 Amy L. Kenter
Amy L. Kenter Corey T. Watson
Corey T. Watson Jan-Hendrik Spille
Jan-Hendrik Spille
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 18 May 2021
Sec. B Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.682589
This article is part of the Research Topic Multi-Dimensional Genomic Structures in the Immune System View all 12 articles
Vast repertoires of unique antigen receptors are created in developing B and T lymphocytes. The antigen receptor loci contain many variable (V), diversity (D) and joining (J) gene segments that are arrayed across very large genomic expanses and are joined to form variable-region exons of expressed immunoglobulins and T cell receptors. This process creates the potential for an organism to respond to large numbers of different pathogens. Here, we consider the possibility that genetic polymorphisms with alterations in a vast array of regulatory elements in the immunoglobulin heavy chain (IgH) locus lead to changes in locus topology and impact immune-repertoire formation.
The adaptive immune response has evolved to recognize pathogens using antigen-specific receptors expressed on B and T lymphocytes. Two identical immunoglobulin (Ig) heavy chains (IgH) and two identical light chains (Igκ or Igλ) constitute the B-cell receptor (BCR). The two lineages of T cells are distinguished by the type of T-cell receptor (TCR) expressed. TCRαβ is encoded by the Tcra and Tcrb loci, whereas TCRγδ is encoded by the Tcrg and Tcrd loci. Developing B and T cells undergo an ordered set of DNA rearrangements termed V(D)J recombination, using RAG recombinase (RAG1/2) and thereby creating a diverse repertoire of antigen receptors (1). The assembly of antigen receptors involves the juxtaposition of variable (V), diversity (D) and joining (J) gene segments into a V gene exon that encodes the antigen binding domain of antigen receptors. However, there are several barriers which must be overcome to enable a suitably diverse Ig repertoire to emerge. Many of the concepts discussed here are applicable to TCR loci.
Formation of a diverse Ig repertoire is critically dependent on proficient pro-B and pre-B cell function since it is in these cells that IgH and IgL chain genes are assembled through V(D)J recombination, respectively. V(D)J recombination requires that antigen receptor genes undergo ordered rearrangement with DH to JH joining preceding VH to DHJH recombination (Figure 1A). There are ~100 functional Igh locus VH gene exons that must recombine with one rearranged DJH element, that is assembled from one of 8-12 DH and one of 4 JH gene segments in C57BL/6 mice (Figure 1) (1). The introduction of RAG dependent DNA breaks at recombination signal sequences (RSSs) adjacent to each rearranging gene segment initiates Igh gene assembly (1). RAG1/2 loads at the recombination center (RC) situated in the region spanning Eµ and the most 3’ DH segment, DQ52 (2). RAG1/2 has been proposed to track from the RC to locate a suitable RSS for synapsis and DNA cleavage (3).
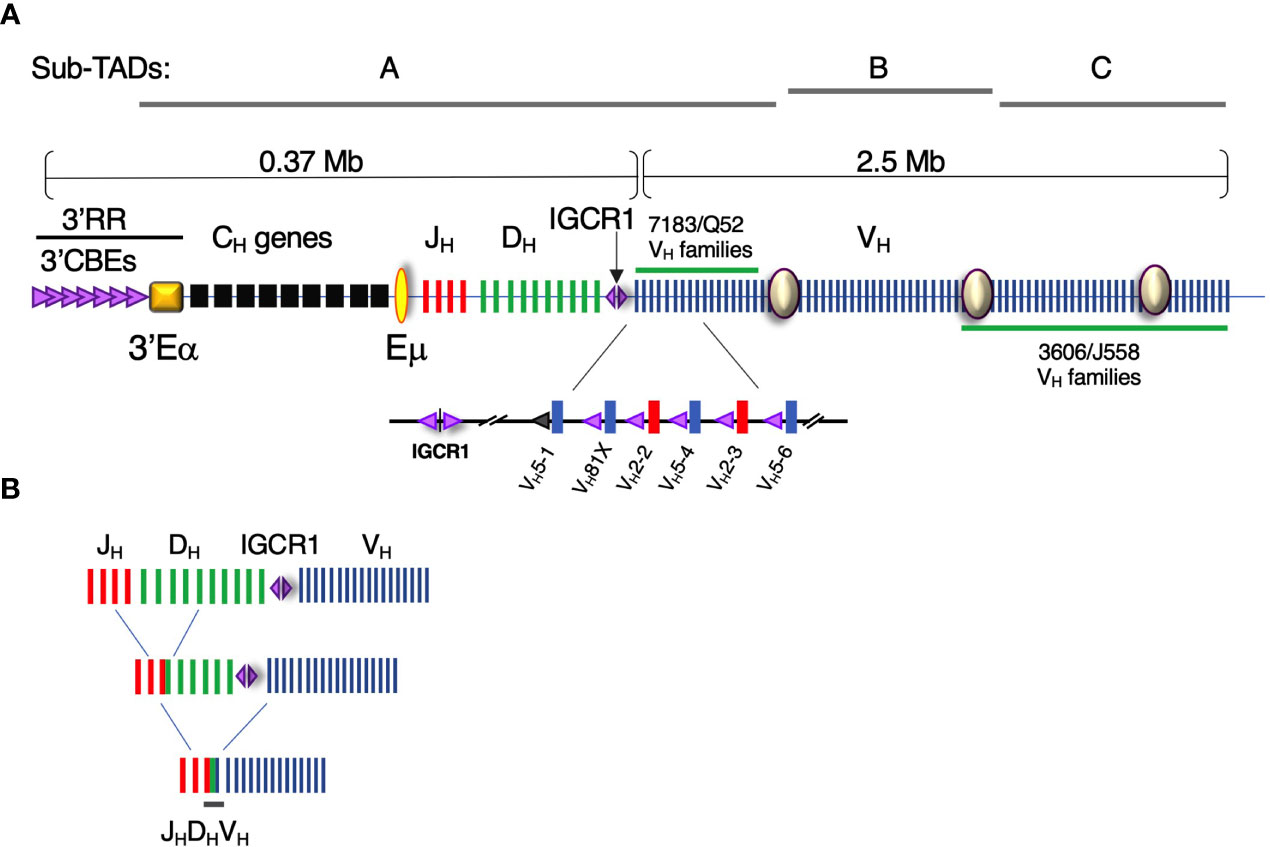
Figure 1 The Igh locus contains ~100 VH gene segments over an almost 3Mb genomic interval. (A) (Upper panel) Diagram of the Igh locus indicating VH, D, JH, and CH exons and regulatory elements (not to scale). The intronic Eμ and 3′Eα super-enhancers and intergenic control region 1 (IGCR1), composed of two divergent CBEs, are critical regulatory elements. CBE orientation is indicated by (purple) triangle direction. The 3′ regulatory region (3′RR) is a composite of nine CBEs located at the 3′ boundary of the Igh locus adjacent to 3′Eα super-enhancer. Sites I, II, and III (purple circles) anchor the sub-topologically associating domain (Sub-TADs) A, B, and C. The VHS107 family along with nine smaller VH families comprise the intermediate VH segments. The interspersed distal VH gene segments are composed of the VHJ558 and VH3609 families and are located at the 5’ end of the locus. (Lower panel) The VH7183 (blue bars) and VHQ52 families (red bars) are located at the DHJH-proximal end of the locus. Each DHJH -proximal VH exon is paired with a recombination signal sequence (not shown) and a CBE (purple triangle). The CBE associated with VH5-1 exon is non-functional (gray triangle). VH81X (VH5-2) is the second VH exon gene relative to IGCR1. (B) Schematic of the stepwise process of V(D)J recombination. DH-JH rearrangement precedes VH-DHJH recombination.
