 Yan-mei Qiu
Yan-mei Qiu Chun-lin Zhang
Chun-lin Zhang An-qi Chen†
An-qi Chen† Hai-ling Wang
Hai-ling Wang Ya-nan Li
Ya-nan Li Bo Hu
Bo Hu- Department of Neurology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Blood-Brain Barrier (BBB) disruption is an important pathophysiological process of acute ischemic stroke (AIS), resulting in devastating malignant brain edema and hemorrhagic transformation. The rapid activation of immune cells plays a critical role in BBB disruption after ischemic stroke. Infiltrating blood-borne immune cells (neutrophils, monocytes, and T lymphocytes) increase BBB permeability, as they cause microvascular disorder and secrete inflammation-associated molecules. In contrast, they promote BBB repair and angiogenesis in the latter phase of ischemic stroke. The profound immunological effects of cerebral immune cells (microglia, astrocytes, and pericytes) on BBB disruption have been underestimated in ischemic stroke. Post-stroke microglia and astrocytes can adopt both an M1/A1 or M2/A2 phenotype, which influence BBB integrity differently. However, whether pericytes acquire microglia phenotype and exert immunological effects on the BBB remains controversial. Thus, better understanding the inflammatory mechanism underlying BBB disruption can lead to the identification of more promising biological targets to develop treatments that minimize the onset of life-threatening complications and to improve existing treatments in patients. However, early attempts to inhibit the infiltration of circulating immune cells into the brain by blocking adhesion molecules, that were successful in experimental stroke failed in clinical trials. Therefore, new immunoregulatory therapeutic strategies for acute ischemic stroke are desperately warranted. Herein, we highlight the role of circulating and cerebral immune cells in BBB disruption and the crosstalk between them following acute ischemic stroke. Using a robust theoretical background, we discuss potential and effective immunotherapeutic targets to regulate BBB permeability after acute ischemic stroke.
Introduction
Stroke is the second leading cause of death worldwide and is characterized by a high rate of morbidity, mortality, and disability. Therefore, there is a substantial social and economic burden associated with stroke (1). Stroke is classified into two types: ischemic stroke and hemorrhagic stroke. Acute ischemic stroke (AIS) accounts for 87% of the total incidence of stroke, and is characterized by a sudden cessation of oxygen and blood supply due to arterial occlusion in local cerebral tissue (2). One of the hallmark pathophysiological features of ischemic stroke is blood-brain barrier (BBB) disruption, which is characterized by an increased permeability due to the degradation of tight junctions (TJs) and an enhancement in endothelial vesicle transport. As a result, there is an uncontrolled influx of blood-borne cells, macromolecules, and fluid, resulting in devastating cytotoxic and vasogenic edema and life-threatening hemorrhagic transformation (HT) (3–5). Ischemic stroke patients with severe BBB disruption present worse National Institutes of Health Stroke Scale (NIHSS) scores, functional prognosis, and increased mortality rates compared to those with mild BBB disruption (6, 7). As such, ameliorating BBB damage can improve neurologic outcomes in stroke patients (8).
In recent years, the understanding of the inflammatory mechanism of BBB disruption during AIS has improved. Ischemia manifests weakened inflammation-inhibiting signals, such as fractalkine [CX3C motif chemokine ligand 1 (CX3CL1)] as well as enhanced “help me” signals, such as damage-associated molecular patterns (DAMPs), from dying or necrotic neurons and glia, which may activate quiescent resident microglia, astrocytes, and pericytes by pattern recognition receptors (PRR) (9, 10). Activated cerebral immune cells upregulate the expression of pro-inflammatory factors and chemokines, as well as activate matrix metalloproteinases (MMPs) to compromise BBB integrity and recruit peripheral immune cells to the injured areas, leading to secondary BBB damage. In comparison, the transformation of immune cells to alternative phenotypes (N2 neutrophils, M2 macrophages, and A2 astrocytes) may protect the BBB against inflammatory injury by promoting the resolution of inflammation and angiogenesis, thereby re-establishing the integrity of the BBB in the latter phase of stroke. In addition, immune cells in the central nervous system (CNS) engage in crosstalk with infiltrated peripheral immune cells, forming a complicated inflammatory network that may indirectly influence BBB integrity. Therefore, circulating and cerebral immune cells play a profound and dual role in BBB disruption following ischemic stroke.
Currently, the first-line AIS therapies include intravenous alteplase(rt-PA) administration and mechanical thrombectomy (MT) (11). Medication-induced thrombolysis is the only approved thrombolytic therapy for AIS. However, MT is highly beneficial to AIS patients with large vessel occlusion. AIS reperfusion therapies by rt-PA and MT are limited to only 5 - 10% of patients because of the narrow window in which they are effective. There are only 4.5 hours after AIS onset for intravenous thrombolysis and up to 24 hours for mechanical thrombectomy, depending on the availability of imaging techniques to diagnosis AIS (4, 5). Because of the limited options for treatment of AIS, the identification of novel candidate drugs for treating AIS is an urgent challenge. Given the critical role of immune cells in BBB disruption, an in-depth exploration of the inflammatory mechanism of BBB disruption is crucial to identifying valuable therapeutic targets for treating ischemic stroke.
In this review, we first introduce the structure and function of the BBB. Then, we discuss the role of circulating and cerebral immune cells in BBB disruption and the crosstalk between them following acute ischemic stroke. Finally, we summarize the current immunoregulation targets to find potential and effective immunotherapy targets to regulate BBB destruction after acute ischemic stroke.
Physiological Structure and Function of Blood-Brain Barrier
The BBB is a unique and tightly regulated anatomical interface between circulating blood and the CNS and is collectively formed by endothelial cells (ECs) the end-feet of astrocytes, and pericytes embedded in the basement membrane of capillary vessel (12). Continuous non-fenestrated ECs sealed by TJs constitute the innermost luminal side of the BBB (13). The ECs are surrounded by pericytes embedded in the capillaries and the end-feet of astrocyte that ensheathe the basement membrane. The intimate interactions between the ECs, pericytes, astrocytes, microglia, and neurons form the neurovascular unit, which is necessary for ensuring the functional integrity of the CNS (14).
The BBB plays a vital role in maintaining homeostasis in the neuronal microenvironment of the CNS through a number of different mechanisms. First, the BBB is a solid and highly regulated physical barrier that prevents exogenous neurotoxic components from entering the CNS. Distinguished from peripheral ECs, cerebral ECs form a continuous monolayer without fenestrations characterized by a low transcytosis rate and specified TJs (15, 16). TJs encompass three transmembrane proteins: claudins, occludin, and junction adhesion molecules (JAMs) (13, 17). Adherens junctions comprise transmembrane proteins, vascular endothelial (VE)-cadherin, with extracellular segments homotypically interacting and cytoplasmic domains binding to the plaque proteins, such as β-catenin, γ-catenin, and p120-catenin (18). Tight junctions (TJs) and adherens junctions form a circumferential zipper-like seal between adjacent ECs, positioning them as a gatekeeper for limiting paracellular permeability. The coverage ratio of pericytes in the brain is the highest among the vasculatures throughout the whole body (19). Due to the low permeability of the BBB, only small lipophilic molecules (<400kD) and gaseous molecules (e.g., N2, CO2, and O2) are capable of entering the cerebral parenchyma, while the delivery of blood-derived macromolecules into the cerebral parenchyma is severely limited (12).
The BBB is also an efficient and tightly regulated transport barrier that enables the delivery of essential nutrients to the CNS to meet the high-energy demands of neuronal activity. Cerebral ECs feature much higher numbers of mitochondria than peripheral ECs (12). Cerebral ECs also express thousands of ion channels, receptors, transporters, and active efflux pumps to selectively regulate molecular transport between the blood and the brain (20). In addition, the BBB is a molecular metabolic barrier. The unique vascular metabolism of the BBB ECs can change the solubility, reactivity, and transport properties of molecules. Last but not least, BBB is an immunologic barrier that blocks a variety of circulating leukocytes from entering the CNS and prevents CNS-specific antigens from infiltrating into the peripheral immune system. The extravasation of peripheral immune cells is dependent on adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1) and intercellular cell adhesion molecule-1 (ICAM-1), which are expressed at extremely low levels in ECs and pericytes (21). Under normal physiological conditions, ECs suppress pro-inflammatory gene expression and quiesce circulating leukocytes (12). Therefore, the BBB has a direct role in regulating immune reactions within the CNS rather than act as a neutral and passive barrier. The BBB can modulate the function and fate of infiltrating immune cells in healthy states (22). The junctions of ECs, low expression region of the extracellular matrix(ECM), and gaps between pericytes are situated in parallel longitudinally at the venule of capillaries, forming a preferential pathway for enabling the diapedesis of peripheral leukocytes (23). Under systemic inflammatory conditions, such as in septic encephalopathy and neurodegenerative diseases, excessive immune responses damage TJs and ECs (24). Above all, the BBB acts as a physical barrier, transport barrier, metabolic barrier, and immunological barrier, all of which allow it to maintain delicate and narrow homeostasis in CNS.
Role of Peripheral Immune Cells in the Disruption of BBB in Stroke
The Temporal Trend of Peripheral Immune Cells Infiltrating the Brain
Numerous peripheral immune cells infiltrate into the ischemic hemisphere in successive order after acute ischemic stroke. Circulating neutrophil counts displayed an exponential increase within a few hours after symptom onset and remained elevated for one week (25). Experimental studies have shown that neutrophil accumulation in the ischemic hemisphere increased significantly after 3 hours, reaching maximum accumulation after 24 hours, followed by a steady dissipation over 7 days (Figure 1) (26). The levels of strong neutrophil chemoattractants, such as chemokine-like factor 1 (CKLF-1), C-X-C motif chemokine ligand 1 (CXCL1), C-X-C motif chemokine ligand 2 (CXCL2), C-X-C motif chemokine ligand 5 (CXCL5), monocyte chemoattractant protein-1/C-C chemokine ligand 2 (MCP-1/CCL2), C-C chemokine ligand 3(CCL3), and C-C chemokine ligand 5 (CCL5), all increased dramatically after stroke. These chemoattractants enable neutrophils to be the first blood-borne immune cells to migrate to the injured brain tissue by binding to C-C chemokine receptor 5 (CCR5) and C-X-C chemokine receptor 1 (CXCR1) on the surface of neutrophils (27). CKLF-1 increased significantly at 3 hours poststroke, which bind to CCR5 on the surface of neutrophils. The binding of CKLF-1 to CCR5 mediate neutrophils migration through the Akt/GSK-3β and mitogen-activated protein kinase (MAPK) pathways (28).
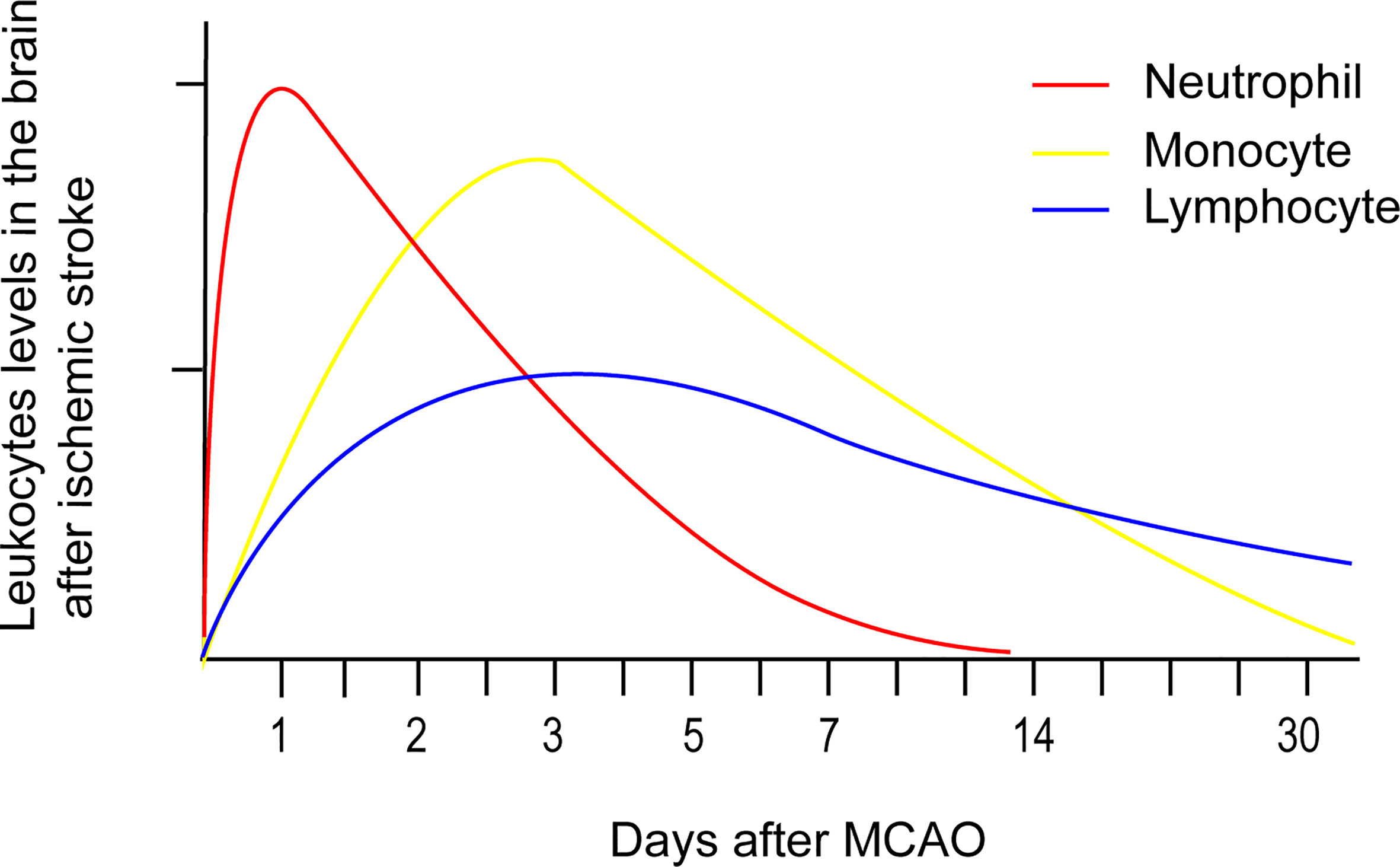
Figure 1 Temporal profile of peripheral immune cells accumulation after stroke onset based on experimental data. Neutrophil accumulation in the ischemic hemisphere increased significantly after 3 hours, reaching maximum accumulation after 24 hours, followed by a steady dissipation over 7 days. Monocyte counts in the ipsilateral hemisphere robustly increased after 1 day, peaked after 3 - 7 days, and then returned to baseline levels after 14 days. Lymphocytes extravasate into the injured hemisphere in smaller counts and longer persistence compared to the former two immune cells. The accumulation of T cells in the ischemic hemisphere significantly increased as early as 24 hours after AIS, peaked at 3 days, and persisted for 1 month.
Overall monocyte counts were dramatically increased in the blood of patients with acute ischemic stroke over the course of 16 days after onset. In addition, monocyte counts (Figure 1) in the ipsilateral hemisphere robustly increased after 1 day, peaked after 3 - 7 days, and then returned to baseline levels after 14 days (29). Monocyte recruitment is greatly dependent on CCR2, the receptor on surface of the classical monocytes that undergoes ligation with its ligand CCL2. T lymphocyte counts in the blood of acute ischemic stroke patients exponentially declined over 7 days, with the lowest count observed after 12 hours (25). Lymphocytes extravasate into the injured hemisphere in smaller counts and longer persistence compared to the former two immune cells. The accumulation of T cells (Figure 1) in the ischemic hemisphere significantly increased as early as 24 hours after AIS, peaked at 3 days, and persisted for 1 month (30). Double negative T cell (DNT) and CD8+T cell infiltration increased significantly for 3 - 24 hours following ischemic insult (26), while infiltrating CD4+T cells did not increase until 24 hours in a permanent middle cerebral artery occlusion (pMCAO) model (31). Regulatory CD4+ T cells (Tregs) were found to increase in the lesion area in a delayed manner after several days and persisted 30 days after stroke (32). Natural killer (NK) cells infiltrated the ischemic region as early as 3 hours after stroke, peaked at 12 hours, and remained elevated for at least 4 days (33). The attraction of Treg and gammadeltaT cells (γδT cell) were mediated by the CCL5/CCR5 and CCL6/CCR6 axes separately (34, 35). NK cells were recruited to the ischemic region through the IP-10/CXCR3 and CX3CL1/CX3CR1 axes (36).
Intravascular Immune Cells Exert Deleterious Effects on BBB Disruption After Acute Ischemic Stroke
Neutrophils roll and adhere to ECs at venules to occlude blood flow, a process known as no-reflow phenomenon. Myeloid-specific α9-deficiency, which is a method to uniquely block neutrophil adhesion, remarkedly decreased the infarct volume and neurological deficit in a murine transient middle cerebral artery occlusion (tMCAO) model (37). Polymorphonuclear cells were detected in capillaries and post-capillary venules in the ipsilateral hemisphere as early as 30 minutes after stroke and reached a peak at 12 hours (38). P-selectin glycoprotein ligand-1 (PSGL-1) and macrophage-1 antigen (Mac-1) were elevated on the surface of neutrophils within a few hours after ischemic stroke (39). Meanwhile, P-selectin and ICAM-1 were up-regulated on ECs. PSGL-1/P-selectin and Mac-1/ICAM-1 interactions between neutrophils and ECs not only caused no-reflow but also increased paracellular permeability (40, 41). Upon adhesion, ICAM-1 induced a series of dedicated downstream pathways within ECs, including transient increases in the concentration of intracellular free calcium, subsequent myosin light chain kinase (MLCK) activation, focal adhesion kinase phosphorylation, and small GTPase Rho/ROCK activation, further enhancing actin polymerization and the breakdown of adherens junctions (42). Thus, neutrophil adhesion resulted in the contraction of ECs and paracellular hyperpermeability. In addition, neutrophil-platelet aggregations mediated by transcellular interactions between Mac-1and the GPIb counter receptor also increased thrombus formation and BBB permeability in experimental stroke (43).
T cells infiltrated the human brain profoundly for as long as 14 days following stroke (30). Upon arriving at the microvascular interface of artery occlusion, T cell capture, roll, arrest, crawl, and across the BBB through interactions with ECs via PSGL/P-selectin, very late antigen 4 (a4β1)/VCAM-1, function-associated antigen-1 (LFA-1)/ICAM-1 signaling (44). Receptor responsible for recruiting and extravasating including A kinase anchoring protein 7 (AKAP7) and CD74, were upregulated in T cells in patients with AIS. The increased expression of AKAP7 and CD74 was associated with enhanced hyperintense acute reperfusion marker (HARM), infarction size, and NIHSS score (45, 46). HARM is a delayed enhancement of the cerebrospinal fluid space that is observed on post-contrast MRI and is indicative of BBB disruption. Intravascular T lymphocytes interplay with ECs and platelets to facilitate the adhesion of platelets and leukocytes leading to microthrombus formation, which is known as thrombo-inflammation. Intravascular Tregs were found to induce enhanced microvascular dysfunction via the LFA-1/ICAM-1 pathway, which increased the infarction damage within 24 hours in a tMCAO model (47). When penetrating the cerebral parenchyma, T lymphocytes produced MMPs, reactive oxygen species (ROS), and pro-inflammatory factors to degrade the extracellular matrix (ECM) and damage ECs (48). However, T lymphocytes may also protect BBB integrity and prevent HT by binding to P-selectin on the surface of platelets (49).
Neutrophil
Neutrophils and BBB Disruption in Human AIS
Neutrophils are well-known for their destructive role in BBB disruption after ischemic stroke. Clinical observations support the detrimental effects of neutrophils on the integrity of the BBB after ischemic stroke. The elevated neutrophil-to-lymphocyte ratio (NLR) in circulation at admission is an essential predictor of HT, especially parenchymal hematoma and symptomatic intracranial hemorrhage (sICH) (50, 51). Higher NLR was independently correlated to early neurological deterioration 24 hours post-stroke, as well as poor functional outcome and mortality after 3 months in patients with AIS (52, 53). Higher neutrophil counts and myeloperoxidase (MPO) plasma concentration in plasma were positively correlated with stroke severity and worsened outcomes (54).
Evaluating the function of circulating neutrophils in patients with AIS can reveal the overwhelming intracellular change in neutrophils in AIS. The percentage of the overactive senescent neutrophil subpopulation increased following ischemic stroke, which was characterized by elevated adhesion molecule and elastase expression as well as ROS production (55). Transcriptomic analysis revealed that mammalian target of rapamycin (mTOR), oxidative phosphorylation, growth factor signaling, and calpain proteases signaling pathways were generally up-regulated in circulating neutrophils of ischemic stroke patients (56). MMP-9-positive neutrophils were detected extensively surrounding the cerebral microvessels in the hemorrhagic and infarcted regions of patients with fatal HT after ischemic stroke, which was associated with high MMP9 expression levels and severe basal lamina collagen IV degradation (57). Neutrophil extracellular traps (NETs), whose components are mainly derived from neutrophils, were significantly increased in the plasma of ischemic stroke patients, associated with stroke severity and mortality (58). By immunostaining analysis, NET constituted more than 13% of the whole thrombi from patients subjected to endovascular thrombectomy (59).
Role of Neutrophils in BBB Disruption in AIS: Preclinical Evidence
Targeting neutrophils in rodents subjected to experimental stroke significantly decreased the infarct volume. Myeloid Mcl1 ablation protected mice from ischemic injury by selectively inhibiting the presence and infiltration of neutrophils (60). Doxorubicin (DOX)-conjugated protein nanoparticles (NPs) selectively targeted inflammatory neutrophils to induce apoptosis. Administration of DOX-NPs notably restored behavioral outcome in a cerebral ischemic/reperfusion model (61). These clinical and experimental stroke results suggested that, not only were there strong links between neutrophils and BBB disruption, but also targeting neutrophils may protect the BBB against inflammatory injury to improve stroke outcomes.
Neutrophils potentiate BBB disruption in many ways (Figure 2). First, it is well-known that neutrophils produce ROS, proteases (MMPs, proteinase 3, elastase), lipocalin-2(LCN-2), and NETs to degrade the BBB structure. Excessive ROS (e.g., superoxide anions, peroxynitrite, and hydrogen peroxide) production engenders damages to junction proteins (mainly cadherin-β-catenin complex, occludin, ZO-1, and claudin-5) and a reorganization of the endothelial cytoskeleton through hyperpermeability associated signaling pathways (e.g., MLCK, PKC, MAPK, and Rho GTPases) (62). Proteases, including MMPs, proteinase 3, and elastase were also released during neutrophil degranulation during ischemic stroke. MMPs directly facilitate the degradation of the BBB structure by cleaving glycocalyx, VE-cadherin, focal adhesion components, and basement membrane proteins, such as collagen IV (63). LCN-2, also termed neutrophil gelatinase-associated lipocalin, was detected in the serum of tMCAO mice as early as 1 hour following occlusion. The level of LCN-2 peaked after 24 hours and then subsided after 48 hours in the ipsilateral hemispheres (64). LCN-2 exacerbateed Evans blue (EB) extravasation and brain edema by reducing the expression of junction proteins, namely claudin-5 and β-catenin, during AIS. Therefore, LCN-2 neutralization can attenuate BBB disruption (65). However, another study found that LCN-1 reversed the decreased endothelial translocation of ZO-1 and VE-cadherin from the membrane to the cytoplasm of ECs after tumor necrosis factor-alpha (TNF-α) treatment, which mitigated BBB disruption (66). This suggested that LCN influences BBB permeability through various mechanisms under different situations.
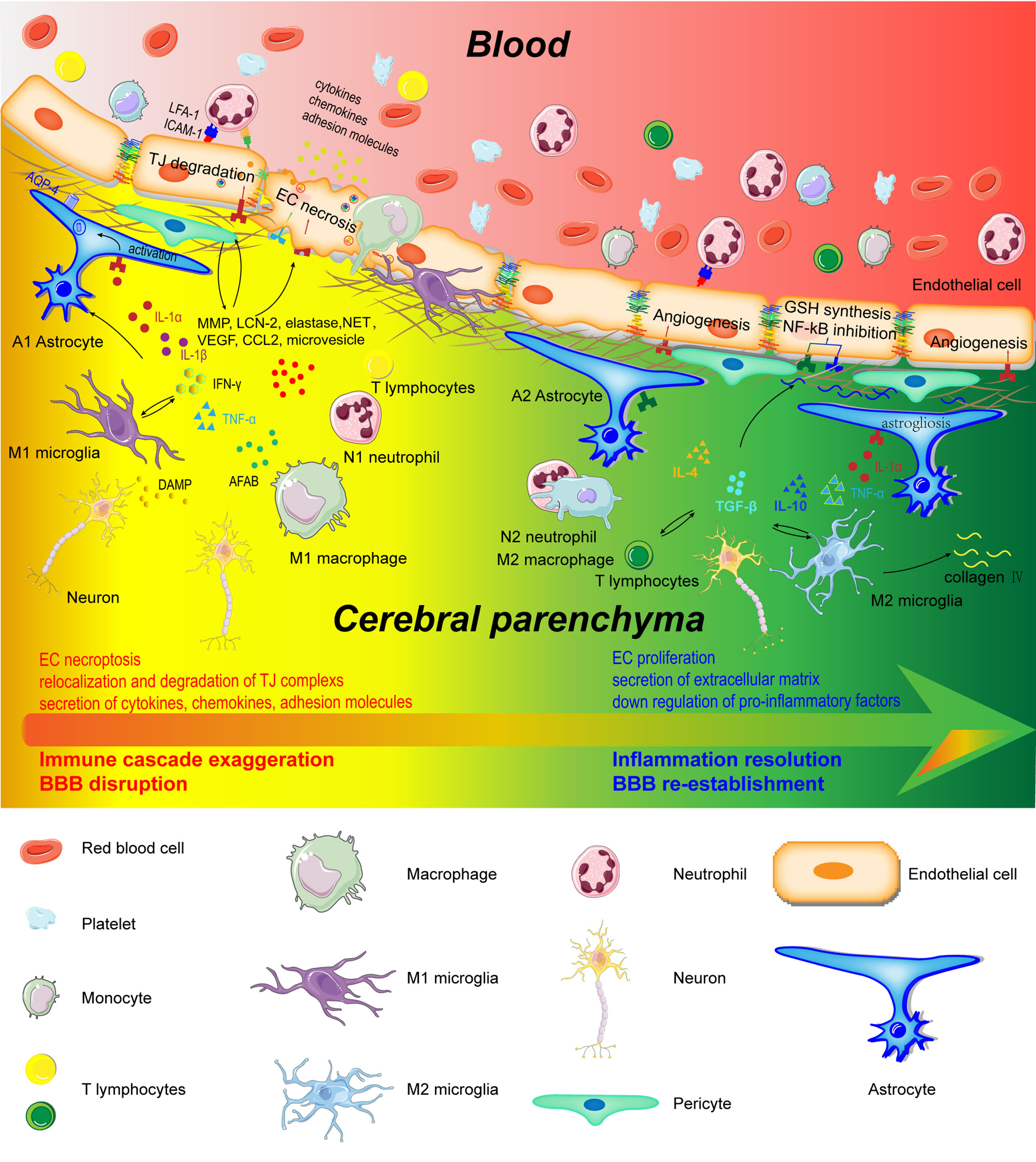
Figure 2 Schematic representation of cerebral and peripheral immune cells regulating BBB integrity during early and later phases of ischemic stroke. Infiltrated neutrophils produce proteases (MMPs, proteinase 3, and elastase), lipocalin-2, NET, microvesicles, cytokines, and chemokines to destroy the BBB structure. M1-type monocytes secret cytokines and chemokines to degrade TJs. Perivascular microglia phagocyte ECs, which directly lead to endothelial dysfunction and BBB disintegration. In addition, M1 microglia disrupt BBB integrity through the production and secretion of pro-inflammatory factors (IL-1α, IL-1β, IL-6, TNF-α, IFN-γ, and CCL2), MMP9, and VEGF. A1 astrocytes directly exert deleterious effects on BBB through increasing VEGF, cytokines (IL-1β, IL-6, and TNF-α), chemokines (CCL2 and CCL5), MMP, and LCN-2. However, in the recovery phase of AIS, these immune cells contribute to inflammation resolution and BBB re-establishment. N2 neutrophils promote engulfment of neutrophils by macrophages and inflammation resolution. Monocyte-derived M2 macrophages facilitate the expression of collagen IV and efferocytosis. Microglia directly protect BBB integrity through the secretion of IL-10 and TGF-β. A2 astrocytes are capable of secreting IL-2, IL-10, and TGF-β to accelerate inflammation resolution.
