
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 28 May 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.683803
This article is part of the Research Topic NLRP3 Activation and Regulation in Innate Immune Responses View all 12 articles
 Patrick Münzer1,2,3,4
Patrick Münzer1,2,3,4 Roberto Negro1,5
Roberto Negro1,5 Shoichi Fukui1,2
Shoichi Fukui1,2 Lucas di Meglio1,4,6
Lucas di Meglio1,4,6 Karen Aymonnier1,2,4
Karen Aymonnier1,2,4 Long Chu1,2Deya Cherpokova1,2
Long Chu1,2Deya Cherpokova1,2 Sarah Gutch1,2Nicoletta Sorvillo1,2
Sarah Gutch1,2Nicoletta Sorvillo1,2 Lai Shi1,2Venkat Giri Magupalli1,5Alexander N. R. Weber7
Lai Shi1,2Venkat Giri Magupalli1,5Alexander N. R. Weber7 Rüdiger E. Scharf1,2,8
Rüdiger E. Scharf1,2,8 Clare M. Waterman4,9Hao Wu1,5
Clare M. Waterman4,9Hao Wu1,5 Denisa D. Wagner1,2,4,10*
Denisa D. Wagner1,2,4,10*Neutrophil extracellular trap formation (NETosis) and the NLR family pyrin domain containing 3 (NLRP3) inflammasome assembly are associated with a similar spectrum of human disorders. While NETosis is known to be regulated by peptidylarginine deiminase 4 (PAD4), the role of the NLRP3 inflammasome in NETosis was not addressed. Here, we establish that under sterile conditions the cannonical NLRP3 inflammasome participates in NETosis. We show apoptosis-associated speck-like protein containing a CARD (ASC) speck assembly and caspase-1 cleavage in stimulated mouse neutrophils without LPS priming. PAD4 was needed for optimal NLRP3 inflammasome assembly by regulating NLRP3 and ASC protein levels post-transcriptionally. Genetic ablation of NLRP3 signaling resulted in impaired NET formation, because NLRP3 supported both nuclear envelope and plasma membrane rupture. Pharmacological inhibition of NLRP3 in either mouse or human neutrophils also diminished NETosis. Finally, NLRP3 deficiency resulted in a lower density of NETs in thrombi produced by a stenosis-induced mouse model of deep vein thrombosis. Altogether, our results indicate a PAD4-dependent formation of the NLRP3 inflammasome in neutrophils and implicate NLRP3 in NETosis under noninfectious conditions in vitro and in vivo.
Initially described as part of the innate immune response to microbes (1), there is now increasing evidence that neutrophil extracellular traps (NETs) are produced under sterile conditions. They are implicated in a wide variety of inflammatory, (auto)immune, and thrombo-occlusive disorders. In particular, NETs are known to foster thrombosis (2, 3), contribute to ischemia/reperfusion injury (4), and age-related tissue fibrosis (5). NETs also likely contribute to the severe side effects of a COVID-19 infection (6). Moreover, NET formation is stimulated by diseases, such as diabetes (7) and cancer (8), and contributes to cancer progression (9, 10).
NETs are decondensed chromatin meshworks ejected by neutrophils upon inflammatory stimulation or hypoxia. A key characteristic of the extracellular neutrophil chromatin is the inclusion of pro-thrombotic, pro-inflammatory, and cytotoxic components, in particular histones and microbicidal proteases (3, 11). While the clinical relevance of NETs is recognized, the underlying cellular mechanisms of their induction are poorly defined. Recently, NET formation (NETosis) was determined to be a well-orchestrated sequence of cellular events, including disassembly of the cellular cytoskeletons, endomembrane fragmentation, nuclear rounding, plasma membrane permeabilization, and finally nuclear and plasma membrane rupture (12).
A major prerequisite for NETosis is the peptidylarginine deiminase 4 (PAD4)-dependent post-translational modification of histones (13, 14). In general, PAD enzymes are calcium-dependent enzymes which deiminate the positively charged arginine residue of proteins, thus transforming arginine to a neutral citrulline. PAD4 is mainly expressed in granulocytes and transferred into an enzymatic active conformation upon calcium binding. Moreover, since PAD4 is the only PAD isoform that contains a nuclear localization sequence, it is required for nuclear histone citrullination (12, 15). During the course of NETosis, it is thought that citrullination of histones reduces their DNA/histone binding ability which causes chromatin decondensation and subsequently culminates in chromatin expulsion.
Effective inducers of NETosis in vitro are the calcium ionophore ionomycin, the protein kinase C activator phorbol 12-myristate 13-acetate (PMA), and the potassium ionophore nigericin (16, 17). Interestingly, nigericin is widely used in macrophages to induce assembly of the NLR family pyrin domain containing 3 (NLRP3) inflammasome, which is also expressed in neutrophils (18, 19). In neutrophils the NLRP3 inflammasome was found to be activated after bacterial infection (20, 21) or after lipopolysaccharide (LPS) pretreatment with subsequent ATP stimulation (22). In addition, an activating mutation (A352V) in NLRP3 leading to Muckle Wells syndrome is associated with excessive neutrophil granule exocytosis (23) and a gain-of-function mutation in NLRP3, which results in Familial Mediterranean Fever (FMF) is subsequently linked to augmented NETosis (24, 25).
Inflammasomes are multiprotein signaling platforms, described mainly in macrophages, that mediate pivotal responses of innate immunity after activation by pathogen- and danger-associated molecular patterns (PAMPs and DAMPs). The most prominent and best studied inflammasome is NLRP3 (26, 27). Assembly of the NLRP3 inflammasome in macrophages in vitro requires a two-step mechanism. First, protein expression levels of NLRP3 have to be increased transcriptionally by priming macrophages with lipopolysaccharide (LPS) and, second, subsequent stimulation for example with pore-forming toxins (nigericin), results in a prominent NIMA (never in mitosis gene a)-related kinase 7 (NEK7)-dependent oligomerization of NLRP3 (28, 29). Successively, apoptosis-associated speck-like protein containing a CARD (ASC) is recruited, polymerized, and crosslinked with pro-caspase-1 leading to the formation of a macromolecular multiprotein structure designated ASC speck (30). ASC speck formation results in activation of caspase-1 (31), which in turn allows processing of the pro-inflammatory cytokines IL-1ß and IL-18, as well as the pore-forming protein gasdermin D (GSDMD) (32). Specific inhibition by the small molecule MCC950, an established NLRP3 inhibitor that binds to NLRP3 (33), results in impaired ATP hydrolysis with a subsequent blockade of NLRP3 inflammasome formation (34, 35).
Interestingly, neutrophils also have been described as a source of NLRP3/ASC-dependent IL-1ß production after Staphylococcus aureus infection (36). NLRP3-linked disorders, like hypoxia-induced venous thromboembolism (37), atherosclerosis (27), and tissue damage after ischemia/reperfusion (38) have an inflammatory thrombo-occlusive pathology in common that is associated with PAD4 and NETosis. So far, NLRP3 assembly and ASC speck formation have been described in neutrophils only after pathogen-induced infections (19, 20) or in the presence of LPS (22). Nothing is known about NLRP3 inflammasome assembly in neutrophils in sterile inflammation or their potential role in NETosis.
Here, we demonstrate that the formation of the NLRP3 inflammasome supports NETosis in the absence of LPS both in vitro and in vivo, and that PAD4, in addition to its known role in chromatin decondensation, also regulates NLRP3 inflammasome assembly in neutrophils. Our studies provide an important link between NETosis and the NLRP3 inflammasome, explaining, at least in part, the overlapping features of disorders in which both components are involved.
A detailed list of used material and corresponding ordering informations can be found in the Supplementary Informations.
Nlrp3–/– (stock no. #021302) and corresponding wild-type (C57BL/6J; stock no. #000664) mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA). Padi4–/– mice were originally generated by Y. Wang (13) and back-crossed with C57BL/6J in the Wagner laboratory. All mouse lines were housed in the animal facility of Boston Children’s Hospital.
Padi4fl/fl mice (stock no. #026708), previously described by Hemmers and colleagues (39), and Vav1-iCre mice (stock no #008610) were purchased from Jackson Laboratory and intercrossed by the Wagner laboratory to generate mice lacking PAD4, specifically in the hematopoietic lineage (Padi4Vav1Cre/+). ASC-deficient mice (C57BL/6J background) used for antibody validation were a kind gift of A. Yazdi (Aachen University, Germany) and were previously described (40). All offsprings were housed in the according institutional animal facility, and mice of both sexes were randomly assigned for experiments. Data analysis was blinded to the identity of the sample.
All experimental animal procedures in this study were approved by the Institutional Animal Care and Use Committee of Boston Children’s Hospital under the protocol numbers 20-01-4096R or 20-02-4097R or the Regierungspräsidium Tübingen and were performed under the ARRIVE guidelines.
Flow restriction of the inferior vena cava (IVC) was performed as described elsewhere (41). Briefly, the IVC of 8-week-old male Nlrp3–/– and corresponding wild-type mice was exposed, and the renal and iliac veins were ligated. Subsequently, the IVC was partially (90%) ligated with a 7-0 polypropylene suture using a 30-gauge needle as a spacer. After removal of the spacer, the peritoneum and skin were closed by monofilament sutures, and mice were euthanized 6 or 48 hours after surgery. Formed thrombi were harvested for weight and length measurements and cryo-embedded in Tissue-Tek® O.C.T.™.
Cryo-embedded thrombi were cryo-sectioned into 10 µm sections and fixed in 4% paraformaldehyde (PFA) overnight at 4°C. After being washed once with phosphate-buffered saline (PBS), thrombi sections were permeabilized (0.1% Triton X-100, 0.1% sodium citrate) for 10 minutes at 4°C and subsequently incubated with blocking buffer (2.5% BSA, 0.5% Tween-20 in 1x PBS) at 37°C for 1 hour. Following incubation with the primary antibodies H4Cit (1:250) and Ly6G (1:500) at 4°C overnight, the sections were washed 3 times with PBS and incubated with the secondary antibodies (1:1,500) for 2 hours at room temperature (RT). After another 3 washes with PBS, the coverslips were mounted using mounting medium containing 4′,6-diamidin-2-phenylindol (DAPI) and visualized on an Olympus confocal laser scanning microscope (FluoView FV1000) using a 20x air objective with a tile and stitching mode. Images were identically acquired and processed with Fiji/ImageJ to calculate the percentage of H4Cit and Ly6G positive area.
Blood was collected from the retro-orbital plexus of anesthetized mice in 1 mL of ethylenediaminetetraacetic acid (EDTA) anticoagulated buffer supplemented with 1% endotoxin-free BSA in sterile PBS, and peripheral blood neutrophils were subsequently isolated. Bone marrow–derived neutrophils were obtained by flushing the mouse femur 3-4 times with phenol red-free RPMI 1640 medium supplemented with 10 mM HEPES. The bone marrow–cell suspension was strained using a 40 µm cell strainer, and cells were pelleted by 10 minutes of 500 x g centrifugation before finally being resuspended in PBS.
Subsequently, peripheral or bone marrow–derived neutrophils were isolated by Percoll gradient centrifugation, as described elsewhere (7). Neutrophils were then resuspended in phenol red-free RPMI 1640 medium supplemented with 10 mM HEPES, and cell purity was assessed by Wright-Giemsa stain. After the neutrophil count was determined, the required cell density was adjusted by adding HEPES supplemented RPMI 1640 medium.
The experimental procedure was approved by the Office of Clinical Investigations at Boston Children’s Hospital (protocol number IRB-P00003283). Informed consent was provided by donors. Blood was drawn from healthy donors in EDTA-coated vacutainers, and blood samples were de-identified prior to isolation. Neutrophils were isolated using gradient centrifugation, as described elsewhere (7). Cells were resuspended in phenol red free RPMI 1640 medium supplemented with HEPES, assessed for purity by Wright-Giemsa stain, and adjusted to the desired cell density.