The number of V, D, and J gene segments and the availability of those segments for rearrangement determines the composition and complexity of antigen receptor repertoires. The Igh locus is quite large in linear genomic distance, extending 2.9 Mb and containing ~100 functional VH gene segments. V gene usage is only quasi-random in the pre-selected Igh repertoire as V genes rearrange at very different intrinsic frequencies (4–11). In studies of V germline transcript levels, transcription factor (TF) binding, RSS quality, and the distribution of a variety of epigenetic marks each make a contribution, but no one variable, or combination of variables, fully accounts for unequal V gene usage (4–6, 8, 12). Although the V gene accessibility hypothesis (13–15) offered an attractive model to explain V gene usage, recent studies have made plain that V gene accessibility is a necessary but insufficient condition for participation in V->DJ rearrangement (16, 17). Therefore, the factors underpinning unequal V gene rearrangement frequencies remain to be determined.
It is important to note that the potential contribution of Ig haplotype diversity in these processes has been underappreciated (18). Despite the fact that the Ig loci of natural outbred organisms are known to be extremely diverse, much of our understanding of the mechanisms dictating V(D)J recombination have come from studies of inbred models. However, even in inbred models, it has been demonstrated that Ig genetic diversity is more extensive than initially appreciated, in many cases mirroring (or even exceeding) what has been observed in human populations and other more outbred organisms (19–28). In mouse, for example, a comparison of germline VH sequences between C57BL/6 and BALB/c revealed surprisingly little overlap in the germline repertoires of these two strains. Of the 99 C57BL/6 and 164 BALB/c VH alleles compared, only 5 were found to be identical (23), likely the result of both allelic sequence divergence and structural variation associated with differences in V gene content between the two strains. Extended comparisons of VH germline alleles across additional inbred wild-derived strains, thought to represent diverse mouse sub-species origins, revealed even greater diversity, suggesting that Ig germline variation across commonly used mouse inbred strains is likely to be vast (24). Similar inter-strain diversity has been observed within the mouse DH gene loci as well (19, 20). These and other data clearly demonstrate the presence of extensive polymorphism within the Igh locus, and highlight the potential influence of both sequence diversity and Ig gene segment number as significant contributors to Ig repertoire diversity.
There is a growing body of evidence supporting the potential impact of genetic polymorphism on V(D)J recombination. First, multiple studies of the naïve repertoire in human monozygotic twins have demonstrated that VH, DH, and JH usage is highly heritable (29, 30). Second, genetic variants within the Ig loci associate with variation in gene usage observed between individuals, including examples of Ig gene coding and non-coding single nucleotide polymorphisms (SNPs) (31, 32), as well as large structural variants (32–34). Here we examine the confluence of VH gene identity with locus polymorphism and locus architecture as determinants for VH gene usage in V(D)J recombination and ultimately the diversity of the preselected Ig repertoire.
It is essential that all VH genes achieve spatial proximity with the RC located at the Eµ-DHJH domain to produce a fully representative Ig repertoire (Figure 1B). In C57BL/6, VH exons are segregated into three clusters, the proximal, intermediate and distal VH regions, that collectively span ~2.4 Mb. Recent studies have established that chromosomes are folded into hierarchical domains of various length scales. Chromosomes are nonrandomly located in nuclei within chromosomal territories (35, 36) which are subdivided into chromosomal compartments (36) that are further partitioned into topologically associating domains (TADs) (36, 37). TADs are zones in which intraregional interactions are more frequent than those traversing the boundaries between TADs (37–39). TAD organization reflects the functional partition of chromatin regions by transcriptional activity (37, 39), histone modifications (37–40), and replication timing (41) implying a link between function and genome structure.
The Igh locus is contained within a 2.9-Mb TAD in pro-B cells (42). 5C studies demonstrate that the murine Igh TAD is subdivided into two highly structured sub-TADs A and C, corresponding to the DH-proximal and DH-distal VH gene families, respectively, while the less structured sub-TAD B includes the intermediate VH gene segments (42). Correspondingly, live pro-B cell imaging indicates that Igh locus topology is organized as a series of three large, intermingled chromatin loops anchored close to the DJH RC, that provide comparable access between distal VH gene segments with rearranged 3’ DHJH (43). Hence, Igh locus topology is best described as a series of three large chromatin loops that are anchored at sub-TAD boundaries.
