 Stephanie L. Schell
Stephanie L. Schell Ziaur S. M. Rahman
Ziaur S. M. Rahman- Department of Microbiology and Immunology, Pennsylvania State University College of Medicine, Hershey, PA, United States
Loss of B cell tolerance is central to autoimmune diseases such as systemic lupus erythematosus (SLE). As such, the mechanisms involved in B cell development, maturation, activation, and function that are aberrantly regulated in SLE are of interest in the design of targeted therapeutics. While many factors are involved in the generation and regulation of B cell responses, miRNAs have emerged as critical regulators of these responses within the last decade. To date, miRNA involvement in B cell responses has largely been studied in non-autoimmune, immunization-based systems. However, miRNA profiles have also been strongly associated with SLE in human patients and these molecules have proven critical in both the promotion and regulation of disease in mouse models and in the formation of autoreactive B cell responses. Functionally, miRNAs are small non-coding RNAs that bind to complementary sequences located in target mRNA transcripts to mediate transcript degradation or translational repression, invoking a post-transcriptional level of genetic regulation. Due to their capacity to target a diverse range of transcripts and pathways in different immune cell types and throughout the various stages of development and response, targeting miRNAs is an interesting potential therapeutic avenue. Herein, we focus on what is currently known about miRNA function in both normal and SLE B cell responses, primarily highlighting miRNAs with confirmed functions in mouse models. We also discuss areas that should be addressed in future studies and whether the development of miRNA-centric therapeutics may be a viable alternative for the treatment of SLE.
Introduction
B cell development and function is critical for the establishment of a B cell repertoire that can respond to a diversity of foreign antigens (1). Antigenic exposure initiates B cell responses that target invading pathogens and leads to the formation of long-lived plasma cell and memory B cell responses that protect the host against future reinfection (2). While B cells are critical for the establishment of normal immune responses against pathogens, they can become dysregulated under certain circumstances, leading to the development of autoimmunity (3). Systemic lupus erythematosus (SLE) is a complex autoimmune disease that causes multi-organ dysfunction. The onset of SLE is dependent on both the possession of susceptibility genes and the environmental triggers (e.g. infection, chemicals, retroviral elements) (4, 5). Genetic and environmental factors synergize to cause aberrantly regulated immune activation which leads to the loss of B cell tolerance to self-antigens and high-affinity anti-nuclear antibody (ANA) production (6, 7). ANAs generated by B cells form immune complexes that enter the circulation and deposit in peripheral tissues, leading to the recruitment of myeloid cells, which promote local inflammation (8–10). Inflammation in the kidneys, termed lupus nephritis (10), and various cardiovascular disease manifestations (11) are common causes of morbidity and mortality in individuals living with SLE.
Due to the fact that much still remains unclear about the development of lupus, only one FDA approved therapy specifically developed and approved for SLE, belimumab, has emerged (12). Belimumab is a monoclonal antibody that targets B cell survival by binding to and sequestering Blys, an essential B cell survival factor. Belimumab has an encouraging efficacy in dampening disease manifestations, however it also leaves patients susceptible to infection, as it non-specifically suppresses the immune system (13, 14). A better understanding of the mechanisms involved in SLE development is required to develop novel therapeutics for SLE that may avoid some of the negative immunosuppressive effects of current therapies. The development of microRNA (miRNA) therapeutics has started to gain traction for the treatment of other diseases (15), but their implementation in autoimmunity is still lacking as more studies are required to fully elucidate the contribution of these factors to disease development and progression.
As such, efforts to understand the role of miRNA function in the development of normal B cell responses and dysregulation of these miRNAs in SLE represents a growing field. In general, miRNAs have been implicated as causative agents and biomarkers in a number of diseases (16, 17). In regard to SLE, miRNA centric studies have focused on the differences in miRNA expression between the healthy and diseased states, what cell types are altered by aberrant miRNA expression, and what genes and processes these miRNAs target to either promote or prevent autoimmunity. Many studies have focused on profiling the miRNAs that are expressed in healthy individuals versus those with SLE, with some of these studies determining the miRNAs expressed during the active versus inactive stages of disease (18–23). Cells and tissues used for miRNA profiling in SLE vary, but most studies have profiled the expression of miRNAs in peripheral blood mononuclear cells (PBMCs), B cells, T cells, and blood. Additionally, many studies have assessed miRNAs associated with lupus nephritis through the analysis of urine (24, 25). In addition to profiling miRNAs in human patients, miRNA profiling has been performed in animal models of SLE, demonstrating that there is a conserved profile between several different lupus mouse models and human patients (19, 26, 27).
Broad miRNA profiling has divulged a large amount of information about miRNA expression patterns in normal and SLE B cell responses and has opened the door for mechanistic studies. These mechanistic studies are required to determine how individual or combinations of miRNAs are specifically involved in the loss of B cell tolerance. Additionally, understanding if similar mechanisms are involved in normal protective B cell responses is important for shaping any future therapeutic pursuits. First, we will briefly outline how miRNAs function and the different stages of B cell development and response to antigen. We will then discuss key studies in non-autoimmune and autoimmune systems that frame our understanding of miRNAs in these responses and the implications for therapeutic targeting in the future.
miRNA Processing and Targeting Mechanisms
miRNAs are small non-coding RNAs, approximately 22 nucleotides in length, that mediate post-transcriptional gene regulation. miRNAs are transcribed from the genome by RNA Polymerase II via dedicated promoters or are processed from intronic or exonic sequences located in other transcription units (28–32). This generates a primary miRNA transcript (pri-miRNA). While still in the nucleus, the pri-miRNA is cleaved into the hairpin shaped pre-miRNA by the Microprocessor complex, which contains the RNAse III enzyme Drosha and RNA binding protein DGCR8 (33–36). The pre-miRNA, which is approximately 60-70 nucleotides, is exported from the nucleus into the cytosol via the activity of exportin-5 and ran-GTP (37). Once in the cytosol, Dicer cleaves the pre-miRNA into a duplex structure (38, 39). The mature miRNA duplex associates with Argonaute and is dissociated into the 5’ guide strand, which is preferentially retained by Argonaute, and the 3’ passenger (or star) strand, which is preferentially degraded (40). Binding to Argonaute and association with additional proteins that comprise the RNA Induced Silencing Complex (RISC) stabilizes the miRNA from degradation (40–42).
Once incorporated into the RISC, the miRNA has been traditionally thought to exert genetic control by base pairing with complementary sequences found in the 3’ UTR of gene transcripts (43–45). However, more recently binding has also been observed within coding regions and 5’ UTRs (44–46). While the 5’ guide strand is typically incorporated into the RISC, the passenger strand can also be incorporated to target its own set of genes, though usually at a reduced level compared to the guide strand (47, 48). The targeting efficiency achieved by the miRNA can depend on the binding strength of the interaction, as miRNA-transcript interaction can occur through perfect or slightly imperfect complementarity with 6-8 base pair motifs located in the target transcript (49). Once the miRNA interacts with its target, negative regulation of gene expression can occur through both degradation of the transcript and translational repression (50–54). Regardless of the regulatory mechanism employed, ultimately the effect is the impediment of protein being translated from target transcripts. This process is summarized in Figure 1.
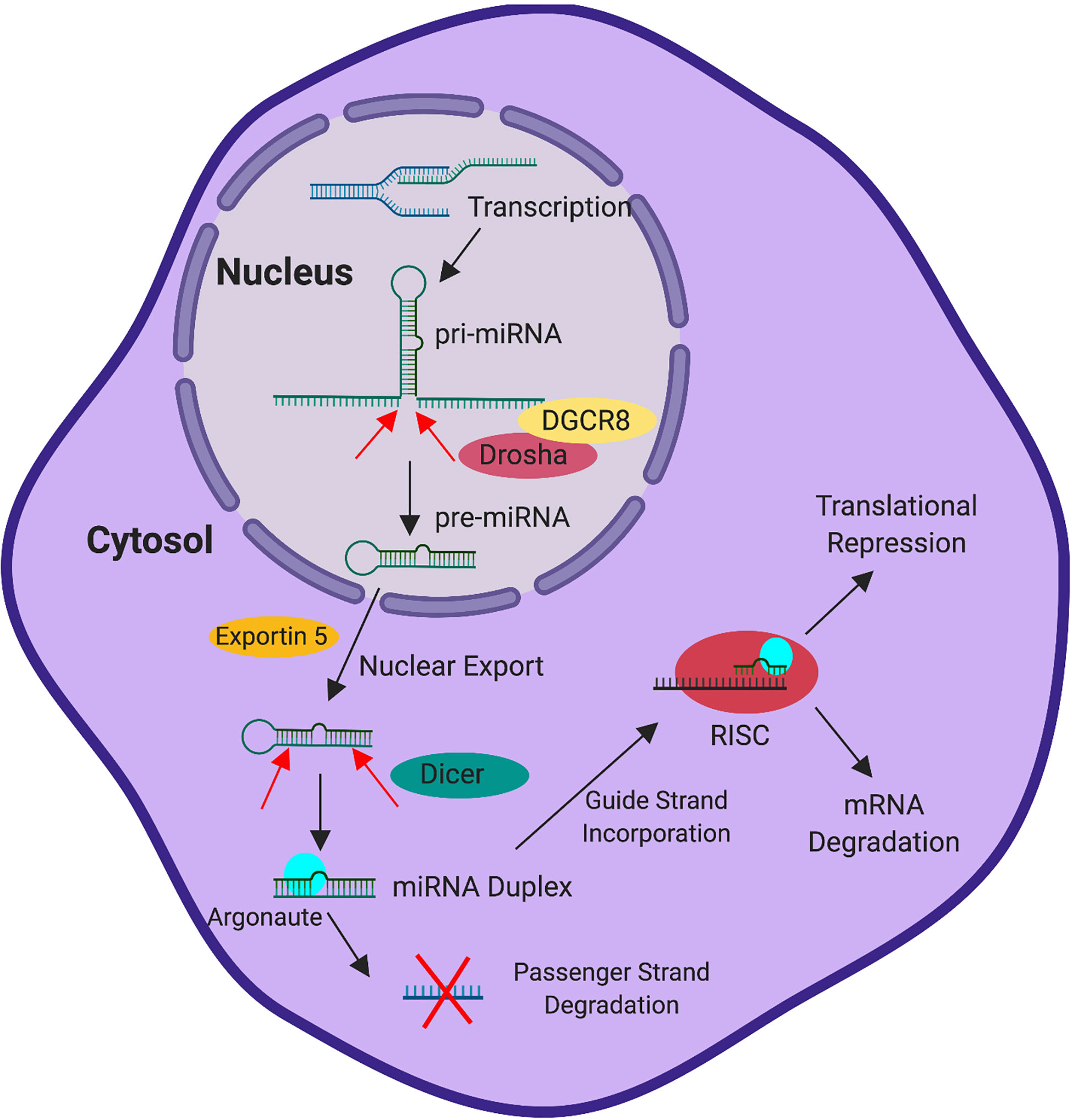
Figure 1 miRNA Processing and Activity. Transcription induced in the nucleus generates a pri-miRNA transcript. The pri-miRNA is cleaved by Drosha, with the aid of co-factor DGCR8, into the pre-miRNA while still in the nucleus. Subsequently, exportin 5 exports the pre-miRNA into the cytoplasm. Following delivery into the cytoplasm, Dicer cleaves the pre-miRNA into the mature miRNA duplex. The mature miRNA duplex (associated with Argonaute) is then dissociated into two strands, the guide strand and the passenger strand. The guide strand preferentially associates with Argonaute in the RNA-induced silencing complex (RISC) and the passenger strand is preferentially degraded. Following association of the miRNA and target transcript, the RISC drives the degradation of the mRNA or mediates translational repression to control gene expression.
To date, there have been over 1000 miRNAs discovered, with each miRNA capable of targeting hundreds of genes. A significant portion of miRNAs are found in clusters in the genome, further adding to the sophistication with which they can impart genetic control (55). Importantly, miRNAs are highly conserved among species, making their functional study in mouse models relevant to developing an understanding of their function in human (56, 57). In addition to binding sequence and strength of interaction, the ability of a miRNA to target complementary transcripts relies on the level of miRNA expression in the cell type of interest as well as the number and expression level of target genes in the same cell (58). Accordingly, the expression of the miRNAs and target genes vary in different tissues and cell types, and at different stages of development, making miRNA-mediated regulation a fluid process that is extremely specific to conditions and outside stimuli (59). This applies to the immune response where miRNA function is critically important at various stages (60). The profile of miRNAs among immune cell subsets and their functions in these cells confers the ability to specifically fine-tune the activity of many diverse signaling pathways associated with the activation and regulation of immune cell functions. As such, one miRNA can have vastly different gene targets and effects among different immune cell subsets (61). In this review, we focus on how this concept can be applied to miRNA function in normal B cell responses and B cell tolerance in the context of SLE.
Key Stages of the B Cell Life Cycle in Development and Tolerance
B cell development begins in the bone marrow with commitment of the common lymphocyte progenitor (CLP) to the B cell lineage (62), followed by further differentiation through the stages of pro B cell, pre B cell, and immature B cell (1). In the bone marrow, B cells undergo VDJ recombination to produce a diverse array of BCR specificities and processes exist to negatively select autoreactive B cells that form during this process (central tolerance) (63, 64). Functional, non-autoreactive B cells then egress to the secondary lymphoid organs where they acquire a transitional phenotype. Transitional B cells consist of three independent fractions, the T1 fraction, the T2 fraction, and the T3 fraction (65). Transitional B cells receiving the appropriate levels of stimulation and survival signals eventually differentiate into marginal zone or follicular B cells, whereas autoreactive B cells can be regulated at the T1 and T3 stages through apoptosis or anergy (66, 67) (Figure 2). In SLE, defects have been observed in early stages of B cell tolerance and loss of tolerance at this stage is usually linked to the possession of certain genetic susceptibility loci (68–72).
