 Xuewei Ding
Xuewei Ding Yi Ren
Yi Ren Xiaojie He
Xiaojie He- 1Institute of Pediatrics, The Second Xiangya Hospital, Central South University, Changsha, China
- 2Laboratory of Pediatric Nephrology, Institute of Pediatrics, The Second Xiangya Hospital, Central South University, Changsha, China
- 3Pediatric Internal Medicine Department, Haikou Maternal and Child Health Hospital, Haikou, China
Lupus nephritis (LN) is a common complication of systemic lupus erythematosus (SLE) and a major risk factor for morbidity and mortality. The abundant cell-free nucleic (DNA/RNA) in SLE patients, especially dsDNA, is a key substance in the pathogenesis of SLE and LN. The deposition of DNA/RNA-immune complexes (DNA/RNA-ICs) in the glomerulus causes a series of inflammatory reactions that lead to resident renal cell disturbance and eventually renal fibrosis. Cell-free DNA/RNA is the most effective inducer of type I interferons (IFN-I). Resident renal cells (rather than infiltrating immune cells) are the main source of IFN-I in the kidney. IFN-I in turn damages resident renal cells. Not only are resident renal cells victims, but also participants in this immunity war. However, the mechanism for generation of IFN-I in resident renal cells and the pathological mechanism of IFN-I promoting renal fibrosis have not been fully elucidated. This paper reviews the latest epidemiology of LN and its development process, discusses the mechanism for generation of IFN-I in resident renal cells and the role of IFN-I in the pathogenesis of LN, and may open a new perspective for the treatment of LN.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease in which immune complexes (ICs) form and deposit in many organs. The kidney is one of the main target organs. Lupus nephritis (LN) is present in at least 30% to 60% of SLE patients, and almost all patients have pathologic renal involvement. The incidence of SLE and LN varies widely between regions of the world and between ethnic groups (1). Although SLE is more prevalent in women than men across all age groups and populations, several studies have shown that men with lupus more get LN than women with lupus and patients with LN are younger, mostly of African, Asian, and Hispanic race/ethnicity (2–5). LN has a mortality rate six times higher than that of the general population (6). LN is a major risk factor for SLE mortality, with 10% of patients with LN developing the end-stage renal disease (ESRD) (1, 7). Compared with SLE patients without LN, LN patients had a higher standard mortality rate (6-6.8 versus 2.4) and earlier time of death (6, 8–10). In recent years, early diagnosis, standardized treatment, and new immunosuppressants such as mycophenolate mofetil, anti-CD20 monoclonal antibody, belimumab, and other drugs have significantly improved LN prognosis. However, the 5-year mortality rate in patients with severe refractory LN remains high (1, 3, 11, 12). Therefore, elucidating its pathogenesis can provide a theoretical basis for the screening of effective therapeutic targets for LN.
IFN-I is a central factor in the occurrence and development of SLE. Recent studies suggest that IFN-I may play a role at the level of terminal organs in SLE, especially LN. IFN-I is a response to the activation of most immune cells. At present, studies on the relationship between IFN-I and LN mainly focus on immune cells in serum and kidney. Resident renal cells also have immune functions and are involved in the immune war. Previous literature has shown that resident renal cells (rather than infiltrating immune cells) are the major source of IFN-I in the kidney and that IFN-I can cause renal injury. However, there are few studies on the production of IFN-I in the kidney and the damage of IFN-I to resident renal cells. This paper reviews the relationship between IFN-I and LN resident renal cells and explores the related pathways of IFN-I promoting the pathogenesis of LN.
Pathogenesis of LN
IC Deposition
Nucleic acid exposure, the production of nephrogenic pathogenic antibodies and the formation of ICs are the key links leading to LN. Three mechanisms have been proposed for ICs formation or deposition on the glomerulus, and they include (1) the deposition of preformed circulating immune complexes (CICs) in the kidney, (2) the formation of in-situ ICs in the glomerulus, and (3) binding of anti-dsDNA antibodies to cross-reactive antigens present either on the surface of resident renal cells or in the extracellular environment (13–17).
Circulating autoantigens and antibodies form CICs, which are deposited in the kidney. Due to improper clearance of necrotic, apoptotic cells and/or abnormal increase in cell death in SLE patients, undegraded nucleosomes (complexes of DNA and histone-containing pairs of histone peptides) are released into the bloodstream, increasing circulating autoantigens and subsequent antibodies, which form CICs. They evade recognition by the immune system and are deposited in the kidney.
ICs can also be formed in situ. Electron-dense structures (EDS) associated with glomerular basement membrane (GBM) and the mesangial matrix constitute the main target for in situ-bound antibodies in both murine and human lupus nephritis. Nucleosomes and chromatin fragments accumulate due to the loss of intrarenal and extrarenal deoxyribonuclease 1 (Dnase-1) activity. Then nucleosomes and chromatin fragments readily stimulate TLR9 in infiltrating macrophages and dendritic cells, triggering the secretion of local MMPs (18, 19). MMPs degrades the membrane barrier, allowing nucleosomes and chromatin fragments to bind to GBM (20, 21). Exposure to glomerular chromatin in situ induces anti-chromatin (anti-dsDNA and anti-nucleosome) antibodies to become nephrogenic and pathogenic, secondary to the formation of in-situ ICs (15).
In addition to binding to DNA fragments, they also bind to cross-reactive antigens on the surface of renal cells to activate cell proliferation, apoptosis, inflammation and fibrosis pathways (13, 14, 17). Anti-dsDNA antibodies bind to renal mesangial cells (RMCs) through cross-reacting with the cell surface annexin II (22), α-actinin (23, 24), and ribosomal P protein (25). Anti-dsDNA antibodies bind to glomerular endothelial cells (GECs) through cross-reacting with membrane proteins with M.W. of 30–35, 44, 68, 110, and 180 kDa (26). Anti-dsDNA antibodies bind to renal tubular epithelial cells (TECs) through cross-reacting with A and D SnRNP polypeptides (27). The polyreactivity of anti-dsDNA antibodies may be related to structural/conformational similarity or molecular simulation (28). Upon binding to the cell surface, anti-dsDNA antibodies migrate to the cytoplasm and/or nucleus, promoting cell growth and proliferation, or in turn inducing apoptosis (29). Recent studies have reported that the RG2 extract from intestinal symbiotic bacterium R. gnavus cross-reacts with anti-dsDNA antibodies to trigger or exacerbate the immune pathogenesis of LN (30, 31).
Depending on the type, duration, and severity of LN, ICs can be found in the subendothelial, subepithelial, mesangial, and tubulointerstitial regions (Figure 1). The distribution, quantity, and proinflammatory properties of ICs in renal parenchyma determine complement activation, inflammation, cell proliferation, and the severity of glomerular and tubulointerstitial injuries (3, 5, 32, 33).
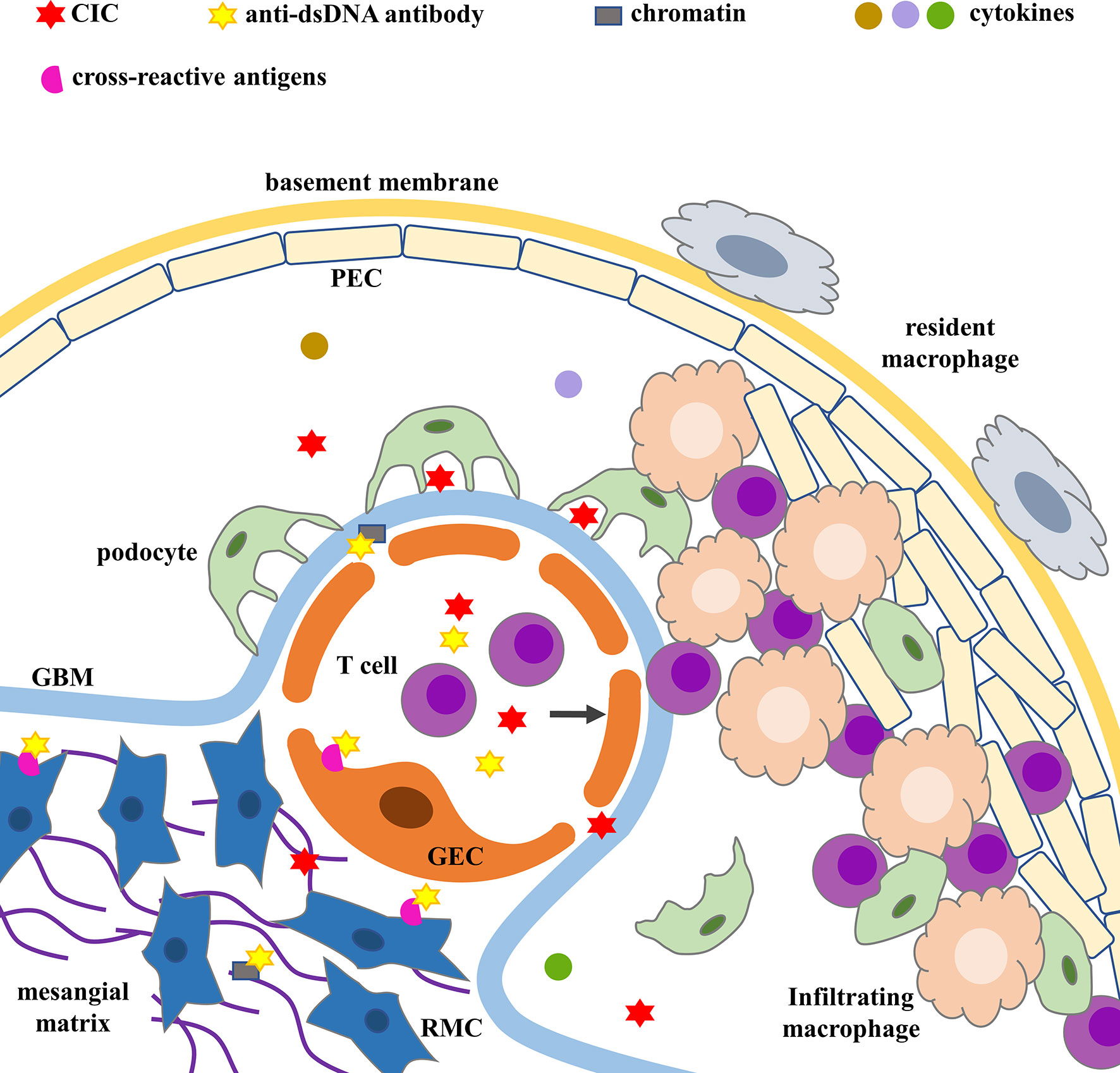
Figure 1 ICs deposition and glomerular injury. Cross-reactive antigens include annexin II, α-actinin, ribosomal P protein on RMCs’ surface and membrane proteins with M.W. of 30–35, 44, 68, 110, and 180 kDa on GECs’ surface. CIC, circulating immune complex; GBM, glomerular basement membrane; GEC, glomerular endothelial cell; PEC, parietal epithelial cell; RMC, renal mesangial cell.
Glomerulus Loss
ICs are mainly deposited in the glomerulus. The main mediator of IC-induced glomerular injury is the complement system, especially the formation of the C5b-9 membrane attack complex. C5b-9 is inserted into the glomerular membrane in extremely low amounts, transforming normal cells into inflammatory effector cells (34). Immunostimulatory glomerular cells produce large amounts of pro-inflammatory cytokines (35, 36), accelerating cell damage/aging, which may be one of the mechanisms of glomerular injury in LN (37).
The initial IC-mediated glomerular injury varies with the location of the IC deposition. IC subendothelial deposition leads to the accumulation of proinflammatory cells, causing proliferative disease and glomerular crescent (38). The GEC and the GEC surface layer (also known as the glycocalyx) are the first points of contact with the components of the circulating immune system. T cells are recruited to the glomerulus via the direct binding of their CD44 to the hyaluronic acid (HA) component of GEC glycocalyx (39). ICs alter cell morphology, up-regulate active caspase-3’ expression, inhibit angiogenesis, and increase NO production in GECs (40). Autophagy is a conserved metabolism that plays a protective role in many cell types and diseases. ICs inhibit the autophagy activity of GECs through Akt/mTOR-dependent pathway (41). LN antibodies promote increased secretion of endothelin-1 by GECs, leading to disruption of tight intercellular junctions (42). IC subepithelial deposition leads to podocytes damage and varying degrees of proteinuria. Podocyte injury is characterized by the foot process effacement (FPE), loss of podocyte-specific markers and cell detachment (43). Podocytes also contribute to glomerular crescent formation. Dedifferentiated podocytes migrate to cellular crescents. Podocyte injury ultimately leads to the activation and proliferation of parietal epithelial cells (PECs) through the JAK/STAT pathway, the production of HB-EGF and IL-6, and/or absence of (C-X-C motif) ligand (CXCL) 12, jointly contributing to glomerular crescent formation (43). LN IgG stimulates cellular cytoskeletal rearrangement and decreases vascular endothelial growth factor (VEGF) levels in podocytes (42). IC mesangial deposition leads to RMC proliferation and mesangial matrix increase. The inflammatory environment of LN induces RMCs to produce pro-inflammatory cytokines, which recruit leukocytes (44); promotes RMCs to express higher levels of matrix proteins and regulate matrix degradation enzymes, which lead to mesangial matrix deposition (44, 45); regulate the cell cycle and promote RMC proliferation (46).
Podocytes, GECs, and RMCs in the glomerulus interact with and support each other. Podocytes produce VEGF needed for GECs survival (47, 48); GECs produce platelet-derived growth factor (PDGF) needed for RMCs survival; RMCs isolate the potential transforming growth factor-β (TGF-β), thereby protecting GECs from apoptosis (49). Progressive injury to one cell type can eventually lead to damage of the other cell types. The activation, dedifferentiation, or proliferation of glomerular cells leads to the loss of structural integrity of the glomerular cluster and ultimately to glomerular death.
Tubulointerstitial Fibrosis
Renal tubulointerstitium blood supply is provided by glomerular runoff. Glomerular loss affects tubulointerstitial survival. Changes resulting from loss of tubular interstitial viability, such as tubular atrophy, fibrosis, and interstitial infiltration. Injury of renal tubular epithelial cells (TECs) is an important cause of renal fibrosis (50, 51). The severity and frequency of TECs injury determine whether this repair mechanism leads to recovery or progression to fibrosis (52). TECs performs the repair mechanism to restore normal function when the injury is minor or for a short time. TECs experience maladaptive repair when severe and persistent injury exceeds the normal repair mechanism. The maladaptive repair is manifested in two aspects: cell cycle arresting in the G2/M phase, which is characterized by the expression of p53, p21 and p16INK4a; aging-associated secretory phenotypes, which is characterized by the secretion of pro-inflammatory factors and pro-fibrosis factors, including TGF-β1, connective tissue growth factor (CTGF), CXCL1, IL-6, IL-8 (50, 53–56). These factors promote a chronic inflammatory microenvironment conducive to fibrous tissue (53). TECs secret pro-inflammatory cytokines to recruit and activate different inflammatory cells. And these recruited cells further produce cytokines that drive the transformation of TECs, fibroblasts, and pericytes to myofibroblast type (50, 57, 58). Eventually, TECs, fibroblasts, and pericytes express α-smooth muscle actin (α-SMA) and promoting the deposition of extracellular matrix (ECM), contributing to the final process of renal fibrosis.
Although ICs are predominantly detected in the glomerulus affecting glomerular and tubulointerstitial capacity, about 70% of LN patients also have ICs aggregates along the tubular basement membrane resulting in tubulointerstitial inflammation and fibrosis. A study of LN biopsy found that tubular ICs are independent of circulatory and glomerular ICs (59). Anti-dsDNA antibodies have been shown to bind A and D SnRNP in TECs, causing them to be internalized and transported to the cytoplasmic and nuclear subcellular compartments, or they can remain at the cell surface where interaction with complement results in cell lysis (27). The binding of anti-dsDNA antibodies to TECs induces phenotypic changes in TECs that may promote the epithelial-to-mesenchymal transition (EMT) (60). Another study has shown that anti-dsDNA antibodies induce TECs secretion of soluble fibronectin and increase downstream TGF-β1 and collagen synthesis by prior activation of ERK, p38 MAPK, JNK, PKC-α and PKC-βII (61).
