 Jiaye Liu
Jiaye Liu Zhanjun Jia
Zhanjun Jia Wei Gong
Wei Gong- 1Nanjing Key Lab of Pediatrics, Children’s Hospital of Nanjing Medical University, Nanjing, China
- 2Jiangsu Key Laboratory of Pediatrics, Nanjing Medical University, Nanjing, China
- 3Department of Nephrology, State Key Laboratory of Reproductive Medicine, Children’s Hospital of Nanjing Medical University, Nanjing, China
Mitochondrial dysfunction is increasingly considered as a critical contributor to the occurrence and progression of acute kidney injury (AKI). However, the mechanisms by which damaged mitochondria mediate AKI progression are multifactorial and complicated. Mitochondrial DNA (mtDNA) released from damaged mitochondria could serve as a danger-associated molecular pattern (DAMP) and activate the innate immune system through STING, TLR9, NLRP3, and some other adaptors, and further mediate tubular cell inflammation and apoptosis. Accumulating evidence has demonstrated the important role of circulating mtDNA and its related pathways in the progression of AKI, and regulating the proteins involved in these pathways may be an effective strategy to reduce renal tubular injury and alleviate AKI. Here, we aim to provide a comprehensive overview of recent studies on mtDNA-mediated renal pathological events to provide new insights in the setting of AKI.
Introduction
Acute kidney injury (AKI) is a cluster of clinical syndromes characterized by a rapid decline in renal function over short period of time, such as hours or days (1). The incidence of AKI is increasing with the aging population and leads to high mortality and disability, especially in patients in intensive care units (ICUs) (2, 3). Unfortunately, AKI is now still monitored by urine volume and serum creatinine level; indexes for early detection remain to be discovered. Additionally, there is a lack of effective therapeutic strategies for AKI (4). Therefore, studies on new diagnostic markers and treatment approaches are urgently needed (5).
Mitochondria are well known as energy-producing organelles. In addition to their canonical function to meet the energy requirements of cells, mitochondria also control the innate immune responses to sterile and infectious insults (6). In particular, mitochondria are essential for maintaining renal tubular cell survival and normal function but are highly susceptible to damage (7). Mitochondrial dysfunction is a crucial pathogenic factor which leads to tubular injury (8). The multiple pathological features related to tubular cell injury, such as oxidative stress, immune cell recruitment, inflammatory cytokine accumulation, and apoptosis, could all be caused by mitochondrial dysfunction (9). Pathological analysis of renal biopsy samples obtained from septic patients in the ICU supported the view that mitochondrial injury contributes to the pathogenesis of sepsis-causing AKI (10). Recent evidence suggests that mitochondrial damage occurs at early stage of AKI and even before the tubular cell apoptosis is detectable (11–13). Strategies for mitochondrial protection could effectively protect against AKI (14, 15). However, the mechanisms by which mitochondrial dysfunction promotes tubular cell injury and AKI progression are complex and need further study (16).
Circulating mtDNA Serves as a Universal Danger-Associated Molecular Pattern in AKI
In addition to nuclei, mitochondria are the only organelles that contain DNA in eukaryotic cells. Mitochondrial DNA (mtDNA) is a circular double-stranded DNA (dsDNA) that encodes enzyme proteins, related ribosomal RNA, and transfer RNA required for various steps of oxidative phosphorylation (17). Normally, mtDNA is present in the mitochondrial matrix, but in cases of membrane potential reduction and/or mitochondrial membrane integrity damage, mtDNA can be translocated from the mitochondria to the cytoplasm (18). Free in the cytoplasm, mtDNA can serve as a danger-associated molecular pattern (DAMP) to trigger the innate immune system to initiate a non-infection-related inflammatory response, and that could mediate local inflammation, cell death, tissue injury, and dysfunction (6, 19). Moreover, biological behaviors, such as cell necrosis and pyroptosis that damage the structure of the cell membrane, can cause mtDNA to be released from the intracellular and enter the extracellular environment, such as blood or urine, which results in systemic inflammation and injury (20). As a newly identified mitochondrial DAMP, mtDNA has attracted growing interest in clinical and basic research, including studies on AKI in recent years.
In early AKI, pathological changes in the mitochondria are found in multiple kinds of AKI, including the septic, ischemic and toxic insults in origin (8, 21, 22). Circulating mtDNA in AKI patients’ blood or urine has been investigated in numerous studies. As reported, the plasma mtDNA quantity was enhanced in circulation after trauma and was associated with the mortality of patients in ICUs (23, 24). MtDNA in plasma has been suggested to be a promising biomarker in the clinical context of AKI and severe lung injury (25, 26). Evidence from clinical samples showed that in addition to the circulation, mtDNA in urine is an important marker of renal injury and AKI progression (27–30). Urine mtDNA levels were significantly enhanced in patients with AKI and positively correlated with serum creatinine levels, urine neutrophil gelatinase associated lipocalin (NGAL), kidney injury molecule 1 (Kim1), and the levels of inflammatory factors (30). Animal experiments also confirmed that in the AKI mouse model, urine mtDNA was derived from mitochondria-damaged renal tubular cells and significantly and positively associated with serum creatinine and urea nitrogen levels (27). Moreover, in vitro experiments showed that mtDNA derived from necrotic cells induced inflammation in renal tubular epithelial cells (31–33). The above evidence suggests that free mtDNA is a significant damage factor during the occurrence and progression of AKI, and could serve as a novel predictor in AKI. Furthermore, strategies of targeting mtDNA-related pathways can be potentially applied in the treatment of AKI.
The innate immune system plays a crucial role in mediating tubular injury and renal dysfunction. During AKI, elevated cytokines produced through innate immune pathways are closely related to the renal pathological damage (34, 35). Moreover, mtDNA, as an identified DAMP, can trigger the activation of the innate immune system in numerous pathological conditions such as sepsis, AKI, liver failure, and lung injury (36–38). Considering the important contribution of mtDNA in AKI and its related innate immune pathways, we summarize the major signaling pathways which are triggered by mtDNA and the potential therapies based on these studies.
Mechanisms Contributing to the mtDNA-Associated Pathogenesis of AKI
As a DAMP, free DNA is recognized and consequently elicits the related signals to transfer downstream through pattern recognition receptors (PRRs). MtDNA has been reported to be captured by different PRRs (6); various PRRs which could recognize mtDNA and their related pathways are reported to play vital roles in the progression of AKI, as discussed below (Figure 1).