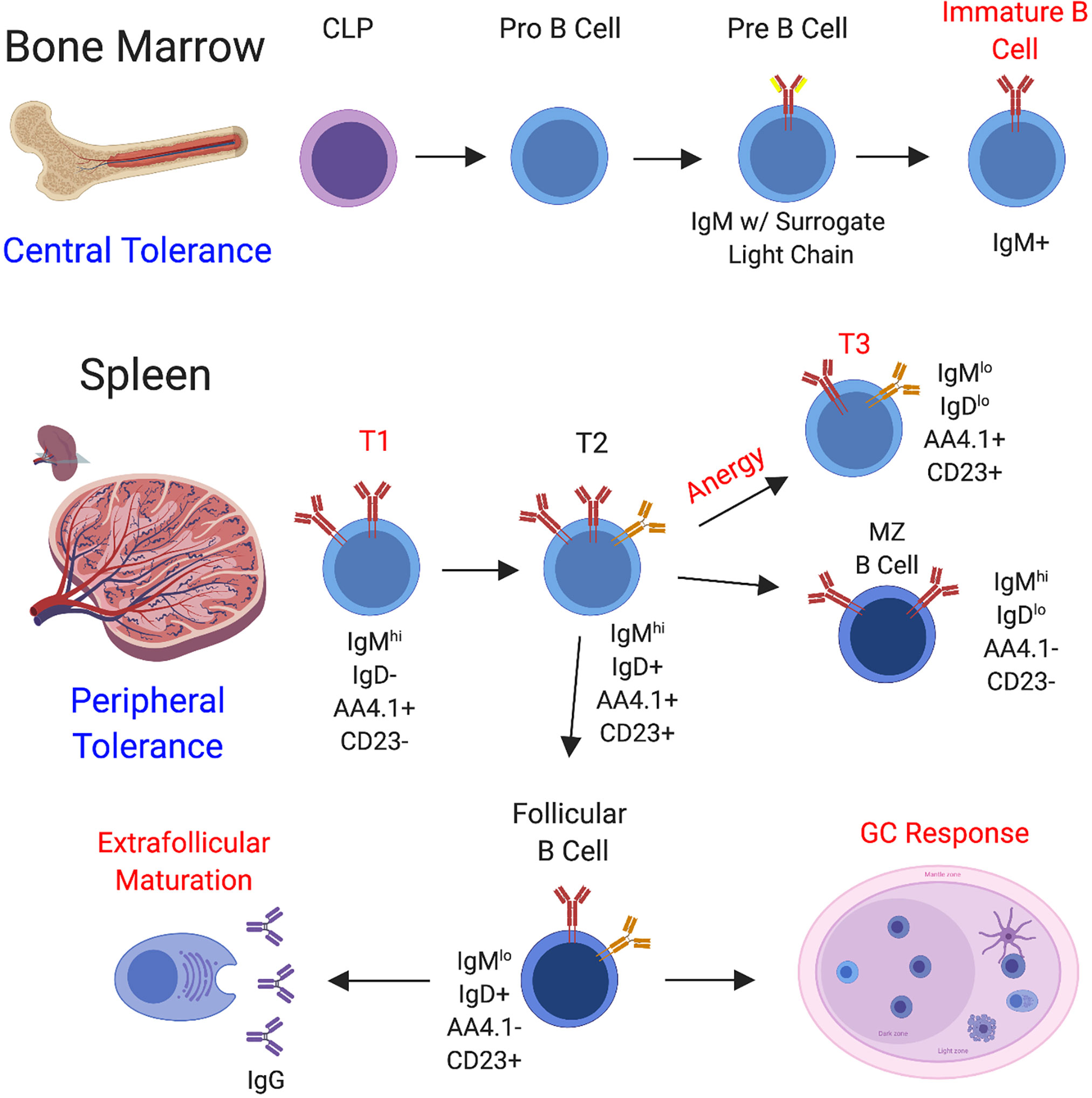
Figure 2 B Cell Development and Sites of B Cell Tolerance. B cell development begins in the bone marrow through a series of steps. Hematopoietic progenitor stem cells (HPSCs) undergo a series of differentiation steps leading to the generation of the common lymphocyte progenitor (CLP), from which B cell differentiation can proceed. Throughout the stages of B cell development in the bone marrow, a functional BCR is assembled and central tolerance is employed, whereby self-reactive B cells are directed to undergo receptor editing or cell death. B cells that pass this checkpoint migrate to the secondary lymphoid organs, including the spleen (depicted here) and lymph nodes. In the spleen, B cells continue to mature and peripheral B cell tolerance is enacted at several different stages of development. B cells enter the spleen at the T1 stage and strong BCR engagement can drive apoptosis at this stage. T1 B cells can progress on to the T2 stage, and from there can be induced to seed the marginal zone and follicle. In addition to regulation at the T1 stage, transitional B cells can be directed to undergo anergy and assume the T3 phenotype, whereby the BCR is downregulated to promote hyporesponsiveness. Marginal zone B cells generally become activated to provide a source of IgM. Alternatively, the follicular B cell subset directs the generation of IgG-secreting plasmablasts through both the follicular germinal center pathway and the extrafollicular pathway. Regulation is employed through both of these pathways. To summarize, well-documented regulatory stages are highlighted in red.
Mature B cells enter two major pathways following antigenic challenge to generate antibody responses, the germinal center (GC) and the extrafollicular pathway. During pathogen-driven immune responses, B cell development through the GC is critical for the generation of plasma cells that secrete high-affinity, class-switched antibodies and the differentiation of memory B cells (73, 74). However, GCs can become enlarged and dysregulated in SLE, leading to the production of high-affinity, class-switched autoantibodies that cause downstream pathology (74, 75). Many reviews have extensively detailed the mechanisms involved in the initiation and maintenance of GC responses driven by foreign antigen and in autoimmunity (73, 74, 76).
Alternatively, extrafollicular foci form in the red pulp and can occur rapidly in response to T-independent and T-dependent antigens (77). Activation of B cells through the extrafollicular pathway leads to rapid plasmablast formation, from which a select number of plasma cells will develop. Responses generated through the extrafollicular pathway can also undergo class-switching and somatic hypermutation independent of the GC, although at a lower frequency (77). In SLE, significant maturation of autoreactive B cells can occur outside of the GC (78–82). Ultimately, dysregulation of B cell responses at any stage of development and response to antigen can lead to autoimmunity.
miRNA Function in Protective B Cell Responses
While the goal for therapeutic development is to ultimately understand how miRNA expression and function is dysregulated in SLE, in order to achieve this, we must also understand how miRNAs function during normal B cell responses. This is important because miRNA expression level heavily impacts its function. miRNAs may drive aberrant B cell regulation due to overexpression, underexpression, or novel expression in B cells or other cell types that affect B cell responses. Additionally, any potential therapeutic design will ideally leave miRNA function involved in normal B cell responses intact to prevent host susceptibility to infection. The studies that have shaped our current understanding of miRNA function in B cell responses are discussed below. While the focus is predominantly on the B cell and T cell intrinsic expression of these molecules, it is important to note that their expression in innate immune cell types can also shape B cell responses through the regulation of cytokines and other factors.
Technically, studies of miRNA contribution to B cell responses are comprised of multiple approaches which collectively help build a full picture of miRNA involvement. miRNA expression profiling studies establish a starting point by identifying specific miRNAs for further functional analysis. The following phenotypic studies that narrow down on individual miRNAs have implemented a combination of in vitro and in vivo approaches. While in vitro approaches cannot determine the absolute requirement for specific miRNAs in the generation of B cell responses that require specific signals and interactions in vivo, such as GC responses, they can identify and confirm mRNA targets in some cell types of interest. On the other hand, mouse models that implement overexpression (lentiviral or genetic), miRNA antagomir administration, or knockouts of individual miRNAs are valuable tools for determining the absolute and non-redundant requirements for these factors in generating specific B cell responses and highlight the function of miRNAs in the presence of stimuli that are specific to in vivo conditions (Tables 1, 2).

Table 1 Methods to Study or Therapeutically Modulate miRNAs in Mouse Models.
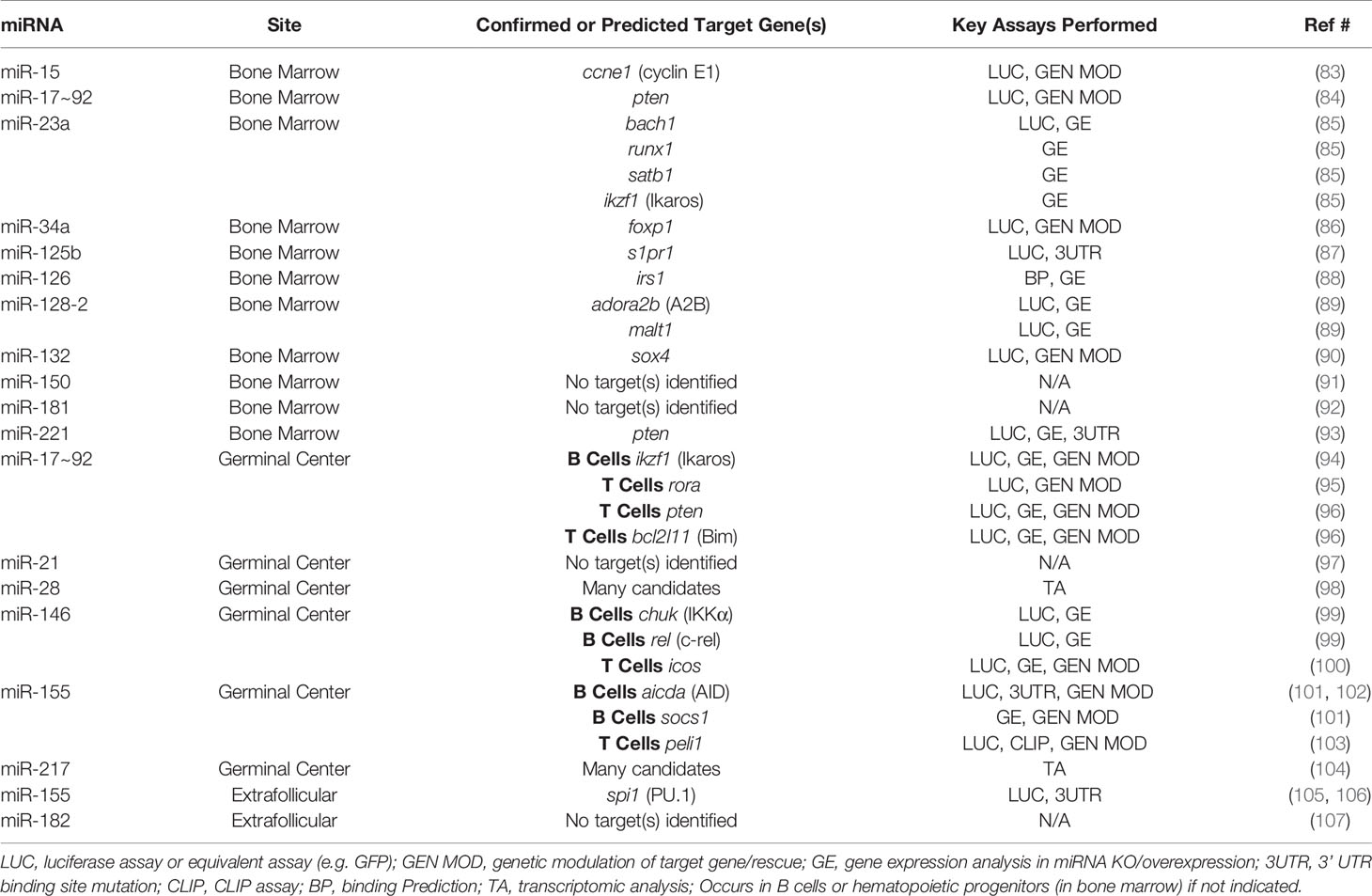
Table 2 miRNAs and Direct Target Genes in Protective B Cell Responses.
Regulation of Multiple miRNAs Is Involved in Early B Cell Development
Multiple steps, outlined earlier, are involved in B cell development. miRNAs have been shown to fluctuate in expression throughout the different stages of B cell development in the bone marrow, supporting the idea that their expression is important for guiding B cells through this process (108). Broadly, deletion of DGCR8 in B cells, which inhibits miRNA processing, caused a block in B cell development from the pro-B cell to pre-B cell stage (109). This was due to increased apoptosis of pro-B cells and resulted in a severe loss of B cells in the periphery of these mice (109), indicating that global miRNA expression is indispensable for B cell development.
Specifically, multiple studies have found important roles for several miRNAs during early fate decisions that polarize progenitors to the B cell lineage, or alternatively the T cell or myeloid cell lineages (Figure 3). Early expression of miR-181 or miR-126 in hematopoietic progenitor cells resulted in increased commitment to the B cell lineage (88, 92). miR-126 was shown to target IRS-1 to drive this commitment decision (88). Alternatively, expression of miR-132 or miR-23a in hematopoietic progenitors or miR-128-2 in common lymphoid progenitors (CLPs), resulted in reduced B cell lineage commitment, indicating that these miRNAs negatively regulate differentiation into the B cell lineage (85, 89, 90, 110). Mechanistically, miR-23a was able to regulate multiple transcription factors, including Ik2f1, Bach1, Satb1, and Runx1 (85), whereas miR-132 was shown to target Sox4 which was previously implicated in B cell development (90). Additionally, apoptosis modulation was responsible for a miR-128-2 dependent increase in CLPs, with A2B and Malt1 identified as candidate targets of miR-128-2 (89). The exact mechanisms of miRNA targeting involved in the activity of miR-181 in this process remains an open question.
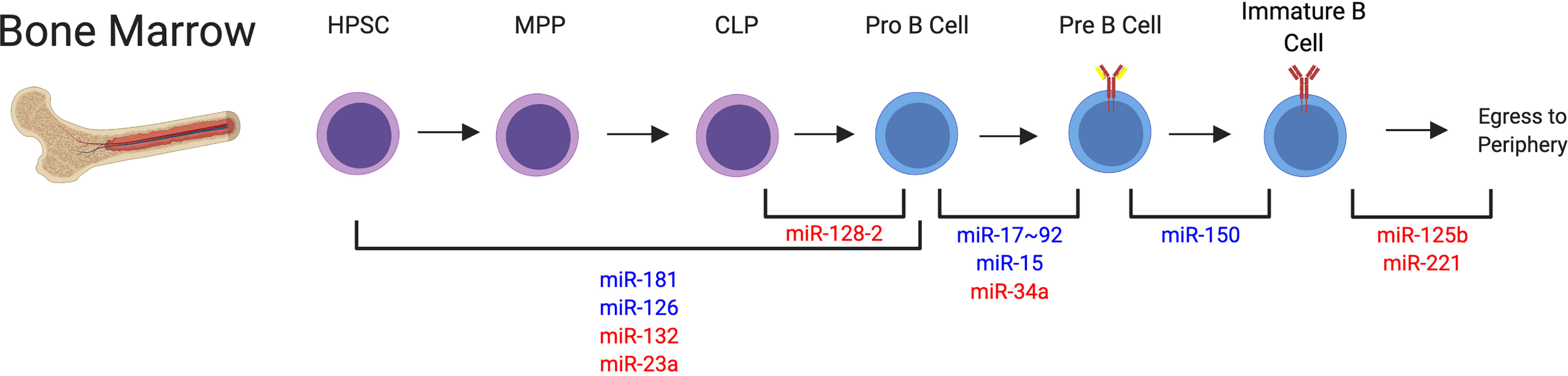
Figure 3 miRNAs that Impact B Cell Development in the Bone Marrow. Many miRNAs have been identified in the regulation of B cell development through each of the individual stages. This indicates a dynamic regulation of miRNA expression is required for proper developmental programs. miRNA regulation involved at these different stages is depicted. miRNAs that positively regulate these specific steps are depicted in blue. miRNAs that negatively regulate these specific steps are depicted in red. miRNAs are indicated at their confirmed or predicted stages of activity. HPSC, hematopoietic progenitor stem cell; MPP, multipotent progenitor; CLP, common lymphocyte progenitor.
Following lineage commitment, miR-17~92 was shown to increase PI3K activity in pro-B cells to regulate RAG expression and allow for transition to the pre-B cell stage (84). Further, miR-15 was shown to be involved in the induction of transcriptional programming required for the differentiation to the pre-B cell stage, with cyclin E1 identified a direct target gene of miR-15 and cyclin D3 identified as an indirect target of this miRNA (83). Additionally, differentiation from the pro to pre-B cell stage was revealed to be sensitive to the levels of miR-34a expression, which must be reduced to allow expression of Foxp1, a direct target of miR-34a, to occur (86).
Multiple studies also support the activity of miRNAs in later stages of B cell development in the bone marrow. miR-150 is likely involved in the transition from the pre-B cell to immature B cell stage, although aberrant premature expression can block development at earlier stages in the bone marrow (91). Downregulation of miR-125b and miR-221 have been shown to promote egress of B cells to the spleen, with S1PR1 and PI3K signaling regulation involved in this process (87, 93, 111). It is less clear which miRNAs are alternatively upregulated to promote B cell egress from the bone marrow. Ultimately, these studies indicate that there is a dynamic regulation of miRNA expression that controls the multiple stages of B cell development in the bone marrow, with a delicate balance of miRNAs providing both positive and negative regulation of these responses (Figure 3). Accordingly, improper miRNA expression can generate excessive B cell responses, with clinical manifestations of malignancy (112) or autoimmunity (to be discussed in detail). Mouse studies also suggest that miRNAs could play a role in clinical immunodeficiency syndromes such as severe combined immunodeficiency (SCID), since the discussed studies indicate that miRNA function is required for proper B cell development. However, the exact role of miRNAs in clinical immunodeficiency observed in human patients requires further study.
Identifying and Determining the Requirement for miRNAs in GC Response
The contributions of miRNAs to the GC response have also been extensively documented. Similar to analyses assessing the overall importance of miRNAs in early stages of B cell development in the bone marrow, the loss of Dicer function (and thus the inability to generate mature miRNAs) in B cells undergoing class-switching via an AID-Cre based system effectively ablated the GC response and class-switched antibody production in mice (113). Likewise, the loss of DGCR8 function in T cells prevented the differentiation and function of follicular helper T cells (Tfh) and in turn GC B cells (95). These studies indicate that miRNA function is generally indispensable for the establishment of GC responses.
The hindrance of miRNA processing machinery represents a global loss of miRNA function. Additional studies that profile the expression of miRNAs in GC B cells and Tfh, as compared to their respective precursor cells, have identified specific miRNAs that may be absolutely crucial for mediating the differentiation and function of these specific cell types (100, 103, 114–116). We will further discuss those that have been studied in mouse models (Figure 4).