NETs are web-like structure comprising nuclear and granular contents, mainly double-stranded DNA and granule proteins, such as MPO and elastase. H3Cit is a common biomarker of NET formation. The concentration of H3Cit began to increase as early as 6 hours after AIS onset and was present its highest concentration after 3 - 5 days, with distribution predominantly in the peri-infarct cortex. In fact, 78.7% of H3Cit was derived from neutrophils. NETs increased BBB permeability and extravascular IgG deposits, and decreased pericyte coverage in experimental stroke (67).
Second, neutrophils play a detrimental role in BBB integrity during AIS through the production of neutrophils-derived cytokines [e.g., interleukin-1β (IL-1β), interleukin-6 (IL-6), interleukin-8 (IL-8), and TNF-α) and chemokines (CCL2, CCL3, and CCL5)] (68). CCL2, also known as MCP-1, reduced the transendothelial electrical resistance and EB extravasation through TJ disassembly via the internalization of occludin and claudin-5 in the p-p38 MAPK signaling pathway (69, 70).
Furthermore, neutrophil-derived microvesicles were found to alter the transcriptomic profile of human cerebral microvascular ECs to dysregulate TJ and versicle transport in vitro (71).
Interestingly, recent evidence has suggested that neutrophils may adopt an anti-inflammatory phenotype by expressing of Ym1 and CD206 (72). Accelerated by peroxisome proliferator-activated receptor gamma (PPAR-γ) agonists while being hampered by Toll-like-receptor (TLR) activation, the N2 polarization of neutrophils promoted the engulfment of neutrophils by microglia/macrophages, which led to reductions in brain edema and infarct volume (73, 74). Neutrophils may also have a dual function in the evolution of cerebral ischemic damage by accelerating inflammatory response or the resolution of inflammation. Skewing neutrophils toward the N2 phenotype rather than comprehensively suppressing the infiltration and function of neutrophils may be a novel and promising strategy for treating ischemic stroke in the future.
Monocytes
Monocytes and BBB Disruption in Human AIS
The exact role of monocytes in BBB disruption remains unclear, since clinical observations are controversial. Some studies suggested that their roles were harmful to BBB disruption. Because increased numbers of circulating monocytes were associated with HARM, larger AIS volume, worsened NIHSS score, and poorer outcomes compared to lower numbers of circulating monocytes (75). Higher monocyte-to-HDL (high-density lipoprotein) ratios (MHR) and monocyte-to-lymphocyte ratios (MLR) were associated with higher HT risk and poorer outcomes compared to lower ratios (76, 77). Monocytes were found to be accumulated and randomly distributed in erythrocytic thrombi, engendering a higher NIHSS score (78). However, other investigations argued for the opposite view. Because monocyte counts did not change during AIS, the counts cannot predict long-term mortality (79, 80). A recent investigation demonstrated that lower MHRs were independently associated with increased HT risk, especially sICH in AIS patients (81). The possible explanation for these paradoxical results may be the functional heterogeneity. In human, there are three monocyte subsets: classical CD14++CD16−, intermediate CD14+CD16+, and alternative/non-classical CD14+CD16++. However, in mice, there are two subpopulations: classical Ly6C+CCR2highCX3CR1low and alternative/non-classic Ly6C−CCR2lowCX3CR1high. Elevated levels of classical monocytes were associated with early clinical worsening and higher mortality in ischemic stroke, while non-classical monocytes were significantly decreased in stroke patients. Decreased levels of non-classical monocytes was inversely related to poor prognosis (82). Thus, distinct subpopulations may exert different roles in BBB integrity and stroke outcome.
Role of Monocytes in BBB Disruption in AIS: Preclinical Evidence
In experimental stroke, inhibiting monocyte recruitment by blocking or depleting CCR2 significantly protected against brain edema but impaired long-term recovery (83, 84). Pro-inflammatory monocytes were the predominant monocytes subsets infiltrating into the injured brain, while alternative monocytes tend to be redundant in AIS (85, 86). However, it remains unclear what role non-classical monocytes play in BBB disruption after AIS.
This counterintuitive result might be explained by the phenotype transformation of monocytes. The phenotype and function of blood-derived monocytes were not univariable during AIS, depending on the inflammatory context. The phenotype of monocyte shifted from pro-inflammatory M1 dominant day 3 to anti-inflammatory M2 dominant 7 days after stroke, indicating a functional transformation of the monocytes from amplifying the immune response to the resolution of inflammation (87). Infiltrating monocytes subsequently down-regulated Ly6C, up-regulated F4/80, and acquired macrophage features. Interestingly, M2 macrophages can be further subdivided into M2a, M2b, and M2c (88, 89), and some authors also classify the M2d subtype (90). M2a macrophages were typically induced by IL-4 and IL-13. IL-13 administration effectively promoted the transformation of macrophages from the M1 state to the M2a state at 3 days following permanent ischemia in mice (91). M2b macrophages were induced by the immunoglobulin Fcγ receptor, lipopolysaccharide and IL-1β complex. M2c macrophages were induced by the anti-inflammatory cytokine IL-10 and glucocorticoids, while M2d macrophages were induced by the stimulation of IL-6 and adenosine.
Monocytes destroy BBB integrity in the early phase of AIS (Figure 2). Monocytes were first observed within the injured region of capillaries and venules 4-6 hours after occlusion in rats (38). M1-type monocytes secrete ROS, cytokines, and chemokines through the activation of the inflammasome to degrade TJs between ECs in the BBB. The polarization of the M1 pro-inflammatory phenotype was promoted through the activation of acute purinergic receptor P2X4 (P2X4R) (92). Blood-borne monocytes exacerbated secondary inflammatory damage to the BBB by up-regulating triggering receptor expressed on myeloid cells 1, assisting other innate immune receptors (93). In addition, monocytes upregulated adipocyte fatty acid-binding protein expression to potentiate the MMP9-mediated degradation of TJs by enhancing JNK/c-Jun signaling (94). Thus, monocytes can damage BBB in a paracrine manner.
On the other hand, monocyte-derived M2 macrophages may protect the BBB against ischemic damage by vascular remodeling, physical attachment, and inflammation resolution. RNA sequencing revealed the overactivation of JAK1, JAK3, and STAT3, as well as angiogenesis in monocytes (56, 95). Monocytes and macrophages with elongated morphologies were observed lining the vessels in the infarct core and peri-infarction regions (85). The adhesion of macrophages to ECs directly pulled the ruptured ends, narrowing the lesion through the polymerization of microfilaments and phosphatidylinositide 3-kinase- or Rac1- mediated mechanical traction forces (96). All four M2 macrophages subtypes acquired enhanced phagocytic ability and expressed IL-10, contributing to the resolution of inflammation. Once differentiated into mature phagocytes, monocytes were found to promote the expression of collagen IV to prevent HT in the smad2/TGF-β signaling pathway (97). Furthermore, efferocytosis-related genes were robustly up-regulated in macrophages (98). Therefore, hematogenous macrophages contribute to BBB recovery indirectly through inflammation resolution and efferocytosis.
Despite being an M2 subtype, the function of macrophages may be diverse. M2a macrophages express various anti-inflammatory and neurotrophic factors such as arginase 1 (Arg1) and insulin-like growth factor-1. M2c macrophages increase the expression of TGF-β, CD163, and sphingosine kinase. IL-13 induced transition from M1 macrophage to M2a macrophages attenuated the pro-inflammatory cascade and neurological disorders (91). However, M2b macrophages increase the production of pro-inflammatory factors including IL-1β, IL-6, TNF-a, which may potentiate inflammation and increase the BBB permeability in the early phase of AIS (99). M2d macrophages secrete VEGF-A and TNF-α, all of which were found to be deleterious to BBB integrity in AIS. Investigations on the specific roles of these different M2 macrophages in BBB disruption during AIS are relatively lacking. Therefore, more in-depth studies are warranted to develop more insights into these processes.
T Lymphocytes
The Gut-Brain Axis Contributes to the Peripheral T Lymphocytes Subpopulation Disorder and BBB Disruption
Clinical investigations revealed that the differentiation of T lymphocyte pools skewed to pro-inflammatory subpopulations after AIS. T lymphocytes represent a conglomeration of different subpopulations with tremendous heterogeneity. The percentage of immunosuppressive Tregs was significantly decreased in response to AIS, which was associated with poor stroke outcome (100). CD39+ Tregs are typically representative of functionally active Tregs and are the most strongly reduced subpopulation of T lymphocytes after stroke, indicating that the immunosuppressive function of Tregs was largely impaired during AIS (101). Thus, Tregs-derived anti-inflammatory factors, including transforming growth factor-beta (TGF-β) and interleukin-10(IL-10), were reduced after stroke (102). In contrast, the increased proportion of pro-inflammatory T helper 17 (Th17) cells and γδT cells in the serum of AIS patients was accompanied by elevated levels of pro-inflammatory factors, such as interleukin-17A (IL-17A), interleukin-23 (IL-23), IL-6, and IL-1β (102, 103). CD4+CD28- T cells have the potential to amplify immune responses and tissue damage. The significantly increase in CD4+CD28- T cells was associated with a higher recurrence/death rate in ischemic stroke (104). The levels of Tim-3, a specific marker of T-helper 1 (Th1) cells, were elevated, along with TNF-α and IL-17, in circulating blood (105). These Th1 cells may participate in the pathogenesis of AIS by enhancing the immune cascade.
Interestingly, the gut-brain axis, which represents the interaction between the brain and the gut, was recently shown to demonstrate a causal relationship between abnormalities in T cell subpopulation and BBB disruption during ischemic stroke. Acute infarction in the brain induced microbiota dysbiosis in the gut. Patients with severe ischemic stroke showed a more significantly reduced intestinal diversity compared to those without AIS, with the enrichment of trimethylamine-N-oxide (TMAO)-producing bacteria, opportunistic pathogens, and loss of bacteria implicated in butyrate production (106, 107). In turn, gut microbiota dysbiosis resulted in deteriorated stroke outcomes. The plasma TMAO levels of AIS patients increased significantly post-stroke, and these elevated levels were associated with a worse modified Rankin Scale (mRS) score after 3 months (108). However, other studies demonstrated that TMAO levels in blood declined or did not change in the plasma of AIS patients (109). TMAO intensified ischemic damage through three main mechanisms: by increasing the proportion of proinflammatory monocytes; by activating the nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome to amplify the immune response, and by stimulating ROS production in the mitochondria to damage ECs (110, 111).
The mechanism underlying the attenuation of peripheral T lymphocytes and BBB disruption was explored in experimental stroke. More than 70% of immune cells were pooled in the gut, where the balance of the peripheral T lymphocytes was skewed toward pro-inflammatory cells after ischemic stroke, from where the lymphocytes migrate to the cerebral infarction area. The histamine (HA)/gut histamine receptor mediated the elevation of pro-inflammatory factors [IL-6, TNF-α, Interferon-gamma (IFN-γ), and granulocyte-colony stimulating factor (G-CSF)] in the gut, facilitating the differentiation into pro-inflammatory T cells (112). The gut microbiota dysbiosis also abolished the ability of Dendritic cells (DCs) to drive Treg differentiation and promoted the ability of DCs to induce γδT cell differentiation (113). Therefore, targeting the gut microbiota may be a promising treatment for ischemic stroke.
The Disturbed Balance Between Pro-Inflammatory Subsets and Anti-Inflammatory Subsets Contributes to BBB Disruption: Preclinical Evidence
The enhanced and elevated pro-inflammatory T cells accompanying AIS directly degrade the BBB through the production of cytokines (Figure 3). Th1 and Th17 cells degraded TJs and compromised BBB integrity by secreting IFN-γ, IL-17, and IL-21 in the acute phase of ischemic stroke (114, 115). γδ T cells have been reported to be pathogenic in stroke outcomes through IL-17. IL-17 deteriorated BBB integrity by decreasing the expression of occludin and ZO-1, and it facilitated the recruitment of monocytes and neutrophils by increasing CCL2 and CXCL1 expression in ECs (116). IL-17 also reduced the expression of TJs in ECs by increasing ROS production in an NADPH oxidase- or xanthine oxidase-dependent manner (117). Interestingly, Th17 cells produced IL-26, a BBB-protective cytokine, that up-regulated the expression of occludin, claudins, and ZO-1 in ECs in an in vitro BBB model (118). However, whether or not IL-17 protects intact BBB in ischemic stroke remains unknown. NK cells significantly increased cerebral microvascular EC permeability under oxygen-glucose deprivation (OGD) conditions (33). NK cells aggregated the ischemic lesion through the production of IFN-γ and ROS (36). Previous studies confirmed that CD8+T cells potentiated the progression of ischemic stroke by granzyme-b and FasL induced cytotoxicity and by TNF-α and IFN-γ (119). Recently, Fudong Shi’s lab reported that CD8+T cells engendered an increased EC permeability by degrading claudin-5 as early as 1 hour after OGD treatment (120). However, the exact and comprehensive effects of CD8+T cells on BBB integrity remain much to be explored.
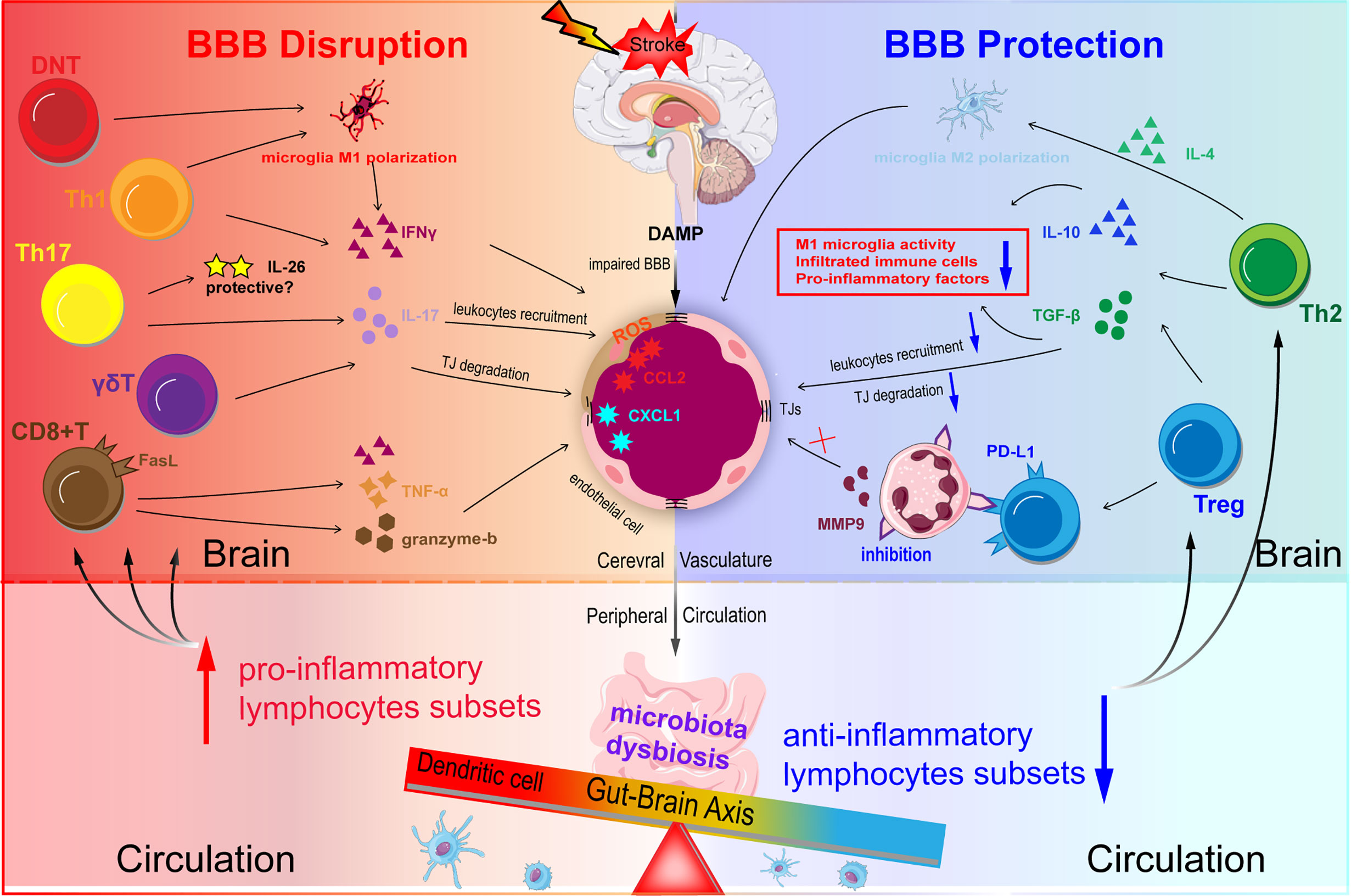
Figure 3 Schematic representation of T lymphocytes homeostasis disorder regulating BBB integrity in ischemic stroke. DAMP and chemokines are released from the ischemic brain via impaired BBB. Acute infarction in the brain can induce microbiota dysbiosis in the gut. Microbiota dysbiosis abolishes the ability of Dendritic cells (DC) to drive the Treg differentiation and promotes the power of DC to induce γδT cell differentiation. The disturbed balance between pro-inflammatory subsets and anti-inflammatory subsets contributes to BBB disruption during ischemic stroke. Th1 and Th17 may degrade TJs and destroy BBB integrity through secreting IFN-γ, IL-17, and IL-21 in the acute phase. CD8+T cells potentiate the ischemic stroke progression by two main methods: one is granzyme-b and FasL induced cytotoxicity through FasL-PDPK1 pathway and the other is TNF-α and IFN-γ. Treg suppresses the overactivation of resident microglia, infiltrated T cells, and neutrophils, and decrease pro-inflammatory factors (TNF-α, IFN-γ, IL-1β) levels mainly through the action of IL-10 and TGF-β.
In contrast to pro-inflammatory T cells, the immunosuppressive functions of anti-inflammatory T cells were impaired in ischemic stroke. Tregs and Th2 shield the ischemic hemisphere from causing an overactive immune response and aggravating secondary injury progression after AIS. Tregs suppressed the overactivation of resident microglia and infiltrated T cells and decreased the levels of pro-inflammatory factors (TNF-α, IFN-γ, IL-1β) mainly via upregulated expression of IL-10 and TGF-β (121). Interestingly, Tregs directly inhibited the production of MMP9 in neutrophils by transcellularly binding to neutrophils via the program death 1-ligand 1 (PD-L1), which resulted in an improved BBB integrity and neurological performance (122). Tregs also significantly attenuated BBB damage and functional deficits by downregulating the expression of endothelial CCL2 and MMP9 (123).
Role of Cerebral Immune Cells in the BBB Disruption in AIS: Preclinical Evidence
Microglia form the largest immune cell population in the brain and feature small cell soma that are highly ramified, enabling them to continually scan the surrounding environment (124). The activation of microglia was restrained by neurons via CX3CL1/CX3CR1 signaling pathway in healthy brains (125). Astrocytes are another type of cerebral immune cells and express significant amounts of PRRs, and TLRs, as well as mannose, scavenrer, and NOD-like receptors. Astrocytes also secrete cytokines, chemokines, adhesion molecules, and proteases to regulate BBB integrity and form glial scars in response to danger signals (126). In contrast, the role of pericytes in BBB disruption has been investigated from a different perspective recently. Pericytes consistently express PRRs on their surface to induce IL-1β secretion when activated by danger signals and pro-inflammatory factors (127). Pericytes also regulate the diapedesis and migration of leukocytes at the post-capillary venule of the BBB and engage in crosstalk with extraverted immune cells to modulate their function (23). An increasing number of studies have identified the critical immunological function of pericytes in the CNS, but more investigations are needed to better understand the role of pericytes in BBB structure and function.
Microglia
The Spatiotemporal and Phenotypic Dynamic of Microglia After AIS
The number of microglia in the infarct core decreased immediately after stroke. In addition, the number of microglia in the penumbra increased within hours after AIS and peaked at 48 - 72 hours, but microglia persisted in this region for several weeks (128). Microglia in the peri-infarct area were activated within minutes by DAMP, and these activated microglia in the penumbra became amoeboid- or round-like in morphology 12~24 hours after tMCAO (129). Proliferated microglia in the penumbra began migrating to the infarct core 1 day after stroke onset, which was mediated by the annexin-1/casein kinase II pathway (130).
Following morphological changes of microglia, the progression of intracellular signaling pathways and functions are differentiated between M1 pro-inflammatory microglia versus M2 anti-inflammatory microglia. Resident M2 microglia shifted to the M1 phenotype in the peri-infarct region in the early phase (within 48 hours) of ischemic stroke (131). High mobility group box-1 (HMGB1) proteins, heat shock proteins, and purines activated microglia by binding to TLR4 and purinergic receptor P2Y12. Activated microglia primed the nuclear transcription factor-kappa B (NF-kB) for translocation to the nucleus and to formation of the NLRP3 inflammasome, resulting in elevated levels of IL-1β (132). The triggering receptor expressed on the surface of myeloid cells 1 became elevated on the surface of microglia during ischemic stroke, activating CARD9/NF-κB, and NLRP3/caspase-1 proinflammatory pathway (133). M1-like microglia are characterized by the activation of inducible nitric oxide synthase (iNOS) and NF-кB, which produce NO, pro-inflammatory cytokines, and chemokines. The polarization of M2 microglia commenced several days after ischemic stroke and was induced by IL-4, IL-10, vascular endothelial-derived growth factor (VEGF), and GSF secreted from neurons (134). M2 type microglia acquire a phagocytic phenotype, which is indistinguishable from the phenotype of infiltrating macrophages due to the similarity in morphology and lack of specific markers between the two types of immune cells. The majority of macrophages present in the ischemic hemisphere 4-7 days following ischemic stroke derived from microglia (135). Recently, the comparative analysis of microglia-specific and monocytes-derived macrophage-specific transcripts revealed that microglia were more likely skewed toward transition to the M1 harmful phenotype compared to infiltrating macrophages in response to stroke (136).
Microglia Play a Dual Role in BBB Disruption in AIS
Microglia actively participate in the degradation of the BBB after ischemia (Figure 2). Following ischemic stroke, activated microglia in the penumbra engulfed blood vessels by expanding cellular protrusions toward vessels, enabling the extravasation of blood-borne macrophages. In addition, perivascular microglia engulfed ECs, leading to endothelial dysfunction and BBB disintegration (137).
M1 microglia enabled BBB disruption through the production and secretion of pro-inflammatory factors (IL-1α, IL-1β, IL-6, TNF-α, IFN-γ, CCL2), MMP9, VEGF, and ROS. IL-1α and IL-1β mediate their biological functions through binding to IL-1R1 expressed on nearly every cell in the brain. The expression of both IL-1α and IL-1β was dramatically elevated in the ischemic hemisphere 6-24 hours after stroke onset (138). Depletion of IL-1α or IL-1β attenuated BBB disruption and decreased infarct size in experimental stroke. IL-1α is a vital mediator of the activation of ECs to induce CXCL1 and IL-6 expression, and it induced AQP4 expression in astrocytes in ischemic tissue, which engendered both the deterioration of BBB and brain edema (139). Meanwhile, IL-1β increased the degradation and relocation of occluding and ZO-1 in ECs (140), and activated astrocytes with the over expressed GFAP by phosphorylating NF-кB p65. The astrocytes then upregulated VEGF expression after IL-1β stimulation (141). IL-1β also caused an increased secretion of CCL2, CCL20, and CXCL2, and it downregulated the expression of Sonic hedgehog, an important signal in maintaining BBB integrity in astrocytes (142).
Microglia-derived TNF-α induced endothelial necroptosis and the downregulation of occludin through binding to TNF receptor 1 (143). Meanwhile, TNF-α induced MMP9 expression but decreased the expression of collagen IV in ECs, both of which significantly increased BBB permeability in vitro (144). IFN-γ damaged the integrity of BBB by promoting the relocation of ZO-1 and VE-cadherin to cytoplasm in ECs (145). IL-1β, TNF-α, IFN-γ, and IL-6 increased the endothelial expression of ICAM-1 and VCAM-1, facilitating peripheral immune cell infiltration (145). Therefore, inhibition of microglial activation or blocking detrimental cytokines post-stroke may attenuate primary and secondary mechanism of BBB breakdown.