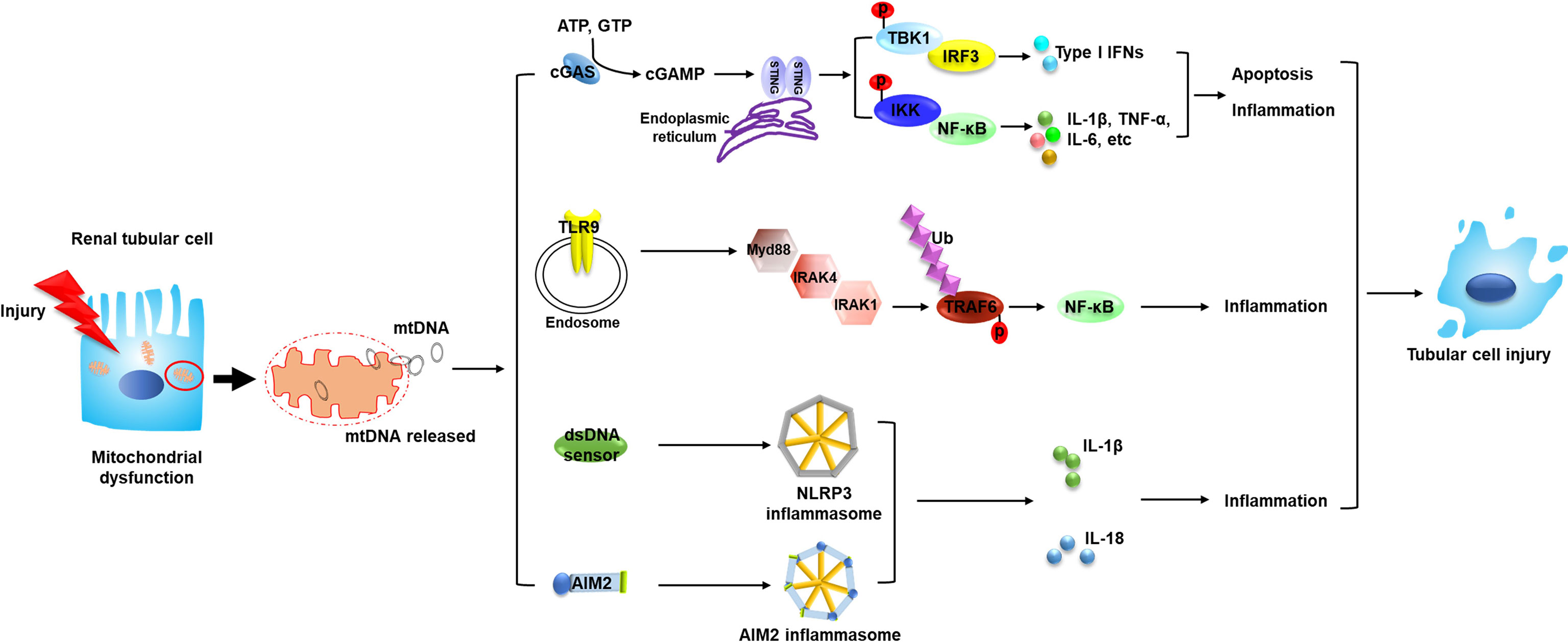
Figure 1 Circulating mtDNA triggers the innate immune system through several mechanisms and mediates the pathogenesis of AKI. The occurrence of AKI is closely associated with mitochondrial dysfunction in renal tubular cells. Tubular mitochondrial damage leads to mtDNA leakage into cytosol and extracellular space. Circulating mtDNA binds with different DNA sensors and activates several innate immune signaling pathways such as cGAS–STING, TLR9-Myd88-NF-κB and NLRP3 inflammasome, leading to tubular inflammation and injury, which contributes to the progression of AKI. MtDNA, mitochondrial DNA; STING, stimulator of interferon genes; TBK1, TANK binding kinase 1; IKK, the IκB kinase; p, phosphorylation; IRF3, interferon regulatory factor 3; NF-κB, nuclear factor kappa B; TNF, tumor necrosis factor; TLR9, toll-like receptor 9; Myd88, myeloid differentiation factor 88; IRAK, interleukin 1 receptor associated kinase; Ub, ubiquitylation; TRAF6, TNF receptor associated factor 6; AIM2, absent in melanoma 2; IL, interleukin.
The cGAS–STING System
Stimulator of interferon genes (STING), also called mediator of IRF3 activation (MITA) or transmembrane protein 173 (TMEM173), was found to be a novel DNA recognition receptor. Since STING was discovered in 2012, an increasing number of studies have confirmed the existence of the cGAS–cGAMP–STING pathway in different conditions and its vital role in the removal of DNA viruses (39, 40).
The cGAS–STING system starts with the recognition of DNA. DNA is bound to cyclic GMP-AMP synthase (cGAS) in a non-sequence-dependent way through its phosphate ribose skeleton to form a dimer, which can catalyze the synthesis of 2′-3′ cyclic AMP-GMP (2′–3′ cGAMP), a secondary messenger in cells. Then, cGAMP is transferred to the joint protein STING and binds it through hydrophobic interaction and hydrogen (41, 42). Activated STING transfers to the Golgi apparatus, where two cysteine residues of STING (Cys88 and Cys91) undergo palmitoylation, which leads to the exposure of the active C-tail domain (CTD). The structure of CTD is similar to the substrate of TANK-bound kinase 1 (TBK1), and the combination with TBK1 activates the downstream pathways. Two main downstream pathways of STING have been recently described: 1) TBK1 mediates IRF3 phosphorylation, then initiates the transcription of type 1 interferon to produce IFN-α and IFN-β, and also induces many other target genes such as interleukin (IL)-6 and IL-12; 2) Stimulation of cGAS–STING activates the classical NF-κB inflammatory responses, which is not fully dependent on the CTD of STING, to generate TNF-α, IL-Iβ, IL-6, and so on (43–48). Previous studies mainly focused on the antiviral effect of STING in removing DNA viruses. In fact, the cGAS–cGAMP–STING pathway plays many other roles in addition to its antiviral ability. For example, STING is important for many non-infection-related inflammatory states by mediating acute and chronic inflammatory injury and participating in the onset and progression of auto-inflammatory diseases (49). Strikingly, many studies have confirmed that not nuclear DNA but mtDNA is the key factor for triggering the cGAS–cGAMP–STING pathway, possibly because mtDNA has fewer DNA repair systems than nuclear DNA and is more prone to damage (50–54).
