 Iwan G. A. Raza
Iwan G. A. Raza Alexander J. Clarke
Alexander J. Clarke- 1Medical Sciences Division, University of Oxford, Oxford, United Kingdom
- 2Kennedy Institute of Rheumatology, University of Oxford, Oxford, United Kingdom
B cells are central to the pathogenesis of multiple autoimmune diseases, through antigen presentation, cytokine secretion, and the production of autoantibodies. During development and differentiation, B cells undergo drastic changes in their physiology. It is emerging that these are accompanied by equally significant shifts in metabolic phenotype, which may themselves also drive and enforce the functional properties of the cell. The dysfunction of B cells during autoimmunity is characterised by the breaching of tolerogenic checkpoints, and there is developing evidence that the metabolic state of B cells may contribute to this. Determining the metabolic phenotype of B cells in autoimmunity is an area of active study, and is important because intervention by metabolism-altering therapeutic approaches may represent an attractive treatment target.
Introduction
B lymphocytes play a crucial role in immune responses against pathogens and tumours through the production of protective antibodies. They are also implicated in the development and control of autoimmunity, in which the immune response is directed towards self-antigens. Various B cell populations display the capacity for both protective and self-destructive activity. The best understood B lymphocytes are conventional bone marrow-derived B2 cells, which mediate adaptive humoral responses. In various autoimmune diseases, including rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), B2 cells are responsible for autoantibody production, the presentation of endogenous peptides to self-reactive T cells, and the secretion of proinflammatory cytokines (1–5). Additional to B2 cells are a distinct population of innate–like antibody–secreting lymphocytes, known as B1 cells. Generated in the foetal liver, B1 cells are found with the peritoneum and are capable of resting self–renewal. B1 cells produce natural IgM against bacterial polysaccharide antigens and exhibit a significant degree of self–reactivity (6, 7). The autoantibodies and cytokines produced by B1 cells can mediate autoimmunity (8, 9). However, B1 cell–derived autoantibodies also facilitate the clearance of apoptotic cells, a major source of autoantigens, and appear to promote intestinal homeostasis (10–12). Meanwhile, regulatory B cells (Bregs) play a well–defined role in tolerogenesis, secreting immunoregulatory cytokines such as IL–10 and TGF–β, and enacting contact–dependent suppression of self–reactive lymphocytes (13).
The causes of B cell dysfunction in autoimmunity remain incompletely defined. Metabolism, although a well–established regulator of cellular activity, has only relatively recently been appreciated as a determinant of B cell function in health and disease. Physiologically, metabolism enables proper B cell development, differentiation and antibody secretion (14, 15). Unsurprisingly, the contrasting phenotypes of B cells at different stages of development and maturity are reflected in significant variations in metabolic activity (16, 17). Moreover, different B cell subsets, particularly B1 and B2 cells, utilise divergent metabolic pathways (18). Metabolism forms an important component of deletional and anergic checkpoints against autoimmunity, and metabolic dysregulation is associated with the escape from tolerogenic checkpoints and the enhanced functionality of self–reactive B cells (19–22). As well as traditional aspects of cellular metabolism, B cells depend on autophagy, a mechanism of degradative processing, principally of damaged cellular components. Autophagy maintains metabolic homeostasis during nutrient deprivation and supports long–term plasma cell (PC) viability (23). Pathologically, autophagy appears to support self–reactive B cells in subverting autoimmune checkpoints, to undergo activation by innate immune signals, and present autoantigens to T lymphocytes (24–26).
In this review, we describe B cell metabolism and autophagy in health, highlighting their roles in immune homeostasis. We then discuss the metabolic and autophagic abnormalities seen in autoimmune B cells, and how these may promote self–destructive responses. Finally, the potential of B cell metabolism and autophagy as therapeutic targets in autoimmunity will be explored. This review focuses primarily on conventional B2 cells, which are relatively well defined metabolically, although comparisons are made to B1 cells and Bregs where possible.
Metabolism and Autophagy in B Cell Development
The development of conventional B2 B cells must generate a vast and hugely diverse repertoire of cells capable of antibody secretion, whilst purging cells with self–reactive antigen specificities. Moreover, rapid growth and proliferation must be achieved in the metabolically challenging environment of the bone marrow, requiring the careful balancing of anabolic and catabolic signalling. The former is largely mediated by c–Myc and mitochondrial target of rapamycin complex (mTORC) signalling, which fuel protein synthesis and cell growth by upregulating glycolysis and oxidative phosphorylation (OXPHOS) (14, 27, 28). mTORC signalling plays a crucial role in successful B cell development (27, 28). In mice, deletion of the mTORC1–associated protein Raptor prevents the interleukin (IL)–7–driven development of pro–B cells (14, 29). Without mTORC1 signalling, pro–B cells are less able to transition into pre–B cells, the precursors in which the pre–B cell receptor (BCR), consisting of mature immunoglobulin heavy chains and surrogate light chains, is expressed (14, 29). Unlike mTORC1, the role of mTORC2 in early B cell development has been contested, although it appears to facilitate peripheral B cell maturation (14, 30). Specifically, mTORC2 has been implicated in regulating mTORC1 and c–Myc activity during the terminal stages of B cell development (31).
While mTORC signalling is important during B cell development, excessive anabolic activity is detrimental. Indeed, B cell development is compromised at the large pre–B cell stage following the deletion of Fnip1 (32). Fnip1 interacts with 5’ adenosine monophosphate–activated protein kinase (AMPK), an energy stress sensor and catabolic regulator which opposes mTORC1. Although AMPK can promote catabolism in the absence of Fnip1, its ability to inhibit mTORC signalling is impaired (32). The metabolic stress resulting from unrestrained anabolism renders pre–B cells more vulnerable to apoptosis following pre–BCR cross–linking (32). The concept of metabolism as a regulator of cell death, controlling B cell precursor viability in response to antigen stimulation, has implications for tolerogenic checkpoints against autoimmunity (33).
The large pre–B cell stage, during which the pre–BCR is expressed on the cell surface and tested for affinity towards self–antigens in the bone marrow, represents both an autoimmune checkpoint and a period of metabolic vulnerability (34). Although its necessity during B cell development is controversial, glucose metabolism represents one example of this vulnerability (20, 32, 35). The rapid proliferation of large pre–B cells is believed to be sustained through upregulated glucose metabolism, given that large pre–B cells import more glucose than other precursor populations (16, 35). Large pre–B cells experience significant oxidative stress and are vulnerable to glycolytic inhibition, which impairs their transition into small pre–B cells (16, 35). Signalling through an autoreactive pre–BCR drives hyperactivation of the phosphoinositide 3–kinase (PI3K)–protein kinase B (Akt)–mTORC1 pathway, with the resulting metabolic stress inducing negative selection (19). Activation of a non–autoreactive pre–BCR does not affect Akt activation or viability in pre–B or leukemic pre–B (pre–B ALL) cells (19, 36). In contrast, activation of an autoreactive pre–BCR induces rapid, Akt–dependent cell death (19). In pre–B ALL cells, deletion of the PI3K inhibitor phosphatase and tensin homologue (PTEN) increases glycolytic flux, although elevated anabolism results in ATP depletion and cell death (19). These changes are reversed by the mTORC1 inhibitor rapamycin, suggesting that hyperactivation of the PI3K–Akt–mTORC1 pathway downstream of an autoreactive pre–BCR results in an energy crisis (19). PTEN plays a vital role in the development of pro–B cells, reducing their susceptibility to apoptosis (14). While PTEN deletion affects non–metabolic features of B cell precursors, such as B lymphoid transcription factor expression, these results further highlight the importance of balanced metabolic programmes during B cell development (14).
Alongside controlling other aspects of development, B lymphoid transcriptional factors themselves impose metabolic restriction upon B cell precursors, perhaps to enable hyperactivation–induced cell death (37). Mutations in the transcription factors PAX5 and IKZF1 are commonly seen in acute lymphoblastic leukaemia, suggesting that their expression may confer a selective disadvantage (37). The inducible reconstitution of PAX5 and IKZF1 in pre–B ALL cells reduces glucose uptake and ATP synthesis, promoting cell death (37). Notably, the B cell–specific expression of a non–functional IKZF1 predisposes mice to the development of autoimmunity, supporting the idea that these transcription factors may play a tolerogenic role during B cell development (38). Metabolic restriction is also a prominent feature of B cell anergy. Along with apoptosis and receptor editing, tolerogenesis can be exerted on self–reactive B cells and their precursors through the induction of anergy, rendering B cells hyporesponsive to antigenic stimulation. Anergy is a major mechanism of tolerising early transitional B cells following egress from the bone marrow. Anergic B cells are characterised by suppressed PI3K signalling and impaired metabolic reprogramming in response to BCR or Toll–like receptor (TLR) 4 stimulation (20, 39). Presumably, metabolic suppression increases the activation threshold of anergic self–reactive B cells.
Given its role in promoting metabolic homeostasis, the role of autophagy in B2 cell development has been explored (Figure 1) (40). Reconstitution of the foetal livers of Rag1–/–mice with cells lacking the key autophagy gene Atg5 demonstrates a developmental block at the pre–B cell stage (41). When Atg5 deletion was restricted to mature B cells, splenic and lymph node B cell populations were unaffected, implying that autophagy is necessary for the development, but not peripheral maintenance, of B2 cells (41). However, pro– to pre–B cell transition occurs in the absence of Atg5 expression following conditional deletion using Cd79a–cre (40). In contrast, autophagy was necessary to maintain populations of mature B cells in the periphery (40). That autophagy may be dispensable in B2 cell precursors is perhaps unsurprising, given the crucial developmental role of the autophagy inhibitor mTORC1 (14).
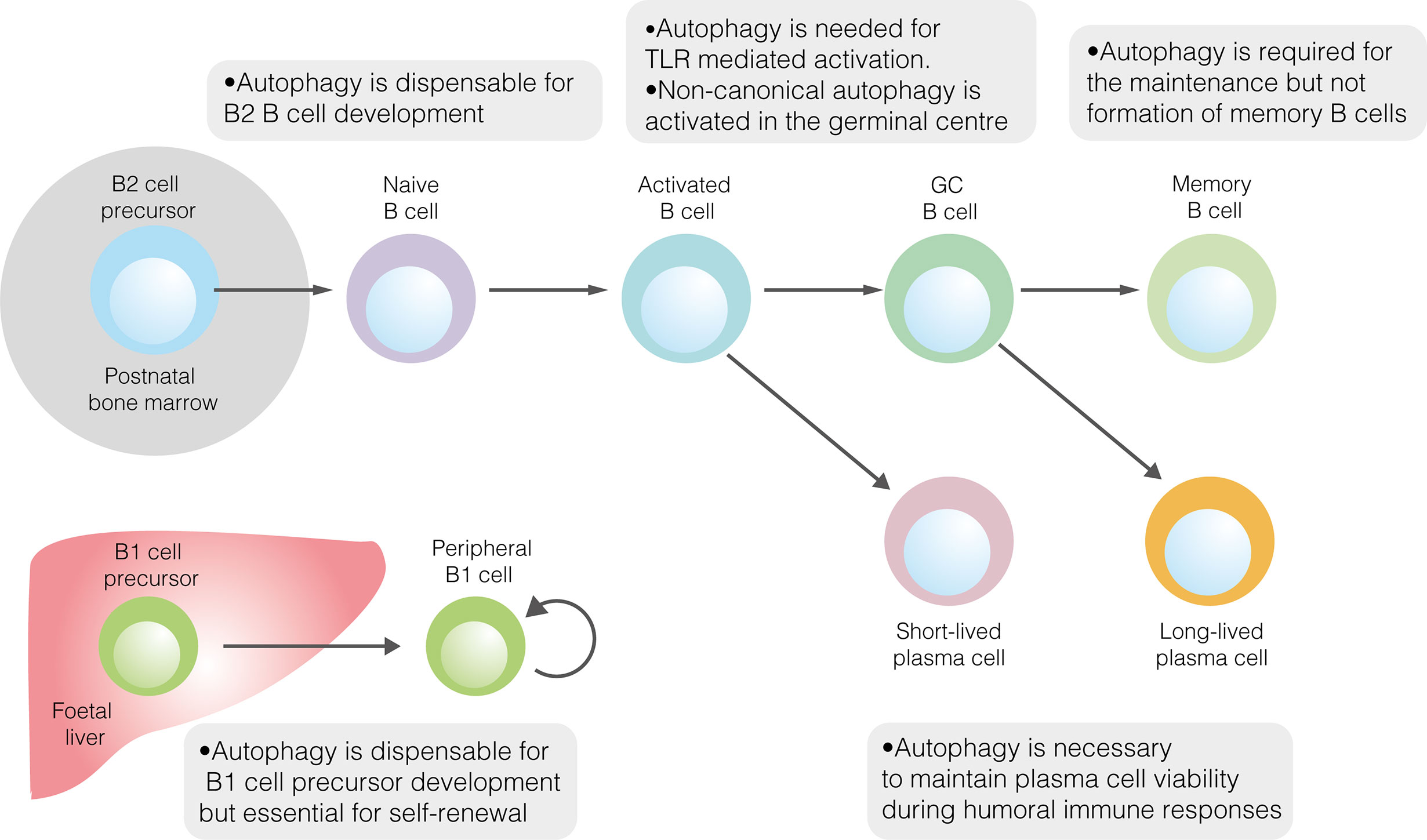
Figure 1 The role of autophagy in developmental and mature B cells. Autophagy performs homeostatic roles in various B cell subsets. It is dispensable for the development of both B1 and B2 cells but is required for the peripheral self–renewal of B1 cells. Regarding B2 cells, autophagy is necessary for TLR–mediated, but not antigen–driven, activation. Autophagy also plays important roles in sustaining plasma cell immunoglobulin production and ensuring memory B cell survival.