TAD boundaries are frequently marked by CBEs in a convergent orientation (40, 44) which participate in loop extrusion (45, 46). The loop extrusion model posits that chromatin loops are formed when cohesin is loaded onto and reels in DNA in an ATP-dependent process (45–48). Architectural “stripes”, visualized within Hi-C maps (49) may form when one subunit cohesin stalls near a strong CTCF loop anchor while the second one slides along the chromatin to form multiple interactions. The extrusion model explains how enhancers can processively track along arrays of promoters separated by long genomic intervals (45, 46, 50) and has been proposed as the mechanism that enforces deletional CSR (51, 52) and creates Igh locus contraction during V(D)J recombination (52). However, while sharp TAD boundaries are lost upon CTCF inactivation, compartment organization as well as TAD-like globular chromatin domain structures are preserved in single cell experiments (53) and the impact of CTCF inactivation on the transcriptome is small (54). Cohesin has been shown to promote clustering of enhancer elements in 3D spatial hubs (55). Intra-TAD contacts between regulatory elements facilitated by cohesin loop extrusion can be stabilized by other mechanisms such as homo-dimerization of the structural regulator YY1 (56). The inter-relationship of loop extrusion and a putative promoter-enhancer interactome in the Igh locus remains largely undefined.
New unpublished work from the Kenter group has identified highly transcribed VH gene promoters and a series of novel enhancers (NEs) that are pro-B cell specific, are involved in anchoring Igh subTAD loops and influence Igh repertoire formation in pro-B cells through formation of a promoter-promoter-enhancer hub. This is an interesting proposition as hundreds of VH exon promoters and newly recognized enhancers could participate in an intricate contact interactome that spatially organizes VH segments within the previously defined large chromatin loops and defines access probability to the RC and DJ segments. The presence of intra-TAD promoter-promoter-enhancer interactomes has been documented in several genetic loci and in different developmental and differentiation systems. Here we consider evidence that enhancers and promoters initiate specific interactions in nuclear space and propose that this interactome influences repertoire diversity.
Multiple lines of evidence support the existence of enhancer interactomes in different genomic contexts (57). While some studies link chromatin contacts between regulatory elements to transcriptional activity (58), in other examples these contacts precede gene activation (59). Most prominently, enhancers in olfactory sensory neurons form a large inter-chromosomal hub (60). In other systems, super-enhancers, clustered arrays of enhancer elements in close spatial proximity that can span several kilobases and are linked to the regulation of cell-identity genes with high transcriptional activity (61, 62), are highly involved in the formation of specific chromatin contacts. Genome architecture mapping identified abundant three-way contacts between super-enhancers and highly transcribed chromatin regions beyond the pairwise interactions detectable by 3C techniques (58). Similarly, the enhancer elements within a super-enhancer and target promoters can cluster spatially to form a hub structure with simultaneous multi-way interactions as demonstrated by multi-contact 4C for the locus control region of the beta-globin locus (63).
When considering the Igh locus it is important to note that genetic differences between inbred strains extend beyond coding variation into intergenic regions. To date, the mouse IgH locus has only been fully characterized in C57BL/6, which, as we noted above, has served as the primary model for characterizing the functional regions and mechanisms that dictate V(D)J recombination. However, a partial assembly, including the proximal region of Igh in the 129S1/SvImJ mouse strain was published in 2007 (20). A comparison of these haplotypes revealed evidence of both local sequence conservation as well as divergence, including examples of structural variation and single nucleotide differences (20) (Figure 2). For example, several complex regions (Figure 2A) represent insertions of Ig genes in the 129S1/SvImJ strain that are absent in C57BL/6. In addition, the degree of sequence identity between these two strains varies considerably across the locus, with sequence identities ranging between 88% and 98%. Even in regions characterized by high degrees of homology between 129S1/SvImJ and C57BL/6, SNPs occur at relatively high densities in both coding and intergenic regions (Figure 2B). The impact of such inter-strain haplotype diversity on V(D)J recombination has not been investigated.
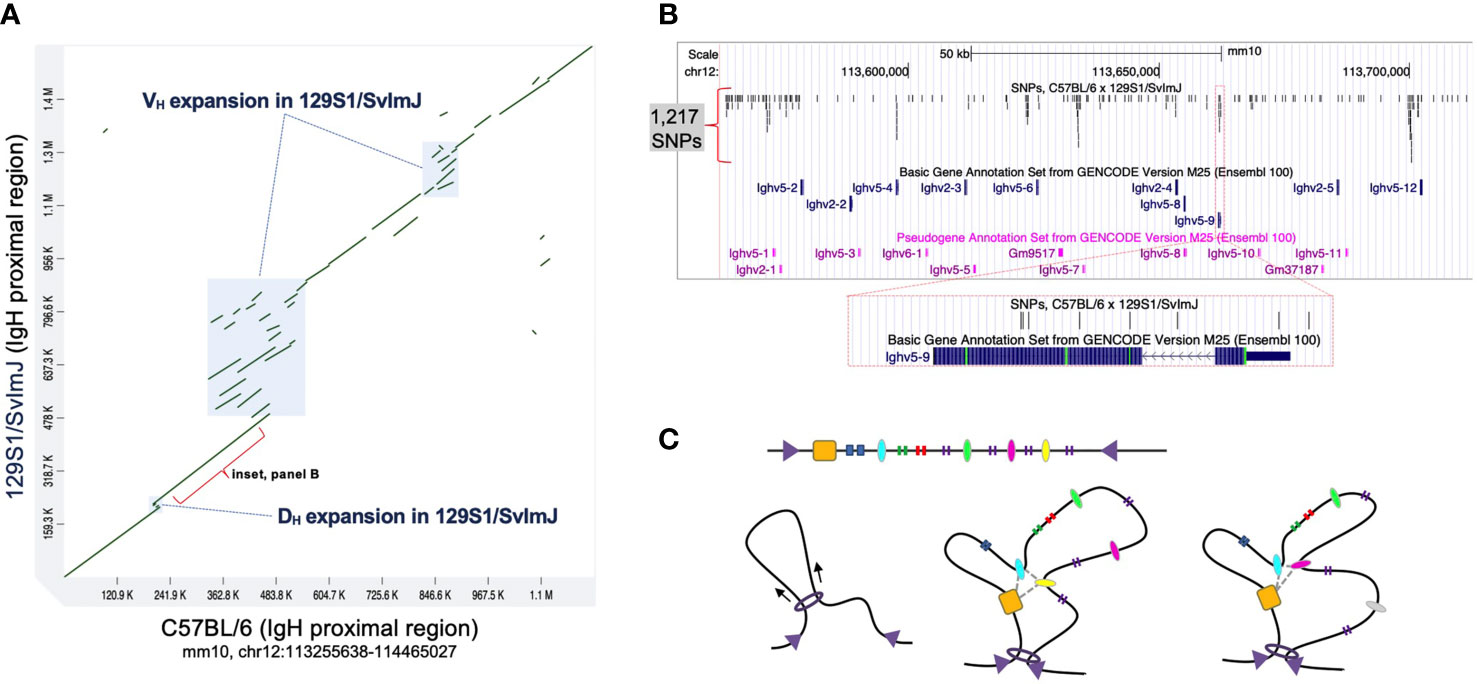