Immortalized mouse bone marrow–derived macrophages (iBMDM) (42) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 1% penicillin/streptomycin and supplemented with L-glutamine and sodium pyruvate. Cells were split every 3 days in a 1:10 ratio by detaching them in PBS (pH 7.4) containing 2 mM EDTA.
PAD4 overexpressing construct was generated using a murine PAD4-mScarlet vector, which was produced by inserting mScarlet cDNA into the pLV-eGFP plasmid (a gift from Pantelis Tsoulfas, Addgene no: 36083) between XbaI and BamHI sites. The mouse full-length PAD4 insert was amplified and ligated between the AgeI and SalI sites rom he cDNA using the following primers: forward primer of 5’-ACCTCCATAGAAGACACCGACTCTAGAATGGCCCAAGGCGCGGTGATCCA-3’, and reverse primer of 5’-CTTGCTCACCATTGAGCCGCTACCGGTGGGCACCATGTGCCACCACTTGA-3’.
Stable PAD4-mScarlet overexpressing iBMDM cell lines were generated by a lentiviral transfection approach. To this end, HEK293T cells were co-transfected with 1 μg of pLV plasmid containing the corresponding gene, 750 ng psPAX2 packaging plasmid, and 250 ng pMD2.G envelope plasmid (both plasmids were a gift from Didier Trono, Addgene no: 12260 and 12259, respectively) on day 0 and incubated overnight. On day 1, the medium was removed, replenished with 1 mL fresh medium, and the cells were incubated for another day. On day 2, the supernatant containing the virus was filtered using a 0.45 μm filter and used directly to infect iBMDM cells by spinfection at 2,500 x g for 90 minutes at RT using 8 μg/mL polybrene. Subsequently, cells were incubated in the corresponding culture medium for 24 hours, and positively infected cells were sorted by flow cytometry using mScarlet (PAD4) wavelengths. Positive cell colonies were validated at protein and functional level.
For western blot analysis, cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitors, according to the manufacturer’s protocol, and incubated for 30 minutes on ice. After centrifugation at 20,000 x g for 15 minutes at 4°C, the protein concentration in the supernatant was measured using Bradford reagent. The protein samples were then denatured in LDS buffer and reducing agent for 5 minutes at 95°C. Lysates were separated by 4-12% Bis-Tris gradient gels, and proteins were electrotransferred on a PVDF membrane using the iBlot system. Membranes were blocked with 5% BSA in TBS-T buffer (0.05% Tween-20 in 1x TBS) for 1 hour at RT and incubated overnight at 4°C with anti ASC (1:800), anti NLRP3 (1:1,000), anti caspase-1 (1:1,000), anti PAD4 (1:200) or anti GAPDH (1:5,000) antibodies. For probing with the PAD4 antibody, the membranes were stripped for 20 minutes at RT using 0.5 M NaOH solution, blocked with 5% BSA TBS-T buffer, and incubated for 4 hours at RT using a custom-made mouse-specific PAD4 antibody (Thermo Fisher Scientific) directed against mouse PAD4 peptide DKEDPQASGMDFEDDKILD that does not cross react with mouse PAD2. After incubation with primary antibodies, membranes were washed 3 times with TBS-T buffer before incubation with HRP-conjugated secondary antibodies (1:10,000) and were incubated for 2 hours at RT. Then the membranes were washed 3 more times with TBS-T and subsequently probed with enhanced chemiluminescence (ECL) detection solution.
IL-1ß was measured according to the manufacturer’s instruction. A 96-well plate was coated with the capturing antibody overnight at 4°C. The following day, the plate was washed 3 times with buffer (PBS + 0.05% Tween-20) and then blocked at RT for 1 hour under gentle shaking. Subsequently, after 3 further washing steps, 100 µL of iBMDM supernatant or standard solution was added per well and incubated for 2 hours at RT under gentle shaking. The plate was again washed 3 times before the addition of the detection antibody. After incubation of the detection antibody for 1 hour at RT, avidin-HRP was added and incubated for 0.5 hours at RT. After final washing steps, TMB solution was supplemented, and 2N H2SO4 was used as stop solution. The absorbance was read at 450 and 570 nm. To obtain final values, the 570 nm values were subtracted from the 450 nm values.
Total RNA was extracted using the PureLink™ RNA Mini Kit (ThermoFisher) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized using All-In-One RT MasterMix following the manufacturer’s instructions. Quantitative PCR of specific genes was performed using SYBR Green SuperMix in the StepOnePlus RealTime PCR System. Cycling conditions were as follows: initial denaturation at 95°C for 2 minutes, followed by 40 cycles of 95°C for 15 sec, 55°C for 30 sec, and 70°C for 15 sec. For amplification, the following primer pairs were used (5`- >3`orientation): β-actin: fwd: CGGTTCCGATGCCCTGAGGCTCTT; rev: CGTCACACTTCATGATGGAATTGA for isolated peripheral neutrophils and fwd: CATTGCTGACAGGATGCAGAAGG; rev: TGCTGGAAGGTGGACAGTGAGG for iBMDM cells; ASC: fwd: CAGAGTACAGCCAGAACAGGACAC, rev: GTGGTCTCTGCACGAACTGCCTG; NLRP3: fwd: GTTCTGAGCTCCAACCATTCT, rev: CACTGTGGGTCCTTCATCTTT; IL1ß: fwd: TGGACCTTCCAGGATGAGGACA; rev: GTTCATCTCGGAGCCTGTAGTG.
To confirm the equal RNA input, β-actin mRNA expression and the relative expression of inflammasome mRNA were calculated with the △△Ct method. Specificity of the amplification was checked by melting curve analysis, and data were recorded and analyzed using StepOne Software v2.1.
1.5x104 mouse or human neutrophils per well were resuspended in HEPES supplemented phenol red-free RPMI 1640 medium and plated in a 96-well plate. After allowing the cells to adhere for 30 minutes at 37°C and 5% CO2 in the absence or presence of 1 µM MCC950, the cells were stimulated with vehicle control, nigericin (15 µM), or ionomycin (4 µM) for 4 hours. Fixation was performed in 2% PFA containing Hoechst 33342 (1:10,000) at 4°C overnight. Cells were washed 3 times with PBS the next day before imaging on a Zeiss Axiovert 200M microscope. The percentage of NETs was analyzed from 8 non-overlapping and randomized visual fields per well by quantifying cells with a web-like chromatin structure and positive citrullinated histone H4 staining. The average percentage of NETing cells was taken from duplicates in each experiment.
6x104 mouse or 1x105 human neutrophils per condition were plated on a sterilized coverslip in a 6-well plate and allowed to adhere for 30 min at 37°C and 5% CO2 before stimulating the cells with nigericin (15 µM), ionomycin (4 µM), or PMA (50 nM) for 4 hours. The cells were fixed with 4% PFA for 1 hour at RT, washed once with PBS, permeabilized (0.1% Triton X-100, 0.1% sodium citrate) for 10 minutes at 4°C, and incubated with blocking buffer (2.5% BSA, 0.5% Tween-20 in 1x PBS) at 37°C for 1 hour. Samples were incubated at 4°C ON with the primary antibodies against ASC (1:800, mouse neutrophils or 1:200, human neutrophils) and subsequently washed 3 times with PBS before incubation with the secondary antibody (1:1,500) for 2 hours at RT. After another 3 washing steps with PBS, the coverslips were mounted using mounting medium containing DAPI and visualized on a confocal Nikon Eclipse Ti2 microscope using a 60x oil immersion objective (mouse neutrophils) or an Olympus confocal laser scanning microscope FluoView (FV1000) using a 40x air objective (human neutrophils). Images were identically acquired and processed with Fiji/ImageJ. ASC speck frequency was determined by capturing 37 Z-stacks of 0.1625 µm size from 6 by 6 tiles on a Nikon Eclipse Ti2 A1R confocal microscope (mouse neutrophils) and by 5 non-overlapping and randomized visuals fields on an Olympus FluoView (FV1000) confocal microscope (human neutrophils) in the center of the coverslip. The percentage of neutrophils forming ASC speck was quantified.
iBMDMs were plated on a 35 mm glass-bottom dish and primed for 4 hours with LPS from E.coli (1 µg/mL) at 37°C and 5% CO2 before stimulating the cells with nigericin (20 µM) for 30 minutes. iBMDMs were fixed in 3% PFA containing 1:10,000 Hoechst 33342 for 30 minutes at RT, washed twice with PBS for 5 minutes, and permeabilized (0.1% Triton X-100, 0.1% sodium citrate) for 7 minutes at RT, followed by a washing step. Afterwards, cells were blocked for 1 hour at RT with blocking buffer (3% BSA in PBS) and subsequently incubated with the primary antibody ASC (1:1,000) overnight. Followed by extensive washing steps, cells were incubated with the corresponding secondary antibody (1:2,000) the next day for 2 hours at RT. Finally, cells were visualized with an Olympus confocal laser scanning microscope FluoView (FV1000). Images were captured using a 60x water immersion objective with Olympus FluoView version 3.0 viewer software. The images were identically acquired and processed using ImageJ software, and the percentage of iBMDMs developing an ASC speck was quantified.
Time-lapse microscopy was performed using isolated peripheral neutrophils from Nlrp3+/+ and Nlrp3–/– mice. To this end, 1x106 mouse neutrophils were stained for 30 min at 37°C and 5% CO2 using 2 µM SiR-DNA to visualize chromatin and 1 µM ER-tracker red dye to visualize the endoplasmic reticulum and nuclear envelope. Subsequently, cells were washed and resuspended in 300 µL of imaging media (phenol red-free RPMI 1640, 25 mM HEPES, 1% penicillin/streptomycin) before the cell suspension was added and allowed to adhere for 5 min in a non-coated, 24-well glass-bottom plate located on a 37°C pre-warmed microscope stage. 3-10 random fields per well were visualized using a Nikon Eclipse Ti2 microscope equipped with Perfect Focus™, a Yokogawa CSU-X1 spinning disc scanhead, a Nikon motorized stage with XY linear encoders containing a Nano-Z100 piezo insert, and a Hamamatsu Orca-flash 4.0 v3 camera with a Plan Apo Tirf 60x oil 1.49 NA DIC Nikon objective lens. Confocal and DIC images were acquired every 2 minutes for the first 80 minutes and every 5 minutes for the rest of the visualization up to 4 hours. Three images were acquired of unstimulated cells, followed by addition of imaging medium containing ionomycin to achieve a final ionomycin concentration of 4 µM.
All data are presented as mean ± standard error of the mean (SEM). Statistical analysis was performed using GraphPad Prism. Significance was tested with unpaired t-test or, for experiments with more than two groups, with two-way ANOVA multiple comparison test. p<0.05 was considered statistically significant.
NLRP3 is expressed in human and mouse neutrophils (22, 23). To investigate whether NLRP3 inflammasome assembles in neutrophils under sterile conditions, human peripheral neutrophils were activated with PMA or nigericin for 4 hours. Both agents induced inflammasome assembly, observed in a subset of cells by immunostaining for ASC speck formation, which was absent in unstimulated neutrophils (Figure 1A). Like human neutrophils, isolated peripheral mouse neutrophils displayed ASC speck formation when stimulated with ionomycin or nigericin (Figure 1B) and stained with an antibody specific for ASC (Figure S1) subsequently. Interestingly, ASC speck formation occurred in NET-forming human or mouse neutrophils upon stimulation with nigericin or ionomycin, respectively (Figure 1C). After ionomycin- or nigericin- stimulated NET release, a fraction of neutrophils undergoing NETosis also showed ASC speck or its fragments associated with the expelled extracellular chromatin (Figure 1C; red arrowheads).
