 James Alexander Pearson
James Alexander Pearson F. Susan Wong
F. Susan Wong Li Wen
Li Wen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 09 June 2021
Sec. Immunological Tolerance and Regulation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.686956
This article is part of the Research TopicImmunopathology of Type 1 DiabetesView all 21 articles
Microbiota have been identified as an important modulator of susceptibility in the development of Type 1 diabetes in both animal models and humans. Collectively these studies highlight the association of the microbiota composition with genetic risk, islet autoantibody development and modulation of the immune responses. However, the signaling pathways involved in mediating these changes are less well investigated, particularly in humans. Importantly, understanding the activation of signaling pathways in response to microbial stimulation is vital to enable further development of immunotherapeutics, which may enable enhanced tolerance to the microbiota or prevent the initiation of the autoimmune process. One such signaling pathway that has been poorly studied in the context of Type 1 diabetes is the role of the inflammasomes, which are multiprotein complexes that can initiate immune responses following detection of their microbial ligands. In this review, we discuss the roles of the inflammasomes in modulating Type 1 diabetes susceptibility, from genetic associations to the priming and activation of the inflammasomes. In addition, we also summarize the available inhibitors for therapeutically targeting the inflammasomes, which may be of future use in Type 1 diabetes.
Inflammasomes, a term first coined by Dr. Jurg Tschopp in 2002, are multiprotein complexes found in the cytosol, which mediate the activation of inflammatory caspases (1). Inflammasome formation is driven (“primed”) by activation of the pattern-recognition receptors (PRRs) in response to pathogen-associated molecular patterns (PAMPs) or damage signals (e.g. damage-associated molecular patterns that are also known as danger-associated molecular patterns, DAMPs) in the cytosol (2–4) (Figure 1). In some inflammasomes, the inflammasome adaptor protein designated as Apoptosis-associated Speck-like protein, containing a Caspase activation and recruitment domain (ASC), aids in the oligomerization of the inflammasome components and links the upstream inflammasome sensor molecules to procaspase 1 (21). In ASC-independent inflammasomes, interactions occur between inflammasome components, which can alter the protein structure e.g. NLRC4 can be activated by Neuronal apoptosis inhibitory proteins (NAIPs), resulting in the formation of the disk-like inflammasome (22, 23). In both ASC-dependent and -independent inflammasomes, procaspase 1 becomes dimerized and through autoproteolysis forms catalytically-active caspase 1, which subsequently induces IL-1β and IL-18 cytokine release, as well as inducing pyroptosis, a form of lytic cell death. There are many different types of proteins involved in the formation of the inflammasomes, including the NBD leucine-rich repeat-containing receptor (NLR) family (e.g. NLRP1) and the PYHIN protein families [e.g. absent in melanoma 2 (AIM2)]. In humans, there are 22 NLRs but only NLRP1, NLRP3, NLRP6, NLRP7, NLRP12 and NLRC4 have been shown to form inflammasomes (24–30). Structural and functional differences between the inflammasome proteins result in differences in their ability to bind their respective ligands, and thus each can be activated by different mechanisms (Figure 2). In the case of NLRP3, multiple types of ligands can be recognized, which induce disassembly of the trans-Golgi network, leading to the recruitment and binding of NLRP3 via its lysine motif (between the PYRIN and NACHT domain) to the phosphatidylinositol-4-phosphate on the disassembled trans face of the golgi (39). However, it is unclear whether there are additional mechanisms, including the question of whether other factors contribute to the Golgi network disassembly, or protection from disassembly, or whether similar mechanisms exist for other inflammasomes.
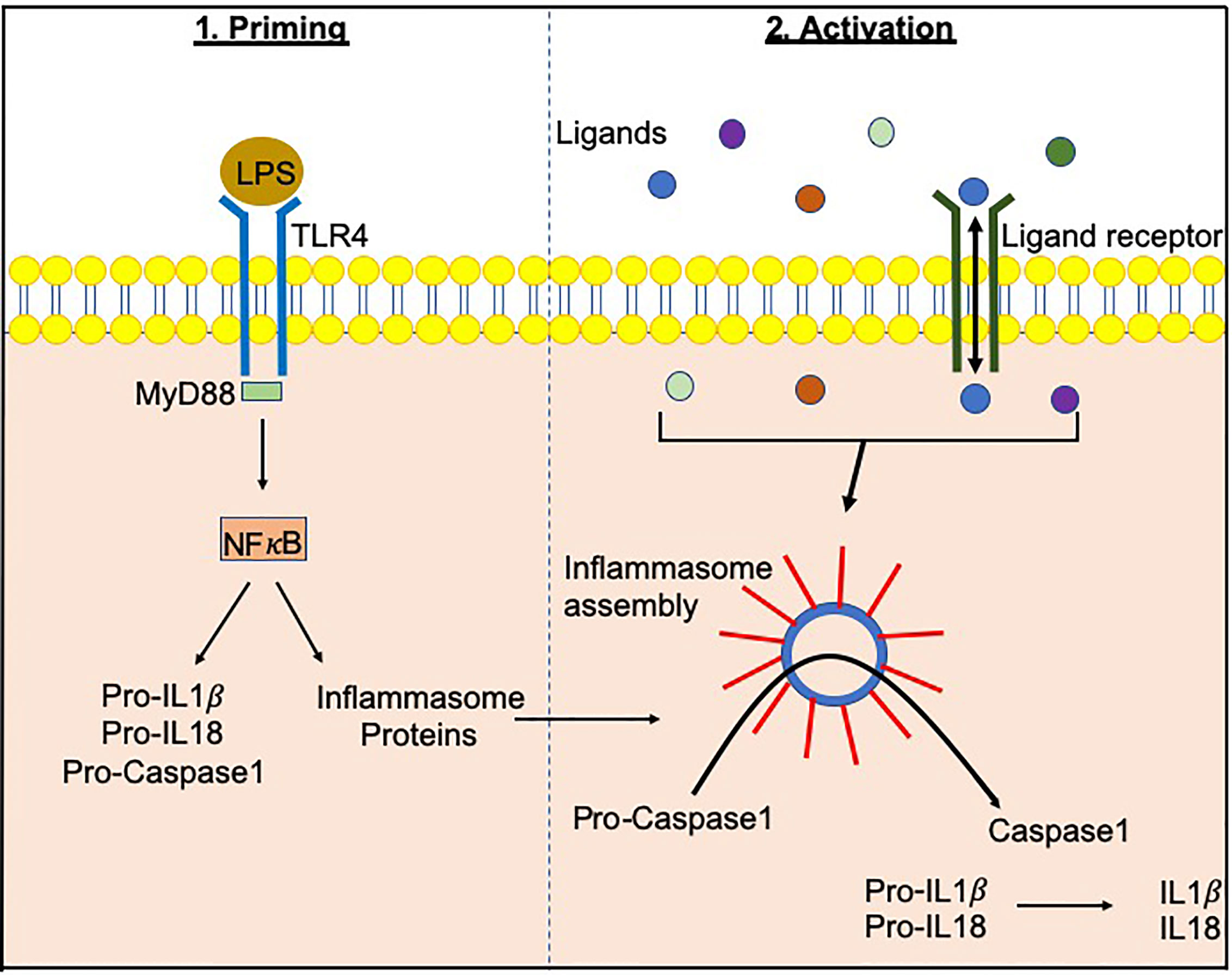
Figure 1 Inflammasome priming and activationInflammasome-related genes e.g. NLRP3, NLRC4 are transcribed following PAMP/DAMP recognition by their respective receptors e.g. bacterial Lipopolysachharide (LPS) recognition by TLR4 pathogen-associated molecular patterns. This “priming” step alerts the cells to potential dangers and prepares the inflammasome machinery to be translated. Upon recognition of additional activating signals (Figure 2), the inflammasome proteins oligomerize and form a wheel/disk-like structure. The formation of these inflammasome complexes enables the activation of caspase 1 from its precursor form (procaspase 1), which in turn activates other cytokines including IL-1β and IL-18 (5, 6). Inflammasome-associated proteins can also activate other caspases including caspase 4, 5, 8 and 11 (7–20).
