 Huimei Huang
Huimei Huang Shisheng Li
Shisheng Li Gangcai Zhu
Gangcai Zhu- Department of Otolaryngology-Head and Neck Surgery, The Second Xiangya Hospital, Central South University, Changsha, China
Nasopharyngeal carcinoma (NPC) is a malignant tumor of the nasopharynx mainly characterized by geographic distribution and EBV infection. Metabolic reprogramming, one of the cancer hallmarks, has been frequently reported in NPCs to adapt to internal energy demands and external environmental pressures. Inevitably, the metabolic reprogramming within the tumor cell will lead to a decreased pH value and diverse nutritional supplements in the tumor-infiltrating micro-environment incorporating immune cells, fibroblasts, and endothelial cells. Accumulated evidence indicates that metabolic reprogramming derived from NPC cells may facilitate cancer progression and immunosuppression by cell-cell communications with their surrounding immune cells. This review presents the dysregulated metabolism processes, including glucose, fatty acid, amino acid, nucleotide metabolism, and their mutual interactions in NPC. Moreover, the potential connections between reprogrammed metabolism, tumor immunity, and associated therapy would be discussed in this review. Accordingly, the development of targets on the interactions between metabolic reprogramming and immune cells may provide assistances to overcome the current treatment resistance in NPC patients.
Introduction
Nasopharyngeal carcinoma (NPC) is a unique epithelial malignancy with the highest incidence in distinctive ethnic and geographic populations in Southeast Asia, and it is characterized by lymphoepithelial-like histological features and Epstein-Barr virus (EBV) infection (1). More than 70% of the 130,000 new global cases reported each year are diagnosed in East and Southeast Asia, especially southern China (2). GBD Diseases and Injuries Collaborators reported that, by 2019, the fatality rate and disability-adjusted life years of patients with NPC had increased annually (3). Although the 5-year overall survival rate in patients with early-stage NPC is approximately 95%, it is only 54.2% in early-staged NPC patients (4). Notably, the nasopharynx, enriched with lymphoid tissues, is a unique inductive site for B cell responses, plasma cell generation, and naïve T cell maturation and differentiation into the effector and memory T cells that eliminate inciting antigen cells, mounting a defense against NPC initiation and development (5). The mechanism by which NPC cells avoid immune surveillance remains unclear. Somatic alternations in chromatin modification and metabolism-associated genes, identified by two whole-exome sequencing studies (6, 7), imply the importance of reprogramming energy metabolism in NPCs. Reprogramming energy metabolism and evading immune destruction have been extensively investigated in NPC (8). Recently, emerging strategies have been avidly explored to target metabolic pathways that enhance anti-tumor immunity. However, no review has focused on these two features or their mutual connections in NPC; here, we review the dysregulated metabolic processes, including glucose, fatty acid, amino acid, and nucleotide metabolism, and the mutual interplay of these processes in NPC. Moreover, the potential connections between reprogrammed metabolism, tumor immunity, and the associated therapies are discussed in this review.
Aerobic Glycolysis
In correlative studies of tumor metabolism, the earliest and most focused point is the Warburg effect, through which cancer cells consume glucose to produce lactate even when the oxygen level is sufficient and do not enter the TCA cycle (9). The glucose metabolism of NPC (as shown in Figure 1) mostly depends on aerobic glycolysis, and it is frequently accompanied by the abnormal expression of glucose transport and metabolic enzymes (hexokinase (HK), phosphofructokinase-1, lactate dehydrogenase (LDH), etc.) (10).
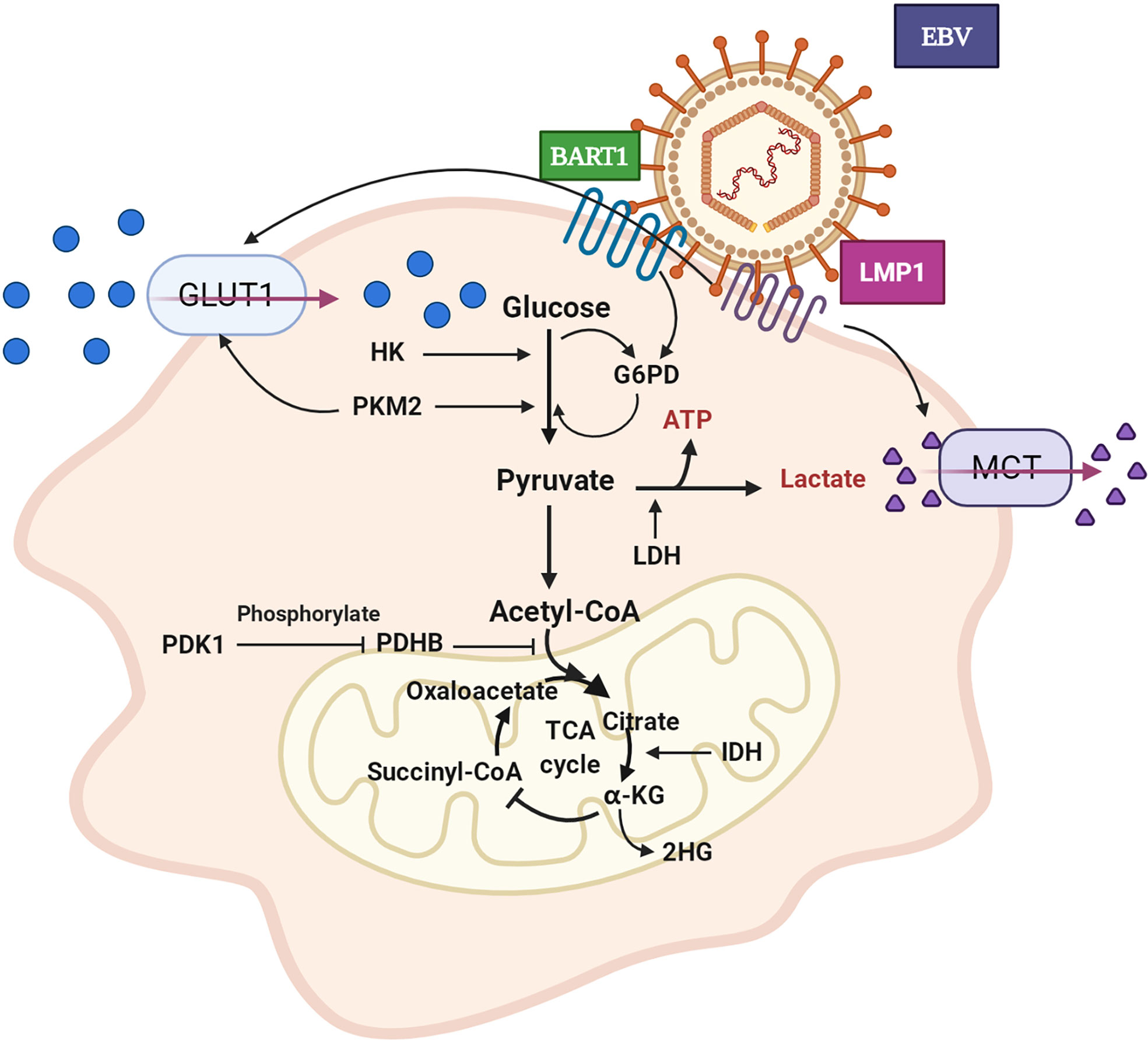
Figure 1 Aerobic glycolysis of NPC cell. GLUT1 expression is up-regulated and glucose uptake is increased. The expression level of HK is increased and promotes glycolysis. PKM2 over-expression promotes GLUT1 expression and glucose absorption. The expression of PDHB is down-regulated, which inhibits the conversion of pyruvate to acetyl CoA. PDK1 is highly expressed, which leads to the inactivation of PDHB. The over-expression of IDH2 promoted the transformation of α - KG to 2-HG. LDH expression and lactate production increased. EBV secreted-protein LMP1 promotes glucose uptake and lactate production. In pentose phosphate pathway, EBV-miR-BART1 promotes G6PD over-expression. GLUT1: glucose transporter 1, HK: hexokinase, PKM2: pyruvate kinase M2, PDHB: pyruvate dehydrogenase B, PDK1: pyruvate dehydrogenase kinase 1, IDH2: isocitrate dehydrogenase, α-KG: α–ketoglutarate, 2-HG: 2-hydroxyglutaric acid, LDH: lactate dehydrogenase, LMP1: EBV latent membrane protein 1, BART1: EBV-encoded microRNA BART1, G6PD: glucose-6-phosphate dehydrogenase.
GLUT1 for Glucose Transport
The application of [18F]-fluorodeoxyglucose positron emission tomography (PET-CT) in NPC patient imaging in clinical practice has provided the best evidence to support the increased uptake of glucose in NPC (11). Several investigations have shown that total lesion glycolysis (TLG), a prognostic indicator, is in consistent with metabolic tumor volume (MTV) NPC patients, as shown by [F18]-PET-CT (11–13). GLUT1 has a high affinity for glucose, facilitating glucose uptake from outside cancer cells (14). GLUT-1 expression is relevant to clinical stage and lymph node metastasis in NPC (15). EBV exists in almost all NPCs. LMP1, an EBV-encoded specific protein, promotes GLUT-1 transcription by activating mTORC1/NF-κB signalling, resulting in the regulation of aerobic glycolysis and the growth of NPC cells (16). Effective inhibitors of the GLUT family can inhibit glycolysis flux and tumor growth (17). Regulatory enzymes of glucose metabolism such as pyruvate kinase affect the process of GLUT1-induced glucose transportation. Aberration of M2 pyruvate kinase (PKM2) is notable in NPC cells. Inhibition of PKM2 decreases GLUT1 activity and the Warburg effect in NPC cells (18). As summarized in Table 1, GLUT1 and PKM2 show hypomethylation in their promoter regions in NPC.

Table 1 The Aberrant Methylation of the Key Metabolic Genes in NPC Microarrays.
From Glucose to Pyruvate
Aerobic and anaerobic oxidation of glucose proceed through three and two stages, respectively. These processes share the same glycolysis stage in which glucose is decomposed into two molecules of pyruvate. Three vital enzymes, hexokinase (HK) (also known as glucokinase), phosphofructokinase 1 (PFK1), and pyruvate kinase (PK), determine glycolytic flow speed and direction (19). HK is associated with NPC patient prognosis and outcomes, and its expression has been shown to be promoted by FOXC2-YAP signalling in NPC cells (20, 21). HK inhibitors can limit tumor glycolysis and increase free radicals that result in cell apoptosis (22, 23). The abnormal expression of PK enhances glucose uptake and increases the rate of phosphoenolpyruvate to pyruvate (18). Pyruvate kinase M2 (PKM2), a subtype of PK, is overexpressed in NPC cells and upregulated significantly by activation of the phosphatidylinositol 3 kinase (PIK3)/AKT signalling pathway, leading to increased glucose uptake, lactate, and ATP levels (24). In addition, PKM2 promotes cancer metastasis by activating EGFR-stimulated nuclear translocation of PKM2, and PKM2 regulates the transcription of FOSL1 and ANTXR2 (25). Ten-eleven translocation protein 2 (TET2) can block the process of PKM nuclear translocation and thus inhibit the glycolysis, proliferation, and invasion of NPC cells (26). To date, PFK1 has not been reported in NPC. However, early studies revealed that upregulation of hypoxia-inducible factor 1α (HIF-1α) in carcinoma can increase PFK1 protein expression and promote aerobic glycolysis, providing some clues for future NPC research (27, 28). Taken together, the regulation of these key enzymes in NPC cells may control aerobic glycolysis and lead to NPC development.
From Pyruvate to ATP and Carbon Dioxide in Mitochondria
In the second stage of aerobic oxidation, pyruvate is transported to mitochondria where it is converted to acetyl-CoA. The reaction that converts pyruvate into acetyl-CoA is catalysed by the pyruvate dehydrogenase complex (PDHC), which includes pyruvate dehydrogenase (E1), dihydrolipoamide transacetylase (E2), and dihydrolipoamide transdehydrogenase (E3) (19). Previous studies have found that the expression of pyruvate dehydrogenase B (PDHB, a member of PDHC-E1) was downregulated in the context of NPC, which inhibited the conversion of pyruvate to acetyl-CoA (29). In addition, genes related to PDHB are abnormally expressed in cancer stem cell-like cells (30). Moreover, PDH action is regulated by PDK. PDK1 is a pyruvate dehydrogenase kinase isoenzyme located in the mitochondria and overexpressed in NPC cells, and it mediates the conversion of pyruvate to acetyl-CoA, inactivates PDH, playing an important role in the regulation of glucose and fatty acid metabolism. The frequent overexpression of PDK1 in primary NPC is associated with poor patient prognosis, clinical stage, and metastasis (31, 32). Chibby, a β-Catenin-related antagonist, has been shown to arrest aerobic glycolysis in human NPC via inhibition of PDK1 (33). In addition, in head and neck squamous cell carcinoma, mitochondrial gene mutations promote the accumulation of HIF-1α because PDH is downregulated by PDK2 (34). In general, PDK exerts a crucial influence on the metabolic regulation of NPC.
In the third stage of aerobic oxidation, acetyl-CoA enters the TCA cycle and is coupled with oxidative phosphorylation to form ATP. This process requires catalysis of at least seven metabolic enzymes, of which the three most important rate-limiting enzymes are citrate synthase (CS), isocitrate dehydrogenase (IDH), and the α-ketoglutarate dehydrogenase complex (α-KGDH) (19). Isocitrate dehydrogenase 1 (IDH1) exhibits hypomethylation in NPC (Table 1). Isocitrate dehydrogenase 2 (IDH2) is involved in EBV-dependent metabolic reprogramming and tumorigenesis. The expression of IDH2 is upregulated in NPC, leading to increased intracellular α-ketoglutaric acid (α-KG)-catalysed 2-hydroxyglutaric acid (2-HG). The level of 2-HG is positively correlated with local lymph node metastasis in NPC (35). However, recent studies have found that IDH2 is differentially mutated in different NPC subtypes (36, 37). IDH mutation endows the enzyme product with the novel ability to catalyse α-ketoglutaric acid (α-KG) to 2-hydroxyglutaric acid (2HG). Ultimately, it can be inferred that IDH blocks the TCA cycle by catalysing the conversion of α-KG to 2HG. Unfortunately, the roles of the other two rate-limiting enzymes, α-KGDH and CS, have not been reported in NPC. However, mounting evidence suggests that high expression of succinate dehydrogenase B (SDHB) in recurrent local NPC is beneficial for prolonging patient survival time, indirectly implying roles for α-KGDH and CS in NPC cells (38).
From Pyruvate to Lactate and the Pentose Phosphate Pathway
Lactate is the largest glucose metabolite in carcinoma tissue. The reduction of pyruvate to lactate, which is crucial to the Warburg effect in cancer cells, is catalysed by lactate dehydrogenase (LDH). High serum LDH levels are associated with poor survival in NPC (39). Inhibition of LDH can induce G2/M cell cycle arrest by downregulating the CDK1/cyclinB1 pathway, consequently promoting cell apoptosis by increasing mitochondrial ROS production (40). The expression of lactate dehydrogenase 5 (LDH-5) is also increased in oral squamous cell carcinoma (OSCC) (41). Currently, LDH-5, a subtype of lactate dehydrogenase, is considered a promising anticancer target (42). Hence, efforts to reprogramme LDH have led to a significant change in the production of lactate.
