 Rebecca Dowey
Rebecca Dowey Ahmed Iqbal
Ahmed Iqbal Simon R. Heller
Simon R. Heller Ian Sabroe2
Ian Sabroe2 Lynne R. Prince
Lynne R. Prince
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 03 June 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.678771
This article is part of the Research TopicNeutrophil Functions in Host Immunity, Inflammation and Tissue RepairView all 24 articles
Chronic and recurrent infections occur commonly in both type 1 and type 2 diabetes (T1D, T2D) and increase patient morbidity and mortality. Neutrophils are professional phagocytes of the innate immune system that are critical in pathogen handling. Neutrophil responses to infection are dysregulated in diabetes, predominantly mediated by persistent hyperglycaemia; the chief biochemical abnormality in T1D and T2D. Therapeutically enhancing host immunity in diabetes to improve infection resolution is an expanding area of research. Individuals with diabetes are also at an increased risk of severe coronavirus disease 2019 (COVID-19), highlighting the need for re-invigorated and urgent focus on this field. The aim of this review is to explore the breadth of previous literature investigating neutrophil function in both T1D and T2D, in order to understand the complex neutrophil phenotype present in this disease and also to focus on the development of new therapies to improve aberrant neutrophil function in diabetes. Existing literature illustrates a dual neutrophil dysfunction in diabetes. Key pathogen handling mechanisms of neutrophil recruitment, chemotaxis, phagocytosis and intracellular reactive oxygen species (ROS) production are decreased in diabetes, weakening the immune response to infection. However, pro-inflammatory neutrophil pathways, mainly neutrophil extracellular trap (NET) formation, extracellular ROS generation and pro-inflammatory cytokine generation, are significantly upregulated, causing damage to the host and perpetuating inflammation. Reducing these proinflammatory outputs therapeutically is emerging as a credible strategy to improve infection resolution in diabetes, and also more recently COVID-19. Future research needs to drive forward the exploration of novel treatments to improve infection resolution in T1D and T2D to improve patient morbidity and mortality.
The number of people with diabetes (PWD) globally will exceed 500 million by 2035. Type 1 diabetes (T1D) is an autoimmune condition characterised by T-cell mediated pancreatic β cell destruction and absolute insulin deficiency (1). T1D represents up to 10% of all diabetes worldwide and a small percentage (<10% type 1B) of affected individuals have no evidence of autoimmunity with the pathogenesis being idiopathic (2, 3). A complex interplay of genetic, epigenetic, environmental, and immunologic factors is thought to contribute to the pathogenesis of T1D. Genome-wide association studies have identified more than 50 genetic risk loci to date but the main genes predisposing to T1D are located within the human leukocyte antigen (HLA) on chromosome 6 (4, 5). Alleles at the HLA locus account for up to 50% of cases with familial clustering (6–8). Epidemiological studies have implicated a number of environmental factors in the pathogenesis of T1D, including viruses and nutrients such as cow’s milk protein (4, 5). These factors are thought to trigger an autoimmune response consequent upon molecular mimicry in that pancreatic autoantigens that resemble viral or dietary epitopes undergo cellular destruction (9, 10). Pancreatic β cell destruction involves both cellular and humoral immunity. Autoreactive T-cells are thought to induce apoptosis in a pancreatic islet milieu rich in pro-inflammatory cytokines including IL-1, TNF-α, and IFN-γ (11). The presence of circulating autoantibodies against proinsulin and other autoantigens in β cells highlights the role of humoral immunity in disease pathogenesis. Indeed, circulating autoantibodies in T1D can occur before the biochemical and clinical manifestations and the presence of two or more autoantibodies in first-degree relatives strongly predicts clinical progression to T1D (12).
In type 2 diabetes (T2D), which accounts for 90-95% of all diabetes, a combined resistance to insulin both in skeletal muscle and the liver, in addition to defective insulin production by pancreatic β cells is present (13). In contrast to T1D, no predominant genetic locus has been found to increase susceptibility to T2D. Genomic studies reveal over 40 genetic variants that increase the risk of T2D, however, overall these genes account for 10% heritability (14, 15). A positive family history is important nonetheless with a 38% life-time risk of T2D in individuals who have one parent with T2D with this risk increasing to 60% if both parents have T2D (16, 17). In addition to multiple genes, environmental factors play a critical role in the pathogenesis of T2D. A sedentary lifestyle in addition to consumption of high-fat, high-calorie diets means the majority of individuals with T2D are overweight (6). Obesity related insulin resistance together with hypertension, dyslipidaemia, glucose intolerance, and eventually frank hyperglycaemia defines the metabolic syndrome and this clinical phenotype is commonly encountered in many people with T2D (18). Several mechanisms have been proposed to explain both insulin resistance in T2D which occurs early in the disease and pancreatic β cell dysfunction which is typically a late phenomenon. Increased levels of non-esterified fatty acids, pro-inflammatory cytokines, adipokines, and mitochondrial dysfunction are thought to drive insulin resistance (19). Progressive β cell failure is thought to occur due to glucotoxicity, lipotoxicity and direct cytotoxic effects from deposition of islet amyloid polypeptide (19). There is accumulating evidence that many of these mechanisms work in concert and are underpinned by low-grade activation of the innate immune system (20). This not only plays a part in the pathogenesis of T2D but is also causally linked to associated complications including dyslipidaemia and atherosclerosis (20). Elevated levels of pro-inflammatory cytokines including IL-6 and TNF-α and acute phase markers such as C-reactive protein are thought to disrupt insulin signalling although effects on glucose metabolism remain less clear (21). Humoral immunity may also play a part in the pathogenesis of T2D. Elevated serum gamma globulin levels, a nonspecific marker of humoral immune activation, have been associated with an increased risk of T2D in certain populations although the wider significance remains to be elucidated (22). Despite differences in pathophysiology, chronic hyperglycaemia is a fundamental biochemical abnormality present in both T1D and T2D, which is a key driver of aberrant neutrophil function. Increased susceptibility to infection is found in both types of diabetes, therefore this review will explore neutrophil function in the context of both T1D and T2D simultaneously.
PWD are at an increased risk of infection at various sites including skin and soft tissue (SSIs), urinary tract and the respiratory system (23, 24). Abscesses can be the first clinical presentation of diabetes in undiagnosed individuals, which occur before later vascular and neuropathic symptoms (25, 26). Infection resolution is often delayed and can lead to limb amputation in the lower extremities (27, 28). Reliance on antibiotic therapy means PWD receive increased prescriptions of antibiotics (29, 30). Antibiotic resistance is a global health concern and methicillin resistant Staphylococcus aureus (MRSA) was isolated in 15-30% of cases of diabetic foot disease (DFD), highlighting the importance of conservative antibiotic usage in this cohort and a need for new therapeutic strategies (31–33).
The innate immune system is dysregulated in both T1D and T2D (34, 35). Neutrophils are professional phagocytes of the host immune system and are critical in the clearance of pathogens, in particular S. aureus, which is the most common pathogen isolated in SSIs in PWD (36–38). Neutrophils are equipped with an arsenal of microbicidal effector functions. Upon activation in the circulation neutrophils migrate to sites of infection and inflammation by chemotaxis and respond to infection by engulfing pathogens via phagocytosis for intracellular killing by the release of cytotoxic granules and reactive oxygen species (ROS) (39). Neutrophils also release ROS, neutrophil extracellular traps (NETs) and granule proteins extracellularly in response to pathogens, all of which can damage host tissues (39, 40). Neutrophils are also central in co-ordinating the immune response to infection and produce a range of pro-inflammatory and anti-inflammatory cytokines which have autocrine and paracrine actions (39). Neutrophils rely predominantly on glucose as the sole energy source for the cell, and the impacts of hyperglycaemia and associated advanced glycation end products (AGEs) are the key causes of altered neutrophil function in T1D and T2D (41–45). T2D and obesity are also inherently linked, with increased circulating saturated fatty acids and the associated pro-inflammatory milieu also having immune-modulatory roles (46, 47).
Previous research investigating neutrophil function in T1D and T2D covers an expansive body of literature spanning 60 years, with every function of the neutrophil shown to be dysregulated in T1D or T2D. Early research in the field focused on neutrophil chemotaxis and phagocytosis, with the weight of evidence demonstrating a reduction of these functions in those with diabetes (48–51). There were some conflicting findings between early studies, perhaps caused by variations in participant selection and early experimental designs (52, 53). More recent research has predominantly focused on neutrophil ROS generation, pro-inflammatory cytokine production and aberrant neutrophil cell death mechanisms, which are proving to be critical mediators in the weakened response to infection in diabetes (54–57). Extracellular ROS production, pro-inflammatory cytokine release and NET formation are increased in diabetes, whereas neutrophil migration, apoptosis and intracellular ROS production are reduced, which ultimately impairs bacterial killing and inflammation (56, 58–61). Phenotypic variations in neutrophil function are supported by transcriptomic data, showcasing a fundamentally altered profile in key pro-inflammatory genes in neutrophils in PWD (62, 63).
Research aiming to therapeutically modify neutrophil function in response to infection in T1D or T2D lags compared to the volume of studies reporting observational differences between those with and without diabetes. However, research aiming to restore aberrant neutrophil function in diabetes is gaining momentum in the field, with a focus on modifying neutrophil ROS production and NETosis to improve infection outcomes (64–67). The enhanced susceptibility in PWD to COVID-19 infection has garnered global interest during the ongoing COVID-19 pandemic and approaches to improve neutrophil responses in people with diabetes might have important therapeutic potential (68–70). Furthermore, powerful stress responses during COVID-19 lead to hyperglycaemia and diabetic ketoacidosis among people with T2D, perhaps explaining in part their increased susceptibility to severe disease. Here, we will first explore the key drivers of neutrophil function in the diabetic microenvironment and then review key aspects of neutrophil function and how these critical functions are modified in diabetes. We then explore how these pathways have been therapeutically targeted to enhance infection clearance in diabetes, and highlight future important directions for research.
Hyperglycaemia is a key mediator of neutrophil dysfunction in T1D and T2D. Elevated blood glucose concentrations resulting from insulin insufficiency and tolerance is a core pathology of the disease. The impacts of hyperglycaemia on neutrophils are multi-factorial and present a complex interplay of dysregulated cellular mechanisms. Neutrophil metabolism is altered in response to excess glucose, to ensure intracellular glucose levels do not become toxic (42). Molecular shunting of glucose from glycolysis into the polyol and hexosamine pathway occurs (42, 71, 72). Metabolism via these pathways decreases levels of the intracellular ROS scavenger, glutathione and modifies transcription factors regulating pro-inflammatory genes (NF-κB, TGF-α, TGF-β) (42, 43, 71). Enhanced generation of cytokines further activates subsequent neutrophils, causing a feed forward loop of excessive inflammation in diabetes (73). Furthermore, hyperglycaemia causes de novo synthesis of the protein kinase C (PKC) activator, diacylglycerol (DAG), upregulating the formation of NADPH oxidase complex at the plasma membrane and leading to oxidative stress and NET formation (44, 71). Hyperglycaemia alters the osmolarity of the body fluids and hyperosmotic stress causes cell shrinkage and calcium influx into neutrophils, leading to derangements in phagocytosis and upregulation of pro-inflammatory cytokines (74, 75). High intracellular calcium concentrations deplete available ATP, impacting key energy dependant functions such as phagocytosis (74, 76). High glucose also impacts maturing neutrophils in the bone marrow. Hyperglycaemia induced myelopoiesis and leucocytosis in streptozotocin (STZ) and Akita mice (murine models of T1D) is mediated by the production of neutrophil alarmins s100 calcium proteins 8 and 9 (S1008/9) (77).
Hyperglycaemia upregulates the receptor for advanced glycation end products (RAGE) on the neutrophil cell surface (78). Advanced glycation end products (AGE) are formed from the non-enzymatic glycation of proteins (79). The pro-inflammatory impacts of AGE, which are extensively reviewed elsewhere, are of particular importance in mediating cardiovascular sequalae in diabetes (79–81). In brief, AGEs induce oxidative stress and pro-inflammatory gene expression (NF-κB) in multiple cell types, including neutrophils (42, 82, 83). AGE signals via the RAGE receptor on the neutrophil cell surface, which importantly is a multi-ligand receptor also for the alarmins S1008/9 and high-mobility group box 1 (HMGB1), further perpetuating inflammation (84, 85). Epigenetic modifications, which are the enzymatic alterations of chromatin to manipulate gene expression, were found in healthy murine macrophages co-incubated with AGE (86, 87). Increased methylation of NF-κB and enhanced cytokine transcription was subsequently found (86). Neutrophils display ‘metabolic memory’ in PWD, whereby modified cell phenotypes are maintained after the restoration of normoglycaemia, further prolonging deleterious effects (88–90). Investigation of epigenetic alterations of neutrophils in T1D and T2D is warranted to provide additional mechanistic understanding of the persisting neutrophil phenotype. Furthermore, whether hypoglycaemia or the oscillations between high and low blood glucose concentrations promotes neutrophil dysfunction is not yet known.
Glucose is not the only pro-inflammatory mediator increased in T1D and T2D. Lipid metabolism is altered in response to insulin deficiency and resistance, which increases lipogenesis and adipose tissue metabolism (91, 92). Circulating levels of free fatty acids and lipoproteins are increased in T1D and T2D, which can be further exacerbated by obesity and poor diet (93). The pro-inflammatory impacts of lipids and neutrophils are reviewed elsewhere and have shown to upregulate key pro-inflammatory neutrophil functions including cytokine generation and ROS production (91, 92, 94, 95). The negative impacts of ageing on neutrophil function is well documented, and also contributes to the neutrophil phenotype in older individuals with diabetes (96, 97).
Despite what is already known about the influence of the diabetic microenvironment on neutrophil function, there are still potential drivers yet to be explored. Complement protein, C5a, is a potent anaphylatoxin and critical mediator of inflammation (98). Recent research demonstrates C5a was increased in the plasma of PWD and in murine models of both diabetes types (99). The impact of C5a on neutrophil function has not been directly investigated in the context of diabetes previously. However, there is a strong rationale to further investigate the role of C5a as neutrophil phagocytosis, phagosomal maturation, ROS production and apoptosis were impacted by C5a mediated signalling in patients with critical illness and sepsis and similar mechanisms may be of significant importance in diabetes (98, 100–102). Key mediators of neutrophil function in diabetes are summarised in Figure 1.
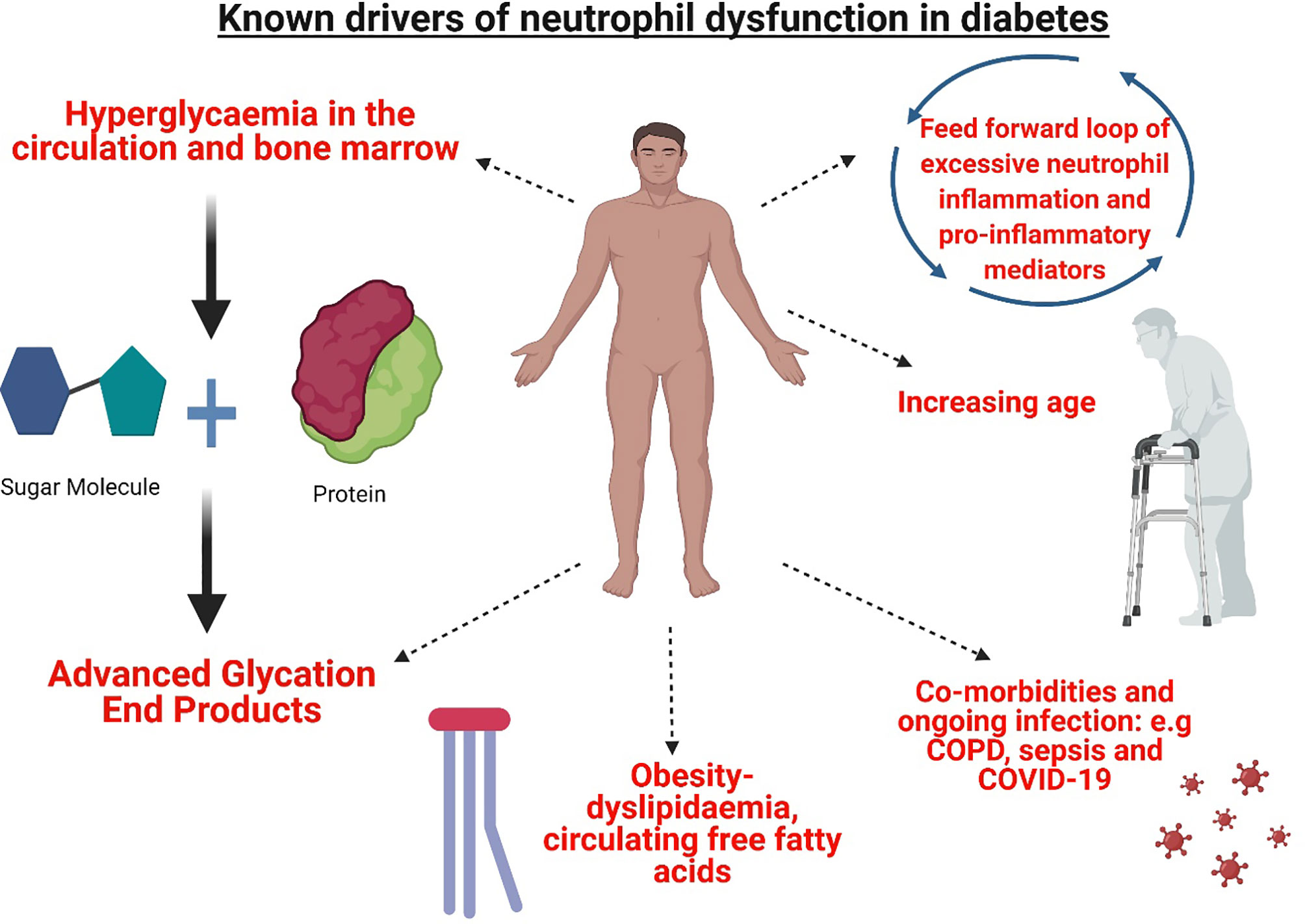
Figure 1 Mediators of neutrophil dysfunction present in T1D and T2D. The microenvironment of T1D and T2D presents a complex interplay of mediators of neutrophil dysfunction. Hyperglycaemia and the formation of advanced glycation end products in the circulation and the bone marrow modify circulating neutrophils and myeloid precursors. Metabolic perturbations in lipid metabolism and increased synthesis of circulating free fatty acids further contribute to aberrant dysfunction. Resulting activated neutrophils produce pro-inflammatory mediators adding to a cycle of inflammation. Increased age further impacts neutrophil function, in addition to co-morbidities and infection, where altered neutrophil functions are previously shown e.g chronic obstructive pulmonary disease (COPD) sepsis and COVID-19. Figure created with BioRender.com.