Figure 2 Comparison of the C57BL/6 and 129S1/SvlmJ Igh loci reveals significant polymorphisms. (A) A dot plot representing a sequence comparison of the proximal region of IgH (mm10, chr12:113255638-114465027) between the mouse strains 129S1/SvlmJ and C57BL/6; sequences from each strain were compared using MashMap (64, 65). Annotated boxes represent regions of complex structural variants, in which Ig gene segments vary in copy number between the two strains. Sequence identities within homologous regions (green diagonal lines) range between 84% and 98%. (B) A map of a 150 Kb region within the Igh proximal region (mm10, chr12:113562489-113712488). Shown are the positions of functional Ighv genes and pseudogenes, and single nucleotide polymorphisms (SNPs; indels not included) that differentiate the 129S1/SvlmJ and C57BL/6 haplotypes. In total, 1,217 SNPs are present, including variants in both coding and non-coding sequences, as illustrated in the inset panel centered on the functional gene Ighv5-9. SNPs in this region were identified by mapping the corresponding Igh sequence from 129S1/SvlmJ to mm10 using BLASR. (C) Top: Linear sequence of a chromatin neighborhood flanked by converging CTCF sites (purple triangles). Regulatory elements (ovals) and gene elements (vertical bars) are dispersed throughout the neighborhood. Left: Cohesin (ring) reels the chromatin fiber in to extrude a loop, facilitating interactions between regulatory elements. Middle: A promoter-enhancer interactome (gray dashed line) can confer locus structure with a distinct set of interactions. Right: Loss of association with the interactome results in topological changes and affects access to genetic elements.
Elsewhere in the genome, deletions or mutations in enhancer sequences can lead to aberrant gene expression and disease phenotypes (66). Genome-wide association studies show that the vast majority of sequence variants associated with common diseases and traits are located in such non-coding parts of the genome (67). In line with long-range enhancer interactions discussed above, misregulation of target gene expression due to variation in enhancers can occur tens of kilobases away in linear sequence space (68).
Promoters and enhancers are characterized by a high density of sometimes overlapping TF binding sites. Active enhancers are established through the recruitment of TFs to those binding sites which opens chromatin (69). Mechanistically, TFs can mediate promoter–enhancer contacts in a variety of ways directly or indirectly through binding of additional factors and structural proteins (reviewed in (57)). For example, in mouse embryonic stem cells, deletion of KLF4 binding sites or KLF4 ablation results in reduced contact frequency in enhancer hubs and diminished expression of multiple target genes (70).
Single nucleotide changes in regulatory sequences can impact the affinity for TF binding (71). It can tip the balance in sites where different factors compete for the same space (72, 73) or regulatory regions have multiple functions (74). Since TF binding is essential for establishing an active enhancer, SNPs detected in Igh intergenic regulatory regions could potentially ensue in a cascade of downstream effects. They can alter TF binding in enhancers and promoters, impact long-range enhancer-promoter interactions, and thereby change the composition of promoter-promoter-enhancer interactomes (Figure 2C).
In this context it is significant that variation of intergenic CBE sequence can have a profound effect on VH gene usage. VH gene access to the RC was recently shown to be dependent on the quality of the flanking CTCF binding element (CBE) and related ability of the gene to loop with IGCR1 (16, 17). VH81X is the second gene in the locus and is most prominently used. VH5-1 is the most DH proximal VH gene in the locus and is very rarely used even though its promoter and recombination signal sequence are intact and similar to that found for VH81X. However, the quality of the flanking CBE for VH5-1 is poor and when replaced with a functional motif directs looping with IGCR1 and high frequency recombination. Thus, the quality of CBEs within the Igh locus highlights the importance of the integrity of similar regulatory sequences which can be altered by Igh polymorphisms.
Genetic differences, such as those observed between 129S1/SvImJ and C57BL/6 (Figures 2A, B), when considered alongside observations that have been made for regulatory elements elsewhere in the genome, raise important questions about the potential for Ig genetic diversity to impact V(D)J recombination. First, there are numerous examples for which germline VH variants have been shown to contribute to antigen specificity (75–79) and associate with disease and clinical phenotypes in the context of infection, inflammation, and vaccination (31, 32, 80–83). Second, both large structural variants and single nucleotide polymorphisms could modify key regulatory elements, such as CTCF sites and in promoters and enhancers, either through the disruption of these elements (e.g. sequence deletions or loss-of-functions SNPs) or the creation of novel elements (e.g. through sequence duplication or gain-of-function SNPs). In addition, structural variants could also be expected to change the spatial organization of interacting regulatory elements by increasing or decreasing the genomic distance between particular elements, or by changing their orientation. These modifications would in turn be expected to impact promoter/enhancer interactomes, lead to changes in the epigenetic landscape, and influence the overall locus architecture and TAD structure, and ultimately affect the selection of particular VH, DH, and JH segments into the repertoire. We expect the discovery of such examples to continue as the inclusion of genetic variation in the study of repertoire diversity and dynamics becomes more commonplace.