In contrast to B2 cell development in bone marrow, innate–like B1 cells develop in the foetal liver before migrating principally to the peritoneum and pleura (42). Given that B1 cells occupy different niches and rely upon self–renewal for population maintenance, their metabolic phenotype unsurprisingly differs from that of conventional B2 cells (42). Compared to follicular B2 cells, peritoneal B1 cells are characterised by greater glucose uptake, higher rates of glycolysis and OXPHOS and heightened sensitivity to glycolytic inhibition (18). Mechanistically, elevated glucose metabolism is likely driven by high levels of c–Myc expression (18, 43). In keeping with their localisation in the lipid–rich environment of the peritoneum, B1a cells extensively acquire exogenous fatty acids, while also utilising endogenous fatty acid synthesis (18).
Autophagy has also been explored in B1 cells (Figure 1). The loss of autophagic flux in mature B cells leads to significant depletion of B1a, though not B1b, cells (41). The specific role of autophagy in B1 cell development has only recently been explored (18). In mice, the B1 progenitor population is unaffected by the absence of autophagy, suggesting that autophagy is not required for B1 cell development (18). This conclusion is supported by temporal changes in the murine B1a cell population in the absence of autophagy. While the B1a cell compartment is normal at two weeks of age, by 12 weeks it is dramatically smaller than in wildtype mice (18). Together, these results suggest that autophagy is needed for peripheral self–renewal but not the differentiation of B1a cells. In peritoneal B1a cells, autophagy appears to be important for controlling the expression of metabolic genes, fatty acid uptake and degradation of lipid droplets (lipophagy) (18).
B Cell Metabolism Following Activation
In the periphery, naïve B cells are maintained in a state of metabolic quiescence, which likely promotes their long–term viability (44). The maturation of transitional B cells, which egress from the bone marrow and are peripherally tolerised, to follicular cells, is associated with a reduction in OXPHOS, glycolysis and protein synthesis (44). An increased ratio of AMPK: mTORC1 activity suppresses anabolism and exposure to oxidative stress (44). However, the survival of naïve B cells requires some degree of metabolic activity. Their maintenance depends on tonic signalling via the BCR and B cell activating factor (BAFF) receptor, both of which activate PI3K (45, 46). Notably, BAFF signalling upregulates the expression of enzymes involved in glucose metabolism (46). IL–4, another extrinsic signal required by naïve B cells, upregulates glucose uptake and glycolysis in murine B cells in a PI3K–independent manner (47). Although these signals promote glucose metabolism, naïve B cells appear to rely heavily on fatty acid oxidation to generate ATP (20).
During B cell activation, this relatively inactive metabolic state is rapidly reversed, enabling growth and proliferation. Stimulation of murine B cells via their BCR increases mitochondrial mass and PI3K–dependent glucose uptake (20, 48). In mice, elevated glucose import is enabled by Glut1 upregulation, although human B cells express relatively little Glut1 and may rely on other transporters (15, 48). However, the nature of glucose metabolism in activated B cells has been contested. Activation via TLR4 or the BCR had been thought to upregulate both glycolysis and OXPHOS in a balanced manner, with only the former considered essential for successful antibody responses (20, 48, 49). A recent analysis, combining RNA sequencing and stable isotope tracing, has instead suggested that mitochondrial metabolism, but not glycolysis, is upregulated during B cell activation (17). While glucose did flux through glycolysis, much was diverted towards the pentose phosphate pathway, providing substrates for nucleotide synthesis and the management of oxidative stress (17, 48). While excessive oxidative stress compromises cell viability, reactive oxygen species (ROS) represent important signalling molecules in B cells. ROS levels, determined in part by mTORC1 activity, provide instructive signals during differentiation through the regulation of haem synthesis (50, 51). As well as feeding the pentose phosphate pathway, imported glucose is used for de novo lipogenesis (17, 52). Fatty acid oxidation, which is extensively utilised by naïve B cells, is downregulated following activation (20). Instead, glutamine appears to represent a major substrate for mitochondrial respiration following B cell activation (17). In activated B cells, mitochondrial respiratory capacity and homeostasis are maintained by AMPK (53).
Successful antigen–driven B cell activation requires T cell costimulation, creating a checkpoint against autoimmunity. T cells are subject to stringent tolerogenesis and play an important role in preventing the activation of self–reactive B cells which have escaped other tolerance mechanisms. Metabolism contributes to the function of this post–activation checkpoint (54). In murine B cells, metabolic activation following anti–IgM stimulation is not sufficiently supported by increased mitochondrial biogenesis or glucose uptake (54). This results in ROS accumulation and apoptosis driven by mitochondrial dysfunction, which is averted by the provision of T cell help (54). Overall, antigen engagement appears to create a brief window within which B cells must obtain costimulation to avoid activation–induced cell death.
Following B cell activation, antibody–secreting cells are generated through PC differentiation. The vast quantity of immunoglobulin secreted by PCs necessitates an increase in protein production capacity. During PC differentiation, the transcription factors Blimp1 and Xbp1 mediate substantial expansion of the endoplasmic reticulum (ER) (55). However, high levels of immunoglobulin production result in the accumulation of misfolded proteins and ER stress. To maintaining metabolic homeostasis, PCs utilise both antioxidant responses and the unfolded protein response (UPR), which limits mRNA translation while enhancing protein folding capacity and the ability of the ER to degrade misfolded proteins (56, 57). While Xbp1 and Blimp1 control the UPR in PCs, it has recently been shown that mTORC1 mediates a predictive UPR, which precedes antibody secretion (58, 59). Misfolded proteins are also degraded via the proteasome. Surprisingly, proteasome capacity decreases progressively during PC differentiation in mice, with proteasome inhibition reducing PC viability (60). Excessive ER stress has been suggested as a factor limiting the lifespan of short–lived PCs (SLPCs), although the expression ER stress response genes is equivalent in SLPCs and long–lived PCs (LLPCs) (61, 62).
PCs require a high level of metabolic activity to fuel extensive immunoglobulin production. Metabolic remodelling occurs during PC differentiation, with Blimp1 promoting oxidative metabolism (49). Basal OXPHOS is fed by long–chain fatty acids, with glucose–derived pyruvate acting as a biosynthetic substrate and providing spare respiratory capacity in LLPCs (15). The majority of glucose taken up by PCs is diverted towards antibody glycosylation via the hexosamine pathway, although glycolysis can supplement ATP production (15, 49). Predictably, PCs utilise amino acid metabolism: the expression of CD98, a component of many amino acid transporters, is induced by Blimp1, and is upregulated in LLPCs compared to SLPCs (58, 61). Amino acids are used during antibody synthesis, glutamine is used to generate several of these amino acids, while also acting as a substrate for oxidative metabolism (61). The activation of mTORC1, which is crucial for PC differentiation and optimising antibody output, is supported by high levels of amino acids (58, 63). In mice, AMPK is dispensable for LLPC persistence but restrains antibody synthesis, promoting metabolic homeostasis (53). As in other biological processes, a delicate balance of mTORC1 and AMPK signalling appear to provide an optimal metabolic environment for PC function.
SLPCs are generated following initial B cell activation, while LLPCs, characterised by affinity maturation and class–switch recombination, are produced within germinal centres (GC) of secondary lymphoid organs (Figure 2). Each GC consists of light and dark zones, defined by histological appearance. Within the light zone, B cells of different antigen specificities compete for pro–survival signals provided by follicular helper T (TFH) cells. B cells with high antigen affinities are more likely to receive sufficient T cell help, enabling them to migrate to the dark zone, where they undergo clonal expansion and somatic hypermutation. To fuel this proliferation, light zone B cells, known as centrocytes, adopt an anabolic phenotype mediated by mTORC1 (64). Centrocytes which receive CD40–mediated signals, representing TFH cell help, activate mTORC1 to promote glucose uptake, cell growth and ribosomal biogenesis (64). mTORC1 activation, specifically in the light zone, is essential for GC function (64). The activity of centrocytes also requires PI3K signalling (65). Upstream of the PI3K–Akt–mTORC1 pathway, the GTPase R–Ras2 couples TFH–derived signals to mitochondrial and glycolytic metabolism (66). Recently, however, it has been shown that GC B cells rely on fatty acid oxidation for ATP production, utilising glycolysis minimally (67). Despite being highly active, GCs are characterised by nutrient deprivation and hypoxia, particularly within the light zone (68, 69). Within the GC, mTORC1 signalling is opposed by HIF–1α and glycogen synthase kinase 3, which promotes B cell viability during nutrient deprivation (68, 69). Interestingly, B cell metabolism changes as the GC reaction progresses (70). Over time, the GC goes from producing predominantly memory cells to LLPCs, with this switch thought to be partly mediated by metabolic changes (70). Indeed, a subset of B cells in the GC, characterised by reduced mTORC1 and c–Myc signalling, display a propensity for memory B cell differentiation (71).
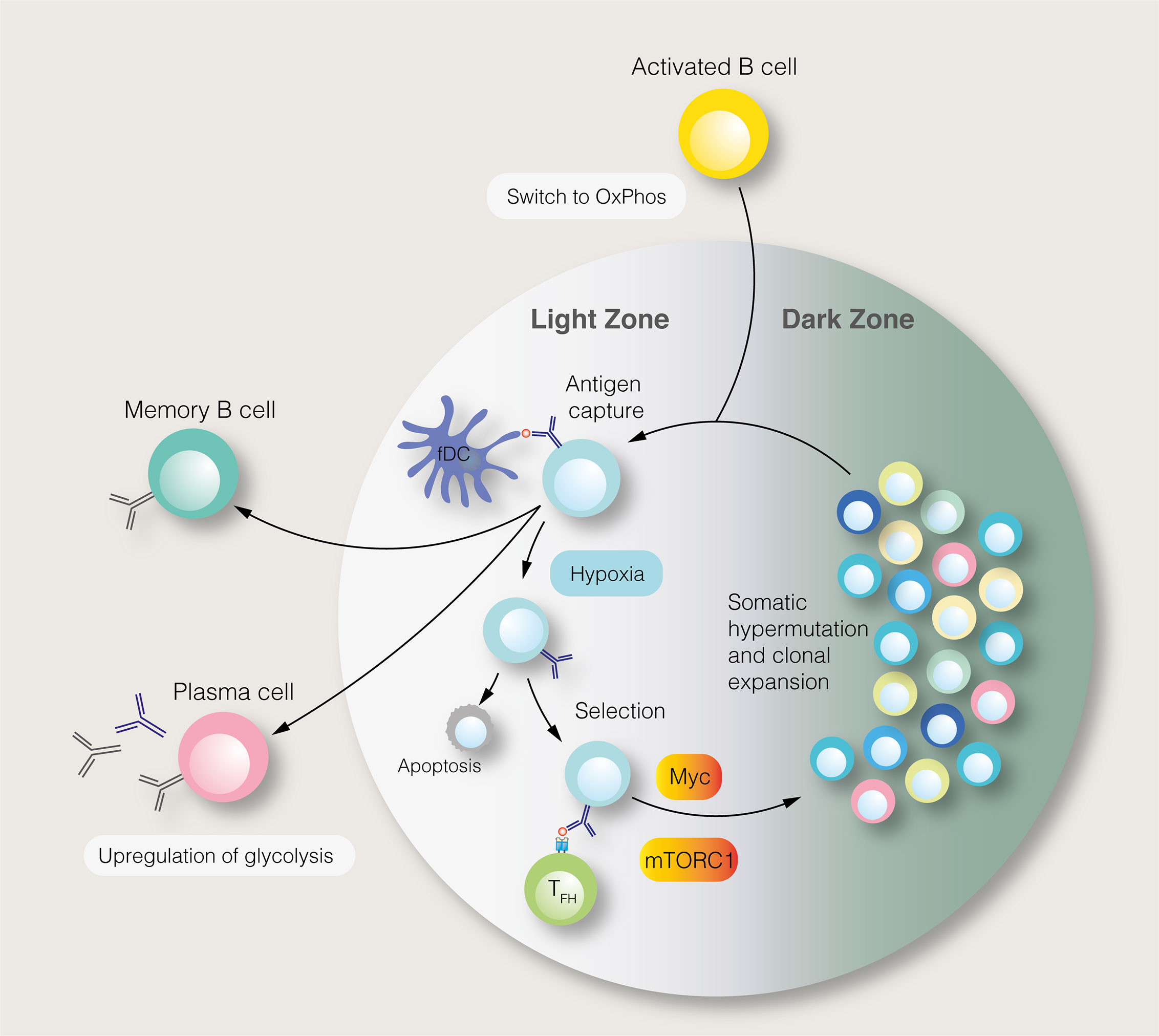
Figure 2 The germinal centre reaction and its regulation by metabolic signals. In the germinal centre light zone, centrocytes capture antigens from follicular dendritic cells (fDC). Centrocytes with the highest antigen affinities are most likely to acquire antigens and present antigen–derived peptides to follicular helper T (TFH) cells. Centrocytes which receive pro–survival signals from TFH cells may migrate to the dark zone, where, as centroblasts (CB), they undergo somatic hypermutation and clonal expansion. This process generates B cells with new antigen specificities. The proliferation of centroblasts is enabled by mTORC1 and c–Myc signalling in the light zone, although this anabolic phenotype is restrained by germinal centre hypoxia. Instead of entering the dark zone, centrocytes, may differentiate into long–lived plasma cells and memory B cells.