Like hematogenous macrophages, microglia develop an alternatively protective phenotype in the later phase of AIS. M2 microglia promoted inflammation resolution through the secretion of IL-4, IL-10, and TGF-β and the engulfment of immune cells, indirectly protecting against inflammation-induced BBB disruption. IL-4 and IL-10 restricted the expression of IFNγ, TNFα, and IL1β while elevating the levels of anti-inflammatory factors in the ischemic brain by inhibiting the activation of NF-κB (146). TGF-β decreased the levels of TNFα and MCP-1 through the ALK5-p-Smad2/3 signaling pathway (147). IL-10 decreased the expression of ICAM-1 and VCAM-1 in ECs, limiting the infiltration of immune cell into the brain (148). Microglia also secreted zinc finger E-box binding homeobox 1, which downregulated the expression of CXCL1 in astrocytes, thereby attenuating the infiltration of infiltration (149).
Microglia directly protect the BBB from damage through secretion of IL-10 and TGF-β in ischemic stroke. IL-10 protected ECs against ischemic injury by increasing the expression of γ‐glutamylcysteine synthase, a key enzyme in the synthesis of GSH and therefore, a critical player in maintaining the antioxidative status of cells (146). IL-10 also attenuated EC apoptosis by downregulating caspase-3 in the STAT3 pathway (150). Moreover, IL-10 limited the expression and activity of MMPs and enhanced the activity of tissue inhibitors of metalloproteinase (151). Meanwhile, TGF-β significantly prevented BBB leakage and HT by preventing the activity of MMPs and the degradation of ECM (152). Thus, IL-10 and TGF-β contribute to maintaining BBB structure and function in ischemic stroke.
Interestingly, IL-1 and TNF-α may also be participated in long-term BBB repair and angiogenesis in the chronic phase of AIS. IL-1 upregulated the expression of pentraxin-3 in the ischemic brain to promote glial scar formation and ECM production, maintainin BBB integrity and contributing to the resolution of brain edema (153). IL-1α promoted the proliferation and migration of ECs as well as the formation of tube-like structure (154). Microglia-derived TNF-α upregulated the expression of ephrin-A3 and ephrin-A4 in ECs to facilitate angiogenesis in vitro (155). In addition, TGF-β/ALK5 signaling attenuated infarction injury by promoting angiogenesis (156). Therefore, promoting the polarization of M2 microglia in the ischemic brain may accelerate the resolution of inflammation, BBB repair, and functional recovery.
Role of Astrocytes in BBB Disruption in AIS
Emerging studies have suggested that astrocytes are not merely bystanders in immune responses after ischemic stroke. The activation of astrocytes during AIS, which is identified by GFAP positive immunostaining, is widespread and long-lasting. DAMP, including cytochrome c and ATP, activated NF-кB as well as induced the formation of the NLRP3 inflammasome and the release of IL-1β from astrocytes (157). A1 astrocytes were induced by pro-inflammatory factors, such as IL-1α and TNF-α, and the polarization of A1 astrocytes was characterized by the expression of iNOS, which could be modulated by LCN-2 in an autocrine manner (158). Microglia-derived anti-inflammatory factors transformed astrocytes into the protective A2 phenotype by downregulating the expression of P2Y1R in a brain trauma model (159). This indicated that A2 astrocytes might also be induced in the subacute phase of AIS. However, the spatiotemporal dynamic and the mechanism underlying the activation and transformation of astrocytes under acute ischemic injury have not been clarified, so more investigations are required in the future.
The terminology of ischemia-mediated two different phenotypes of A1 and A2 astrocytes parallel the terminology of M1 and M2 microglia in the CNS injury. Thus, similar to microglia, reactive astrocytes play dual roles in BBB breakdown after ischemic stroke.
On the one hand, astrocytes accelerate BBB disruption by amplifying inflammation injury and the secretion of soluble factors (Figure 2). Transcriptome analysis in reactive astrocytes found that genes involved in inflammation, leukocyte transendothelial migration, and JAK/STAT3 signaling were upregulated (160). Astrocytes directly exerted deleterious effects on the integrity of BBB by increasing the expression of VEGF, cytokines (IL-1β, IL-6, TNF-α, and IL-15), chemokines (CCL2 and CCL5), ROS, MMP, and LCN-2. Astrocyte-derived VEGF decreased the expression of TJs in ECs, which aggravated the BBB damage, infarction progression, and neurological deficits (161). Polymerase δ-interacting protein 2 was also upregulated in astrocytes following a stroke, which caused an increases in the extravasation of Evans blue by inducing the expression of TNF-α, IL-6, MCP-1, VEGF, and MMP (162).
Astrocytes also produce soluble factors to activate microglia and recruit peripheral immune cells, which indirectly potentiate the inflammation-induced BBB disruption. Lysophosphatidylcholine was found to increase CCL2 and CCR2 expression in microglia through G protein-coupled receptor 132 and P2X7R (163). Astrocyte-derived chemokines influenced microglia polarization in a paracrine manner through diverse chemokine receptors expressed on the microglia in AIS. In addition, astrocyte-derived VEGF aggregated the infiltration of peripheral immune cells by increasing the expression of ICAM-1 and VCAM-1 in ECs (161).
In contrast, astrocytes promote BBB repair by enabling the resolution of inflammation. A2 astrocytes are capable of secreting IL-2, IL-10, and TGF-β, leading to accelerated inflammation resolution. Pentraxin 3, released by astrocytes, attenuated IgG staining in ischemic cerebral tissue by inhibiting VEGF (164). Astrocytic insulin-like growth factor-1 (IGF-1) protected post-stroke BBB integrity and neurological function by shifting immune cells toward an anti-inflammatory profile in the ischemic environment (165). Astrocytes restricted microglia overactivation by upregulating the expression of CX3CR1 and IL-4Ra on the surface of microglia through TGF-β (166). However, TGF-β overexpression in astrocytes led to mural cell degeneration and dropout long-term (167).
Recent studies revealed that A2-specific transcripts were dominant over A1-specific transcripts 3 days following occlusion. Reactive A2 astrocytes expressed genes associated with scar formation and the regulation of ECM integrity, known as astrogliosis. Astrogliosis restricted the migration of infiltrating immune cells and limited the immune reaction within the infarct region (160). Surprisingly, growing evidence has shown that A2 astrocytes acquire the ability to engulf other cells under pathological situations. Reactive astrocytes in the penumbra were found to engulf and degrade cellular debris to assist with the resolution of inflammation via the ATP binding cassette A1 (ABCA1) after stroke (168). Depletion of ABCA1 in astrocytes profoundly increased BBB permeability and white matter lesions after ischemic stroke (169). Astrocytes exert such phagocytic activities in a delayed and prolonged manner. The phagocytotic activity of astrocytes typically starts 3 days post-ischemic stroke, peaks at 7 days, and persists for as long as 14 days.
Role of Pericytes in BBB Disruption in AIS
Recently, a significant urge in attention has been paid to investigating the immunological role of pericytes in BBB disruption in ischemic stroke. In one investigation, TLRs were found to be constitutively expressed on the surface of pericytes, indicating that ischemia-induced DAMP might trigger NF-кB nuclear translocation and the upregulation of pro-inflammatory factors in pericytes (170). The sphingosine monophosphate receptor 2 was found to be involved in the NF-кB activation in pericytes during ischemic stroke by decreasing microRNA-149-5p levels, which was associated with increased BBB permeability (171). The activation of pericytes activation caused an increase in the expression of STAT3 binding genes as early as 2 hours after OGD (172). The immunological function of pericytes can also be modulated. For example, pericytes were capable of increasing the expression and secretion of cytokines (IL-6 and IL-8), chemokines (CX3CL1 and MCP-1), and adhesion molecules (VCAM-1) in response to TNF-a, IL-1β, IFN-γ, or TGF-β stimulation in vitro and in an inflammatory environment in vivo (173–175). In ischemic stroke, pericytes acquired a CD11b-positive inflammatory phenotype and upregulated the expression of the pro-inflammatory cytokine due to astrocyte-derived sema4D binding to plexinB1 receptor on the surface of pericytes, which contributed to increased BBB permeability after ischemic stroke (176). In addition, IL-1β-induced expression of MCP-1, IL-8, and ICAM-1 in pericytes was inhibited by the activation of transcription factor CCAAT/enhancer binding protein delta (CEBPD) (177). Thus, pericytes may adopt a biphasic phenotype during ischemic stroke, but this remains much to be explored.
Functioning as potential immune cells, pericytes mitigate BBB disruption in ischemic insult through the secretion of MMP, pro-inflammatory mediators, ROS, and laminin 5a. in an in vitro coculture of ECs and pericytes, the TEER value was significantly reduced under OGD conditions (178). In areas where pericytes adjoined ECs, a three-fold higher frequency of IgG leakage was observed in the photothrombotic occlusion model compared to areas without pericytes adjoined ECs. This rapidly increased BBB permeability might have been mediated by the activation of MMPs in pericytes (179). Pericytes strongly overexpressed inflammation-associated molecules, chemokines (CCL2 and CXCL10), and cytokines (IL-6), contributing to a worse outcome. The expression of pro-inflammatory molecules in pericytes is regulated by the transcription factor RBPJ (180). NADPH oxidase 4, an enzyme involved in the production of ROS, was overexpressed in pericytes in ischemic stroke, potentiating the degradation of TJs by increasing NF-кB and MMP activation (181). Laminin 5a in pericytes was detrimental to BBB disruption by amplifying uncontrolled infiltration of inflammatory cell in AIS (182). However, pericytes also improved BBB integrity by producing TGF-β (183).
Interestingly, pericytes, which feature Fc receptors, M2-marker (ED2), and scavenger receptors, behave like macrophages (184). The accumulation of lysosome-like granules in pericytes was observed in the human heart and animal brain, indicating that the pericytes were capable of phagocytizing other cells during AIS (185, 186). In addition, pericytes manifested a microglial-like phenotype that was abundant in activated-microglia-specific mRNA in ischemic stroke (187, 188). The phagocytic function of pericytes may promote inflammation resolution and BBB repair during ischemic stroke. However, whether pericytes are capable of demonstrating the microglia phenotype remains controversial (189). Nerve/glia antigen 2 (NG2) is a single membrane-spanning proteoglycan expressed by pericytes but not microglia in the healthy brain. Interestingly, NG2 immuno-positive microglia were detected in experimental stroke, which may support the possibility of pericytes differentiating into microglia. However, using three knock-in mouse lines, Huang et al. identified that activated microglia were expressed NG2 in an ischemic-dependent manner, indicating that pericytes could not differentiate into microglia in ischemic stroke.
The Crosstalk Between Peripheral Immune Cells and Cerebral Immune Cells
Immune cells play an important role in BBB disruption during ischemic stroke. Moreover, the crosstalk between cerebral immune cells (CICs) and peripheral immune cells (PICs) forms a delicate and sophisticated network, the disturbance of which may indirectly influence BBB integrity during ischemic stroke.
CICs regulate the recruitment, extravasation, and function of PICs. For example, microglia, astrocytes, and pericytes upregulate and release diverse chemokines to motivate neutrophils, monocytes, and lymphocytes to the injured region after ischemic insult. Astrocytes attracted neutrophils in ischemic stroke by increasing the secretion of CXCL1 in response to the synergistic effects of TNF-α and IL-17A (114). CICs also secreted IL-1β and VEGF to stimulate the expression of VCAM-1 and ICAM-1 in the endothelium, which facilitates the attachment and infiltration of PICs. Owning to the unique location adjacent to microvascular ECs, astrocytes, and pericytes directly guide the extravasation of PICs. Both the chemoattractant MIF and ICAM-1 were upregulated in pericytes, which instructed the leukocytes infiltration pattern (190). Upon the interaction of pericytes with neutrophils, the cytoskeleton of pericytes became relaxed by inhibiting the RhoA/ROCK signaling pathway, which resulted in an enlargement of gaps between pericytes and low expression regions (LERs) of matrix proteins, thereby facilitating leukocyte extravasation (23, 191). Astrocytes provided the route guidance to infiltrated monocytes and macrophages through the interaction of fractalkine and CX3CR1 (192). After recognizing the invasion of neutrophil, microglia were observed to trap, contact, uptake, and finally engulf infiltrated neutrophils, diminishing the accumulation of neutrophils (193).
CICs also modulate the pro- or anti-inflammatory status of infiltrated PICs after ischemic stroke. M1 type microglia accelerate the polarization of Th1 cells by the secretion of IL-12 and TNF-α in the early stages of stroke. Meanwhile, microglia strongly induced the overexpression of HIF-1a/Sirtuin2 in Tregs through cell-to-cell contact, which inhibited the immunosuppressive function of Tregs (194). IL-15 derived from microglia and astrocytes increased the levels and activation of CD8+T cell and NK cell to potentiate BBB disruption in AIS (195). Astrocytes also polarized CD4+T cells into Th1 cells in vitro (196). Primary astrocytes were capable of maintaining FOXP3 expression in Tregs through IL-2/STAT5 signaling (197). Pericytes significantly inhibited the proliferation in and the secretion of pro-inflammatory factors from active T cells in the retina (198). Therefore, although there was no direct evidence in experimental stroke, astrocytes and pericytes may influence peripheral immune cells in ischemic conditions.
In turn, infiltrating PICs influence the polarization of CICs in ischemic stroke. Hematogenous monocytes and macrophages potentiated the polarization and process extension of astrocytes, promoting the BBB re-establishment (199). Microphage-derived exosomes polarized the microglia from the M1 pro-inflammatory phenotype to the M2 anti-inflammatory phenotype (200). Bone marrow-derived macrophages produced amphiregulin under ATP stimulation to change the status of pericytes by inducing TGF-β activation in acute lung injury, corroborating that the interactions between macrophages and pericytes could contribute to the BBB restoration in ischemic stroke (201). T lymphocyte subsets mainly exerted immunoregulatory effects in the ischemic brain. For instance, the Th1 subpopulation promoted M1 polarization of microglia through secretion of pro-inflammatory factors, such as IFNγ, which accelerated the amplification of inflammation in ischemic stroke. DNT increased the percentage of M1 pro-inflammatory microglia in the ischemic brain through the FasL/PTPN2/TNF-α pathway (202). Th2 cell-derived IL-4 inhibited the HMGB-1-induced expression of NF-κB and the formation of NLRP3 in astrocytes through STAT6/PPARγ singling (203). Th2 cells also potentiated the M2 polarization of microglia and the phagocytic capability of microglia in ischemic stroke through secreting IL-4 (204). Tregs inhibited the activation of microglia but promote the M2 polarization of microglia through recreation of IL-10 (197).
Given the complicated relationship between PICs and CICs, therapies targeting only one type of immune cell may prove deleterious or offset the benefit from another type of immune cell, resulting in an unsatisfied stroke prognosis.
Immunotherapies Targeting BBB Disruption During Ischemic Stroke
Therapies Targeting Neutrophils
Preclinical Attempts to Target Neutrophils for Treating AIS
Experimental investigations into targeting neutrophils for treating ischemic stroke have made tremendous breakthroughs in the past few decades. Modulating crucial neutrophil courses, including recruitment, adhesion, transmigration, and polarization, can significantly reverse severe BBB disruption and worse infarct damage (Table 1).
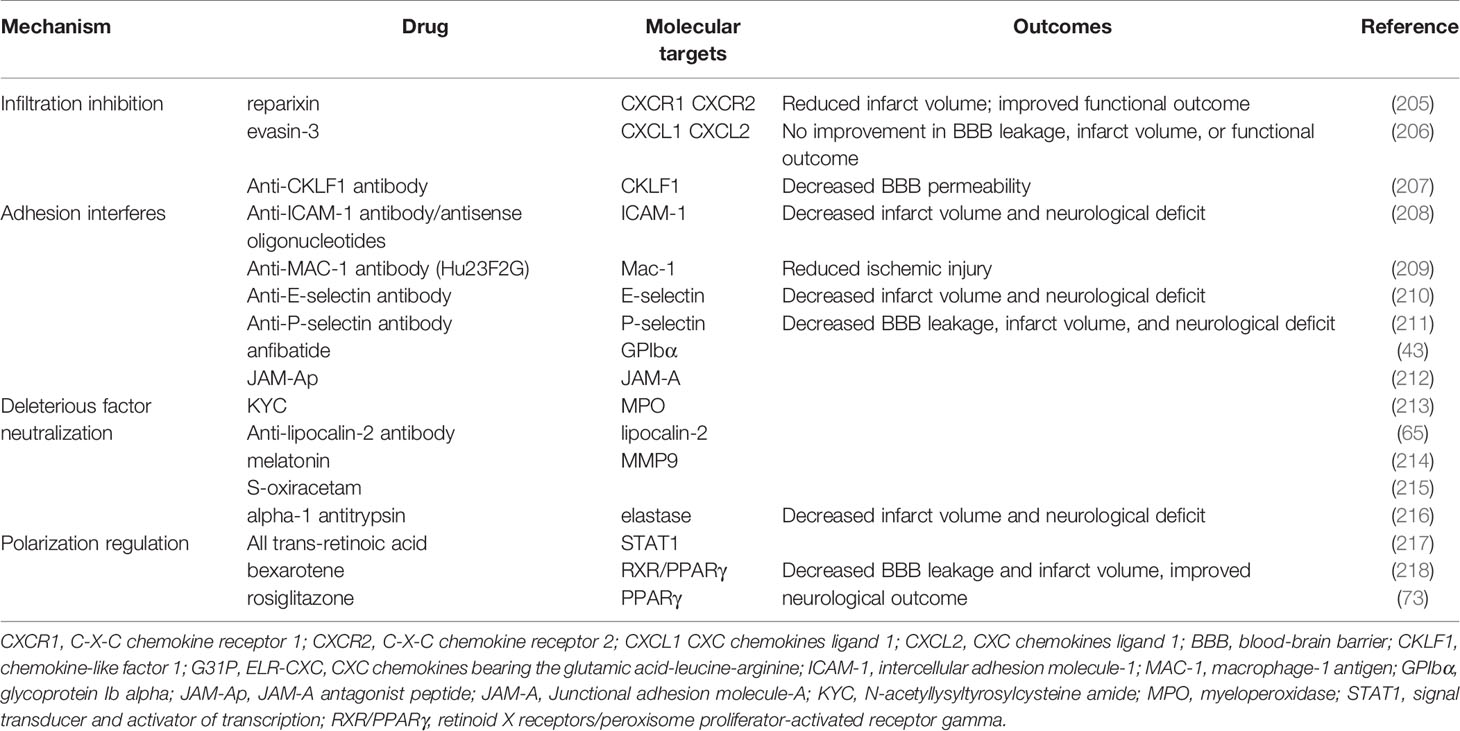
Table 1 Preclinical attempts to target neutrophils for treating AIS.
Targeting neutrophil activation and recruitment may significantly reduce the infiltration of neutrophils into the ischemic hemisphere, but whether the treatment can improve BBB integrity and the functional outcome remains controversial. One study showed that CKLF1 inhibition alleviated MMP9 activity, ZO-1, and occludin disruption, as well as EB leakage after stroke (207). Reparixin, an inhibitor of CXCR1 and CXCR2, significantly decreased neutrophil extravasation and infarct volume and improved functional outcomes following ischemia-reperfusion (205). However, inhibiting CXCR2 alone did not attenuate infarction and motor impairment progression (60). Neutralizing the bioactivity of CXCL1 and CXCL2 by evasin-3 decreased neutrophil infiltration but did not reduce infarct volume, neurological deficit, or BBB permeability (206).
Recently, a novel strategy to redirect neutrophils from the ischemic brain to the periphery by peripherally implanting a CXCL1-soaked sponge led to improved neurological performance and infarction by reducing neutrophil-induced inflammation (219). Platelet-mimetic nanoparticles (PTNPs) were co-loaded with superparamagnetic iron oxide (SPIO), a T2 contrast agent. Piceatannol is a selective tyrosine kinase inhibitor. Like the CXCL1-soaked sponge, piceatannol, a selective tyrosine kinase inhibitor, can successfully recognize, intervene, monitor, and detach neutrophils into circulation, limiting infiltration of neutrophils in ischemia (220). Small extracellular vesicles derived from mesenchymal stromal cells protected against BBB disruption, ischemic injury, and neurological dysfunction, specifically by modulating the entry and accumulation of neutrophils (221).
Interfering with neutrophil rolling and adhesion by anti ICAM-1 (208) Mac-1 (209, 222), P-selectin/E-selectin (210, 211) antibody were effective in limiting the accumulation of neutrophils, BBB leakage, and ischemic injury in animals. The blocking of platelet glycoprotein receptor Ib decreased ischemic-induced BBB hyperpermeability through inactivation and downregulation of Mac-1 and P-selectin (43). In addition, inhibiting neutrophil-endothelia interactions with the JAM-A antagonist peptide reduced the expression of inflammatory mediators, BBB leakage, infarct size, and functional deficit (212).
Ameliorating the function of deleterious effectors derived from neutrophils protected the ischemic hemisphere from brain edema, infarction, and neurological deterioration. Blood substitution therapy profoundly improved stroke outcomes through the robust reduction of neutrophil, cytokine storm, and MMP-9 production. The benefits of blood substitution were diminished by the supplementation of MMP9 to the blood (223). Supplementing high-density lipoproteins or alpha-1 antitrypsin (an elastase inhibitor) inhibited neutrophil degranulation and the proteolysis of ECM (224). The diminishment of LCN-2 by antibodies significantly reduced brain edema, BBB permeability, and functional outcome after stroke injury (65). Inhibition of MPO by N-acetyl lysyltyrosylcysteine amide significantly decreases IgG extravasation and neurological severity score in stroke (213). Administration of either melatonin or S-oxiracetam maintained BBB integrity, which was partly attributed to inhibiting the activity of deleterious factors from neutrophils in the MCAO model (214, 215).
Lastly, targeting the polarization of neutrophils offers a promising target to attenuate ischemic damage to BBB. All-trans-retinoic acid prevented BBB disruption, infarction, and behavior deficits by skewing neutrophils toward the N2 phenotype by suppressing STAT1 (217). Retinoid X receptor (RXR)/PPARγ agonists, including bexarotene and rosiglitazone, elevated the number of Ym1 positive neutrophils by inhibiting NF-κB transactivation, which was also associated with the attenuation of BBB leakage, infarction enlargement, and neurological deficits (73, 218).
Clinical Trials Targeting Neutrophils in AIS
Inspired by the positive results from inhibiting adhesion molecules in animals, targeting neutrophil adhesion in patients suffering ischemic stroke has been widely evaluated in clinical trials (Table 2). Unfortunately, they all failed. Enlimomab (anti-ICAM-1 antibody)-treated patients had worse functional outcomes and more adverse events than placebo-treated patients in AIS (235). Administration of UK-279,276, a Mac-1 inhibitor, did not improve recovery from AIS (236). In the LeukArrest study, clinical trials of Hu23F23F2G, which targeted Mac-1 and LFA-1, terminated early without final reports (237). Clinical trial to evaluate the safety and effectiveness of E-Selectin tolerance induction in patients who have had a transient ischemic attack or stroke was suspended without available results (238).
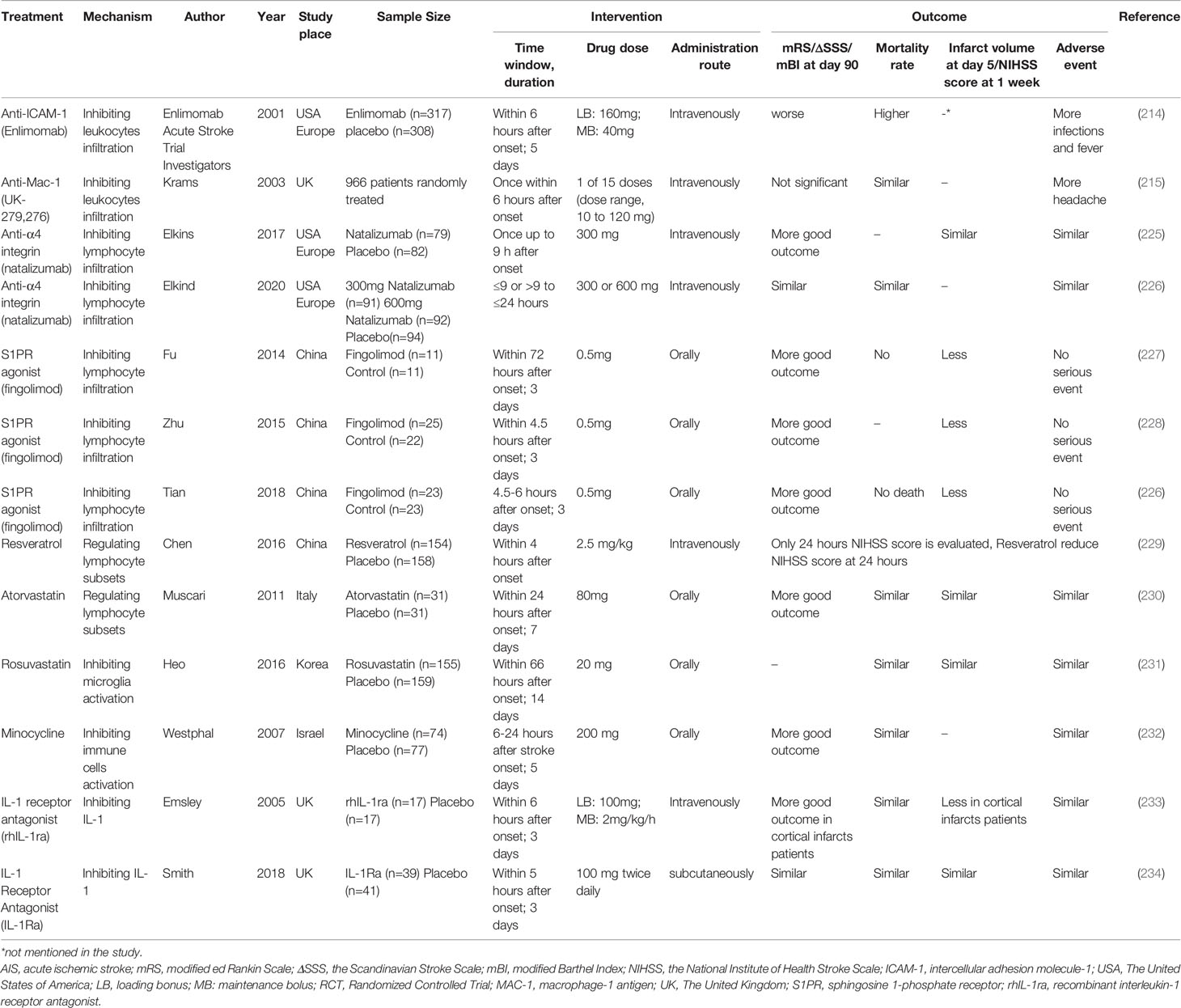
Table 2 Randomized controlled trials targeting immune cells in human ischemic stroke.