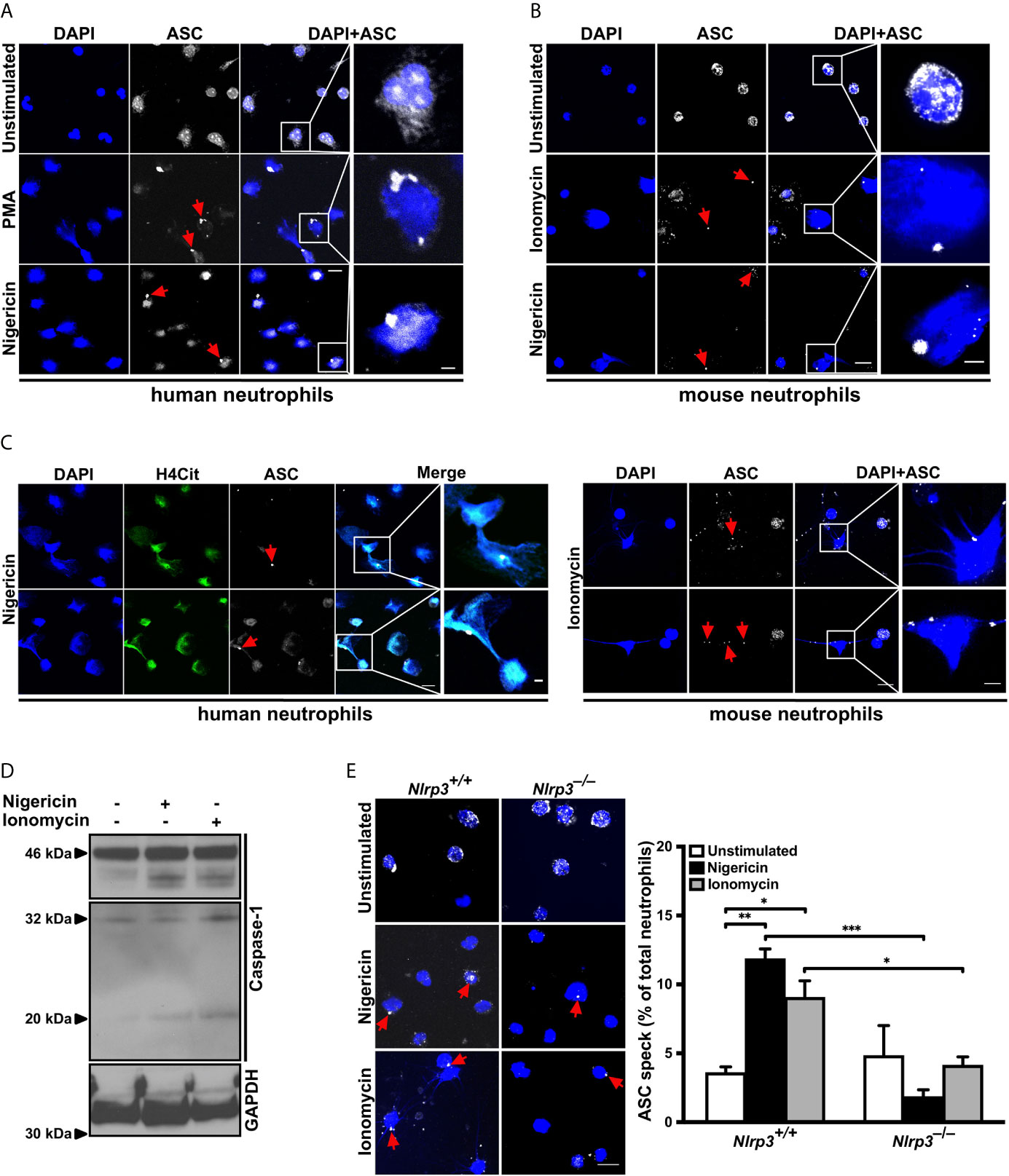
Figure 1 NLRP3 inflammasome-dependent ASC speck formation and release on NETs by stimulated neutrophils (A) Confocal microscopy images of immunostained human neutrophils in the absence (unstimulated) or presence of PMA (50 nM) or nigericin (15 µM) for 4 hours. Blue, DNA (DAPI); grey, ASC antibody staining. Red arrows indicate ASC speck. Scale bar equals 10 µm in the overview and 2.5 µm in the zoom panel. Representative of n=4 experiments. (B) Confocal microscopy images of immunostained mouse neutrophils in the absence (unstimulated) or presence of ionomycin (4 µM) or nigericin (15 µM) for 4 hours. Blue, DNA (DAPI); grey, ASC antibody staining. Red arrows indicate ASC speck. Scale bar equals 10 µm in the overview and 2.5 µm in the zoom panel. Representative of n=5 experiments. (C) Confocal microscopy images of ASC speck formation in NETting cells and associated with NETs in human (left panel) or mouse neutrophils (right panel) in the presence of nigericin (15 µM) or ionomycin (4 µM). Blue, DNA (DAPI); green, H4Cit antibody stain; grey, ASC antibody staining. Red arrows indicate ASC speck. Scale bar equals 10 µm in the overview and 2.5 µm in the zoom panel. Representative of n=4-5 experiments. (D) Western blot of caspase-1 cleavage (antibody clone: Casper-1) in neutrophils from wild-type mice in the absence or presence of nigericin (15 µM) or ionomycin (4 µM) for 0.5 hours. Representative of n=3 experiments. (E) Representative confocal microscopy images (left panel) and arithmetic means ± SEM (right panel; n=4-5 mice) of ASC speck formation in neutrophils from wild-type (Nlrp3+/+) or Nlrp3–/– mice in the absence (unstimulated, open bars) or presence of nigericin (15 µM, black bars) or ionomycin (4 µM, grey bars) for 4 hours. Blue, DNA (DAPI); grey, ASC antibody staining. Red arrows indicate ASC speck. Scale bar equals 10 µm. *p<0.05, **p<0.01 and ***p<0.001.
Next, since activation of pro-caspase-1 upon ASC speck formation is an established hallmark of inflammasome activation, caspase-1 cleavage products were investigated in mouse neutrophils. Stimulation with nigericin or ionomycin caused generation of the characteristic p32 and p20 caspase-1 fragments (Figure 1D).
To confirm that the observed ASC speck formation in neutrophils is due to NLRP3 assembly, we treated circulating neutrophils from wild-type (Nlrp3+/+) and NLRP3-deficient (Nlrp3–/–) mice with nigericin or ionomycin. As shown in Figure 1E, nigericin or ionomycin stimulation induced ASC speck formation in approximately 10% of wild-type neutrophils. By contrast, this activation-dependent increase in ASC speck formation was significantly reduced in neutrophils from Nlrp3–/– mice, indicating that the majority of observed ASC speck in the mouse neutrophils was part of the NLRP3 inflammasome (Figure 1E).
Taken together, these results demonstrate that neutrophils can assemble a physiologically active inflammasome/ASC speck in the absence of bacteria or LPS.
ASC speck forms in stimulated neutrophils, where PAD4 is a prerequisite for NET formation (15). We wondered whether PAD4 may promote ASC speck assembly as well. Since PAD4 can be synthesized by many cell types and can be found in plasma, a hematopoietic cell-specific Padi4 knockout mouse was generated using Cre-Lox recombination by intercrossing Padi4fl/fl with Vav1-iCre mice (Figure S2A). The resulting PAD4 wild-type (Padi4fl/fl) and hematopoietic cell-specific knockout (Padi4Vav1Cre/+) mice showed no differences in blood cell counts (Figure S2B) and, in agreement with global Padi4–/– mice, Padi4Vav1Cre/+ mice displayed significantly reduced NETosis (Figure S2C).
Immunostaining of unstimulated Padi4fl/fl or Padi4Vav1Cre/+ neutrophils detected ASC speck formation in approximately 2% of the cells. However, nigericin or ionomycin stimulation resulted in ASC speck formation in about 15% of Padi4fl/fl cells, while only 6-8% of neutrophils from Padi4Vav1Cre/+ mice showed ASC speck formation (Figure 2A), which shows that neutrophil PAD4 is needed to fully stimulate ASC speck assembly. Since no LPS pretreatment was needed for induction of ASC speck formation in neutrophils, we decided to compare the protein levels of NLRP3 and ASC in these cells. Interestingly, peripheral neutrophils lacking PAD4 (Padi4Vav1Cre/+) displayed decreased ASC and NLRP3 protein levels when compared with PAD4-positive neutrophils from Padi4fl/fl mice (Figure 2B), while LPS pretreatment equalized the ASC and NLRP3 protein levels in these neutrophils (Figure S2D). Although protein levels of the NLRP3 inflammasome are transcriptionally regulated by NFκB in macrophages (43), neutrophils from Padi4fl/fl and Padi4Vav1Cre/+ mice showed no difference in NLRP3 and ASC mRNA levels (Figure 2C).
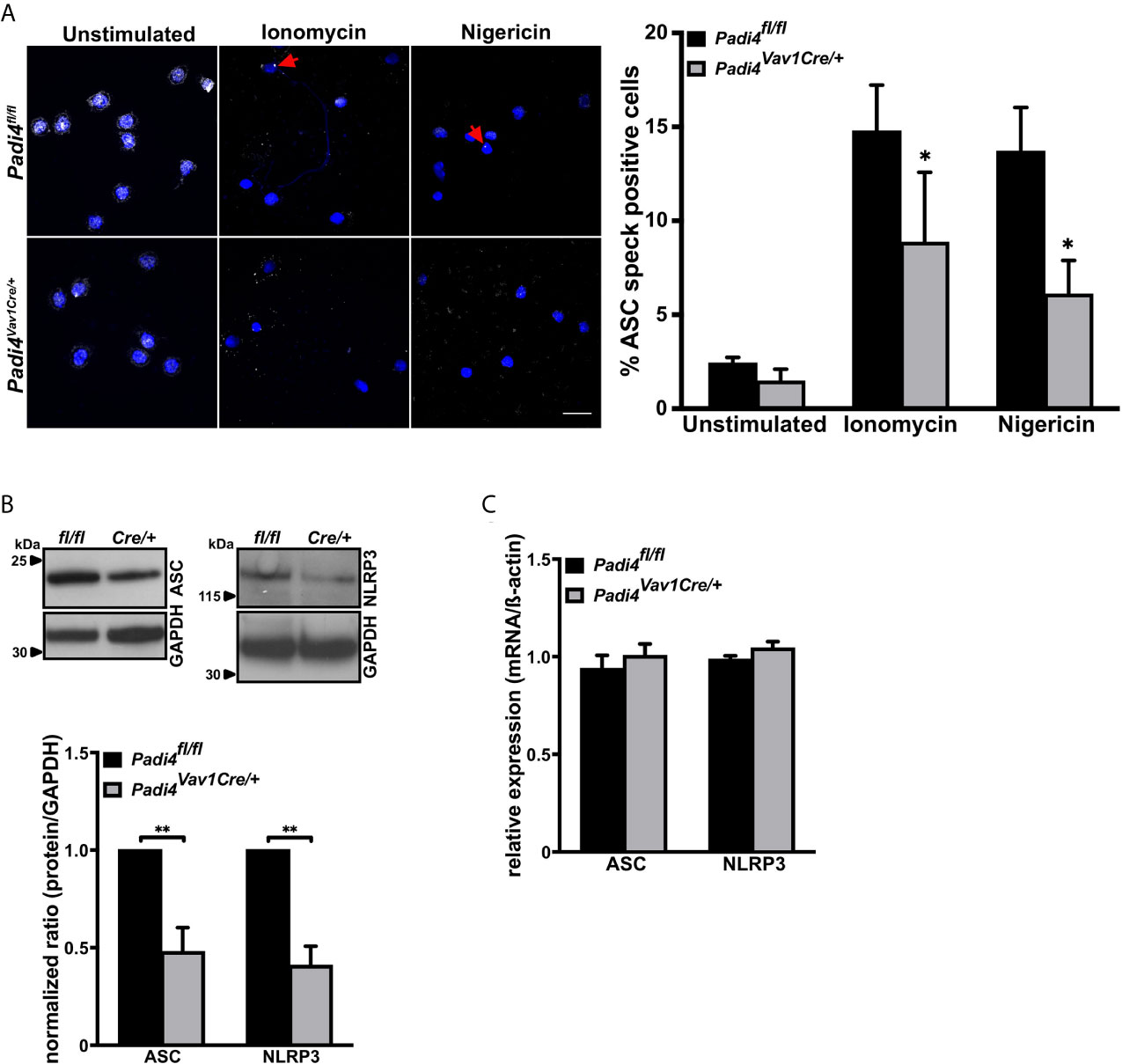
Figure 2 ASC speck formation in neutrophils is, in part, directed by PAD4-dependent regulation of ASC and NLRP3 protein levels (A) Representative confocal microscopy images (left panel) and arithmetic means ± SEM (right panel, n=4-6 mice) of ASC speck formation in neutrophils from wild-type (Padi4fl/fl, black bars) or hematopoietic cell-specific Padi4-deficient mice (Padi4Vav1Cre/+, grey bars) in the absence (unstimulated) or presence of ionomycin (4 µM) or nigericin (15 µM) for 4 hours. Blue, DNA (DAPI); grey, ASC antibody staining. Red arrows indicate ASC speck. Scale bar equals 10 µm. *p<0.05. (B) Representative western blots (upper panel) and arithmetic means ± SEM (lower panel, n=4 mice) of ASC and NLRP3 protein levels in naive neutrophils from wild-type (Padi4fl/fl, black bars) or hematopoietic specific Padi4-deficient mice (Padi4Vav1Cre/+, grey bars). **p<0.01. (C) Arithmetic means ± SEM (n=3 mice) of relative mRNA levels in naive neutrophils from wild-type (Padi4fl/fl, black bars) or hematopoietic specific Padi4-deficient mice (Padi4Vav1Cre/+, grey bars).