As in AKI, when the kidney suffers from cisplatin-induced injury, mitochondrial damage occurs accompanied by the increased mitochondrial membrane permeability, which causes the mtDNA to leak into the cytoplasm through the BAX pore on the outer mitochondrial membrane. After mtDNA leakage, the cGAS–STING pathway is activated, which leads to the phosphorylation of transcription factors, promotion of inflammatory factor secretion, and AKI progression. Activation of the cGAS–STING pathway has been observed in multiple AKI mice models and AKI patients (37, 55, 56). Moreover, STING knockout mice showed attenuated renal function, tubular injury, and inflammation when subjected to cisplatin treatment (37). In addition, STING also mediates the secondary renal inflammation and tubular injury (57). Although few studies focused specifically on the kidney injury in sepsis, a role for the cGAS–STING pathway in sepsis has been identified. Li N et al. found that STING can activate the expression of the NOD-like receptor family pyrin domain-containing-3 (NLRP3) inflammasome by promoting the nuclear translocation of phosphorylated IRF3, stimulating the expression of inflammatory factors in myocardial cells in LPS-induced septic mice and aggravating myocardial injury (58). Qiongyuan Hu et al. reported that STING signaling pathway in the intestinal tract was significantly activated, and the expression level of STING in the human intestinal lamina was related to intestinal inflammation in patients with sepsis and cecal ligation and puncture (CLP)-induced septic mice. Moreover, STING-knockout mice showed reduced bacterial translocation, decreased intestinal permeability, and a weaker inflammatory response, indicating that modulation of the mtDNA–STING pathway might facilitate healing of mucosa and defend the intestinal barrier in septic patients (59). However, STING-mediated specific downstream mechanism in AKI needs to be further explored.
As STING pathway is involved in the pathogenesis of AKI, STING inhibitors have been employed to explore their effects on AKI. To date, two kinds of STING antagonists have been tested in AKI mouse models and displayed beneficial effects towards ameliorating acute tubular injury and renal dysfunction (37, 60). Of note, there is a species difference between human STING and murine STING which is possibly due to the difference in the amino acid sequences between species and the different binding activities to specific cyclic dinucleotides (61, 62). Therefore, it is required to take this into account for the design of STING antagonists.
TLR9 Signaling
Toll-like receptors (TLRs) are important pattern recognition receptors that regulate adaptive and innate immunity and mediate resistance to microbial invasion. To date, 12 TLRs have been found in mice, and 10 have been found in humans (63). TLRs can be categorized into cell-surface TLRs (TLR1, 2, 4, 5, and 6, which recognize fungal or bacterial products) and intracellular TLRs (TLR3, 7, 8, and 9, which recognize RNA and DNA products) (64). TLR9 is a cytoplasmic receptor for unmethylated CpG motif containing DNA (CpG-DNA) found in DNA viruses and microbial DNA (65). Generally, TLR9 is localized to intracellular membrane compartments, such as the endoplasmic reticulum, endosome, and lysosome. Therefore, clathrin-mediated endocytosis is needed for the DNA translocating from the cell surface to the intracellular compartment and binding with TLR9 (66–68). Recent studies suggest that mtDNA can be recognized by TLR9, which triggers MYD88-dependent NF-κB-mediated gene transcription and leads to inflammation and apoptosis in acute lung injury, non-alcoholic steatohepatitis, and heart injury (26, 69, 70). Besides, mtDNA is also recognized following endocytosis and binding to TLR9 (71, 72).
In AKI, in vivo studies have illustrated that TLR9 contributes to the development of septic and ischemic renal tubular injury by promoting inflammation, apoptosis, and necrosis (73–76). Meanwhile, enhanced levels of mtDNA were found in the peritoneal cavity and plasma of septic mice induced by CLP or LPS. Furthermore, renal function injury, abnormal mitochondrial dysfunction and oxidative stress damage in the proximal tubules can be reversed by TLR9 knockout. More importantly, intravenous injection of mitochondrial debris (MTD) including substantial amounts of mtDNA in mice induced the similar inflammatory response to that of mice subjected to CLP; while knocking out TLR9 or DNase pretreatment attenuated this effect, which provided evidence that free mtDNA could mediate kidney injury through TLR9 (77). All these studies imply the role of mtDNA in the TLR9-mediated pathogenesis in AKI.
However, it is inconsistent whether the TLR9 pathway specifically causes tubular dysfunction in sepsis. Results from a previous study indicated that TLR9 was predominant on dendritic cells (DCs) in the interstitium but not on tubular epithelial cells (78). Consistent with this observation, Liu et al. documented that TLR9 expression was weakly detected in glomerular cells or renal tubular epithelial cells in control mice, although it was upregulated in tubular epithelial cells and glomerular cells in the mice subjected to CLP (79). Another study suggested that only the activation of TLR9 on renal tubular epithelial cells exacerbated the ischemic AKI; the activation of TLR9 on other cell types was renoprotective during AKI (80). These conflicting results suggest a necessity for further experimentation to clarify the mechanism of TLR9 activation in AKI. However, the endogenous mtDNA-mediated activation of TLR9 should contribute to the pathogenesis of AKI, and targeting TLR9 using siRNA or selective antagonist through targeting tubular delivery could protect against AKI (79, 81, 82).