Pericytes are potential sources of myofibroblasts (50, 57, 58). Loss of pericytes leads to thinning of capillaries. Capillary thinning induced anoxia in TECs, which increases interstitial oxidative stress. Injured or hypoxic TECs secrete hypoxia-inducing factor-1α (HIF-1α) and subsequent VEGF to promote endothelial cells (ECs) survival and proliferation, increasing perivascular capillary density (62, 63). However, excessive production of VEGF promotes the formation of leaky and nonfunctional vessels, thus resulting in a hypoxic and highly oxidative environment (64). Besides, VEGF can be used as a pro-inflammatory factor to aggravate fibrosis response (64). Hypoxia has been shown to promote EMT as an important microenvironmental factor (65–68). Increased matrix strength also aggravates tubular hypoxia and the progression of EMT. The above factors form a vicious circle.
Renal Microvascular Lesions
Renal microvascular lesions are common in LN and are increasingly being recognized as a marker of LN. Five pathological types of LN renal microvascular lesions have been proposed and they are vascular immune complex deposits (ICD), arteriosclerosis (AS), thrombotic microangiopathy (TMA), non-inflammatory necrotizing vasculopathy (NNV), and true renal vasculitis (TRV) (69). Up to one-third of LN patients have two or more vascular lesions at the same time. Although each lesion type may exhibit its unique factors, there are some common mechanisms among different vascular lesions. Damaged TECs induce loss of pericytes leading to thinning of capillaries (50, 57, 58, 62–64). The activation and dysfunction of vascular ECs, as well as immune system dysfunction, are key mechanisms of LN renal microvascular lesions, especially IC-induced vascular inflammation and anti-phospholipid antibody (APL)-related thrombotic events (69). The binding of autoantibodies to vascular ECs and the deposition of CICs on the microvessels lead to changes in the connections between ECs, thus activating complement, increasing the expression of adhesion molecules, inflammatory cytokines and chemokines, and increasing the permeability of ECs. The activation and dysfunction of ECs further recruit monocytes through adhesion molecules and chemokines, which induces platelet aggregation, resulting in procoagulant activity and microthrombosis (70, 71). APL-induced thrombotic events are the important mechanism of renal LN TMA (72). Patients with TMA had the worst renal outcomes (73). Renal microvascular lesions adversely affect long-term renal outcomes and may determine the selection of treatment strategies (73, 74) (Figure 2).
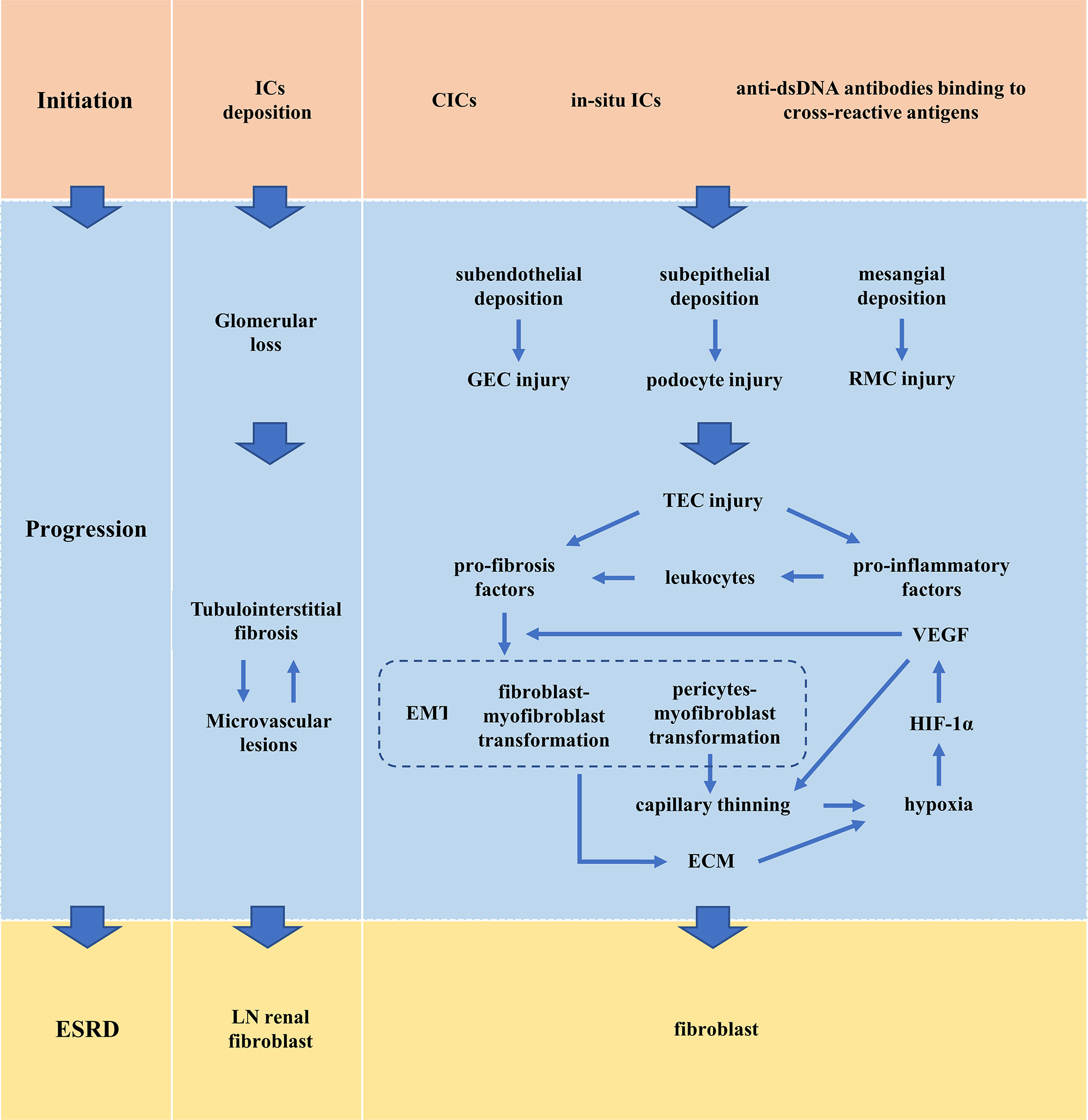
Figure 2 Pathogenesis of LN. CIC, circulating immune complex; ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition; ESRD, end-stage renal disease; GEC, glomerular endothelial cell; HIF-1α, hypoxia-inducing factor-1α; IC, immune complex; LN, lupus nephritis; RMC, renal mesangial cell; TEC, renal tubular epithelial cell; VEGF, vascular endothelial growth factor.
The Mechanisms for Generation of IFN-I in the Kidney
Clinical studies have found that LN patients overexpress IFN-I, and IFN-I activity is closely related to inflammation of LN (75–77). Experimental animal studies have shown that exposure to IFN-I in NZB/W mice or C57BL/6J mice accelerates glomerulonephritis, glomerular crescent, and renal tubular interstitial nephritis (78–80); reducing the biological activity of IFN-I in NZB/W mice alleviated renal pathology and improved survival rate (81). Although a study has shown that Toll-like receptor 7 (TLR7) -mediated LN is independent of IFN-I signaling, it is not enough to mask the ultimate role of IFN-I in nephritis acceleration (82). IFN-I includes IFN-α and IFN-β, which play a biological role by binding to type I interferon receptor (IFNAR).
The Mechanisms for Generation of IFN-I
Cell-free nucleic acid (DNA/RNA) is the most effective inducer of IFN-I. They are recognized by intracellular nucleic acid sensors, which activate the signaling pathway that produces IFN-I (Figure 3). DNA sensors include the endosome TLR9, DNA-dependent activator of IFN-regulatory factors (DAI), interferon-inducible protein 16 (IFI16), and cyclic GMP-AMP (cGAMP) synthase (cGAS). RNA sensors include TLR3, TLR7, TLR8, retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5). TLR7/8 binding with ssRNA and TLR9 binding with CpG DNA activates downstream signaling pathways—adaptor protein MyD88 and transcription factors such as IRAKs, TRAF6 and IRF7, then leading to secretion of IFN-α (83, 84).TLR3 binding with dsRNA induces IFN-β mainly through the TRIF-TBK1-IRF3 signaling pathway. cGAS (85, 86), DAI (87), IFI16 (88) recognize dsDNA and then activate the stimulator of interferon genes (STING)- TANK-binding kinase 1 (TBK1)-IRF3 signaling pathway to regulate transcription of IFN-β and IFN-induced genes. RIG-I and MDA5 recognize dsRNA and undergo conformational changes to induce mitochondrial antiviral signaling (MAVS), then activate IRF3/7 by TRAF6/3, resulting in the production of IFN-I (89).
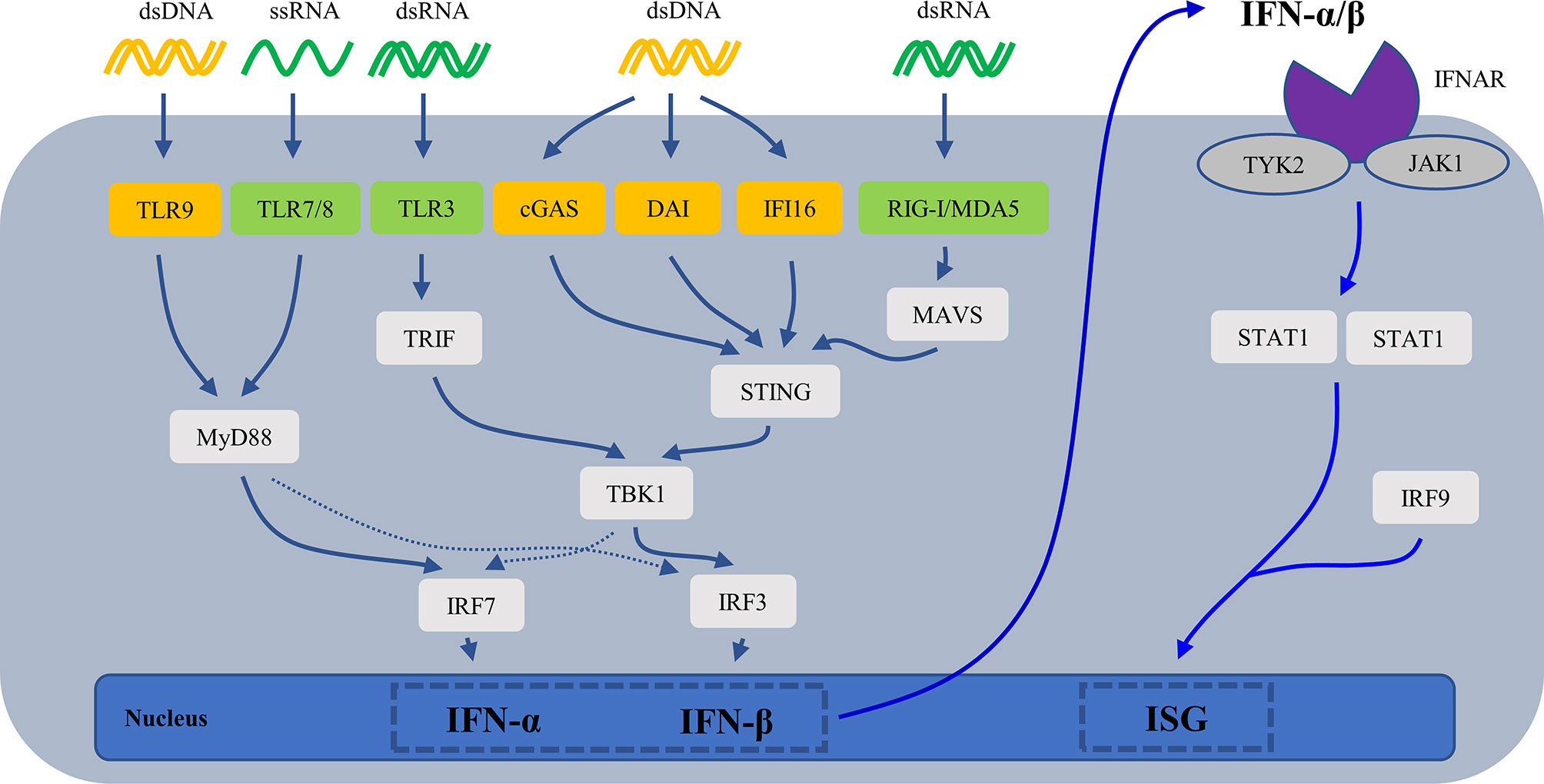
Figure 3 The mechanisms for generation of IFN-I. cGAS, cyclic GMP-AMP (cGAMP) synthase; DAI, DNA-dependent activator of IFN-regulatory factors; IFI16, interferon-inducible protein 16; IFN, interferons; IFNAR, type I interferon receptor; ISG, IFN-stimulated gene; MAVS, mitochondrial antiviral signaling; MDA5, melanoma differentiation-associated protein 5; RIG-I, retinoic acid-inducible gene I; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; TLR, Toll-like receptor.
SLE patients are rich in chromatin or cell-free nucleic acids, especially dsDNA, due to defective clearance of apoptotic cells and necrotic cells and increased neutrophil extracellular traps (NETs). These cell-free DNA/RNA acids activate above signaling pathways through intracellular DNA/RNA sensors to trigger the production of IFN-I (90). Studies have shown that there are several SLE-related susceptibility gene loci in the above signaling pathways, and their gene variants contribute to the production of IFN-I and the progression of LN (Table 1).
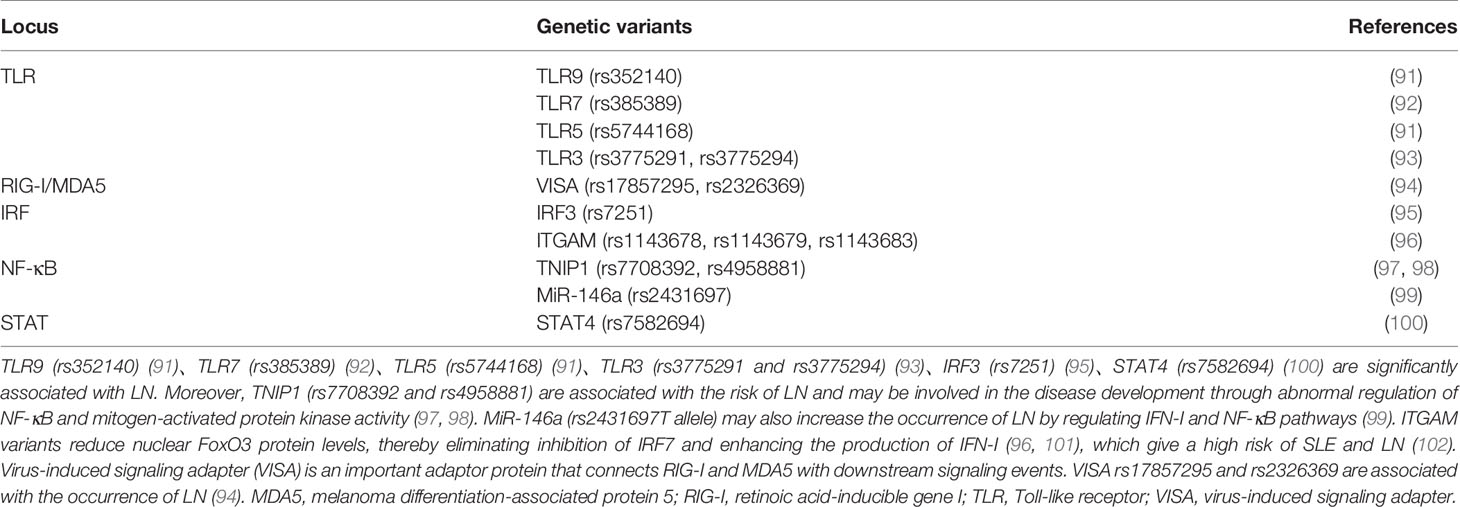
Table 1 Genetic variants in the DNA/RNA-IFN signaling pathway that contributes to the progression of LN.