Neutrophil transmigration from the circulation to the tissues in response to infection and inflammation is a well described process (103). In brief, circulating neutrophils respond to tissue derived signals for infection and injury which includes chemokines, damage associated molecular patterns (DAMPs) and bacterial products. This in turn triggers their interaction with the blood vessel wall via surface ligands to endothelial cell P and L selectins, facilitating the tethering and rolling of the neutrophil across the surface of the endothelium (104, 105). Sequential activation of neutrophils stimulates expression of integrins, slowing neutrophil rolling and facilitating neutrophil crawling along the endothelial cell surface, guided by chemoattractant gradients to the source of infection (104, 105). Neutrophils transmigrate predominantly through endothelial cell junctions into the tissue interstitial space and onwards to the site of injury or infection (103).
Chronically inflamed tissue such as in peripheral arterial disease, a common sequela of T2D, notably modifies the dynamics of neutrophil migration (106). Neutrophil recruitment to infection sites was reduced in multiple animal studies exploring infections caused by a range of pathogens, thus demonstrating migration to be a fundamental defect in neutrophil function in diabetes. They found neutrophil infiltration to the peritoneal cavity was reduced in alloxan treated mice (T1D model) with polymicrobial sepsis, in addition to reduced migration to the bladder in STZ treated mice with a UTI, caused by uropathogenic Escherichia coli (107, 108). Furthermore, reduced infiltration of neutrophils was demonstrated in a S. aureus hind paw infection in leptin deficient mice (murine T2D model) (109). Reduced neutrophil migration was associated with poor infection resolution and increased mortality across a number of studies (107–109).
There are multiple causative mechanisms for aberrant neutrophil migration with both the neutrophil and the endothelium shown to be altered in the diabetic microenvironment. Internalisation of the chemokine receptor CXCR2 was associated with reduced migration in a study of sepsis in mice with alloxan induced diabetes, which is a shared neutrophil dysfunction mechanism common to non-diabetes sepsis models (107, 110). CXCR2 expression is downregulated by TLR2 signalling, which involves G protein coupled receptor kinase-2 (GRK2) (111). TLR2 is activated by high glucose concentrations, AGEs, lipoproteins and DAMPS, which are released at increased levels in diabetes (55, 112, 113). Furthermore, the serum derived acute phase protein, α1-acid glycoprotein, upregulates GRK2 and further contributes to perturbations in neutrophil migration in vivo (107). Increased α1-acid glycoprotein concentration and glycosylation were found in people with T2D or with sepsis (114, 115). Administration of insulin reduced concentrations of α1-acid glycoprotein and restored neutrophil migration in a rodent model of sepsis and alloxan induced diabetes in vivo (107). Interestingly, Perieria et al. found an unknown serum protein from alloxan treated rats which inhibited chemotaxis in vitro and the activity of such was abolished by insulin, which one could speculate was α1-acid glycoprotein, although further studies are needed to corroborate this (116).
Neutrophil chemotaxis has been widely investigated in vitro in numerous studies using animal models and volunteers with T1D or T2D, with most of the research conducted in the 1970s - 1990s (Table 1). Despite some contradictory findings, which may be accountable by variations in study and experimental design, the burden of evidence suggests that neutrophil chemotaxis in diabetes is reduced (49, 51, 122). Early research using human volunteers found no correlation between increased blood glucose concentration and aberrant neutrophil chemotaxis, suggesting a reliance on existing blood glucose lowering agents may not be sufficient to restore chemotaxis in all individuals (48, 51, 118).

Table 1 Studies investigating neutrophil chemotaxis in diabetes.
Despite a reduction in neutrophil migration and chemotaxis to infection, neutrophil activation measured by CD11b cell surface marker expression, and adhesion to the endothelium, were increased in both rodent models of diabetes and people with T2D in vitro (128–130). High glucose mediates increased neutrophil adhesion, by increasing expression of endothelial adhesion molecules (intracellular adhesion molecule-1, P-selectin and E-selectin), which was dependant on PKC signalling and nitric oxide production (131, 132). Increased neutrophil adhesion in diabetes has predominantly been investigated in the context of vascular sequalae and is therefore outside the scope of this review (129, 130, 133).
In health, ROS production in neutrophils is tightly regulated, since reduced or increased production impacts infection resolution and tissue integrity respectively (134, 135). Neutrophil ROS production, via the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, is significantly impacted by hyperglycaemia in diabetes and has been extensively studied previously in animal models, healthy volunteers, and those with type 1 or T2D (Table 2). Formation of the NADPH oxidase complex occurs at the phagosomal membrane for intracellular killing of pathogens and at the plasma membrane for extracellular ROS release (161, 162).

Table 2 Studies Investigating Neutrophil ROS production in Diabetes.
Although laboratory assays that detect extracellular ROS in isolation, such as the ferricytochrome c assay, are hampered by technical limitations, this technique has shown that superoxide production was significantly higher in those with diabetes and poor glycaemic control, compared to well controlled diabetes and healthy controls (Table 2) (59, 163). This was concomitant with a significant increase in PKC activity and DAG (59). There are previous studies showing no difference or reduced extracellular ROS production in diabetes and due to large differences in experimental design between studies it is difficult to discern the cause of conflicting evidence in the field, but may be due to differences in the ROS inducer employed and the variations in animal and human subjects used (123, 144, 145). Overall, the weight of evidence supports increased ROS production in neutrophils, as additional studies using chemiluminescent based assays that detect both intracellular and extracellular ROS, found increased levels in human subjects with T1D or T2D (151, 152, 158, 159).
Increased ROS, together with decreased activity of the ROS scavenging enzyme superoxide dismutase, is not uncommon in diabetes and the use of antioxidants to reduce excess ROS in experimental models of diabetes has been explored (158, 164, 165). The antioxidant, astaxanthin, significantly reduced extracellular ROS from neutrophils in alloxan treated rats at baseline, but not when stimulated with artificial ROS inducer Phorbol 12-myristate 13-acetate (PMA) (158). Astaxanthin also significantly lowered ROS in retinal and pancreatic cells in rats with STZ-induced diabetes and is currently undergoing clinical trials for treating diabetic retinopathy (NCT03702374) (166–168). A further antioxidant Captopril, an angiotensin-converting enzyme inhibitor, was shown to be effective in reducing ROS in human subjects with T2D and STZ-treated rats in vitro (155, 169).
Unlike extracellular ROS, intracellular ROS is most often reported to be significantly decreased in neutrophils in studies of T1D or T2D, which is thought to contribute to susceptibility to infection (Table 2) (49, 154, 170). This was demonstrated in both a model of S. aureus hind paw infection in leptin deficient diabetic mice, and a model of polymicrobial bacterial sepsis in obese diet-induced diabetic mice (model of T2D) (65, 109). Reduced levels of ROS were associated with lower bacterial clearance and increased mortality (65, 109).
Hyperglycaemia causes reduced ROS production due to the molecular shunting of excess glucose from glycolysis to the polyol pathway, which increases the requirement for NADPH and thereby reducing the availability to produce ROS (171, 172). Multiple approaches to increase neutrophil ROS production have been studied including via Tolrestat and Epalrestat, which are inhibitors of aldose reductase, a key enzyme in the polyol pathway. The inhibitors significantly increased ROS in neutrophils in both human and rodent models of diabetes (148, 171, 172). Also, enhancing ROS using granulocyte-colony stimulating factor (G-CSF) was effective in increasing ROS in multiple studies of patients with DFD (145, 173, 174). Interestingly, a Cochrane review of 5 randomised controlled trials (RCTs) with a total of 167 patients concluded that G-CSF should not be recommended as an adjuvant to current therapies for treating DFD, as it did not improve infection resolution (175). However, G-CSF treatment did reduce the need for surgical interventions and length of hospital stay in some studies. The RCTs reviewed were graded as low quality and were not statistically powered to robustly explore treatment differences. Further, large scale RCTs were recommended (175). An alternative therapeutic approach to increasing neutrophil ROS production was demonstrated in a placebo-controlled clinical study of 30 patients with T2D, using the NADPH precursor nicotinamide (149). However, until recently, there has been little, if any, further research in this area, perhaps as a result of an increased focus on reducing oxidative stress to manage diabetic complications as opposed to increasing ROS to aid pathogen handling. Nonetheless, enhancing neutrophil ROS production in sepsis is a novel clinical context where this therapeutic approach may be of value. The administration of granulocyte macrophage- colony stimulating factor (GM-CSF), significantly increased ROS production and survival in obese diabetic mice with sepsis (65). People with diabetes and sepsis have a worse prognosis than those with sepsis alone, and enhancing ROS production for acute infections, may outweigh the negative impacts of oxidative stress long term (176).
As a last resort in the anti-microbial defence strategy neutrophils undergo a programmed cell death known as NETosis, in order to capture extracellular bacteria. The release of NETs, which are extruded DNA networks decorated with antimicrobial proteins and histones, can occur via ROS dependant or ROS independent pathways (177–180). Chromatin decondensation, a key part of NETosis, is mediated by protein arginine deiminase 4 (PAD-4), which citrullinates DNA, as well as myeloperoxidase and neutrophil elastase (181–183). Elevated NETosis damages host tissue and exacerbates inflammation and is widely investigated in the pathology of multiple chronic diseases including chronic obstructive pulmonary disease (COPD) (184, 185). In diabetes, NETs are implicated in disease complications, contributing to poor infection resolution in DFD, retinopathy and cardiovascular sequalae, as well in the early pathophysiology of T1D (186–189). NETosis is increased in the presence of high glucose concentrations and is consistently shown to be upregulated in diabetes (56, 190–193). The mechanism of hyperglycaemia induced ROS production, as discussed above, also drives enhanced NETosis in diabetes and the impacts on NETosis are durable, with elevated NET levels persisting for up to a year post normalisation of blood glucose in study of people with T2D (90, 193). Membrane bound and intracellular proteases, such as neutrophil elastase, that are externalised during the process of NETosis have greater activity in people with diabetes, further contributing to the excessive inflammation observed (194). The strong association between increased NETosis and diabetes means it is a leading area in the field for the investigation of immune-modulating therapies.
Wong et al., demonstrated for the first time that neutrophils isolated from people with T1D, T2D or STZ-induced diabetic mice were primed to undergo NETosis (56). Furthermore, wound healing was impaired in STZ treated mice, which was reversed in PAD-4 knockout mice, providing a rationale for targeting PAD-4 therapeutically to reduce NETosis (56). Fadini et al. (195) showed evidence of NETosis occurring in skin lysates of diabetic mice, and that the PAD-4 inhibitor, cl-amidine, improved wound healing in STZ-treated mice (195). However, ROS dependant NETosis does not rely exclusively on PAD-4 for NETosis, therefore different approaches to reduce ROS production have also shown positive outcomes in lowering levels of NETosis (182). Targeting PKCβ2, using ruboxistaurin improved wound healing and reduced NETosis in STZ treated mice with sterile injury (64). Anti–vascular endothelial growth factor therapy was also shown to be effective in reducing ROS-dependant NETosis in retinas from STZ-treated rats, in the context of diabetic retinopathy and has yet to be explored in the context of active infection in diabetes (Wang et al., 2019).
Pathogen, rather than host therapeutic targets also show promise. A monoclonal blocking antibody to S. aureus pore-forming alpha toxin (MEDI4893), significantly reduced NETosis and S. aureus wound burden, and increased wound resolution in TALLYHO/JngJ mice (a polygenic T2D mouse model) (196). The efficacy of MEDI4893 was supported in subsequent research and provided a novel mechanism of NETosis inhibition. Low density neutrophils (LDNs) are a sub-population of neutrophils, which have an immature nuclear structure and are associated with increased NETosis in other chronic diseases such systemic lupus erythematosus (197, 198). A significant increase in the number of LDNs and neutrophils undergoing NETosis were detected in diabetic mice (both db/db, a T2D model, and STZ treated mice) following systemic S. aureus infection compared to non-diabetic control animals (66). Interestingly, neutralising S. aureus alpha-toxin with MEDI4893 inhibited TGF-β-mediated induction of LDNs and NET production, and increased animal survival (66). Improving the bactericidal capacity of NETs to improve infection resolution has also been explored. Clarithromycin increased the killing capability of NETs from people with T2D by increasing the antimicrobial cathelicidin peptide, LL-37 (199). Manipulating NETosis is an expanding area of research in the field and provides a promising future avenue for immune-modulating therapies to help reduce complications of T1D and T2D and improve handling of infections.
COVID-19 caused by the novel SARS-CoV-2 virus is a complex respiratory and multi-organ syndrome characterised by respiratory distress, hyper inflammation, and coagulation. COVID-19 disproportionately impacts the elderly and those with underlying health issues, and both T1D and T2D are associated with severe COVID-19 disease and increased mortality (200–204). In those with diabetes, a high HbA1c, suggesting chronic hyperglycaemia, upon hospital admission is an independent risk factor for poor prognosis and mortality (205–208). An increased neutrophil/lymphocyte ratio predicts poor clinical outcomes in COVID-19 patients and increased NETosis is considered a key mechanism driving airway inflammation and lung damage in this disease (68, 209–211). Serum from people with COVID-19, as well as live SARS-CoV-2, induce NETosis in neutrophils isolated from healthy donors, which in turn have the capacity to cause lung epithelial cell death (69, 209). Furthermore, circulating NET markers are high in people with COVID-19 and NETs are visualised in both lung aspirates and tissue specimens (69, 212). Developing new therapies to reduce NETosis in COVID-19 is an active area of research in the midst of the global COVID-19 pandemic (69, 213). It is possible that successful anti-NETosis therapies for COVID-19 may help other chronic diseases where NETs are implicated in the pathogenesis, with diabetes being a key candidate for this.
Neutrophils produce a range of pro-inflammatory and anti-inflammatory cytokines, which are integral to effective innate and adaptive immune responses (214). Neutrophils isolated from people with T2D generated significantly increased levels of pro-inflammatory cytokines; IL-8, TNF-α and IL-1β at both basal levels and when stimulated with LPS in vitro (215). Increased gene expression of pro-inflammatory cytokines IL-6, TNF-α and IFN-β, were demonstrated in a subsequent in vitro study of neutrophils isolated from people with T2D and good glucose control (HbA1c 6-7.5%) (216). However, there was not an elevated cytokine profile in the sub-group of patients with complications of diabetes such as DFD, neuropathy and nephropathy, irrespective of glucose control, with the authors suggesting a ‘burnt out’ neutrophil phenotype in those with severe complications, although a mechanism for this phenotype was not explored (216). Furthermore, there were no differences in serum cytokine levels between the T2D group and healthy controls (216). This finding is at odds with previous studies showing elevated pro-inflammatory cytokines (IL-1α, IL-4, IL-6) in serum and whole blood from both children and adults with either T1D or T2D (217, 218).
Increased pro-inflammatory cytokine generation can result from hyperglycaemia and AGEs, which drive ROS production and intracellular calcium concentration, activating NF-κB, and promoting the transcription of pro-inflammatory cytokines (219–221). Blood glucose lowering therapies; insulin, metformin and glibenclamide, were demonstrated to reduce neutrophil cytokine production in a rodent model and from neutrophils isolated from people with T2D (222–224). However, suppression of IL-1β production by neutrophils in patients receiving glibenclamide, was associated with enhanced susceptibility to Burkholderia pseudomallei infection in people with T2D, highlighting the need for careful consideration of unwanted side effects when seeking to modify excessive inflammation in diabetes (224). Nevertheless, the usefulness of targeting cytokine production in response to infection and inflammation is demonstrated by the efficacy of Tocilizumab, a receptor inhibitor of IL-6, used for treating rheumatoid arthritis (RA) and more recently shown to increase patient survival in severe COVID-19 (225, 226). However, limited data suggests the effectiveness of Tocilizumab was confounded in hyperglycaemic patients with COVID-19 (both with and without diabetes) and warrants further investigation to understand the potential efficacy in treating inflammation in patients with diabetes (227).
Neutrophils produce anti-inflammatory cytokines in order to downregulate inflammation. IL-1 receptor antagonist (IL-1ra) is upregulated in people with T2D compared to healthy controls, despite pro-inflammatory cytokines also being significantly increased (214, 216, 228). A RCT of 39 patients with RA and T2D were treated with anakinra, a recombinant IL-1ra (229). The primary endpoint of the study was a reduction of HbA1c, which was met with no adverse events (229). Anakinra is also under exploration for treating COVID-19 associated inflammation, with small scale trials showing clinical improvements in patients (229–231). Anakinra was demonstrated to reduce IL-1 induction of NETs in vitro, using cells isolated from people with pyogenic arthritis, pyoderma gangrenosum and acne (PAPA) syndrome and in human bronchial epithelial cells (232, 233). Owing to the importance of NETs in the pathology of diabetes complications, detailed exploration on the efficacy on anakinra in treating diabetes associated neutrophil dysfunction would be an important novel addition to the field.