Activation of NLRP3 Inflammasomes
NLRP3 inflammasomes are mainly composed of the receptor protein NLRP3, adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD) and downstream caspase-1 (83). Studies have verified the vital roles of activated inflammasomes in numerous pathological conditions, such as metabolic, autoimmune, autoinflammatory and infectious diseases (84–88). NLRP3 inflammasomes are mainly expressed in monocytes, macrophages, neutrophils, dendritic cells, and many non-hematopoietic cells (89, 90). Once activated, NLRP3 can trigger the self-cleavage and maturation of caspase-1, then activated caspase-1 not only promotes the maturation and secretion of various pro-inflammatory cytokines, including IL-1β and IL-18, but also triggers pyroptosis, which can eliminate pathogens and damaged cells (91, 92). Notably, NLRP3 inflammasomes are able to recognize various patterns that are closely related to many diseases, such as bacterial polypeptide antibiotics, bacterial RNA, and influenza virus ion channel proteins, ATP, and mtDNA (93–99). Although NLRP3 inflammasome activation triggered by cytosolic mtDNA has been demonstrated in many cases (98, 100, 101), direct evidence of mtDNA triggering NLRP3 inflammasomes in AKI is lacking. However, studies found that patients in the ICU had high levels of free mtDNA and IL-18 in the plasma, which were positively associated with the severity and mortality of diseases (25, 102), which suggested that cytosolic mtDNA might drive inflammasome activation and IL-18 secretion in an indirect or possibly a direct manner.
Others
In addition to the aforementioned sensors and adaptors, studies have found other kinds of DNA sensors. However, not all receptors can combine with mtDNA directly. Absent in melanoma 2 (AIM2), a member of the ALR family, is composed of the DNA-binding HIN domain and pyrin-signaling domain (PYD). Cytoplasmic dsDNA can bind to the HIN domain of AIM2 non-specifically, which stimulates the assembly of AIM2 in inflammasomes. Activation of the AIM2 inflammasome promotes apoptosis and the maturation of pro-inflammatory cytokines IL-18 and IL-1β (103, 104). However, the minimum number of base pairs of DNA that can be recognized by AIM2 is 80 kb (105), while mtDNA is a short strand of nucleic acid, at 17 kb (47), which cannot effectively bind to AIM2. AIM2 has a protective effect against microbial infection, but it is pathogenic to sterile inflammatory diseases, such as cardiovascular diseases, skin diseases, neuroinflammatory diseases, and CKD. During the pathogenesis of CKD, DNA from necrotic cells in the kidney was recognized by AIM2, which induced the assembly of the inflammasome complex and activated macrophages to produce IL-18 and IL-1β (106). Therefore, we hypothesize that AIM2, although unable to bind with mtDNA, may mediate the similar inflammatory effect in renal injury by responding to nuclear DNA. In addition, there are some other sensors that might recognize cytosolic mtDNA, such as DNA-dependent activator of IFN-regulatory factors (DAI) and RNA polymerase III. However, further studies are needed to explore their binding with mtDNA and/or the function in the pathogenesis of AKI (107, 108).
Summary and Future Perspectives
Mitochondrial injury is a common pathological phenomenon in different types of AKI models, which leads to the release of mtDNA into the cytoplasm. Currently, circulating mtDNA from damaged mitochondria has been reported to be involved in the pathogenesis of AKI. Clinical studies demonstrated that level of free mtDNA was elevated in AKI patients and was associated with the disease severity and prognosis (28, 29), suggesting that circulating mtDNA has the potential to serve as a predictor for AKI. As one kind of mitochondrial DAMPs, mtDNA could promote inflammatory response through binding with the DNA sensors and triggering the innate immune activation. Although the identified sensors of mtDNA could also recognize other kinds of nucleic acids, studies have mentioned the importance of mtDNA, but not nuclear DNA, in the occurrence and the development of AKI (38). Furthermore, depletion of mtDNA markedly attenuated the acute renal tubular cell injury (37), indicating that strategies targeting both mtDNA-mediated pathways and mtDNA clearance mechanisms need to be considered in AKI therapy. However, the detailed mechanism underlying mtDNA-mediated pathogenesis in AKI and the strategies antagonizing mtDNA-associated kidney injury need to be further explored.
Author Contributions
WG and ZJ conducted a review of articles. JL and WG wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81873599, 81970581 and 82070701), and the Nanjing Medical Science and Technique Development Foundation (QRX17166).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Makris K, Spanou L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin Biochem Rev (2016) 37(2):85–98.
2. Ronco C, Bellomo R, Kellum JA. Acute Kidney Injury. Lancet (2019) 394(10212):1949–64. doi: 10.1016/S0140-6736(19)32563-2
3. Qian JY, Wang B, Liu BC. Acute Kidney Injury in the 2019 Novel Coronavirus Disease. Kidney Dis (2020) 6:6. doi: 10.1159/000509086
4. Jamme M, Legrand M, Geri G. Outcome of Acute Kidney Injury: How to Make a Difference? Ann Intensive Care (2021) 11(1):60. doi: 10.1186/s13613-021-00849-x