Some of these potential drugs discussed were safe and efficacious against other diseased, but they require more clinical trials for determining their efficacy in treating AIS. For instance, the safety and efficacy of reparixin were demonstrated in treating breast cancer recently (239). Melatonin supplementation reduced myocardial ischemic-reperfusion injury in a dose-dependent manner (240). In addition, all trans-retinoic acid might be an alternative therapy for leukemia (241).
Therapies Targeting Monocytes
Preclinical Attempts to Target Monocytes for Treating AIS
Skewing monocyte-derived macrophages toward the alternatively activated M2 phenotype can be a potential and effective target for AIS treatment (Table 3). Administration of P2X4R antagonist 5-BDBD was found to significantly reduce BBB leakage, infarct size, and neurological deficit (242). Propane-2-sulfonic acid octadec-9-enyl-amide (243), 1, 25-dihydroxyvitamin D3 (244), rosiglitazone (245), and recombinant human fibroblast growth factor 21 (270) all upregulated the expression of Arg-1,Ym1, and CD206 and downregulated the expression of iNOS in microglia and macrophages by activating PPARγ. These PPARγ agonists profoundly maintained the TJs and BBB integrity, improved infarction injury and neurological score. The activation of GPR35 by pamoic acid increased the number of alternative monocytes and macrophages in the ischemic hemisphere and reduced the infarct volume and functional disorder through the protein kinase B (Akt)/p38 MAPK signaling pathway (247). Other modulators, such as DHA (248), 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (250), miR-669c (251), significantly elevated the expression of Arg1 and reduced infarct volume and neurological disorder caused by cerebral ischemia.
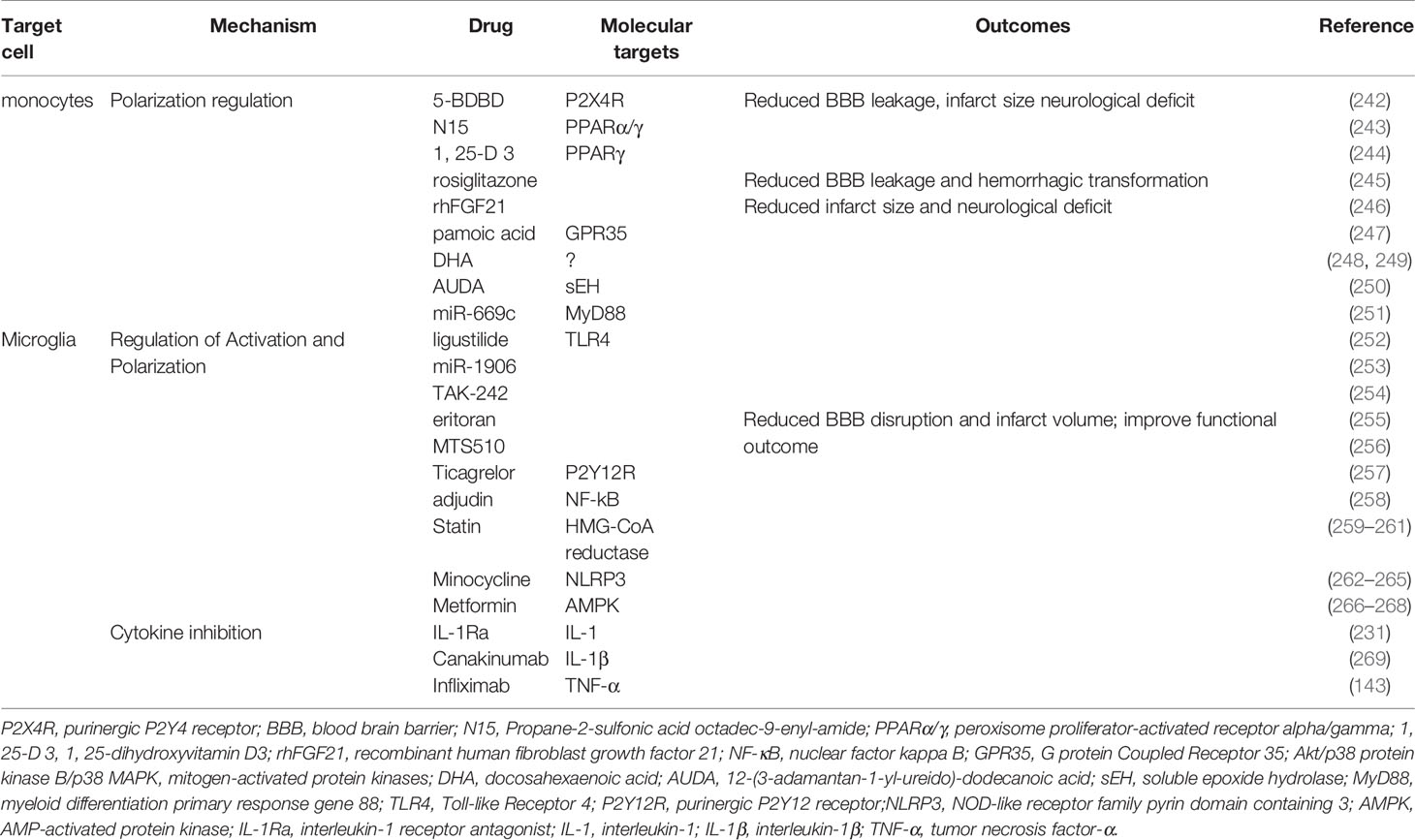
Table 3 Preclinical attempts to target monocyte and microglia for treating AIS.
Clinical Trials Targeting Monocytes in AIS
There is no drug that directly targets monocytes in clinical ischemic stroke currently. Due to similar polarization mechanism and function with microglia, drugs that regulate microglia activation and polarization may also interfere monocytes status in AIS (see Part 5.4.2 Clinical Trials Targeting Microglia in AIS). Further studies are warranted for developing strategies to modulate monocytes in AIS.
Therapies Targeting T Lymphocytes
Preclinical Attempts to Target T Lymphocytes for Treating AIS
The therapeutic options for AIS that targets T lymphocytes involve three pathways (Table 4). The first entails decreasing the infiltration of T lymphocytes.

Table 4 Preclinical attempts to target T lymphocytes for treating AIS.
Blocking the infiltration of T lymphocytes by inhibiting CD49d (integrin α4) reduced the overall infiltrated T cell count, while the terminal stroke outcome is controversial (271, 286). FTY720 (Fingolimod), a sphingosine-1-phosphate (S1P) receptor agonist, is a strong candidate for AIS treatment because it blocks the motivation of lymphocytes from lymph nodes to the bloodstream (287). Fingolimod administration improved the BBB integrity, infarct injury, and functional outcome (272, 273).
The second pathway entails manipulating the imbalance between pro-inflammatory subsets and anti-inflammatory subsets. Changing the T cell subpopulation disorder rescued BBB hyperpermeability after ischemic stroke. Poly (ADP-ribose) polymerase-1 (PARP-1) is an abundant nuclear protein involved in DNA repair. Inhibitors of PARP-1, namely JPI-289, FR247304, and MP-124, significantly maintained the TJs and reduced the MMP9 levels, brain swelling, HT occurrence, infarction size, and functional deficiency in stroke (274, 276, 277). The post-stroke suppression of acetyl coenzyme A carboxylase 1(ACC1), a critical metabolic enzyme involved in fatty acid synthesis, maintained the ratios of Treg to Th17, which reduced the inflammation and ischemic lesion (288). Injection of recombinant IL-33 into mice dramatically attenuated the deterioration of brain edema and neurological deficit by maintaining the levels and activities of Th2 cells and Tregs and dampening the counts and immune responses of Th1 and Th17 cells (278, 279). IL-33 induced TGF-β and IL-10 elevation via ligation to the ST2 receptor (279). Supplementing vitamin D 3, resveratrol, or atorvastatin prevented infarction and neurological dysfunction by shifting immune balance to enhanced Treg reaction and depressed Th17 and γδT response (281–283). The effect of atorvastatin on Treg expansion was concentration-dependent, as Treg immunosuppression was hindered at a high concentration of atorvastatin in vitro (289).
The third pathway involves regulating the composition of gut microbiota. Targeting the gut microbiota to protect the BBB and the brain against ischemic insult has demonstrated strong potential in animals. Resveratrol robustly attenuated the inflammation-induced degradation of BBB by regulating the intestinal flora (284).
The post stroke transplantation of beneficial fecal microbiota, abundant in butyric acid and short-chain fatty acids, namely Clostridium butyricum, Bifidobacterium longum, Lactobacillus fermentum, and Faecalibacterium prausnitzii, significantly alleviated post-stroke BBB disruption, infarct size, and neurological deficits in mice (225, 285). In addition, supplementing microbial metabolites, such as short-chain fatty acids (SCFAs), in ischemic stroke maintained the balance of pro- and anti-inflammatory subsets by inhibiting the activity of histone deacetylase (HDAC). Furthermore, HDAC inhibitors, including sodium butyrate and valproic acid, protected against stroke-induced BBB disruption and brain edema by inhibiting MMP9 production, TJ degradation, and NF-kB activation (227, 228).
Clinical Trials Targeting T Lymphocytes in AIS
Natalizumab is a monoclonal antibody targeting α4 integrin to block the adhesion and infiltration of lymphocytes. Natalizumab administration in a prior phase 2 trial (ACTION) failed to reduce infarct volume but improved functional outcome 90 days after AIS (290). However, the benefits on functional outcomes were not seen in ACTION II (226). Combining results of ACTION and ACTION II cannot support the evidence of the benefit of natalizumab for AIS (Table 2). FTY720 is a promising drug for improving stroke outcomes by its ability to decrease the infiltration of lymphocytes. Several pilot trials and randomized control trials have verified the safety and efficacy of FTY720 in patients with AIS as a single therapy that exceeded the therapeutic window. FTY720 treatment combined with alteplase thrombolysis also improved stroke outcomes by decreasing microvascular permeability (229, 291–293). The mechanisms underlying the efficacy of fingolimod in treating AIS are multifaced and are reviewed in (287).
Regulating the homeostasis of T lymphocyte subsets in patients might be a hopeful treatment for AIS. One study discovered that inhibiting PARP-1 increased the proportion of Treg and Treg-relevant transcription factors (i.e., FoxP3 and CTLA-4 mRNA) in the blood of ischemic patients (100). Meanwhile, PARP-1 inhibition reversed the disbalance between pro-inflammatory factor (IL-17, IFN-γ, and TNF-α), and anti-inflammatory factors (TGF-β, IL-4, and IL-10). The safety and tolerance of JPI-289, a PPAR-1 inhibitor, have been successfully demonstrated in healthy volunteers recently, which served as a foundation for further related clinical trials (294). Resveratrol administration significantly improved the functional outcome in stroke patients (230). However, oral supplementation with vitamin D 3 failed to decrease the risk of stroke occurrence and did not improve stroke outcomes in patients long-term (295, 296). Randomized placebo-controlled trials revealed that atorvastatin did not decrease the infarct volume or improve functional behavior in AIS patients (297, 298).
Therapies Targeting Microglia
Preclinical Attempts to Target Microglia for Treating AIS
The discovery of therapies targeting microglia for the treatment of AIS is of great interest for inhibiting microglial activation and regulating microglia polarization (Table 3).
PRR ablation shielded microglia from activation after ischemic stroke. Blocking TLR4 with eritoran, MTS510, ligustilide, miR-1906, and TAK-242 reduced the number of activated microglia and the production of iNOS, ROS, and pro-inflammatory factors, thereby protecting the BBB against inflammatory injury in stroke (252–256). Ticagrelor, a reversible antagonist of P2Y12R, significantly attenuated BBB damage by decreasing the activation of microglial and lowering the level of IL-1β, CCL2/MCP-1, and iNOS (257). Teriflunomide reserved pericytes coverage and TJ expression by reducing microglial activation and downregulating cyclooxygenase-2 and IL-1β 3 days following tMCAO (299). Interfering with NF-kB signaling through supplementation with adjudin significantly inhibited the activation of microglia, overexpression of TNF-α, IL-1β and IL-6, and the disruption of ZO-1, occluding, and JAM-A. Adjudin decreased the IgG extravasation, infarct size, and functional disorder in experimental stroke (258). In addition, statin therapy significantly reduced BBB breakdown, brain swelling, and functional deficit by decreasing microglial activation (259, 300).
Suppressing the NLRP3 inflammasome promoted the M2 polarization of microglia. Minocycline, a tetracycline antibiotic that demonstrated anti-inflammatory effects in neurodegenerative diseases inhibited microglial activation and promoted microglia M2 polarization by suppressing the NLRP3 inflammasome (262). Minocycline increased the expression of IL-10, TGF-β, Ym-1, and Arg-1 but decreased the expression of IL-1β, IL-6 andTNF-a (263). Thus, minocycline administration significantly reduced BBB leakage, brain edema, infarct volume, and neurological dysfunction (264). Metformin, which is widely used in clinical for treating diabetes, activated adenosine 5′monophosphate-activated protein kinase (AMPK). Metformin not only attenuated post-stroke BBB damage but also promoted angiogenesis and recovery in a microglia M2 polarization-dependent manner (266–268). In addition, PPARγ agonist discussed preciously potentiated polarization of microglia to M2 phenotype.
Blocking the production of harmful cytokines during ischemic stroke may also be a promising therapeutic target for treating AIS. IL-1R antagonist (IL-1Ra) is a natural inhibitor of IL-1 signaling. IL-1Ra administration reduced inflammation-induced BBB disruption, infarct size, and neurological deficit in rats (231). Cell therapy based on IL-1Ra was also effective in treating ischemic stroke (301). Canakinumab, an inhibitor of IL-1β, significantly decreased brain edema, infarct volume, and functional deficit in a murine tMCAO model (269). Anti TNF-α (infliximab) therapy significantly enabled the preservation of BBB structure and behavioral performance in experimental stroke (143). However, further investigation is required to evaluate the efficacy and safety of biomimetics and antibodies against pro-inflammatory cytokines in treating AIS.
Clinical Trials Targeting Microglia in AIS
Regulating the activation and polarization of microglia in AIS patients has been identified as an effective strategy to improve the prognosis of AIS (Table 2). The use of statin in patients with AIS was found to be safe and well-tolerated. Patients receiving statins after ischemic stroke onset displayed reduced microemboli and HT (233, 234). Minocycline treatment in patients with AIS markedly improved the patients’ functional outcome after 3 months, which was based on mRS scores of 0-2 (302). Patients who were administered metformin before ischemic stroke showed significantly lower stroke severity, mRS score, and mortality compared to those who were not given metformin (303).
Inhibiting pro-inflammatory cytokines is also helpful for clinical intervention of ischemic stroke. Supplementing with IL-1Ra was determined to be safe and well-tolerated in ischemic stroke patients. Recombinant human IL-1ra (rhIL-1ra) administered intravenously in ischemic stroke patients displayed profoundly lower inflammatory biomarkers in their serum and better clinical outcomes 3 months after ischemic stroke (304). IL-1Ra administered subcutaneously was not associated with an improved functional outcome; however, it significantly reduced the presence of circulating inflammatory markers (305). Antibodies against IL-1β and TNF-α were not evaluated in ischemic stroke patients. CANTOS Trial demonstrated that canakinumab significantly reduced atherothrombotic cardiovascular events in 10,061 patients (306). Lastly, infliximab and its biosimilar were widely investigated in patients with autoimmune diseases (307).
Therapies Targeting Astrocytes
Preclinical Attempts to Target Astrocytes for Treating AIS
Given the essential role of astrocytes, whether deleterious or beneficial, in BBB disruption during AIS, astrocytes can be potential therapeutic targets. Treatments targeting astrocytes involve decreasing astrocyte activation and inhibiting the detrimental factors from astrocytes (Table 5).
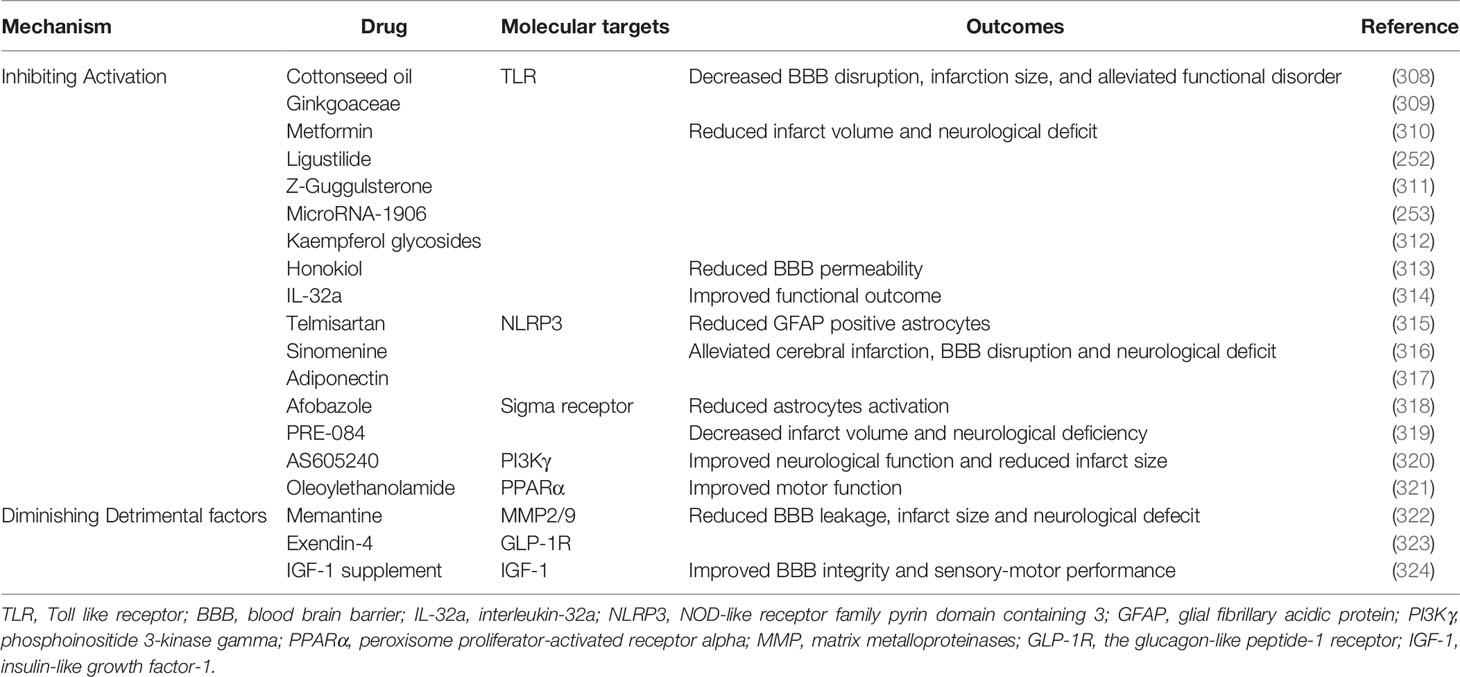
Table 5 Preclinical attempts to target astrocytes for treating AIS.
Blocking the activation of astrocytes in ischemic stroke may prevent BBB disruption and promote proper neurological performance. Given the similarities in activation signaling between astrocytes and microglia, some drugs that function to inhibit immune cell activation are efficacious against both microglia and astrocytes. TLR-NF-kB signaling is the initial step in astrocyte activation. By inhibiting TLR and NF-kB expression, cottonseed oil (308), Ginkgoaceae (309), metformin (310), ligustilide (252), Z-guggulsterone (311), IL-32a (314), microRNA-1906 (253), honokiol (313), and kaempferol glycosides (312), all reduced the number of reactive astrocytes and the levels of IL-1β, IL-6, and TNF-α, which significantly attenuated BBB disruption, brain edema, and functional neurological performance. NLRP3 inflammasome is an important signaling mechanism involved in the activation of A1 astrocytes. Thus, many drugs, such as telmisartan (315), sinomenine (316), and adiponectin (317) alleviated A1 astrocytes-induced BBB disruption and neurological deficit by inhibiting NLRP3. In addition to afobazole and PRE-084, animals treated with the sigma receptor agonist showed elevated astrocyte numbers but decreased proportion of reactive astrocytes in the ischemic hemisphere, leading to a small infarct volume (318, 319). AS605240, a PI3Kγ inhibitor, significantly reduced the astrocyte activation and morphological changes as well as the expression of pro-inflammatory factors, resulting in improved stroke outcome (320). Oleoylethanolamide promoted the expression and nuclear transportation of PPARα in astrocytes to inhibit astrocyte activation and neurological deficit in the ischemic hemisphere (321).
Diminishing astrocytic detrimental factors also protected the BBB against inflammatory injury. Vagus nerve stimulation and memantine administration attenuated BBB leakage and infarct size by reducing astroglia MMP2/9 expression (322, 325). Exendin-4, a glucagon-like peptide-1 receptor agonist, decreased the HT by reducing the expression of astrocyte-derived MCP-1, CXCL1, VEGF, and MMP9 through the reduction of highly glycosylated CD147, an MMP inducer (323, 326) Supplementing with IGF-1 also improved BBB integrity and sensory-motor performance in a murine model of tMCAO (324).
Clinical Trials Targeting Astrocytes in AIS
Some drugs mentioned above have already been approved by the FDA or the Chinese government for other diseases. For example, telmisartan is widely used as an angiotensin-receptor blocker for the treatment of hypertension. Sinomenine is the main active ingredient in the plant Sinomenium acutum and has been used to treat rheumatic and arthritic diseases in China for over 1000 years (327). Afobazole, an anxiolytic drug, used by patients with generalized anxiety disorder. Memantine is an N-methyl-D-aspartate (NMDA) receptor antagonist approved for the treatment of Alzheimer’s disease. Exenatide is a synthetic product of the glucagon-like peptide-1 analogue exendin-4 and is clinically utilized for patients with type 2 diabetes.
Among these candidates, the safety and efficiency of telmisartan and exenatide in treating AIS have been evaluated through clinical trials. Exenatide behaved well in controlling post-stroke hyperglycemia without worsening neurological outcomes (328). Currently, the superiority of exenatide treatment compared to standard treatment is being evaluated in a phase 2 trial as a potential treatment for AIS (329). In the Prevention Regimen for Effectively Avoiding Second Strokes (PRoFESS) trial, telmisartan utilization did not alter the neurological deficiencies in patients with recurrent stroke (330). Considering these recent advancements, while drug repositioning or repurposing may be relatively inexpensive and reliable in AIS compared to the development of new, specific therapeutics, more investigations related to the discovery of new drugs for treating AIS are warranted.
Therapies Targeting Pericytes
Potential immunological therapies targeting pericytes in ischemic stroke have not been explored deeply due to the uncomprehensive understanding of their role. From the information discussed above, it has been hypothesized that modulating the inflammatory status of pericytes against the secretion of detrimental factors and promoting the phagocytotic properties of pericytes may limit inflammation-induced BBB leakage to promote functional recovery in ischemic stroke.
Discussion
Currently, only intravenous thrombolysis with rt-PA and MT are effective treatments for acute ischemic stroke (11). However, they are limited by only being efficacious in a narrow treatment window. Neuron-centric neuroprotective therapies for treating AIS have failed. Therefore, it is crucial to discover new therapeutic targets for ischemic stroke. BBB disruption often engenders a poor prognosis and mortality in patients with AIS. Because both cerebral and circulating immune cells play a complex and lasting role in BBB destruction, immunotherapies targeting BBB disruption can be adjusted accordingly at various time points during the treatment of AIS.
Peripheral immune cells are recruited and infiltrate into the brain after ischemic stroke. Neutrophils have long been considered a destructive effector in BBB disruption. Interestingly, the other protective classification of neutrophils was discovered recently. Monocytes have a dual role in BBB destruction after cerebral infarction through the ability to differentiate into M1 and M2 macrophages. Promoting the differentiation into M2 macrophages has become a hot spot and potential target for therapeutic exploration. T lymphocytes have a tendency to differentiate into pro-inflammatory subgroups after cerebral infarction, which damage BBB integrity. Thus, identifying the driver and regulator of T lymphocytes pool differentiation after cerebral infarction is advantageous to the discovery of novel and effective therapeutic targets.
Cerebral immune cells are activated rapidly after artery occlusion. Perivascular immune cells can directly influence BBB integrity in stroke. Similar to monocytes, microglia polarization has gained significant attention over the years and may be a promising target in treating ischemic stroke. Astrocytes are also innate immune cells and regulate the course of inflammation and BBB disruption through the secretion of diverse soluble molecules. Recently, researchers have discovered that the immunological effects of pericytes after AIS is beyond previous thoughts. However, it remains unclear how astrocytes and pericytes regulate BBB stability using the immune system.
There are two main bottlenecks that impede the clinical development of immunoregulatory therapies targeting BBB disruption. One is the mismatch between clinical stroke and experimental stroke. The efficacy of immunoregulatory treatments in animals (experimental stroke) was based on the tMCAO model (331). However, the majority of AIS patients underwent permanent ischemia without reperfusion. The other is the difficulty of knowing when to start and terminate immune intervention in ischemic stroke. Most immune cells do not have a single role during the course of AIS. Treatment with immunoregulatory drugs must be easy to stop to prevent further offsetting the benefit from an early stage of AIS. Moreover, circulating and cerebral immune cells form a delicate and sophisticated inflammatory network. Therapies targeting only one type of immune cell may prove deleterious or offset the benefit from another type of immune cell, resulting in an unsatisfied stroke prognosis. However, further exploration is needed to identify effective treatment measures.
Overall, targeting immune cells in the BBB disruption in AIS is a promising strategy to improve stroke prognosis and improve existing treatments. More investigations using animal models that better simulate human stroke are warranted. In addition, detailed mechanisms underlying the inflammatory mechanism of BBB disruption remain to be uncovered.
