 Jiachen Liu
Jiachen Liu Lirong Liu
Lirong Liu Xiaoyu Wang
Xiaoyu Wang Rundong Jiang1
Rundong Jiang1 Qinqin Bai
Qinqin Bai Gaiqing Wang
Gaiqing Wang- 1Xiangya Medical College of Central South University, Changsha, China
- 2Department of Neurology, Shanxi Medical University, Taiyuan, China
- 3Department of Neurology, Sanya Central Hospital (Hainan Third People’s Hospital), Sanya, China
Microglia are the resident immune cells of the central nervous system (CNS). It is well established that microglia are activated and polarized to acquire different inflammatory phenotypes, either pro-inflammatory or anti-inflammatory phenotypes, which act as a critical component in the neuroinflammation following intracerebral hemorrhage (ICH). Microglia produce pro-inflammatory mediators at the early stages after ICH onset, anti-inflammatory microglia with neuroprotective effects appear to be suppressed. Previous research found that driving microglia towards an anti-inflammatory phenotype could restrict inflammation and engulf cellular debris. The principal objective of this review is to analyze the phenotypes and dynamic profiles of microglia as well as their shift in functional response following ICH. The results may further the understanding of the body’s self-regulatory functions involving microglia following ICH. On this basis, suggestions for future clinical development and research are provided.
Introduction
Microglia constitute 5% to 10% of adult brain cells and form the largest group of immune cells in the CNS (1). The primary source of microglia is yolk sac erythromyeloid precursors (EMPs) that migrate into the brain rudiment during embryo development (2). Under physiological conditions, microglia self-renew for the entire lifespan of the organism and interact with numerous other cells in the brain, such as astrocytes, neurons, and oligodendrocytes (3). Mounting evidence suggests that microglia, as brain resident immune cells, play an essential role in maintaining normal brain function. When pathologic changes disrupt homeostasis in the brain, microglia are activated to exert regulatory effects (4). Microglia are highly diverse, and their phenotype depends on the context and type of stressor or pathology (5). Specifically, during the different periods after intracerebral hemorrhage (ICH), microglia may polarize to produce pro-inflammatory mediators or acquire a more anti-inflammatory phenotype, which has a decisive influence on ICH progression (Figure 1) (6).
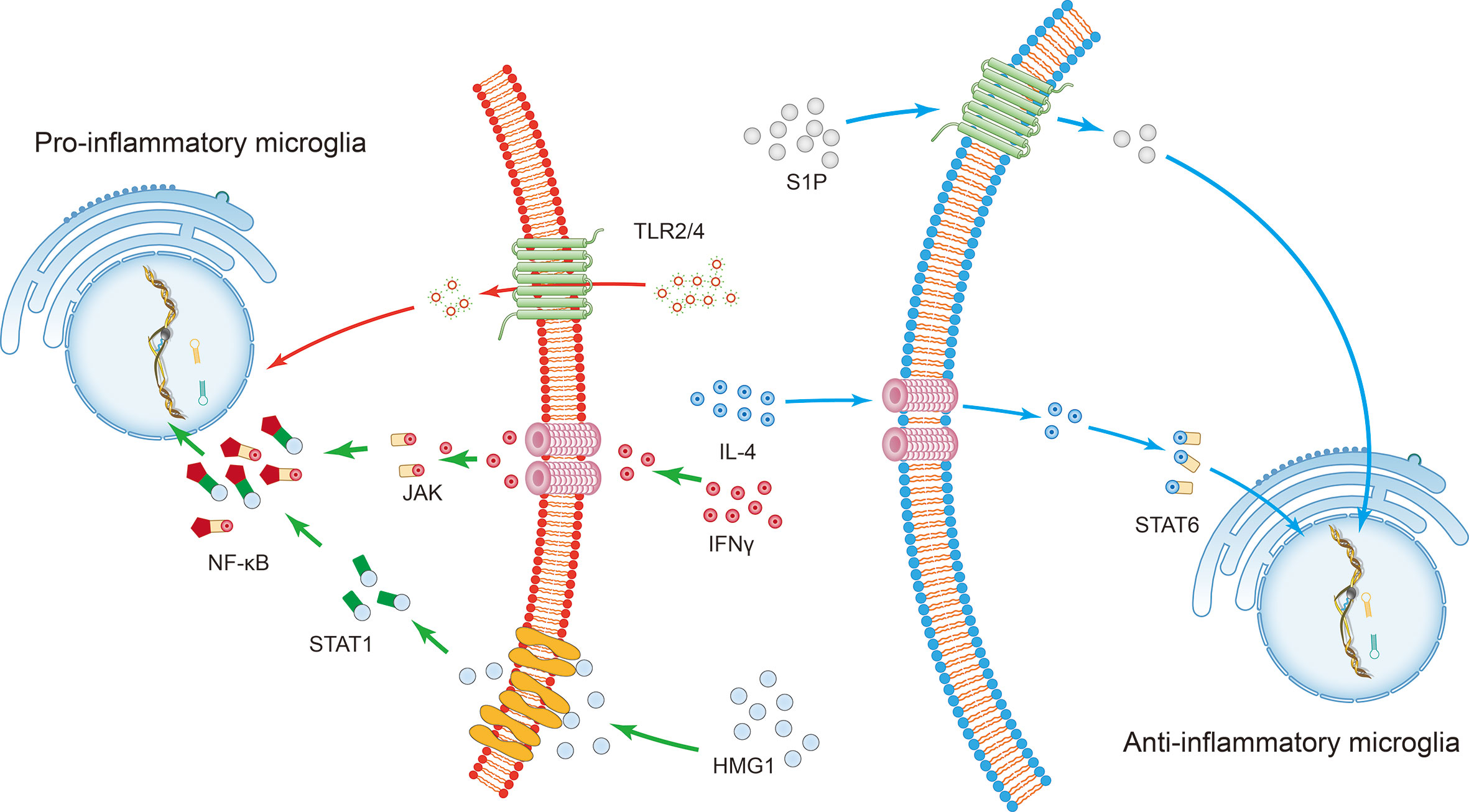
