, Koliopoulos MG, Rittinger K. Structural Determinants of TRIM Protein Function. Biochem Soc Trans (2017) 45:183–91. doi: 10.1042/BST20160325
16. Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, et al. The Tripartite Motif Family Identifies Cell Compartments. EMBO J (2001) 20:2140–51. doi: 10.1093/emboj/20.9.2140
17. James LC, Keeble AH, Khan Z, Rhodes DA, Trowsdale J. Structural Basis for PRYSPRY-mediated Tripartite Motif (TRIM) Protein Function. Proc Natl Acad Sci USA (2007) 104:6200–5. doi: 10.1073/pnas.0609174104
18. Choudhury NR, Heikel G, Trubitsyna M, Kubik P, Nowak JS, Webb S, et al. RNA-Binding Activity of TRIM25 is Mediated by its PRY/SPRY Domain and is Required for Ubiquitination. BMC Biol (2017) 15:105. doi: 10.1186/s12915-017-0444-9
19. Wolska N, Rybakowska P, Rasmussen A, Brown M, Montgomery C, Klopocki A, et al. Brief Report: Patients With Primary Sjögren’s Syndrome Who Are Positive for Autoantibodies to Tripartite Motif-Containing Protein 38 Show Greater Disease Severity. Arthritis Rheumatol (2016) 68:724–9. doi: 10.1002/art.39497
20. Aljabban J, Syed S, Syed S, Rohr M, Weisleder N, McElhanon KE, et al. Investigating Genetic Drivers of Dermatomyositis Pathogenesis Using Meta-Analysis. Heliyon (2020) 6:e04866. doi: 10.1016/j.heliyon.2020.e04866
21. Hu MM, Shu HB. Multifaceted Roles of TRIM38 in Innate Immune and Inflammatory Responses. Cell Mol Immunol (2017) 14:331–8. doi: 10.1038/cmi.2016.66
22. Xue Q, Zhou Z, Lei X, Liu X, He B, Wang J, et al. TRIM38 Negatively Regulates TLR3-mediated Ifn-β Signaling by Targeting TRIF for Degradation. PloS One (2012) 7:e46825. doi: 10.1371/journal.pone.0046825
23. Zhao W, Wang L, Zhang M, Wang P, Yuan C, Qi J, et al. Tripartite Motif-Containing Protein 38 Negatively Regulates TLR3/4- and RIG-I-mediated Ifn-β Production and Antiviral Response by Targeting NAP1. J Immunol (2012) 188:5311–8. doi: 10.4049/jimmunol.1103506
24. Zhao W, Wang L, Zhang M, Yuan C, Gao C. E3 Ubiquitin Ligase Tripartite Motif 38 Negatively Regulates TLR-mediated Immune Responses by Proteasomal Degradation of TNF Receptor-Associated Factor 6 in Macrophages. J Immunol (2012) 188:2567–74. doi: 10.4049/jimmunol.1103255
25. Hu MM, Yang Q, Zhang J, Liu SM, Zhang Y, Lin H, et al. TRIM38 Inhibits Tnfα- and IL-1β-Triggered NF-κb Activation by Mediating Lysosome-Dependent Degradation of TAB2/3. Proc Natl Acad Sci USA (2014) 111:1509–14. doi: 10.1073/pnas.1318227111
26. Lin Y, Luo Z. NLRP6 Facilitates the Interaction Between TAB2/3 and TRIM38 in Rheumatoid Arthritis Fibroblast-Like Synoviocytes. FEBS Lett (2017) 591:1141–9. doi: 10.1002/1873-3468.12622
27. Hu MM, Liao CY, Yang Q, Xie XQ, Shu HB. Innate Immunity to RNA Virus is Regulated by Temporal and Reversible SUMOylation of RIG-I and MDA5. J Exp Med (2017) 214:973–89. doi: 10.1084/jem.20161015
28. Liu X, Lei X, Zhou Z, Sun Z, Xue Q, Wang J, et al. Enterovirus 71 Induces Degradation of TRIM38, a Potential E3 Ubiquitin Ligase. Virol J (2011) 8:61. doi: 10.1186/1743-422X-8-61
29. Hu MM, Yang Q, Xie XQ, Liao CY, Lin H, Liu TT, et al. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity (2016) 45:555–69. doi: 10.1016/j.immuni.2016.08.014