These results indicate that PAD4 has the ability to upregulate NLRP3 inflammasome components in a post-transcriptional manner without de novo mRNA synthesis in neutrophils.
To study whether PAD4 also plays a role in ASC speck formation in other cell types, we prepared primary bone marrow–derived macrophages (BMDM) from wild-type and Padi4Vav1Cre/+ mice. While inhibition of citrullinating activity of several PAD enzymes by Cl-amidine almost completely abrogated ASC speck formation in primary wild-type BMDMs (Figures S3A, B), there was no statistically significant difference in ASC speck formation in primary bone marrow–derived macrophages from Padi4Vav1Cre/+ mice when compared with Padi4fl/fl BMDMs after LPS and nigericin exposure (Figure S3C). This observation confirms a previous report (44) and indicates that in mouse macrophages, as in neutrophils, citrullination is necessary for ASC speck formation. However, other PAD enzymes may compensate for the lack of PAD4 in macrophages.
To further investigate the role of PAD4 in NLRP3 inflammasome assembly in macrophages, we generated PAD4 overexpressing immortalized bone marrow–derived macrophages (iBMDM). While these cells exhibited a 4-fold increased PAD4 protein level compared with wild-type iBMDM (Figures S4A, B), PAD4 overexpression had no effect on IL-1ß mRNA levels in iBMDM (Figure S4C). Although in vitro stimulation of NLRP3 assembly in iBMDMs requires pretreatment with LPS and subsequent stimulation with nigericin, we observed that nigericin alone was able to induce ample ASC speck formation and IL-1ß production in PAD4 overexpressing iBMDMs (Figures 3A, B). Stimulation of control iBMDMs (empty vector-treated cells) with nigericin or LPS alone did not induce ASC speck formation or IL-1ß production (Figures 3A, B). In agreement with the observation in neutrophils, PAD4 overexpression in iBMDMs resulted in significantly increased NLRP3 protein levels and a small but significant increase in ASC levels when compared with empty vector expressing cells, without a significant change in the corresponding mRNA levels (Figures 3C, D).
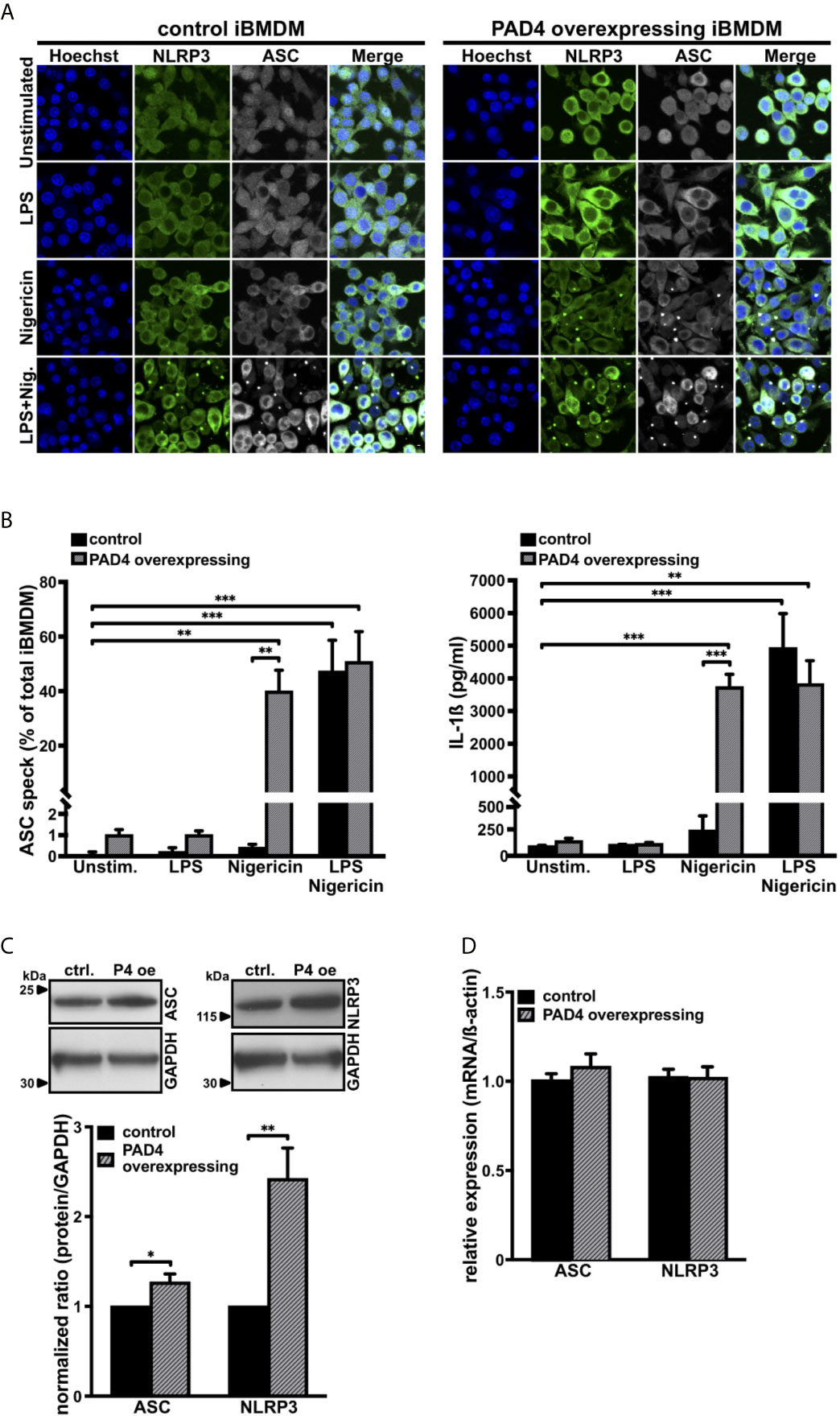
Figure 3 PAD4 overexpression leads to priming-independent ASC speck formation and IL-1ß secretion with an increase in ASC and NLRP3 protein levels in immortalized bone marrow–derived murine macrophages (A) Confocal microscopy images of immunostained native mouse immortalized bone marrow–derived macrophages (iBMDM; control, left panel) and PAD4 overexpressing iBMDM (right panel) in the absence (unstimulated) or presence of LPS only (1 µg/mL), nigericin only (15 µM), or LPS and nigericin (15 µM, 0.5 hours). Blue, DNA (Hoechst); green, NLRP3 antibody staining; grey, ASC antibody staining. Scale bar equals 5 µm. Representative of n=4 experiments. (B) Arithmetic means ± SEM (n=4) of percentage of ASC speck formation (left panel) and IL-1ß production (right panel) in native iBMDM (control, black bars) or PAD4 overexpressing iBMDM (grey shaded bars) in the absence (unstim.) or presence of LPS only (1 µg/mL), nigericin only (15 µM), or LPS (1 µg/mL) and nigericin (15 µM, 0.5 hours). **p<0.01 and ***p<0.001. (C) Representative western blots (upper panel) and arithmetic means ± SEM (lower panel, n=4) of ASC and NLRP3 protein levels in native iBMDM (ctrl.) or PAD4 overexpressing (P4 oe) iBMDM. *p<0.05 and **p<0.01. (D) Arithmetic means ± SEM (n=4) of relative mRNA levels in native iBMDM (control) or PAD4 overexpressing iBMDM.
These results show the importance of PAD4 activity in inflammasome/ASC speck assembly in iBMDM cells under sterile conditions.
Since ASC speck formation was observed in neutrophils forming NETs, we addressed the importance of the NLRP3 inflammasome assembly on NETosis. To this end, we examined the effects of inflammasome perturbations on NETosis in in vitro assays. Neutrophils isolated from Nlrp3–/– mice showed significantly decreased NETosis after nigericin or ionomycin stimulation when compared with neutrophils from wild-type mice (Figure 4A). The role of NLRP3 was further substantiated both in mouse and human neutrophils by pharmacological inhibition of the NLRP3 inflammasome. Pretreatment for 30 minutes with the specific small molecule NLRP3 inhibitor MCC950 (33) resulted in significantly decreased NETosis after nigericin or ionomycin stimulation than observed in vehicle-treated neutrophils (Figures 4B, C). Thereby, the inhibition was more pronounced in mouse than human neutrophils, but was statistically significant in both cases. In line with results from NLRP3 perturbations, we found that pharmacological inhibition of the NLRP3 inflammasome effector molecule caspase-1 also revealed markedly reduced NETosis in human neutrophils (Figure S5), indicating that NLRP3-mediated caspase-1 activation is needed for efficient NETosis.