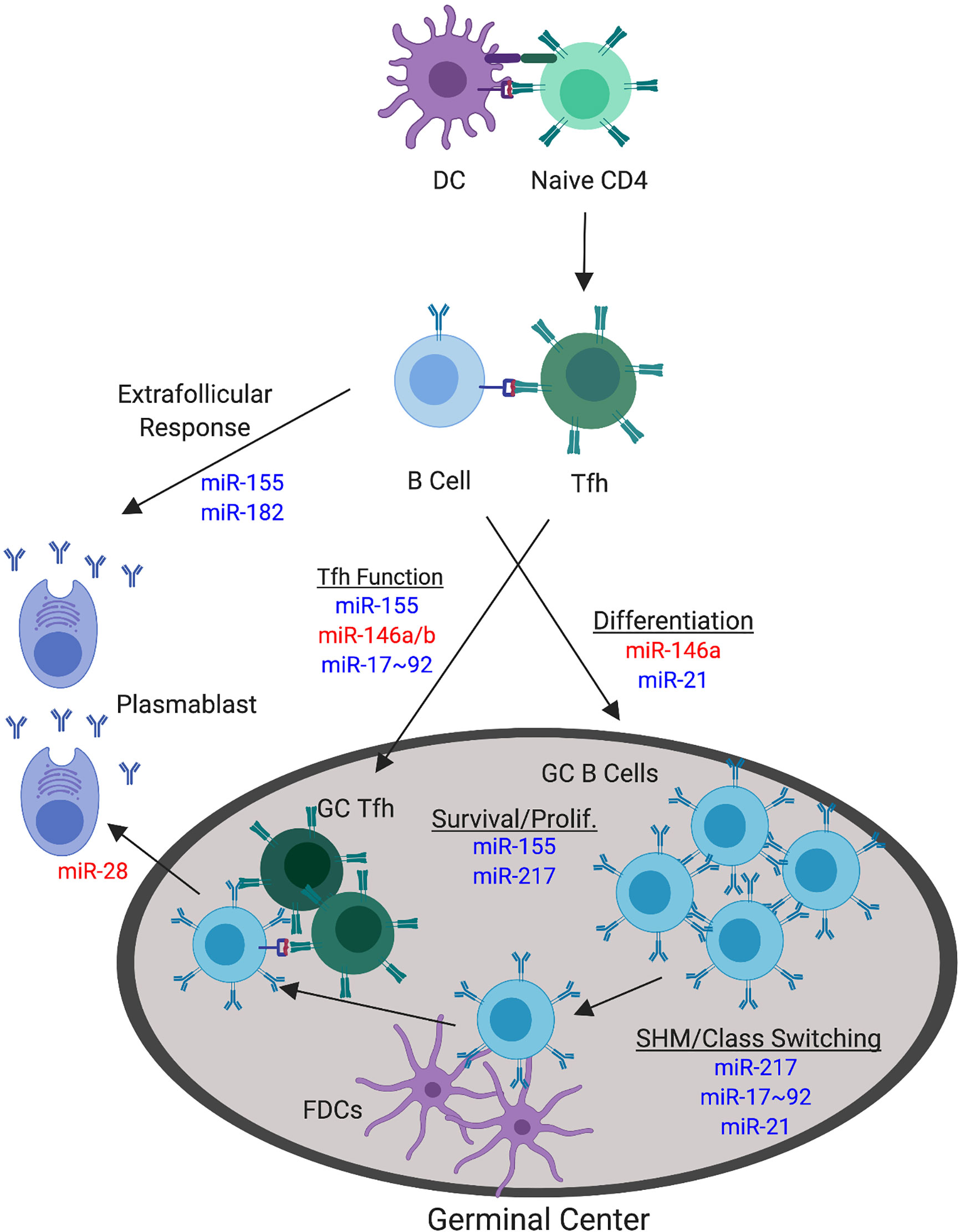
Figure 4 miRNAs with Confirmed Functions in the Non-autoimmune GC Response. The germinal center (GC) response involves a number of processes that can be targeted by miRNA function. The focus herein pertains to direct modulation of GC B cell and Tfh responses during normal, non-autoimmune GC responses. Major processes involved in the GC response are underlined. miRNAs that positively regulate these specific GC processes are depicted in blue. miRNAs that negatively regulate these specific GC processes are depicted in red. miRNAs are indicated at their confirmed or predicted stages of activity.
miR-155 Is a Positive Master Regulator of the GC Response
miR-155 has emerged as arguably the most important and well-studied miRNA during the GC response, acting as a master regulator, with both B and T cell intrinsic functions demonstrated to date. The requirement for miR-155 in GC responses and subsequent antibody production was first described in 2007 through the implementation of both overexpression and knockout systems in mice (117, 118). The absence of miR-155 resulted in reduced GC responses in both the lymph nodes and Peyer’s patches, whereas overexpression enhanced these responses following immunization. A downstream effect on antigen-specific antibody production following immunization in this system was also observed (117). Subsequent studies of miR-155 function separated its B and T cell-intrinsic functions. B cell-intrinsic miR-155 expression was required for optimal GC responses and Ab production, with a primary effect on IgG1, following immunization (105). miR-155 was found to functionally target AID (aicda), as well as SOCS1 expression to promote cell survival via control of p53 (101, 102). The targeting of AID is somewhat counterintuitive as AID is required for somatic hypermutation and class-switching. However, despite AID being a verified target of miR-155, miR-155 deficiency did not overtly affect somatic hypermutation or class-switching processes when measured directly (102, 117). Therefore, the deficiency in class-switched antibody production in the absence of miR-155 is more likely associated with reduced differentiation and the survival of plasmablasts, which is further discussed later in regard to the effects of miR-155 on the extrafollicular B cell response (119). In addition to B cell-intrinsic effects of miR-155, miR-155 deficiency in T cells resulted in significantly blunted Tfh and GC B cell responses, as well as primary and memory antibody responses, demonstrating non-redundant roles for miR-155 in B and T cells that lead to similar phenotypic effects (103, 120). Mechanistically, miR-155 was found to target peli1 in T cells to increase c-Rel expression during Tfh development, resulting phenotypically in the modulation of proliferation and CD40L expression (103). Collectively, miR-155 targets an array of genes and processes in B and T cells during the formation and activity of the GC response and can be considered a master regulatory miRNA during this process.
The miR-146 Family Negatively Regulates the GC Response
In addition to the well-described and multifaceted function of miR-155 in promoting GC responses, both miR-146a and miR-146b have emerged as negative regulators of the GC response. Further, miR-146 modulation also occurs within both B and T cells. Carola Vinuesa’s group first showed that miR-146a loss in T cells through a mixed bone marrow chimeric approach resulted in the spontaneous expansion of Tfh and GC B cells, with some added effect of T cell extrinsic factors (100). As modulation of ICOS-ICOSL signaling through blockade or genetic methods could rescue Tfh and GC B cell accumulation, miR-146a modulation of this signaling axis was determined as a significant form of action utilized to spontaneously control Tfh numbers (100). Another study later clarified that B cell-intrinsic miR-146a deficiency following immunization-induced response does indeed result in increased GC B cell, Tfh, and antibody responses in part due to control of CD40 signaling and control of the GC B cell differentiation process (99). However, this study did not find an effect of miR-146a alone on the modulation of Tfh responses both spontaneously and following immunization when using a Cre-flox system (99), exhibiting contrasting results to previous study. Instead, they found a cooperative T cell-intrinsic role of miR-146a and miR-146b in this process (99). While these results are slightly divergent, it is clear that the miR-146 family is collectively responsible for the negative regulation of the GC, demonstrating that miRNAs can both positively and negatively regulate GC responses.
The miR-17~92 Cluster Modulates Tfh Responses
While miR-146 is critical in negative regulation of Tfh responses, the miR-17~92 cluster has conversely emerged as a critical T cell-intrinsic positive regulator of Tfh, GC, and antibody responses as detailed by multiple studies employing immunization, viral infection, and spontaneous systems in non-autoimmune mice (95, 96, 121, 122). PTEN and Bim (bcl2l11) were the first identified targets of the miR-17~92 cluster in CD4 T cells and loss of one allele each of Pten and Bim (bcl2l11) could produce a similar phenotype to the overexpression of miR-17~92 (96). More convincingly, the loss of one copy of Pten in mice lacking miR-17~92 in T cells could rescue the response, further suggesting that PI3K signaling is critical for miR-17~92 function (121). In addition to the promotion of Tfh function, the activity of miR-17~92 inhibits the expression of factors associated with other CD4 T cell subsets, such as the direct target of rora (95). A separate study tested the B cell-intrinsic requirement for miR-17~92 and did not observe a difference in GC formation, though miR-17~92 had a drastic effect specifically on the production of IgG2c (94). These data suggest that miR-17~92 primarily acts in a T cell-intrinsic manner to modulate GC responses through promoting Tfh differentiation. Conversely, its B cell intrinsic functions appear less pertinent, or may only be involved in specific types of responses.
Other miRNAs Explored in the GC Response
While the study of miR-155, miR-146, and miR-17~92 has been of primary focus in the GC field, additional studies utilizing mouse models are emerging to both support and exclude the role of other miRNAs in this process. In one study, the overexpression of miR-217 in mice resulted in enhanced GC B cell and antibody responses, including enhanced somatic hypermutation events, during primary and secondary responses following immunization (104). Conversely, dampening miR-217 function resulted in reduced GC B cell and antibody responses, indicating that miR-217 promotes these events (104). These responses were associated with the prevention of Bcl6 degradation in GC B cells (104). In a separate study, miR-28 modulation did not affect the magnitude of the GC B cell response, but the employment of a miR-28 sponge in transferred B cells resulted in enhanced memory formation and plasma cell differentiation, indicating a cell-intrinsic negative regulation during GC B cell terminal differentiation into plasmablasts (98). Lastly, we identified a role for miR-21 in driving GC responses to foreign antigen. miR-21 deficient mice exhibited a two-fold reduction in the magnitude of the GC response and reduced class-switched IgG antibody responses after immunization, indicating that miR-21 is required for optimal GC response to foreign antigen (97).
Additionally, some miRNAs are highly expressed in GC B cells or Tfh, but are dispensable for in vivo responses. Among these miRNAs are those contained in the miR-183 cluster (miR-182, miR-183, and miR-92) which are upregulated in GC B cells and Tfh (103, 123, 124), as well as miR-22 which is specifically upregulated in Tfh (103). However, it is worth noting that miRNA requirement may be specific to the type of ongoing B cell and GC response, so these results may not hold true for all conditions. miRNAs that have similar seed sequences may also exhibit some redundancy, meaning that loss of both miRNAs may be required to observe a phenotype in vivo. In summary, the miRNAs that contribute at different stages of the GC response are depicted in Figure 4.
miR-182 and miR-155 Are Implicated in Extrafollicular B Cell Responses
Overall, very little is currently known about the miRNAs involved in any form of extrafollicular B cell response, beyond a role for miR-182 and miR-155 (105, 107). miR-182KO mice immunized with T-dependent antigen (TD-Ag) exhibited intact GC responses, but showed reduced antibody responses at early time points following immunization, indicating an impairment in early extrafollicular B cell responses (107). This reduced early response did not affect the ability to elicit memory responses (107). Similar to miR-182KO mice, B cell intrinsic deficiency of miR-155 resulted in reduced antibody forming cell (AFC) responses at 7d post-immunization with TD-Ag, indicating impaired extrafollicular responses (105). This was shown to be dependent on miR-155 mediated regulation of PU.1 expression, which regulates the formation of plasmablasts (105, 106). Study of miR-155 indicates that some miRNA function in B cells can impact both the GC and extrafollicular B cell responses, likely because there is much crosstalk in the factors that must be activated to support both of these processes. It is likely that additional miRNAs are involved in extrafollicular B cell responses against T-dependent foreign antigens, but further study is required to identify these factors.
Considerations for Future Study of miRNAs in Protective B Cell Responses
While multiple miRNA profiling studies have been performed on B cells undergoing developmental processes and GC responses, these profiling studies are lacking to identify miRNAs which may be involved during the extrafollicular B cell response. In general, many mechanistic questions remain in regard to extrafollicular B cell function and identifying critical miRNAs and their targets can help speed up discovery of additional important pathways that mediate this response. Further, while many studies have illustrated that modulated expression of multiple B cell intrinsic miRNAs is crucial for B cell development in the bone marrow, miRNA expression in other cell types during this process such as stromal cells may also significantly impact B cell development but remain unexplored. Similarly, in addition to the B cell and T cell-intrinsic effects of miRNAs on GC responses, miRNA expression in other cell types involved in the establishment of GC responses, (i.e. follicular dendritic cells (FDCs)), Tfh-priming DCs, and T follicular regulatory cells) may significantly affect their optimal formation. Some studies have started to profile miRNA expression using FDC and FDC-like cell lines (125, 126), however further study is required to determine if this accurately represents what is observed in vivo. Additionally, many miRNAs modulate the cytokine microenvironment, providing a potential indirect mechanism of GC control.
The availability of mouse models and reagents for miRNAs of interest is critical in the continued study of their contribution to this process. Additionally, most of the targets identified thus far were discovered using immunization-based systems, meaning that it is unclear which miRNAs are similarly or differentially modulated in spontaneous GC and B cell responses in the periphery. Some studies have started to address this and will be mentioned in the next section. Notably, some miRNAs such as miR-17~92 and miR-155 appear to be involved throughout multiple stages of B cell development and are critically tied to many important B cell fate decisions. Thus far, other miRNAs have only been described at one stage of B cell response. Establishing a complete picture of miRNA responses at all stages of the B cell response is an important consideration in regard to any therapeutic pursuits for autoimmune or other B cell dependent diseases.
miRNA Regulation of B Cell Responses in SLE
Due to the critical involvement of miRNAs at many stages of the B cell response, it is not surprising that dysregulated miRNA expression can lead to the loss of B cell tolerance at multiple stages of B cell development and effector function. As such, global miRNA expression in B cells, studied with a B cell specific knockout of Dicer, was shown to skew the B cell repertoire and result in high titers of serum autoreactive antibodies and kidney pathology (127). This study illustrated that miRNAs can collectively impact B cell tolerance. We will further discuss which individual miRNAs have been characterized in this process and what is known about the mechanisms of their activity (Figure 5 and Table 3).