Publicly available datasets were analyzed in this study. Sequence data and related gene annotations for the Igh locus in C57BL/6 were extracted from the mm10 genome reference assembly, available at https://genome.ucsc.edu. The sequence of the proximal Igh region of 129S1/SvlmJ was previously published by Retter et al. (20) available on GenBank under the accession number AJ851868.3.
Conceptualization, and Writing: AK, CW, and J-HS. Funding Acquisition, AK. All authors contributed to the article and approved the submitted version.
This work was supported by grants to AK from the NIH (RO1AI121286, R21AI151892).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Oscar Rodriguez for help conducting the analysis presented in Figure 2B.
1. Schatz DG, Ji Y. Recombination Centres and the Orchestration of V(D)J Recombination. Nat Rev Immunol (2011) 11(4):251–63. doi: 10.1038/nri2941
2. Ji Y, Resch W, Corbett E, Yamane A, Casellas R, Schatz DG. The In Vivo Pattern of Binding of RAG1 and RAG2 to Antigen Receptor Loci. Cell (2010) 141(3):419–31. doi: 10.1016/j.cell.2010.03.010
3. Hu J, Zhang Y, Zhao L, Frock RL, Du Z, Meyers RM, et al. Chromosomal Loop Domains Direct the Recombination of Antigen Receptor Genes. Cell (2015) 163(4):947–59. doi: 10.1016/j.cell.2015.10.016
4. Choi NM, Loguercio S, Verma-Gaur J, Degner SC, Torkamani A, Su AI, et al. Deep Sequencing of the Murine IgH Repertoire Reveals Complex Regulation of Nonrandom V Gene Rearrangement Frequencies. J Immunol (2013) 191(5):2393–402. doi: 10.4049/jimmunol.1301279
5. Bolland DJ, Koohy H, Wood AL, Matheson LS, Krueger F, Stubbington MJ, et al. Two Mutually Exclusive Local Chromatin States Drive Efficient V(D)J Recombination. Cell Rep (2016) 15(11):2475–87. doi: 10.1016/j.celrep.2016.05.020
6. Kleiman E, Loguercio S, Feeney AJ. Epigenetic Enhancer Marks and Transcription Factor Binding Influence Vkappa Gene Rearrangement in Pre-B Cells and Pro-B Cells. Front Immunol (2018) 9:2074. doi: 10.3389/fimmu.2018.02074
7. Aoki-Ota M, Torkamani A, Ota T, Schork N, Nemazee D. Skewed Primary Igkappa Repertoire and V-J Joining in C57BL/6 Mice: Implications for Recombination Accessibility and Receptor Editing. J Immunol (2012) 188(5):2305–15. doi: 10.4049/jimmunol.1103484
8. Matheson LS, Bolland DJ, Chovanec P, Krueger F, Andrews S, Koohy H, et al. Local Chromatin Features Including PU.1 and IKAROS Binding and H3K4 Methylation Shape the Repertoire of Immunoglobulin Kappa Genes Chosen for V(D)J Recombination. Front Immunol (2017) 8:1550. doi: 10.3389/fimmu.2017.01550
9. Williams GS, Martinez A, Montalbano A, Tang A, Mauhar A, Ogwaro KM, et al. Unequal VH Gene Rearrangement Frequency Within the Large VH7183 Gene Family is Not Due to Recombination Signal Sequence Variation, and Mapping of the Genes Shows a Bias of Rearrangement Based on Chromosomal Location. J Immunol (2001) 167(1):257–63. doi: 10.4049/jimmunol.167.1.257
10. Feeney AJ, Goebel P, Espinoza CR. Many Levels of Control of V Gene Rearrangement Frequency. Immunol Rev (2004) 200:44–56. doi: 10.1111/j.0105-2896.2004.00163.x
12. Degner-Leisso SC, Feeney AJ. Epigenetic and 3-Dimensional Regulation of V(D)J Rearrangement of Immunoglobulin Genes. Semin Immunol (2010) 22(6):346–52. doi: 10.1016/j.smim.2010.08.002
13. Yancopoulos GD, DePinho RA, Zimmerman KA, Lutzker SG, Rosenberg N, Alt FW. Secondary Genomic Rearrangement Events in Pre-B Cells: VHDJH Replacement by a LINE-1 Sequence and Directed Class Switching. EMBO J (1986) 5(12):3259–66. doi: 10.1002/j.1460-2075.1986.tb04637.x
14. Yancopoulos GD, Alt FW. Developmentally Controlled and Tissue-Specific Expression of Unrearranged VH Gene Segments. Cell (1985) 40(2):271–81. doi: 10.1016/0092-8674(85)90141-2
15. Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of Programmed DNA Lesions and Genomic Instability in the Immune System. Cell (2013) 152(3):417–29. doi: 10.1016/j.cell.2013.01.007
16. Qiu X, Kumari G, Gerasimova T, Du H, Labaran L, Singh A, et al. Sequential Enhancer Sequestration Dysregulates Recombination Center Formation at the IgH Locus. Mol Cell (2018) 70(1):21–33 e6. doi: 10.1016/j.molcel.2018.02.020
17. Jain S, Ba Z, Zhang Y, Dai HQ, Alt FW. Ctcf-Binding Elements Mediate Accessibility of RAG Substrates During Chromatin Scanning. Cell (2018) 174(1):102–116 e14. doi: 10.1016/j.cell.2018.04.035
18. Watson CT, Glanville J, Marasco WA. The Individual and Population Genetics of Antibody Immunity. Trends Immunol (2017) 38(7):459–70. doi: 10.1016/j.it.2017.04.003
19. Trepicchio W Jr., Barrett KJ. The Igh-V Locus of MRL Mice: Restriction Fragment Length Polymorphism in Eleven Strains of Mice as Determined With VH and D Gene Probes. J Immunol (1985) 134(4):2734–9.