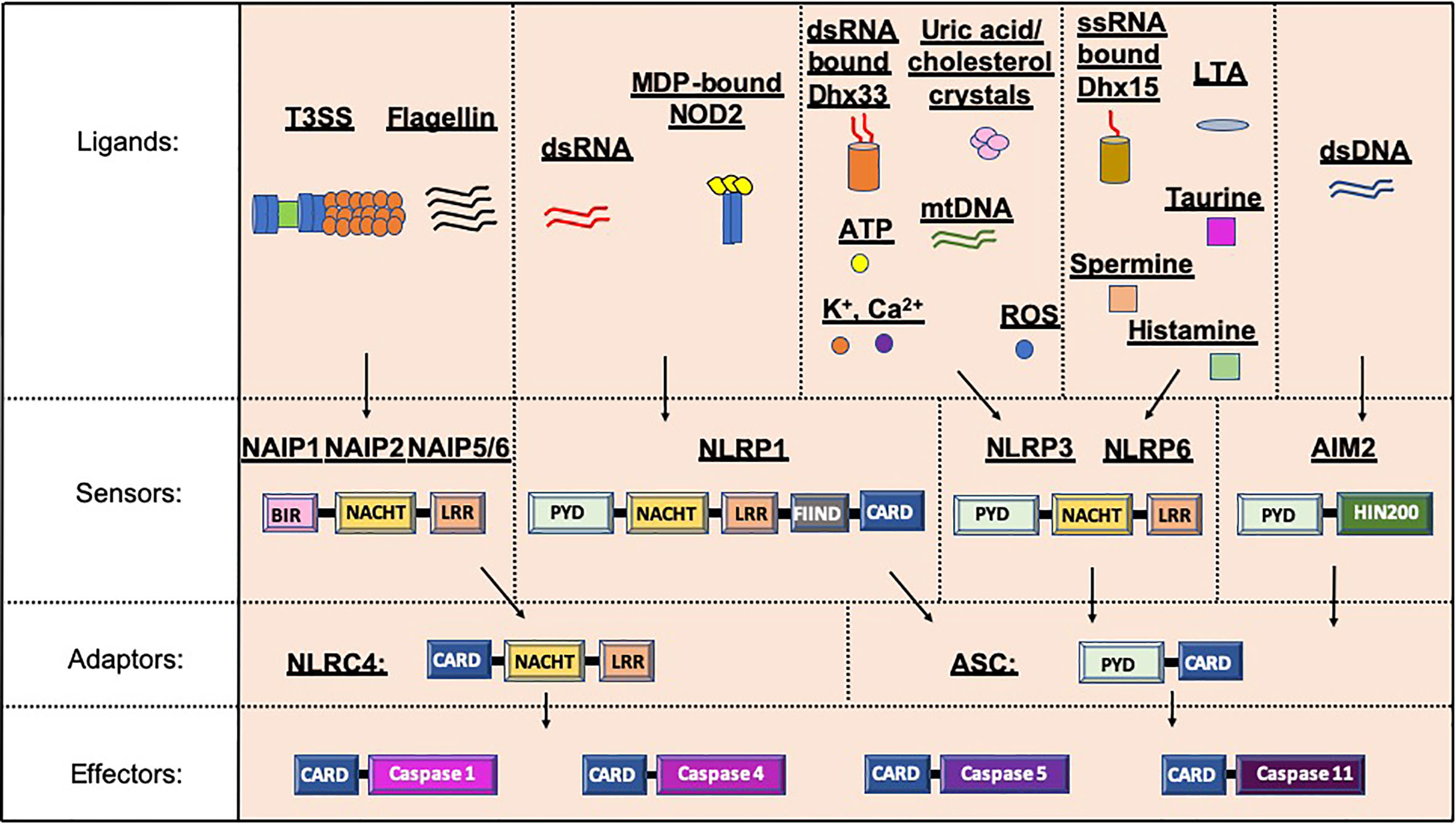
Figure 2 Inflammasome protein sensors and adaptors recognize a variety of ligands, either directly or indirectly. Upon ligand binding, the sensors and adaptors interact via PYD-PYD domain interactions to form the oligomers prior to ASC-mediated recruitment of the Procaspase via CARD-CARD interactions (5–11, 21, 25, 31, 32). NAIP1, 2 and 5/6 bind bacterial-derived Type 3 Secretion system (T3SS) rod or needle proteins or flagellin respectively, prior to activation of the NLRC4 inflammasome (12, 13). NLRP1 can be activated by double stranded RNA (dsRNA; human only) or muramyl dipeptide (MDP) bound to the Nuclear oligomerization domain-containing 2 (NOD2) protein (14, 33). Numerous ligands for NLRP3 have been found including K+, Ca2+, reactive oxygen species (ROS), Adenosine triphosphate (ATP), uric acid crystals, cholesterol crystals, double-stranded RNA (dsRNA) bound by DExD/H-box helicase (Dhx) 33 and mitochondrial DNA (mtDNA) (7, 16, 34–37). Single stranded RNA (ssRNA) bound to Dhx15, lipoteichoic acid (LTA) as well as spermine, taurine and histamine can all activate the NLRP6 inflammasome (32, 35, 38). To date, double stranded DNA is the only ligand known for AIM2 (10, 19, 20). PYD, Pyrin domain; HIN200, Hematopoietic expression, interferon-inducible nature, and nuclear localization 200 domain; NACHT, Nucleotide binding and oligomerization domain; LRR, Leucine-rich repeat; FIIND, function to find domain; CARD, Caspase recruitment domain; BIR, Baculovirus IAP-repeat domains.
Inflammasomes can be activated by a number of components released during cell/tissue damage, metabolism, infection or by commensal bacteria. Microbial ligands from host commensals or infectious organisms e.g. type 3 secretion system proteins, flagellin, and DNA/RNA can all activate inflammasome proteins. Furthermore, aggregates of Lipopolysaccharides (LPS; specifically, the Lipid A component), an endotoxin present in the outer membrane of gram-negative bacteria, can directly bind to and activate non-canonical inflammasome caspases 4 and 5 (humans) and 11 (mice) (40–43). Importantly, this process 1) is independent of Toll-like receptor (TLR) 4, which can also bind LPS (40, 42), and 2) promotes protection from cytosolic invading pathogens (40–43). Together, these suggest an important role for microbial modulation of inflammasome responses.
Studies using inflammasome-deficient mice have demonstrated that inflammasomes can influence disease susceptibility to inflammatory bowel disease (IBD) (27, 44), cancer (44, 45), obesity (46, 47), viral/bacterial infection (38, 48–53) and type 1 diabetes (T1D) (34, 54, 55). To date, few studies have functionally investigated the mechanistic role of inflammasomes in T1D; however, there are studies indicating a link to inflammasomes and susceptibility to T1D. As susceptibility to T1D can be modulated by microbial components, as discussed later, we highlight the role of inflammasomes as important microbial sensors in the context of T1D.
Genetic analyses often provide important insight into genes or mutations that may be associated with disease susceptibility in humans. Gene mutations in NLRP3, resulting in a gain of function and thus increased IL-1β secretion, were initially linked to a number of inherited autosomal dominant inflammatory diseases e.g. Muckle-Wells syndrome and familial cold autoinflammatory syndrome and chronic infantile neurological cutaneous articular syndrome (56). Since then, single nucleotide polymorphisms (SNPs) in NLRP1, NLRP3 and NLRC4 have been associated with many autoimmune diseases including IBD (57), celiac disease (58), multiple sclerosis (59) and autoimmune diabetes (60–64). Table 1 summarizes the SNPs in NLRP1, NLRP3 and NLRC4 genes that have been investigated in individuals with Type 1 diabetes. Of these SNPs, only 2 are within the coding region of NLRP1 and NLRP3 genes (rs12150220 and rs35829419 respectively) and both have been linked to a gain of function and excessive IL-1β and IL-18 secretion in other disease settings (67, 68). The other SNPs that are located in the promoter region may influence gene regulation, but this has not yet been fully elucidated. As Table 1 illustrates, not all populations studied show the same SNP associations in individuals with Type 1 diabetes. For example, the SNP rs12150220, located in the NLRP1 gene region, was increased in a Norwegian population with T1D (60); however, no associations were identified in either a Polish (65) or Brazilian (62) population with T1D, compared to their controls. There may be many reasons for this, including population-based genetic differences, the presence of other comorbidities or the microbiota composition. Two studies conducted in the Han Chinese population also showed SNP associations in NLRP3 and NLRC4 gene regions with clinical characteristics, including the age of diabetes onset, 2-hour postprandial c-peptide and the presence of anti-glutamic acid decarboxylase (GAD) autoantibodies (63, 66). These suggest a potential link to altered immunity; however, larger scale studies are needed to help us to better understand the association of different allelic variants and combinations of haplotypes in the inflammasome-related genes and susceptibility to Type 1 diabetes. Studies using knock-in mice, in which the SNPs can be introduced into the gene, may provide valuable tools to elucidate the functional consequences of these SNPs.

Table 1 SNPs in inflammasome genes that have been investigated for associations with autoimmune diabetes in humans.
Environmental factors, e.g. the microbiota (referring to all microorganisms including bacteria, viruses, fungi, protozoa and archaea), have gained significant traction as modulators of susceptibility to T1D. In turn, it is clear that genes involved in the genetic susceptibility to T1D are important modulators of the bacterial composition in humans and animal models (69, 70). Furthermore, altered gut bacterial composition has been found in individuals diagnosed with T1D (71–75), in Bio-breeding (BB) rats (76), and in Non-obese diabetic (NOD) mice (77, 78), compared to non-diabetic controls. In addition, in individuals who are at genetic risk of developing T1D, changes in gut bacteria are associated with the early development of β-cell autoimmunity (74, 75, 79–81). As mentioned, microbial ligands are one activator of the inflammasomes; changes in the microbial composition and thus the availability of microbial ligands may alter inflammasome activation (Figure 3), and this may be one way in which microbes influence pathogenesis of type 1 diabetes.
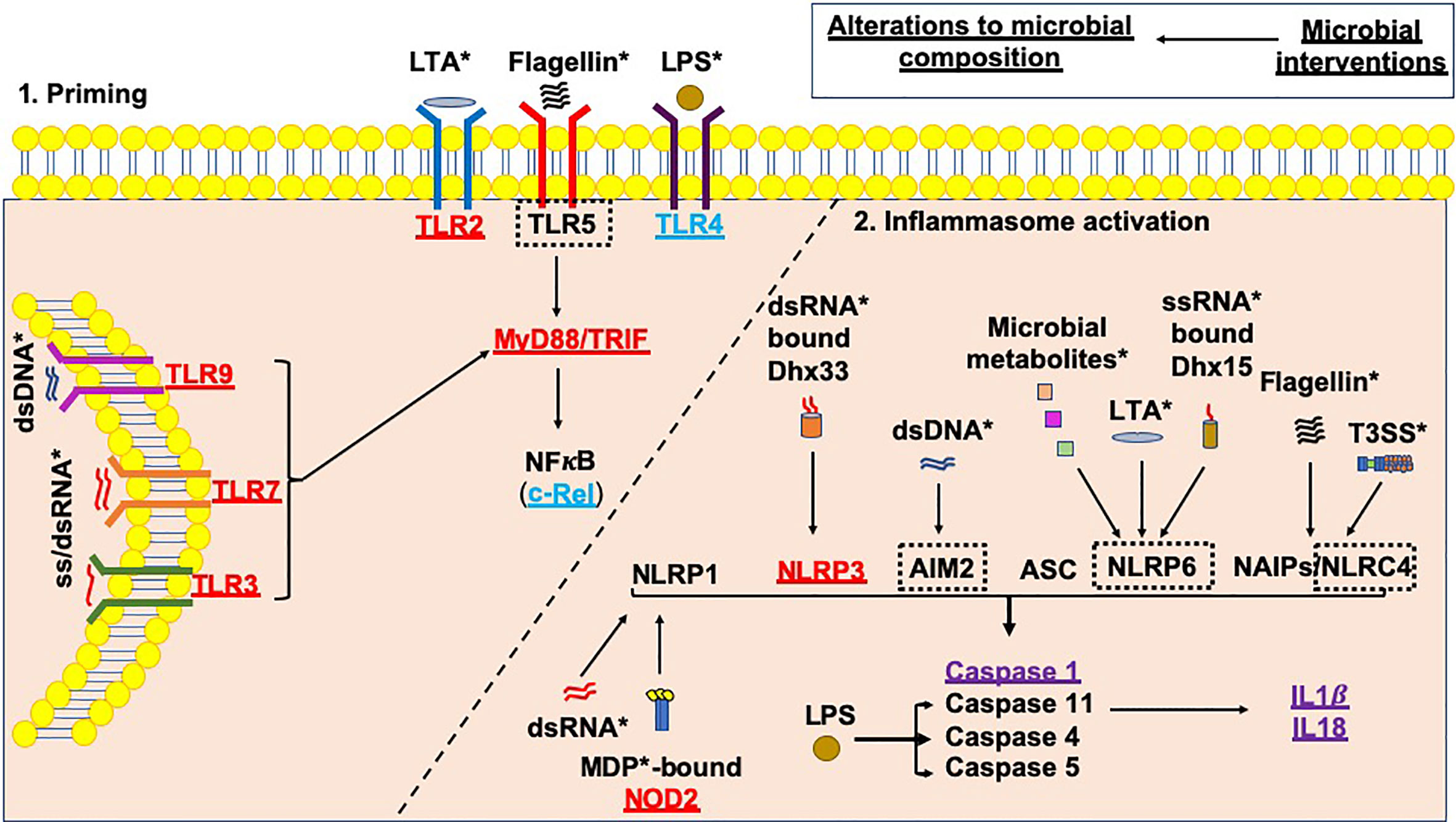
Figure 3 Microbial influences on inflammasome priming and activation in type 1 diabetes. Microbial interventions e.g. fecal microbiota transplants, antibiotic, probiotic and prebiotic usage can all influence the microbial composition, subsequently altering the availability of microbial ligands involved in both the priming, and canonical and non-canonical activation of inflammasomes (as shown by *). Studies of single PRR or inflammasome (nlrp3) gene-deficient mice have shown that these proteins would be needed to promote the development of T1D (shown in red); however, Tlr4-deficient and c-Rel-deficient NOD mice (c-Rel is a subunit of the NFкB protein) promote tolerance and limit the development of T1D (shown in blue). In addition, some gene-deficient mice showed no significant effect on mediating susceptibility to T1D (shown in purple). A number of planned studies are currently underway using a number of gene-deficient mice to assess their ability to alter susceptibility to T1D development, as shown by the black dotted boxes. Paradoxically, the gene-deficient mice are also likely to have altered microbial composition, contributing to the protection against/susceptibility to disease. Studies of these gene-deficient mice will need to evaluate the contribution of the gene independently from any alterations to the microbial composition.
