 C. Zografou
C. Zografou A. G. Vakrakou
A. G. Vakrakou P. Stathopoulos
P. Stathopoulos
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 17 June 2021
Sec. B Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.686466
This article is part of the Research TopicNew Insights into B cell Subsets in Health and DiseaseView all 18 articles
As B cells differentiate into antibody-secreting cells (ASCs), short-lived plasmablasts (SLPBs) are produced by a primary extrafollicular response, followed by the generation of memory B cells and long-lived plasma cells (LLPCs) in germinal centers (GCs). Generation of IgG4 antibodies is T helper type 2 (Th2) and IL-4, -13, and -10-driven and can occur parallel to IgE, in response to chronic stimulation by allergens and helminths. Although IgG4 antibodies are non-crosslinking and have limited ability to mobilize complement and cellular cytotoxicity, when self-tolerance is lost, they can disrupt ligand-receptor binding and cause a wide range of autoimmune disorders including neurological autoimmunity. In myasthenia gravis with predominantly IgG4 autoantibodies against muscle-specific kinase (MuSK), it has been observed that one-time CD20+ B cell depletion with rituximab commonly leads to long-term remission and a marked reduction in autoantibody titer, pointing to a short-lived nature of autoantibody-secreting cells. This is also observed in other predominantly IgG4 autoantibody-mediated neurological disorders, such as chronic inflammatory demyelinating polyneuropathy and autoimmune encephalitis with autoantibodies against the Ranvier paranode and juxtaparanode, respectively, and extends beyond neurological autoimmunity as well. Although IgG1 autoantibody-mediated neurological disorders can also respond well to rituximab induction therapy in combination with an autoantibody titer drop, remission tends to be less long-lasting and cases where titers are refractory tend to occur more often than in IgG4 autoimmunity. Moreover, presence of GC-like structures in the thymus of myasthenic patients with predominantly IgG1 autoantibodies against the acetylcholine receptor and in ovarian teratomas of autoimmune encephalitis patients with predominantly IgG1 autoantibodies against the N‐methyl‐d‐aspartate receptor (NMDAR) confers increased the ability to generate LLPCs. Here, we review available information on the short-and long-lived nature of ASCs in IgG1 and IgG4 autoantibody-mediated neurological disorders and highlight common mechanisms as well as differences, all of which can inform therapeutic strategies and personalized medical approaches.
B cells are the major components of the humoral adaptive immune system. Prior to antigenic stimulation, B cells develop in the bone marrow, where V (variable), D (diversity), and J (joining) gene recombination occurs, leading to the formation of the immunoglobulin antigen binding domains and the naïve B cell receptor repertoire. During this process of development and diversity generation, autoreactive clones are physiologically cleared by mechanisms imposed by two tolerance checkpoints: one central and one peripheral (1). Upon antigenic challenge, B cells of secondary lymphoid tissue are exposed to the antigen and form antibody-secreting B cells (ASC) by two complementary pathways: the first extrafollicular, and the second involving a germinal center (GC) reaction (follicular pathway) (2–4). A canonical response to a foreign antigen involves a switch from the first pathway to the second within approximately a week. The products of B cell development and differentiation—ASCs—can be divided into short-lived plasmablasts (SLPBs) and long-lived plasma cells (LLPCs). SLPBs express unswitched or isotype-switched immunoglobulin (Ig), and their formation can indicate a rapid antigen clearance response (5). In contrast, precursors of LLPCs are typically, but not always, isotype switched and upon exit from GCs either become peripheral memory B cells or enter a survival niche—such as the bone marrow—and become LLPCs.
Both SLPBs and LLPCs may contribute to the pathogenesis of neurological autoimmune diseases. Moreover, pathogenic autoantibodies produced by autoreactive ASCs and directed against neurologic antigens can either be predominantly of the IgG1 or the IgG4 subclass, or in rarer cases can be of both subclasses. Interestingly, the predominant subclass seems to be connected to whether autoantibody-secreting cells are short- or long-lived. A specific example relates to myasthenia gravis (MG) associated with predominantly IgG4 autoantibodies against muscle-specific kinase (MuSK), where autoreactive ASCs appear to be short-lived (6). This short-lived nature is supported by the observation that MuSK autoantibody titers decrease rapidly after CD20+ B cell depletion with rituximab (7–9). As most of the ASCs are CD20- and are not directly targeted by rituximab, titer reduction can be explained by depletion of the CD20+ ASC-progenitor cells in combination with the short-lived nature of MuSK ASCs. In MG, however, with predominantly IgG1 autoantibodies against the nicotinic acetylcholine receptor (AChR), titer decline post rituximab shows that B cell depletion varies from minimal (9, 10) to less pronounced in comparison to MuSK MG (11–13). Hence the AChR ASCs are presumed to be more long-lived (14). Of note, clinical responses to rituximab resemble —to some extent—autoantibody titers and comprise dramatic improvement in most cases of MuSK MG (8, 9, 15, 16) but are less pronounced (yet favorable in many cases) in AChR MG (NCT02110706) (17–24).
In this review, we aim to examine whether the MG paradigm extends to other autoimmune neurologic disorders with pathogenic autoantibodies of the IgG1 and IgG4 subclass. Further, we discuss how antigen-experienced B cells differentiate into predominantly IgG1- or IgG4- secreting ASCs and how IgG1 and IgG4 B cell responses generate short- and long-lived autoantibody-producing cells differently. While focusing on human data, we give an overview of how the different subclasses and ASCs contribute to different autoimmune neurological diseases, and in parallel, highlight advances in B cell biology that relate to the development of pathogenic autoantibodies.
With the exception of IgM autoantibodies against myelin-associated glycoprotein (MAG) (25), all pathogenic autoantibodies against neurologic cell surface protein antigens are class-switched immunoglobulins (IgG). In turn, presence of IgG is typically, but not always, associated with GC responses and the formation of long-lasting immunological memory. With the current wealth of information about the development of humoral responses, we can better appreciate the cellular and molecular mechanisms that lead to the generation of such responses to pathogens, as well as to allergens and autoantigens. In the following sections, we will briefly summarize current concepts of B cell responses, general rules and their exceptions, and further explain how these might be relevant to the formation of neurologic autoantibodies of the IgG1 and IgG4 subclass.
Observations of follicular and extrafollicular responses rely mainly on rodent models. Shortly after exposure to a T-cell-dependent antigen, responding B cells and T cells appear at the B cell follicle/T cell zone border of the lymph node (26). The initial humoral response involves B cells differentiating into SLPBs with the help of T follicular helper cells (Tfh) at extrafollicular sites (2, 27) (Figure 1). These extrafollicular B-cell responses generate many of the early-induced antibodies approximately four days to one week after exposure to the antigen (2, 28). In canonical responses, the extrafollicular response reverts after the first week and gives its place to the GC responses. However, extrafollicular responses persist in non-canonical responses [e.g. in the setting of Salmonella (29) and Borrelia infection (30) or certain types of autoimmunity such as rheumatoid arthritis patients’ synovia (31) and lupus-prone mice (32)]. Although characteristics of the GC response (somatic hypermutation, class-switch recombination and generation of B cell memory) can also be found in canonical and non-canonical extrafollicular responses (29, 31–37), they are present to a lesser extent. One exception is the ability to establish LLPCs, which is absent in the extrafollicular response (38–41). Classical views regard the extrafollicular response as a response that is promiscuous and of low, yet detectable specificity (29, 36, 42).

Figure 1 Differentiation of B cells into short- and long-lived antibody-secreting cells. In the initial phase of the immune response to a T cell-dependent antigen, responding naïve B cells appear in the T cell zone of the lymph node (upper left), where their development and differentiation is facilitated by T cell-secreted cytokines. T helper type 2 (Th2) type cytokine secretion, such as IL-4, -10, and -13 favors the induction of an IgG4 response. B cells enter the extrafollicular pathway and undergo B cell receptor (BCR) activation by encountering antigens on follicular dendritic cells (FDCs), which they then present to T follicular helper (Tfh) cells through MHC-II. The extrafollicular pathway gives rise to (i) short-lived plasmablasts (SLPBs) that enter the periphery, and (ii) germinal center (GC)-independent memory B cells. In a second phase, activated B cells enter the GC dark zone, where they mutate (a process called somatic hypermutation) and clonally expand (therefore termed centroblasts). B cells cycle between the dark and the light zone (where they are termed centrocytes). The dynamic cycle of the GC allows centrocytes that entered the light zone to be chosen based on the affinity of their BCRs to the antigen. Low-affinity B cells that are not presenting antigen on their BCRs will eventually become apoptotic and die. B cells that do present the antigen receive help from Tfh through CD40L and IL21 survival signals. The end-products of the GC reaction are (i) memory B cells, and (ii) long-lived plasma cells (LLPCs). GC memory B cells will enter the periphery and re-enter the GC upon BCR stimulation. LLPCs exit the GC and find a survival niche, typically the bone marrow.
Development of B cell immunologic memory and differentiation of B cells into high-affinity LLPCs primarily occurs within secondary lymphoid tissue—specifically the lymphoid follicles (B cell follicles) of the lymph nodes or the spleen, which harbor structures known as GCs. After the initial B cell response, the initiation of GCs is orchestrated by various immune cells, including B cells, helper T cells (such as Tfh cells driven by bcl-6), follicular dendritic cells (FDCs), and macrophages, as well as cytokines such as IL-6 (43–46). Since the discovery of GCs as affinity-maturation entities (47), microscopy-visualized GC reactions (48, 49) have shed light onto GC structure and dynamics. Two zones comprise the GC: the light zone and the dark zone; the light zone facilitates interaction with the antigen via Tfh and FDCs in order to select higher-affinity B cell clones while the dark zone facilitates B cell proliferation and somatic hypermutation in order to generate candidates for clonal selection in the light zone (50) (Figure 1). Accordingly, dark-zone B cells (centroblasts) engage mitosis-related genes and activation-induced cytidine deaminase (AID), thus enabling somatic hypermutation and class-switch recombination (51). Mature GCs allow entry to naïve B cells in addition to re-entry of higher-affinity clones, as repeated involvement of memory B cells in GC reactions has been observed in human lymph nodes (52). As a consequence, exported LLPCs display higher affinity and more somatic mutations compared to memory B cells (53). Overall, higher-affinity clones that mature within the dark zone differentiate into either LLPCs that migrate to the bone marrow or memory B cells, while lower-affinity clones undergo apoptosis.
ASCs exiting extrafollicular or follicular maturation processes are grossly divided into SLPBs and LLPCs and are connected to specific immunophenotypes measured with flow cytometry. More specifically, SLPBs are often described as CD20- CD19med/+ IgD- CD27hi CD38hi (of those, some but not all are also CD138+), while LLPCs are described as CD20- CD19- CD38hi CD138+ (54). It should be taken into account that as B cells differentiate toward high-affinity LLPCs, a phenotypic continuum is formed that does not entirely fit into the two immunophenotypes. Accordingly, a minority of presumably immature circulating SLPBs retain CD20 expression (55), and a minority of bone marrow LLPCs retain CD19 expression (56, 57). Both markers (CD20 and CD19) are ultimately lost as CD38 and CD138 expressions peak in Blimp-1-driven LLPCs.
While IgG1 is the most predominant IgG subclass in healthy adults (more than 60% of total IgG) and IgG4 by far the rarest (approximately 5% of total IgG) (58–61), IgG4 is of special interest not only because it is related to autoimmunity but also because of its anti-inflammatory properties and its coexistence with Th2-driven IgE (allergic) responses (62). In beekeepers, chronic exposure to allergen leads to upregulation of both IgE and IgG4 (63); IgG4 competes with IgE for the same antigen but has weak effector functions [mobilization of complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC)]. Moreover, IgG4 cannot crosslink the antigen as it exchanges its Fab arms to become functionally monovalent (64). Consequently, production of antigen-specific IgG4 protects beekeepers from IgE-mediated allergy. Allergen tolerization therapy (e.g., grass pollen) is clinically effective and induces an IgG4 antigen-specific response. Of note, repetitive or prolonged antigenic stimulation seems to elicit an IgG4 response in other settings as well, such as in chronic biologic therapeutics administration (e.g. clotting factor VIII, natalizumab, adalimumab) (65–67), repeated immunization (68), and helminth infection (69–73).
Both IgG4 and IgE class-switch and production are induced by IL-4 and IL-13 (74–76), however additional IL-10 signals divert Ig production in favor of IgG4 (77–79). Allergic sensitization and immediate hypersensitivity points to the presence of IgE memory and total IgE titers persist in actively atopic patients (10, 62, 80). Data from murine models point to IgE B cells showing difficulty remaining in the GC and generating memory B cells and LLPCs (81, 82). This is corroborated by a post-seasonal total IgG4/IgE titer drop (62) and transient-only IgE increases seen in non-atopic children (83). IgG4 B cells seem to be equally ineffective in producing LLPCs. Compared to IgG1 B cells, human IgG4 B cells express less CXCR4, a chemokine important for bone marrow chemotaxis, and low numbers of IgG4 cells are observed in human secondary lymphoid organs (84). Data from IgG4 related disease (IgG4-RD) also support the notion that the generation of LLPCs is diminished in IgG4 responses as levels of circulating IgG4 SLPBs correlate with total IgG4 levels. In addition, rituximab treatment affects a significant drop in IgG4 (and IgE) levels (85), but the drop in IgG1 titers is not as pronounced (86). It should be noted though that as the response to rituximab treatment can be partial, some LLPCs most likely do exist.
Regulatory T cell (Treg) involvement may be different in IgG4 and IgG1 responses. Apart from extrafollicular Tregs that could control GC initiation, follicular regulatory T cells (Tfr) may balance Tfh cells and participate in determining the fate of B cells. Tfr cells may either directly suppress GC B cells through CTLA-4-mediated inhibition of CD80/CD86 co-stimulatory signaling or indirectly do so through IL-10 secretion acting on Tfh cells (87). Importantly, allergen-specific Tregs from healthy individuals can suppress IgE and induce IgG4 production ex vivo via IL-10 and TGF-β (88, 89). During human helminth infection (which causes IgE and IgG4 elevation), Tregs that produce IL-10 and inhibit effector T cells can be found in the peripheral blood and may play a role in limiting inflammation (90), while in murine models of helminth infection, Tregs expand, produce IL-10, and can limit the Th1 more than the Th2 response (91). Similarly, Tregs are expanded in the peripheral blood of IgG4-RD patients (along with IgG4 and Th2 cells) (92, 93) and infiltrate target organs (along with IgG4 cells) (94). Conversely, in MG mediated by predominantly IgG1 autoantibodies against AChR, patients have been shown to harbor dysfunctional Tregs (95), and further, induction of Tregs via GM-CSF effectively suppressed experimental autoimmune MG (96). In pemphigus vulgaris (PV) mediated by predominantly IgG4 autoantibodies against desmoglein, contradicting data that Tregs have both not been able to suppress (97) and have suppressed autoimmune responses (98) are reported. These results underline the need for further investigations into the role of Tregs in autoantibody-mediated autoimmunity.
Class-switching recombination (CSR) is a fundamental change connected to the maturation of B cells as they evolve towards antibody secretion in response to antigens. CSR is facilitated by AID and materialized by the excision of DNA fragments and the subsequent joining of previously distant regions. As a consequence, CSR can only happen in the 5’-3’ direction of chromosomal topology of the different (corresponding to different classes and subclasses) constant region fragments (Figure 2). The use of high-throughput, next-generation sequencing (NGS) of the B cell receptor variable region and the beginning of the constant region (which can determine class and subclass) offers direct insight into human CSR mechanisms. Mutational analysis allows for the construction of B cell lineage trees, and at the same time, subclass is assigned to clonal family members. That way, one can pinpoint CSR with the use of somatic mutations as a ‘molecular clock’ (99). NGS analysis shows that the majority of naïve IgD/M switch to proximal classes (IgG3, IgG1, IgA1), and that the proximal classes then (secondarily, or indirectly) switch to more distal classes (IgG2, IgG4, IgA2). Indeed, more distal subclasses display greater mutational load, on average (61, 100–105).
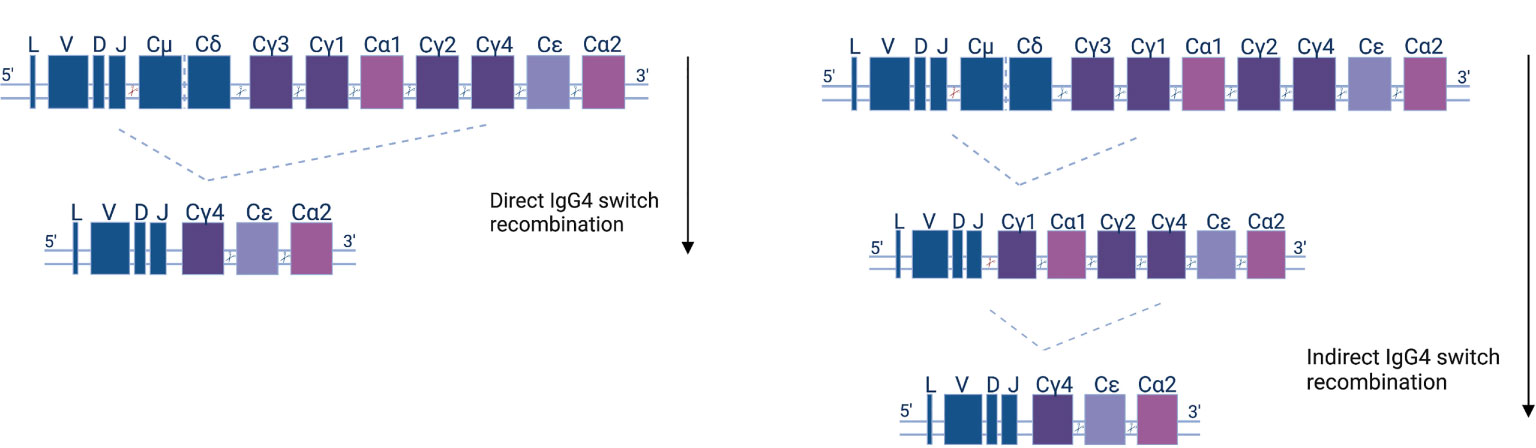
Figure 2 Human antibody isotype and class-switch recombination; diagram of the organization of the heavy chain gene locus. The constant genes are shown as squares and their width represents the relative gene size. Constant μ and δ (Cμ &Cδ) genes, preceded by the leader (L) and variable (V), diversity (D), or joining (J) gene regions, are expressed early in the B cell development. Recombination events (indicated as cutting sites) replace the Cμ and Cδ genes with other isotypes and subclasses (Cγ1-4, Cα1-2, and Cϵ) according to the depicted downstream order. IgG4 subclass antibodies can either be generated directly (left panel)—by IgM or IgD recombination and loss of the respective IGHC genes—or indirectly in two steps (right panel).
