 Sergio Castro-Gonzalez
Sergio Castro-Gonzalez Sydney Simpson
Sydney Simpson Yuhang Shi
Yuhang Shi Yuexuan Chen
Yuexuan Chen Jared Benjamin
Jared Benjamin Ruth Serra-Moreno
Ruth Serra-Moreno- Microbiology and Immunology, University of Rochester Medical Center, Rochester, NY, United States
Ubiquitination is a process that acts upon every step of the HIV replication cycle. The activity, subcellular localization, and stability of HIV dependency factors as well as negative modulators can be affected by ubiquitination. These modifications consequently have an impact on the progression and outcome of infection. Additionally, recent findings suggest new roles for ubiquitination in the interplay between HIV and the cellular environment, specifically in the interactions between HIV, autophagy and apoptosis. On one hand, autophagy is a defense mechanism against HIV that promotes the degradation of the viral protein Gag, likely through ubiquitination. Gag is an essential structural protein that drives virion assembly and release. Interestingly, the ubiquitination of Gag is vital for HIV replication. Hence, this post-translational modification in Gag represents a double-edged sword: necessary for virion biogenesis, but potentially detrimental under conditions of autophagy activation. On the other hand, HIV uses Nef to circumvent autophagy-mediated restriction by promoting the ubiquitination of the autophagy inhibitor BCL2 through Parkin/PRKN. Although the Nef-promoted ubiquitination of BCL2 occurs in both the endoplasmic reticulum (ER) and mitochondria, only ER-associated ubiquitinated BCL2 arrests the progression of autophagy. Importantly, both mitochondrial BCL2 and PRKN are tightly connected to mitochondrial function and apoptosis. Hence, by enhancing the PRKN-mediated ubiquitination of BCL2 at the mitochondria, HIV might promote apoptosis. Moreover, this effect of Nef might account for HIV-associated disorders. In this article, we outline our current knowledge and provide perspectives of how ubiquitination impacts the molecular interactions between HIV, autophagy and apoptosis.
Ubiquitination in the Mutual Antagonism Between HIV and Autophagy
Ubiquitination post-translational modifications are used for multiple purposes including signaling transduction, enhancing protein function, driving protein subcellular localization and targeting proteins for degradation (1–3). One of these degradation pathways is autophagy, which targets components in the cytosol, including subcellular organelles and microbial pathogens, for lysosomal degradation. Whereas Lys-48 polyubiquitination commonly directs substrates to the proteasome (4–7), autophagy cargos are usually tagged with Lys-63 ubiquitin chains (4, 7, 8). Specifically, Lys-63 polyubiquitinated molecules are recognized by the ubiquitin binding domain (UBD) of different specialized autophagic receptors, including SQSTM1/p62. These receptors can simultaneously bind to ubiquitinated cargo and autophagosomal markers (i.e., LC3), allowing for the encapsulation of substrates into elongating autophagosomes (9–11).
Autophagy itself is highly regulated, in part through ubiquitination, as well as other post-translational modifications (1). For instance, ULK1, a serine/threonine kinase responsible for inducing autophagy under conditions of amino acid withdrawal, is ubiquitinated by the E3 ubiquitin ligase TRAF6, which enhances ULK1’s function and stability (12). Moreover, TRAF6 ubiquitinates Beclin1/BECN1, which promotes autophagy induced by Toll-like receptor 4 signaling (13). p62 activity can similarly be regulated through E2-supported ubiquitination by UB2D2/UB2D3, which allows for this receptor to recognize cargo (14) (Figure 1). In addition to triggering autophagy, ubiquitination can also down-regulate this pathway by targeting components of this cascade for degradation (15). Particularly, the ubiquitination of (i) ULK1 and VPS34 by the E3 ligase Cul3-KLHL20, (ii) BECN1 by RNF216, and (iii) AMBRA1 by Cullin-4, promotes the proteasomal degradation of these autophagy regulators and, thus, reduces autophagy flux (16–19) (Figure 1). Additionally, many members of the tripartite motif (TRIM) family of E3 ligases regulate autophagy, although their mode of action does not always involve ubiquitination. Among the TRIM proteins that positively modulate autophagy we find TRIM5α, TRIM6, TRIM16, TRIM17, TRIM20, TRIM21, TRIM22, TRIM23, TRIM49 and TRIM50. These molecules trigger autophagy in response to stimuli such as viral infections, cell damage, IFNγ stimulation and pattern recognition receptor (PRR) engagement (20–28). Examples of TRIM members that down-regulate autophagy are TRIM17, TRIM28, TRIM37 and TRIM59. Despite its ability to promote autophagy of midbodies, TRIM17 can also hinder autophagy by stabilizing the autophagy inhibitor MCL1. In an analogous manner, TRIM37 increases the stability of mTOR, which naturally keeps autophagy off (29–31). By contrast, rather than enhancing the activity of autophagy inhibitors, TRIM28 and TRIM59 prevent autophagy through the ubiquitination and subsequent degradation of the positive regulators AMPK1α and TRAF6, respectively (32, 33) (Figure 1). Although not through ubiquitination, LC3 – a key player in autophagy initiation, progression and execution – is regulated through a ubiquitin-like process. Particularly, LC3 becomes lipidated and this post-translational modification requires the concerted action of enzymes that mimic the function of the E1, E2 and E3 enzymes involved in ubiquitination, and they are often referred to as E3-like ligases (34–36).