Unlike NETosis, neutrophil apoptosis is an anti-inflammatory form of programmed cell death. There are two main routes to apoptosis; the extrinsic (initiated by membrane bound death receptors) and intrinsic (regulated at the mitochondrial level) pathways, which share a common mechanism of caspase mediated cell shrinkage, cytoskeleton breakdown and nuclear fragmentation (234–236). Accumulation of neutrophils at sites of inflammation, without undergoing apoptosis and clearance, causes host tissue damage and release of pro-inflammatory cytokines (237–241). Delayed neutrophil apoptosis is reported in chronic respiratory diseases such as COPD (242–244). Research in diabetes thus far presents a complex dysregulation of neutrophil apoptosis, whereby apoptosis is reduced but there is a weak response to anti-apoptotic (pro-survival) signals.
Manosudprasit et al. demonstrated reduced spontaneous apoptosis in peripheral blood neutrophils from people with T2D and those with T2D and periodontitis; a common oral infection in PWD (57). Down regulation of the key proteases involved in neutrophil apoptosis, caspases 3 and 9 were reported. Within the patient group, apoptosis was delayed significantly in those with a high HbA1C (>7.5%). However, this phenotype could not be recreated using healthy donor neutrophils incubated in high glucose media (25 mM) in vitro. Delayed neutrophil apoptosis was observed in non-obese diabetic mice (a T1D model) infected with S. aureus, which was associated with enhanced production of TNF-α (58). Elevated levels of TNF-α are implicated in the aetiology of chronic wounds in diabetes and periodontitis (245, 246). In contrast, neutrophils from people with diabetes do not have a cell survival advantage in response to lipopolysaccharide (LPS), potentially enhancing susceptibility to infection in humans (47, 247, 248). LPS is a cell wall component of Gram-negative bacteria and is a well characterised pro-survival stimulus in neutrophils (239, 249). LPS tolerance, in which cells become less responsive to LPS, is observed in Goto-Kakizaki rats (a T2D model), and mediated by impaired Toll-like-receptor 4 (TLR4) signalling (47, 250) which may have profound consequences on immune responses to infection.
Furthermore, the impact of delayed apoptosis is exacerbated in diabetes due to reduced macrophage efferocytosis (251, 252). Promoting neutrophil apoptosis as an anti-inflammatory strategy has been successfully demonstrated in experimental models of other chronic diseases including COPD and in human studies in vitro (150, 244, 253–256). However, this therapeutic approach has not been widely investigated in diabetes. Limited data demonstrates 1,25-dihydroxy-vitamin-D3 (1,25VitD3) increased apoptosis in vitro in people with T2D and periodontitis and presents an area where additional research is warranted (257).
Neutrophil phagocytosis is the engulfment and internalisation of organisms into membrane bound compartments (phagosomes) prior to pathogen killing. Phagocytosis phenotypes have been widely investigated in diabetes previously, predominantly in small scale studies using volunteers with T1D or T2D and rodent models (Table 3). The weight of evidence demonstrates a reduction in neutrophil phagocytosis in response to a variety of stimuli (123, 147, 264, 267). However, some studies report no difference in phagocytosis by neutrophils from PWD in comparison to healthy controls (49, 145, 268). The majority of studies reviewed do not recruit treatment naïve PWD, and therefore it must be assumed that some participants will be prescribed standard anti-hyperglycaemic therapies. Insulin therapy restored blood glucose and neutrophil phagocytosis, in leptin-deficient mice, highlighting the importance of accounting for current treatments when designing studies using human volunteers (271). Hyperglycaemia elevates intracellular calcium levels as a result of cell shrinkage in response to osmotic stress, which in turn dysregulates cellular signalling mechanisms required for actin rearrangement in phagocytosis (74). Inhibiting uptake of calcium, using the calcium ion channel blocker, amlodipine (a treatment for hypertension and coronary heart disease) increased neutrophil phagocytosis in patients with T2D (263, 272). Furthermore, high glucose interferes with complement protein C3 mediated opsonisation of S. aureus and Candida albicans, which could further add to reduced neutrophil phagocytosis and pathogen handling in T1D and T2D (273, 274).
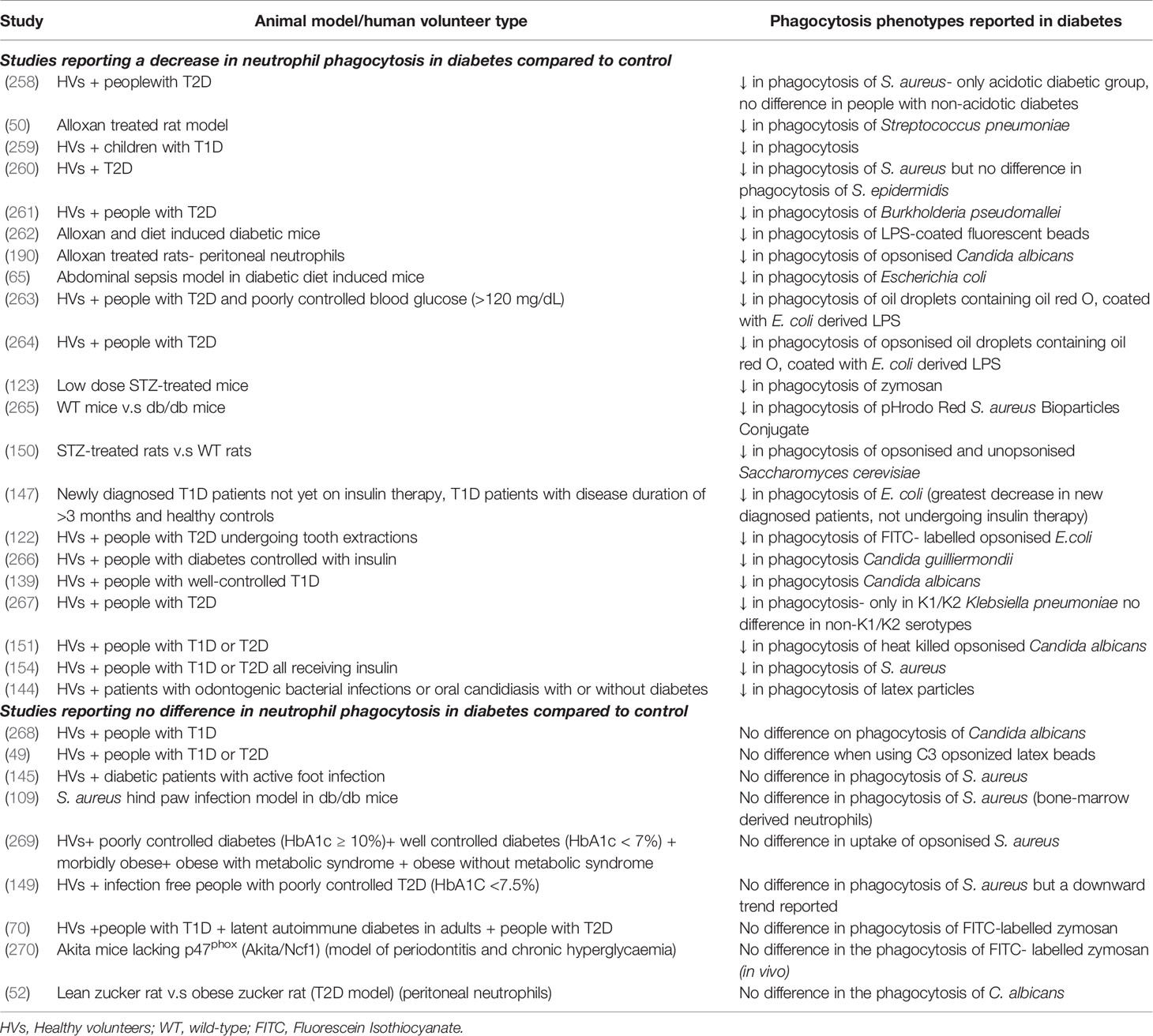
Table 3 Studies investigating neutrophil phagocytosis in diabetes.
Once phagocytosis is complete and bacteria are contained within the phagosome, phagosomal maturation occurs in order to conclude the killing process. Here the phagosome fuses with intracellular granules to form a phaglysosome, into which antimicrobial granule contents are discharged. Furthemore, the phagolysosome becomes acidified, which is required for effective pathogen killing (275). Phagosomal maturation has not been studied widely in the context of diabetes previously. Limited data in a db/db mice model, showed a significant reduction in phagosome maturation and killing of S. aureus compared to control mice and maturation was augmented by insulin treatment (271). Phagosomal maturation relies on glycolysis, and perturbations of glycolysis, mediated by pathogens including Salmonella typhimurium have been demonstrated to reduce acidification and bacterial killing in macrophages from healthy volunteers (276, 277). Reduced activities of key glycolytic enzymes (G6PDH and glutaminase) were demonstrated in STZ treated rats, and whether aberrant glycolysis is an important factor in phagosomal maturation is yet to be investigated in T1D or T2D (278). Furthermore, complement protein C5a, was shown to impact phagosomal maturation via phosphoinositide 3-kinase (PI3K) signalling in neutrophils from critically ill patients in response to S. aureus challenge in vitro (279). Further research is required to explore whether phagosomal maturation is a fundamental defect in those with T1D or T2D and to elucidate the causative mechanisms.
Iatrogenic hypoglycaemia remains one of the major challenges in the treatment of T1D and T2D (280). Data from self-reporting studies, which are likely to be underestimates, suggest people with T1D have approximately two hypoglycaemic episodes per week, with an annual incidence of severe hypoglycaemia, where third party assistance is needed, being 1.15 events per person per year in T1D versus 0.35 events per person per year in T2D (281, 282). Mechanistic studies employing the hyperinsulinaemic-hypoglycaemia clamps in both healthy individuals and those with T1D and T2D, demonstrate that acute moderate hypoglycaemia initiates a pro-longed pro-inflammatory state with upregulation of C-reactive-protein (CRP), increased platelet reactivity and mobilisation of pro-inflammatory leukocyte subsets (283–286). Additionally, in response to low endotoxin challenge in healthy volunteers, neutrophil counts were significantly increased in those allocated to experimental hypoglycaemia 48 hours earlier when compared to euglycaemic controls (284). However, whether neutrophils released into the circulation in response to hypoglycaemia have an altered function has not been widely investigated. A small-scale study compared the neutrophil oxidative burst in response to S. aureus in people with T1D versus healthy controls, after an insulin induced hypoglycaemic episode (287). A greater reduction in oxidative burst was shown in the healthy control group compared to those with T1D (287). Sub-populations of PWD are more prone to hypoglycaemic events; including older people with multiple co-morbidities such as chronic kidney and liver disease, those with a long disease duration, people treated with insulin and sulfonylureas, those with impaired awareness of hypoglycaemia and individuals with low c-peptide levels (288–290). Investigating neutrophil function in observational cohorts susceptible to hypoglycaemia in both T1D and T2D, could provide novel insights into the impacts of hypoglycaemia on neutrophil function in the free-living condition. Notwithstanding potential confounding factors from unmeasured variables, these data could be highly relevant in understanding the effects of hypoglycaemia on neutrophil function in a ‘real-world’ setting. This is because existing literature on neutrophil function from hyperinsulinaemic-clamp studies is limited by supraphysiological doses of intravenous insulin used that are almost never encountered in routine clinical practice and insulin at these levels is known to exert strong inflammatory effects (291, 292).
Increased susceptibility to recurrent and chronic infections is a key clinical characteristic of both T1D and T2D. This literature review has highlighted that there is an abundance of small-scale studies observing phenotypic changes between either human volunteers or animal models with and without diabetes, conducted over the last 60 years (summarised in Figure 2). Analysis of the breadth of literature demonstrates that neutrophils in those with T1D or T2D are fundamentally altered compared to neutrophils from healthy donors. Neutrophil effector mechanisms pertinent to infection and inflammation are aberrant in diabetes. Key neutrophil pathways critical in the response to infection (recruitment, chemotaxis, phagocytosis and intracellular ROS production) are impaired in diabetes, whereas pro-inflammatory cytokine production, extracellular ROS production, cell survival and NETosis, are upregulated, and are emerging as critical mediators of diabetic complications (47, 56, 122, 140, 147, 192). An avenue of under explored research is the investigation of tissue resident neutrophils. This is particularly important in DFD which is characterised by vascular and infectious tissue pathologies. Skin lesions from in vivo diabetes models have provided evidence for the presence of NETs in the tissue, which supports a role for the diabetic tissue environment in modifying neutrophil function (56, 195). Tissue neutrophils in other diseases have demonstrated to have tissue specific phenotypes (293, 294). For example, increased release of neutrophil elastase was shown in neutrophils isolated from bronchial lavage fluid but not in circulating neutrophils from children with cystic fibrosis (295). Phenotyping tissue neutrophils is challenging, particularly in light of their sensitivity to ex vivo manipulation and short lifespan, but doing so would allow us a greater understanding of the specific role of the tissue microenvironment in modifying neutrophil function in diabetes.
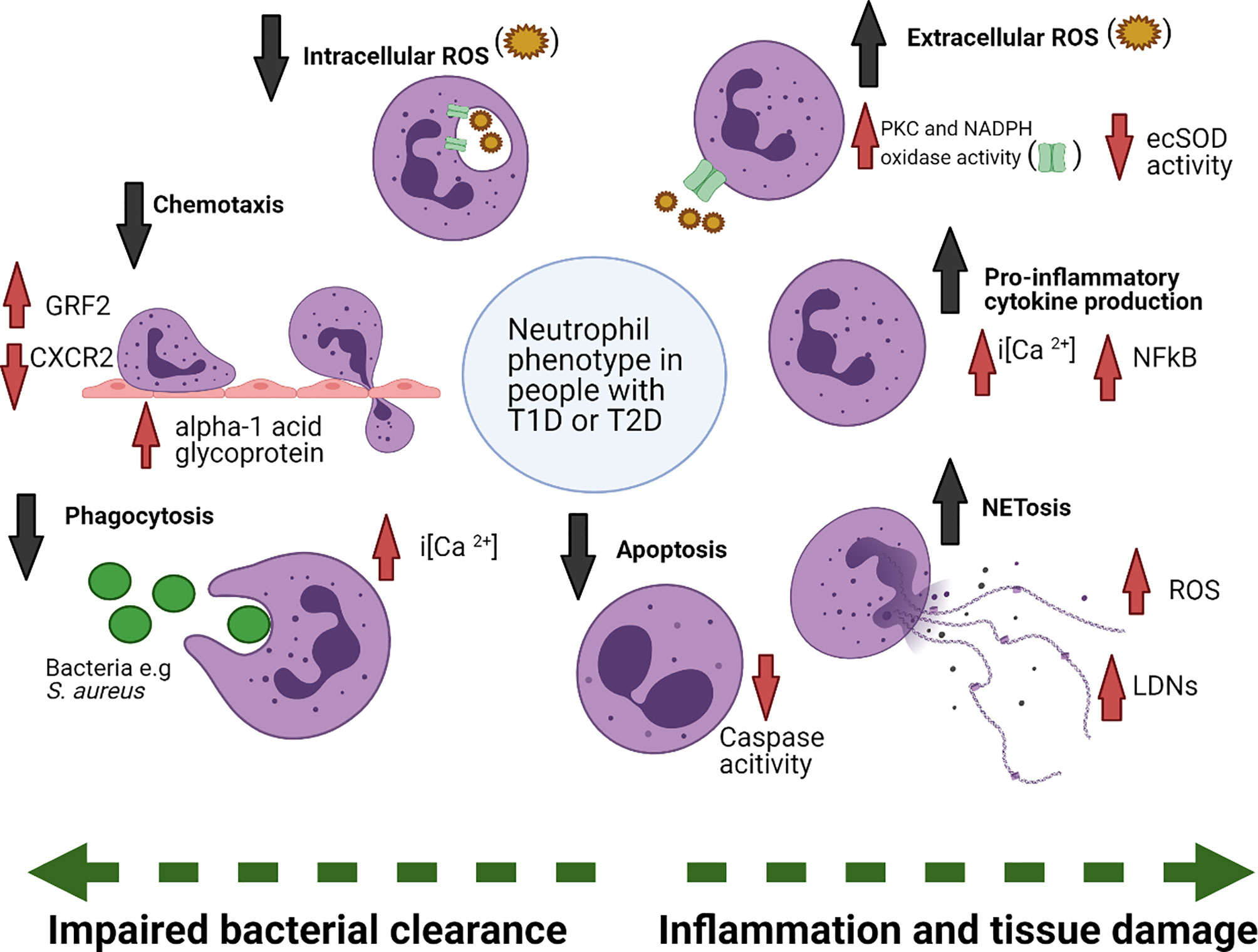
Figure 2 Summary of changes in neutrophil function in diabetes. Neutrophils in diabetes are functionally altered, due to exposure to the diabetic microenvironment, including changes to blood glucose as well as other factors. Phagocytosis, chemotaxis, intracellular ROS production and apoptosis are reduced in diabetes, whereas extracellular ROS, cytokines and NETosis are increased. Examples of mechanisms underpinning the functional changes are also noted. extracellular superoxide dismutase (ecSOD), protein kinase C (PKC), nicotinamide adenine dinucleotide phosphate (NADPH), reactive oxygen species (ROS), Nuclear factor-κB (NF-κB), low-density neutrophils (LDNs), G protein coupled receptor kinase-2 (GRK2). Figure created with BioRender.com.
Hyperglycaemia and AGEs are the key drivers of altered neutrophil function in diabetes, with dysregulated neutrophil function found in patients despite anti-hyperglycaemic therapies (90, 192). However, there are gaps in the mechanistic understanding yet to be explored. Epigenetic modification of neutrophils in T1D or T2D has not been addressed previously and could provide further insight as to how hyperglycaemia may impact neutrophil function beyond what is already understood, potentially identifying new therapeutic targets to treat dysfunction. Also, whether hypoglycaemia in diabetes can alter neutrophil function is not known. Furthermore, a pro-inflammatory neutrophil phenotype is not unique to diabetes and aberrant neutrophil function is implicated in multiple contexts including COPD and critical illness (296–298). PWD often have comorbidities which may also contribute to neutrophil dysfunction and understanding common drivers of neutrophil function maybe a useful approach for future research.