Figure 1 The induction of microglial polarization by several transcription factors. Activation of high mobility histone 1 (HMG1) and Toll-like receptor (TLR) 2 or TLR4 promotes the pro-inflammatory phenotypic polarization of microglia. Interferon-gamma (IFN-γ) promotes the pro-inflammatory phenotypic polarization of microglia through the signaling sensors signal transducer and activator of transcription (STAT) 1 and Janus kinase (JAK). This series of processes involves the nuclear factor kappa B (NF-κB) signaling pathway. On the other hand, STAT6 accumulates under the action of IL-4 and is responsible for the transcription of M2 phenotype-related genes. The sphingosine-1-phosphate (S1P) receptor signaling pathway downregulates the expression of pro-inflammatory cytokines and enhances anti-inflammatory responses following intracerebral hemorrhage.
Pathological analysis of microglial activity during ICH has revealed that microglia induce potent immune responses after extravasation of blood into the brain (7). The strong inflammatory response in microglia is caused by the rapid accumulation of blood-derived products (e.g., hemoglobin, heme, and iron) after ICH (8), which can directly damage the brain parenchyma, and a sustained microglia-mediated inflammatory response results in neurologic deterioration (9). Interestingly, Chang et al. (10) have experimentally proven that as early as 1 to 1.5 h after ICH, microglia respond to hemorrhagic injury and exhibit a protective alternative activation phenotype.
The role of microglia following ICH is complex, and the entirely different action of different phenotypes of microglia plays an essential role in the development of cerebral inflammatory injury and recovery of the brain after ICH (Figure 2). Besides, the neuroprotective effect of microglia may serve as promising targets for ICH treatment. For example, the primary role of activated microglia is to phagocytose the hematoma, thereby reducing ICH-induced brain swelling and neuronal loss and improving neurological deficits (11). Microglial depletion can lead to more severe brain swelling, neuronal loss, and functional defects following ICH (12).
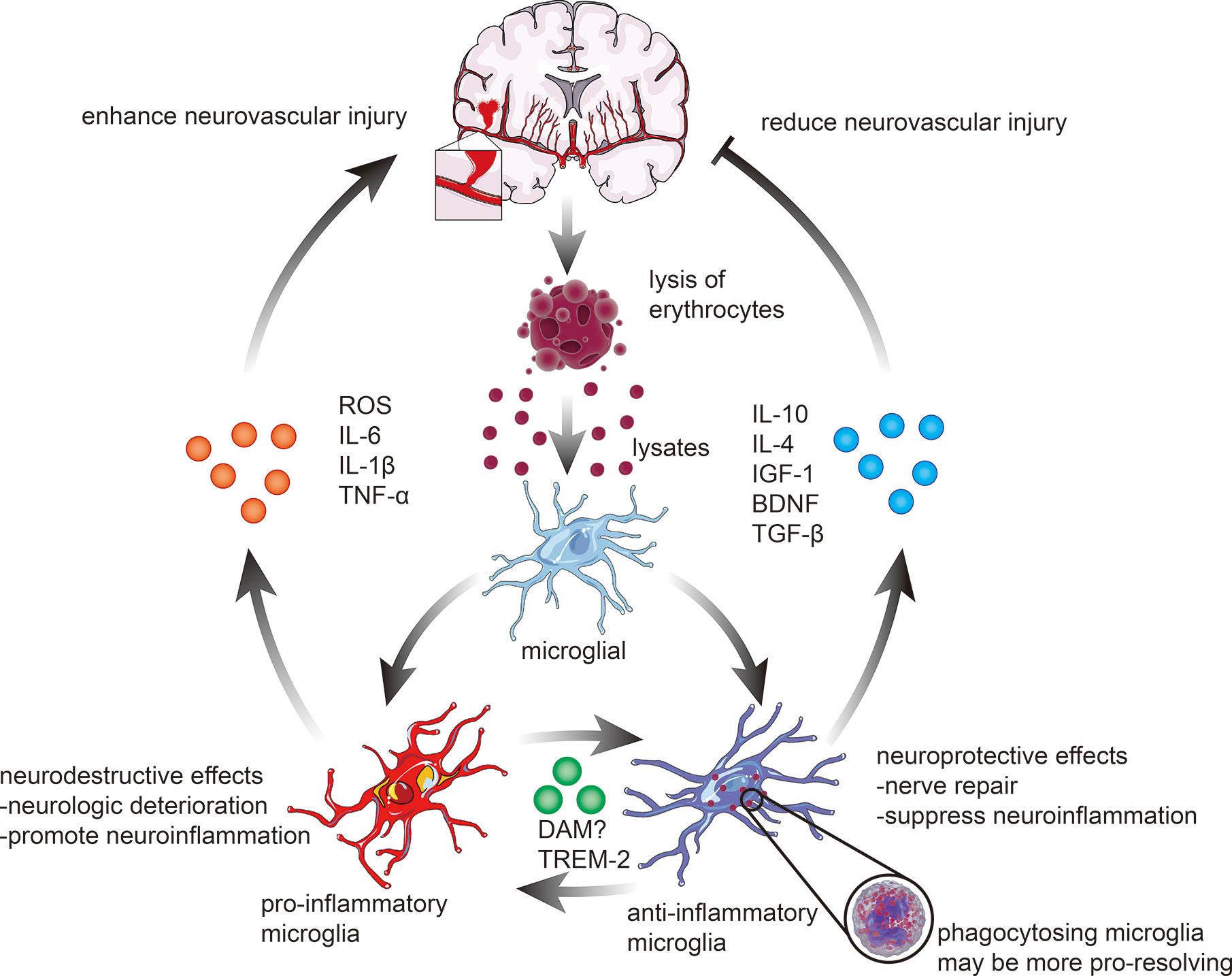
Figure 2 Microglial activation and polarization following ICH. The polarization of microglia in response to stimulation with erythrocyte lysates after ICH can be broadly classified into two categories. 1) pro-inflammatory microglia elevate the levels of ROS, interleukin-6 (IL-6), interleukin-1β (IL-1β), and tumor necrosis factor-α (TNF-α), hence enhancing the pro-inflammatory and destructive effects of ICH on the brain. 