
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 13 May 2021
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.676301
This article is part of the Research Topic Cancer Immunotherapies: From Efficacy to Resistance Mechanisms View all 42 articles
 Miranda W. Yu1,2
Miranda W. Yu1,2 Daniela F. Quail1,2,3*
Daniela F. Quail1,2,3*Glioblastoma is a highly lethal brain cancer with a median survival rate of less than 15 months when treated with the current standard of care, which consists of surgery, radiotherapy and chemotherapy. With the recent success of immunotherapy in other aggressive cancers such as advanced melanoma and advanced non-small cell lung cancer, glioblastoma has been brought to the forefront of immunotherapy research. Resistance to therapy has been a major challenge across a multitude of experimental candidates and no immunotherapies have been approved for glioblastoma to-date. Intra- and inter-tumoral heterogeneity, an inherently immunosuppressive environment and tumor plasticity remain barriers to be overcome. Moreover, the unique tissue-specific interactions between the central nervous system and the peripheral immune system present an additional challenge for immune-based therapies. Nevertheless, there is sufficient evidence that these challenges may be overcome, and immunotherapy continues to be actively pursued in glioblastoma. Herein, we review the primary ongoing immunotherapy candidates for glioblastoma with a focus on immune checkpoint inhibitors, myeloid-targeted therapies, vaccines and chimeric antigen receptor (CAR) immunotherapies. We further provide insight on mechanisms of resistance and how our understanding of these mechanisms may pave the way for more effective immunotherapeutics against glioblastoma.
Glioblastomas are grade IV gliomas of the central nervous system (CNS) and are the most common and most aggressive type of brain maligancy (1). Patient prognosis is extremely poor, with a median survival of less than 15 months with the current standard of care (SOC), which consists of surgical debulking followed by radiation and chemotherapy (temozolomide) (2). Glioblastomas are currently considered incurable, and all patients inevitably experience and succumb to tumor recurrence, highlighting the urgent need to identify new therapeutic options (3).
The 2016 World Health Organization (WHO) classification of CNS tumors broadly groups glioblastomas based on the mutational status of isocitrate dehydrogenase 1/2 (IDH) (4). Most glioblastomas are IDH-wildtype (wt), which typically arise in older patients (age >50) and are associated with poor prognosis (4). A small subset of glioblastomas (~10%) are IDH-mutant (mut), which are often secondary tumors that arise from the progression of lower grade gliomas and are associated with better survival compared to IDH-wt (4). Glioblastomas can be further classified into classical, mesenchymal, and proneural subtypes based on unique molecular signatures (5, 6). Classical tumors are characterized by EGFR amplification as well as lack of TP53 mutations and homozygous deletion of CDKN2A (5, 6). Mesenchymal tumors have the worst prognosis and are characterized by expression of NF1, often co-mutated with PTEN (5, 6). Proneural tumors have the best prognosis and are characterized by PDGFRA expression (5, 6). Whilst it was previously thought that a fourth subtype (neural) existed, this notion was revised after the neural signature could not be found in tumor cells (5, 6). Glioblastoma tumors are highly heterogenous, with multiple subtypes making up different regions of a single tumor (7, 8). Moreover, each subtype is functionally distinct with unique immunological landscapes including differences in T cell infiltration and macrophage/microglia composition (9). For example, loss of NF1 (i.e. mesenchymal subtype) is associated with a characteristic increase in tumor-associated macrophages (TAMs) (9). Recurrent glioblastomas tend to accumulate macrophages and resemble a mesenchymal state as they become increasingly aggressive and treatment-resistant (10). The immense heterogeneity and microenvironmental evolution of glioblastoma tumors must be considered when developing potential therapies.
Since the addition of temozolomide to glioblastoma SOC in 2005 (2), substantial research efforts and hundreds of clinical trials have been initiated to in an effort to further improve SOC, with very little success. Anti-angiogenic drugs such as bevacizumab, an inhibitor of vascular endothelial growth factor-A (VEGF-A), and cilengitide, an inhibitor of ⍺Vβ3 and ⍺Vβ5 integrin, have been highly pursued in glioblastoma clinical trials, however both of these compounds failed to improve survival of newly diagnosed and recurrent glioblastoma (11–13). In fact, out of the hundreds of clinical trials that have been initiated for glioblastoma in the last decade, few have improved overall survival. Among those that have been moderately successful is the tumor-treating fields (TTF) device, which was approved by the U.S. Food and Drug Administration (FDA) in 2011 for recurrent or refractory glioblastoma (14). TTF involves the local delivery of low-intensity electric fields to disrupt mitosis of glioblastoma cells. In phase III clinical trials, patients with newly diagnosed glioblastoma treated with TTFs in combination with maintenance chemotherapy had a median overall survival of 20.9 months compared to 16 months with maintenance chemotherapy alone (14). Despite this modest success, TTFs have not been incorporated into SOC due to ongoing skepticism amongst the medical community regarding the unblinded nature of TTF trials, as well as issues with patient compliance, which is critical for treatment efficacy (15).
Overall, the failure of past therapeutic candidates to improve glioblastoma SOC is in part a reflection of the rapid and aggressive progression of this disease. Therefore, major research efforts are being made to better understand the brain tumor microenvironment (TME), which holds untapped potential for novel cancer therapies. The immune compartment of glioblastomas is quite substantial, with the majority of cells coming from the myeloid lineage (16). Despite this, glioblastomas are effective at escaping host immune surveillance. Indeed, one of the hallmarks of cancer is the ability to evade cellular immunity (17). Immunotherapies seek to re-direct immune cells against a tumor by exploiting a patient’s immune system. Many immunotherapies such as immune checkpoint inhibitors (ICIs) and chimeric antigen receptor (CAR) T cell therapy have been enormously successful for other aggressive cancers and are now being investigated as potential therapies for glioblastoma (18–22). Herein, we review several ongoing immunotherapeutic approaches for glioblastoma with a focus on ICIs, myeloid-targeted therapies, tumor vaccines, and CAR immunotherapies. We further discuss some key challenges facing immunotherapy in glioblastoma including mechanisms of resistance, which must be overcome in order for the next generation of immunotherapeutics to bring meaningful benefit to patients.
The unique relationship between the brain and the immune system is central to the use of immunotherapy in brain diseases such as glioblastoma. Historically, the brain has been viewed as a tightly sealed organ, guarded by a closely regulated blood brain barrier (BBB), and devoid of any lymphatics or immune surveillance. However, this notion of “immune privilege” was disputed when it was discovered that allo-antigens could illicit an immunological response in the brain (23). Several subsequent isograft versus allograft studies further substantiated this field-shifting discovery (24, 25). As a result of technological advances such as intravital imaging, it is now known that immune surveillance and specifically, the priming and activation of T cells, largely takes place in the meningeal compartment of the CNS (26). However, it only became clear in the last decade how the CNS connects to the peripheral immune system. In 2015, two seminal studies showed for the first time a network of functional lymphatic vessels that line the dural sinuses, which drain into the deep cervical lymph nodes, and serve as a gateway for T cell trafficking between the periphery and the cerebrospinal fluid (CSF) of the CNS (27, 28). While once thought to be immune privileged, it is now appreciated that the brain receives constant immune surveillance and communication with the peripheral immune system, allowing the possibility of immunotherapy as a means of treating diseases of the CNS.
Despite these potential opportunities, one remaining challenge for glioblastoma treatment efficacy is overcoming the BBB. This tightly regulated barrier between the peripheral blood and CNS functions to facilitate the movement of ions, neurotransmitters, and nutrients while shielding the CNS from neurotoxins and most macromolecules (29). Thus, while small (<400Da), lipid-soluble (<8 hydrogen bonds) drugs may be able to passively diffuse across the BBB (30), large or water-soluble drugs are largely excluded by a network of extremely tight junctions (29). This presents a significant challenge for systemic immune-based therapies that rely on effective antibody delivery into tumors or peripheral transfer of cells. Interestingly, one of the hallmarks of brain tumors is a loss of BBB integrity and subsequent increased tight junction permeability (31). In glioblastoma, this characteristic is attributed to loss of claudin-3 and altered levels of claudin-1 and claudin-5, which are the major structural proteins that regulate BBB tight junction permeability (32, 33). While disruption of the BBB may seemingly be advantageous for drug delivery, especially for drugs that depend on the recruitment of peripheral immune cells, loss of BBB integrity may also enhance tumorigenicity by enabling the infiltration of pro-tumorigenic cells such as peripherally-derived immunosuppressive macrophages (34). This double-edged sword is further complicated by the fact that the BBB is not ubiquitously disrupted, and in fact remains completely intact within specific regions of glioblastoma tumors (35). Therefore, systemic therapies for glioblastoma must be able to overcome these complex limitations in order to be effective.