To build upon the wealth of phenotypic data already collected, future research should focus on conducting RCTs. Limited previous research shows that therapeutic modulation of dysregulated neutrophil functions can restore host immunity and improve infection resolution in diabetes (64, 65, 196). The therapeutic reduction of pro-inflammatory ROS production and NETosis is the main direction of emerging research, with positive effects on infection resolution demonstrated in small scale animal and patient research (64, 66, 158). Furthermore, the exploration of investigative therapies shown to be useful in modulating neutrophil function in other diseases such as sepsis and RA should be prioritised in diabetes. For example, anakinra, the IL-1ra antagonist targeting neutrophils in RA could be useful in reducing chronic inflammation in diabetes (232, 233). The urgency and necessity of continued research and development in the field is exemplified by the susceptibility of PWD to develop life-threatening acute respiratory distress syndrome (ARDS) with COVID-19 (299). Increased ROS and NETosis are drivers of alveolar oedema, which is characteristic of ARDS and therapies reducing these mechanisms would be of huge value (300). To conclude, future research should focus on driving forward investigation of novel experimental treatments targeting neutrophil induced oxidative stress and increased NETosis in diabetes, with the aim of conducting RCTs to translate the abundance of previous phenotypic research into effective treatments to improve the lives of people with T1D and T2D.
The following search strategy was used for this review. Literature searches were conducted using the PubMed database (1964–2020). Key word searches included ‘Diabetes’ and ‘Neutrophil’ and then either ‘Recruitment’, ‘Cytokines’ ‘Chemotaxis’,‘Phagocytosis’, ‘Reactive oxygen species’, ‘ROS’, ‘NETosis’, ‘Apoptosis’, ‘hypoglycaemia’, ‘hyperglycaemia. or ‘epigenetics’. All articles that were found using the search terms were included.
RD performed extensive literature searches, created tables and figures and wrote the first draft of the review. AI, SH, IS, and LP wrote individual sections as well as reviewed and edited the review drafts. All authors contributed to the article and approved the submitted version.
PhD Studentship (The University of Sheffield) and Medical Research Council AMR cross-council funding to the SHIELD consortium “Optimising Innate Host Defence to Combat Antimicrobial Resistance” (MRNO2995X/1). AI is supported by a National Institute for Health Research (NIHR) Academic Clinical Lectureship.
SH undertakes consultancy for Eli Lilly, Sanofi Aventis, NovoNordisk, Zealand Pharma, and have been on speaker panels for NovoNordisk and Astra Zeneca. These companies’ products have effects on hypoglycaemia when treating individuals with diabetes and are therefore related to this paper. AI has consulted for OrbiMed LLC and received educational grant support from Sanofi S.A.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Atkinson MA, Bluestone JA, Eisenbarth GS, Hebrok M, Herold KC, Accili D, et al. How Does Type 1 Diabetes Develop? The Notion of Homicide or β-Cell Suicide Revisited. Diabetes (2011) 60:1370–9. doi: 10.2337/db10-1797
2. Paschou SA, Petsiou A, Chatzigianni K, Tsatsoulis A, Papadopoulos GK. Type 1 Diabetes as an Autoimmune Disease: The Evidence. Diabetologia (2014) 57:1500–1. doi: 10.1007/s00125-014-3229-5
3. Epstein FH, Atkinson MA, Maclaren NK. The Pathogenesis of Insulin-Dependent Diabetes Mellitus. N Engl J Med (1994) 331:1428–36. doi: 10.1056/NEJM199411243312107
4. Paschou SA, Papadopoulou-Marketou N, Chrousos GP, Kanaka-Gantenbein C. On Type 1 Diabetes Mellitus Pathogenesis. Endocr Connect (2018) 7:R38–46. doi: 10.1530/EC-17-0347
5. Størling J, Pociot F. Type 1 Diabetes Candidate Genes Linked to Pancreatic Islet Cell Inflammation and Beta-Cell Apoptosis. Genes (Basel) (2017) 8:1–12. doi: 10.3390/genes8020072
6. Polonsky KS. The Past 200 Years in Diabetes. N Engl J Med (2012) 367:1332–40. doi: 10.1056/NEJMra1110560
7. Nerup J, Platz P, Andersen OO, Christy M, Lyngs o, Poulsen JE, et al. HL-a Antigens and Diabetes Mellitus. Lancet (1974) 304:864–6. doi: 10.1016/S0140-6736(74)91201-X
8. Ounissi-Benkalha H, Polychronakos C. The Molecular Genetics of Type 1 Diabetes: New Genes and Emerging Mechanisms. Trends Mol Med (2008) 14:268–75. doi: 10.1016/j.molmed.2008.04.002
9. Karlsson MGE, Ludvigsson J. The ABBOS-peptide From Bovine Serum Albumin Causes an IFN-γ and IL-4 mRNA Response in Lymphocytes From Children With Recent Onset of Type 1 Diabetes. Diabetes Res Clin Pract (2000) 47:199–207. doi: 10.1016/S0168-8227(99)00127-8
10. Szopa TM, Titchener PA, Portwood ND, Taylor KW. Diabetes Mellitus Due to Viruses - Some Recent Developments. Diabetologia (1993) 36:687–95. doi: 10.1007/BF00401138
11. Eizirik DL, Colli ML, Ortis F. The Role of Inflammation in Insulitis and B-Cell Loss in Type 1 Diabetes. Nat Rev Endocrinol (2009) 5:219–26. doi: 10.1038/nrendo.2009.21
12. Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Chase HP, et al. Prediction of Type I Diabetes in First-Degree Relatives Using a Combination of Insulin, GAD, and ICA512bdc/IA-2 Autoantibodies. Diabetes (1996) 45:926–33. doi: 10.2337/diab.45.7.926
13. DeFronzo RA. Pathogenesis of Type 2 Diabetes Mellitus. Med Clin North Am (2004) 88:787–835. doi: 10.1016/j.mcna.2004.04.013
14. Stolerman ES, Florez JC. Genomics of Type 2 Diabetes Mellitus: Implications for the Clinician. Nat Rev Endocrinol (2009) 5:429–36. doi: 10.1038/nrendo.2009.129
15. Ahlqvist E, Ahluwalia TS, Groop L. Genetics of Type 2 Diabetes. Clin Chem (2011) 57:241–54. doi: 10.1373/clinchem.2010.157016
16. Pierce M, Keen H, Bradley C. Risk of Diabetes in Offspring of Parents With Non-insulin-dependent Diabetes. Diabetes Med (1995) 12:6–13. doi: 10.1111/j.1464-5491.1995.tb02054.x
17. Tattersall RB, Fajans SS. Prevalence of Diabetes and Glucose Intolerance in 199 Offspring of Thirty Seven Conjugal Diabetic Parents. Diabetes (1975) 24:452–62. doi: 10.2337/diab.24.5.452
18. Reaven GM. Why Syndrome X? From Harold Himsworth to the Insulin Resistance Syndrome. Cell Metab (2005) 1:9–14. doi: 10.1016/j.cmet.2004.12.001
19. Stumvoll M, Goldstein BJ, Van Haeften TW. Type 2 Diabetes: Principles of Pathogenesis and Therapy. Lancet (Elsevier BV) (2005) 365(9467):1333–46. doi: 10.1016/S0140-6736(05)61032-X
20. Pickup JC. Inflammation and Activated Innate Immunity in the Pathogenesis of Type 2 Diabletes. Diabetes Care (2004) 27:813–23. doi: 10.2337/diacare.27.3.813
21. Steensberg A, Fischer CP, Sacchetti M, Keller C, Osada T, Schjerling P, et al. Acute Interleukin-6 Administration Does Not Impair Muscle Glucose Uptake or Whole-Body Glucose Disposal in Healthy Humans. J Physiol (2003) 548:631–8. doi: 10.1113/jphysiol.2002.032938
22. Lindsay RS, Krakoff J, Hanson RL, Bennett PH, Knowler WC. Gamma Globulin Levels Predict Type 2 Diabetes in the Pima Indian Population. Diabetes (2001) 50:1598–603. doi: 10.2337/diabetes.50.7.1598
23. Carey IM, Critchley JA, DeWilde S, Harris T, Hosking FJ, Cook DG. Risk of Infection in Type 1 and Type 2 Diabetes Compared With the General Population: A Matched Cohort Study. Diabetes Care (2018) 41:513–21. doi: 10.2337/dc17-2131
24. Ndosi M, Wright-Hughes A, Brown S, Backhouse M, Lipsky BA, Bhogal M, et al. Prognosis of the Infected Diabetic Foot Ulcer: A 12-Month Prospective Observational Study. Diabetes Med (2018) 35:78–88. doi: 10.1111/dme.13537
25. Wei PL, Keller JJ, Kuo LJ, Lin HC. Increased Risk of Diabetes Following Perianal Abscess: A Population-Based Follow-Up Study. Int J Color Dis (2013) 28:235–40. doi: 10.1007/s00384-012-1519-2
26. Alagl AS. Periodontal Abscess as a Possible Oral Clinical Sign in the Diagnosis of Undiagnosed Diabetes Mellitus of Elderly in a Dental Clinic Set Up – A 7-Year Cross-Sectional Study. J Investig Clin Dent (2017) 8:e12217. doi: 10.1111/jicd.12217
27. Rodrigues BT, Vangaveti VN, Malabu UH. Prevalence and Risk Factors for Diabetic Lower Limb Amputation: A Clinic-Based Case Control Study. J Diabetes Res (2016), 1–7. doi: 10.1155/2016/5941957
28. Frydrych LM, Fattahi F, He K, Ward PA, Delano MJ. Diabetes and Sepsis: Risk, Recurrence, and Ruination. Front Endocrinol (Lausanne) (2017) 8:271. doi: 10.3389/fendo.2017.00271
29. Venmans LMAJ, Hak E, Gorter KJ, Rutten GEHM. Incidence and Antibiotic Prescription Rates for Common Infections in Patients With Diabetes in Primary Care Over the Years 1995 to 2003. Int J Infect Dis (2009) 13:e344–51. doi: 10.1016/j.ijid.2008.12.003
30. Barwell ND, Devers MC, Kennon B, Hopkinson HE, McDougall C, Young MJ, et al. Diabetic Foot Infection: Antibiotic Therapy and Good Practice Recommendations. Int J Clin Pract (2017) 71:e13006. doi: 10.1111/ijcp.13006
31. Trivedi U, Parameswaran S, Armstrong A, Burgueno-Vega D, Griswold J, Dissanaike S, et al. Prevalence of Multiple Antibiotic Resistant Infections in Diabetic Versus Nondiabetic Wounds. J Pathog (2014) 2014:173053. doi: 10.1155/2014/173053
32. Reveles KR, Duhon BM, Moore RJ, Hand EO, Howell CK. Epidemiology of Methicillin-Resistant Staphylococcus Aureus Diabetic Foot Infections in a Large Academic Hospital: Implications for Antimicrobial Stewardship. PloS One (2016) 11:e0161658. doi: 10.1371/journal.pone.0161658
33. Stacey HJ, Clements CS, Welburn SC, Jones JD. The Prevalence of Methicillin-Resistant Staphylococcus Aureus Among Diabetic Patients: A Meta-Analysis. Acta Diabetol (2019) 56:907–21. doi: 10.1007/s00592-019-01301-0
34. Donath MY, Dinarello CA, Mandrup-Poulsen T. Targeting Innate Immune Mediators in Type 1 and Type 2 Diabetes. Nat Rev Immunol (2019) 19:734–46. doi: 10.1038/s41577-019-0213-9
35. Drucker DJ. Coronavirus Infections and Type 2 Diabetes-Shared Pathways With Therapeutic Implications. Endocr Rev (2020) 41:457–70. doi: 10.1210/endrev/bnaa011
36. Jenkins TC, Knepper BC, Moore SJ, Saveli CC, Pawlowski SW, Perlman DM, et al. Comparison of the Microbiology and Antibiotic Treatment Among Diabetic and Nondiabetic Patients Hospitalized for Cellulitis or Cutaneous Abscess. J Hosp Med (2014) 9:788–94. doi: 10.1002/jhm.2267
37. Lipsky BA, Tabak YP, Johannes RS, Vo L, Hyde L, Weigelt JA. Skin and Soft Tissue Infections in Hospitalised Patients With Diabetes: Culture Isolates and Risk Factors Associated With Mortality, Length of Stay and Cost. Diabetologia (2010) 53:914–23. doi: 10.1007/s00125-010-1672-5
38. Buvelot H, Posfay-Barbe KM, Linder P, Schrenzel J, Krause KH. Staphylococcus Aureus, Phagocyte NADPH Oxidase and Chronic Granulomatous Disease. FEMS Microbiol Rev (2017) 41:139–57. doi: 10.1093/femsre/fuw042
39. Liew PX, Kubes P. The Neutrophil’s Role During Health and Disease. Physiol Rev (2019) 99:1223–48. doi: 10.1152/physrev.00012.2018
40. Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular Mechanisms of Netosis. Annu Rev Cell Dev Biol (2020) 3:41. doi: 10.1146/annurev-cellbio-020520-111016
41. Burns JS, Manda G. Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance. Int J Mol Sci (2017) 18:2755. doi: 10.3390/ijms18122755
42. Giacco F, Brownlee M. Oxidative Stress and Diabetic Complications. Circ Res (2010) 107:1058–70. doi: 10.1161/circresaha.110.223545
43. James LR, Tang D, Ingram A, Ly H, Thai K, Cai L, et al. Flux Through the Hexosamine Pathway Is a Determinant of Nuclear Factor κB– Dependent Promoter Activation. Diabetes (2002) 51:1146–56. doi: 10.2337/diabetes.51.4.1146
44. Xia P, Inoguchi T, Kern TS, Engerman RL, Oates PJ, King GL. Characterization of the Mechanism for the Chronic Activation of Diacylglycerol-Protein Kinase C Pathway in Diabetes and Hypergalactosemia. Diabetes (1994) 43:1122–9. doi: 10.2337/diab.43.9.1122
45. Yan SDFDF, Yan SDFDF, Ramasamy R, Schmidt AM. Tempering the Wrath of RAGE: An Emerging Therapeutic Strategy Against Diabetic Complications, Neurodegeneration, and Inflammation. Ann Med (2009) 41:408–22. doi: 10.1080/07853890902806576
46. Rogero MM, Calder PC. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients (2018) 10:432. doi: 10.3390/nu10040432
47. Kuwabara WMT, Yokota CNF, Curi R, Alba-Loureiro TC. Obesity and Type 2 Diabetes Mellitus Induce Lipopolysaccharide Tolerance in Rat Neutrophils. Sci Rep (2018) 8:17534. doi: 10.1038/s41598-018-35809-2
48. Mowat AG, Baum J. Chemotaxis of Polymorphonuclear Leukocytes From Patients With Diabetes Mellitus. N Engl J Med (1971) 284:621–. doi: 10.1056/nejm197103252841201
49. Delamaire M, Maugendre D, Moreno M, Le Goff M-C, Allannic H, Genetet B. Impaired Leucocyte Functions in Diabetic Patients. Diabetes Med (1997) 14:29–34. doi: 10.1002/(SICI)1096-9136(199701)14:1<29::AID-DIA300>3.0.CO;2-V
50. Drachman RH, Root RK, Wood WB Jr. Studies on the Effect of Experimental Nonketotic Diabetes Mellitus on Antibacterial Defense. I. Demonstration of a Defect in Phagocytosis. J Exp Med (1966) 124:227–40. doi: 10.1084/jem.124.2.227
51. Gustke CJ, Stein SH, Hart TC, Hoffman WH, Hanes PJ, Russell CM, et al. HLA-DR Alleles are Associated With IDDM, But Not With Impaired Neutrophil Chemotaxis in IDDM. J Dent Res (1998) 77:1497–503. doi: 10.1177/00220345980770070401
52. Plotkin BJ, Paulson D, Chelich A, Jurak D, Cole J, Kasimos J, et al. Immune Responsiveness in a Rat Model for Type II Diabetes (Zucker Rat, fa/fa): Susceptibility to Candida Albicans Infection and Leucocyte Function. J Med Microbiol (1996) 44:277–83. doi: 10.1099/00222615-44-4-277
53. Donovan RM, Goldstein E, Kim Y, Lippert W, Kailath E, Aoki TT, et al. A Computer- Assisted Image-Analysis System for Analyzing Polymorphonuclear Leukocyte Chemotaxis in Patients With Diabetes-Mellitus. J Infect Dis (1987) 155:737–41. doi: 10.1093/infdis/155.4.737
54. Hand WL, Hand DL, Vasquez Y. Increased Polymorphonuclear Leukocyte Respiratory Burst Function in Type 2 Diabetes. Diabetes Res Clin Pract (2007) 76:44–50. doi: 10.1016/j.diabres.2006.07.015
55. Gyurko R, Siqueira CC, Caldon N, Gao L, Kantarci A, Van Dyke TE. Chronic Hyperglycemia Predisposes to Exaggerated Inflammatory Response and Leukocyte Dysfunction in Akita Mice. J Immunol (2006) 177:7250–6. doi: 10.4049/jimmunol.177.10.7250
56. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes Primes Neutrophils to Undergo NETosis, Which Impairs Wound Healing. Nat Med (2015) 21:815–9. doi: 10.1038/nm.3887
57. Manosudprasit A, Kantarci A, Hasturk H, Stephens D, Van Dyke TE. Spontaneous PMN Apoptosis in Type 2 Diabetes and the Impact of Periodontitis. J Leukoc Biol (2017) 102:1431–40. doi: 10.1189/jlb.4A0416-209RR
58. Hanses F, Park S, Rich J, Lee JC. Reduced Neutrophil Apoptosis in Diabetic Mice During Staphylococcal Infection Leads to Prolonged Tnfα Production and Reduced Neutrophil Clearance. PloS One (2011) 6:e23633. doi: 10.1371/journal.pone.0023633
59. Karima M, Kantarci A, Ohira T, Hasturk H, Jones VL, Nam BH, et al. Enhanced Superoxide Release and Elevated Protein Kinase C Activity in Neutrophils From Diabetic Patients: Association With Periodontitis. J Leukoc Biol (2005) 78:862–70. doi: 10.1189/jlb.1004583
60. Javid A, Zlotnikov N, Pětrošová H, Tang TT, Zhang Y, Bansal AK, et al. Hyperglycemia Impairs Neutrophil-Mediated Bacterial Clearance in Mice Infected With the Lyme Disease Pathogen. PloS One (2016) 11:1–20. doi: 10.1371/journal.pone.0158019