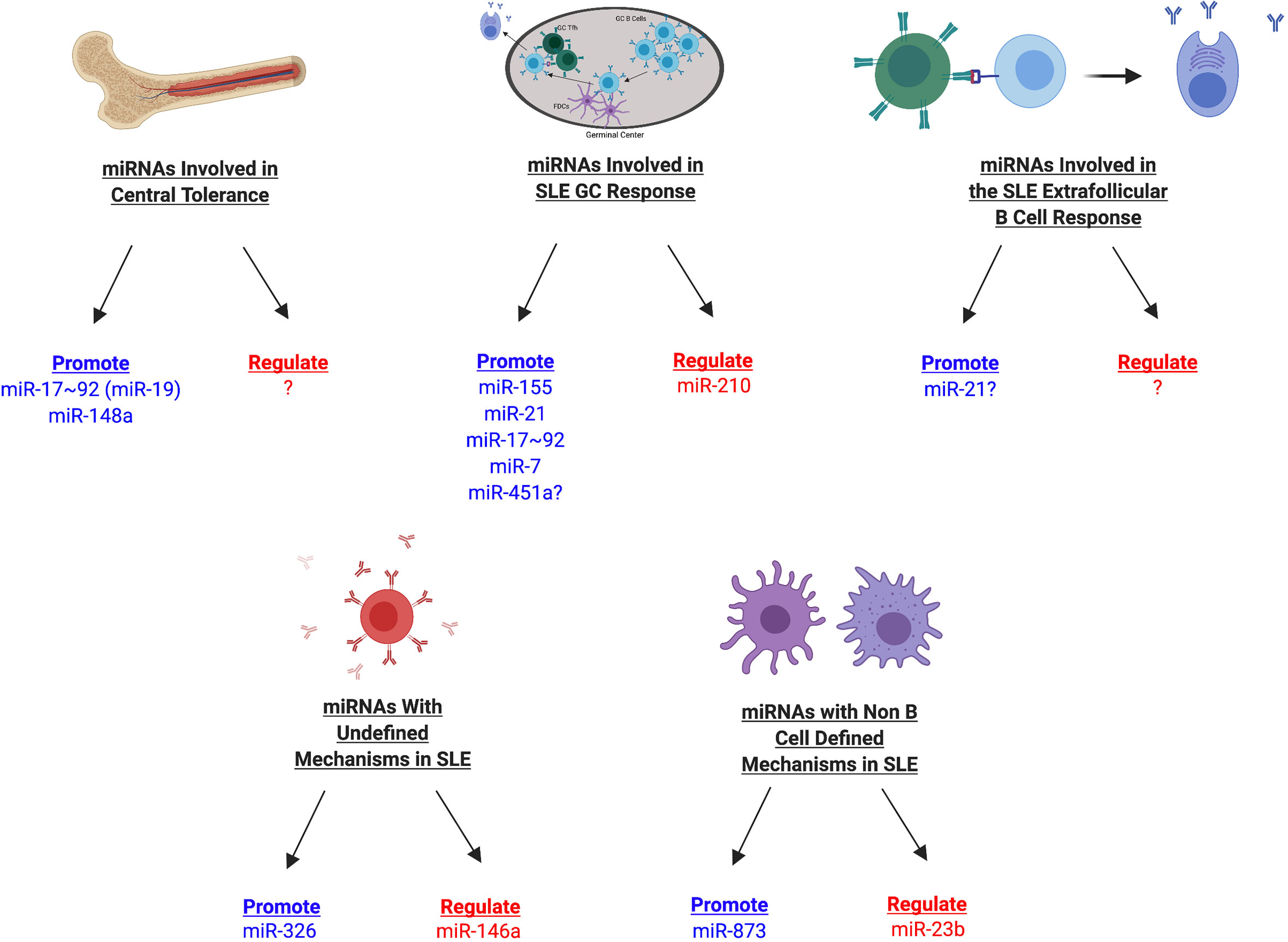
Figure 5 miRNAs Involved in SLE Development in Mice. miRNAs involved in SLE development can be divided into multiple categories depending on their association with different stages of the B cell response (central tolerance, germinal center, extrafollicular response, other mechanisms), in addition to whether they promote or regulate disease manifestations. Those miRNAs that promote disease are shown in blue whereas those that regulate disease are shown in red. miRNAs with currently undefined mechanisms in SLE may later be identified during a specific stage of the B cell response or may have B cell independent mechanisms during SLE.
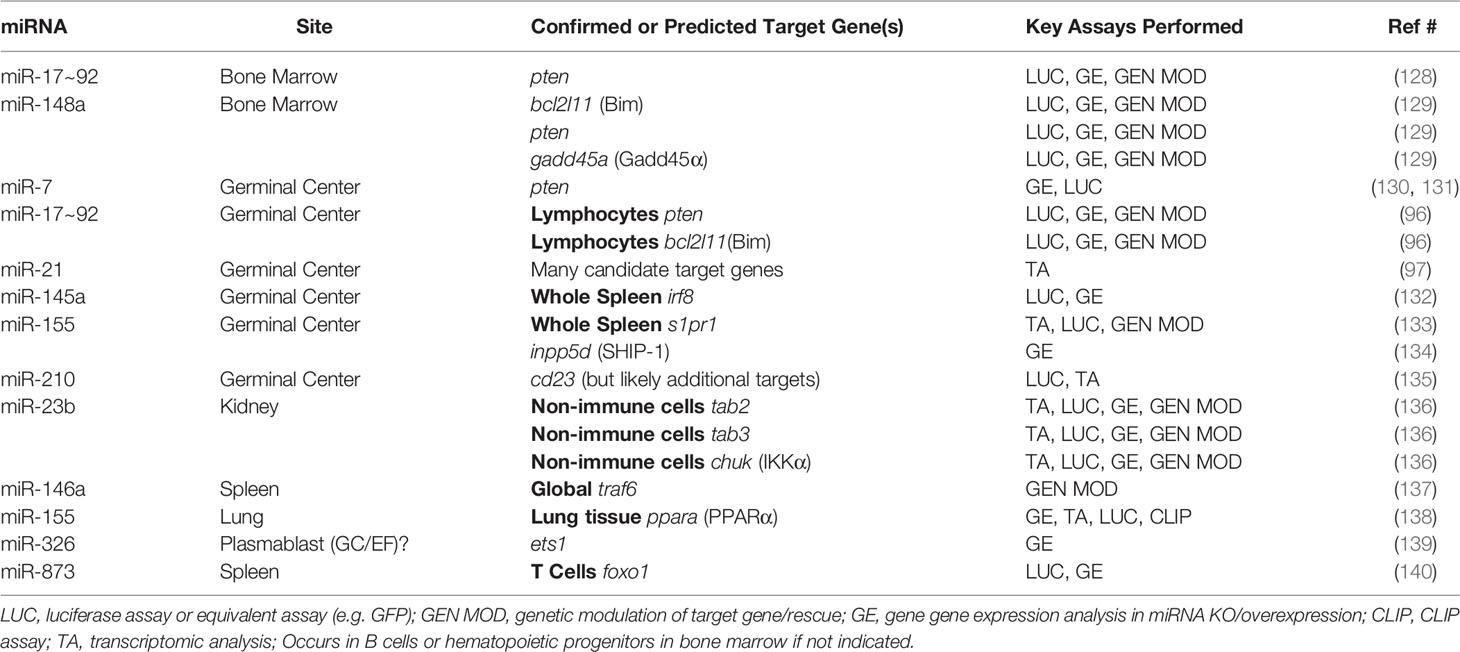
Table 3 miRNAs and Direct Target Genes in SLE Responses.
miR-17~92 and miR-148a Break Central B Cell Tolerance
Several miRNAs have been identified as key mediators of central B cell tolerance. The implementation of the IgMb-macroself mouse model has recently been adopted as a mechanism to study central tolerance (141). IgMb-macroself mice express an IgMb superantigen and editing of the BCR in the bone marrow is unable to remedy this reactivity, resulting in deletion of nearly all B cells in the bone marrow and consequent loss of B cells in the periphery. Therefore, this model allows for the assessment of B cell tolerance by screening for B cells that have escaped this method of tolerance and entered the periphery. In one study utilizing this model, IgMb-macroself mice reconstituted with bone marrow from CD19-Cre; miR-17~92 mice revealed that overexpression of this miRNA cluster allows developing B cells to escape central tolerance in the bone marrow (128). Most of this effect was attributed specifically to the miR-19 subfamily suppressing Pten activity (128). Further, normal levels of miR-17~92 were shown to regulate the degree of receptor editing occurring at this stage of tolerance, indicating that some level of miR-17~92 expression is important to mediate normal processes that occur during B cell development (128).
A separate study performed a functional screen of 113 miRNAs separated into 4 pools in the regulation of central B cell tolerance, also employing the IgMb-macroself mouse model. From this screen, they identified 7 miRNAs that may be involved in escape of central tolerance. Using this model to study the effect of specific miRNAs, they determined that miR-26a, miR-26b, miR-342, miR-423, and miR-182 have modest effects on central tolerance, whereas miR-148a was identified as a key regulator of central tolerance, with increased levels promoting escape of tolerance (129). Mechanistically, miR-148a was found to regulate immature B cell apoptosis through targeting Bim, PTEN, and Gadd45α, which were also verified to regulate central tolerance in this mouse model (129). Consequently, MRL/lpr mice overexpressing miR-148a developed accelerated autoimmune manifestations and higher autoantibody titers (129).
While these studies have made significant progress toward our understanding of miRNAs that regulate central B cell tolerance, some questions still remain. Gonzalez-Martin et al. screened a subset of 113 miRNAs in hematopoietic progenitor stem cells (HPSCs). Downstream determination of miRNAs involved in loss of central B cell tolerance was dependent on their enhanced expression in B cells that seeded the spleen in their system. However, expression of these miRNAs in other cell types may indirectly impact B cell escape of central tolerance. Additionally, other miRNAs that were not screened in this study may have a significant contribution to this process. Once B cells are in the spleen, they are also regulated at the transitional stage. Little is currently known about the involvement of miRNAs in transitional B cell tolerance, but miRNAs have been identified in regulating BCR signaling induced growth and apoptosis, indicating that these may be prime candidates for the study of tolerance at the transitional stage of B cell development (142).
miRNAs in SLE-Associated GC Responses
Based on the master regulatory function of miR-155 in the GC responses, it is not surprising that miR-155 has a great importance in the onset of SLE in two different mouse models. Faslpr mice with a deficiency in miR-155 have reduced spleen size, proteinuria, and kidney pathology with age (133, 134). This was associated with reduced serum autoantibody titers and GC responses in this model (133, 134), illustrating that miR-155 is also important in promoting the formation of GCs in autoimmune-prone mice. In one of these studies, miR-155 was shown to target expression of SHIP-1 (inpp5d), a previously characterized miR-155 target gene (143), following BCR activation to promote B cell proliferation (134). In the other study, S1PR1 was validated as a miR-155 target. Antagonizing S1PR1 expression lead to increased Tfh responses (133), revealing another mechanism of miR-155 activity. In the pristane-induced lupus model, knockout or antagonism of miR-155 dampened diffuse alveolar hemorrhage, reduced kidney pathology, and reduced autoantibody titers, demonstrating common effects among multiple lupus models (138, 144).
Another miRNA which has been studied using multiple SLE models is miR-21. Antagonism of miR-21 was initially shown to dampen splenomegaly and autoantibody titers in the Sle1.2.3 lupus model although no detailed mechanism of action was suggested in this seminal study (145). This reduction in splenomegaly and autoantibody production was also observed in the bm12 adoptive transfer chronic graft versus host disease (cGVHD) model, which exhibits SLE-like manifestations (146). We recently addressed more concerning the mechanism of miR-21 activity and showed that miR-21 has context dependent effects in different SLE models, which is likely related to the level of miR-21 activity (97). In a TLR7 induced model where Sle1b mice are treated with imiquimod, miR-21 modulated plasma cell formation and autoreactive B cell selection, which was independent of the magnitude of the GC response and was associated with the modulation of a number of miR-21 target genes in B cells. In addition to differences in the B cell response, myeloid cell infiltration and proinflammatory cytokine production was blunted in the absence of miR-21, suggesting that miR-21 can also control the cytokine environment (97). In contrast, in a spontaneous TLR7 overexpression model, Sle1b.Yaa, miR-21 was required for increased GC responses and autoimmune B cell responses (97). These data indicate that miR-21 has multifaceted effects at several different stages of the B cell response depending on the SLE model. Additional study is required to determine the cell-intrinsic contributions to these distinct responses.
While the impact of miR-17~92 on autoimmunity has not yet been tested in a standard mouse model of SLE, preliminary - study indicates that there will likely be broad effects among several SLE models. Overexpression of miR-17~92 in lymphocytes results in multiorgan autoimmune manifestations, including lymphoproliferation, kidney pathology, and the development of autoantibody titers (96), hallmarks of SLE. These mice exhibited increased GC responses (96), but the study of central tolerance suggests that miR-17~92 can affect tolerance at multiple stages of B cell development and effector function (128).
In addition to these miRNAs, multiple other studies have illustrated roles for additional miRNAs in GC responses and autoimmunity. miR-7 was studied in the MRL/lpr mouse model and was found to enhance GC responses, drive proinflammatory cytokine secretion, and impact autoantibody production and kidney pathology (130). Treatment of MRL/lpr mice with antagomir-7 for 5 weeks was able to reduce all of these responses significantly and normalized PTEN expression in B cells (130, 131). Another study addressed miR-451a deficiency in the Faslpr model and found a partial reduction in Tfh responses and a significant reduction in IL-21 production that correlated with the loss of autoantibody production and kidney pathology (132). IRF8 was verified as a gene target (132). While IRF8 is important in the GC response (147), it is unclear to what extent IRF8 modulation produces the observed effects and that what extent other mechanisms may be at play. Lastly, another study hinted that miR-210 may be involved in regulating the loss of tolerance through the GC, as the loss of miR-210 resulted in spontaneous development of autoimmunity and enhanced spontaneous GC B cell responses with age (135). The exact mechanisms of miR-451a and miR-210 in the GC responses in autoimmune-prone mice remain to be fully elucidated.
miRNAs That Modulate B Cell Tolerance via Other or Undefined Mechanisms
Another well studied miRNA in autoimmunity is miR-146a, which was found to prevent the development of autoimmunity, similar to its regulatory effects on the GC responses to TD-antigen immunization or spontaneous responses in non-autoimmune mouse models. This was determined by two studies employing opposite approaches. First, in the more classical BXSB model, administration of miR-146a expressing virus like particles (VLPs) reduced autoantibody production (148). Second, even in the absence of susceptibility loci, miR-146aKO mice develop a chronic inflammatory disease and autoantibody production with age (149). While it is evident that non-autoimmune spontaneous-GC responses are affected in miR-146aKO mice through a separate study previously discussed (100), analysis in autoimmune-prone mice did not directly address this mechanism and thus further study of GC responses in autoimmune-prone mice is required to determine if alteration of the GC response may be a causative factor under these conditions. However, it is difficult to directly attribute autoimmunity solely to lymphocyte dysregulation in the absence of miR-146, as these mice also exhibit a profound amount of myeloproliferation when miR-146a is globally deficient, which can be significantly reduced by attenuation of the miR-146a target gene TRAF6 (137, 149). It is likely that a combination of these factors may be involved in contributing to the overall phenotype observed, which requires further study. Additional study of human SLE genetics has revealed that the rs2431697 SNP is critically tied to miR-146a expression levels and the T/T variant at this locus correlates with disease activity, renal involvement, and autoantibody production (150). This is linked to its alterations in cell type specific enhancer activity that can control miR-146a expression by promoter interactions (151). These studies indicate that miR-146a activity has relevance in human SLE and that genetics can control miR-146a expression in SLE patients.
miR-326 is also intriguing due to its modulation of B cell responses. Enhancing miR-326 expression via lentiviral vector in MRL/lpr mice resulted in enhanced autoantibody production and immune complex deposition in the kidneys, which was associated with increased plasmablast responses (139). Ets-1 expression was modulated in B cells, with Ets-1 previously identified as a miR-326 target gene (152). However, the source of plasmablast generation (GC versus extrafollicular pathway) was not characterized and reverse experiments implementing lentivirus expressing miR-326 sponge showed only minimal effects on these processes, implying the need for further inquiry (139).
Two additional miRNAs have detailed roles in the development of autoimmunity but are associated with other divergent mechanisms focused on T cell and cytokine responses. miR-873 inhibition could modestly reduce autoantibody responses and proteinuria in MRL/lpr mice, but was associated with Foxo1 targeting and Th17 responses (140). MRL/lpr mice infected with a viral vector expressing a miR-23b sponge or virus engineered to overexpress miR-23b demonstrated that miR-23b has a suppressive effect on the development of kidney pathology (136). This study attributed miR-23b mediated protection from autoimmunity to its negative regulation of proinflammatory cytokine production in resident non-immune cells (136).
Considerations for Further Study
Altogether, miRNAs crucially regulate autoimmune responses, in both negative and positive manners (Figure 5). While these miRNAs have already been established in this context, much study remains to identify additional miRNAs involved in autoimmunity, as well as to further determine the mechanisms involved in these processes. The list of miRNAs that modulate autoimmunity through regulation of the B cell responses is likely to grow as the tools and reagents to study these factors expand. For example, recently miR-152-3p was identified as an important miRNA in isolated human SLE B cells, which controls their activation level and production of BAFF (153). Modeling this miRNA in mouse models may reveal more information about the importance of this miRNA in driving disease progression.
Interestingly, none of the miRNAs listed above are located on the X chromosome (154). However, it would be interesting to determine if aberrant miRNA expression caused by incomplete X chromosome inactivation may contribute to the pathogenesis of SLE. Additionally, the study of miR-146a indicates that genetic susceptibility to SLE may involve mutations that lead to aberrant regulation of miRNA expression that predisposes one to SLE development (150, 151). Additional studies addressing this potential phenomenon in regard to the expression of other miRNAs will be informative.
Of further consideration for future study is the level of miRNA expression that is required to promote or restrain disease, as many of these miRNAs are expressed during normal B cell processes, but at different levels. Many of these models focus on extreme alterations in expression, either by knocking out expression entirely or significantly overexpressing the miRNA by viral vector. Approaches that fine-tune miRNA expression, such as using heterozygous mice or miRNA sponges, should be considered in future studies.