The discovery of immune checkpoint molecules PD-1 and CTLA-4 has undoubtedly transformed the field of cancer immunotherapy (36, 37). Anti-CTLA-4 and anti-PD-1/PD-L1 ICIs have been extremely successful for aggressive cancers such as advanced melanoma and non-small cell lung cancer (NSCLC) (18–20), and there is growing interest in the utility of ICIs as a potential treatment for glioblastoma. In chronic inflammatory conditions such as cancer, prolonged T cell activation leads to increased CTLA-4-expressing Tregs and upregulation of CTLA-4 on cytotoxic T lymphocytes (CTLs), which interacts with the B7 family of receptors and leads to reduced T cell proliferation and survival (Figure 1) (38). In gliomas, this immunosuppression is bolstered by the upregulation of PD-L1 on tumor cells and circulating monocytes/macrophages, which further inhibits CD8+ and CD4+ T cell activation (39, 40). Prolonged T cell activation also causes upregulation of PD-1, which recognizes PD-L1 on antigen-presenting cells (APCs) and tumor cells, and results in T cell exhaustion and reduced survival (Figure 1) (38). These immune signatures, including the upregulation of multiple immune checkpoints and an increased fraction of Tregs, are highly characteristic of the glioblastoma TME, and warrant investigation of ICIs as a potential means of restoring T cell responses (41–44).
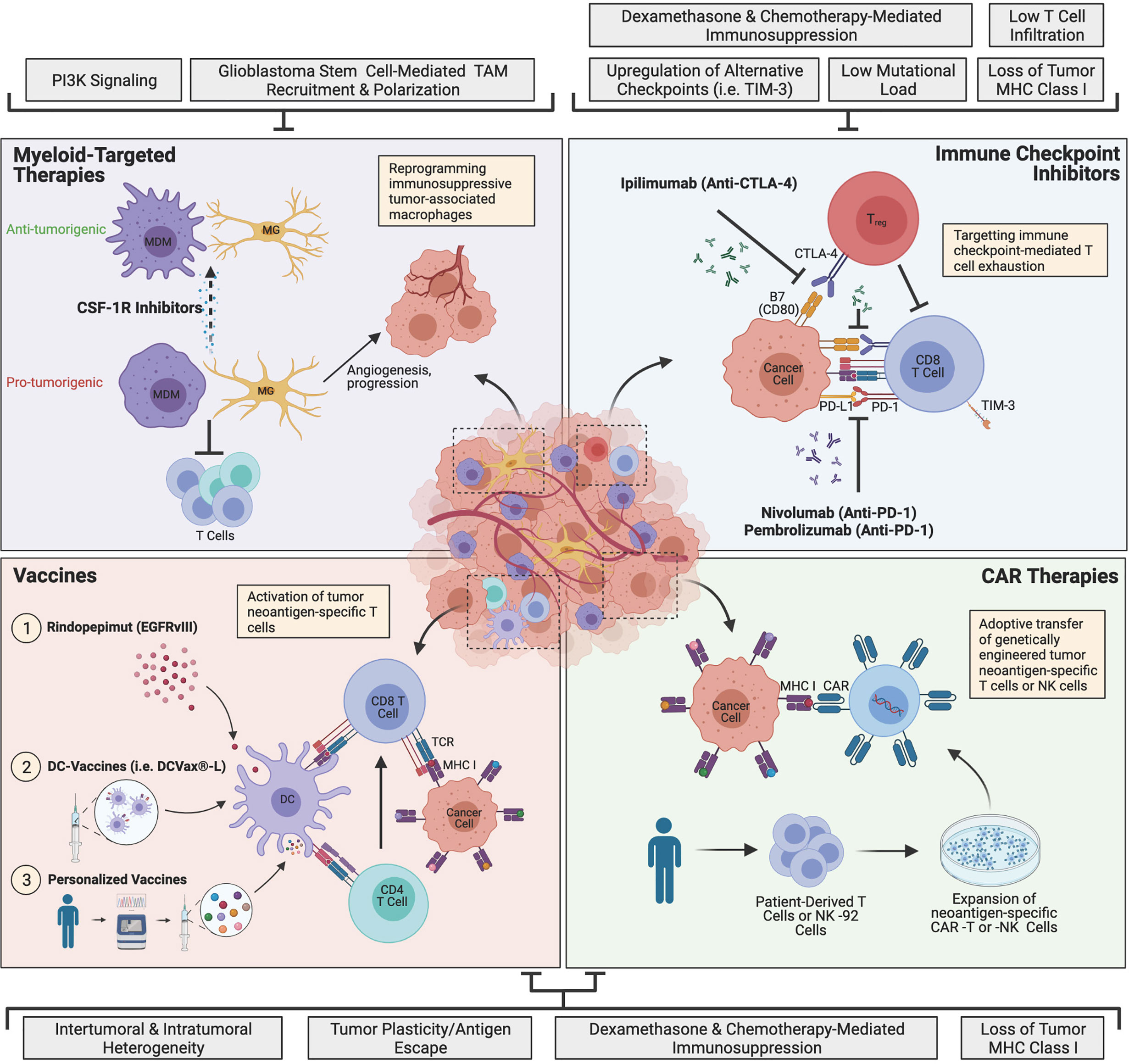
Figure 1 The current landscape of major glioblastoma immunotherapies and mechanisms of resistance. Immune checkpoint inhibitors (ICIs) target T cell exhaustion through blockade of immune checkpoints PD-1 and CTLA-4 to restore T cell function and antitumor activity. Myeloid-targeted therapies such as CSF-1R inhibitors reprogram immunosuppressive microglia (MG) or monocyte-derived macrophages (MDMs) (pro-tumorigenic) to become more anti-tumorigenic. Peptide vaccines, dendritic cell (DC)-vaccines and personalized vaccines educate T cells to target tumor neoantigen(s). Chimeric antigen receptor (CAR) immunotherapies involve genetically engineering a patient’s own T cells or non-patient NK-92 cells to express neoantigen-specific CARs, which are expanded in culture and adoptively transferred to the patient. Glioblastoma is highly resistant to therapy, and currently, none of the depicted immunotherapies have succeeded in improving treatment, although many clinical trials are currently ongoing. The grey boxes outline major mechanisms of resistance that are barriers to each immunotherapeutic approach, including intrinsic, adaptive and iatrogenic mechanisms. Image made with BioRender.com.
Accordingly, several studies have explored the use of ICIs in experimental models of glioma and results have been promising (45–49). For example, in an implanted mouse model of glioma using SMA-560 cells, anti-CTLA-4 conferred long-term survival in 80% of mice, and reduced the fraction of infiltrating Tregs (49). Additionally, anti-PD-1 eradicated 44% of orthotopic GL261 tumors when used alone, and 100% when combined with temozolomide (45). In a glioblastoma stem cell (GSC) mouse model, triple combination therapy with anti-CTLA-4, anti-PD-1 and an IL-12 expressing oncolytic virus (G47Δ-mIL12) cured 89% of mice, with 100% of the cured mice alive at 96 days post-tumor re-challenge, suggesting establishment of immunological memory with this combination therapy (50).
Although preclinical work has been promising, ICI efficacy in glioblastoma patients has been limited. There have been a number of case studies reporting dramatic responses in glioblastoma patients receiving nivolumab (anti-PD-1) (51, 52), most striking of which is the case of a 60-year-old patient with recurrent glioblastoma who received nivolumab for 2 years without any progression, toxicity or need for corticosteroid treatment (52). Despite these exceptional cases, overall, ICI clinical trials in glioblastoma have been disappointing. Checkmate 143 trial was the first randomized trial testing ICIs for recurrent cases of glioblastoma. The initial phase I study assessed the safety of nivolumab (anti-PD-1) and ipilumamab (anti-CTLA-4) in 40 patients with recurrent disease, and results showed that nivolumab alone was better tolerated compared to the dual therapy, with adverse advents associated with ipilumumab (53). Unfortunately, the subsequent open-label randomized phase 3 trial comparing nivolumab to bevacizumab failed to improve overall survival in 369 patients with recurrent glioblastoma (54). Additionally, a recent phase II clinical trial assessing pembrolizumab (anti-PD-1) with or without bevacizumab in recurrent glioblastoma patients failed to meet the primary endpoint of 6 months progression-free survival (PFS) with either therapeutic approach (55). Attention has since shifted to newly diagnosed glioblastoma, where a pre-surgical dose of nivolumab followed by post-surgical continuation of treatment was reported to provide long-term survival benefit in two patients with newly diagnosed glioblastoma, who were alive at 33 and 28 months post-surgery (56). However, all clinical studies to-date evaluating nivolumab in primary glioblastoma, including Checkmate 498 and Checkmate 548 trials, have failed to meet primary endpoints.