The pentose phosphate pathway is another pathway of glucose metabolism that substantially contributes to biosynthesis and antioxidation. The most important catalytic enzyme in the pentose phosphate pathway is glucose-6-phosphate dehydrogenase (G6PD). G6PD participates in the metabolic pathway of pentose phosphate to produce NADPH and ribose phosphate (43). Through oxidation and group transfer, fructose-6-phosphate and glyceraldehyde 3-phosphate are formed and return to the glycolysis metabolic pathway. NADPH activity induced by G6PD can maintain reduced levels of glutathione, which has been found to be an important antioxidant in vivo (44). NPC patients with low serum levels of G6PD tend to relapse and have a poor prognosis. To promote tumor cell antioxidation and prevent oxidative damage, increased expression of G6PD may be attenuated by EBV-miR-BART1 signalling in NPC cells (45, 46).
Monocarboxylic Acid Transporter
Lactate produced by cancer cells is conveyed to the extracellular environment through monocarboxylic acid transporters (MCTs), especially monocarboxylic acid transporter 4 (MCT4), thus forming an acidic tumor microenvironment (47). Moreover, cancer cells within the tumor cooperate to establish metabolic “symbiosis”. Cancer cells in hypoxic regions consume glucose and release lactate through anaerobic glycolysis, and similar to a lactate shuttle, this lactate is used as fuel for the TCA cycle by cells in adjacent oxygen-containing tumor areas (48). This movement of lactate shows interplay with monocarboxylic acid transporters (MCTs), which are differentially expressed: Anoxic cancer cells overexpress MCT4 as the main lactate export transporter, and oxygen-rich cancer cells overexpress MCT1 as a lactate input transporter (49). MCT1 is highly expressed in NPC and may promote cell invasion and migration through the PI3K/Akt signalling pathway (50).
Effect of LMP1 on Aerobic Glycolysis
EBV latent membrane protein 1 (LMP1) can promote aerobic glycolysis in NPC cells. LMP1 significantly increases glucose consumption, lactate production, and LDH activity. The content of hypoxia-inducible factor 1 (HIF-1α) and that of its targets PDK1 and PKM2 also continuously increase (51). LMP1 activates a variety of cellular signalling pathways (52). Studies have suggested that LMP1 is associated with EBV-driven NPC pathogenesis by mediating the FGF2/FGFR1 signalling pathway. It increases glucose and glutamine uptake, LDHA activity, and lactate production by activating the FGFR1 signalling pathway and increases the phosphorylation of PKM2 and LDHA, as well as the expression of PDHK1, c-Myc, and HIF-1α (53). In summary, these findings suggest that LMP1 promotes aerobic glycolysis by regulating changes in metabolic enzymes and related genes such as c-Myc and HIF-1α.
Fatty Acid Metabolism
Lipid Droplet Transport
The high demand for nutrients in tumor cells increases the uptake and synthesis of fatty acids. In addition to the de novo synthesis of fatty acids, dietary fatty acids in the blood are decomposed into triglycerides by lipoprotein lipase (LPL) and transported into tumor cells by CD36 (a fatty acid transferase) (54, 55). Serum lipid levels constitute a risk factor for cancer metastasis. In contrast to the level in normal nasopharyngeal epithelium, the accumulation of lipid droplets in NPC increases greatly, which may be associated with downregulation of the lipolysis gene adipose triglyceride lipase (ATGL) via EBV-encoded LMP2A (56). Lipid-lowering drugs, such as lovastatin, can effectively inhibit nasopharyngeal oncogenesis (57, 58).
Fatty Acid Synthesis
Under normal circumstances, the sources of fatty acids are mainly exogenous fatty acids digested and absorbed in the small intestine and then synthesized in the liver. However, malignant cells, including NPCs, can independently synthesize endogenous fatty acids due to the high expression of fatty acid synthase (FASN) (59, 60). As shown in Table 1, FASN is hypomethylated in the TSS200 region. Silencing or targeting FASN makes NPC cells more sensitive to radiation (61, 62). In addition, quercetin, a polyphenolic flavonoid interferes with the activity of FASN and reduces NPC cell proliferation (63). Consistently, the expression of fatty acid synthase-related protein (FADD) in nasopharyngeal cells has been found to be higher than that in noncancerous tissues (64). Accordingly, acetyl-CoA transported to the cytoplasm via the citric acid-pyruvate cycle allows NPC cells to synthesize fatty acids de novo and provides rich substrates for the β-oxidation of fatty acids. Malic enzyme 1 (ME1) overexpressed in NPC cells catalyses the malic acid to pyruvate conversion and then promotes NADPH production to satisfy the increasing demand for de novo fatty acid synthesis (65).
Fatty Acid Oxidation
β-oxidation of fatty acids (FAO) produces a large amount of ATP to meet cellular needs. Fatty acids are oxidized into acyl CoA and then transported to mitochondria by carnitine acyltransferase 1 (CPT1). Acetyl-CoA, FADH2, and NADH are formed by stepwise processes involving dehydrogenation, hydration, dehydrogenation, and thiolysis in mitochondria (19). CPT1 is a rate-limiting enzyme that affects the role played by acyl-CoA in mitochondria. Studies suggested that FAO is highly activated due to the upregulated expression of CPT1A in radiation-tolerant NPC cells, which supplies energy for the proliferation and progression of tumor cells (66). PROX1 promotes β-oxidation of fatty acids by upregulating CPT1A expression, and acetyl-CoA cooperates with histone acetyltransferase p300 in the acetylation of histones on lymphangiogenic genes. Thus, the PROX1-p300 interaction facilitates preferential histone acetylation (67, 68). This indicates that CPT1A may mediate epigenetic changes in oncogenes by enhancing fatty acid availability for acetyl-CoA. Taken together, fatty acid β-oxidation can promote the proliferation and differentiation of lymphatic endothelial cells, repair pathologically damaged lymphatic vessels, and contribute to favourable conditions for lymphatic metastasis of malignant cells. Thus, blocking CPT1A activity inhibits lymphangiogenesis, implying a potential therapeutic role for targeting FAO in NPC lymphatic metastasis (67, 68). To prevent lipid droplet accumulation and leverage this reduced accumulation, SOD1, a regulator of antioxidants, is highly expressed in NPCs. Therefore, blocking the activity of SOD1 can cause lipid accumulation and probably inhibit NPC cell growth (69). When the energy demand of tumor cells increases, peroxisome proliferator-activated receptor coactivator 1 (PGC-1α) can promote FAO in NPC cells via the CEBPB/CPT1A signalling axis to protect mitochondria from toxic lipid overload and resist radiation (70). Monoacylglycerol lipase (MAGL) is a serine hydrolase that can hydrolyse monoacylglycerol esters into free fatty acids and glycerol (71). MAGL is highly expressed in NPC and may affect CPT1-mediated β-oxidation through the FFA pathway, promote the oxidative decomposition of fatty acids, and promote metastasis through the epithelial-mesenchymal transition (EMT) (71, 72). Moreover, the CPT1A expression level is significantly correlated with the prognosis of NPC patients.
The acetyl-CoA produced by fatty acid β-oxidation is partially converted into ketone bodies (73). Inhibition of ketone body metabolism has been found in NPC in recent years. HMG-CoA lyase (HMGCL), a vital enzyme in the metabolic pathway of ketone bodies, is inactivated in NPC cells, which boosts their proliferation and metastasis by reducing the production of β-hydroxybutyric acid (β-HB) and inhibiting reactive oxygen species (ROS) generation (74). Therefore, agents that target pathways that regulate the homeostasis of fatty acid metabolism in NPCs, such as Pueraria lobata extract, may be effective because they disrupt lipid metabolic reprogramming (75, 76).
In conclusion, lipid metabolism changes in NPC include alterations to lipid transport and absorption, fatty acid synthesis, and fatty acid oxidation. As shown in Figure 2, LMP2A encoded by EBV downregulates ATGL to accumulate lipid droplets; CD36 transports lipids to the cytoplasm. The malate-pyruvate cycle and transfer of acetyl-CoA from mitochondria to the cytoplasm for fatty acid synthesis are accelerated by ME1. FASN notably upregulates palmitic acid expression, catalysing palmitic acid biosynthesis to produce free palmitic acid. CPT1A is regulated by various genes (PROX1, SOD, PGC1α, and MAGL), leading to active fatty acid oxidation in mitochondria, which supplies abundant ATP and acetyl-CoA for the body and meets the material and energy needs of proliferating, growing, and metastasis of NPC cells. Furthermore, the metabolism of fatty acids into ketone bodies is inhibited by inactivated HMGCL and reduced ROS levels.
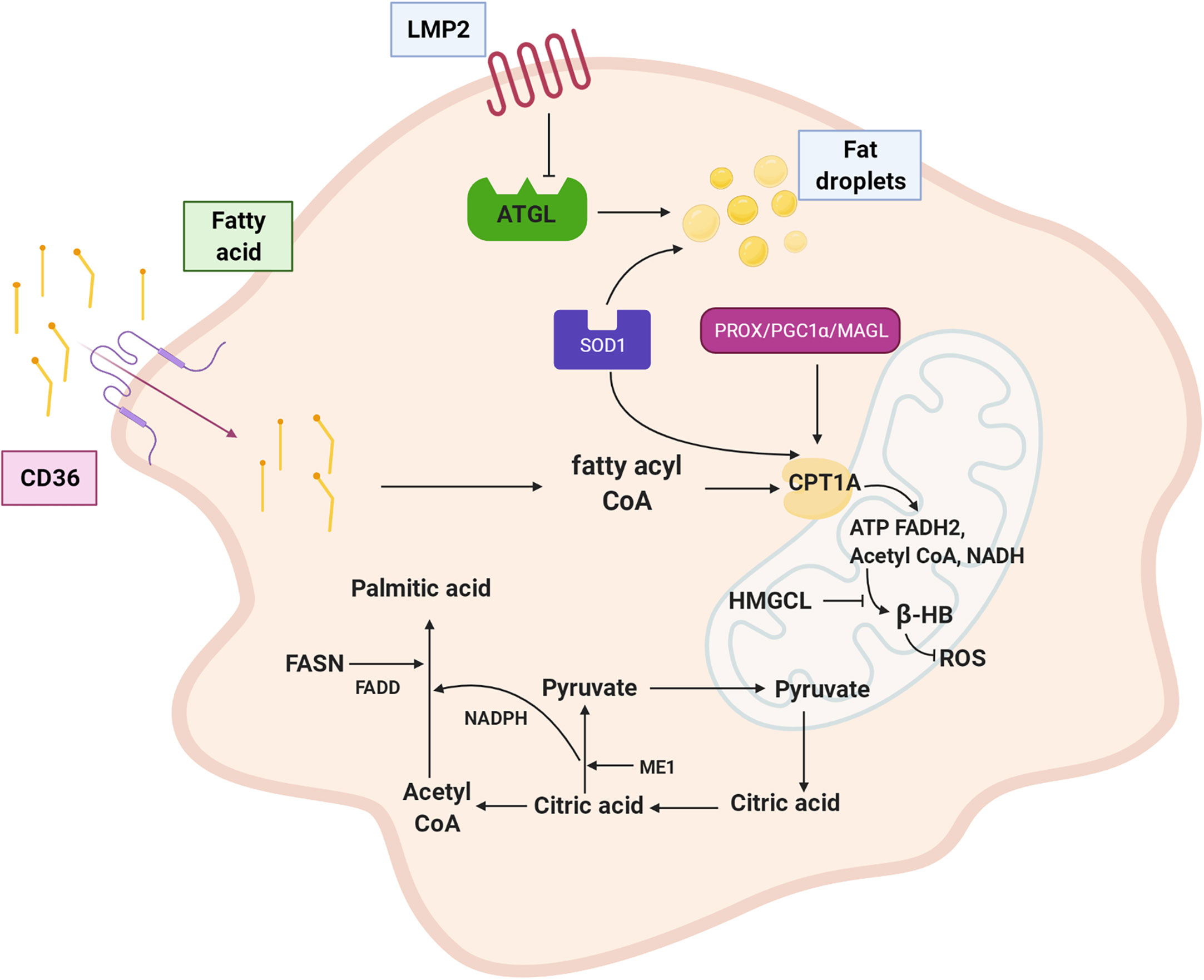
Figure 2 Fatty acid metabolism of NPC cell. LMP2A down-regulates lipolysis gene ATGL and promotes fat droplet accumulation; CD36 transports fatty acid to the cytoplasm. ME1 enhances malate pyruvate cycle, promotes transfer of acetyl CoA from mitochondria to cytoplasm, and participates in palmitic acid synthesis. FASN is up-regulated expression and palmitic acid synthesis is vigorous. CPT1A is positively regulated by PROX1、SOD1、PGC1α and MAGL. Inactivation of HMGCL inhibits ketone like β-HB metabolism of fatty acid and reduced production of ROS. LMP2A: EBV encoded latent membrane protein 2A, ATGL: adipotriglyceride lipase, CD36: fatty acid translocase, ME1: malic enzyme 1, FASN: fatty acid synthase, CPT1A: carnitine acyltransferase 1, PROX1: Prospero homeobox protein-1, SOD1: superoxide dismutase 1, PGC1α: peroxisome proliferator-activated receptor coactivator 1, MAGL: monoacylglycerol lipase, HMGCL: HMG-CoA lyase, β-HB: β-hydroxybutyric acid,ROS: reactive oxygen species.
Amino Acid Metabolism
Exogenous proteins are hydrolysed into amino acids and oligopeptides in the stomach and small intestine and then absorbed by different transporters or in the γ-glutamyl cycle. The activation of corresponding α-ketoacids, induced by glucose metabolism or amino acid catabolism, generates certain nonessential amino acids (alanine, aspartic acid, and glutamic acid). A deaminated amino acid can be converted to α-keto acid, while the coenzyme pyridoxal phosphate is converted to pyridoxamine phosphate and transfers amino groups to the corresponding residue. The excess ammonia produced during amino acid metabolism enters the urea cycle and excreted as urea out of the body (19). Arginine succinate synthase (ASS1) is a key enzyme of urea metabolism that inhibits the removal of ammonia in NPC (77).
Glutamine provides carbon and nitrogen for biosynthesis (78). Glutamine synthetase catalyses the conversion of glutamate with ammonia into glutamine (79). Glutamine synthetase genes are highly expressed in radiation-resistant cancer cells, promoting recovery of malignant cells from G2/M blockades (80). This may be the mechanism by which NPC cells exhibit resistance to radiotherapy. Moreover, the expression of GLS1-encoded glutaminase K (KGA) and glutaminase C (GAC) is increased in EBV-infected NPC cells. In addition, the expression of glutamate dehydrogenase 1 (GLUD1) and glutamate dehydrogenase 2 (GLUD2) is also upregulated in NPC cells, and they catalyse the conversion of glutamate to α–ketoglutarate (81). Mounting evidence suggests that the protein levels of KGA and GAC are controlled by c-MYC expression, promoting glutamine catabolism (82). Glutamine metabolites, namely, glutamic acid, α-ketoglutaric acid, and aspartic acid, can mediate the metabolism, epigenetic, nucleotide synthesis, and redox balance of tumor cells. Hence, a variety of compounds targeting glutamine metabolism have been developed for anticancer therapy (47).
The metabolism of branched-chain amino acid (leucine, isoleucine, and valine) affects diverse biological processes in protein synthesis and epigenetic regulation. The metabolic pathways of branched-chain amino acids are altered in many solid tumors, such as melanoma, NPC, and breast cancer (76, 83). Phosphoserine transaminase 1 (PSAT1), with a role in serine biosynthesis, is associated with NPC outcomes (84).
In summary (Figure 3), the synthesis and catabolism of glutamine play very prominent roles in NPC cells. Glutamine synthetase promotes a combination of glutamate and amino acids to form glutamine, which provides energy and scavenges ammonia. The upregulated expression of the KGA and GAC isoforms GLUD1 and GLUD2 promotes glutamine and glutamate decomposition and generates increasing levels of ammonia and α-ketoglutarate, which provides additional raw materials for energy generation and biosynthesis. When the ammonia pathway is blocked, more nitrogen becomes available. Downregulated ASS1 inhibits the ornithine cycle and boosts tumor cell proliferation. The metabolic pathway of branched-chain amino acids in NPC is dysregulated, although the detailed mechanism needs to be further explored.