Author Contributions
Y-mQ and C-lZ wrote the manuscript. Y-nL and BH conceived this article. A-qC and H-lW drew the chart. Y-fZ helped revise the manuscript and check the grammar. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82071336 to YNL), Natural Science Foundation of Hubei Province (No. 2020CF763 to YNL), National Key R&D Program of China (No. 2018YFC1312200), National Natural Science Foundation of China (No. 82090044 to BH, No. 81820108010 to BH, No. 81901212 to YFZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Owens Johnson C, Nguyen M, Roth GA, Nichols E, Alam T, Abate D. Global, Regional, and National Burden of Stroke, 1990-2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol (2019) 18(5):439–58. doi: 10.1016/s1474-4422(19)30034-1
2. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation (2016) 133(4):e38–360. doi: 10.1161/cir.0000000000000350
3. Jiang X, Andjelkovic AV, Zhu L, Yang T, Bennett MVL, Chen J, et al. Blood-Brain Barrier Dysfunction and Recovery After Ischemic Stroke. Prog Neurobiol (2018) 163-164:144–71. doi: 10.1016/j.pneurobio.2017.10.001
4. Leigh R, Christensen S, Campbell BC, Marks MP, Albers GW, Lansberg MG. Pretreatment Blood-Brain Barrier Disruption and Post-Endovascular Intracranial Hemorrhage. Neurology (2016) 87(3):263–9. doi: 10.1212/wnl.0000000000002862
5. Leigh R, Jen SS, Hillis AE, Krakauer JW, Barker PB. Pretreatment Blood-Brain Barrier Damage and Post-Treatment Intracranial Hemorrhage in Patients Receiving Intravenous Tissue-Type Plasminogen Activator. Stroke (2014) 45(7):2030–5. doi: 10.1161/strokeaha.114.005249
6. Desilles JP, Rouchaud A, Labreuche J, Meseguer E, Laissy JP, Serfaty JM, et al. Blood-Brain Barrier Disruption Is Associated With Increased Mortality After Endovascular Therapy. Neurology (2013) 80(9):844–51. doi: 10.1212/WNL.0b013e31828406de
7. Villringer K, Sanz Cuesta BE, Ostwaldt AC, Grittner U, Brunecker P, Khalil AA, et al. DCE-MRI Blood-Brain Barrier Assessment in Acute Ischemic Stroke. Neurology (2017) 88(5):433–40. doi: 10.1212/wnl.0000000000003566
8. Ji B, Zhou F, Han L, Yang J, Fan H, Li S, et al. Sodium Tanshinone Iia Sulfonate Enhances Effectiveness Rt-PA Treatment in Acute Ischemic Stroke Patients Associated With Ameliorating Blood-Brain Barrier Damage. Transl Stroke Res (2017) 8(4):334–40. doi: 10.1007/s12975-017-0526-6
9. He HY, Ren L, Guo T, Deng YH. Neuronal Autophagy Aggravates Microglial Inflammatory Injury by Downregulating CX3CL1/Fractalkine After Ischemic Stroke. Neural Regener Res (2019) 14(2):280–8. doi: 10.4103/1673-5374.244793
10. Liesz A, Dalpke A, Mracsko E, Antoine DJ, Roth S, Zhou W, et al. DAMP Signaling Is a Key Pathway Inducing Immune Modulation After Brain Injury. J Neurosci (2015) 35(2):583–98. doi: 10.1523/jneurosci.2439-14.2015
11. Warner JJ, Harrington RA, Sacco RL, Elkind MSV. Guidelines for the Early Management of Patients With Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke. Stroke (2019) 50(12):3331–2. doi: 10.1161/strokeaha.119.027708
12. Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol Rev (2019) 99(1):21–78. doi: 10.1152/physrev.00050.2017
13. Tietz S, Engelhardt B. Brain Barriers: Crosstalk Between Complex Tight Junctions and Adherens Junctions. J Cell Biol (2015) 209(4):493–506. doi: 10.1083/jcb.201412147
14. McConnell HL, Kersch CN, Woltjer RL, Neuwelt EA. The Translational Significance of the Neurovascular Unit. J Biol Chem (2017) 292(3):762–70. doi: 10.1074/jbc.R116.760215
15. Crone C, Olesen SP. Electrical Resistance of Brain Microvascular Endothelium. Brain Res (1982) 241(1):49–55. doi: 10.1016/0006-8993(82)91227-6
16. Craig LE, Spelman JP, Strandberg JD, Zink MC. Endothelial Cells From Diverse Tissues Exhibit Differences in Growth and Morphology. Microvasc Res (1998) 55(1):65–76. doi: 10.1006/mvre.1997.2045
17. Lv J, Hu W, Yang Z, Li T, Jiang S, Ma Z, et al. Focusing on Claudin-5: A Promising Candidate in the Regulation of BBB to Treat Ischemic Stroke. Prog Neurobiol (2018) 161:79–96. doi: 10.1016/j.pneurobio.2017.12.001
18. Sandoval KE, Witt KA. Blood-Brain Barrier Tight Junction Permeability and Ischemic Stroke. Neurobiol Dis (2008) 32(2):200–19. doi: 10.1016/j.nbd.2008.08.005
19. Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, et al. Capillary Pericytes Regulate Cerebral Blood Flow in Health and Disease. Nature (2014) 508(7494):55–60. doi: 10.1038/nature13165
20. Moura RP, Martins C, Pinto S, Sousa F, Sarmento B. Blood-Brain Barrier Receptors and Transporters: An Insight on Their Function and How to Exploit Them Through Nanotechnology. Expert Opin Drug Delivery (2019) 16(3):271–85. doi: 10.1080/17425247.2019.1583205
21. Wettschureck N, Strilic B, Offermanns S. Passing the Vascular Barrier: Endothelial Signaling Processes Controlling Extravasation. Physiol Rev (2019) 99(3):1467–525. doi: 10.1152/physrev.00037.2018
22. Ifergan I, Kébir H, Bernard M, Wosik K, Dodelet-Devillers A, Cayrol R, et al. The Blood-Brain Barrier Induces Differentiation of Migrating Monocytes Into Th17-Polarizing Dendritic Cells. Brain (2008) 131(Pt3):785–99. doi: 10.1093/brain/awm295
23. Rudziak P, Ellis CG, Kowalewska PM. Role and Molecular Mechanisms of Pericytes in Regulation of Leukocyte Diapedesis in Inflamed Tissues. Mediators Inflamm (2019) 2019:4123605. doi: 10.1155/2019/4123605
24. Varatharaj A, Galea I. The Blood-Brain Barrier in Systemic Inflammation. Brain Behav Immun (2017) 60:1–12. doi: 10.1016/j.bbi.2016.03.010
25. Gill D, Sivakumaran P, Aravind A, Tank A, Dosh R, Veltkamp R. Temporal Trends in the Levels of Peripherally Circulating Leukocyte Subtypes in the Hours After Ischemic Stroke. J Stroke Cerebrovasc Dis (2018) 27(1):198–202. doi: 10.1016/j.jstrokecerebrovasdis.2017.08.023
26. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and Spatial Dynamics of Cerebral Immune Cell Accumulation in Stroke. Stroke (2009) 40(5):1849–57. doi: 10.1161/strokeaha.108.534503
27. Chen C, Chu SF, Liu DD, Zhang Z, Kong LL, Zhou X, et al. Chemokines Play Complex Roles in Cerebral Ischemia. Neurochem Int (2018) 112:146–58. doi: 10.1016/j.neuint.2017.06.008
28. Chen C, Chu SF, Ai QD, Zhang Z, Chen NH. CKLF1/CCR5 Axis Is Involved in Neutrophils Migration of Rats With Transient Cerebral Ischemia. Int Immunopharmacol (2020) 85:106577. doi: 10.1016/j.intimp.2020.106577
29. Kovacs-Litman A, Vonderwalde I. Monocyte-Derived Macrophages Modulate Inflammation and Promote Long-Term Functional Recovery in a Mouse Model of Ischemia. J Neurosci (2016) 36(38):9757–9. doi: 10.1523/jneurosci.1906-16.2016
30. Feng Y, Liao S, Wei C, Jia D, Wood K, Liu Q, et al. Infiltration and Persistence of Lymphocytes During Late-Stage Cerebral Ischemia in Middle Cerebral Artery Occlusion and Photothrombotic Stroke Models. J Neuroinflamm (2017) 14(1):248. doi: 10.1186/s12974-017-1017-0
31. Chu HX, Kim HA, Lee S, Moore JP, Chan CT, Vinh A, et al. Immune Cell Infiltration in Malignant Middle Cerebral Artery Infarction: Comparison With Transient Cerebral Ischemia. J Cereb Blood Flow Metab (2014) 34(3):450–9. doi: 10.1038/jcbfm.2013.217
32. Stubbe T, Ebner F, Richter D, Engel O, Klehmet J, Royl G, et al. Regulatory T Cells Accumulate and Proliferate in the Ischemic Hemisphere for Up to 30 Days After MCAO. J Cereb Blood Flow Metab (2013) 33(1):37–47. doi: 10.1038/jcbfm.2012.128
33. Zhang Y, Gao Z, Wang D, Zhang T, Sun B, Mu L, et al. Accumulation of Natural Killer Cells in Ischemic Brain Tissues and the Chemotactic Effect of IP-10. J Neuroinflamm (2014) 11:79. doi: 10.1186/1742-2094-11-79
34. Arunachalam P, Ludewig P, Melich P, Arumugam TV, Gerloff C, Prinz I, et al. Ccr6 (Cc Chemokine Receptor 6) Is Essential for the Migration of Detrimental Natural Interleukin-17-Producing γδ T Cells in Stroke. Stroke (2017) 48(7):1957–65. doi: 10.1161/strokeaha.117.016753
35. Li P, Wang L, Zhou Y, Gan Y, Zhu W, Xia Y, et al. C-C Chemokine Receptor Type 5 (CCR5)-Mediated Docking of Transferred Tregs Protects Against Early Blood-Brain Barrier Disruption After Stroke. J Am Heart Assoc (2017) 6(8):e006387. doi: 10.1161/jaha.117.006387
36. Gan Y, Liu Q, Wu W, Yin JX, Bai XF, Shen R, et al. Ischemic Neurons Recruit Natural Killer Cells That Accelerate Brain Infarction. Proc Natl Acad Sci USA (2014) 111(7):2704–9. doi: 10.1073/pnas.1315943111
37. Dhanesha N, Jain M, Tripathi AK, Doddapattar P, Chorawala M, Bathla G, et al. Targeting Myeloid-Specific Integrin α9β1 Improves Short- and Long-Term Stroke Outcomes in Murine Models With Preexisting Comorbidities by Limiting Thrombosis and Inflammation. Circ Res (2020) 126(12):1779–94. doi: 10.1161/circresaha.120.316659
38. Garcia JH, Liu KF, Yoshida Y, Lian J, Chen S, del Zoppo GJ. Influx of Leukocytes and Platelets in an Evolving Brain Infarct (Wistar Rat). Am J Pathol (1994) 144(1):188–99.
39. Tsai NW, Chang WN, Shaw CF, Jan CR, Huang CR, Chen SD, et al. The Value of Leukocyte Adhesion Molecules in Patients After Ischemic Stroke. J Neurol (2009) 256(8):1296–302. doi: 10.1007/s00415-009-5117-3
40. Kataoka H, Kim SW, Plesnila N. Leukocyte-Endothelium Interactions During Permanent Focal Cerebral Ischemia in Mice. J Cereb Blood Flow Metab (2004) 24(6):668–76. doi: 10.1097/01.wcb.0000117812.35136.5b
41. Ma Y, Yang X, Chatterjee V, Meegan JE, Beard RS Jr., Yuan SY. Role of Neutrophil Extracellular Traps and Vesicles in Regulating Vascular Endothelial Permeability. Front Immunol (2019) 10:1037. doi: 10.3389/fimmu.2019.01037
42. Meegan JE, Yang X, Coleman DC, Jannaway M, Yuan SY. Neutrophil-Mediated Vascular Barrier Injury: Role of Neutrophil Extracellular Traps. Microcirculation (2017) 24(3). doi: 10.1111/micc.12352
43. Chen C, Li T, Zhao Y, Qian Y, Li X, Dai X, et al. Platelet Glycoprotein Receptor Ib Blockade Ameliorates Experimental Cerebral Ischemia-Reperfusion Injury by Strengthening the Blood-Brain Barrier Function and Anti-Thrombo-Inflammatory Property. Brain Behav Immun (2018) 69:255–63. doi: 10.1016/j.bbi.2017.11.019
44. Ao LY, Yan YY, Zhou L, Li CY, Li WT, Fang WR, et al. Immune Cells After Ischemic Stroke Onset: Roles, Migration, and Target Intervention. J Mol Neurosci (2018) 66(3):342–55. doi: 10.1007/s12031-018-1173-4
45. O’Connell GC, Treadway MB, Petrone AB, Tennant CS, Lucke-Wold N, Chantler PD, et al. Peripheral Blood AKAP7 Expression as an Early Marker for Lymphocyte-Mediated Post-Stroke Blood Brain Barrier Disruption. Sci Rep (2017) 7(1):1172. doi: 10.1038/s41598-017-01178-5
46. Yang L, Kong Y, Ren H, Li M, Wei CJ, Shi E, et al. Upregulation of CD74 and Its Potential Association With Disease Severity in Subjects With Ischemic Stroke. Neurochem Int (2017) 107:148–55. doi: 10.1016/j.neuint.2016.11.007
47. Schuhmann MK, Kraft P, Stoll G, Lorenz K, Meuth SG, Wiendl H, et al. CD28 Superagonist-Mediated Boost of Regulatory T Cells Increases Thrombo-Inflammation and Ischemic Neurodegeneration During the Acute Phase of Experimental Stroke. J Cereb Blood Flow Metab (2015) 35(1):6–10. doi: 10.1038/jcbfm.2014.175
48. Brait VH, Arumugam TV, Drummond GR, Sobey CG. Importance of T Lymphocytes in Brain Injury, Immunodeficiency, and Recovery After Cerebral Ischemia. J Cereb Blood Flow Metab (2012) 32(4):598–611. doi: 10.1038/jcbfm.2012.6
49. Salas-Perdomo A, Miró-Mur F, Urra X, Justicia C, Gallizioli M, Zhao Y, et al. T Cells Prevent Hemorrhagic Transformation in Ischemic Stroke by P-Selectin Binding. Arterioscler Thromb Vasc Biol (2018) 38(8):1761–71. doi: 10.1161/atvbaha.118.311284
50. Maestrini I, Strbian D, Gautier S, Haapaniemi E, Moulin S, Sairanen T, et al. Higher Neutrophil Counts Before Thrombolysis for Cerebral Ischemia Predict Worse Outcomes. Neurology (2015) 85(16):1408–16. doi: 10.1212/wnl.0000000000002029
51. Pikija S, Sztriha LK, Killer-Oberpfalzer M, Weymayr F, Hecker C, Ramesmayer C, et al. Neutrophil to Lymphocyte Ratio Predicts Intracranial Hemorrhage After Endovascular Thrombectomy in Acute Ischemic Stroke. J Neuroinflamm (2018) 15(1):319. doi: 10.1186/s12974-018-1359-2
52. Lux D, Alakbarzade V, Bridge L, Clark CN, Clarke B, Zhang L, et al. The Association of Neutrophil-Lymphocyte Ratio and Lymphocyte-Monocyte Ratio With 3-Month Clinical Outcome After Mechanical Thrombectomy Following Stroke. J Neuroinflamm (2020) 17(1):60. doi: 10.1186/s12974-020-01739-y
53. Aly M, Abdalla RN, Batra A, Shaibani A, Hurley MC, Jahromi BS, et al. Follow-Up Neutrophil-Lymphocyte Ratio After Stroke Thrombectomy Is an Independent Biomarker of Clinical Outcome. J Neurointerv Surg (2020) 2020:neurintsurg-2020-016342. doi: 10.1136/neurintsurg-2020-016342
54. Maestrini I, Tagzirt M, Gautier S, Dupont A, Mendyk AM, Susen S, et al. Analysis of the Association of MPO and MMP-9 With Stroke Severity and Outcome: Cohort Study. Neurology (2020) 95(1):e97–e108. doi: 10.1212/wnl.0000000000009179
55. Weisenburger-Lile D, Dong Y, Yger M, Weisenburger G, Polara GF, Chaigneau T, et al. Harmful Neutrophil Subsets in Patients With Ischemic Stroke: Association With Disease Severity. Neurol Neuroimmunol Neuroinflamm (2019) 6(4):e571. doi: 10.1212/nxi.0000000000000571
56. Carmona-Mora P, Ander BP, Jickling GC, Dykstra-Aiello C, Zhan X, Ferino E, et al. Distinct Peripheral Blood Monocyte and Neutrophil Transcriptional Programs Following Intracerebral Hemorrhage and Different Etiologies of Ischemic Stroke. J Cereb Blood Flow Metab (2021) 41(6):1398–416. doi: 10.1177/0271678x20953912 271678x20953912
57. Rosell A, Cuadrado E, Ortega-Aznar A, Hernández-Guillamon M, Lo EH, Montaner J. Mmp-9-positive Neutrophil Infiltration Is Associated to Blood-Brain Barrier Breakdown and Basal Lamina Type IV Collagen Degradation During Hemorrhagic Transformation After Human Ischemic Stroke. Stroke (2008) 39(4):1121–6. doi: 10.1161/strokeaha.107.500868
58. Vallés J, Lago A, Santos MT, Latorre AM, Tembl JI, Salom JB, et al. Neutrophil Extracellular Traps Are Increased in Patients With Acute Ischemic Stroke: Prognostic Significance. Thromb Haemost (2017) 117(10):1919–29. doi: 10.1160/th17-02-0130
59. Laridan E, Denorme F, Desender L, François O, Andersson T, Deckmyn H, et al. Neutrophil Extracellular Traps in Ischemic Stroke Thrombi. Ann Neurol (2017) 82(2):223–32. doi: 10.1002/ana.24993
60. Frieler RA, Chung Y, Ahlers CG, Gheordunescu G, Song J, Vigil TM, et al. Genetic Neutrophil Deficiency Ameliorates Cerebral Ischemia-Reperfusion Injury. Exp Neurol (2017) 298(Pt A):104–11. doi: 10.1016/j.expneurol.2017.08.016
61. Zhang CY, Dong X, Gao J, Lin W, Liu Z, Wang Z. Nanoparticle-Induced Neutrophil Apoptosis Increases Survival in Sepsis and Alleviates Neurological Damage in Stroke. Sci Adv (2019) 5(11):eaax7964. doi: 10.1126/sciadv.aax7964
62. Krizbai IA, Bauer H, Bresgen N, Eckl PM, Farkas A, Szatmári E, et al. Effect of Oxidative Stress on the Junctional Proteins of Cultured Cerebral Endothelial Cells. Cell Mol Neurobiol (2005) 25(1):129–39. doi: 10.1007/s10571-004-1378-7
63. Turner RJ, Sharp FR. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front Cell Neurosci (2016) 10:56. doi: 10.3389/fncel.2016.00056
64. Wang G, Weng YC, Han X, Whaley JD, McCrae KR, Chou WH. Lipocalin-2 Released in Response to Cerebral Ischaemia Mediates Reperfusion Injury in Mice. J Cell Mol Med (2015) 19(7):1637–45. doi: 10.1111/jcmm.12538
65. Wang G, Weng YC, Chiang IC, Huang YT, Liao YC, Chen YC, et al. Neutralization of Lipocalin-2 Diminishes Stroke-Reperfusion Injury. Int J Mol Sci (2020) 21(17):6253. doi: 10.3390/ijms21176253
66. Du Y, Li W, Lin L, Lo EH, Xing C. Effects of Lipocalin-2 on Brain Endothelial Adhesion and Permeability. PloS One (2019) 14(7):e0218965. doi: 10.1371/journal.pone.0218965
67. Kang L, Yu H, Yang X, Zhu Y, Bai X, Wang R, et al. Neutrophil Extracellular Traps Released by Neutrophils Impair Revascularization and Vascular Remodeling After Stroke. Nat Commun (2020) 11(1):2488. doi: 10.1038/s41467-020-16191-y
68. Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, Sharp FR. Targeting Neutrophils in Ischemic Stroke: Translational Insights From Experimental Studies. J Cereb Blood Flow Metab (2015) 35(6):888–901. doi: 10.1038/jcbfm.2015.45
69. Stamatovic SM, Keep RF, Wang MM, Jankovic I, Andjelkovic AV. Caveolae-Mediated Internalization of Occludin and Claudin-5 During CCL2-Induced Tight Junction Remodeling in Brain Endothelial Cells. J Biol Chem (2009) 284(28):19053–66. doi: 10.1074/jbc.M109.000521
70. Guo F, Xu D, Lin Y, Wang G, Wang F, Gao Q, et al. Chemokine CCL2 Contributes to BBB Disruption Via the P38 MAPK Signaling Pathway Following Acute Intracerebral Hemorrhage. FASEB J (2020) 34(1):1872–84. doi: 10.1096/fj.201902203RR
71. Ajikumar A, Long MB, Heath PR, Wharton SB, Ince PG, Ridger VC, et al. Neutrophil-Derived Microvesicle Induced Dysfunction of Brain Microvascular Endothelial Cells In Vitro. Int J Mol Sci (2019) 20(20). doi: 10.3390/ijms20205227
72. Hou Y, Yang D, Xiang R, Wang H, Wang X, Zhang H, et al. N2 Neutrophils may Participate in Spontaneous Recovery After Transient Cerebral Ischemia by Inhibiting Ischemic Neuron Injury in Rats. Int Immunopharmacol (2019) 77:105970. doi: 10.1016/j.intimp.2019.105970
73. Cuartero MI, Ballesteros I, Moraga A, Nombela F, Vivancos J, Hamilton JA, et al. N2 Neutrophils, Novel Players in Brain Inflammation After Stroke: Modulation by the Pparγ Agonist Rosiglitazone. Stroke (2013) 44(12):3498–508. doi: 10.1161/strokeaha.113.002470
74. García-Culebras A, Durán-Laforet V, Peña-Martínez C, Moraga A, Ballesteros I, Cuartero MI, et al. Role of TLR4 (Toll-Like Receptor 4) in N1/N2 Neutrophil Programming After Stroke. Stroke (2019) 50(10):2922–32. doi: 10.1161/strokeaha.119.025085
75. Dong X, Nao J, Gao Y. Peripheral Monocyte Count Predicts Outcomes in Patients With Acute Ischemic Stroke Treated With rtPA Thrombolysis. Neurotox Res (2020) 37(2):469–77. doi: 10.1007/s12640-019-00103-0
76. Liu H, Liu K, Pei L, Gao Y, Zhao L, Sun S, et al. Monocyte-to-High-Density Lipoprotein Ratio Predicts the Outcome of Acute Ischemic Stroke. J Atheroscler Thromb (2020) 27(9):959–68. doi: 10.5551/jat.51151
77. Liu H, Zhan F, Wang Y. Evaluation of Monocyte-to-High-Density Lipoprotein Cholesterol Ratio and Monocyte-to-Lymphocyte Ratio in Ischemic Stroke. J Int Med Res (2020) 48(7):300060520933806. doi: 10.1177/0300060520933806
78. Schuhmann MK, Gunreben I, Kleinschnitz C, Kraft P. Immunohistochemical Analysis of Cerebral Thrombi Retrieved by Mechanical Thrombectomy From Patients With Acute Ischemic Stroke. Int J Mol Sci (2016) 17(3):298. doi: 10.3390/ijms17030298
79. Fan L, Gui L, Chai EQ, Wei CJ. Routine Hematological Parameters are Associated With Short- and Long-Term Prognosis of Patients With Ischemic Stroke. J Clin Lab Anal (2018) 32(2):e22244. doi: 10.1002/jcla.22244
80. Kraft P, Drechsler C, Schuhmann MK, Gunreben I, Kleinschnitz C. Characterization of Peripheral Immune Cell Subsets in Patients With Acute and Chronic Cerebrovascular Disease: A Case-Control Study. Int J Mol Sci (2015) 16(10):25433–49. doi: 10.3390/ijms161025433
81. Wang Y, Cheng Y, Song Q, Wei C, Liu J, Wu B, et al. The Association Between Monocyte to High-Density Lipoprotein Ratio and Hemorrhagic Transformation in Patients With Acute Ischemic Stroke. Aging (Albany NY) (2020) 12(3):2498–506. doi: 10.18632/aging.102757
82. Urra X, Villamor N, Amaro S, Gómez-Choco M, Obach V, Oleaga L, et al. Monocyte Subtypes Predict Clinical Course and Prognosis in Human Stroke. J Cereb Blood Flow Metab (2009) 29(5):994–1002. doi: 10.1038/jcbfm.2009.25
83. Fang W, Zhai X, Han D, Xiong X, Wang T, Zeng X, et al. CCR2-Dependent Monocytes/Macrophages Exacerbate Acute Brain Injury But Promote Functional Recovery After Ischemic Stroke in Mice. Theranostics (2018) 8(13):3530–43. doi: 10.7150/thno.24475
84. Pedragosa J, Miró-Mur F, Otxoa-de-Amezaga A, Justicia C, Ruíz-Jaén F, Ponsaerts P, et al. CCR2 Deficiency in Monocytes Impairs Angiogenesis and Functional Recovery After Ischemic Stroke in Mice. J Cereb Blood Flow Metab (2020) 40(1_suppl):S98–s116. doi: 10.1177/0271678x20909055
85. Garcia-Bonilla L, Faraco G, Moore J, Murphy M, Racchumi G, Srinivasan J, et al. Spatio-Temporal Profile, Phenotypic Diversity, and Fate of Recruited Monocytes Into the Post-Ischemic Brain. J Neuroinflamm (2016) 13(1):285. doi: 10.1186/s12974-016-0750-0
86. Michaud JP, Pimentel-Coelho PM, Tremblay Y, Rivest S. The Impact of Ly6Clow Monocytes After Cerebral Hypoxia-Ischemia in Adult Mice. J Cereb Blood Flow Metab (2014) 34(7):e1–9. doi: 10.1038/jcbfm.2014.80