Overall, ICIs have failed to demonstrate a significant benefit in glioblastoma thus far and several explanations have been proposed (Figure 1). Glioblastomas are inherently immunologically “cold”, containing few T cells and predominantly occupied by pro-tumorigenic TAMs, particularly in IDH-wt tumors (57, 58). While ICIs may initially restore T cell function, the overwhelming presence of immunosuppressive myeloid cells remains a prevailing source of resistance to treatment (59). Immunologically “hot” tumors, characterized by high T cell infiltration and immune activation, have generally been more responsive to ICIs, and there is ongoing research aimed at understanding how to turn immunologically cold tumors, like glioblastoma, into hot tumors, in order to improve ICI efficacy (60, 61). Moreover, only 3.5% of glioblastomas exhibit a high tumor mutational load (62), which influences sensitivity to ICIs (63), suggesting that a very small minority of glioblastoma patients are likely to benefit from this treatment.
Another potentially overlooked mechanism of resistance to ICIs is iatrogenic resistance in response to chemotherapy or steroids. The combination of ICIs with chemotherapy is receiving widespread attention as a mechanism to induce tumor mutations (neo-antigens) (64). However, systemic chemotherapy, including temozolomide, is inherently immunosuppressive and causes lymphodepletion and myelotoxicity in preclinical models and in cancer patients (64). This may be particularly harmful for glioblastoma patients as tumor-infiltrating lymphocytes are already rare. Studies have explored the possibility of local chemotherapy using implanted slow-release polymers (65, 66), which avoids systemic lymphodepletion and significantly enhances response to ICIs in preclinical models by increasing tumor antigen-specific T cells (67). In addition, corticosteroids are routinely prescribed for cancer patients to manage symptoms, including dexamethasone, which is given to glioblastoma patients to manage cerebral edema. However, corticosteroids are anti-inflammatory, and may antagonize the therapeutic effects of ICIs; in fact, they are used to treat immune-related adverse events from ICIs (68). Alternative therapies for cerebral edema have been proposed, such as bevacizumab or mannitol. However, both agents come with significant drawbacks, including the need for repeated intravenous infusions, elevated bleeding risk (69), impaired perioperative healing (69), hypertension (70), and diminished efficacy with prolonged use (71). Therefore, it is unclear how to effectively integrate ICIs with current SOC treatments that are critical for glioblastoma management.
Finally, glioblastoma tumors can adapt to immune checkpoint blockade by upregulating alternative checkpoints such as TIM-3 following ICI treatment (72). Combining anti-PD-1 with TIM-3 blockade may potentially overcome this acquired resistance. For example, combining anti-PD-1 with anti-TIM-3 improved overall survival from 28% (anti-PD-1 alone) to 60% (dual therapy) in preclinical GL261 models, and this was further enhanced to 100% when combined as a triple therapy with stereotactic radiosurgery (SRS) (73). In addition to the PD-1 pathway, recent work has identified expression of the inhibitory receptor CD161 on intratumoral T cells in glioblastoma, and blockade of CD161 enhanced T cell anti-tumor activity both in vitro and in GL261 transplantable mouse models (74). Interestingly, CD161 is encoded by the NK cell gene, KLRB1, highlighting NK cell receptors as potential targets for immunotherapy. Taken together, future studies should explore novel targets and combination therapies to improve ICI efficacy.
Macrophages are the most abundant cell type in glioblastoma, accounting for up to 30% of the tumor, and are highly associated with disease progression (16, 75). In glioblastoma, macrophages can be either yolk sac-derived tissue-resident microglia (MG) or monocyte-derived macrophages (MDMs) from the periphery (34, 76, 77), with infiltrating MDMs representing the majority of TAMs (78). In addition to having distinct ontogenies, TAMs also adopt a variety of activation states that are not restricted to the conventional M1/M2 designations (77, 79). Interestingly, glioblastoma stem cells (GSCs) have been shown to recruit TAMs by secreting periostin and cytokines associated with alternative activation (80, 81). Once recruited, TAMs further drive disease progression by enhancing the invasion of GSCs through TGF-β1 signaling (82). In addition to the direct protumorigenic effects of TAMs, they can also indirectly mediate tumor progression by promoting T cell exhaustion via the PD-L1/PD-1 pathway (Figure 1) (83). Moreover, infiltrating TAMs in glioblastoma lack essential costimulatory molecules for T cell activation (CD80, CD86, CD40), which further contributes to an immunologically inactive tumor (84). Finally, TAMs play an important role in tumor angiogenesis and have been associated with resistance to anti-angiogenic therapies such as bevacizumab (Figure 1) (85–87). Angiogenic factors not only facilitate tumor progression, but also suppress APCs, DCs and T cells, while augmenting the effects of TAMs and Tregs, resulting in a continuous cycle of immunosuppression (88). Taken together, therapies that target the myeloid compartment may be an effective approach to reversing active immunosuppression in the TME and preventing tumor progression.
There are many approaches to targeting TAMs in glioblastoma, one of which is inhibition of colony stimulating factor 1 receptor (CSF-1R), an important receptor for macrophage differentiation and survival (89, 90). In mice, CSF-1R inhibition re-educates macrophages to adopt an anti-tumor phenotype, leading to tumor regression and increased survival, with a particularly profound effect in proneural glioblastoma (89, 90). However, despite dramatic improvements in survival, drug resistance eventually develops via alternative pathways such as PI3K signaling (Figure 1) (91). In a phase II clinical study, treatment with CSF-1R inhibitors in recurrent glioblastoma patients failed to meet primary endpoint of 6 months PFS (92), which may be attributable to the high frequency of PTEN and PI3K pathway mutations among glioblastoma patients (5, 93). Although CSF-1R inhibitors have generated little clinical success as monotherapies, emerging studies have suggested that TAM-targeted therapies may be synergistic with radiotherapy, which may serve as a more effective approach for targeting the myeloid compartment (94, 95). In GL261-implanted glioblastoma mice, irradiation enhanced survival when combined with local delivery of lipid nanoparticles directed against PD-L1-expressing TAMs and dinaciclib, a cyclin-dependent kinase 5 inhibitor (95). Moreover, in preclinical mouse models of glioblastoma driven by PDGFB overexpression and/or p53 knockdown, irradiation combined with daily CSF-1R inhibition drastically increased survival compared to either treatment alone (94). Despite these promising preclinical studies, a phase 1b/2 clinical trial evaluating CSF-1R inhibition in combination with radiotherapy and temozolomide for newly diagnosed glioblastoma did not improve median PFS or overall survival compared to historical controls (NCT01790503) (96). Although a comprehensive review of why this clinical trial failed is currently ongoing, preclinical studies demonstrated that daily dosing was critical to the efficacy of CSF-1R inhibition and unfortunately, patient tolerability restricted dosing to 5 days/week in the clinical setting (94).
In contrast to CSF-1R inhibitors, which target bulk macrophages, little is known about the potential benefit of targeting specific macrophage phenotypes and/or their recruitment. New studies have enabled the investigation of MG and MDMs and their distinct contributions to glioblastoma based on identifying distinguishing markers such as MDM-specific expression of CD49d and expression of Tmem119, CX3CR1 and SiglecH on MG (34, 97, 98). In accordance with these findings, anti-CD49d has been shown to selectively reduce tumor MDM numbers in preclinical glioblastoma models (94). Interestingly, while anti-CD49d monotherapy had no impact on survival, combining this treatment with irradiation prolonged survival in both mouse models, warranting further investigation (94). In the GL261 mouse model of glioblastoma, histological analyses have shown that MDMs are more readily recruited to perivascular tumor regions compared to MG, which is a niche for GSCs (78). Moreover, selectively limiting MDM infiltration through genetic Ccl2 reduction prolongs survival of GL261 tumor-bearing mice (78). Although targeting CCL2-mediated recruitment of MDMs has not yet been clinically explored, combining CCL2 inhibition with anti-PD-1 treatment prolonged survival in GSC glioblastoma-bearing mice, and may be a potential candidate for future studies (99). Interestingly, Tie2-expressing MDMs have been identified as a distinct hematopoietic lineage of cells that are actively recruited to glioblastoma tumors and were shown to drive tumor angiogenesis in an orthotopic xenograft model of human glioblastoma (87). Remarkably, loss of Tie2-expressing MDMs completely abrogated neovascularization in human glioblastoma-derived tumor-bearing mice, suggesting that selectively targeting Tie2-expressing MDMs may be another potential therapeutic avenue (87). Taken together, reprogramming macrophage phenotypes and targeting specific TAM recruitment may be a more effective approach to disease control that has yet to be clinically explored.
Oncogenic driver mutations and passenger mutations can give rise to new proteins (neoantigens), which contain unique sequences (neoepitopes) that can be recognized by T cells when presented by major histocompatibility complex (MHC) molecules on the surface of cancer cells or APCs (100). Vaccine-based therapeutics facilitate the education of tumor-specific CTLs by soliciting highly expressed tumor neoepitopes (Figure 1) (101). The most rudimentary approach to therapeutic vaccines is to directly administer one or more peptides that mimic the tumor neoepitope(s) of interest, although dendritic cell (DC)-based vaccines and personalized vaccines are also being explored as potential therapies in glioblastoma.