2) anti-inflammatory microglia, on the contrary, mainly exert neuroprotective effects, including actions related to nerve repair and anti-inflammatory effects, through phagocytosis of lysates, which are always associated with higher expression of IL-10, IL-4, insulin-like growth factor-1 (IGF-1), brain-derived neurotrophic factor (BDNF), and transforming growth factor-β (TGF-β). In recent years, an increasing number of studies have revealed that microglia can also be polarized into neuroprotective and neurodestructive phenotypes, such as disease-associated microglia (DAM) and triggering receptor expressed on myeloid cells 2 (TREM2) phenotypes, which remain to be further investigated.
The dual role of reactive microglia in inflammation processes has a biphasic influence on the brain, which acts as a double‐edged offensive and defensive sword in brain injury. This paper reviews recent empirical studies on microglial activity following ICH to identify the most critical factors that influence this process, offering a fresh perspective for developing novel therapeutic strategies.
The Dual Role Of Microglia After Ich
The Effects of Microglia on the Blood-Brain Barrier Following ICH
Destruction of the blood-brain barrier (BBB) and consequent brain edema is the most common secondary causes of life-threatening events after ICH (13). One recent study by Chen et al. showed that microglia-derived TNF-α mediates endothelial necroptosis contributing to blood-brain-barrier disruption (14). Besides, activated microglia cause an imbalance between endogenous vasodilators and vasoconstrictors, further leading to edema formation after ICH (15).
Anti-inflammatory actions of microglia may mediate BBB protection and neural repair by producing anti-inflammatory cytokines, extracellular matrix proteins, glucocorticoids, and other substances (16). As signaling via IL-4 and IL-10 can induce an anti-inflammatory phenotype of microglia, it can be targeted for ICH treatment through modulating BBB physiology (17).
The Functions of Microglia in Secondary Brain Damage Following ICH
Intracranial hematoma is a crucial factor contributing to brain injury after ICH. Mechanical damage is induced in adjacent tissues due to compression and dissection. Simultaneously, iron, heme, and cytotoxic hemoglobin can be passively released due to the lysis of erythrocytes adjacent to the hematoma (18). Microglial phagocytosis of hematoma occurs before erythrocytes lysis to protect the brain (19, 20). This process can be regulated by alternative activation of microglia via activating the CCR4/ERK/Nrf2 pathway and peroxisome proliferator-activated receptor γ (PPAR-γ) (21–23) (Figure 3).