The Main Producers of IFN-I in the Kidney
The IFN-I system in SLE is in a long-term activation state. All nucleated cell types can produce IFN-I during pathogenic infection. Under the background of SLE, immune cells are abnormally activated. For example, plasmacytoid dendritic cells (pDCs) massively produce IFN-α (103); neutrophils secrete IFN-I in the early stages of the disease (104). Early T1 B cells in SLE produce IFN-I, especially IFN-β (105). A previous study has shown that renal resident cells (rather than infiltrating immune cells) were the main source of IFN-I in the kidney (80). Besides the circulating cell-free nucleic acid and the nucleic acid component of CICs, renal immunostimulatory nucleic acids are an important source of pathogenic nucleic acid. Large chromatin fragments in the kidney are exposed due to selective down-regulation of Dnase1 activity in the kidney (16, 106, 107). Lupus nephrogenic autoantibodies enter renal cells, damaging cell structure, enhancing DNA cleavage, and inducing cell death (29, 108). Another potential source of renal immunostimulatory nucleic acids is NETs released by neutrophils in the glomerulus and renal tubule, which are not fully degraded and are made of DNA, histones, and neutrophil proteins (109–111). NETs activate the cGAS-STING pathway or the TLR9 pathway to produce IFN-I (111, 112). The IFN-I subtype secreted by renal resident cells and DNA/RNA receptors’ expression in renal resident cells varied (Table 2).
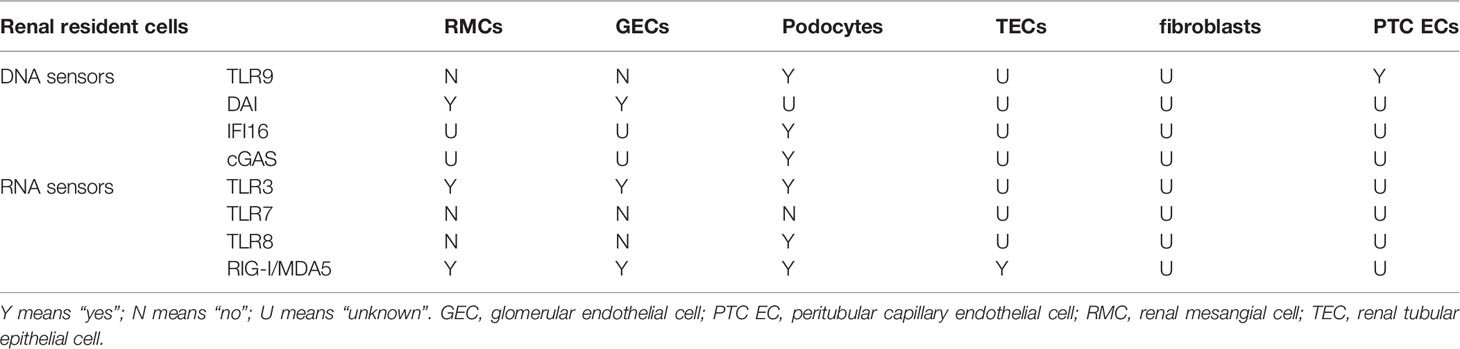
Table 2 Distribution and expression of DNA/RNA sensors in renal resident cells.
Podocyte
DNA/RNA-ICs induce IFN-β production in podocytes. Podocytes treated with TLR3 ligand—polyIC—expressed IFN-I. And podocytes express TLR1-6 and TLR9 (113). Masum MA et al. found that TLR9 is overexpressed in podocytes in mice with autoimmune glomerulonephritis (AGN), which is associated with glomerular podocyte injury (114). However, Machida H et al. found that TLR9 was expressed only in podocytes from active LN patients and disappeared during remission (115). cGAS and IFI16 are the main DNA sensors in podocytes and trigger the expression of IFN-β by activating the cGAS/IFI16-STING pathway, thereby promoting the progression of LN in SLE patients (116). Besides, Kimura J et al. analyzed BXSB/MPJ-YAa lupus model mice and found that the expression of TLR8 and its downstream cytokines was significantly increased in lupus mice, and TLR8 was localized in podocytes (117).
RMC
DNA/RNA-ICs induce IFN-β production in RMCs. In the context of SLE, RMCs express TLR1-4 and TLR6, especially highly express TLR3 (118).TLR3 belongs to the nucleic acid-specific TLR subgroup that activates the IFN-β production by recognizing dsRNA (119). However, RMCs do not express other members of the TLR subgroup—TLR7-9 (118, 119). Besides, LN patients’ RMCs show high levels of MDA5 expression (120). dsRNA induces RMCs to release IFN-α/β by MDA5 (rather than RIG-I); IFN-α/β can activate RMCs in an autocrine-paracrine loop (121). Although RMCs do not express TLR9 (118, 119), DNA-ICs also induce RMCs activation. Qing X et al. found that IgG anti-dsDNA antibodies up-regulate RMCs pro-inflammatory genes in MRL/LPR mice (122). Allam R et al. found that viral dsDNA stimulated RMCs to produce IFN-β and IFN-induced genes which are independent of DAI (123).
GEC
DNA/RNA-ICs induce IFN-β production in GECs. GECs express TLR1-6 (124). dsRNA activates TLR3 and induces GECs to express IFN-β (125, 126). Liu Q et al. found that dsRNA induced GECs expression of RIG-I and MDA5 through the TLR3/IFN-β signaling pathway (127). At the same time, dsRNA activates GECs through RIG-I to secrete IFN-α/β, while IFN-α/β cannot activate GECs in an autocrine-paracrine loop (128). GECs lacks a unique DNA-specific TLR—TLR9 (124). However, Hagele H et al. stimulated GECs with viral dsDNA and found that viral dsDNA entered GECs through endocytosis and then activated GECs to produce IFN-α/β in a TLR-independent manner (129). IFN-β can induce DAI expression and IRF3 phosphorylation, but IFN-β cannot activate GECs in an autocrine-paracrine loop (129).
Others
Resident renal cells also include TECs, renal interstitial fibroblasts, and peritubular capillary endothelial cells (PTC ECs). Castellano G et al. found that TECs were the main producer of IFN-α (130). Recent studies have found that TECs express RIG-I, an intracellular pattern-recognition receptor that participates in the production of IFN-β by recognizing RNA (131). It is unknown whether renal interstitial fibroblasts produce IFN-I and their intracellular DNA/RNA receptor expression. The TLR9 expression level was significantly increased in PTC ECs in lupus-prone AGN model mice and was associated with peritubular capillary and renal tubular interstitial injury (132).
The Damage Effects of IFN-I in LN
Renal resident cells are the main source of IFN-I in the kidney (80). Renal resident cell-induced IFN-I, in turn, promotes the inflammatory state of glomerular cells, leading to renal fibrosis, scarring and renal loss (80). The damage of IFN-I is manifested in three aspects: (1) IFN-I induces the production of nuclear antigen and autoantibodies, promoting the formation of ICs; (2) IFN-I recruits leukocytes to promote proliferative lesions; (3) IFN-I acts on resident renal cells, leading to cell activation, injury, apoptosis, and progression to renal fibrosis (Figure 4).
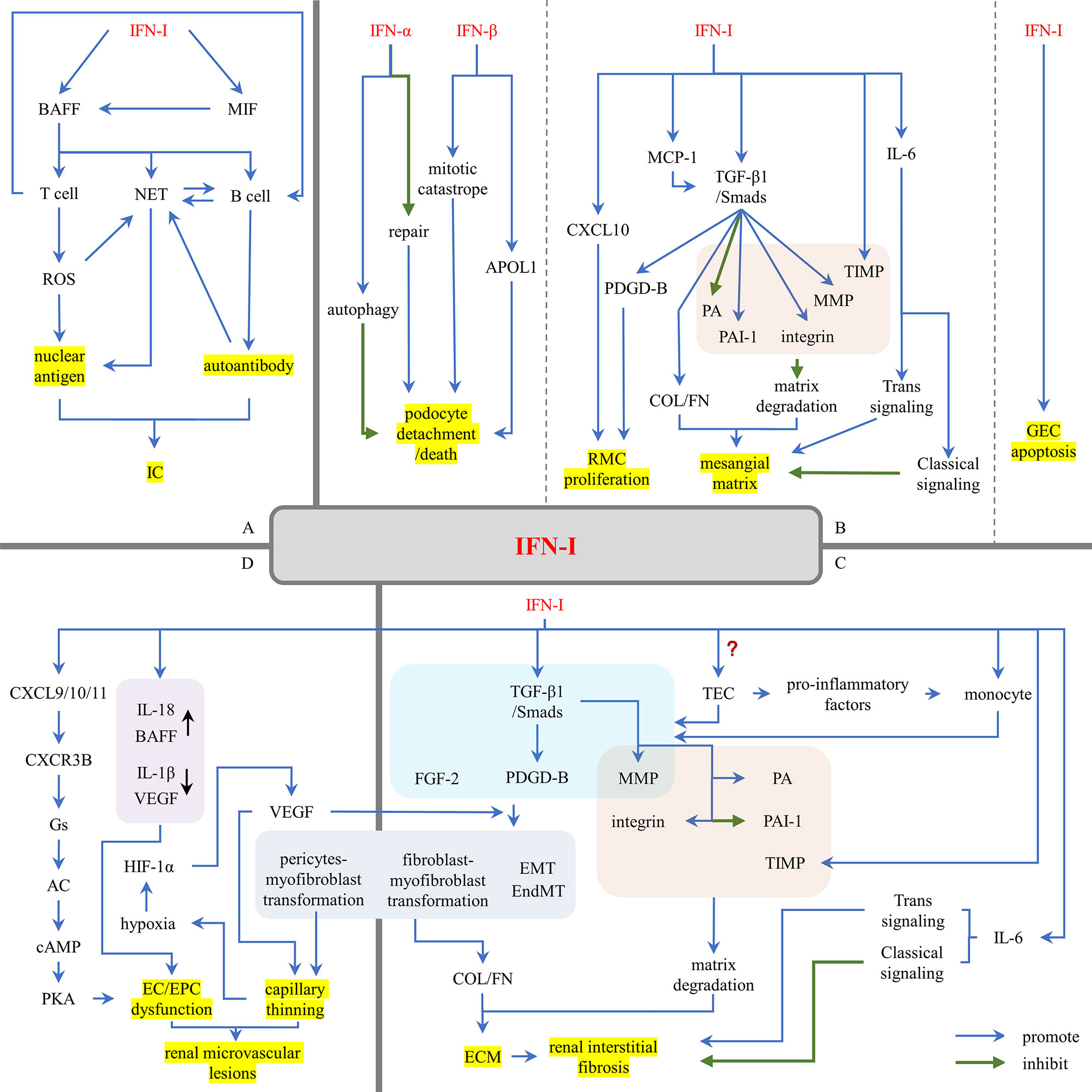
Figure 4 The damage effects of IFN-I in LN. (A) IFN-I promotes the formation of nuclear antigens and autoantibodies. (B) IFN-I promotes glomerular sclerosis. (C) IFN-I promotes renal interstitial fibrosis. (D) IFN-I promotes renal microvascular lesions. AC, adenylyl cyclase; APOL1, apolipoprotein L1; BAFF, B cell activating factor; cAMP, cyclic adenosine monophosphate; COL, collagen; CXCL, (C-X-C motif) ligand; CXCR3B, chemokine-receptor-3B; EC, endothelial cell; ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition; EndMT, endothelial-to-mesenchymal transition; EPC, endothelial progenitor cell; FGF-2, fibroblast growth factor 2; FN, fibronectin; GEC, glomerular endothelial cell; HIF-1α, hypoxia-inducing factor-1α; IC, immune complex; IFN-I, type I interferons; MCP-1, monocyte chemotaxis proteins 1; MIF, macrophage inhibitory factor; MMP, matrix metalloproteinases; NET, neutrophil extracellular trap; PA, plasminogen activator; PAI-1, plasminogen activator inhibitor 1; PDGD, PDGF, platelet-derived growth factor; PKA, protein kinase A; RMC, renal mesangial cell; ROS, reactive oxygen species; TEC, renal tubular epithelial cell; TIMP, tissue inhibitors of metalloproteinases; TGF-β1, transforming growth factor-β1.
IFN-I Promotes the Formation of Nuclear Antigens and Autoantibodies
IFN-I promotes the formation of nuclear antigens. IFN-I can induce B cell activating factor (BAFF) expression and mobilization (133, 134). BAFF promotes the activation of T cells (135) and the production of NETs (136). Overactivity of SLE T cells leads to mitochondrial hyperpolarization, which ultimately leads to increased production of reactive oxygen species (ROS) (137). ROS can modify cellular components and metabolites, giving them immunogenicity (138). ROS contributes to the formation of NETs (112). NETs trigger a concerted activation of TLR9 and B-cell receptor (BCR) leading to autoantibodies production in lupus (139). Follicular helper T cells (TFHs) (140, 141), CXCR5-CXCR3+PD1hiCD4+T helper cells (142), and peripheral helper T cells (TPHs) (143) promote B cells differentiation and antibody production in different ways.
IFN-I promotes the formation of autoantibodies. BAFF is a key factor in the maturation, survival and function of SLE pathogenic B cells (144, 145), which are responsible for the production of autoantibodies. IFN-I not only directly mobilized BAFF (133, 134) but also indirectly regulated the BAFF pathway by promoting the production of macrophage inhibitory factor (MIF) (146–148). BAFF also promotes the activation of B cells by IFN (149). Besides, SLE-related autoantibodies and ICs can induce the strong release of NETs (150), increasing nucleic acid exposure.
Then increased nuclear antigens and autoantibodies induced by IFN-I enhance the chance of IC formation and triggers LN.
IFN-I Promotes Leukocyte Infiltration
IFN-I strongly induces chemokine CXCL9/10/11, then recruiting leukocytes into the inflammatory site through the CXCR3A-Gi-PI3K-MAPK signaling pathway (151, 152). Several studies have found that renal IFN-I induced leukocytes into the kidney in LN patients. Increased leukocyte recruitment might be an operative mechanism that IFN-I drives immune-mediated nephritis (80). Triantafyllopoulou A et al. induced IFN-β overexpression in NZB/W mice using TLR3 ligand poly (I: C) and found that IFN-β induced macrophagic infiltration in renal tissue (78). Yoshikawa M et al. found that IFN-β down-regulates CXCR5 expression in B cells and IFN-γ upregulates CXCR3 expression in B cells, which induces B cell infiltration in renal tissue of LN patients (153). Besides, IFN-I regulates these immune cells. Kishimoto D et al. found that IFN-I inhibited the anti-inflammatory properties of M2-like macrophages in the glomerulus by up-regulating Bach1 and down-regulating ho-1 expression, thus promoting glomerular inflammation (154).
IFN-I Promotes Renal Tissue Injury
IFN-I Promotes Glomerular Sclerosis
Podocyte
Impair to podocyte structure is one of the early symptoms of glomerular injury and is a characteristic of LN (155–157). Podocytes are highly differentiated epithelial cells that are fixed on the basement membrane through the extension of the foot process and interact with the surrounding podocytes to form a slit diaphragm and eventually a filtration barrier. The slit diaphragm is a unique cellular connection formed by podocin-specific proteins such as nephrin and podocin, which interact with the actin cytoskeleton (158). The actin cytoskeleton is the main structure of podocytes. Disorders of actin cytoskeleton play a major role in FPE and mitotic catastrophe, leading to podocytes detachment and proteinuria (159–161).