87. Wattananit S, Tornero D, Graubardt N, Memanishvili T, Monni E, Tatarishvili J, et al. Monocyte-Derived Macrophages Contribute to Spontaneous Long-Term Functional Recovery After Stroke in Mice. J Neurosci (2016) 36(15):4182–95. doi: 10.1523/jneurosci.4317-15.2016
88. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
89. Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and Functional Characterization of Three Activated Macrophage Populations. J Leukoc Biol (2006) 80(6):1298–307. doi: 10.1189/jlb.0406249
90. Rőszer T. Understanding the Mysterious M2 Macrophage Through Activation Markers and Effector Mechanisms. Mediators Inflamm (2015) 2015:816460. doi: 10.1155/2015/816460
91. Kolosowska N, Keuters MH, Wojciechowski S, Keksa-Goldsteine V, Laine M, Malm T, et al. Peripheral Administration of IL-13 Induces Anti-Inflammatory Microglial/Macrophage Responses and Provides Neuroprotection in Ischemic Stroke. Neurotherapeutics (2019) 16(4):1304–19. doi: 10.1007/s13311-019-00761-0
92. Verma R, Cronin CG, Hudobenko J, Venna VR, McCullough LD, Liang BT. Deletion of the P2X4 Receptor Is Neuroprotective Acutely, But Induces a Depressive Phenotype During Recovery From Ischemic Stroke. Brain Behav Immun (2017) 66:302–12. doi: 10.1016/j.bbi.2017.07.155
93. Liu Q, Johnson EM, Lam RK, Wang Q, Bo Ye H, Wilson EN, et al. Peripheral TREM1 Responses to Brain and Intestinal Immunogens Amplify Stroke Severity. Nat Immunol (2019) 20(8):1023–34. doi: 10.1038/s41590-019-0421-2
94. Liao B, Geng L, Zhang F, Shu L, Wei L, Yeung PKK, et al. Adipocyte Fatty Acid-Binding Protein Exacerbates Cerebral Ischaemia Injury by Disrupting the Blood-Brain Barrier. Eur Heart J (2020) 41(33):3169–80. doi: 10.1093/eurheartj/ehaa207
95. Wang R, Liu Y, Ye Q, Hassan SH, Zhao J, Li S, et al. RNA Sequencing Reveals Novel Macrophage Transcriptome Favoring Neurovascular Plasticity After Ischemic Stroke. J Cereb Blood Flow Metab (2020) 40(4):720–38. doi: 10.1177/0271678x19888630
96. Liu C, Wu C, Yang Q, Gao J, Li L, Yang D, et al. Macrophages Mediate the Repair of Brain Vascular Rupture Through Direct Physical Adhesion and Mechanical Traction. Immunity (2016) 44(5):1162–76. doi: 10.1016/j.immuni.2016.03.008
97. Gliem M, Mausberg AK, Lee JI, Simiantonakis I, van Rooijen N, Hartung HP, et al. Macrophages Prevent Hemorrhagic Infarct Transformation in Murine Stroke Models. Ann Neurol (2012) 71(6):743–52. doi: 10.1002/ana.23529
98. Zhang W, Zhao J, Wang R, Jiang M, Ye Q, Smith AD, et al. Macrophages Reprogram After Ischemic Stroke and Promote Efferocytosis and Inflammation Resolution in the Mouse Brain. CNS Neurosci Ther (2019) 25(12):1329–42. doi: 10.1111/cns.13256
99. Xue Y, Nie D, Wang LJ, Qiu HC, Ma L, Dong MX, et al. Microglial Polarization: Novel Therapeutic Strategy Against Ischemic Stroke. Aging Dis (2021) 12(2):466–79. doi: 10.14336/ad.2020.0701
100. Noh MY, Lee WM, Lee SJ, Kim HY, Kim SH, Kim YS. Regulatory T Cells Increase After Treatment With Poly (ADP-Ribose) Polymerase-1 Inhibitor in Ischemic Stroke Patients. Int Immunopharmacol (2018) 60:104–10. doi: 10.1016/j.intimp.2018.04.043
101. Ruhnau J, Schulze J, von Sarnowski B, Heinrich M, Langner S, Pötschke C, et al. Reduced Numbers and Impaired Function of Regulatory T Cells in Peripheral Blood of Ischemic Stroke Patients. Mediators Inflammation (2016) 2016:2974605. doi: 10.1155/2016/2974605
102. Hu Y, Zheng Y, Wu Y, Ni B, Shi S. Imbalance Between IL-17A-Producing Cells and Regulatory T Cells During Ischemic Stroke. Mediators Inflamm (2014) 2014:813045. doi: 10.1155/2014/813045
103. Dolati S, Ahmadi M, Khalili M, Taheraghdam AA, Siahmansouri H, Babaloo Z, et al. Peripheral Th17/Treg Imbalance in Elderly Patients With Ischemic Stroke. Neurol Sci (2018) 39(4):647–54. doi: 10.1007/s10072-018-3250-4
104. Nadareishvili ZG, Li H, Wright V, Maric D, Warach S, Hallenbeck JM, et al. Elevated Pro-Inflammatory CD4+CD28- Lymphocytes and Stroke Recurrence and Death. Neurology (2004) 63(8):1446–51. doi: 10.1212/01.wnl.0000142260.61443.7c
105. Zhao D, Hou N, Cui M, Liu Y, Liang X, Zhuang X, et al. Increased T Cell Immunoglobulin and Mucin Domain 3 Positively Correlate With Systemic IL-17 and TNF-α Level in the Acute Phase of Ischemic Stroke. J Clin Immunol (2011) 31(4):719–27. doi: 10.1007/s10875-011-9534-6
106. Haak BW, Westendorp WF, van Engelen TSR, Brands X, Brouwer MC, Vermeij JD, et al. Disruptions of Anaerobic Gut Bacteria Are Associated With Stroke and Post-Stroke Infection: A Prospective Case-Control Study. Transl Stroke Res (2020). doi: 10.1007/s12975-020-00863-4
107. Li N, Wang X, Sun C, Wu X, Lu M, Si Y, et al. Change of Intestinal Microbiota in Cerebral Ischemic Stroke Patients. BMC Microbiol (2019) 19(1):191. doi: 10.1186/s12866-019-1552-1
108. Zhai Q, Wang X, Chen C, Tang Y, Wang Y, Tian J, et al. Prognostic Value of Plasma Trimethylamine N-Oxide Levels in Patients With Acute Ischemic Stroke. Cell Mol Neurobiol (2019) 39(8):1201–6. doi: 10.1007/s10571-019-00714-3
109. Tan C, Wang H, Gao X, Xu R, Zeng X, Cui Z, et al. Dynamic Changes and Prognostic Value of Gut Microbiota-Dependent Trimethylamine-N-Oxide in Acute Ischemic Stroke. Front Neurol (2020) 11:29. doi: 10.3389/fneur.2020.00029
110. Haghikia A, Li XS, Liman TG, Bledau N, Schmidt D, Zimmermann F, et al. Gut Microbiota-Dependent Trimethylamine N-Oxide Predicts Risk of Cardiovascular Events in Patients With Stroke and Is Related to Proinflammatory Monocytes. Arterioscler Thromb Vasc Biol (2018) 38(9):2225–35. doi: 10.1161/atvbaha.118.311023
111. Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. Trimethylamine-N-Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome Through the SIRT3-SOD2-mtROS Signaling Pathway. J Am Heart Assoc (2017) 6(9):e006347. doi: 10.1161/jaha.117.006347
112. Blasco MP, Chauhan A, Honarpisheh P, Ahnstedt H, d’Aigle J, Ganesan A, et al. Age-Dependent Involvement of Gut Mast Cells and Histamine in Post-Stroke Inflammation. J Neuroinflamm (2020) 17(1):160. doi: 10.1186/s12974-020-01833-1
113. Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al. Commensal Microbiota Affects Ischemic Stroke Outcome by Regulating Intestinal γδ T Cells. Nat Med (2016) 22(5):516–23. doi: 10.1038/nm.4068
114. Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al. Neutralization of the IL-17 Axis Diminishes Neutrophil Invasion and Protects From Ischemic Stroke. Blood (2012) 120(18):3793–802. doi: 10.1182/blood-2012-02-412726
115. Clarkson BD, Ling C, Shi Y, Harris MG, Rayasam A, Sun D, et al. T Cell-Derived Interleukin (IL)-21 Promotes Brain Injury Following Stroke in Mice. J Exp Med (2014) 211(4):595–604. doi: 10.1084/jem.20131377
116. Wojkowska DW, Szpakowski P, Glabinski A. Interleukin 17a Promotes Lymphocytes Adhesion and Induces CCL2 and CXCL1 Release From Brain Endothelial Cells. Int J Mol Sci (2017) 18(5):1000. doi: 10.3390/ijms18051000
117. Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, et al. Cellular Mechanisms of IL-17-Induced Blood-Brain Barrier Disruption. FASEB J (2010) 24(4):1023–34. doi: 10.1096/fj.09-141978
118. Broux B, Zandee S, Gowing E, Charabati M, Lécuyer MA, Tastet O, et al. Interleukin-26, Preferentially Produced by T(H)17 Lymphocytes, Regulates CNS Barrier Function. Neurol Neuroimmunol Neuroinflamm (2020) 7(6):e870. doi: 10.1212/nxi.0000000000000870
119. Fan L, Zhang CJ, Zhu L, Chen J, Zhang Z, Liu P, et al. FasL-PDPK1 Pathway Promotes the Cytotoxicity of CD8(+) T Cells During Ischemic Stroke. Transl Stroke Res (2020) 11(4):747–61. doi: 10.1007/s12975-019-00749-0
120. Shi K, Zou M, Jia DM, Shi S, Yang X, Liu Q, et al. tPA Mobilizes Immune Cells That Exacerbate Hemorrhagic Transformation in Stroke. Circ Res (2021) 128(1):62–75. doi: 10.1161/circresaha.120.317596
121. Na SY, Mracsko E, Liesz A, Hünig T, Veltkamp R. Amplification of Regulatory T Cells Using a CD28 Superagonist Reduces Brain Damage After Ischemic Stroke in Mice. Stroke (2015) 46(1):212–20. doi: 10.1161/strokeaha.114.007756
122. Li P, Mao L, Liu X, Gan Y, Zheng J, Thomson AW, et al. Essential Role of Program Death 1-Ligand 1 in Regulatory T-Cell-Afforded Protection Against Blood-Brain Barrier Damage After Stroke. Stroke (2014) 45(3):857–64. doi: 10.1161/strokeaha.113.004100
123. Mao L, Li P, Zhu W, Cai W, Liu Z, Wang Y, et al. Regulatory T Cells Ameliorate Tissue Plasminogen Activator-Induced Brain Haemorrhage After Stroke. Brain (2017) 140(7):1914–31. doi: 10.1093/brain/awx111
124. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting Microglial Cells are Highly Dynamic Surveillants of Brain Parenchyma In Vivo. Science (2005) 308(5726):1314–8. doi: 10.1126/science.1110647
125. Lauro C, Chece G, Monaco L, Antonangeli F, Peruzzi G, Rinaldo S, et al. Fractalkine Modulates Microglia Metabolism in Brain Ischemia. Front Cell Neurosci (2019) 13:414. doi: 10.3389/fncel.2019.00414
126. Colombo E, Farina C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol (2016) 37(9):608–20. doi: 10.1016/j.it.2016.06.006
127. Nyúl-Tóth Á., Kozma M, Nagyőszi P, Nagy K, Fazakas C, Haskó J, et al. Expression of Pattern Recognition Receptors and Activation of the Non-Canonical Inflammasome Pathway in Brain Pericytes. Brain Behav Immun (2017) 64:220–31. doi: 10.1016/j.bbi.2017.04.010
128. Nowicka D, Rogozinska K, Aleksy M, Witte OW, Skangiel-Kramska J. Spatiotemporal Dynamics of Astroglial and Microglial Responses After Photothrombotic Stroke in the Rat Brain. Acta Neurobiol Exp (Wars) (2008) 68(2):155–68.
129. Perego C, Fumagalli S, De Simoni MG. Temporal Pattern of Expression and Colocalization of Microglia/Macrophage Phenotype Markers Following Brain Ischemic Injury in Mice. J Neuroinflamm (2011) 8:174. doi: 10.1186/1742-2094-8-174
130. Liu S, Gao Y, Yu X, Zhao B, Liu L, Zhao Y, et al. Annexin-1 Mediates Microglial Activation and Migration Via the CK2 Pathway During Oxygen-Glucose Deprivation/Reperfusion. Int J Mol Sci (2016) 17(10):1770. doi: 10.3390/ijms17101770
131. Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, et al. Microglia/Macrophage Polarization Dynamics Reveal Novel Mechanism of Injury Expansion After Focal Cerebral Ischemia. Stroke (2012) 43(11):3063–70. doi: 10.1161/strokeaha.112.659656
132. Gülke E, Gelderblom M, Magnus T. Danger Signals in Stroke and Their Role on Microglia Activation After Ischemia. Ther Adv Neurol Disord (2018) 11:1756286418774254. doi: 10.1177/1756286418774254
133. Xu P, Zhang X, Liu Q, Xie Y, Shi X, Chen J, et al. Microglial TREM-1 Receptor Mediates Neuroinflammatory Injury Via Interaction With SYK in Experimental Ischemic Stroke. Cell Death Dis (2019) 10(8):555. doi: 10.1038/s41419-019-1777-9
134. Esposito E, Hayakawa K, Ahn BJ, Chan SJ, Xing C, Liang AC, et al. Effects of Ischemic Post-Conditioning on Neuronal VEGF Regulation and Microglial Polarization in a Rat Model of Focal Cerebral Ischemia. J Neurochem (2018) 146(2):160–72. doi: 10.1111/jnc.14337
135. Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial Activation Precedes and Predominates Over Macrophage Infiltration in Transient Focal Cerebral Ischemia: A Study in Green Fluorescent Protein Transgenic Bone Marrow Chimeric Mice. Exp Neurol (2003) 183(1):25–33. doi: 10.1016/s0014-4886(03)00082-7
136. Kronenberg G, Uhlemann R, Richter N, Klempin F, Wegner S, Staerck L, et al. Distinguishing Features of Microglia- and Monocyte-Derived Macrophages After Stroke. Acta Neuropathol (2018) 135(4):551–68. doi: 10.1007/s00401-017-1795-6
137. Jolivel V, Bicker F, Binamé F, Ploen R, Keller S, Gollan R, et al. Perivascular Microglia Promote Blood Vessel Disintegration in the Ischemic Penumbra. Acta Neuropathol (2015) 129(2):279–95. doi: 10.1007/s00401-014-1372-1
138. Clausen BH, Wirenfeldt M, Høgedal SS, Frich LH, Nielsen HH, Schrøder HD, et al. Characterization of the TNF and IL-1 Systems in Human Brain and Blood After Ischemic Stroke. Acta Neuropathol Commun (2020) 8(1):81. doi: 10.1186/s40478-020-00957-y
139. Murata Y, Sugimoto K, Yang C, Harada K, Gono R, Harada T, et al. Activated Microglia-Derived Macrophage-Like Cells Exacerbate Brain Edema After Ischemic Stroke Correlate With Astrocytic Expression of Aquaporin-4 and Interleukin-1 Alpha Release. Neurochem Int (2020) 140:104848. doi: 10.1016/j.neuint.2020.104848
140. Kangwantas K, Pinteaux E, Penny J. The Extracellular Matrix Protein Laminin-10 Promotes Blood-Brain Barrier Repair After Hypoxia and Inflammation In Vitro. J Neuroinflamm (2016) 13:25. doi: 10.1186/s12974-016-0495-9
141. Argaw AT, Zhang Y, Snyder BJ, Zhao ML, Kopp N, Lee SC, et al. Il-1beta Regulates Blood-Brain Barrier Permeability Via Reactivation of the Hypoxia-Angiogenesis Program. J Immunol (2006) 177(8):5574–84. doi: 10.4049/jimmunol.177.8.5574
142. Wang Y, Jin S, Sonobe Y, Cheng Y, Horiuchi H, Parajuli B, et al. Interleukin-1β Induces Blood-Brain Barrier Disruption by Downregulating Sonic Hedgehog in Astrocytes. PloS One (2014) 9(10):e110024. doi: 10.1371/journal.pone.0110024
143. Chen AQ, Fang Z, Chen XL, Yang S, Zhou YF, Mao L, et al. Microglia-Derived TNF-α Mediates Endothelial Necroptosis Aggravating Blood Brain-Barrier Disruption After Ischemic Stroke. Cell Death Dis (2019) 10(7):487. doi: 10.1038/s41419-019-1716-9
144. Ding XW, Sun X, Shen XF, Lu Y, Wang JQ, Sun ZR, et al. Propofol Attenuates TNF-α-Induced MMP-9 Expression in Human Cerebral Microvascular Endothelial Cells by Inhibiting Ca(2+)/CAMK II/Erk/Nf-κb Signaling Pathway. Acta Pharmacol Sin (2019) 40(10):1303–13. doi: 10.1038/s41401-019-0258-0
145. Sonar SA, Shaikh S, Joshi N, Atre AN, Lal G. Ifn-γ Promotes Transendothelial Migration of CD4(+) T Cells Across the Blood-Brain Barrier. Immunol Cell Biol (2017) 95(9):843–53. doi: 10.1038/icb.2017.56
146. de Bilbao F, Arsenijevic D, Moll T, Garcia-Gabay I, Vallet P, Langhans W, et al. In Vivo Over-Expression of Interleukin-10 Increases Resistance to Focal Brain Ischemia in Mice. J Neurochem (2009) 110(1):12–22. doi: 10.1111/j.1471-4159.2009.06098.x
147. Yang E, Cai Y, Yao X, Liu J, Wang Q, Jin W, et al. Tissue Plasminogen Activator Disrupts the Blood-Brain Barrier Through Increasing the Inflammatory Response Mediated by Pericytes After Cerebral Ischemia. Aging (Albany NY) (2019) 11(22):10167–82. doi: 10.18632/aging.102431
148. Kang H, Yang PY, Rui YC. Adenovirus Viral Interleukin-10 Inhibits Adhesion Molecule Expressions Induced by Hypoxia/Reoxygenation in Cerebrovascular Endothelial Cells. Acta Pharmacol Sin (2008) 29(1):50–6. doi: 10.1111/j.1745-7254.2008.00718.x
149. Li D, Lang W, Zhou C, Wu C, Zhang F, Liu Q, et al. Upregulation of Microglial Zeb1 Ameliorates Brain Damage After Acute Ischemic Stroke. Cell Rep (2018) 22(13):3574–86. doi: 10.1016/j.celrep.2018.03.011
150. Lin R, Chen F, Wen S, Teng T, Pan Y, Huang H. Interleukin-10 Attenuates Impairment of the Blood-Brain Barrier in a Severe Acute Pancreatitis Rat Model. J Inflamm (Lond) (2018) 15:4. doi: 10.1186/s12950-018-0180-0
151. Waehre T, Halvorsen B, Damås JK, Yndestad A, Brosstad F, Gullestad L, et al. Inflammatory Imbalance Between IL-10 and TNFalpha in Unstable Angina Potential Plaque Stabilizing Effects of IL-10. Eur J Clin Invest (2002) 32(11):803–10. doi: 10.1046/j.1365-2362.2002.01069.x
152. Cai Y, Liu X, Chen W, Wang Z, Xu G, Zeng Y, et al. TGF-β1 Prevents Blood-Brain Barrier Damage and Hemorrhagic Transformation After Thrombolysis in Rats. Exp Neurol (2015) 266:120–6. doi: 10.1016/j.expneurol.2015.02.013
153. Rodriguez-Grande B, Swana M, Nguyen L, Englezou P, Maysami S, Allan SM, et al. The Acute-Phase Protein PTX3 Is an Essential Mediator of Glial Scar Formation and Resolution of Brain Edema After Ischemic Injury. J Cereb Blood Flow Metab (2014) 34(3):480–8. doi: 10.1038/jcbfm.2013.224
154. Salmeron K, Aihara T, Redondo-Castro E, Pinteaux E, Bix G. Il-1alpha Induces Angiogenesis in Brain Endothelial Cells In Vitro: Implications for Brain Angiogenesis After Acute Injury. J Neurochem (2016) 136(3):573–80. doi: 10.1111/jnc.13422
155. Li Y, Liu DX, Li MY, Qin XX, Fang WG, Zhao WD, et al. Ephrin-A3 and ephrin-A4 Contribute to Microglia-Induced Angiogenesis in Brain Endothelial Cells. Anat Rec (Hoboken) (2014) 297(10):1908–18. doi: 10.1002/ar.22998
156. Meng H, Song Y, Zhu J, Liu Q, Lu P, Ye N, et al. LRG1 Promotes Angiogenesis Through Upregulating the TGF−β1 Pathway in Ischemic Rat Brain. Mol Med Rep (2016) 14(6):5535–43. doi: 10.3892/mmr.2016.5925
157. Wenzel TJ, Bajwa E, Klegeris A. Cytochrome C can be Released Into Extracellular Space and Modulate Functions of Human Astrocytes in a Toll-Like Receptor 4-Dependent Manner. Biochim Biophys Acta Gen Subj (2019) 1863(11):129400. doi: 10.1016/j.bbagen.2019.07.009
158. Zhao N, Xu X, Jiang Y, Gao J, Wang F, Xu X, et al. Lipocalin-2 may Produce Damaging Effect After Cerebral Ischemia by Inducing Astrocytes Classical Activation. J Neuroinflamm (2019) 16(1):168. doi: 10.1186/s12974-019-1556-7
159. Shinozaki Y, Shibata K, Yoshida K, Shigetomi E, Gachet C, Ikenaka K, et al. Transformation of Astrocytes to a Neuroprotective Phenotype by Microglia Via P2Y(1) Receptor Downregulation. Cell Rep (2017) 19(6):1151–64. doi: 10.1016/j.celrep.2017.04.047
160. Rakers C, Schleif M, Blank N, Matušková H, Ulas T, Händler K, et al. Stroke Target Identification Guided by Astrocyte Transcriptome Analysis. Glia (2019) 67(4):619–33. doi: 10.1002/glia.23544
161. Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, et al. Astrocyte-Derived VEGF-A Drives Blood-Brain Barrier Disruption in CNS Inflammatory Disease. J Clin Invest (2012) 122(7):2454–68. doi: 10.1172/jci60842
162. Hernandes MS, Lassègue B, Hilenski LL, Adams J, Gao N, Kuan CY, et al. Polymerase Delta-Interacting Protein 2 Deficiency Protects Against Blood-Brain Barrier Permeability in the Ischemic Brain. J Neuroinflamm (2018) 15(1):45. doi: 10.1186/s12974-017-1032-1
163. Inose Y, Kato Y, Kitagawa K, Uchiyama S, Shibata N. Activated Microglia in Ischemic Stroke Penumbra Upregulate MCP-1 and CCR2 Expression in Response to Lysophosphatidylcholine Derived From Adjacent Neurons and Astrocytes. Neuropathology (2015) 35(3):209–23. doi: 10.1111/neup.12182
164. Shindo A, Maki T, Mandeville ET, Liang AC, Egawa N, Itoh K, et al. Astrocyte-Derived Pentraxin 3 Supports Blood-Brain Barrier Integrity Under Acute Phase of Stroke. Stroke (2016) 47(4):1094–100. doi: 10.1161/strokeaha.115.012133
165. Okoreeh AK, Bake S, Sohrabji F. Astrocyte-Specific Insulin-Like Growth Factor-1 Gene Transfer in Aging Female Rats Improves Stroke Outcomes. Glia (2017) 65(7):1043–58. doi: 10.1002/glia.23142
166. Norden DM, Fenn AM, Dugan A, Godbout JP. TGFβ Produced by IL-10 Redirected Astrocytes Attenuates Microglial Activation. Glia (2014) 62(6):881–95. doi: 10.1002/glia.22647
167. Kato T, Sekine Y, Nozaki H, Uemura M, Ando S, Hirokawa S, et al. Excessive Production of Transforming Growth Factor β1 Causes Mural Cell Depletion From Cerebral Small Vessels. Front Aging Neurosci (2020) 12:151. doi: 10.3389/fnagi.2020.00151
168. Morizawa YM, Hirayama Y, Ohno N, Shibata S, Shigetomi E, Sui Y, et al. Reactive Astrocytes Function as Phagocytes After Brain Ischemia Via ABCA1-Mediated Pathway. Nat Commun (2017) 8(1):28. doi: 10.1038/s41467-017-00037-1
169. Cui X, Chopp M, Zacharek A, Karasinska JM, Cui Y, Ning R, et al. Deficiency of Brain ATP-binding Cassette Transporter A-1 Exacerbates Blood-Brain Barrier and White Matter Damage After Stroke. Stroke (2015) 46(3):827–34. doi: 10.1161/strokeaha.114.007145
170. Guijarro-Muñoz I, Compte M, Álvarez-Cienfuegos A, Álvarez-Vallina L, Sanz L. Lipopolysaccharide Activates Toll-Like Receptor 4 (TLR4)-Mediated NF-κb Signaling Pathway and Proinflammatory Response in Human Pericytes. J Biol Chem (2014) 289(4):2457–68. doi: 10.1074/jbc.M113.521161
171. Wan Y, Jin HJ, Zhu YY, Fang Z, Mao L, He Q, et al. MicroRNA-149-5p Regulates Blood-Brain Barrier Permeability After Transient Middle Cerebral Artery Occlusion in Rats by Targeting S1PR2 of Pericytes. FASEB J (2018) 32(6):3133–48. doi: 10.1096/fj.201701121R
172. Carlsson R, Özen I, Barbariga M, Gaceb A, Roth M, Paul G. STAT3 Precedes HIF1α Transcriptional Responses to Oxygen and Oxygen and Glucose Deprivation in Human Brain Pericytes. PloS One (2018) 13(3):e0194146. doi: 10.1371/journal.pone.0194146
173. Matsumoto J, Takata F, Machida T, Takahashi H, Soejima Y, Funakoshi M, et al. Tumor Necrosis Factor-α-Stimulated Brain Pericytes Possess a Unique Cytokine and Chemokine Release Profile and Enhance Microglial Activation. Neurosci Lett (2014) 578:133–8. doi: 10.1016/j.neulet.2014.06.052