5. Klouche K, Amigues L, Morena M, Brunot V, Dupuy AM, Jaussent A, et al. On-Line Hemodiafiltration did Not Induce an Overproduction of Oxidative Stress and Inflammatory Cytokines in Intensive Care Unit-Acute Kidney Injury. BMC Nephrol (2017) 18(1):371. doi: 10.1186/s12882-017-0785-1
6. Banoth B, Cassel SL. Mitochondria in Innate Immune Signaling. Transl Res (2018) 202:52–68. doi: 10.1016/j.trsl.2018.07.014
7. Martín-Hernández E, García-Silva MT, Vara J, Campos Y, Cabello A, Muley R, et al. Renal Pathology in Children With Mitochondrial Diseases. Pediatr Nephrol (2005) 20(9):1299–305. doi: 10.1007/s00467-005-1948-z
8. Emma F, Montini G, Parikh SM, Salviati L. Mitochondrial Dysfunction in Inherited Renal Disease and Acute Kidney Injury. Nat Rev Nephrol (2016) 12(5):267–80. doi: 10.1038/nrneph.2015.214
9. Bhatia D, Capili A, Choi ME. Mitochondrial Dysfunction in Kidney Injury, Inflammation, and Disease: Potential Therapeutic Approaches. Kidney Res Clin Pract (2020) 39(3):244–58. doi: 10.23876/j.krcp.20.082
10. van der Slikke EC, Star BS, van Meurs M, Henning RH, Moser J, Bouma HR. Sepsis Is Associated With Mitochondrial DNA Damage and a Reduced Mitochondrial Mass in the Kidney of Patients With Sepsis-AKI. Crit Care (2021) 25(1):36. doi: 10.1186/s13054-020-03424-1
11. Funk JA, Schnellmann RG. Persistent Disruption of Mitochondrial Homeostasis After Acute Kidney Injury. Am J Physiol Renal Physiol (2012) 302(7):F853–64. doi: 10.1152/ajprenal.00035.2011
12. Aparicio-Trejo OE, Avila-Rojas SH, Tapia E, Rojas-Morales P, León-Contreras JC, Martínez-Klimova E, et al. Chronic Impairment of Mitochondrial Bioenergetics and β-Oxidation Promotes Experimental AKI-to-CKD Transition Induced by Folic Acid. Free Radic Biol Med (2020) 154:18–32. doi: 10.1016/j.freeradbiomed.2020.04.016
13. Hall AM, Rhodes GJ, Sandoval RM, Corridon PR, Molitoris BA. In Vivo Multiphoton Imaging of Mitochondrial Structure and Function During Acute Kidney Injury. Kidney Int (2013) 83(1):72–83. doi: 10.1038/ki.2012.328
14. Brooks C, Wei Q, Cho SG, Dong Z. Regulation of Mitochondrial Dynamics in Acute Kidney Injury in Cell Culture and Rodent Models. J Clin Invest (2009) 119(5):1275–85. doi: 10.1172/JCI37829
15. Perry HM, Huang L, Wilson RJ, Bajwa A, Sesaki H, Yan Z, et al. Dynamin-Related Protein 1 Deficiency Promotes Recovery From AKI. J Am Soc Nephrol (2018) 29(1):194–206. doi: 10.1681/ASN.2017060659
16. Duann P, Lin PH. Mitochondria Damage and Kidney Disease. Adv Exp Med Biol (2017) 982:529–51. doi: 10.1007/978-3-319-55330-6_27
17. Yan C, Duanmu X, Zeng L, Liu B, Song Z. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells (2019) 8(4):379. doi: 10.3390/cells8040379
18. McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, et al. BAK/BAX Macropores Facilitate Mitochondrial Herniation and mtDNA Efflux During Apoptosis. Science (2018) 359(6378):eaao6047. doi: 10.1126/science.aao6047
19. Mihm S. Danger-Associated Molecular Patterns (DAMPS): Molecular Triggers for Sterile Inflammation in the Liver. Int J Mol Sci (2018) 19(10):3104. doi: 10.3390/ijms19103104
20. Cecchino GN, Garcia-Velasco JA. Mitochondrial DNA Copy Number as a Predictor of Embryo Viability. Fertil Steril (2019) 111(2):205–11. doi: 10.1016/j.fertnstert.2018.11.021
21. Szeto HH. Pharmacologic Approaches to Improve Mitochondrial Function in AKI and CKD. J Am Soc Nephrol (2017) 28(10):2856–65. doi: 10.1681/ASN.2017030247
22. Song SJ, Kim SM, Lee SH, Moon JY, Hwang HS, Kim JS, et al. Rhabdomyolysis-Induced AKI was Ameliorated in NLRP3 Ko Mice Via Alleviation of Mitochondrial Lipid Peroxidation in Renal Tubular Cells. Int J Mol Sci (2020) 21(22):8564. doi: 10.3390/ijms21228564
23. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating Mitochondrial DAMPs Cause Inflammatory Responses to Injury. Nature (2010) 464(7285):104–7. doi: 10.1038/nature08780
24. Saad A, Herrmann SMS, Eirin A, Ferguson CM, Glockner JF, Bjarnason H, et al. Phase 2a Clinical Trial of Mitochondrial Protection (Elamipretide) During Stent Revascularization in Patients With Atherosclerotic Renal Artery Stenosis. Circ Cardiovasc Interv (2017) 10(9):e005487. doi: 10.1161/CIRCINTERVENTIONS.117.005487
25. Nakahira K, Kyung SY, Rogers AJ, Gazourian L, Youn S, Massaro AF, et al. Circulating Mitochondrial DNA in Patients in the ICU as a Marker of Mortality: Derivation and Validation. PloS Med (2013) 10(12):e1001577; discussion e. doi: 10.1371/journal.pmed.1001577
26. Gan L, Chen X, Sun T, Li Q, Zhang R, Zhang J, et al. Significance of Serum Mtdna Concentration in Lung Injury Induced by Hip Fracture. Shock (2015) 44(1):52–7. doi: 10.1097/SHK.0000000000000366
27. Ho PW, Pang WF, Luk CC, Ng JK, Chow KM, Kwan BC, et al. Urinary Mitochondrial DNA Level as a Biomarker of Acute Kidney Injury Severity. Kidney Dis (Basel) (2017) 3(2):78–83. doi: 10.1159/000475883
28. Whitaker RM, Stallons LJ, Kneff JE, Alge JL, Harmon JL, Rahn JJ, et al. Urinary Mitochondrial DNA Is a Biomarker of Mitochondrial Disruption and Renal Dysfunction in Acute Kidney Injury. Kidney Int (2015) 88(6):1336–44. doi: 10.1038/ki.2015.240
29. Hu Q, Ren J, Wu J, Li G, Wu X, Liu S, et al. Urinary Mitochondrial DNA Levels Identify Acute Kidney Injury in Surgical Critical Illness Patients. Shock (2017) 48(1):11–7. doi: 10.1097/SHK.0000000000000830