Viruses have also been implicated in the pathogenesis of T1D. Coxsackie viruses and Rotaviruses have been implicated in the development of T1D due to 1) their association with the development of autoantibodies (82, 83), which are predictive biomarkers for immune progression and T1D development (84); 2) viral proteins e.g. enteroviral capsid protein vp1 can be identified in the islets (85–89); 3) susceptibility to T1D in animal models can be modulated by viral infections (90–98); and 4) an oral Rotavirus vaccine has shown potential to protect individuals at risk of developing T1D from future development of the disease (99). We recently demonstrated that a mouse norovirus infection in NOD mice modulated susceptibility to T1D, mediated through changes in the gut microbiota (100), highlighting the necessity for increased understanding of broader microbial community interactions. Changes in the viral DNA and RNA abundance, alongside any virus-induced bacterial changes, would also potentially alter inflammasome activation.
Both fungal glucans and parasite/helminth antigens can also stimulate inflammasomes and these may modulate susceptibility to T1D in animal models (101–104); however, few studies have been conducted in humans. Individuals with T1D have greater fungal species diversity compared with healthy controls (105). Others demonstrated that individuals with islet autoimmunity, who later progressed to T1D, had a higher abundance of Sacchromyces and Candida, compared to those who did not progress to T1D over the 8-9 years of follow up (106). There has been much debate about whether parasitic infection modulates autoimmunity in T1D. One study in Norwegian children showed fewer Enterobius vermicularis (a pinworm) infections in children at high genetic risk for T1D (107), while another study in Sweden, suggested no association with worms and the development of T1D in children (108). It is possible that parasites may contribute to the reduction in autoimmunity, as parasite-endemic areas have lower incidences of T1D in their populations, compared to non-parasite endemic areas (109). Whilst this may be because parasitic infections promote Th2 immune responses, other factors are likely to be involved including the lower genetic susceptibility to T1D of the populations living in parasite endemic areas. Thus far, although work in animal models has suggested that helminths, and other parasites like schistosomes or their antigenic products (101, 102, 110) could have a beneficial effect on autoimmunity, these have not yet been translated into therapeutics for humans with type 1 diabetes.
Most of the studies mentioned above focus on the microbiota composition and association with the development of either islet autoimmunity or T1D; however, understanding the mechanisms by which the immune system is activated by the microbiota is important. Furthermore, all of these changes in microbial composition may have profound impacts on inflammasome activation (Figure 3).
Microbial recognition by PRRs expressed by immune cells are key to regulating crosstalk between immune cells and the microbiota. PRRs such as Toll-like receptors (TLRs), of which there are 10 in humans (TLR1-10) and 12 in mice (TLR1-9, 11-13), selectively bind to their unique microbial ligands, leading to the downstream activation of proinflammatory cytokines (111). These TLRs can be found on different immune and non-immune cells, including the islet β-cells in both humans and mice (112). Studies using TLR-deficient NOD mice have identified that signaling through TLR2, 3, 7 and 9 (97, 113–116) are important for promoting disease, while TLR4 signaling prevents disease development (117). These TLRs signal through one of two key adaptor proteins: Myeloid differentiation primary response 88 (MyD88, which all TLRs utilize except TLR3) or TIR domain-containing adaptor inducing IFN-β (TRIF, which only TLRs 3 and 4 utilize). Deficiencies in either (118, 119), or both (120), of these two key genes results in significant protection of the NOD mice from the development of diabetes, indicating a reliance on downstream-mediated signaling to induce the proinflammatory immune response. Interestingly, only MyD88-deficient mice, but not MyD88 and TRIF double-deficient mice, were protected from immune infiltration in the islets, suggesting that TRIF-mediated signaling, most likely due to TLR4 signaling, was responsible for inducing tolerance (120). TLR4 signaling in human monocyte-derived DCs, stimulated by E.coli lipopolysaccharide [LPS; a TLR4 ligand (121)], induced immune tolerance, unlike the effect seen from stimulation with LPS derived from B.dorei (122). As Finnish children have a higher abundance of B.dorei, and a higher incidence of Type 1 diabetes, compared to their genetically-similar Russian neighbors, it is likely that LPS-induced tolerance is important for modulating susceptibility to T1D in humans (122). TLR activation is also important for priming the inflammasome proteins and thus, changes to the TLR stimulation highlighted above are likely to modulate inflammasome activation as well. It is unclear, at present, whether any of these studies of TLR-deficient mice, or studies of TLR stimulation of cells from individuals with Type 1 diabetes, will differentially influence the activation of the inflammasome and how the functional consequences of this could influence susceptibility to T1D.
In addition to the TLRs, there are also other microbial sensors that can prime the inflammasome complexes, including the cytosolic Nucleotide-binding oligomerization domain (NOD) proteins, NOD1 and NOD2. NOD1 and NOD2 both recognize bacterial peptidoglycan moieties (123, 124) and upon binding, oligomerize and signal through the Receptor-interacting-serine/threonine-protein kinase 2 (RIP2) resulting in the activation of NFκB and production of inflammatory cytokines (125). Using a streptozotocin (STZ)-induced type 1 diabetes model, NOD2 deficiency, but not NOD1 or RIP2 deficiency, protected the mice from disease development (126). These findings were also supported by other studies in NOD mice, demonstrating that NOD2-deficient NOD mice were protected from type 1 diabetes development, and this was dependent on the gut microbiota composition (127), whereas RIP2-deficient NOD mice were not protected (120). Interestingly, both NOD1 and NOD2 appear to have RIP2 independent functions; NOD2 binds CARD9 to mediate downstream signaling independent of RIP2 (128), while NOD1 regulates MAPK signaling independent of RIP2 (129). It is still unclear what the role, if any, NOD1 has in the immunopathogenesis of autoimmune Type 1 diabetes. Importantly, following muramyl dipeptide (ligand) binding, NOD2, complexed with NLRP1, promotes inflammasome activation (33), independent of NOD1 activation (25). Furthermore, in NOD2-deficient mice, induction of intestinal inflammation by dextran sodium sulfate (DSS) resulted in elevated NLRP3 inflammasome formation, suggesting that NOD2 may interact with and/or modulate NLRP3 inflammasome formation (130). Thus, understanding NOD2 activation and its role in modulating inflammasome formation in relation to T1D pathogenesis will need further mechanistic investigation.
It should be noted that in most studies using PRR-deficient NOD mice, the microbiome can be altered by the gene deficiency, which promotes a tolerizing influence and suppression of type 1 diabetes development, as in the case with NOD2-deficient NOD mice (127). Thus, in evaluating studies using these models, it is vital to control for environmental variables such as cage effects (i.e. comparisons between mice in different cages) and legacy effects (i.e. comparisons between mice bred from different breeders), both of which can substantially alter the bacterial composition (131, 132). Failure to consider these variables can promote non-reproducible data and thus future studies need to 1. be transparent in the reporting of these elements in their animal experiments, and 2. Control for these variables.
To date, only two inflammasome-associated proteins (NLRP3 and AIM2) have been studied for their role in modulating susceptibility to T1D using gene-deficient mice (34, 54, 55). NLRP3-deficient NOD mice were protected from the development of T1D compared to wild-type littermates, as were wild-type NOD mice treated with an NLRP3 inhibitor (parthenolide; 10mg/kg body weight, twice a week for 4 weeks from 10-12 weeks of age) (54). NLRP3-deficient C57BL/6 mice were also protected from diabetes development following STZ treatment, whereas ASC-deficient C57BL/6 mice were not (34). NLRP3 deficiency in NOD mice was found to reduce T cell activation and Th1 differentiation, as well as reducing T cell expression of both the chemokines CCR5 and CXCR3, and ccl5 and cxcl10 gene expression from the islet β-cells, resulting in poor T cell chemotaxis into the islets and protection from T1D development (54). Furthermore, diabetic NOD mice exhibited increased Nlrp3 and pro-il-1β gene expression in the pancreatic lymph nodes, compared to pre-diabetic NOD mice, suggesting an increasing role for inflammasome activation (shown to be mediated by circulating mitochondrial DNA) with disease progression (34). In contrast to NLRP3-deficient C57BL/6 mice, AIM2-deficient C57BL/6 mice had accelerated STZ-induced diabetes development, compared to wild-type control mice (55), implying that ASC regulates inflammasome activation. This acceleration in STZ-induced diabetes development in AIM2-deficient mice occurred through enhanced gut permeability and increased bacterial translocation to the pancreatic lymph nodes. These findings were similar to those from the STZ-induced NOD2-deficient mouse study (126), with the inference that NOD2 activation of inflammasomes may be ASC-dependent. In humans, Aim2 gene expression was increased in the pancreas but not in peripheral blood mononuclear cells (PBMCs) in individuals with T1D compared to healthy controls (55); however, the data from the pancreas was only available in a small group (n=4-8) and thus needs to be confirmed in larger cohorts, ideally separating infiltrating immune cells from the islet β-cells. Another study in humans found that NLRP1 and NLRP3 gene expression was reduced in PBMCs and granulocytes in individuals with newly diagnosed T1D (less than 6 months), compared to healthy controls (133). While these studies indicate an important involvement of two of the inflammasome proteins in the development of T1D, further studies are needed to evaluate the other inflammasome-related proteins and how different types of stimulation may influence their function. More studies both in animal models, particularly those developing spontaneous autoimmune diabetes, and in humans, are needed to better understand inflammasome involvement and modulation during diabetes development. Finally, identifying the role of inflammasomes in individual cell types will be pivotal for understanding the key players in inflammasome activation and regulation. Thus, cell-specific gene knock out mice may be valuable tools for such studies.