Little is known about the metabolism of memory B cells themselves, although they are thought to be relatively quiescent metabolically. Unlike naïve B cells and PCs, memory cells are capable of BAFF–independent survival (72). AMPK appears to play a crucial role in maintaining metabolic homeostasis in memory B cells, supporting mitochondrial function and preventing excessive oxidative stress (53). Memory B cell inactivity is rapidly reversed upon antigen re–exposure, enabling PC differentiation. During secondary humoral immune responses, the balance between mTORC1 and AMPK signalling controls the nature of this differentiation (22, 53).
As with memory B cells, the metabolic phenotype of unconventional B cell subsets is largely unexplored. Recently, the production of the immunoregulatory cytokine IL–10 by Bregs has been shown to depend on both cholesterol metabolism and HIF–1α (73, 74). However, more work is needed to metabolically characterise Bregs, as well as B1 cells.
Autophagy in Activated B Cells
As with other aspects of metabolism, autophagy plays a crucial role in B cell function post–activation. In the initial stages of B cell activation, the differential regulation of autophagy by BCR engagement and T cell costimulation has been proposed to create an autoimmune checkpoint (75). While BCR stimulation of primary B cells promotes autophagy and triggers apoptosis, concomitant stimulation via the CD40 coreceptor induces a more modest rise in autophagy levels, limiting cell death (75). Although the amelioration of B cell autophagy by CD40 engagement has been disputed, the non–optimal induction of autophagy in the absence of costimulation may sensitise self–reactive B cells to cell death pathways (75, 76). Acutely, increased autophagic flux following BCR engagement is driven by the upregulation of non–canonical autophagy, which is resistant to inhibition by bafilomycin A1 (76). In contrast, bafilomycin–sensitive canonical autophagy is temporarily inhibited following BCR activation, likely due to increased mTORC1 activity (76).
While autophagy is induced by BCR signalling, it appears to be dispensable for BCR–driven B cell activation, given that proliferation following anti–IgM stimulation is unaffected in the absence of autophagy (Figure 1) (40). B cell autophagy is also induced by stimulation with TLR ligands, although in this context it appears to be necessary for plasmablast differentiation and survival (Figure 1) (23, 76). It was initially demonstrated that plasmablast differentiation did occur in autophagy–deficient B cells following stimulation with the TLR4 ligand lipopolysaccharide (LPS), although significant cell death was incurred (23). Furthermore, cells in which Atg5 deletion was not penetrant were selectively enriched during differentiation (23). It has been subsequently reported that stimulation with LPS or the TLR9 ligand CpG leads to impaired plasmablast differentiation and viability among B cells lacking the autophagy genes Atg5 or Atg7, characterised by the accumulation of damaged mitochondria and reduced expression of B cell transcriptional regulators (25, 40, 77). The effect of autophagy on plasmablast differentiation likely varies between studies according to the extent of metabolic stress induced during different protocols.
In the context of T cell–dependent (TD) antigens, costimulation requires antigen presentation on class II major histocompatibility complexes (MHC–II) to cognate T cells. Several early studies demonstrated that the ability of B cells to present antigens on MHC–II was altered following pharmacological inhibition of autophagy or its induction through starvation (78–80). Of note, these studies were conducted with Epstein–Barr virus (EBV)–infected B cells. EBV infection modifies B cell antigen presentation, limiting the physiological relevance of this model (24). More recently, autophagy has been implicated in the polarisation of internalised antigens following BCR engagement (81). In the absence of autophagy, the colocalisation of antigen–BCR complexes with MHC–II–containing vesicles is disrupted, particularly in the case of particulate antigens (81). While partial relocalisation of the BCR–antigen complex is sufficient for B cells to present peptides from soluble antigens on MHC–II, the presentation of particulate antigen epitopes to cognate T cells is compromised (81). Overall, autophagy appears to be necessary for the presentation of some, but not all, antigens by B cells.
The GC reaction, which enables the generation of memory B cells and LLPCs, is also characterised by extensive B cell autophagy. Of note, levels of canonical autophagy are relatively low, likely due to the upregulation of mTORC1 by TFH cell–derived costimulation (76). The high overall levels of autophagy in GC B cells are skewed towards non–canonical flux, regulated by WIPI2 (76). The role of autophagy in GC function has been questioned, as neither its macroscopic appearance nor the extent of B cell affinity maturation is affected by B–cell–specific Atg5 deletion (Figure 1) (23, 82). However, while it has only been shown that class III PI3K–independent non–canonical autophagy occurs in the GC, some forms of non–canonical autophagy proceed in the absence of Atg5 itself (76, 83). It is possible that non–canonical autophagy occurs in the GC, and in other B cell subsets, following the deletion of core autophagy genes.
Given its roles in metabolic homeostasis and the degradation of misfolded proteins, it is unsurprising that autophagy appears to play an important role in the maintenance of antibody responses in both SLPCs and LLPCs (Figure 1). However, the precise effects of its inhibition are contested. As discussed in the context of plasmablast differentiation, there is disagreement as to the necessity of autophagy for initial antibody production following LPS–induced activation. Atg7 deletion has been shown to compromise short–term IgM production, in line with failed plasmablast differentiation (25). However, it has also been reported that B cells lacking Atg5 display elevated short–term antibody production following LPS stimulation, although in this study plasmablasts were characterised by ATP deficiency and elevated ER stress (23). This discrepancy, as in differences seen in plasmablast differentiation, may be caused by differences in the degree and timing of metabolic stress experienced by activated B cells. The idea that autophagy may restrain short–term immunoglobulin output suggests that it instead promotes long–term PC viability (23). In support of this conclusion, IgM and IgG responses to T cell independent (TI) pneumococcal polysaccharide antigens and the TD antigen NP–CGG were diminished in B cell autophagy–deficient mice, as was long–term LLPC survival (23). In contrast, humoral immunity against the TI hapten NP–Ficoll was unaffected by the loss of B cell autophagy in mice, perhaps due to continual B cell activation by this antigen (23). At the same time, another report found that early antibody responses to hapten–conjugated TD and TI antigens, as well as helminths, were diminished in B–autophagy–deficient mice (77). Subsequently, B cell autophagy was suggested to be important to antigen–specific IgM but not IgG responses against the TD antigen ovalbumin in mice (40). Autophagy was concluded to be important for PC survival, with LLPCs displaying greater resistance to a short–term loss of autophagy than SLPCs (40). Meanwhile, a separate study found that the murine humoral response to the TD antigen NP–KLH was largely unaffected by the loss of B cell autophagy, although anti–NP IgG1 trended towards a decrease six weeks post–immunisation (84). Clearly, there is significant heterogeneity in results, likely arising from differences in immunisation protocols. However, these results together suggest an important role for autophagy in maintaining PC viability over time.
To enable their longevity, LLPCs would logically utilise autophagy to a greater degree than SLPCs. Accordingly, the extent to which PCs utilise autophagy varies according to cellular lifespan. Among adult human bone marrow PCs, the expression of autophagy–associated genes and the prevalence of autophagic LC3B–II punctae are greater among LLPCs than SLPCs (85). In agreement with human data, autophagosomes are more prevalent in murine PCs with longer half–lives (61). Autophagy also appears to support the long–term survival of murine memory B cells, thus promoting the maintenance of immunological memory (Figure 1) (82, 84). Expression of autophagy–associated genes is higher in memory B cells than other B cell subsets, suggesting an important role in their function (84). In B cell autophagy–deficient mice, secondary humoral immunity against NP–KLH is affected to a much greater degree than the primary response, indicating a failure of B cell memory (84). While the number of memory cells formed two weeks post–immunisation was normal in mice lacking B cell autophagy, their number was markedly diminished by eight weeks, indicating that autophagy is not necessary for memory B cell formation, but is needed to maintain this population (82). Autophagy–deficient early, but not late, memory B cells are able to mount functional immune responses following antigen re–exposure (82). The survival of autophagy–deficient memory B cell survival is partially restored by NecroX–2, a necrosis inhibitor which reduces oxidative stress (84). Recently, it has been shown that mitochondrial autophagy, which helps to limit oxidative stress, is regulated in memory B cells by AMPK (53). Together, these results implicate autophagy as an important pro–survival mechanism in memory B cells, as well as PCs.
B Cell Metabolism in Autoimmunity
The past decade has seen an explosion in research exploring the association between immunometabolism and autoimmunity (86). Within this field, B cells have received relatively little attention. Nevertheless, there are clear indications that the metabolic profile of B cells is disturbed in autoimmunity, particularly in SLE. Importantly, dysregulated B cell metabolism has been implicated in promoting and exacerbating disease pathology.
The dysfunction of tolerogenic B cell checkpoints is seen in many autoimmune diseases (87, 88). As has been discussed, these checkpoints, which play a crucial role in restricting the self–reactive potential of the B cell repertoire, have a significant metabolic component (19, 54). It has therefore been investigated whether metabolic dysregulation could compromise the function of these checkpoints. One apparent mechanism underlying defective tolerogenesis is increased exposure to BAFF. Serum BAFF levels are elevated compared to health in several autoimmune diseases, including RA, IgA nephropathy and SLE (89–91). In otherwise healthy mice, autoimmune manifestations are seen following transgenic BAFF overexpression (92). Exposure to excess BAFF rescues self–reactive B cells from deletional checkpoints and supports the survival of anergic B cells (21, 93). Anergic B cells show an increased reliance on BAFF signalling and fail to compete with non–anergic B cells for the cytokine under normal conditions (94). Elevated BAFF may promote their survival, increasing the likelihood of peripheral activation. Regarding metabolism, chronic exposure to BAFF increases the glycolytic and oxidative metabolism of B cells (20). Prolonged exposure to high levels of BAFF may allow self–reactive B cells to avoid the metabolic restriction and energy crisis which contribute to anergy and deletion, respectively.
The autoimmune manifestations of TRAF3–deficient mice further evidence the importance of BAFF in autoimmunity. TRAF3 inhibits BAFF–induced activation of nuclear factor (NF)–κB2 signalling (95, 96). In mice, B cell–specific loss of TRAF3 enhances the survival of resting B cells in a BAFF–independent manner. This results in an expanded B cell compartment, increased spontaneous GC formation and autoimmunity (95). As with exposure to elevated BAFF, TRAF3 deficiency in B cells increases glucose uptake, glycolysis and OXPHOS, in an NF–κB–dependent manner (97).
Another important mechanism underlying B cell anergy is the suppression of PI3K signalling by PTEN, Src homology region 2 domain–containing phosphatase (SHP)–1 and Src homology 2 domain–containing inositol polyphosphate 5–phosphatase (SHIP)–1 (39, 98–100). In mice, PTEN deletion alters the responsiveness of B cells to tolerogenic signals, with activation and proliferation favoured over anergy (39). The loss of either PTEN or SHP–1 in B cells results in autoimmunity (39, 101). Moreover, the expression of PTEN by human B cells is reduced in patients with type 1 diabetes mellitus, autoimmune thyroiditis and SLE compared to healthy controls (98, 102). Together, these results suggest that unrestrained PI3K signalling, through the loss of negative regulators, enables self–reactive B cells to escape tolerogenesis.