61. Repine JE, Clawson CC, Goetz FC. Bactericidal Function of Neutrophils From Patients With Acute Bacterial Infections and From Diabetics. J Infect Dis (1980) 142:869–75. doi: 10.1093/infdis/142.6.869
62. Vecchio F, Buono N, Stabilini A, Nigi L, Dufort MJ, Geyer S, et al. Abnormal Neutrophil Signature in the Blood and Pancreas of Presymptomatic and Symptomatic Type 1 Diabetes Federica Vecchio, …, the Type 1 Diabetes Trialnet Study Group, Manuela Battaglia Find the Latest Version : Abnormal Neutrophil Signature in the Bl. JCI Insight (2018) 3:1–17. doi: 10.1172/jci.insight.122146
63. Fang X, Dorcely B, Ding X-P, Yin S, Son N-H, Hu S-L, et al. Glycemic Reduction Alters White Blood Cell Counts and Inflammatory Gene Expression in Diabetes. J Diabetes Complications (2018) 32:1027–34. doi: 10.1016/j.jdiacomp.2018.08.003
64. Das SK, Yuan YF, Li MQ. Specific PKC betaII Inhibitor: One Stone Two Birds in the Treatment of Diabetic Foot Ulcers. Biosci Rep (2018) 38:1573–4935. doi: 10.1042/BSR20171459
65. Frydrych LM, Bian G, Fattahi F, Morris SB, O’Rourke RW, Lumeng CN, et al. Gm-Csf Administration Improves Defects in Innate Immunity and Sepsis Survival in Obese Diabetic Mice. J Immunol (2018) 202:931–42. doi: 10.4049/jimmunol.1800713
66. Cohen TS, Takahashi V, Bonnell J, Tovchigrechko A, Chaerkady R, Yu W, et al. Staphylococcus Aureus Drives Expansion of Low-Density Neutrophils in Diabetic Mice. J Clin Invest (2019) 129:2133–44. doi: 10.1172/JCI126938
67. Wang L, Zhou X, Yin Y, Mai Y, Wang D, Zhang X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front Immunol (2019) 10:3076. doi: 10.3389/fimmu.2018.03076
68. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting Potential Drivers of COVID-19: Neutrophil Extracellular Traps. J Exp Med (2020) 217:e20200652. doi: 10.1084/jem.20200652
69. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. Sars-CoV-2-triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J Exp Med (2020) 217:1–12. doi: 10.1084/jem.20201129
70. Huang J, Xiao Y, Zheng PL, Zhou WZ, Wang YF, Huang G, et al. Distinct Neutrophil Counts and Functions in Newly Diagnosed Type 1 Diabetes, Latent Autoimmune Diabetes in Adults, and Type 2 Diabetes. Diabetes Metab Res Rev (2019) 35:10. doi: 10.1002/dmrr.3064
71. Kashiwagi A, Asahina T, Ikebuchi M, Tanaka Y, Takagi Y, Nishio Y, et al. Abnormal Glutathione Metabolism and Increased Cytotoxicity Caused by H2O2 in Human Umbilical Vein Endothelial Cells Cultured in High Glucose Medium. Diabetologia (1994) 37:264–9. doi: 10.1007/BF00398053
72. Berrone E, Beltramo E, Solimine C, Ape AU, Porta M. Regulation of Intracellular Glucose and Polyol Pathway by Thiamine and Benfotiamine in Vascular Cells Cultured in High Glucose. J Biol Chem (2006) 281:9307–13. doi: 10.1074/jbc.M600418200
73. Guest CB, Park MJ, Johnson DR, Freund GG. The Implication of Proinflammatory Cytokines in Type 2 Diabetes. Front Biosci (2008) 13:5187–94. doi: 10.2741/3074
74. Demerdash TM, Seyrek N, Smogorzewski M, Marcinkowski W, Nasser-Moadelli S, Massry SG. Pathways Through Which Glucose Induces a Rise in [Ca2+]i of Polymorphonuclear Leukocytes of Rats. Kidney Int (1996) 50:2032–40. doi: 10.1038/ki.1996.526
75. Otto NM, Schindler R, Lun A, Boenisch O, Frei U, Oppert M. Hyperosmotic Stress Enhances Cytokine Production and Decreases Phagocytosis In Vitro. Crit Care (2008) 12:R107. doi: 10.1186/cc6989
76. Guerrero-Hernandez A, Verkhratsky A. Calcium Signalling in Diabetes. Cell Calcium (2014) 56:297–301. doi: 10.1016/j.ceca.2014.08.009
77. Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, et al. Hyperglycemia Promotes Myelopoiesis and Impairs the Resolution of Atherosclerosis. Cell Metab (2013) 17:695–708. doi: 10.1016/j.cmet.2013.04.001
78. Lee RH, Bergmeier W. Sugar Makes Neutrophils RAGE: Linking Diabetesassociated Hyperglycemia to Thrombocytosis and Platelet Reactivity. J Clin Invest (2017) 127:2040–3. doi: 10.1172/JCI94494
79. Yan Shi F, Ramasamy R, Naka Y, Schmidt Ann M. Glycation, Inflammation, and RAGE. Circ Res (2003) 93:1159–69. doi: 10.1161/01.RES.0000103862.26506.3D
80. Nowotny K, Jung T, Höhn A, Weber D, Grune T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules (2015) 5:194–222. doi: 10.3390/biom5010194
81. Hudson BI, Lippman ME. Targeting RAGE Signaling in Inflammatory Disease. Annu Rev Med (2018) 69:349–64. doi: 10.1146/annurev-med-041316-085215
82. Collison KS, Parhar RS, Saleh SS, Meyer BF, Kwaasi AA, Hammami MM, et al. RAGE-Mediated Neutrophil Dysfunction is Evoked by Advanced Glycation End Products (Ages). J Leukoc Biol (2002) 71:433–44. doi: 10.1189/jlb.71.3.433
83. Wong RKM, Pettit AI, Quinn PA, Jennings SC, Davies JE, Ng LL. Advanced Glycation End Products Stimulate an Enhanced Neutrophil Respiratory Burst Mediated Through the Activation of Cytosolic Phospholipase A2 and Generation of Arachidonic Acid. Circulation (2003) 108:1858–64. doi: 10.1161/01.CIR.0000089372.64585.3B
84. Ramasamy R, Yan SF, Schmidt AM, Receptor for AGE. (RAGE): Signaling Mechanisms in the Pathogenesis of Diabetes and its Complications. Ann N Y Acad Sci (2011) 1243:88–102. doi: 10.1111/j.1749-6632.2011.06320.x
85. Egaña-Gorroño L, López-Díez R, Yepuri G, Ramirez LS, Reverdatto S, Gugger PF, et al. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights From Human Subjects and Animal Models. Front Cardiovasc Med (2020) 7:37. doi: 10.3389/fcvm.2020.00037
86. Jin X, Yao T, Zhou Z, Zhu J, Zhang S, Hu W, et al. Advanced Glycation End Products Enhance Macrophages Polarization Into M1 Phenotype Through Activating RAGE/NF-B Pathway. BioMed Res Int (2015) 2015:1–12. doi: 10.1155/2015/732450
87. Ahmed M, de Winther MPJ, Van den Bossche J. Epigenetic Mechanisms of Macrophage Activation in Type 2 Diabetes. Immunobiology (2017) 222:937–43. doi: 10.1016/j.imbio.2016.08.011
88. Corgnali M, Piconi L, Ihnat M, Ceriello A. Evaluation of Gliclazide Ability to Attenuate the Hyperglycaemic “Memory” Induced by High Glucose in Isolated Human Endothelial Cells. Diabetes Metab Res Rev (2008) 24:301–9. doi: 10.1002/dmrr.804
89. Ceriello A, Ihnat MA, Thorpe JE. The “Metabolic Memory”: Is More Than Just Tight Glucose Control Necessary to Prevent Diabetic Complications? J Clin Endocrinol Metab (2009) 94:410–5. doi: 10.1210/jc.2008-1824
90. Carestia A, Frechtel G, Cerrone G, Linari MA, Gonzalez CD, Casais P, et al. Netosis Before and After Hyperglycemic Control in Type 2 Diabetes Mellitus Patients. PloS One (2016) 11:e0168647–e0168647. doi: 10.1371/journal.pone.0168647
91. Rodrigues HG, Takeo Sato F, Curi R, Vinolo MAR. Fatty Acids as Modulators of Neutrophil Recruitment, Function and Survival. Eur J Pharmacol (2016) 785:50–8. doi: 10.1016/j.ejphar.2015.03.098
92. Eid S, Sas KM, Abcouwer SF, Feldman EL, Gardner TW, Pennathur S, et al. New Insights Into the Mechanisms of Diabetic Complications: Role of Lipids and Lipid Metabolism. Diabetologia (2019) 62:1539–49. doi: 10.1007/s00125-019-4959-1
93. Radzikowska U, Rinaldi AO, Sözener ZÇ, Karaguzel D, Wojcik M, Cypryk K, et al. The Influence of Dietary Fatty Acids on Immune Responses. Nutrients (2019) 11:2990. doi: 10.3390/nu11122990
94. Jüttner B, Kröplin J, Coldewey SM, Witt L, Osthaus WA, Weilbach C, et al. Unsaturated Long-Chain Fatty Acids Induce the Respiratory Burst of Human Neutrophils and Monocytes in Whole Blood. Nutr Metab (2008) 5:19. doi: 10.1186/1743-7075-5-19
95. Palvinskaya T, Antkowiak M, Burg E, Lenox CC, Ubags N, Cramer A, et al. Effects of Acute and Chronic Low Density Lipoprotein Exposure on Neutrophil Function. Pulm Pharmacol Ther (2013) 26:405–11. doi: 10.1016/j.pupt.2012.10.002
96. Wenisch C, Patruta S, Daxböck F, Krause R, Hörl W. Effect of Age on Human Neutrophil Function. J Leukoc Biol (2000) 67:40–5. doi: 10.1002/jlb.67.1.40
97. Sapey E, Patel JM, Greenwood HL, Walton GM, Hazeldine J, Sadhra C, et al. Pulmonary Infections in the Elderly Lead to Impaired Neutrophil Targeting, Which is Improved by Simvastatin. Am J Respir Crit Care Med (2017) 196:1325–36. doi: 10.1164/rccm.201704-0814OC
98. Wood AJT, Vassallo A, Summers C, Chilvers ER, Conway-Morris A. C5a Anaphylatoxin and its Role in Critical Illness-Induced Organ Dysfunction. Eur J Clin Invest (2018) 48:e13028. doi: 10.1111/eci.13028
99. Tan SM, Ziemann M, Thallas-Bonke V, Snelson M, Kumar V, Laskowski A, et al. Complement C5a Induces Renal Injury in Diabetic Kidney Disease by Disrupting Mitochondrial Metabolic Agility. Diabetes (2020) 69:83–98. doi: 10.2337/db19-0043
100. Huber-Lang MS, Younkin EM, Sarma JV, McGuire SR, Lu KT, Guo RF, et al. Complement-Induced Impairment of Innate Immunity During Sepsis. J Immunol (2002) 169:3223–31. doi: 10.4049/jimmunol.169.6.3223
101. Conway Morris A, Kefala K, Wilkinson TS, Dhaliwal K, Farrell L, Walsh T, et al. C5a Mediates Peripheral Blood Neutrophil Dysfunction in Critically Ill Patients. Am J Respir Crit Care Med (2009) 180:19–28. doi: 10.1164/rccm.200812-1928OC
102. Unnewehr H, Rittirsch D, Sarma JV, Zetoune F, Flierl MA, Perl M, et al. Changes and Regulation of the C5a Receptor on Neutrophils During Septic Shock in Humans. J Immunol (2013) 190:4215–25. doi: 10.4049/jimmunol.1200534
103. Nourshargh S, Alon R. Leukocyte Migration Into Inflamed Tissues. Immunity (2014) 41:694–707. doi: 10.1016/j.immuni.2014.10.008
104. Hyun Y, Hong C. Deep Insight Into Neutrophil Trafficking in Various Organs. J Leukoc Biol (2017) 102:617–29. doi: 10.1189/jlb.1ru1216-521r
105. Maas SL, Soehnlein O, Viola JR. Organ-Specific Mechanisms of Transendothelial Neutrophil Migration in the Lung, Liver, Kidney, and Aorta. Front Immunol (2018) 9:2739. doi: 10.3389/fimmu.2018.02739
106. Ma B, Whiteford JR, Nourshargh S, Woodfin A. Underlying Chronic Inflammation Alters the Profile and Mechanisms of Acute Neutrophil Recruitment. J Pathol (2016) 240:291–303. doi: 10.1002/path.4776
107. Spiller F, Carlos D, Souto FO, De Freitas A, Soares FS, Vieira SM, et al. α1-Acid Glycoprotein Decreases Neutrophil Migration and Increases Susceptibility to Sepsis in Diabetic Mice. Diabetes (2012) 61:1584–91. doi: 10.2337/db11-0825
108. Ozer A, Altuntas CZ, Bicer F, Izgi K, Hultgren SJ, Liu G, et al. Impaired Cytokine Expression, Neutrophil Infiltration and Bacterial Clearance in Response to Urinary Tract Infection in Diabetic Mice. Pathog Dis (2015) 73:2. doi: 10.1093/femspd/ftv002
109. Park S, Rich J, Hanses F, Lee JC. Defects in Innate Immunity Predispose C57BL/6J-Lepr(Db)/Lepr(Db) Mice to Infection by Staphylococcus Aureus. Infect Immun (2009) 77:1008–14. doi: 10.1128/iai.00976-08
110. Seree-aphinan C, Vichitkunakorn P, Navakanitworakul R, Khwannimit B. Distinguishing Sepsis From Infection by Neutrophil Dysfunction: A Promising Role of CXCR2 Surface Level. Front Immunol (2020) 11:608696. doi: 10.3389/fimmu.2020.608696
111. Alves-Filho JC, Freitas A, Souto FO, Spiller F, Paula-Neto H, Silva JS, et al. Regulation of Chemokine Receptor by Toll-like Receptor 2 is Critical to Neutrophil Migration and Resistance to Polymicrobial Sepsis. Proc Natl Acad Sci U.S.A. (2009) 106:4018–23. doi: 10.1073/pnas.0900196106
112. Dasu MR, Devaraj S, Zhao L, Hwang DH, Jialal I. High Glucose Induces Toll-Like Receptor Expression in Human Monocytes Mechanism of Activation. Diabetes (2008) 57:3090–8. doi: 10.2337/db08-0564
113. Dasu MR, Devaraj S, Park S, Jialal I. Increased Toll-Like Receptor (TLR) Activation and TLR Ligands in Recently Diagnosed Type 2 Diabetic Subjects. Diabetes Care (2010) 33:861–8. doi: 10.2337/dc09-1799
114. Higai K, Azuma Y, Aoki Y, Matsumoto K. Altered Glycosylation of α1-Acid Glycoprotein in Patients With Inflammation and Diabetes Mellitus. Clin Chim Acta (2003) 329:117–25. doi: 10.1016/S0009-8981(02)00427-8
115. Mestriner FLAC, Spiller F, Laure HJ, Souto FO, Tavares-Murta BM, Rosa JC, et al. Acute-Phase Protein α-1-acid Glycoprotein Mediates Neutrophil Migration Failure in Sepsis by a Nitric Oxide-Dependent Mechanism. Proc Natl Acad Sci U.S.A. (2007) 104:19595–600. doi: 10.1073/pnas.0709681104
116. Pereira MAA, Sannomiya P, Leme JG. Inhibition of Leukocyte Chemotaxis by Factor in Alloxan-Induced Diabetic Rat Plasma. Diabetes (1987) 36:1307–14. doi: 10.2337/diab.36.11.1307
117. Miller M, Baker L. Leukocyte Functions in Juvenile Diabetes Mellitus: Humoral and Cellular Aspects. J Pediatr (1972) 81:979–82. doi: 10.1016/S0022-3476(72)80555-9
118. Manouchehr-Pour M, Spagnuolo PJ, Rodman HM, Bissada NF. Comparison of Neutrophil Chemotactic Response in Diabetic Patients With Mild and Severe Periodontal Disease. J Periodontol (1981) 52:410–5. doi: 10.1902/jop.1981.52.8.410
119. Wierusz-Wysocka B, Wysocki H, Siekierka H, Wykretowicz A, Szczepanik A, Klimas R. Evidence of Polymorphonuclear Neutrophils (PMN) Activation in Patients With Insulin-Dependent Diabetes Mellitus. J Leukoc Biol (1987) 42:519–23. doi: 10.1002/jlb.42.5.519
120. Özsoy N, Bostancı H, Ayvalı C. The Investigation of the Ultrastructural Neutrophil Changes in Alloxan-Induced Diabetes in Rats: Response to a Chemotactic Challenge. Cell Biochem Funct (2004) 22:81–7. doi: 10.1002/cbf.1059
121. Sannomiya P, Pereira MAA, Garcia-Leme J. Inhibition of Leukocyte Chemotaxis by Serum Factor in Diabetes Mellitus: Selective Depression of Cell Responses Mediated by Complement-Derived Chemoattractants. Agents Actions (1990) 30:369–76. doi: 10.1007/BF01966301
122. Fernandes KS, Glick M, De Souza MS, Kokron CM, Gallottini M. Association Between Immunologic Parameters, Glycemic Control, and Postextraction Complications in Patients With Type 2 Diabetes. J Am Dent Assoc (2015) 146:592–9. doi: 10.1016/j.adaj.2015.02.014
123. Kannan Y, Tokunaga M, Moriyama M, Kinoshita H, Nakamura Y. Beneficial Effects of Troglitazone on Neutrophil Dysfunction in Multiple Low-Dose Streptozotocin-Induced Diabetic Mice. Clin Exp Immunol (2004) 137:263–71. doi: 10.1111/j.1365-2249.2004.02532.x
124. Fikrig SM, Reddy CM, Orti E, Herod L, Suntharalingam K. Diabetes and Neutrophil Chemotaxis. Diabetes (1977) 26:466–8. doi: 10.2337/diab.26.5.466
125. De Toni S, Piva E, Lapolla A, Fontana G, Fedele D, Plebani M. Respiratory Burst of Neutrophils in Diabetic Patients With Periodontal Disease. Ann New York Acad Sci (1997) 832:363–7. doi: 10.1111/j.1749-6632.1997.tb46264.x
126. Fontana G, Lapolla A, Sanzari M, Piva E, Mussap M, De Toni S, et al. An Immunological Evaluation of Type II Diabetic Patients With Periodontal Disease. J Diabetes Complications (1999) 13:23–30. doi: 10.1016/S1056-8727(98)00021-X
127. Naghibi M, Smith RP, Baltch AL, Gates SA, Wu DH, Hammer MC, et al. The Effect of Diabetes Mellitus on Chemotactic and Bactericidal Activity of Human Polymorphonuclear Leukocytes. Diabetes Res Clin Pract (1987) 4:27–35. doi: 10.1016/S0168-8227(87)80030-X
128. Chello M, Mastroroberto P, Cirillo F, Bevacqua E, Carrano A, Perticone F, et al. Neutrophil-Endothelial Cells Modulation in Diabetic Patients Undergoing Coronary Artery Bypass Grafting. Eur J Cardio Thoracic Surg (1998) 14:373–9. doi: 10.1016/S1010-7940(98)00222-X
129. Barouch FC, Miyamoto K, Allport JR, Fujita K, Bursell SE, Aiello LP, et al. Integrin-Mediated Neutrophil Adhesion and Retinal Leukostasis in Diabetes. Investig Ophthalmol Vis Sci (2000) 41:1153–8.