CSR and origins of IgE and IgG4 have been extensively exanimated in allergy and autoimmunity. In allergy, whole-repertoire NGS data from healthy and allergic individuals indicate that indirect switching from IgG1 is the primary source of IgE, while indirect switching from IgG4 is also significant (given overall rarity of IgG4) (103, 106). In accordance, examination of antigen-specific clonal families in allergic individuals showed common presence of IgE with predominantly IgG1, and to a lesser degree, IgG4 B cells within the same clonal family (107, 108). Most importantly, in pemphigus vulgaris (PV), a bullous skin disease mediated by pathogenic autoantibodies (mostly IgG4) against desmoglein, the NGS approach showed that IgG4 antigen-specific cells are not predominantly formed from IgG1 (or IgA) precursors. In fact, IgG1 and IgG4 desmoglein responses evolve in parallel along the same lineage tree, and it is plausible that the IgM-to-IgG4 direct switch is predominant for antigen-specific cells (109).
IgG1 and IgG4 autoantibodies bear different mechanisms of pathogenicity because of their molecular characteristics (e.g. P331S, L234F, P228S, A327G, and R409K amino acid substitutions in the IgG4 Fc region) (59, 79). Pro-inflammatory IgG1 autoantibodies activate CDC and ADCC through their constant fragment (Fc) and moreover can crosslink the antigen and affect its internalization. In contrast, IgG4 antibodies constitute a principally anti-inflammatory IgG subclass and have limited ability to mobilize CDC and ADCC due to a low affinity for C1q complement components and Fcγ receptors (110), however lgG4 antibodies can block the ligand-receptor interaction of the target antigen. One cardinal feature of IgG4 antibodies is that they undergo Fab-arm exchange, which results in bivalent binding properties of different specificities for each arm and thereby lose their ability to crosslink (64). This Fab-arm exchange effectively leads to functional monovalency of IgG4 antibodies. In addition to Fab-arm exchange, the lower mobility of IgG4s due to their shorter—compared to IgG1—hinge region complicates their structure, making IgG4 even less likely to be crosslinking antigens (111). Overall, the IgG4 subclass remains relatively poorly studied yet plays a fundamental role in neurological and non-neurological autoimmunity (58–61).
In many neurologic diseases, some of them newly defined, pathology is mediated by autoantibodies against surface/extracellular protein antigens. Among those are MG, chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), neuromyelitis optica and neuromyelitis optica spectrum disorders (NMOSD), acute disseminated encephalomyelitis (ADEM), relapsing optic neuritis (ON), pediatric acquired demyelinating syndromes, and autoimmune encephalitis (AE) (112–116). Such neuronal autoantibodies can affect multiple facets of the CNS and PNS as they target a wide spectrum of antigens, neurotransmitter receptors, ion channels and glycoproteins (Table 1). Clinical manifestations of neurological diseases mediated by autoantibodies are diverse and include fatigability, weakness, sensory and visual disturbances, movement and sleep disorders, epileptic seizures, decreased consciousness, and various cognitive symptoms. Serum and CSF autoantibodies have transitioned from being mere diagnostic tools to being defining factors of the clinical spectrum; and moreover, autoantibody titers can sometimes serve as markers of disease activity and more often of response to treatment. Of note, neuronal autoantigens can be classified into two categories depending on their localization: surface or intracellular. Pathogenicity of the latter is questionable, and as accessibility of intracellular targets to autoantibodies is limited in intact cells, the humoral response could be secondary to a primary cellular-damage event. In these cases, the primary response could involve a T cell (162, 212) or other cytotoxic immune cell-mediated mechanism. Here, we focus on cell surface protein antigens, where humoral responses are directly implicated in disease pathology.
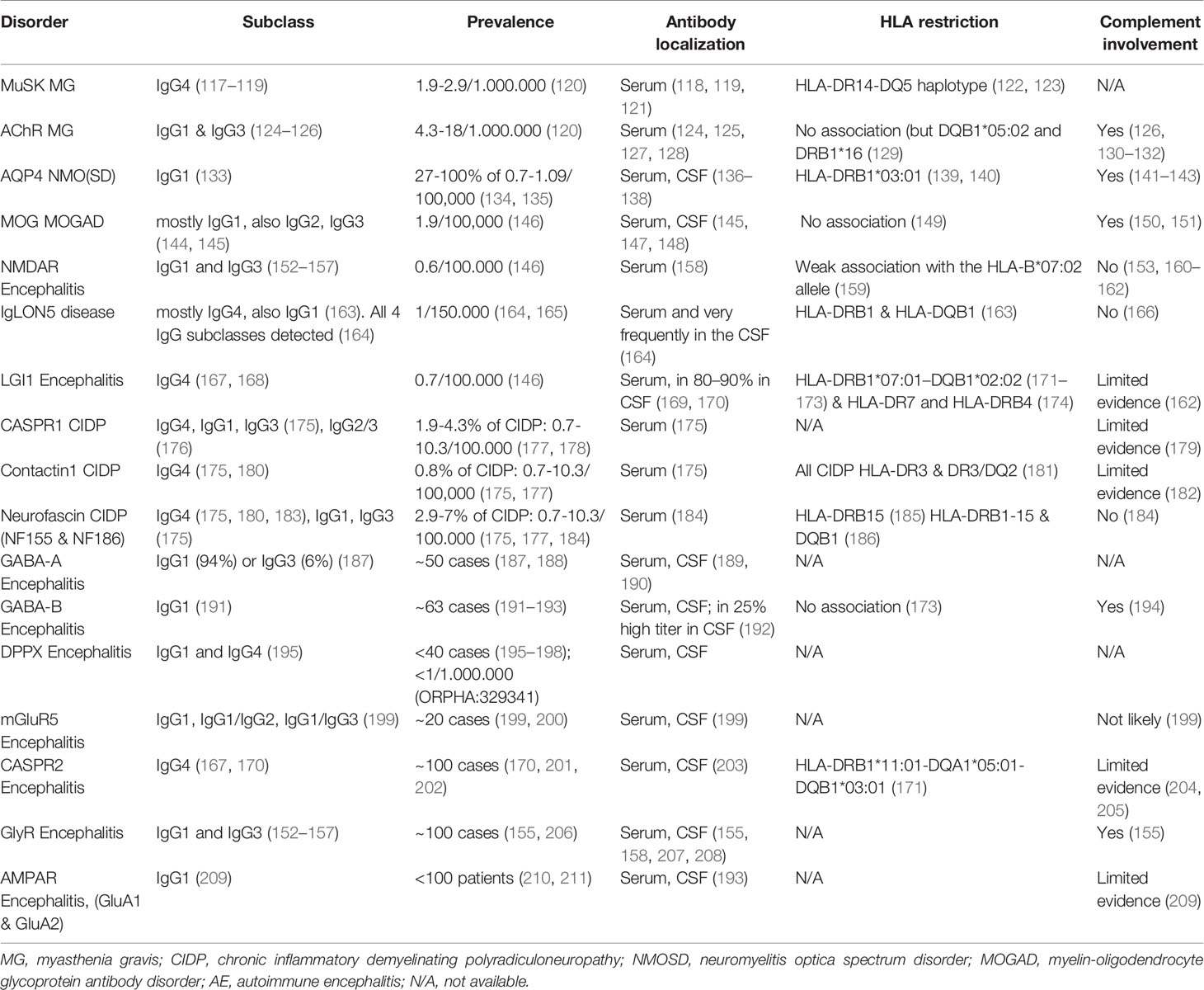
Table 1 Autoimmune neurological diseases mediated by IgG1 and IgG4 autoantibodies.
In addition to IgG1 autoantibodies, several different CNS or PNS antigens have been identified as targets of IgG4 autoantibodies, and this discovery has enabled us to categorize autoimmune diseases by pathophysiological mechanism. IgG4 autoantibody-mediated diseases are not driven by antibody effector functions but by direct and mechanical disruption of ligand-receptor interactions (175). Examples include antibodies specific to MuSK, a protein that is instrumental in the agrin-LRP4 pathway leading to AChR clustering (213); LGI1, a secreted protein that stabilizes the transsynaptic complex between the pre and postsynaptic receptors, ADAM23 and ADAM22 (169, 214, 215); and the protein complex of contactin-1, neurofascin-155 and caspr-1, which anchors myelin loops to the axon at the Ranvier paranode (216). A further interesting aspect of autoantibody-mediated neurological diseases is HLA restriction seen in patients as compared to healthy controls, likely meaning that specific antigenic peptides are better presented to T cells by specific HLA alleles (217, 218). This is relevant to B cell function, as B cells can pick up antigens with the B cell receptor (BCR) and process them and effectively present them via MHC II to T cells (219). Such a restriction has been shown both in diseases mediated by IgG1 and by IgG4 autoantibodies, and strong associations to specific—mainly class II—HLA haplotypes have been detected, with odds ratios exceeding 8 in disorders such as encephalitis with autoantibodies against leucine-rich glioma-inactivated protein 1 (LGI1) and contactin-associated protein 2 (caspr2) (171), chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) with autoantibodies against neurofascin 155 (NF155) (185) and IgLON5 disease (220).
The short- and long-lived nature of pathogenic autoantibodies is directly relevant to immunotherapeutic strategies. There are indications from the study of MG—the prototype of autoantibody-mediated diseases—that there is a systemic difference between predominantly IgG1 and predominantly IgG4 autoantibody responses regarding the longevity of ASCs. We have therefore examined the relevant evidence in the more common neurologic autoantibody-mediated disease entities. Data can be divided into two categories. First, we examined autoantibody titer response to rituximab, the CD20+ B cell depleting monoclonal antibody that has revolutionized neurologic therapeutics, and a rapid decline in autoantibody titer post-rituximab supports the short-lived nature of ASCs. Second, we reviewed studies that have directly examined ASCs by employing an array of techniques, from immunohistochemistry and bulk B cell culture to single B cell cloning and production of monoclonal antibodies. In particular, immune phenotypes of B cells from which antigen-specific monoclonal antibodies are derived provide information about the short- or long-lived nature of ASCs. These two groups of data are summarized in Table 2.
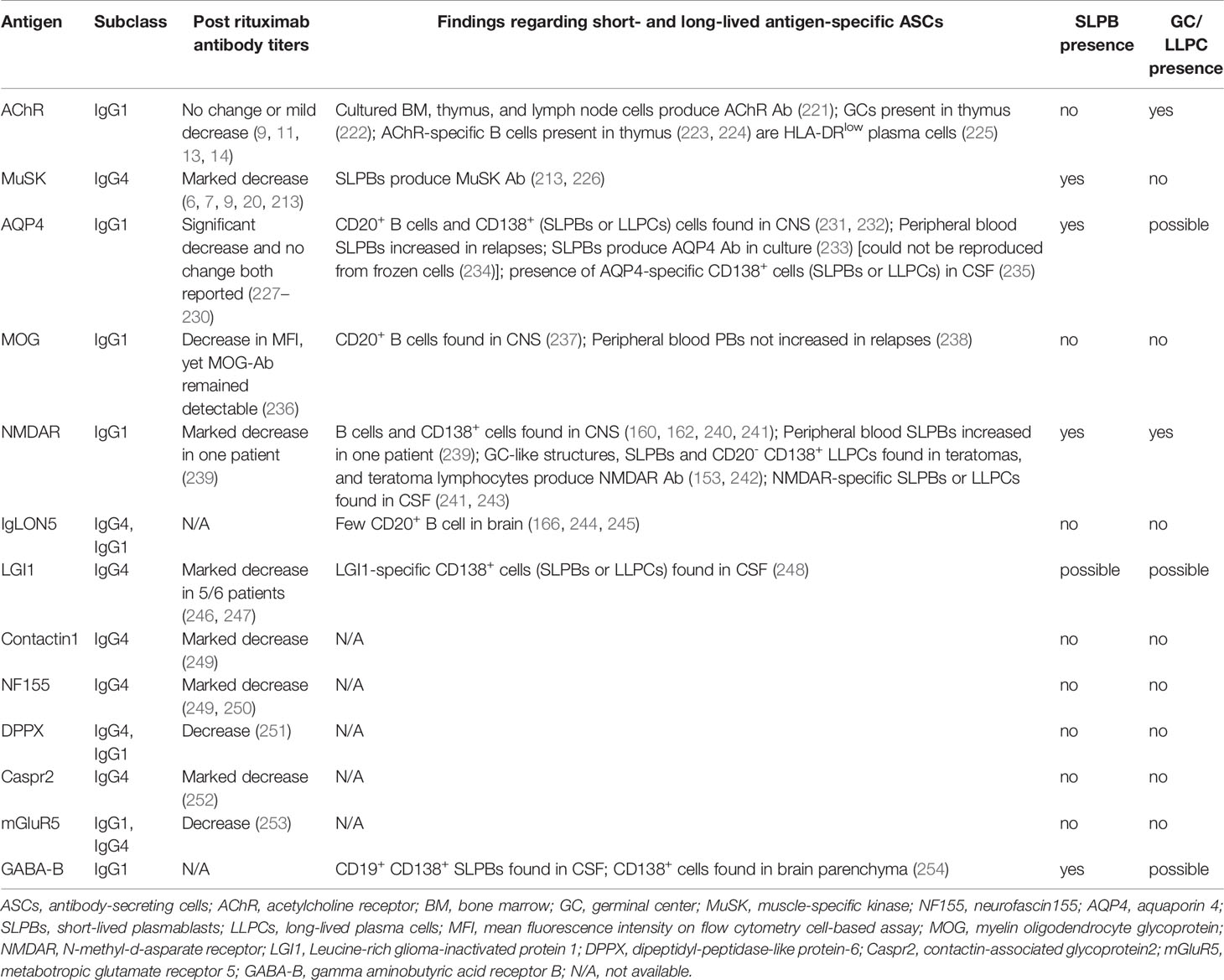
Table 2 Summary of data supporting presence of short and long-lived ASCs in neurological disorders.
It should be noted that rituximab-induced B cell depletion is materialized by ADCC, CDC, and antibody-dependent cellular phagocytosis (ADCP) (255). Circulating CD20+ B cells become undetectable almost immediately after rituximab treatment, and levels remain low for at least 6 months. Repeated dosing every 6 months can affect longer (than 6 months) B cell depletion after treatment ends (256–259). Moreover, rituximab affects a reduction but not a compete depletion in B cells of lymph nodes (260). Finally, repeated rituximab dosing results in gradual total serum IgM and IgG reduction (261).
Evidence from several case series suggests that rituximab is clinically effective in the majority of MG patients; especially in patients with MuSK MG the improvement is more pronounced compared to AChR MG (8, 9, 13, 16–24). Despite the many reports of good efficacy of rituximab in AChR MG, a one-year phase 2 trial of rituximab (NCT02110706) did not meet the primary endpoint, which was achievement of a 75% reduction in mean daily prednisone. This was possibly due to the study duration and a high percentage of patients meeting the endpoint in the placebo arm. The secondary endpoints, improvement in quantitative MG scales, was greater in the rituximab arm but the difference was not significant. This clinical difference between MuSK and AChR MG is reflected in autoantibody titers post-rituximab. Most patients with MuSK MG receiving rituximab show marked decline in MuSK autoantibody titer (7–9, 20). In contrast, titer decline in AChR MG is variable and in many patients is less pronounced (9, 11–14). Interestingly, the intensity of rituximab induction seems to be proportionate to the durability of the response of MuSK MG patients (262).
In parallel, cellular approaches have also contributed to deciphering of the short- or long-lived nature of ASCs in MG. In AChR MG, cultured bone marrow cells produced higher concentrations of AChR autoantibodies compared to peripheral blood, thymus, and lymph node lymphocytes, thus providing direct evidence for LLPC involvement in autoantibody production (221). Moreover, the presence of GCs in the thymus of early onset AChR MG patients underscores the ability to produce LLPCs (222). Of note, such thymic hyperplasia is not present in MuSK MG (263). The ability of thymic cells to produce AChR autoantibodies is well documented with different approaches (221, 223–225) and the contribution of LLPCs to this production is based on indirect observations, such as the absence of HLA-DR expression of some ASCs (225). Thymic ASCs can potentially survive in the thymus through constitutive stimulation by autoreactive T cells, and AChR-specific T cells have been found in the periphery of MG patients (264). In MuSK MG, evidence points to the presence of antigen-specific ASCs within the peripheral SLPB compartment. First, cultures of CD3- CD14- CD19med/+ IgD- CD27hi CD38hi SLPBs from the peripheral blood of MuSK MG patients produced MuSK autoantibodies (226). Second, IgG4 and IgG3 MuSK-specific monoclonal autoantibodies were able to be produced from the CD3- CD14- CD19med/+ IgD- CD27hi CD38hi SLPB fraction with the use of complementary single-cell sorting strategies (213, 226, 265). While this evidence for the presence of MuSK-specific SLPBs in MuSK MG is strong, the presence of MuSK-specific LLPCs cannot be excluded. Taken together, data point to autoantibody production that relies more on SLPBs in the predominantly IgG4 MuSK MG than in the predominantly IgG1 AChR MG, where the presence of antigen-specific LLPCs is well documented.
In NMO and NMOSD with IgG1 autoantibodies against aquaporin-4, rituximab has demonstrated remarkable clinical efficacy (229, 230). Titer response to B cell depletion with rituximab, however, has been variable. In a report about three patients, rituximab infusions led to parallel decreases in CD19+ count and AQP4 autoantibody titer. The response did not lead to total eradication of autoantibodies and was not particularly prolonged, since titers increased again along with CD19+ B cells after approximately one to one-and-a-half years later (227). This pattern was corroborated in a single patient where AQP4 antibodies were tested in nine different centers for validation purposes (266). In subsequent reports, titers for seven to thirteen patients were reported as a function of time, and responses to rituximab were mixed: autoantibody titers were refractory to rituximab in some patients, dropping in response to rituximab in others, and autoantibodies were beneath the level of detection in a third group (229, 230). A further study of the titers of five AQP4+ patients demonstrated a transient and incomplete response of titers to rituximab in three patients and a complete lack of response in the remaining two (228). Collectively these data point to variable titer responses and therefore to the presence of both AQP4-specific SLPBs and LLPCs.
Cellular approaches, both histopathological and ex vivo bulk and single-cell, have added important pieces of information on the nature of ASCs. First, biopsy and autopsy CNS studies have shown (i) perivascular B cells to a varying degree in addition to T cells (231, 267, 268); (ii) perivascular CD138+ cell infiltrates in one patient (232); and (iii) IL-6 transcripts in another patient (269). Second, peripheral blood CD19+ CD27hi CD38hi SLPBs were shown to increase in AQP4 autoantibody-positive NMO patients, more so during relapses. Importantly, these cells were able to produce AQP4 autoantibodies when cultured in the presence of IL-6, a cytokine also known to be increased in NMO relapses (270). These results were not reproducible in a follow up study, but it employed frozen cells, which could have impacted the viability of ASCs (234). Third, in a study of single CSF B cells from an early NMO patient, 3.7% of the CSF lymphocyte population was CD19+ CD138– B cells, and 0.9% were CD138+ ASCs; most of the CD138+ CSF ASCs (70.5%) were CD19+ CD138+ SLPBs, the rest were CD138+.CD19- LLPCs. Production of monoclonal antibodies from these mostly IgG1 (and rarely IgG2) CD138+ SLPBs or LLPCs demonstrated AQP4 specificity and somatic mutations (235). Moreover, AQP4 CSF SLPBs were found to be clonally related to peripheral plasmablasts as well as peripheral memory cells (271, 272). Of note, the involvement of IL-6, a cytokine that promotes GC formation as well as ASC survival, is further supported by the use of the anti-IL-6 receptor antibody tocilizumab in rituximab-resistant, aggressive cases of NMO (273). Interestingly, treatment of an AQP4 NMO patient with tocilizumab led to reduction in the frequency of CD19int CD27hi CD38hi SLPBs and anti-AQP4 antibody titer within one month of treatment (274). Taken together, data from serological and cellular approaches support a role for both short- and long-lived ASCs in AQP4 NMOSD pathology.