Downstream of PI3K, hyperactive B cell mTORC1 signalling is seen in autoimmunity. Given its roles in B cell development, GC reactions and PC function, an association between altered mTORC signalling and autoimmunity is perhaps unsurprising (14, 64). mTORC1 hyperactivity is seen in animal models of autoimmunity, including murine models of SLE and RA (103, 104). In the latter, mTORC1 hyperactivation in B cells was associated with increased glucose metabolism, although disturbed B cell metabolism was deemed to occur downstream of T cell dysregulation (104). In human autoimmunity, elevated mTORC signalling is seen in the B cells of SLE patients, where it is correlated with disease activity, and in the salivary gland B cells of patients with Sjögren’s syndrome (22, 105).
The precise metabolic consequences of dysregulated B cell signalling networks in autoimmunity have not been well defined. However, there is clear evidence to suggest that the tight metabolic control of B cell function is lost in autoimmunity and that this has consequences for pathophysiology. More work is needed to directly investigate the nature and consequences of metabolic dysregulation seen in autoimmune B cells. Furthermore, a more complete characterisation of metabolism in different B cell subsets is needed. Comparatively little is known about the metabolism of B1 cells, Bregs and memory B cells in both health and disease. Given the different phenotypes and functions of different B cells, it is likely that they affected differently by metabolic disturbances. Finally, most research has focussed on B cells in SLE, with the dysfunction of B metabolism in other diseases mediated by autoantibodies and B cell cytokines warranting further investigation.
B Cell Autophagy in Autoimmunity
As discussed above, autophagy promotes metabolic homeostasis of B cells, particularly following peripheral activation and in memory cell maintenance (23, 84). It has also been implicated in antigen presentation and deletional autoimmune checkpoints (75, 81). This has fuelled considerable interest in the role that autophagy may play in expanding the repertoire, functionality and survival of self–reactive B cells in diseases such as SLE, RA, and multiple sclerosis (MS). Although not cell–specific, early evidence of perturbed autophagy in autoimmunity came from genome–wide association studies (GWAS). Specifically, several reports linked single nucleotide polymorphisms (SNPs) in the Atg5 gene and Prdm1–Atg5 intergenic region to the development of SLE and, in a European population, to RA (106–109). While GWAS are unable to resolve associations in a cell–specific manner, it was subsequently demonstrated that the B cells of patients with SLE displayed elevated Atg5 expression compared to healthy controls (110). Furthermore, an SLE–associated SNP in the Prdm1–Atg5 intergenic region was associated with elevated expression of autophagy–associated genes in the B cells of SLE patients and healthy individuals (110). In parallel to these early GWAS, autophagy was implicated in SLE through therapeutic studies. The 21–mer peptide rigerimod (P140), which ameliorates lupus pathology, was shown to inhibit B cell autophagy (111). Specifically, rigerimod targets chaperone–mediated autophagy, which is upregulated in lupus–prone mice, suggesting that inhibiting dysregulated autophagy may underlie the efficacy of rigerimod (112). The apparent association between B cell autophagy and autoimmunity appears to be mediated by several distinct mechanisms, which may occur in a disease–specific manner.
As with metabolism, dysregulated autophagy has been implicated in the loss of tolerogenic checkpoint function in autoimmunity. While autophagy is dispensable for B2 cell development, this does not preclude a role for altered autophagy in enabling the subversion of metabolic or apoptotic checkpoints by self–reactive B cell precursors (Figure 3) (25, 40). Indeed, while autophagy is upregulated in lupus–prone mice compared to healthy controls, this difference is restricted to bone marrow–resident pre–B, immature and mature B cells (25). No difference is seen in among splenic B cells, including PCs. In patients with SLE, elevated autophagy compared to healthy controls is more notable in naïve B cells than PCs or memory cells (25). Both pre–B and naïve B cells are subject to tolerogenic checkpoints. Upregulated autophagy may protect self–reactive B cells against apoptotic or metabolically damaging stimuli. Similarly, the induction of autophagy has been proposed to mediate T cell escape from tolerogenic checkpoints (113).
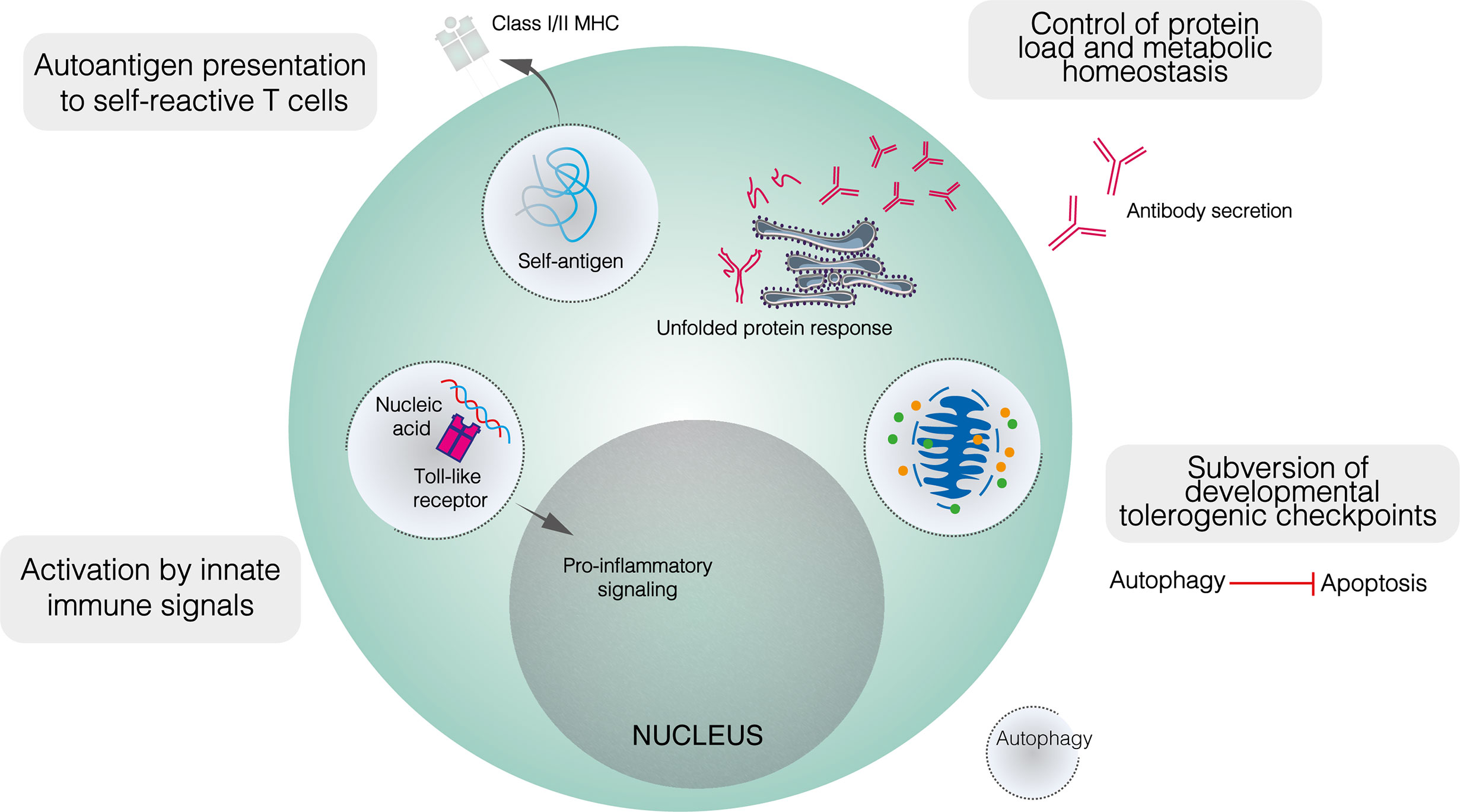
Figure 3 The role of autophagy in the function of self–reactive B cells. Developmentally, upregulated autophagy is thought to enable autoimmune B cell precursors to subvert apoptotic checkpoints. In mature B cells, autophagy appears to mediate both the presentation of self–peptides by B cells, and the recognition of innate immune ligands such as nucleic acids. It is also likely that autophagy contributes to metabolic homeostasis and control of the misfolded protein load in plasma cells.
Autophagy also appears to play a role in enabling B cells to process and present peptides derived from self–antigens to cognate T cells (Figure 3). Physiologically, autophagy has been shown to mediate the presentation of peptides derived from particular antigens (81). In autoimmunity, autophagy enables B cells to present citrullinated peptides on MHC–II, antibodies against citrullinated antigens feature prominently in autoimmune diseases, most notably in RA (114, 115). In vitro, the presentation of endogenous citrullinated peptides by B lymphoma cells was enabled by serum starvation, a state which activates autophagy, and suppressed by both the class III PI3K inhibitor 3–MA and Atg5 knockdown (115). In contrast, neither starvation nor 3–MA affected the presentation of non–citrullinated antigens. Furthermore, stimulation of primary B cells with anti–IgM antibodies induced significant citrullinated peptide presentation and increased LC3–II levels (115). Given the frequency of anti–Ig antibodies in RA and the apparent induction of autophagy following BCR engagement, such antibodies have been proposed to trigger the presentation of endogenous citrullinated antigens on MHC–II (115). Although the exact relationship between autophagy and RA remains unclear, B cell autophagosome density is not upregulated in patients with RA compared to healthy controls, yet autophagic activity appears necessary for the presentation of certain autoantigens (116).
B cell autophagy has also been associated with the presentation of citrullinated self–antigens to cytotoxic T lymphocytes in MS. EBV, a well–established risk factor for development of MS, induces autophagy in B cells, and has recently been linked to the class I MHC (MHC–I) presentation of myelin oligodendrocyte glycoprotein (MOG)–derived peptides (24, 117–119). The role of autophagy in this process is poorly defined, although autophagosomes may protect citrullinated MOG peptides from cathepsin–mediated degradation, directing them towards MHC–I cross–presentation machinery (24).
As well as BCR ligands, autophagy has been implicated in B cell recognition of innate immune triggers, specifically nucleic acid antigens, by TLRs 7 and 9. These receptors recognise pathogen–derived ssRNA and CpG–containing dsDNA respectively, with their cellular expression restricted to endosomes to prevent cross–recognition of self–nucleic acids. The recognition of self–nucleic acids may directly activate B cells or sensitise them to BCR–mediated activation: the transgenic overexpression of Tlr7 or Tlr9 generates autoreactive PCs and triggers lupus–like disease in mice (26, 120, 121). Murine experimental lupus induced by transgenic Tlr7 overexpression is ameliorated in the absence of B cell autophagy (122). Autophagy mediates the transport of endocytosed RNA–containing immune complexes to TLR7–containing endosomes in dendritic cells and may play a similar role in B cells (Figure 3) (123). Autophagy has also been implicated in TLR9 signalling (124). Following BCR engagement, TLR9–containing endosomes are recruited to autophagosomes which contain the internalised BCR–antigen complex, although this co–localisation is abolished by 3–MA (124). Anti–IgM conjugated to CpG DNA hyperactivates B cells compared to anti–IgM alone, and this additive effect was abolished when TLR9 recruitment to autophagosomes was blocked by disrupting microtubule function (124). Collectively, autophagy seems to play an important role in delivering nucleic acid antigens to endosomal TLRs, potentially impairing the discrimination between self– and non–self–nucleic acids.
Intuitively, the role of autophagy in maintaining PC viability should extend to self–reactive PCs (Figure 3) (23, 40, 60). While PC autophagy does not appear to be upregulated in lupus–prone mice compared to healthy controls, it contributes demonstrably to the pathology of murine experimental lupus (25, 40). In mice with Atg5 conditionally deleted in mature B cells, anti–dsDNA IgM levels were similar to those seen in wildtype mice, whereas anti–dsDNA IgG was significantly lower (40). The splenic B cell repertoire, including the proportions of different B cell subpopulations and the appearance of germinal centres, was largely unaffected by the absence of autophagy, although bone marrow LLPCs were depleted (40). Together, these results suggest that autophagy makes little contribution to early B cell activation in autoimmunity but is important in ensuring the long–term survival of autoreactive PCs. It was proposed that SLPC survival may have been compromised in the absence of autophagy but was compensated for by increased replenishment (40). Importantly, autophagy deficiency likely also reduces the memory B cell compartment (84).