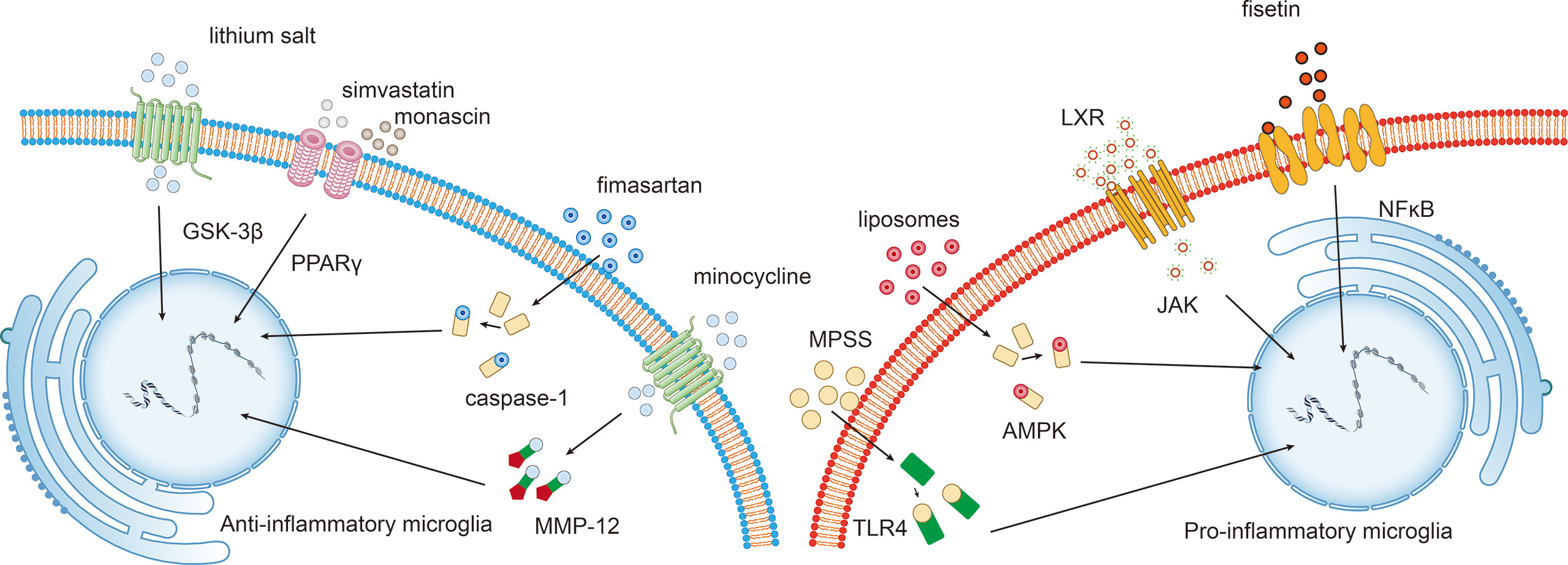
Figure 3 Potential intervention strategies targeting the microglial phenotypic shift following ICH. Interventions that induce the polarization of pro-inflammatory microglia towards the anti-inflammatory phenotype exert beneficial effects following ICH. For example, fisetin mediates the NFκB pathway, and liver X receptor (LXR)-mediated JAK, liposome-mediated adenosine monophosphate-activated protein kinase (AMPK), and methylprednisolone sodium succinate (MPSS)-mediated TLR4 inhibit M1 microglia. While lithium salt mediates glycogen synthase kinase-3beta (GSK-3β) expression, simvastatin and monascin mediate peroxisome proliferator-activated receptor gamma (PPARγ) levels, fimasartan mediates caspase-1 levels, and minocycline upregulates matrix metalloproteinase 12 (MMP-12) expression.
There is ample evidence that microglial activation is essential for secondary damage to the brain after ICH (24). For example, microglia also produce pro-inflammatory factors (TNF-α, IL-1β, IL-6) and chemokines (CXCL2), which promote neuroinflammation (25, 26). Besides, the absorption of hematoma may also trigger a series of inflammatory reactions leading to primary and secondary brain damage (27).
Microglial Polarization Following Ich
Although the terminology of “microglia polarization” is still widely used in the literature, most commonly in the M1 and M2 phenotypes. Complex high-content experiment and multi-omics technologies, including transcriptomic, epigenomic, and proteomics, have found novel microglia polarization states beyond the standard M1/M2 dichotomy, which leads to a fierce debate in microglial M1/M2 polarization in recent years (28).
As early as 2013, Chiu and colleagues utilized flow cytometry and deep RNA sequencing of acutely isolated spinal cord microglia. The study aimed to prove that microglial reactions must be interpreted in light of the tissue in which the activating stimulus is present (29). However, the M1/M2 model is not a pure phenomenon in vivo, as previously proven by Butovsky, by profiling CNS cells with an MG400 microglial chip (30), which ignores the crucial concept demonstrated by Chiu et al. (30). Besides, transcriptionally distinguishable subpopulations of microglia that appear to be a transcriptional continuum of the local population of microglia can be detected during homeostasis (31, 32), representing a transcriptional basis for the microglia phenotype diversity (33).
Single-cell RNA-sequence analysis of microglia suggested converged expression of M1 and M2 markers due to the influence of disease-related inflammatory processes (34). Furthermore, precise categorization of different microglia or monocyte subtypes based on specific types and stages of pathology or their relation to specific tissue injury types is also possible (35). In conclusion, a precise definition of microglial polarization has proven elusive, and the description of M1 or M2 phenotypes is an oversimplification of the complex biology of microglia.