Inflammasome activation induces IL-1β and IL-18 cytokine release following Caspase activation. Both IL-1β and IL-18 cytokines increase with progression to diabetes and destruction of the islet β-cells (134–136). To further investigate whether blocking these pathways could be therapeutically useful, studies targeting the IL-1 pathway were conducted in individuals with recent-onset T1D. Two Phase 2a randomized, multicenter, double-blind, placebo-controlled trials were carried out in which Canakinumab (a human monoclonal anti-IL-1 antibody), or Anakinra (a human IL-1 receptor antagonist), were administered (137). Contrary to expectations, these single immunotherapy interventions failed to prevent the ongoing autoimmunity. This result was concordant with data from NOD mouse models that included IL-1 receptor- (138), Caspase-1- (139, 140), IL-1β- (140) and IL-18- (141)-deficient NOD mice, where no significant changes to diabetes protection were observed with any of these mutations. However, a study combining anti-CD3 treatment with either Anakinra or an anti-IL-1β antibody resulted in reversal of diabetes in recent-onset T1D NOD mice (142), suggesting that combined therapy may also improve clinical efficacy in humans. Given the success of Teplizumab (anti-CD3) in delaying the development of T1D in relatives at risk (143, 144), a combined study evaluating the role of Teplizumab with IL-1 blockade may further enhance clinical efficacy. It is intriguing that NLRP3-deficient NOD mice were protected from T1D, while IL-1 receptor-, Caspase-1/11-, IL-1β- and IL-18-deficient NOD mice were not. There could be multiple reasons for this including: 1. Altered microbiota caused by the gene deficiency, influencing priming/activation of inflammasomes, 2. Promotion of other inflammasome signaling when Nlpr3 is deficient, 3. Effects on other caspases, for example Caspase 8 can also regulate inflammasome activation (145, 146), 4. Effects on other proteases which can process IL-1β (147, 148), and 5. Other unknown protein interactions may be involved. It is clear that further study of multiple pathways of influence is needed to fully comprehend and understand these differences.
Modulation of inflammasomes has had some therapeutic success in autoimmune diseases. A small-molecule inhibitor (MCC950), specifically targeting NLRP3 inflammasome activation (ASC oligomerization) but not AIM2, NLRC4 or NLRP1 inflammasomes, was able to attenuate mouse models of multiple sclerosis (149) and Parkinson’s disease (150). Additional NLRP3 selective inhibitors have been developed, which inhibit ATPase activity (151, 152), or oligomerization of NLRP3 (153), and these inhibitors prevented or ameliorated the development of joint inflammation in arthritis (154), metabolic perturbation in high fat diet-fed mice (151, 153), and autoinflammatory syndromes (151–153). There are also less selective natural inflammasome inhibitors including Genepin, a component of Gardenis fruits (155), which can inhibit NLRP3 and NLRC4 inflammasome activation via inhibiting autophagy, the eicosanoid 15-deoxy-Δ(12,14)-PGJ2 (15d-PGJ2) and related cyclopentenone prostaglandins (156), which inhibit the NLRP1 and NLRP3 inflammasomes and thence conversion of procaspase 1 to caspase 1. Parthenolide inhibits NLRP1, NLRP3 and NLRC4 inflammasomes (but not AIM2) (157–160), by alkylating the cysteine residues in Caspase 1 and in the ATPase domain of NLRP3 and inhibiting IκB kinase function required for NF-κB activation. As previously mentioned, Parthenolide prevented the development of T1D in 10-12-week old prediabetic NOD mice after 4 weeks of treatment (54). Thus, further investigation of inflammasome inhibitors as a potential therapeutic intervention in T1D is needed. More inflammasome regulators and inhibitors have been studied in different diseases, and which have been reviewed elsewhere (161–163). Future studies should focus on the more selective inflammasome inhibitors, as these will likely have minimal effects on other inflammasome pathways, thereby minimizing detrimental impacts on host defense. Initiating these studies will be vital to fully determine their potential clinical benefits and long-term safety.
Microbes contain multiple ligands that can promote inflammasome activation, thus, therapies targeting the microbiome may also modulate inflammasome responses. Therapies employing microbes or their metabolites have shown some promise in modulating T1D development in animal models (164–168). While supplementation with bacterial-derived short chain fatty acids (SCFAs) protected NOD mice from the development of T1D (164, 168), a human intervention study in which butyrate was administered to longstanding T1D participants was found to have minimal immunological or metabolic effects compared to placebo-treated individuals (169). The human studies were not comparable with the NOD mouse studies however, and further investigation of SCFA administration including dose, duration and timing of treatment should be conducted in those at risk of developing T1D, if the human and mouse investigations are to be compared. In children, early probiotic administration (at the age of 0-27 days) was associated with reduced islet autoimmunity (autoantibodies), compared with children receiving probiotics later than 27 days of age, or those who had never received them (170). A recent study showed that β-cell function could be preserved in newly diagnosed T1D patients, who were recipients of an autologous fecal microbiota transplant, when compared to recipients of an allogeneic (healthy donors) fecal microbiota transplant (171). Together, these studies highlight the potential of harnessing the microbiota as a therapy to modulate ongoing immunity in T1D; however, these studies have not yet evaluated the involvement of the microbial-sensing pathways such as inflammasomes for their ability to modulate the development of diabetes or improved β-cell survival and function.
Inflammasomes are important activators of the innate immune response, leading to subsequent adaptive immune responses, particularly in response to microbial ligands. There has been a clear knowledge gap in understanding these inflammasomes in the context of Type 1 diabetes, but more studies are emerging highlighting the importance of the following areas - 1) single nucleotide polymorphisms in inflammasome genes; 2) priming of the inflammasome and 3) the function of the inflammasome proteins in modulating susceptibility to Type 1 diabetes. Together these studies indicate a need to better understand the role of inflammasomes in responding to the microbiota in Type 1 diabetes. At present, to achieve this would require investigators to 1) enlarge the sample sizes for the SNP association studies and investigate the mechanisms behind their association with disease; 2) decipher TLR signaling and inflammasome crosstalk in disease development; 3) investigate how inflammasomes specifically modulate microbial composition and 4) further evaluate inflammasome inhibitors in disease development and how these may be used therapeutically. While this is a new area of investigation, the evidence suggests that studying the inflammasome may provide another possible set of involved pathways that may be amenable to therapeutic targeting to prevent or delay Type 1 diabetes development. Finally, while inflammasomes may have a role in modulating susceptibility to T1D, we should not forget that they are likely to form a part of a multi-mechanistic pathway contributing to the development of T1D. Thus, assessing inflammasome activation in conjunction with other mechanisms of immune activation and regulation may be important to determine a broader picture for clinical interventions.
JAP wrote the review. FSW and LW edited the review. All authors contributed to the article and approved the submitted version.
This work was funded by a Medical Research Council Career Development Award (MR/T010525/1) to JP, MRC research grant (MR/K021141/1) to SW, and National Institutes of Health (DK 045735, HD 097808), Diabetes Action Research and Education Foundation and Diabetes Research Connection to LW.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Martinon F, Burns K, Tschopp J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of Proil-Beta. Mol Cell (2002) 10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3
2. Matzinger P. Tolerance, Danger, and the Extended Family. Annu Rev Immunol (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
3. Matzinger P. The Danger Model: A Renewed Sense of Self. Science (2002) 296(5566):301–5. doi: 10.1126/science.1071059
4. von Moltke J, Ayres JS, Kofoed EM, Chavarría-Smith J, Vance RE. Recognition of Bacteria by Inflammasomes. Annu Rev Immunol (2013) 31:73–106. doi: 10.1146/annurev-immunol-032712-095944
5. Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, et al. Activation of Interferon-Gamma Inducing Factor Mediated by Interleukin-1beta Converting Enzyme. Science (1997) 275(5297):206–9. doi: 10.1126/science.275.5297.206
6. Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 Processes IFN-Gamma-Inducing Factor and Regulates LPS-Induced IFN-Gamma Production. Nature (1997) 386(6625):619–23. doi: 10.1038/386619a0
7. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
8. Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, et al. AIM2 and NLRP3 Inflammasomes Activate Both Apoptotic and Pyroptotic Death Pathways Via ASC. Cell Death Differ (2013) 20(9):1149–60. doi: 10.1038/cdd.2013.37
9. Viganò E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A. Human Caspase-4 and Caspase-5 Regulate the One-Step Non-Canonical Inflammasome Activation in Monocytes. Nat Commun (2015) 6:8761. doi: 10.1038/ncomms9761
10. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 Recognizes Cytosolic dsDNA and Forms a Caspase-1-Activating Inflammasome With ASC. Nature (2009) 458(7237):514–8. doi: 10.1038/nature07725
11. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic Flagellin Activates Caspase-1 and Secretion of Interleukin 1beta Via Ipaf. Nat Immunol (2006) 7(6):569–75. doi: 10.1038/ni1344
12. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential Activation of the Inflammasome by Caspase-1 Adaptors ASC and Ipaf. Nature (2004) 430(6996):213–8. doi: 10.1038/nature02664
13. Kofoed EM, Vance RE. Innate Immune Recognition of Bacterial Ligands by NAIPs Determines Inflammasome Specificity. Nature (2011) 477(7366):592–5. doi: 10.1038/nature10394
14. Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE, Hornung V. Human NLRP1 Is a Sensor for Double-Stranded RNA. Science (2021) 29371(6528):eabd081. doi: 10.1126/science.abd0811
15. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-Associated Uric Acid Crystals Activate the NALP3 Inflammasome. Nature (2006) 440(7081):237–41. doi: 10.1038/nature04516
16. Mitoma H, Hanabuchi S, Kim T, Bao M, Zhang Z, Sugimoto N, et al. The DHX33 RNA Helicase Senses Cytosolic RNA and Activates the NLRP3 Inflammasome. Immunity (2013) 39(1):123–35. doi: 10.1016/j.immuni.2013.07.001
17. Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell (2015) 163(6):1428–43. doi: 10.1016/j.cell.2015.10.048
18. Hara H, Seregin SS, Yang D, Fukase K, Chamaillard M, Alnemri ES, et al. The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell (2018) 175(6):1651–64.e14. doi: 10.1016/j.cell.2018.09.047
19. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 Activates the Inflammasome and Cell Death in Response to Cytoplasmic DNA. Nature (2009) 458(7237):509–13. doi: 10.1038/nature07710
20. Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, et al. An Orthogonal Proteomic-Genomic Screen Identifies AIM2 as a Cytoplasmic DNA Sensor for the Inflammasome. Nat Immunol (2009) 10(3):266–72. doi: 10.1038/ni.1702
21. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes. Cell (2014) 156(6):1193–206. doi: 10.1016/j.cell.2014.02.008
22. Halff EF, Diebolder CA, Versteeg M, Schouten A, Brondijk TH, Huizinga EG. Formation and Structure of a NAIP5-NLRC4 Inflammasome Induced by Direct Interactions With Conserved N- and C-Terminal Regions of Flagellin. J Biol Chem (2012) 287(46):38460–72. doi: 10.1074/jbc.M112.393512
23. Hu Z, Yan C, Liu P, Huang Z, Ma R, Zhang C, et al. Crystal Structure of NLRC4 Reveals Its Autoinhibition Mechanism. Science (2013) 341(6142):172–5. doi: 10.1126/science.1236381
24. Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, et al. The NLR Gene Family: A Standard Nomenclature. Immunity (2008) 28(3):285–7. doi: 10.1016/j.immuni.2008.02.005
25. Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted NALP1 Inflammasome Reveals Two-Step Mechanism of Caspase-1 Activation. Mol Cell (2007) 25(5):713–24. doi: 10.1016/j.molcel.2007.01.032
26. Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, et al. GBP5 Promotes NLRP3 Inflammasome Assembly and Immunity in Mammals. Science (2012) 336(6080):481–5. doi: 10.1126/science.1217141
27. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 Inflammasome Regulates Colonic Microbial Ecology and Risk for Colitis. Cell (2011) 145(5):745–57. doi: 10.1016/j.cell.2011.04.022
28. Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L, et al. An NLRP7-Containing Inflammasome Mediates Recognition of Microbial Lipopeptides in Human Macrophages. Immunity (2012) 36(3):464–76. doi: 10.1016/j.immuni.2012.02.001
29. Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, et al. The NLRP12 Inflammasome Recognizes Yersinia Pestis. Immunity (2012) 37(1):96–107. doi: 10.1016/j.immuni.2012.07.006
30. Qu Y, Misaghi S, Izrael-Tomasevic A, Newton K, Gilmour LL, Lamkanfi M, et al. Phosphorylation of NLRC4 Is Critical for Inflammasome Activation. Nature (2012) 490(7421):539–42. doi: 10.1038/nature11429
31. Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, et al. Noncanonical Inflammasome Activation of Caspase-4/Caspase-11 Mediates Epithelial Defenses Against Enteric Bacterial Pathogens. Cell Host Microbe (2014) 16(2):249–56. doi: 10.1016/j.chom.2014.07.002
32. Grenier JM, Wang L, Manji GA, Huang WJ, Al-Garawi A, Kelly R, et al. Functional Screening of Five PYPAF Family Members Identifies PYPAF5 as a Novel Regulator of NF-kappaB and Caspase-1. FEBS Lett (2002) 530(1-3):73–8. doi: 10.1016/s0014-5793(02)03416-6
33. Hsu LC, Ali SR, McGillivray S, Tseng PH, Mariathasan S, Humke EW, et al. A NOD2-NALP1 Complex Mediates Caspase-1-Dependent IL-1beta Secretion in Response to Bacillus Anthracis Infection and Muramyl Dipeptide. Proc Natl Acad Sci USA (2008) 105(22):7803–8. doi: 10.1073/pnas.0802726105
34. Carlos D, Costa FR, Pereira CA, Rocha FA, Yaochite JN, Oliveira GG, et al. Mitochondrial DNA Activates the NLRP3 Inflammasome and Predisposes to Type 1 Diabetes in Murine Model. Front Immunol (2017) 8:164. doi: 10.3389/fimmu.2017.00164
35. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical Role for Calcium Mobilization in Activation of the NLRP3 Inflammasome. Proc Natl Acad Sci USA (2012) 109(28):11282–7. doi: 10.1073/pnas.1117765109
36. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K⁺ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity (2013) 38(6):1142–53. doi: 10.1016/j.immuni.2013.05.016
37. Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial Reactive Oxygen Species Induces NLRP3-Dependent Lysosomal Damage and Inflammasome Activation. J Immunol (2013) 191(10):5230–8. doi: 10.4049/jimmunol.1301490
38. Ghimire L, Paudel S, Jin L, Baral P, Cai S, Jeyaseelan S. NLRP6 Negatively Regulates Pulmonary Host Defense in Gram-Positive Bacterial Infection Through Modulating Neutrophil Recruitment and Function. PloS Pathog (2018) 14(9):e1007308. doi: 10.1371/journal.ppat.1007308
39. Chen J, Chen ZJ. PtdIns4P on Dispersed Trans-Golgi Network Mediates NLRP3 Inflammasome Activation. Nature (2018) 564(7734):71–6. doi: 10.1038/s41586-018-0761-3
40. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science (2013) 341(6151):1250–3. doi: 10.1126/science.1240988
41. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 Protects Against Bacteria That Escape the Vacuole. Science (2013) 339(6122):975–8. doi: 10.1126/science.1230751
42. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science (2013) 341(6151):1246–9. doi: 10.1126/science.1240248
43. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory Caspases Are Innate Immune Receptors for Intracellular LPS. Nature (2014) 514(7521):187–92. doi: 10.1038/nature13683
44. Chen GY, Liu M, Wang F, Bertin J, Núñez G. A Functional Role for NLRP6 in Intestinal Inflammation and Tumorigenesis. J Immunol (2011) 186(12):7187–94. doi: 10.4049/jimmunol.1100412
45. Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, et al. Nod-Like Receptor Pyrin Domain-Containing Protein 6 (NLRP6) Controls Epithelial Self-Renewal and Colorectal Carcinogenesis Upon Injury. Proc Natl Acad Sci USA (2011) 108(23):9601–6. doi: 10.1073/pnas.1100981108
46. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-Mediated Dysbiosis Regulates Progression of NAFLD and Obesity. Nature (2012) 482(7384):179–85. doi: 10.1038/nature10809
47. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat Med (2011) 17:179–88. doi: 10.1038/nm.2279
48. Wang P, Zhu S, Yang L, Cui S, Pan W, Jackson R, et al. NLRP6 Regulates Intestinal Antiviral Innate Immunity. Science (2015) 350(6262):826–30. doi: 10.1126/science.aab3145
49. Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 Negatively Regulates Innate Immunity and Host Defence Against Bacterial Pathogens. Nature (2012) 488(7411):389–93. doi: 10.1038/nature11250
50. Amer A, Franchi L, Kanneganti TD, Body-Malapel M, Ozören N, Brady G, et al. Regulation of Legionella Phagosome Maturation and Infection Through Flagellin and Host Ipaf. J Biol Chem (2006) 281(46):35217–23. doi: 10.1074/jbc.M604933200
51. Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozören N, Jagirdar R, et al. Cytosolic Flagellin Requires Ipaf for Activation of Caspase-1 and Interleukin 1beta in Salmonella-Infected Macrophages. Nat Immunol (2006) 7(6):576–82. doi: 10.1038/ni1346
52. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced Pyroptosis is an Innate Immune Effector Mechanism Against Intracellular Bacteria. Nat Immunol (2010) 11(12):1136–42. doi: 10.1038/ni.1960
53. Miao EA, Warren SE. Innate Immune Detection of Bacterial Virulence Factors Via the NLRC4 Inflammasome. J Clin Immunol (2010) 30(4):502–6. doi: 10.1007/s10875-010-9386-5