While the experimental focus on miRNA has largely pertained to their classical mechanisms of function, other mechanisms of miRNA activity are emerging that require consideration and may alter the manner in which we think about targeting them. There is evidence that miRNAs can also act as ligands by binding to and inducing pattern recognition receptor (PRR) signaling, predominately by TLR7 and TLR8, which recognize ssRNA ligands (155–157). The idea of miRNAs being able to stimulate TLRs is intriguing as this opens the door for miRNAs produced by one cell to control the genetic profile and function of surrounding cells. In fact, it has already been shown that T cells can transfer exosomes and their miRNA cargo to APCs through the immunological synapse to mediate fusion and release into the recipient cell (158). Additionally, DCs can shuttle miRNAs via exosomes to the cytosol of other DCs to mediate recipient gene expression (159). Interestingly, exosomes derived from SLE patients can stimulate the production of IFNα from pDCs in vitro through miRNA stimulation of TLRs, indicating that exosomal delivery of miRNAs can also alter the cytokine profile in disease (160). Realistically, this means that the overall phenotype observed for specific miRNAs may actually be a balance between miRNAs produced intrinsically and those internalized from surrounding cells, which may result in TLR stimulation as well as gene regulation in the recipient cell (Figure 6).
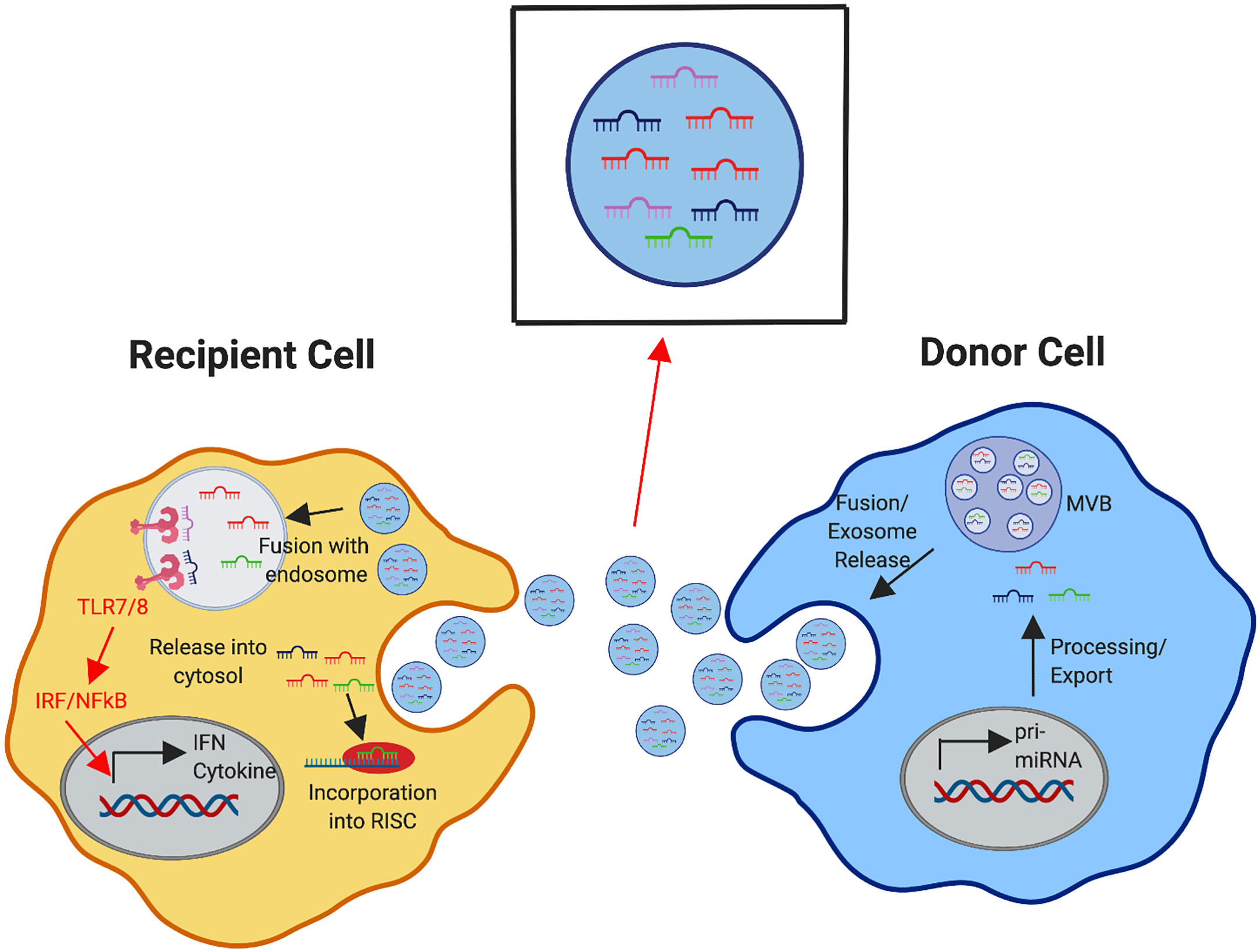
Figure 6 Exosomal Delivery of miRNA to Modulate Recipient Cell Response. Donor immune cells produce and process miRNAs (as depicted in detail in Figure 1). The formation of multivesicular bodies (MVBs) with intraluminal vesicles containing mature miRNAs in the donor cell leads to fusion of the MVBs with the plasma membrane, releasing exosomes containing these miRNAs. Recipient immune cells can receive exosomal cargo into the cytosol by direct fusion of the exosome with the plasma membrane or exosomes can be internalized and delivered to the endosomal compartment. Within the cytosol, these received miRNAs may compete with the internally generated miRNAs for incorporation in the RNA-induced silencing complex (RISC) to mediate gene expression in the recipient cell. miRNAs that are directed into the endosomal compartment may bind to TLR7/8 to initiate downstream signaling cascades, triggering the induction of cytokines, interferons, and other interferon stimulated genes.
An initial study recently applied this concept to the T and B cell interactions required for B cell class-switching and GC formation. This study found that during in vitro culture, T cells can transfer multiple miRNAs, including miR-155, via exosomes to B cells upon formation of immune synapses. Blocking exosome release from T cells to B cells in vivo, and thus reducing miRNA transfer from T cells to B cells, resulted in slightly reduced B cell class-switching and GC formation. This indicates that miRNA transfer contributes to these B cell responses in vivo, with greater contribution from intrinsic miRNA responses (161). It is unclear if this mechanism may be amplified during autoimmune responses and which cell types may participate in this mechanism in a context dependent manner.
Lastly, while miRNAs target individual genes in the cell, they have been implicated in targeting the chromatin remodeling machinery to confer epigenetic and transcriptional changes in the genome that have broader effects on cellular function. This includes targeting factors involved in methylation, acetylation, and the SWI/SNF complex (162–168). A few studies have started to address miRNA targeting of DNA methyltransferases (DMNTs) in CD4 T cells in SLE (19, 169, 170). However, the study of miRNAs in the modulation of the chromatin remodeling machinery has not been pursued extensively. This concept is intriguing though, because SLE has been associated with epigenetic modulation, particularly hypomethylation (171). Altogether, miRNA function may not be limited to a single mode of action, but rather a combination of effects that alter the functionality of a cell and surrounding cells. This has broad implications for the mechanistic control of both non-autoimmune and autoimmune responses.
Considerations for miRNAs as Therapeutic Targets
miRNAs are an attractive therapeutic option for SLE and other diseases because of the ability to dampen their expression rather than completely ablate it, allowing for the ability to fine-tune gene expression to a desired level. In the context of SLE, this is an interesting concept because current therapeutics globally target B cells or employ other approaches that result in broad immunosuppression, thus ablating both anti-pathogen and autoimmune responses (172). In turn, this leaves patients on these therapies susceptible to infection. If miRNA expression can be fine-tuned to a level that allows for adequate anti-pathogen response, while significantly dampening autoimmune manifestations, miRNA targeting would be effective. Alternatively, finding miRNAs that promote autoimmunity but are not overtly involved in anti-pathogen response provides another attractive avenue of attaining specificity.
Methods for Targeting miRNAs and miRNAs in Clinical Trials for Other Diseases
In regard to the specific methods of miRNA targeting, many groups have already worked with miRNA targeting in cell culture and in animal models through the introduction of antagomirs, which are 23 nucleotide antisense 2’-O’-methyl-modified oligonucleotide sequences bound to cholesterol (173) (Table 1). The cholesterol linkage allows for cellular entry while the antisense sequence promotes pairing to and neutralization of the target miRNA. Another form of anti-miRNA oligonucleotide is the locked nucleic acid (LNA) anti-miR agent. LNA anti-miRs are another form of targeted oligoribonucleotide that are stabilized due to locked nucleic acids (LNAs) (174). More recently LNA anti-miRs have been designed that are as short as 8 mer sequences (tiny LNAs) that specifically target the 5’ seed region of the miRNA (175). Of note, while mature miRNA can be targeted, the pri-miRNA and pre-miRNA stages of miRNA processing can be targeted by miRNA therapies as well, preventing generation of the mature miRNA (176). While anti-miRNA oligonucleotides are intended to dampen miRNA function, some miRNAs (i.e. miR-146a) have a regulatory role in disease onset and progression. Bearing this in mind, a separate approach is to heighten the expression of these miRNAs to restore tolerance, such as through the administration of a miRNA mimic (or agomir), which is a chemically modified double stranded oligonucleotide sequence designed to mimic the miRNA of interest.
While simple injection based approaches are limited to systemic effects, targeted delivery may be achieved through viral vectors utilizing different expression promoters (177). Also advantageously, viral vector based delivery methods can promote long-term modulation of the response through forming episomes or integrating into the genome (177). Anti-miRNA sequences expressed under the control of a viral vector are referred to as miRNA sponges (178). Alternatively, viral vectors can be engineered to express miRNAs. Lastly, nanoparticles are an attractive cell-specific targeting option that continue to be studied and optimized for a variety of purposes, including as small RNA delivery vehicles (179).
Despite miRNA therapeutics being a relatively newer pursuit, some miRNA-centric therapeutics are being tested in clinical trials for the treatment of diseases. The first miRNA-centric therapeutic to advance to Phase II clinical trials was Miraversin, which dampens miR-122 expression during hepatitis C infection to prevent viral replication (176, 180). As such, many companies have emerged with the goal of designing miRNA-based therapeutics for a variety of purposes (181). miRagen has already directed a miR-155 targeting oligonucleotide (Cobomarsen) into a Phase II clinical trial in an effort to treat blood cancer (182, 183). Additionally, therapeutics targeting other miRNAs, such as miR-21, miR-29, and miR-92, have completed or are currently active for early stage trials in the treatment of fibrotic diseases, cardiovascular disease, kidney disease, and solid organ cancers (181, 184). While the implementation of miRNA-centric therapeutics is steadily increasing, to our knowledge there are no trials to date that have focused on testing miRNA targeted therapeutics for autoimmune diseases. However, the technology exists for potential implementation.
Considerations for Implementing miRNA Therapeutics in SLE
While miRNA targeting is an interesting therapeutic option, there are some points of consideration pertaining to off-target effects and the potential stimulatory ability of miRNAs, the necessity for long-term intervention in autoimmune diseases, and miRNA redundancy. Other points of consideration will likely emerge as more groups begin testing miRNA targeting in animal models. While antagomirs/anti-miRs are, by nature, targeted therapeutics due to their base-pairing properties, design must be careful and specific to avoid off target RNA binding. Despite this concern, study suggests that off-target effects on mRNAs are minimal when using LNAs in animal models (175). In addition to the generation of sequence specific off target effects, there is evidence that miRNAs can also act as ligands by binding to and inducing PRR signaling, predominately by TLR7 and TLR8, which recognize ssRNA ligands (155–157). If miRNAs can stimulate some PRRs, we must exercise caution in therapeutically enhancing miRNA doses that could trigger downstream proinflammatory cytokine production. Conversely, some antagomirs have been shown to block TLR7 and 8 signaling in a sequence specific manner in addition to their miRNA binding effects, possibly introducing other unintended effects (185). Careful modeling, design, and testing are required to overcome these barriers to specific therapeutic design.
Another point in regard to therapeutic development is how often antagomirs would need to be introduced into the system, as miRNAs are constantly transcribed from the genome in response to immune stimuli and SLE is chronic disease. Removal of the miRNA targeting therapy will likely result in a relapse of symptoms as the original expression levels are restored following therapy discontinuation. As discussed above, the use of viral vectors that mediate long-term expression is an option but requires further study. In addition to the frequency of miRNA dosage, it is unclear which miRNAs can be targeted therapeutically or only prophylactically in regard to SLE and other autoimmune diseases. Ultimately, this likely depends on the specific miRNA of interest and whether that miRNA is dysregulated in response to genetic susceptibility gene possession or triggered by ongoing immune responses during the course of SLE progression. To determine when miRNA therapeutics would be most effective, miRNA dampening or loss of miRNAs at specific stages of disease progression in mouse models can begin to clarify therapeutic approaches and effectiveness through different stages of disease development. Lastly, while those miRNAs that produce strong effects in mouse models are of obvious consideration for targeting, miRNA redundancy does exist. Targeting multiple miRNAs, or miRNA families with conserved target sequences, may produce additive effects compared to single miRNA targeting. After weighing both the advantages and possible unintended consequences in the development of miRNA therapeutics, targeting miRNAs can be deemed as a promising option, but much proof of concept and testing remains to be addressed.
Conclusions
The studies reviewed herein demonstrate the essential roles of miRNAs in B cell responses and the development of autoimmune disease. Not only do miRNAs promote these responses, but some miRNAs also possess critical negative regulatory functions. While the studies to date have begun to elucidate the role of these factors, much work remains to fully characterize their impact during autoimmunity, as well as the capacity to effectively target them to dampen disease. Additionally, many miRNAs have yet to be studied using in vivo mouse systems but with increasing access to mouse models, the list of miRNAs involved in the processes discussed herein will likely expand significantly. Extensive miRNA expression profiling in cell types and specific stages of the B cell response will aid in identifying additional miRNAs worth studying in mouse models in the future. While simply identifying the miRNAs and their target genes that contribute to disease is important, further attention to the level of miRNA expression that modulates the observed effects and the stage of disease at which the miRNA of interest is involved has important therapeutic ramifications that must be addressed more heavily in future studies. Lastly, traditional miRNA mediated regulation has been of focus, but non-traditional regulation by miRNAs is an interesting avenue of research that deserves significant attention and may alter how we approach targeting miRNA function. Altogether, the further study and modeling of miRNAs in relevant animal systems will inform future therapeutic design and identify those miRNAs that may provide the most promising targets for potential miRNA-centric therapeutic development to treat SLE.
Author Contributions
SS and ZR wrote the article. SS designed figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Institutes of Health R21AI128111 to ZR.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
All figures were designed with Biorender software (Biorender.com).