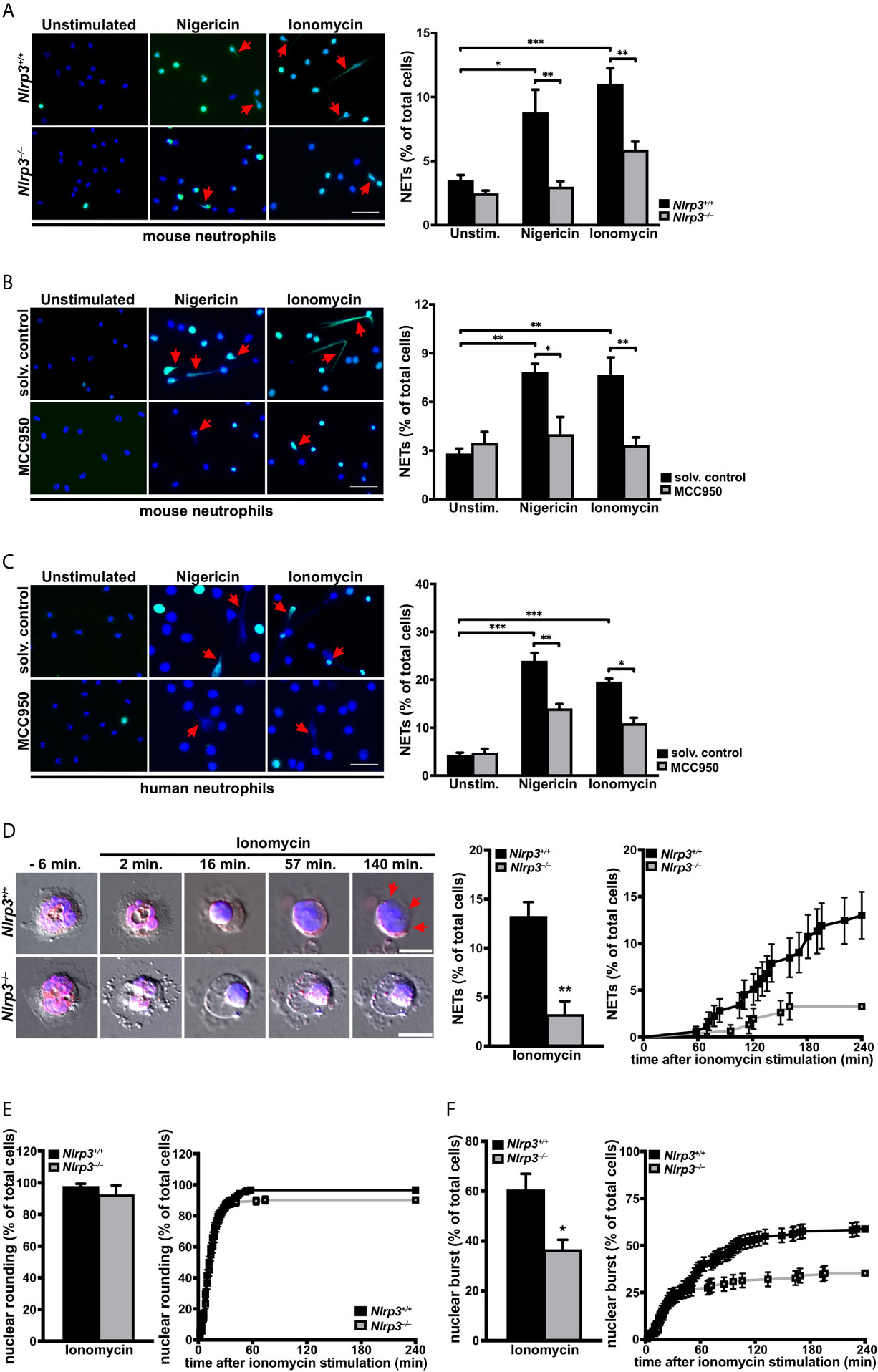
Figure 4 The NLRP3 inflammasome promotes activation-dependent NET formation through nuclear envelope and plasma membrane breakdown in primary neutrophils. (A) Overview (left panel) and arithmetic means ± SEM (right, n=5-7 mice) of NET formation by neutrophils from wild-type (Nlrp3+/+, black bars) or Nlrp3–/– mice (grey bars) in the absence (unstimulated) or presence of nigericin (15 µM) or ionomycin (4 µM) for 4 hours. Blue, DNA (DAPI); green, H4Cit antibody stain. Red arrows indicate NETs. Scale bar equals 50 µm. *p<0.05, **p<0.01 and ***p<0.001. (B) Overview (left panel) and arithmetic means ± SEM (right, n=5-6 mice) of NET formation in untreated (solvent control, black bars) or MCC950 pretreated (1 µM, grey bars) mouse neutrophils in the absence (unstimulated) or presence of nigericin (15 µM) or ionomycin (4 µM) for 4 hours. Blue, DNA (DAPI); green, H4Cit antibody stain. Red arrows indicate NETs. Scale bar equals 50 µm. *p<0.05 and **p<0.01. (C) Overview (left panel) and arithmetic means ± SEM (right, n=4-8 donors) of NET formation in untreated (solvent control, black bars) or MCC950 pretreated (1µM, grey bars) human neutrophils in the absence (unstimulated) or presence of nigericin (15 µM) or ionomycin (4 µM) for 4 hours. Blue, DNA (DAPI); green, H4Cit antibody stain. Red arrows indicate NETs. Scale bar equals 50 µm. *p<0.05, **p<0.01 and ***p<0.001. (D) Representative time-lapse differential interference contrast (DIC) spinning-disk confocal microscopy images at indicated time intervals (left panel) and arithmetic means ± SEM (center/right panel; n=5 mice) of percentage of total (middle) and time course (right) of plasma membrane rupture (NETosis) in neutrophils from wild-type (Nlrp3+/+, black bars) or Nlrp3–/– mice (grey bars) in the presence of ionomycin (4 µM). Blue, DNA (siR-DNA); red, nuclear envelope (ER-tracker). Red arrows indicate area of plasma membrane rupture. Scale bar equals 5 µm. **p<0.01. (E) Arithmetic means ± SEM (n=5 mice) of percentage of total (left panel) and time course (right panel) of nuclear rounding in neutrophils from wild-type (Nlrp3+/+, black bars) or Nlrp3–/– mice (grey bars) in the presence of ionomycin (4 µM). (F) Arithmetic means ± SEM (n=5 mice) of percentage of total (left panel) and time course (right panel) of nuclear envelope rupture in neutrophils from wild-type (Nlrp3+/+, black bars) or Nlrp3–/– mice (grey bars) in the presence of ionomycin (4 µM). *p<0.05.
To determine the underlying NLRP3-dependent cellular processes in NETosis, we performed time-lapse microscopy of ionomycin-stimulated neutrophils from Nlrp3+/+ and Nlrp3–/– mice. Spinning disk confocal and DIC microscopy of cells stained with SiR-DNA as a marker of chromatin and ER-tracker as a marker of the endoplasmic reticulum (ER) and nuclear envelope confirmed a significantly impaired NET formation in neutrophils from Nlrp3–/– mice when compared with wild-type neutrophils. While neutrophils from Nlrp3+/+ mice showed a robust breakage of the plasma membrane (NETosis) starting 60 minutes after stimulation, Nlrp3-deficient neutrophils displayed four-fold reduced plasma membrane rupture (Figure 4D). While nuclear rounding, another described cellular characteristic of NETosis (45), occurred both in neutrophils from Nlrp3+/+ and Nlrp3–/– mice, most Nlrp3–/– neutrophils arrested the NETosis process at this stage (Figure 4E and Videos S1, S2). Accordingly, the rupture of the nuclear envelope was significantly impaired in neutrophils from Nlrp3-/- mice when compared with wild-type controls (Figure 4F).
The above observations demonstrate an important role of the NLRP3 inflammasome in nuclear envelope and plasma membrane breakdown after sterile inflammation, thus pointing to a central role of the NLRP3 inflammasome in NETosis.
We next sought to verify that NLRP3 also regulates NETosis in deep vein thrombosis (DVT) to unravel the physiological importance of our findings. To this end, we applied a mouse model of stenosis-induced DVT, which is an acknowledged murine model for sterile thrombo-inflammation, in wild-type (Nlrp3+/+) and Nlrp3–/– mice. In our previous work with PAD4-deficient mice, we noted that NETs likely stabilize the thrombus, since lack of NETosis displayed a more important phenotype at later timepoints of venous thrombus progression (14). Therefore, we induced vascular stenosis for 6 or 48 hours to monitor thrombus progression. While the incidence of formed thrombi in Nlrp3+/+ and Nlrp3–/– mice was similar at both time points (Figure 5A), there was a significant NLRP3-dependent difference in thrombus progression. Interestingly, thrombus length and weight did not differ between Nlrp3+/+ and Nlrp3–/– mice 6 hours after stenosis. By contrast, a significant reduction in thrombus size was observed in Nlrp3–/– mice after 48 hours when compared with Nlrp3+/+ mice. Furthermore, although the thrombi from Nlrp3+/+ mice increased in weight and length over the 48-hour period, thrombi from NLRP3-deficient mice reached their final small size already after only 6 hours, resulting in smaller thrombi in NLRP3–/– mice when compared with thrombi from wild-type mice (Figure 5B). This observation points to a role of NLRP3 in venous thrombosis progression under sterile conditions.
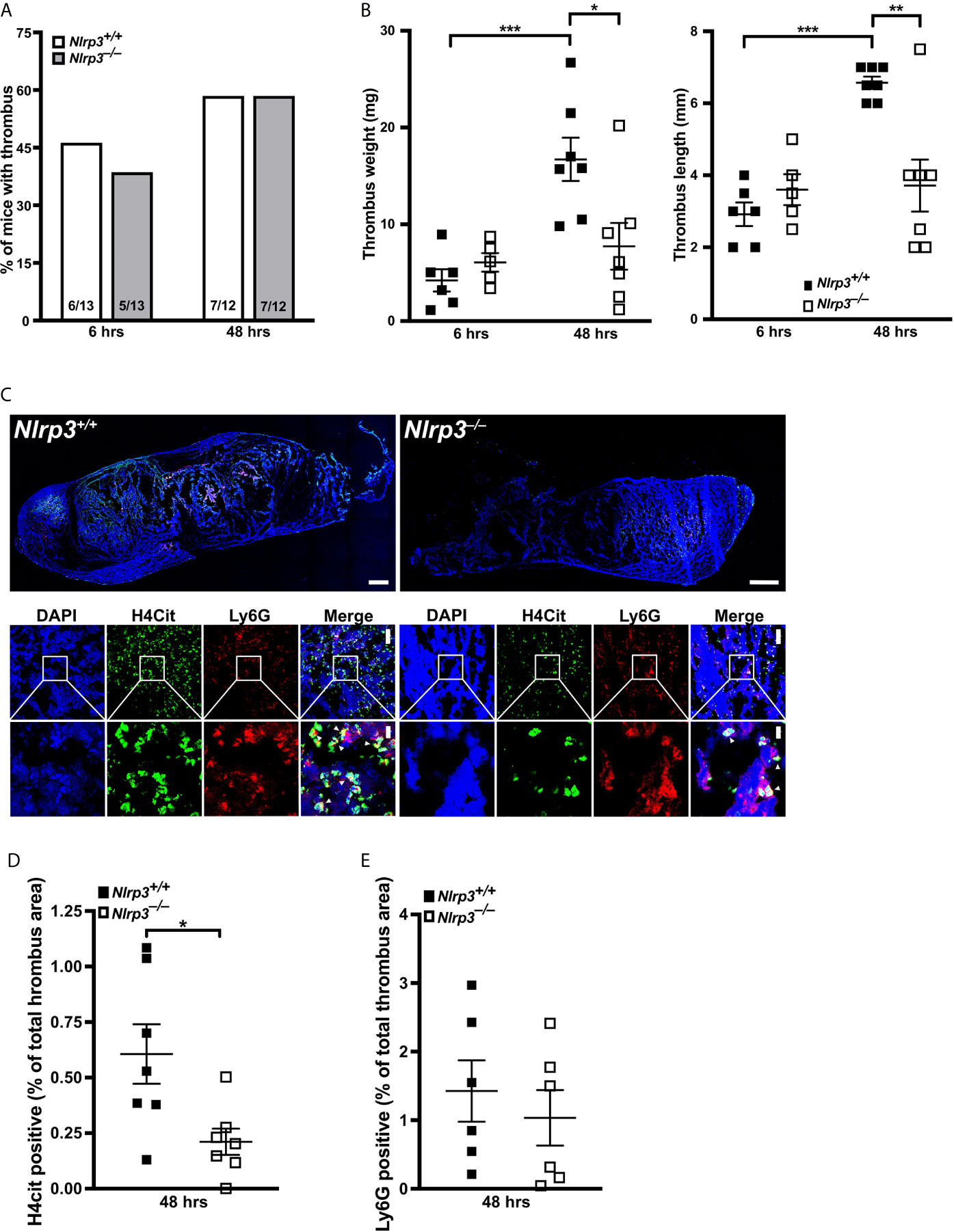
Figure 5 NLRP3 deficiency decreases NETosis and thrombus growth in a stenosis-induced model of deep vein thrombosis. (A) Thrombus incidence in wild-type (Nlrp3+/+, open bars) or Nlrp3–/– mice (grey bars) after 6 or 48 hours of stenosis of the inferior vena cava (IVC). (B) Thrombus weight (left panel) and thrombus length (right panel) of thrombi from wild-type (Nlrp3+/+, black) or Nlrp3–/– mice (white) after 6 or 48 hours of stenosis of the IVC. Each dot represents a thrombus. *p<0.05, **p<0.01 and ***p<0.001. (C) Representative composite images of thrombi by confocal microscopy (upper panels) and zoom images (lower panels) of thrombi from wild-type (Nlrp3+/+) or Nlrp3–/– mice after 48 hours of stenosis of the IVC. Blue, DNA (DAPI); green, H4Cit antibody stain; red, Ly6G antibody staining. Overview images were composed of several photographs. Scale bar equals 300 µm (thrombus overview), 50 µm (upper panel) or 10 µm (lower panel) in the zoom panels. (D) Percentage of thrombus area covered by H4Cit in thrombi from wild-type (Nlrp3+/+, black) or Nlrp3–/– mice (white) after 48 hours of stenosis of the IVC. Each dot represents a thrombus. *p<0.05. (E) Percentage of thrombus area covered by Ly6G positive cells in thrombi from wild-type (Nlrp3+/+, black) or Nlrp3–/– mice (white) after 48 hours of stenosis of the IVC. Each dot represents a thrombus.