The pathological roles of anti-MOG IgG1 antibodies are not fully understood, and some MOG antibody disease (MOGAD) patients exhibit high titers of autoantibodies with pathogenic properties, while other patients—along with healthy and disease controls—exhibit lower titers (275, 276). Of note and in contrast to AQP4 NMOSD, clinical response of MOGAD patients to rituximab is modest, with up to a third of patients relapsing despite full depletion of peripheral B cells (236, 277–279). Currently there are no reliable predictors of inadequate response to rituximab in MOGAD patients, and the post-rituximab pattern of memory B-cell compartment population did not differ between responders and non-responders (236). In a case series of 16 MOGAD patients who received rituximab, mean fluorescence intensity (MFI) in the flow cytometric autoantibody detection cell-based assay decreased in most patients, while MOG antibodies remained detectable, which suggests a role for both SLPBs and LLPCs in MOG-IgG production (236). Of note, MFI could be viewed as a correlate of autoantibody titer. In pathological investigations, CD20+ B cells have been identified in the brain of MOGAD patients (237). Moreover, unlike patients with AQP4-NMOSD, where SLPBs were elevated, peripheral blood SLPBs were not elevated in the active phase of MOGAD patients (238). Taken together, serological and cellular data point to the presence of a mixed (both short- and long-lived) population of MOG autoantibody-secreting cells.
Antibodies against the N‐methyl‐d‐aspartate receptor (NMDAR) are predominately of the IgG1 subclass (280). Patients with NMDAR encephalitis generally respond to rituximab, and in one case titers were undetectable post-treatment (152, 239, 280–282). In an effort to enhance the effect of rituximab induction treatment, two non-randomized trials observed clinical benefit from repeated monthly dosing of rituximab in addition to induction dosing (283); and the addition of tocilizumab to rituximab (240). Several investigations of cellular immunopathology have been applied in NMDAR encephalitis. First, a histopathological study of autopsy and biopsy material revealed the presence of B cells and CD138+ cells in perivascular regions and interstitial spaces that could provide a local source of antibody production, as well as the presence of T cells and the absence of complement deposits and neuronal loss (153, 160, 162, 241). Moreover, a histopathological study of teratomas (seen in 20% of patients with NMDAR encephalitis) has shown GC-like structures harboring CD3+ T cells, CD20+ B cells, CD19+ CD27hi CD38hi SLPBs, and CD20- CD138+ plasma cells (284, 285). Importantly, NR1 NMDAR subunit expression was high in teratoma B cells, and teratoma-derived lymphocytes were able to produce NMDAR autoantibodies when stimulated in culture.
Two studies analyzed the intrathecal cellular response in NMDAR encephalitis with the use of flow cytometric cell sorting and construction of monoclonal antibodies from single cells (241, 243). Cells that were NMDAR-specific were identified as IgG3 CD3- CD14- CD16- CD20+ IgD- CD27+ memory B cells, IgG1 and IgG2 CD3- CD14- CD16- CD27+ CD38+ ASCs (could be both SLPBs or LLPCs) (243) and CD19+ CD138+ SLPBs (241). NMDA specificity was associated in most, but not all, cases with somatic mutations, which indicates some degree of affinity maturation. Interestingly, CD19+ CD138+ SLPBs disappeared from the CSF after immunotherapy with methylprednisolone, mycophenolate and azathioprine (241). These collective data suggest involvement of IgG1, IgG2, and IgG3, both SLPBs and LLPCs, in the production of the pathogenic NMDAR autoantibodies.
In IgLON5 disease, patients harbor autoantibodies of all four IgG subclasses, with some studies reporting a predominance of IgG4 and others of both IgG1 and IgG4 (163, 164, 286–288). In vitro experiments have shown that IgG1 (not IgG4) antibody binding to IgLON5 results in protein internalization and an overall decrease of neuron surface IgLON5 levels. This was not reversed when IgLON5 antibodies were removed, thereby suggesting permanent destruction of the protein’s biological function (286). In accordance, clinical data demonstrate better effectiveness of early compared to late immunotherapy (164, 288–290). Different series report rituximab use in 5–80% of IgLON5 patients (163–165, 288, 289, 291), and the response rate was calculated by a recent meta-analysis at 37.5% (292). More insight is needed about whether this variable response to rituximab is associated to the depletion of certain subclasses and whether prompt administration positively impacts patient outcome. In isolated cases where brain pathology was performed, few brain-infiltrating B cells were detected and no CD138 staining was reported (166, 244, 245).
Leucine-rich glioma-inactivated protein 1 (LGI1) antibodies are mainly of IgG4 subclass (168). A study of rituximab treatment in six patients with LGI1 encephalitis resulted in clear improvement in only two patients. However, the treatment might have been applied too late in the course of the disease in refractory cases (247, 293). The response of autoantibody titers though was a marked reduction in all cases but one, where the decline was mild. These data are in agreement with titer responses in other IgG4 autoantibody-mediated disorders. Immunopathology studies in brain samples from human patients and cats with LGI1 encephalitis indicate participation of CD20+ B cells in CNS inflammatory infiltrates, as well as marked IgG and complement deposition (162, 294). The presence of complement deposition points to the putative role of other, coexisting, non-IgG4 antibodies as complement activating factors, or alternatively, points to the ability of IgG4 antibodies to mobilize complement despite classical views, possibly via altered IgG4 glycosylation and the lectin pathway (295–299), or IgG4 aggregation (79).
Single-cell approaches from the CSF of patients with long-lasting progressive LGI1 encephalitis identified IgG1, IgG2, and IgG4, LGI1-specific CD3- CD14- CD16- CD20+ CD27+ memory B cells and CD3- CD14- CD16- CD138+ ASCs (could be both SLPBs or LLPCs); V gene sequences of the LGI1-specific B cells were mutated (248). In a separate study, application of BCR NGS on both sides of the blood-brain barrier provided strong evidence in favor of GC reactions within the CNS. However, this was not shown for LGI1-specific B cells (300). Taken together, the serological evidence points to a predominance of short-lived LGI1 autoantibody-producing cells over LLPCs, while cellular data are not conclusive for the presence of a particular cell type.
In CIDP with predominantly IgG4 autoantibodies against the paranodal components neurofascin-155 (NF155), contactin-1, or contactin-associated protein 1 (Caspr1), rituximab has been applied to corticosteroid and IVIg-refractory cases (249). Albeit limited by the low N given the rarity of the disease, antibody titers dropped significantly and rapidly in two patients (one with NF155 and one with contactin1 autoantibodies) after rituximab treatment, which correlated with marked clinical improvement. In a third patient (with NF155 autoantibodies), titers dropped but remained high, and a second rituximab infusion was required, after which autoantibodies were undetectable. In a larger cohort of 13 patients (8 with NF155 and 5 with contactin1 antibodies), rituximab administration resulted in a dramatic reduction of autoantibody titer that correlated with clinical improvement (250). One further study reported on titer drop (from 1:32,000 to 1:4,000) and clinical improvement after rituximab administration in a NF155 patient, even though the titers remained high (301). These studies provide evidence for a similar pattern of response in autoantibody-mediated CIDP that resembles MuSK MG.
In progressive encephalopathy with rigidity and myoclonus (PERM), where IgG1 autoantibodies against the glycine receptor have been found, rituximab has been associated with some improvement and lack of relapse (155, 302). In encephalitis with predominantly IgG1 autoantibodies against the GABA-B receptor (191), both partial and full response to rituximab have been noted in two patients (303). In encephalitis with predominantly IgG1 autoantibodies against the GABA-A receptor (187), rituximab has been administered (189), and a range of responses from full recovery to death has been documented (188, 304). Of interest, in a study of three patients with GABA-B encephalitis, CD19+ CD138+ SLPBs were seen in the CSF, along with CD138+ cells (that could be SLPBs or LLPCs) perivascularly in the brain parenchyma (254).
In dipeptidyl-peptidase-like protein-6 (DPPX) encephalitis, autoantibodies are of both the IgG1 and IgG4 subclass, but pathogenicity might be linked to IgG1 and its cross-linking functions (195, 196). In contactin-associated protein-2 (caspr2) encephalitis, autoantibodies are predominantly of the IgG4 subclass (170). In a metabotropic glutamate receptor type 5 (mGluR) encephalitis case series of 11 patients (199), autoantibodies were predominantly IgG1, but the co-existence of IgG1 with IgG4 autoantibodies has been noted in one case (253). Autoantibody titer data in response to rituximab treatment are available in all three disorders. In DPPX encephalitis, a significant clinical response to rituximab (195, 305) along with titer reduction (251) has been observed. In caspr2 encephalitis, a dramatic improvement along with autoantibody elimination has been observed (252), however post rituximab relapse has also been noted (306). Favorable clinical response to rituximab in Caspr2 encephalitis has further been observed in larger case series (170, 307). In mGluR encephalitis, rituximab has been associated with an improved course in two patients with IgG1 and IgG3 autoantibodies (199), with relapse upon rituximab discontinuation in a patient where the subclass was not specified (308), and with significant improvement along with titer reduction in a patient who originally harbored IgG1 and IgG4 autoantibodies (253). Overall, both clinical and titer responses seem to be more favorable in the case of IgG4 autoantibodies as in the case of Caspr2, and good responses in cases with IgG1 and mixed subclass autoantibodies have also been frequently noted; however, the limited number of patients given the rarity of the syndromes makes general conclusions difficult to draw.
The paradigm we discuss for neurological diseases associated with or mediated by predominantly IgG4 autoantibodies is not confined to the nervous system. CD20+ B cell depletion therapy has been applied in a plethora of non-neurological diseases associated with IgG4 autoantibodies such as pemphigus vulgaris (PV), membranous nephropathy (MN), and thrombotic thrombocytopenic purpura (TTP). In all these diseases, B cell depletion has resulted in marked clinical improvement and rapid decrease in autoantibody titers, similar to IgG4 autoantibody-mediated neurological disorders (309–313). A specific mention should be given to PV, which was the first autoimmune disease described as predominantly IgG4 autoantibody-mediated and has been extensively studied. High quality data from a prospective, multicenter, open-label, randomized trial of continued rituximab administration in PV demonstrated complete and sustained remission at the end of the second year of follow-up in 89% of 46 patients who received rituximab; rapid normalization of anti-desmoglein-3 (DSG-3) antibody titers post B cell depletion was also shown, thereby underscoring the short-lived nature of ASCs producing anti-DSG-3 autoantibodies (314, 315). In TTP, rituximab induction therapy of 40 patients resulted in a rapid and sustained recovery of platelet counts and, in parallel, a rapid and sustained decrease of pathogenic, predominantly IgG4 anti- A disintegrin and metalloproteinase with thromboSpondin‐1 motifs; 13th member of the family (ADAMTS13) autoantibody titers (310). In membranous nephropathy, rituximab administration resulted in significant decline or disappearance of the predominantly IgG4 (likely pathogenic) autoantibodies against phospholipase A2 receptor in 68% of 35 patients within 12 months, correlating with partial or complete clinical remission (311). Taken together, these studies show that the short-lived nature of autoantibody-secreting cells (as evidenced by post-rituximab autoantibody titer data) in diseases associated with IgG4 autoantibodies is not a phenomenon restricted to the nervous system.
When collectively examining the titer of pathogenic autoantibodies against extracellular antigens (or correlates of an antibody titer on a flow cytometric cell-based assay like MFI), one can conclude that in disorders where IgG4 autoantibodies are prevalent, there is a marked decrease post-rituximab administration. This is the case in MuSK MG, in CIDP with antibodies against NF155 or contactin1, and in autoimmune encephalitis with LGI1 or Caspr2 antibodies, and also extends beyond the nervous system to disorders such as PV, TTP and MN. This is a clear indication that in these diseases, autoantigen-specific ASCs are short-lived. Further, decreases in post-rituximab titers are seen in disorders where IgG1 and IgG4 autoantibodies coexist, such as DPPX and mGluR5 encephalitis. In disorders where IgG1 autoantibodies are prevalent, rituximab treatment affects a variable titer response, meaning that in some patients, titers are refractory, in some patients, titers mildly decline, and in other patients the reduction of titer or MFI is more pronounced. This variable response is noted in both AChR MG and AQP4 NMO and NMOSD, but in MOGAD MFI decrease seems to be consistent. These results point to the presence of antigen-specific LLPCs in a significant number of patients harboring IgG1 autoantibodies, but also the presence of SLPBs. It should be noted that these studies are complicated by the fact that the disorders in question are rare and therefore the N is low. Moreover, titers are not systematically recorded pre- and post-rituximab. As titers offer valuable information about treatment responses, every effort should be made to record titers more frequently.
When collectively interpreting data from experiments that more directly examine autoantigen-specific ASCs, one can conclude that in disorders with IgG4 autoantibodies, the presence of LLPCs is not definitively shown. In MuSK MG, peripheral blood antigen-specific ASCs have been shown to have a SLPB phenotype, whereas in LGI1 encephalitis, CSF antigen-specific ASCs can express CD138. However, it was not specified whether these cells retain CD19 expression. Therefore, these cells could be either SLPBs or LLPCs, which means that the presence of some LLPCs in IgG4-mediated disorders cannot be excluded. In contrast, in diseases with IgG1 autoantibodies there is more definitive evidence for the presence of LLPCs. In AChR MG, production of autoantibodies from bone marrow cells and the presence of thymic GCs and HLA-DRlow antigen-specific cells all point to the existence and ability to generate autoantigen-specific LLPCs. In NMDAR encephalitis, the presence of GC-like structures and CD20- CD138+ ASCs in ovarian teratomas also point to the existence of and ability to produce LLPCs. Moreover, CD138+ ASCs have been observed in the CNS and the CSF of AQP4 NMOSD, NMDAR and GABA-B encephalitis patients, and in the case of AQP4 and NMDAR, the antigen specificity of the CSF ASCs was demonstrated. Unfortunately, and similarly to the case of the IgG4 LGI1 encephalitis, a clear absence of CD19/CD20 staining and negativity prevents the definitive characterization of these cells as LLPCs. In such instances, use of an additional flow cytometric marker in combination with index sorting and the use of an additional immunohistochemical stain would permit capture of this mechanistically valuable piece of information.
In conclusion, it seems that the paradigm of the predominantly IgG4 MuSK and predominantly IgG1 AChR MG can be extrapolated to other autoimmune neurological (and non-neurological) disorders. Moreover, in disorders with IgG1 autoantibodies, the generation of antigen-specific LLPCs seems to occur to a greater extent as compared to IgG4 disorders. It should be noted, however, that a significant degree of variability exists and that both antigen-specific LLPCs can be generated in some patients with IgG4 autoantibody-mediated disorders, and SLPBs—perhaps more frequently—can be significant producers of autoantibodies in some patients with IgG1 disorders. Variability in relation to the nature of autoantibody-producing cells could also occur at different times in the same patient. This variability underscores the need for personalized medical approaches. Exceptions aside, a generally reduced ability to establish LLPCs in IgG4 responses is strongly supported by immunological observations on the longevity of ACSs from the field of allergy (both in humans and animal models) and IgG4-related disease. It could be the case that IgG4 autoantibody-mediated autoimmunity constitutes a mainly extrafollicular response, but follicular hyperplasia (in the absence of pathogenic antigen-specificity) has been observed in IgG4-RD (316). The tendency to generate predominantly IgG1 or IgG4 autoimmune responses may stem from HLA and/or non-HLA genetic differences (317, 318), but incomplete GWAS data (due to disease rarity) would have to be complemented by functional studies to better support such an argument. On the other hand, many aspects of IgG1 and IgG4 autoimmunity are similar, such as B cell tolerance defects resulting in autoreactive naïve B cells (234, 319–321) and T cell-assisted autoantigen affinity maturation, as evidenced by the presence of somatic mutations in most ASCs. In further support of the role of T cell help, autoreactive T cells have been observed in both IgG1 and IgG4 autoantibody-mediated disorders (264, 322, 323).
Differences between IgG4 and IgG1 autoimmune responses are not clinically trivial and can inform therapeutic decisions, especially since IgG4 autoantibody pathology responds impressively well to rituximab induction. More specifically, in IgG4 autoantibody-mediated disorders, prompt induction with rituximab 375 mg/m2 once a week for 4 to 6 weeks can result in a long-lasting favorable response and is highly recommended. The same induction can be applied in IgG1 disorders. It is important to obtain a pre-rituximab baseline and a post-B cell depletion autoantibody titer in all patients at regular intervals. In the case of a persistence of high titers of either IgG1 or, less frequently, IgG4 autoantibodies, which indicates the presence of LLPCs, repeated rituximab (or other CD20-depleting drug) dosing can be applied to enforce a deeper depletion of lymph node B cells and prevent the formation of new LLPCs, while existing ones slowly wane. An alternative strategy in such refractory cases is the administration of inebilizumab (or other CD19 depleting drug), which would neutralize CD19+ CD20- ASCs that lie more towards the LLPC end of the ASC spectrum, or the application of daratumumab, an anti-CD38 agent more broadly targeting LLPCs. In the case that follicular or extrafollicular reactions within the CNS are suspected (e.g., based on advanced 7T MRI), it would be reasonable to apply an agent that, in contrast to monoclonal antibodies, can penetrate the blood-brain barrier and target B cells, such as a Bruton tyrosine kinase inhibitor. Finally, the application of new therapeutic avenues such as blockade of B-T cell interaction (CD40L) or IL-4 in polyrefractory cases warrants investigation. Ultimately and ideally, all these novel approaches should be tested in clinical trials prior to routine application.
Our review is not without limitations. First, many of the studies we reference were biased by the use of other immunosuppressants in addition to rituximab and did not have appropriate controls groups, since it is extremely hard to perform randomized controlled trials for rare disorders. Moreover, many of the diseases are aggressive and life-threatening and justify use of more than one immunosuppressant. Second, many of the studies presented and discussed relied on peripheral blood samples, which are easily accessible but not always representative of immunopathological procedures within secondary lymphoid organs or potential tertiary lymphoid structures within the CNS and PNS.
Overall, the differences between IgG1 and IgG4 autoimmune responses lead to many interesting new questions that could be explored in future investigations. Is the IgG4 autoimmune response a purely extrafollicular one? Can secondary or tertiary lymphoid structures be located in IgG4 autoantibody-mediated disorders? What are the different features of IgG1 and IgG4 response in the human lymph node? Does the memory cell compartment differ in IgG1 and IgG4 disorders? Is chronic antigenic stimulation necessary for emergence of autoimmunity of the IgG4 type and if yes, is it amenable to tolerization strategies? In IgG1 autoimmune responses, can LLPCs survive in the brain as they do in the bone marrow, and if yes, how can one target all LLPC niches therapeutically? Answering these questions would involve a detailed investigation of IgG1 and IgG4 differential maturation pathways in either extrafollicular spaces or in GC and would improve our understanding of disease mechanisms as well as facilitate development of new therapeutic avenues. New concepts could involve drugs that target crucial cellular interactions that are perceived to be responsible for B cell differentiation and maturation—in particular the T cell and B cell interaction, always keeping in mind that it is the aberrant and not the physiological response that needs to be stopped.