While B2 cell autophagy plays an important role in autoimmunity, its relevance in other B cell subsets remains poorly defined. Although B1 cells display great sensitivity to changes in autophagic flux, it is not clear whether changes in B cell autophagy seen in autoimmunity apply to these cells and what the effects might be (18). This is important to understand, given that self–reactive IgM produced by B1 cells is implicated both in autoimmune pathogenicity and in the clearance of apoptotic cells (8, 11). Moreover, B1 cells in gut–associated lymphoid tissue promote intestinal homeostasis through IgA and IgM production (12). The loss of B cell autophagy reduces mucosal IgA and impairs B cell responses to intestinal inflammation (77). Overall, B1 cell autophagy may be relevant to inflammatory and autoimmune diseases, particularly within the gastrointestinal tract. As in B1 cells, the effects of perturbed autophagy on Breg function have yet to be defined, although their unique phenotype suggests that they may be affected differently to antibody–secreting cells. Further investigation is required to understand the role of autophagy in unconventional B cells during autoimmunity.
B Cell Metabolism and Autophagy as Therapeutic Targets
The dysregulation of B cell metabolism and autophagy in autoimmune diseases has raised the prospect of targeting these processes therapeutically. While traditional immunosuppressive drugs such as glucocorticoids affect immunometabolism, recent efforts have focussed on disrupting metabolic pathways more specifically (125). Several drugs being explored or approved for the treatment of autoimmune diseases target B cell immunometabolism or its regulators, likely contributing to their efficacy.
As discussed above, elevated BAFF levels are seen in several autoimmune diseases (89–91). B cells exposed to high levels of BAFF have enhanced metabolic capacity and can escape from tolerogenic checkpoints (20, 21). Efforts to inhibit this dysregulated signalling culminated in the approval of the BAFF–specific monoclonal antibody belimumab as an add–on therapy in SLE, having demonstrated efficacy, including ameliorating B cell dysfunction, in phase 3 clinical trials (126). The treatment of SLE patients with belimumab induces anergy in autoreactive B cells (93). Notably, the BAFF–independent survival of memory B cells means that humoral immune responses to vaccine antigens are left intact by belimumab (127). The ability to target self–reactive B cells without compromising physiological B cell responses is clearly desirable. Excessive BAFF signalling may also be attenuated through blockade of its receptor. Recently, the BAFF receptor inhibitor ianalumab was shown to effectively deplete B cells and improve clinical parameters in patients with Sjögren’s syndrome (128).
Hyperactive mTORC1 signalling, a feature of both T and B lymphocytes in autoimmunity, can be inhibited with rapamycin. In murine models of SLE, rapamycin attenuates pathology, including decreasing anti–dsDNA antibody titres (129). Although its effects have been largely attributed to changes in T cells, rapamycin inhibits BAFF–mediated mTORC1 signalling in B cells, limiting proliferation and survival (130, 131). mTORC1 inhibition also results in the induction of autophagy. Although self–reactive B cells appear to benefit from a degree of autophagy upregulation, increased autophagic flux may be detrimental to B cell survival (75). In a single–arm, open–label Phase 1/2 trial in SLE patients, rapamycin was associated with decreased disease activity and reduced levels of some autoantibodies (132).
mTORC dysregulation is also targeted by metformin. Used to treat type 2 diabetes mellitus, metformin activates AMPK and is, therefore, able to downregulate mTORC1 activity. Metformin has shown efficacy in the treatment of lupus–like autoimmunity in mice (133). As well as decreasing TFH and TH17 populations, metformin suppressed PC differentiation and GC formation, resulting in lower autoantibody levels (133). In a separate study, metformin given alongside the glycolytic inhibitor 2–DG was shown to ameliorate autoimmune pathology in lupus–prone mice (134). A recent clinical trial into the efficacy of metformin in SLE patients found no efficacy in reducing the incidence of disease flares, although pooled analysis with a previous trial suggested a modest reduction in flare incidence was achieved, warranting further investigation (135, 136).
While some of the therapeutic effects of rapamycin and metformin may derive from targeting B cell autophagy, this process is affected more clearly by rigerimod (111). Rigerimod disrupts chaperone–mediated autophagy, likely affecting the MHC–II–restricted presentation of endogenous antigens to autoreactive T cells (112, 137). In mice, rigerimod suppresses autoimmune responses without compromising anti–viral immunity (138, 139). Although rigerimod showed efficacy in early clinical trials in SLE patients, it failed to meet its primary endpoint in a recent phase 3 clinical trial (140, 141). However, promising therapeutic potential has meant a new phase 3 trial is scheduled to commence in 2021.
Owing to their limited proteasome capacity and high levels of immunoglobulin production, PCs display exquisite sensitivity to proteasome inhibition (60). Proteasome inhibition has been investigated as a potential therapeutic strategy in diseases characterised by autoantibody production. Efforts to target the proteasome have focussed on bortezomib, which was originally developed for the treatment of multiple myeloma. In multiple myeloma, B cells with high immunoglobulin production are disproportionately vulnerable to proteasome inhibition (142). The high immunoglobulin output of self–reactive B cells in autoimmune diseases will likely confer elevated sensitivity to proteasome inhibitors. In a murine model of lupus, bortezomib alleviated autoantibody–driven pathology (143). In small numbers of human participants, bortezomib has shown promise in treating autoantibody–driven diseases, such as anti–NMDA receptor encephalitis and SLE (144, 145).
Although the metabolism and autophagy of self–reactive B cells represent promising therapeutic avenues, there are several challenges associated with such targets. Firstly, a full understanding of the metabolic regulation of B cell function is lacking in both health and disease. Moreover, much of our current knowledge comes from studying B cell metabolism in mice, not humans. A critical goal in the field is uncovering cell–specific metabolic pathways which may be targeted more selectively. Similarly, immune cells differ considerably in their metabolic plasticity and therefore resistance to metabolic pathway inhibition. However, many of the most commonly used drugs in medicine (e.g. metformin, statins, and methotrexate) all target broadly active metabolic processes. This observation suggests that tolerability of metabolic inhibition be better than supposed. As always when treating autoimmunity, excessive immunosuppression must be avoided, and the risk of infection will remain an important consideration. Finally, the extent to which specific metabolic pathways are disturbed in patients with autoimmune disease is likely to vary significantly between individuals. The use of metabolomic biomarkers may be needed to identify patients who are likely to respond to treatments targeting metabolism.
Conclusion
Although metabolism has emerged relatively recently as a regulator of immunological function, it is clear that it has far–reaching consequences in both the physiological state and autoimmunity. Arguably, efforts have been made to exploit B cell metabolism and autophagy therapeutically before these processes have been well characterised. Nevertheless, it appears that they do offer real translational potential. While much focus to date has been rightly placed on understanding the role of metabolic regulators such as mTORC1 and AMPK, metabolism itself, as well as its impact on B cell function, require greater investigation. Finally, it is imperative that metabolic interactions between B cells and other leukocyte populations are explored. While this review has focussed only on B cells, exploiting the full therapeutic potential of B cell metabolism in autoimmune diseases will require it to be understood within the context of wider immune dysfunction.
Author Contributions
IR wrote and edited the manuscript AC supervised and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
AC is supported by a Wellcome Trust award (211072/Z/18/Z).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Yaniv G, Twig G, Shor DB, Furer A, Sherer Y, Mozes O, et al. A Volcanic Explosion of Autoantibodies in Systemic Lupus Erythematosus: A Diversity of 180 Different Antibodies Found in SLE Patients. Autoimmun Rev (2015) 14(1):75–9. doi: 10.1016/j.autrev.2014.10.003
2. Molnarfi N, Schulze–Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin–Doyer M, et al. MHC Class II–Dependent B Cell APC Function is Required for Induction of CNS Autoimmunity Independent of Myelin–Specific Antibodies. J Exp Med (2013) 210(13):2921–37. doi: 10.1084/jem.20130699
3. Bar–Or A, Fawaz L, Fan B, Darlington PJ, Rieger A, Ghorayeb C, et al. Abnormal B–Cell Cytokine Responses a Trigger of T–Cell–Mediated Disease in MS? Ann Neurol (2010) 67(4):452–61. doi: 10.1002/ana.21939
4. Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, et al. B Cell Depletion Therapy Ameliorates Autoimmune Disease Through Ablation of IL–6–Producing B Cells. J Exp Med (2012) 209(5):1001–10. doi: 10.1084/jem.20111675
5. Meednu N, Zhang H, Owen T, Sun W, Wang V, Cistrone C, et al. Production of RANKL by Memory B Cells: A Link Between B Cells and Bone Erosion in Rheumatoid Arthritis. Arthritis Rheumatol (2016) 68(4):805–16. doi: 10.1002/art.39489
6. Haas KM, Poe JC, Steeber DA, Tedder TF. B–1a and B–1b Cells Exhibit Distinct Developmental Requirements and Have Unique Functional Roles in Innate and Adaptive Immunity to S. Pneumoniae. Immunity (2005) 23(1):7–18. doi: 10.1016/j.immuni.2005.04.011
7. Chou MY, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, et al. Oxidation–Specific Epitopes are Dominant Targets of Innate Natural Antibodies in Mice and Humans. J Clin Invest (2009) 119(5):1335–49. doi: 10.1172/JCI36800
8. Zhong X, Lau S, Bai C, Degauque N, Holodick NE, Steven SJ, et al. A Novel Subpopulation of B–1 Cells Is Enriched With Autoreactivity in Normal and Lupus–Prone Mice. Arthritis Rheum (2009) 60(12):3734–43. doi: 10.1002/art.25015
9. Deng J, Wang X, Chen Q, Sun X, Xiao F, Ko KH, et al. B1a Cells Play a Pathogenic Role in the Development of Autoimmune Arthritis. Oncotarget (2016) 7(15):19299–311. doi: 10.18632/oncotarget.8244