Podocyte cells are induced to produce IFN-β, which in turn stimulates podocyte B7-1 expression and actin remodeling (162). IFN-β specifically promotes podocyte detachment or death by inducing mitotic catastrophe in podocytes. IFN-α prevents podocyte repair by causing cell-cycle arrest and inhibiting proliferation and migration of PECs. And both of the above IFNs suppress renal progenitor differentiation into mature podocytes, which conducive to focal scar formation but not to glomerular repair (163). dsDNA induces podocytes to secret IFN-β. IFN-β expression activates IFNAR. IFNAR-associated JAK1 and TYK2 kinases then phosphorylate STAT1, which promotes transcription of apolipoprotein L1 (APOL1). And activated STAT1 up-regulates IFI16, which triggers a positive feedback mechanism promoting APOL1’s expression (116). Overexpression of APOL1 in podocytes is highly toxic. The APOL1 allele G1 and G2 are risk factors for LN and end-stage renal disease associated with lupus nephritis (LN-ESRD) in African Americans (164, 165). The observed injury of glomerular podocytes in LN suggests that the increase of APOL1 risk variant in podocytes of SLE patients may promote the faster progression of LN and LN-ESRD (155–157). Recent studies have shown that IFN-α is also associated with damage to podocyte structure and function. IFN-α has a significant effect on the filtration barrier function of podocytes. At the same time, IFN-α attenuates the mTORC1 signal and induces podocyte autophagy. However, increased autophagy ameliorates IFN-α-induced podocyte injury (166). This seems to show a protective negative feedback regulation.
GEC
GECs are also the component of the glomerular filtration barrier. Previous studies have shown that IFN-I, especially IFN-α, mediated endothelial dysfunction and caused the EC apoptosis (167), which increases GECs permeability and results in a loss of glomerular filtration barrier function.
RMC
RMCs are the key factor in LN glomerular fibrosis in LN. They play an important role in homeostasis by maintaining glomerular structure, producing and maintaining mesangial matrix, regulating filtration surface area and phagocytizing apoptotic cells or ICs (49). In response to ICs deposition and cytokine-induced injury, RMCs promote glomerular fibrosis through hypertrophy and proliferation (168). PDGF-B is a proliferation/migration-inducing growth factor that induces RMC proliferation in glomerulonephritis (169). TGF-β1 activates the downstream Smads signaling pathway by autocrine/paracrine, inducing the production of PDGF-B. The IFN-β autocrine/paracrine loop activates Smad7 which inhibits Smad3/4 activation and prevents induction of PDGF-B (170). However, studies have shown that IFN-α/β stimulation increases TGF-β1 expression (44, 78), which may enhance the expression of PDGF-B and promote the proliferation of RMCs. Moreover, CXCL10 induced by IFN-I not only recruit leukocytes but also aggravates RMC proliferation by activating ERK signaling pathway (171).
In addition to overproliferation, RMCs are one of the major stromal generating cells, secreting mesangial matrix components, such as type I collagen (COL I), type III collagen (COL III), and fibronectin (FN). TGF-β1/Smads signaling pathway plays a major role in the excess extracellular matrix (ECM) (172, 173). First, the TGF-β1/Smad signaling pathway up-regulated matrix protein synthesis, including COL I and COL III. Second, the TGF-β1/Smad signaling pathway inhibits matrix degradation. The addition of TGF-β1 to normal glomerulus significantly reduced the activity of plasminogen activator (PA) and increased the synthesis of plasminogen activator inhibitor 1 (PAI-1) (174). TGF-β1 regulates the expression of MMP-9 (44); the main function of MMPs is to degrade ECM components, so it seems that TGF-β1 enhances matrix degradation. However, a large number of studies have shown that the levels of MMPs and tissue inhibitors of metalloproteinases (TIMPs) in serum, urine and glomerulus of LN patients are increased, accompanied by the deposition of mesangial matrix (78, 175–179). Overexpressed MMPs interact with TIMPs, changing matrix composition to promote mesangial matrix expansion (178). IFN-α/β induces high expression of MMP-9 and TIMP-1 in the kidney (78). Besides, TGF-β1 alters the expression of mesangial α1β1 and α5β1 integrins and their ligands (such as laminin, collagen, and FN), promoting matrix adhesion (180).
IFN-I can indirectly induce TGF-β1 expression in RMCs. In addition to CXCL10, IFN-I induced RMCs expression of monocyte chemotaxis proteins 1(MCP-1/CCL2) and IL6. Increased MCP-1 levels stimulate the formation of TGF-β1 in renal resident cells (181) and induce Col IV mRNA expression, collagen deposition, and FN expression (182). The role of IL-6 in renal fibrosis remains controversial. Previous studies have shown that IL-6 does not play an important role in the development of renal fibrosis (183). Recent studies have shown that overexpression of IL-6 and its receptor reduces the abundance of FN and Col IV in RMCs (184); IL-6 trans signal transduction may be involved in the occurrence and development of renal fibrosis (185). This is consistent with the theory that IL-6 signaling is mediated through two main pathways. The anti-inflammatory activity of IL-6 is mediated through classical signaling pathways, whereas the pro-inflammatory property is mediated through trans-signaling pathways (186). Moreover, IFN-I autocrine/paracrine loops largely induce RMCs death (121). On the whole, IFN-I shows a significant damaging effect on RMCs (187, 188).
IFN-I Promotes Renal Interstitial Fibrosis
Renal interstitial fibrosis is the result of the chronic inflammatory process. During chronic inflammation, different cell components and complex signaling networks interact to lead to the development of renal myofibroblasts, which lead to excessive accumulation of ECM, a major and common feature of different chronic kidney diseases. The possible origin of myofibroblasts from renal epithelial/endothelial cells, fibroblasts, or pericytes remains a subject of debate (189–195). LeBleu VS et al. showed that proliferative myofibroblasts account for 50%, derived from resident fibroblasts; non-proliferative myofibroblasts derive through differentiation from bone marrow (35%), the endothelial-to-mesenchymal transition (EndMT) program (10%) and the epithelial-to-mesenchymal transition (EMT) program (5%) (193). TGF-β1 still plays a central role in many fibrotic factors (196). First, TGF-β1 promotes the proliferation of fibroblasts. fibroblast growth factor 2 (FGF-2) is a powerful mitogen of fibroblasts, promoting the autocrine growth of fibroblasts (197). TGF-β1, PDGF-B and FGF-2 jointly promote the proliferation of fibroblasts (197–199). Second, TGF-β1 promotes the transformation of other cells into myofibroblasts. TGF-β1 induces the functional transformation of TECs and GECs into myofibroblasts, which are responsible for ECM deposition (193, 200–204). MMP-9 is involved in EndMT and EMT through Notch signaling up-regulation, and its activation is located downstream of TGF-β1 (205, 206). FGF-2 also plays an important role in EMT (207–209). TGF-β1 is also involved in fibroblast-myofibroblast transformation through TGFR1 phosphorylation and subsequent Smad2/3 pathway mediating α-SMA transcription and myofibroblast differentiation (210). TGF-β1 and PDGF transform fibroblasts into myofibroblasts (211, 212), which together with fibroblasts produce ECM (213, 214). In addition to the TGF-β1 signaling pathway, the PDGF signaling induces pericytes proliferation and differentiation into myofibroblasts (64, 215–218). Moreover, TGF-β1 regulates PA, PAI-1, MMP-9, and integrin to inhibit matrix degradation and promote ECM accumulation and interstitial fibrosis (44, 174, 178, 180).
TGF-β1 is mainly produced by TECs. Whether IFN-I induces TECs to secrete TGF-β1 remains to be seen. IFN-α induces barrier instability and apoptosis of TECs (219–221), which may activate TECs. In the recent single-cell RNA sequencing studies of renal biopsies from LN patients, the expression of IFN-I response genes in TECs from LN patients was significantly higher than those of healthy control subjects (222) and correlates with clinical scores and with the response to treatment (223). Activated TECs secrete a series of pro-inflammatory mediators and absorb more circulating monocytes into the renal tubulointerstitium; infiltrated monocytes become activated macrophages (224). IFN-I also recruited macrophages to infiltrate (78). Activated macrophages secrete PDGF, TGF-β1, MMP and TIMP, which are involved in the regulation of tissue fibrosis (224). Similarly, IFN-I can enhance the process of renal interstitial fibrosis through MCP-1/CCl2 and IL-6 (181, 184, 185).
IFN-I Promotes Renal Microvascular Lesions
The imbalance between vascular endothelial injury and repair is a key event in vascular lesions. IFN-I breaks this balance (225). Endothelial progenitor cells (EPCs) are the main repair mechanism. IFN-I induces CXCL9/10/11 expression. CXCL9/10/11 activates chemokine-receptor-3B (CXCR3B)-Gs-adenylyl cyclase (AC)-cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA) signaling pathways, directly promoting ECs and EPCs dysfunction (225). It also up-regulates the function of other pro-EPC dysfunction factors (IL-18 (226), BAFF (133, 134, 227)) and down-regulates the function of pro-angiogenic molecules (IL-1β and VEGF (167)), which indirectly leads to EPC dysfunction.
IFN-I promotes vascular imperfection by affecting pericytes. IFN-I regulates TGF-β1 and PDGF expression (44, 78, 170). TGF-β1 and PDGF signaling pathways induce pericytes to proliferate and differentiate into myofibroblasts (64, 215–218). Pericytes are attached to the surface of capillary wall and share a developmental origin with fibroblasts. Normal pericytes stabilize the walls of blood vessels and maintain vessel tranquility and integrity. Activated pericytes are shed from the vascular wall and transformed into myofibroblasts (195, 228–232). Loss of pericytes leads to the formation of fragile capillaries and unstable, pathological blood vessels, ultimately resulting in renal vascular thinning (233). The loss of capillaries around renal tubules is closely related to renal fibrosis.
Conclusion
Cell-free DNA/RNA accumulation is the initial step of lupus and LN. Cell-free DNA/RNA and the nucleic acid components of ICs trigger the DNA/RNA sensors in renal resident cells, thus activating the signaling pathway for IFN-I production. IFN-I in turn induces nucleic acid exposure and the formation of autoantibodies. IFN-I acts on renal resident cells and is involved in the whole process of renal injury, especially the activation of the TGF-β1/Smads signaling pathway. Also, IFN-I recruits leukocytes into renal tissues through the CXCL9/10/11-CXCR3A-Gi-PI3K-MAPK signaling pathway, enhancing renal fibrosis response. Moreover, IFN-I promotes renal microvascular lesions, further damaging renal function. IFN-I is found in almost every link of the pathogenesis of LN. Therefore, IFN-I plays an important role in the pathogenesis of LN. Targeting IFN-I systems in the kidney has potential therapeutic effects on the premature emergence of LN in SLE patients. It also suggests that the immune function of renal resident cells is greater than that of the renal immune cells in LN and renal resident cells are the dominant player and acceptor in the occurrence and development of LN. The study on renal resident cells will further deepen the understanding of LN and contribute to the targeted therapy of LN in the future.
Author Contributions
XD and YR did the literature search and drafted the article. XH gave insight. XH revised the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (No. 61562021 and No. 81560275, No. 81960885, No. 81260139, No. 81060073, No. 30560161), Hainan Major Science and Technology Projects (ZDKJ2039010), Hainan Association for academic excellence Youth Science and Technology Innovation Program (201515), Hainan special projects of Social Development (ZDYF2018103 and 2015SF39).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thanked Xiayang Chen for reviewing and revising this paper.
Glossary

References
1. Almaani S, Meara A, Rovin BH. Update on Lupus Nephritis. Clin J Am Soc Nephrol (2017) 12:825–35. doi: 10.2215/CJN.05780616
2. Hanly JG, O’Keeffe AG, Su L, Urowitz MB, Romero-Diaz J, Gordon C, et al. The Frequency and Outcome of Lupus Nephritis: Results From an Internationalinception Cohort Study. Rheumatol (Oxford) (2016) 55:252–62. doi: 10.1093/rheumatology/kev311
3. Parikh SV, Almaani S, Brodsky S, Rovin BH. Update on Lupus Nephritis: Core Curriculum 2020. Am J Kidney Dis (2020) 76:265–81. doi: 10.1053/j.ajkd.2019.10.017
4. Maningding E, Dall’Era M, Trupin L, Murphy LB, Yazdany J. Racial and Ethnic Differences in the Prevalence and Time to Onset of Manifestationsof Systemic Lupus Erythematosus: the California Lupus Surveillance Project. Arthritis Care Res (Hoboken) (2020) 72:622–9. doi: 10.1002/acr.23887
5. Pinheiro S, Dias RF, Fabiano R, Araujo SA, Silva A. Pediatric Lupus Nephritis. J Bras Nefrol (2019) 41:252–65. doi: 10.1590/2175-8239-jbn-2018-0097
6. Yap DY, Tang CS, Ma MK, Lam MF, Chan TM. Survival Analysis and Causes of Mortality in Patients With Lupus Nephritis. Nephrol Dial Transplant (2012) 27:3248–54. doi: 10.1093/ndt/gfs073
7. Tektonidou MG, Dasgupta A, Ward MM. Risk of End-Stage Renal Disease in Patients With Lupus Nephritis, 1971-2015: Asystematic Review and Bayesian Meta-Analysis. Arthritis Rheumatol (2016) 68:1432–41. doi: 10.1002/art.39594
8. Faurschou M, Dreyer L, Kamper AL, Starklint H, Jacobsen S. Long-Term Mortality and Renal Outcome in a Cohort of 100 Patients With Lupusnephritis. Arthritis Care Res (Hoboken) (2010) 62:873–80. doi: 10.1002/acr.20116
9. Lerang K, Gilboe IM, Steinar TD, Gran JT. Mortality and Years of Potential Life Loss in Systemic Lupus Erythematosus: Apopulation-Based Cohort Study. Lupus (2014) 23:1546–52. doi: 10.1177/0961203314551083
10. Bernatsky S, Boivin JF, Joseph L, Manzi S, Ginzler E, Gladman DD, et al. Mortality in Systemic Lupus Erythematosus. Arthritis Rheum (2006) 54:2550–7. doi: 10.1002/art.21955
11. Furie R, Rovin BH, Houssiau F, Malvar A, Teng Y, Contreras G, et al. Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis. N Engl J Med (2020) 383:1117–28. doi: 10.1056/NEJMoa2001180
12. Zhang H, Zhou M, Han X, Yang Y, Yu X. Mycophenolate Mofetil in the Treatment of Chinese Patients With Lupus Nephritis: APRISMA-Compliant Meta-Analysis. Med (Baltimore) (2020) 99:e21121. doi: 10.1097/MD.0000000000021121
13. Yung S, Chan TM. Autoantibodies and Resident Renal Cells in the Pathogenesis of Lupus Nephritis:Getting to Know the Unknown. Clin Dev Immunol (2012) 2012:139365. doi: 10.1155/2012/139365
14. Lech M, Anders HJ. The Pathogenesis of Lupus Nephritis. J Am Soc Nephrol (2013) 24:1357–66. doi: 10.1681/ASN.2013010026
15. Mortensen ES, Rekvig OP. Nephritogenic Potential of Anti-DNA Antibodies Against Necrotic Nucleosomes. J Am Soc Nephrol (2009) 20:696–704. doi: 10.1681/ASN.2008010112
16. Fismen S, Mortensen ES, Rekvig OP. Nuclease Deficiencies Promote End-Stage Lupus Nephritis But Not Nephritogenicautoimmunity in (NZB × NZW) F1 Mice. Immunol Cell Biol (2011) 89:90–9. doi: 10.1038/icb.2010.75
17. Yung S, Chan TM. Anti-Dsdna Antibodies and Resident Renal Cells - Their Putative Roles Inpathogenesis of Renal Lesions in Lupus Nephritis. Clin Immunol (2017) 185:40–50. doi: 10.1016/j.clim.2016.09.002
18. Lim EJ, Lee SH, Lee JG, Kim JR, Yun SS, Baek SH, et al. Toll-Like Receptor 9 Dependent Activation of MAPK and NF-Kb is Required for the Cpgodn-Induced Matrix Metalloproteinase-9 Expression. Exp Mol Med (2007) 39:239–45. doi: 10.1038/emm.2007.27
19. Merrell MA, Ilvesaro JM, Lehtonen N, Sorsa T, Gehrs B, Rosenthal E, et al. Toll-Like Receptor 9 Agonists Promote Cellular Invasion by Increasing Matrixmetalloproteinase Activity. Mol Cancer Res (2006) 4:437–47. doi: 10.1158/1541-7786.MCR-06-0007
20. Overall CM. Molecular Determinants of Metalloproteinase Substrate Specificity: Matrixmetalloproteinase Substrate Binding Domains, Modules, and Exosites. Mol Biotechnol (2002) 22:51–86. doi: 10.1385/MB:22:1:051
21. Overall CM, Butler GS. Protease Yoga: Extreme Flexibility of a Matrix Metalloproteinase. Structure (2007) 15:1159–61. doi: 10.1016/j.str.2007.10.001
22. Yung S, Cheung KF, Zhang Q, Chan TM. Anti-Dsdna Antibodies Bind to Mesangial Annexin II in Lupus Nephritis. J Am Soc Nephrol (2010) 21:1912–27. doi: 10.1681/ASN.2009080805
23. Zhao Z, Deocharan B, Scherer PE, Ozelius LJ, Putterman C. Differential Binding of Cross-Reactive Anti-DNA Antibodies to Mesangial Cells: Therole of Alpha-Actinin. J Immunol (2006) 176:7704–14. doi: 10.4049/jimmunol.176.12.7704
24. Deocharan B, Qing X, Lichauco J, Putterman C. Alpha-Actinin is a Cross-Reactive Renal Target for Pathogenic Anti-DNA Antibodies. J Immunol (2002) 168:3072–8. doi: 10.4049/jimmunol.168.6.3072
25. Sun KH, Liu WT, Tsai CY, Tang SJ, Han SH, Yu CL. Anti-Dsdna Antibodies Cross-React With Ribosomal P Proteins Expressed on the Surfaceof Glomerular Mesangial Cells to Exert a Cytostatic Effect. Immunology (1995) 85:262–9.