CZ: drafting and editing. AV: drafting and editing. PS: concept, design, drafting, and editing. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Prof. C. Kilidireas, Prof. M.C. Dalakas, and Prof. L. Stefanis of the University of Athens, Greece, Prof. K. C. O’Connor of Yale University, USA, and the Onassis Foundation for their continued support, as well as Ms. K. Boss for textual editing. Figures were created with BioRender.com.
1. Wardemann H. Predominant Autoantibody Production by Early Human B Cell Precursors. Science (2003) 301:1374–7. doi: 10.1126/science.1086907
2. Elsner RA, Shlomchik MJ. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity (2020) 53:1136–50. doi: 10.1016/j.immuni.2020.11.006
3. Cyster JG, Allen CDC. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell (2019) 177:524–40. doi: 10.1016/j.cell.2019.03.016
4. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The Generation of Antibody-Secreting Plasma Cells. Nat Rev Immunol (2015) 15:160–71. doi: 10.1038/nri3795
5. McHeyzer-Williams MG, Ahmed R. B Cell Memory and the Long-Lived Plasma Cell. Curr Opin Immunol (1999) 11:172–9. doi: 10.1016/S0952-7915(99)80029-6
6. Stathopoulos P, Kumar A, Heiden JAV, Pascual-Goñi E, Nowak RJ, O’Connor KC. Mechanisms Underlying B Cell Immune Dysregulation and Autoantibody Production in MuSK Myasthenia Gravis: B Cell Abnormalities in MuSK Myasthenia Gravis. Ann NY Acad Sci (2018) 1412:154–65. doi: 10.1111/nyas.13535
7. Marino M, Basile U, Spagni G, Napodano C, Iorio R, Gulli F, et al. Long-Lasting Rituximab-Induced Reduction of Specific-But Not Total-IgG4 in MuSK-Positive Myasthenia Gravis. Front Immunol (2020) 11:613. doi: 10.3389/fimmu.2020.00613
8. Keung B, Robeson KR, DiCapua DB, Rosen JB, O’Connor KC, Goldstein JM, et al. Long-Term Benefit of Rituximab in MuSK Autoantibody Myasthenia Gravis Patients: Table 1. J Neurol Neurosurg Psychiatry (2013) 84:1407–9. doi: 10.1136/jnnp-2012-303664
9. Diaz-Manera J, Martinez-Hernandez E, Querol L, Klooster R, Rojas-Garcia R, Suarez-Calvet X, et al. Long-Lasting Treatment Effect of Rituximab in MuSK Myasthenia. Neurology (2012) 78:189–93. doi: 10.1212/WNL.0b013e3182407982
10. Wong C-Y, Yeh K-W, Huang J-L, Su K-W, Tsai M-H, Hua M-C, et al. Longitudinal Analysis of Total Serum IgE Levels With Allergen Sensitization and Atopic Diseases in Early Childhood. Sci Rep (2020) 10:21278. doi: 10.1038/s41598-020-78272-8
11. Nowak RJ, DiCapua DB, Zebardast N, Goldstein JM. Response of Patients With Refractory Myasthenia Gravis to Rituximab: A Retrospective Study. Ther Adv Neurol Disord (2011) 4:259–66. doi: 10.1177/1756285611411503
12. Jing S, Song Y, Song J, Pang S, Quan C, Zhou L, et al. Responsiveness to Low-Dose Rituximab in Refractory Generalized Myasthenia Gravis. J Neuroimmunol (2017) 311:14–21. doi: 10.1016/j.jneuroim.2017.05.021
13. Robeson KR, Kumar A, Keung B, DiCapua DB, Grodinsky E, Patwa HS, et al. Durability of the Rituximab Response in Acetylcholine Receptor Autoantibody-Positive Myasthenia Gravis. JAMA Neurol (2017) 74:60–6. doi: 10.1001/jamaneurol.2016.4190
14. Illa I, Diaz-Manera J, Rojas-Garcia R, Pradas J, Rey A, Blesa R, et al. Sustained Response to Rituximab in Anti-AChR and Anti-MuSK Positive Myasthenia Gravis Patients. J Neuroimmunol (2008) 201–202:90–4. doi: 10.1016/j.jneuroim.2008.04.039
15. Hehir MK, Hobson-Webb LD, Benatar M, Barnett C, Silvestri NJ, Howard JF, et al. Rituximab as Treatment for Anti-MuSK Myasthenia Gravis: Multicenter Blinded Prospective Review. Neurology (2017) 89:1069–77. doi: 10.1212/WNL.0000000000004341
16. Chan F, Swayne A, Gillis D, Walsh M, Henderson RD, McCombe PA, et al. Long-Term Follow-Up of Patients With Myasthenia Gravis Treated With Low-Dose Rituximab. J Neurol Neurosurg Psychiatry (2019) 90:955–6. doi: 10.1136/jnnp-2018-319410
17. Di Stefano V, Lupica A, Rispoli MG, Di Muzio A, Brighina F, Rodolico C. Rituximab in AChR Subtype of Myasthenia Gravis: Systematic Review. J Neurol Neurosurg Psychiat (2020) 91:392–5. doi: 10.1136/jnnp-2019-322606
18. Dos Santos A, Noury J -B., Genestet S, Nadaj-Pakleza A, Cassereau J, Baron C, et al. Efficacy and Safety of Rituximab in Myasthenia Gravis: A French Multicentre Real-Life Study. Eur J Neurol (2020) 27:2277–85. doi: 10.1111/ene.14391
19. Sahai SK, Maghzi AH, Lewis RA. Rituximab in Late-Onset Myasthenia Gravis Is Safe and Effective. Muscle Nerve (2020) 62:377–80. doi: 10.1002/mus.26876
20. Litchman T, Roy B, Kumar A, Sharma A, Njike V, Nowak RJ. Differential Response to Rituximab in Anti-AChR and Anti-MuSK Positive Myasthenia Gravis Patients: A Single-Center Retrospective Study. J Neurol Sci (2020) 411:116690. doi: 10.1016/j.jns.2020.116690
21. Lu J, Zhong H, Jing S, Wang L, Xi J, Lu J, et al. Low-Dose Rituximab Every 6 Months for the Treatment of Acetylcholine Receptor-Positive Refractory Generalized Myasthenia Gravis. Muscle Nerve (2020) 61:311–5. doi: 10.1002/mus.26790
22. Topakian R, Zimprich F, Iglseder S, Embacher N, Guger M, Stieglbauer K, et al. High Efficacy of Rituximab for Myasthenia Gravis: A Comprehensive Nationwide Study in Austria. J Neurol (2019) 266:699–706. doi: 10.1007/s00415-019-09191-6
23. Landon-Cardinal O, Friedman D, Guiguet M, Laforêt P, Heming N, Salort-Campana E, et al. Efficacy of Rituximab in Refractory Generalized Anti-Achr Myasthenia Gravis. J Neuromuscul Dis (2018) 5:241–9. doi: 10.3233/JND-180300
24. Beecher G, Anderson D, Siddiqi ZA. Rituximab in Refractory Myasthenia Gravis: Extended Prospective Study Results. Muscle Nerve (2018) 58:452–5. doi: 10.1002/mus.26156
25. Braun PE, Frail DE, Latov N. Myelin-Associated Glycoprotein Is the Antigen for a Monoclonal IgM in Polyneuropathy. J Neurochem (1982) 39:1261–5. doi: 10.1111/j.1471-4159.1982.tb12563.x
26. Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, Jenkins MK. Visualization of Specific B and T Lymphocyte Interactions in the Lymph Node. Science (1998) 281:96–9. doi: 10.1126/science.281.5373.96
27. Weisel F, Shlomchik M. Memory B Cells of Mice and Humans. Annu Rev Immunol (2017) 35:255–84. doi: 10.1146/annurev-immunol-041015-055531
28. Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, et al. Rapid Cloning of High-Affinity Human Monoclonal Antibodies Against Influenza Virus. Nature (2008) 453:667–71. doi: 10.1038/nature06890
29. Di Niro R, Lee S-J, Vander Heiden JA, Elsner RA, Trivedi N, Bannock JM, et al. Salmonella Infection Drives Promiscuous B Cell Activation Followed by Extrafollicular Affinity Maturation. Immunity (2015) 43:120–31. doi: 10.1016/j.immuni.2015.06.013
30. Hastey CJ, Elsner RA, Barthold SW, Baumgarth N. Delays and Diversions Mark the Development of B Cell Responses to Borrelia Burgdorferi Infection. J Immunol (2012) 188:5612–22. doi: 10.4049/jimmunol.1103735
31. Schröder AE, Greiner A, Seyfert C, Berek C. Differentiation of B Cells in the Nonlymphoid Tissue of the Synovial Membrane of Patients With Rheumatoid Arthritis. Proc Natl Acad Sci USA (1996) 93:221–5. doi: 10.1073/pnas.93.1.221
32. William J, Euler C, Christensen S, Shlomchik MJ. Evolution of Autoantibody Responses Via Somatic Hypermutation Outside of Germinal Centers. Science (2002) 297:2066–70. doi: 10.1126/science.1073924
33. Cunningham AF, Gaspal F, Serre K, Mohr E, Henderson IR, Scott-Tucker A, et al. Salmonella Induces a Switched Antibody Response Without Germinal Centers That Impedes the Extracellular Spread of Infection. J Immunol (2007) 178:6200–7. doi: 10.4049/jimmunol.178.10.6200
34. Marshall JL, Zhang Y, Pallan L, Hsu M-C, Khan M, Cunningham AF, et al. Early B Blasts Acquire a Capacity for Ig Class Switch Recombination That is Lost as They Become Plasmablasts. Eur J Immunol (2011) 41:3506–12. doi: 10.1002/eji.201141762
35. Toellner KM, Luther SA, Sze DM, Choy RK, Taylor DR, MacLennan IC, et al. T Helper 1 (Th1) and Th2 Characteristics Start to Develop During T Cell Priming and Are Associated With an Immediate Ability to Induce Immunoglobulin Class Switching. J Exp Med (1998) 187:1193–204. doi: 10.1084/jem.187.8.1193
36. Trivedi N, Weisel F, Smita S, Joachim S, Kader M, Radhakrishnan A, et al. Liver Is a Generative Site for the B Cell Response to Ehrlichia Muris. Immunity (2019) 51:1088–101.e5. doi: 10.1016/j.immuni.2019.10.004
37. Yates JL, Racine R, McBride KM. Winslow Gm. T Cell–Dependent IgM Memory B Cells Generated During Bacterial Infection Are Required for IgG Responses to Antigen Challenge. J Immunol (2013) 191:1240–9. doi: 10.4049/jimmunol.1300062
38. Toyama H, Okada S, Hatano M, Takahashi Y, Takeda N, Ichii H, et al. Memory B Cells Without Somatic Hypermutation Are Generated From Bcl6-Deficient B Cells. Immunity (2002) 17:329–39. doi: 10.1016/S1074-7613(02)00387-4
39. Takahashi T, Miyazawa I, Misu T, Takano R, Nakashima I, Fujihara K, et al. Intractable Hiccup and Nausea in Neuromyelitis Optica With Anti-Aquaporin-4 Antibody: A Herald of Acute Exacerbations. J Neurol Neurosurg Psychiatry (2008) 79:1075–8. doi: 10.1136/jnnp.2008.145391
40. Elsner RA, Hastey CJ, Olsen KJ, Baumgarth N. Suppression of Long-Lived Humoral Immunity Following Borrelia Burgdorferi Infection. PloS Pathog (2015) 11:e1004976. doi: 10.1371/journal.ppat.1004976
41. Racine R, Jones DD, Chatterjee M, McLaughlin M, MacNamara KC, Winslow GM. Impaired Germinal Center Responses and Suppression of Local IgG Production During Intracellular Bacterial Infection. J Immunol (2010) 184:5085–93. doi: 10.4049/jimmunol.0902710
42. Dal Porto JM, Haberman AM, Shlomchik MJ, Kelsoe G. Antigen Drives Very Low Affinity B Cells to Become Plasmacytes and Enter Germinal Centers. J Immunol (1998) 161:5373–81.
43. Hunter CA, Jones SA. IL-6 as a Keystone Cytokine in Health and Disease. Nat Immunol (2015) 16:448–57. doi: 10.1038/ni.3153
44. González-García I, Ocaña E, Jiménez-Gómez G, Campos-Caro A, Brieva JA. Immunization-Induced Perturbation of Human Blood Plasma Cell Pool: Progressive Maturation, Il-6 Responsiveness, and High Prdi-Bf1/Blimp1 Expression Are Critical Distinctions Between Antigen-Specific and Nonspecific Plasma Cells. J Immunol (2006) 176:4042–50. doi: 10.4049/jimmunol.176.7.4042
45. Wols HAM, Underhill GH, Kansas GS, Witte PL. The Role of Bone Marrow-Derived Stromal Cells in the Maintenance of Plasma Cell Longevity. J Immunol (2002) 169:4213–21. doi: 10.4049/jimmunol.169.8.4213
46. Crotty S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity (2019) 50:1132–48. doi: 10.1016/j.immuni.2019.04.011
47. Nieuwenhuis P, Opstelten D. Functional Anatomy of Germinal Centers. Am J Anat (1984) 170:421–35. doi: 10.1002/aja.1001700315
48. Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, et al. In Vivo Imaging of Germinal Centres Reveals a Dynamic Open Structure. Nature (2007) 446:83–7. doi: 10.1038/nature05573
49. Hauser AE, Junt T, Mempel TR, Sneddon MW, Kleinstein SH, Henrickson SE, et al. Definition of Germinal-Center B Cell Migration In Vivo Reveals Predominant Intrazonal Circulation Patterns. Immunity (2007) 26:655–67. doi: 10.1016/j.immuni.2007.04.008
50. Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, Nussenzweig MC. Identification of Human Germinal Center Light and Dark Zone Cells and Their Relationship to Human B-Cell Lymphomas. Blood (2012) 120:2240–8. doi: 10.1182/blood-2012-03-415380
51. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class Switch Recombination and Hypermutation Require Activation-Induced Cytidine Deaminase (AID), a Potential RNA Editing Enzyme. Cell (2000) 102:553–63. doi: 10.1016/s0092-8674(00)00078-7
52. Bende RJ, van Maldegem F, Triesscheijn M, Wormhoudt TAM, Guijt R, van Noesel CJM. Germinal Centers in Human Lymph Nodes Contain Reactivated Memory B Cells. J Exp Med (2007) 204:2655–65. doi: 10.1084/jem.20071006
53. Viant C, Weymar GHJ, Escolano A, Chen S, Hartweger H, Cipolla M, et al. Antibody Affinity Shapes the Choice Between Memory and Germinal Center B Cell Fates. Cell (2020) 183:1298–311.e11. doi: 10.1016/j.cell.2020.09.063
54. Wei C, Jenks S, Sanz I. Polychromatic Flow Cytometry in Evaluating Rheumatic Disease Patients. Arthritis Res Ther (2015) 17:46. doi: 10.1186/s13075-015-0561-1
55. Quách TD, Rodríguez-Zhurbenko N, Hopkins TJ, Guo X, Hernández AM, Li W, et al. Distinctions Among Circulating Antibody-Secreting Cell Populations, Including B-1 Cells, in Human Adult Peripheral Blood. J Immunol (2016) 196:1060–9. doi: 10.4049/jimmunol.1501843
56. Mei HE, Wirries I, Frölich D, Brisslert M, Giesecke C, Grün JR, et al. A Unique Population of IgG-expressing Plasma Cells Lacking CD19 is Enriched in Human Bone Marrow. Blood (2015) 125:1739–48. doi: 10.1182/blood-2014-02-555169
57. Halliley JL, Tipton CM, Liesveld J, Rosenberg AF, Darce J, Gregoretti IV, et al. Long-Lived Plasma Cells Are Contained Within the CD19–CD38hiCD138+ Subset in Human Bone Marrow. Immunity (2015) 43:132–45. doi: 10.1016/j.immuni.2015.06.016
58. Koneczny I. Update on IgG4-mediated Autoimmune Diseases: New Insights and New Family Members. Autoimmun Rev (2020) 19:102646. doi: 10.1016/j.autrev.2020.102646
59. Koneczny I. A New Classification System for IgG4 Autoantibodies. Front Immunol (2018) 9:97. doi: 10.3389/fimmu.2018.00097
60. Huijbers MG, Plomp JJ, van der Maarel SM, Verschuuren JJ. IgG4-Mediated Autoimmune Diseases: A Niche of Antibody-Mediated Disorders: IgG4-Mediated Autoimmune Diseases. Ann NY Acad Sci (2018) 1413:92–103. doi: 10.1111/nyas.13561
61. Huijbers MG, Querol LA, Niks EH, Plomp JJ, van der Maarel SM, Graus F, et al. The Expanding Field of IgG4-Mediated Neurological Autoimmune Disorders. Eur J Neurol (2015) 22:1151–61. doi: 10.1111/ene.12758
62. Aalberse RC, Platts-Mills TA, Rispens T. The Developmental History of IgE and IgG4 Antibodies in Relation to Atopy, Eosinophilic Esophagitis, and the Modified Th2 Response. Curr Allergy Asthma Rep (2016) 16:45. doi: 10.1007/s11882-016-0621-x
63. Aalberse RC, van der Gaag R, van Leeuwen J. Serologic Aspects of IgG4 Antibodies. I. Prolonged Immunization Results in an IgG4-Restricted Response. J Immunol (1983) 130:722–6.
64. van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martínez-Martínez P, Vermeulen E, et al. Anti-Inflammatory Activity of Human IgG4 Antibodies by Dynamic Fab Arm Exchange. Science (2007) 317:1554–7. doi: 10.1126/science.1144603
65. Hofbauer CJ, Whelan SFJ, Hirschler M, Allacher P, Horling FM, Lawo J-P, et al. Affinity of FVIII-Specific Antibodies Reveals Major Differences Between Neutralizing and Nonneutralizing Antibodies in Humans. Blood (2015) 125:1180–8. doi: 10.1182/blood-2014-09-598268
66. van Schouwenburg PA, Krieckaert CL, Nurmohamed M, Hart M, Rispens T, Aarden L, et al. IgG4 Production Against Adalimumab During Long Term Treatment of RA Patients. J Clin Immunol (2012) 32:1000–6. doi: 10.1007/s10875-012-9705-0
67. Lundkvist M, Engdahl E, Holmén C, Movérare R, Olsson T, Hillert J, et al. Characterization of Anti-Natalizumab Antibodies in Multiple Sclerosis Patients. Mult Scler (2013) 19:757–64. doi: 10.1177/1352458512462920
68. Devey ME, Bleasdale-Barr KM, Bird P, Amlot PL. Antibodies of Different Human IgG Subclasses Show Distinct Patterns of Affinity Maturation After Immunization With Keyhole Limpet Haemocyanin. Immunology (1990) 70:168–74.