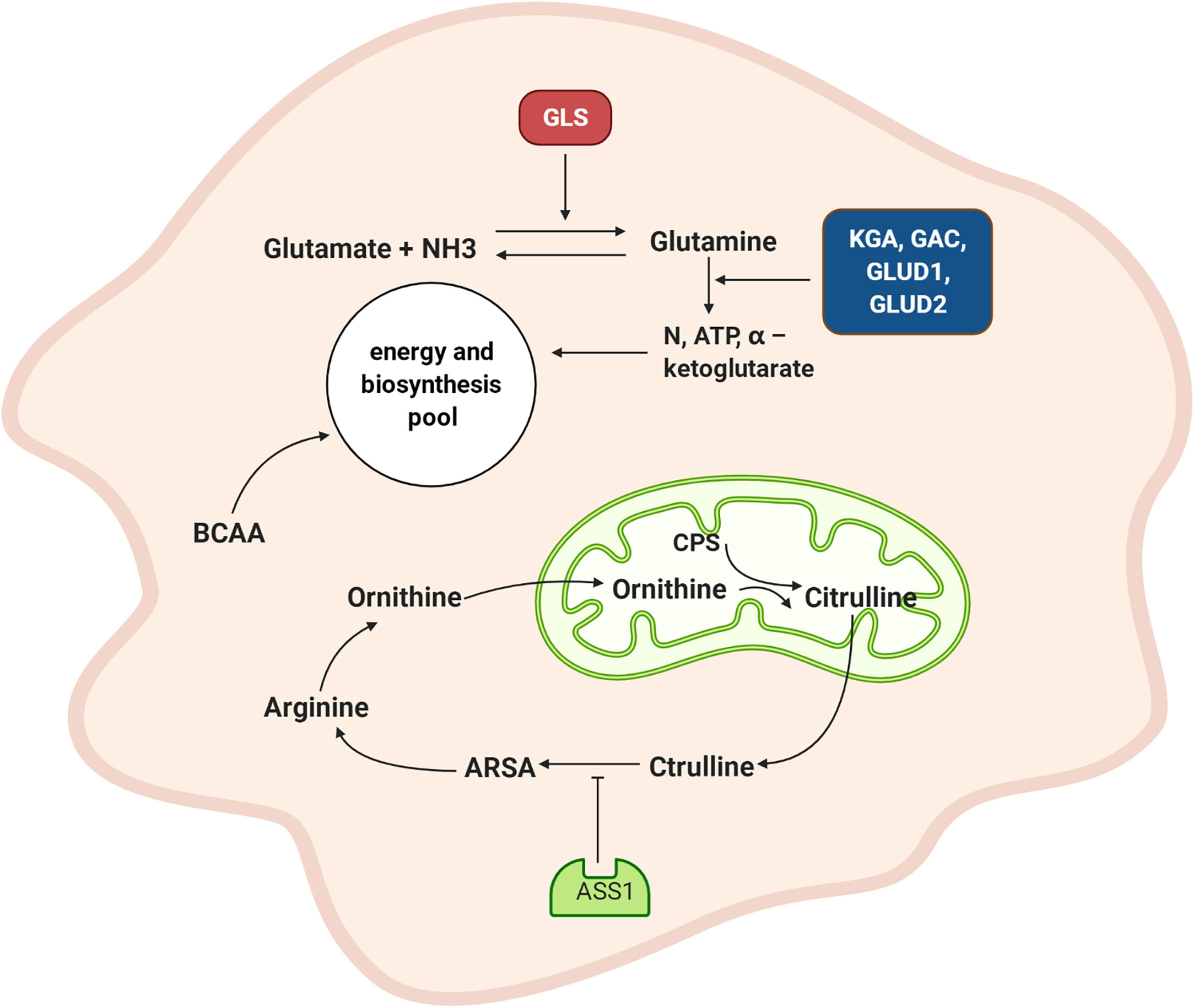
Figure 3 Amino acid metabolism of NPC cell. GLS promotes the combination of glutamate and amino groups to produce glutamine, which not only supplies energy but also scavenges ammonia. The expression of KGA and GAC subtypes are increased, and GLUD1 and GLUD2 are also up-regulated and promote the decomposition of glutamine and glutamic acid and produced more ammonia and α – ketoglutarate. This provides raw materials for energy generation and biosynthesis. ASS1 is down-regulated, which inhibited the ornithine cycle. GLS: glutamine synthetase, KGA: K-glutaminase GAC: glutaminase C, GLUD1: glutamate dehydrogenase 1, GLUD2: glutamate dehydrogenase 2, CPS: carbamoyl phosphate, ARSA: arginine succinate, ASS1: arginine succinate synthase, BCAA: branched-chain amino acid.
In three important metabolic pathways, key genes generally induce changes in metabolic processes. In order to further explore these genes, retrieval for transcriptional sequencing and somatic mutation was performed. A summary of the expression deregulation and somatic mutation of certain genes is presented in Tables 2 and 3, respectively. Overview of somatic mutation for key metabolic genes in NPC and other head and neck cancer is exhibited in Figure 4.

Figure 4 The landscape of key metabolic gene mutations, variants and copy number alternations in NPC and other head and neck cancer from cBioPortal (https://www.cbioportal.org/).

Table 2 The Expression of the Key Metabolic Genes in NPC Microarrays.
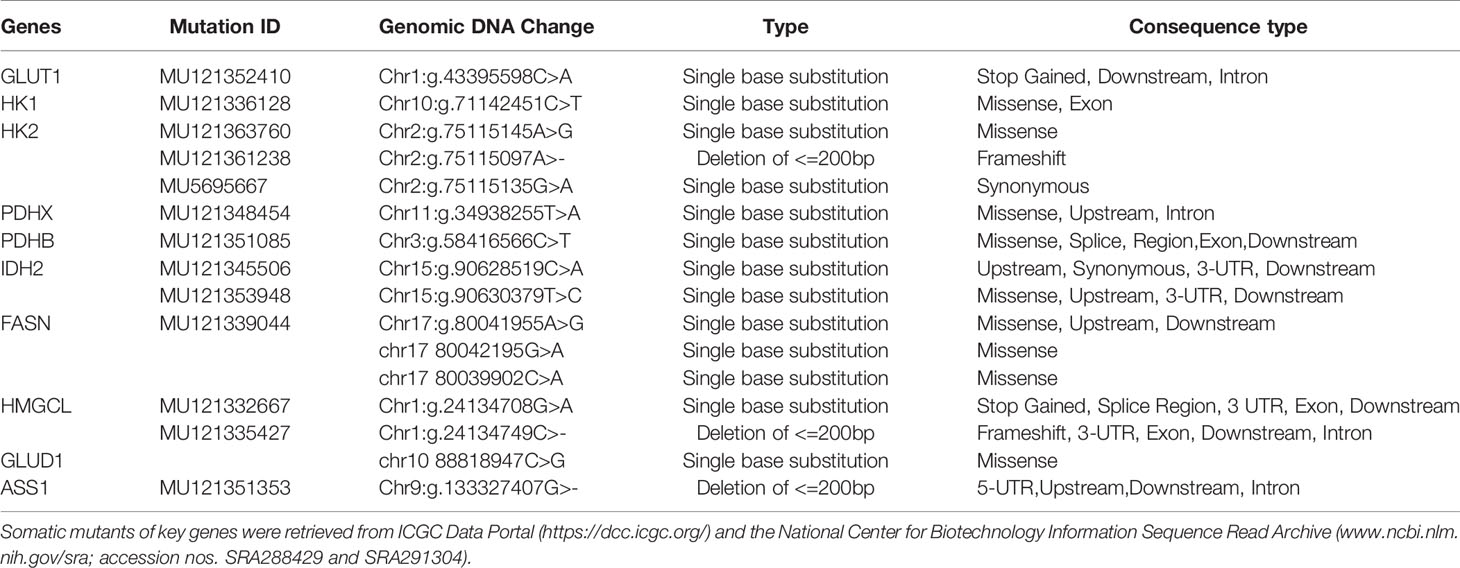
Table 3 Characteristics of Somatic Mutation of the Key Metabolic Genes in NPC.
Reprogramming of Other Metabolic Pathways
Nitric Oxide
In addition to the abovementioned metabolic patterns, NPC exhibits other metabolic reprogramming mechanisms. The central role of NOS is catalysis of the arginine precursor into nitric oxide (NO). Increased NO release has been detected in the tumor cells of recurring NPC (85, 86). The role of NO in inducing p53 mutation and angiogenesis has been established. Interestingly, tumor cell infiltration by NO can lead to the upregulation of DNA-dependent protein kinase catalytic subunit (DNA-PKcs); however, DNA-PKcs is typically directed to the repair of double-strand DNA breaks. Increased expression of DNA-PKcs protects cancer cells from the toxic effects of NO and the consequences of DNA damage (such as DNA-damaging chemotherapeutic drugs and X-ray irradiation) (87). In contrast to NO derived from tumor cells, NO derived from macrophages shows latent cytotoxicity to adjoining tumor cells (88).
Currently, radiotherapy is a fundamental treatment for NPC. Radiation-resistant tumor cells lead to residual and recurring cancer (89, 90). In radiation-resistant NPC cells, the production of NO is increased by upregulating the expression of CLIC4 (chloride ion intracellular channel 4), promoting the nuclear translocation of iNOS, reducing ROS levels, and inhibiting the cell death induced by ROS oxidative stress (85, 91). The internal tumor environment itself is anoxic (92). According to these characteristics, it can be hypothesized that radiation-resistant NPC cells create a stable microenvironment and acquire tolerance to stress. In general, NO production is increased in NPC, especially in radiation-resistant cancer cells. In the tumor microenvironment infiltrated by NO, DNA-PKcs is overexpressed in tumor cells, strengthening the resistance of tumor cells to NO toxicity and reducing tumor sensitivity to radiotherapy and chemotherapy on DNA damage.
Fe Metabolism
Lactoferrin is an iron-binding protein, and its N-terminus is a serine protease with an immunological effect against bacteria and viruses (93). However, downregulated lactoferrin has been found to confer immune defence against EBV infection and to increase PDK1-induced glycolysis (94, 95). Furthermore, lactoferrin can inhibit NPC development and metastasis through the MAPK and PI3K/AKT pathways (94, 96).
Ferroptosis is a cell death mode that differs from apoptosis, necrosis, and autophagy and is caused by iron-dependent oxidative damage and associated with cytoplasmic and lipid peroxidation, smaller mitochondria, and higher mitochondrial membrane density (97). Ferroptosis is inhibited in the absence of glutamine or glutamine catabolism blockade. The decomposition product of glutamine, α-KG, is necessary for ferroptosis induction (98). The speed of glutathione synthesis is limited by cysteine level. Early studies found that exogenous cysteine is essential for the growth of HeLa and other cells (99). Cells cultured in cysteine-deficient medium die of glutathione depletion, which can be recovered by supplementing the medium with iron chelators (100, 101). Hence, ferroptosis depends on amino acid metabolism.
Iron is a trace element necessary for the accumulation of lipid peroxide and induction of ferroptosis. Transferrin and transferrin receptors, which enable iron internalization from the extracellular environment, are necessary for ferroptosis (98, 102). A low level of BDH2 (3-hydroxybutyrate dehydrogenase 2) amplifies NPC cell malignant behaviours by promoting intracellular iron overload, indicating that reduced intracellular iron might be favourable for NPC treatment (103). Fe is a cofactor in a variety of intracellular synthesis or decomposition reactions, and it contributes to ferroptosis induction and decreased NPC cell proliferation (104).
Epigenetic Changes in Nucleotide Metabolism, DNA, and Histones
Nucleotide metabolism refers to the synthesis and catabolism of purine nucleotides and pyrimidine nucleotides, whose de novo synthesis requires glutamine, aspartic acid, one-carbon units of tetrahydrofolic acid, and CO2 (105). The expression of thymine synthase (TYMS) and dihydrofolate reductase (DHFR) in NPC is remarkably increased (106), which can lead to increased tetrahydrofolate expression for rapid nucleotide synthesis and to promoted pyrimidine nucleotides for facilitated tumor DNA replication and repair.
Epigenetic changes, especially DNA methylation abnormalities caused by EBV, play important roles in EBV-associated cancers such as NPC (107). DNA methylation relies on S-adenosylmethionine (SAM) to provide methyl groups, and the formation of SAM depends on extracellular folic acid, which can be increased in NPC cells by α-folate receptor activity (108). Isocitrate dehydrogenase (IDH1) mutation is associated with increased histone and DNA methylation because it decreases α-KG and α-KG-dependent prolyl hydroxylase (PHD) activity and increases HIF-1α activity (109–111). The expression of hypermethylated genes such as UBE2L6 is silenced in NPC, and this silencing has been found to stimulate ATGL degradation in adipocytes, causing abnormal catabolism (112). Similarly, ASS1, a rate-limiting enzyme in the ornithine cycle, is downregulated in NPC due to DNA methylation and contributes to lymphatic metastasis and invasion of malignant cells (77).
The Relationship Between Three Major Metabolic Pathways
Glucose, lipids, proteins, and nucleic acids do not function in isolation; they are interrelated and transformed by common metabolic intermediates, the TCA cycle, and biological oxidation in NPC cells. In each metabolic process, the activity of enzymes is regulated by mutant genes or anomalous signalling pathways. For example, activation of the mutant p53 gene and the MAPK, Wnt/β-catenin, and PIK3/AKT signalling pathways primarily involves three major metabolic processes (113). Metabolic intermediates can prevent tumor cell apoptosis by affecting the levels of free radicals or ROS. Notably, in addition to regulating enzymes, these intermediates can be induced to modulate nutrient intake and transportation. The increased intake of glucose, fat, and protein meets the fast-growing energy needs of tumors. Amino acid synthesis, such as glutamine, provides substrates for purine and pyrimidine nucleotides. Fatty glycerol can be converted into dihydroxyacetone phosphate. Amino acids (alanine, tryptophan, serine, etc.) can be converted into pyruvic acid, which enhances glycolysis to a certain extent, produces lactate and provides energy quickly. NADPH provides acetyl-CoA with the reducing equivalents necessary for fatty acid synthesis (19).
NPC Immune Evasion
Tumor cells resist or escape immune clearance by the host immune system through various mechanisms (114). The NPC tumor microenvironment (TME) is complex due to enriched lymphoid tissues and direct connection with the natural respiratory channels. Metabolic reprogramming involves metabolites and abnormally expressed metabolism-related proteins to cause immunosuppression. Here, we elaborate on the relationship between metabolic reprogramming and tumor-related immune escape.
The coexistence of tumor-infiltrating lymphocytes and EBV-infected NPC cells creates a unique TME that supports immune escape in the early stage of EBV infection (115). EBV-infected NPC cells can secrete cytokines and exosomes containing viral products to modulate the function of stromal cells in the NPC tumor microenvironment and thus facilitate disease progression and prevent host immune attacks (1). Next, we describe the immune evasion mechanism of NPC cells (Figure 5).