130. Mastej K, Adamiec R. Neutrophil Surface Expression of CD11b and CD62L in Diabetic Microangiopathy. Acta Diabetol (2008) 45:183–90. doi: 10.1007/s00592-008-0040-0
131. Omi H, Okayama N, Shimizu M, Okouchi M, Ito S, Fukutomi T, et al. Participation of High Glucose Concentrations in Neutrophil Adhesion and Surface Expression of Adhesion Molecules on Cultured Human Endothelial Cells - Effect of Antidiabetic Medicines. J Diabetes Complications (2002) 16:201–8. doi: 10.1016/S1056-8727(01)00163-5
132. Takeuchi Y, Okayama N, Imaeda K, Okouchi M, Omi H, Imai S, et al. Effects of Histamine 2 Receptor Antagonists on Endothelial-Neutrophil Adhesion and Surface Expression of Endothelial Adhesion Molecules Induced by High Glucose Levels. J Diabetes Complications (2007) 21:50–5. doi: 10.1016/j.jdiacomp.2006.02.002
133. Galkina E, Ley K. Leukocyte Recruitment and Vascular Injury in Diabetic Nephropathy. J Am Soc Nephrol (2006) 17:368–77. doi: 10.1681/ASN.2005080859
134. Pizzino G, Irrera N, Cucinotta M, Pallio G, Mannino F, Arcoraci V, et al. Oxidative Stress: Harms and Benefits for Human Health. Oxid Med Cell Longev (2017) 2017:8416763. doi: 10.1155/2017/8416763
136. Shurtz-Swirski R, Sela S, Herskovits AT, Shasha SM, Shapiro G, Nasser L, et al. Involvement of Peripheral Polymorphonuclear Leukocytes in Oxidative Stress and Inflammation in Type 2 Diabetic Patients. Diabetes Care (2001) 24:104–10. doi: 10.2337/diacare.24.1.104
137. Lin X, Candlish JK, Thai AC. Superoxide Production by Neutrophils From Diabetics and Normal Subjects in Response to Glucose and Galactose. Exp Mol Pathol (1993) 58:229–36. doi: 10.1006/exmp.1993.1020
138. Ortmeyer J, Mohsenin V. Inhibition of Phospholipase D and Superoxide Generation by Glucose in Diabetic Neutrophils. Life Sci (1996) 59:255–62. doi: 10.1016/0024-3205(96)00284-6
139. Serlenga E, Garofalo AR, De Pergola G, Ventura MT, Tortorella C, Antonaci S. Polymorphonuclear Cell-Mediated Phagocytosis and Superoxide Anion Release in Insulin-Dependent Diabetes Mellitus. Cytobios (1993) 74:189–95. doi: 10.1016/j.jdiacomp.2018.08.003
140. Ayilavarapu S, Kantarci A, Fredman G, Turkoglu O, Omori K, Liu H, et al. Diabetes-Induced Oxidative Stress Is Mediated by Ca 2+ -Independent Phospholipase A2 in Neutrophils. J Immunol (2010) 184:1507–15. doi: 10.4049/jimmunol.0901219
141. Freedman SF, Hatchell DL. Enhanced Superoxide Radical Production by Stimulated Polymorphonuclear Leukocytes in a Cat Model of Diabetes. Exp Eye Res (1992) 55:767–73. doi: 10.1016/0014-4835(92)90181-Q
142. Shah SV, Wallin JD, Eilenj SD. Chemiluminescence and Superoxide Anion Production by Leukocytes From Diabetic Patients. J Clin Endocrinol Metab (1983) 57:402–9. doi: 10.1210/jcem-57-2-402
143. Sato N, Shimizu H, Shimomura Y, Uehara Y, Takahashi M, Kobayashi I. Reduced Ability of Neutrophils to Produce Active Oxygen Species in STreptozotocin-induced Diabetic Rates. Exp Clin Endocrinol (1992) 99:31–3. doi: 10.1055/s-0029-1211128
144. Ueta E, Osaki T, Yoneda K, Yamamoto T. Prevalence of Diabetes Mellitus in Odontogenic Infections and Oral Candidiasis: An Analysis of Neutrophil Suppression. J Oral Pathol Med (1993) 22:168–74. doi: 10.1111/j.1600-0714.1993.tb01051.x
145. Peck KR, Son DW, Song JH, Kim S, Oh MD, Choe KW. Enhanced Neutrophil Functions by Recombinant Human Granulocyte Colony-Stimulating Factor in Diabetic Patients With Foot Infections In Vitro. J Korean Med Sci (2001) 16:39–44. doi: 10.3346/jkms.2001.16.1.39
146. Watanabe A, Tomino Y, Yokoyama K -I, Koide H. Production of Hydrogen Peroxide by Neutrophilic Polymorphonuclear Leukocytes in Patients With Diabetic Nephropathy. J Clin Lab Anal (1993) 7:209–13. doi: 10.1002/jcla.1860070404
147. Bilgic S, Aktas E, Salman F, Ersahin G, Erten G, Yilmaz MT, et al. Intracytoplasmic Cytokine Levels and Neutrophil Functions in Early Clinical Stage of Type 1 Diabetes. Diabetes Res Clin Pract (2008) 79:31–6. doi: 10.1016/j.diabres.2007.06.011
148. Ihm S-H, Yoo HJ, Park SW, Park CJ. Effect of Tolrestat, an Aldose Reductase Inhibitor, on Neutrophil Respiratory Burst Activity in Diabetic Patients. Metabolism (1997) 46:634–8. doi: 10.1016/S0026-0495(97)90005-6
149. Osar Z, Samanci T, Demirel GY, Damci T, Ilkova H. Nicotinamide Effects Oxidative Burst Activity of Neutrophils in Patients With Poorly Controlled Type 2 Diabetes Mellitus. Exp Diabesity Res (2004) 5:155–62. doi: 10.1080/15438600490424244
150. Nabi AHMN, Islam LN, Rahman MM, Biswas KB. Polymorphonuclear Neutrophil Dysfunctions in Streptozotocin-Induced Type 1 Diabetic Rats. J Biochem Mol Biol (2005) 38:661–7. doi: 10.5483/bmbrep.2005.38.6.661
151. Barañao RI, Rumi LS, Tesone PA, Foglia VG. Some Characteristics of Neutrophils From Diabetic Patients and Their Relation to the Levels of Circulating Immune Complexes. Acta Diabetol Lat (1988) 25:13–23. doi: 10.1007/BF02581241
152. Tikhonova IV, Grinevich AA, Guseva IE, Safronova VG. Modified Kinetics of Generation of Reactive Species in Peripheral Blood of Patients With Type 2 Diabetes. Free Radic Biol Med (2020) 159:76–86. doi: 10.1016/j.freeradbiomed.2020.06.014
153. Kashima K, Sato N, Sato K, Shimizu H, Mori M. Effect of Epalrestat, an Aldose Reductase Inhibitor, on the Generation of Oxygen-Derived Free Radicals in Neutrophils From Streptozotocin-Induced Diabetic Rats. Endocrinology (1998) 139:3404–8. doi: 10.1210/endo.139.8.6152
154. Marhoffer W, Stein M, Maeser E, Federlin K. Impairment of Polymorphonuclear Leukocyte Function and Metabolic Control of Diabetes. Diabetes Care (1992) 15:256–60. doi: 10.2337/diacare.15.2.256
155. de Araujo GR, de Faria KG, Lima WG, da Pádua BC, Rossoni JV, Souza AA, et al. Effect of Captopril and the Bradykinin-PKC Pathway on ROS Production in Type 1 Diabetic Rats. Can J Physiol Pharmacol (2011) 89:923–33. doi: 10.1139/Y11-097
156. Sato N, Kashima K, Uehara Y, Ohtani K, Shimizu H, Mori M. Epalrestat, an Aldose Reductase Inhibitor, Improves an Impaired Generation of Oxygen-Derived Free Radicals by Neutrophils From Poorly Controlled NIDDM Patients. Diabetes Care (1997) 20:995–8. doi: 10.2337/diacare.20.6.995
157. Nogueira-Machado JA, Silva F, Cunha EP, Calsolari MR, Costa DC, Perilo CS, et al. Modulation of the Production of Reactive Oxygen Species (ROS) by cAMP-elevating Agents in Granulocytes From Diabetic Patients: An Akt/PKB-dependent Phenomenon. Diabetes Metab (2006) 32:331–5. doi: 10.1016/s1262-3636(07)70287-2
158. Marin DP, Bolin AP, Macedo RDS, Sampaio SC, Otton R. ROS Production in Neutrophils From Alloxan-Induced Diabetic Rats Treated In Vivo With Astaxanthin. Int Immunopharmacol (2011) 11:103–9. doi: 10.1016/j.intimp.2010.10.013
159. Ridzuan N, John CM, Sandrasaigaran P, Maqbool M, Liew LC, Lim J, et al. Basic Study Preliminary Study on Overproduction of Reactive Oxygen Species by Neutrophils in Diabetes Mellitus. World J Diabetes (2016) 7:271–8. doi: 10.4239/wjd.v7.i13.271
160. Marhoffer W, Stein M, Schleinkofer L, Federlin K. Tive Burst and Phagocytic Capacity in Type-1 Diabetes-Mellitus. Diabetes Res Clin Pract (1993) 19:183–8. doi: 10.1016/0168-8227(93)90112-i
161. Winterbourn CC, Kettle AJ, Hampton MB. Reactive Oxygen Species and Neutrophil Function. Annu Rev Biochem (2016) 85:765–92. doi: 10.1146/annurev-biochem-060815-014442
162. Belambri SA, Rolas L, Raad H, Hurtado-Nedelec M, Dang PM-C, El-Benna J. NADPH Oxidase Activation in Neutrophils: Role of the Phosphorylation of its Subunits. Eur J Clin Invest (2018) 48:e12951. doi: 10.1111/eci.12951
163. Dikalov SI, Harrison DG. Methods for Detection of Mitochondrial and Cellular Reactive Oxygen Species. Antioxid Redox Signal (2014) 20:372–82. doi: 10.1089/ars.2012.4886
164. Macedo RC, Bolin AP, Marin DP, Otton R. Astaxanthin Addition Improves Human Neutrophils Function: In Vitro Study. Eur J Nutr (2010) 49:447–57. doi: 10.1007/s00394-010-0103-1
165. Guerra BA, Otton R. Impact of the Carotenoid Astaxanthin on Phagocytic Capacity and ROS/RNS Production of Human Neutrophils Treated With Free Fatty Acids and High Glucose. Int Immunopharmacol (2011) 11:2220–6. doi: 10.1016/j.intimp.2011.10.004
166. Yeh P-T, Huang H-W, Yang C-M, Yang W-S, Yang C-H. Astaxanthin Inhibits Expression of Retinal Oxidative Stress and Inflammatory Mediators in Streptozotocin-Induced Diabetic Rats. PloS One (2016) 11:e0146438. doi: 10.1371/journal.pone.0146438
167. Penislusshiyan S, Chitra L, Ancy I, Kumaradhas P, Palvannan T. Novel Antioxidant Astaxanthin-s-Allyl Cysteine Biconjugate Diminished Oxidative Stress and Mitochondrial Dysfunction to Triumph Diabetes in Rat Model. Life Sci (2020) 245:117367. doi: 10.1016/j.lfs.2020.117367
168. Chen Q, Tao J, Li GP, Zheng DX, Tan Y, Li RB, et al. Astaxanthin Ameliorates Experimental Diabetes-Induced Renal Oxidative Stress and Fibronectin by Upregulating connexin43 in Glomerular Mesangial Cells and Diabetic Mice. Eur J Pharmacol (2018) 840:33–43. doi: 10.1016/j.ejphar.2018.09.028
169. Inukai T, Yoshida N, Wakabayashi S, Inukai Y, Matsutomo R, Takanashi K, et al. Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers Effectively and Directly Potentiate Superoxide Scavenging by Polymorphonuclear Leukocytes From Patients With Type 2 Diabetes Mellitus. Am J Med Sci (2005) 329:222–7. doi: 10.1097/00000441-200505000-00002
170. Daoud AK, Tayyar MA, Fouda IM, Harfeil NA. Effects of Diabetes Mellitus vs. In Vitro Hyperglycemia on Select Immune Cell Functions. J Immunotoxicol (2009) 6:36–41. doi: 10.1080/15476910802604564
171. Tebbs SE, Gonzalez AM, Wilson RM. The Role of Aldose Reductase Inhibition in Diabetic Neutrophil Phagocytosis and Killing. Clin Exp Immunol (1991) 84:482–7.