69. Carrasco B, Garcia de la Torre J, Davis KG, Jones S, Athwal D, Walters C, et al. Crystallohydrodynamics for Solving the Hydration Problem for Multi-Domain Proteins: Open Physiological Conformations for Human IgG. Biophys Chem (2001) 93:181–96. doi: 10.1016/s0301-4622(01)00220-4
70. Lu Y, Harding SE, Michaelsen TE, Longman E, Davis KG, Ortega A, et al. Solution Conformation of Wild-Type and Mutant IgG3 and IgG4 Immunoglobulins Using Crystallohydrodynamics: Possible Implications for Complement Activation. Biophys J (2007) 93:3733–44. doi: 10.1529/biophysj.107.108993
71. Abe Y, Gor J, Bracewell DG, Perkins SJ, Dalby PA. Masking of the Fc Region in Human IgG4 by Constrained X-Ray Scattering Modelling: Implications for Antibody Function and Therapy. Biochem J (2010) 432:101–11. doi: 10.1042/BJ20100641
72. Adjobimey T, Hoerauf A. Induction of Immunoglobulin G4 in Human Filariasis: An Indicator of Immunoregulation. Ann Trop Med Parasitol (2010) 104:455–64. doi: 10.1179/136485910X12786389891407
73. Grogan JL, Kremsner PG, van Dam GJ, Metzger W, Mordmüller B, Deelder AM, et al. Antischistosome IgG4 and IgE Responses Are Affected Differentially by Chemotherapy in Children Versus Adults. J Infect Dis (1996) 173:1242–7. doi: 10.1093/infdis/173.5.1242
74. Lebman DA, Coffman RL. Interleukin 4 Causes Isotype Switching to IgE in T Cell-Stimulated Clonal B Cell Cultures. J Exp Med (1988) 168:853–62. doi: 10.1084/jem.168.3.853
75. Agresti A, Vercelli D. c-Rel Is a Selective Activator of a Novel IL-4/CD40 Responsive Element in the Human Ig Gamma4 Germline Promoter. Mol Immunol (2002) 38:849–59. doi: 10.1016/s0161-5890(01)00121-3
76. Punnonen J, Aversa G, Cocks BG, McKenzie AN, Menon S, Zurawski G, et al. Interleukin 13 Induces Interleukin 4-Independent IgG4 and IgE Synthesis and CD23 Expression by Human B Cells. Proc Natl Acad Sci (1993) 90:3730–4. doi: 10.1073/pnas.90.8.3730
77. Jeannin P, Lecoanet S, Delneste Y, Gauchat JF, Bonnefoy JY. Ige Versus IgG4 Production Can Be Differentially Regulated by IL-10. J Immunol (1998) 160:3555–61.
78. Francis JN, James LK, Paraskevopoulos G, Wong C, Calderon MA, Durham SR, et al. Grass Pollen Immunotherapy: IL-10 Induction and Suppression of Late Responses Precedes IgG4 Inhibitory Antibody Activity. J Allergy Clin Immunol (2008) 121:1120–5.e2. doi: 10.1016/j.jaci.2008.01.072
79. Lighaam L, Rispens T. The Immunobiology of Immunoglobulin G4. Semin Liver Dis (2016) 36:200–15. doi: 10.1055/s-0036-1584322
80. Kawamoto N, Fukao T, Kaneko H, Hirayama K, Sakurai S, Arai T, et al. Total IgE at 6 Months Predicts Remittance or Persistence of Atopic Dermatitis at 14 Months. Allergy Asthma Proc (2013) 34:362–9. doi: 10.2500/aap.2013.34.3678
81. Davies JM, Platts-Mills TA, Aalberse RC. The Enigma of IgE+ B-Cell Memory in Human Subjects. J Allergy Clin Immunol (2013) 131:972–6. doi: 10.1016/j.jaci.2012.12.1569
82. Gould HJ, Ramadani F. Ige Responses in Mouse and Man and the Persistence of IgE Memory. Trends Immunol (2015) 36:40–8. doi: 10.1016/j.it.2014.11.002
83. Hattevig G, Kjellman B, Björkstén B. Appearance of IgE Antibodies to Ingested and Inhaled Allergens During the First 12 Years of Life in Atopic and Non-Atopic Children. Pediatr Allergy Immunol (1993) 4:182–6. doi: 10.1111/j.1399-3038.1993.tb00089.x
84. Unger P-PA, Lighaam LC, Vermeulen E, Kruithof S, Makuch M, Culver EL, et al. Divergent Chemokine Receptor Expression and the Consequence for Human IgG4 B Cell Responses. Eur J Immunol (2020) 50:1113–25. doi: 10.1002/eji.201948454
85. Mattoo H, Mahajan VS, Della-Torre E, Sekigami Y, Carruthers M, Wallace ZS, et al. De Novo Oligoclonal Expansions of Circulating Plasmablasts in Active and Relapsing IgG4-Related Disease. J Allergy Clin Immunol (2014) 134:679–87. doi: 10.1016/j.jaci.2014.03.034
86. Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab Therapy Leads to Rapid Decline of Serum IgG4 Levels and Prompt Clinical Improvement in IgG4-Related Systemic Disease. Arthritis Rheum (2010) 62:1755–62. doi: 10.1002/art.27435
87. Vanderleyden I, Linterman MA, Smith KGC. Regulatory T Cells and Control of the Germinal Centre Response. Arthritis Res Ther (2014) 16:471. doi: 10.1186/s13075-014-0471-7
88. Meiler F, Klunker S, Zimmermann M, Akdis CA, Akdis M. Distinct Regulation of IgE, IgG4 and IgA by T Regulatory Cells and Toll-Like Receptors. Allergy (2008) 63:1455–63. doi: 10.1111/j.1398-9995.2008.01774.x
89. Satoguina JS, Adjobimey T, Arndts K, Hoch J, Oldenburg J, Layland LE, et al. Tr1 and Naturally Occurring Regulatory T Cells Induce IgG4 in B Cells Through GITR/GITR-L Interaction, IL-10 and TGF-Beta. Eur J Immunol (2008) 38:3101–13. doi: 10.1002/eji.200838193
90. Satoguina J, Mempel M, Larbi J, Badusche M, Löliger C, Adjei O, et al. Antigen-Specific T Regulatory-1 Cells Are Associated With Immunosuppression in a Chronic Helminth Infection (Onchocerciasis). Microbes Infect (2002) 4:1291–300. doi: 10.1016/s1286-4579(02)00014-x
91. Taylor JJ, Mohrs M, Pearce EJ. Regulatory T Cell Responses Develop in Parallel to Th Responses and Control the Magnitude and Phenotype of the Th Effector Population. J Immunol (2006) 176:5839–47. doi: 10.4049/jimmunol.176.10.5839
92. Miyoshi H, Uchida K, Taniguchi T, Yazumi S, Matsushita M, Takaoka M, et al. Circulating Naïve and CD4+CD25high Regulatory T Cells in Patients With Autoimmune Pancreatitis. Pancreas (2008) 36:133–40. doi: 10.1097/MPA.0b013e3181577553
93. Heeringa JJ, Karim AF, van Laar JAM, Verdijk RM, Paridaens D, van Hagen PM, et al. Expansion of Blood IgG 4 + B, T H 2, and Regulatory T Cells in Patients With IgG 4 -Related Disease. J Allergy Clin Immunol (2018) 141:1831–43.e10. doi: 10.1016/j.jaci.2017.07.024
94. Kusuda T, Uchida K, Miyoshi H, Koyabu M, Satoi S, Takaoka M, et al. Involvement of Inducible Costimulator- and Interleukin 10-Positive Regulatory T Cells in the Development of IgG4-Related Autoimmune Pancreatitis. Pancreas (2011) 40:1120–30. doi: 10.1097/MPA.0b013e31821fc796
95. Thiruppathi M, Rowin J, Ganesh B, Sheng JR, Prabhakar BS, Meriggioli MN. Impaired Regulatory Function in Circulating CD4(+)CD25(High)CD127(Low/-) T Cells in Patients With Myasthenia Gravis. Clin Immunol (2012) 145:209–23. doi: 10.1016/j.clim.2012.09.012
96. Sheng JR, Li L, Ganesh BB, Vasu C, Prabhakar BS, Meriggioli MN. Suppression of Experimental Autoimmune Myasthenia Gravis by Granulocyte-Macrophage Colony-Stimulating Factor Is Associated With an Expansion of FoxP3+ Regulatory T Cells. J Immunol (2006) 177:5296–306. doi: 10.4049/jimmunol.177.8.5296
97. Su W, Zhou Q, Ke Y, Xue J, Shen J. Functional Inhibition of Regulatory CD4+CD25+T Cells in Peripheral Blood of Patients With Pemphigus Vulgaris. Clin Exp Dermatol (2020) 45:1019–26. doi: 10.1111/ced.14309
98. Bieber K, Sun S, Witte M, Kasprick A, Beltsiou F, Behnen M, et al. Regulatory T Cells Suppress Inflammation and Blistering in Pemphigoid Diseases. Front Immunol (2017) 8:1628. doi: 10.3389/fimmu.2017.01628
99. Horns F, Vollmers C, Croote D, Mackey SF, Swan GE, Dekker CL, et al. Lineage Tracing of Human B Cells Reveals the In Vivo Landscape of Human Antibody Class Switching. Elife (2016) 5. doi: 10.7554/eLife.16578
100. Jackson KJL, Liu Y, Roskin KM, Glanville J, Hoh RA, Seo K, et al. Human Responses to Influenza Vaccination Show Seroconversion Signatures and Convergent Antibody Rearrangements. Cell Host Microbe (2014) 16:105–14. doi: 10.1016/j.chom.2014.05.013
101. Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: An Odd Antibody. Clin Exp Allergy (2009) 39:469–77. doi: 10.1111/j.1365-2222.2009.03207.x
102. Aalberse RC, Dieges PH, Knul-Bretlova V, Vooren P, Aalbers M, van Leeuwen J. IgG4 as a Blocking Antibody. Clin Rev Allergy (1983) 1:289–302. doi: 10.1007/BF02991163
103. Looney TJ, Lee J-Y, Roskin KM, Hoh RA, King J, Glanville J, et al. Human B-Cell Isotype Switching Origins of Ige. J Allergy Clin Immunol (2016) 137:579–86.e7. doi: 10.1016/j.jaci.2015.07.014
104. Jackson KJL, Wang Y, Collins AM. Human Immunoglobulin Classes and Subclasses Show Variability in VDJ Gene Mutation Levels. Immunol Cell Biol (2014) 92:729–33. doi: 10.1038/icb.2014.44
105. Wang Y, Jackson KJL, Chen Z, Gaëta BA, Siba PM, Pomat W, et al. Ige Sequences in Individuals Living in an Area of Endemic Parasitism Show Little Mutational Evidence of Antigen Selection. Scand J Immunol (2011) 73:496–504. doi: 10.1111/j.1365-3083.2011.02525.x
106. Wu Y-CB, James LK, Vander Heiden JA, Uduman M, Durham SR, Kleinstein SH, et al. Influence of Seasonal Exposure to Grass Pollen on Local and Peripheral Blood IgE Repertoires in Patients With Allergic Rhinitis. J Allergy Clin Immunol (2014) 134:604–12. doi: 10.1016/j.jaci.2014.07.010
107. Hoh RA, Joshi SA, Liu Y, Wang C, Roskin KM, Lee J-Y, et al. Single B-Cell Deconvolution of Peanut-Specific Antibody Responses in Allergic Patients. J Allergy Clin Immunol (2016) 137:157–67. doi: 10.1016/j.jaci.2015.05.029
108. Levin M, King JJ, Glanville J, Jackson KJL, Looney TJ, Hoh RA, et al. Persistence and Evolution of Allergen-Specific IgE Repertoires During Subcutaneous Specific Immunotherapy. J Allergy Clin Immunol (2016) 137:1535–44. doi: 10.1016/j.jaci.2015.09.027
109. Ellebrecht CT, Mukherjee EM, Zheng Q, Choi EJ, Reddy SG, Mao X, et al. Autoreactive IgG and Iga B Cells Evolve Through Distinct Subclass Switch Pathways in the Autoimmune Disease Pemphigus Vulgaris. Cell Rep (2018) 24:2370–80. doi: 10.1016/j.celrep.2018.07.093
110. van der Zee JS, van Swieten P, Aalberse RC. Inhibition of Complement Activation by IgG4 Antibodies. Clin Exp Immunol (1986) 64:415–22.
111. Aalberse RC, Schuurman J. IgG4 Breaking the Rules. Immunology (2002) 105:9–19. doi: 10.1046/j.0019-2805.2001.01341.x
112. Verschuuren J, Strijbos E, Vincent A. Neuromuscular Junction Disorders. In: Handbook of Clinical Neurology. Elsevier (2016). vol. 133, p. 447–66. doi: 10.1016/B978-0-444-63432-0.00024-4
113. Hinson SR, Lennon VA, Pittock SJ. Autoimmune AQP4 Channelopathies and Neuromyelitis Optica Spectrum Disorders. In: Handbook of Clinical Neurology. Elsevier (2016). vol. 133, p. 377–403. doi: 10.1016/B978-0-444-63432-0.00021-9
114. O’Connor KC, McLaughlin KA, De Jager PL, Chitnis T, Bettelli E, Xu C, et al. Self-Antigen Tetramers Discriminate Between Myelin Autoantibodies to Native or Denatured Protein. Nat Med (2007) 13:211–7. doi: 10.1038/nm1488
115. Chalmoukou K, Alexopoulos H, Akrivou S, Stathopoulos P, Reindl M, Dalakas MC. Anti-MOG Antibodies Are Frequently Associated With Steroid-Sensitive Recurrent Optic Neuritis. Neurol Neuroimmunol Neuroinflamm (2015) 2:e131. doi: 10.1212/NXI.0000000000000131
116. Linnoila J, Rosenfeld M, Dalmau J. Neuronal Surface Antibody-Mediated Autoimmune Encephalitis. Semin Neurol (2014) 34:458–66. doi: 10.1055/s-0034-1390394
117. Niks EH, van Leeuwen Y, Leite MI, Dekker FW, Wintzen AR, Wirtz PW, et al. Clinical Fluctuations in MuSK Myasthenia Gravis Are Related to Antigen-Specific IgG4 Instead of Igg1. J Neuroimmunol (2008) 195:151–6. doi: 10.1016/j.jneuroim.2008.01.013
118. Huijbers MG, Zhang W, Klooster R, Niks EH, Friese MB, Straasheijm KR, et al. Musk IgG4 Autoantibodies Cause Myasthenia Gravis by Inhibiting Binding Between MuSK and Lrp4. Proc Natl Acad Sci (2013) 110:20783–8. doi: 10.1073/pnas.1313944110
119. McConville J, Farrugia ME, Beeson D, Kishore U, Metcalfe R, Newsom-Davis J, et al. Detection and Characterization of MuSK Antibodies in Seronegative Myasthenia Gravis. Ann Neurol (2004) 55:580–4. doi: 10.1002/ana.20061
120. Carr AS, Cardwell CR, McCarron PO, McConville J. A Systematic Review of Population Based Epidemiological Studies in Myasthenia Gravis. BMC Neurol (2010) 10:46. doi: 10.1186/1471-2377-10-46
121. Huda S, Waters P, Woodhall M, Leite MI, Jacobson L, De Rosa A, et al. IgG-specific Cell-Based Assay Detects Potentially Pathogenic MuSK-Abs in Seronegative MG. Neurol Neuroimmunol Neuroinflamm (2017) 4:e357. doi: 10.1212/NXI.0000000000000357
122. Bartoccioni E, Scuderi F, Augugliaro A, Chiatamone Ranieri S, Sauchelli D, Alboino P, et al. HLA Class II Allele Analysis in MuSK-Positive Myasthenia Gravis Suggests a Role for DQ5. Neurology (2009) 72:195–7. doi: 10.1212/01.wnl.0000339103.08830.86
123. Niks EH, Kuks JBM, Roep BO, Haasnoot GW, Verduijn W, Ballieux BEPB, et al. Strong Association of MuSK Antibody-Positive Myasthenia Gravis and HLA-DR14-DQ5. Neurology (2006) 66:1772–4. doi: 10.1212/01.wnl.0000218159.79769.5c
124. Lefvert AK, Cuénoud S, Fulpius BW. Binding Properties and Subclass Distribution of Anti-Acetylcholine Receptor Antibodies in Myasthenia Gravis. J Neuroimmunol (1981) 1:125–35. doi: 10.1016/0165-5728(81)90015-1
125. Rødgaard A, Nielsen FC, Djurup R, Somnier F, Gammeltoft S. Acetylcholine Receptor Antibody in Myasthenia Gravis: Predominance of IgG Subclasses 1 and 3. Clin Exp Immunol (1987) 67:82–8.
126. Leite MI, Jacob S, Viegas S, Cossins J, Clover L, Morgan BP, et al. IgG1 Antibodies to Acetylcholine Receptors in “Seronegative” Myasthenia Gravis. Brain (2008) 131:1940–52. doi: 10.1093/brain/awn092
127. Vincent A, Newsom Davis J. Anti-Acetylcholine Receptor Antibodies. J Neurol Neurosurg Psychiatry (1980) 43:590–600. doi: 10.1136/jnnp.43.7.590
128. Kapinas K, Tzartos S, Kokkas B, Divanoglou D, Tsolaki M, Anogiannakis G, et al. Absence of Oligoclonal IgG Bands and Anti-Achr Antibodies in the Cerebrospinal Fluid of Patients With Myasthenia Gravis. Int J Immunopathol Pharmacol (1998) 11:87–90. doi: 10.1177/039463209801100206
129. Testi M, Terracciano C, Guagnano A, Testa G, Marfia GA, Pompeo E, et al. Association of HLA-DQB1∗05:02 and DRB1∗16 Alleles With Late-Onset, Nonthymomatous, AChR-Ab-Positive Myasthenia Gravis. Autoimmune Dis (2012) 2012:541760. doi: 10.1155/2012/541760
130. Romi F, Kristoffersen EK, Aarli JA, Gilhus NE. The Role of Complement in Myasthenia Gravis: Serological Evidence of Complement Consumption In Vivo. J Neuroimmunol (2005) 158:191–4. doi: 10.1016/j.jneuroim.2004.08.002
131. Howard JF. Myasthenia Gravis: The Role of Complement at the Neuromuscular Junction. Ann N Y Acad Sci (2018) 1412:113–28. doi: 10.1111/nyas.13522
132. Vincent A, Leite MI, Farrugia ME, Jacob S, Viegas S, Shiraishi H, et al. Myasthenia Gravis Seronegative for Acetylcholine Receptor Antibodies. Ann N Y Acad Sci (2008) 1132:84–92. doi: 10.1196/annals.1405.020
133. Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, et al. Pathogenic Potential of IgG Binding to Water Channel Extracellular Domain in Neuromyelitis Optica. Neurology (2007) 69:2221–31. doi: 10.1212/01.WNL.0000289761.64862.ce
134. Papp V, Magyari M, Aktas O, Berger T, Broadley SA, Cabre P, et al. Worldwide Incidence and Prevalence of Neuromyelitis Optica: A Systematic Review. Neurology (2021) 96:59–77. doi: 10.1212/WNL.0000000000011153
135. Flanagan EP, Cabre P, Weinshenker BG, Sauver JS, Jacobson DJ, Majed M, et al. Epidemiology of Aquaporin-4 Autoimmunity and Neuromyelitis Optica Spectrum: Aquaporin-4-IgG Seroprevalence. Ann Neurol (2016) 79:775–83. doi: 10.1002/ana.24617
136. Jarius S, Franciotta D, Paul F, Ruprecht K, Bergamaschi R, Rommer PS, et al. Cerebrospinal Fluid Antibodies to Aquaporin-4 in Neuromyelitis Optica and Related Disorders: Frequency, Origin, and Diagnostic Relevance. J Neuroinflamm (2010) 7:52. doi: 10.1186/1742-2094-7-52
137. Jarius S, Wildemann B. Aquaporin-4 Antibodies (Nmo-IgG) as a Serological Marker of Neuromyelitis Optica: A Critical Review of the Literature: Aquaporin-4 Antibodies as Diagnostic Marker of Neuromyelitis Optica. Brain Pathol (2013) 23:661–83. doi: 10.1111/bpa.12084
138. Waters P, Vincent A. Detection of Anti-Aquaporin-4 Antibodies in Neuromyelitis Optica: Current Status of the Assays. Int MS J (2008) 15:99–105.