20. Retter I, Chevillard C, Scharfe M, Conrad A, Hafner M, Im TH, et al. Sequence and Characterization of the Ig Heavy Chain Constant and Partial Variable Region of the Mouse Strain 129S1. J Immunol (2007) 179(4):2419–27. doi: 10.4049/jimmunol.179.4.2419
21. Kidd MJ, Chen Z, Wang Y, Jackson KJ, Zhang L, Boyd SD, et al. The Inference of Phased Haplotypes for the Immunoglobulin H Chain V Region Gene Loci by Analysis of VDJ Gene Rearrangements. J Immunol (2012) 188(3):1333–40. doi: 10.4049/jimmunol.1102097
22. Gonzalez-Garay ML, Cranford SM, Braun MC, Doris PA. Diversity in the Preimmune Immunoglobulin Repertoire of SHR Lines Susceptible and Resistant to End-Organ Injury. Genes Immun (2014) 15(8):528–33. doi: 10.1038/gene.2014.40
23. Collins AM, Wang Y, Roskin KM, Marquis CP, Jackson KJ. The Mouse Antibody Heavy Chain Repertoire is Germline-Focused and Highly Variable Between Inbred Strains. Philos Trans R Soc Lond B Biol Sci (2015) 370(1676). doi: 10.1098/rstb.2014.0236
24. Watson CT, Kos JT, Gibson WS, Newman L, Deikus G, Busse CE, et al. A Comparison of Immunoglobulin IGHV, IGHD and IGHJ Genes in Wild-Derived and Classical Inbred Mouse Strains. Immunol Cell Biol (2019) 97(10):888–901. doi: 10.1111/imcb.12288
25. Vazquez Bernat N, Corcoran M, Nowak I, Kaduk M, Castro Dopico X, Narang S, et al. Rhesus and Cynomolgus Macaque Immunoglobulin Heavy-Chain Genotyping Yields Comprehensive Databases of Germline VDJ Alleles. Immunity (2021) 54(2):355–366 e4. doi: 10.1016/j.immuni.2020.12.018
26. Rodriguez OL, Gibson WS, Parks T, Emery M, Powell J, Strahl M, et al. A Novel Framework for Characterizing Genomic Haplotype Diversity in the Human Immunoglobulin Heavy Chain Locus. Front Immunol (2020) 11:2136. doi: 10.3389/fimmu.2020.02136
27. Gidoni M, Snir O, Peres A, Polak P, Lindeman I, Mikocziova I, et al. Mosaic Deletion Patterns of the Human Antibody Heavy Chain Gene Locus Shown by Bayesian Haplotyping. Nat Commun (2019) 10(1):628. doi: 10.1038/s41467-019-08489-3
28. Scheepers C, Shrestha RK, Lambson BE, Jackson KJ, Wright IA, Naicker D, et al. Ability to Develop Broadly Neutralizing HIV-1 Antibodies Is Not Restricted by the Germline Ig Gene Repertoire. J Immunol (2015) 194(9):4371–8. doi: 10.4049/jimmunol.1500118
29. Glanville J, Kuo TC, von Budingen HC, Guey L, Berka J, Sundar PD, et al. Naive Antibody Gene-Segment Frequencies are Heritable and Unaltered by Chronic Lymphocyte Ablation. Proc Natl Acad Sci USA (2011) 108(50):20066–71. doi: 10.1073/pnas.1107498108
30. Rubelt F, Bolen CR, McGuire HM, Vander Heiden JA, Gadala-Maria D, Levin M, et al. Individual Heritable Differences Result in Unique Cell Lymphocyte Receptor Repertoires of Naive and Antigen-Experienced Cells. Nat Commun (2016) 7:11112. doi: 10.1038/ncomms11112
31. Feeney AJ, Atkinson MJ, Cowan MJ, Escuro G, Lugo G. A Defective Vkappa A2 Allele in Navajos Which may Play a Role in Increased Susceptibility to Haemophilus Influenzae Type B Disease. J Clin Invest (1996) 97(10):2277–82. doi: 10.1172/JCI118669
32. Avnir Y, Watson CT, Glanville J, Peterson EC, Tallarico AS, Bennett AS, et al. Ighv1-69 Polymorphism Modulates Anti-Influenza Antibody Repertoires, Correlates With IGHV Utilization Shifts and Varies by Ethnicity. Sci Rep (2016) 6:20842. doi: 10.1038/srep20842
33. Sasso EH, Johnson T, Kipps TJ. Expression of the Immunoglobulin VH Gene 51p1 is Proportional to its Germline Gene Copy Number. J Clin Invest (1996) 97(9):2074–80. doi: 10.1172/JCI118644
34. Kidd MJ, Jackson KJ, Boyd SD, Collins AM. Dj Pairing During VDJ Recombination Shows Positional Biases That Vary Among Individuals With Differing Ighd Locus Immunogenotypes. J Immunol (2016) 196(3):1158–64. doi: 10.4049/jimmunol.1501401
35. Cremer T, Cremer M. Chromosome Territories. Cold Spring Harb Perspect Biol (2010) 2(3):a003889. doi: 10.1101/cshperspect.a003889
36. Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science (2009) 326(5950):289–93. doi: 10.1126/science.1181369
37. Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature (2012) 485(7398):376–80. doi: 10.1038/nature11082
38. Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, et al. Spatial Partitioning of the Regulatory Landscape of the X-inactivation Centre. Nature (2012) 485(7398):381–5. doi: 10.1038/nature11049
39. Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, et al. Three-Dimensional Folding and Functional Organization Principles of the Drosophila Genome. Cell (2012) 148(3):458–72. doi: 10.1016/j.cell.2012.01.010
40. Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell (2014) 159(7):1665–80. doi: 10.1016/j.cell.2014.11.021
41. Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, et al. Topologically Associating Domains are Stable Units of Replication-Timing Regulation. Nature (2014) 515(7527):402–5. doi: 10.1038/nature13986