References
1. LeBien TW, Tedder TF. B Lymphocytes: How They Develop and Function. Blood (2008) 112:1570–80. doi: 10.1182/blood-2008-02-078071
2. Cyster JG, Allen CDC. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell (2019) 177:524–40. doi: 10.1016/j.cell.2019.03.016
3. Shlomchik MJ. Sites and Stages of Autoreactive B Cell Activation and Regulation. Immunity (2008) 28:18–28. doi: 10.1016/j.immuni.2007.12.004
4. Gulati G, Brunner HI. Environmental Triggers in Systemic Lupus Erythematosus. Semin Arthritis Rheum (2018) 47:710–7. doi: 10.1016/j.semarthrit.2017.10.001
5. Balada E, Ordi-Ros J, Vilardell-Tarrés M. Molecular Mechanisms Mediated by Human Endogenous Retroviruses (HERVs) in Autoimmunity. Rev Med Virol (2009) 19:273–86. doi: 10.1002/rmv.622
6. Papp K, Végh P, Hóbor R, Szittner Z, Vokó Z, Podani J, et al. Immune Complex Signatures of Patients With Active and Inactive SLE Revealed by Multiplex Protein Binding Analysis on Antigen Microarrays. PloS One (2012) 7:e44824. doi: 10.1371/journal.pone.0044824
7. Budde P, Zucht HD, Vordenbäumen S, Goehler H, Fischer-Betz R, Gamer M, et al. Multiparametric Detection of Autoantibodies in Systemic Lupus Erythematosus. Lupus (2016) 25:812–22. doi: 10.1177/0961203316641770
8. Tsokos GC. Systemic Lupus Erythematosus. N Engl J Med (2011) 365:2110–21. doi: 10.1056/NEJMra1100359
9. Gottschalk TA, Tsantikos E, Hibbs ML. Pathogenic Inflammation and Its Therapeutic Targeting in Systemic Lupus Erythematosus. Front Immunol (2015) 6:550. doi: 10.3389/fimmu.2015.00550
10. Flores-Mendoza G, Sansón SP, Rodríguez-Castro S, Crispín JC, Rosetti F. Mechanisms of Tissue Injury in Lupus Nephritis. Trends Mol Med (2018) 24:364–78. doi: 10.1016/j.molmed.2018.02.003
11. Zeller CB, Appenzeller S. Cardiovascular Disease in Systemic Lupus Erythematosus: The Role of Traditional and Lupus Related Risk Factors. Curr Cardiol Rev (2008) 4:116–22. doi: 10.2174/157340308784245775
12. Dubey AK, Handu SS, Dubey S, Sharma P, Sharma KK, Ahmed QM. Belimumab: First Targeted Biological Treatment for Systemic Lupus Erythematosus. J Pharmacol Pharmacother (2011) 2:317–9. doi: 10.4103/0976-500X.85930
13. Pasoto SG, Ribeiro AC, Bonfa E. Update on Infections and Vaccinations in Systemic Lupus Erythematosus and Sjögren’s Syndrome. Curr Opin Rheumatol (2014) 26:528–37. doi: 10.1097/BOR.0000000000000084
14. Leblanc-Trudeau C, Masetto A, Bocti C. Progressive Multifocal Leukoencephalopathy Associated With Belimumab in a Patient With Systemic Lupus Erythematosus. J Rheumatol (2015) 42:551–2. doi: 10.3899/jrheum.140577
15. Rupaimoole R, Slack FJ. MicroRNA Therapeutics: Towards a New Era for the Management of Cancer and Other Diseases. Nat Rev Drug Discov (2017) 16:203–22. doi: 10.1038/nrd.2016.246
16. Jin F, Hu H, Xu M, Zhan S, Wang Y, Zhang H, et al. Serum Microrna Profiles Serve as Novel Biomarkers for Autoimmune Diseases. Front Immunol (2018) 9:2381. doi: 10.3389/fimmu.2018.02381
17. Condrat CE, Thompson DC, Barbu MG, Bugnar OL, Boboc A, Cretoiu D, et al. miRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells (2020) 9(2):276. doi: 10.3390/cells9020276
18. Carlsen AL, Schetter AJ, Nielsen CT, Lood C, Knudsen S, Voss A, et al. Circulating microRNA Expression Profiles Associated With Systemic Lupus Erythematosus. Arthritis Rheum (2013) 65:1324–34. doi: 10.1002/art.37890
19. Pan W, Zhu S, Yuan M, Cui H, Wang L, Luo X, et al. MicroRNA-21 and microRNA-148a Contribute to DNA Hypomethylation in Lupus CD4+ T Cells by Directly and Indirectly Targeting DNA Methyltransferase 1. J Immunol (2010) 184:6773–81. doi: 10.4049/jimmunol.0904060
20. Stagakis E, Bertsias G, Verginis P, Nakou M, Hatziapostolou M, Kritikos H, et al. Identification of Novel microRNA Signatures Linked to Human Lupus Disease Activity and Pathogenesis: miR-21 Regulates Aberrant T Cell Responses Through Regulation of PDCD4 Expression. Ann Rheum Dis (2011) 70:1496–506. doi: 10.1136/ard.2010.139857
21. Guo G, Wang H, Shi X, Ye L, Wu K, Lin K, et al. NovelmiRNA-25 Inhibits AMPD2 in Peripheral Blood Mononuclear Cells of Patients With Systemic Lupus Erythematosus and Represents a Promising Novel Biomarker. J Transl Med (2018) 16:370. doi: 10.1186/s12967-018-1739-5
22. Zeng L, Wu JL, Liu LM, Jiang JQ, Wu HJ, Zhao M, et al. Serum miRNA-371b-5p and miRNA-5100 Act as Biomarkers for Systemic Lupus Erythematosus. Clin Immunol (2018) 196:103–9. doi: 10.1016/j.clim.2018.10.004
23. Duroux-Richard I, Cuenca J, Ponsolles C, Piñeiro AB, Gonzalez F, Roubert C, et al. MicroRNA Profiling of B Cell Subsets From Systemic Lupus Erythematosus Patients Reveals Promising Novel Biomarkers. Int J Mol Sci (2015) 16:16953–65. doi: 10.3390/ijms160816953
24. Tangtanatakul P, Klinchanhom S, Sodsai P, Sutichet T, Promjeen C, Avihingsanon Y, et al. Down-Regulation of let-7a and miR-21 in Urine Exosomes From Lupus Nephritis Patients During Disease Flare. Asian Pac J Allergy Immunol (2018) 37(4):189–97. doi: 10.12932/AP-130318-0280.
25. Wang G, Tam LS, Li EK, Kwan BC, Chow KM, Luk CC, et al. Serum and Urinary Free microRNA Level in Patients With Systemic Lupus Erythematosus. Lupus (2011) 20:493–500. doi: 10.1177/0961203310389841
26. Wang Z, Heid B, Dai R, Ahmed SA. Similar Dysregulation of Lupus-Associated miRNAs in Peripheral Blood Mononuclear Cells and Splenic Lymphocytes in MRL/lpr Mice. Lupus Sci Med (2018) 5:e000290. doi: 10.1136/lupus-2018-000290
27. Dai R, Zhang Y, Khan D, Heid B, Caudell D, Crasta O, et al. Identification of a Common Lupus Disease-Associated microRNA Expression Pattern in Three Different Murine Models of Lupus. PloS One (2010) 5:e14302. doi: 10.1371/journal.pone.0014302
28. Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of Mammalian microRNA Host Genes and Transcription Units. Genome Res (2004) 14:1902–10. doi: 10.1101/gr.2722704
29. Godnic I, Zorc M, Jevsinek Skok D, Calin GA, Horvat S, Dovc P, et al. Genome-Wide and Species-Wide in Silico Screening for Intragenic MicroRNAs in Human, Mouse and Chicken. PloS One (2013) 8:e65165. doi: 10.1371/journal.pone.0065165
30. Ramalingam P, Palanichamy JK, Singh A, Das P, Bhagat M, Kassab MA, et al. Biogenesis of Intronic miRNAs Located in Clusters by Independent Transcription and Alternative Splicing. RNA (2014) 20:76–87. doi: 10.1261/rna.041814.113
31. Saini HK, Griffiths-Jones S, Enright AJ. Genomic Analysis of Human microRNA Transcripts. Proc Natl Acad Sci USA (2007) 104:17719–24. doi: 10.1073/pnas.0703890104
32. Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA Genes are Transcribed by RNA Polymerase II. EMBO J (2004) 23:4051–60. doi: 10.1038/sj.emboj.7600385
33. Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 Complex in Primary microRNA Processing. Genes Dev (2004) 18:3016–27. doi: 10.1101/gad.1262504
34. Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, et al. Molecular Basis for the Recognition of Primary microRNAs by the Drosha-DGCR8 Complex. Cell (2006) 125:887–901. doi: 10.1016/j.cell.2006.03.043
35. Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, et al. The Microprocessor Complex Mediates the Genesis of Micrornas. Nature (2004) 432:235–40. doi: 10.1038/nature03120
36. Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, et al. The Nuclear RNase Iii Drosha Initiates microRNA Processing. Nature (2003) 425:415–9. doi: 10.1038/nature01957
37. Lund E, Güttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear Export of microRNA Precursors. Science (2004) 303:95–8. doi: 10.1126/science.1090599
38. Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH. Dicer Functions in RNA Interference and in Synthesis of Small RNA Involved in Developmental Timing in C. Elegans. Genes Dev (2001) 15:2654–9. doi: 10.1101/gad.927801
39. Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA Maturation: Stepwise Processing and Subcellular Localization. EMBO J (2002) 21:4663–70. doi: 10.1093/emboj/cdf476
40. Hammond SM, Boettcher S, Caudy AA, Kobayashi R, Hannon GJ. Argonaute2, a Link Between Genetic and Biochemical Analyses of RNAi. Science (2001) 293:1146–50. doi: 10.1126/science.1064023
41. Elkayam E, Kuhn CD, Tocilj A, Haase AD, Greene EM, Hannon GJ, et al. The Structure of Human Argonaute-2 in Complex With miR-20a. Cell (2012) 150:100–10. doi: 10.1016/j.cell.2012.05.017
42. Mourelatos Z, Dostie J, Paushkin S, Sharma A, Charroux B, Abel L, et al. miRNPs: A Novel Class of Ribonucleoproteins Containing Numerous Micrornas. Genes Dev (2002) 16:720–8. doi: 10.1101/gad.974702
43. Lewis BP, Burge CB, Bartel DP. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates That Thousands of Human Genes are microRNA Targets. Cell (2005) 120:15–20. doi: 10.1016/j.cell.2004.12.035
44. Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, et al. Transcriptome-Wide Identification of RNA-binding Protein and microRNA Target Sites by PAR-CLIP. Cell (2010) 141:129–41. doi: 10.1016/j.cell.2010.03.009
45. Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP Decodes microRNA-mRNA Interaction Maps. Nature (2009) 460:479–86. doi: 10.1038/nature08170
46. Lee I, Ajay SS, Yook JI, Kim HS, Hong SH, Kim NH, et al. New Class of microRNA Targets Containing Simultaneous 5’-UTR and 3’-UTR Interaction Sites. Genome Res (2009) 19:1175–83. doi: 10.1101/gr.089367.108
47. Okamura K, Phillips MD, Tyler DM, Duan H, Chou YT, Lai EC. The Regulatory Activity of microRNA* Species has Substantial Influence on microRNA and 3’ UTR Evolution. Nat Struct Mol Biol (2008) 15:354–63. doi: 10.1038/nsmb.1409
48. Guo L, Lu Z. The Fate of miRNA* Strand Through Evolutionary Analysis: Implication for Degradation as Merely Carrier Strand or Potential Regulatory Molecule? PloS One (2010) 5:e11387. doi: 10.1371/journal.pone.0011387
49. Lai EC. Micro RNAs are Complementary to 3’ UTR Sequence Motifs That Mediate Negative Post-Transcriptional Regulation. Nat Genet (2002) 30:363–4. doi: 10.1038/ng865
50. Jin HY, Xiao C. MicroRNA Mechanisms of Action: What Have We Learned From Mice? Front Genet (2015) 6:328. doi: 10.3389/fgene.2015.00328
51. Eulalio A, Huntzinger E, Nishihara T, Rehwinkel J, Fauser M, Izaurralde E. Deadenylation is a Widespread Effect of miRNA Regulation. RNA (2009) 15:21–32. doi: 10.1261/rna.1399509
52. Wakiyama M, Takimoto K, Ohara O, Yokoyama S. Let-7 microRNA-mediated mRNA Deadenylation and Translational Repression in a Mammalian Cell-Free System. Genes Dev (2007) 21:1857–62. doi: 10.1101/gad.1566707
53. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs Predominantly Act to Decrease Target mRNA Levels. Nature (2010) 466:835–40. doi: 10.1038/nature09267
54. Shin C, Nam JW, Farh KK, Chiang HR, Shkumatava A, Bartel DP. Expanding the microRNA Targeting Code: Functional Sites With Centered Pairing. Mol Cell (2010) 38:789–802. doi: 10.1016/j.molcel.2010.06.005
55. Olena AF, Patton JG. Genomic Organization of Micrornas. J Cell Physiol (2010) 222:540–5. doi: 10.1002/jcp.21993
56. Wheeler BM, Heimberg AM, Moy VN, Sperling EA, Holstein TW, Heber S, et al. The Deep Evolution of Metazoan Micrornas. Evol Dev (2009) 11:50–68. doi: 10.1111/j.1525-142X.2008.00302.x
57. Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, et al. Mammalian microRNAs: Experimental Evaluation of Novel and Previously Annotated Genes. Genes Dev (2010) 24:992–1009. doi: 10.1101/gad.1884710
58. Denzler R, McGeary SE, Title AC, Agarwal V, Bartel DP, Stoffel M. Impact of MicroRNA Levels, Target-Site Complementarity, and Cooperativity on Competing Endogenous RNA-Regulated Gene Expression. Mol Cell (2016) 64:565–79. doi: 10.1016/j.molcel.2016.09.027
59. Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, et al. A Mammalian microRNA Expression Atlas Based on Small RNA Library Sequencing. Cell (2007) 129:1401–14. doi: 10.1016/j.cell.2007.04.040
60. Mehta A, Baltimore D. MicroRNAs as Regulatory Elements in Immune System Logic. Nat Rev Immunol (2016) 16:279–94. doi: 10.1038/nri.2016.40
61. Hsin JP, Lu Y, Loeb GB, Leslie CS, Rudensky AY. The Effect of Cellular Context on miR-155-mediated Gene Regulation in Four Major Immune Cell Types. Nat Immunol (2018) 19:1137–45. doi: 10.1038/s41590-018-0208-x
62. Ramírez J, Lukin K, Hagman J. From Hematopoietic Progenitors to B Cells: Mechanisms of Lineage Restriction and Commitment. Curr Opin Immunol (2010) 22:177–84. doi: 10.1016/j.coi.2010.02.003
63. Bassing CH, Swat W, Alt FW. The Mechanism and Regulation of Chromosomal V(D)J Recombination. Cell (2002) 109 Suppl:S45–55. doi: 10.1016/S0092-8674(02)00675-X
64. Pelanda R, Torres RM. Central B-cell Tolerance: Where Selection Begins. Cold Spring Harb Perspect Biol (2012) 4:a007146. doi: 10.1101/cshperspect.a007146
65. Chung JB, Silverman M, Monroe JG. Transitional B Cells: Step by Step Towards Immune Competence. Trends Immunol (2003) 24:343–9. doi: 10.1016/S1471-4906(03)00119-4
66. Petro JB, Gerstein RM, Lowe J, Carter RS, Shinners N, Khan WN. Transitional Type 1 and 2 B Lymphocyte Subsets are Differentially Responsive to Antigen Receptor Signaling. J Biol Chem (2002) 277:48009–19. doi: 10.1074/jbc.M200305200
67. Merrell KT, Benschop RJ, Gauld SB, Aviszus K, Decote-Ricardo D, Wysocki LJ, et al. Identification of Anergic B Cells Within a Wild-Type Repertoire. Immunity (2006) 25:953–62. doi: 10.1016/j.immuni.2006.10.017
68. Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, et al. Defective B Cell Tolerance Checkpoints in Systemic Lupus Erythematosus. J Exp Med (2005) 201:703–11. doi: 10.1084/jem.20042251