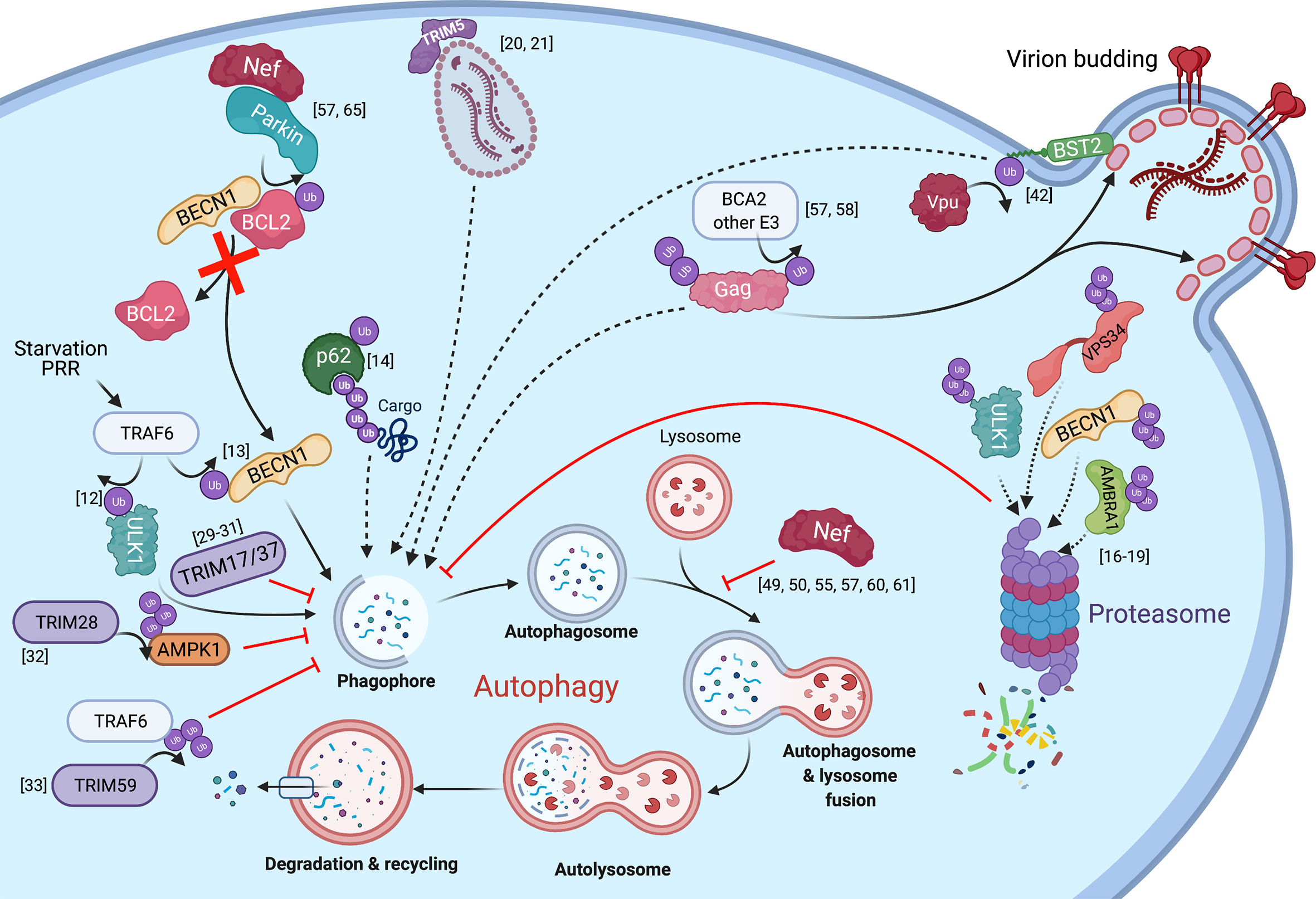
Figure 1 The intersections between HIV infection and autophagy mediated by ubiquitination. The autophagy pathway and known interactions with HIV proteins are shown with regards to ubiquitination. Solid arrows represent the transfer of a ubiquitin group, direct contribution to the autophagy pathway, or show the steps of the autophagy cycle. Dotted lines represent substrates targeted for elimination. Red lines or arrows represent autophagy inhibition at corresponding steps in the pathway.
Ubiquitination also plays a role in viral infections, including HIV. On one hand, the HIV accessory proteins Vif, Vpr, Vpx and Vpu usurp ubiquitination signaling to target host factors – which would otherwise limit virion production – for proteasomal or lysosomal degradation (37–41), and also for autophagy-mediated clearance (42). For instance, Vpu has been reported to re-route the restriction factor Tetherin/BST2 to autophagosomes in a non-canonical manner (Figure 1). On the other hand, ubiquitination can cause the degradation of HIV proteins through the proteasomal, lysosomal and/or autophagy pathways. For example, the ubiquitination of the HIV core by TRIM5α promotes the proteasomal degradation of the capsid and premature termination of reverse transcription. In addition to this well-known activity, TRIM5α also triggers autophagy in response to HIV infection and leads to the autophagic degradation of the HIV core (20, 21) (Figure 1). Another example of ubiquitin-dependent degradation is that of the HIV protein Tat, which is responsible for switching from a state of viral latency to one of productive replication. Tat is ubiquitinated by the E3 ligase CHIP, which consequently causes its proteasomal degradation, and thus, negatively impacts viral transcription (43). However, Tat is also susceptible to ubiquitination in a non-degradative manner by the E3 ligase PJA2. Specifically, PJA2 adds atypical ubiquitin chains to Tat that increase its transactivating potential (44). Besides being degraded at the proteasome, Tat has also been found as an autophagy target in CD4+ T cells, although its ubiquitination is not required to route it to autophagosomes (45). Conversely, Tat has been reported to down-regulate autophagy in neurons (46). Similar to Tat, Vif is also directed for autophagy-mediated clearance in a ubiquitin-independent manner through an association with the HDAC6 deacetylase (47). Paradoxically, Vif has been reported to inhibit canonical autophagy by associating with LC3 and preventing its incorporation into autophagosomal structures. Although the underlying mechanism by which Vif achieves this is not well understood, it does not seem to rely on LC3 ubiquitination or degradation (48).
The dichotomy of Tat and Vif in their relationship with the autophagy machinery underscores that the autophagy-HIV interactions are complex. Actually, conflicting reports exist regarding this interplay, with studies supporting that autophagy enhances or inhibits the progression of infection, depending on cell type (20, 45, 49–56). However, our group recently found that the HIV Gag polyprotein, the immature precursor of several structural proteins required for virion maturation, is degraded through autophagy regardless of cell type (57). Since Gag is susceptible to ubiquitination, which is part of its role in facilitating virion budding, it is likely that the targeting of Gag to autophagosomal membranes requires ubiquitination as well. In fact, our previous work on breast cancer-associated gene 2 (BCA2), a RING finger E3 ubiquitin ligase, showed that this protein promotes the ubiquitination and subsequent lysosomal degradation of HIV Gag (58). Hence, Gag ubiquitination could similarly tag this protein for autophagy-mediated clearance (Figure 1). Despite the antiviral potential of autophagy against HIV, our work also revealed that the virus has evolved mechanisms to counteract the autophagy-mediated destruction of viral elements needed for replication, including Gag. In particular, the HIV protein Nef counteracts the antiviral effects of autophagy. Importantly, while Nef is not required for replication in vitro, it enhances infection and contributes to pathogenesis in vivo by affording immune evasion through multiple mechanisms, including MHC-I down-regulation and counteraction of the restriction factors SERINC3 and SERINC5 [reviewed in (59)]. Besides these roles, Nef was also known to prevent viral degradation caused by autophagy through (i) a physical association with BECN1, inhibiting in turn autophagolysosomal biogenesis, and (ii) by promoting the cytosolic sequestration of the transcription factor TFEB, a master regulator of autophagy genes (49, 50, 55, 60, 61). In both models, the ultimate outcome is a defect in the fusion between autophagosomes and lysosomes. In support of these findings, we also found that Nef blocks autophagy maturation. However, we uncovered an additional mechanism by which Nef blocks autophagy at early stages of the cascade, and that requires a physical association between Nef and the E3 ubiquitin ligase Parkin/PRKN. Specifically, Nef recruits PRKN to promote the mono-ubiquitination of BCL2, an autophagy inhibitor (Figure 1) (57). Under normal conditions, BCL2 interacts with the autophagy initiator BECN1 to prevent autophagosome formation. However, under conditions of stress, BECN1 dissociates from BCL2 to initiate autophagosome biogenesis (62–64). Remarkably, the sequestration of BECN1 by BCL2 is enhanced if BCL2 is mono-ubiquitinated (65). In fact, we found that the Nef-mediated recruitment of PRKN not only increases BCL2 ubiquitination but also BECN1-BCL2 association. As a consequence of this, autophagosome formation is severely inhibited, while Gag levels and virion production are restored (Figure 1) (57). Based on these observations, we conclude that, besides its already described roles in autophagy maturation, Nef circumvents the autophagy block by intersecting with an early event in the autophagy cascade.