In a recent transcriptional single-cell study, Keren-Shaul et al. found that microglial phenotypes other than the M1/M2 phenotypes exist. For example, a new subpopulation named disease-associated microglia (DAM) has been discovered through genome-wide transcriptomic analyses of microglia under different disease conditions. Although the gene profile of DAM and M1 microglia partially overlapped, the molecular signatures have shown apparent differences (36). Interestingly, DAM also exhibits anti-inflammatory/phagocytic and pro-inflammatory profiles (37). Activation of DAM depends on triggering receptors expressed on myeloid cells 2 (TREM2), a receptor located mainly on the surface of microglia. TREM2 promotes the phagocytosis of apoptotic neurons producing tiny quantities of pro-inflammatory cytokines (38). Research has also shown that TREM2 is activated in perihematomal areas, which improved attenuated neuroinflammation and neuronal apoptosis after ICH (39). Besides, according to a study done by Gao et al. in 2019, CEBPα, IRF1, and LXRβ are likely regulators of pro-inflammatory and anti-inflammatory DAM states. Based on emerging findings, it is possible to conclude that DAM represents a switch that substantially alters microglial function (40). While the DAM concept has been widely used in neurodegenerative diseases such as Alzheimer’s disease rather than ICH, it is clear that microglia is in a constant flux state and exquisitely sensitive to their environment.
Many studies have investigated the spatially and temporally restricted subsets of microglia during development and disease, further identifying the distinct molecular hallmarks and diverse cellular kinetics using massively parallel single-cell analysis and computational modeling (32, 41). For example, using single-cell RNA sequencing from human cerebral cortex samples, Olah et al. confirm the presence of four microglial subsets and elucidate the significance of subsets, such as the association with Alzheimer’s disease (AD) (42). More recently, Ochocka et al. demonstrate cellular and functional heterogeneity of microglia using flow cytometry and scRNAseq. In this experiment, multiple microglial clusters were obtained, and gene expression profiles underlying a specific cluster could reflect different functions. Hom-MG and activated microglia (Act-MG) were identified, which shows the distinct spatial distribution in experimental gliomas (43). Furthermore, an environment-dependent transcriptional network specifying microglia-specific programs have been developed, which identified substantial subsets of microglia associate with neurodegenerative and behavioral diseases (44).
In summary, microglia are activated by various pathologic events or changes in brain homeostasis, which are highly diverse and depend on the context and type of stressor or pathology. The complicated functional roles of microglia support the existence of distinct pro-inflammatory and anti-inflammatory functional states following ICH. The significance of defining microglial subtypes is to identify novel microglial functional conditions; determine the impact of molecules on microglia types, and discover ways to mediate functions in healthy physiology or disease (45). During the ICH progress, most investigators continue to use the expression of M1/M2 markers and microglia polarization as a surrogate for a genuine mechanistic understanding of how microglial function changes (46). Therefore, it would be interesting to identify the regulators and influencing factors that contribute to the polarization of microglia towards a neuroprotective or neurodestructive phenotype, which may shed new light on the pathogenetic role of microglia following ICH.
Endogenous Mechanisms of Microglia
Much of the literature has emphasized the autoregulation of microglia during ICH progress. For example, studies by Wu et al. showed that soluble epoxide hydrolase expression is upregulated in microglia after ICH, which causes neuroinflammatory responses by degrading anti-inflammatory epoxyeicosatrienoic acid (47). Other studies have provided further evidence that microglial recruitment is associated with TWIK-related K+ channel 1 (TREK-1), which also triggers the secretion of pro-inflammatory factors such as IL-1β and TNF-α as well as cell adhesion molecules following ICH (48). Besides, low-density LRP1 in the neurovascular unit interacts with Mac-1 expressed by microglia to promote tPA-mediated activation of platelet-derived growth factor-cc (PDGF-cc). Activation of potential PDGF-cc and PDGF receptor-α signals can increase the permeability of the blood-brain barrier and deterioration following ICH (13).
Microglia have similarly been shown to be involved in anti-inflammatory and phagocytic effects on the hematoma, contributing to neurologic recovery after ICH. The correlation between regulatory T lymphocytes (Tregs) and neuroinflammatory response after ICH has been defined. In vitro experiments have demonstrated that Tregs modulate microglia polarization toward the anti-inflammation phenotype through the IL-10/GSK3β/PTEN axis in this regulatory process (49, 50).
The Regulatory Effect of miRNAs
As gene expression is regulated through genetic and epigenetic regulatory networks, there is growing evidence that miRNAs play essential roles in the microglial effects after ICH (51). For example, miRNA-7 (miR-7) can inhibit the expression of Toll-like receptor 4 (TLR4) and provoke a secondary microglia-mediated inflammatory response after ICH (52). Further studies have confirmed that agents that target TLR4 and miR-7, such as ligustilide (LIG) and senkyunolide H (SH), can exert neuroprotective effects against ICH by inhibiting Prx1/TLR4/NF-κB signaling via activation of microglia and astrocytes (53).
Recent studies have found that inhibition of miRNA-222 suppresses microglia-mediated inflammatory responses and improves neurological functions in a preclinical mouse model of ICH. Integrin subunit β8 (ITGB8) was identified as a directly negatively regulated target of miR−222 in microglial cells, leading to the attenuation of inflammation and apoptosis (54). Besides, miR-132 enhances the cholinergic blockade of the inflammatory response by targeting acetylcholinesterase (AChE), which also inhibits the activation of pro-inflammatory microglia and provides protection against neuronal death caused by ischemia (55).