69. Kanta H, Mohan C. Three Checkpoints in Lupus Development: Central Tolerance in Adaptive Immunity, Peripheral Amplification by Innate Immunity and End-Organ Inflammation. Genes Immun (2009) 10:390–6. doi: 10.1038/gene.2009.6
70. Dieudonné Y, Gies V, Guffroy A, Keime C, Bird AK, Liesveld J, et al. Transitional B Cells in Quiescent SLE: An Early Checkpoint Imprinted by IFN. J Autoimmun (2019) 102:150–8. doi: 10.1016/j.jaut.2019.05.002
71. Wang T, Marken J, Chen J, Tran VB, Li QZ, Li M, et al. High TLR7 Expression Drives the Expansion of CD19(+)CD24(hi)CD38(hi) Transitional B Cells and Autoantibody Production in SLE Patients. Front Immunol (2019) 10:1243. doi: 10.3389/fimmu.2019.01243
72. Giltiay NV, Chappell CP, Sun X, Kolhatkar N, Teal TH, Wiedeman AE, et al. Overexpression of TLR7 Promotes Cell-Intrinsic Expansion and Autoantibody Production by Transitional T1 B Cells. J Exp Med (2013) 210:2773–89. doi: 10.1084/jem.20122798
73. Victora GD, Nussenzweig MC. Germinal Centers. Annu Rev Immunol (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
74. Domeier PP, Schell SL, Rahman ZS. Spontaneous Germinal Centers and Autoimmunity. Autoimmunity (2017) 50:4–18. doi: 10.1080/08916934.2017.1280671
75. Wong EB, Khan TN, Mohan C, Rahman ZS. The Lupus-Prone NZM2410/NZW Strain-Derived Sle1b Sublocus Alters the Germinal Center Checkpoint in Female Mice in a B Cell-Intrinsic Manner. J Immunol (2012) 189:5667–81. doi: 10.4049/jimmunol.1201661
76. Shlomchik MJ, Luo W, Weisel F. Linking Signaling and Selection in the Germinal Center. Immunol Rev (2019) 288:49–63. doi: 10.1111/imr.12744
77. Jenks SA, Cashman KS, Woodruff MC, Lee FE, Sanz I. Extrafollicular Responses in Humans and SLE. Immunol Rev (2019) 288:136–48. doi: 10.1111/imr.12741
78. William J, Euler C, Christensen S, Shlomchik MJ. Evolution of Autoantibody Responses Via Somatic Hypermutation Outside of Germinal Centers. Science (2002) 297:2066–70. doi: 10.1126/science.1073924
79. Luzina IG, Atamas SP, Storrer CE, daSilva LC, Kelsoe G, Papadimitriou JC, et al. Spontaneous Formation of Germinal Centers in Autoimmune Mice. J Leukoc Biol (2001) 70:578–84 doi: 10.1189/jlb.70.4.578
80. Mandik-Nayak L, Seo SJ, Sokol C, Potts KM, Bui A, Erikson J. MRL-Lpr/Lpr Mice Exhibit a Defect in Maintaining Developmental Arrest and Follicular Exclusion of Anti-Double-Stranded DNA B Cells. J Exp Med (1999) 189:1799–814. doi: 10.1084/jem.189.11.1799
81. Morel L, Blenman KR, Croker BP, Wakeland EK. The Major Murine Systemic Lupus Erythematosus Susceptibility Locus, Sle1, is a Cluster of Functionally Related Genes. Proc Natl Acad Sci USA (2001) 98:1787–92. doi: 10.1073/pnas.031336098
82. Jenks SA, Cashman KS, Zumaquero E, Marigorta UM, Patel AV, Wang X, et al. Distinct Effector B Cells Induced by Unregulated Toll-Like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity (2018) 49:725–39.e726. doi: 10.1016/j.immuni.2018.08.015
83. Lindner SE, Lohmüller M, Kotkamp B, Schuler F, Knust Z, Villunger A, et al. The miR-15 Family Reinforces the Transition From Proliferation to Differentiation in Pre-B Cells. EMBO Rep (2017) 18:1604–17. doi: 10.15252/embr.201643735
84. Benhamou D, Labi V, Getahun A, Benchetrit E, Dowery R, Rajewsky K, et al. The C-Myc/miR17-92/PTEN Axis Tunes PI3K Activity to Control Expression of Recombination Activating Genes in Early B Cell Development. Front Immunol (2018) 9:2715. doi: 10.3389/fimmu.2018.02715
85. Kurkewich JL, Hansen J, Klopfenstein N, Zhang H, Wood C, Boucher A, et al. The miR-23a~27a~24-2 microRNA Cluster Buffers Transcription and Signaling Pathways During Hematopoiesis. PloS Genet (2017) 13:e1006887. doi: 10.1371/journal.pgen.1006887
86. Rao DS, O’Connell RM, Chaudhuri AA, Garcia-Flores Y, Geiger TL, Baltimore D. MicroRNA-34a Perturbs B Lymphocyte Development by Repressing the Forkhead Box Transcription Factor Foxp1. Immunity (2010) 33:48–59. doi: 10.1016/j.immuni.2010.06.013
87. Li G, So AY, Sookram R, Wong S, Wang JK, Ouyang Y, et al. Epigenetic Silencing of miR-125b is Required for Normal B-cell Development. Blood (2018) 131:1920–30. doi: 10.1182/blood-2018-01-824540
88. Okuyama K, Ikawa T, Gentner B, Hozumi K, Harnprasopwat R, Lu J, et al. MicroRNA-126-mediated Control of Cell Fate in B-cell Myeloid Progenitors as a Potential Alternative to Transcriptional Factors. Proc Natl Acad Sci USA (2013) 110:13410–5. doi: 10.1073/pnas.1220710110
89. Yang Y, Xu J, Chen H, Fei X, Tang Y, Yan Y, et al. MiR-128-2 Inhibits Common Lymphoid Progenitors From Developing Into Progenitor B Cells. Oncotarget (2016) 7:17520–31. doi: 10.18632/oncotarget.8161
90. Mehta A, Mann M, Zhao JL, Marinov GK, Majumdar D, Garcia-Flores Y, et al. The microRNA-212/132 Cluster Regulates B Cell Development by Targeting Sox4. J Exp Med (2015) 212:1679–92. doi: 10.1084/jem.20150489
91. Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF. miR-150, a microRNA Expressed in Mature B and T Cells, Blocks Early B Cell Development When Expressed Prematurely. Proc Natl Acad Sci USA (2007) 104:7080–5. doi: 10.1073/pnas.0702409104
92. Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs Modulate Hematopoietic Lineage Differentiation. Science (2004) 303:83–6. doi: 10.1126/science.1091903
93. Petkau G, Kawano Y, Wolf I, Knoll M, Melchers F. MiR221 Promotes Precursor B-cell Retention in the Bone Marrow by Amplifying the PI3K-signaling Pathway in Mice. Eur J Immunol (2018) 48:975–89. doi: 10.1002/eji.201747354
94. Xu S, Ou X, Huo J, Lim K, Huang Y, Chee S, et al. Mir-17-92 Regulates Bone Marrow Homing of Plasma Cells and Production of Immunoglobulin G2c. Nat Commun (2015) 6:6764. doi: 10.1038/ncomms7764
95. Baumjohann D, Kageyama R, Clingan JM, Morar MM, Patel S, de Kouchkovsky D, et al. The microRNA Cluster miR-17∼92 Promotes TFH Cell Differentiation and Represses Subset-Inappropriate Gene Expression. Nat Immunol (2013) 14:840–8. doi: 10.1038/ni.2642
96. Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, et al. Lymphoproliferative Disease and Autoimmunity in Mice With Increased miR-17-92 Expression in Lymphocytes. Nat Immunol (2008) 9:405–14. doi: 10.1038/ni1575
97. Schell SL, Bricker KN, Fike AJ, Chodisetti SB, Domeier PP, Choi NM, et al. Context-Dependent miR-21 Regulation of TLR7-mediated Autoimmune and Foreign Antigen Driven Antibody-Forming Cell and Germinal Center Responses. bioRxiv (2021). doi: 10.1101/2021.03.12.435182
98. Bartolomé-Izquierdo N, de Yébenes VG, Álvarez-Prado AF, Mur SM, Lopez Del Olmo JA, Roa S, et al. miR-28 Regulates the Germinal Center Reaction and Blocks Tumor Growth in Preclinical Models of non-Hodgkin Lymphoma. Blood (2017) 129:2408–19. doi: 10.1182/blood-2016-08-731166
99. Cho S, Lee HM, Yu IS, Choi YS, Huang HY, Hashemifar SS, et al. Differential Cell-Intrinsic Regulations of Germinal Center B and T Cells by miR-146a and miR-146b. Nat Commun (2018) 9:2757. doi: 10.1038/s41467-018-05196-3
100. Pratama A, Srivastava M, Williams NJ, Papa I, Lee SK, Dinh XT, et al. MicroRNA-146a Regulates ICOS-ICOSL Signalling to Limit Accumulation of T Follicular Helper Cells and Germinal Centres. Nat Commun (2015) 6:6436. doi: 10.1038/ncomms7436
101. Bouamar H, Jiang D, Wang L, Lin AP, Ortega M, Aguiar RC. MicroRNA 155 Control of p53 Activity is Context Dependent and Mediated by Aicda and Socs1. Mol Cell Biol (2015) 35:1329–40. doi: 10.1128/MCB.01446-14
102. Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, et al. MicroRNA-155 is a Negative Regulator of Activation-Induced Cytidine Deaminase. Immunity (2008) 28:621–9. doi: 10.1016/j.immuni.2008.03.015
103. Liu WH, Kang SG, Huang Z, Wu CJ, Jin HY, Maine CJ, et al. A miR-155-Peli1-c-Rel Pathway Controls the Generation and Function of T Follicular Helper Cells. J Exp Med (2016) 213:1901–19. doi: 10.1084/jem.20160204
104. de Yébenes VG, Bartolomé-Izquierdo N, Nogales-Cadenas R, Pérez-Durán P, Mur SM, Martínez N, et al. miR-217 is an Oncogene That Enhances the Germinal Center Reaction. Blood (2014) 124:229–39. doi: 10.1182/blood-2013-12-543611
105. Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, et al. microRNA-155 Regulates the Generation of Immunoglobulin Class-Switched Plasma Cells. Immunity (2007) 27:847–59. doi: 10.1016/j.immuni.2007.10.009
106. Lu D, Nakagawa R, Lazzaro S, Staudacher P, Abreu-Goodger C, Henley T, et al. The miR-155-PU.1 Axis Acts on Pax5 to Enable Efficient Terminal B Cell Differentiation. J Exp Med (2014) 211:2183–98. doi: 10.1084/jem.20140338
107. Li YF, Ou X, Xu S, Jin ZB, Iwai N, Lam KP. Loss of miR-182 Affects B-cell Extrafollicular Antibody Response. Immunology (2016) 148:140–9. doi: 10.1111/imm.12592
108. Jensen K, Brusletto BS, Aass HC, Olstad OK, Kierulf P, Gautvik KM. Transcriptional Profiling of mRNAs and microRNAs in Human Bone Marrow Precursor B Cells Identifies Subset- and Age-Specific Variations. PloS One (2013) 8:e70721. doi: 10.1371/journal.pone.0070721
109. Brandl A, Daum P, Brenner S, Schulz SR, Yap DY, Bösl MR, et al. The Microprocessor Component, DGCR8, is Essential for Early B-cell Development in Mice. Eur J Immunol (2016) 46:2710–8. doi: 10.1002/eji.201646348
110. Kong KY, Owens KS, Rogers JH, Mullenix J, Velu CS, Grimes HL, et al. MIR-23A microRNA Cluster Inhibits B-cell Development. Exp Hematol (2010) 38:629–40.e621. doi: 10.1016/j.exphem.2010.04.004
111. Knoll M, Simmons S, Bouquet C, Grün JR, Melchers F. miR-221 Redirects Precursor B Cells to the BM and Regulates Their Residence. Eur J Immunol (2013) 43:2497–506. doi: 10.1002/eji.201343367
112. Mazan-Mamczarz K, Gartenhaus RB. Role of microRNA Deregulation in the Pathogenesis of Diffuse Large B-cell Lymphoma (DLBCL). Leuk Res (2013) 37:1420–8. doi: 10.1016/j.leukres.2013.08.020
113. Xu S, Guo K, Zeng Q, Huo J, Lam KP. The RNase III Enzyme Dicer is Essential for Germinal Center B-cell Formation. Blood (2012) 119:767–76. doi: 10.1182/blood-2011-05-355412
114. Tan LP, Wang M, Robertus JL, Schakel RN, Gibcus JH, Diepstra A, et al. miRNA Profiling of B-cell Subsets: Specific miRNA Profile for Germinal Center B Cells With Variation Between Centroblasts and Centrocytes. Lab Invest (2009) 89:708–16. doi: 10.1038/labinvest.2009.26
115. Basso K, Sumazin P, Morozov P, Schneider C, Maute RL, Kitagawa Y, et al. Identification of the Human Mature B Cell Mirnome. Immunity (2009) 30:744–52. doi: 10.1016/j.immuni.2009.03.017
116. Ripamonti A, Provasi E, Lorenzo M, De Simone M, Ranzani V, Vangelisti S, et al. Repression of miR-31 by BCL6 Stabilizes the Helper Function of Human Follicular Helper T Cells. Proc Natl Acad Sci USA (2017) 114:12797–802. doi: 10.1073/pnas.1705364114
117. Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, et al. Regulation of the Germinal Center Response by microRNA-155. Science (2007) 316:604–8. doi: 10.1126/science.1141229
118. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of Bic/microRNA-155 for Normal Immune Function. Science (2007) 316:608–11. doi: 10.1126/science.1139253
119. Arbore G, Henley T, Biggins L, Andrews S, Vigorito E, Turner M, et al. MicroRNA-155 is Essential for the Optimal Proliferation and Survival of Plasmablast B Cells. Life Sci Alliance (2019) 2(3):e201800244. doi: 10.26508/lsa.201800244
120. Hu R, Kagele DA, Huffaker TB, Runtsch MC, Alexander M, Liu J, et al. miR-155 Promotes T Follicular Helper Cell Accumulation During Chronic, Low-Grade Inflammation. Immunity (2014) 41:605–19. doi: 10.1016/j.immuni.2014.09.015
121. Kang SG, Liu WH, Lu P, Jin HY, Lim HW, Shepherd J, et al. MicroRNAs of the miR-17∼92 Family are Critical Regulators of T(FH) Differentiation. Nat Immunol (2013) 14:849–57. doi: 10.1038/ni.2648
122. Wu T, Wieland A, Lee J, Hale JS, Han JH, Xu X, et al. Cutting Edge: miR-17-92 Is Required for Both CD4 Th1 and T Follicular Helper Cell Responses During Viral Infection. J Immunol (2015) 195:2515–9. doi: 10.4049/jimmunol.1500317
123. Pucella JN, Yen WF, Kim MV, van der Veeken J, Luo CT, Socci ND, et al. miR-182 is Largely Dispensable for Adaptive Immunity: Lack of Correlation Between Expression and Function. J Immunol (2015) 194:2635–42. doi: 10.4049/jimmunol.1402261
124. Pucella JN, Cols M, Yen WF, Xu S, Chaudhuri J. The B Cell Activation-Induced miR-183 Cluster Plays a Minimal Role in Canonical Primary Humoral Responses. J Immunol (2019) 202:1383–96. doi: 10.4049/jimmunol.1800071
125. Lin J, Lwin T, Zhao JJ, Tam W, Choi YS, Moscinski LC, et al. Follicular Dendritic Cell-Induced microRNA-mediated Upregulation of PRDM1 and Downregulation of BCL-6 in non-Hodgkin’s B-cell Lymphomas. Leukemia (2011) 25:145–52. doi: 10.1038/leu.2010.230
126. Aungier SR, Ohmori H, Clinton M, Mabbott NA. MicroRNA-100-5p Indirectly Modulates the Expression of Il6, Ptgs1/2 and Tlr4 mRNA in the Mouse Follicular Dendritic Cell-Like Cell Line, FL-Y. Immunology (2015) 144:34–44. doi: 10.1111/imm.12342
127. Belver L, de Yébenes VG, Ramiro AR. MicroRNAs Prevent the Generation of Autoreactive Antibodies. Immunity (2010) 33:713–22. doi: 10.1016/j.immuni.2010.11.010
128. Lai M, Gonzalez-Martin A, Cooper AB, Oda H, Jin HY, Shepherd J, et al. Regulation of B-cell Development and Tolerance by Different Members of the miR-17∼92 Family Micrornas. Nat Commun (2016) 7:12207. doi: 10.1038/ncomms12207
129. Gonzalez-Martin A, Adams BD, Lai M, Shepherd J, Salvador-Bernaldez M, Salvador JM, et al. The microRNA miR-148a Functions as a Critical Regulator of B Cell Tolerance and Autoimmunity. Nat Immunol (2016) 17:433–40. doi: 10.1038/ni.3385