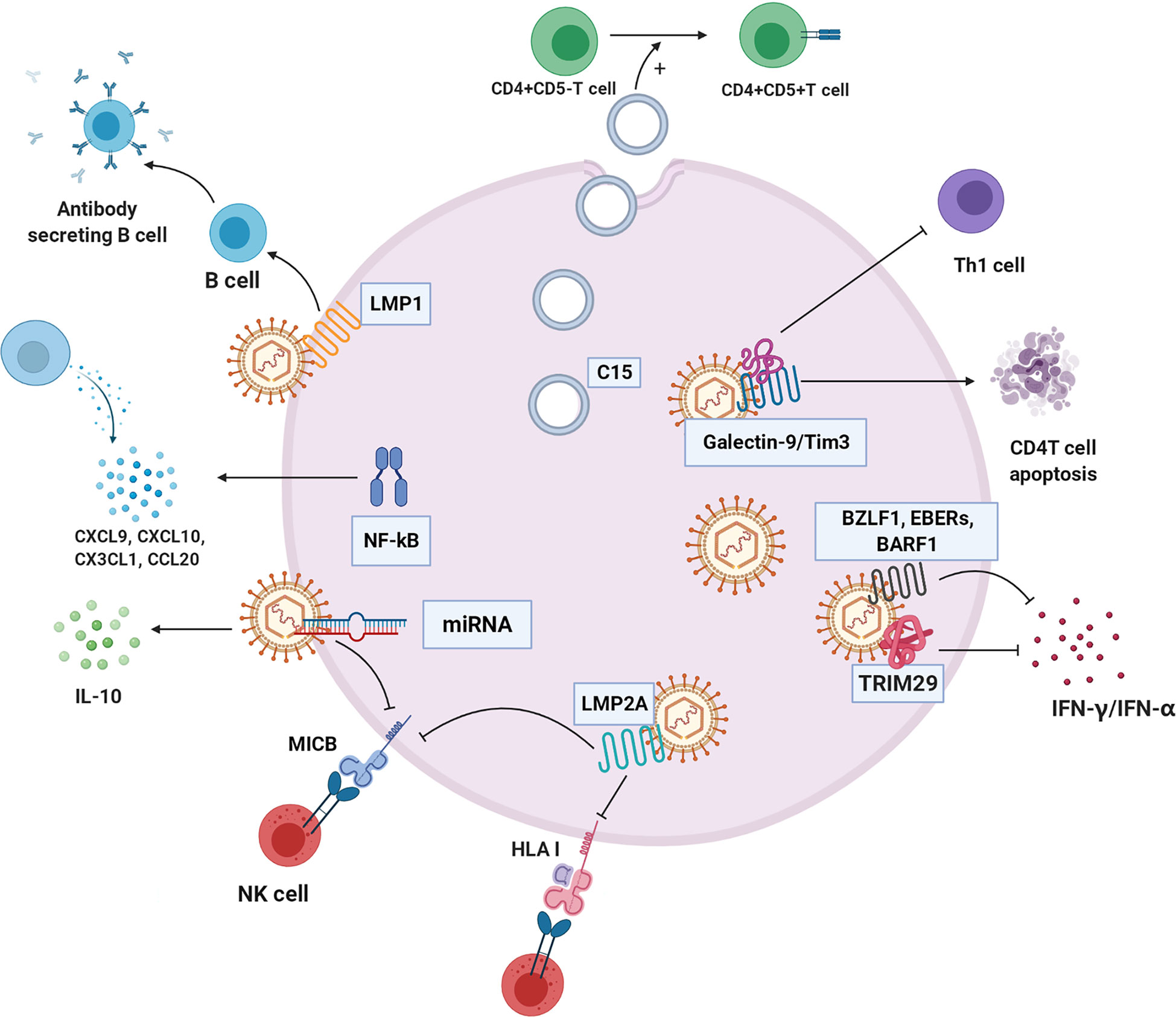
Figure 5 The immune evasion mechanism of NPC cell. C15 exosomes induced transformation and aggregation of CD4+CD25+Treg cells. NF-κB regulates a variety of chemokines CXCL9, CXCL10, CX3CL1, and CCL20 to reshape the tumor immune microenvironment. TRIM29 blocks interferon production. Galectin-9/Tim3 can induce apoptosis of CD4 T cells and inhibit Th1. LMP2A changes HLA-I, block recognition and presentation of cancer cell antigen peptide, and inhibit killing effect of NK cells. EBV-related products, such as BZLF1, EBERS AND BARF1, inhibit IFN - γ and IFN - α to escape immune clearance, respectively. LMP1 inhibits the differentiation of B cells into antibody-secreting cells. Some microRNAs of EBV down-regulate immune ligand MICB, which hinders recognition of NK cells.C15:C15 exosomes, CXCL9, CXCL10, CX3CL1, CCL20: chemokines of different subfamilies, TRIM29: EBV-induced tripartite motif-containing 29 protein, LMP2A: EBV latent membrane protein 2A, HLA-I: human leukocyte antigen I, BZLF1, EBERs, BARF1:EBV-related production, LMP1: EBV latent membrane protein 1, MICB: MHC class I chain-related MIC.
Exosomes
NPC cells imbue exosomes with LMP1, viral miRNAs, C15, and other molecules and secrete them into the immune environment (116). NPC-derived exosomes carrying C15 can transform CD4+CD25- T lymphocytes into Treg cells (CD4+CD25+) and stimulate Treg cell aggregation and immunosuppressive effects, allowing tumor cells to escape immune clearance (117). Interestingly, studies have indicated that this immunosuppressive mechanism probably can be unleashed by treatment with an anti-CCL20 blocking monoclonal antibody (117, 118). LMP1 in exosomes effectively stimulates MAPK and Akt activation, promotes cell proliferation and inhibits host immunity (119). In addition, LMP1 can induce B cell activation but severely inhibits B cell differentiation into antibody-secreting cells (ASCs) (120).
Cytokines and Chemokines
EBV disrupts the immunological response by stimulating regulatory T cells that secrete IL-10, an inhibitory cytokine that reduces the infiltration of cytotoxic T cells (121, 122).
NPC is abnormally regulated through four cancer-related pathways (the EGFR-PI3K-Akt-mTOR, NOTCH, and NF-κB pathways) (123). EBNA1, a gene encoded by EBV, inhibits NF-κB activity in the epidermis, a situation that can be exploited for immune escape and can cause cells to be continuously infected (124, 125). In contrast, LMP1 is an effective activator of the NF-κB signalling pathway (126). In EBV-positive NPC cells, activated NF-κB regulates the production of many chemokines, such as CXCL9, CXCL10, CX3CL1, and CCL20, leading to T lymphocyte infiltration into the cancer environment of NPC (1). Regardless, certain viral oncoproteins lead to the stimulation of the NF-κB pathway; the constitutive activation of NF-κB may be part of an important mechanism by which tumor cells escape immune surveillance and resist immunotherapy (127).
EBV induces members of the TRIM29 protein family to inhibit innate immunity in human airway epithelial cells. TRIM29 knockout accelerates type-I interferon (IFN1) and almost eliminates EBV and lethal herpes simplex virus-1 in human NPC cells (128). Therefore, EBV and possibly other double-stranded DNA viruses drive TRIM29 to suppress local innate immunity, resulting in persistent DNA viral infection. In addition, exosomes carrying galectin-9 released by EBV-infected NPC cells inhibit Th1 activity and induce the apoptosis of EBV-specific CD4 T cells (114). Blocking galectin-9/Tim-3 activity in vitro can release Th1 inhibition and lead to a maintained antitumor T cell response, thus improving the clinical efficacy of NPC immunotherapy (129).
Mutation of HLA Epitopes Associated With EBV
EBV strains carrying LMP1 and HLA-A2-restricting epitopes are prevalent in NPC in populations in South China and Taiwan (130). This EBV strain resists immune recognition, evades CTL recognition, weakens the IFN-γ response, and may lead to NPC prevalence to some extent (131). LMP2A downregulates the expression of HLA-ABC and MIC-A/B through promoter hypermethylation, hinders recognition and presentation of endogenous antigenic peptides, and inhibits T and NK cell action (132).
Products Related to EBV
EBV affects interferon expression to support EBV-positive NPC growth in vivo (133). The early EBV protein BZLF1 decreases IFN-γ receptor expression, which further affects IFN-γ-induced MHC-II expression, implying that EBV can evade the antiviral immune response in early infection (134, 135). Another EBV secretory protein, BARF1, directly inhibits α-interferon secretion from monocytes, reducing the innate response to the virus (136). Similarly, BDLF3, encoded by EBV, is an early factor that can target the cell surface and intracellular MHC-I molecules for ubiquitin-mediated proteasome degradation and induce immune cell apoptosis (137).
LMP1 not only affects NPC cell metabolism but also participates in host antitumor immunity. LMP1-mediated glycolysis regulates IL-1β, IL-6, and GM-CSF expression through the NLRP3 inflammatory body, COX-2, and NF-kB signalling pathways, which enhances the expansion of tumor-related bone marrow-derived suppressor cells (MDSCs) (138).
The immune evasion of cancer is also related to virus-related microRNAs (139). In many viruses, such as HCMV and EBV, miRNAs target the immune ligand MICB and downregulate MICB, resulting in inhibited recognition of natural killer cells (140). In addition, EBV miR-BART15 decreases IL-1β expression (141).
Effects of Metabolites Derived From Tumor Cells on Immune Cells
Lactate is the metabolite in cancer cells with the most profound effect. A high concentration of lactate interferes with T cell metabolism (142), damages dendritic cell and tumor-associated macrophage function, and inhibits monocyte migration and cytokine release, which promote cancer cell development (143–145). Lactate mediates M2-like polarization of tumor-associated macrophages (TAMs) through hypoxia-inducible factor-1α (HIF-1α), activates the VEGF pathway and augments cancer cell perfusion in blood (146–148). Acidification of the tumor environment caused by lactate inhibits monocyte-secreted TNF, protecting malignant cells from immune clearance (149).
In addition to immunosuppression by environmental acidification, other tumor metabolites, such as adenosine, kynurenine, and tryptophan, can play roles in tumor immunity. Extracellular adenosine weakens the adhesion and cytotoxicity of T cells via A2a and A3 adenosine receptors (150). Indoleamine-2,3-dioxygenase (IDO) is highly expressed in tumor cells, catalysing tryptophan, which negatively regulates the immune response, inhibits T cell proliferation, and induces T cell apoptosis, thus causing immune tolerance (151, 152). As shown in Table 2, IDO1 is overexpressed in NPC. Kynurenine catabolized from tryptophan within tumor cells can promote Treg cell function and numbers by activating kynurenine aryl-hydrocarbon receptor (AHR) in CD4+ T cells (47). Furthermore, IDO directly activates Treg cells. IDO inhibitors are being actively studied as single agent drugs or for use in combination with other treatments against cancer (153–155). Lipids obtained in part from adjacent cancer cells with enhanced fatty acid synthesis accumulate in tumor-infiltrating myeloid cells, including myeloid-derived suppressor cells (MDSCs) and TAMs, and can promote metabolic reprogramming and promote the acquisition of immunosuppressive phenotypes (47). MAGL induces TAMs to M2-like polarization, helping tumor cells evade immune clearance (156).
Nutritional Competition Between Cancer Cells and Immune Cells
Energy tumor cells and immune cells compete within the tumor microenvironment. Metabolic reprogramming helps tumor cells increase their competitive advantage by enabling greater absorption of nutrients and acquisition of energy than their surrounding immune cells, which greatly weakens the tumor cell recognition and clearance by host immunity (157). Glutamine deprivation in cancer cells inhibits T cell proliferation and cytokine production (47). The increased utilization of glucose in tumor cells leads to limited extracellular levels for use by tumor-infiltrating immune cells. Similarly, upregulation of GLUT1 expression in NPC cells accelerates glucose consumption, which inhibits T cell effector functions. Thus, the T cell response against NPC might be restored by blocking cancer cell glucose and arginine consumption and increasing their levels in infiltrating T cells (49). These two aforementioned mechanisms are depicted in Figure 6.
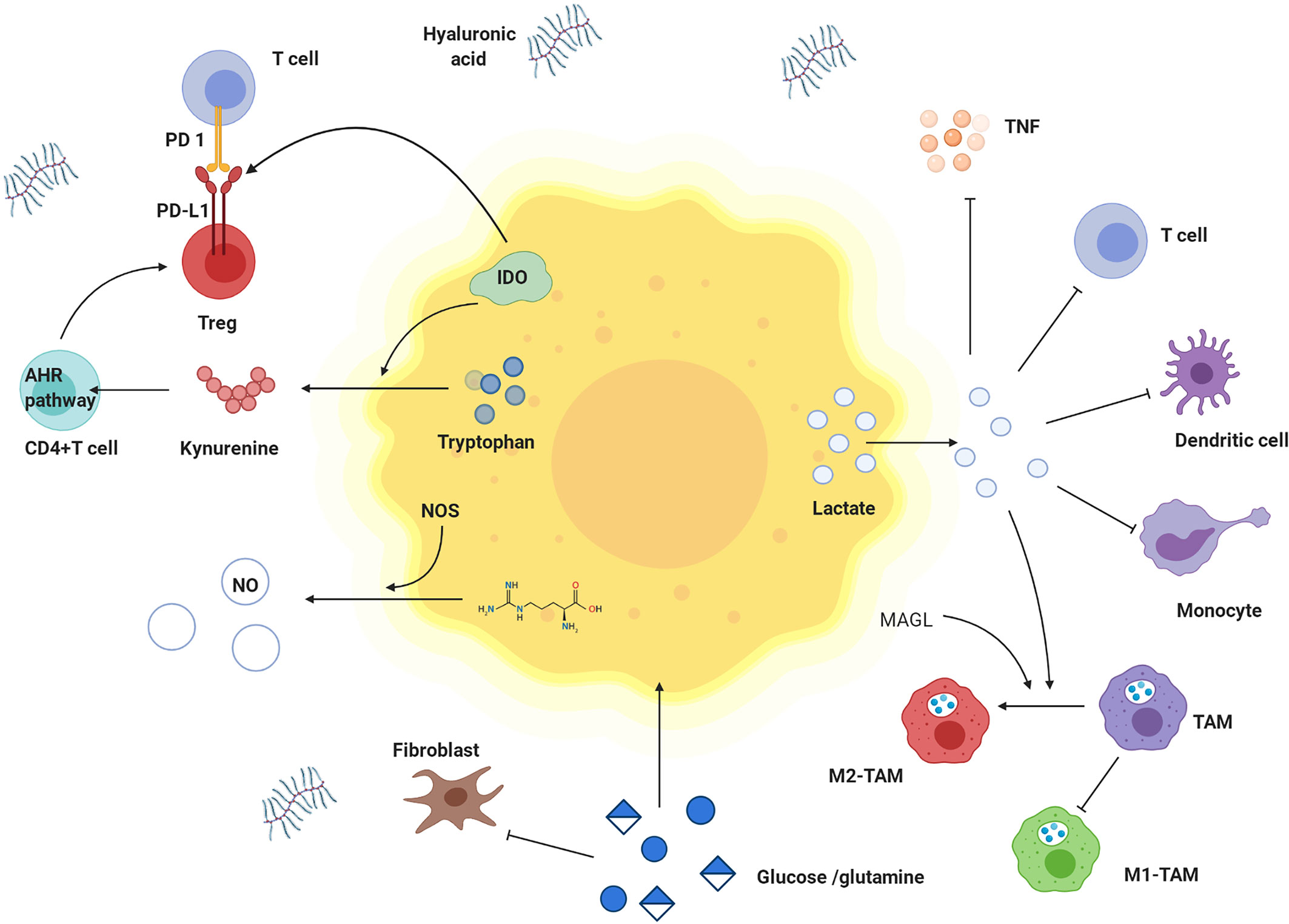
Figure 6 Effects of metabolites derived from tumor cells on immune cells and nutritional competition between cancer cells and immune cells. Tumor cells have competitive advantages over glutamine and glucose. Tumor cells produce a lot of lactate, which is transported to extracellular environment. High concentration of lactate hinders transfer of lactate from T cells and interferes with metabolism and function of T cells. Lactate damages dendritic cells and inhibits monocyte migration and cytokine release. Lactate inhibits the effect of TNF. In tumor microenvironment, tryptophan is decreased, while kynurenine and tryptophan is increased. IDO is highly expressed in tumor cells and catalyzes tryptophan degradation. Tryptophan is metabolized to produce kynurenine, which promotes the differentiation of CD4 + T cells into Treg cells by activating kynurenine AHR signal axis. IDO also activates Treg cells to up-regulate the expression of PD-L1 and inhibited the proliferation of T cells through PD-1/PD-L1 pathway. M1-TAM decreases but M2- TAM increases. Lactate and MAGL induced M2 like polarization of TAMs. In addition, hyaluronic acid is increased in matrix, and Treg cells are highly expressed. TNF: tumor necrosis factor, IDO: indoleamine 2,3-dioxygenase, AHR pathway: kynurenine aryl hydrocarbon receptor signal axis, TAM: tumor-associated macrophage, M1-TAM: M1 type tumor-associated macrophage, M2-TAM: M2 type tumor-associated macrophage.