54. Hu C, Ding H, Li Y, Pearson JA, Zhang X, Flavell RA, et al. NLRP3 Deficiency Protects From Type 1 Diabetes Through the Regulation of Chemotaxis Into the Pancreatic Islets. Proc Natl Acad Sci USA (2015) 112(36):11318–23. doi: 10.1073/pnas.1513509112
55. Leite JA, Pessenda G, Guerra-Gomes IC, de Santana AKM, André Pereira C, Ribeiro Campos Costa F, et al. The DNA Sensor AIM2 Protects Against Streptozotocin-Induced Type 1 Diabetes by Regulating Intestinal Homeostasis Via the IL-18 Pathway. Cells (2020) 9(4). doi: 10.3390/cells9040959
56. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a New Gene Encoding a Putative Pyrin-Like Protein Causes Familial Cold Autoinflammatory Syndrome and Muckle-Wells Syndrome. Nat Genet (2001) 29(3):301–5. doi: 10.1038/ng756
57. Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, et al. Common Variants in the NLRP3 Region Contribute to Crohn’s Disease Susceptibility. Nat Genet (2009) 41(1):71–6. doi: 10.1038/ng.285
58. Pontillo A, Vendramin A, Catamo E, Fabris A, Crovella S. The Missense Variation Q705K in CIAS1/NALP3/NLRP3 Gene and an NLRP1 Haplotype are Associated With Celiac Disease. Am J Gastroenterol (2011) 106(3):539–44. doi: 10.1038/ajg.2010.474
59. Soares JL, Oliveira EM, Pontillo A. Variants in NLRP3 and NLRC4 Inflammasome Associate With Susceptibility and Severity of Multiple Sclerosis. Mult Scler Relat Disord (2019) 29:26–34. doi: 10.1016/j.msard.2019.01.023
60. Magitta NF, Bøe Wolff AS, Johansson S, Skinningsrud B, Lie BA, Myhr KM, et al. A Coding Polymorphism in NALP1 Confers Risk for Autoimmune Addison’s Disease and Type 1 Diabetes. Genes Immun (2009) 10(2):120–4. doi: 10.1038/gene.2008.85
61. Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, et al. NALP1 in Vitiligo-Associated Multiple Autoimmune Disease. N Engl J Med (2007) 356(12):1216–25. doi: 10.1056/NEJMoa061592
62. Pontillo A, Brandao L, Guimaraes R, Segat L, Araujo J, Crovella S. Two SNPs in NLRP3 Gene Are Involved in the Predisposition to Type-1 Diabetes and Celiac Disease in a Pediatric Population From Northeast Brazil. Autoimmunity (2010) 43(8):583–9. doi: 10.3109/08916930903540432
63. Sun X, Xia Y, Liu Y, Wang Y, Luo S, Lin J, et al. Polymorphisms in NLRP1 Gene Are Associated With Type 1 Diabetes. J Diabetes Res (2019) 2019:7405120. doi: 10.1155/2019/7405120
64. Casteels KM, Gysemans CA, Waer M, Bouillon R, Laureys JM, Depovere J, et al. Sex Difference in Resistance to Dexamethasone-Induced Apoptosis in NOD Mice: Treatment With 1,25(OH)2D3 Restores Defect. Diabetes (1998) 47(7):1033–7. doi: 10.2337/diabetes.47.7.1033
65. Zurawek M, Fichna M, Fichna P, Januszkiewicz D, Nowak J. No Evidence for Association of the Polymorphisms in NLRP1 Gene With Type 1 Diabetes in Poland. Diabetes Res Clin Pract (2011) 92(3):e49–51. doi: 10.1016/j.diabres.2011.02.004
66. Xu L, Sun X, Xia Y, Luo S, Lin J, Xiao Y, et al. Polymorphisms of the NLRC4 Gene Are Associated With the Onset Age, Positive Rate of GADA and 2-H Postprandial C-Peptide in Patients With Type 1 Diabetes. Diabetes Metab Syndr Obes (2020) 13:811–8. doi: 10.2147/DMSO.S244882
67. Verma D, Särndahl E, Andersson H, Eriksson P, Fredrikson M, Jönsson JI, et al. The Q705K Polymorphism in NLRP3 is a Gain-of-Function Alteration Leading to Excessive Interleukin-1β and IL-18 Production. PloS One (2012) 7(4):e34977. doi: 10.1371/journal.pone.0034977
68. Levandowski CB, Mailloux CM, Ferrara TM, Gowan K, Ben S, Jin Y, et al. NLRP1 Haplotypes Associated With Vitiligo and Autoimmunity Increase Interleukin-1β Processing Via the NLRP1 Inflammasome. Proc Natl Acad Sci USA (2013) 110(8):2952–6. doi: 10.1073/pnas.1222808110
69. Mullaney JA, Stephens JE, Costello ME, Fong C, Geeling BE, Gavin PG, et al. Type 1 Diabetes Susceptibility Alleles are Associated With Distinct Alterations in the Gut Microbiota. Microbiome (2018) 6(1):35. doi: 10.1186/s40168-018-0417-4
70. Russell JT, Roesch LFW, Ördberg M, Ilonen J, Atkinson MA, Schatz DA, et al. Genetic Risk for Autoimmunity Is Associated With Distinct Changes in the Human Gut Microbiome. Nat Commun (2019) 10(1):3621. doi: 10.1038/s41467-019-11460-x
71. Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, et al. Toward Defining the Autoimmune Microbiome for Type 1 Diabetes. ISME J (2011) 5(1):82–91. doi: 10.1038/ismej.2010.92
72. Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, et al. Gut Microbiota in Children With Type 1 Diabetes Differs From That in Healthy Children: A Case-Control Study. BMC Med (2013) 11:46. doi: 10.1186/1741-7015-11-46
73. de Groot PF, Belzer C, Aydin Ö, Levin E, Levels JH, Aalvink S, et al. Distinct Fecal and Oral Microbiota Composition in Human Type 1 Diabetes, An Observational Study. PloS One (2017) 12(12):e0188475. doi: 10.1371/journal.pone.0188475
74. Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The Human Gut Microbiome in Early-Onset Type 1 Diabetes From the TEDDY Study. Nature (2018) 562(7728):589–94. doi: 10.1038/s41586-018-0620-2
75. Stewart CJ, Ajami NJ, O’Brien JL, Hutchinson DS, Smith DP, Wong MC, et al. Temporal Development of the Gut Microbiome in Early Childhood From the TEDDY Study. Nature (2018) 562(7728):583–8. doi: 10.1038/s41586-018-0617-x
76. Roesch LF, Lorca GL, Casella G, Giongo A, Naranjo A, Pionzio AM, et al. Culture-independent Identification of Gut Bacteria Correlated With The Onset of Diabetes in A Rat Model. ISME J (2009) 3(5):536–48. doi: 10.1038/ismej.2009.5
77. Daft JG, Ptacek T, Kumar R, Morrow C, Lorenz RG. Cross-Fostering Immediately After Birth Induces a Permanent Microbiota Shift That Is Shaped by the Nursing Mother. Microbiome (2015) 3:17. doi: 10.1186/s40168-015-0080-y
78. Hu Y, Peng J, Li F, Wong FS, Wen L. Evaluation of Different Mucosal Microbiota Leads to Gut Microbiota-Based Prediction of Type 1 Diabetes in NOD Mice. Sci Rep (2018) 8(1):15451. doi: 10.1038/s41598-018-33571-z
79. de Goffau MC, Luopajärvi K, Knip M, Ilonen J, Ruohtula T, Härkönen T, et al. Fecal Microbiota Composition Differs Between Children With β-Cell Autoimmunity and Those Without. Diabetes (2013) 62(4):1238–44. doi: 10.2337/db12-0526
80. Endesfelder D, zu Castell W, Ardissone A, Davis-Richardson AG, Achenbach P, Hagen M, et al. Compromised Gut Microbiota Networks in Children With Anti-Islet Cell Autoimmunity. Diabetes (2014) 63(6):2006–14. doi: 10.2337/db13-1676
81. Kostic AD, Gevers D, Siljander H, Vatanen T, Hyötyläinen T, Hämäläinen AM, et al. The Dynamics of the Human Infant Gut Microbiome in Development and in Progression Toward Type 1 Diabetes. Cell Host Microbe (2015) 17(2):260–73. doi: 10.1016/j.chom.2015.01.001
82. Honeyman MC, Coulson BS, Stone NL, Gellert SA, Goldwater PN, Steele CE, et al. Association Between Rotavirus Infection and Pancreatic Islet Autoimmunity in Children At Risk of Developing Type 1 Diabetes. Diabetes (2000) 49(8):1319–24. doi: 10.2337/diabetes.49.8.1319
83. Laitinen OH, Honkanen H, Pakkanen O, Oikarinen S, Hankaniemi MM, Huhtala H, et al. Coxsackievirus B1 Is Associated With Induction of β-Cell Autoimmunity That Portends Type 1 Diabetes. Diabetes (2014) 63(2):446–55. doi: 10.2337/db13-0619
84. Bingley PJ, Bonifacio E, Williams AJ, Genovese S, Bottazzo GF, Gale EA. Prediction of IDDM in the General Population: Strategies Based on Combinations of Autoantibody Markers. Diabetes (1997) 46(11):1701–10. doi: 10.2337/diab.46.11.1701
85. Onodera T, Jenson AB, Yoon JW, Notkins AL. Virus-Induced Diabetes Mellitus: Reovirus Infection of Pancreatic Beta Cells in Mice. Science (1978) 201(4355):529–31. doi: 10.1126/science.208156
86. Yoon JW, Onodera T, Notkins AL. Virus-Induced Diabetes Mellitus. XV. Beta Cell Damage and Insulin-Dependent Hyperglycemia in Mice Infected With Coxsackie Virus B4. J Exp Med (1978) 148(4):1068–80. doi: 10.1084/jem.148.4.1068
87. Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The Prevalence of Enteroviral Capsid Protein Vp1 Immunostaining in Pancreatic Islets in Human Type 1 Diabetes. Diabetologia (2009) 52(6):1143–51. doi: 10.1007/s00125-009-1276-0
88. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Immunohistochemical Analysis of the Relationship Between Islet Cell Proliferation and the Production of the Enteroviral Capsid Protein, VP1, in the Islets of Patients With Recent-Onset Type 1 Diabetes. Diabetologia (2011) 54(9):2417–20. doi: 10.1007/s00125-011-2192-7
89. Busse N, Paroni F, Richardson SJ, Laiho JE, Oikarinen M, Frisk G, et al. Detection and Localization of Viral Infection in the Pancreas of Patients With Type 1 Diabetes Using Short Fluorescently-Labelled Oligonucleotide Probes. Oncotarget (2017) 8(8):12620–36. doi: 10.18632/oncotarget.14896
90. Graham KL, O’Donnell JA, Tan Y, Sanders N, Carrington EM, Allison J, et al. Rotavirus Infection of Infant and Young Adult Nonobese Diabetic Mice Involves Extraintestinal Spread and Delays Diabetes Onset. J Virol (2007) 81(12):6446–58. doi: 10.1128/JVI.00205-07
91. Graham KL, Sanders N, Tan Y, Allison J, Kay TW, Coulson BS. Rotavirus Infection Accelerates Type 1 Diabetes in Mice With Established Insulitis. J Virol (2008) 82(13):6139–49. doi: 10.1128/JVI.00597-08
92. Pane JA, Fleming FE, Graham KL, Thomas HE, Kay TW, Coulson BS. Rotavirus Acceleration of Type 1 Diabetes in non-Obese Diabetic Mice Depends on Type I Interferon Signalling. Sci Rep (2016) 6:29697. doi: 10.1038/srep29697