30. Zurek B, Schoultz I, Neerincx A, Napolitano LM, Birkner K, Bennek E, et al. TRIM27 Negatively Regulates NOD2 by Ubiquitination and Proteasomal Degradation. PloS One (2012) 7:e41255. doi: 10.1371/journal.pone.0041255
31. Chen SY, Zhang HP, Li J, Shi JH, Tang HW, Zhang Y, et al. Tripartite Motif-Containing 27 Attenuates Liver Ischemia/Reperfusion Injury by Supressing Transforming Growth Factor β-Activated Kinase 1 (TAK1) by TAK1 Binding Protein 2/3 Degradation. Hepatology (2020) 73:738–58. doi: 10.1002/hep.31295
32. Zha J, Han KJ, Xu LG, He W, Zhou Q, Chen D, et al. The Ret Finger Protein Inhibits Signaling Mediated by the Noncanonical and Canonical IkappaB Kinase Family Members. J Immunol (2006) 176:1072–80. doi: 10.4049/jimmunol.176.2.1072
33. Zhang HX, Xu ZS, Lin H, Li M, Xia T, Cui K, et al. TRIM27 Mediates STAT3 Activation At Retromer-Positive Structures to Promote Colitis and Colitis-Associated Carcinogenesis. Nat Commun (2018) 9:3441. doi: 10.1038/s41467-018-05796-z
34. Miao X, Xiang Y, Mao W, Chen Y, Li Q, Fan B. TRIM27 Promotes IL-6-induced Proliferation and Inflammation Factor Production by Activating STAT3 Signaling in HaCaT Cells. Am J Physiol Cell Physiol (2020) 318:C272–81. doi: 10.1152/ajpcell.00314.2019
35. Wang J, Teng JL, Zhao D, Ge P, Li B, Woo PC, et al. The Ubiquitin Ligase TRIM27 Functions as a Host Restriction Factor Antagonized by Mycobacterium Tuberculosis PtpA During Mycobacterial Infection. Sci Rep (2016) 6:34827. doi: 10.1038/srep34827
36. Chen Y, Cao S, Sun Y, Li C. Gene Expression Profiling of the TRIM Protein Family Reveals Potential Biomarkers for Indicating Tuberculosis Status. Microb Pathog (2018) 114:385–92. doi: 10.1016/j.micpath.2017.12.008
37. Zheng Q, Hou J, Zhou Y, Yang Y, Xie B, Cao X. Siglec1 Suppresses Antiviral Innate Immune Response by Inducing TBK1 Degradation Via the Ubiquitin Ligase TRIM27. Cell Res (2015) 25:1121–36. doi: 10.1038/cr.2015.108
38. Zheng F, Xu N, Zhang Y. Trim27 Promotes Hepatitis C Virus Replication by Suppressing Type I Interferon Response. Inflammation (2019) 42:1317–25. doi: 10.1007/s10753-019-00992-5
39. Wan W, Wang L, Chen X, Zhu S, Shang W, Xiao G, et al. A Subcellular Quantitative Proteomic Analysis of Herpes Simplex Virus Type 1-Infected HEK 293t Cells. Molecules (2019) 24:4215. doi: 10.3390/molecules24234215
40. Uchil PD, Quinlan BD, Chan WT, Luna JM, Mothes W. Trim E3 Ligases Interfere With Early and Late Stages of the Retroviral Life Cycle. PloS Pathog (2008) 4:e16. doi: 10.1371/journal.ppat.0040016
41. Conwell SE, White AE, Harper JW, Knipe DM. Identification of TRIM27 as a Novel Degradation Target of Herpes Simplex Virus 1 ICP0. J Virol (2015) 89:220–9. doi: 10.1128/JVI.02635-14
42. Onoufriadis A, Stone K, Katsiamides A, Amar A, Omar Y, de Lange KM, et al. Exome Sequencing and Genotyping Identify a Rare Variant in NLRP7 Gene Associated With Ulcerative Colitis. J Crohns Colitis (2018) 12:321–6. doi: 10.1093/ecco-jcc/jjx157
43. Yucesoy B, Talzhanov Y, Michael Barmada M, Johnson VJ, Kashon ML, Baron E, et al. Association of MHC Region SNPs With Irritant Susceptibility in Healthcare Workers. J Immunotoxicol (2016) 13:738–44. doi: 10.3109/1547691X.2016.1173135
44. Ning L, Ko JM, Yu VZ, Ng HY, Chan CK, Tao L, et al. Nasopharyngeal Carcinoma MHC Region Deep Sequencing Identifies HLA and Novel non-HLA TRIM31 and TRIM39 Loci. Commun Biol (2020) 3:759. doi: 10.1038/s42003-020-01487-y