26. Chan TM, Cheng IK. Identification of Endothelial Cell Membrane Proteins That Bind Anti-DNA Antibodiesfrom Patients With Systemic Lupus Erythematosus by Direct or Indirect Mechanisms. J Autoimmun (1997) 10:433–9. doi: 10.1006/jaut.1997.9998
27. Koren E, Koscec M, Wolfson-Reichlin M, Ebling FM, Tsao B, Hahn BH, et al. Murine and Human Antibodies to Native DNA That Cross-React With the a and D Snrnppolypeptides Cause Direct Injury of Cultured Kidney Cells. J Immunol (1995) 154:4857–64.
28. Waldman M, Madaio MP. Pathogenic Autoantibodies in Lupus Nephritis. Lupus (2005) 14:19–24. doi: 10.1191/0961203305lu2054oa
29. Tang S, Lui SL, Lai KN. Pathogenesis of Lupus Nephritis: an Update. Nephrol (Carlton) (2005) 10:174–9. doi: 10.1111/j.1440-1797.2005.00392.x
30. Kim JW, Kwok SK, Choe JY, Park SH. Recent Advances in Our Understanding of the Link Between the Intestinal Microbiotaand Systemic Lupus Erythematosus. Int J Mol Sci (2019) 20:4871. doi: 10.3390/ijms20194871
31. Azzouz D, Omarbekova A, Heguy A, Schwudke D, Gisch N, Rovin BH, et al. Lupus Nephritis is Linked to Disease-Activity Associated Expansions and Immunity Toa Gut Commensal. Ann Rheum Dis (2019) 78:947–56. doi: 10.1136/annrheumdis-2018-214856
32. Imran TF, Yick F, Verma S, Estiverne C, Ogbonnaya-Odor C, Thiruvarudsothy S, et al. Lupus Nephritis: an Update. Clin Exp Nephrol (2016) 20:1–13. doi: 10.1007/s10157-015-1179-y
33. Maria NI, Davidson A. Protecting the Kidney in Systemic Lupus Erythematosus: From Diagnosis to Therapy. Nat Rev Rheumatol (2020) 16:255–67. doi: 10.1038/s41584-020-0401-9
34. Nangaku M, Couser WG. Mechanisms of Immune-Deposit Formation and the Mediation of Immune Renal Injury. Clin Exp Nephrol (2005) 9:183–91. doi: 10.1007/s10157-005-0357-8
35. Kwok SK, Tsokos GC. New Insights Into the Role of Renal Resident Cells in the Pathogenesis of Lupusnephritis. Korean J Intern Med (2018) 33:284–9. doi: 10.3904/kjim.2017.383
36. Dimou P, Wright RD, Budge KL, Midgley A, Satchell SC, Peak M, et al. The Human Glomerular Endothelial Cells are Potent Pro-Inflammatory Contributors Inan In Vitro Model of Lupus Nephritis. Sci Rep (2019) 9:8348. doi: 10.1038/s41598-019-44868-y
37. Yang C, Xue J, An N, Huang XJ, Wu ZH, Ye L, et al. Accelerated Glomerular Cell Senescence in Experimental Lupus Nephritis. Med Sci Monit (2018) 24:6882–91. doi: 10.12659/MSM.909353
38. Fujita E, Nagahama K, Shimizu A, Aoki M, Higo S, Yasuda F, et al. Glomerular Capillary and Endothelial Cell Injury is Associated With the Formation Ofnecrotizing and Crescentic Lesions in Crescentic Glomerulonephritis. J Nippon Med Sch (2015) 82:27–35. doi: 10.1272/jnms.82.27
39. Kadoya H, Yu N, Schiessl IM, Riquier-Brison A, Gyarmati G, Desposito D, et al. Essential Role and Therapeutic Targeting of the Glomerular Endothelial Glycocalyx Inlupus Nephritis. JCI Insight (2020) 5:131252. doi: 10.1172/jci.insight.131252
40. Wang L, Law H. Immune Complexes Impaired Glomerular Endothelial Cell Functions in Lupus Nephritis. Int J Mol Sci (2019) 20:5281. doi: 10.3390/ijms20215281
41. Wang L, Law H. Immune Complexes Suppressed Autophagy in Glomerular Endothelial Cells. Cell Immunol (2018) 328:1–8. doi: 10.1016/j.cellimm.2018.02.013
42. Yuan M, Tan Y, Wang Y, Wang SX, Yu F, Zhao MH. The Associations of Endothelial and Podocyte Injury in Proliferative Lupusnephritis: From Observational Analysis to In Vitro Study. Lupus (2019) 28:347–58. doi: 10.1177/0961203319828509
43. Sakhi H, Moktefi A, Bouachi K, Audard V, Hénique C, Remy P, et al. Podocyte Injury in Lupus Nephritis. J Clin Med (2019) 8:1340. doi: 10.3390/jcm8091340
44. Wright RD, Dimou P, Northey SJ, Beresford MW. Mesangial Cells are Key Contributors to the Fibrotic Damage Seen in the Lupusnephritis Glomerulus. J Inflammation (Lond) (2019) 16:22. doi: 10.1186/s12950-019-0227-x
45. Feng X, Yang R, Tian Y, Miao X, Guo H, Gao F, et al. HMGB1 Protein Promotes Glomerular Mesangial Matrix Deposition Via TLR2 in Lupusnephritis. J Cell Physiol (2020) 235:5111–9. doi: 10.1002/jcp.29379
46. Zhang CX, Chen J, Cai L, Wu J, Wang JY, Cao LF, et al. DNA Induction of MDM2 Promotes Proliferation of Human Renal Mesangial Cells Andalters Peripheral B Cells Subsets in Pediatric Systemic Lupus Erythematosus. Mol Immunol (2018) 94:166–75. doi: 10.1016/j.molimm.2018.01.003
47. Jourde-Chiche N, Fakhouri F, Dou L, Bellien J, Burtey S, Frimat M, et al. Endothelium Structure and Function in Kidney Health and Disease. Nat Rev Nephrol (2019) 15:87–108. doi: 10.1038/s41581-018-0098-z
48. Long DA, Norman JT, Fine LG. Restoring the Renal Microvasculature to Treat Chronic Kidney Disease. Nat Rev Nephrol (2012) 8:244–50. doi: 10.1038/nrneph.2011.219
49. Abboud HE. Mesangial Cell Biology. Exp Cell Res (2012) 318:979–85. doi: 10.1016/j.yexcr.2012.02.025
50. Liu BC, Tang TT, Lv LL, Lan HY. Renal Tubule Injury: a Driving Force Toward Chronic Kidney Disease. Kidney Int (2018) 93:568–79. doi: 10.1016/j.kint.2017.09.033
51. Qi R, Yang C. Renal Tubular Epithelial Cells: the Neglected Mediator of Tubulointerstitialfibrosis After Injury. Cell Death Dis (2018) 9:1126. doi: 10.1038/s41419-018-1157-x
52. Takaori K, Nakamura J, Yamamoto S, Nakata H, Sato Y, Takase M, et al. Severity and Frequency of Proximal Tubule Injury Determines Renal Prognosis. J Am Soc Nephrol (2016) 27:2393–406. doi: 10.1681/ASN.2015060647
53. Ferenbach DA, Bonventre JV. Mechanisms of Maladaptive Repair After AKI Leading to Accelerated Kidney Ageing Andckd. Nat Rev Nephrol (2015) 11:264–76. doi: 10.1038/nrneph.2015.3
54. Bonventre JV. Primary Proximal Tubule Injury Leads to Epithelial Cell Cycle Arrest, Fibrosis,Vascular Rarefaction, and Glomerulosclerosis. Kidney Int Suppl (2011) (2014) 4:39–44. doi: 10.1038/kisup.2014.8
55. Yang L, Humphreys BD, Bonventre JV. Pathophysiology of Acute Kidney Injury to Chronic Kidney Disease: Maladaptiverepair. Contrib Nephrol (2011) 174:149–55. doi: 10.1159/000329385
56. Andrade L, Rodrigues CE, Gomes SA, Noronha IL. Acute Kidney Injury as a Condition of Renal Senescence. Cell Transplant (2018) 27:739–53. doi: 10.1177/0963689717743512
57. Gewin LS. Renal Fibrosis: Primacy of the Proximal Tubule. Matrix Biol (2018) 68-69:248–62. doi: 10.1016/j.matbio.2018.02.006
58. Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, et al. Epithelial-to-Mesenchymal Transition Induces Cell Cycle Arrest and Parenchymaldamage in Renal Fibrosis. Nat Med (2015) 21:998–1009. doi: 10.1038/nm.3902
59. Satoskar AA, Brodsky SV, Nadasdy G, Bott C, Rovin B, Hebert L, et al. Discrepancies in Glomerular and Tubulointerstitial/Vascular Immune Complex Iggsubclasses in Lupus Nephritis. Lupus (2011) 20:1396–403. doi: 10.1177/0961203311416533
60. Yung S, Tsang RC, Sun Y, Leung JK, Chan TM. Effect of Human Anti-DNA Antibodies on Proximal Renal Tubular Epithelial Cellcytokine Expression: Implications on Tubulointerstitial Inflammation in Lupusnephritis. J Am Soc Nephrol (2005) 16:3281–94. doi: 10.1681/ASN.2004110917
61. Yung S, Ng CY, Ho SK, Cheung KF, Chan KW, Zhang Q, et al. Anti-Dsdna Antibody Induces Soluble Fibronectin Secretion by Proximal Renal Tubularepithelial Cells and Downstream Increase of TGF-β1 and Collagen Synthesis. J Autoimmun (2015) 58:111–22. doi: 10.1016/j.jaut.2015.01.008
62. Rodríguez-Romo R, Benítez K, Barrera-Chimal J, Pérez-Villalva R, Gómez A, Aguilar-León D, et al. AT1 Receptor Antagonism Before Ischemia Prevents the Transition of Acute Kidneyinjury to Chronic Kidney Disease. Kidney Int (2016) 89:363–73. doi: 10.1038/ki.2015.320
63. Chen Y, Jiang S, Zou J, Zhong Y, Ding X. Silencing HIF-1α Aggravates Growth Inhibition and Necrosis of Proximal Renal Tubularepithelial Cell Under Hypoxia. Ren Fail (2016) 38:1726–34. doi: 10.1080/0886022X.2016.1229994
64. Gewin L, Zent R, Pozzi A. Progression of Chronic Kidney Disease: Too Much Cellular Talk Causes Damage. Kidney Int (2017) 91:552–60. doi: 10.1016/j.kint.2016.08.025
65. Zhang L, Liu L, Bai M, Liu M, Wei L, Yang Z, et al. Hypoxia-Induced HE4 in Tubular Epithelial Cells Promotes Extracellular Matrixaccumulation and Renal Fibrosis Via NF-κb. FASEB J (2020) 34:2554–67. doi: 10.1096/fj.201901950R
66. Liu M, Liu L, Bai M, Zhang L, Ma F, Yang X, et al. Hypoxia-Induced Activation of Twist/Mir-214/E-Cadherin Axis Promotes Renal Tubularepithelial Cell Mesenchymal Transition and Renal Fibrosis. Biochem Biophys Res Commun (2018) 495:2324–30. doi: 10.1016/j.bbrc.2017.12.130
67. Du R, Xia L, Ning X, Liu L, Sun W, Huang C, et al. Hypoxia-Induced Bmi1 Promotes Renal Tubular Epithelial Cell-Mesenchymal Transitionand Renal Fibrosis Via Pi3k/Akt Signal. Mol Biol Cell (2014) 25:2650–9. doi: 10.1091/mbc.e14-01-0044
68. Liu T, Liu L, Liu M, Du R, Dang Y, Bai M, et al. Microrna-493 Targets STMN-1 and Promotes Hypoxia-Induced Epithelial Cell Cyclearrest in G(2)/M and Renal Fibrosis. FASEB J (2019) 33:1565–77. doi: 10.1096/fj.201701355RR
69. Ding Y, Tan Y, Qu Z, Yu F. Renal Microvascular Lesions in Lupus Nephritis. Ren Fail (2020) 42:19–29. doi: 10.1080/0886022X.2019.1702057
70. Prechl J, Czirják L. The Endothelial Deprotection Hypothesis for Lupus Pathogenesis: the Dual Role of C1qas a Mediator of Clearance and Regulator of Endothelial Permeability. F1000res (2015) 4:24. doi: 10.12688/f1000research.6075.1
71. Atehortúa L, Rojas M, Vásquez GM, Castaño D. Endothelial Alterations in Systemic Lupus Erythematosus and Rheumatoid Arthritis:Potential Effect of Monocyte Interaction. Mediators Inflammation (2017) 2017:9680729. doi: 10.1155/2017/9680729
72. Petri M. Epidemiology of the Antiphospholipid Antibody Syndrome. J Autoimmun (2000) 15:145–51. doi: 10.1006/jaut.2000.0409
73. Wu LH, Yu F, Tan Y, Qu Z, Chen MH, Wang SX, et al. Inclusion of Renal Vascular Lesions in the 2003 ISN/RPS System for Classifying Lupusnephritis Improves Renal Outcome Predictions. Kidney Int (2013) 83:715–23. doi: 10.1038/ki.2012.409
74. Barber C, Herzenberg A, Aghdassi E, Su J, Lou W, Qian G, et al. Evaluation of Clinical Outcomes and Renal Vascular Pathology Among Patients Withlupus. Clin J Am Soc Nephrol (2012) 7:757–64. doi: 10.2215/CJN.02870311
75. Dall’Era MC, Cardarelli PM, Preston BT, Witte A, Davis JJ. Type I Interferon Correlates With Serological and Clinical Manifestations of SLE. Ann Rheum Dis (2005) 64:1692–7. doi: 10.1136/ard.2004.033753
76. Oke V, Gunnarsson I, Dorschner J, Eketjäll S, Zickert A, Niewold TB, et al. High Levels of Circulating Interferons Type I, Type II and Type III Associate Withdistinct Clinical Features of Active Systemic Lupus Erythematosus. Arthritis Res Ther (2019) 21:107. doi: 10.1186/s13075-019-1878-y
77. Newling M, Fiechter RH, Sritharan L, Hoepel W, van Burgsteden JA, Hak AE, et al. Dysregulated Fcγ Receptor Iia-Induced Cytokine Production in Dendritic Cells Oflupus Nephritis Patients. Clin Exp Immunol (2020) 199:39–49. doi: 10.1111/cei.13371
78. Triantafyllopoulou A, Franzke CW, Seshan SV, Perino G, Kalliolias GD, Ramanujam M, et al. Proliferative Lesions and Metalloproteinase Activity in Murine Lupus Nephritismediated by Type I Interferons and Macrophages. Proc Natl Acad Sci USA (2010) 107:3012–7. doi: 10.1073/pnas.0914902107
79. Fairhurst AM, Mathian A, Connolly JE, Wang A, Gray HF, George TA, et al. Systemic IFN-Alpha Drives Kidney Nephritis in B6.Sle123 Mice. Eur J Immunol (2008) 38:1948–60. doi: 10.1002/eji.200837925
80. Fairhurst AM, Xie C, Fu Y, Wang A, Boudreaux C, Zhou XJ, et al. Type I Interferons Produced by Resident Renal Cells may Promote End-Organ Disease Inautoantibody-Mediated Glomerulonephritis. J Immunol (2009) 183:6831–8. doi: 10.4049/jimmunol.0900742
81. Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, et al. Type-I Interferon Receptor Deficiency Reduces Lupus-Like Disease in NZB Mice. J Exp Med (2003) 197:777–88. doi: 10.1084/jem.20021996
82. Wolf SJ, Theros J, Reed TJ, Liu J, Grigorova IL, Martínez-Colón G, et al. TLR7-Mediated Lupus Nephritis is Independent of Type I IFN Signaling. J Immunol (2018) 201:393–405. doi: 10.4049/jimmunol.1701588
83. Barber GN. Cytoplasmic Dna Innate Immune Pathways. Immunol Rev (2011) 243:99–108. doi: 10.1111/j.1600-065X.2011.01051.x
84. Laska MJ, Troldborg A, Hansen B, Stengaard-Pedersen K, Junker P, Nexø BA, et al. Polymorphisms Within Toll-Like Receptors are Associated With Systemic Lupuserythematosus in a Cohort of Danish Females. Rheumatol (Oxford) (2014) 53:48–55. doi: 10.1093/rheumatology/ket316
85. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP Synthase is a Cytosolic DNA Sensor That Activates the Type Iinterferon Pathway. Science (2013) 339:786–91. doi: 10.1126/science.1232458
86. Kato K, Omura H, Ishitani R, Nureki O. Cyclic GMP-AMP as an Endogenous Second Messenger in Innate Immune Signaling Bycytosolic Dna. Annu Rev Biochem (2017) 86:541–66. doi: 10.1146/annurev-biochem-061516-044813
87. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a Cytosolic DNA Sensor and an Activator of Innate Immuneresponse. Nature (2007) 448:501–5. doi: 10.1038/nature06013
88. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an Innate Immune Sensor for Intracellular DNA. Nat Immunol (2010) 11:997–1004. doi: 10.1038/ni.1932
89. West AP, Shadel GS, Ghosh S. Mitochondria in Innate Immune Responses. Nat Rev Immunol (2011) 11:389–402. doi: 10.1038/nri2975
90. Bai Y, Tong Y, Liu Y, Hu H. Self-Dsdna in the Pathogenesis of Systemic Lupus Erythematosus. Clin Exp Immunol (2018) 191:1–10. doi: 10.1111/cei.13041
91. Elloumi N, Fakhfakh R, Abida O, Ayadi L, Marzouk S, Hachicha H, et al. Relevant Genetic Polymorphisms and Kidney Expression of Toll-Like Receptor (TLR)-5and TLR-9 in Lupus Nephritis. Clin Exp Immunol (2017) 190:328–39. doi: 10.1111/cei.13022
92. Raafat II, El GN, Shahin R, Samy LA, El RR. Toll-Like Receptor 7 Gene Single Nucleotide Polymorphisms and the Risk for Systemiclupus Erythematosus: a Case-Control Study. Z Rheumatol (2018) 77:416–20. doi: 10.1007/s00393-017-0283-7
93. Elloumi N, Fakhfakh R, Abida O, Hachicha H, Marzouk S, Fourati M, et al. RNA Receptors, TLR3 and TLR7, are Potentially Associated With SLE Clinical Features. Int J Immunogenet (2021). doi: 10.1111/iji.12531
94. Liu X, Jiao Y, Wen X, Wang L, Ma C, Gao X, et al. Possible Association of VISA Gene Polymorphisms With Susceptibility to Systemiclupus Erythematosus in Chinese Population. Mol Biol Rep (2011) 38:4583–8. doi: 10.1007/s11033-010-0590-4
95. Zhang F, Wang YF, Zhang Y, Lin Z, Cao Y, Zhang H, et al. Independent Replication on Genome-Wide Association Study Signals Identifies IRF3 Asa Novel Locus for Systemic Lupus Erythematosus. Front Genet (2020) 11:600. doi: 10.3389/fgene.2020.00600
96. Faridi MH, Khan SQ, Zhao W, Lee HW, Altintas MM, Zhang K, et al. CD11b Activation Suppresses TLR-Dependent Inflammation and Autoimmunity in Systemiclupus Erythematosus. J Clin Invest (2017) 127:1271–83. doi: 10.1172/JCI88442
97. Caster DJ, Korte EA, Nanda SK, McLeish KR, Oliver RK, G’Sell RT, et al. ABIN1 Dysfunction as a Genetic Basis for Lupus Nephritis. J Am Soc Nephrol (2013) 24:1743–54. doi: 10.1681/ASN.2013020148
98. Rizk MM, Elsayed ET, ElKeraie AF, Ramzy I. Association of Tumor Necrosis Factor Alpha-Induced Protein 3 Interacting Protein 1(TNIP1) Gene Polymorphism (Rs7708392) With Lupus Nephritis in Egyptian Patients. Biochem Genet (2018) 56:478–88. doi: 10.1007/s10528-018-9855-8
99. Fouda ME, Nour EDD, Mahgoub MY, Elashkar AE, Abdel HW. Genetic Variants of Microrna-146a Gene: an Indicator of Systemic Lupus Erythematosussusceptibility, Lupus Nephritis, and Disease Activity. Mol Biol Rep (2020) 47:7459–66. doi: 10.1007/s11033-020-05802-y
100. Nghiem TD, Do GT, Luong LH, Nguyen QL, Dang HV, Viet AN, et al. Association of the STAT4, CDKN1A, and IRF5 Variants With Risk of Lupus Nephritis Andrenal Biopsy Classification in Patients in Vietnam. Mol Genet Genomic Med (2021), e1648. doi: 10.1002/mgg3.1648
101. Litvak V, Ratushny AV, Lampano AE, Schmitz F, Huang AC, Raman A, et al. A FOXO3-IRF7 Gene Regulatory Circuit Limits Inflammatory Sequelae of Antiviralresponses. Nature (2012) 490:421–5. doi: 10.1038/nature11428
102. Khan SQ, Khan I, Gupta V. CD11b Activity Modulates Pathogenesis of Lupus Nephritis. Front Med (Lausanne) (2018) 5:52. doi: 10.3389/fmed.2018.00052
103. Swiecki M, Colonna M. The Multifaceted Biology of Plasmacytoid Dendritic Cells. Nat Rev Immunol (2015) 15:471–85. doi: 10.1038/nri3865
104. Lindau D, Mussard J, Rabsteyn A, Ribon M, Kotter I, Igney A, et al. TLR9 Independent Interferon Alpha Production by Neutrophils on Netosis in Response to Circulating Chromatin, a Key Lupus Autoantigen. Ann Rheum Dis (2014) 73:2199–207. doi: 10.1136/annrheumdis-2012-203041
105. Hamilton JA, Wu Q, Yang P, Luo B, Liu S, Hong H, et al. Cutting Edge: Endogenous IFN-Beta Regulates Survival and Development of Transitional B Cells. J Immunol (2017) 199:2618–23. doi: 10.4049/jimmunol.1700888
106. Seredkina N, Zykova SN, Rekvig OP. Progression of Murine Lupus Nephritis is Linked to Acquired Renal Dnase1 Deficiencyand Not to Up-Regulated Apoptosis. Am J Pathol (2009) 175:97–106. doi: 10.2353/ajpath.2009.080943
107. Fenton K, Fismen S, Hedberg A, Seredkina N, Fenton C, Mortensen ES, et al. Anti-Dsdna Antibodies Promote Initiation, and Acquired Loss of Renal Dnase1 Promotesprogression of Lupus Nephritis in Autoimmune (Nzbxnzw)F1 Mice. PloS One (2009) 4:e8474. doi: 10.1371/journal.pone.0008474
108. Bhargava R, Tsokos GC. The Immune Podocyte. Curr Opin Rheumatol (2019) 31:167–74. doi: 10.1097/BOR.0000000000000578
109. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of Neutrophil Extracellular Trap Degradation is Associated With Lupusnephritis. Proc Natl Acad Sci USA (2010) 107:9813–8. doi: 10.1073/pnas.0909927107
110. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Exposeimmunostimulatory Molecules in Systemic Lupus Erythematosus. J Immunol (2011) 187:538–52. doi: 10.4049/jimmunol.1100450
111. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting Neutrophils are Major Inducers of Type I IFN Production in Pediatric Systemic Lupus Erythematosus. Sci Transl Med (2011) 3:20r–73r. doi: 10.1126/scitranslmed.3001201
112. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil Extracellular Traps Enriched in Oxidized Mitochondrial DNA are Interferogenic and Contribute to Lupus-Like Disease. Nat Med (2016) 22:146–53. doi: 10.1038/nm.4027
113. Shimada M, Ishimoto T, Lee PY, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, et al. Toll-Like Receptor 3 Ligands Induce CD80 Expression in Human Podocytes Via Annf-κb-Dependent Pathway. Nephrol Dial Transplant (2012) 27:81–9. doi: 10.1093/ndt/gfr271
114. Masum MA, Ichii O, Hosny AEY, Nakamura T, Otani Y, Hosotani M, et al. Overexpression of Toll-Like Receptor 9 Correlates With Podocyte Injury in a Murinemodel of Autoimmune Membranoproliferative Glomerulonephritis. Autoimmunity (2018) 51:386–98. doi: 10.1080/08916934.2018.1549234
115. Machida H, Ito S, Hirose T, Takeshita F, Oshiro H, Nakamura T, et al. Expression of Toll-Like Receptor 9 in Renal Podocytes in Childhood-Onset Active Andinactive Lupus Nephritis. Nephrol Dial Transplant (2010) 25:2530–7. doi: 10.1093/ndt/gfq058
116. Davis SE, Khatua AK, Popik W. Nucleosomal Dsdna Stimulates APOL1 Expression in Human Cultured Podocytes Byactivating the CGAS/IFI16-STING Signaling Pathway. Sci Rep (2019) 9:15485. doi: 10.1038/s41598-019-51998-w
117. Kimura J, Ichii O, Miyazono K, Nakamura T, Horino T, Otsuka-Kanazawa S, et al. Overexpression of Toll-Like Receptor 8 Correlates With the Progression of Podocyteinjury in Murine Autoimmune Glomerulonephritis. Sci Rep (2014) 4:7290. doi: 10.1038/srep07290
118. Patole PS, Pawar RD, Lech M, Zecher D, Schmidt H, Segerer S, et al. Expression and Regulation of Toll-Like Receptors in Lupus-Like Immune Complexglomerulonephritis of MRL-Fas(Lpr) Mice. Nephrol Dial Transplant (2006) 21:3062–73. doi: 10.1093/ndt/gfl336
119. Patole PS, Gröne HJ, Segerer S, Ciubar R, Belemezova E, Henger A, et al. Viral Double-Stranded RNA Aggravates Lupus Nephritis Through Toll-Like Receptor 3 Onglomerular Mesangial Cells and Antigen-Presenting Cells. J Am Soc Nephrol (2005) 16:1326–38. doi: 10.1681/ASN.2004100820
120. Imaizumi T, Aizawa-Yashiro T, Tsuruga K, Tanaka H, Matsumiya T, Yoshida H, et al. Melanoma Differentiation-Associated Gene 5 Regulates the Expression of a Chemokinecxcl10 in Human Mesangial Cells: Implications for Chronic Inflammatory Renaldiseases. Tohoku J Exp Med (2012) 228:17–26. doi: 10.1620/tjem.228.17
121. Flür K, Allam R, Zecher D, Kulkarni OP, Lichtnekert J, Schwarz M, et al. Viral RNA Induces Type I Interferon-Dependent Cytokine Release and Cell Death Inmesangial Cells Via Melanoma-Differentiation-Associated Gene-5: Implications Forviral Infection-Associated Glomerulonephritis. Am J Pathol (2009) 175:2014–22. doi: 10.2353/ajpath.2009.080585
122. Qing X, Zavadil J, Crosby MB, Hogarth MP, Hahn BH, Mohan C, et al. Nephritogenic Anti-DNA Antibodies Regulate Gene Expression in MRL/Lpr Mouseglomerular Mesangial Cells. Arthritis Rheum (2006) 54:2198–210. doi: 10.1002/art.21934
123. Allam R, Lichtnekert J, Moll AG, Taubitz A, Vielhauer V, Anders HJ. Viral RNA and DNA Trigger Common Antiviral Responses in Mesangial Cells. J Am Soc Nephrol (2009) 20:1986–96. doi: 10.1681/ASN.2008101067
124. Allam R, Pawar RD, Kulkarni OP, Hornung V, Hartmann G, Segerer S, et al. Viral 5’-Triphosphate RNA and non-Cpg DNA Aggravate Autoimmunity and Lupus Nephritisvia Distinct TLR-Independent Immune Responses. Eur J Immunol (2008) 38:3487–98. doi: 10.1002/eji.200838604
125. Liu Q, Imaizumi T, Kawaguchi S, Aizawa T, Matsumiya T, Watanabe S, et al. Toll-Like Receptor 3 Signaling Contributes to Regional Neutrophil Recruitment Incultured Human Glomerular Endothelial Cells. Nephron (2018) 139:349–58. doi: 10.1159/000489507
126. Hirono K, Imaizumi T, Aizawa T, Watanabe S, Tsugawa K, Shiratori T, et al. Endothelial Expression of Fractalkine (CX3CL1) is Induced by Toll-Like Receptor 3signaling in Cultured Human Glomerular Endothelial Cells. Mod Rheumatol (2019) 30:1074–81. doi: 10.1080/14397595.2019.1682768
127. Liu Q, Imaizumi T, Aizawa T, Hirono K, Kawaguchi S, Watanabe S, et al. Cytosolic Sensors of Viral RNA are Involved in the Production of Interleukin-6 Viatoll-Like Receptor 3 Signaling in Human Glomerular Endothelial Cells. Kidney Blood Press Res (2019) 44:62–71. doi: 10.1159/000498837
128. Hägele H, Allam R, Pawar RD, Anders HJ. Double-Stranded RNA Activates Type I Interferon Secretion in Glomerular Endothelialcells Via Retinoic Acid-Inducible Gene (RIG)-1. Nephrol Dial Transplant (2009) 24:3312–8. doi: 10.1093/ndt/gfp339
129. Hägele H, Allam R, Pawar RD, Reichel CA, Krombach F, Anders HJ. Double-Stranded DNA Activates Glomerular Endothelial Cells and Enhances Albuminpermeability Via a Toll-Like Receptor-Independent Cytosolic DNA Recognition Pathway. Am J Pathol (2009) 175:1896–904. doi: 10.2353/ajpath.2009.090182