Effects of the Nef-Promoted Ubiquitination of BCL2 on Autophagy and Apoptosis
The increased levels of ubiquitinated BCL2 promoted by Nef may have further implications for infected cells. BCL2 is found in multiple subcellular compartments, including the endoplasmic reticulum (ER) and the mitochondria (66–68). ER-associated BCL2 is mainly involved in the regulation of autophagy through its interaction with BECN1, as stated above. Specifically, BCL2 binds to the BCL2 homology domain 3 (BH-3 domain) of BECN1 and sequesters this molecule, which in turn impairs autophagy at both initiation and maturation stages. Autophagy initiation and maturation are dependent on the respective formation of the protein complexes PI3K-C1 and PI3K-C2 (class III PI 3-kinase complex 1 and complex 2). The core structure of PI3K-C1 consists of VPS34, BECN1, VPS15, and ATG14. PI3K-C2 core structure differs from C1 by the absence of ATG14 and the addition of UVRAG. The presence of ATG14 or UVRAG targets each complex towards phagophore membranes or autophagosomal membranes, which helps modulate autophagy initiation and maturation, respectively (69–71). Besides its role in sequestering BECN1, BCL2 may additionally intersect with C1/C2 and inhibit VPS34 kinase activity by blocking BECN1 interactions with ATG14 and UVRAG in their respective complexes (57, 69, 72, 73). The resulting effect of BCL2 binding to BECN1 is an overall antagonization of autophagy. Although this phenomenon has been described in more detail for C1 formation, the sequestration of free BECN1 might also impact the biogenesis of C2 and, thus, autophagy maturation (Figure 2A). Therefore, a major point of regulation in autophagy is the promotion and reduction of BECN1-BCL2 complexes.
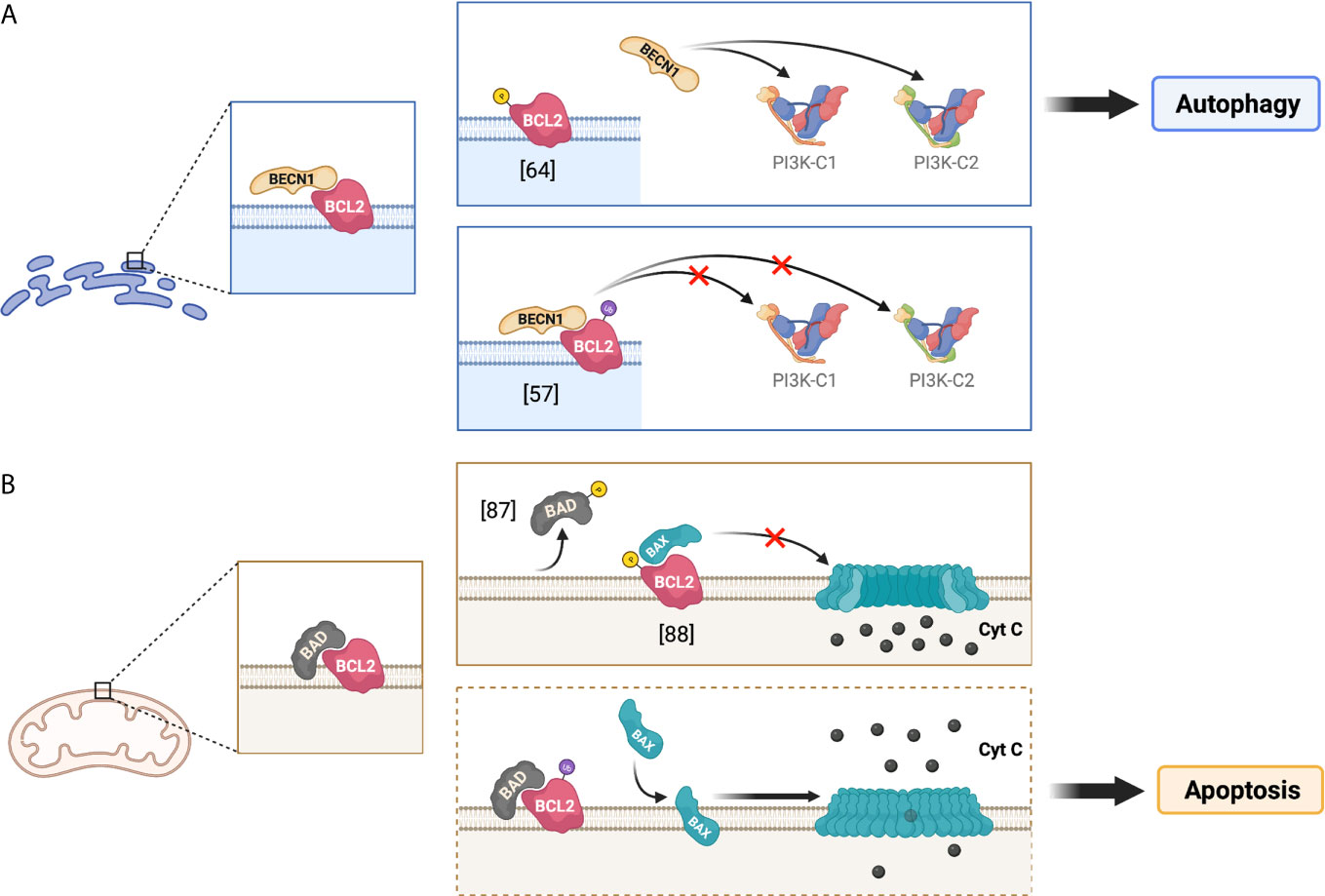
Figure 2 Implications of the phosphorylation and ubiquitination of BCL2 at the ER and mitochondria. The figure illustrates the regulation of BCL2 functions through its post-translational modifications at different subcellular localizations, depicting the potential effect of BCL2 ubiquitination on autophagy and apoptosis. (A) presents the roles of BCL2 in the endoplasmic reticulum (ER), whereas (B) depicts the roles of BCL2 at the mitochondria. The dotted frame in (B) represents putative roles.