174. Rustenhoven J, Aalderink M, Scotter EL, Oldfield RL, Bergin PS, Mee EW, et al. TGF-Beta1 Regulates Human Brain Pericyte Inflammatory Processes Involved in Neurovasculature Function. J Neuroinflamm (2016) 13:37. doi: 10.1186/s12974-016-0503-0
175. Smyth LCD, Rustenhoven J, Park TI, Schweder P, Jansson D, Heppner PA, et al. Unique and Shared Inflammatory Profiles of Human Brain Endothelia and Pericytes. J Neuroinflamm (2018) 15(1):138. doi: 10.1186/s12974-018-1167-8
176. Zhou YF, Li YN, Jin HJ, Wu JH, He QW, Wang XX, et al. Sema4d/PlexinB1 Inhibition Ameliorates Blood-Brain Barrier Damage and Improves Outcome After Stroke in Rats. FASEB J (2018) 32(4):2181–96. doi: 10.1096/fj.201700786RR
177. Rustenhoven J, Scotter EL, Jansson D, Kho DT, Oldfield RL, Bergin PS, et al. An Anti-Inflammatory Role for C/Ebpδ in Human Brain Pericytes. Sci Rep 5:12132. doi: 10.1038/srep12132
178. Gerhartl A, Pracser N, Vladetic A, Hendrikx S, Friedl HP, Neuhaus W. The Pivotal Role of Micro-Environmental Cells in a Human Blood-Brain Barrier In Vitro Model of Cerebral Ischemia: Functional and Transcriptomic Analysis. Fluids Barriers CNS (2020) 17(1):19. doi: 10.1186/s12987-020-00179-3
179. Underly RG, Levy M, Hartmann DA, Grant RI, Watson AN, Shih AY. Pericytes as Inducers of Rapid, Matrix Metalloproteinase-9-Dependent Capillary Damage During Ischemia. J Neurosci (2017) 37(1):129–40. doi: 10.1523/jneurosci.2891-16.2016
180. Diéguez-Hurtado R, Kato K, Giaimo BD, Nieminen-Kelhä M, Arf H, Ferrante F, et al. Loss of the Transcription Factor RBPJ Induces Disease-Promoting Properties in Brain Pericytes. Nat Commun (2019) 10(1):2817. doi: 10.1038/s41467-019-10643-w
181. Nishimura A, Ago T, Kuroda J, Arimura K, Tachibana M, Nakamura K, et al. Detrimental Role of Pericyte Nox4 in the Acute Phase of Brain Ischemia. J Cereb Blood Flow Metab (2016) 36(6):1143–54. doi: 10.1177/0271678x15606456
182. Nirwane A, Johnson J, Nguyen B, Miner JH, Yao Y. Mural Cell-Derived Laminin-α5 Plays a Detrimental Role in Ischemic Stroke. Acta Neuropathol Commun (2019) 7(1):23. doi: 10.1186/s40478-019-0676-8
183. Dohgu S, Takata F, Yamauchi A, Nakagawa S, Egawa T, Naito M, et al. Brain Pericytes Contribute to the Induction and Up-Regulation of Blood-Brain Barrier Functions Through Transforming Growth Factor-Beta Production. Brain Res (2005) 1038(2):208–15. doi: 10.1016/j.brainres.2005.01.027
184. Balabanov R, Washington R, Wagnerova J, Dore-Duffy P. CNS Microvascular Pericytes Express Macrophage-Like Function, Cell Surface Integrin Alpha M, and Macrophage Marker ED-2. Microvasc Res (1996) 52(2):127–42. doi: 10.1006/mvre.1996.0049
185. Jeynes B. Reactions of Granular Pericytes in a Rabbit Cerebrovascular Ischemia Model. Stroke (1985) 16(1):121–5. doi: 10.1161/01.str.16.1.121
186. Katare RG, Madeddu P. Pericytes From Human Veins for Treatment of Myocardial Ischemia. Trends Cardiovasc Med (2013) 23(3):66–70. doi: 10.1016/j.tcm.2012.09.002
187. Özen I, Deierborg T, Miharada K, Padel T, Englund E, Genové G, et al. Brain Pericytes Acquire a Microglial Phenotype After Stroke. Acta Neuropathol (2014) 128(3):381–96. doi: 10.1007/s00401-014-1295-x
188. Sakuma R, Kawahara M, Nakano-Doi A, Takahashi A, Tanaka Y, Narita A, et al. Brain Pericytes Serve as Microglia-Generating Multipotent Vascular Stem Cells Following Ischemic Stroke. J Neuroinflamm (2016) 13(1):57. doi: 10.1186/s12974-016-0523-9
189. Huang W, Bai X, Meyer E, Scheller A. Acute Brain Injuries Trigger Microglia as an Additional Source of the Proteoglycan NG2. Acta Neuropathol Commun (2020) 8(1):146. doi: 10.1186/s40478-020-01016-2
190. Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von Brühl ML, et al. Capillary and Arteriolar Pericytes Attract Innate Leukocytes Exiting Through Venules and ‘Instruct’ Them With Pattern-Recognition and Motility Programs. Nat Immunol (2013) 14(1):41–51. doi: 10.1038/ni.2477
191. Wang S, Cao C, Chen Z, Bankaitis V, Tzima E, Sheibani N, et al. Pericytes Regulate Vascular Basement Membrane Remodeling and Govern Neutrophil Extravasation During Inflammation. PloS One (2012) 7(9):e45499. doi: 10.1371/journal.pone.0045499
192. Tei N, Tanaka J, Sugimoto K, Nishihara T, Nishioka R, Takahashi H, et al. Expression of MCP-1 and Fractalkine on Endothelial Cells and Astrocytes may Contribute to the Invasion and Migration of Brain Macrophages in Ischemic Rat Brain Lesions. J Neurosci Res (2013) 91(5):681–93. doi: 10.1002/jnr.23202
193. Otxoa-de-Amezaga A, Miró-Mur F, Pedragosa J, Gallizioli M, Justicia C, Gaja-Capdevila N, et al. Microglial Cell Loss After Ischemic Stroke Favors Brain Neutrophil Accumulation. Acta Neuropathol (2019) 137(2):321–41. doi: 10.1007/s00401-018-1954-4
194. Shu L, Xu CQ, Yan ZY, Yan Y, Jiang SZ, Wang YR. Post-Stroke Microglia Induce Sirtuin2 Expression to Suppress the Anti-Inflammatory Function of Infiltrating Regulatory T Cells. Inflammation (2019) 42(6):1968–79. doi: 10.1007/s10753-019-01057-3
195. Lee GA, Lin TN, Chen CY, Mau SY, Huang WZ, Kao YC, et al. Interleukin 15 Blockade Protects the Brain From Cerebral Ischemia-Reperfusion Injury. Brain Behav Immun (2018) 73:562–70. doi: 10.1016/j.bbi.2018.06.021
196. Beurel E, Harrington LE, Buchser W, Lemmon V, Jope RS. Astrocytes Modulate the Polarization of CD4+ T Cells to Th1 Cells. PloS One (2014) 9(1):e86257. doi: 10.1371/journal.pone.0086257
197. Xie L, Choudhury GR, Winters A, Yang SH, Jin K. Cerebral Regulatory T cells Restrain Microglia/Macrophage-Mediated Inflammatory Responses Via IL-10. Eur J Immunol (2015) 45(1):180–91. doi: 10.1002/eji.201444823
198. Tu Z, Li Y, Smith DS, Sheibani N, Huang S, Kern T, et al. Retinal Pericytes Inhibit Activated T Cell Proliferation. Invest Ophthalmol Vis Sci (2011) 52(12):9005–10. doi: 10.1167/iovs.11-8008
199. Gliem M, Krammes K, Liaw L, van Rooijen N, Hartung HP, Jander S. Macrophage-Derived Osteopontin Induces Reactive Astrocyte Polarization and Promotes Re-Establishment of the Blood Brain Barrier After Ischemic Stroke. Glia (2015) 63(12):2198–207. doi: 10.1002/glia.22885
200. Zheng Y, He R, Wang P, Shi Y, Zhao L, Liang J. Exosomes From LPS-stimulated Macrophages Induce Neuroprotection and Functional Improvement After Ischemic Stroke by Modulating Microglial Polarization. Biomater Sci (2019) 7(5):2037–49. doi: 10.1039/c8bm01449c
201. Minutti CM, Modak RV, Macdonald F, Li F, Smyth DJ, Dorward DA, et al. A Macrophage-Pericyte Axis Directs Tissue Restoration Via Amphiregulin-Induced Transforming Growth Factor Beta Activation. Immunity (2019) 50(3):645–654.e6. doi: 10.1016/j.immuni.2019.01.008
202. Meng H, Zhao H, Cao X, Hao J, Zhang H, Liu Y, et al. Double-Negative T Cells Remarkably Promote Neuroinflammation After Ischemic Stroke. Proc Natl Acad Sci USA (2019) 116(12):5558–63. doi: 10.1073/pnas.1814394116
203. Yao X, Jiang Q, Ding W, Yue P, Wang J, Zhao K, et al. Interleukin 4 Inhibits High Mobility Group Box-1 Protein-Mediated NLRP3 Inflammasome Formation by Activating Peroxisome Proliferator-Activated Receptor-γ in Astrocytes. Biochem Biophys Res Commun (2019) 509(2):624–31. doi: 10.1016/j.bbrc.2018.11.145
204. Liu X, Liu J, Zhao S, Zhang H, Cai W, Cai M, et al. Interleukin-4 Is Essential for Microglia/Macrophage M2 Polarization and Long-Term Recovery After Cerebral Ischemia. Stroke (2016) 47(2):498–504. doi: 10.1161/strokeaha.115.012079
205. Villa P, Triulzi S, Cavalieri B, Di Bitondo R, Bertini R, Barbera S, et al. The Interleukin-8 (IL-8/CXCL8) Receptor Inhibitor Reparixin Improves Neurological Deficits and Reduces Long-Term Inflammation in Permanent and Transient Cerebral Ischemia in Rats. Mol Med (2007) 13(3-4):125–33. doi: 10.2119/2007–00008.Villa
206. Copin JC, da Silva RF, Fraga-Silva RA, Capettini L, Quintao S, Lenglet S, et al. Treatment With Evasin-3 Reduces Atherosclerotic Vulnerability for Ischemic Stroke, But Not Brain Injury in Mice. J Cereb Blood Flow Metab (2013) 33(4):490–8. doi: 10.1038/jcbfm.2012.198
207. Kong LL, Wang ZY, Hu JF, Yuan YH, Li H, Chen NH. Inhibition of Chemokine-Like Factor 1 Improves Blood-Brain Barrier Dysfunction in Rats Following Focal Cerebral Ischemia. Neurosci Lett (2016) 627:192–8. doi: 10.1016/j.neulet.2016.06.003
208. Vemuganti R, Dempsey RJ, Bowen KK. Inhibition of Intercellular Adhesion Molecule-1 Protein Expression by Antisense Oligonucleotides Is Neuroprotective After Transient Middle Cerebral Artery Occlusion in Rat. Stroke (2004) 35(1):179–84. doi: 10.1161/01.str.0000106479.53235.3e
209. Soriano SG, Coxon A, Wang YF, Frosch MP, Lipton SA, Hickey PR, et al. Mice Deficient in Mac-1 (CD11b/CD18) Are Less Susceptible to Cerebral Ischemia/Reperfusion Injury. Stroke (1999) 30(1):134–9. doi: 10.1161/01.str.30.1.134
210. Huang J, Choudhri TF, Winfree CJ, McTaggart RA, Kiss S, Mocco J, et al. Postischemic Cerebrovascular E-Selectin Expression Mediates Tissue Injury in Murine Stroke. Stroke (2000) 31(12):3047–53. doi: 10.1161/01.STR.31.12.3047
211. Jin AY, Tuor UI, Rushforth D, Kaur J, Muller RN, Petterson JL, et al. Reduced Blood Brain Barrier Breakdown in P-selectin Deficient Mice Following Transient Ischemic Stroke: A Future Therapeutic Target for Treatment of Stroke. BMC Neurosci (2010) 11:12. doi: 10.1186/1471-2202-11-12
212. Sladojevic N, Stamatovic SM, Keep RF, Grailer JJ, Sarma JV, Ward PA, et al. Inhibition of Junctional Adhesion molecule-A/LFA Interaction Attenuates Leukocyte Trafficking and Inflammation in Brain Ischemia/Reperfusion Injury. Neurobiol Dis (2014) 67:57–70. doi: 10.1016/j.nbd.2014.03.010
213. Yu G, Liang Y, Huang Z, Jones DW, Pritchard KA Jr, Zhang H. Inhibition of Myeloperoxidase Oxidant Production by N-acetyl Lysyltyrosylcysteine Amide Reduces Brain Damage in a Murine Model of Stroke. J Neuroinflamm (2016) 13(1):119. doi: 10.1186/s12974-016-0583-x
214. Hung YC, Chen TY, Lee EJ, Chen WL, Huang SY, Lee WT, et al. Melatonin Decreases Matrix Metalloproteinase-9 Activation and Expression and Attenuates Reperfusion-Induced Hemorrhage Following Transient Focal Cerebral Ischemia in Rats. J Pineal Res (2008) 45(4):459–67. doi: 10.1111/j.1600-079X.2008.00617.x
215. Huang L, Shang E, Fan W, Li X, Li B, He S, et al. S-Oxiracetam Protect Against Ischemic Stroke Via Alleviating Blood Brain Barrier Dysfunction in Rats. Eur J Pharm Sci (2017) 109:40–7. doi: 10.1016/j.ejps.2017.07.029
216. Moldthan HL, Hirko AC, Thinschmidt JS, Grant MB, Li Z, Peris J, et al. Alpha 1-Antitrypsin Therapy Mitigated Ischemic Stroke Damage in Rats. J Stroke Cerebrovasc Dis (2014) 23(5):e355–63. doi: 10.1016/j.jstrokecerebrovasdis.2013.12.029
217. Cai W, Wang J, Hu M, Chen X, Lu Z, Bellanti JA, et al. All Trans-Retinoic Acid Protects Against Acute Ischemic Stroke by Modulating Neutrophil Functions Through STAT1 Signaling. J Neuroinflamm (2019) 16(1):175. doi: 10.1186/s12974-019-1557-6
218. Certo M, Endo Y, Ohta K, Sakurada S, Bagetta G, Amantea D. Activation of RXR/Pparγ Underlies Neuroprotection by Bexarotene in Ischemic Stroke. Pharmacol Res (2015) 102:298–307. doi: 10.1016/j.phrs.2015.10.009
219. Stamatovic SM, Phillips CM, Keep RF, Andjelkovic AV. A Novel Approach to Treatment of Thromboembolic Stroke In Mice: Redirecting Neutrophils Toward a Peripherally Implanted CXCL1-Soaked Sponge. Exp Neurol (2020) 330:113336. doi: 10.1016/j.expneurol.2020.113336
220. Tang C, Wang C, Zhang Y, Xue L, Li Y, Ju C, et al. Recognition, Intervention, and Monitoring of Neutrophils in Acute Ischemic Stroke. Nano Lett (2019) 19(7):4470–7. doi: 10.1021/acs.nanolett.9b01282
221. Wang C, Börger V, Sardari M, Murke F, Skuljec J, Pul R, et al. Mesenchymal Stromal Cell-Derived Small Extracellular Vesicles Induce Ischemic Neuroprotection by Modulating Leukocytes and Specifically Neutrophils. Stroke (2020) 51(6):1825–34. doi: 10.1161/strokeaha.119.028012
222. Yenari MA, Kunis D, Sun GH, Onley D, Watson L, Turner S, et al. Hu23F2G, an Antibody Recognizing the Leukocyte CD11/CD18 Integrin, Reduces Injury in a Rabbit Model of Transient Focal Cerebral Ischemia. Exp Neurol (1998) 153(2):223–33. doi: 10.1006/exnr.1998.6876
223. Ren X, Hu H, Farooqi I, Simpkins JW. Blood Substitution Therapy Rescues the Brain of Mice From Ischemic Damage. Nat Commun (2020) 11(1):4078. doi: 10.1038/s41467-020-17930-x
224. Bao Dang Q, Lapergue B, Tran-Dinh A, Diallo D, Moreno JA, Mazighi M, et al. High-Density Lipoproteins Limit Neutrophil-Induced Damage to the Blood-Brain Barrier In Vitro. J Cereb Blood Flow Metab (2013) 33(4):575–82. doi: 10.1038/jcbfm.2012.206
225. Chen R, Xu Y, Wu P, Zhou H, Lasanajak Y, Fang Y, et al. Transplantation of Fecal Microbiota Rich in Short Chain Fatty Acids and Butyric Acid Treat Cerebral Ischemic Stroke by Regulating Gut Microbiota. Pharmacol Res (2019) 148:104403. doi: 10.1016/j.phrs.2019.104403
226. Elkind MSV, Veltkamp R, Montaner J, Johnston SC, Singhal AB, Becker K, et al. Natalizumab in Acute Ischemic Stroke (ACTION II): A Randomized, Placebo-Controlled Trial. Neurology (2020) 95(8):e1091–104. doi: 10.1212/wnl.0000000000010038
227. Wang Z, Leng Y, Tsai LK, Leeds P, Chuang DM. Valproic Acid Attenuates Blood-Brain Barrier Disruption in a Rat Model of Transient Focal Cerebral Ischemia: The Roles of HDAC and MMP-9 Inhibition. J Cereb Blood Flow Metab (2011) 31(1):52–7. doi: 10.1038/jcbfm.2010.195
228. Park MJ, Sohrabji F. The Histone Deacetylase Inhibitor, Sodium Butyrate, Exhibits Neuroprotective Effects for Ischemic Stroke in Middle-Aged Female Rats. J Neuroinflamm (2016) 13(1):300. doi: 10.1186/s12974-016-0765-6
229. Zhu Z, Fu Y, Tian D, Sun N, Han W, Chang G, et al. Combination of the Immune Modulator Fingolimod With Alteplase in Acute Ischemic Stroke: A Pilot Trial. Circulation (2015) 132(12):1104–12. doi: 10.1161/circulationaha.115.016371
230. Chen J, Bai Q, Zhao Z, Sui H, Xie X. Resveratrol Improves Delayed r-tPA Treatment Outcome by Reducing Mmps. Acta Neurol Scand (2016) 134(1):54–60. doi: 10.1111/ane.12511
231. Pradillo JM, Denes A, Greenhalgh AD, Boutin H, Drake C, McColl BW, et al. Delayed Administration of Interleukin-1 Receptor Antagonist Reduces Ischemic Brain Damage and Inflammation in Comorbid Rats. J Cereb Blood Flow Metab (2012) 32(9):1810–9. doi: 10.1038/jcbfm.2012.101
232. Lampl Y, Boaz M, Gilad R, Lorberboym M, Dabby R, Rapoport A, et al. Minocycline Treatment in Acute Stroke: An Open-Label, Evaluator-Blinded Study. Neurology (2007) 69(14):1404–10. doi: 10.1212/01.wnl.0000277487.04281.db
233. Chen X, Zhuang X, Peng Z, Yang H, Chen L, Yang Q. Intensive Statin Therapy for Acute Ischemic Stroke to Reduce the Number of Microemboli: A Preliminary, Randomized Controlled Study. Eur Neurol (2018) 80(3-4):163–70. doi: 10.1159/000494989
234. Heo JH, Song D, Nam HS, Kim EY, Kim YD, Lee KY, et al. Effect and Safety of Rosuvastatin in Acute Ischemic Stroke. J Stroke (2016) 18(1):87–95. doi: 10.5853/jos.2015.01578
235. Sherman DG, Bes A, Easton JD, Hacke W, Kaste M, Polmar SH. Use of anti-ICAM-1 Therapy in Ischemic Stroke: Results of the Enlimomab Acute Stroke Trial. Neurology (2001) 57(8):1428–34. doi: 10.1212/wnl.57.8.1428
236. Krams M, Lees KR, Hacke W, Grieve AP, Orgogozo JM, Ford GA. Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN): An Adaptive Dose-Response Study of UK-279,276 in Acute Ischemic Stroke. Stroke (2003) 34(11):2543–8. doi: 10.1161/01.str.0000092527.33910.89
237. Becker KJ. Anti-Leukocyte Antibodies: LeukArrest (Hu23F2G) and Enlimomab (R6.5) in Acute Stroke. Curr Med Res Opin (2002) 18(Suppl 2):s18–22. doi: 10.1185/030079902125000688
238. Shekhar S, Cunningham MW, Pabbidi MR, Wang S, Booz GW, Fan F. Targeting Vascular Inflammation in Ischemic Stroke: Recent Developments on Novel Immunomodulatory Approaches. Eur J Pharmacol (2018) 833:531–44. doi: 10.1016/j.ejphar.2018.06.028
239. Goldstein LJ, Perez RP, Yardley D, Han LK, Reuben JM, Gao H, et al. A Window-of-Opportunity Trial of the CXCR1/2 Inhibitor Reparixin in Operable HER-2-Negative Breast Cancer. Breast Cancer Res (2020) 22(1):4. doi: 10.1186/s13058-019-1243-8
240. Dwaich KH, Al-Amran FG, Al-Sheibani BI, Al-Aubaidy HA. Melatonin Effects on Myocardial Ischemia-Reperfusion Injury: Impact on the Outcome in Patients Undergoing Coronary Artery Bypass Grafting Surgery. Int J Cardiol (2016) 221:977–86. doi: 10.1016/j.ijcard.2016.07.108
241. Abaza Y, Kantarjian H, Garcia-Manero G, Estey E, Borthakur G, Jabbour E, et al. Long-Term Outcome of Acute Promyelocytic Leukemia Treated With All-Trans-Retinoic Acid, Arsenic Trioxide, and Gemtuzumab. Blood (2017) 129(10):1275–83. doi: 10.1182/blood-2016-09-736686
242. Srivastava P, Cronin CG, Scranton VL, Jacobson KA, Liang BT, Verma R. Neuroprotective and Neuro-Rehabilitative Effects of Acute Purinergic Receptor P2X4 (P2X4R) Blockade After Ischemic Stroke. Exp Neurol (2020) 329:113308. doi: 10.1016/j.expneurol.2020.113308
243. Li Y, Xu L, Zeng K, Xu Z, Suo D, Peng L, et al. Propane-2-sulfonic Acid octadec-9-enyl-amide, a Novel Pparα/γ Dual Agonist, Protects Against Ischemia-Induced Brain Damage in Mice by Inhibiting Inflammatory Responses. Brain Behav Immun (2017) 66:289–301. doi: 10.1016/j.bbi.2017.07.015
244. Guo T, Wang Y, Guo Y, Wu S, Chen W, Liu N, et al. 1, 25-D(3) Protects From Cerebral Ischemia by Maintaining Bbb Permeability Via PPAR-γ Activation. Front Cell Neurosci (2018) 12:480. doi: 10.3389/fncel.2018.00480
245. Li Y, Zhu ZY, Lu BW, Huang TT, Zhang YM, Zhou NY, et al. Rosiglitazone Ameliorates Tissue Plasminogen Activator-Induced Brain Hemorrhage After Stroke. CNS Neurosci Ther (2019) 25(12):1343–52. doi: 10.1111/cns.13260
246. Wang D, Liu F, Zhu L, Lin P, Han F, Wang X, et al. FGF21 Alleviates Neuroinflammation Following Ischemic Stroke by Modulating the Temporal and Spatial Dynamics of Microglia/Macrophages. J Neuroinflamm (2020) 17(1):257. doi: 10.1186/s12974-020-01921-2
247. Sharmin O, Abir AH, Potol A, Alam M, Banik J, Rahman A, et al. Activation of GPR35 Protects Against Cerebral Ischemia by Recruiting Monocyte-Derived Macrophages. Sci Rep (2020) 10(1):9400. doi: 10.1038/s41598-020-66417-8
248. Cai W, Liu S, Hu M, Sun X, Qiu W, Zheng S, et al. Post-Stroke DHA Treatment Protects Against Acute Ischemic Brain Injury by Skewing Macrophage Polarity Toward the M2 Phenotype. Transl Stroke Res (2018) 9(6):669–80. doi: 10.1007/s12975-018-0662-7
249. Belayev L, Hong SH, Menghani H, Marcell SJ, Obenaus A, Freitas RS, et al. Docosanoids Promote Neurogenesis and Angiogenesis, Blood-Brain Barrier Integrity, Penumbra Protection, and Neurobehavioral Recovery After Experimental Ischemic Stroke. Mol Neurobiol (2018) 55(8):7090–106. doi: 10.1007/s12035-018-1136-3
250. Yeh CF, Chuang TY, Hung YW, Lan MY, Tsai CH, Huang HX, et al. Inhibition of Soluble Epoxide Hydrolase Regulates Monocyte/Macrophage Polarization and Improves Neurological Outcome in a Rat Model of Ischemic Stroke. Neuroreport (2019) 30(8):567–72. doi: 10.1097/wnr.0000000000001248
251. Kolosowska N, Gotkiewicz M, Dhungana H, Giudice L, Giugno R, Box D, et al. Intracerebral Overexpression of miR-669c Is Protective in Mouse Ischemic Stroke Model by Targeting MyD88 and Inducing Alternative Microglial/Macrophage Activation. J Neuroinflamm (2020) 17(1):194. doi: 10.1186/s12974-020-01870-w
252. Kuang X, Wang LF, Yu L, Li YJ, Wang YN, He Q, et al. Ligustilide Ameliorates Neuroinflammation and Brain Injury in Focal Cerebral Ischemia/Reperfusion Rats: Involvement of Inhibition of TLR4/peroxiredoxin 6 Signaling. Free Radic Biol Med (2014) 71:165–75. doi: 10.1016/j.freeradbiomed.2014.03.028
253. Xu X, Wen Z, Zhao N, Xu X, Wang F, Gao J, et al. MicroRNA-1906, a Novel Regulator of Toll-Like Receptor 4, Ameliorates Ischemic Injury After Experimental Stroke in Mice. J Neurosci (2017) 37(43):10498–515. doi: 10.1523/jneurosci.1139-17.2017
254. Hua F, Tang H, Wang J, Prunty MC, Hua X, Sayeed I, et al. Tak-242, an Antagonist for Toll-like Receptor 4, Protects Against Acute Cerebral Ischemia/Reperfusion Injury in Mice. J Cereb Blood Flow Metab (2015) 35(4):536–42. doi: 10.1038/jcbfm.2014.240
255. Parada E, Casas AI, Palomino-Antolin A, Gómez-Rangel V, Rubio-Navarro A, Farré-Alins V, et al. Early Toll-Like Receptor 4 Blockade Reduces ROS and Inflammation Triggered by Microglial Pro-Inflammatory Phenotype in Rodent and Human Brain Ischaemia Models. Br J Pharmacol (2019) 176(15):2764–79. doi: 10.1111/bph.14703