93. Serreze DV, Ottendorfer EW, Ellis TM, Gauntt CJ, Atkinson MA. Acceleration of Type 1 Diabetes by a Coxsackievirus Infection Requires a Preexisting Critical Mass of Autoreactive T-Cells in Pancreatic Islets. Diabetes (2000) 49(5):708–11. doi: 10.2337/diabetes.49.5.708
94. Drescher KM, Kono K, Bopegamage S, Carson SD, Tracy S. Coxsackievirus B3 Infection and Type 1 Diabetes Development in NOD Mice: Insulitis Determines Susceptibility of Pancreatic Islets to Virus Infection. Virology (2004) 329(2):381–94. doi: 10.1016/j.virol.2004.06.049
95. Serreze DV, Wasserfall C, Ottendorfer EW, Stalvey M, Pierce MA, Gauntt C, et al. Diabetes Acceleration or Prevention by a Coxsackievirus B4 Infection: Critical Requirements for Both Interleukin-4 and Gamma Interferon. J Virol (2005) 79(2):1045–52. doi: 10.1128/JVI.79.2.1045-1052.2005
96. Richer MJ, Lavallée DJ, Shanina I, Horwitz MS. Toll-Like Receptor 3 Signaling on Macrophages Is Required for Survival Following Coxsackievirus B4 Infection. PloS One (2009) 4(1):e4127. doi: 10.1371/journal.pone.0004127
97. McCall KD, Thuma JR, Courreges MC, Benencia F, James CB, Malgor R, et al. Toll-Like Receptor 3 Is Critical for Coxsackievirus B4-Induced Type 1 Diabetes in Female NOD Mice. Endocrinology (2015) 156(2):453–61. doi: 10.1210/en.2013-2006
98. Sioofy-Khojine AB, Lehtonen J, Nurminen N, Laitinen OH, Oikarinen S, Huhtala H, et al. Coxsackievirus B1 Infections Are Associated With the Initiation of Insulin-Driven Autoimmunity That Progresses to Type 1 Diabetes. Diabetologia (2018) 61(5):1193–202. doi: 10.1007/s00125-018-4561-y
99. Perrett KP, Jachno K, Nolan TM, Harrison LC. Association of Rotavirus Vaccination With the Incidence of Type 1 Diabetes in Children. JAMA Pediatr (2019) 173(3):280–2. doi: 10.1001/jamapediatrics.2018.4578
100. Pearson JA, Tai N, Ekanayake-Alper DK, Peng J, Hu Y, Hager K, et al. Norovirus Changes Susceptibility to Type 1 Diabetes by Altering Intestinal Microbiota and Immune Cell Functions. Front Immunol (2019) 10:2654. doi: 10.3389/fimmu.2019.02654
101. Cooke A, Tonks P, Jones FM, O’Shea H, Hutchings P, Fulford AJ, et al. Infection With Schistosoma Mansoni Prevents Insulin Dependent Diabetes Mellitus in non-Obese Diabetic Mice. Parasite Immunol (1999) 21(4):169–76. doi: 10.1046/j.1365-3024.1999.00213.x
102. Zaccone P, Burton O, Miller N, Jones FM, Dunne DW, Cooke A. Schistosoma Mansoni Egg Antigens Induce Treg That Participate in Diabetes Prevention in NOD Mice. Eur J Immunol (2009) 39(4):1098–107. doi: 10.1002/eji.200838871
103. Karumuthil-Melethil S, Gudi R, Johnson BM, Perez N, Vasu C. Fungal β-Glucan, a Dectin-1 Ligand, Promotes Protection From Type 1 Diabetes by Inducing Regulatory Innate Immune Response. J Immunol (2014) 193(7):3308–21. doi: 10.4049/jimmunol.1400186
104. Karumuthil-Melethil S, Sofi MH, Gudi R, Johnson BM, Perez N, Vasu C. TLR2- and Dectin 1-Associated Innate Immune Response Modulates T-cell Response to Pancreatic β-Cell Antigen and Prevents Type 1 Diabetes. Diabetes (2015) 64(4):1341–57. doi: 10.2337/db14-1145
105. Kowalewska B, Zorena K, Szmigiero-Kawko M, Wąż P, Myśliwiec M. Higher Diversity in Fungal Species Discriminates Children With Type 1 Diabetes Mellitus From Healthy Control. Patient Prefer Adherence (2016) 10:591–9. doi: 10.2147/PPA.S97852
106. Honkanen J, Vuorela A, Muthas D, Orivuori L, Luopajärvi K, Tejesvi MVG, et al. Fungal Dysbiosis and Intestinal Inflammation in Children With Beta-Cell Autoimmunity. Front Immunol (2020) 11:468. doi: 10.3389/fimmu.2020.00468
107. Bøås H, Tapia G, Sødahl JA, Rasmussen T, Rønningen KS. Enterobius Vermicularis and Risk Factors in Healthy Norwegian Children. Pediatr Infect Dis J (2012) 31(9):927–30. doi: 10.1097/INF.0b013e318258cdb5
108. Ludvigsson J, Jones MP, Faresjö Å. Worm Infestations and Development of Autoimmunity in Children - The ABIS Study. PloS One (2017) 12(3):e0173988. doi: 10.1371/journal.pone.0173988
109. Mobasseri M, Shirmohammadi M, Amiri T, Vahed N, Hosseini Fard H, Ghojazadeh M. Prevalence and Incidence of Type 1 Diabetes in the World: A Systematic Review and Meta-Analysis. Health Promot Perspect (2020) 10(2):98–115. doi: 10.34172/hpp.2020.18
110. Zaccone P, Burton OT, Gibbs SE, Miller N, Jones FM, Schramm G, et al. The S. Mansoni Glycoprotein ω-1 Induces Foxp3 Expression in NOD Mouse CD4⁺ T Cells. Eur J Immunol (2011) 41(9):2709–18. doi: 10.1002/eji.201141429
111. Iwasaki A, Medzhitov R. Toll-Like Receptor Control of the Adaptive Immune Responses. Nat Immunol (2004) 5(10):987–95. doi: 10.1038/ni1112
112. Wen L, Peng J, Li Z, Wong FS. The Effect of Innate Immunity on Autoimmune Diabetes and the Expression of Toll-Like Receptors on Pancreatic Islets. J Immunol (2004) 172(5):3173–80. doi: 10.4049/jimmunol.172.5.3173
113. Kim HS, Han MS, Chung KW, Kim S, Kim E, Kim MJ, et al. Toll-Like Receptor 2 Senses Beta-Cell Death and Contributes to the Initiation of Autoimmune Diabetes. Immunity (2007) 27(2):321–33. doi: 10.1016/j.immuni.2007.06.010
114. Wong FS, Hu C, Zhang L, Du W, Alexopoulou L, Flavell RA, et al. The Role of Toll-like Receptors 3 and 9 in the Development of Autoimmune Diabetes in NOD Mice. Ann NY Acad Sci (2008) 1150:146–8. doi: 10.1196/annals.1447.039
115. Zhang Y, Lee AS, Shameli A, Geng X, Finegood D, Santamaria P, et al. TLR9 Blockade Inhibits Activation of Diabetogenic CD8+ T Cells and Delays Autoimmune Diabetes. J Immunol (2010) 184(10):5645–53. doi: 10.4049/jimmunol.0901814
116. Tai N, Wong FS, Wen L. TLR9 Deficiency Promotes CD73 Expression in T Cells and Diabetes Protection in Nonobese Diabetic Mice. J Immunol (2013) 191(6):2926–37. doi: 10.4049/jimmunol.1300547
117. Gülden E, Ihira M, Ohashi A, Reinbeck AL, Freudenberg MA, Kolb H, et al. Toll-Like Receptor 4 Deficiency Accelerates the Development of Insulin-Deficient Diabetes in Non-Obese Diabetic Mice. PloS One (2013) 8(9):e75385. doi: 10.1371/journal.pone.0075385
118. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate Immunity and Intestinal Microbiota in the Development of Type 1 Diabetes. Nature (2008) 455(7216):1109–13. doi: 10.1038/nature07336
119. Gülden E, Chao C, Tai N, Pearson JA, Peng J, Majewska-Szczepanik M, et al. TRIF Deficiency Protects Non-Obese Diabetic Mice From Type 1 Diabetes by Modulating the Gut Microbiota and Dendritic Cells. J Autoimmun (2018) 93:57–65. doi: 10.1016/j.jaut.2018.06.003
120. Burrows MP, Volchkov P, Kobayashi KS, Chervonsky AV. Microbiota Regulates Type 1 Diabetes Through Toll-like Receptors. Proc Natl Acad Sci USA (2015) 112(32):9973–7. doi: 10.1073/pnas.1508740112
121. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS Signaling in C3H/HeJ and C57BL/10ScCr Mice: Mutations in Tlr4 Gene. Science (1998) 282(5396):2085–8. doi: 10.1126/science.282.5396.2085
122. Vatanen T, Kostic AD, d’Hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in Microbiome Lps Immunogenicity Contributes to Autoimmunity in Humans. Cell (2016) 165(4):842–53. doi: 10.1016/j.cell.2016.04.007
123. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a General Sensor of Peptidoglycan Through Muramyl Dipeptide (MDP) Detection. J Biol Chem (2003) 278(11):8869–72. doi: 10.1074/jbc.C200651200
124. Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, et al. An Essential Role for NOD1 in Host Recognition of Bacterial Peptidoglycan Containing Diaminopimelic Acid. Nat Immunol (2003) 4(7):702–7. doi: 10.1038/ni945
125. Yoshino H, Ueda T, Kawahata M, Kobayashi K, Ebihara Y, Manabe A, et al. Natural Killer Cell Depletion by Anti-Asialo GM1 Antiserum Treatment Enhances Human Hematopoietic Stem Cell Engraftment in NOD/Shi-scid Mice. Bone Marrow Transplant (2000) 26(11):1211–6. doi: 10.1038/sj.bmt.1702702
126. Costa FR, Françozo MC, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al. Gut Microbiota Translocation to the Pancreatic Lymph Nodes Triggers NOD2 Activation and Contributes to T1D Onset. J Exp Med (2016) 213(7):1223–39. doi: 10.1084/jem.20150744
127. Li YY, Pearson JA, Chao C, Peng J, Zhang X, Zhou Z, et al. Nucleotide-Binding Oligomerization Domain-Containing Protein 2 (Nod2) Modulates T1DM Susceptibility by Gut Microbiota. J Autoimmun (2017) 82:85–95. doi: 10.1016/j.jaut.2017.05.007
128. Parkhouse R, Boyle JP, Mayle S, Sawmynaden K, Rittinger K, Monie TP. Interaction Between NOD2 and CARD9 Involves the NOD2 NACHT and the Linker Region Between the NOD2 Cards and NACHT Domain. FEBS Lett (2014) 588(17):2830–6. doi: 10.1016/j.febslet.2014.06.035
129. Wu XM, Chen WQ, Hu YW, Cao L, Nie P, Chang MX. Rip2 Is a Critical Regulator for NLRs Signaling and MHC Antigen Presentation But Not for MAPK and PI3K/Akt Pathways. Front Immunol (2018) 9:726. doi: 10.3389/fimmu.2018.00726
130. Umiker B, Lee HH, Cope J, Ajami NJ, Laine JP, Fregeau C, et al. The NLRP3 Inflammasome Mediates DSS-Induced Intestinal Inflammation in Nod2 Knockout Mice. Innate Immun (2019) 25(2):132–43. doi: 10.1177/1753425919826367
131. Wullaert A, Lamkanfi M, McCoy KD. Defining the Impact of Host Genotypes on Microbiota Composition Requires Meticulous Control of Experimental Variables. Immunity (2018) 48(4):605–7. doi: 10.1016/j.immuni.2018.04.001
132. Khan AA, Yurkovetskiy L, O’Grady K, Pickard JM, de Pooter R, Antonopoulos DA, et al. Polymorphic Immune Mechanisms Regulate Commensal Repertoire. Cell Rep (2019) 29(3):541–50.e4. doi: 10.1016/j.celrep.2019.09.010