Furthermore, as the critical factors in autophagy, miRNAs negatively regulate gene expression and autophagic activity of microglia. For example, miRNA-144 targets mTOR by directly interacting with 3′ untranslated regions (UTRs), which are involved in hemoglobin-mediated activation of microglial autophagy and inflammatory responses (56). The specific function of autophagy is dualistic and has been difficult to assess whether it has harmful or beneficial effects following ICH thus far. Despite many studies demonstrated that autophagy could enhance the protection of endoplasmic reticulum stress and reducing oxidative damage after ICH via clearing up the cell rubbish and oxidative-stress products (57, 58), recent studies showed autophagy positively regulates inflammation following ICH (59, 60).
Regulation of Microglia Function by Intracellular Signaling Following ICH
Anti-inflammatory microglia functions are accomplished by combining various signaling pathways that compose a complex network involved in multiple biological processes. Exploring the network of biological signaling pathways and its molecular basis contributes to novel interventions targeting signaling pathways that block the pathological progression of ICH (Figure 3).
The Roles of the AMPK Pathway and AdipoR1 in Microglia Function Following ICH
It has been demonstrated that adenosine monophosphate-activated protein kinase (AMPK) can drive the phenotypic shift from a pro-inflammatory state to an anti-inflammatory state (61). The expression of endogenous C1q/TNF-related protein 9 (CTRP9), an upstream trigger of the AMPK signaling pathway and an agonist of AdipoR1, is increased after ICH in animal models of long-term neurobehavior, peaking at 24 h after ICH. Further experiments have confirmed that the expression of AdipoR1 and p-AMPK can reduce the expression of inflammatory cytokines after ICH (62). Besides, the activation of MC4R also alleviates neurological deficits through the AMPK pathway following ICH, and interventions targeting MC4R, such as RO27-3225 administration, have been proven to be effective in animal experiments (63).
The Roles of the JNK Pathway in Microglia Function Following ICH
As mentioned above, Treg cells inhibit microglia-mediated inflammatory responses and improve neurological function in vivo, mainly by activate NF-κB through the JNK pathway (49, 64). There are controversies regarding the role of the JNK signaling pathway following ICH, which has received increased attention in the clinic in recent years. For example, the synthesis of the liver X receptor (LXR) agonist TO901317 was shown to exert specific effects in an ICH model by inhibiting JNK signaling. The hyperbaric oxygen preconditioning (HBOP) model of ICH has demonstrated the potential relevance between JNK phosphorylation and the immunological activity of anti-inflammatory microglia (65). As current practical limitations include drug side effects, uncertainties regarding efficacy, surgical injuries, and complications, there are no standardized clinical interventions for ICH except intracranial pressure-lowering therapies. Therefore, hyperbaric oxygen therapy provides a feasible alternative intervention with mild adverse effects against ICH, and the mechanism of HBOP in ICH needs further exploration and verification.
The Impact of the Toll-like Receptor 4 (TLR-4) Pathway on Microglia Function
Toll-like receptor 4 (TLR4) plays a crucial role in the innate immune response. It can be concluded that loss of TLR4 reduces the recruitment of pro-inflammatory microglia and markedly alleviates inflammation around the hematoma in the animal model (66). Further studies have shown that TLR4 also inhibits the phagocytosis of microglia on the surface of red blood cells, resulting in hematoma absorption delay and severe neurological deficits in ICH patients (67). TLR4-mediated autophagy of microglial activation contributes to secondary brain injury and brain recovery and inflammatory damage following ICH (68).
The role of TLR4 in secondary brain damage following ICH has been elaborated in detail, and therapeutic strategies targeting TLR4 are relatively well-developed. Therefore, TLR4 remains a promising target for inhibiting undesired microglial responses, and interventions targeting TLR4-related pathways may represent future candidates for ICH therapy.
Regulation of Microglia Function by Extracellular Signals Following ICH
As the regulatory effects of intracellular signaling pathways on microglia after ICH have been discussed, the final section of this paper addresses how extracellular signaling regulators influence microglial morphology and function. Compared with intracellular signals, extracellular signals are more complicated and vulnerable to disruption, which means they have the potential to be translated into clinically effective targeted therapies for ICH.
Interventions Targeting Microglia Functions for ICH
Interleukins
The interleukins (ILs) level is intimately associated with the development and progression of ICH, which may be achieved by modulation of microglia functions. For example, activation of the IL-4/transcription 6 (STAT6) axis improved long-term functional recovery in a mouse model of ICH (69). Conversely, expression of IL-15 exacerbates brain injury following ICH by mediate the crosstalk between microglia and astrocytes (70).