42. Montefiori L, Wuerffel R, Roqueiro D, Lajoie B, Guo C, Gerasimova T, et al. Extremely Long-Range Chromatin Loops Link Topological Domains to Facilitate a Diverse Antibody Repertoire. Cell Rep (2016) 14(4):896–906. doi: 10.1016/j.celrep.2015.12.083
43. Lucas JS, Zhang Y, Dudko OK, Murre C. 3D Trajectories Adopted by Coding and Regulatory DNA Elements: First-Passage Times for Genomic Interactions. Cell (2014) 158(2):339–52. doi: 10.1016/j.cell.2014.05.036
44. Vietri Rudan M, Barrington C, Henderson S, Ernst C, Odom DT, Tanay A, et al. Comparative Hi-C Reveals That CTCF Underlies Evolution of Chromosomal Domain Architecture. Cell Rep (2015) 10(8):1297–309. doi: 10.1016/j.celrep.2015.02.004
45. Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, et al. Chromatin Extrusion Explains Key Features of Loop and Domain Formation in Wild-Type and Engineered Genomes. Proc Natl Acad Sci USA (2015) 112(47):E6456–65. doi: 10.1073/pnas.1518552112
46. Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep (2016) 15(9):2038–49. doi: 10.1016/j.celrep.2016.04.085
47. Nasmyth K. Disseminating the Genome: Joining, Resolving, and Separating Sister Chromatids During Mitosis and Meiosis. Annu Rev Genet (2001) 35:673–745. doi: 10.1146/annurev.genet.35.102401.091334
48. Nichols MH, Corces VG. A CTCF Code for 3D Genome Architecture. Cell (2015) 162(4):703–5. doi: 10.1016/j.cell.2015.07.053
49. Vian L, Pekowska A, Rao SSP, Kieffer-Kwon KR, Jung S, Baranello L, et al. The Energetics and Physiological Impact of Cohesin Extrusion. Cell (2018) 173(5):1165–1178 e20. doi: 10.1016/j.cell.2018.03.072
50. Dekker J, Mirny L. The 3d Genome as Moderator of Chromosomal Communication. Cell (2016) 164(6):1110–21. doi: 10.1016/j.cell.2016.02.007
51. Dong J, Panchakshari RA, Zhang T, Zhang Y, Hu J, Volpi SA, et al. Orientation-Specific Joining of AID-initiated DNA Breaks Promotes Antibody Class Switching. Nature (2015) 525(7567):134–9. doi: 10.1038/nature14970
52. Zhang X, Zhang Y, Ba Z, Kyritsis N, Casellas R, Alt FW. Fundamental Roles of Chromatin Loop Extrusion in Antibody Class Switching. Nature (2019) 575(7782):385–9. doi: 10.1038/s41586-019-1723-0
53. Bintu B, Mateo LJ, Su JH, Sinnott-Armstrong NA, Parker M, Kinrot S, et al. Super-Resolution Chromatin Tracing Reveals Domains and Cooperative Interactions in Single Cells. Science (2018) 362(6413). doi: 10.1126/science.aau1783
54. Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, et al. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains From Genomic Compartmentalization. Cell (2017) 169(5):930–944 e22. doi: 10.1016/j.cell.2017.05.004
55. Ing-Simmons E, Seitan VC, Faure AJ, Flicek P, Carroll T, Dekker J, et al. Spatial Enhancer Clustering and Regulation of Enhancer-Proximal Genes by Cohesin. Genome Res (2015) 25(4):504–13. doi: 10.1101/gr.184986.114
56. Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, et al. Yy1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell (2017) 171(7):1573–1588 e28. doi: 10.1016/j.cell.2017.11.008
57. Di Giammartino DC, Polyzos A, Apostolou E. Transcription Factors: Building Hubs in the 3D Space. Cell Cycle (2020) 19(19):2395–410. doi: 10.1080/15384101.2020.1805238
58. Beagrie RA, Scialdone A, Schueler M, Kraemer DC, Chotalia M, Xie SQ, et al. Complex Multi-Enhancer Contacts Captured by Genome Architecture Mapping. Nature (2017) 543(7646):519–24. doi: 10.1038/nature21411
59. Ghavi-Helm Y, Klein FA, Pakozdi T, Ciglar L, Noordermeer D, Huber W, et al. Enhancer Loops Appear Stable During Development and are Associated With Paused Polymerase. Nature (2014) 512(7512):96–100. doi: 10.1038/nature13417
60. Markenscoff-Papadimitriou E, Allen WE, Colquitt BM, Goh T, Murphy KK, Monahan K, et al. Enhancer Interaction Networks as a Means for Singular Olfactory Receptor Expression. Cell (2014) 159(3):543–57. doi: 10.1016/j.cell.2014.09.033
61. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell (2013) 153(2):307–19. doi: 10.1016/j.cell.2013.03.035
62. Parker SC, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, et al. A. National Institutes of Health Intramural Sequencing Center Comparative Sequencing Program and N. C. S. P. Authors: Chromatin Stretch Enhancer States Drive Cell-Specific Gene Regulation and Harbor Human Disease Risk Variants. Proc Natl Acad Sci USA (2013) 110(44):17921–6. doi: 10.1073/pnas.1317023110
63. Allahyar A, Vermeulen C, Bouwman BAM, Krijger PHL, Verstegen M, Geeven G, et al. Enhancer Hubs and Loop Collisions Identified From Single-Allele Topologies. Nat Genet (2018) 50(8):1151–60. doi: 10.1038/s41588-018-0161-5