Since thrombus growth in the DVT model is substantially dependent on NETosis, we finally investigated the density of citrullinated histone H4 (H4Cit) in thrombi from Nlrp3+/+ and Nlrp3–/– mice after 48 hours of stenosis as a direct marker for the presence of NETs. As shown in Figures 5C, D, NET density, and thus their formation, was significantly reduced in thrombi from Nlrp3–/– mice when compared with wild-type controls (Figures 5C, D). Neutrophil density in the corresponding thrombi were comparable between Nlrp3+/+ and Nlrp3–/– mice (Figure 5E), emphasizing an NLRP3-dependent enhancement of NETosis with subsequent increase in venous thrombus growth in vivo.
Our results demonstrate that, at least in the mouse, NLRP3 inflammasome is critical for NETosis in vivo.
NETs and NLRP3 inflammasome are formed in a similar set of human disorders and infectious diseases. Among these pathologies, noninfectious diseases such as cancer, immunothrombosis, myocardial infarction, and stroke are the main causes of death (46). In particular, it is known that neutrophils and NETosis are major inducers of venous thrombosis (47) and hypoxia-induced venous thrombosis is linked to elevated IL-1ß and NLRP3 levels in thrombi (37, 48). However, it was not known if canonical inflammasome assembly takes place in neutrophils under these noninfectious conditions, and if inflammasome assembly contributes to NETosis.
In 2018, two groups of investigators clearly demonstrated that GSDMD pore formation is implicated in NETosis in an NLRP3 inflammasome–independent manner. They proposed that GSDMD is activated either by neutrophil proteases that play a role in NETosis and can cleave GSDMD to its active fragments (49) or by caspase-11-mediated GSDMD cleavage after cytosolic infection by gram-negative bacteria (50). However, as our experiments were exclusively performed under sterile conditions, the observed effects are most likely mediated by caspase-1-dependent mechanisms. In macrophages the cleaved N-terminal GSDMD fragment is established as pore-forming compound (51), while in neutrophils, the elastase (NE)-dependent activation of GSDMD can lead to alternative GSDMD cleavage and localization resulting in pyroptosis-independent signaling (52). Nevertheless, caspase-11 as well as NE-dependent GSDMD processing affect nuclear extension (49) and nuclear permeabilization (50) in neutrophils. In this context, the importance of caspase-1 or 11 in NETosis is consistent with our collaborative observations showing that caspase-1/11-deficient mice do not expel NETs (53) and that pharmacological perturbation of caspase-1 activity using a specific caspase-1 inhibitor resulted in impaired NET formation (Figure S5). Interestingly, all types of sterile stimulation that we tested produced significant ASC speck/inflammasome assembly (Figure 1). However, further research is needed to clarify the exact roles of caspase-1 and GSDMD in neutrophil activation. Since occasional speck formation was observed in resting neutrophils and not in naive mononuclear cells, it appears that neutrophils are already primed for inflammasome formation (Figure 6).
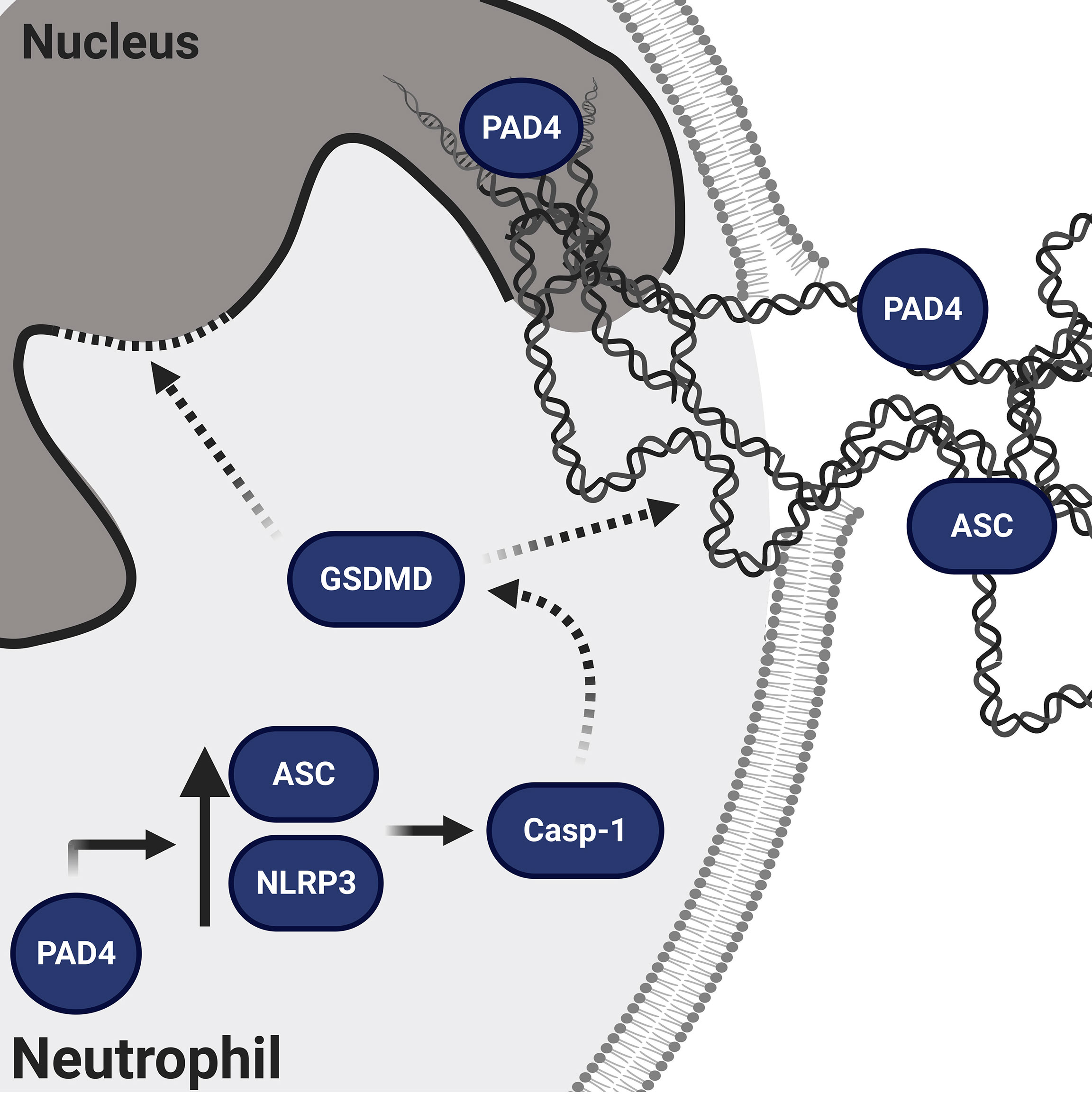
Figure 6 Schematic representation of the proposed mechanism of NLRP3/PAD4-induced NETosis in the absence of infection. PAD4 is present both in the nucleus and cytoplasm (15). In the nucleus, PAD4 orchestrates chromatin decondensation, whereas in the cytoplasm, PAD4 increases NLRP3 and ASC protein levels post-transcriptionally, thus favoring NLRP3 inflammasome/ASC speck assembly. The NLRP3 inflammasome activates caspase-1, which is known to generate the N-terminal fragment of GSDMD pore that facilitates nuclear expansion (49) and nuclear permeabilization (50). In addition to GSDMD, caspase-1 has many other intracellular substrates (54) and therefore its activation could support the cytoskeletal and nuclear disassembly necessary for NETosis (12).
Apart from NLRP3, other inflammasomes present in neutrophils, such as the NLRC4, NLRP1 and AIM2 inflammasomes, are also known to induce ASC speck-dependent caspase-1 activation (19, 26, 55). NLRP3 and NLRP1 expression levels in neutrophils are higher than in macrophages (56) and especially NLRP1 was recently described as mediator of anthrax lethal toxin induced neutrophil activation (55). Additionally, the AIM2 inflammasome is vastly activated by cytosolic dsDNA, which is a characteristic of NETosis. These observations by others may explain why residual ASC speck formation is still observed in Nlrp3–/– (Figure 1E) or MCC950-treated neutrophils (Figures 4B, C). Further investigations are needed to determine the role of inflammasomes other than NLRP3 in NETosis.
PAD4-dependent citrullination events are important in NETosis (12, 15). PAD4 deficiency protects mice from thrombosis, ischemia reperfusion injury, and age-related tissue fibrosis (5, 15). The observation that NLRP3 is linked to similar conditions and functional decline in aging (57, 58) suggested to us that PAD4 could also be regulating the NLRP3 inflammasome. Indeed, we found an impaired ASC speck formation in neutrophils from PAD4-deficient mice when compared with wild-type neutrophils (Figure 2A). Recently, a study was published showing that PAD enzymes were also necessary for NLRP3/ASC speck formation in macrophages. While Cl-amidine treatment fully inhibited ASC speck formation, PAD4 deficiency alone, however, had no effect on ASC speck formation (44) (Figure S3), showing that PAD4 is more important in neutrophil inflammasome assembly than in macrophages. These observations point to a compensatory effect of other PAD enzymes in macrophages, such as PAD2, which is the main PAD isoform in these leukocytes (59). Since post-translational modifications such as phosphorylation, ubiquitination, or the change in the charge of a single amino acid are known to modify the oligomerization or stability abilities of the NLRP3 inflammasome (60, 61), citrullination of arginine residues by PAD4 could regulate NLRP3 inflammasome oligomerization. Particularly, treatment with the calcium ionophore ionomycin increases intracellular calcium levels and thus likely induce the enzymatic activity of PAD4 with subsequent citrullination of inflammasome components. The same mechanisms is assumed for the potassium ionophore nigericin, by indirectly increasing cytosolic calcium concentrations with subsequent inflammasome activation (62). Another way by which PAD4 could promote inflammasome assembly is the upregulation of NLRP3 production (26, 43). PAD4-dependent citrullination of the NFκB subunit p65 has been described as a mediator of its nuclear translocation (63). Thus, PAD4 elevation in disease (15) could regulate NLRP3 and ASC protein levels in neutrophils and, when overexpressed, also in iBMDMs, as we observed in our study (Figures 2B, 3C). Although nigericin rarely induces caspase-1 activation in unprimed macrophages and monocytes (64, 65), PAD4 overexpression results in LPS-independent inflammasome activation upon stimulation with nigericin (Figures 3A, B) without affecting IL-1ß mRNA levels (Figure S4C). Remarkably, relative mRNA levels of ASC or NLRP3 were not different in wild-type or PAD4-deficient neutrophils as well as in empty vector or PAD4 overexpressing iBMDMs (Figures 2C, 3D). The post-transcriptional effect of PAD4 on NLRP3 and ASC protein levels was further substantiated, as induction of transcription by LPS priming showed similar ASC and NLRP3 protein levels between wild-type and PAD4-deficient neutrophils (Figure S2D). Consequently, these findings show that the inflammasome protein levels are most likely regulated by an increase in translation or a reduced clearance of NLRP3. Indeed, it will be interesting to investigate the mechanism by which citrullination controls NLRP3 inflammasome in the future.