10. Grönwall C, Akhter E, Oh C, Burlingame RW, Petri M, Silverman GJ. Igm Autoantibodies to Distinct Apoptosis–Associated Antigens Correlate With Protection From Cardiovascular Events and Renal Disease in Patients With SLE. Clin Immunol (2012) 142(3):390–8. doi: 10.1016/j.clim.2012.01.002
11. Chen Y, Park YB, Patel E, Silverman GJ. Igm Antibodies to Apoptosis–Associated Determinants Recruit C1q and Enhance Dendritic Cell Phagocytosis of Apoptotic Cells. J Immunol (2009) 182(10):6031–43. doi: 10.4049/jimmunol.0804191
12. Shimomura Y, Mizoguchi E, Sugimoto K, Kibe R, Benno Y, Mizoguchi A, et al. Regulatory Role of B–1 B Cells in Chronic Colitis. Int Immunol (2008) 20(6):729–37. doi: 10.1093/intimm/dxn031
13. Yang M, Rui K, Wang S, Lu L. Regulatory B Cells in Autoimmune Diseases. Cell Mol Immunol (2013) 10(2):122–32. doi: 10.1038/cmi.2012.60
14. Zeng H, Yu M, Tan H, Li Y, Su W, Shi H, et al. Discrete Roles and Bifurcation of PTEN Signaling and mTORC1–Mediated Anabolic Metabolism Underlie IL–7–Driven B Lymphopoiesis. Sci Adv (2018) 4(1):eaar5701. doi: 10.1126/sciadv.aar5701
15. Lam WY, Becker AM, Kennerly KM, Wong R, Curtis JD, Llufrio EM, et al. Mitochondrial Pyruvate Import Promotes Long–Term Survival of Antibody–Secreting Plasma Cells. Immunity (2016) 45(1):60–73. doi: 10.1016/j.immuni.2016.06.011
16. Stein M, Dütting S, Mougiakakos D, Bösl M, Fritsch K, Reimer D, et al. A Defined Metabolic State in Pre B Cells Governs B–Cell Development and Is Counterbalanced by Swiprosin–2/Efhd1. Cell Death Differ (2017) 24(7):1239–52. doi: 10.1038/cdd.2017.52
17. Waters LR, Ahsan FM, Wolf DM, Shirihai O, Teitell MA. Initial B Cell Activation Induces Metabolic Reprogramming and Mitochondrial Remodeling. iScience (2018) 5:99–109. doi: 10.1016/j.isci.2018.07.005
18. Clarke AJ, Riffelmacher T, Braas D, Cornall RJ, Simon AK. B1a B Cells Require Autophagy for Metabolic Homeostasis and Self–Renewal. J Exp Med (2018) 215(2):399–413. doi: 10.1084/jem.20170771
19. Shojaee S, Chan LN, Buchner M, Cazzaniga V, Cosgun KN, Geng H, et al. PTEN Opposes Negative Selection and Enables Oncogenic Transformation of Pre–B Cells. Nat Med (2016) 22(4):379–87. doi: 10.1038/nm.4062
20. Caro–Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, et al. Metabolic Reprogramming Is Required for Antibody Production That Is Suppressed in Anergic But Exaggerated in Chronically BAFF–Exposed B Cells. J Immunol (2014) 192(8):3626–36. doi: 10.4049/jimmunol.1302062
21. Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, et al. Excess BAFF Rescues Self–Reactive B Cells From Peripheral Deletion and Allows Them to Enter Forbidden Follicular and Marginal Zone Niches. Immunity (2004) 20(6):785–98. doi: 10.1016/j.immuni.2004.05.010
22. Torigoe M, Iwata S, Nakayamada S, Sakata K, Zhang M, Hajime M, et al. Metabolic Reprogramming Commits Differentiation of Human CD27(+)Igd(+) B Cells to Plasmablasts or CD27(–)IgD(–) Cells. J Immunol (2017) 199(2):425–34. doi: 10.4049/jimmunol.1601908
23. Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, et al. Plasma Cells Require Autophagy for Sustainable Immunoglobulin Production. Nat Immunol (2013) 14(3):298–305. doi: 10.1038/ni.2524
24. Morandi E, Jagessar SA, Hart BA, Gran B. Ebv Infection Empowers Human B Cells for Autoimmunity: Role of Autophagy and Relevance to Multiple Sclerosis. J Immunol (2017) 199(2):435–48. doi: 10.4049/jimmunol.1700178
25. Clarke AJ, Ellinghaus U, Cortini A, Stranks A, Simon AK, Botto M, et al. Autophagy Is Activated in Systemic Lupus Erythematosus and Required for Plasmablast Development. Ann Rheum Dis (2015) 74(5):912–20. doi: 10.1136/annrheumdis-2013-204343
26. Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, et al. Control of Toll–Like Receptor 7 Expression Is Essential to Restrict Autoimmunity and Dendritic Cell Proliferation. Immunity (2007) 27(5):801–10. doi: 10.1016/j.immuni.2007.09.009
27. Zhang S, Readinger JA, DuBois W, Janka–Junttila M, Robinson R, Pruitt M, et al. Constitutive Reductions in mTOR Alter Cell Size, Immune Cell Development, and Antibody Production. Blood (2011) 117(4):1228–38. doi: 10.1182/blood-2010-05-287821
28. Zhang S, Pruitt M, Tran D, Du Bois W, Zhang K, Patel R, et al. B Cell–Specific Deficiencies in mTOR Limit Humoral Immune Responses. J Immunol (2013) 191(4):1692–703. doi: 10.4049/jimmunol.1201767
29. Iwata TN, Ramírez JA, Tsang M, Park H, Margineantu DH, Hockenbery DM, et al. Conditional Disruption of Raptor Reveals an Essential Role for mTORC1 in B Cell Development, Survival, and Metabolism. J Immunol (2016) 197(6):2250–60. doi: 10.4049/jimmunol.1600492
30. Zhang Y, Hu T, Hua C, Gu J, Zhang L, Hao S, et al. Rictor Is Required for Early B Cell Development in Bone Marrow. PloS One (2014) 9(8):e103970. doi: 10.1371/journal.pone.0103970
31. Li M, Lazorchak AS, Ouyang X, Zhang H, Liu H, Arojo OA, et al. Sin1/mTORC2 Regulate B Cell Growth and Metabolism by Activating mTORC1 and Myc. Cell Mol Immunol (2019) 16(9):757–69. doi: 10.1038/s41423-018-0185-x
32. Park H, Staehling K, Tsang M, Appleby MW, Brunkow ME, Margineantu D, et al. Disruption of Fnip1 Reveals a Metabolic Checkpoint Controlling B Lymphocyte Development. Immunity (2012) 36(5):769–81. doi: 10.1016/j.immuni.2012.02.019
33. Green DR, Galluzzi L, Kroemer G. Cell Biology. Metabolic Control of Cell Death. Science (2014) 345(6203):1250256. doi: 10.1126/science.1250256
34. Keenan RA, De Riva A, Corleis B, Hepburn L, Licence S, Winkler TH, et al. Censoring of Autoreactive B Cell Development by the Pre–B Cell Receptor. Science (2008) 321(5889):696–9. doi: 10.1126/science.1157533
35. Kojima H, Kobayashi A, Sakurai D, Kanno Y, Hase H, Takahashi R, et al. Differentiation Stage–Specific Requirement in Hypoxia–Inducible factor–1alpha–Regulated Glycolytic Pathway During Murine B Cell Development in Bone Marrow. J Immunol (2010) 184(1):154–63. doi: 10.4049/jimmunol.0800167
36. Ochiai K, Maienschein–Cline M, Mandal M, Triggs JR, Bertolino E, Sciammas R, et al. A Self–Reinforcing Regulatory Network Triggered by Limiting IL–7 Activates Pre–BCR Signaling and Differentiation. Nat Immunol (2012) 13(3):300–7. doi: 10.1038/ni.2210
37. Chan LN, Chen Z, Braas D, Lee JW, Xiao G, Geng H, et al. Metabolic Gatekeeper Function of B–Lymphoid Transcription Factors. Nature (2017) 542(7642):479–83. doi: 10.1038/nature21076
38. Wojcik H, Griffiths E, Staggs S, Hagman J, Winandy S. Expression of a non–DNA–Binding Ikaros Isoform Exclusively in B Cells Leads to Autoimmunity But Not Leukemogenesis. Eur J Immunol (2007) 37(4):1022–32. doi: 10.1002/eji.200637026
39. Browne CD, Del Nagro CJ, Cato MH, Dengler HS, Rickert RC. Suppression of Phosphatidylinositol 3,4,5–Trisphosphate Production Is a Key Determinant of B Cell Anergy. Immunity (2009) 31(5):749–60. doi: 10.1016/j.immuni.2009.08.026
40. Arnold J, Murera D, Arbogast F, Fauny JD, Muller S, Gros F. Autophagy is Dispensable for B–Cell Development But Essential for Humoral Autoimmune Responses. Cell Death Differ (2016) 23(5):853–64. doi: 10.1038/cdd.2015.149
41. Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, et al. The Autophagy Gene ATG5 Plays an Essential Role in B Lymphocyte Development. Autophagy (2008) 4(3):309–14. doi: 10.4161/auto.5474
42. Krop I, de Fougerolles AR, Hardy RR, Allison M, Schlissel MS, Fearon DT. Self–Renewal of B–1 Lymphocytes Is Dependent on CD19. Eur J Immunol (1996) 26(1):238–42. doi: 10.1002/eji.1830260137
43. Hayakawa K, Formica AM, Brill–Dashoff J, Shinton SA, Ichikawa D, Zhou Y, et al. Early Generated B1 B Cells With Restricted BCRs Become Chronic Lymphocytic Leukemia With Continued c–Myc and Low Bmf Expression. J Exp Med (2016) 213(13):3007–24. doi: 10.1084/jem.20160712
44. Farmer JR, Allard–Chamard H, Sun N, Ahmad M, Bertocchi A, Mahajan VS, et al. Induction of Metabolic Quiescence Defines the Transitional to Follicular B Cell Switch. Sci Signal (2019) 12(604):eaaw5573. doi: 10.1126/scisignal.aaw5573
45. Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, et al. PI3 Kinase Signals BCR–Dependent Mature B Cell Survival. Cell (2009) 139(3):573–86. doi: 10.1016/j.cell.2009.08.041
46. Patke A, Mecklenbräuker I, Erdjument–Bromage H, Tempst P, Tarakhovsky A. BAFF Controls B Cell Metabolic Fitness Through a PKC Beta– and Akt–Dependent Mechanism. J Exp Med (2006) 203(11):2551–62. doi: 10.1084/jem.20060990
47. Dufort FJ, Bleiman BF, Gumina MR, Blair D, Wagner DJ, Roberts MF, et al. Cutting Edge: IL–4–Mediated Protection of Primary B Lymphocytes From Apoptosis Via Stat6–Dependent Regulation of Glycolytic Metabolism. J Immunol (2007) 179(8):4953–7. doi: 10.4049/jimmunol.179.8.4953
48. Doughty CA, Bleiman BF, Wagner DJ, Dufort FJ, Mataraza JM, Roberts MF, et al. Antigen Receptor–Mediated Changes in Glucose Metabolism in B Lymphocytes: Role of Phosphatidylinositol 3–Kinase Signaling in the Glycolytic Control of Growth. Blood (2006) 107(11):4458–65. doi: 10.1182/blood-2005-12-4788
49. Price MJ, Patterson DG, Scharer CD, Boss JM. Progressive Upregulation of Oxidative Metabolism Facilitates Plasmablast Differentiation to a T–Independent Antigen. Cell Rep (2018) 23(11):3152–9. doi: 10.1016/j.celrep.2018.05.053
50. Jang KJ, Mano H, Aoki K, Hayashi T, Muto A, Nambu Y, et al. Mitochondrial Function Provides Instructive Signals for Activation–Induced B–Cell Fates. Nat Commun (2015) 6:6750. doi: 10.1038/ncomms7750
51. Tsui C, Martinez–Martin N, Gaya M, Maldonado P, Llorian M, Legrave NM, et al. Protein Kinase C–β Dictates B Cell Fate by Regulating Mitochondrial Remodeling, Metabolic Reprogramming, and Heme Biosynthesis. Immunity (2018) 48(6):1144–59.e5. doi: 10.1016/j.immuni.2018.04.031
52. Dufort FJ, Gumina MR, Ta NL, Tao Y, Heyse SA, Scott DA, et al. Glucose–Dependent De Novo Lipogenesis in B Lymphocytes: A Requirement for Atp–Citrate Lyase in Lipopolysaccharide–Induced Differentiation. J Biol Chem (2014) 289(10):7011–24. doi: 10.1074/jbc.M114.551051
53. Brookens SK, Cho SH, Basso PJ, Boothby MR. Ampkα1 in B Cells Dampens Primary Antibody Responses Yet Promotes Mitochondrial Homeostasis and Persistence of B Cell Memory. J Immunol (2020) 205(11):3011–22. doi: 10.4049/jimmunol.1901474
54. Akkaya M, Traba J, Roesler AS, Miozzo P, Akkaya B, Theall BP, et al. Second Signals Rescue B Cells From Activation–Induced Mitochondrial Dysfunction and Death. Nat Immunol (2018) 19(8):871–84. doi: 10.1038/s41590-018-0156-5
55. Shaffer AL, Shapiro–Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, Downstream of Blimp–1, Expands the Secretory Apparatus and Other Organelles, and Increases Protein Synthesis in Plasma Cell Differentiation. Immunity (2004) 21(1):81–93. doi: 10.1016/j.immuni.2004.06.010
56. Bertolotti M, Yim SH, Garcia–Manteiga JM, Masciarelli S, Kim YJ, Kang MH, et al. B– to Plasma–Cell Terminal Differentiation Entails Oxidative Stress and Profound Reshaping of the Antioxidant Responses. Antioxid Redox Signal (2010) 13(8):1133–44. doi: 10.1089/ars.2009.3079
57. Gass JN, Gunn KE, Sriburi R, Brewer JW. Stressed–Out B Cells? Plasma–Cell Differentiation and the Unfolded Protein Response. Trends Immunol (2004) 25(1):17–24. doi: 10.1016/j.it.2003.11.004