64. Cabanettes F, Klopp C. D-GENIES: Dot Plot Large Genomes in an Interactive, Efficient and Simple Way. PeerJ (2018) 6:e4958. doi: 10.7717/peerj.4958
65. Jain C, Koren S, Dilthey A, Phillippy AM, Aluru S. A Fast Adaptive Algorithm for Computing Whole-Genome Homology Maps. Bioinformatics (2018) 34(17):i748–56. doi: 10.1093/bioinformatics/bty597
66. Schoenfelder S, Fraser P. Long-Range Enhancer-Promoter Contacts in Gene Expression Control. Nat Rev Genet (2019) 20(8):437–55. doi: 10.1038/s41576-019-0128-0
67. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science (2012) 337(6099):1190–5. doi: 10.1126/science.1222794
68. Smith E, Shilatifard A. Enhancer Biology and Enhanceropathies. Nat Struct Mol Biol (2014) 21(3):210–9. doi: 10.1038/nsmb.2784
69. Calo E, Wysocka J. Modification of Enhancer Chromatin: What, How, and Why? Mol Cell (2013) 49(5):825–37. doi: 10.1016/j.molcel.2013.01.038
70. Di Giammartino DC, Kloetgen A, Polyzos A, Liu Y, Kim D, Murphy D, et al. KLF4 is Involved in the Organization and Regulation of Pluripotency-Associated Three-Dimensional Enhancer Networks. Nat Cell Biol (2019) 21(10):1179–90. doi: 10.1038/s41556-019-0390-6
71. Pai AA, Pritchard JK, Gilad Y. The Genetic and Mechanistic Basis for Variation in Gene Regulation. PloS Genet (2015) 11(1):e1004857. doi: 10.1371/journal.pgen.1004857
72. Glasmacher E, Agrawal S, Chang AB, Murphy TL, Zeng W, Vander Lugt B, et al. A Genomic Regulatory Element That Directs Assembly and Function of Immune-Specific AP-1-IRF Complexes. Science (2012) 338(6109):975–80. doi: 10.1126/science.1228309
73. Chang YK, Zuo Z, Stormo GD. Quantitative Profiling of BATF Family Proteins/JUNB/IRF Hetero-Trimers Using Spec-Seq. BMC Mol Biol (2018) 19(1):5. doi: 10.1186/s12867-018-0106-7
74. Hua JT, Ahmed M, Guo H, Zhang Y, Chen S, Soares F, et al. Risk SNP-Mediated Promoter-Enhancer Switching Drives Prostate Cancer Through Lncrna PCAT19. Cell (2018) 174(3):564–575 e18. doi: 10.1016/j.cell.2018.06.014
75. Sui J, Hwang WC, Perez S, Wei G, Aird D, Chen LM, et al. Structural and Functional Bases for Broad-Spectrum Neutralization of Avian and Human Influenza A Viruses. Nat Struct Mol Biol (2009) 16(3):265–73. doi: 10.1038/nsmb.1566
76. Yeung YA, Foletti D, Deng X, Abdiche Y, Strop P, Glanville J, et al. Germline-Encoded Neutralization of a Staphylococcus Aureus Virulence Factor by the Human Antibody Repertoire. Nat Commun (2016) 7:1–14. doi: 10.1038/ncomms13376
77. Yacoob C, Pancera M, Vigdorovich V, Oliver BG, Glenn JA, Feng J, et al. Differences in Allelic Frequency and CDRH3 Region Limit the Engagement of HIV Env Immunogens by Putative Vrc01 Neutralizing Antibody Precursors. Cell Rep (2016) 17(6):1560–70. doi: 10.1016/j.celrep.2016.10.017
78. Liu L, Lucas AH. Igh V3-23*01 and its Allele V3-23*03 Differ in Their Capacity to Form the Canonical Human Antibody Combining Site Specific for the Capsular Polysaccharide of Haemophilus Influenzae Type B. Immunogenetics (2003) 55(5):336–8. doi: 10.1007/s00251-003-0583-8
79. Ying T, Prabakaran P, Du L, Shi W, Feng Y, Wang Y, et al. Junctional and Allele-Specific Residues are Critical for MERS-CoV Neutralization by an Exceptionally Potent Germline-Like Antibody. Nat Commun (2015) 6:8223. doi: 10.1038/ncomms9223
80. Johnson TA, Mashimo Y, Wu JY, Yoon D, Hata A, Kubo M, et al. Association of an IGHV3-66 Gene Variant With Kawasaki Disease. J Hum Genet (2020). doi: 10.1038/s10038-020-00864-z
81. Parks T, Mirabel MM, Kado J, Auckland K, Nowak J, Rautanen A, et al. Association Between a Common Immunoglobulin Heavy Chain Allele and Rheumatic Heart Disease Risk in Oceania. Nat Commun (2017) 8:14946. doi: 10.1038/ncomms14946
82. Dhande IS, Kneedler SC, Joshi AS, Zhu Y, Hicks MJ, Wenderfer SE, et al. Germ-Line Genetic Variation in the Immunoglobulin Heavy Chain Creates Stroke Susceptibility in the Spontaneously Hypertensive Rat. Physiol Genomics (2019) 51(11):578–85. doi: 10.1152/physiolgenomics.00054.2019
83. Raposo B, Dobritzsch D, Ge C, Ekman D, Xu B, Lindh I, et al. Epitope-Specific Antibody Response is Controlled by Immunoglobulin V(H) Polymorphisms. J Exp Med (2014) 211(3):405–11. doi: 10.1084/jem.20130968
Keywords: Chromatin, B cell, immunoglobulin, VDJ recombination, VDJ repertoire
Citation: Kenter AL, Watson CT and Spille J-H (2021) Igh Locus Polymorphism May Dictate Topological Chromatin Conformation and V Gene Usage in the Ig Repertoire. Front. Immunol. 12:682589. doi: 10.3389/fimmu.2021.682589
Received: 18 March 2021; Accepted: 26 April 2021;
Published: 18 May 2021.
Edited by:
Hermann Eibel, University of Freiburg Medical Center, GermanyCopyright © 2021 Kenter, Watson and Spille. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amy L. Kenter, c3RhcjFAdWljLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.