130. Wang M, Chen H, Qiu J, Yang HX, Zhang CY, Fei YY, et al. Antagonizing miR-7 Suppresses B Cell Hyperresponsiveness and Inhibits Lupus Development. J Autoimmun (2020) 109:102440. doi: 10.1016/j.jaut.2020.102440
131. Wu XN, Ye YX, Niu JW, Li Y, Li X, You X, et al. Defective PTEN Regulation Contributes to B Cell Hyperresponsiveness in Systemic Lupus Erythematosus. Sci Transl Med (2014) 6:246ra299. doi: 10.1126/scitranslmed.3009131
132. Cheng J, Wu R, Long L, Su J, Liu J, Wu XD, et al. miRNA-451a Targets IFN Regulatory Factor 8 for the Progression of Systemic Lupus Erythematosus. Inflammation (2017) 40:676–87. doi: 10.1007/s10753-017-0514-8
133. Xin Q, Li J, Dang J, Bian X, Shan S, Yuan J, et al. miR-155 Deficiency Ameliorates Autoimmune Inflammation of Systemic Lupus Erythematosus by Targeting S1pr1 in Faslpr/lpr Mice. J Immunol (2015) 194:5437–45. doi: 10.4049/jimmunol.1403028
134. Thai TH, Patterson HC, Pham DH, Kis-Toth K, Kaminski DA, Tsokos GC. Deletion of microRNA-155 Reduces Autoantibody Responses and Alleviates Lupus-Like Disease in the Fas(lpr) Mouse. Proc Natl Acad Sci USA (2013) 110:20194–9. doi: 10.1073/pnas.1317632110
135. Mok Y, Schwierzeck V, Thomas DC, Vigorito E, Rayner TF, Jarvis LB, et al. MiR-210 is Induced by Oct-2, Regulates B Cells, and Inhibits Autoantibody Production. J Immunol (2013) 191:3037–48. doi: 10.4049/jimmunol.1301289
136. Zhu S, Pan W, Song X, Liu Y, Shao X, Tang Y, et al. The microRNA miR-23b Suppresses IL-17-associated Autoimmune Inflammation by Targeting TAB2, TAB3 and IKK-α. Nat Med (2012) 18:1077–86. doi: 10.1038/nm.2815
137. Magilnick N, Reyes EY, Wang WL, Vonderfecht SL, Gohda J, Inoue JI, et al. miR-146a-Traf6 Regulatory Axis Controls Autoimmunity and Myelopoiesis, but is Dispensable for Hematopoietic Stem Cell Homeostasis and Tumor Suppression. Proc Natl Acad Sci USA (2017) 114:E7140–9. doi: 10.1073/pnas.1706833114
138. Zhou S, Wang Y, Meng Y, Xiao C, Liu Z, Brohawn P, et al. In Vivo Therapeutic Success of MicroRNA-155 Antagomir in a Mouse Model of Lupus Alveolar Hemorrhage. Arthritis Rheumatol (2016) 68:953–64. doi: 10.1002/art.39485
139. Xia Y, Tao JH, Fang X, Xiang N, Dai XJ, Jin L, et al. MicroRNA-326 Upregulates B Cell Activity and Autoantibody Production in Lupus Disease of MRL/lpr Mice. Mol Ther Nucleic Acids (2018) 11:284–91. doi: 10.1016/j.omtn.2018.02.010
140. Liu L, Liu Y, Yuan M, Xu L, Sun H. Elevated Expression of microRNA-873 Facilitates Th17 Differentiation by Targeting Forkhead Box O1 (Foxo1) in the Pathogenesis of Systemic Lupus Erythematosus. Biochem Biophys Res Commun (2017) 492:453–60. doi: 10.1016/j.bbrc.2017.08.075
141. Duong BH, Ota T, Aoki-Ota M, Cooper AB, Ait-Azzouzene D, Vela JL, et al. Negative Selection by IgM Superantigen Defines a B Cell Central Tolerance Compartment and Reveals Mutations Allowing Escape. J Immunol (2011) 187:5596–605. doi: 10.4049/jimmunol.1102479
142. Kluiver JL, Chen CZ. MicroRNAs Regulate B-cell Receptor Signaling-Induced Apoptosis. Genes Immun (2012) 13:239–44. doi: 10.1038/gene.2012.1
143. Costinean S, Sandhu SK, Pedersen IM, Tili E, Trotta R, Perrotti D, et al. Src Homology 2 Domain-Containing inositol-5-phosphatase and CCAAT Enhancer-Binding Protein Beta are Targeted by miR-155 in B Cells of Emicro-MiR-155 Transgenic Mice. Blood (2009) 114:1374–82. doi: 10.1182/blood-2009-05-220814
144. Leiss H, Salzberger W, Jacobs B, Gessl I, Kozakowski N, Blüml S, et al. MicroRNA 155-Deficiency Leads to Decreased Autoantibody Levels and Reduced Severity of Nephritis and Pneumonitis in Pristane-Induced Lupus. PloS One (2017) 12:e0181015. doi: 10.1371/journal.pone.0181015
145. Garchow BG, Bartulos Encinas O, Leung YT, Tsao PY, Eisenberg RA, Caricchio R, et al. Silencing of microRNA-21 In Vivo Ameliorates Autoimmune Splenomegaly in Lupus Mice. EMBO Mol Med (2011) 3:605–15. doi: 10.1002/emmm.201100171
146. Garchow B, Kiriakidou M. MicroRNA-21 Deficiency Protects From Lupus-Like Autoimmunity in the Chronic Graft-Versus-Host Disease Model of Systemic Lupus Erythematosus. Clin Immunol (2016) 162:100–6. doi: 10.1016/j.clim.2015.11.010
147. Lee CH, Melchers M, Wang H, Torrey TA, Slota R, Qi CF, et al. Regulation of the Germinal Center Gene Program by Interferon (IFN) Regulatory Factor 8/IFN Consensus Sequence-Binding Protein. J Exp Med (2006) 203:63–72. doi: 10.1084/jem.20051450
148. Pan Y, Jia T, Zhang Y, Zhang K, Zhang R, Li J, et al. MS2 VLP-based Delivery of microRNA-146a Inhibits Autoantibody Production in Lupus-Prone Mice. Int J Nanomedicine (2012) 7:5957–67. doi: 10.2147/IJN.S37990
149. Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, et al. miR-146a is a Significant Brake on Autoimmunity, Myeloproliferation, and Cancer in Mice. J Exp Med (2011) 208:1189–201. doi: 10.1084/jem.20101823
150. Fouda ME, Nour El Din DM, Mahgoub MY, Elashkar AE, Abdel Halim WA. Genetic Variants of microRNA-146a Gene: An Indicator of Systemic Lupus Erythematosus Susceptibility, Lupus Nephritis, and Disease Activity. Mol Biol Rep (2020) 47:7459–66. doi: 10.1007/s11033-020-05802-y
151. Hou G, Harley ITW, Lu X, Zhou T, Xu N, Yao C, et al. SLE non-Coding Genetic Risk Variant Determines the Epigenetic Dysfunction of an Immune Cell Specific Enhancer That Controls Disease-Critical microRNA Expression. Nat Commun (2021) 12:135. doi: 10.1101/2020.05.13.092932
152. Du C, Liu C, Kang J, Zhao G, Ye Z, Huang S, et al. MicroRNA miR-326 Regulates TH-17 Differentiation and is Associated With the Pathogenesis of Multiple Sclerosis. Nat Immunol (2009) 10:1252–9. doi: 10.1038/ni.1798
153. Luo S, Ding S, Liao J, Zhang P, Liu Y, Zhao M, et al. Excessive miR-152-3p Results in Increased BAFF Expression in SLE B-Cells by Inhibiting the KLF5 Expression. Front Immunol (2019) 10:1127. doi: 10.3389/fimmu.2019.01127
154. Pinheiro I, Dejager L, Libert C. X-Chromosome-Located microRNAs in Immunity: Might They Explain Male/Female Differences? The X Chromosome-Genomic Context may Affect X-located miRNAs and Downstream Signaling, Thereby Contributing to the Enhanced Immune Response of Females. Bioessays (2011) 33:791–802. doi: 10.1002/bies.201100047
155. Zhang ZJ, Guo JS, Li SS, Wu XB, Cao DL, Jiang BC, et al. TLR8 and its Endogenous Ligand miR-21 Contribute to Neuropathic Pain in Murine DRG. J Exp Med (2018) 215:3019–37. doi: 10.1084/jem.20180800
156. Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, et al. MicroRNAs Bind to Toll-like Receptors to Induce Prometastatic Inflammatory Response. Proc Natl Acad Sci USA (2012) 109:E2110–2116. doi: 10.1073/pnas.1209414109
157. Wang Y, Liang H, Jin F, Yan X, Xu G, Hu H, et al. Injured Liver-Released miRNA-122 Elicits Acute Pulmonary Inflammation Via Activating Alveolar Macrophage TLR7 Signaling Pathway. Proc Natl Acad Sci USA (2019) 116:6162–71. doi: 10.1073/pnas.1814139116
158. Mittelbrunn M, Gutiérrez-Vázquez C, Villarroya-Beltri C, González S, Sánchez-Cabo F, González M, et al. Unidirectional Transfer of microRNA-loaded Exosomes From T Cells to Antigen-Presenting Cells. Nat Commun (2011) 2:282. doi: 10.1038/ncomms1285
159. Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM, et al. Mechanism of Transfer of Functional microRNAs Between Mouse Dendritic Cells Via Exosomes. Blood (2012) 119:756–66. doi: 10.1182/blood-2011-02-338004
160. Salvi V, Gianello V, Busatto S, Bergese P, Andreoli L, D’Oro U, et al. Exosome-Delivered microRNAs Promote IFN-α Secretion by Human Plasmacytoid DCs Via TLR7. JCI Insight (2018) 3(10):e98204. doi: 10.1172/jci.insight.98204
161. Fernández-Messina L, Rodríguez-Galán A, de Yébenes VG, Gutiérrez-Vázquez C, Tenreiro S, Seabra MC, et al. Transfer of Extracellular vesicle-microRNA Controls Germinal Center Reaction and Antibody Production. EMBO Rep (2020) 21:e48925. doi: 10.15252/embr.201948925
162. Seeley JJ, Baker RG, Mohamed G, Bruns T, Hayden MS, Deshmukh SD, et al. Induction of Innate Immune Memory Via microRNA Targeting of Chromatin Remodelling Factors. Nature (2018) 559:114–9. doi: 10.1038/s41586-018-0253-5
163. Duursma AM, Kedde M, Schrier M, le Sage C, Agami R. miR-148 Targets Human DNMT3b Protein Coding Region. RNA (2008) 14:872–7. doi: 10.1261/rna.972008
164. Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, et al. MicroRNA-29 Family Reverts Aberrant Methylation in Lung Cancer by Targeting DNA Methyltransferases 3A and 3B. Proc Natl Acad Sci USA (2007) 104:15805–10. doi: 10.1073/pnas.0707628104
165. Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, et al. MicroRNA-29b Induces Global DNA Hypomethylation and Tumor Suppressor Gene Reexpression in Acute Myeloid Leukemia by Targeting Directly DNMT3A and 3B and Indirectly DNMT1. Blood (2009) 113:6411–8. doi: 10.1182/blood-2008-07-170589
166. Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, et al. Genomic Loss of microRNA-101 Leads to Overexpression of Histone Methyltransferase EZH2 in Cancer. Science (2008) 322:1695–9. doi: 10.1126/science.1165395
167. Sander S, Bullinger L, Klapproth K, Fiedler K, Kestler HA, Barth TF, et al. MYC Stimulates EZH2 Expression by Repression of its Negative Regulator miR-26a. Blood (2008) 112:4202–12. doi: 10.1182/blood-2008-03-147645
168. Sakurai K, Furukawa C, Haraguchi T, Inada K, Shiogama K, Tagawa T, et al. MicroRNAs miR-199a-5p and -3p Target the Brm Subunit of SWI/SNF to Generate a Double-Negative Feedback Loop in a Variety of Human Cancers. Cancer Res (2011) 71:1680–9. doi: 10.1158/0008-5472.CAN-10-2345
169. Zhao S, Wang Y, Liang Y, Zhao M, Long H, Ding S, et al. MicroRNA-126 Regulates DNA Methylation in CD4+ T Cells and Contributes to Systemic Lupus Erythematosus by Targeting DNA Methyltransferase 1. Arthritis Rheum (2011) 63:1376–86. doi: 10.1002/art.30196
170. Qin H, Zhu X, Liang J, Wu J, Yang Y, Wang S, et al. MicroRNA-29b Contributes to DNA Hypomethylation of CD4+ T Cells in Systemic Lupus Erythematosus by Indirectly Targeting DNA Methyltransferase 1. J Dermatol Sci (2013) 69:61–7. doi: 10.1016/j.jdermsci.2012.10.011
171. Hedrich CM. Epigenetics in SLE. Curr Rheumatol Rep (2017) 19:58. doi: 10.1007/s11926-017-0685-1
172. Touma Z, Gladman DD. Current and Future Therapies for SLE: Obstacles and Recommendations for the Development of Novel Treatments. Lupus Sci Med (2017) 4:e000239. doi: 10.1136/lupus-2017-000239
173. Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of microRNAs In Vivo With ‘Antagomirs’. Nature (2005) 438:685–9. doi: 10.1038/nature04303
174. Ørom UA, Kauppinen S, Lund AH. LNA-Modified Oligonucleotides Mediate Specific Inhibition of microRNA Function. Gene (2006) 372:137–41. doi: 10.1016/j.gene.2005.12.031
175. Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, et al. Silencing of microRNA Families by Seed-Targeting Tiny LNAs. Nat Genet (2011) 43:371–8. doi: 10.1038/ng.786
176. Gebert LF, Rebhan MA, Crivelli SE, Denzler R, Stoffel M, Hall J. Miravirsen (SPC3649) can Inhibit the Biogenesis of miR-122. Nucleic Acids Res (2014) 42:609–21. doi: 10.1093/nar/gkt852
177. Herrera-Carrillo E, Liu YP, Berkhout B. Improving Mirna Delivery by Optimizing miRNA Expression Cassettes in Diverse Virus Vectors. Hum Gene Ther Methods (2017) 28:177–90. doi: 10.1089/hgtb.2017.036
178. Ebert MS, Sharp PA. MicroRNA Sponges: Progress and Possibilities. RNA (2010) 16:2043–50. doi: 10.1261/rna.2414110
179. Chaudhary V, Jangra S, Yadav NR. Nanotechnology Based Approaches for Detection and Delivery of microRNA in Healthcare and Crop Protection. J Nanobiotechnology (2018) 16:40. doi: 10.1186/s12951-018-0368-8
180. Ottosen S, Parsley TB, Yang L, Zeh K, van Doorn LJ, van der Veer E, et al. In Vitro Antiviral Activity and Preclinical and Clinical Resistance Profile of Miravirsen, a Novel Anti-Hepatitis C Virus Therapeutic Targeting the Human Factor miR-122. Antimicrob Agents Chemother (2015) 59:599–608. doi: 10.1128/AAC.04220-14
181. Chakraborty C, Sharma AR, Sharma G, Doss CGP, Lee SS. Therapeutic miRNA and Sirna: Moving From Bench to Clinic as Next Generation Medicine. Mol Ther Nucleic Acids (2017) 8:132–43. doi: 10.1016/j.omtn.2017.06.005
182. Seto AG, Beatty X, Lynch JM, Hermreck M, Tetzlaff M, Duvic M, et al. Cobomarsen, an Oligonucleotide Inhibitor of miR-155, Co-Ordinately Regulates Multiple Survival Pathways to Reduce Cellular Proliferation and Survival in Cutaneous T-cell Lymphoma. Br J Haematol (2018) 183:428–44. doi: 10.1111/bjh.15547
183. Anastasiadou E, Seto AG, Beatty X, Hermreck M, Gilles ME, Stroopinsky D, et al. Cobomarsen, an Oligonucleotide Inhibitor of miR-155, Slows DLBCL Tumor Cell Growth in Vitro and In Vivo. Clin Cancer Res (2021) 27:1139–49. doi: 10.1158/1078-0432.CCR-20-3139
184. Hanna J, Hossain GS, Kocerha J. The Potential for Microrna Therapeutics and Clinical Research. Front Genet (2019) 10:478. doi: 10.3389/fgene.2019.00478
Keywords: miRNA, autoimmunity, B cells, germinal center, systemic lupus erythematosus
Citation: Schell SL and Rahman ZSM (2021) miRNA-Mediated Control of B Cell Responses in Immunity and SLE. Front. Immunol. 12:683710. doi: 10.3389/fimmu.2021.683710
Received: 22 March 2021; Accepted: 04 May 2021;
Published: 17 May 2021.
Edited by:
Zhenming Xu, The University of Texas Health Science Center at San Antonio, United StatesReviewed by:
Robert A. Eisenberg, University of Pennsylvania, United StatesNan Shen, Shanghai JiaoTong University, China
Copyright © 2021 Schell and Rahman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ziaur S. M. Rahman, enJhaG1hbkBwZW5uc3RhdGVoZWFsdGgucHN1LmVkdQ==