139. Alvarenga MP, do Carmo LF, Vasconcelos CCF, Alvarenga MP, Alvarenga-Filho H, de Melo Bento CA, et al. Neuromyelitis Optica Is an HLA Associated Disease Different From Multiple Sclerosis: A Systematic Review With Meta-Analysis. Sci Rep (2021) 11:152. doi: 10.1038/s41598-020-80535-3
140. Brum DG, Barreira AA, dos Santos AC, Kaimen-Maciel DR, Matiello M, Costa RM, et al. HLA-DRB Association in Neuromyelitis Optica Is Different From That Observed in Multiple Sclerosis. Mult Scler (2010) 16:21–9. doi: 10.1177/1352458509350741
141. Nytrova P, Potlukova E, Kemlink D, Woodhall M, Horakova D, Waters P, et al. Complement Activation in Patients With Neuromyelitis Optica. J Neuroimmunol (2014) 274:185–91. doi: 10.1016/j.jneuroim.2014.07.001
142. Soltys J, Liu Y, Ritchie A, Wemlinger S, Schaller K, Schumann H, et al. Membrane Assembly of Aquaporin-4 Autoantibodies Regulates Classical Complement Activation in Neuromyelitis Optica. J Clin Invest (2019) 129:2000–13. doi: 10.1172/JCI122942
143. Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF, et al. Eculizumab in AQP4-IgG-Positive Relapsing Neuromyelitis Optica Spectrum Disorders: An Open-Label Pilot Study. Lancet Neurol (2013) 12:554–62. doi: 10.1016/S1474-4422(13)70076-0
144. Mariotto S, Ferrari S, Monaco S, Benedetti MD, Schanda K, Alberti D, et al. Clinical Spectrum and IgG Subclass Analysis of Anti-Myelin Oligodendrocyte Glycoprotein Antibody-Associated Syndromes: A Multicenter Study. J Neurol (2017) 264:2420–30. doi: 10.1007/s00415-017-8635-4
145. Reindl M, Waters P. Myelin Oligodendrocyte Glycoprotein Antibodies in Neurological Disease. Nat Rev Neurol (2019) 15:89–102. doi: 10.1038/s41582-018-0112-x
146. Dubey D, Pittock SJ, Kelly CR, McKeon A, Lopez-Chiriboga AS, Lennon VA, et al. Autoimmune Encephalitis Epidemiology and a Comparison to Infectious Encephalitis. Ann Neurol (2018) 83:166–77. doi: 10.1002/ana.25131
147. Mariotto S, Gajofatto A, Batzu L, Delogu R, Sechi G, Leoni S, et al. Relevance of Antibodies to Myelin Oligodendrocyte Glycoprotein in CSF of Seronegative Cases. Neurology (2019) 93(20):e1867-72. doi: 10.1212/WNL.0000000000008479
148. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. Mog-IgG in NMO and Related Disorders: A Multicenter Study of 50 Patients. Part 1: Frequency, Syndrome Specificity, Influence of Disease Activity, Long-Term Course, Association With AQP4-IgG, and Origin. J Neuroinflamm (2016) 13:279. doi: 10.1186/s12974-016-0717-1
149. Bruijstens AL, Wong YYM, van Pelt DE, van der Linden PJE, Haasnoot GW, Hintzen RQ, et al. HLA Association in MOG-IgG- and AQP4-IgG-Related Disorders of the CNS in the Dutch Population. Neurol Neuroimmunol Neuroinflamm (2020) 7. doi: 10.1212/NXI.0000000000000702
150. Mader S, Gredler V, Schanda K, Rostasy K, Dujmovic I, Pfaller K, et al. Complement Activating Antibodies to Myelin Oligodendrocyte Glycoprotein in Neuromyelitis Optica and Related Disorders. J Neuroinflamm (2011) 8:184. doi: 10.1186/1742-2094-8-184
151. Peschl P, Bradl M, Höftberger R, Berger T, Reindl M. Myelin Oligodendrocyte Glycoprotein: Deciphering a Target in Inflammatory Demyelinating Diseases. Front Immunol (2017) 8:529. doi: 10.3389/fimmu.2017.00529
152. Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, et al. Anti-NMDA-Receptor Encephalitis: Case Series and Analysis of the Effects of Antibodies. Lancet Neurol (2008) 7:1091–8. doi: 10.1016/S1474-4422(08)70224-2
153. Tüzün E, Zhou L, Baehring JM, Bannykh S, Rosenfeld MR, Dalmau J. Evidence for Antibody-Mediated Pathogenesis in Anti-NMDAR Encephalitis Associated With Ovarian Teratoma. Acta Neuropathol (2009) 118:737–43. doi: 10.1007/s00401-009-0582-4
154. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and Synaptic Mechanisms of Anti-NMDA Receptor Encephalitis. J Neurosci (2010) 30:5866–75. doi: 10.1523/JNEUROSCI.0167-10.2010
155. Carvajal-González A, Leite MI, Waters P, Woodhall M, Coutinho E, Balint B, et al. Glycine Receptor Antibodies in PERM and Related Syndromes: Characteristics, Clinical Features and Outcomes. Brain (2014) 137:2178–92. doi: 10.1093/brain/awu142
156. Balint B, Bhatia KP. Stiff Person Syndrome and Other Immune-Mediated Movement Disorders - New Insights. Curr Opin Neurol (2016) 29:496–506. doi: 10.1097/WCO.0000000000000351
157. Chefdeville A, Honnorat J, Hampe CS, Desestret V. Neuronal Central Nervous System Syndromes Probably Mediated by Autoantibodies. Eur J Neurosci (2016) 43:1535–52. doi: 10.1111/ejn.13212
158. Ekizoglu E, Tuzun E, Woodhall M, Lang B, Jacobson L, Icoz S, et al. Investigation of Neuronal Autoantibodies in Two Different Focal Epilepsy Syndromes. Epilepsia (2014) 55:414–22. doi: 10.1111/epi.12528
159. Mueller SH, Färber A, Prüss H, Melzer N, Golombeck KS, Kümpfel T, et al. Genetic Predisposition in Anti-LGI1 and Anti-NMDA Receptor Encephalitis. Ann Neurol (2018) 83:863–9. doi: 10.1002/ana.25216
160. Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y, Sangha N, Martinez-Lage M, Dalmau J. Analysis of Complement and Plasma Cells in the Brain of Patients With Anti-NMDAR Encephalitis. Neurology (2011) 77:589–93. doi: 10.1212/WNL.0b013e318228c136
161. Dalmau J, Tüzün E, Wu H, Masjuan J, Rossi JE, Voloschin A, et al. Paraneoplastic Anti-N-methyl-D-aspartate Receptor Encephalitis Associated With Ovarian Teratoma. Ann Neurol (2007) 61:25–36. doi: 10.1002/ana.21050
162. Bien CG, Vincent A, Barnett MH, Becker AJ, Blümcke I, Graus F, et al. Immunopathology of Autoantibody-Associated Encephalitides: Clues for Pathogenesis. Brain (2012) 135:1622–38. doi: 10.1093/brain/aws082
163. Gaig C, Graus F, Compta Y, Högl B, Bataller L, Brüggemann N, et al. Clinical Manifestations of the Anti-IgLON5 Disease. Neurology (2017) 88:1736–43. doi: 10.1212/WNL.0000000000003887
164. Honorat JA, Komorowski L, Josephs KA, Fechner K, St Louis EK, Hinson SR, et al. IgLON5 Antibody: Neurological Accompaniments and Outcomes in 20 Patients. Neurol Neuroimmunol Neuroinflamm (2017) 4:e385. doi: 10.1212/NXI.0000000000000385
165. Nissen MS, Blaabjerg M. Anti-IgLON5 Disease: A Case With 11-Year Clinical Course and Review of the Literature. Front Neurol (2019) 10:1056. doi: 10.3389/fneur.2019.01056
166. Gelpi E, Höftberger R, Graus F, Ling H, Holton JL, Dawson T, et al. Neuropathological Criteria of Anti-IgLON5-related Tauopathy. Acta Neuropathol (2016) 132:531–43. doi: 10.1007/s00401-016-1591-8
167. Bastiaansen AEM, van Sonderen A, Titulaer MJ. Autoimmune Encephalitis With Anti-Leucine-Rich Glioma-Inactivated 1 or Anti-Contactin-Associated Protein-Like 2 Antibodies (Formerly Called Voltage-Gated Potassium Channel-Complex Antibodies). Curr Opin Neurol (2017) 30:302–9. doi: 10.1097/WCO.0000000000000444
168. Thompson J, Bi M, Murchison AG, Makuch M, Bien CG, Chu K, et al. The Importance of Early Immunotherapy in Patients With Faciobrachial Dystonic Seizures. Brain (2018) 141:348–56. doi: 10.1093/brain/awx323
169. Lehmann-Horn K, Irani SR, Wang S, Palanichamy A, Jahn S, Greenfield AL, et al. Intrathecal B-Cell Activation in LGI1 Antibody Encephalitis. Neurol Neuroimmunol Neuroinflamm (2020) 7:e669. doi: 10.1212/NXI.0000000000000669
170. van Sonderen A, Ariño H, Petit-Pedrol M, Leypoldt F, Körtvélyessy P, Wandinger K-P, et al. The Clinical Spectrum of Caspr2 Antibody-Associated Disease. Neurology (2016) 87:521–8. doi: 10.1212/WNL.0000000000002917
171. Binks S, Varley J, Lee W, Makuch M, Elliott K, Gelfand JM, et al. Distinct HLA Associations of LGI1 and CASPR2-Antibody Diseases. Brain (2018) 141:2263–71. doi: 10.1093/brain/awy109
172. Kim T-J, Lee S-T, Moon J, Sunwoo J-S, Byun J-I, Lim J-A, et al. Anti-LGI1 Encephalitis Is Associated With Unique HLA Subtypes. Ann Neurol (2017) 81:183–92. doi: 10.1002/ana.24860
173. Hu F, Liu X, Zhang L, Chen C, Gong X, Lin J, et al. Novel Findings of HLA Association With Anti-LGI1 Encephalitis: HLA-DRB1*03:01 and HLA-DQB1*02:01. J Neuroimmunol (2020) 344:577243. doi: 10.1016/j.jneuroim.2020.577243
174. van Sonderen A, Roelen DL, Stoop JA, Verdijk RM, Haasnoot GW, Thijs RD, et al. Anti-LGI1 Encephalitis Is Strongly Associated With HLA-DR7 and HLA-DRB4. Ann Neurol (2017) 81:193–8. doi: 10.1002/ana.24858
175. Cortese A, Lombardi R, Briani C, Callegari I, Benedetti L, Manganelli F, et al. Antibodies to Neurofascin, contactin-1, and Contactin-Associated Protein 1 in CIDP: Clinical Relevance of IgG Isotype. Neurol Neuroimmunol Neuroinflamm (2020) 7. doi: 10.1212/NXI.0000000000000639
176. Appeltshauser L, Brunder A-M, Heinius A, Körtvélyessy P, Wandinger K-P, Junker R, et al. Antiparanodal Antibodies and IgG Subclasses in Acute Autoimmune Neuropathy. Neurol Neuroimmunol Neuroinflamm (2020) 7. doi: 10.1212/NXI.0000000000000817
177. Broers MC, Bunschoten C, Nieboer D, Lingsma HF, Jacobs BC. Incidence and Prevalence of Chronic Inflammatory Demyelinating Polyradiculoneuropathy: A Systematic Review and Meta-Analysis. Neuroepidemiology (2019) 52:161–72. doi: 10.1159/000494291
178. Pascual-Goñi E, Fehmi J, Lleixà C, Martín-Aguilar L, Devaux J, Höftberger R, et al. Antibodies to the Caspr1/Contactin-1 Complex in Chronic Inflammatory Demyelinating Polyradiculoneuropathy. Brain (2021) 144:1183–96. doi: 10.1093/brain/awab014
179. Quast I, Keller CW, Hiepe F, Tackenberg B, Lünemann JD. Terminal Complement Activation Is Increased and Associated With Disease Severity in CIDP. Ann Clin Transl Neurol (2016) 3:730–5. doi: 10.1002/acn3.331
180. Querol L, Nogales-Gadea G, Rojas-Garcia R, Martinez-Hernandez E, Diaz-Manera J, Suárez-Calvet X, et al. Antibodies to Contactin-1 in Chronic Inflammatory Demyelinating Polyneuropathy. Ann Neurol (2013) 73:370–80. doi: 10.1002/ana.23794
181. Cotti Piccinelli S, Carella G, Frassi M, Caria F, Gallo Cassarino S, Baldelli E, et al. Human Leukocyte Antigens Class II in CIDP Spectrum Neuropathies. J Neurol Sci (2019) 407:116533. doi: 10.1016/j.jns.2019.116533
182. Appeltshauser L, Weishaupt A, Sommer C, Doppler K. Complement Deposition Induced by Binding of Anti-Contactin-1 Auto-Antibodies Is Modified by Immunoglobulins. Exp Neurol (2017) 287:84–90. doi: 10.1016/j.expneurol.2016.10.006
183. Ogata H, Yamasaki R, Hiwatashi A, Oka N, Kawamura N, Matsuse D, et al. Characterization of IgG4 Anti-Neurofascin 155 Antibody-Positive Polyneuropathy. Ann Clin Transl Neurol (2015) 2:960–71. doi: 10.1002/acn3.248
184. Devaux JJ, Miura Y, Fukami Y, Inoue T, Manso C, Belghazi M, et al. Neurofascin-155 IgG4 in Chronic Inflammatory Demyelinating Polyneuropathy. Neurology (2016) 86:800–7. doi: 10.1212/WNL.0000000000002418
185. Martinez-Martinez L, Lleixà MC, Boera-Carnicero G, Cortese A, Devaux J, Siles A, et al. Anti-NF155 Chronic Inflammatory Demyelinating Polyradiculoneuropathy Strongly Associates to HLA-DRB15. J Neuroinflamm (2017) 14:224. doi: 10.1186/s12974-017-0996-1
186. Ogata H, Isobe N, Zhang X, Yamasaki R, Fujii T, Machida A, et al. Unique HLA Haplotype Associations in IgG4 Anti-Neurofascin 155 Antibody-Positive Chronic Inflammatory Demyelinating Polyneuropathy. J Neuroimmunol (2020) 339:577139. doi: 10.1016/j.jneuroim.2019.577139
187. Pettingill P, Kramer HB, Coebergh JA, Pettingill R, Maxwell S, Nibber A, et al. Antibodies to GABAA Receptor α1 and γ2 Subunits: Clinical and Serologic Characterization. Neurology (2015) 84:1233–41. doi: 10.1212/WNL.0000000000001326
188. Petit-Pedrol M, Armangue T, Peng X, Bataller L, Cellucci T, Davis R, et al. Encephalitis With Refractory Seizures, Status Epilepticus, and Antibodies to the GABAA Receptor: A Case Series, Characterisation of the Antigen, and Analysis of the Effects of Antibodies. Lancet Neurol (2014) 13:276–86. doi: 10.1016/S1474-4422(13)70299-0
189. Spatola M, Petit-Pedrol M, Simabukuro MM, Armangue T, Castro FJ, Barcelo Artigues MI, et al. Investigations in GABAA Receptor Antibody-Associated Encephalitis. Neurology (2017) 88:1012–20. doi: 10.1212/WNL.0000000000003713
190. Zhang X, Lang Y, Sun L, Zhang W, Lin W, Cui L. Clinical Characteristics and Prognostic Analysis of Anti-Gamma-Aminobutyric Acid-B (GABA-B) Receptor Encephalitis in Northeast China. BMC Neurol (2020) 20:1. doi: 10.1186/s12883-019-1585-y
191. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, et al. Antibodies to the GABA(B) Receptor in Limbic Encephalitis With Seizures: Case Series and Characterisation of the Antigen. Lancet Neurol (2010) 9:67–76. doi: 10.1016/S1474-4422(09)70324-2
192. Lin J, Li C, Li A, Liu X, Wang R, Chen C, et al. Encephalitis With Antibodies Against the GABAB Receptor: High Mortality and Risk Factors. Front Neurol (2019) 10:1030. doi: 10.3389/fneur.2019.01030
193. Höftberger R, van Sonderen A, Leypoldt F, Houghton D, Geschwind M, Gelfand J, et al. Encephalitis and AMPA Receptor Antibodies: Novel Findings in a Case Series of 22 Patients. Neurology (2015) 84:2403–12. doi: 10.1212/WNL.0000000000001682
194. Batocchi AP, Marca GD, Frisullo G. Complement-Mediated Cytotoxicity of Antibodies to the GABAB Receptor. Lancet Neurol (2010) 9:343. doi: 10.1016/S1474-4422(10)70070-3
195. Hara M, Ariño H, Petit-Pedrol M, Sabater L, Titulaer MJ, Martinez-Hernandez E, et al. DPPX Antibody-Associated Encephalitis: Main Syndrome and Antibody Effects. Neurology (2017) 88:1340–8. doi: 10.1212/WNL.0000000000003796
196. Piepgras J, Höltje M, Michel K, Li Q, Otto C, Drenckhahn C, et al. Anti-DPPX Encephalitis: Pathogenic Effects of Antibodies on Gut and Brain Neurons. Neurology (2015) 85:890–7. doi: 10.1212/WNL.0000000000001907
197. Boronat A, Gelfand JM, Gresa-Arribas N, Jeong H-Y, Walsh M, Roberts K, et al. Encephalitis and Antibodies to Dipeptidyl-Peptidase-Like Protein-6, a Subunit of Kv4.2 Potassium Channels. Ann Neurol (2013) 73:120–8. doi: 10.1002/ana.23756
198. Tobin WO, Lennon VA, Komorowski L, Probst C, Clardy SL, Aksamit AJ, et al. DPPX Potassium Channel Antibody: Frequency, Clinical Accompaniments, and Outcomes in 20 Patients. Neurology (2014) 83:1797–803. doi: 10.1212/WNL.0000000000000991
199. Spatola M, Sabater L, Planagumà J, Martínez-Hernandez E, Armangué T, Prüss H, et al. Encephalitis With mGluR5 Antibodies: Symptoms and Antibody Effects. Neurology (2018) 90:e1964–72. doi: 10.1212/WNL.0000000000005614
200. Lancaster E, Martinez-Hernandez E, Titulaer MJ, Boulos M, Weaver S, Antoine J-C, et al. Antibodies to Metabotropic Glutamate Receptor 5 in the Ophelia Syndrome. Neurology (2011) 77:1698–701. doi: 10.1212/WNL.0b013e3182364a44
201. Joubert B, Saint-Martin M, Noraz N, Picard G, Rogemond V, Ducray F, et al. Characterization of a Subtype of Autoimmune Encephalitis With Anti-Contactin-Associated Protein-Like 2 Antibodies in the Cerebrospinal Fluid, Prominent Limbic Symptoms, and Seizures. JAMA Neurol (2016) 73:1115–24. doi: 10.1001/jamaneurol.2016.1585