Besides its ability to associate with the ER, BCL2 and other members of the BCL2 family can also be found at the mitochondria, where they regulate apoptosis by controlling the permeabilization of the outer mitochondrial membrane. Apoptosis is an essential process that removes damaged or infected cells in an orderly fashion. Under conditions of extreme stress or damage, pro-apoptotic effector proteins, such as BCL2-associated X protein (BAX) or BCL2 antagonist/killer-1 (BAK), homo-oligomerize to form pores on the outer mitochondrial membrane (74–77). The formation of these pores allows for cytochrome C to escape from the mitochondrial lumen. The release of cytochrome C to the cytosol enables its binding to the apoptotic protease activating factor (Apaf-1) and consequently triggers multiple caspase cascades that lead to this controlled death fate (77–79). Conversely, under healthy conditions, BCL2 pro-survival proteins directly bind to the BH3 domain in BAX to prevent pore formation on the outer mitochondrial membrane, which in turn prevents apoptosis (80, 81). Due to the importance of maintaining a healthy cell survival/cell death equilibrium, there are other relevant cellular factors involved in this apoptotic regulation. In particular, the BH3 domain of BCL2-associated agonist of cell death (BAD) is able to directly bind to mitochondria-associated BCL2 to hamper its anti-apoptotic function (82–84) (Figure 2B). Therefore, the BCL2-mediated inactivation of BAX, as well as its own regulation via BAD interaction, are crucial to control mitochondrial membrane integrity and consequently cell survival.
Whereas its subcellular localization dictates which cellular pathway will be susceptible for BCL2-mediated inhibition (autophagy vs. apoptosis), post-translational modifications of BCL2 will regulate the degree of restriction exerted over these two pathways. From this perspective, BCL2 is susceptible to become phosphorylated at Ser70 by the JNK1 kinase (85–87). When this phosphorylation occurs at the ER, it causes a reduction in the association between BCL2 and BECN1 and thus, it facilitates autophagy activation and progression by increasing the availability of BECN1 (62, 64, 85) (Figure 2A; top). Alternatively, JNK1 as well as other kinases have been reported to mediate the phosphorylation of both BCL2 and its inhibitor BAD at the outer mitochondrial membrane (86, 88). This phosphorylation can promote the dissociation of BCL2 from BAD, which enables BCL2 anti-apoptotic function by increasing the ratio BCL2 to BAX (86, 88) (Figure 2B; top).
As noted above, in addition to phosphorylation, BCL2 is mono-ubiquitinated by PRKN. Of note, PRKN primarily localizes at the mitochondria where it helps regulate mitochondrial quality control, namely through mitophagy – the autophagy of mitochondria – when these organelles are impaired (89–91). The Nef-promoted mono-ubiquitination of BCL2 at the ER increases its stability and enhances its association with BECN1 to further restrict the early stages and progression of the autophagy pathway (57, 65) (Figure 2A; bottom). Besides facilitating BCL2 mono-ubiquitination at the ER, HIV Nef also increases mono-ubiquitination and stability of the BCL2 isoform in the mitochondria, although it does not cause an enhancement in its association with BECN1 (57). However, the potential impacts of mitochondrial BCL2 mono-ubiquitination on mitophagy and apoptosis are yet to be determined. One could speculate in this matter that, analogously to what applies for the ER in terms of BCL2-BECN1 interactions, the mono-ubiquitination of BCL2 at the mitochondria might promote BCL2-BAD association and consequently, facilitate cell death through apoptosis (Figure 2B; bottom).
Discussion and Concluding Remarks
The effects of the Nef-dependent mono-ubiquitination of BCL2 could have strong implications in our understanding of the role of Nef in the development of pathologies associated with HIV+ individuals. First of all, the fact that not only Nef impacts autophagy restriction but also might facilitate apoptotic events through the PRKN-dependent mono-ubiquitination of BCL2 could provide mechanistic support to previous studies that connect Nef expression and apoptosis in different cell types, such as cardiomyocytes, brain endothelial cells or CD4+ T cells (92–94). Importantly, this Nef-mediated apoptosis may be responsible, at least in part, for the depletion of infected as well as bystander CD4+ T cells, which is the main cause of the immunodeficiency observed in infected individuals. Additionally, the effect of the Nef-enhanced ubiquitination of BCL2 on autophagy could also account for the development of other HIV-associated pathologies, including HIV-associated neurological disorders (HAND) or HIV-associated pulmonary hypertension (HIV-PH) (95–97). These conditions are not only relevant because of their undesired symptomatology and prognosis, but also because they are usually associated with persons living with HIV, even those controlling the infection due to their adherence to the antiretroviral regimens. Whereas Nef expression has been linked to the development of these pathologies, the underlying mechanisms still remain unknown. Remarkably, several studies have demonstrated that autophagic dysfunction is directly associated with pulmonary hypertension as well as numerous neurological and cognitive conditions, including HAND (96, 98, 99). In fact, autophagy dysregulation by HIV Tat has been linked with neuronal degeneration (46), so the effect of Nef on BCL2 ubiquitination and its role in autophagy might exacerbate neuronal damage. In addition, malfunction of the ubiquitin ligase PRKN seems to be associated with neurological disorders (100, 101). Therefore, Nef’s capacity to intersect with autophagy through the recruitment of PRKN to drive the mono-ubiquitination of BCL2 might be a key driver in the development of these HIV-associated conditions.
Ubiquitin has a relevant position in the arms-race between HIV and the host cell environment. Despite being required for viral particle assembly, the ubiquitination of HIV Gag might be tagging this protein for the so-called ‘kiss of death’ fate. Conversely, HIV has evolved a strategy to hijack the cellular ubiquitin machinery in order to overcome this hurdle. This mechanism involves the mono-ubiquitination of BCL2 that, in addition to preventing the degradation of Gag, might also be lifting the endogenous control over apoptosis. This in turn could have a wide range of implications for the pathogenesis and prognosis of HIV+ individuals. Therefore, further elucidation of the role of ubiquitin within the interplay between HIV and the host could be promising for the identification of new therapeutic targets against this virus.
Author Contributions
SC-G, SS, and RS-M outlined the perspectives paper. SC-G, SS, YS, YC, JB, and RS-M researched specific sections of the perspectives article and provided the literature referenced here. SC-G wrote the abstract. SC-G and SS designed the figures. SC-G, SS and RS-M wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
We thank the University of Rochester Medical Center (URMC) for supporting our work. SC-G is supported by a NIH HARC collaboration grant (P50AI150476; subcontract award 12310sc; Serra-Moreno PI), YS is supported by NIH R21AI138589 (Serra-Moreno, PI), YC is supported by NSF RAPID MCB-2032518 (Serra-Moreno, PI). JB and SS are supported by RS-M startup funds. RS-M is supported by URMC, P50AI150476, R21AI138589, and MCB-2032518.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor has declared a past collaboration with one of the authors [RS-M].