30. Hu Q, Ren J, Ren H, Wu J, Wu X, Liu S, et al. Urinary Mitochondrial DNA Identifies Renal Dysfunction and Mitochondrial Damage in Sepsis-Induced Acute Kidney Injury. Oxid Med Cell Longevity (2018) 2018:1–14. doi: 10.1155/2018/8074936
31. Kim K, Moon H, Lee YH, Seo JW, Kim YG, Moon JY, et al. Clinical Relevance of Cell-Free Mitochondrial DNA During the Early Postoperative Period in Kidney Transplant Recipients. Sci Rep (2019) 9(1):18607. doi: 10.1038/s41598-019-54694-x
32. Homolova J, Janovicova L, Konecna B, Vlkova B, Celec P, Tothova L, et al. Plasma Concentrations of Extracellular DNA in Acute Kidney Injury. Diag (Basel) (2020) 10(3):152. doi: 10.3390/diagnostics10030152
33. Jansen MPB, Pulskens WP, Butter LM, Florquin S, Juffermans NP, Roelofs J, et al. Mitochondrial DNA Is Released in Urine of SIRS Patients With Acute Kidney Injury and Correlates With Severity of Renal Dysfunction. Shock (2018) 49(3):301–10. doi: 10.1097/SHK.0000000000000967
34. Singbartl K, Formeck CL, Kellum JA. Kidney-Immune System Crosstalk in AKI. Semin Nephrol (2019) 39(1):96–106. doi: 10.1016/j.semnephrol.2018.10.007
35. Singbartl K, Joannidis M. Short-Term Effects of Acute Kidney Injury. Crit Care Clin (2015) 31(4):751–62. doi: 10.1016/j.ccc.2015.06.010
36. Benmerzoug S, Rose S, Bounab B, Gosset D, Duneau L, Chenuet P, et al. STING-Dependent Sensing of Self-DNA Drives Silica-Induced Lung Inflammation. Nat Commun (2018) 9(1):5226. doi: 10.1038/s41467-018-07425-1
37. Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H, et al. Mitochondrial Damage Causes Inflammation Via Cgas-STING Signaling in Acute Kidney Injury. Cell Rep (2019) 29(5):1261–73.e6. doi: 10.1016/j.celrep.2019.09.050
38. Yu Y, Liu Y, An W, Song J, Zhang Y, Zhao X. STING-Mediated Inflammation in Kupffer Cells Contributes to Progression of Nonalcoholic Steatohepatitis. J Clin Invest (2019) 129(2):546–55. doi: 10.1172/JCI121842
39. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science (2013) 339(6121):786–91. doi: 10.1126/science.1232458
40. Phelan T, Little MA, Brady G. Targeting of the cGAS-STING System by DNA Viruses. Biochem Pharmacol (2020) 174:113831. doi: 10.1016/j.bcp.2020.113831
41. Kato K, Omura H, Ishitani R, Nureki O. Cyclic GMP-AMP as an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Annu Rev Biochem (2017) 86:541–66. doi: 10.1146/annurev-biochem-061516-044813
42. Ishikawa H, Barber GN. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature (2008) 455(7213):674–8. doi: 10.1038/nature07317
43. Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. Sting Activation by Translocation From the ER Is Associated With Infection and Autoinflammatory Disease. Cell Host Microbe (2015) 18(2):157–68. doi: 10.1016/j.chom.2015.07.001
44. Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, et al. Activation of STING Requires Palmitoylation at the Golgi. Nat Commun (2016) 7:11932. doi: 10.1038/ncomms11932
45. Shu C, Yi G, Watts T, Kao CC, Li P. Structure of STING Bound to Cyclic di-GMP Reveals the Mechanism of Cyclic Dinucleotide Recognition by the Immune System. Nat Struct Mol Biol (2012) 19(7):722–4. doi: 10.1038/nsmb.2331
46. Luteijn RD, Zaver SA, Gowen BG, Wyman SK, Garelis NE, Onia L, et al. SLC19A1 Transports Immunoreactive Cyclic Dinucleotides. Nature (2019) 573(7774):434–8. doi: 10.1038/s41586-019-1553-0
47. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature (2015) 520(7548):553–7. doi: 10.1038/nature14156
48. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat Rev Immunol (2021). doi: 10.1038/s41577-021-00524-z
49. Motwani M, Pesiridis S, Fitzgerald KA. DNA Sensing by the cGAS-STING Pathway in Health and Disease. Nat Rev Genet (2019) 20(11):657–74. doi: 10.1038/s41576-019-0151-1
50. Scheibye-Knudsen M, Fang EF, Croteau DL, Wilson DM 3rd, Bohr VA. Protecting the Mitochondrial Powerhouse. Trends Cell Biol (2015) 25(3):158–70. doi: 10.1016/j.tcb.2014.11.002
51. Shadel GS, Clayton DA. Mitochondrial DNA Maintenance in Vertebrates. Annu Rev Biochem (1997) 66:409–35. doi: 10.1146/annurev.biochem.66.1.409
52. Bai J, Liu F. The cGAS-cGAMP-STING Pathway: A Molecular Link Between Immunity and Metabolism. Diabetes (2019) 68(6):1099–108. doi: 10.2337/dbi18-0052
53. Qiao JT, Cui C, Qing L, Wang LS, He TY, Yan F, et al. Activation of the STING-IRF3 Pathway Promotes Hepatocyte Inflammation, Apoptosis and Induces Metabolic Disorders in Nonalcoholic Fatty Liver Disease. Metabolism (2018) 81:13–24. doi: 10.1016/j.metabol.2017.09.010
54. Ma R, Ortiz Serrano TP, Davis J, Prigge AD, Ridge KM. The cGAS-STING Pathway: The Role of Self-DNA Sensing in Inflammatory Lung Disease. FASEB J (2020) 34(10):13156–70. doi: 10.1096/fj.202001607R
55. Inoue T, Abe C, Sung SS, Moscalu S, Jankowski J, Huang L, et al. Vagus Nerve Stimulation Mediates Protection From Kidney Ischemia-Reperfusion Injury Through Alpha7nAChR+ Splenocytes. J Clin Invest (2016) 126(5):1939–52. doi: 10.1172/JCI83658
56. Kojima I, Tanaka T, Inagi R, Kato H, Yamashita T, Sakiyama A, et al. Protective Role of Hypoxia-Inducible Factor-2alpha Against Ischemic Damage and Oxidative Stress in the Kidney. J Am Soc Nephrol (2007) 18(4):1218–26. doi: 10.1681/ASN.2006060639
57. Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab (2019) 30(4):784–99 e5. doi: 10.1016/j.cmet.2019.08.003
58. Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W, et al. Sting-IRF3 Contributes to Lipopolysaccharide-Induced Cardiac Dysfunction, Inflammation, Apoptosis and Pyroptosis by Activating NLRP3. Redox Biol (2019) 24:101215. doi: 10.1016/j.redox.2019.101215
59. Hu Q, Ren H, Li G, Wang D, Zhou Q, Wu J, et al. STING-Mediated Intestinal Barrier Dysfunction Contributes to Lethal Sepsis. EBioMedicine (2019) 41:497–508. doi: 10.1016/j.ebiom.2019.02.055
60. Gong W, Lu L, Zhou Y, Liu J, Ma H, Fu L, et al. The Novel STING Antagonist H151 Ameliorates Cisplatin-Induced Acute Kidney Injury and Mitochondrial Dysfunction. Am J Physiol Renal Physiol (2021) 320(4):F608–F16. doi: 10.1152/ajprenal.00554.2020
61. Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, et al. Structure-Function Analysis of STING Activation by C[G(2’,5’)Pa(3’,5’)P] and Targeting by Antiviral DMXAA. Cell (2013) 154(4):748–62. doi: 10.1016/j.cell.2013.07.023
62. Skouboe MK, Knudsen A, Reinert LS, Boularan C, Lioux T, Perouzel E, et al. STING Agonists Enable Antiviral Cross-Talk Between Human Cells and Confer Protection Against Genital Herpes in Mice. PloS Pathog (2018) 14(4):e1006976. doi: 10.1371/journal.ppat.1006976
63. Gong T, Liu L, Jiang W, Zhou R. DAMP-Sensing Receptors in Sterile Inflammation and Inflammatory Diseases. Nat Rev Immunol (2020) 20(2):95–112. doi: 10.1038/s41577-019-0215-7
64. Cui J, Chen Y, Wang HY, Wang RF. Mechanisms and Pathways of Innate Immune Activation and Regulation in Health and Cancer. Hum Vaccin Immunother (2014) 10(11):3270–85. doi: 10.4161/21645515.2014.979640
65. Fitzgerald KA, Kagan JC. Toll-Like Receptors and the Control of Immunity. Cell (2020) 180(6):1044–66. doi: 10.1016/j.cell.2020.02.041
66. Gallucci S, Maffei ME. DNA Sensing Across the Tree of Life. Trends Immunol (2017) 38(10):719–32. doi: 10.1016/j.it.2017.07.012
67. Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, et al. TLR9 Signals After Translocating From the ER to CpG DNA in the Lysosome. Nat Immunol (2004) 5(2):190–8. doi: 10.1038/ni1028
68. Kumagai Y, Takeuchi O, Akira S. TLR9 as a Key Receptor for the Recognition of DNA. Adv Drug Delivery Rev (2008) 60(7):795–804. doi: 10.1016/j.addr.2007.12.004
69. Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously Oxidized Mitochondrial DNA Induces In Vivo and In Vitro Inflammatory Responses. J Leukoc Biol (2004) 75(6):995–1000. doi: 10.1189/jlb.0703328
70. Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, et al. Hepatocyte Mitochondrial DNA Drives Nonalcoholic Steatohepatitis by Activation of TLR9. J Clin Invest (2016) 126(3):859–64. doi: 10.1172/JCI83885
71. Bao W, Xia H, Liang Y, Ye Y, Lu Y, Xu X, et al. Toll-Like Receptor 9 Can be Activated by Endogenous Mitochondrial DNA to Induce Podocyte Apoptosis. Sci Rep (2016) 6:22579. doi: 10.1038/srep22579
72. Bueno M, Zank D, Buendia-Roldan I, Fiedler K, Mays BG, Alvarez D, et al. PINK1 Attenuates mtDNA Release in Alveolar Epithelial Cells and TLR9 Mediated Profibrotic Responses. PloS One (2019) 14(6):e0218003. doi: 10.1371/journal.pone.0218003
73. Endre ZH, Erlich JH. Targeted Protection of Proximal Tubular Cells by Nanoparticle-Enhanced Delivery of a TLR9-Antagonist. Kidney Int (2020) 98(1):48–50. doi: 10.1016/j.kint.2020.04.024
74. Naito Y, Tsuji T, Nagata S, Tsuji N, Fujikura T, Ohashi N, et al. IL-17A Activated by Toll-Like Receptor 9 Contributes to the Development of Septic Acute Kidney Injury. Am J Physiol Renal Physiol (2020) 318(1):F238–F47. doi: 10.1152/ajprenal.00313.2019
75. Han SJ, Li H, Kim M, D’Agati V, Lee HT. Intestinal Toll-Like Receptor 9 Deficiency Leads to Paneth Cell Hyperplasia and Exacerbates Kidney, Intestine, and Liver Injury After Ischemia/Reperfusion Injury. Kidney Int (2019) 95(4):859–79. doi: 10.1016/j.kint.2018.10.035
76. Alikhan MA, Summers SA, Gan PY, Chan AJ, Khouri MB, Ooi JD, et al. Endogenous Toll-Like Receptor 9 Regulates AKI by Promoting Regulatory T Cell Recruitment. J Am Soc Nephrol (2016) 27(3):706–14. doi: 10.1681/ASN.2014090927
77. Tsuji N, Tsuji T, Ohashi N, Kato A, Fujigaki Y, Yasuda H. Role of Mitochondrial DNA in Septic AKI Via Toll-Like Receptor 9. J Am Soc Nephrol (2016) 27(7):2009–20. doi: 10.1681/ASN.2015040376
78. Anders H, Banas B, Schlöndorff D. Signaling Danger: Toll-Like Receptors and Their Potential Roles in Kidney Disease. J Am Soc Nephrol: JASN (2004) 15(4):854–67. doi: 10.1097/01.ASN.0000121781.89599.16
79. Liu L, Li Y, Hu Z, Su J, Huo Y, Tan B, et al. Small Interfering RNA Targeting Toll-Like Receptor 9 Protects Mice Against Polymicrobial Septic Acute Kidney Injury. Nephron Exp Nephrol (2012) 122(1-2):51–61. doi: 10.1159/000346953
80. Han SJ, Li H, Kim M, Shlomchik MJ, Lee HT. Kidney Proximal Tubular TLR9 Exacerbates Ischemic Acute Kidney Injury. J Immunol (2018) 201(3):1073–85. doi: 10.4049/jimmunol.1800211
81. Han SJ, Williams RM, D’Agati V, Jaimes EA, Heller DA, Lee HT. Selective Nanoparticle-Mediated Targeting of Renal Tubular Toll-Like Receptor 9 Attenuates Ischemic Acute Kidney Injury. Kidney Int (2020) 98(1):76–87. doi: 10.1016/j.kint.2020.01.036