Besides, antibodies against IL-17A can prevent ICH-induced expression of TNF-α, IL-1β, and IL-6 and inhibit microglial activation (71). Further examination revealed that IL-17A promotes autophagy in pro-inflammatory microglia, thus maintaining the body’s normal immune response and alleviating brain edema after ICH (60). Notably, recent studies have found that intraventricular infusion of IL-33 can alleviate neurological deficits following ICH by promoting the transformation of pro-inflammatory microglia (72). Deferroxamine (DFA) can also inhibit the activation of pro-inflammatory microglia by downregulating IL-1β and TNF expression, reducing secondary brain insult following ICH.
Nuclear Factor-κB
Evidence has suggested that NF-κB translocates to the nucleus, and pro-inflammatory mediators (NO, TNF-a, and IL-6) are produced following inflammatory response after ICH. These results suggest that combined targeting of NF-κB signaling pathway inhibition may be a more effective anti-neuroinflammatory strategy following ICH (53).
Regulation of NF-κB activity may also have promising clinical benefits following ICH. Analysis of thrombin toxicity in vitro shows that has thrombin release after ICH led to the increased expression of NF-κB in microglia (73, 74). Treatment modalities disrupting this harmful process, such as miR-181c mimic therapy, are expected to regulate thrombin-driven inflammation after cerebral hemorrhage (75).
Glycogen Synthase Kinase-3β
It is widely acknowledged that glycogen synthase kinase-3beta (GSK-3β) exerts a potent pro-inflammatory effect following ICH (76). Studies have shown that the hematoma volume is significantly decreased by GSK-3β inhibition after ICH due to enhanced microglia-mediated phagocytosis (77). Consistently, the GSK-3β inhibitor 6-bromoindirubin-3′-oxime (BIO) has been shown to relieve inflammation by blocking GSK-3β Tyr216 phosphorylation/activation following ICH. BIO may exert a protective effect against ICH by increasing the number of anti-inflammatory microglia through inactivating GSK-3β (78).
It is interesting to note that the molecular mechanism by which lithium salt can treat ICH in clinical practice has already been elucidated. Recently, it has been shown that LiCl treatment decreased the death of mature oligodendrocytes (OLGs) in ICH mice, which may be regulated by the LiCl-induced inhibition of glycogen synthase kinase-3β (GSK-3β) (79).
Peroxisome Proliferator-Activated Receptor Gamma (PPAR-γ)
The phagocytic activity of microglia is required to remove the hematoma after ICH; however, the pro-inflammatory mediators and free radicals released as a result of microglial activation and phagocytosis are toxic to neighboring cells and lead to secondary brain damage following ICH (80). ICH mouse model demonstrated that peroxisome proliferator-activated receptor gamma (PPAR-γ) prevents LPS‐induced pro-inflammatory microglial activation while facilitating microglial polarization towards the anti-inflammatory phenotype (81). Besides, PPAR-γ promotes phagocytosis in a timely and effective manner, limiting the toxic effects of hemolysis by facilitating hematoma clearance following ICH (82).
Studies have demonstrated that PPAR-γ activation is imperative for enhancing the phagocytic ability of anti-inflammatory microglia by CD36 (18). Furthermore, 15(S)-hydroxyeicosatetraenoic acid, an exogenous PPAR-γ agonist, improves functional recovery following ICH and exerts neuroprotective effects (83).
Based on in-depth basic research, PPAR-γ agonists have been widely used in clinical treatment. For example, the neuroprotective effects of statins following ICH through PPAR-γ activation and enhancement of microglia-induced erythrocyte phagocytosis have been established (84). Besides, monascin, as a novel dual agonist of PPAR-γ and Nrf2, facilitates microglial phagocytosis of the hematoma and exerts neuroprotective effects following ICH (85, 86).
Caspase Family
Caspase-mediated cascades play an essential role in mediating anti-inflammatory microglial death (87). For example, AC-YVAD-CMK can alleviate brain edema by inhibiting the activation of pro-caspase-1 and downregulating the expression of inflammation-related factors, which is accompanied by decreasing activated microglia at 24 h post-ICH (88).
Clinical studies have found that fimasartan (an angiotensin II receptor blocker) significantly reduces the activation of the caspase-1 pathway after ICH (89), suggesting that it is effective in ICH by regulating caspase-1–mediated microglial autophagy.
Matrix Metalloproteinases (MMPs)
The first serious discussions of MMP-12, which is harmful and contributes to secondary damage after ICH, emerged in 2005 (90). Subsequent studies found that MMP-9 binds to injured neurons in culture, activates pro-inflammatory microglia, and exerts neurotoxic effects after ICH (91). Based on this, further research proposed that inhibition of MMP-9 improves prognosis following ICH (92).
MMP-mediated microglial activation has become a potential therapeutic target for ICH. For example, minocycline, a widely available drug that alleviates brain damage, effectively reduces early upregulation of MMP-12 expression (93, 94)and induces anti-inflammation microglial polarization, which reduces the levels of inflammatory cytokines and the number of microglia surrounding the hematoma after ICH (86). It should be noted that although the molecular mechanism is unclear, MMP-12 expression and microglial infiltration around the hematoma are significantly reduced after stem cell transplantation following ICH (95, 96).