Progress of Immunotherapy and Metabolism-Targeted Therapy in NPC
Recently, increasing evidence has shown that anti-PD-1 and anti-PD-L1 monoclonal antibodies are effective in the treatment of head and neck squamous cell carcinoma, especially for recurrent or metastatic head and neck squamous cell carcinoma (158–162). Treating NPC patients with PD-L1 blockade is an effective strategy (163, 164); however, the anti-PD1 monoclonal antibody induces an immune response in NPC patients but shows no substantial effect in reducing tumor burden (165).
Clinical studies have suggested that EBV-specific cytotoxic T cell (EBV-CTL) infusion is safe and effective because it targets the EBV antigens EBNA1 and LMP1 and LMP2 (166). Interestingly, Fas gene silencing in EBV-CTL cells might facilitate cytotoxic T cells clearance of NPC cells (167). Given that almost all EBV-positive nasopharyngeal carcinomas encode LMP1, the Ad-Δ LMP1-LMP2 gene was used to transduce dendritic cells in patients with advanced nasopharyngeal carcinoma and achieved satisfactory results in a clinical trial (168). However, immunotherapy alone may have only limited effects on constrained cancers. Small-molecule drugs or antibody interventions targeting metabolic processes can be used as pharmacological adjuncts with checkpoint blockade, as suggested by their roles as reported in preclinical models and ongoing or closed clinical trials (summarized in Table 4). Metabolic inhibitors such as 3-bromopyruvate and oxamate have been reported to suppress HK and LHDA activity in tumors and promote macrophage and T cell activation in a preclinical NPC model (22, 40). Other metabolic inhibitors, including 2-DG, dichloroacetate (DCA), and epacadostat, have entered or passed different stages of clinical trials, showing promising therapeutic effects on metabolic targets by reinforcing T cells and suppressing tumor metabolism. Given the impacts of metabolism on immunity as mentioned above and the history of therapy development, the combination of metabolic drugs with immunotherapy reagents may help overcome immunotherapy resistance and may soon be a potential clinical option for cancer patients.

Table 4 Metabolic Inhibitors with Reported Immunomodulatory Roles in Cancer.
Limitation and Perspective
Despite great growing reports on metabolic rewiring and immune escape in NPC-associated cancer research, most of them are limited to tell one side (either metabolic rewiring or immune escape) over both sides’ story. The mutual interactions between metabolic reprogramming and immune escape in NPC are crucial to understanding the comprehensive mechanism of treatment-resistant, which should be further investigated in the future. Although metabolic interventions that enhance anti-tumor immunity have been developed and trialed in cancers, few enroll NPC patients. Given the influence of EBV in NPCs, another direction could be studying the mutual connection of metabolic reprogramming with host immunity in an EBV-associated virologic environment.
Author Contributions
Conceptualization: HH and GZ. Writing—original draft preparation: HH. Writing—review and editing: HH and GZ. Supervision: GZ, SL and QT. Funding acquisition: GZ and HH. All authors contributed to the article and approved the submitted version.
Funding
This review was supported by National Natural Science Foundation of China (Nos. 82173341 and 81602389), Natural Science Foundation of Hunan Province (Nos. 2020JJ4827 and 2017JJ3456), Graduate Independent Exploration and Innovation Project of Central South University (Nos.2021zzts1069), and the Project of Hunan Health Commission (B2019165).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tsang CM, Lui VWY, Bruce JP, Pugh TJ, Lo KW. Translational Genomics of Nasopharyngeal Cancer. Semin Cancer Biol (2020) 61:84–100. doi: 10.1016/j.semcancer.2019.09.006
2. Chen YP, Chan ATC, Le QT, Blanchard P, Sun Y, Ma J. Nasopharyngeal Carcinoma. Lancet (2019) 394(10192):64–80. doi: 10.1016/S0140-6736(19)30956-0
3. Diseases GBD, Injuries C. Global Burden of 369 Diseases and Injuries in 204 Countries and Territories, 1990-2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet (2020) 396(10258):1204–22. doi: 10.1016/S0140-6736(20)30925-9
4. Sun XS, Liu SL, Luo MJ, Li XY, Chen QY, Guo SS, et al. The Association Between the Development of Radiation Therapy, Image Technology, and Chemotherapy, and the Survival of Patients With Nasopharyngeal Carcinoma: A Cohort Study From 1990 to 2012. Int J Radiat Oncol Biol Phys (2019) 105(3):581–90. doi: 10.1016/j.ijrobp.2019.06.2549
5. Lu J, Liu QH, Wang F, Tan JJ, Deng YQ, Peng XH, et al. Exosomal miR-9 Inhibits Angiogenesis by Targeting MDK and Regulating PDK/AKT Pathway in Nasopharyngeal Carcinoma. J Exp Clin Cancer Res (2018) 37(1):147. doi: 10.1186/s13046-018-0814-3
6. Zheng H, Dai W, Cheung AK, Ko JM, Kan R, Wong BW, et al. Whole-Exome Sequencing Identifies Multiple Loss-of-Function Mutations of NF-kappaB Pathway Regulators in Nasopharyngeal Carcinoma. Proc Natl Acad Sci USA (2016) 113(40):11283–8. doi: 10.1073/pnas.1607606113
7. Lin DC, Meng X, Hazawa M, Nagata Y, Varela AM, Xu L, et al. The Genomic Landscape of Nasopharyngeal Carcinoma. Nat Genet (2014) 46(8):866–71. doi: 10.1038/ng.3006
8. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
9. Warburg O. On Respiratory Impairment in Cancer Cells. Science (1956) 124(3215):269–70. doi: 10.1126/science.124.3215.267
10. Huang PY, Zeng TT, Li MQ, Ban X, Zhu YH, Zhang BZ, et al. Proteomic Analysis of a Nasopharyngeal Carcinoma Cell Line and a Nasopharyngeal Epithelial Cell Line. Tumori (2015) 101(6):676–83. doi: 10.5301/tj.5000345
11. Zschaeck S, Li Y, Lin Q, Beck M, Amthauer H, Bauersachs L, et al. Prognostic Value of Baseline [18F]-Fluorodeoxyglucose Positron Emission Tomography Parameters MTV, TLG and Asphericity in an International Multicenter Cohort of Nasopharyngeal Carcinoma Patients. PloS One (2020) 15(7):e0236841. doi: 10.1371/journal.pone.0236841
12. Lv W, Yuan Q, Wang Q, Ma J, Feng Q, Chen W, et al. Radiomics Analysis of PET and CT Components of PET/CT Imaging Integrated With Clinical Parameters: Application to Prognosis for Nasopharyngeal Carcinoma. Mol Imaging Biol (2019) 21(5):954–64. doi: 10.1007/s11307-018-01304-3
13. Chan SC, Ng SH, Chang JT, Lin CY, Chen YC, Chang YC, et al. Advantages and Pitfalls of 18F-Fluoro-2-Deoxy-D-Glucose Positron Emission Tomography in Detecting Locally Residual or Recurrent Nasopharyngeal Carcinoma: Comparison With Magnetic Resonance Imaging. Eur J Nucl Med Mol Imaging (2006) 33(9):1032–40. doi: 10.1007/s00259-005-0054-6
14. Macheda ML, Rogers S, Best JD. Molecular and Cellular Regulation of Glucose Transporter (GLUT) Proteins in Cancer. J Cell Physiol (2005) 202(3):654–62. doi: 10.1002/jcp.20166
15. Zhou JC, Zhang JJ, Zhang W, Ke ZY, Ma LG, Liu M. Expression of GLUT-1 in Nasopharyngeal Carcinoma and its Clinical Significance. Eur Rev Med Pharmacol Sci (2017) 21(21):4891–5.
16. Zhang J, Jia L, Lin W, Yip YL, Lo KW, Lau VMY, et al. Epstein-Barr Virus-Encoded Latent Membrane Protein 1 Upregulates Glucose Transporter 1 Transcription via the Mtorc1/NF-kappaB Signaling Pathways. J Virol (2017) 91(6):e02168–16. doi: 10.1128/JVI.02168-16
17. Reckzeh ES, Karageorgis G, Schwalfenberg M, Ceballos J, Nowacki J, Stroet MCM, et al. Inhibition of Glucose Transporters and Glutaminase Synergistically Impairs Tumor Cell Growth. Cell Chem Biol (2019) 26(9):1214–1228 e1225. doi: 10.1016/j.chembiol.2019.06.005
18. Zhao P, Zhou M, Chen R, Su R. Suppressed "Warburg Effect" in Nasopharyngeal Carcinoma Via the Inhibition of Pyruvate Kinase Type M2-Mediated Energy Generation Pathway. Technol Cancer Res Treat (2020) 19:1533033820945804. doi: 10.1177/1533033820945804
19. Lundblad RL. Biochemistry and Molecular Biology Compendium. Boca Raton, Florida, the United States: CRC Press (2019).
20. Song L, Tang H, Liao W, Luo X, Li Y, Chen T, et al. FOXC2 Positively Regulates YAP Signaling and Promotes the Glycolysis of Nasopharyngeal Carcinoma. Exp Cell Res (2017) 357(1):17–24. doi: 10.1016/j.yexcr.2017.04.019
21. Zhang MX, Hua YJ, Wang HY, Zhou L, Mai HQ, Guo X, et al. Long-Term Prognostic Implications and Therapeutic Target Role of Hexokinase II in Patients With Nasopharyngeal Carcinoma. Oncotarget (2016) 7(16):21287–97. doi: 10.18632/oncotarget.7116
22. Zou X, Zhang M, Sun Y, Zhao S, Wei Y, Zhang X, et al. Inhibitory Effects of 3-Bromopyruvate in Human Nasopharyngeal Carcinoma Cells. Oncol Rep (2015) 34(4):1895–904. doi: 10.3892/or.2015.4147
23. Deng Q, Yu X, Xiao L, Hu Z, Luo X, Tao Y, et al. Neoalbaconol Induces Energy Depletion and Multiple Cell Death in Cancer Cells by Targeting PDK1-PI3-K/Akt Signaling Pathway. Cell Death Dis (2013) 4:e804. doi: 10.1038/cddis.2013.324
24. Zhang J, Zhou J, Xiao S. Shikonin Inhibits Growth, Invasion and Glycolysis of Nasopharyngeal Carcinoma Cells Through Inactivating the Phosphatidylinositol 3 Kinase/AKT Signal Pathway. Anti-cancer Drugs (2020) 31(9):932–41. doi: 10.1097/CAD.0000000000000920
25. Chen S, Youhong T, Tan Y, He Y, Ban Y, Cai J, et al. EGFR-PKM2 Signaling Promotes the Metastatic Potential of Nasopharyngeal Carcinoma Through Induction of FOSL1 and ANTXR2. Carcinogenesis (2020) 41(6):723–33. doi: 10.1093/carcin/bgz180
26. Zhang X, Yang J, Shi D, Cao Z. TET2 Suppresses Nasopharyngeal Carcinoma Progression by Inhibiting Glycolysis Metabolism. Cancer Cell Int (2020) 20:363. doi: 10.1186/s12935-020-01456-9
27. Hu, Zeng Z, Xia Q, Liu Z, Feng X, Chen J, et al. Metformin Attenuates Hepatoma Cell Proliferation by Decreasing Glycolytic Flux Through the HIF-1alpha/PFKFB3/PFK1 Pathway. Life Sci (2019) 239:116966. doi: 10.1016/j.lfs.2019.116966
28. Sun R, Meng X, Pu Y, Sun F, Man Z, Zhang J, et al. Overexpression of HIF-1a Could Partially Protect K562 Cells From 1,4-Benzoquinone Induced Toxicity by Inhibiting ROS, Apoptosis and Enhancing Glycolysis. Toxicol In Vitro (2019) 55:18–23. doi: 10.1016/j.tiv.2018.11.005
29. Tang H, Luo X, Li J, Zhou Y, Li Y, Song L, et al. Pyruvate Dehydrogenase B Promoted the Growth and Migration of the Nasopharyngeal Carcinoma Cells. Tumor Biol (2016) 37(8):10563–9. doi: 10.1007/s13277-016-4922-4
30. Cao JX, Cui YX, Long ZJ, Dai ZM, Lin JY, Liang Y, et al. Pluripotency-Associated Genes in Human Nasopharyngeal Carcinoma CNE-2 Cells Are Reactivated by a Unique Epigenetic Sub-Microenvironment. BMC Cancer (2010) 10:68. doi: 10.1186/1471-2407-10-68
31. Xiang G, Li X, Cao L, Zhu C, Dai Z, Pan S, et al. Frequent Overexpression of PDK1 in Primary Nasopharyngeal Carcinoma is Associated With Poor Prognosis. Pathol Res Pract (2016) 212(12):1102–7. doi: 10.1016/j.prp.2016.10.006
32. Dai Z, Pan S, Chen C, Cao L, Li X, Chen X, et al. Down-Regulation of Succinate Dehydrogenase Subunit B and Up-Regulation of Pyruvate Dehydrogenase Kinase 1 Predicts Poor Prognosis in Recurrent Nasopharyngeal Carcinoma. Tumor Biol (2016) 37(4):5145–52. doi: 10.1007/s13277-015-4107-6
33. Cai CF, Ye GD, Shen DY, Zhang W, Chen ML, Chen XX, et al. Chibby Suppresses Aerobic Glycolysis and Proliferation of Nasopharyngeal Carcinoma via the Wnt/beta-Catenin-Lin28/let7-PDK1 Cascade. J Exp Clin Cancer Res (2018) 37(1):104. doi: 10.1186/s13046-018-0769-4
34. Sun W, Zhou S, Chang SS, McFate T, Verma A, Califano JA. Mitochondrial Mutations Contribute to HIF1alpha Accumulation via Increased Reactive Oxygen Species and Up-Regulated Pyruvate Dehydrogenease Kinase 2 in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res (2009) 15(2):476–84. doi: 10.1158/1078-0432.CCR-08-0930
35. Shi F, He Y, Li J, Tang M, Li Y, Xie L, et al. Wild-Type IDH2 Contributes to Epstein-Barr Virus-Dependent Metabolic Alterations and Tumorigenesis. Mol Metab (2020) 36:100966. doi: 10.1016/j.molmet.2020.02.009
36. Mito JK, Bishop JA, Sadow PM, Stelow EB, Faquin WC, Mills SE, et al. Immunohistochemical Detection and Molecular Characterization of IDH-Mutant Sinonasal Undifferentiated Carcinomas. Am J Surg Pathol (2018) 42(8):1067–75. doi: 10.1097/PAS.0000000000001064
37. Ali SM, Yao M, Yao J, Wang J, Cheng Y, Schrock AB, et al. Comprehensive Genomic Profiling of Different Subtypes of Nasopharyngeal Carcinoma Reveals Similarities and Differences to Guide Targeted Therapy. Cancer (2017) 123(18):3628–37. doi: 10.1002/cncr.30781
38. Liu CX, Wang H, Qian XM, Lu FX, Zhuang SF, Yang MX, et al. The Effect of Three-Dimensional Conformal Radiotherapy on Locally Recurrent Nasopharyngeal Carcinoma and on the Expression of Succinate Dehydrogenase B. Eur Rev Med Pharmacol Sci (2016) 20(23):4852–7.