133. Liu H, Xu R, Kong Q, Liu J, Yu Z, Zhao C. Downregulated NLRP3 and NLRP1 Inflammasomes Signaling Pathways in the Development and Progression of Type 1 Diabetes Mellitus. BioMed Pharmacother (2017) 94:619–26. doi: 10.1016/j.biopha.2017.07.102
134. Rothe H, Jenkins NA, Copeland NG, Kolb H. Active Stage of Autoimmune Diabetes Is Associated With the Expression of a Novel Cytokine, IGIF, Which Is Located Near Idd2. J Clin Invest (1997) 99(3):469–74. doi: 10.1172/JCI119181
135. Amrani A, Verdaguer J, Thiessen S, Bou S, Santamaria P. IL-1alpha, IL-1beta, and IFN-Gamma Mark Beta Cells for Fas-Dependent Destruction by Diabetogenic CD4(+) T Lymphocytes. J Clin Invest (2000) 105(4):459–68. doi: 10.1172/JCI8185
136. Harms RZ, Yarde DN, Guinn Z, Lorenzo-Arteaga KM, Corley KP, Cabrera MS, et al. Increased Expression of IL-18 in the Serum and Islets of Type 1 Diabetics. Mol Immunol (2015) 64(2):306–12. doi: 10.1016/j.molimm.2014.12.012
137. Moran A, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Interleukin-1 Antagonism in Type 1 Diabetes of Recent Onset: Two Multicentre, Randomised, Double-Blind, Placebo-Controlled Trials. Lancet (2013) 381(9881):1905–15. doi: 10.1016/S0140-6736(13)60023-9
138. Thomas HE, Irawaty W, Darwiche R, Brodnicki TC, Santamaria P, Allison J, et al. IL-1 Receptor Deficiency Slows Progression to Diabetes in the NOD Mouse. Diabetes (2004) 53(1):113–21. doi: 10.2337/diabetes.53.1.113
139. Schott WH, Haskell BD, Tse HM, Milton MJ, Piganelli JD, Choisy-Rossi CM, et al. Caspase-1 Is Not Required for Type 1 Diabetes in the NOD Mouse. Diabetes (2004) 53(1):99–104. doi: 10.2337/diabetes.53.1.99
140. Wen L, Green EA, Stratmann T, Panosa A, Gomis R, Eynon EE, et al. In Vivo Diabetogenic Action of CD4+ T Lymphocytes Requires FAS Expression and Is Independent of IL-1 and IL-18. Eur J Immunol (2011) 41(5):1344–51. doi: 10.1002/eji.201041216
141. Marleau AM, Sarvetnick NE. Il-18 is Required for Self-Reactive T Cell Expansion in NOD Mice. J Autoimmun (2011) 36(3-4):263–77. doi: 10.1016/j.jaut.2011.02.005
142. Ablamunits V, Henegariu O, Hansen JB, Opare-Addo L, Preston-Hurlburt P, Santamaria P, et al. Synergistic Reversal of Type 1 Diabetes in NOD Mice With anti-CD3 and Interleukin-1 Blockade: Evidence of Improved Immune Regulation. Diabetes (2012) 61(1):145–54. doi: 10.2337/db11-1033
143. Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, et al. An Anti-CD3 Antibody, Teplizumab, in Relatives At Risk for Type 1 Diabetes. N Engl J Med (2019) 381:603–13. doi: 10.1056/NEJMoa1902226
144. Sims EK, Bundy BN, Stier K, Serti E, Lim N, Long SA, et al. Teplizumab Improves and Stabilizes Beta Cell Function in Antibody-Positive High-Risk Individuals. Sci Transl Med (2021) 13(583). doi: 10.1126/scitranslmed.abc8980
145. Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T, et al. Dectin-1 Is an Extracellular Pathogen Sensor for the Induction and Processing of IL-1β Via a Noncanonical Caspase-8 Inflammasome. Nat Immunol (2012) 13(3):246–54. doi: 10.1038/ni.2222
146. Antonopoulos C, Russo HM, El Sanadi C, Martin BN, Li X, Kaiser WJ, et al. Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling. J Biol Chem (2015) 290(33):20167–84. doi: 10.1074/jbc.M115.652321
147. Kono H, Orlowski GM, Patel Z, Rock KL. The IL-1-dependent Sterile Inflammatory Response Has a Substantial Caspase-1-Independent Component That Requires Cathepsin C. J Immunol (2012) 189(7):3734–40. doi: 10.4049/jimmunol.1200136
148. Karmakar M, Sun Y, Hise AG, Rietsch A, Pearlman E. Cutting Edge: IL-1β Processing During Pseudomonas Aeruginosa Infection Is Mediated by Neutrophil Serine Proteases and is Independent of NLRC4 and Caspase-1. J Immunol (2012) 189(9):4231–5. doi: 10.4049/jimmunol.1201447
149. Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A Small-Molecule Inhibitor of the NLRP3 Inflammasome for the Treatment of Inflammatory Diseases. Nat Med (2015) 21(3):248–55. doi: 10.1038/nm.3806
150. Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, et al. Inflammasome Inhibition Prevents α-Synuclein Pathology and Dopaminergic Neurodegeneration in Mice. Sci Transl Med (2018) 10(465). doi: 10.1126/scitranslmed.aah4066
151. Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, et al. Identification of a Selective and Direct NLRP3 Inhibitor to Treat Inflammatory Disorders. J Exp Med (2017) 214(11):3219–38. doi: 10.1084/jem.20171419
152. Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, et al. OLT1177, A β-Sulfonyl Nitrile Compound, Safe in Humans, Inhibits the NLRP3 Inflammasome and Reverses the Metabolic Cost of Inflammation. Proc Natl Acad Sci USA (2018) 115(7):E1530–E9. doi: 10.1073/pnas.1716095115
153. Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, et al. Tranilast Directly Targets NLRP3 to Treat Inflammasome-Driven Diseases. EMBO Mol Med (2018) 10(4). doi: 10.15252/emmm.201708689
154. Marchetti C, Swartzwelter B, Koenders MI, Azam T, Tengesdal IW, Powers N, et al. NLRP3 Inflammasome Inhibitor OLT1177 Suppresses Joint Inflammation in Murine Models of Acute Arthritis. Arthritis Res Ther (2018) 20(1):169. doi: 10.1186/s13075-018-1664-2
155. Yu SX, Du CT, Chen W, Lei QQ, Li N, Qi S, et al. Genipin Inhibits NLRP3 and NLRC4 Inflammasome Activation Via Autophagy Suppression. Sci Rep (2015) 5:17935. doi: 10.1038/srep17935
156. Maier NK, Leppla SH, Moayeri M. The Cyclopentenone Prostaglandin 15d-PGJ2 Inhibits the NLRP1 and NLRP3 Inflammasomes. J Immunol (2015) 194(6):2776–85. doi: 10.4049/jimmunol.1401611
157. Saadane A, Masters S, DiDonato J, Li J, Berger M. Parthenolide Inhibits IkappaB Kinase, NF-kappaB Activation, and Inflammatory Response in Cystic Fibrosis Cells and Mice. Am J Respir Cell Mol Biol (2007) 36(6):728–36. doi: 10.1165/rcmb.2006-0323OC
158. Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW, et al. Anti-Inflammatory Compounds Parthenolide and Bay 11-7082 Are Direct Inhibitors of the Inflammasome. J Biol Chem (2010) 285(13):9792–802. doi: 10.1074/jbc.M109.082305
159. Coll RC, Robertson A, Butler M, Cooper M, O’Neill LA. The Cytokine Release Inhibitory Drug CRID3 Targets ASC Oligomerisation in the NLRP3 and AIM2 Inflammasomes. PloS One (2011) 6(12):e29539. doi: 10.1371/journal.pone.0029539
160. Honda H, Nagai Y, Matsunaga T, Okamoto N, Watanabe Y, Tsuneyama K, et al. Isoliquiritigenin Is a Potent Inhibitor of NLRP3 Inflammasome Activation and Diet-Induced Adipose Tissue Inflammation. J Leukoc Biol (2014) 96(6):1087–100. doi: 10.1189/jlb.3A0114-005RR
161. Karki R, Kanneganti TD. Diverging Inflammasome Signals in Tumorigenesis and Potential Targeting. Nat Rev Cancer (2019) 19(4):197–214. doi: 10.1038/s41568-019-0123-y
162. Swanson KV, Deng M, Ting JP. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
163. Zheng D, Liwinski T, Elinav E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell Discovery (2020) 6:36. doi: 10.1038/s41421-020-0167-x
164. Mariño E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, et al. Gut Microbial Metabolites Limit the Frequency of Autoimmune T Cells and Protect Against Type 1 Diabetes. Nat Immunol (2017) 18(5):552–62. doi: 10.1038/ni.3713
165. Valladares R, Sankar D, Li N, Williams E, Lai KK, Abdelgeliel AS, et al. Lactobacillus Johnsonii N6.2 Mitigates the Development of Type 1 Diabetes in BB-DP Rats. PloS One (2010) 5(5):e10507. doi: 10.1371/journal.pone.0010507
166. Calcinaro F, Dionisi S, Marinaro M, Candeloro P, Bonato V, Marzotti S, et al. Oral Probiotic Administration Induces Interleukin-10 Production and Prevents Spontaneous Autoimmune Diabetes in the Non-Obese Diabetic Mouse. Diabetologia (2005) 48(8):1565–75. doi: 10.1007/s00125-005-1831-2
167. Pearson JA, Agriantonis A, Wong FS, Wen L. Modulation of the Immune System by the Gut Microbiota in the Development of Type 1 Diabetes. Hum Vaccin Immunother (2018) 14:2580–96. doi: 10.1080/21645515.2018.1514354
168. Huang J, Pearson JA, Peng J, Hu Y, Sha S, Xing Y, et al. Gut Microbial Metabolites Alter IgA Immunity in Type 1 Diabetes. JCI Insight (2020) 5(10):e135718. doi: 10.1172/jci.insight.135718
169. de Groot PF, Nikolic T, Imangaliyev S, Bekkering S, Duinkerken G, Keij FM, et al. Oral Butyrate Does Not Affect Innate Immunity and Islet Autoimmunity in Individuals With Longstanding Type 1 Diabetes: A Randomised Controlled Trial. Diabetologia (2020) 63(3):597–610. doi: 10.1007/s00125-019-05073-8
170. Uusitalo U, Liu X, Yang J, Aronsson CA, Hummel S, Butterworth M, et al. Association of Early Exposure of Probiotics and Islet Autoimmunity in the TEDDY Study. JAMA Pediatr (2016) 170(1):20–8. doi: 10.1001/jamapediatrics.2015.2757
Keywords: inflammasomes, microbiota, type 1 diabetes, NOD mice, humans
Citation: Pearson JA, Wong FS and Wen L (2021) Inflammasomes and Type 1 Diabetes. Front. Immunol. 12:686956. doi: 10.3389/fimmu.2021.686956
Received: 28 March 2021; Accepted: 17 May 2021;
Published: 09 June 2021.
Edited by:
Lucienne Chatenoud, Université Paris Descartes, FranceReviewed by:
Marika Falcone, San Raffaele Hospital, Scientific Institute for Research, Hospitalization and Healthcare (IRCCS), ItalyCopyright © 2021 Pearson, Wong and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James Alexander Pearson, cGVhcnNvbmoxQGNhcmRpZmYuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.