58. Tellier J, Shi W, Minnich M, Liao Y, Crawford S, Smyth GK, et al. Blimp–1 Controls Plasma Cell Function Through the Regulation of Immunoglobulin Secretion and the Unfolded Protein Response. Nat Immunol (2016) 17(3):323–30. doi: 10.1038/ni.3348
59. Gaudette BT, Jones DD, Bortnick A, Argon Y, Allman D. mTORC1 Coordinates an Immediate Unfolded Protein Response–Related Transcriptome in Activated B Cells Preceding Antibody Secretion. Nat Commun (2020) 11(1):723–1. doi: 10.1038/s41467-019-14032-1
60. Cenci S, Mezghrani A, Cascio P, Bianchi G, Cerruti F, Fra A, et al. Progressively Impaired Proteasomal Capacity During Terminal Plasma Cell Differentiation. EMBO J (2006) 25(5):1104–13. doi: 10.1038/sj.emboj.7601009
61. Lam WY, Jash A, Yao CH, D’Souza L, Wong R, Nunley RM, et al. Metabolic and Transcriptional Modules Independently Diversify Plasma Cell Lifespan and Function. Cell Rep (2018) 24(9):2479–92.e6. doi: 10.1016/j.celrep.2018.07.084
62. Auner HW, Beham–Schmid C, Dillon N, Sabbattini P. The Life Span of Short–Lived Plasma Cells Is Partly Determined by a Block on Activation of Apoptotic Caspases Acting in Combination With Endoplasmic Reticulum Stress. Blood (2010) 116(18):3445–55. doi: 10.1182/blood-2009-10-250423
63. Jones DD, Gaudette BT, Wilmore JR, Chernova I, Bortnick A, Weiss BM, et al. mTOR Has Distinct Functions in Generating Versus Sustaining Humoral Immunity. J Clin Invest (2016) 126(11):4250–61. doi: 10.1172/JCI86504
64. Ersching J, Efeyan A, Mesin L, Jacobsen JT, Pasqual G, Grabiner BC, et al. Germinal Center Selection and Affinity Maturation Require Dynamic Regulation of mTORC1 Kinase. Immunity (2017) 46(6):1045–58.e6. doi: 10.1016/j.immuni.2017.06.005
65. Sander S, Chu VT, Yasuda T, Franklin A, Graf R, Calado DP, et al. Pi3 Kinase and FOXO1 Transcription Factor Activity Differentially Control B Cells in the Germinal Center Light and Dark Zones. Immunity (2015) 43(6):1075–86. doi: 10.1016/j.immuni.2015.10.021
66. Mendoza P, Martínez–Martín N, Bovolenta ER, Reyes–Garau D, Hernansanz–Agustín P, Delgado P, et al. R–Ras2 Is Required for Germinal Center Formation to Aid B Cells During Energetically Demanding Processes. Sci Signal (2018) 11(532):eaal1506. doi: 10.1126/scisignal.aal1506
67. Weisel FJ, Mullett SJ, Elsner RA, Menk AV, Trivedi N, Luo W, et al. Germinal Center B Cells Selectively Oxidize Fatty Acids for Energy While Conducting Minimal Glycolysis. Nat Immunol (2020) 21(3):331–42. doi: 10.1038/s41590-020-0598-4
68. Cho SH, Raybuck AL, Stengel K, Wei M, Beck TC, Volanakis E, et al. Germinal Centre Hypoxia and Regulation of Antibody Qualities by a Hypoxia Response System. Nature (2016) 537(7619):234–8. doi: 10.1038/nature19334
69. Jellusova J, Cato MH, Apgar JR, Ramezani–Rad P, Leung CR, Chen C, et al. Gsk3 Is a Metabolic Checkpoint Regulator in B Cells. Nat Immunol (2017) 18(3):303–12. doi: 10.1038/ni.3664
70. Weisel FJ, Zuccarino–Catania GV, Chikina M, Shlomchik MJ. A Temporal Switch in the Germinal Center Determines Differential Output of Memory B and Plasma Cells. Immunity (2016) 44(1):116–30. doi: 10.1016/j.immuni.2015.12.004
71. Inoue T, Shinnakasu R, Kawai C, Ise W, Kawakami E, Sax N, et al. Exit From Germinal Center to Become Quiescent Memory B Cells Depends on Metabolic Reprograming and Provision of a Survival Signal. J Exp Med (2021) 218(1):e20200866. doi: 10.1084/jem.20200866
72. Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, et al. Cutting Edge: The Dependence of Plasma Cells and Independence of Memory B Cells on BAFF and APRIL. J Immunol (2008) 180(6):3655–9. doi: 10.4049/jimmunol.180.6.3655
73. Bibby JA, Purvis HA, Hayday T, Chandra A, Okkenhaug K, Rosenzweig S, et al. Cholesterol Metabolism Drives Regulatory B Cell IL–10 Through Provision of Geranylgeranyl Pyrophosphate. Nat Commun (2020) 11(1):3412–4. doi: 10.1038/s41467-020-17179-4
74. Meng X, Grötsch B, Luo Y, Knaup KX, Wiesener MS, Chen XX, et al. Hypoxia–Inducible Factor–1α Is a Critical Transcription Factor for IL–10–Producing B Cells in Autoimmune Disease. Nat Commun (2018) 9(1):251–x. doi: 10.1038/s41467-017-02683-x
75. Watanabe K, Ichinose S, Hayashizaki K, Tsubata T. Induction of Autophagy by B Cell Antigen Receptor Stimulation and Its Inhibition by Costimulation. Biochem Biophys Res Commun (2008) 374(2):274–81. doi: 10.1016/j.bbrc.2008.07.013
76. Martinez–Martin N, Maldonado P, Gasparrini F, Frederico B, Aggarwal S, Gaya M, et al. A Switch From Canonical to Noncanonical Autophagy Shapes B Cell Responses. Science (2017) 355(6325):641–7. doi: 10.1126/science.aal3908
77. Conway KL, Kuballa P, Khor B, Zhang M, Shi HN, Virgin HW, et al. ATG5 Regulates Plasma Cell Differentiation. Autophagy (2013) 9(4):528–37. doi: 10.4161/auto.23484
78. Nimmerjahn F, Milosevic S, Behrends U, Jaffee EM, Pardoll DM, Bornkamm GW, et al. Major Histocompatibility Complex Class II–Restricted Presentation of a Cytosolic Antigen by Autophagy. Eur J Immunol (2003) 33(5):1250–9. doi: 10.1002/eji.200323730
79. Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Müller M, et al. Autophagy Promotes MHC Class II Presentation of Peptides From Intracellular Source Proteins. Proc Natl Acad Sci USA (2005) 102(22):7922–7. doi: 10.1073/pnas.0501190102
80. Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, et al. Endogenous MHC Class II Processing of a Viral Nuclear Antigen After Autophagy. Science (2005) 307(5709):593–6. doi: 10.1126/science.1104904
81. Arbogast F, Arnold J, Hammann P, Kuhn L, Chicher J, Murera D, et al. ATG5 is Required for B Cell Polarization and Presentation of Particulate Antigens. Autophagy (2019) 15(2):280–94. doi: 10.1080/15548627.2018.1516327
82. Chen M, Kodali S, Jang A, Kuai L, Wang J. Requirement for Autophagy in the Long–Term Persistence But Not Initial Formation of Memory B Cells. J Immunol (2015) 194(6):2607–15. doi: 10.4049/jimmunol.1403001
83. Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, et al. Discovery of Atg5/Atg7–Independent Alternative Macroautophagy. Nature (2009) 461(7264):654–8. doi: 10.1038/nature08455
84. Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, et al. Essential Role for Autophagy in the Maintenance of Immunological Memory Against Influenza Infection. Nat Med (2014) 20(5):503–10. doi: 10.1038/nm.3521
85. Halliley JL, Tipton CM, Liesveld J, Rosenberg AF, Darce J, Gregoretti IV, et al. Long–Lived Plasma Cells Are Contained Within the CD19(–)CD38(hi)CD138(+) Subset in Human Bone Marrow. Immunity (2015) 43(1):132–45. doi: 10.1016/j.immuni.2015.06.016
86. Galgani M, Bruzzaniti S, Matarese G. Immunometabolism and Autoimmunity. Curr Opin Immunol (2020) 67:10–7. doi: 10.1016/j.coi.2020.07.002
87. Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, et al. Defective B Cell Tolerance Checkpoints in Systemic Lupus Erythematosus. J Exp Med (2005) 201(5):703–11. doi: 10.1084/jem.20042251
88. Samuels J, Ng YS, Coupillaud C, Paget D, Meffre E. Impaired Early B Cell Tolerance in Patients With Rheumatoid Arthritis. J Exp Med (2005) 201(10):1659–67. doi: 10.1084/jem.20042321
89. Bosello S, Youinou P, Daridon C, Tolusso B, Bendaoud B, Pietrapertosa D, et al. Concentrations of BAFF Correlate With Autoantibody Levels, Clinical Disease Activity, and Response to Treatment in Early Rheumatoid Arthritis. J Rheumatol (2008) 35(7):1256–64.
90. Xin G, Shi W, Xu LX, Su Y, Yan LJ, Li KS. Serum BAFF is Elevated in Patients With IgA Nephropathy and Associated With Clinical and Histopathological Features. J Nephrol (2013) 26(4):683–90. doi: 10.5301/jn.5000218
91. Salazar–Camarena DC, Ortiz–Lazareno PC, Cruz A, Oregon–Romero E, Machado–Contreras JR, Muñoz–Valle JF, et al. Association of BAFF, APRIL Serum Levels, BAFF–R, TACI and BCMA Expression on Peripheral B–Cell Subsets With Clinical Manifestations in Systemic Lupus Erythematosus. Lupus (2016) 25(6):582–92. doi: 10.1177/0961203315608254
92. Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, et al. Mice Transgenic for BAFF Develop Lymphocytic Disorders Along With Autoimmune Manifestations. J Exp Med (1999) 190(11):1697–710. doi: 10.1084/jem.190.11.1697
93. Malkiel S, Jeganathan V, Wolfson S, Manjarrez Orduño N, Marasco E, Aranow C, et al. Checkpoints for Autoreactive B Cells in the Peripheral Blood of Lupus Patients Assessed by Flow Cytometry. Arthritis Rheumatol (2016) 68(9):2210–20. doi: 10.1002/art.39710
94. Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, et al. Reduced Competitiveness of Autoantigen–Engaged B Cells Due to Increased Dependence on BAFF. Immunity (2004) 20(4):441–53. doi: 10.1016/S1074-7613(04)00079-2
95. Xie P, Stunz LL, Larison KD, Yang B, Bishop GA. Tumor Necrosis Factor Receptor–Associated Factor 3 Is a Critical Regulator of B Cell Homeostasis in Secondary Lymphoid Organs. Immunity (2007) 27(2):253–67. doi: 10.1016/j.immuni.2007.07.012
96. Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 Signal Adapters Act Cooperatively to Control the Maturation and Survival Signals Delivered to B Cells by the BAFF Receptor. Immunity (2008) 28(3):391–401. doi: 10.1016/j.immuni.2008.01.009
97. Mambetsariev N, Lin WW, Wallis AM, Stunz LL, Bishop GA. TRAF3 Deficiency Promotes Metabolic Reprogramming in B Cells. Sci Rep (2016) 6:35349. doi: 10.1038/srep35349
98. Smith MJ, Ford BR, Rihanek M, Coleman BM, Getahun A, Sarapura VD, et al. Elevated PTEN Expression Maintains Anergy in Human B Cells and Reveals Unexpectedly High Repertoire Autoreactivity. JCI Insight (2019) 4(3):e123384. doi: 10.1172/jci.insight.123384
99. O’Neill SK, Getahun A, Gauld SB, Merrell KT, Tamir I, Smith MJ, et al. Monophosphorylation of CD79a and CD79b ITAM Motifs Initiates a SHIP–1 Phosphatase–Mediated Inhibitory Signaling Cascade Required for B Cell Anergy. Immunity (2011) 35(5):746–56. doi: 10.1016/j.immuni.2011.10.011
100. Getahun A, Beavers NA, Larson SR, Shlomchik MJ, Cambier JC. Continuous Inhibitory Signaling by Both SHP–1 and SHIP–1 Pathways Is Required to Maintain Unresponsiveness of Anergic B Cells. J Exp Med (2016) 213(5):751–69. doi: 10.1084/jem.20150537
101. Pao LI, Lam KP, Henderson JM, Kutok JL, Alimzhanov M, Nitschke L, et al. B Cell–Specific Deletion of Protein–Tyrosine Phosphatase Shp1 Promotes B–1a Cell Development and Causes Systemic Autoimmunity. Immunity (2007) 27(1):35–48. doi: 10.1016/j.immuni.2007.04.016
102. Wu XN, Ye YX, Niu JW, Li Y, Li X, You X, et al. Defective PTEN Regulation Contributes to B Cell Hyperresponsiveness in Systemic Lupus Erythematosus. Sci Transl Med (2014) 6(246):246ra99. doi: 10.1126/scitranslmed.3009131
103. Wu T, Qin X, Kurepa Z, Kumar KR, Liu K, Kanta H, et al. Shared Signaling Networks Active in B Cells Isolated From Genetically Distinct Mouse Models of Lupus. J Clin Invest (2007) 117(8):2186–96. doi: 10.1172/JCI30398
104. Abboud G, Choi SC, Kanda N, Zeumer–Spataro L, Roopenian DC, Morel L. Inhibition of Glycolysis Reduces Disease Severity in an Autoimmune Model of Rheumatoid Arthritis. Front Immunol (2018) 9:1973. doi: 10.3389/fimmu.2018.01973