45. Song H, Liu B, Huai W, Yu Z, Wang W, Zhao J, et al. The E3 Ubiquitin Ligase TRIM31 Attenuates NLRP3 Inflammasome Activation by Promoting Proteasomal Degradation of NLRP3. Nat Commun (2016) 7:13727. doi: 10.1038/ncomms13727
46. Huang P, Liu W, Chen J, Hu Y, Wang Y, Sun J, et al. TRIM31 Inhibits NLRP3 Inflammasome and Pyroptosis of Retinal Pigment Epithelial Cells Through Ubiquitination of NLRP3. Cell Biol Int (2020) 44:2213–19. doi: 10.1002/cbin.11429
47. Wu X, Lu M, Ding S, Zhong Q. Tripartite Motif 31 Alleviates IL-1ß Secretion Via Promoting the Ubiquitination of Pyrin Domain Domains-Containing Protein 3 in Human Periodontal Ligament Fibroblasts. Odontology (2020) 108:424–32. doi: 10.1007/s10266-020-00519-7
48. Yang X, Sun J, Sun F, Yao Y, Tian T, Zhou J, et al. TRIM31 Promotes Apoptosis Via TAK1-mediated Activation of NF-κb Signaling in Sepsis-Induced Myocardial Dysfunction. Cell Cycle (2020) 19:2685–700. doi: 10.1080/15384101.2020.1826235
49. Wang H, Yao L, Gong Y, Zhang B. TRIM31 Regulates Chronic Inflammation Via NF-κb Signal Pathway to Promote Invasion and Metastasis in Colorectal Cancer. Am J Transl Res (2018) 10:1247–59. doi: 10.1080/15384101.2020.1826235
50. Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G, et al. The Ubiquitin E3 Ligase TRIM31 Promotes Aggregation and Activation of the Signaling Adaptor MAVS Through Lys63-linked Polyubiquitination. Nat Immunol (2017) 18:214–24. doi: 10.1038/ni.3641
51. Cheung PH, Lee TT, Kew C, Chen H, Yuen KY, Chan CP, et al. Virus Subtype-Specific Suppression of MAVS Aggregation and Activation by PB1-F2 Protein of Influenza A (H7N9) Virus. PloS Pathog (2020) 16:e1008611. doi: 10.1371/journal.ppat.1008611
52. Dai T, Wu L, Wang S, Wang J, Xie F, Zhang Z, et al. Faf1 Regulates Antiviral Immunity by Inhibiting MAVS But Is Antagonized by Phosphorylation Upon Viral Infection. Cell Host Microbe (2018) 24:776–90. doi: 10.1016/j.chom.2018.10.006
53. Yang S, Harding AT, Sweeney C, Miao D, Swan G, Zhou C, et al. Control of Antiviral Innate Immune Response by Protein Geranylgeranylation. Sci Adv (2019) 5:eaav7999. doi: 10.1126/sciadv.aav7999
54. Tan G, Yi Z, Song H, Xu F, Li F, Aliyari R, et al. Type-I-Ifn-Stimulated Gene Trim5γ Inhibits Hbv Replication by Promoting Hbx Degradation. Cell Rep (2019) 29:3551–63.e3. doi: 10.1016/j.celrep.2019.11.041
55. Cagliani R, Riva S, Pozzoli U, Fumagalli M, Comi GP, Bresolin N, et al. Balancing Selection is Common in the Extended MHC Region But Most Alleles With Opposite Risk Profile for Autoimmune Diseases are Neutrally Evolving. BMC Evol Biol (2011) 11:171. doi: 10.1186/1471-2148-11-171
56. Noguchi K, Okumura F, Takahashi N, Kataoka A, Kamiyama T, Todo S, et al. TRIM40 Promotes Neddylation of Ikkγ and is Downregulated in Gastrointestinal Cancers. Carcinogenesis (2011) 32:995–1004. doi: 10.1093/carcin/bgr068
57. Yang H, Meng L, Ai D, Hou N, Li H, Shuai X, et al. Acetic Acid Alleviates the Inflammatory Response and Liver Injury in Septic Mice by Increasing the Expression of TRIM40. Exp Ther Med (2019) 17:2789–98. doi: 10.3892/etm.2019.7274
58. Zhao C, Jia M, Song H, Yu Z, Wang W, Li Q, et al. The E3 Ubiquitin Ligase TRIM40 Attenuates Antiviral Immune Responses by Targeting MDA5 and RIG-I. Cell Rep (2017) 21:1613–23. doi: 10.1016/j.celrep.2017.10.020
59. Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare Variants of IFIH1, a Gene Implicated in Antiviral Responses, Protect Against Type 1 Diabetes. Science (2009) 324:387–9. doi: 10.1126/science.1167728
60. Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, et al. Aicardi-Goutières Syndrome is Caused by IFIH1 Mutations. Am J Hum Genet (2014) 95:121–5. doi: 10.1016/j.ajhg.2014.06.007
61. Jang MA, Kim EK, Now H, Nguyen NT, Kim WJ, Yoo JY, et al. Mutations in DDX58, Which Encodes RIG-I, Cause Atypical Singleton-Merten Syndrome. Am J Hum Genet (2015) 96:266–74. doi: 10.1016/j.ajhg.2014.11.019
62. Nyaga DM, Vickers MH, Jefferies C, Perry JK, O’Sullivan JM. Type 1 Diabetes Mellitus-Associated Genetic Variants Contribute to Overlapping Immune Regulatory Networks. Front Genet (2018) 9:535. doi: 10.3389/fgene.2018.00535
63. Lyu XM, Zhu XW, Zhao M, Zuo XB, Huang ZX, Liu X, et al. A Regulatory Mutant on TRIM26 Conferring the Risk of Nasopharyngeal Carcinoma by Inducing Low Immune Response. Cancer Med (2018) 7:3848–61. doi: 10.1002/cam4.1537
64. Cree BA, Rioux JD, McCauley JL, Gourraud PA, Goyette P, McElroy J, et al. A Major Histocompatibility Class I Locus Contributes to Multiple Sclerosis Susceptibility Independently From HLA-DRB1*15:01. PloS One (2010) 5:e11296. doi: 10.1371/journal.pone.0011296
65. Wang P, Zhao W, Zhao K, Zhang L, Gao C. TRIM26 Negatively Regulates Interferon-β Production and Antiviral Response Through Polyubiquitination and Degradation of Nuclear IRF3. PloS Pathog (2015) 11:e1004726. doi: 10.1371/journal.ppat.1004726
66. Ran Y, Zhang J, Liu LL, Pan ZY, Nie Y, Zhang HY, et al. Autoubiquitination of TRIM26 Links TBK1 to NEMO in RLR-mediated Innate Antiviral Immune Response. J Mol Cell Biol (2016) 8:31–43. doi: 10.1093/jmcb/mjv068
67. Liang Y, Zhang G, Li Q, Han L, Hu X, Guo Y, et al. TRIM26 is a Critical Host Factor for HCV Replication and Contributes to Host Tropism. Sci Adv (2021) 7:eabd9732. doi: 10.1126/sciadv.abd9732
68. Kurata R, Nakaoka H, Tajima A, Hosomichi K, Shiina T, Meguro A, et al. TRIM39 and RNF39 are Associated With Behçet’s Disease Independently of HLA-B∗51 and -a∗26. Biochem Biophys Res Commun (2010) 401:533–7. doi: 10.1016/j.bbrc.2010.09.088
69. Lu Y, Cheng Y, Yan W, Nardini C. Exploring the Molecular Causes of Hepatitis B Virus Vaccination Response: An Approach With Epigenomic and Transcriptomic Data. BMC Med Genomics (2014) 7:12. doi: 10.1186/1755-8794-7-12
70. Renauer P, Coit P, Jeffries MA, Merrill JT, McCune WJ, Maksimowicz-McKinnon K, et al. DNA Methylation Patterns in Naïve CD4+ T Cells Identify Epigenetic Susceptibility Loci for Malar Rash and Discoid Rash in Systemic Lupus Erythematosus. Lupus Sci Med (2015) 2:e000101. doi: 10.1136/lupus-2015-000101
71. Maltby VE, Lea RA, Sanders KA, White N, Benton MC, Scott RJ, et al. Differential Methylation At MHC in CD4+ T Cells is Associated With Multiple Sclerosis Independently of HLA-DRB1. Clin Epigenet (2017) 9:71. doi: 10.1186/s13148-017-0371-1
72. Morin A, Laviolette M, Pastinen T, Boulet LP, Laprise C. Combining Omics Data to Identify Genes Associated With Allergic Rhinitis. Clin Epigenet (2017) 9:3. doi: 10.1186/s13148-017-0310-1
73. Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, et al. A Whole-Genome Association Study of Major Determinants for Host Control of HIV-1. Science (2007) 317:944–7. doi: 10.1126/science.1143767
74. Fellay J, Ge D, Shianna KV, Colombo S, Ledergerber B, Cirulli ET, et al. Common Genetic Variation and the Control of HIV-1 in Humans. PloS Genet (2009) 5:e1000791. doi: 10.1371/journal.pgen.1000791
75. Lin YJ, Chen CY, Jeang KT, Liu X, Wang JH, Hung CH, et al. Ring Finger Protein 39 Genetic Variants Associate With HIV-1 Plasma Viral Loads and its Replication in Cell Culture. Cell Biosci (2014) 4:40. doi: 10.1186/2045-3701-4-40
76. Wang W, Jia M, Zhao C, Yu Z, Song H, Qin Y, et al. RNF39 Mediates K48-linked Ubiquitination of DDX3X and Inhibits RLR-dependent Antiviral Immunity. Sci Adv (2021) 77:eabe5877. doi: 10.1126/sciadv.abe5877
77. Lanata CM, Nititham J, Taylor KE, Chung SA, Torgerson DG, Seldin MF, et al. Genetic Contributions to Lupus Nephritis in a Multi-Ethnic Cohort of Systemic Lupus Erythematous Patients. PloS One (2018) 13:e0199003. doi: 10.1371/journal.pone.0199003
78. Uchil PD, Hinz A, Siegel S, Coenen-Stass A, Pertel T, Luban J, et al. TRIM Protein-Mediated Regulation of Inflammatory and Innate Immune Signaling and its Association With Antiretroviral Activity. J Virol (2013) 87:257–72. doi: 10.1128/JVI.01804-12
79. Suzuki M, Watanabe M, Nakamaru Y, Takagi D, Takahashi H, Fukuda S, et al. TRIM39 Negatively Regulates the Nfκb-Mediated Signaling Pathway Through Stabilization of Cactin. Cell Mol Life Sci (2016) 73:1085–101. doi: 10.1007/s00018-015-2040-x
80. Kisiel B, Kisiel K, Szymański K, Mackiewicz W, Biało-Wójcicka E, Uczniak S, et al. The Association Between 38 Previously Reported Polymorphisms and Psoriasis in a Polish Population: High Predicative Accuracy of a Genetic Risk Score Combining 16 Loci. PloS One (2017) 12:e0179348. doi: 10.1371/journal.pone.0179348
81. Kunz M, König IR, Schillert A, Kruppa J, Ziegler A, Grallert H, et al. Genome-Wide Association Study Identifies New Susceptibility Loci for Cutaneous Lupus Erythematosus. Exp Dermatol (2015) 24:510–5. doi: 10.1111/exd.12708
82. McDermott E, Ryan EJ, Tosetto M, Gibson D, Burrage J, Keegan D, et al. Dna Methylation Profiling in Inflammatory Bowel Disease Provides New Insights Into Disease Pathogenesis. J Crohns Colitis (2016) 10:77–86. doi: 10.1093/ecco-jcc/jjv176
83. Kurata R, Tajima A, Yonezawa T, Inoko H. TRIM39R, But Not TRIM39B, Regulates Type I Interferon Response. Biochem Biophys Res Commun (2013) 436:90–5. doi: 10.1016/j.bbrc.2013.05.064
Keywords: MHC class I region, E3 ubiquitin ligases, innate immunity, post-translational modifications, autoimmune diseases
Citation: Jia X, Zhao C and Zhao W (2021) Emerging Roles of MHC Class I Region-Encoded E3 Ubiquitin Ligases in Innate Immunity. Front. Immunol. 12:687102. doi: 10.3389/fimmu.2021.687102
Received: 29 March 2021; Accepted: 27 May 2021;
Published: 10 June 2021.
Edited by:
Konstantin Sparrer, Ulm University Medical Center, GermanyReviewed by:
Katrin Rittinger, Francis Crick Institute, United KingdomSébastien Nisole, UMR9004 Institut de Recherche en Infectiologie de Montpellier (IRIM), France
Copyright © 2021 Jia, Zhao and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Zhao, d3poYW9Ac2R1LmVkdS5jbg==