39. Petrelli F, Cabiddu M, Coinu A, Borgonovo K, Ghilardi M, Lonati V, et al. Prognostic Role of Lactate Dehydrogenase in Solid Tumors: A Systematic Review and Meta-Analysis of 76 Studies. Acta Oncol (2015) 54(7):961–70. doi: 10.3109/0284186X.2015.1043026
40. Zhai X, Yang Y, Wan J, Zhu R, Wu Y. Inhibition of LDH-A by Oxamate Induces G2/M Arrest, Apoptosis and Increases Radiosensitivity in Nasopharyngeal Carcinoma Cells. Oncol Rep (2013) 30(6):2983–91. doi: 10.3892/or.2013.2735
41. Saluja TS, Spadigam A, Dhupar A, Syed S. Equating Salivary Lactate Dehydrogenase (LDH) With LDH-5 Expression in Patients With Oral Squamous Cell Carcinoma: An Insight Into Metabolic Reprogramming of Cancer Cell as a Predictor of Aggressive Phenotype. Tumor Biol (2016) 37(4):5609–20. doi: 10.1007/s13277-015-4415-x
42. Augoff K, Hryniewicz-Jankowska A, Tabola R. Lactate Dehydrogenase 5: An Old Friend and a New Hope in the War on Cancer. Cancer Lett (2015) 358(1):1–7. doi: 10.1016/j.canlet.2014.12.035
43. Ghergurovich JM, Garcia-Canaveras JC, Wang J, Schmidt E, Zhang Z, TeSlaa T, et al. A Small Molecule G6PD Inhibitor Reveals Immune Dependence on Pentose Phosphate Pathway. Nat Chem Biol (2020) 16(7):731–9. doi: 10.1038/s41589-020-0533-x
44. Stanton RC. Glucose-6-Phosphate Dehydrogenase, NADPH, and Cell Survival. IUBMB Life (2012) 64(5):362–9. doi: 10.1002/iub.1017
45. Guo Q, Ren XH, Zhao CP, Zhang BH. MicroRNA-206 Inhibits Tumor Metastasis of Nasopharyngeal Carcinoma Through Targeting G6PD. J Biol Regul Homeost Agents (2020) 34(2). doi: 10.23812/20-36A
46. Ye Y, Zhou Y, Zhang L, Chen Y, Lyu X, Cai L, et al. EBV-miR-BART1 Is Involved in Regulating Metabolism-Associated Genes in Nasopharyngeal Carcinoma. Biochem Biophys Res Commun (2013) 436(1):19–24. doi: 10.1016/j.bbrc.2013.05.008
47. Li X, Wenes M, Romero P, Huang SC, Fendt SM, Ho PC. Navigating Metabolic Pathways to Enhance Antitumor Immunity and Immunotherapy. Nat Rev Clin Oncol (2019) 16(7):425–41. doi: 10.1038/s41571-019-0203-7
48. Nakajima EC, Van Houten B. Metabolic Symbiosis in Cancer: Refocusing the Warburg Lens. Mol Carcinog (2013) 52(5):329–37. doi: 10.1002/mc.21863
49. Li F, Simon MC. Cancer Cells Don't Live Alone: Metabolic Communication Within Tumor Microenvironments. Dev Cell (2020) 54(2):183–95. doi: 10.1016/j.devcel.2020.06.018
50. Zhang P, Ma J, Gao J, Liu F, Sun X, Fang F, et al. Downregulation of Monocarboxylate Transporter 1 Inhibits the Invasion and Migration Through Suppression of the PI3K/Akt Signaling Pathway in Human Nasopharyngeal Carcinoma Cells. J Bioenerg Biomembr (2018) 50(4):271–81. doi: 10.1007/s10863-018-9763-y
51. Sung WW, Chen PR, Liao MH, Lee JW. Enhanced Aerobic Glycolysis of Nasopharyngeal Carcinoma Cells by Epstein-Barr Virus Latent Membrane Protein 1. Exp Cell Res (2017) 359(1):94–100. doi: 10.1016/j.yexcr.2017.08.005
52. Dawson CW, Port RJ, Young LS. The Role of the EBV-Encoded Latent Membrane Proteins LMP1 and LMP2 in the Pathogenesis of Nasopharyngeal Carcinoma (NPC). Semin Cancer Biol (2012) 22(2):144–53. doi: 10.1016/j.semcancer.2012.01.004
53. Lo AK, Dawson CW, Young LS, Ko CW, Hau PM, Lo KW. Activation of the FGFR1 Signalling Pathway by the Epstein-Barr Virus-Encoded LMP1 Promotes Aerobic Glycolysis and Transformation of Human Nasopharyngeal Epithelial Cells. J Pathol (2015) 237(2):238–48. doi: 10.1002/path.4575
54. Goldberg IJ, Eckel RH, Abumrad NA. Regulation of Fatty Acid Uptake Into Tissues: Lipoprotein Lipase- and CD36-Mediated Pathways. J Lipid Res (2009) 50(Suppl):S86–90. doi: 10.1194/jlr.R800085-JLR200
55. Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, et al. Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP Proteins. Cancer Discov (2018) 8(8):1006–25. doi: 10.1158/2159-8290.CD-17-1371
56. Zheng S, Matskova L, Zhou X, Xiao X, Huang G, Zhang Z, et al. Downregulation of Adipose Triglyceride Lipase by EB Viral-Encoded LMP2A Links Lipid Accumulation to Increased Migration in Nasopharyngeal Carcinoma. Mol Oncol (2020) 14(12):3234–52. doi: 10.1002/1878-0261.12824
57. Xie Z, Shao Y. The Predictive Value of Serum Lipids for Eye Metastases in Male Nasopharyngeal Carcinoma Patients. Biosci Rep (2020) 40(6):BSR20201082. doi: 10.1042/BSR20201082
58. Mo Y, Wang Y, Xiong F, Ge X, Li Z, Li X, et al. Proteomic Analysis of the Molecular Mechanism of Lovastatin Inhibiting the Growth of Nasopharyngeal Carcinoma Cells. J Cancer (2019) 10(10):2342–9. doi: 10.7150/jca.30454
59. Daker M, Bhuvanendran S, Ahmad M, Takada K, Khoo AS. Deregulation of Lipid Metabolism Pathway Genes in Nasopharyngeal Carcinoma Cells. Mol Med Rep (2013) 7(3):731–41. doi: 10.3892/mmr.2012.1253
60. Liu F, Wei J, Hao Y, Tang F, Jiao W, Qu S, et al. Long Noncoding RNAs and Messenger RNAs Expression Profiles Potentially Regulated by ZBTB7A in Nasopharyngeal Carcinoma. BioMed Res Int (2019) 2019:7246491. doi: 10.1155/2019/7246491
61. Chen J, Zhang F, Ren X, Wang Y, Huang W, Zhang J, et al. Targeting Fatty Acid Synthase Sensitizes Human Nasopharyngeal Carcinoma Cells to Radiation via Downregulating Frizzled Class Receptor 10. Cancer Biol Med (2020) 17(3):740–52. doi: 10.20892/j.issn.2095-3941.2020.0219
62. Kao YC, Lee SW, Lin LC, Chen LT, Hsing CH, Hsu HP, et al. Fatty Acid Synthase Overexpression Confers an Independent Prognosticator and Associates With Radiation Resistance in Nasopharyngeal Carcinoma. Tumor Biol (2013) 34(2):759–68. doi: 10.1007/s13277-012-0605-y
63. Daker M, Ahmad M, Khoo AS. Quercetin-Induced Inhibition and Synergistic Activity With Cisplatin - a Chemotherapeutic Strategy for Nasopharyngeal Carcinoma Cells. Cancer Cell Int (2012) 12(1):34. doi: 10.1186/1475-2867-12-34
64. Li J, Wen Q, Xu L, Wang W, Luo J, Chu S, et al. Fatty Acid Synthase-Associated Protein With Death Domain: A Prognostic Factor for Survival in Patients With Nasopharyngeal Carcinoma. Hum Pathol (2014) 45(12):2447–52. doi: 10.1016/j.humpath.2014.08.010
65. Zheng FJ, Ye HB, Wu MS, Lian YF, Qian CN, Zeng YX. Repressing Malic Enzyme 1 Redirects Glucose Metabolism, Unbalances the Redox State, and Attenuates Migratory and Invasive Abilities in Nasopharyngeal Carcinoma Cell Lines. Chin J Cancer (2012) 31(11):519–31. doi: 10.5732/cjc.012.10088
66. Tan Z, Xiao L, Tang M, Bai F, Li J, Li L, et al. Targeting CPT1A-Mediated Fatty Acid Oxidation Sensitizes Nasopharyngeal Carcinoma to Radiation Therapy. Theranostics (2018) 8(9):2329–47. doi: 10.7150/thno.21451
67. Wong BW, Wang X, Zecchin A, Thienpont B, Cornelissen I, Kalucka J, et al. The Role of Fatty Acid Beta-Oxidation in Lymphangiogenesis. Nature (2017) 542(7639):49–54. doi: 10.1038/nature21028
68. Draoui N, de Zeeuw P, Carmeliet P. Angiogenesis Revisited From a Metabolic Perspective: Role and Therapeutic Implications of Endothelial Cell Metabolism. Open Biol (2017) 7(12):170219. doi: 10.1098/rsob.170219
69. Li S, Fu L, Tian T, Deng L, Li H, Xia W, et al. Disrupting SOD1 Activity Inhibits Cell Growth and Enhances Lipid Accumulation in Nasopharyngeal Carcinoma. Cell Commun Signal (2018) 16(1):28. doi: 10.1186/s12964-018-0240-3
70. Du Q, Tan Z, Shi F, Tang M, Xie L, Zhao L, et al. PGC1alpha/CEBPB/CPT1A Axis Promotes Radiation Resistance of Nasopharyngeal Carcinoma Through Activating Fatty Acid Oxidation. Cancer Sci (2019) 110(6):2050–62. doi: 10.1111/cas.14011
71. Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol Lipase Regulates a Fatty Acid Network That Promotes Cancer Pathogenesis. Cell (2010) 140(1):49–61. doi: 10.1016/j.cell.2009.11.027
72. Hu WR, Lian YF, Peng LX, Lei JJ, Deng CC, Xu M, et al. Monoacylglycerol Lipase Promotes Metastases in Nasopharyngeal Carcinoma. Int J Clin Exp Pathol (2014) 7(7):3704–13. doi: 10.1172/JCI69589
73. Fukao T, Mitchell G, Sass JO, Hori T, Orii K, Aoyama Y. Ketone Body Metabolism and its Defects. J Inherit Metab Dis (2014) 37(4):541–51. doi: 10.1007/s10545-014-9704-9
74. Luo W, Qin L, Li B, Liao Z, Liang J, Xiao X, et al. Inactivation of HMGCL Promotes Proliferation and Metastasis of Nasopharyngeal Carcinoma by Suppressing Oxidative Stress. Sci Rep (2017) 7(1):11954. doi: 10.1038/s41598-017-11025-2
75. Lee CM, Yoon MS, Kim YC. Effects of Pueraria lobata Root Ethanol Extract on Adipogenesis and Lipogenesis During 3T3-L1 Differentiation Into Adipocytes. Toxicol Res (2015) 31(2):191–201. doi: 10.5487/TR.2015.31.2.191
76. Zheng C, Han L, Wu S. A Metabolic Investigation of Anticancer Effect of G. Glabra Root Extract on Nasopharyngeal Carcinoma Cell Line, C666-1. Mol Biol Rep (2019) 46(4):3857–64. doi: 10.1007/s11033-019-04828-1
77. Lan J, Tai HC, Lee SW, Chen TJ, Huang HY, Li CF. Deficiency in Expression and Epigenetic DNA Methylation of ASS1 Gene in Nasopharyngeal Carcinoma: Negative Prognostic Impact and Therapeutic Relevance. Tumor Biol (2014) 35(1):161–9. doi: 10.1007/s13277-013-1020-8
78. Altman BJ, Stine ZE, Dang CV. From Krebs to Clinic: Glutamine Metabolism to Cancer Therapy. Nat Rev Cancer (2016) 16(10):619–34. doi: 10.1038/nrc.2016.71
79. Fu S, Li Z, Xiao L, Hu W, Zhang L, Xie B, et al. Glutamine Synthetase Promotes Radiation Resistance via Facilitating Nucleotide Metabolism and Subsequent DNA Damage Repair. Cell Rep (2019) 28(5):1136–1143 e1134. doi: 10.1016/j.celrep.2019.07.002
80. Peng Y, Fu S, Hu W, Qiu Y, Zhang L, Tan R, et al. Glutamine Synthetase Facilitates Cancer Cells to Recover From Irradiation-Induced G2/M Arrest. Cancer Biol Ther (2020) 21(1):43–51. doi: 10.1080/15384047.2019.1665394
81. Krishna G, Soman Pillai V, Valiya Veettil M. Upregulation of GLS1 Isoforms KGA and GAC Facilitates Mitochondrial Metabolism and Cell Proliferation in Epstein-Barr Virus Infected Cells. Viruses (2020) 12(8):811. doi: 10.3390/v12080811
82. Szeliga M, Albrecht J. Opposing Roles of Glutaminase Isoforms in Determining Glioblastoma Cell Phenotype. Neurochem Int (2015) 88:6–9. doi: 10.1016/j.neuint.2014.11.004
83. Sivanand S, Vander Heiden MG. Emerging Roles for Branched-Chain Amino Acid Metabolism in Cancer. Cancer Cell (2020) 37(2):147–56. doi: 10.1016/j.ccell.2019.12.011
84. Liao KM, Chao TB, Tian YF, Lin CY, Lee SW, Chuang HY, et al. Overexpression of the PSAT1 Gene in Nasopharyngeal Carcinoma Is an Indicator of Poor Prognosis. J Cancer (2016) 7(9):1088–94. doi: 10.7150/jca.15258
85. Zhu L, Chen Q, Zhang L, Hu S, Zheng W, Wang C, et al. CLIC4 Regulates Radioresistance of Nasopharyngeal Carcinoma by iNOS After Gamma-Rays But Not Carbon Ions Irradiation. Am J Cancer Res (2020) 10(5):1400–15.