202. Patterson KR, Dalmau J, Lancaster E. Mechanisms of Caspr2 Antibodies in Autoimmune Encephalitis and Neuromyotonia. Ann Neurol (2018) 83:40–51. doi: 10.1002/ana.25120
203. Muñiz-Castrillo S, Joubert B, Elsensohn M-H, Pinto A-L, Saint-Martin M, Vogrig A, et al. Anti-CASPR2 Clinical Phenotypes Correlate With HLA and Immunological Features. J Neurol Neurosurg Psychiatry (2020) 91:1076–84. doi: 10.1136/jnnp-2020-323226
204. Körtvelyessy P, Bauer J, Stoppel CM, Brück W, Gerth I, Vielhaber S, et al. Complement-Associated Neuronal Loss in a Patient With CASPR2 Antibody-Associated Encephalitis. Neurol Neuroimmunol Neuroinflamm (2015) 2:e75. doi: 10.1212/NXI.0000000000000075
205. Doppler K, Appeltshauser L, Villmann C, Martin C, Peles E, Krämer HH, et al. Auto-Antibodies to Contactin-Associated Protein 1 (Caspr) in Two Patients With Painful Inflammatory Neuropathy. Brain (2016) 139:2617–30. doi: 10.1093/brain/aww189
206. Turner MR, Irani SR, Leite MI, Nithi K, Vincent A, Ansorge O. Progressive Encephalomyelitis With Rigidity and Myoclonus: Glycine and NMDA Receptor Antibodies. Neurology (2011) 77:439–43. doi: 10.1212/WNL.0b013e318227b176
207. Crisp SJ, Balint B, Vincent A. Redefining Progressive Encephalomyelitis With Rigidity and Myoclonus After the Discovery of Antibodies to Glycine Receptors. Curr Opin Neurol (2017) 30:310–6. doi: 10.1097/WCO.0000000000000450
208. Zuliani L, Ferlazzo E, Andrigo C, Casano A, Cianci V, Zoccarato M, et al. Glycine Receptor Antibodies in 2 Cases of New, Adult-Onset Epilepsy. Neurol Neuroimmunol Neuroinflamm (2014) 1:e16. doi: 10.1212/NXI.0000000000000016
209. Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, et al. AMPA Receptor Antibodies in Limbic Encephalitis Alter Synaptic Receptor Location. Ann Neurol (2009) 65:424–34. doi: 10.1002/ana.21589
210. Graus F, Boronat A, Xifro X, Boix M, Svigelj V, Garcia A, et al. The Expanding Clinical Profile of Anti-AMPA Receptor Encephalitis. Neurology (2010) 74:857–9. doi: 10.1212/WNL.0b013e3181d3e404
211. Laurido-Soto O, Brier MR, Simon LE, McCullough A, Bucelli RC, Day GS. Patient Characteristics and Outcome Associations in AMPA Receptor Encephalitis. J Neurol (2019) 266:450–60. doi: 10.1007/s00415-018-9153-8
212. Roberts WK, Deluca IJ, Thomas A, Fak J, Williams T, Buckley N, et al. Patients With Lung Cancer and Paraneoplastic Hu Syndrome Harbor HuD-Specific Type 2 CD8+ T Cells. J Clin Invest (2009) 119(7):2042–51. doi: 10.1172/JCI36131
213. Takata K, Stathopoulos P, Cao M, Mané-Damas M, Fichtner ML, Benotti ES, et al. Characterization of Pathogenic Monoclonal Autoantibodies Derived From Muscle-Specific Kinase Myasthenia Gravis Patients. JCI Insight (2019) 4:e127167. doi: 10.1172/jci.insight.127167
214. Ramberger M, Berretta A, Tan JMM, Sun B, Michael S, Yeo T, et al. Distinctive Binding Properties of Human Monoclonal LGI1 Autoantibodies Determine Pathogenic Mechanisms. Brain (2020) 143:1731–45. doi: 10.1093/brain/awaa104
215. van Sonderen A, Thijs RD, Coenders EC, Jiskoot LC, Sanchez E, de Bruijn MAAM, et al. Anti-LGI1 Encephalitis: Clinical Syndrome and Long-Term Follow-Up. Neurology (2016) 87:1449–56. doi: 10.1212/WNL.0000000000003173
216. Stathopoulos P, Alexopoulos H, Dalakas MC. Autoimmune Antigenic Targets at the Node of Ranvier in Demyelinating Disorders. Nat Rev Neurol (2015) 11:143–56. doi: 10.1038/nrneurol.2014.260
217. Koneczny I, Yilmaz V, Lazaridis K, Tzartos J, Lenz TL, Tzartos S, et al. Common Denominators in the Immunobiology of IgG4 Autoimmune Diseases: What Do Glomerulonephritis, Pemphigus Vulgaris, Myasthenia Gravis, Thrombotic Thrombocytopenic Purpura and Autoimmune Encephalitis Have in Common? Front Immunol (2021) 11:605214. doi: 10.3389/fimmu.2020.605214
218. Muñiz-Castrillo S, Vogrig A, Honnorat J. Associations Between HLA and Autoimmune Neurological Diseases With Autoantibodies. Autoimmun Highlights (2020) 11:2. doi: 10.1186/s13317-019-0124-6
219. Lanzavecchia A. Antigen-Specific Interaction Between T and B Cells. Nature (1985) 314:537–9. doi: 10.1038/314537a0
220. Gaig C, Ercilla G, Daura X, Ezquerra M, Fernández-Santiago R, Palou E, et al. HLA and Microtubule-Associated Protein Tau H1 Haplotype Associations in Anti-IgLON5 Disease. Neurol Neuroimmunol Neuroinflamm (2019) 6. doi: 10.1212/NXI.0000000000000605
221. Fujii Y, Monden Y, Hashimoto J, Nakahara K, Kawashima Y. Acetylcholine Receptor Antibody Production by Bone Marrow Cells in a Patient With Myasthenia Gravis. Neurology (1985) 35:577–9. doi: 10.1212/wnl.35.4.577
222. Sambrook MA, Reid H, Mohr PD, Boddie HG. Myasthenia Gravis: Clinical and Histological Features in Relation to Thymectomy. J Neurol Neurosurg Psychiatry (1976) 39:38–43. doi: 10.1136/jnnp.39.1.38
223. Vrolix K, Fraussen J, Losen M, Stevens J, Lazaridis K, Molenaar PC, et al. Clonal Heterogeneity of Thymic B Cells From Early-Onset Myasthenia Gravis Patients With Antibodies Against the Acetylcholine Receptor. J Autoimmun (2014) 52:101–12. doi: 10.1016/j.jaut.2013.12.008
224. Graus YF, de Baets MH, Parren PW, Berrih-Aknin S, Wokke J, van Breda Vriesman PJ, et al. Human Anti-Nicotinic Acetylcholine Receptor Recombinant Fab Fragments Isolated From Thymus-Derived Phage Display Libraries From Myasthenia Gravis Patients Reflect Predominant Specificities in Serum and Block the Action of Pathogenic Serum Antibodies. J Immunol (1997) 158:1919–29.
225. Willcox HN, Newsom-Davis J, Calder LR. Cell Types Required for Anti-Acetylcholine Receptor Antibody Synthesis by Cultured Thymocytes and Blood Lymphocytes in Myasthenia Gravis. Clin Exp Immunol (1984) 58:97–106.
226. Stathopoulos P, Kumar A, Nowak RJ, O’Connor KC. Autoantibody-Producing Plasmablasts After B Cell Depletion Identified in Muscle-Specific Kinase Myasthenia Gravis. JCI Insight (2017) 2:e94263. doi: 10.1172/jci.insight.94263
227. Jarius S, Aboul-Enein F, Waters P, Kuenz B, Hauser A, Berger T, et al. Antibody to Aquaporin-4 in the Long-Term Course of Neuromyelitis Optica. Brain (2008) 131:3072–80. doi: 10.1093/brain/awn240
228. Pellkofer HL, Krumbholz M, Berthele A, Hemmer B, Gerdes LA, Havla J, et al. Long-Term Follow-Up of Patients With Neuromyelitis Optica After Repeated Therapy With Rituximab. Neurology (2011) 76:1310–5. doi: 10.1212/WNL.0b013e3182152881
229. Kim S-H, Kim W, Li XF, Jung I-J, Kim HJ. Repeated Treatment With Rituximab Based on the Assessment of Peripheral Circulating Memory B Cells in Patients With Relapsing Neuromyelitis Optica Over 2 Years. Arch Neurol (2011) 68:1412–20. doi: 10.1001/archneurol.2011.154
230. Kim S-H, Huh S-Y, Lee SJ, Joung A, Kim HJ. A 5-Year Follow-up of Rituximab Treatment in Patients With Neuromyelitis Optica Spectrum Disorder. JAMA Neurol (2013) 70:1110. doi: 10.1001/jamaneurol.2013.3071
231. Guo Y, Weigand SD, Popescu BF, Lennon VA, Parisi JE, Pittock SJ, et al. Pathogenic Implications of Cerebrospinal Fluid Barrier Pathology in Neuromyelitis Optica. Acta Neuropathol (2017) 133:597–612. doi: 10.1007/s00401-017-1682-1
232. Ohara S, Miyahira T-A, Oguchi K, Takei Y-I, Yanagimura F, Kawachi I, et al. Neuromyelitis Optica Spectrum Disorder With Massive Basal Ganglia Involvement: A Case Report. BMC Neurol (2019) 19:351. doi: 10.1186/s12883-019-1580-3
233. Chihara N, Aranami T, Sato W, Miyazaki Y, Miyake S, Okamoto T, et al. Interleukin 6 Signaling Promotes Anti-Aquaporin 4 Autoantibody Production From Plasmablasts in Neuromyelitis Optica. Proc Natl Acad Sci USA (2011) 108:3701–6. doi: 10.1073/pnas.1017385108
234. Wilson R, Makuch M, Kienzler A-K, Varley J, Taylor J, Woodhall M, et al. Condition-Dependent Generation of Aquaporin-4 Antibodies From Circulating B Cells in Neuromyelitis Optica. Brain (2018) 141:1063–74. doi: 10.1093/brain/awy010
235. Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, et al. Intrathecal Pathogenic Anti-Aquaporin-4 Antibodies in Early Neuromyelitis Optica. Ann Neurol (2009) 66:617–29. doi: 10.1002/ana.21802
236. Durozard P, Rico A, Boutiere C, Maarouf A, Lacroix R, Cointe S, et al. Comparison of the Response to Rituximab Between Myelin Oligodendrocyte Glycoprotein and Aquaporin-4 Antibody Diseases. Ann Neurol (2020) 87:256–66. doi: 10.1002/ana.25648
237. Takai Y, Misu T, Kaneko K, Chihara N, Narikawa K, Tsuchida S, et al. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: An Immunopathological Study. Brain (2020) 143:1431–46. doi: 10.1093/brain/awaa102
238. Tanaka S, Hashimoto B, Izaki S, Oji S, Fukaura H, Nomura K. Clinical and Immunological Differences Between MOG Associated Disease and Anti AQP4 Antibody-Positive Neuromyelitis Optica Spectrum Disorders: Blood-Brain Barrier Breakdown and Peripheral Plasmablasts. Mult Scler Relat Disord (2020) 41:102005. doi: 10.1016/j.msard.2020.102005
239. Hachiya Y, Uruha A, Kasai-Yoshida E, Shimoda K, Satoh-Shirai I, Kumada S, et al. Rituximab Ameliorates Anti-N-Methyl-D-Aspartate Receptor Encephalitis by Removal of Short-Lived Plasmablasts. J Neuroimmunol (2013) 265:128–30. doi: 10.1016/j.jneuroim.2013.09.017
240. Lee W-J, Lee S-T, Shin Y-W, Lee HS, Shin H-R, Kim D-Y, et al. Rituximab and Tocilizumab (T-SIRT) in Anti-NMDAR Encephalitis. Neurotherapeutics (2021) 18(1):474–87. doi: 10.1007/s13311-020-00921-7
241. Malviya M, Barman S, Golombeck KS, Planagumà J, Mannara F, Strutz-Seebohm N, et al. NMDAR Encephalitis: Passive Transfer From Man to Mouse by a Recombinant Antibody. Ann Clin Transl Neurol (2017) 4:768–83. doi: 10.1002/acn3.444
242. Makuch M, Wilson R, Al-Diwani A, Varley J, Kienzler A-K, Taylor J, et al. N-Methyl-D-Aspartate Receptor Antibody Production From Germinal Center Reactions: Therapeutic Implications. Ann Neurol (2018) 83:553–61. doi: 10.1002/ana.25173
243. Kreye J, Wenke NK, Chayka M, Leubner J, Murugan R, Maier N, et al. Human Cerebrospinal Fluid Monoclonal N -Methyl-D-Aspartate Receptor Autoantibodies Are Sufficient for Encephalitis Pathogenesis. Brain (2016) 139:2641–52. doi: 10.1093/brain/aww208
244. Erro ME, Sabater L, Martínez L, Herrera M, Ostolaza A, García de Gurtubay I, et al. Anti-IGLON5 Disease: A New Case Without Neuropathologic Evidence of Brainstem Tauopathy. Neurol Neuroimmunol Neuroinflamm (2020) 7:e651. doi: 10.1212/NXI.0000000000000651
245. Montagna M, Amir R, De Volder I, Lammens M, Huyskens J, Willekens B. Iglon5-Associated Encephalitis With Atypical Brain Magnetic Resonance Imaging and Cerebrospinal Fluid Changes. Front Neurol (2018) 9:329. doi: 10.3389/fneur.2018.00329
246. Irani SR, Lehmann-Horn K, Geschwind M, Wang S, Vincent A, von Büdingen H-C. The Active Intrathecal B-Cell Response in LGI1-Antibody Encephalitis. Lancet (2015) 385(Suppl 1):S46. doi: 10.1016/S0140-6736(15)60361-0
247. Markovic I, Basic S, Devedjija S. Aggressive Anti-LGI1 Encephalitis Defeated by One Cycle of Intravenous Rituximab-A Case Report. Neurol Sci (2020) 41:1949–50. doi: 10.1007/s10072-020-04264-1
248. Kornau H-C, Kreye J, Stumpf A, Fukata Y, Parthier D, Sammons RP, et al. Human Cerebrospinal Fluid Monoclonal Lgi1 Autoantibodies Increase Neuronal Excitability. Ann Neurol (2020) 87:405–18. doi: 10.1002/ana.25666
249. Querol L, Rojas-García R, Diaz-Manera J, Barcena J, Pardo J, Ortega-Moreno A, et al. Rituximab in Treatment-Resistant CIDP With Antibodies Against Paranodal Proteins. Neurol Neuroimmunol Neuroinflamm (2015) 2:e149. doi: 10.1212/NXI.0000000000000149
250. Delmont E, Brodovitch A, Kouton L, Allou T, Beltran S, Brisset M, et al. Antibodies Against the Node of Ranvier: A Real-Life Evaluation of Incidence, Clinical Features and Response to Treatment Based on a Prospective Analysis of 1500 Sera. J Neurol (2020) 267:3664–72. doi: 10.1007/s00415-020-10041-z
251. Ye L, Schnegelsberg M, Obermann M. Dipeptidyl-Peptidase-Like Protein 6 Encephalitis Treated With Immunotherapy. Proc (Bayl Univ Med Cent) (2020) 34:114–5. doi: 10.1080/08998280.2020.1822132
252. Ong E, Viaccoz A, Ducray F, Pérol M, Cavillon G, Rogemond V, et al. Dramatic Improvement After Rituximab in a Patient With Paraneoplastic Treatment-Refractory Morvan Syndrome Associated With Anti-CASPR2 Antibodies. Eur J Neurol (2013) 20:e96–97. doi: 10.1111/ene.12164
253. Christ M, Müller T, Bien C, Hagen T, Naumann M, Bayas A. Autoimmune Encephalitis Associated With Antibodies Against the Metabotropic Glutamate Receptor Type 1: Case Report and Review of the Literature. Ther Adv Neurol Disord (2019) 12:1756286419847418. doi: 10.1177/1756286419847418
254. Golombeck KS, Bönte K, Mönig C, van Loo KM, Hartwig M, Schwindt W, et al. Evidence of a Pathogenic Role for CD8 + T Cells in Anti-GABA B Receptor Limbic Encephalitis. Neurol Neuroimmunol Neuroinflamm (2016) 3:e232. doi: 10.1212/NXI.0000000000000232
255. Smith MR. Rituximab (Monoclonal Anti-CD20 Antibody): Mechanisms of Action and Resistance. Oncogene (2003) 22:7359–68. doi: 10.1038/sj.onc.1206939
256. Edwards JCW, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, et al. Efficacy of B-Cell-Targeted Therapy With Rituximab in Patients With Rheumatoid Arthritis. N Engl J Med (2004) 350:2572–81. doi: 10.1056/NEJMoa032534
257. Yi JS, DeCroos EC, Sanders DB, Weinhold KJ, Guptill JT. Prolonged B-Cell Depletion in MuSK Myasthenia Gravis Following Rituximab Treatment: Letters to the Editor. Muscle Nerve (2013) 48:992–3. doi: 10.1002/mus.24063
258. Mouquet H, Musette P, Gougeon M-L, Jacquot S, Lemercier B, Lim A, et al. B-Cell Depletion Immunotherapy in Pemphigus: Effects on Cellular and Humoral Immune Responses. J Invest Dermatol (2008) 128:2859–69. doi: 10.1038/jid.2008.178
259. Popa C, Leandro MJ, Cambridge G, Edwards JCW. Repeated B Lymphocyte Depletion With Rituximab in Rheumatoid Arthritis Over 7 Yrs. Rheumatology (2006) 46:626–30. doi: 10.1093/rheumatology/kel393
260. Schröder C, Azimzadeh AM, Wu G, Price JO, Atkinson JB, Pierson RN. Anti-CD20 Treatment Depletes B-Cells in Blood and Lymphatic Tissue of Cynomolgus Monkeys. Transpl Immunol (2003) 12:19–28. doi: 10.1016/S0966-3274(03)00059-5
261. De La Torre I, Leandro MJ, Valor L, Becerra E, Edwards JCW, Cambridge G. Total Serum Immunoglobulin Levels in Patients With RA After Multiple B-Cell Depletion Cycles Based on Rituximab: Relationship With B-Cell Kinetics. Rheumatology (2012) 51:833–40. doi: 10.1093/rheumatology/ker417
262. Cortés-Vicente E, Rojas-Garcia R, Díaz-Manera J, Querol L, Casasnovas C, Guerrero-Sola A, et al. The Impact of Rituximab Infusion Protocol on the Long-Term Outcome in Anti-MuSK Myasthenia Gravis. Ann Clin Transl Neurol (2018) 5:710–6. doi: 10.1002/acn3.564
263. Evoli A. Clinical Correlates With Anti-MuSK Antibodies in Generalized Seronegative Myasthenia Gravis. Brain (2003) 126:2304–11. doi: 10.1093/brain/awg223