We examined how NLRP3 deficiency impaired NETosis by using time-lapse microscopy. This revealed that initiation of NETosis appeared normal, including plasma membrane vesiculation and nuclear rounding (45); however, following rounding there was diminished rupture of the nuclear envelope and, importantly, almost absent plasma membrane rupture (Figures 4D, F). While GSDMD was shown to induce nuclear membrane permeability in neutrophils, it has been suggested that this could promote nuclear membrane breakdown (50). However, plasma membrane rupture during pyroptotic cell death is a well-established event of GSDMD activity after inflammasome activation (51). Since we observed that plasma membrane permeabilization occurs prior to plasma membrane rupture in NETosis (12), inflammasome-dependent GSDMD pore likely prepares the plasma membrane for rupture. In neutrophils, subjected to sterile stimulation, this process is NLRP3 dependent.
Our data point to an important connection between inflammasome/ASC speck and NETosis (Figure 6) and thereby possibly promoting inflammatory noninfectious disorders. Diseases that are associated with increased PAD4 expression in neutrophils and elevated susceptibility to NETosis could possibly be linked to elevated NLRP3 inflammasome assembly in both neutrophils and macrophages, as we observed. Neutrophils from diabetic patients for instance showed a four-fold increase in PAD4 levels leading to augmented NETosis (7), and NLRP3 inflammasome is long well-known in type 2 diabetes (66). PAD4-dependent NETosis is also a crucial part of venous thrombosis (14). Thus it is not surprising that we observed that NLRP3 deficiency also diminished venous thrombosis (Figure 5). Moreover, considering the clinical importance of excessive IL-1ß generation in a wide variety of thrombo-inflammatory disorders, as shown by the CANTOS trial (67, 68), PAD-dependent regulation of NLRP3 protein levels could be an important mechanism in inflammasome-driven diseases and makes PADs promising new targets in the therapy of cardiovascular diseases (15, 69, 70).
It is likely that inflammasome-driven chronic diseases could be promoted further by neutrophilic inflammasome, as we found ASC speck expelled with extracellular chromatin on NETs (Figure 1C). These ASC specks could be taken up by other cells and propagate inflammation. Indeed, ASC has been described as a “prionoid”, that, when phagocytosed by macrophages, induce inflammasome assembly (27, 71, 72).
To conclude, here we have evaluated the link between canonical inflammasome and NETosis in sterile environment (Figure 6). Our results show inflammasome-dependent signaling is part of NETosis and PAD4 regulates ASC speck formation. These observations open many new avenues that may provide a base for new approaches in the prevention and treatment of inflammatory diseases. Finally, this work revealed additional positive effects of known drugs in development. We show that an inhibitor of NLRP3 will also reduce the toxic effects of NETs and propose that PAD inhibitors may improve inflammasome-driven human disorders, including cardiovascular disease and thrombosis.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Office of Clinical Investigations at Boston Children’s Hospital. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Institutional Animal Care and Use Committee of Boston Children’s Hospital or the Regierungspräsidium Tübingen.
PM, RN, SF, LM, KA, LC, DC, SG, NS, LS, VM, and ANRW performed experiments and analyzed the data. ANRW provided reagents. PM, RN, RES, CMW, HW, and DDW designed the research. PM and DDW designed the project and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by a grant from National Heart, Lung, and Blood Institute of the National Institutes of Health (grant R35 HL135765) and a Steven Berzin family support to DDW, an Individual Erwin Deutsch fellowship by the German, Austrian and Swiss Society of Thrombosis and Hemostasis Research to RES, a Whitman fellowship (MBL) to DDW, and an Individual Marie Skłodowska-Curie Actions fellowship by the European Commission (796365 - COAGULANT) to PM. ANRW was funded by the Deutsche Forschungsgemeinschaft (TRR156/2 –246807620) and a research grant (We-4195/15-19). CMW was supported by the Division of Intramural Research, NHLBI, NIH.
DDW is on the Scientific Advisory Board of Neutrolis, a preclinical-stage biotech company focused on DNases.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledge the meticulous help in preparation of the manuscript by Tiffany Frary. We thank the Marine Biological Laboratory (MBL) for support through the Whitman Fellows program and Nikon Instruments for loan of microscopy equipment and support through the Nikon Fellows program at MBL. In particular, we want to acknowledge the outstanding help by John Allen (Nikon Instruments).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.683803/full#supplementary-material
1. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
2. Martinod K, Wagner DD. Thrombosis: Tangled Up in Nets. Blood (2014) 123(18):2768–76. doi: 10.1182/blood-2013-10-463646
3. Sorvillo N, Mizurini DM, Coxon C, Martinod K, Tilvawala R, Cherpokova D, et al. Plasma Peptidylarginine Deiminase IV Promotes VWF-Platelet String Formation and Accelerates Thrombosis After Vessel Injury. Circ Res (2019) 125(5):507–19. doi: 10.1161/CIRCRESAHA.118.314571
4. Savchenko AS, Borissoff JI, Martinod K, De Meyer SF, Gallant M, Erpenbeck L, et al. VWF-Mediated Leukocyte Recruitment With Chromatin Decondensation by PAD4 Increases Myocardial Ischemia/Reperfusion Injury in Mice. Blood (2014) 123(1):141–8. doi: 10.1182/blood-2013-07-514992
5. Martinod K, Witsch T, Erpenbeck L, Savchenko A, Hayashi H, Cherpokova D, et al. Peptidylarginine Deiminase 4 Promotes Age-Related Organ Fibrosis. J Exp Med (2017) 214(2):439–58. doi: 10.1084/jem.20160530
6. Wang J, Li Q, Yin Y, Zhang Y, Cao Y, Lin X, et al. Excessive Neutrophils and Neutrophil Extracellular Traps in COVID-19. Front Immunol (2020) 11:2063. doi: 10.3389/fimmu.2020.02063
7. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes Primes Neutrophils to Undergo NETosis, Which Impairs Wound Healing. Nat Med (2015) 21(7):815–9. doi: 10.1038/nm.3887
8. Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, et al. Cancers Predispose Neutrophils to Release Extracellular DNA Traps That Contribute to Cancer-Associated Thrombosis. Proc Natl Acad Sci USA (2012) 109(32):13076–81. doi: 10.1073/pnas.1200419109
9. Demers M, Wagner DD. Netosis: A New Factor in Tumor Progression and Cancer-Associated Thrombosis. Semin Thromb Hemost (2014) 40(3):277–83. doi: 10.1055/s-0034-1370765
10. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil Extracellular Traps Produced During Inflammation Awaken Dormant Cancer Cells in Mice. Science (2018) 361(6409). doi: 10.1126/science.aao4227
11. Yipp BG, Kubes P. Netosis: How Vital is it? Blood (2013) 122(16):2784–94. doi: 10.1182/blood-2013-04-457671
12. Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, Goldman AE, et al. Netosis Proceeds by Cytoskeleton and Endomembrane Disassembly and PAD4-mediated Chromatin Decondensation and Nuclear Envelope Rupture. Proc Natl Acad Sci USA (2020) 117(13):7326–37. doi: 10.1073/pnas.1909546117
13. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is Essential for Antibacterial Innate Immunity Mediated by Neutrophil Extracellular Traps. J Exp Med (2010) 207(9):1853–62. doi: 10.1084/jem.20100239
14. Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, et al. Neutrophil Histone Modification by Peptidylarginine Deiminase 4 is Critical for Deep Vein Thrombosis in Mice. Proc Natl Acad Sci USA (2013) 110(21):8674–9. doi: 10.1073/pnas.1301059110
15. Wong SL, Wagner DD. Peptidylarginine Deiminase 4: A Nuclear Button Triggering Neutrophil Extracellular Traps in Inflammatory Diseases and Aging. FASEB J (2018) 32:6358–70. doi: 10.1096/fj.201800691R
16. Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, et al. Diverse Stimuli Engage Different Neutrophil Extracellular Trap Pathways. Elife (2017) 6. doi: 10.7554/eLife.24437
17. Gupta S, Chan DW, Zaal KJ, Kaplan MJ. A High-Throughput Real-Time Imaging Technique To Quantify NETosis and Distinguish Mechanisms of Cell Death in Human Neutrophils. J Immunol (2018) 200(2):869–79. doi: 10.4049/jimmunol.1700905
18. Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, et al. Differential Expression of NLRP3 Among Hematopoietic Cells. J Immunol (2011) 186(4):2529–34. doi: 10.4049/jimmunol.1002720
19. Chen KW, Gross CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, et al. The Neutrophil NLRC4 Inflammasome Selectively Promotes IL-1beta Maturation Without Pyroptosis During Acute Salmonella Challenge. Cell Rep (2014) 8(2):570–82. doi: 10.1016/j.celrep.2014.06.028
20. Tzeng TC, Schattgen S, Monks B, Wang D, Cerny A, Latz E, et al. A Fluorescent Reporter Mouse for Inflammasome Assembly Demonstrates an Important Role for Cell-Bound and Free Asc Specks During In Vivo Infection. Cell Rep (2016) 16(2):571–82. doi: 10.1016/j.celrep.2016.06.011
21. Zhang T, Du H, Feng S, Wu R, Chen T, Jiang J, et al. NLRP3/ASC/Caspase-1 Axis and Serine Protease Activity are Involved in Neutrophil IL-1beta Processing During Streptococcus Pneumoniae Infection. Biochem Biophys Res Commun (2019) 513(3):675–80. doi: 10.1016/j.bbrc.2019.04.004
22. Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E. Neutrophil P2X7 Receptors Mediate NLRP3 Inflammasome-Dependent IL-1beta Secretion in Response to ATP. Nat Commun (2016) 7:10555. doi: 10.1038/ncomms10555
23. Johnson JL, Ramadass M, Haimovich A, McGeough MD, Zhang J, Hoffman HM, et al. Increased Neutrophil Secretion Induced by NLRP3 Mutation Links the Inflammasome to Azurophilic Granule Exocytosis. Front Cell Infect Microbiol (2017) 7:507. doi: 10.3389/fcimb.2017.00507
24. Skendros P, Papagoras C, Mitroulis I, Ritis K. Autoinflammation: Lessons From the Study of Familial Mediterranean Fever. J Autoimmun (2019) 104:102305. doi: 10.1016/j.jaut.2019.102305
25. Stoler I, Freytag J, Orak B, Unterwalder N, Henning S, Heim K, et al. Gene-Dose Effect of MEFV Gain-of-Function Mutations Determines Ex Vivo Neutrophil Activation in Familial Mediterranean Fever. Front Immunol (2020) 11:716. doi: 10.3389/fimmu.2020.00716
26. Schroder K, Tschopp J. The Inflammasomes. Cell (2010) 140(6):821–32. doi: 10.1016/j.cell.2010.01.040