86. Keshet R, Erez A. Arginine and the Metabolic Regulation of Nitric Oxide Synthesis in Cancer. Dis Model Mech (2018) 11(8):dmm033332. doi: 10.1242/dmm.033332
87. Xu W, Liu L, Smith GC, Charles l G. Nitric Oxide Upregulates Expression of DNA-PKcs to Protect Cells From DNA-Damaging Anti-Tumor Agents. Nat Cell Biol (2000) 2(6):339–45. doi: 10.1038/35014028
88. Lechner M, Lirk P, Rieder J. Inducible Nitric Oxide Synthase (iNOS) in Tumor Biology: The Two Sides of the Same Coin. Semin Cancer Biol (2005) 15(4):277–89. doi: 10.1016/j.semcancer.2005.04.004
89. Lu J, Tang M, Li H, Xu Z, Weng X, Li J, et al. EBV-LMP1 Suppresses the DNA Damage Response Through DNA-PK/AMPK Signaling to Promote Radioresistance in Nasopharyngeal Carcinoma. Cancer Lett (2016) 380(1):191–200. doi: 10.1016/j.canlet.2016.05.032
90. Rottey S, Madani I, Deron P, Van Belle S. Modern Treatment for Nasopharyngeal Carcinoma: Current Status and Prospects. Curr Opin Oncol (2011) 23(3):254–8. doi: 10.1097/CCO.0b013e328344f527
91. Moloney JN, Cotter TG. ROS Signalling in the Biology of Cancer. Semin Cell Dev Biol (2018) 80:50–64. doi: 10.1016/j.semcdb.2017.05.023
92. Graham K, Unger E. Overcoming Tumor Hypoxia as a Barrier to Radiotherapy, Chemotherapy and Immunotherapy in Cancer Treatment. Int J Nanomed (2018) 13:6049–58. doi: 10.2147/IJN.S140462
93. Kell DB, Heyden EL, Pretorius E. The Biology of Lactoferrin, an Iron-Binding Protein That Can Help Defend Against Viruses and Bacteria. Front Immunol (2020) 11:1221. doi: 10.3389/fimmu.2020.01221
94. Deng M, Zhang W, Tang H, Ye Q, Liao Q, Zhou Y, et al. Lactotransferrin Acts as a Tumor Suppressor in Nasopharyngeal Carcinoma by Repressing AKT Through Multiple Mechanisms. Oncogene (2013) 32(36):4273–83. doi: 10.1038/onc.2012.434
95. Zheng Y, Zhang W, Ye Q, Zhou Y, Xiong W, He W, et al. Inhibition of Epstein-Barr Virus Infection by Lactoferrin. J Innate Immun (2012) 4(4):387–98. doi: 10.1159/000336178
96. Zhou Y, Zeng Z, Zhang W, Xiong W, Wu M, Tan Y, et al. Lactotransferrin: A Candidate Tumor Suppressor-Deficient Expression in Human Nasopharyngeal Carcinoma and Inhibition of NPC Cell Proliferation by Modulating the Mitogen-Activated Protein Kinase Pathway. Int J Cancer (2008) 123(9):2065–72. doi: 10.1002/ijc.23727
97. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
98. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell (2015) 59(2):298–308. doi: 10.1016/j.molcel.2015.06.011
99. Eagle H. Nutrition Needs of Mammalian Cells in Tissue Culture. Science (1955) 122(3168):501–14. doi: 10.1126/science.122.3168.501
100. Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate Toxicity in a Neuronal Cell Line Involves Inhibition of Cystine Transport Leading to Oxidative Stress. Neuron (1989) 2(6):1547–58. doi: 10.1016/0896-6273(89)90043-3
101. Bannai S, Tsukeda H, Okumura H. Effect of Antioxidants on Cultured Human Diploid Fibroblasts Exposed to Cystine-Free Medium. Biochem Biophys Res Commun (1977) 74(4):1582–8. doi: 10.1016/0006-291X(77)90623-4
102. Yang WS, Stockwell BR. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem Biol (2008) 15(3):234–45. doi: 10.1016/j.chembiol.2008.02.010
103. Li B, Liao Z, Mo Y, Zhao W, Zhou X, Xiao X, et al. Inactivation of 3-Hydroxybutyrate Dehydrogenase Type 2 Promotes Proliferation and Metastasis of Nasopharyngeal Carcinoma by Iron Retention. Br J Cancer (2020) 122(1):102–10. doi: 10.1038/s41416-019-0638-8
104. Li Y, Chen F, Chen J, Chan S, He Y, Liu W, et al. Disulfiram/Copper Induces Antitumor Activity Against Both Nasopharyngeal Cancer Cells and Cancer-Associated Fibroblasts Through ROS/MAPK and Ferroptosis Pathways. Cancers (Basel) (2020) 12(1):138. doi: 10.3390/cancers12010138
105. Hove-Jensen B, Andersen KR, Kilstrup M, Martinussen J, Switzer RL, Willemoes M. Phosphoribosyl Diphosphate (PRPP): Biosynthesis, Enzymology, Utilization, and Metabolic Significance. Microbiol Mol Biol Rev (2017) 81(1):e00040–16. doi: 10.1128/MMBR.00040-16
106. Lee SW, Chen TJ, Lin LC, Li CF, Chen LT, Hsing CH, et al. Overexpression of Thymidylate Synthetase Confers an Independent Prognostic Indicator in Nasopharyngeal Carcinoma. Exp Mol Pathol (2013) 95(1):83–90. doi: 10.1016/j.yexmp.2013.05.006
107. Zhao W, Mo Y, Wang S, Midorikawa K, Ma N, Hiraku Y, et al. Quantitation of DNA Methylation in Epstein-Barr Virus-Associated Nasopharyngeal Carcinoma by Bisulfite Amplicon Sequencing. BMC Cancer (2017) 17(1):489. doi: 10.1186/s12885-017-3482-3
108. Jhaveri MS, Wagner C, Trepel JB. Impact of Extracellular Folate Levels on Global Gene Expression. Mol Pharmacol (2001) 60(6):1288–95. doi: 10.1124/mol.60.6.1288
109. Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Glioma-Derived Mutations in IDH1 Dominantly Inhibit IDH1 Catalytic Activity and Induce HIF-1alpha. Science (2009) 324(5924):261–5. doi: 10.1126/science.1170944
110. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-Associated IDH1 Mutations Produce 2-Hydroxyglutarate. Nature (2009) 462(7274):739–44. doi: 10.1038/nature08617
111. Loenarz C, Schofield CJ. Expanding Chemical Biology of 2-Oxoglutarate Oxygenases. Nat Chem Biol (2008) 4(3):152–6. doi: 10.1038/nchembio0308-152
112. Zhou X, Wei J, Chen F, Xiao X, Huang T, He Q, et al. Epigenetic Downregulation of the ISG15-Conjugating Enzyme UbcH8 Impairs Lipolysis and Correlates With Poor Prognosis in Nasopharyngeal Carcinoma. Oncotarget (2015) 6(38):41077–91. doi: 10.18632/oncotarget.6218
113. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
114. Gourzones C, Barjon C, Busson P. Host-Tumor Interactions in Nasopharyngeal Carcinomas. Semin Cancer Biol (2012) 22(2):127–36. doi: 10.1016/j.semcancer.2012.01.002
115. Huang SCM, Tsao SW, Tsang CM. Interplay of Viral Infection, Host Cell Factors and Tumor Microenvironment in the Pathogenesis of Nasopharyngeal Carcinoma. Cancers (Basel) (2018) 10(4):106. doi: 10.3390/cancers10040106
116. Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, et al. Glioblastoma Microvesicles Transport RNA and Proteins That Promote Tumor Growth and Provide Diagnostic Biomarkers. Nat Cell Biol (2008) 10(12):1470–6. doi: 10.1038/ncb1800
117. Mrizak D, Martin N, Barjon C, Jimenez-Pailhes AS, Mustapha R, Niki T, et al. Effect of Nasopharyngeal Carcinoma-Derived Exosomes on Human Regulatory T Cells. J Natl Cancer Inst (2015) 107(1):363. doi: 10.1093/jnci/dju363
118. Thomson AS, Mai SH, Bouma G, Herdman M, Byrne M, Hottenstein CS, et al. Structure and Functional Characterization of a Humanized Anti-CCL20 Antibody Following Exposure to Serum Reveals the Formation of Immune Complex That Leads to Toxicity. J Immunol (2021) 206(5):1067–76. doi: 10.4049/jimmunol.2000336
119. Meckes DG Jr., Shair KH, Marquitz AR, Kung CP, Edwards RH, Raab-Traub N. Human Tumor Virus Utilizes Exosomes for Intercellular Communication. Proc Natl Acad Sci USA (2010) 107(47):20370–5. doi: 10.1073/pnas.1014194107
120. Tsai CY, Sakakibara S, Yasui T, Minamitani T, Okuzaki D, Kikutani H. Bystander Inhibition of Humoral Immune Responses by Epstein-Barr Virus LMP1. Int Immunol (2018) 30(12):579–90. doi: 10.1093/intimm/dxy053
121. Yao M, Ohshima K, Suzumiya J, Kume T, Shiroshita T, Kikuchi M. Interleukin-10 Expression and Cytotoxic-T-Cell Response in Epstein-Barr-Virus-Associated Nasopharyngeal Carcinoma. Int J Cancer (1997) 72(3):398–402. doi: 10.1002/(SICI)1097-0215(19970729)72:3<398::AID-IJC4>3.0.CO;2-K
122. Marshall NA, Vickers MA, Barker RN. Regulatory T Cells Secreting IL-10 Dominate the Immune Response to EBV Latent Membrane Protein 1. J Immunol (2003) 170(12):6183–9. doi: 10.4049/jimmunol.170.12.6183
123. Chow YP, Tan LP, Chai SJ, Abdul Aziz N, Choo SW, Lim PV, et al. Exome Sequencing Identifies Potentially Druggable Mutations in Nasopharyngeal Carcinoma. Sci Rep (2017) 7:42980. doi: 10.1038/srep42980
124. Valentine R, Dawson CW, Hu C, Shah KM, Owen TJ, Date KL, et al. Epstein-Barr Virus-Encoded EBNA1 Inhibits the Canonical NF-kappaB Pathway in Carcinoma Cells by Inhibiting IKK Phosphorylation. Mol Cancer (2010) 9:1. doi: 10.1186/1476-4598-9-1
125. Yi M, Cai J, Li J, Chen S, Zeng Z, Peng Q, et al. Rediscovery of NF-kappaB Signaling in Nasopharyngeal Carcinoma: How Genetic Defects of NF-kappaB Pathway Interplay With EBV in Driving Oncogenesis? J Cell Physiol (2018) 233(8):5537–49. doi: 10.1002/jcp.26410
126. Li YY, Chung GT, Lui VW, To KF, Ma BB, Chow C, et al. Exome and Genome Sequencing of Nasopharynx Cancer Identifies NF-kappaB Pathway Activating Mutations. Nat Commun (2017) 8:14121. doi: 10.1038/ncomms14121
127. Ravi R, Bedi A. NF-kappaB in Cancer–a Friend Turned Foe. Drug Resist Update (2004) 7(1):53–67. doi: 10.1016/j.drup.2004.01.003
128. Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng MS, et al. TRIM29 Promotes DNA Virus Infections by Inhibiting Innate Immune Response. Nat Commun (2017) 8(1):945. doi: 10.1038/s41467-017-00101-w
129. Klibi J, Niki T, Riedel A, Pioche-Durieu C, Souquere S, Rubinstein E, et al. Blood Diffusion and Th1-Suppressive Effects of Galectin-9-Containing Exosomes Released by Epstein-Barr Virus-Infected Nasopharyngeal Carcinoma Cells. Blood (2009) 113(9):1957–66. doi: 10.1182/blood-2008-02-142596
130. Lin JC, Cherng JM, Lin HJ, Tsang CW, Liu YX, Lee SP. Amino Acid Changes in Functional Domains of Latent Membrane Protein 1 of Epstein-Barr Virus in Nasopharyngeal Carcinoma of Southern China and Taiwan: Prevalence of an HLA A2-Restricted 'Epitope-Loss Variant'. J Gen Virol (2004) 85(Pt 7):2023–34. doi: 10.1099/vir.0.19696-0
131. Lin HJ, Cherng JM, Hung MS, Sayion Y, Lin JC. Functional Assays of HLA A2-Restricted Epitope Variant of Latent Membrane Protein 1 (LMP-1) of Epstein-Barr Virus in Nasopharyngeal Carcinoma of Southern China and Taiwan. J BioMed Sci (2005) 12(6):925–36. doi: 10.1007/s11373-005-9017-y
132. Singh S, Banerjee S. Downregulation of HLA-ABC Expression Through Promoter Hypermethylation and Downmodulation of MIC-A/B Surface Expression in LMP2A-Positive Epithelial Carcinoma Cell Lines. Sci Rep (2020) 10(1):5415. doi: 10.1038/s41598-020-62081-0
133. Tsao SW, Tsang CM, Lo KW. Epstein-Barr Virus Infection and Nasopharyngeal Carcinoma. Philos Trans R Soc Lond B Biol Sci (2017) 372(1732):20160270. doi: 10.1098/rstb.2016.0270
134. Bentz GL, Liu R, Hahn AM, Shackelford J, Pagano JS. Epstein-Barr Virus BRLF1 Inhibits Transcription of IRF3 and IRF7 and Suppresses Induction of Interferon-Beta. Virology (2010) 402(1):121–8. doi: 10.1016/j.virol.2010.03.014
135. Morrison TE, Mauser A, Wong A, Ting JP, Kenney SC. Inhibition of IFN-Gamma Signaling by an Epstein-Barr Virus Immediate-Early Protein. Immunity (2001) 15(5):787–99. doi: 10.1016/S1074-7613(01)00226-6
136. Cohen JI, Lekstrom K. Epstein-Barr Virus BARF1 Protein is Dispensable for B-Cell Transformation and Inhibits Alpha Interferon Secretion From Mononuclear Cells. J Virol (1999) 73(9):7627–32. doi: 10.1128/JVI.73.9.7627-7632.1999
137. Ersing I, Nobre L, Wang LW, Soday L, Ma Y, Paulo JA, et al. A Temporal Proteomic Map of Epstein-Barr Virus Lytic Replication in B Cells. Cell Rep (2017) 19(7):1479–93. doi: 10.1016/j.celrep.2017.04.062
138. Cai TT, Ye SB, Liu YN, He J, Chen QY, Mai HQ, et al. LMP1-Mediated Glycolysis Induces Myeloid-Derived Suppressor Cell Expansion in Nasopharyngeal Carcinoma. PloS Pathog (2017) 13(7):e1006503. doi: 10.1371/journal.ppat.1006503
139. Zuo L, Yue W, Du S, Xin S, Zhang J, Liu L, et al. An Update: Epstein-Barr Virus and Immune Evasion via microRNA Regulation. Virol Sin (2017) 32(3):175–87. doi: 10.1007/s12250-017-3996-5
140. Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O. Diverse Herpesvirus microRNAs Target the Stress-Induced Immune Ligand MICB to Escape Recognition by Natural Killer Cells. Cell Host Microbe (2009) 5(4):376–85. doi: 10.1016/j.chom.2009.03.003
141. Haneklaus M, Gerlic M, Kurowska-Stolarska M, Rainey AA, Pich D, McInnes IB, et al. Cutting Edge: miR-223 and EBV miR-BART15 Regulate the NLRP3 Inflammasome and IL-1beta Production. J Immunol (2012) 189(8):3795–9. doi: 10.4049/jimmunol.1200312
142. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory Effect of Tumor Cell-Derived Lactic Acid on Human T Cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
143. Goetze K, Walenta S, Ksiazkiewicz M, Kunz-Schughart LA, Mueller-Klieser W. Lactate Enhances Motility of Tumor Cells and Inhibits Monocyte Migration and Cytokine Release. Int J Oncol (2011) 39(2):453–63. doi: 10.3892/ijo.2011.1055
144. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-Derived Lactic Acid Modulates Dendritic Cell Activation and Antigen Expression. Blood (2006) 107(5):2013–21. doi: 10.1182/blood-2005-05-1795
145. Carmona-Fontaine C, Bucci V, Akkari L, Deforet M, Joyce JA, Xavier JB. Emergence of Spatial Structure in the Tumor Microenvironment Due to the Warburg Effect. Proc Natl Acad Sci USA (2013) 110(48):19402–7. doi: 10.1073/pnas.1311939110