172. Shimizu H, Sato K, Kashima K, Mori M, Sato N, Sato K, et al. Effect of Epalrestat, an Aldose Reductase Inhibitor, on the Generation of Oxygen-Derived Free Radicals in Neutrophils From Streptozotocin-Induced Diabetic Rats*. Endocrinology (1998) 139:3404–8. doi: 10.1210/endo.139.8.6152
173. Gough A, Clapperton M, Rolando N, Foster AVM, PhilpottHoward J, Edmonds ME. Randomised Placebo-Controlled Trial of Granulocyte-Colony Stimulating Factor in Diabetic Foot Infection. Lancet (1997) 350:855–9. doi: 10.1016/s0140-6736(97)04495-4
174. Yonem A, Cakir B, Guler S, Azal O, Corakci A. Effects of Granulocyte-Colony Stimulating Factor in the Treatment of Diabetic Foot Infection. Diabetes Obes Metab (2001) 3:332–7. doi: 10.1046/j.1463-1326.2001.00142.x
175. Cruciani M, Lipsky BA, Mengoli C, de Lalla F. Granulocyte-colony Stimulating Factors as Adjunctive Therapy for Diabetic Foot Infections. Cochrane Database Syst Rev (2013) 8:1469–93. doi: 10.1002/14651858.CD006810.pub3
176. Tiwari S, Pratyush DD, Gahlot A, Singh SK. Sepsis in Diabetes: A Bad Duo. Diabetes Metab Syndr Clin Res Rev (2011) 5:222–7. doi: 10.1016/j.dsx.2012.02.026
177. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Sci (80- ) (2004) 303:1532–5. doi: 10.1126/science.1092385
178. Neubert E, Meyer D, Rocca F, Günay G, Kwaczala-Tessmann A, Grandke J, et al. Chromatin Swelling Drives Neutrophil Extracellular Trap Release. Nat Commun (2018) 9:3767. doi: 10.1038/s41467-018-06263-5
179. Kenny EF, Herzig A, Krüger R, Muth A, Mondal S, Thompson PR, et al. Diverse Stimuli Engage Different Neutrophil Extracellular Trap Pathways. Elife (2017) 6:e24437. doi: 10.7554/eLife.24437
180. Khan MA, D’Ovidio A, Tran H, Palaniyar N. Anthracyclines Suppress Both NADPH Oxidase-Dependent and -Independent NETosis in Human Neutrophils. Cancers (Basel) (2019) 11:1328. doi: 10.3390/cancers11091328
181. Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, Goldman AE, et al. Netosis Proceeds by Cytoskeleton and Endomembrane Disassembly and PAD4-mediated Chromatin Decondensation and Nuclear Envelope Rupture. Proc Natl Acad Sci (2020) 117:7326. doi: 10.1073/pnas.1909546117
182. Holmes CL, Shim D, Kernien J, Johnson CJ, Nett JE, Shelef MA. Insight Into Neutrophil Extracellular Traps Through Systematic Evaluation of Citrullination and Peptidylarginine Deiminases. J Immunol Res (2019) 2019:2160192. doi: 10.1155/2019/2160192
183. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil Elastase and Myeloperoxidase Regulate the Formation of Neutrophil Extracellular Traps. J Cell Biol (2010) 191:677–91. doi: 10.1083/jcb.201006052
184. Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, et al. Activated Endothelial Cells Induce Neutrophil Extracellular Traps and are Susceptible to NETosis-mediated Cell Death. FEBES Lett (2010) 584:3193–7. doi: 10.1016/j.febslet.2010.06.006
185. Pedersen F, Marwitz S, Goldmann T, Kirsten A, Magnussen H, Rabe KF, et al. Enhanced Neutrophil Extracellular Trap (NET) Formation in Sputum of Stable COPD Patients. Eur Respir J (2015) 46:PA379. doi: 10.1183/13993003.congress-2015.PA379
186. Njeim R, Azar WS, Fares AH, Azar ST, Kfoury Kassouf H, Eid AA. Netosis Contributes to the Pathogenesis of Diabetes and its Complications. J Mol Endocrinol (2020) 65:R65–76. doi: 10.1530/JME-20-0128
187. Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, et al. Crosstalk Between Neutrophils, B-1a Cells and Plasmacytoid Dendritic Cells Initiates Autoimmune Diabetes. Nat Med (2013) 19:65–73. doi: 10.1038/nm.3042
188. Wang LZY, Zhou X, Yin YZ, Mai YX, Wang DS, Zhang XD. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front Immunol (2019) 9:3076. doi: 10.3389/fimmu.2018.03076
189. Berezin A. Neutrophil Extracellular Traps: The Core Player in Vascular Complications of Diabetes Mellitus. Diabetes Metab Syndr Clin Res Rev (2019) 13:3017–23. doi: 10.1016/j.dsx.2018.07.010
190. Ferreira CD, Araujo TH, Angelo ML, Pennacchi PC, Okada SS, Paula FBD, et al. Neutrophil Dysfunction Induced by Hyperglycemia: Modulation of Myeloperoxidase Activity. Cell Biochem Funct (2012) 30:604–10. doi: 10.1002/cbf.2840
191. Joshi MB, Baipadithaya G, Balakrishnan A, Hegde M, Vohra M, Ahamed R, et al. Elevated Homocysteine Levels in Type 2 Diabetes Induce Constitutive Neutrophil Extracellular Traps. Sci Rep (2016) 6:36362. doi: 10.1038/srep36362
192. Menegazzo L, Ciciliot S, Poncina N, Mazzucato M, Persano M, Bonora B, et al. Netosis is Induced by High Glucose and Associated With Type 2 Diabetes. Acta Diabetol (2015) 52:497–503. doi: 10.1007/s00592-014-0676-x
193. Rodríguez-Espinosa O, Rojas-Espinosa O, Moreno-Altamirano MMB, López-Villegas EO, Sánchez-García FJ. Metabolic Requirements for Neutrophil Extracellular Traps Formation. Immunology (2015) 145:213–24. doi: 10.1111/imm.12437
194. Zurawska-Płaksej E, Piwowar A, Knapik-Kordecka M, Warwas M. Activities of Neutrophil Membrane-Bound Proteases in Type 2 Diabetic Patients. Arch Med Res (2014) 45:36–43. doi: 10.1016/j.arcmed.2013.10.003
195. Fadini GP, Menegazzo L, Rigato M, Scattolini V, Poncina N, Bruttocao A, et al. Netosis Delays Diabetic Wound Healing in Mice and Humans. Diabetes (2016) 65:1061–71. doi: 10.2337/db15-0863
196. Ortines RV, Liu HY, Cheng LI, Cohen TS, Lawlor H, Gami A, et al. Neutralizing Alpha-Toxin Accelerates Healing of Staphylococcus Aureus-Infected Wounds in Nondiabetic and Diabetic Mice. Antimicrob Agents Chemother (2018) 62:14. doi: 10.1128/aac.02288-17
197. Yu Y, Su K. Neutrophil Extracellular Traps and Systemic Lupus Erythematosus. J Clin Cell Immunol (2013) 4:1–7. doi: 10.4172/2155-9899.1000139
198. Kumagai Y, Ohzawa H, Miyato H, Horie H, Hosoya Y, Lefor AK, et al. Surgical Stress Increases Circulating Low-Density Neutrophils Which May Promote Tumor Recurrence. J Surg Res (2020) 246:52–61. doi: 10.1016/j.jss.2019.08.022
199. Arampatzioglou A, Papazoglou D, Konstantinidis T, Chrysanthopoulou A, Mitsios A, Angelidou I, et al. Clarithromycin Enhances the Antibacterial Activity and Wound Healing Capacity in Type 2 Diabetes Mellitus by Increasing LL-37 Load on Neutrophil Extracellular Traps. Front Immunol (2018) 9:2064. doi: 10.3389/fimmu.2018.02064
200. Guisado-Vasco P, Valderas-Ortega S, Carralón-González MM, Roda-Santacruz A, González-Cortijo L, Sotres-Fernández G, et al. Clinical Characteristics and Outcomes Among Hospitalized Adults With Severe COVID-19 Admitted to a Tertiary Medical Center and Receiving Antiviral, Antimalarials, Glucocorticoids, or Immunomodulation With Tocilizumab or Cyclosporine: A Retrospective Obser. EClinicalMedicine (2020) 28:100591. doi: 10.1016/j.eclinm.2020.100591
201. Khan M, Khan H, Khan S, Nawaz M. Epidemiological and Clinical Characteristics of Coronavirus Disease (COVID-19) Cases at a Screening Clinic During the Early Outbreak Period: A Single-Centre Study. J Med Microbiol (2020) 69:1114–23. doi: 10.1099/jmm.0.001231
202. Yan Y, Yang Y, Wang F, Ren H, Zhang S, Shi X, et al. Clinical Characteristics and Outcomes of Patients With Severe covid-19 With Diabetes. BMJ Open Diabetes Res Care (2020) 8:1–9. doi: 10.1136/bmjdrc-2020-001343
203. Barron E, Bakhai C, Kar P, Weaver A, Bradley D, Ismail H, et al. Associations of Type 1 and Type 2 Diabetes With COVID-19-related Mortality in England: A Whole-Population Study. Lancet Diabetes Endocrinol (2020) 8:813–22. doi: 10.1016/S2213-8587(20)30272-2
204. De Almeida-Pititto B, Dualib PM, Zajdenverg L, Dantas JR, De Souza FD, Rodacki M, et al. Severity and Mortality of COVID 19 in Patients With Diabetes, Hypertension and Cardiovascular Disease: A Meta-Analysis. Diabetol Metab Syndr (2020) 12:75. doi: 10.1186/s13098-020-00586-4
205. Holman N, Knighton P, Kar P, O’Keefe J, Curley M, Weaver A, et al. Risk Factors for COVID-19-related Mortality in People With Type 1 and Type 2 Diabetes in England: A Population-Based Cohort Study. Lancet Diabetes Endocrinol (2020) 8:823–33. doi: 10.1016/S2213-8587(20)30271-0
206. Coppelli A, Giannarelli R, Aragona M, Penno G, Falcone M, Tiseo G, et al. Hyperglycemia at Hospital Admission is Associated With Severity of the Prognosis in Patients Hospitalized for COVID-19: The Pisa COVID-19 Study. Diabetes Care (2020) 43:2345–8. doi: 10.2337/dc20-1380
207. Wang S, Ma P, Zhang S, Song S, Wang Z, Ma Y, et al. Fasting Blood Glucose at Admission is an Independent Predictor for 28-Day Mortality in Patients With COVID-19 Without Previous Diagnosis of Diabetes: A Multi-Centre Retrospective Study. Diabetologia (2020) 63:2102–11. doi: 10.1007/s00125-020-05209-1
208. Sardu C, D’Onofrio N, Balestrieri ML, Barbieri M, Rizzo MR, Messina V, et al. Outcomes in Patients With Hyperglycemia Affected by COVID-19: can We do More on Glycemic Control? Diabetes Care (2020) 43:1408–15. doi: 10.2337/dc20-0723
209. Arcanjo A, Logullo J, Menezes CCB, de Souza Carvalho Giangiarulo TC, dos Reis MC, de Castro GMM, et al. The Emerging Role of Neutrophil Extracellular Traps in Severe Acute Respiratory Syndrome Coronavirus 2 (COVID-19). Sci Rep (2020) 10:1–11. doi: 10.1038/s41598-020-76781-0
210. Imran MM, Ahmed U, Usman U, Ali M, Shaukat A, Gul N. Neutrophil/Lymphocyte Ratio – A Marker of COVID-19 Pneumonia Severity. Int J Clin Pract (2020) 75(4):e13698. doi: 10.1111/ijcp.13698
211. Ruiz SJ, Ventura PS, Vázquez JMC, García-Adasme SI, García MM, Pol PT, et al. Prognostic Implications of Neutrophil-Lymphocyte Ratio in COVID-19. Eur J Clin Invest (2020) 51(1):1–9. doi: 10.1111/eci.13404
212. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood (2020) 136:1169–79. doi: 10.1182/blood.2020007008
213. Lee YY, Park HH, Park W, Kim H, Jang JG, Hong KS, et al. Long-Acting Nanoparticulate DNase-1 for Effective Suppression of SARS-CoV-2-mediated Neutrophil Activities and Cytokine Storm. Biomaterials (2021) 267:1–10. doi: 10.1016/j.biomaterials.2020.120389
214. Tecchio C, Micheletti A, Cassatella MA. Neutrophil-Derived Cytokines: Facts Beyond Expression. Front Immunol (2014) 5:508. doi: 10.3389/fimmu.2014.00508
215. Hatanaka E, Monteagudo PT, Marrocos MSM, Campa A. Neutrophils and Monocytes as Potentially Important Sources of Proinflammatory Cytokines in Diabetes. Clin Exp Immunol (2006) 146:443–7. doi: 10.1111/j.1365-2249.2006.03229.x
216. Gupta S, Maratha A, Siednienko J, Natarajan A, Gajanayake T, Hoashi S, et al. Analysis of Inflammatory Cytokine and TLR Expression Levels in Type 2 Diabetes With Complications. Sci Rep (2017) 7:1–10. doi: 10.1038/s41598-017-07230-8
217. Sekizuka K, Tomino Y, Sei C, Kurusu A, Tashiro K, Yamaguchi Y, et al. Detection of Serum IL-6 in Patients With Diabetic Nephropathy [11]. Nephron (1994) 68:284–5. doi: 10.1159/000188281
218. Rosa JS, Flores RL, Oliver SR, Pontello AM, Zaldivar FP, Galassetti PR. Sustained IL-1α, IL-4, and IL-6 Elevations Following Correction of Hyperglycemia in Children With Type 1 Diabetes Mellitus. Pediatr Diabetes (2008) 9:9–16. doi: 10.1111/j.1399-5448.2007.00243.x
219. Kuhns DB, Young HA, Gallin EK, Gallin JI. Ca2+-Dependent Production and Release of IL-8 in Human Neutrophils. J Immunol (1998) 161:4332–9.
220. Seo JY, Kim H, Seo JT, Kim KH. Oxidative Stress Induced Cytokine Production in Isolated Rat Pancreatic Acinar Cells: Effects of Small-Molecule Antioxidants. Pharmacology (2002) 64:63–70. doi: 10.1159/000056152
221. Crapo JD. Oxidative Stress as an Initiator of Cytokine Release and Cell Damage. In: European Respiratory Journal, Supplement. Lausanne, Switzerland: European Respiratory Society. (2003) p. 4s–6s. doi: 10.1183/09031936.03.00000203a
222. Wang H, Meng QH, Gordon JR, Khandwala H, Wu L. Proinflammatory and Proapoptotic Effects of Methylglyoxal on Neutrophils From Patients With Type 2 Diabetes Mellitus. Clin Biochem (2007) 40:1232–9. doi: 10.1016/j.clinbiochem.2007.07.016
223. Martins JO, Campos CAL, Cruz JWMC, Manzolli S, Alves VAF, Vianna EO, et al. Insulin Modulates Cytokine Release and Selectin Expression in the Early Phase of Allergic Airway Inflammation in Diabetic Rats. BMC Pulm Med (2010) 10:1–9. doi: 10.1186/1471-2466-10-39
224. Kewcharoenwong C, Rinchai D, Utispan K, Suwannasaen D, Bancroft GJ, Ato M, et al. Glibenclamide Reduces Pro-Inflammatory Cytokine Production by Neutrophils of Diabetes Patients in Response to Bacterial Infection. Sci Rep (2013) 3:1–8. doi: 10.1038/srep03363
225. Hennigan S, Kavanaugh A. Interleukin-6 Inhibitors in the Treatment of Rheumatoid Arthritis. Ther Clin Risk Manag (2008) 4:767–75. doi: 10.2147/tcrm.s3470
226. Horby PW, Pessoa-Amorim G, Peto L, Brightling CE, Sarkar R, Thomas K, et al. Tocilizumab in Patients Admitted to Hospital With COVID-19 (RECOVERY): Preliminary Results of a Randomised, Controlled, Open-Label, Platform Trial. medRxiv (2021) 396(10259):1345–52. doi: 10.1101/2021.02.11.21249258
227. Marfella R, Paolisso P, Sardu C, Bergamaschi L, D’Angelo EC, Barbieri M, et al. Negative Impact of Hyperglycaemia on Tocilizumab Therapy in Covid-19 Patients. Diabetes Metab (2020) 46:403–5. doi: 10.1016/j.diabet.2020.05.005
228. McColl SR, Paquin R, Ménard C, Beaulieu AD. Human Neutrophils Produce High Levels of the Interleukln 1 Receptor Antagonist in Response to Granulocyte/Macrophage Colony-Stimulating Factor and Tumor Necrosis Factor α. J Exp Med (1992) 176:593–8. doi: 10.1084/jem.176.2.593
229. Ruscitti P, Masedu F, Alvaro S, Airò P, Battafarano N, Cantarini L, et al. Anti-Interleukin-1 Treatment in Patients With Rheumatoid Arthritis and Type 2 Diabetes (TRACK): A Multicentre, Open-Label, Randomised Controlled Trial. PloS Med (2019) 16:e1002901. doi: 10.1371/journal.pmed.1002901
230. Cavalli G, De Luca G, Campochiaro C, Della-Torre E, Ripa M, Canetti D, et al. Interleukin-1 Blockade With High-Dose Anakinra in Patients With COVID-19, Acute Respiratory Distress Syndrome, and Hyperinflammation: A Retrospective Cohort Study. Lancet Rheumatol (2020) 2:e325–31. doi: 10.1016/S2665-9913(20)30127-2
231. Langer-Gould A, Smith JB, Gonzales EG, Castillo RD, Figueroa JG, Ramanathan A, et al. Early Identification of COVID-19 Cytokine Storm and Treatment With Anakinra or Tocilizumab. Int J Infect Dis (2020) 99:291–7. doi: 10.1016/j.ijid.2020.07.081
232. Mistry P, Carmona-Rivera C, Ombrello AK, Hoffmann P, Seto NL, Jones A, et al. Dysregulated Neutrophil Responses and Neutrophil Extracellular Trap Formation and Degradation in PAPA Syndrome. Ann Rheum Dis (2018) 77:1825–33. doi: 10.1136/annrheumdis-2018-213746
233. Hudock KM, Collins MS, Imbrogno M, Snowball J, Kramer EL, Brewington JJ, et al. Neutrophil Extracellular Traps Activate IL-8 and IL-1 Expression in Human Bronchial Epithelia. Am J Physiol - Lung Cell Mol Physiol (2020) 319:L137–47. doi: 10.1152/ajplung.00144.2019
234. Fox S, Leitch AE, Duffin R, Haslett C, Rossi AG. Neutrophil Apoptosis: Relevance to the Innate Immune Response and Inflammatory Disease. J Innate Immun (2010) 2:216–27. doi: 10.1159/000284367
235. Prince LR, Prosseda SD, Higgins K, Carlring J, Prestwich EC, Ogryzko NV, et al. NR4A Orphan Nuclear Receptor Family Members, NR4A2 and NR4A3, Regulate Neutrophil Number and Survival. Blood (2017) 130:1014–25. doi: 10.1182/blood-2017-03-770164
236. McCracken JM, Allen LAH. Regulation of Human Neutrophil Apoptosis and Lifespan in Health and Disease. J Cell Death (2014) 7:15–23. doi: 10.4137/JCD.S11038
237. Dibbert B, Weber M, Nikolaizik WH, Vogt P, Schöni MH, Blaser K, et al. Cytokine-Mediated Bax Deficiency and Consequent Delayed Neutrophil Apoptosis: A General Mechanism to Accumulate Effector Cells in Inflammation. Proc Natl Acad Sci (1999) 96:13330. doi: 10.1073/pnas.96.23.13330
238. Koedel U, Frankenberg T, Kirschnek S, Obermaier B, Häcker H, Paul R, et al. Apoptosis Is Essential for Neutrophil Functional Shutdown and Determines Tissue Damage in Experimental Pneumococcal Meningitis. PloS Pathog (2009) 5:e1000461. doi: 10.1371/journal.ppat.1000461
239. Sabroe I, Jones EC, Usher LR, Whyte MKB, Dower SK. Toll-Like Receptor (TLR)2 and TLR4 in Human Peripheral Blood Granulocytes: A Critical Role for Monocytes in Leukocyte Lipopolysaccharide Responses. J Immunol (2002) 168:4701. doi: 10.4049/jimmunol.168.9.4701
240. Rowe SJ, Allen L, Ridger VC, Hellewell PG, Whyte MKB. Caspase-1-Deficient Mice Have Delayed Neutrophil Apoptosis and a Prolonged Inflammatory Response to Lipopolysaccharide-Induced Acute Lung Injury. J Immunol (2002) 169:6401–7. doi: 10.4049/jimmunol.169.11.6401
241. Haslett C. Granulocyte Apoptosis and its Role in the Resolution and Control of Lung Inflammation. In: American Journal of Respiratory and Critical Care Medicine. Chicago, USA: American Lung Association (1999). p. S5–S11. doi: 10.1164/ajrccm.160.supplement_1.4
242. Weinmann P, Moura RA, Caetano-Lopes JR, Pereira PA, Canhão H, Queiroz MV, et al. Delayed Neutrophil Apoptosis in Very Early Rheumatoid Arthritis Patients is Abrogated by Methotrexate Therapy. Clin Exp Rheumatol (2007) 25:885–7.