264. Cao Y, Amezquita RA, Kleinstein SH, Stathopoulos P, Nowak RJ, O’Connor KC. Autoreactive T Cells From Patients With Myasthenia Gravis Are Characterized by Elevated IL-17, Ifn-γ, and GM-CSF and Diminished IL-10 Production. J Immunol (2016) 196:2075–84. doi: 10.4049/jimmunol.1501339
265. Huijbers MG, Vergoossen DL, Fillié-Grijpma YE, van Es IE, Koning MT, Slot LM, et al. MuSK Myasthenia Gravis Monoclonal Antibodies: Valency Dictates Pathogenicity. Neurol Neuroimmunol Neuroinflamm (2019) 6:e547. doi: 10.1212/NXI.0000000000000547
266. Waters PJ, McKeon A, Leite MI, Rajasekharan S, Lennon VA, Villalobos A, et al. Serologic Diagnosis of NMO: A Multicenter Comparison of Aquaporin-4-IgG Assays. Neurology (2012) 78:665–71. doi: 10.1212/WNL.0b013e318248dec1
267. Ringelstein M, Metz I, Ruprecht K, Koch A, Rappold J, Ingwersen J, et al. Contribution of Spinal Cord Biopsy to Diagnosis of Aquaporin-4 Antibody Positive Neuromyelitis Optica Spectrum Disorder. Mult Scler (2014) 20:882–8. doi: 10.1177/1352458513510981
268. Takai Y, Misu T, Suzuki H, Takahashi T, Okada H, Tanaka S, et al. Staging of Astrocytopathy and Complement Activation in Neuromyelitis Optica Spectrum Disorders. Brain (2021) awab102. doi: 10.1093/brain/awab102
269. Almekhlafi MA, Clark AW, Lucchinetti CF, Zhang Y, Power C, Bell RB. Neuromyelitis Optica With Extensive Active Brain Involvement: An Autopsy Study. Arch Neurol (2011) 68:508–12. doi: 10.1001/archneurol.2011.62
270. Chihara N, Matsumoto R, Yamamura T. Plasmablasts and Neuroimmunological Disorders. Immunol Med (2019) 42:103–7. doi: 10.1080/25785826.2019.1659476
271. Chihara N, Aranami T, Oki S, Matsuoka T, Nakamura M, Kishida H, et al. Plasmablasts as Migratory IgG-Producing Cells in the Pathogenesis of Neuromyelitis Optica. PloS One (2013) 8:e83036. doi: 10.1371/journal.pone.0083036
272. Kowarik MC, Astling D, Gasperi C, Wemlinger S, Schumann H, Dzieciatkowska M, et al. CNS Aquaporin-4-Specific B Cells Connect With Multiple B-cell Compartments in Neuromyelitis Optica Spectrum Disorder. Ann Clin Transl Neurol (2017) 4:369–80. doi: 10.1002/acn3.418
273. Ayzenberg I, Kleiter I, Schröder A, Hellwig K, Chan A, Yamamura T, et al. Interleukin 6 Receptor Blockade in Patients With Neuromyelitis Optica Nonresponsive to Anti-CD20 Therapy. JAMA Neurol (2013) 70:394–7. doi: 10.1001/jamaneurol.2013.1246
274. Araki M, Aranami T, Matsuoka T, Nakamura M, Miyake S, Yamamura T. Clinical Improvement in a Patient With Neuromyelitis Optica Following Therapy With the Anti-IL-6 Receptor Monoclonal Antibody Tocilizumab. Mod Rheumatol (2013) 23:827–31. doi: 10.1007/s10165-012-0715-9
275. Stathopoulos P, Chastre A, Waters P, Irani S, Fichtner ML, Benotti ES, et al. Autoantibodies Against Neurologic Antigens in Nonneurologic Autoimmunity. J Immunol (2019) 202:2210–9. doi: 10.4049/jimmunol.1801295
276. Spadaro M, Winklmeier S, Beltrán E, Macrini C, Höftberger R, Schuh E, et al. Pathogenicity of Human Antibodies Against Myelin Oligodendrocyte Glycoprotein: MOG Antibody Pathogenicity. Ann Neurol (2018) 84:315–28. doi: 10.1002/ana.25291
277. Cobo-Calvo A, Sepúlveda M, Rollot F, Armangué T, Ruiz A, Maillart E, et al. Evaluation of Treatment Response in Adults With Relapsing MOG-Ab-Associated Disease. J Neuroinflamm (2019) 16:134. doi: 10.1186/s12974-019-1525-1
278. Whittam DH, Cobo-Calvo A, Lopez-Chiriboga AS, Pardo S, Gornall M, Cicconi S, et al. Treatment of MOG-IgG-Associated Disorder With Rituximab: An International Study of 121 Patients. Multiple Sclerosis Relat Disord (2020) 44:102251. doi: 10.1016/j.msard.2020.102251
279. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. Mog-IgG in NMO and Related Disorders: A Multicenter Study of 50 Patients. Part 2: Epidemiology, Clinical Presentation, Radiological and Laboratory Features, Treatment Responses, and Long-Term Outcome. J Neuroinflamm (2016) 13:280. doi: 10.1186/s12974-016-0718-0
280. Irani SR, Bera K, Waters P, Zuliani L, Maxwell S, Zandi MS, et al. N-Methyl-D-Aspartate Antibody Encephalitis: Temporal Progression of Clinical and Paraclinical Observations in a Predominantly Non-Paraneoplastic Disorder of Both Sexes. Brain (2010) 133:1655–67. doi: 10.1093/brain/awq113
281. Kong S-S, Chen Y-J, Su I-C, Lin J-J, Chou I-J, Chou M-L, et al. Immunotherapy for Anti-NMDA Receptor Encephalitis: Experience From a Single Center in Taiwan. Pediatr Neonatology (2019) 60:417–22. doi: 10.1016/j.pedneo.2018.10.006
282. Dou X, Li D, Wu Y, Wang Z, Yang L, Ma N, et al. Efficacy and Safety of Rituximab in Chinese Children With Refractory Anti-NMDAR Encephalitis. Front Neurol (2020) 11:606923. doi: 10.3389/fneur.2020.606923
283. Lee W-J, Lee S-T, Byun J-I, Sunwoo J-S, Kim T-J, Lim J-A, et al. Rituximab Treatment for Autoimmune Limbic Encephalitis in an Institutional Cohort. Neurology (2016) 86:1683–91. doi: 10.1212/WNL.0000000000002635
284. Chefdeville A, Treilleux I, Mayeur M-E, Couillault C, Picard G, Bost C, et al. Immunopathological Characterization of Ovarian Teratomas Associated With Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Acta Neuropathol Commun (2019) 7:38. doi: 10.1186/s40478-019-0693-7
285. Makuch M, Wilson R, Al-Diwani A, Varley J, Kienzler A-K, Taylor J, et al. N-Methyl-D-Aspartate Receptor Antibody Production From Germinal Center Reactions: Therapeutic Implications: NMDAR-Antibody Production From Germinal Centers. Ann Neurol (2018) 83:553–61. doi: 10.1002/ana.25173
286. Sabater L, Planagumà J, Dalmau J, Graus F. Cellular Investigations With Human Antibodies Associated With the Anti-IgLON5 Syndrome. J Neuroinflamm (2016) 13:226. doi: 10.1186/s12974-016-0689-1
287. Balint B, Bhatia KP. Friend or Foe? IgLON5 Antibodies in a Novel Tauopathy With Prominent Sleep Movement Disorder, Ataxia, and Chorea. Mov Disord (2014) 29:989. doi: 10.1002/mds.25926
288. Sabater L, Gaig C, Gelpi E, Bataller L, Lewerenz J, Torres-Vega E, et al. A Novel Non-Rapid-Eye Movement and Rapid-Eye-Movement Parasomnia With Sleep Breathing Disorder Associated With Antibodies to IgLON5: A Case Series, Characterisation of the Antigen, and Post-Mortem Study. Lancet Neurol (2014) 13:575–86. doi: 10.1016/S1474-4422(14)70051-1
289. Grüter T, Behrendt V, Bien CI, Gold R, Ayzenberg I. Early Immunotherapy Is Highly Effective in IgG1/IgG4 Positive IgLON5 Disease. J Neurol (2020) 267:2151–3. doi: 10.1007/s00415-020-09924-y
290. Simabukuro MM, Sabater L, Adoni T, Cury RG, Haddad MS, Moreira CH, et al. Sleep Disorder, Chorea, and Dementia Associated With IgLON5 Antibodies. Neurol Neuroimmunol Neuroinflamm (2015) 2:e136. doi: 10.1212/NXI.0000000000000136
291. Werner J, Jelcic I, Schwarz EI, Probst-Müller E, Nilsson J, Schwizer B, et al. Anti-IgLON5 Disease: A New Bulbar-Onset Motor Neuron Mimic Syndrome. Neurol Neuroimmunol Neuroinflamm (2021) 8:e962. doi: 10.1212/NXI.0000000000000962
292. Cabezudo-García P, Mena-Vázquez N, Estivill Torrús G, Serrano-Castro P. Response to Immunotherapy in Anti-IgLON5 Disease: A Systematic Review. Acta Neurol Scand (2020) 141:263–70. doi: 10.1111/ane.13207
293. Irani SR, Gelfand JM, Bettcher BM, Singhal NS, Geschwind MD. Effect of Rituximab in Patients With Leucine-Rich, Glioma-Inactivated 1 Antibody–Associated Encephalopathy. JAMA Neurol (2014) 71:896. doi: 10.1001/jamaneurol.2014.463
294. Klang A, Schmidt P, Kneissl S, Bagó Z, Vincent A, Lang B, et al. IgG and Complement Deposition and Neuronal Loss in Cats and Humans With Epilepsy and Voltage-Gated Potassium Channel Complex Antibodies. J Neuropathol Exp Neurol (2014) 73:403–13. doi: 10.1097/NEN.0000000000000063
295. Haddad G, Lorenzen JM, Ma H, de Haan N, Seeger H, Zaghrini C, et al. Altered Glycosylation of IgG4 Promotes Lectin Complement Pathway Activation in Anti-PLA2R1–Associated Membranous Nephropathy. J Clin Invest (2021) 131:e140453. doi: 10.1172/JCI140453
296. Lhotta K, Würzner R, König P. Glomerular Deposition of Mannose-Binding Lectin in Human Glomerulonephritis. Nephrol Dial Transplant (1999) 14:881–6. doi: 10.1093/ndt/14.4.881
297. Yang Y, Wang C, Jin L, He F, Li C, Gao Q, et al. IgG4 Anti-Phospholipase A2 Receptor Might Activate Lectin and Alternative Complement Pathway Meanwhile in Idiopathic Membranous Nephropathy: An Inspiration From a Cross-Sectional Study. Immunol Res (2016) 64:919–30. doi: 10.1007/s12026-016-8790-1
298. Segawa Y, Hisano S, Matsushita M, Fujita T, Hirose S, Takeshita M, et al. Igg Subclasses and Complement Pathway in Segmental and Global Membranous Nephropathy. Pediatr Nephrol (2010) 25:1091–9. doi: 10.1007/s00467-009-1439-8
299. Hayashi N, Okada K, Matsui Y, Fujimoto K, Adachi H, Yamaya H, et al. Glomerular Mannose-Binding Lectin Deposition in Intrinsic Antigen-Related Membranous Nephropathy. Nephrol Dial Transplant (2018) 33:832–40. doi: 10.1093/ndt/gfx235
300. Lehmann-Horn K, Irani SR, Wang S, Palanichamy A, Jahn S, Greenfield AL, et al. Intrathecal B-Cell Activation in LGI1 Antibody Encephalitis. Neurol Neuroimmunol Neuroinflamm (2020) 7:e669. doi: 10.1212/NXI.0000000000000669
301. Garg N, Park SB, Yiannikas C, Vucic S, Howells J, Noto Y-I, et al. Neurofascin-155 IGG4 Neuropathy: Pathophysiological Insights, Spectrum of Clinical Severity and Response to Treatment. Muscle Nerve (2018) 57:848–51. doi: 10.1002/mus.26010
302. Gluck L, Hernandez AL, Wesley SF, Fulbright RK, Longbrake EE, Stathopoulos P. Therapeutic Considerations in a Case of Progressive Encephalomyelitis With Rigidity and Myoclonus. J Neurol Sci (2020) 416:116993. doi: 10.1016/j.jns.2020.116993
303. van Coevorden-Hameete MH, de Bruijn MAAM, de Graaff E, Bastiaansen DAEM, Schreurs MWJ, Demmers JAA, et al. The Expanded Clinical Spectrum of Anti-GABABR Encephalitis and Added Value of KCTD16 Autoantibodies. Brain (2019) 142:1631–43. doi: 10.1093/brain/awz094
304. O’Connor K, Waters P, Komorowski L, Zekeridou A, Guo C-Y, Mgbachi VC, et al. Gaba A Receptor Autoimmunity: A Multicenter Experience. Neurol Neuroimmunol Neuroinflamm (2019) 6:e552. doi: 10.1212/NXI.0000000000000552
305. Boronat A, Gelfand JM, Gresa-Arribas N, Jeong H-Y, Walsh M, Roberts K, et al. Encephalitis and Antibodies to Dipeptidyl-Peptidase-Like protein-6, a Subunit of Kv4.2 Potassium Channels. Ann Neurol (2013) 73:120–8. doi: 10.1002/ana.23756
306. Sveinsson O, Al Nimer F, Piehl F. Morvan’s Syndrome Treated Successfully With Rituximab and Lacosamide. BMJ Case Rep (2019) 12. doi: 10.1136/bcr-2018-226832
307. Lancaster E, Huijbers MGM, Bar V, Boronat A, Wong A, Martinez-Hernandez E, et al. Investigations of Caspr2, an Autoantigen of Encephalitis and Neuromyotonia. Ann Neurol (2011) 69:303–11. doi: 10.1002/ana.22297
308. Lopez-Chiriboga AS, Komorowski L, Kümpfel T, Probst C, Hinson SR, Pittock SJ, et al. Metabotropic Glutamate Receptor Type 1 Autoimmunity: Clinical Features and Treatment Outcomes. Neurology (2016) 86:1009–13. doi: 10.1212/WNL.0000000000002476
309. Joly P, Mouquet H, Roujeau J-C, D’Incan M, Gilbert D, Jacquot S, et al. A Single Cycle of Rituximab for the Treatment of Severe Pemphigus. N Engl J Med (2007) 357:545–52. doi: 10.1056/NEJMoa067752
310. Scully M, McDonald V, Cavenagh J, Hunt BJ, Longair I, Cohen H, et al. A Phase 2 Study of the Safety and Efficacy of Rituximab With Plasma Exchange in Acute Acquired Thrombotic Thrombocytopenic Purpura. Blood (2011) 118:1746–53. doi: 10.1182/blood-2011-03-341131
311. Beck LH, Fervenza FC, Beck DM, Bonegio RGB, Malik FA, Erickson SB, et al. Rituximab-Induced Depletion of Anti-PLA2R Autoantibodies Predicts Response in Membranous Nephropathy. J Am Soc Nephrol (2011) 22:1543–50. doi: 10.1681/ASN.2010111125
312. Kasperkiewicz M, Shimanovich I, Ludwig RJ, Rose C, Zillikens D, Schmidt E. Rituximab for Treatment-Refractory Pemphigus and Pemphigoid: A Case Series of 17 Patients. J Am Acad Dermatol (2011) 65:552–8. doi: 10.1016/j.jaad.2010.07.032
313. Dupuy A, Viguier M, Bédane C, Cordoliani F, Blaise S, Aucouturier F, et al. Treatment of Refractory Pemphigus Vulgaris With Rituximab (Anti-Cd20 Monoclonal Antibody). Arch Dermatol (2004) 140. doi: 10.1001/archderm.140.1.91
314. Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al. First-Line Rituximab Combined With Short-Term Prednisone Versus Prednisone Alone for the Treatment of Pemphigus (Ritux 3): A Prospective, Multicentre, Parallel-Group, Open-Label Randomised Trial. Lancet (2017) 389:2031–40. doi: 10.1016/S0140-6736(17)30070-3
315. Lee J, Lundgren DK, Mao X, Manfredo-Vieira S, Nunez-Cruz S, Williams EF, et al. Antigen-Specific B Cell Depletion for Precision Therapy of Mucosal Pemphigus Vulgaris. J Clin Invest (2020) 130:6317–24. doi: 10.1172/JCI138416
316. Sato Y, Yoshino T. IgG4-Related Lymphadenopathy. Int J Rheumatol (2012) 2012:572539. doi: 10.1155/2012/572539
317. Renton AE, Pliner HA, Provenzano C, Evoli A, Ricciardi R, Nalls MA, et al. A Genome-Wide Association Study of Myasthenia Gravis. JAMA Neurol (2015) 72:396–404. doi: 10.1001/jamaneurol.2014.4103
318. Vodo D, Sarig O, Sprecher E. The Genetics of Pemphigus Vulgaris. Front Med (Lausanne) (2018) 5:226. doi: 10.3389/fmed.2018.00226
319. Lee J-Y, Stathopoulos P, Gupta S, Bannock JM, Barohn RJ, Cotzomi E, et al. Compromised Fidelity of B-Cell Tolerance Checkpoints in AChR and MuSK Myasthenia Gravis. Ann Clin Transl Neurol (2016) 3:443–54. doi: 10.1002/acn3.311
320. Cotzomi E, Stathopoulos P, Lee CS, Ritchie AM, Soltys JN, Delmotte FR, et al. Early B Cell Tolerance Defects in Neuromyelitis Optica Favour Anti-AQP4 Autoantibody Production. Brain (2019) 142:1598–615. doi: 10.1093/brain/awz106
321. Janssen M, Bruijstens AL, van Langelaar J, Wong Y, Wierenga-Wolf AF, Melief M-J, et al. Naive B Cells in Neuromyelitis Optica Spectrum Disorders: Impact of Steroid Use and Relapses. Brain Commun (2020) 2:fcaa197. doi: 10.1093/braincomms/fcaa197
322. Varrin-Doyer M, Spencer CM, Schulze-Topphoff U, Nelson PA, Stroud RM, Cree BAC, et al. Aquaporin 4-Specific T Cells in Neuromyelitis Optica Exhibit a Th17 Bias and Recognize Clostridium ABC Transporter. Ann Neurol (2012) 72:53–64. doi: 10.1002/ana.23651
Keywords: short-lived, long-lived plasma cells, IgG4, autoantibody-mediated disorders, rituximab, neurological autoimmunity
Citation: Zografou C, Vakrakou AG and Stathopoulos P (2021) Short- and Long-Lived Autoantibody-Secreting Cells in Autoimmune Neurological Disorders. Front. Immunol. 12:686466. doi: 10.3389/fimmu.2021.686466
Received: 26 March 2021; Accepted: 28 May 2021;
Published: 17 June 2021.
Edited by:
Mirjam van der Burg, Leiden University Medical Center, NetherlandsReviewed by:
Maartje G. Huijbers, Leiden University Medical Center, NetherlandsCopyright © 2021 Zografou, Vakrakou and Stathopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: P. Stathopoulos, cG1zdGF0aG9wb3Vsb3NAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.