Approximately 40% of glioblastomas overexpress EGFR, with the most common variant being EGFRvIII, arising from the loss of exons 2-7 from the EGFR coding sequence (102–104). The high frequency of EGFRvIII across glioblastoma patients has led to the development of Rindopepimut (CDX-110), a synthetic 14-amino acid peptide that mimics the EGFRvIII mutational site coupled to keyhole limpet hemocyanin (KLH), an immunogenic carrier protein (105). In 2015, the FDA granted rindopepimut the “Breakthrough Therapy Designation”, supporting the expedition of its approval for glioblastoma, given that clinical studies demonstrate substantial benefit over other available therapies. The single arm multicenter phase II trial (ACT III), which administered rindopepimut and adjuvant chemotherapy for newly diagnosed EGFRvIII+ glioblastoma patients, had promising results with a median overall survival of 21.8 months compared to matched historical controls treated with SOC (106). However, the subsequent randomized double-blinded phase III trial (ACT IV) failed to demonstrate any increase in survival and was terminated (107). Loss of EGFRvIII expression following vaccination suggests that the recurrent tumor can become resistant to EGFRvIII-targeting memory T cells (Figure 1) (106). In fact, half of all glioblastomas that are initially EGFRvIII+ lose EGFRvIII expression upon recurrence (108). While overexpression of EGFRvIII was once believed to be predictive of poor prognosis (103), a recent study assessing the EGFR status of 106 patients found no association between EGFRvIII and overall survival or progression-free survival in either newly diagnosed or recurrent glioblastoma (104). Taken together, these observations may explain why EGFRvIII-targeted vaccines have failed to control disease and improve survival.
DCs are an essential component of vaccination because of their role in antigen presentation and the priming and activation of T cells (101). It was once thought that DCs played little to no role in the active immunity of the brain, with MG assumed to be the predominating APCs (109, 110). However, DCs are increasingly being recognized for their functional role in the brain as APCs and it has been reported that they can even arise from MG differentiation (111, 112). Interestingly, MG exhibit a great amount of plasticity and can be skewed towards macrophage-like or DC-like cells by M-CSF or GM-CSF, respectively (111). While traditional vaccines rely on the activation of DCs and other APCs in vivo, DC-based vaccines deliver DCs pre-loaded with antigen by pulsing patient-derived DCs ex vivo with either tumor lysate or predetermined neoantigens (101). For glioblastoma, DC-based vaccines have shown promise in early clinical studies (113, 114). A phase 1 clinical study investigating the dose-escalation of DCs pulsed with tumor peptides in 12 newly diagnosed glioblastoma patients demonstrated safety and tolerability of this therapy (113). The double-blinded randomized phase II trial of ICT-107, involving DCs pulsed with six synthetic peptides, increased overall survival of newly diagnosed glioblastoma patients by 2 months compared to placebo control, although it was not statistically significant (114). Another DC vaccine, DCVax®-L, demonstrated safety and tolerability in early studies and recently underwent phase 3 evaluation, but was unfortunately prematurely suspended due to lack of funds (115). Interestingly, there appears to be subtype-specific benefits of DC-based vaccines, whereby the mesenchymal subtype is associated with heightened responsiveness, including increased infiltration of CD3+ and CD8+ T cells compared to other glioblastoma subtypes, and increased survival compared to historical controls of the same molecular subtype (116). Therefore, molecular subtyping may be an important consideration for future study enrollment and design.
Neoantigen-targeted vaccines for glioblastoma are extremely limited by the high level of inter- and intra-tumoral heterogeneity of these tumors (Figure 1) (7, 8). Tumor cells also actively evade T cell immunosurveillance by altering surface MHC expression and antigen presentation pathways (Figure 1) (117). Thus, while the identification of neoantigens is critical, immunization against a single molecular target, such as EGFRvIII (rindopepimut), selectively eliminates neoantigen-expressing cells, leaving the remaining tumor resistant to the activated T cells (106, 118). As an alternative approach, personalized vaccines may be more appropriate in highly heterogenous tumors like glioblastoma (100). The personalized vaccine pipeline involves first characterizing the mutational profile of an individual’s tumor through comparative sequencing, followed by selection of patient-specific targets and finally, vaccine production (100). This personalized approach effectively circumvents patient-to-patient variability and seeks to maximize the affected tumor area by generating T cell immunity against many targets. Preliminary studies using personalized vaccines in newly diagnosed glioblastoma patients have been generally positive (119, 120). In a phase I/Ib trial, patients were immunized post-radiation with up to 20 synthetic long peptides generated based on tumor DNA/RNA sequencing, and given an immunostimulant, poly-ICLC. Neoantigen-specific T cell responses were observed in patients who did not receive dexamethasone and multiplex immunofluorescent staining of tumor specimens revealed increased CD8+ and CD4+ T cell infiltration in these responsive patients (119). Combining personalized neoantigen vaccines with vaccination against unmutated antigen (GAPVAC) have shown similarly promising results where immunization generated sustained central memory CD8+ T cell responses against unmutated antigen, as well as neoepitope-specific Th1 responses in CD4+ T cells (120). There are currently over 50 ongoing clinical trials for various forms of vaccines against glioblastoma, with results expected to be rolled out in the coming years.
CAR T cell therapy is a highly personalized form of adoptive T cell therapy that takes advantage of a patient’s own T cells and strategically engineers them to express CARs, which target cancer cells (Figure 1). CARs consist of an intracellular T cell activation domain and an extracellular antigen-recognition domain, which are joined together by a transmembrane domain connected to a hinge (121). For refractory hematologic cancers such as acute lymphoblastic leukemia (ALL) and diffuse large-B-cell lymphoma (DLBCL), CAR T cell therapy has been transformational (21, 22), however translating this therapy to solid tumors comes with a unique set of challenges and no CAR T cells have been approved for solid cancers to-date (122). Since their inception, CARs have quickly evolved from basic CD3ζ-signaling in the first-generation, to incorporating co-stimulatory domains such as CD28, 4-1BB, OX40 and ICOS in second and third-generations, followed by the addition of cytokine-expressing domains in fourth-generation CARs (TRUCKs) and most recently, cytokine receptor-expressing domains in fifth-generation CARs (121–123). Despite the successful engineering of more potent and immunogenic CAR-T cells, off-target effects, poor tumor infiltration and a highly immunosuppressive TME remain major barriers to the clinical efficacy of CAR T cells for solid tumors (121).
There are several ongoing CAR T cell candidates for glioblastoma including CARs directed against EGFRvIII, IL13Rα2 and HER2. In an orthotopically transplanted human glioblastoma xenograft model, third generation EGFRvIII-specific CAR T cells prolonged survival of tumor-bearing mice by up to 55 days compared to untreated mice (124). However, clinical benefit has yet to be observed in patients where tumor adaptations, including loss of EGFRvIII expression and post-treatment infiltration of Tregs, invariably leads to resistance against EGFRvIII-directed CARs (118, 125). Alternatively, CAR T cell therapies can target IL13Rα2, which is overexpressed in 58% of glioblastomas and is associated with poor prognosis and a mesenchymal gene signature (126). IL13Rα2-specific CAR T cells have been clinically well-tolerated, and structurally optimized to prevent off-target Fc interactions (127, 128). This therapeutic candidate, which is currently being clinically evaluated (NCT02208362) (129), was reported to cause dramatic tumor reduction and a sustained complete clinical response (7.5 months) in a patient bearing seven highly aggressive recurrent glioblastoma tumors (128). HER2-targeted CAR T cells have demonstrated similar promise in early phase clinical trials, where careful engineering has improved tumor-specificity and reduced off-target effects (130, 131).
The propensity for glioblastoma tumors to quickly adapt through antigen escape remains a major barrier to CAR T cell therapy (Figure 1) (132). To minimize the risk of treatment resistance, it is likely that CAR T cells should target multiple antigens or be combined with a synergistic therapy. For example, a bispecific CAR molecule directed against both IL13Rα2 and HER2 (TanCAR) has been shown to promote tumor regression and increase survival in mice xenografted with a HER2+ IL13Rα2+ human glioblastoma cell line compared to CAR T cells against either target alone (133). IL13Rα2 CAR T cells are also currently being clinically evaluated in combination with nivolumab and ipilimumab for recurrent and refractory glioblastoma (NCT04003649) (134). Synergistic combinatorial approaches will be instrumental in improving CAR T cell efficacy, since CAR T cells alone have shown limited utility against solid tumors, including glioblastoma, thus far.