243. Zhang J, He J, Xia J, Chen Z, Chen X. Delayed Apoptosis by Neutrophils From COPD Patients is Associated With Altered Bak, Bcl-Xl, and Mcl-1 mRNA Expression. Diagn Pathol (2012) 7:65. doi: 10.1186/1746-1596-7-65
244. Zhu M, Yuan K, Lu Q, Zhu Q, Zhang S, Li X, et al. Emodin Ameliorates Rheumatoid Arthritis by Promoting Neutrophil Apoptosis and Inhibiting Neutrophil Extracellular Trap Formation. Mol Immunol (2019) 112:188–97. doi: 10.1016/j.molimm.2019.05.010
245. Goren I, Müller E, Schiefelbein D, Christen U, Pfeilschifter J, Mühl H, et al. Systemic Anti-Tnfα Treatment Restores Diabetes-Impaired Skin Repair in Ob/Ob Mice by Inactivation of Macrophages. J Invest Dermatol (2007) 127:2259–67. doi: 10.1038/sj.jid.5700842
246. Mayer Y, Balbir-Gurman A, Machtei EE. Anti-Tumor Necrosis Factor-Alpha Therapy and Periodontal Parameters in Patients With Rheumatoid Arthritis. J Periodontol (2009) 80:1414–20. doi: 10.1902/jop.2009.090015
247. Glowacka E, Banasik M, Lewkowicz P, Tchorzewski H. The Effect of LPS on Neutrophils From Patients With High Risk of Type 1 Diabetes Mellitus in Relation to IL-8, Il-10 and IL-12 Production and Apoptosis In Vitro. Scand J Immunol (2002) 55:210–7. doi: 10.1046/j.1365-3083.2002.01046.x
248. Tennenberg SD, Finkenauer R, Dwivedi A. Absence of Lipopolysaccharide-Induced Inhibition of Neutrophil Apoptosis in Patients With Diabetes. Arch Surg (1999) 134:1229–34. doi: 10.1001/archsurg.134.11.1229
249. Du X, Poltorak A, Silva M, Beutler B. Analysis of Tlr4-mediated LPS Signal Transduction in Macrophages by Mutational Modification of the Receptor. Blood Cells Mol Dis (1999) 25:328–38. doi: 10.1006/bcmd.1999.0262
250. Kim JJ, Sears DD. TLR4 and Insulin Resistance. Gastroenterol Res Pract (2010) 2010:1–11. doi: 10.1155/2010/212563
251. Ward MG, Li G, Hao M. Apoptotic β-Cells Induce Macrophage Reprogramming Under Diabetic Conditions. J Biol Chem (2018) 293:16160–73. doi: 10.1074/jbc.RA118.004565
252. Pavlou S, Lindsay J, Ingram R, Xu H, Chen M. Sustained High Glucose Exposure Sensitizes Macrophage Responses to Cytokine Stimuli But Reduces Their Phagocytic Activity. BMC Immunol (2018) 19:24. doi: 10.1186/s12865-018-0261-0
253. Yang H, Long F, Zhang Y, Yu R, Zhang P, Li W, et al. 1α,25-Dihydroxyvitamin D3 Induces Neutrophil Apoptosis Through the P38 MAPK Signaling Pathway in Chronic Obstructive Pulmonary Disease Patients. PloS One (2015) 10:e0120515. doi: 10.1371/journal.pone.0120515
254. Sousa LP, Lopes F, Silva DM, Tavares LP, Vieira AT, Rezende BM, et al. PDE4 Inhibition Drives Resolution of Neutrophilic Inflammation by Inducing Apoptosis in a PKA-PI3K/Akt-dependent and NF-κb-Independent Manner. J Leukoc Biol (2010) 87:895–904. doi: 10.1189/jlb.0809540
255. Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, et al. Cyclin-Dependent Kinase Inhibitors Enhance the Resolution of Inflammation by Promoting Inflammatory Cell Apoptosis. Nat Med (2006) 12:1056–64. doi: 10.1038/nm1468
256. Rahman A, Henry KM, Herman KD, R Thompson AA, Isles HM, Tulotta C, et al. Inhibition of ErbB Kinase Signalling Promotes Resolution of Neutrophilic Inflammation. Elife (2019) 8:e50990. doi: 10.7554/eLife.50990
257. Tang YP, Liu JY, Yan YM, Fang H, Guo CW, Xie RD, et al. 1,25-dihydroxyvitamin-D-3 Promotes Neutrophil Apoptosis in Periodontitis With Type 2 Diabetes Mellitus Patients Via the p38/MAPK Pathway. Med (Baltimore) (2018) 97:13. doi: 10.1097/md.0000000000013903
258. Bybee JD, Rogers DE. The Phagoctyic Activity of Polymorphonuclear Leukocytes Obtained From Patients With Diabetes Mellitus. J Lab Clin Med (1964) 64:1–13. doi: 10.5555/uri:pii:0022214364901337
259. Sabioncello A, Rabatic S, Kadrnka-Lovrencic M, Oberiter V, Dekaris D. Decreased Phagocytosis and Antibody-Dependent Cellular Cytotoxicity (ADCC) in Type-1 Diabetes. Biomedicine (1981) 35:227–9.
260. Musclow CE, Farkashimsley H, Durbahn G, Spragg L. Fluorescence Assay to Monitor Phagocytosis by Blood-Clot Derived Polymorphonuclear Leukocytes. Cytobios (1991) 65:15–24.
261. Chanchamroen S, Kewcharoenwong C, Susaengrat W, Ato M, Lertmemongkolchai G. Human Polymorphonuclear Neutrophil Responses to Burkholderia Pseudomallei in Healthy and Diabetic Subjects. Infect Immun (2009) 77:456–63. doi: 10.1128/IAI.00503-08
262. Pettersson US, Christoffersson G, Massena S, Ahl D, Jansson L, Henriksnäs J, et al. Increased Recruitment But Impaired Function of Leukocytes During Inflammation in Mouse Models of Type 1 and Type 2 Diabetes. PloS One (2011) 6:e22480–0. doi: 10.1371/journal.pone.0022480
263. Krol E, Agueel R, Banue S, Smogorzewski M, Kumar D, Massry SG. Amlodipine Reverses the Elevation in [Ca2+]i and the Impairment of Phagocytosis in PMNLs of NIDDM Patients. Kidney Int (2003) 64:2188–95. doi: 10.1046/j.1523-1755.2003.00311.x
264. Alexiewicz JM, Kumar D, Smogorzewski M, Klin M, Massry SG. Polymorphonuclear Leukocytes in non-Insulin-Dependent Diabetes Mellitus: Abnormalities in Metabolism and Function. Ann Intern Med (1995) 123:919–24. doi: 10.7326/0003-4819-123-12-199512150-00004
265. Soto M, Gaffney KJ, Rodgers KE. Improving the Innate Immune Response in Diabetes by Modifying the Renin Angiotensin System. Front Immunol (2019) 10:2885. doi: 10.3389/fimmu.2019.02885
266. Davidson NJ, Sowden JM, Fletcher J. Defective Phagocytosis in Insulin Controlled Diabetics: Evidence for a Reaction Between Glucose and Opsonising Proteins. J Clin Pathol (1984) 37:783–6. doi: 10.1136/jcp.37.7.783
267. Lin JC, Siu LK, Fung CP, Tsou HH, Wang JJ, Chen CT, et al. Impaired Phagocytosis of Capsular Serotypes K1 or K2 Klebsiella Pneumoniae in Type 2 Diabetes Mellitus Patients With Poor Glycemic Control. J Clin Endocrinol Metab (2006) 91:3084–7. doi: 10.1210/jc.2005-2749
268. Wilson RM, Reeves WG. Neutrophil Phagocytosis and Killing in Insulin-Dependant Diabetes. Clin Exp Immunol (1986) 63:478–84. doi: 10.1016/0278-2316(86)90030-7
269. Scully IL, McNeil LK, Pathirana S, Singer CL, Liu YD, Mullen S, et al. Neutrophil Killing of Staphylococcus Aureus in Diabetes, Obesity and Metabolic Syndrome: A Prospective Cellular Surveillance Study. Diabetol Metab Syndr (2017) 9:13. doi: 10.1186/s13098-017-0276-3
270. Zamakhchari MF, Sima C, Sama K, Fine N, Glogauer M, Van Dyke TE, et al. Lack of p47phox in Akita Diabetic Mice is Associated With Interstitial Pneumonia, Fibrosis, and Oral Inflammation. Am J Pathol (2016) 186:659–70. doi: 10.1016/j.ajpath.2015.10.026
271. Yano H, Kinoshita M, Fujino K, Nakashima M, Yamamoto Y, Miyazaki H, et al. Insulin Treatment Directly Restores Neutrophil Phagocytosis and Bactericidal Activity in Diabetic Mice and Thereby Improves Surgical Site Staphylococcus Aureus Infection. Infect Immun (2012) 80:4409–16. doi: 10.1128/IAI.00787-12
272. Seyrek N, Marcinkowski W, Smogorzewski M, Demerdash TM, Massry SG. Amlodipine Prevents and Reverses the Elevation in [Ca2+](i) and the Impaired Phagocytosis of PMNL of Diabetic Rats. Nephrol Dial Transplant (1997) 12:265–72. doi: 10.1093/ndt/12.2.265
273. Hostetter MK. Handicaps to Host Defense. Effects of Hyperglycemia on C3 and Candida Albicans. Diabetes (1990) 39:271–5. doi: 10.2337/diab.39.3.271
274. Hair PS, Echague CG, Rohn RD, Krishna NK, Nyalwidhe JO, Cunnion KM. Hyperglycemic Conditions Inhibit C3-mediated Immunologic Control of Staphylococcus Aureus. J Transl Med (2012) 10:35. doi: 10.1186/1479-5876-10-35
275. Pauwels AM, Trost M, Beyaert R, Hoffmann E. Patterns, Receptors, and Signals: Regulation of Phagosome Maturation. Trends Immunol (2017) 38:407–22. doi: 10.1016/j.it.2017.03.006
276. Burlak C, Whitney AR, Mead DJ, Hackstadt T, DeLeo FR. Maturation of Human Neutrophil Phagosomes Includes Incorporation of Molecular Chaperones and Endoplasmic Reticulum Quality Control Machinery. Mol Cell Proteomics (2006) 5:620–34. doi: 10.1074/mcp.M500336-MCP200
277. Gutiérrez S, Fischer J, Ganesan R, Cildir G, Wolke M, Pessia A, et al. Typhimurium Impairs Glycolysis-Mediated Acidification of Phagosomes to Evade Macrophage Defense. BioRxiv (pre-print) (2021). doi: 10.1101/2021.01.14.426635
278. Alba-Loureiro TC, Hirabara SM, Mendonça JR, Curi R, Pithon-Curi TC, Mendonca JR, et al. Diabetes Causes Marked Changes in Function and Metabolism of Rat Neutrophils. J Endocrinol (2006) 188:295–303. doi: 10.1677/joe.1.06438
279. Wood AJT, Vassallo AM, Ruchaud-Sparagano MH, Scott J, Zinnato C, Gonzalez-Tejedo C, et al. C5a Impairs Phagosomal Maturation in the Neutrophil Through Phosphoproteomic Remodeling. JCI Insight (2020) 5:e137029. doi: 10.1172/jci.insight.137029
280. Chaudhury A, Duvoor C, Dendi VSR, Kraleti S, Chada A, Ravilla R, et al. Clinical Review of Antidiabetic Drugs: Implications for Type 2 Diabetes Mellitus Management. Front Endocrinol (Lausanne) (2017) 8:6. doi: 10.3389/fendo.2017.00006
281. Goto A, Goto M, Terauchi Y, Yamaguchi N, Noda M. Association Between Severe Hypoglycemia and Cardiovascular Disease Risk in Japanese Patients With Type 2 Diabetes. J Am Heart Assoc (2016) 5:e002875. doi: 10.1161/JAHA.115.002875
282. Cho YS, Chen CH, Hu C, Long JR, Ong RTH, Sim XL, et al. Meta-Analysis of Genome-Wide Association Studies Identifies Eight New Loci for Type 2 Diabetes in East Asians. Nat Genet (2012) 44:67–U97. doi: 10.1038/ng.1019
283. Ratter JA-O, Rooijackers HM, Tack CJ, Hijmans AG, Netea MG, de Galan BE, et al. Proinflammatory Effects of Hypoglycemia in Humans With or Without Diabetes. Diabetes (2017) 66:1052–61. doi: 10.2337/db16-1091
284. Iqbal A, Prince LR, Novodvorsky P, Bernjak A, Thomas MR, Birch L, et al. Effect of Hypoglycemia on Inflammatory Responses and the Response to Low Dose Endotoxemia in Humans. J Clin Endocrinol Metab (2019) 104:1187–99. doi: 10.1210/jc.2018-01168
285. Kahal H, Halama A, Aburima A, Bhagwat AM, Butler AE, Grauman J, et al. Effect of Induced Hypoglycemia on Inflammation and Oxidative Stress in Type 2 Diabetes and Control Subjects. Sci Rep (2020) 10:4750. doi: 10.1038/s41598-020-61531-z
286. Chow E, Iqbal A, Walkinshaw E, Phoenix F, Macdonald IA, Storey RF, et al. Prolonged Prothrombotic Effects of Antecedent Hypoglycemia in Individuals With Type 2 Diabetes. Diabetes Care (2018) 41:2625. doi: 10.2337/dc18-0050
287. Thomson GA, Fisher BM, Gemmell CG, MacCuish AC, Gallacher SJ. Attenuated Neutrophil Respiratory Burst Following Acute Hypoglycaemia in Diabetic Patients and Normal Subjects. Acta Diabetol (1997) 34:253–6. doi: 10.1007/s005920050084
288. Iqbal A, Heller S. Managing Hypoglycaemia. Best Pract Res Clin Endocrinol Metab (2016) 30:413–30. doi: 10.1016/j.beem.2016.06.004
289. Hope SV, Knight BA, Shields BM, Hill AV, Choudhary P, Strain WD, et al. Random non-Fasting C-peptide Testing can Identify Patients With Insulin-Treated Type 2 Diabetes at High Risk of Hypoglycaemia. Diabetologia (2018) 61:66–74. doi: 10.1007/s00125-017-4449-2
290. Gibb FW, McKnight JA, Clarke C, Strachan MWJ. Preserved C-peptide Secretion is Associated With Fewer Low-Glucose Events and Lower Glucose Variability on Flash Glucose Monitoring in Adults With Type 1 Diabetes. Diabetologia (2020) 63:906–14. doi: 10.1007/s00125-020-05099-3
291. Aljada A, Dandona P. Effect of Insulin on Human Aortic Endothelial Nitric Oxide Synthase. Metabolism (2000) 49:147–50. doi: 10.1016/S0026-0495(00)91039-4
292. Dandona P, Ghanim H, Bandyopadhyay A, Korzeniewski K, Sia CL, Dhindsa S, et al. Insulin Suppresses Endotoxin-Induced Oxidative, Nitrosative, and Inflammatory Stress in Humans. Diabetes Care (2010) 33:2416–23. doi: 10.2337/dc10-0929
293. Sapey E, Stockley RA. Getting Stuck or Choosing to Stay Neutrophil Transit Times in the Lung in Acute Inflammation and COPD. Thorax (2019) 74:631–2. doi: 10.1136/thoraxjnl-2018-213000
294. Ballesteros I, Rubio-Ponce A, Genua M, Lusito E, Kwok I, Fernández-Calvo G, et al. Co-Option of Neutrophil Fates by Tissue Environments. Cell (2020) 183:1282–97.e18. doi: 10.1016/j.cell.2020.10.003
295. Margaroli C, Garratt LW, Horati H, Dittrich AS, Rosenow T, Montgomery ST, et al. Elastase Exocytosis by Airway Neutrophils is Associated With Early Lung Damage in Children With Cystic Fibrosis. Am J Respir Crit Care Med (2019) 199:873–81. doi: 10.1164/rccm.201803-0442OC
296. Morris AC, Brittan M, Wilkinson TS, McAuley DF, Antonelli J, McCulloch C, et al. C5a-Mediated Neutrophil Dysfunction is RhoA-dependent and Predicts Infection in Critically Ill Patients. Blood (2011) 117:5178–88. doi: 10.1182/blood-2010-08-304667
297. Hotchkiss RS, Monneret G, Payen D. Sepsis-Induced Immunosuppression: From Cellular Dysfunctions to Immunotherapy. Nat Rev Immunol (2013) 13:862–74. doi: 10.1038/nri3552
298. Jasper AE, McIver WJ, Sapey E, Walton GM. Understanding the Role of Neutrophils in Chronic Inflammatory Airway Disease. F1000Research (2019) 8:1–17. doi: 10.12688/f1000research.18411.1
299. Huang I, Lim MA, Pranata R. Diabetes Mellitus is Associated With Increased Mortality and Severity of Disease in COVID-19 Pneumonia - A Systematic Review, Meta-Analysis, and Meta-Regression. Diabetes Metab Syndr (2020) 14:395–403. doi: 10.1016/j.dsx.2020.04.018
Keywords: type 1 diabetes, type 2 diabetes, neutrophil, inflammation, infection, NETosis, hyperglycaemia, COVID-19
Citation: Dowey R, Iqbal A, Heller SR, Sabroe I and Prince LR (2021) A Bittersweet Response to Infection in Diabetes; Targeting Neutrophils to Modify Inflammation and Improve Host Immunity. Front. Immunol. 12:678771. doi: 10.3389/fimmu.2021.678771
Received: 10 March 2021; Accepted: 10 May 2021;
Published: 03 June 2021.
Edited by:
Sonja Vermeren, University of Edinburgh, United KingdomReviewed by:
Vinicius Frias Carvalho, Oswaldo Cruz Foundation (Fiocruz), BrazilCopyright © 2021 Dowey, Iqbal, Heller, Sabroe and Prince. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rebecca Dowey, UmRvd2V5MUBzaGVmZmllbGQuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.