256. Andresen L, Theodorou K, Grünewald S, Czech-Zechmeister B, Könnecke B, Lühder F, et al. Evaluation of the Therapeutic Potential of Anti-TLR4-Antibody MTS510 in Experimental Stroke and Significance of Different Routes of Application. PloS One (2016) 11(2):e0148428. doi: 10.1371/journal.pone.0148428
257. Gelosa P, Lecca D, Fumagalli M, Wypych D, Pignieri A, Cimino M, et al. Microglia is a Key Player in the Reduction of Stroke Damage Promoted by the New Antithrombotic Agent Ticagrelor. J Cereb Blood Flow Metab (2014) 34(6):979–88. doi: 10.1038/jcbfm.2014.45
258. Liu T, Zhang T, Yu H, Shen H, Xia W. Adjudin Protects Against Cerebral Ischemia Reperfusion Injury by Inhibition of Neuroinflammation and Blood-Brain Barrier Disruption. J Neuroinflamm (2014) 11:107. doi: 10.1186/1742-2094-11-107
259. Lu D, Liu Y, Mai H, Zang J, Shen L, Zhang Y, et al. Rosuvastatin Reduces Neuroinflammation in the Hemorrhagic Transformation After Rt-PA Treatment in a Mouse Model of Experimental Stroke. Front Cell Neurosci (2018) 12:225. doi: 10.3389/fncel.2018.00225
260. Lu D, Shen L, Mai H, Zang J, Liu Y, Tsang CK, et al. HMG-Coa Reductase Inhibitors Attenuate Neuronal Damage by Suppressing Oxygen Glucose Deprivation-Induced Activated Microglial Cells. Neural Plast (2019) 2019:7675496. doi: 10.1155/2019/7675496
261. Saito T, Nito C, Ueda M, Inaba T, Kamiya F, Muraga K, et al. Continuous Oral Administration of Atorvastatin Ameliorates Brain Damage After Transient Focal Ischemia in Rats. Life Sci (2014) 94(2):106–14. doi: 10.1016/j.lfs.2013.11.018
262. Lu Y, Xiao G, Luo W. Minocycline Suppresses NLRP3 Inflammasome Activation in Experimental Ischemic Stroke. Neuroimmunomodulation (2016) 23(4):230–8. doi: 10.1159/000452172
263. Yang Y, Salayandia VM, Thompson JF, Yang LY, Estrada EY, Yang Y. Attenuation of Acute Stroke Injury in Rat Brain by Minocycline Promotes Blood-Brain Barrier Remodeling and Alternative Microglia/Macrophage Activation During Recovery. J Neuroinflamm (2015) 12:26. doi: 10.1186/s12974-015-0245-4
264. Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia Potentiate Damage to Blood-Brain Barrier Constituents: Improvement by Minocycline In Vivo and In Vitro. Stroke (2006) 37(4):1087–93. doi: 10.1161/01.STR.0000206281.77178.ac
265. Yew WP, Djukic ND, Jayaseelan JSP, Walker FR, Roos KAA, Chataway TK, et al. Early Treatment With Minocycline Following Stroke in Rats Improves Functional Recovery and Differentially Modifies Responses of Peri-Infarct Microglia and Astrocytes. J Neuroinflamm (2019) 16(1):6. doi: 10.1186/s12974-018-1379-y
266. Ashabi G, Khalaj L, Khodagholi F, Goudarzvand M, Sarkaki A. Pre-Treatment With Metformin Activates Nrf2 Antioxidant Pathways and Inhibits Inflammatory Responses Through Induction of AMPK After Transient Global Cerebral Ischemia. Metab Brain Dis (2015) 30(3):747–54. doi: 10.1007/s11011-014-9632-2
267. Liu Y, Tang G, Li Y, Wang Y, Chen X, Gu X, et al. Metformin Attenuates Blood-Brain Barrier Disruption in Mice Following Middle Cerebral Artery Occlusion. J Neuroinflamm (2014) 11:177. doi: 10.1186/s12974-014-0177-4
268. Jin Q, Cheng J, Liu Y, Wu J, Wang X, Wei S, et al. Improvement of Functional Recovery by Chronic Metformin Treatment is Associated With Enhanced Alternative Activation of Microglia/Macrophages and Increased Angiogenesis and Neurogenesis Following Experimental Stroke. Brain Behav Immun (2014) 40:131–42. doi: 10.1016/j.bbi.2014.03.003
269. Liberale L, Diaz-Cañestro C, Bonetti NR, Paneni F, Akhmedov A, Beer JH, et al. Post-Ischaemic Administration of the Murine Canakinumab-Surrogate Antibody Improves Outcome in Experimental Stroke. Eur Heart J (2018) 39(38):3511–7. doi: 10.1093/eurheartj/ehy286
270. Jiang Y, Lin L, Liu N, Wang Q, Yuan J, Li Y, et al. Fgf21 Protects Against Aggravated Blood-Brain Barrier Disruption After Ischemic Focal Stroke in Diabetic Db/Db Male Mice Via Cerebrovascular Pparγ Activation. Int J Mol Sci (2020) 21(3):824. doi: 10.3390/ijms21030824
271. Liesz A, Zhou W, Mracskó É., Karcher S, Bauer H, Schwarting S, et al. Inhibition of Lymphocyte Trafficking Shields the Brain Against Deleterious Neuroinflammation After Stroke. Brain (2011) 134(Pt3):704–20. doi: 10.1093/brain/awr008
272. Salas-Perdomo A, Miró-Mur F, Gallizioli M, Brait VH, Justicia C, Meissner A, et al. Role of the S1P Pathway and Inhibition by Fingolimod in Preventing Hemorrhagic Transformation After Stroke. Sci Rep (2019) 9(1):8309. doi: 10.1038/s41598-019-44845-5
273. Kraft P, Göb E, Schuhmann MK, Göbel K, Deppermann C, Thielmann I, et al. FTY720 Ameliorates Acute Ischemic Stroke in Mice by Reducing Thrombo-Inflammation But Not by Direct Neuroprotection. Stroke (2013) 44(11):3202–10. doi: 10.1161/strokeaha.113.002880
274. Kim Y, Kim YS, Kim HY, Noh MY, Kim JY, Lee YJ, et al. Early Treatment With Poly(ADP-Ribose) Polymerase-1 Inhibitor (Jpi-289) Reduces Infarct Volume and Improves Long-Term Behavior in an Animal Model of Ischemic Stroke. Mol Neurobiol (2018) 55(9):7153–63. doi: 10.1007/s12035-018-0910-6
275. Teng F, Beray-Berthat V, Coqueran B, Lesbats C, Kuntz M, Palmier B, et al. Prevention of rt-PA Induced Blood-Brain Barrier Component Degradation by the Poly(ADP-Ribose)Polymerase Inhibitor PJ34 After Ischemic Stroke in Mice. Exp Neurol (2013) 248:416–28. doi: 10.1016/j.expneurol.2013.07.007
276. Iwashita A, Tojo N, Matsuura S, Yamazaki S, Kamijo K, Ishida J, et al. A Novel and Potent Poly(ADP-Ribose) Polymerase-1 Inhibitor, FR247304 (5-chloro-2-[3-(4-phenyl-3,6-dihydro-1(2H)-pyridinyl)propyl]-4(3H)-quinazolinone), Attenuates Neuronal Damage in In Vitro and In Vivo Models of Cerebral Ischemia. J Pharmacol Exp Ther (2004) 310(2):425–36. doi: 10.1124/jpet.104.066944
277. Matsuura S, Egi Y, Yuki S, Horikawa T, Satoh H, Akira T. Mp-124, a Novel Poly(ADP-Ribose) Polymerase-1 (PARP-1) Inhibitor, Ameliorates Ischemic Brain Damage in a non-Human Primate Model. Brain Res (2011) 1410:122–31. doi: 10.1016/j.brainres.2011.05.069
278. Luo Y, Zhou Y, Xiao W, Liang Z, Dai J, Weng X, et al. Interleukin-33 Ameliorates Ischemic Brain Injury in Experimental Stroke Through Promoting Th2 Response and Suppressing Th17 Response. Brain Res (2015) 1597:86–94. doi: 10.1016/j.brainres.2014.12.005
279. Liu X, Hu R, Pei L, Si P, Wang C, Tian X, et al. Regulatory T Cell is Critical for interleukin-33-Mediated Neuroprotection Against Stroke. Exp Neurol (2020) 328:113233. doi: 10.1016/j.expneurol.2020.113233
280. Guo S, Luo Y. Brain Foxp3(+) Regulatory T Cells Can be Expanded by Interleukin-33 in Mouse Ischemic Stroke. Int Immunopharmacol (2020) 81:106027. doi: 10.1016/j.intimp.2019.106027
281. Evans MA, Kim HA, Ling YH, Uong S, Vinh A, De Silva TM, et al. Vitamin D(3) Supplementation Reduces Subsequent Brain Injury and Inflammation Associated With Ischemic Stroke. Neuromolecular Med (2018) 20(1):147–59. doi: 10.1007/s12017-018-8484-z
282. Rodríguez-Perea AL, Gutierrez-Vargas J, Cardona-Gómez GP, Guarin CJ, Rojas M, Hernández PA. Atorvastatin Modulates Regulatory T Cells and Attenuates Cerebral Damage in a Model of Transient Middle Cerebral Artery Occlusion in Rats. J Neuroimmune Pharmacol (2017) 12(1):152–62. doi: 10.1007/s11481-016-9706-5
283. Yang H, Zhang A, Zhang Y, Ma S, Wang C. Resveratrol Pretreatment Protected Against Cerebral Ischemia/Reperfusion Injury in Rats Via Expansion of T Regulatory Cells. J Stroke Cerebrovasc Dis (2016) 25(8):1914–21. doi: 10.1016/j.jstrokecerebrovasdis.2016.04.014
284. Dou Z, Rong X, Zhao E, Zhang L, Lv Y. Neuroprotection of Resveratrol Against Focal Cerebral Ischemia/Reperfusion Injury in Mice Through a Mechanism Targeting Gut-Brain Axis. Cell Mol Neurobiol (2019) 39(6):883–98. doi: 10.1007/s10571-019-00687-3
285. Lee J, d’Aigle J, Atadja L, Quaicoe V, Honarpisheh P, Ganesh BP, et al. Gut Microbiota-Derived Short-Chain Fatty Acids Promote Poststroke Recovery in Aged Mice. Circ Res (2020) 127(4):453–65. doi: 10.1161/circresaha.119.316448
286. Langhauser F, Kraft P, Göb E, Leinweber J, Schuhmann MK, Lorenz K, et al. Blocking of α4 Integrin Does Not Protect From Acute Ischemic Stroke in Mice. Stroke (2014) 45(6):1799–806. doi: 10.1161/strokeaha.114.005000
287. Wang Z, Kawabori M, Houkin K. Fty720 (Fingolimod) Ameliorates Brain Injury Through Multiple Mechanisms and Is a Strong Candidate for Stroke Treatment. Curr Med Chem (2020) 27(18):2979–93. doi: 10.2174/0929867326666190308133732
288. Wang X, Zhou Y, Tang D, Zhu Z, Li Y, Huang T, et al. ACC1 (Acetyl Coenzyme A Carboxylase 1) Is a Potential Immune Modulatory Target of Cerebral Ischemic Stroke. Stroke (2019) 50(7):1869–78. doi: 10.1161/strokeaha.119.024564
289. Rodríguez-Perea AL, Rojas M, Velilla-Hernández PA. High Concentrations of Atorvastatin Reduce In-Vitro Function of Conventional T and Regulatory T Cells. Clin Exp Immunol (2019) 196(2):237–48. doi: 10.1111/cei.13260
290. Elkins J, Veltkamp R, Montaner J, Johnston SC, Singhal AB, Becker K, et al. Safety and Efficacy of Natalizumab in Patients With Acute Ischaemic Stroke (ACTION): A Randomised, Placebo-Controlled, Double-Blind Phase 2 Trial. Lancet Neurol (2017) 16(3):217–26. doi: 10.1016/s1474-4422(16)30357-x
291. Fu Y, Zhang N, Ren L, Yan Y, Sun N, Li YJ, et al. Impact of an Immune Modulator Fingolimod on Acute Ischemic Stroke. Proc Natl Acad Sci USA (2014) 111(51):18315–20. doi: 10.1073/pnas.1416166111
292. Zhang S, Zhou Y, Zhang R, Zhang M, Campbell B, Lin L, et al. Rationale and Design of Combination of an Immune Modulator Fingolimod With Alteplase Bridging With Mechanical Thrombectomy in Acute Ischemic Stroke (FAMTAIS) Trial. Int J Stroke (2017) 12(8):906–9. doi: 10.1177/1747493017710340
293. Tian DC, Shi K, Zhu Z, Yao J, Yang X, Su L, et al. Fingolimod Enhances the Efficacy of Delayed Alteplase Administration in Acute Ischemic Stroke by Promoting Anterograde Reperfusion and Retrograde Collateral Flow. Ann Neurol (2018) 84(5):717–28. doi: 10.1002/ana.25352
294. Han S, Kim YH, Choi HY, Soh DJ, Kim J, Nam J, et al. First-in-Human Evaluation of the Safety, Tolerability, and Pharmacokinetics of a Neuroprotective Poly (ADP-Ribose) Polymerase-1 Inhibitor, JPI-289, in Healthy Volunteers. Drug Des Devel Ther (2020) 14:3189–99. doi: 10.2147/dddt.s235802
295. Manson JE, Cook NR, Lee IM, Christen W, Bassuk SS, Mora S, et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N Engl J Med (2019) 380(1):33–44. doi: 10.1056/NEJMoa1809944
296. Momosaki R, Abo M, Urashima M. Vitamin D Supplementation and Post-Stroke Rehabilitation: A Randomized, Double-Blind, Placebo-Controlled Trial. Nutrients (2019) 11(6):1295. doi: 10.3390/nu11061295
297. Beer C, Blacker D, Bynevelt M, Hankey GJ, Puddey IB. A Randomized Placebo Controlled Trial of Early Treatment of Acute Ischemic Stroke With Atorvastatin and Irbesartan. Int J Stroke (2012) 7(2):104–11. doi: 10.1111/j.1747-4949.2011.00653.x
298. Muscari A, Puddu GM, Santoro N, Serafini C, Cenni A, Rossi V, et al. The Atorvastatin During Ischemic Stroke Study: A Pilot Randomized Controlled Trial. Clin Neuropharmacol (2011) 34(4):141–7. doi: 10.1097/WNF.0b013e3182206c2f
299. Lu Z, Zhang D, Cui K, Fu X, Man J, Lu H, et al. Neuroprotective Action of Teriflunomide in a Mouse Model of Transient Middle Cerebral Artery Occlusion. Neuroscience (2020) 428:228–41. doi: 10.1016/j.neuroscience.2019.12.011
300. Christophe B, Karatela M, Sanchez J, Pucci J, Connolly ES. Statin Therapy in Ischemic Stroke Models: A Meta-Analysis. Transl Stroke Res (2020) 11(4):590–600. doi: 10.1007/s12975-019-00750-7
301. Clausen BH, Lambertsen KL, Dagnæs-Hansen F, Babcock AA, von Linstow CU, Meldgaard M, et al. Cell Therapy Centered on IL-1Ra Is Neuroprotective in Experimental Stroke. Acta Neuropathol (2016) 131(5):775–91. doi: 10.1007/s00401-016-1541-5
302. Malhotra K, Chang JJ, Khunger A, Blacker D, Switzer JA, Goyal N, et al. Minocycline for Acute Stroke Treatment: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. J Neurol (2018) 265(8):1871–9. doi: 10.1007/s00415-018-8935-3
303. Westphal LP, Widmer R, Held U, Steigmiller K, Hametner C, Ringleb P, et al. Association of Prestroke Metformin Use, Stroke Severity, and Thrombolysis Outcome. Neurology (2020) 95(4):e362–73. doi: 10.1212/wnl.0000000000009951
304. Emsley HC, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, et al. A Randomised Phase II Study of Interleukin-1 Receptor Antagonist in Acute Stroke Patients. J Neurol Neurosurg Psychiatry (2005) 76(10):1366–72. doi: 10.1136/jnnp.2004.054882
305. Smith CJ, Hulme S, Vail A, Heal C, Parry-Jones AR, Scarth S, et al. Scil-STROKE (Subcutaneous Interleukin-1 Receptor Antagonist in Ischemic Stroke): A Randomized Controlled Phase 2 Trial. Stroke (2018) 49(5):1210–6. doi: 10.1161/strokeaha.118.020750
306. Everett BM, MacFadyen JG, Thuren T, Libby P, Glynn RJ, Ridker PM. Inhibition of Interleukin-1β and Reduction in Atherothrombotic Cardiovascular Events in the CANTOS Trial. J Am Coll Cardiol (2020) 76(14):1660–70. doi: 10.1016/j.jacc.2020.08.011
307. Ye BD, Pesegova M, Alexeeva O, Osipenko M, Lahat A, Dorofeyev A, et al. Efficacy and Safety of Biosimilar CT-P13 Compared With Originator Infliximab in Patients With Active Crohn’s Disease: An International, Randomised, Double-Blind, Phase 3 non-Inferiority Study. Lancet (2019) 393(10182):1699–707. doi: 10.1016/s0140-6736(18)32196-2
308. Liu M, Xu Z, Wang L, Zhang L, Liu Y, Cao J, et al. Cottonseed Oil Alleviates Ischemic Stroke Injury by Inhibiting the Inflammatory Activation of Microglia and Astrocyte. J Neuroinflamm (2020) 17(1):270. doi: 10.1186/s12974-020-01946-7
309. Li X, Huang L, Liu G, Fan W, Li B, Liu R, et al. Ginkgo Diterpene Lactones Inhibit Cerebral Ischemia/Reperfusion Induced Inflammatory Response in Astrocytes Via TLR4/NF-κb Pathway in Rats. J Ethnopharmacol (2020) 249:112365. doi: 10.1016/j.jep.2019.112365
310. Zhu XC, Jiang T, Zhang QQ, Cao L, Tan MS, Wang HF, et al. Chronic Metformin Preconditioning Provides Neuroprotection Via Suppression of NF-κb-Mediated Inflammatory Pathway in Rats With Permanent Cerebral Ischemia. Mol Neurobiol (2015) 52(1):375–85. doi: 10.1007/s12035-014-8866-7
311. Liu T, Liu M, Zhang T, Liu W, Xu H, Mu F, et al. Z-Guggulsterone Attenuates Astrocytes-Mediated Neuroinflammation After Ischemia by Inhibiting Toll-Like Receptor 4 Pathway. J Neurochem (2018) 147(6):803–15. doi: 10.1111/jnc.14583
312. Yu L, Chen C, Wang LF, Kuang X, Liu K, Zhang H, et al. Neuroprotective Effect of Kaempferol Glycosides Against Brain Injury and Neuroinflammation by Inhibiting the Activation of NF-κb and STAT3 in Transient Focal Stroke. PloS One (2013) 8(2):e55839. doi: 10.1371/journal.pone.0055839
313. Zhang P, Liu X, Zhu Y, Chen S, Zhou D, Wang Y. Honokiol Inhibits the Inflammatory Reaction During Cerebral Ischemia Reperfusion by Suppressing NF-κb Activation and Cytokine Production of Glial Cells. Neurosci Lett (2013) 534:123–7. doi: 10.1016/j.neulet.2012.11.052
314. Hwang CJ, Yun HM, Jung YY, Lee DH, Yoon NY, Seo HO, et al. Reducing Effect of IL-32α in the Development of Stroke Through Blocking of NF-κb, But Enhancement of STAT3 Pathways. Mol Neurobiol (2015) 51(2):648–60. doi: 10.1007/s12035-014-8739-0
315. Kono S, Kurata T, Sato K, Omote Y, Hishikawa N, Yamashita T, et al. Neurovascular Protection by Telmisartan Via Reducing Neuroinflammation in Stroke-Resistant Spontaneously Hypertensive Rat Brain After Ischemic Stroke. J Stroke Cerebrovasc Dis (2015) 24(3):537–47. doi: 10.1016/j.jstrokecerebrovasdis.2014.09.037
316. Qiu J, Wang M, Zhang J, Cai Q, Lu D, Li Y, et al. The Neuroprotection of Sinomenine Against Ischemic Stroke in Mice by Suppressing NLRP3 Inflammasome Via AMPK Signaling. Int Immunopharmacol (2016) 40:492–500. doi: 10.1016/j.intimp.2016.09.024
317. Liu H, Wu X, Luo J, Zhao L, Li X, Guo H, et al. Adiponectin Peptide Alleviates Oxidative Stress and NLRP3 Inflammasome Activation After Cerebral Ischemia-Reperfusion Injury by Regulating AMPK/GSK-3β. Exp Neurol (2020) 329:113302. doi: 10.1016/j.expneurol.2020.113302
318. Katnik C, Garcia A, Behensky AA, Yasny IE, Shuster AM, Seredenin SB, et al. Activation of ς1 and ς2 Receptors by Afobazole Increases Glial Cell Survival and Prevents Glial Cell Activation and Nitrosative Stress After Ischemic Stroke. J Neurochem (2016) 139(3):497–509. doi: 10.1111/jnc.13756
319. Allahtavakoli M, Jarrott B. Sigma-1 Receptor Ligand PRE-084 Reduced Infarct Volume, Neurological Deficits, Pro-Inflammatory Cytokines and Enhanced Anti-Inflammatory Cytokines After Embolic Stroke in Rats. Brain Res Bull (2011) 85(3-4):219–24. doi: 10.1016/j.brainresbull.2011.03.019
320. Shang S, Liu L, Wu X, Fan F, Hu E, Wang L, et al. Inhibition of PI3Kγ by AS605240 Protects Tmcao Mice by Attenuating Pro-Inflammatory Signaling and Cytokine Release in Reactive Astrocytes. Neuroscience (2019) 415:107–20. doi: 10.1016/j.neuroscience.2019.06.001
321. Luo D, Zhang Y, Yuan X, Pan Y, Yang L, Zhao Y, et al. Oleoylethanolamide Inhibits Glial Activation Via Moudulating Pparα and Promotes Motor Function Recovery After Brain Ischemia. Pharmacol Res (2019) 141:530–40. doi: 10.1016/j.phrs.2019.01.027
322. Chen ZZ, Yang DD, Zhao Z, Yan H, Ji J, Sun XL. Memantine Mediates Neuroprotection Via Regulating Neurovascular Unit in a Mouse Model of Focal Cerebral Ischemia. Life Sci (2016) 150:8–14. doi: 10.1016/j.lfs.2016.02.081
323. Shan Y, Tan S, Lin Y, Liao S, Zhang B, Chen X, et al. The Glucagon-Like Peptide-1 Receptor Agonist Reduces Inflammation and Blood-Brain Barrier Breakdown in an Astrocyte-Dependent Manner in Experimental Stroke. J Neuroinflamm (2019) 16(1):242. doi: 10.1186/s12974-019-1638-6
324. De Geyter D, Stoop W, Sarre S, De Keyser J, Kooijman R. Neuroprotective Efficacy of Subcutaneous Insulin-Like Growth Factor-I Administration in Normotensive and Hypertensive Rats With an Ischemic Stroke. Neuroscience (2013) 250:253–62. doi: 10.1016/j.neuroscience.2013.07.016
325. Yang Y, Yang LY, Orban L, Cuylear D, Thompson J, Simon B, et al. Non-Invasive Vagus Nerve Stimulation Reduces Blood-Brain Barrier Disruption in a Rat Model of Ischemic Stroke. Brain Stimul (2018) 11(4):689–98. doi: 10.1016/j.brs.2018.01.034
326. Xie Y, Wang Y, Ding H, Guo M, Wang X, Dong Q, et al. Highly Glycosylated CD147 Promotes Hemorrhagic Transformation After rt-PA Treatment in Diabetes: A Novel Therapeutic Target? J Neuroinflamm (2019) 16(1):72. doi: 10.1186/s12974-019-1460-1
327. Liu W, Zhang Y, Zhu W, Ma C, Ruan J, Long H, et al. Sinomenine Inhibits the Progression of Rheumatoid Arthritis by Regulating the Secretion of Inflammatory Cytokines and Monocyte/Macrophage Subsets. Front Immunol (2018) 9:2228. doi: 10.3389/fimmu.2018.02228
328. Daly SC, Chemmanam T, Loh PS, Gilligan A, Dear AE, Simpson RW, et al. Exenatide in Acute Ischemic Stroke. Int J Stroke (2013) 8(7):E44. doi: 10.1111/ijs.12073
329. Muller C, Cheung NW, Dewey H, Churilov L, Middleton S, Thijs V, et al. Treatment With Exenatide in Acute Ischemic Stroke Trial Protocol: A Prospective, Randomized, Open Label, Blinded End-Point Study of Exenatide vs. Standard Care in Post Stroke Hyperglycemia. Int J Stroke (2018) 13(8):857–62. doi: 10.1177/1747493018784436
330. Diener HC, Sacco RL, Yusuf S, Cotton D, Ounpuu S, Lawton WA, et al. Effects of Aspirin Plus Extended-Release Dipyridamole Versus Clopidogrel and Telmisartan on Disability and Cognitive Function After Recurrent Stroke in Patients With Ischaemic Stroke in the Prevention Regimen for Effectively Avoiding Second Strokes (ProFESS) Trial: A Double-Blind, Active and Placebo-Controlled Study. Lancet Neurol (2008) 7(10):875–84. doi: 10.1016/s1474-4422(08)70198-4
Keywords: immune cells, ischemic stroke, blood-brain barrier, inflammation, immune therapy
Citation: Qiu Y-m, Zhang C-l, Chen A-q, Wang H-l, Zhou Y-f, Li Y-n and Hu B (2021) Immune Cells in the BBB Disruption After Acute Ischemic Stroke: Targets for Immune Therapy? Front. Immunol. 12:678744. doi: 10.3389/fimmu.2021.678744
Received: 11 March 2021; Accepted: 31 May 2021;
Published: 23 June 2021.
Edited by:
Tobias Ruck, Heinrich Heine University of Düsseldorf, GermanyReviewed by:
Dejan Jakimovski, Buffalo Neuroimaging Analysis Center, United StatesTorben Moos, Aalborg University, Denmark
Muge Yemisci, Hacettepe University, Turkey
Copyright © 2021 Qiu, Zhang, Chen, Wang, Zhou, Li and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Hu, aHVib0BodXN0LmVkdS5jbg==; Ya-nan Li, bHluLmh1c3RAcXEuY29t
†These authors have contributed equally to this work