130. Castellano G, Cafiero C, Divella C, Sallustio F, Gigante M, Pontrelli P, et al. Local Synthesis of Interferon-Alpha in Lupus Nephritis is Associated With Type Iinterferons Signature and LMP7 Induction in Renal Tubular Epithelial Cells. Arthritis Res Ther (2015) 17:72. doi: 10.1186/s13075-015-0588-3
131. Zhou Z, Ni J, Li J, Huo C, Miao N, Yin F, et al. RIG-I Aggravates Interstitial Fibrosis Via C-Myc-Mediated Fibroblast Activation Inuuo Mice. J Mol Med (Berl) (2020) 98:527–40. doi: 10.1007/s00109-020-01879-x
132. Masum MA, Ichii O, ELewa Y, Kon Y. Induced Expression of Toll-Like Receptor 9 in Peritubular Capillary Endotheliumcorrelates With the Progression of Tubulointerstitial Lesions in Autoimmunedisease-Prone Mice. Lupus (2019) 28:324–33. doi: 10.1177/0961203319828518
133. Lopez P, Scheel-Toellner D, Rodriguez-Carrio J, Caminal-Montero L, Gordon C, Suarez A. Interferon-Alpha-Induced B-Lymphocyte Stimulator Expression and Mobilization in Healthy and Systemic Lupus Erthymatosus Monocytes. Rheumatol (Oxford) (2014) 53:2249–58. doi: 10.1093/rheumatology/keu249
134. Sjostrand M, Johansson A, Aqrawi L, Olsson T, Wahren-Herlenius M, Espinosa A. The Expression of BAFF is Controlled by IRF Transcription Factors. J Immunol (2016) 196:91–6. doi: 10.4049/jimmunol.1501061
135. Turpeinen H, Raitoharju E, Oksanen A, Oksala N, Levula M, Lyytikainen LP, et al. Proprotein Convertases in Human Atherosclerotic Plaques: the Overexpression of FURIN and its Substrate Cytokines BAFF and APRIL. Atherosclerosis (2011) 219:799–806. doi: 10.1016/j.atherosclerosis.2011.08.011
136. Jeremic I, Djuric O, Nikolic M, Vlajnic M, Nikolic A, Radojkovic D, et al. Neutrophil Extracellular Traps-Associated Markers are Elevated in Patients With Systemic Lupus Erythematosus. Rheumatol Int (2019) 39:1849–57. doi: 10.1007/s00296-019-04426-1
137. Sharabi A, Tsokos GC. T Cell Metabolism: New Insights in Systemic Lupus Erythematosus Pathogenesis Andtherapy. Nat Rev Rheumatol (2020) 16:100–12. doi: 10.1038/s41584-019-0356-x
138. Lightfoot YL, Blanco LP, Kaplan MJ. Metabolic Abnormalities and Oxidative Stress in Lupus. Curr Opin Rheumatol (2017) 29:442–9. doi: 10.1097/BOR.0000000000000413
139. Gestermann N, Di Domizio J, Lande R, Demaria O, Frasca L, Feldmeyer L, et al. Netting Neutrophils Activate Autoreactive B Cells in Lupus. J Immunol (2018) 200:3364–71. doi: 10.4049/jimmunol.1700778
140. Crotty S. T Follicular Helper Cell Biology: a Decade of Discovery and Diseases. Immunity (2019) 50:1132–48. doi: 10.1016/j.immuni.2019.04.011
141. Mountz JD, Hsu HC, Ballesteros-Tato A. Dysregulation of T Follicular Helper Cells in Lupus. J Immunol (2019) 202:1649–58. doi: 10.4049/jimmunol.1801150
142. Caielli S, Veiga DT, Balasubramanian P, Athale S, Domic B, Murat E, et al. A CD4(+) T Cell Population Expanded in Lupus Blood Provides B Cell Help Through Interleukin-10 and Succinate. Nat Med (2019) 25:75–81. doi: 10.1038/s41591-018-0254-9
143. Makiyama A, Chiba A, Noto D, Murayama G, Yamaji K, Tamura N, et al. Expanded Circulating Peripheral Helper T Cells in Systemic Lupus Erythematosus: Association With Disease Activity and B Cell Differentiation. Rheumatol (Oxford) (2019) 58:1861–9. doi: 10.1093/rheumatology/kez077
144. Du SW, Jacobs HM, Arkatkar T, Rawlings DJ, Jackson SW. Integrated B Cell, Toll-Like, and BAFF Receptor Signals Promote Autoantibody Production by Transitional B Cells. J Immunol (2018) 201:3258–68. doi: 10.4049/jimmunol.1800393
145. Theodorou E, Nezos A, Antypa E, Ioakeimidis D, Koutsilieris M, Tektonidou M, et al. B-Cell Activating Factor and Related Genetic Variants in Lupus Related Atherosclerosis. J Autoimmun (2018) 92:87–92. doi: 10.1016/j.jaut.2018.05.002
146. Lapter S, Ben-David H, Sharabi A, Zinger H, Telerman A, Gordin M, et al. A Role for the B-Cell CD74/Macrophage Migration Inhibitory Factor Pathway in Theimmunomodulation of Systemic Lupus Erythematosus by a Therapeutic Tolerogenic Peptide. Immunology (2011) 132:87–95. doi: 10.1111/j.1365-2567.2010.03342.x
147. Schmitz C, Noels H, El BO, Straussfeld E, Megens R, Sternkopf M, et al. Mif-Deficiency Favors an Atheroprotective Autoantibody Phenotype in Atherosclerosis. FASEB J (2018) 32:4428–43. doi: 10.1096/fj.201800058R
148. Feng X, Chen W, Xiao L, Gu F, Huang J, Tsao BP, et al. Artesunate Inhibits Type I Interferon-Induced Production of Macrophage Migrationinhibitory Factor in Patients With Systemic Lupus Erythematosus. Lupus (2017) 26:62–72. doi: 10.1177/0961203316651738
149. Dong G, Yang Y, Li X, Yao X, Zhu Y, Zhang H, et al. Granulocytic Myeloid-Derived Suppressor Cells Contribute to IFN-I Signaling Activation of B Cells and Disease Progression Through the Lncrna NEAT1-BAFF Axisin Systemic Lupus Erythematosus. Biochim Biophys Acta Mol Basis Dis (2020) 1866:165554. doi: 10.1016/j.bbadis.2019.165554
150. Granger V, Peyneau M, Chollet-Martin S, de Chaisemartin L. Neutrophil Extracellular Traps in Autoimmunity and Allergy: Immune Complexes At Work. Front Immunol (2019) 10:2824. doi: 10.3389/fimmu.2019.02824
151. Groom JR, Luster AD. CXCR3 in T Cell Function. Exp Cell Res (2011) 317:620–31. doi: 10.1016/j.yexcr.2010.12.017
152. Datta D, Contreras AG, Grimm M, Waaga-Gasser AM, Briscoe DM, Pal S. Calcineurin Inhibitors Modulate CXCR3 Splice Variant Expression and Mediate Renal Cancer Progression. J Am Soc Nephrol (2008) 19:2437–46. doi: 10.1681/ASN.2008040394
153. Yoshikawa M, Nakayamada S, Kubo S, Nawata A, Kitanaga Y, Iwata S, et al. Type I and II Interferons Commit to Abnormal Expression of Chemokine Receptor on Bcells in Patients With Systemic Lupus Erythematosus. Clin Immunol (2019) 200:1–9. doi: 10.1016/j.clim.2018.12.017
154. Kishimoto D, Kirino Y, Tamura M, Takeno M, Kunishita Y, Takase-Minegishi K, et al. Dysregulated Heme Oxygenase-1(Low) M2-Like Macrophages Augment Lupus Nephritis Viabach1 Induced by Type I Interferons. Arthritis Res Ther (2018) 20:64. doi: 10.1186/s13075-018-1568-1
155. Trivedi S, Zeier M, Reiser J. Role of Podocytes in Lupus Nephritis. Nephrol Dial Transplant (2009) 24:3607–12. doi: 10.1093/ndt/gfp427
156. Rifkin IR, Bonegio RG. Editorial: Podocytes as Active Participants in Lupus Nephritis. Arthritis Rheumatol (2017) 69:1517–20. doi: 10.1002/art.40157
157. Wang Y, Yu F, Song D, Wang SX, Zhao MH. Podocyte Involvement in Lupus Nephritis Based on the 2003 ISN/RPS System: a Largecohort Study From a Single Centre. Rheumatol (Oxford) (2014) 53:1235–44. doi: 10.1093/rheumatology/ket491
158. Nagata M. Podocyte Injury and its Consequences. Kidney Int (2016) 89:1221–30. doi: 10.1016/j.kint.2016.01.012
159. Sever S, Schiffer M. Actin Dynamics At Focal Adhesions: a Common Endpoint and Putative Therapeutic Targetfor Proteinuric Kidney Diseases. Kidney Int (2018) 93:1298–307. doi: 10.1016/j.kint.2017.12.028
160. Kriz W, Lemley KV. Potential Relevance of Shear Stress for Slit Diaphragm and Podocyte Function. Kidney Int (2017) 91:1283–6. doi: 10.1016/j.kint.2017.02.032
161. Perico L, Conti S, Benigni A, Remuzzi G. Podocyte-Actin Dynamics in Health and Disease. Nat Rev Nephrol (2016) 12:692–710. doi: 10.1038/nrneph.2016.127
162. Gurkan S, Cabinian A, Lopez V, Bhaumik M, Chang JM, Rabson AB, et al. Inhibition of Type I Interferon Signalling Prevents TLR Ligand-Mediated Proteinuria. J Pathol (2013) 231:248–56. doi: 10.1002/path.4235
163. Migliorini A, Angelotti ML, Mulay SR, Kulkarni OO, Demleitner J, Dietrich A, et al. The Antiviral Cytokines Ifn-α and IFN-β Modulate Parietal Epithelial Cells Andpromote Podocyte Loss: Implications for IFN Toxicity, Viral Glomerulonephritis, Andglomerular Regeneration. Am J Pathol (2013) 183:431–40. doi: 10.1016/j.ajpath.2013.04.017
164. Freedman BI, Langefeld CD, Andringa KK, Croker JA, Williams AH, Garner NE, et al. End-Stage Renal Disease in African Americans With Lupus Nephritis is Associated Withapol1. Arthritis Rheumatol (2014) 66:390–6. doi: 10.1002/art.38220
165. Larsen CP, Beggs ML, Saeed M, Walker PD. Apolipoprotein L1 Risk Variants Associate With Systemic Lupuserythematosus-Associated Collapsing Glomerulopathy. J Am Soc Nephrol (2013) 24:722–5. doi: 10.1681/ASN.2012121180
166. Qi YY, Zhou XJ, Cheng FJ, Hou P, Ren YL, Wang SX, et al. Increased Autophagy is Cytoprotective Against Podocyte Injury Induced by Antibodyand Interferon-α in Lupus Nephritis. Ann Rheum Dis (2018) 77:1799–809. doi: 10.1136/annrheumdis-2018-213028
167. Thacker SG, Berthier CC, Mattinzoli D, Rastaldi MP, Kretzler M, Kaplan MJ. The Detrimental Effects of IFN-Alpha on Vasculogenesis in Lupus are Mediated by Repression of IL-1 Pathways: Potential Role in Atherogenesis and Renal Vascular Rarefaction. J Immunol (2010) 185:4457–69. doi: 10.4049/jimmunol.1001782
168. Scindia YM, Deshmukh US, Bagavant H. Mesangial Pathology in Glomerular Disease: Targets for Therapeutic Intervention. Adv Drug Delivery Rev (2010) 62:1337–43. doi: 10.1016/j.addr.2010.08.011
169. Floege J, Ostendorf T, Janssen U, Burg M, Radeke HH, Vargeese C, et al. Novel Approach to Specific Growth Factor Inhibition In Vivo: Antagonism Ofplatelet-Derived Growth Factor in Glomerulonephritis by Aptamers. Am J Pathol (1999) 154:169–79. doi: 10.1016/S0002-9440(10)65263-7
170. Chow EK, O’Connell RM, Schilling S, Wang XF, Fu XY, Cheng G. TLR Agonists Regulate PDGF-B Production and Cell Proliferation Through TGF-Beta/Typei IFN Crosstalk. EMBO J (2005) 24:4071–81. doi: 10.1038/sj.emboj.7600867
171. Gao J, Wu L, Wang Y, Cui S, Duan S, Dong Z, et al. Knockdown of Cxcl10 Inhibits Mesangial Cell Proliferation in Murine Habu Nephritisvia ERK Signaling. Cell Physiol Biochem (2017) 42:2118–29. doi: 10.1159/000479914
172. Sun YB, Qu X, Caruana G, Li J. The Origin of Renal Fibroblasts/Myofibroblasts and the Signals That Triggerfibrosis. Differentiation (2016) 92:102–7. doi: 10.1016/j.diff.2016.05.008
173. Isaka Y. Targeting TGF-β Signaling in Kidney Fibrosis. Int J Mol Sci (2018) 19:2532. doi: 10.3390/ijms19092532
174. Tomooka S, Border WA, Marshall BC, Noble NA. Glomerular Matrix Accumulation is Linked to Inhibition of the Plasmin Proteasesystem. Kidney Int (1992) 42:1462–9. doi: 10.1038/ki.1992.442
175. Wang G, Wu L, Su H, Feng X, Shi M, Jin L, et al. Association of Urinary Matrix Metalloproteinase 7 With Incident Renal Flare in Lupusnephritis. Arthritis Rheumatol (2020) 73:265–75. doi: 10.1002/art.41506
176. Lee JM, Kronbichler A, Park SJ, Kim SH, Han KH, Kang HG, et al. Association Between Serum Matrix Metalloproteinase- (MMP-) 3 Levels and Systemiclupus Erythematosus: a Meta-Analysis. Dis Markers (2019) 2019:9796735. doi: 10.1155/2019/9796735
177. Gheita TA, Abdel RD, Kenawy SA, Gheita HA. Clinical Significance of Matrix Metalloproteinase-3 in Systemic Lupus Erythematosuspatients: a Potential Biomarker for Disease Activity and Damage. Acta Reumatol Port (2015) 40:145–9.
178. Tveita AA, Rekvig OP, Zykova SN. Increased Glomerular Matrix Metalloproteinase Activity in Murine Lupus Nephritis. Kidney Int (2008) 74:1150–8. doi: 10.1038/ki.2008.308
179. Adamidis KN, Kopaka ME, Petraki C, Charitaki E, Apostolou T, Christodoulidou C, et al. Glomerular Expression of Matrix Metalloproteinases in Systemic Lupus Erythematosusin Association With Activity Index and Renal Function. Ren Fail (2019) 41:229–37. doi: 10.1080/0886022X.2019.1591998
180. Kagami S, Border WA, Ruoslahti E, Noble NA. Coordinated Expression of Beta 1 Integrins and Transforming Growthfactor-Beta-Induced Matrix Proteins in Glomerulonephritis. Lab Invest (1993) 69:68–76.