Acknowledgments
The figures presented in this manuscript were created with BioRender.com
References
1. Chen RH, Chen YH, Huang TY. Ubiquitin-Mediated Regulation of Autophagy. J BioMed Sci (2019) 26:80. doi: 10.1186/s12929-019-0569-y
2. Kulathu Y, Komander D. Atypical Ubiquitylation - the Unexplored World of Polyubiquitin Beyond Lys48 and Lys63 Linkages. Nat Rev Mol Cell Biol (2012) 13:508–23. doi: 10.1038/nrm3394
3. Yau R, Rape M. The Increasing Complexity of the Ubiquitin Code. Nat Cell Biol (2016) 18:579–86. doi: 10.1038/ncb3358
4. Kwon YT, Ciechanover A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem Sci (2017) 42:873–86. doi: 10.1016/j.tibs.2017.09.002
5. Tai HC, Schuman EM. Ubiquitin, the Proteasome and Protein Degradation in Neuronal Function and Dysfunction. Nat Rev Neurosci (2008) 9:826–38. doi: 10.1038/nrn2499
6. Grice GL, Nathan JA. The Recognition of Ubiquitinated Proteins by the Proteasome. Cell Mol Life Sci (2016) 73:3497–506. doi: 10.1007/s00018-016-2255-5
7. Nathan JA, Kim HT, Ting L, Gygi SP, Goldberg AL. Why do Cellular Proteins Linked to K63-polyubiquitin Chains Not Associate With Proteasomes? EMBO J (2013) 32:552–65. doi: 10.1038/emboj.2012.354
8. Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, et al. Lysine 63-Linked Ubiquitination Promotes the Formation and Autophagic Clearance of Protein Inclusions Associated With Neurodegenerative Diseases. Hum Mol Genet (2008) 17:431–9. doi: 10.1093/hmg/ddm320
9. Wooten MW, Geetha T, Babu JR, Seibenhener ML, Peng J, Cox N, et al. Essential Role of Sequestosome 1/p62 in Regulating Accumulation of Lys63-ubiquitinated Proteins. J Biol Chem (2008) 283:6783–9. doi: 10.1074/jbc.M709496200
10. Wurzer B, Zaffagnini G, Fracchiolla D, Turco E, Abert C, Romanov J, et al. Oligomerization of p62 Allows for Selection of Ubiquitinated Cargo and Isolation Membrane During Selective Autophagy. Elife (2015) 4:e08941. doi: 10.7554/eLife.08941
11. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J Biol Chem (2007) 282:24131–45. doi: 10.1074/jbc.M702824200
12. Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, et al. mTOR Inhibits Autophagy by Controlling ULK1 Ubiquitylation, Self-Association and Function Through AMBRA1 and TRAF6. Nat Cell Biol (2013) 15:406–16. doi: 10.1038/ncb2708
13. Shi CS, Kehrl JH. TRAF6 and A20 Regulate Lysine 63-Linked Ubiquitination of Beclin-1 to Control TLR4-induced Autophagy. Sci Signal (2010) 3:ra42. doi: 10.1126/scisignal.2000751
14. Peng H, Yang J, Li G, You Q, Han W, Li T, et al. Ubiquitylation of p62/sequestosome1 Activates its Autophagy Receptor Function and Controls Selective Autophagy Upon Ubiquitin Stress. Cell Res (2017) 27:657–74. doi: 10.1038/cr.2017.40
15. Kocaturk NM, Gozuacik D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front Cell Dev Biol (2018) 6:128. doi: 10.3389/fcell.2018.00128
16. Liu CC, Lin YC, Chen YH, Chen CM, Pang LY, Chen HA, et al. Cul3-KLHL20 Ubiquitin Ligase Governs the Turnover of ULK1 and VPS34 Complexes to Control Autophagy Termination. Mol Cell (2016) 61:84–97. doi: 10.1016/j.molcel.2015.11.001
17. Gomez-Diaz C, Ikeda F. Roles of Ubiquitin in Autophagy and Cell Death. Semin Cell Dev Biol (2019) 93:125–35. doi: 10.1016/j.semcdb.2018.09.004
18. Xu C, Feng K, Zhao X, Huang S, Cheng Y, Qian L, et al. Regulation of Autophagy by E3 Ubiquitin Ligase RNF216 Through BECN1 Ubiquitination. Autophagy (2014) 10:2239–50. doi: 10.4161/15548627.2014.981792
19. Antonioli M, Albiero F, Nazio F, Vescovo T, Perdomo AB, Corazzari M, et al. AMBRA1 Interplay With Cullin E3 Ubiquitin Ligases Regulates Autophagy Dynamics. Dev Cell (2014) 31:734–46. doi: 10.1016/j.devcel.2014.11.013
20. Mandell MA, Kimura T, Jain A, Johansen T, Deretic V. TRIM Proteins Regulate Autophagy: TRIM5 is a Selective Autophagy Receptor Mediating HIV-1 Restriction. Autophagy (2014) 10:2387–8. doi: 10.4161/15548627.2014.984278
21. Ribeiro CM, Sarrami-Forooshani R, Setiawan LC, Zijlstra-Willems EM, van Hamme JL, Tigchelaar W, et al. Receptor Usage Dictates HIV-1 Restriction by Human TRIM5alpha in Dendritic Cell Subsets. Nature (2016) 540:448–52. doi: 10.1038/nature20567
22. Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, et al. Trims and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-Direct Autophagy in Endomembrane Damage Homeostasis. Dev Cell (2016) 39:13–27. doi: 10.1016/j.devcel.2016.08.003
23. Fusco C, Mandriani B, Di Rienzo M, Micale L, Malerba N, Cocciadiferro D, et al. TRIM50 Regulates Beclin 1 Proautophagic Activity. Biochim Biophys Acta Mol Cell Res (2018) 1865:908–19. doi: 10.1016/j.bbamcr.2018.03.011
24. Kimura T, Jain A, Choi SW, Mandell MA, Schroder K, Johansen T, et al. TRIM-Mediated Precision Autophagy Targets Cytoplasmic Regulators of Innate Immunity. J Cell Biol (2015) 210:973–89. doi: 10.1083/jcb.201503023
25. Mandell MA, Jain A, Arko-Mensah J, Chauhan S, Kimura T, Dinkins C, et al. TRIM Proteins Regulate Autophagy and can Target Autophagic Substrates by Direct Recognition. Dev Cell (2014) 30:394–409. doi: 10.1016/j.devcel.2014.06.013
26. Di Rienzo M, Romagnoli A, Antonioli M, Piacentini M, Fimia GM. TRIM Proteins in Autophagy: Selective Sensors in Cell Damage and Innate Immune Responses. Cell Death Differ (2020) 27:887–902. doi: 10.1038/s41418-020-0495-2
27. Sparrer KMJ, Gableske S, Zurenski MA, Parker ZM, Full F, Baumgart GJ, et al. TRIM23 Mediates Virus-Induced Autophagy Via Activation of TBK1. Nat Microbiol (2017) 2:1543–57. doi: 10.1038/s41564-017-0017-2
28. Lou J, Wang Y, Zheng X, Qiu W. TRIM22 Regulates Macrophage Autophagy and Enhances Mycobacterium Tuberculosis Clearance by Targeting the Nuclear Factor-Multiplicity kappaB/beclin 1 Pathway. J Cell Biochem (2018) 119:8971–80. doi: 10.1002/jcb.27153