82. Yasuda H, Leelahavanichkul A, Tsunoda S, Dear JW, Takahashi Y, Ito S, et al. Chloroquine and Inhibition of Toll-Like Receptor 9 Protect From Sepsis-Induced Acute Kidney Injury. Am J Physiol Renal Physiol (2008) 294(5):F1050–8. doi: 10.1152/ajprenal.00461.2007
83. Swanson KV, Deng M, Ting JP. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
84. Komada T, Muruve DA. The Role of Inflammasomes in Kidney Disease. Nat Rev Nephrol (2019) 15(8):501–20. doi: 10.1038/s41581-019-0158-z
85. Ralston JC, Lyons CL, Kennedy EB, Kirwan AM, Roche HM. Fatty Acids and NLRP3 Inflammasome-Mediated Inflammation in Metabolic Tissues. Annu Rev Nutr (2017) 37:77–102. doi: 10.1146/annurev-nutr-071816-064836
86. Li Z, Guo J, Bi L. Role of the NLRP3 Inflammasome in Autoimmune Diseases. BioMed Pharmacother (2020) 130:110542. doi: 10.1016/j.biopha.2020.110542
87. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent Advances in the Mechanisms of NLRP3 Inflammasome Activation and Its Inhibitors. Cell Death Dis (2019) 10(2):128. doi: 10.1038/s41419-019-1413-8
88. Tartey S, Kanneganti TD. Inflammasomes in the Pathophysiology of Autoinflammatory Syndromes. J Leukoc Biol (2020) 107(3):379–91. doi: 10.1002/JLB.3MIR0919-191R
89. He Y, Hara H, Núñez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
90. Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, et al. Differential Expression of NLRP3 Among Hematopoietic Cells. J Immunol (2011) 186(4):2529–34. doi: 10.4049/jimmunol.1002720
91. Wang Z, Zhang S, Xiao Y, Zhang W, Wu S, Qin T, et al. Nlrp3 Inflammasome and Inflammatory Diseases. Oxid Med Cell Longev (2020) 2020:4063562. doi: 10.1155/2020/4063562
92. Hutton HL, Ooi JD, Holdsworth SR, Kitching AR. The NLRP3 Inflammasome in Kidney Disease and Autoimmunity. Nephrol (Carlton) (2016) 21(9):736–44. doi: 10.1111/nep.12785
93. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 Inflammasome Is Involved in the Innate Immune Response to Amyloid-Beta. Nat Immunol (2008) 9(8):857–65. doi: 10.1038/ni.1636
94. Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin Activates the Inflammasome in Response to Toxins and ATP. Nature (2006) 440(7081):228–32. doi: 10.1038/nature04515
95. Sha W, Mitoma H, Hanabuchi S, Bao M, Weng L, Sugimoto N, et al. Human NLRP3 Inflammasome Senses Multiple Types of Bacterial RNAs. Proc Natl Acad Sci USA (2014) 111(45):16059–64. doi: 10.1073/pnas.1412487111
96. Hafner-Bratkovič I, Pelegrín P. Ion Homeostasis and Ion Channels in NLRP3 Inflammasome Activation and Regulation. Curr Opin Immunol (2018) 52:8–17. doi: 10.1016/j.coi.2018.03.010
97. Ichinohe T, Pang IK, Iwasaki A. Influenza Virus Activates Inflammasomes Via Its Intracellular M2 Ion Channel. Nat Immunol (2010) 11(5):404–10. doi: 10.1038/ni.1861
98. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome During Apoptosis. Immunity (2012) 36(3):401–14. doi: 10.1016/j.immuni.2012.01.009
99. Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 Inflammasome: Its Regulation and Involvement in Atherosclerosis. J Cell Physiol (2018) 233(3):2116–32. doi: 10.1002/jcp.25930
100. West AP, Shadel GS. Mitochondrial DNA in Innate Immune Responses and Inflammatory Pathology. Nat Rev Immunol (2017) 17(6):363–75. doi: 10.1038/nri.2017.21
101. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat Immunol (2011) 12(3):222–30. doi: 10.1038/ni.1980
102. Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, et al. Inflammasome-Regulated Cytokines Are Critical Mediators of Acute Lung Injury. Am J Respir Crit Care Med (2012) 185(11):1225–34. doi: 10.1164/rccm.201201-0003OC
103. Man SM, Karki R, Kanneganti TD. AIM2 Inflammasome in Infection, Cancer, and Autoimmunity: Role in DNA Sensing, Inflammation, and Innate Immunity. Eur J Immunol (2016) 46(2):269–80. doi: 10.1002/eji.201545839
104. Sharma BR, Karki R, Kanneganti TD. Role of AIM2 Inflammasome in Inflammatory Diseases, Cancer and Infection. Eur J Immunol (2019) 49(11):1998–2011. doi: 10.1002/eji.201848070
105. Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, et al. Structures of the HIN Domain:DNA Complexes Reveal Ligand Binding and Activation Mechanisms of the AIM2 Inflammasome and IFI16 Receptor. Immunity (2012) 36(4):561–71. doi: 10.1016/j.immuni.2012.02.014
106. Komada T, Chung H, Lau A, Platnich JM, Beck PL, Benediktsson H, et al. Macrophage Uptake of Necrotic Cell DNA Activates the AIM2 Inflammasome to Regulate a Proinflammatory Phenotype in CKD. J Am Soc Nephrol (2018) 29(4):1165–81. doi: 10.1681/ASN.2017080863
107. Lou H, Pickering MC. Extracellular DNA and Autoimmune Diseases. Cell Mol Immunol (2018) 15(8):746–55. doi: 10.1038/cmi.2017.136
Keywords: acute kidney injury, mitochondrial DNA, STING, TLR9, NLRP3
Citation: Liu J, Jia Z and Gong W (2021) Circulating Mitochondrial DNA Stimulates Innate Immune Signaling Pathways to Mediate Acute Kidney Injury. Front. Immunol. 12:680648. doi: 10.3389/fimmu.2021.680648
Received: 15 March 2021; Accepted: 07 June 2021;
Published: 24 June 2021.
Edited by:
Gilles Kaplanski, Assistance Publique Hôpitaux de Marseille, FranceReviewed by:
Charlotte M. Vines, The University of Texas at El Paso, United StatesEmilia Lecuona, Northwestern University, United States
Copyright © 2021 Liu, Jia and Gong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Gong, Z29uZ3dlaUBuam11LmVkdS5jbg==; Zhanjun Jia, amlhemo3MkBob3RtYWlsLmNvbQ==