181. Wolf G, Jocks T, Zahner G, Panzer U, Stahl RA. Existence of a Regulatory Loop Between MCP-1 and TGF-Beta in Glomerular Immuneinjury. Am J Physiol Renal Physiol (2002) 283:F1075–84. doi: 10.1152/ajprenal.00349.2001
182. Paccosi S, Musilli C, Mangano G, Guglielmotti A, Parenti A. The Monocyte Chemotactic Protein Synthesis Inhibitor Bindarit Prevents Mesangialcell Proliferation and Extracellular Matrix Remodeling. Pharmacol Res (2012) 66:526–35. doi: 10.1016/j.phrs.2012.09.006
183. Yang J, Chen J, Yan J, Zhang L, Chen G, He L, et al. Effect of Interleukin 6 Deficiency on Renal Interstitial Fibrosis. PloS One (2012) 7:e52415. doi: 10.1371/journal.pone.0052415
184. Chaudhari S, Yazdizadeh SP, Tao Y, Davis ME, Mallet RT, Ma R. Inhibition of Interleukin-6 on Matrix Protein Production by Glomerular Mesangialcells and the Pathway Involved. Am J Physiol Renal Physiol (2020) 318:F1478–88. doi: 10.1152/ajprenal.00043.2020
185. Chen W, Yuan H, Cao W, Wang T, Chen W, Yu H, et al. Blocking Interleukin-6 Trans-Signaling Protects Against Renal Fibrosis Bysuppressing Stat3 Activation. Theranostics (2019) 9:3980–91. doi: 10.7150/thno.32352
186. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The Pro- and Anti-Inflammatory Properties of the Cytokine Interleukin-6. Biochim Biophys Acta (2011) 1813:878–88. doi: 10.1016/j.bbamcr.2011.01.034
187. Han X, Wang Y, Zhang X, Qin Y, Qu B, Wu L, et al. Microrna-130b Ameliorates Murine Lupus Nephritis Through Targeting the Type Iinterferon Pathway on Renal Mesangial Cells. Arthritis Rheumatol (2016) 68:2232–43. doi: 10.1002/art.39725
188. Zhang X, Han X, Tang Y, Wu Y, Qu B, Shen N. Mir-744 Enhances Type I Interferon Signaling Pathway by Targeting PTP1B in Primaryhuman Renal Mesangial Cells. Sci Rep (2015) 5:12987. doi: 10.1038/srep12987
189. Liu Y. Renal Fibrosis: New Insights Into the Pathogenesis and Therapeutics. Kidney Int (2006) 69:213–7. doi: 10.1038/sj.ki.5000054
190. Fintha A, Gasparics Á, Fang L, Erdei Z, Hamar P, Mózes MM, et al. Characterization and Role of SCAI During Renal Fibrosis Andepithelial-to-Mesenchymal Transition. Am J Pathol (2013) 182:388–400. doi: 10.1016/j.ajpath.2012.10.009
191. Kim MK, Maeng YI, Sung WJ, Oh HK, Park JB, Yoon GS, et al. The Differential Expression of TGF-β1, ILK and Wnt Signaling Inducing Epithelial Tomesenchymal Transition in Human Renal Fibrogenesis: an Immunohistochemical Study. Int J Clin Exp Pathol (2013) 6:1747–58.
192. Liu Y. Epithelial to Mesenchymal Transition in Renal Fibrogenesis: Pathologic Significance,Molecular Mechanism, and Therapeutic Intervention. J Am Soc Nephrol (2004) 15:1–12. doi: 10.1097/01.ASN.0000106015.29070.E7
193. LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, et al. Origin and Function of Myofibroblasts in Kidney Fibrosis. Nat Med (2013) 19:1047–53. doi: 10.1038/nm.3218
194. Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in Kidney Fibrosis Emerge Via Endothelial-to-Mesenchymal Transition. J Am Soc Nephrol (2008) 19:2282–7. doi: 10.1681/ASN.2008050513
195. Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, et al. Fate Tracing Reveals the Pericyte and Not Epithelial Origin of Myofibroblasts Inkidney Fibrosis. Am J Pathol (2010) 176:85–97. doi: 10.2353/ajpath.2010.090517
196. Sutariya B, Jhonsa D, Saraf MN. Tgf-β: the Connecting Link Between Nephropathy and Fibrosis. Immunopharmacol Immunotoxicol (2016) 38:39–49. doi: 10.3109/08923973.2015.1127382
197. Strutz F, Zeisberg M, Hemmerlein B, Sattler B, Hummel K, Becker V, et al. Basic Fibroblast Growth Factor Expression is Increased in Human Renal Fibrogenesisand may Mediate Autocrine Fibroblast Proliferation. Kidney Int (2000) 57:1521–38. doi: 10.1046/j.1523-1755.2000.00997.x
198. Strutz F, Zeisberg M, Renziehausen A, Raschke B, Becker V, van Kooten C, et al. TGF-Beta 1 Induces Proliferation in Human Renal Fibroblasts Via Induction of Basicfibroblast Growth Factor (FGF-2). Kidney Int (2001) 59:579–92. doi: 10.1046/j.1523-1755.2001.059002579.x
199. Sommer M, Eismann U, Gerth J, Stein G. Interleukin 4 Co-Stimulates the PDGF-BB- and BFGF-Mediated Proliferation Ofmesangial Cells and Myofibroblasts. Nephron (2002) 92:868–80, 880-882. doi: 10.1159/000065451
200. Kalluri R, Neilson EG. Epithelial-Mesenchymal Transition and its Implications for Fibrosis. J Clin Invest (2003) 112:1776–84. doi: 10.1172/JCI200320530
201. Zeisberg M, Maeshima Y, Mosterman B, Kalluri R. Renal Fibrosis. Extracellular Matrix Microenvironment Regulates Migratory Behaviorof Activated Tubular Epithelial Cells. Am J Pathol (2002) 160:2001–8. doi: 10.1016/S0002-9440(10)61150-9
202. Yang J, Liu Y. Dissection of Key Events in Tubular Epithelial to Myofibroblast Transition and Itsimplications in Renal Interstitial Fibrosis. Am J Pathol (2001) 159:1465–75. doi: 10.1016/S0002-9440(10)62533-3
203. Gwon MG, An HJ, Kim JY, Kim WH, Gu H, Kim HJ, et al. Anti-Fibrotic Effects of Synthetic TGF-β1 and Smad Oligodeoxynucleotide on Kidneyfibrosis In Vivo and In Vitro Through Inhibition of Both Epithelialdedifferentiation and Endothelial-Mesenchymal Transitions. FASEB J (2020) 34:333–49. doi: 10.1096/fj.201901307RR
204. Li J, Qu X, Bertram JF. Endothelial-Myofibroblast Transition Contributes to the Early Development Ofdiabetic Renal Interstitial Fibrosis in Streptozotocin-Induced Diabetic Mice. Am J Pathol (2009) 175:1380–8. doi: 10.2353/ajpath.2009.090096
205. Tan TK, Zheng G, Hsu TT, Wang Y, Lee VW, Tian X, et al. Macrophage Matrix Metalloproteinase-9 Mediates Epithelial-Mesenchymal Transition Invitro in Murine Renal Tubular Cells. Am J Pathol (2010) 176:1256–70. doi: 10.2353/ajpath.2010.090188
206. Zhao Y, Qiao X, Wang L, Tan TK, Zhao H, Zhang Y, et al. Matrix Metalloproteinase 9 Induces Endothelial-Mesenchymal Transition Via Notchactivation in Human Kidney Glomerular Endothelial Cells. BMC Cell Biol (2016) 17:21. doi: 10.1186/s12860-016-0101-0
207. Du F, Li S, Wang T, Zhang HY, Li DT, Du ZX, et al. Implication of Bcl-2-Associated Athanogene 3 in Fibroblast Growth Factor-2-Mediatedepithelial-Mesenchymal Transition in Renal Epithelial Cells. Exp Biol Med (Maywood) (2015) 240:566–75. doi: 10.1177/1535370214558023
208. Masola V, Gambaro G, Tibaldi E, Brunati AM, Gastaldello A, D’Angelo A, et al. Heparanase and Syndecan-1 Interplay Orchestrates Fibroblast Growth Factor-2-Inducedepithelial-Mesenchymal Transition in Renal Tubular Cells. J Biol Chem (2012) 287:1478–88. doi: 10.1074/jbc.M111.279836
209. Strutz F, Zeisberg M, Ziyadeh FN, Yang CQ, Kalluri R, Müller GA, et al. Role of Basic Fibroblast Growth Factor-2 in Epithelial-Mesenchymal Transformation. Kidney Int (2002) 61:1714–28. doi: 10.1046/j.1523-1755.2002.00333.x
210. Lawson JS, Syme HM, Wheeler-Jones C, Elliott J. Characterisation of Feline Renal Cortical Fibroblast Cultures and Theirtranscriptional Response to Transforming Growth Factor β1. BMC Vet Res (2018) 14:76. doi: 10.1186/s12917-018-1387-2
211. Vernon MA, Mylonas KJ, Hughes J. Macrophages and Renal Fibrosis. Semin Nephrol (2010) 30:302–17. doi: 10.1016/j.semnephrol.2010.03.004
212. Nikolic-Paterson DJ, Atkins RC. The Role of Macrophages in Glomerulonephritis. Nephrol Dial Transplant (2001) 16 Suppl 5:3–7. doi: 10.1093/ndt/16.suppl_5.3
213. Nakatsuji S, Yamate J, Sakuma S. Macrophages, Myofibroblasts, and Extracellular Matrix Accumulation in Interstitialfibrosis of Chronic Progressive Nephropathy in Aged Rats. Vet Pathol (1998) 35:352–60. doi: 10.1177/030098589803500504
214. Strutz F, Zeisberg M. Renal Fibroblasts and Myofibroblasts in Chronic Kidney Disease. J Am Soc Nephrol (2006) 17:2992–8. doi: 10.1681/ASN.2006050420
215. Kok HM, Falke LL, Goldschmeding R, Nguyen TQ. Targeting CTGF, EGF and PDGF Pathways to Prevent Progression of Kidney Disease. Nat Rev Nephrol (2014) 10:700–11. doi: 10.1038/nrneph.2014.184
216. Chen YT, Chang FC, Wu CF, Chou YH, Hsu HL, Chiang WC, et al. Platelet-Derived Growth Factor Receptor Signaling Activates Pericyte-Myofibroblasttransition in Obstructive and Post-Ischemic Kidney Fibrosis. Kidney Int (2011) 80:1170–81. doi: 10.1038/ki.2011.208
217. Wu CF, Chiang WC, Lai CF, Chang FC, Chen YT, Chou YH, et al. Transforming Growth Factor β-1 Stimulates Profibrotic Epithelial Signaling Toactivate Pericyte-Myofibroblast Transition in Obstructive Kidney Fibrosis. Am J Pathol (2013) 182:118–31. doi: 10.1016/j.ajpath.2012.09.009
218. Wang N, Deng Y, Liu A, Shen N, Wang W, Du X, et al. Novel Mechanism of the Pericyte-Myofibroblast Transition in Renal Interstitialfibrosis: Core Fucosylation Regulation. Sci Rep (2017) 7:16914. doi: 10.1038/s41598-017-17193-5
219. Lechner J, Pfaller W. Interferon Alpha2b Increases Paracellular Permeability of Renal Proximal Tubularllc-PK1 Cells Via a Mitogen Activated Protein Kinase Signaling Pathway. Ren Fail (2001) 23:573–88. doi: 10.1081/JDI-100104739
220. Lechner J, Krall M, Netzer A, Radmayr C, Ryan MP, Pfaller W. Effects of Interferon Alpha-2b on Barrier Function and Junctional Complexes of Renalproximal Tubular LLC-PK1 Cells. Kidney Int (1999) 55:2178–91. doi: 10.1046/j.1523-1755.1999.00487.x
221. Lechner J, Malloth N, Seppi T, Beer B, Jennings P, Pfaller W. IFN-Alpha Induces Barrier Destabilization and Apoptosis in Renal Proximal Tubularepithelium. Am J Physiol Cell Physiol (2008) 294:C153–60. doi: 10.1152/ajpcell.00120.2007
222. Der E, Suryawanshi H, Morozov P, Kustagi M, Goilav B, Ranabothu S, et al. Tubular Cell and Keratinocyte Single-Cell Transcriptomics Applied to Lupus Nephritisreveal Type I IFN and Fibrosis Relevant Pathways. Nat Immunol (2019) 20:915–27. doi: 10.1038/s41590-019-0386-1
223. Der E, Ranabothu S, Suryawanshi H, Akat KM, Clancy R, Morozov P, et al. Single Cell RNA Sequencing to Dissect the Molecular Heterogeneity in Lupusnephritis. JCI Insight (2017) 2:93009. doi: 10.1172/jci.insight.93009
224. Hao W, Rovin BH, Friedman A. Mathematical Model of Renal Interstitial Fibrosis. Proc Natl Acad Sci USA (2014) 111:14193–8. doi: 10.1073/pnas.1413970111
225. Ding X, Xiang W, He X. IFN-I Mediates Dysfunction of Endothelial Progenitor Cells in Atherosclerosis Ofsystemic Lupus Erythematosus. Front Immunol (2020) 11:581385. doi: 10.3389/fimmu.2020.581385
226. Kahlenberg JM, Thacker SG, Berthier CC, Cohen CD, Kretzler M, Kaplan MJ. Inflammasome Activation of IL-18 Results in Endothelial Progenitor Cell Dysfunction in Systemic Lupus Erythematosus. J Immunol (2011) 187:6143–56. doi: 10.4049/jimmunol.1101284
227. Spinelli FR, Barbati C, Cecarelli F, Morello F, Colasanti T, Vomero M, et al. B Lymphocyte Stimulator Modulates Number and Function of Endothelial Progenitor Cells in Systemic Lupus Erythematosus. Arthritis Res Ther (2019) 21:245. doi: 10.1186/s13075-019-2015-7
228. Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and Perivascular Fibroblasts are the Primary Source of Collagen-Producingcells in Obstructive Fibrosis of the Kidney. Am J Pathol (2008) 173:1617–27. doi: 10.2353/ajpath.2008.080433
229. Mack M, Yanagita M. Origin of Myofibroblasts and Cellular Events Triggering Fibrosis. Kidney Int (2015) 87:297–307. doi: 10.1038/ki.2014.287
230. Gomez IG, Roach AM, Nakagawa N, Amatucci A, Johnson BG, Dunn K, et al. TWEAK-Fn14 Signaling Activates Myofibroblasts to Drive Progression of Fibrotickidney Disease. J Am Soc Nephrol (2016) 27:3639–52. doi: 10.1681/ASN.2015111227
231. Bijkerk R, de Bruin RG, van Solingen C, van Gils JM, Duijs JM, van der Veer EP, et al. Silencing of Microrna-132 Reduces Renal Fibrosis by Selectively Inhibitingmyofibroblast Proliferation. Kidney Int (2016) 89:1268–80. doi: 10.1016/j.kint.2016.01.029
232. Stefanska A, Kenyon C, Christian HC, Buckley C, Shaw I, Mullins JJ, et al. Human Kidney Pericytes Produce Renin. Kidney Int (2016) 90:1251–61. doi: 10.1016/j.kint.2016.07.035
Keywords: fibrosis, IFN-I, lupus nephritis, nucleic acid sensors, pathogenesis, renal resident cells
Citation: Ding X, Ren Y and He X (2021) IFN-I Mediates Lupus Nephritis From the Beginning to Renal Fibrosis. Front. Immunol. 12:676082. doi: 10.3389/fimmu.2021.676082
Received: 04 March 2021; Accepted: 06 April 2021;
Published: 20 April 2021.
Edited by:
José Carlos Crispín, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), MexicoReviewed by:
J. Michelle Kahlenberg, University of Michigan, United StatesFlorencia Rosetti, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), Mexico
Tomohiro Koga, Nagasaki University, Japan
Copyright © 2021 Ding, Ren and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojie He, aGV4ajcxNTBAMTYzLmNvbQ==