29. Brigant B, Metzinger-Le Meuth V, Rochette J, Metzinger L. Trimming Down to TRIM37: Relevance to Inflammation, Cardiovascular Disorders, and Cancer in MULIBREY Nanism. Int J Mol Sci (2018) 20:1–14. doi: 10.3390/ijms20010067
30. Wang W, Xia Z, Farre JC, Subramani S. TRIM37 Deficiency Induces Autophagy Through Deregulating the MTORC1-TFEB Axis. Autophagy (2018) 14:1574–85. doi: 10.1080/15548627.2018.1463120
31. Mandell MA, Jain A, Kumar S, Castleman MJ, Anwar T, Eskelinen EL, et al. TRIM17 Contributes to Autophagy of Midbodies While Actively Sparing Other Targets From Degradation. J Cell Sci (2016) 129:3562–73. doi: 10.1242/jcs.190017
32. Pineda CT, Ramanathan S, Fon Tacer K, Weon JL, Potts MB, Ou YH, et al. Degradation of AMPK by a Cancer-Specific Ubiquitin Ligase. Cell (2015) 160:715–28. doi: 10.1016/j.cell.2015.01.034
33. Han T, Guo M, Gan M, Yu B, Tian X, Wang JB. TRIM59 Regulates Autophagy Through Modulating Both the Transcription and the Ubiquitination of BECN1. Autophagy (2018) 14:2035–48. doi: 10.1080/15548627.2018.1491493
34. Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L Complex Specifies the Site of LC3 Lipidation for Membrane Biogenesis in Autophagy. Mol Biol Cell (2008) 19:2092–100. doi: 10.1091/mbc.e07-12-1257
35. Lescouzeres L, Bomont P. E3 Ubiquitin Ligases in Neurological Diseases: Focus on Gigaxonin and Autophagy. Front Physiol (2020) 11:1022. doi: 10.3389/fphys.2020.01022
36. Scrivo A, Codogno P, Bomont P. Gigaxonin E3 Ligase Governs ATG16L1 Turnover to Control Autophagosome Production. Nat Commun (2019) 10:780. doi: 10.1038/s41467-019-08331-w
37. Iwabu Y, Fujita H, Kinomoto M, Kaneko K, Ishizaka Y, Tanaka Y, et al. HIV-1 Accessory Protein Vpu Internalizes Cell-Surface BST-2/Tetherin Through Transmembrane Interactions Leading to Lysosomes. J Biol Chem (2009) 284:35060–72. doi: 10.1074/jbc.M109.058305
38. Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. Vif Overcomes the Innate Antiviral Activity of APOBEC3G by Promoting its Degradation in the Ubiquitin-Proteasome Pathway. J Biol Chem (2004) 279:7792–8. doi: 10.1074/jbc.M313093200
39. Mitchell RS, Katsura C, Skasko MA, Fitzpatrick K, Lau D, Ruiz A, et al. Vpu Antagonizes BST-2-mediated Restriction of HIV-1 Release Via beta-TrCP and Endo-Lysosomal Trafficking. PloS Pathog (2009) 5:e1000450. doi: 10.1371/journal.ppat.1000450
40. Seissler T, Marquet R, Paillart JC. Hijacking of the Ubiquitin/Proteasome Pathway by the HIV Auxiliary Proteins. Viruses (2017) 9:1–21. doi: 10.3390/v9110322
41. Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, et al. Induction of APOBEC3G Ubiquitination and Degradation by an HIV-1 Vif-Cul5-SCF Complex. Science (2003) 302:1056–60. doi: 10.1126/science.1089591
42. Madjo U, Leymarie O, Fremont S, Kuster A, Nehlich M, Gallois-Montbrun S, et al. LC3C Contributes to Vpu-Mediated Antagonism of BST2/Tetherin Restriction on HIV-1 Release Through a Non-canonical Autophagy Pathway. Cell Rep (2016) 17:2221–33. doi: 10.1016/j.celrep.2016.10.045
43. Ali A, Farooqui SR, Banerjea AC. The Host Cell Ubiquitin Ligase Protein CHIP is a Potent Suppressor of HIV-1 Replication. J Biol Chem (2019) 294:7283–95. doi: 10.1074/jbc.RA118.007257
44. Faust TB, Li Y, Jang GM, Johnson JR, Yang S, Weiss A, et al. PJA2 Ubiquitinates the HIV-1 Tat Protein With Atypical Chain Linkages to Activate Viral Transcription. Sci Rep (2017) 7:45394. doi: 10.1038/srep45394
45. Sagnier S, Daussy CF, Borel S, Robert-Hebmann V, Faure M, Blanchet FP, et al. Autophagy Restricts HIV-1 Infection by Selectively Degrading Tat in CD4+ T Lymphocytes. J Virol (2015) 89:615–25. doi: 10.1128/JVI.02174-14
46. Fields J, Dumaop W, Eleuteri S, Campos S, Serger E, Trejo M, et al. Hiv-1 Tat Alters Neuronal Autophagy by Modulating Autophagosome Fusion to the Lysosome: Implications for HIV-associated Neurocognitive Disorders. J Neurosci (2015) 35:1921–38. doi: 10.1523/JNEUROSCI.3207-14.2015
47. Valera MS, de Armas-Rillo L, Barroso-Gonzalez J, Ziglio S, Batisse J, Dubois N, et al. The HDAC6/APOBEC3G Complex Regulates HIV-1 Infectiveness by Inducing Vif Autophagic Degradation. Retrovirology (2015) 12:53. doi: 10.1186/s12977-015-0181-5
48. Borel S, Robert-Hebmann V, Alfaisal J, Jain A, Faure M, Espert L, et al. HIV-1 Viral Infectivity Factor Interacts With Microtubule-Associated Protein Light Chain 3 and Inhibits Autophagy. AIDS (2015) 29:275–86. doi: 10.1097/QAD.0000000000000554
49. Campbell GR, Rawat P, Bruckman RS, Spector SA. Human Immunodeficiency Virus Type 1 Nef Inhibits Autophagy Through Transcription Factor EB Sequestration. PloS Pathog (2015) 11:e1005018. doi: 10.1371/journal.ppat.1005018
50. Campbell GR, Spector SA. Inhibition of Human Immunodeficiency Virus Type-1 Through Autophagy. Curr Opin Microbiol (2013) 16:349–54. doi: 10.1016/j.mib.2013.05.006
51. Daussy CF, Beaumelle B, Espert L. Autophagy Restricts HIV-1 Infection. Oncotarget (2015) 6:20752–3. doi: 10.18632/oncotarget.5123
52. Denizot M, Varbanov M, Espert L, Robert-Hebmann V, Sagnier S, Garcia E, et al. Hiv-1 gp41 Fusogenic Function Triggers Autophagy in Uninfected Cells. Autophagy (2008) 4:998–1008. doi: 10.4161/auto.6880
53. Dinkins C, Pilli M, Kehrl JH. Roles of Autophagy in HIV Infection. Immunol Cell Biol (2015) 93:11–7. doi: 10.1038/icb.2014.88
54. Espert L, Beaumelle B, Vergne I. Autophagy in Mycobacterium Tuberculosis and HIV Infections. Front Cell Infect Microbiol (2015) 5:49. doi: 10.3389/fcimb.2015.00049
55. Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, et al. Autophagy Pathway Intersects With HIV-1 Biosynthesis and Regulates Viral Yields in Macrophages. J Cell Biol (2009) 186:255–68. doi: 10.1083/jcb.200903070
56. Nardacci R, Ciccosanti F, Marsella C, Ippolito G, Piacentini M, Fimia GM. Role of Autophagy in HIV Infection and Pathogenesis. J Intern Med (2017) 281:422–32. doi: 10.1111/joim.12596
57. Castro-Gonzalez S, Shi Y, Colomer-Lluch M, Song Y, Mowery K, Almodovar S, et al. HIV-1 Nef Counteracts Autophagy Restriction by Enhancing the Association Between BECN1 and its Inhibitor BCL2 in a PRKN-dependent Manner. Autophagy (2020) 17(2):1–25. doi: 10.1080/15548627.2020.1725401
58. Nityanandam R, Serra-Moreno R. BCA2/Rabring7 Targets HIV-1 Gag for Lysosomal Degradation in a Tetherin-Independent Manner. PloS Pathog (2014) 10:e1004151. doi: 10.1371/journal.ppat.1004151
59. Buffalo CZ, Iwamoto Y, Hurley JH, Ren X. How HIV Nef Proteins Hijack Membrane Traffic To Promote Infection. J Virol (2019) 93:1–18. doi: 10.1128/JVI.01322-19
60. Chang C, Young LN, Morris KL, von Bulow S, Schoneberg J, Yamamoto-Imoto H, et al. Bidirectional Control of Autophagy by BECN1 Bara Domain Dynamics. Mol Cell (2019) 73:339–353 e6. doi: 10.1016/j.molcel.2018.10.035
61. Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, et al. Identification of a Candidate Therapeutic Autophagy-Inducing Peptide. Nature (2013) 494:201–6. doi: 10.1038/nature11866
62. Decuypere JP, Parys JB, Bultynck G. Regulation of the Autophagic bcl-2/beclin 1 Interaction. Cells (2012) 1:284–312. doi: 10.3390/cells1030284
63. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell (2005) 122:927–39. doi: 10.1016/j.cell.2005.07.002
64. Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates Starvation-Induced Autophagy. Mol Cell (2008) 30:678–88. doi: 10.1016/j.molcel.2008.06.001
65. Chen D, Gao F, Li B, Wang H, Xu Y, Zhu C, et al. Parkin Mono-Ubiquitinates Bcl-2 and Regulates Autophagy. J Biol Chem (2010) 285:38214–23. doi: 10.1074/jbc.M110.101469
66. Akao Y, Otsuki Y, Kataoka S, Ito Y, Tsujimoto Y. Multiple Subcellular Localization of bcl-2: Detection in Nuclear Outer Membrane, Endoplasmic Reticulum Membrane, and Mitochondrial Membranes. Cancer Res (1994) 54:2468–71.
67. Janiak F, Leber B, Andrews DW. Assembly of Bcl-2 Into Microsomal and Outer Mitochondrial Membranes. J Biol Chem (1994) 269:9842–9. doi: 10.1016/S0021-9258(17)36960-0
68. Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigation of the Subcellular Distribution of the Bcl-2 Oncoprotein: Residence in the Nuclear Envelope, Endoplasmic Reticulum, and Outer Mitochondrial Membranes. Cancer Res (1993) 53:4701–14.
69. Hurley JH, Young LN. Mechanisms of Autophagy Initiation. Annu Rev Biochem (2017) 86:225–44. doi: 10.1146/annurev-biochem-061516-044820
70. McKnight NC, Zhenyu Y. Beclin 1, an Essential Component and Master Regulator of PI3K-III in Health and Disease. Curr Pathobiol Rep (2013) 1:231–8. doi: 10.1007/s40139-013-0028-5
71. Brier LW, Ge L, Stjepanovic G, Thelen AM, Hurley JH, Schekman R. Regulation of LC3 Lipidation by the Autophagy-Specific Class III Phosphatidylinositol-3 Kinase Complex. Mol Biol Cell (2019) 30:1098–107. doi: 10.1091/mbc.E18-11-0743
72. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 Network Regulates Autophagy and Apoptosis. Cell Death Different (2011) 18:571–80. doi: 10.1038/cdd.2010.191
73. Chang NC, Nguyen M, Germain M, Shore GC. Antagonism of Beclin 1-Dependent Autophagy by BCL-2 At the Endoplasmic Reticulum Requires NAF-1. EMBO J (2010) 29:606–18. doi: 10.1038/emboj.2009.369
74. Westphal D, Dewson G, Czabotar PE, Kluck RM. Molecular Biology of Bax and Bak Activation and Action. Biochim Biophys Acta (2011) 1813:521–31. doi: 10.1016/j.bbamcr.2010.12.019
75. Westphal D, Kluck RM, Dewson G. Building Blocks of the Apoptotic Pore: How Bax and Bak are Activated and Oligomerize During Apoptosis. Cell Death Differ (2014) 21:196–205. doi: 10.1038/cdd.2013.139
76. Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. And BAK: A Requisite Gateway to Mitochondrial Dysfunction and Death. Science (2001) 292:727–30. doi: 10.1126/science.1059108
77. Eskes R, Desagher S, Antonsson B, Martinou JC. Bid Induces the Oligomerization and Insertion of Bax Into the Outer Mitochondrial Membrane. Mol Cell Biol (2000) 20:929–35. doi: 10.1128/MCB.20.3.929-935.2000
78. Cai J, Yang J, Jones DP. Mitochondrial Control of Apoptosis: The Role of Cytochrome C. Biochim Biophys Acta (1998) 1366:139–49. doi: 10.1016/S0005-2728(98)00109-1
79. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome C and dATP-dependent Formation of Apaf-1/caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell (1997) 91:479–89. doi: 10.1016/S0092-8674(00)80434-1
80. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of Apoptosis by the BCL-2 Protein Family: Implications for Physiology and Therapy. Nat Rev Mol Cell Biol (2014) 15:49–63. doi: 10.1038/nrm3722
81. Skommer J, Brittain T, Raychaudhuri S. Bcl-2 Inhibits Apoptosis by Increasing the Time-to-Death and Intrinsic Cell-to-Cell Variations in the Mitochondrial Pathway of Cell Death. Apoptosis (2010) 15:1223–33. doi: 10.1007/s10495-010-0515-7
82. Howells CC, Baumann WT, Samuels DC, Finkielstein CV. The Bcl-2-associated Death Promoter (BAD) Lowers the Threshold At Which the Bcl-2-interacting Domain Death Agonist (BID) Triggers Mitochondria Disintegration. J Theor Biol (2011) 271:114–23. doi: 10.1016/j.jtbi.2010.11.040
83. Yu Y, Zhong Z, Guan Y. The Downregulation of Bcl-xL/Bcl-2-associated Death Promoter Indicates Worse Outcomes in Patients With Small Cell Lung Carcinoma. Int J Clin Exp Pathol (2015) 8:13075–82.