As CAR T cell therapy continues to advance, CAR-NK cell therapy has also gained attention as a potential tool for cancer immunotherapy. In glioblastoma, NK cells can mediate tumor cell killing and are associated with good prognosis (135). A notable advantage of CAR-NK cell therapy is the ability to be administered to an HLA-mismatched patient, thus allowing the possibility of an off-the-shelf therapy (136). However, the time and cost associated with NK cell expansion and manufacturing remain a barrier for CAR-NK cell therapy (137). Currently, NK-92 cells are the only NK cell line approved by the FDA and are compliant with good manufacturing practices (138). Remarkably, preclinical testing of HER-2-specific NK-92 cells (NK-92/5.28.z) in an orthotopic xenograft mouse model of glioblastoma demonstrated a dramatic increase in survival (200.5 days) compared to mice treated with control NK-92 cells (73 days) (139). Intracranial injection of NK-92/5.28.z cells are being evaluated in the ongoing CAR2BRAIN clinical trial for recurrent glioblastoma, with no toxicities reported thus far at three dose levels (NCT03383978) (140, 141). Although the field of CAR-NK cell therapy is still relatively new, preliminary results have been promising, and the first ever clinical trial of CAR-NK cells for glioblastoma will indeed shed light on whether this immunotherapy can bring benefit to patients.
The field of cancer immunotherapy is rapidly evolving to meet the unique requirements and challenges of diverse cancer types. While immunotherapies have revolutionized the clinical management of NSCLC, melanoma, renal cancer, and several hematological malignancies, it is becoming increasingly apparent that mechanisms of efficacy are not one-size-fits-all. For glioblastoma, conventional therapies provide limited benefit to patients and most attempts to incorporate immunotherapeutics have been futile thus far. Efforts to optimize immunotherapies need to overcome many obstacles to achieve efficacy, including physical barriers to drug delivery (e.g. BBB), prominent tumor heterogeneity, abundant GSC niches, lymphocyte scarcity, and the immunosuppressive effects of SOC treatments. Studying the dynamics of different glioblastoma subtypes, as well as long-term survivors, will be an important resource in understanding aspects of the TME that promote survival. Finally, a prevailing challenge in glioblastoma research is that the effects of immunotherapy in animal models rarely recapitulate clinical observations. Genetically-engineered and transplantable mouse models are the best tools available, however, they fail to fully reflect tumor heterogeneity and host antitumor immunity. Further efforts are needed to generate preclinical models that more accurately recapitulate human disease.
Taken together, there is a desperate need to identify new therapeutic opportunities in glioblastoma in order to improve SOC. While immunotherapies have the potential to transform glioblastoma treatment, many are limited by the unique and challenging characteristics of the tumor. With a better understanding of glioblastoma TME dynamics and improved preclinical tools, we can open doors for more personalized and targeted treatments that ultimately have the potential to have a meaningful impact on patient outcomes.
MY and DQ conceived, wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
DQ is funded by the Brain Tumor Funders’ Collaborative (BTFC), Canada Foundation for Innovation (CFI-JELF, 37488), and a Tier II Canada Research Chair in Tumor Microenvironment Research. MY is supported by the Max E. and Jane K. Childress Fellowship from the Department of Physiology, McGill University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, et al. Cbtrus Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008-2012. Neuro Oncol (2015) 17(Suppl 4):iv1–iv62. doi: 10.1093/neuonc/nov189
2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy Plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
3. Campos B, Olsen LR, Urup T, Poulsen HS. A Comprehensive Profile of Recurrent Glioblastoma. Oncogene (2016) 35(45):5819–25. doi: 10.1038/onc.2016.85
4. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
5. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, Idh1, EGFR, and NF1. Cancer Cell (2010) 17(1):98–110. doi: 10.1016/j.ccr.2009.12.020
6. Sidaway P. CNS Cancer: Glioblastoma Subtypes Revisited. Nat Rev Clin Oncol (2017) 14(10):587. doi: 10.1038/nrclinonc.2017.122
7. Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, et al. Intratumor Heterogeneity in Human Glioblastoma Reflects Cancer Evolutionary Dynamics. Proc Natl Acad Sci U S A (2013) 110(10):4009–14. doi: 10.1073/pnas.1219747110
8. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-Cell RNA-seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science (2014) 344(6190):1396–401. doi: 10.1126/science.1254257
9. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates With Immunological Changes in the Microenvironment. Cancer Cell (2017) 32(1):42–56 e6. doi: 10.1016/j.ccell.2017.06.003
10. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular Subclasses of High-Grade Glioma Predict Prognosis, Delineate a Pattern of Disease Progression, and Resemble Stages in Neurogenesis. Cancer Cell (2006) 9(3):157–73. doi: 10.1016/j.ccr.2006.02.019
11. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab Plus Radiotherapy-Temozolomide for Newly Diagnosed Glioblastoma. N Engl J Med (2014) 370(8):709–22. doi: 10.1056/NEJMoa1308345
12. Wick W, Gorlia T, Bendszus M, Taphoorn M, Sahm F, Harting I, et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N Engl J Med (2017) 377(20):1954–63. doi: 10.1056/NEJMoa1707358
13. Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, et al. Cilengitide Combined With Standard Treatment for Patients With Newly Diagnosed Glioblastoma With Methylated MGMT Promoter (CENTRIC EORTC 26071-22072 Study): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol (2014) 15(10):1100–8. doi: 10.1016/S1470-2045(14)70379-1
14. Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA (2017) 318(23):2306–16. doi: 10.1001/jama.2017.18718
15. Fabian D, Guillermo Prieto Eibl MDP, Alnahhas I, Sebastian N, Giglio P, Puduvalli V, et al. Treatment of Glioblastoma (GBM) With the Addition of Tumor-Treating Fields (Ttf): A Review. Cancers (Basel) (2019) 11(2):174. doi: 10.3390/cancers11020174
16. Hambardzumyan D, Gutmann DH, Kettenmann H. The Role of Microglia and Macrophages in Glioma Maintenance and Progression. Nat Neurosci (2016) 19(1):20–7. doi: 10.1038/nn.4185
17. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
18. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall Survival With Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med (2017) 377(14):1345–56. doi: 10.1056/NEJMoa1709684
19. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-Year Survival With Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med (2019) 381(16):1535–46. doi: 10.1056/NEJMoa1910836
20. Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab Plus Ipilimumab in Advanced non-Small-Cell Lung Cancer. N Engl J Med (2019) 381(21):2020–31. doi: 10.1056/NEJMoa1910231
21. Brudno JN, Kochenderfer JN. Chimeric Antigen Receptor T-cell Therapies for Lymphoma. Nat Rev Clin Oncol (2018) 15(1):31–46. doi: 10.1038/nrclinonc.2017.128
22. Hucks G, Rheingold SR. The Journey to CAR T Cell Therapy: The Pediatric and Young Adult Experience With Relapsed or Refractory B-ALL. Blood Cancer J (2019) 9(2):10. doi: 10.1038/s41408-018-0164-6
23. Medawar PB. Immunity to Homologous Grafted Skin; the Fate of Skin Homografts Transplanted to the Brain, to Subcutaneous Tissue, and to the Anterior Chamber of the Eye. Br J Exp Pathol (1948) 29(1):58–69.
24. Nicholas MK, Antel JP, Stefansson K, Arnason BG. Rejection of Fetal Neocortical Neural Transplants by H-2 Incompatible Mice. J Immunol (1987) 139(7):2275–83.
25. Billingham RE, Brent L, Medawar PB, Sparrow EM. Quantitative Studies on Tissue Transplantation Immunity. I. The Survival Times of Skin Homografts Exchanged Between Members of Different Inbred Strains of Mice. Proc R Soc Lond B Biol Sci (1954) 143(910):43–58. doi: 10.1098/rspb.1954.0053
26. Bartholomaus I, Kawakami N, Odoardi F, Schlager C, Miljkovic D, Ellwart JW, et al. Effector T Cell Interactions With Meningeal Vascular Structures in Nascent Autoimmune CNS Lesions. Nature (2009) 462(7269):94–8. doi: 10.1038/nature08478
27. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and Functional Features of Central Nervous System Lymphatic Vessels. Nature (2015) 523(7560):337–41. doi: 10.1038/nature14432
28. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A Dural Lymphatic Vascular System That Drains Brain Interstitial Fluid and Macromolecules. J Exp Med (2015) 212(7):991–9. doi: 10.1084/jem.20142290
29. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and Function of the Blood-Brain Barrier. Neurobiol Dis (2010) 37(1):13–25. doi: 10.1016/j.nbd.2009.07.030
30. Pardridge WM. Drug Transport Across the Blood-Brain Barrier. J Cereb Blood Flow Metab (2012) 32(11):1959–72. doi: 10.1038/jcbfm.2012.126
31. Larsson HB, Stubgaard M, Frederiksen JL, Jensen M, Henriksen O, Paulson OB. Quantitation of Blood-Brain Barrier Defect by Magnetic Resonance Imaging and gadolinium-DTPA in Patients With Multiple Sclerosis and Brain Tumors. Magn Reson Med (1990) 16(1):117–31. doi: 10.1002/mrm.1910160111
32. Liebner S, Fischmann A, Rascher G, Duffner F, Grote EH, Kalbacher H, et al. Claudin-1 and Claudin-5 Expression and Tight Junction Morphology are Altered in Blood Vessels of Human Glioblastoma Multiforme. Acta Neuropathol (2000) 100(3):323–31. doi: 10.1007/s004010000180
33. Wolburg H, Wolburg-Buchholz K, Kraus J, Rascher-Eggstein G, Liebner S, Hamm S, et al. Localization of Claudin-3 in Tight Junctions of the Blood-Brain Barrier is Selectively Lost During Experimental Autoimmune Encephalomyelitis and Human Glioblastoma Multiforme. Acta Neuropathol (2003) 105(6):586–92. doi: 10.1007/s00401-003-0688-z
34. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep (2016) 17(9):2445–59. doi: 10.1016/j.celrep.2016.10.052
35. Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the Blood-Brain Barrier Really Disrupted in All Glioblastomas? A Critical Assessment of Existing Clinical Data. Neuro Oncol (2018) 20(2):184–91. doi: 10.1093/neuonc/nox175
36. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced Expression of PD-1, a Novel Member of the Immunoglobulin Gene Superfamily, Upon Programmed Cell Death. EMBO J (1992) 11(11):3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
37. Leach DR, Krummel MF, Allison JP. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science (1996) 271(5256):1734–6. doi: 10.1126/science.271.5256.1734
38. Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol (2016) 39(1):98–106. doi: 10.1097/COC.0000000000000239
39. Wintterle S, Schreiner B, Mitsdoerffer M, Schneider D, Chen L, Meyermann R, et al. Expression of the B7-related Molecule B7-H1 by Glioma Cells: A Potential Mechanism of Immune Paralysis. Cancer Res (2003) 63(21):7462–7.
40. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas Promote Immunosuppression Through Induction of B7-H1 Expression in Tumor-Associated Macrophages. Clin Cancer Res (2013) 19(12):3165–75. doi: 10.1158/1078-0432.CCR-12-3314
41. Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased Regulatory T-cell Fraction Amidst a Diminished CD4 Compartment Explains Cellular Immune Defects in Patients With Malignant Glioma. Cancer Res (2006) 66(6):3294–302. doi: 10.1158/0008-5472.CAN-05-3773
42. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, et al. Programmed Death Ligand 1 Expression and Tumor-Infiltrating Lymphocytes in Glioblastoma. Neuro Oncol (2015) 17(8):1064–75. doi: 10.1093/neuonc/nou307
43. Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, et al. Pd-L1 Expression and Prognostic Impact in Glioblastoma. Neuro Oncol (2016) 18(2):195–205. doi: 10.1093/neuonc/nov172
44. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-Cell Exhaustion Signatures Vary With Tumor Type and Are Severe in Glioblastoma. Clin Cancer Res (2018) 24(17):4175–86. doi: 10.1158/1078-0432.CCR-17-1846
45. Park J, Kim CG, Shim JK, Kim JH, Lee H, Lee JE, et al. Effect of Combined anti-PD-1 and Temozolomide Therapy in Glioblastoma. Oncoimmunology (2019) 8(1):e1525243. doi: 10.1080/2162402X.2018.1525243
46. Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol Res (2016) 4(2):124–35. doi: 10.1158/2326-6066.CIR-15-0151
47. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable Therapeutic Efficacy Utilizing Combinatorial Blockade Against IDO, Ctla-4, and PD-L1 in Mice With Brain Tumors. Clin Cancer Res (2014) 20(20):5290–301. doi: 10.1158/1078-0432.CCR-14-0514
48. Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, et al. Anti-PD-1 Blockade and Stereotactic Radiation Produce Long-Term Survival in Mice With Intracranial Gliomas. Int J Radiat Oncol Biol Phys (2013) 86(2):343–9. doi: 10.1016/j.ijrobp.2012.12.025
49. Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, et al. Systemic CTLA-4 Blockade Ameliorates Glioma-Induced Changes to the CD4+ T Cell Compartment Without Affecting Regulatory T-cell Function. Clin Cancer Res (2007) 13(7):2158–67. doi: 10.1158/1078-0432.CCR-06-2070
50. Saha D, Martuza RL, Rabkin SD. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell (2017) 32(2):253–67.e5. doi: 10.1016/j.ccell.2017.07.006
51. Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol (2016) 34(19):2206–11. doi: 10.1200/JCO.2016.66.6552
52. Roth P, Valavanis A, Weller M. Long-Term Control and Partial Remission After Initial Pseudoprogression of Glioblastoma by anti-PD-1 Treatment With Nivolumab. Neuro Oncol (2017) 19(3):454–6. doi: 10.1093/neuonc/now265
53. Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, et al. Nivolumab With or Without Ipilimumab in Patients With Recurrent Glioblastoma: Results From Exploratory Phase I Cohorts of CheckMate 143. Neuro Oncol (2018) 20(5):674–86. doi: 10.1093/neuonc/nox208
54. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol (2020) 6(7):1003–10. doi: 10.1001/jamaoncol.2020.1024
55. Nayak L, Molinaro AM, Peters K, Clarke JL, Jordan JT, de Groot J, et al. Randomized Phase II and Biomarker Study of Pembrolizumab Plus Bevacizumab Versus Pembrolizumab Alone for Patients With Recurrent Glioblastoma. Clin Cancer Res (2020) 27(4):1048–57. doi: 10.1158/1078-0432.CCR-20-2500
56. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, Lopez-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant Nivolumab Modifies the Tumor Immune Microenvironment in Resectable Glioblastoma. Nat Med (2019) 25(3):470–6. doi: 10.1038/s41591-018-0339-5
57. Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell (2020) 181(7):1643–60.e17. doi: 10.1016/j.cell.2020.05.007
58. Quail DF, Joyce JA. The Microenvironmental Landscape of Brain Tumors. Cancer Cell (2017) 31(3):326–41. doi: 10.1016/j.ccell.2017.02.009
59. Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front Immunol (2018) 9:1310. doi: 10.3389/fimmu.2018.01310
60. Liu Z, Han C, Fu YX. Targeting Innate Sensing in the Tumor Microenvironment to Improve Immunotherapy. Cell Mol Immunol (2020) 17(1):13–26. doi: 10.1038/s41423-019-0341-y
61. Duan Q, Zhang H, Zheng J, Zhang L. Turning Cold Into Hot: Firing Up the Tumor Microenvironment. Trends Cancer (2020) 6(7):605–18. doi: 10.1016/j.trecan.2020.02.022
62. Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, et al. Mutational Burden, Immune Checkpoint Expression, and Mismatch Repair in Glioma: Implications for Immune Checkpoint Immunotherapy. Neuro Oncol (2017) 19(8):1047–57. doi: 10.1093/neuonc/nox026
63. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal Neoantigens Elicit T Cell Immunoreactivity and Sensitivity to Immune Checkpoint Blockade. Science (2016) 351(6280):1463–9. doi: 10.1126/science.aaf1490
64. Galluzzi L, Humeau J, Buque A, Zitvogel L, Kroemer G. Immunostimulation With Chemotherapy in the Era of Immune Checkpoint Inhibitors. Nat Rev Clin Oncol (2020) 17(12):725–41. doi: 10.1038/s41571-020-0413-z
65. Brem H, Piantadosi S, Burger PC, Walker M, Selker R, Vick NA, et al. Placebo-Controlled Trial of Safety and Efficacy of Intraoperative Controlled Delivery by Biodegradable Polymers of Chemotherapy for Recurrent Gliomas. The Polymer-Brain Tumor Treatment Group. Lancet (1995) 345(8956):1008–12. doi: 10.1016/s0140-6736(95)90755-6
66. Westphal M, Hilt DC, Bortey E, Delavault P, Olivares R, Warnke PC, et al. A Phase 3 Trial of Local Chemotherapy With Biodegradable Carmustine (BCNU) Wafers (Gliadel Wafers) in Patients With Primary Malignant Glioma. Neuro Oncol (2003) 5(2):79–88. doi: 10.1093/neuonc/5.2.79
67. Mathios D, Kim JE, Mangraviti A, Phallen J, Park CK, Jackson CM, et al. Anti-PD-1 Antitumor Immunity is Enhanced by Local and Abrogated by Systemic Chemotherapy in GBM. Sci Transl Med (2016) 8(370):370ra180. doi: 10.1126/scitranslmed.aag2942
68. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the Immune-Related Adverse Effects of Immune Checkpoint Inhibitors: A Review. JAMA Oncol (2016) 2(10):1346–53. doi: 10.1001/jamaoncol.2016.1051
69. Dietrich J, Rao K, Pastorino S, Kesari S. Corticosteroids in Brain Cancer Patients: Benefits and Pitfalls. Expert Rev Clin Pharmacol (2011) 4(2):233–42. doi: 10.1586/ecp.11.1
70. Meng X, Zhao R, Shen G, Dong D, Ding L, Wu S. Efficacy and Safety of Bevacizumab Treatment for Refractory Brain Edema: Case Report. Med (Baltimore) (2017) 96(44):e8280. doi: 10.1097/MD.0000000000008280
71. Palma L, Bruni G, Fiaschi AI, Mariottini A. Passage of Mannitol Into the Brain Around Gliomas: A Potential Cause of Rebound Phenomenon. A Study on 21 Patients. J Neurosurg Sci (2006) 50(3):63–6.
72. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive Resistance to Therapeutic PD-1 Blockade is Associated With Upregulation of Alternative Immune Checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
73. Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination Therapy With Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin Cancer Res (2017) 23(1):124–36. doi: 10.1158/1078-0432.CCR-15-1535
74. Mathewson ND, Ashenberg O, Tirosh I, Gritsch S, Perez EM, Marx S, et al. Inhibitory CD161 Receptor Identified in Glioma-Infiltrating T Cells by Single-Cell Analysis. Cell (2021) 184(5):1281–98.e26. doi: 10.1016/j.cell.2021.01.022
75. Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible Involvement of the M2 Anti-Inflammatory Macrophage Phenotype in Growth of Human Gliomas. J Pathol (2008) 216(1):15–24. doi: 10.1002/path.2370
76. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-Resident Macrophages Originate From Yolk-Sac-Derived Erythro-Myeloid Progenitors. Nature (2015) 518(7540):547–51. doi: 10.1038/nature13989
77. Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New Insights Into the Multidimensional Concept of Macrophage Ontogeny, Activation and Function. Nat Immunol (2016) 17(1):34–40. doi: 10.1038/ni.3324
78. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res (2017) 77(9):2266–78. doi: 10.1158/0008-5472.CAN-16-2310
79. Mosser DM, Edwards JP. Exploring the Full Spectrum of Macrophage Activation. Nat Rev Immunol (2008) 8(12):958–69. doi: 10.1038/nri2448
80. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin Secreted by Glioblastoma Stem Cells Recruits M2 Tumour-Associated Macrophages and Promotes Malignant Growth. Nat Cell Biol (2015) 17(2):170–82. doi: 10.1038/ncb3090
81. Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, et al. Glioma Cancer Stem Cells Induce Immunosuppressive Macrophages/Microglia. Neuro Oncol (2010) 12(11):1113–25. doi: 10.1093/neuonc/noq082
82. Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, et al. Tumor-Associated Microglia/Macrophages Enhance the Invasion of Glioma Stem-Like Cells Via TGF-beta1 Signaling Pathway. J Immunol (2012) 189(1):444–53. doi: 10.4049/jimmunol.1103248
83. Akins EA, Aghi MK, Kumar S. Incorporating Tumor-Associated Macrophages Into Engineered Models of Glioma. iScience (2020) 23(12):101770. doi: 10.1016/j.isci.2020.101770
84. Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The Role of Human Glioma-Infiltrating Microglia/Macrophages in Mediating Antitumor Immune Responses. Neuro Oncol (2006) 8(3):261–79. doi: 10.1215/15228517-2006-008
85. Piao Y, Liang J, Holmes L, Zurita AJ, Henry V, Heymach JV, et al. Glioblastoma Resistance to anti-VEGF Therapy is Associated With Myeloid Cell Infiltration, Stem Cell Accumulation, and a Mesenchymal Phenotype. Neuro Oncol (2012) 14(11):1379–92. doi: 10.1093/neuonc/nos158
86. Lu-Emerson C, Snuderl M, Kirkpatrick ND, Goveia J, Davidson C, Huang Y, et al. Increase in Tumor-Associated Macrophages After Antiangiogenic Therapy is Associated With Poor Survival Among Patients With Recurrent Glioblastoma. Neuro Oncol (2013) 15(8):1079–87. doi: 10.1093/neuonc/not082
87. De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, et al. Tie2 Identifies a Hematopoietic Lineage of Proangiogenic Monocytes Required for Tumor Vessel Formation and a Mesenchymal Population of Pericyte Progenitors. Cancer Cell (2005) 8(3):211–26. doi: 10.1016/j.ccr.2005.08.002
88. Rahma OE, Hodi FS. The Intersection Between Tumor Angiogenesis and Immune Suppression. Clin Cancer Res (2019) 25(18):5449–57. doi: 10.1158/1078-0432.CCR-18-1543
89. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. Csf-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat Med (2013) 19(10):1264–72. doi: 10.1038/nm.3337
90. Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, et al. Microglial Stimulation of Glioblastoma Invasion Involves Epidermal Growth Factor Receptor (EGFR) and Colony Stimulating Factor 1 Receptor (CSF-1R) Signaling. Mol Med (2012) 18:519–27. doi: 10.2119/molmed.2011.00217
91. Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, et al. The Tumor Microenvironment Underlies Acquired Resistance to CSF-1R Inhibition in Gliomas. Science (2016) 352(6288):aad3018. doi: 10.1126/science.aad3018
92. Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, et al. Orally Administered Colony Stimulating Factor 1 Receptor Inhibitor PLX3397 in Recurrent Glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium Phase II Study. Neuro Oncol (2016) 18(4):557–64. doi: 10.1093/neuonc/nov245
93. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The Somatic Genomic Landscape of Glioblastoma. Cell (2013) 155(2):462–77. doi: 10.1016/j.cell.2013.09.034
94. Akkari L, Bowman RL, Tessier J, Klemm F, Handgraaf SM, de Groot M, et al. Dynamic Changes in Glioma Macrophage Populations After Radiotherapy Reveal CSF-1R Inhibition as a Strategy to Overcome Resistance. Sci Transl Med (2020) 12(552):eaaw7843. doi: 10.1126/scitranslmed.aaw7843
95. Zhang P, Miska J, Lee-Chang C, Rashidi A, Panek WK, An S, et al. Therapeutic Targeting of Tumor-Associated Myeloid Cells Synergizes With Radiation Therapy for Glioblastoma. Proc Natl Acad Sci U S A (2019) 116(47):23714–23. doi: 10.1073/pnas.1906346116
96. A Phase 1b/2 Study of PLX3397 + Radiation Therapy + Temozolomide in Patients With Newly Diagnosed Glioblastoma Nct01790503. Available at: https://clinicaltrials.gov/ct2/show/NCT01790503.
97. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-Expression Profiles and Transcriptional Regulatory Pathways That Underlie the Identity and Diversity of Mouse Tissue Macrophages. Nat Immunol (2012) 13(11):1118–28. doi: 10.1038/ni.2419
98. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New Tools for Studying Microglia in the Mouse and Human CNS. Proc Natl Acad Sci U S A (2016) 113(12):E1738–46. doi: 10.1073/pnas.1525528113
99. Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF, et al. CCR2 Inhibition Reduces Tumor Myeloid Cells and Unmasks a Checkpoint Inhibitor Effect to Slow Progression of Resistant Murine Gliomas. Proc Natl Acad Sci U S A (2020) 117(2):1129–38. doi: 10.1073/pnas.1910856117
100. Sahin U, Tureci O. Personalized Vaccines for Cancer Immunotherapy. Science (2018) 359(6382):1355–60. doi: 10.1126/science.aar7112
101. Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical Use of Dendritic Cells for Cancer Therapy. Lancet Oncol (2014) 15(7):e257–67. doi: 10.1016/S1470-2045(13)70585-0
102. Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical Splicing of Aberrant Epidermal Growth Factor Receptor Transcripts From Amplified Rearranged Genes in Human Glioblastomas. Proc Natl Acad Sci U S A (1990) 87(21):8602–6. doi: 10.1073/pnas.87.21.8602
103. Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, et al. Prognostic Value of Epidermal Growth Factor Receptor in Patients With Glioblastoma Multiforme. Cancer Res (2003) 63(20):6962–70.