27. Grebe A, Hoss F, Latz E. Nlrp3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ Res (2018) 122(12):1722–40. doi: 10.1161/CIRCRESAHA.118.311362
28. Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural Mechanism for NEK7-licensed Activation of NLRP3 Inflammasome. Nature (2019) 570(7761):338–43. doi: 10.1038/s41586-019-1295-z
29. He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an Essential Mediator of NLRP3 Activation Downstream of Potassium Efflux. Nature (2016) 530(7590):354–7. doi: 10.1038/nature16959
30. Franklin BS, Latz E, Schmidt FI. The Intra- and Extracellular Functions of ASC Specks. Immunol Rev (2018) 281(1):74–87. doi: 10.1111/imr.12611
31. Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, et al. Caspase-1 Self-Cleavage is an Intrinsic Mechanism to Terminate Inflammasome Activity. J Exp Med (2018) 215(3):827–40. doi: 10.1084/jem.20172222
32. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
33. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A Small-Molecule Inhibitor of the NLRP3 Inflammasome for the Treatment of Inflammatory Diseases. Nat Med (2015) 21(3):248–55. doi: 10.1038/nm.3806
34. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 Directly Targets the NLRP3 ATP-Hydrolysis Motif for Inflammasome Inhibition. Nat Chem Biol (2019) 15(6):556–9. doi: 10.1038/s41589-019-0277-7
35. Tapia-Abellan A, Angosto-Bazarra D, Martinez-Banaclocha H, de Torre-Minguela C, Ceron-Carrasco JP, Perez-Sanchez H, et al. MCC950 Closes the Active Conformation of NLRP3 to an Inactive State. Nat Chem Biol (2019) 15(6):560–4. doi: 10.1038/s41589-019-0278-6
36. Cho JS, Guo Y, Ramos RI, Hebroni F, Plaisier SB, Xuan C, et al. Neutrophil-Derived IL-1beta is Sufficient for Abscess Formation in Immunity Against Staphylococcus Aureus in Mice. PloS Pathog (2012) 8(11):e1003047. doi: 10.1371/journal.ppat.1003047
37. Gupta N, Sahu A, Prabhakar A, Chatterjee T, Tyagi T, Kumari B, et al. Activation of NLRP3 Inflammasome Complex Potentiates Venous Thrombosis in Response to Hypoxia. Proc Natl Acad Sci USA (2017) 114(18):4763–8. doi: 10.1073/pnas.1620458114
38. Nazir S, Gadi I, Al-Dabet MM, Elwakiel A, Kohli S, Ghosh S, et al. Cytoprotective Activated Protein C Averts Nlrp3 Inflammasome-Induced Ischemia-Reperfusion Injury Via mTORC1 Inhibition. Blood (2017) 130(24):2664–77. doi: 10.1182/blood-2017-05-782102
39. Hemmers S, Teijaro JR, Arandjelovic S, Mowen KA. PAD4-Mediated Neutrophil Extracellular Trap Formation is Not Required for Immunity Against Influenza Infection. PloS One (2011) 6(7):e22043. doi: 10.1371/journal.pone.0022043
40. Drexler SK, Bonsignore L, Masin M, Tardivel A, Jackstadt R, Hermeking H, et al. Tissue-Specific Opposing Functions of the Inflammasome Adaptor ASC in the Regulation of Epithelial Skin Carcinogenesis. Proc Natl Acad Sci USA (2012) 109(45):18384–9. doi: 10.1073/pnas.1209171109
41. Brill A, Fuchs TA, Chauhan AK, Yang JJ, De Meyer SF, Kollnberger M, et al. Von Willebrand Factor-Mediated Platelet Adhesion is Critical for Deep Vein Thrombosis in Mouse Models. Blood (2011) 117(4):1400–7. doi: 10.1182/blood-2010-05-287623
42. Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, et al. HDAC6 Mediates an Aggresome-Like Mechanism for NLRP3 and Pyrin Inflammasome Activation. Science (2020) 369(6510). doi: 10.1126/science.aas8995
43. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting Edge: NF-kappaB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J Immunol (2009) 183(2):787–91. doi: 10.4049/jimmunol.0901363
44. Mishra N, Schwerdtner L, Sams K, Mondal S, Ahmad F, Schmidt RE, et al. Cutting Edge: Protein Arginine Deiminase 2 and 4 Regulate Nlrp3 Inflammasome-Dependent IL-1beta Maturation and ASC Speck Formation in Macrophages. J Immunol (2019) 203(4):795–800. doi: 10.4049/jimmunol.1800720
45. Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular Mechanisms of NETosis. Annu Rev Cell Dev Biol (2020) 36:191–218. doi: 10.1146/annurev-cellbio-020520-111016
46. Mortality GBD. Causes of Death C. Global, Regional, and National Age-Sex Specific All-Cause and Cause-Specific Mortality for 240 Causes of Death, 1990-2013: A Systematic Analysis for the Global Burden of Disease Study 2013. Lancet (2015) 385(9963):117–71. doi: 10.1016/S0140-6736(14)61682-2
47. Brill A, Suidan GL, Wagner DD. Hypoxia, Such as Encountered at High Altitude, Promotes Deep Vein Thrombosis in Mice. J Thromb Haemost (2013) 11(9):1773–5. doi: 10.1111/jth.12310
48. Yadav V, Chi L, Zhao R, Tourdot BE, Yalavarthi S, Jacobs BN, et al. Ectonucleotidase Tri(Di)Phosphohydrolase-1 (ENTPD-1) Disrupts Inflammasome/Interleukin 1beta-Driven Venous Thrombosis. J Clin Invest (2019) 129(7):2872–7. doi: 10.1172/JCI124804
49. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D Plays a Vital Role in the Generation of Neutrophil Extracellular Traps. Sci Immunol (2018) 3(26). doi: 10.1126/sciimmunol.aar6689
50. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical Inflammasome Signaling Elicits Gasdermin D-dependent Neutrophil Extracellular Traps. Sci Immunol (2018) 3(26). doi: 10.1126/sciimmunol.aar6676
51. Lieberman J, Wu H, Kagan JC. Gasdermin D Activity in Inflammation and Host Defense. Sci Immunol (2019) 4(39). doi: 10.1126/sciimmunol.aav1447
52. Karmakar M, Minns M, Greenberg EN, Diaz-Aponte J, Pestonjamasp K, Johnson JL, et al. N-GSDMD Trafficking to Neutrophil Organelles Facilitates IL-1beta Release Independently of Plasma Membrane Pores and Pyroptosis. Nat Commun (2020) 11(1):2212. doi: 10.1038/s41467-020-16043-9
53. Pillai PS, Molony RD, Martinod K, Dong H, Pang IK, Tal MC, et al. Mx1 Reveals Innate Pathways to Antiviral Resistance and Lethal Influenza Disease. Science (2016) 352(6284):463–6. doi: 10.1126/science.aaf3926
54. Denes A, Lopez-Castejon G, Brough D. Caspase-1: Is IL-1 Just the Tip of the Iceberg? Cell Death Dis (2012) 3:e338. doi: 10.1038/cddis.2012.86
55. Greaney AJ, Portley MK, O’Mard D, Crown D, Maier NK, Mendenhall MA, et al. Frontline Science: Anthrax Lethal Toxin-Induced, NLRP1-mediated Il-1beta Release is a Neutrophil and PAD4-dependent Event. J Leukoc Biol (2020) 108(3):773–86. doi: 10.1002/JLB.4HI0320-028R
56. Mankan AK, Dau T, Jenne D, Hornung V. The NLRP3/ASC/Caspase-1 Axis Regulates IL-1beta Processing in Neutrophils. Eur J Immunol (2012) 42(3):710–5. doi: 10.1002/eji.201141921
57. Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, et al. Canonical Nlrp3 Inflammasome Links Systemic Low-Grade Inflammation to Functional Decline in Aging. Cell Metab (2013) 18(4):519–32. doi: 10.1016/j.cmet.2013.09.010
58. Coll RC, O’Neill L, Schroder K. Questions and Controversies in Innate Immune Research: What is the Physiological Role of NLRP3? Cell Death Discovery (2016) 2:16019. doi: 10.1038/cddiscovery.2016.19
59. Vossenaar ER, Radstake TR, van der Heijden A, van Mansum MA, Dieteren C, de Rooij DJ, et al. Expression and Activity of Citrullinating Peptidylarginine Deiminase Enzymes in Monocytes and Macrophages. Ann Rheum Dis (2004) 63(4):373–81. doi: 10.1136/ard.2003.012211
60. Stutz A, Kolbe CC, Stahl R, Horvath GL, Franklin BS, van Ray O, et al. NLRP3 Inflammasome Assembly is Regulated by Phosphorylation of the Pyrin Domain. J Exp Med (2017) 214(6):1725–36. doi: 10.1084/jem.20160933
61. Tang J, Tu S, Lin G, Guo H, Yan C, Liu Q, et al. Sequential Ubiquitination of NLRP3 by RNF125 and Cbl-b Limits Inflammasome Activation and Endotoxemia. J Exp Med (2020) 217(4). doi: 10.1084/jem.20182091
62. Yaron JR, Gangaraju S, Rao MY, Kong X, Zhang L, Su F, et al. K(+) Regulates Ca(2+) to Drive Inflammasome Signaling: Dynamic Visualization of Ion Flux in Live Cells. Cell Death Dis (2015) 6:e1954. doi: 10.1038/cddis.2015.277
63. Sun B, Dwivedi N, Bechtel TJ, Paulsen JL, Muth A, Bawadekar M, et al. Citrullination of NF-kappaB p65 Promotes its Nuclear Localization and TLR-induced Expression of IL-1beta and TNFalpha. Sci Immunol (2017) 2(12). doi: 10.1126/sciimmunol.aal3062
64. Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. RIPK3 Promotes Cell Death and NLRP3 Inflammasome Activation in the Absence of MLKL. Nat Commun (2015) 6:6282. doi: 10.1038/ncomms7282
65. Gritsenko A, Yu S, Martin-Sanchez F, Diaz-Del-Olmo I, Nichols EM, Davis DM, et al. Priming Is Dispensable for NLRP3 Inflammasome Activation in Human Monocytes In Vitro. Front Immunol (2020) 11:565924. doi: 10.3389/fimmu.2020.565924
66. Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 Inflammasome Activation in Patients With Type 2 Diabetes. Diabetes (2013) 62(1):194–204. doi: 10.2337/db12-0420
67. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory Therapy With Canakinumab for Atherosclerotic Disease. N Engl J Med (2017) 377(12):1119–31. doi: 10.1056/NEJMoa1707914
68. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin-1beta Inhibition With Canakinumab on Incident Lung Cancer in Patients With Atherosclerosis: Exploratory Results From a Randomised, Double-Blind, Placebo-Controlled Trial. Lancet (2017) 390(10105):1833–42. doi: 10.1016/S0140-6736(17)32247-X
69. Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ Res (2020) 126(9):1260–80. doi: 10.1161/CIRCRESAHA.120.315937
70. Gomes T, Varady CBS, Lourenco AL, Mizurini DM, Rondon AMR, Leal AC, et al. IL-1beta Blockade Attenuates Thrombosis in a Neutrophil Extracellular Trap-Dependent Breast Cancer Model. Front Immunol (2019) 10:2088. doi: 10.3389/fimmu.2019.02088
71. Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 Inflammasome is Released as a Particulate Danger Signal That Amplifies the Inflammatory Response. Nat Immunol (2014) 15(8):738–48. doi: 10.1038/ni.2919
Keywords: Neutrophils, NETs, NLRP3 inflammasome, MCC950, deep vein thrombosis, PAD4
Citation: Münzer P, Negro R, Fukui S, di Meglio L, Aymonnier K, Chu L, Cherpokova D, Gutch S, Sorvillo N, Shi L, Magupalli VG, Weber ANR, Scharf RE, Waterman CM, Wu H and Wagner DD (2021) NLRP3 Inflammasome Assembly in Neutrophils Is Supported by PAD4 and Promotes NETosis Under Sterile Conditions. Front. Immunol. 12:683803. doi: 10.3389/fimmu.2021.683803
Received: 22 March 2021; Accepted: 10 May 2021;
Published: 28 May 2021.
Edited by:
Chaofeng Han, Second Military Medical University, ChinaReviewed by:
Kaiwen Chen, National University of Singapore, SingaporeCopyright © 2021 Münzer, Negro, Fukui, di Meglio, Aymonnier, Chu, Cherpokova, Gutch, Sorvillo, Shi, Magupalli, Weber, Scharf, Waterman, Wu and Wagner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Denisa D. Wagner, ZGVuaXNhLndhZ25lckBjaGlsZHJlbnMuaGFydmFyZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.