84. Hsu SY, Kaipia A, Zhu L, Hsueh AJ. Interference of BAD (Bcl-xL/Bcl-2-associated Death Promoter)-Induced Apoptosis in Mammalian Cells by 14-3-3 Isoforms and P11. Mol Endocrinol (1997) 11:1858–67. doi: 10.1210/me.11.12.1858
85. Wei Y, Sinha S, Levine B. Dual Role of JNK1-mediated Phosphorylation of Bcl-2 in Autophagy and Apoptosis Regulation. Autophagy (2008) 4:949–51. doi: 10.4161/auto.6788
86. Ruvolo PP, Deng X, May WS. Phosphorylation of Bcl2 and Regulation of Apoptosis. Leukemia (2001) 15:515–22. doi: 10.1038/sj.leu.2402090
87. Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine Phosphorylation of Death Agonist BAD in Response to Survival Factor Results in Binding to 14-3-3 Not BCL-X(L). Cell (1996) 87:619–28. doi: 10.1016/S0092-8674(00)81382-3
88. Ito T, Deng X, Carr B, May WS. Bcl-2 Phosphorylation Required for Anti-Apoptosis Function. J Biol Chem (1997) 272:11671–3. doi: 10.1074/jbc.272.18.11671
89. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is Recruited Selectively to Impaired Mitochondria and Promotes Their Autophagy. J Cell Biol (2008) 183:795–803. doi: 10.1083/jcb.200809125
90. Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, et al. Mitochondrial Pathology and Muscle and Dopaminergic Neuron Degeneration Caused by Inactivation of Drosophila Pink1 is Rescued by Parkin. Proc Natl Acad Sci USA (2006) 103:10793–8. doi: 10.1073/pnas.0602493103
91. Araya J, Tsubouchi K, Sato N, Ito S, Minagawa S, Hara H, et al. PRKN-Regulated Mitophagy and Cellular Senescence During COPD Pathogenesis. Autophagy (2019) 15:510–26. doi: 10.1080/15548627.2018.1532259
92. Rasola A, Gramaglia D, Boccaccio C, Comoglio PM. Apoptosis Enhancement by the HIV-1 Nef Protein. J Immunol (2001) 166:81–8. doi: 10.4049/jimmunol.166.1.81
93. Acheampong EA, Parveen Z, Muthoga LW, Kalayeh M, Mukhtar M, Pomerantz RJ. Human Immunodeficiency Virus Type 1 Nef Potently Induces Apoptosis in Primary Human Brain Microvascular Endothelial Cells Via the Activation of Caspases. J Virol (2005) 79:4257–69. doi: 10.1128/JVI.79.7.4257-4269.2005
94. Lenassi M, Cagney G, Liao M, Vaupotic T, Bartholomeeusen K, Cheng Y, et al. Hiv Nef is Secreted in Exosomes and Triggers Apoptosis in Bystander CD4+ T Cells. Traffic (2010) 11:110–22. doi: 10.1111/j.1600-0854.2009.01006.x
95. Lamers SL, Fogel GB, Liu ES, Barbier AE, Rodriguez CW, Singer EJ, et al. Brain-Specific HIV Nef Identified in Multiple Patients With Neurological Disease. J Neurovirol (2018) 24:1–15. doi: 10.1007/s13365-017-0586-0
96. Cheney L, Guzik H, Macaluso FP, Macian F, Cuervo AM, Berman JW. Hiv Nef and Antiretroviral Therapy Have an Inhibitory Effect on Autophagy in Human Astrocytes That May Contribute to HIV-Associated Neurocognitive Disorders. Cells (2020) 9:1–25. doi: 10.3390/cells9061426
97. Almodovar S, Hsue PY, Morelli J, Huang L, Flores SC, Lung HIVS. Pathogenesis of HIV-associated Pulmonary Hypertension: Potential Role of HIV-1 Nef. Proc Am Thorac Soc (2011) 8:308–12. doi: 10.1513/pats.201006-046WR
98. Zhang CF, Zhao FY, Xu SL, Liu J, Xing XQ, Yang J. Autophagy in Pulmonary Hypertension: Emerging Roles and Therapeutic Implications. J Cell Physiol (2019) 234:16755–67. doi: 10.1002/jcp.28531
99. Dever SM, Rodriguez M, Lapierre J, Costin BN, El-Hage N. Differing Roles of Autophagy in HIV-associated Neurocognitive Impairment and Encephalitis With Implications for Morphine Co-Exposure. Front Microbiol (2015) 6:653. doi: 10.3389/fmicb.2015.00653
100. Ge P, Dawson VL, Dawson TM. PINK1 and Parkin Mitochondrial Quality Control: A Source of Regional Vulnerability in Parkinson’s Disease. Mol Neurodegener (2020) 15:20. doi: 10.1186/s13024-020-00367-7
Keywords: autophagy, apoptosis, HIV, Nef, BCL2, BECN1, PRKN
Citation: Castro-Gonzalez S, Simpson S, Shi Y, Chen Y, Benjamin J and Serra-Moreno R (2021) HIV Nef-mediated Ubiquitination of BCL2: Implications in Autophagy and Apoptosis. Front. Immunol. 12:682624. doi: 10.3389/fimmu.2021.682624
Received: 18 March 2021; Accepted: 21 April 2021;
Published: 06 May 2021.
Edited by:
Konstantin Sparrer, Ulm University Medical Center, GermanyReviewed by:
Daniel Sauter, University Hospital Tübingen, GermanyMichael A. Mandell, University of New Mexico, United States
Copyright © 2021 Castro-Gonzalez, Simpson, Shi, Chen, Benjamin and Serra-Moreno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruth Serra-Moreno, cnV0aF9zZXJyYS1tb3Jlbm9AdXJtYy5yb2NoZXN0ZXIuZWR1; Sergio Castro-Gonzalez, c2VyZ2lvX2dvbnphbGV6QHVybWMucm9jaGVzdGVyLmVkdQ==
†These authors have contributed equally to this work