105. Blokland SLM, Hillen MR, Wichers CGK, Zimmermann M, Kruize AA, Radstake TRDJ, et al. Increased mTORC1 Activation in Salivary Gland B Cells and T Cells From Patients With Sjögren’s Syndrome: mTOR Inhibition as a Novel Therapeutic Strategy to Halt Immunopathology? RMD Open (2019) 5(1):e000701–000701. doi: 10.1136/rmdopen-2018-000701
106. International Consortium for Systemic Lupus Erythematosus Genetics, (SLEGEN), Harley JB, Alarcón–Riquelme ME, Criswell LA, Jacob CO, et al. Genome–Wide Association Scan in Women With Systemic Lupus Erythematosus Identifies Susceptibility Variants in ITGAM, Pxk, KIAA1542 and Other Loci. Nat Genet (2008) 40(2):204–10. doi: 10.1038/ng.81
107. Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, et al. A Large–Scale Replication Study Identifies TNIP1, Prdm1, JAZF1, UHRF1BP1 and IL10 as Risk Loci for Systemic Lupus Erythematosus. Nat Genet (2009) 41(11):1228–33. doi: 10.1038/ng.468
108. Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, et al. Genome–Wide Association Study in a Chinese Han Population Identifies Nine New Susceptibility Loci for Systemic Lupus Erythematosus. Nat Genet (2009) 41(11):1234–7. doi: 10.1038/ng.472
109. Raychaudhuri S, Thomson BP, Remmers EF, Eyre S, Hinks A, Guiducci C, et al. Genetic Variants At CD28, PRDM1 and CD2/CD58 Are Associated With Rheumatoid Arthritis Risk. Nat Genet (2009) 41(12):1313–8. doi: 10.1038/ng.479
110. Zhou XJ, Lu XL, Lv JC, Yang HZ, Qin LX, Zhao MH, et al. Genetic Association of PRDM1–ATG5 Intergenic Region and Autophagy With Systemic Lupus Erythematosus in a Chinese Population. Ann Rheum Dis (2011) 70(7):1330–7. doi: 10.1136/ard.2010.140111
111. Page N, Gros F, Schall N, Décossas M, Bagnard D, Briand JP, et al. HSC70 Blockade by the Therapeutic Peptide P140 Affects Autophagic Processes and Endogenous MHCII Presentation in Murine Lupus. Ann Rheum Dis (2011) 70(5):837–43. doi: 10.1136/ard.2010.139832
112. Macri C, Wang F, Tasset I, Schall N, Page N, Briand JP, et al. Modulation of Deregulated Chaperone–Mediated Autophagy by a Phosphopeptide. Autophagy (2015) 11(3):472–86. doi: 10.1080/15548627.2015.1017179
113. Mocholi E, Dowling SD, Botbol Y, Gruber RC, Ray AK, Vastert S, et al. Autophagy Is a Tolerance–Avoidance Mechanism That Modulates Tcr–Mediated Signaling and Cell Metabolism to Prevent Induction of T Cell Anergy. Cell Rep (2018) 24(5):1136–50. doi: 10.1016/j.celrep.2018.06.065
114. Sakkas LI, Bogdanos DP, Katsiari C, Platsoucas CD. Anti–Citrullinated Peptides as Autoantigens in Rheumatoid Arthritis–Relevance to Treatment. Autoimmun Rev (2014) 13(11):1114–20. doi: 10.1016/j.autrev.2014.08.012
115. Ireland JM, Unanue ER. Autophagy in Antigen–Presenting Cells Results in Presentation of Citrullinated Peptides to CD4 T Cells. J Exp Med (2011) 208(13):2625–32. doi: 10.1084/jem.20110640
116. Chen YM, Chang CY, Chen HH, Hsieh CW, Tang KT, Yang MC, et al. Association Between Autophagy and Inflammation in Patients With Rheumatoid Arthritis Receiving Biologic Therapy. Arthritis Res Ther (2018) 20(1):268–0. doi: 10.1186/s13075-018-1763-0
117. Thacker EL, Mirzaei F, Ascherio A. Infectious Mononucleosis and Risk for Multiple Sclerosis: A Meta–Analysis. Ann Neurol (2006) 59(3):499–503. doi: 10.1002/ana.20820
118. Lee DY, Sugden B. The Latent Membrane Protein 1 Oncogene Modifies B–Cell Physiology by Regulating Autophagy. Oncogene (2008) 27(20):2833–42. doi: 10.1038/sj.onc.1210946
119. Jagessar SA, Holtman IR, Hofman S, Morandi E, Heijmans N, Laman JD, et al. Lymphocryptovirus Infection of Nonhuman Primate B Cells Converts Destructive Into Productive Processing of the Pathogenic Cd8 T Cell Epitope in Myelin Oligodendrocyte Glycoprotein. J Immunol (2016) 197(4):1074–88. doi: 10.4049/jimmunol.1600124
120. Walsh ER, Pisitkun P, Voynova E, Deane JA, Scott BL, Caspi RR, et al. Dual Signaling by Innate and Adaptive Immune Receptors is Required for TLR7–induced B–Cell–Mediated Autoimmunity. Proc Natl Acad Sci USA (2012) 109(40):16276–81. doi: 10.1073/pnas.1209372109
121. Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll–Like Receptor 9 Controls Anti–DNA Autoantibody Production in Murine Lupus. J Exp Med (2005) 202(2):321–31. doi: 10.1084/jem.20050338
122. Weindel CG, Richey LJ, Bolland S, Mehta AJ, Kearney JF, Huber BT. B Cell Autophagy Mediates TLR7–Dependent Autoimmunity and Inflammation. Autophagy (2015) 11(7):1010–24. doi: 10.1080/15548627.2015.1052206
123. Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy–Dependent Viral Recognition by Plasmacytoid Dendritic Cells. Science (2007) 315(5817):1398–401. doi: 10.1126/science.1136880
124. Chaturvedi A, Dorward D, Pierce SK. The B Cell Receptor Governs the Subcellular Location of Toll–Like Receptor 9 Leading to Hyperresponses to DNA–Containing Antigens. Immunity (2008) 28(6):799–809. doi: 10.1016/j.immuni.2008.03.019
125. Kim D, Nguyen QT, Lee J, Lee SH, Janocha A, Kim S, et al. Anti–Inflammatory Roles of Glucocorticoids Are Mediated by Foxp3(+) Regulatory T Cells Via a miR–342–Dependent Mechanism. Immunity (2020) 53(3):581–96.e5. doi: 10.1016/j.immuni.2020.07.002
126. Stohl W, Hiepe F, Latinis KM, Thomas M, Scheinberg MA, Clarke A, et al. Belimumab Reduces Autoantibodies, Normalizes Low Complement Levels, and Reduces Select B Cell Populations in Patients With Systemic Lupus Erythematosus. Arthritis Rheum (2012) 64(7):2328–37. doi: 10.1002/art.34400
127. Chatham WW, Wallace DJ, Stohl W, Latinis KM, Manzi S, McCune WJ, et al. Effect of Belimumab on Vaccine Antigen Antibodies to Influenza, Pneumococcal, and Tetanus Vaccines in Patients With Systemic Lupus Erythematosus in the BLISS–76 Trial. J Rheumatol (2012) 39(8):1632–40. doi: 10.3899/jrheum.111587
128. Dörner T, Posch MG, Li Y, Petricoul O, Cabanski M, Milojevic JM, et al. Treatment of Primary Sjögren’s Syndrome With Ianalumab (VAY736) Targeting B Cells by BAFF Receptor Blockade Coupled With Enhanced, Antibody–Dependent Cellular Cytotoxicity. Ann Rheum Dis (2019) 78(5):641–7. doi: 10.1136/annrheumdis-2018-214720
129. Lui SL, Yung S, Tsang R, Zhang F, Chan KW, Tam S, et al. Rapamycin Prevents the Development of Nephritis in Lupus–Prone NZB/W F1 Mice. Lupus (2008) 17(4):305–13. doi: 10.1177/0961203307088289
130. Zeng Q, Zhang H, Qin J, Xu Z, Gui L, Liu B, et al. Rapamycin Inhibits BAFF–Stimulated Cell Proliferation and Survival by Suppressing Mtor–Mediated PP2A–Erk1/2 Signaling Pathway in Normal and Neoplastic B–Lymphoid Cells. Cell Mol Life Sci (2015) 72(24):4867–84. doi: 10.1007/s00018-015-1976-1
131. Ke Z, Liang D, Zeng Q, Ren Q, Ma H, Gui L, et al. hsBAFF Promotes Proliferation and Survival in Cultured B Lymphocytes Via Calcium Signaling Activation of mTOR Pathway. Cytokine (2013) 62(2):310–21. doi: 10.1016/j.cyto.2013.03.011
132. Lai ZW, Kelly R, Winans T, Marchena I, Shadakshari A, Yu J, et al. Sirolimus in Patients With Clinically Active Systemic Lupus Erythematosus Resistant to, or Intolerant of, Conventional Medications: A Single–Arm, Open–Label, Phase 1/2 Trial. Lancet (2018) 391(10126):1186–96. doi: 10.1016/S0140-6736(18)30485-9
133. Lee SY, Moon SJ, Kim EK, Seo HB, Yang EJ, Son HJ, et al. Metformin Suppresses Systemic Autoimmunity in Roquin(san/san) Mice Through Inhibiting B Cell Differentiation Into Plasma Cells Via Regulation of AMPK/Mtor/STAT3. J Immunol (2017) 198(7):2661–70. doi: 10.4049/jimmunol.1403088
134. Yin Y, Choi SC, Xu Z, Zeumer L, Kanda N, Croker BP, et al. Glucose Oxidation Is Critical for CD4+ T Cell Activation in a Mouse Model of Systemic Lupus Erythematosus. J Immunol (2016) 196(1):80–90. doi: 10.4049/jimmunol.1501537
135. Sun F, Geng S, Wang H, Wang H, Liu Z, Wang X, et al. Effects of Metformin on Disease Flares in Patients With Systemic Lupus Erythematosus: Post Hoc Analyses From Two Randomised Trials. Lupus Sci Med (2020) 7(1):e000429. doi: 10.1136/lupus–000429
136. Sun F, Wang HJ, Liu Z, Geng S, Wang HT, Wang X, et al. Safety and Efficacy of Metformin in Systemic Lupus Erythematosus: A Multicentre, Randomised, Double–Blind, Placebo–Controlled Trial. Lancet Rheumatol (2020) 2(4):e210–6. doi: 10.1016/S2665-9913(20)30004-7
137. Wang F, Tasset I, Cuervo AM, Muller S. In Vivo Remodeling of Altered Autophagy–Lysosomal Pathway by a Phosphopeptide in Lupus. Cells (2020) 9(10):2328. doi: 10.3390/cells9102328
138. Monneaux F, Lozano JM, Patarroyo ME, Briand JP, Muller S. T Cell Recognition and Therapeutic Effect of a Phosphorylated Synthetic Peptide of the 70K snRNP Protein Administered in MR/lpr Mice. Eur J Immunol (2003) 33(2):287–96. doi: 10.1002/immu.200310002
139. Monneaux F, Parietti V, Briand JP, Muller S. Importance of Spliceosomal RNP1 Motif for Intermolecular T–B Cell Spreading and Tolerance Restoration in Lupus. Arthritis Res Ther (2007) 9(5):R111. doi: 10.1186/ar2317
140. Zimmer R, Scherbarth HR, Rillo OL, Gomez–Reino JJ, Muller S. Lupuzor/P140 Peptide in Patients With Systemic Lupus Erythematosus: A Randomised, Double–Blind, Placebo–Controlled Phase IIb Clinical Trial. Ann Rheum Dis (2013) 72(11):1830–5. doi: 10.1136/annrheumdis-2012-202460
141. ImmuPharma. A 52–Week, Randomized, Double–Blind, Parallel–Group, Placebo–Controlled Study to Evaluate the Efficacy and Safety of a 200–Mcg Dose of IPP–201101 Plus Standard of Care in Patients With Systemic Lupus Erythematosus (Lupuzor) (2015). Available at: https://clinicaltrials.gov/ct2/show/study/NCT02504645?id=NCT02504645&draw=2&rank=1&load=cart (Accessed Feb 20, 2021).
142. Meister S, Schubert U, Neubert K, Herrmann K, Burger R, Gramatzki M, et al. Extensive Immunoglobulin Production Sensitizes Myeloma Cells for Proteasome Inhibition. Cancer Res (2007) 67(4):1783–92. doi: 10.1158/0008-5472.CAN-06-2258
143. Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, et al. The Proteasome Inhibitor Bortezomib Depletes Plasma Cells and Protects Mice With Lupus–Like Disease From Nephritis. Nat Med (2008) 14(7):748–55. doi: 10.1038/nm1763
144. Scheibe F, Prüss H, Mengel AM, Kohler S, Nümann A, Köhnlein M, et al. Bortezomib for Treatment of Therapy–Refractory Anti–NMDA Receptor Encephalitis. Neurology (2017) 88(4):366–70. doi: 10.1212/WNL.0000000000003536
Keywords: autophagy, metabolism, autoimmunity, B cell, SLE - systemic lupus erythematosus, B cell development and differentiation
Citation: Raza IGA and Clarke AJ (2021) B Cell Metabolism and Autophagy in Autoimmunity. Front. Immunol. 12:681105. doi: 10.3389/fimmu.2021.681105
Received: 15 March 2021; Accepted: 19 May 2021;
Published: 07 June 2021.
Edited by:
Valentina Pucino, University of Birmingham, United KingdomReviewed by:
Alessandra Bettiol, University of Florence, ItalySimon H. Bridge, Northumbria University, United Kingdom
David Harry Gardner, University of Birmingham, United Kingdom
Copyright © 2021 Raza and Clarke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander J. Clarke, YWxleGFuZGVyLmNsYXJrZUBrZW5uZWR5Lm94LmFjLnVr