146. Zhang J, Muri J, Fitzgerald G, Gorski T, Gianni-Barrera R, Masschelein E, et al. Endothelial Lactate Controls Muscle Regeneration From Ischemia by Inducing M2-Like Macrophage Polarization. Cell Metab (2020) 31(6):1136–53.e1137. doi: 10.1016/j.cmet.2020.05.004
147. Beckert S, Farrahi F, Aslam RS, Scheuenstuhl H, Konigsrainer A, Hussain MZ, et al. Lactate Stimulates Endothelial Cell Migration. Wound Repair Regener (2006) 14(3):321–4. doi: 10.1111/j.1743-6109.2006.00127.x
148. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional Polarization of Tumor-Associated Macrophages by Tumor-Derived Lactic Acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
149. Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, et al. Lactic Acid and Acidification Inhibit TNF Secretion and Glycolysis of Human Monocytes. J Immunol (2010) 184(3):1200–9. doi: 10.4049/jimmunol.0902584
150. Hoskin DW, Mader JS, Furlong SJ, Conrad DM, Blay J. Inhibition of T Cell and Natural Killer Cell Function by Adenosine and its Contribution to Immune Evasion by Tumor Cells (Review). Int J Oncol (2008) 32(3):527–35. doi: 10.3892/ijo.32.3.527
151. Munn DH, Mellor AL. Indoleamine 2,3-Dioxygenase and Tumor-Induced Tolerance. J Clin Invest (2007) 117(5):1147–54. doi: 10.1172/JCI31178
152. Munn DH. Indoleamine 2,3-Dioxygenase, Tumor-Induced Tolerance and Counter-Regulation. Curr Opin Immunol (2006) 18(2):220–5. doi: 10.1016/j.coi.2006.01.002
153. Munn DH. Blocking IDO Activity to Enhance Anti-Tumor Immunity. Front Biosci (Elite Ed) (2012) 4:734–45. doi: 10.2741/e414
154. Roy S, Barik S, Banerjee S, Bhuniya A, Pal S, Basu P, et al. Neem Leaf Glycoprotein Overcomes Indoleamine 2,3 Dioxygenase Mediated Tolerance in Dendritic Cells by Attenuating Hyperactive Regulatory T Cells in Cervical Cancer Stage IIIB Patients. Hum Immunol (2013) 74(8):1015–23. doi: 10.1016/j.humimm.2013.04.022
155. Selvan SR, Dowling JP, Kelly WK, Lin J. Indoleamine 2,3-Dioxygenase (IDO): Biology and Target in Cancer Immunotherapies. Curr Cancer Drug Targets (2016) 16(9):755–64. doi: 10.2174/1568009615666151030102250
156. Yin J, Kim SS, Choi E, Oh YT, Lin W, Kim TH, et al. ARS2/MAGL Signaling in Glioblastoma Stem Cells Promotes Self-Renewal and M2-Like Polarization of Tumor-Associated Macrophages. Nat Commun (2020) 11(1):2978. doi: 10.1038/s41467-020-16789-2
157. Andrejeva G, Rathmell JC. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab (2017) 26(1):49–70. doi: 10.1016/j.cmet.2017.06.004
158. Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med (2016) 375(19):1856–67. doi: 10.1056/NEJMoa1602252
159. Ferris RL, Licitra L, Fayette J, Even C, Blumenschein G Jr., Harrington KJ, et al. Nivolumab in Patients With Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck: Efficacy and Safety in CheckMate 141 by Prior Cetuximab Use. Clin Cancer Res (2019) 25(17):5221–30. doi: 10.1158/1078-0432.CCR-18-3944
160. Harrington KJ, Ferris RL, Blumenschein G Jr., Colevas AD, Fayette J, Licitra L, et al. Nivolumab Versus Standard, Single-Agent Therapy of Investigator's Choice in Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (CheckMate 141): Health-Related Quality-of-Life Results From a Randomised, Phase 3 Trial. Lancet Oncol (2017) 18(8):1104–15. doi: 10.1016/S1470-2045(17)30421-7
161. Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, et al. Safety and Clinical Activity of Pembrolizumab for Treatment of Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-012): An Open-Label, Multicentre, Phase 1b Trial. Lancet Oncol (2016) 17(7):956–65. doi: 10.1016/S1470-2045(16)30066-3
162. Moskovitz J, Moy J, Ferris RL. Immunotherapy for Head and Neck Squamous Cell Carcinoma. Curr Oncol Rep (2018) 20(2):22. doi: 10.1007/s11912-018-0654-5
163. Chen BJ, Chapuy B, Ouyang J, Sun HH, Roemer MG, Xu ML, et al. PD-L1 Expression is Characteristic of a Subset of Aggressive B-Cell Lymphomas and Virus-Associated Malignancies. Clin Cancer Res (2013) 19(13):3462–73. doi: 10.1158/1078-0432.CCR-13-0855
164. Hsu C, Lee SH, Ejadi S, Even C, Cohen RB, Le Tourneau C, et al. Safety and Anti-Tumor Activity of Pembrolizumab in Patients With Programmed Death-Ligand 1-Positive Nasopharyngeal Carcinoma: Results of the KEYNOTE-028 Study. J Clin Oncol (2017) 35(36):4050–6. doi: 10.1200/JCO.2017.73.3675
165. Liu WN, Fong SY, Tan WWS, Tan SY, Liu M, Cheng JY, et al. Establishment and Characterization of Humanized Mouse NPC-PDX Model for Testing Immunotherapy. Cancers (Basel) (2020) 12(4):1025. doi: 10.3390/cancers12041025
166. Louis CU, Straathof K, Bollard CM, Ennamuri S, Gerken C, Lopez TT, et al. Adoptive Transfer of EBV-Specific T Cells Results in Sustained Clinical Responses in Patients With Locoregional Nasopharyngeal Carcinoma. J Immunother (2010) 33(9):983–90. doi: 10.1097/CJI.0b013e3181f3cbf4
167. Dotti G, Savoldo B, Pule M, Straathof KC, Biagi E, Yvon E, et al. Human Cytotoxic T Lymphocytes With Reduced Sensitivity to Fas-Induced Apoptosis. Blood (2005) 105(12):4677–84. doi: 10.1182/blood-2004-08-3337
168. Chia WK, Wang WW, Teo M, Tai WM, Lim WT, Tan EH, et al. A Phase II Study Evaluating the Safety and Efficacy of an Adenovirus-DeltaLMP1-LMP2 Transduced Dendritic Cell Vaccine in Patients With Advanced Metastatic Nasopharyngeal Carcinoma. Ann Oncol (2012) 23(4):997–1005. doi: 10.1093/annonc/mdr341
169. Linke C, Wosle M, Harder A. Anti-Cancer Agent 3-Bromopyruvate Reduces Growth of MPNST and Inhibits Metabolic Pathways in a Representative in-Vitro Model. BMC Cancer (2020) 20(1):896. doi: 10.1186/s12885-020-07397-w
170. Okano T, Saegusa J, Nishimura K, Takahashi S, Sendo S, Ueda Y, et al. 3-Bromopyruvate Ameliorate Autoimmune Arthritis by Modulating Th17/Treg Cell Differentiation and Suppressing Dendritic Cell Activation. Sci Rep (2017) 7:42412. doi: 10.1038/srep42412
171. Yadav S, Pandey SK, Goel Y, Kujur PK, Maurya BN, Verma A, et al. Protective and Recuperative Effects of 3-Bromopyruvate on Immunological, Hepatic and Renal Homeostasis in a Murine Host Bearing Ascitic Lymphoma: Implication of Niche Dependent Differential Roles of Macrophages. BioMed Pharmacother (2018) 99:970–85. doi: 10.1016/j.biopha.2018.01.149
172. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting Glycolytic Metabolism Enhances CD8+ T Cell Memory and Anti-Tumor Function. J Clin Invest (2013) 123(10):4479–88. doi: 10.1172/JCI69589
173. Makita N, Ishiguro J, Suzuki K, Nara F. Dichloroacetate Induces Regulatory T-Cell Differentiation and Suppresses Th17-Cell Differentiation by Pyruvate Dehydrogenase Kinase-Independent Mechanism. J Pharm Pharmacol (2017) 69(1):43–51. doi: 10.1111/jphp.12655
174. Eleftheriadis T, Sounidaki M, Pissas G, Antoniadi G, Liakopoulos V, Stefanidis I. In Human Alloreactive CD4(+) T-Cells, Dichloroacetate Inhibits Aerobic Glycolysis, Induces Apoptosis and Favors Differentiation Towards the Regulatory T-Cell Subset Instead of Effector T-Cell Subsets. Mol Med Rep (2016) 13(4):3370–6. doi: 10.3892/mmr.2016.4912
175. Chen HM, Wang PH, Chen SS, Wen CC, Chen YH, Yang WC, et al. Shikonin Induces Immunogenic Cell Death in Tumor Cells and Enhances Dendritic Cell-Based Cancer Vaccine. Cancer Immunol Immunother (2012) 61(11):1989–2002. doi: 10.1007/s00262-012-1258-9
176. Rajeshkumar NV, Dutta P, Yabuuchi S, de Wilde RF, Martinez GV, Le A, et al. Therapeutic Targeting of the Warburg Effect in Pancreatic Cancer Relies on an Absence of P53 Function. Cancer Res (2015) 75(16):3355–64. doi: 10.1158/0008-5472.CAN-15-0108
177. Souto-Carneiro MM, Klika KD, Abreu MT, Meyer AP, Saffrich R, Sandhoff R, et al. Effect of Increased Lactate Dehydrogenase A Activity and Aerobic Glycolysis on the Proinflammatory Profile of Autoimmune CD8+ T Cells in Rheumatoid Arthritis. Arthritis Rheumatol (2020) 72(12):2050–64. doi: 10.1002/art.41420
178. Qiao T, Xiong Y, Feng Y, Guo W, Zhou Y, Zhao J, et al. Inhibition of LDH-A by Oxamate Enhances the Efficacy of Anti-PD-1 Treatment in an NSCLC Humanized Mouse Model. Front Oncol (2021) 11:632364. doi: 10.3389/fonc.2021.632364
179. Stone SC, Rossetti RAM, Alvarez KLF, Carvalho JP, Margarido PFR, Baracat EC, et al. Lactate Secreted by Cervical Cancer Cells Modulates Macrophage Phenotype. J Leukoc Biol (2019) 105(5):1041–54. doi: 10.1002/JLB.3A0718-274RR
180. Patel CH, Leone RD, Horton MR, Powell JD. Targeting Metabolism to Regulate Immune Responses in Autoimmunity and Cancer. Nat Rev Drug Discov (2019) 18(9):669–88. doi: 10.1038/s41573-019-0032-5
181. Rae C, Fragkoulis GI, Chalmers AJ. Cytotoxicity and Radiosensitizing Activity of the Fatty Acid Synthase Inhibitor C75 Is Enhanced by Blocking Fatty Acid Uptake in Prostate Cancer Cells. Adv Radiat Oncol (2020) 5(5):994–1005. doi: 10.1016/j.adro.2020.06.022
182. Voss K, Luthers CR, Pohida K, Snow AL. Fatty Acid Synthase Contributes to Restimulation-Induced Cell Death of Human CD4 T Cells. Front Mol Biosci (2019) 6:106. doi: 10.3389/fmolb.2019.00106
183. Sawyer BT, Qamar L, Yamamoto TM, McMellen A, Watson ZL, Richer JK, et al. Targeting Fatty Acid Oxidation to Promote Anoikis and Inhibit Ovarian Cancer Progression. Mol Cancer Res (2020) 18(7):1088–98. doi: 10.1158/1541-7786.MCR-19-1057
184. O'Connor RS, Guo L, Ghassemi S, Snyder NW, Worth AJ, Weng L, et al. The CPT1a Inhibitor, Etomoxir Induces Severe Oxidative Stress at Commonly Used Concentrations. Sci Rep (2018) 8(1):6289. doi: 10.1038/s41598-018-24676-6
185. Varghese S, Pramanik S, Williams LJ, Hodges HR, Hudgens CW, Fischer GM, et al. The Glutaminase Inhibitor CB-839 (Telaglenastat) Enhances the Antimelanoma Activity of T-Cell-Mediated Immunotherapies. Mol Cancer Ther (2021) 20(3):500–11. doi: 10.1158/1535-7163.MCT-20-0430
186. Xiang Y, Stine ZE, Xia J, Lu Y, O'Connor RS, Altman BJ, et al. Targeted Inhibition of Tumor-Specific Glutaminase Diminishes Cell-Autonomous Tumorigenesis. J Clin Invest (2015) 125(6):2293–306. doi: 10.1172/JCI75836
187. Kono M, Yoshida N, Maeda K, Suarez-Fueyo A, Kyttaris VC, Tsokos GC. Glutaminase 1 Inhibition Reduces Glycolysis and Ameliorates Lupus-Like Disease in MRL/lpr Mice and Experimental Autoimmune Encephalomyelitis. Arthritis Rheumatol (2019) 71(11):1869–78. doi: 10.1002/art.41019
188. Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine Blockade Induces Divergent Metabolic Programs to Overcome Tumor Immune Evasion. Science (2019) 366(6468):1013–21. doi: 10.1126/science.aav2588
189. Ueda Y, Saegusa J, Okano T, Sendo S, Yamada H, Nishimura K, et al. Additive Effects of Inhibiting Both mTOR and Glutamine Metabolism on the Arthritis in SKG Mice. Sci Rep (2019) 9(1):6374. doi: 10.1038/s41598-019-42932-1
190. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of Arginase by CB-1158 Blocks Myeloid Cell-Mediated Immune Suppression in the Tumor Microenvironment. J Immunother Cancer (2017) 5(1):101. doi: 10.1186/s40425-017-0308-4
191. Li A, Barsoumian HB, Schoenhals JE, Caetano MS, Wang X, Menon H, et al. IDO1 Inhibition Overcomes Radiation-Induced "Rebound Immune Suppression" by Reducing Numbers of IDO1-Expressing Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Int J Radiat Oncol Biol Phys (2019) 104(4):903–12. doi: 10.1016/j.ijrobp.2019.03.022
192. Lu J, Liu X, Liao YP, Salazar F, Sun B, Jiang W, et al. Nano-Enabled Pancreas Cancer Immunotherapy Using Immunogenic Cell Death and Reversing Immunosuppression. Nat Commun (2017) 8(1):1811. doi: 10.1038/s41467-017-01651-9
193. Mediavilla-Varela M, Castro J, Chiappori A, Noyes D, Hernandez DC, Allard B, et al. A Novel Antagonist of the Immune Checkpoint Protein Adenosine A2a Receptor Restores Tumor-Infiltrating Lymphocyte Activity in the Context of the Tumor Microenvironment. Neoplasia (2017) 19(7):530–6. doi: 10.1016/j.neo.2017.02.004
194. Leone RD, Sun IM, Oh MH, Sun IH, Wen J, Englert J, et al. Inhibition of the Adenosine A2a Receptor Modulates Expression of T Cell Coinhibitory Receptors and Improves Effector Function for Enhanced Checkpoint Blockade and ACT in Murine Cancer Models. Cancer Immunol Immunother (2018) 67(8):1271–84. doi: 10.1007/s00262-018-2186-0
Keywords: nasopharyngeal carcinoma, metabolism, immune cell, pH, immunotherapy
Citation: Huang H, Li S, Tang Q and Zhu G (2021) Metabolic Reprogramming and Immune Evasion in Nasopharyngeal Carcinoma. Front. Immunol. 12:680955. doi: 10.3389/fimmu.2021.680955
Received: 15 March 2021; Accepted: 25 August 2021;
Published: 09 September 2021.
Edited by:
Natalia Aptsiauri, University of Granada, SpainReviewed by:
Hong Zheng, Stanford University, United StatesEswar Shankar, Case Western Reserve University, United States
Copyright © 2021 Huang, Li, Tang and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gangcai Zhu, cWlhbmh1ZG9jdG9yQGNzdS5lZHUuY24=