104. Felsberg J, Hentschel B, Kaulich K, Gramatzki D, Zacher A, Malzkorn B, et al. Epidermal Growth Factor Receptor Variant III (Egfrviii) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison Between Primary and Recurrent Tumors. Clin Cancer Res (2017) 23(22):6846–55. doi: 10.1158/1078-0432.CCR-17-0890
105. Humphrey PA, Wong AJ, Vogelstein B, Zalutsky MR, Fuller GN, Archer GE, et al. Anti-Synthetic Peptide Antibody Reacting At the Fusion Junction of Deletion-Mutant Epidermal Growth Factor Receptors in Human Glioblastoma. Proc Natl Acad Sci U S A (1990) 87(11):4207–11. doi: 10.1073/pnas.87.11.4207
106. Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. Multicenter Trial of Rindopepimut (CDX-110) in Newly Diagnosed Glioblastoma: The ACT III Study. Neuro Oncol (2015) 17(6):854–61. doi: 10.1093/neuonc/nou348
107. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut With Temozolomide for Patients With Newly Diagnosed, EGFRvIII-expressing Glioblastoma (ACT IV): A Randomised, Double-Blind, International Phase 3 Trial. Lancet Oncol (2017) 18(10):1373–85. doi: 10.1016/S1470-2045(17)30517-X
108. van den Bent MJ, Gao Y, Kerkhof M, Kros JM, Gorlia T, van Zwieten K, et al. Changes in the EGFR Amplification and EGFRvIII Expression Between Paired Primary and Recurrent Glioblastomas. Neuro Oncol (2015) 17(7):935–41. doi: 10.1093/neuonc/nov013
109. Lowe J, MacLennan KA, Powe DG, Pound JD, Palmer JB. Microglial Cells in Human Brain Have Phenotypic Characteristics Related to Possible Function as Dendritic Antigen Presenting Cells. J Pathol (1989) 159(2):143–9. doi: 10.1002/path.1711590209
110. Hickey WF, Kimura H. Perivascular Microglial Cells of the CNS are Bone Marrow-Derived and Present Antigen In Vivo. Science (1988) 239(4837):290–2. doi: 10.1126/science.3276004
111. Santambrogio L, Belyanskaya SL, Fischer FR, Cipriani B, Brosnan CF, Ricciardi-Castagnoli P, et al. Developmental Plasticity of CNS Microglia. Proc Natl Acad Sci U S A (2001) 98(11):6295–300. doi: 10.1073/pnas.111152498
112. Fischer HG, Reichmann G. Brain Dendritic Cells and Macrophages/Microglia in Central Nervous System Inflammation. J Immunol (2001) 166(4):2717–26. doi: 10.4049/jimmunol.166.4.2717
113. Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, Giovannone AJ, et al. Dendritic Cell Vaccination in Glioblastoma Patients Induces Systemic and Intracranial T-cell Responses Modulated by the Local Central Nervous System Tumor Microenvironment. Clin Cancer Res (2005) 11(15):5515–25. doi: 10.1158/1078-0432.CCR-05-0464
114. Wen PY, Reardon DA, Armstrong TS, Phuphanich S, Aiken RD, Landolfi JC, et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine Ict-107 in Newly Diagnosed Patients With Glioblastoma. Clin Cancer Res (2019) 25(19):5799–807. doi: 10.1158/1078-0432.CCR-19-0261
115. Polyzoidis S, Ashkan K. Dcvax(R)-L–developed by Northwest Biotherapeutics. Hum Vaccin Immunother (2014) 10(11):3139–45. doi: 10.4161/hv.29276
116. Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, et al. Gene Expression Profile Correlates With T-cell Infiltration and Relative Survival in Glioblastoma Patients Vaccinated With Dendritic Cell Immunotherapy. Clin Cancer Res (2011) 17(6):1603–15. doi: 10.1158/1078-0432.CCR-10-2563
117. Dersh D, Holly J, Yewdell JW. A Few Good Peptides: MHC Class I-based Cancer Immunosurveillance and Immunoevasion. Nat Rev Immunol (2021) 21(2):116–28. doi: 10.1038/s41577-020-0390-6
118. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A Single Dose of Peripherally Infused EGFRvIII-directed Car T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients With Recurrent Glioblastoma. Sci Transl Med (2017) 9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984
119. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen Vaccine Generates Intratumoral T Cell Responses in Phase Ib Glioblastoma Trial. Nature. (2019) 565(7738):234–9. doi: 10.1038/s41586-018-0792-9
120. Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, et al. Actively Personalized Vaccination Trial for Newly Diagnosed Glioblastoma. Nature (2019) 565(7738):240–5. doi: 10.1038/s41586-018-0810-y
121. Rafiq S, Hackett CS, Brentjens RJ. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat Rev Clin Oncol (2020) 17(3):147–67. doi: 10.1038/s41571-019-0297-y
122. Martinez M, Moon EK. Car T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front Immunol (2019) 10:128. doi: 10.3389/fimmu.2019.00128
123. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an Old Dog New Tricks: Next-Generation CAR T Cells. Br J Cancer (2019) 120(1):26–37. doi: 10.1038/s41416-018-0325-1
124. Sahin A, Sanchez C, Bullain S, Waterman P, Weissleder R, Carter BS. Development of Third Generation anti-EGFRvIII Chimeric T Cells and EGFRvIII-expressing Artificial Antigen Presenting Cells for Adoptive Cell Therapy for Glioma. PloS One (2018) 13(7):e0199414. doi: 10.1371/journal.pone.0199414
125. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-Transduced T Cells Targeting EgfrvIII in Patients With Glioblastoma. J Immunother (2019) 42(4):126–35. doi: 10.1097/CJI.0000000000000260
126. Brown CE, Warden CD, Starr R, Deng X, Badie B, Yuan YC, et al. Glioma IL13Ralpha2 is Associated With Mesenchymal Signature Gene Expression and Poor Patient Prognosis. PloS One (2013) 8(10):e77769. doi: 10.1371/journal.pone.0077769
127. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor Cd8+ T Cells in Patients With Recurrent Glioblastoma. Clin Cancer Res (2015) 21(18):4062–72. doi: 10.1158/1078-0432.CCR-15-0428
128. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma After Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med (2016) 375(26):2561–9. doi: 10.1056/NEJMoa1610497
129. Genetically Modified T-Cells in Treating Patients With Recurrent or Refractory Malignant Glioma Nct02208362. Available at: https://clinicaltrials.gov/ct2/show/NCT02208362.
130. Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-Specific T Cells Target Primary Glioblastoma Stem Cells and Induce Regression of Autologous Experimental Tumors. Clin Cancer Res (2010) 16(2):474–85. doi: 10.1158/1078-0432.CCR-09-1322
131. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. Her2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol (2017) 3(8):1094–101. doi: 10.1001/jamaoncol.2017.0184
132. Pearson JRD, Cuzzubbo S, McArthur S, Durrant LG, Adhikaree J, Tinsley CJ, et al. Immune Escape in Glioblastoma Multiforme and the Adaptation of Immunotherapies for Treatment. Front Immunol (2020) 11:582106. doi: 10.3389/fimmu.2020.582106
133. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T Cells Targeting HER2 and IL13Ralpha2 Mitigate Tumor Antigen Escape. J Clin Invest (2016) 126(8):3036–52. doi: 10.1172/JCI83416
134. Il13ralpha2-Targeted Chimeric Antigen Receptor (Car) T Cells With or Without Nivolumab and Ipilimumab in Treating Patients With Recurrent or Refractory Glioblastoma Nct04003649. Available at: https://www.clinicaltrials.gov/ct2/show/NCT04003649.
135. Qiu H, Li Y, Cheng S, Li J, He C, Li JA. Prognostic Microenvironment-Related Immune Signature Via ESTIMATE (Promise Model) Predicts Overall Survival of Patients With Glioma. Front Oncol (2020) 10:580263. doi: 10.3389/fonc.2020.580263
136. Burger MC, Zhang C, Harter PN, Romanski A, Strassheimer F, Senft C, et al. Car-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors Into Precision Tools for Cancer Immunotherapy. Front Immunol (2019) 10:2683. doi: 10.3389/fimmu.2019.02683
137. Granzin M, Wagner J, Kohl U, Cerwenka A, Huppert V, Ullrich E. Shaping of Natural Killer Cell Antitumor Activity by Ex Vivo Cultivation. Front Immunol (2017) 8:458. doi: 10.3389/fimmu.2017.00458
138. Nowakowska P, Romanski A, Miller N, Odendahl M, Bonig H, Zhang C, et al. Clinical Grade Manufacturing of Genetically Modified, CAR-expressing Nk-92 Cells for the Treatment of ErbB2-positive Malignancies. Cancer Immunol Immunother (2018) 67(1):25–38. doi: 10.1007/s00262-017-2055-2
139. Zhang C, Burger MC, Jennewein L, Genssler S, Schonfeld K, Zeiner P, et al. Erbb2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J Natl Cancer Inst (2016) 108(5):djv375. doi: 10.1093/jnci/djv375
140. Albinger N, Hartmann J, Ullrich E. Current Status and Perspective of CAR-T and CAR-NK Cell Therapy Trials in Germany. Gene Ther (2021). doi: 10.1038/s41434-021-00246-w
141. Intracranial Injection of NK-92/5.28.z Cells in Patients With Recurrent HER2-Positive Glioblastoma (Car2brain) Nct03383978. Available at: https://clinicaltrials.gov/ct2/show/NCT03383978.
Keywords: glioblastoma, brain cancer, immunotherapy, tumor microenvironment, resistance to therapy
Citation: Yu MW and Quail DF (2021) Immunotherapy for Glioblastoma: Current Progress and Challenges. Front. Immunol. 12:676301. doi: 10.3389/fimmu.2021.676301
Received: 05 March 2021; Accepted: 21 April 2021;
Published: 13 May 2021.
Edited by:
Nicolas Jacquelot, University Health Network, CanadaReviewed by:
Alexander David Barrow, The University of Melbourne, AustraliaCopyright © 2021 Yu and Quail. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela F. Quail, ZGFuaWVsYS5xdWFpbEBtY2dpbGwuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.