Iron Chelators
Iron overload is a significant cause of brain damage because iron toxicity contributes to pro-inflammatory microglial activation following collagenase-induced ICH. Therefore, reducing the accumulation of iron can moderately improve the outcomes after ICH (97). As an iron chelator, minocycline can reduce free iron and iron handling protein levels, thus prevent neuronal death (98). Besides, VK-28, a brain-permeable iron chelator, is superior to and less toxic than DFA following ICH (99, 100).
The evidence from observational studies shows that microglia function is controlled by complex regulatory networks (Table 1), an understanding of which is critical for elucidating phenotypic and genotypic variations in microglia and developing therapeutic interventions for ICH (Figure 4).

Table 1 Potential interventions for microglia polarization after intracerebral hemorrhage.
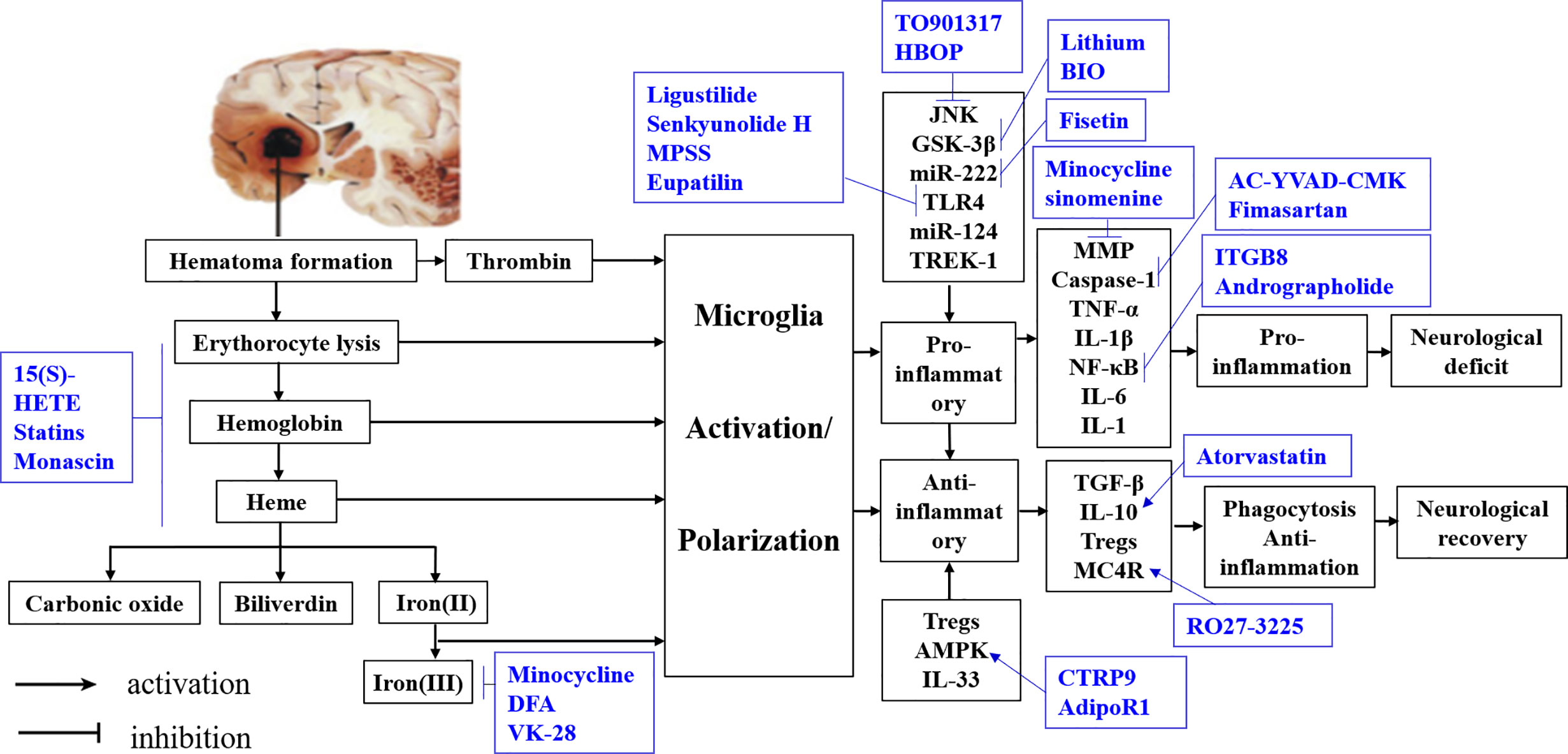
Figure 4 Summary of interventions for ICH targeting microglial polarization. Available intervention strategies are labeled blue.
Potential Therapeutic Strategies Targeting Microglia Function After Ich
Exploring the regulatory mechanism of microglial immunophenotype changes may help identify the hematoma scavenging mechanism and a precise therapeutic target for ICH. The multi-omics technologies have made significant achievements in the research of microglial activation (101). The application of systematic multi-omics approaches to precision medicine and systems biology has great potential to improve the care of patients with ICH. Notably, the target gene identified by multi-omics studies can potentially be used for drug repositioning in ICH, which is approved to be cheaper, quicker, and effective (102).
Conclusion
This review aimed to objectively discuss and assess the role of microglia in regulating neuronal injury after ICH. The findings indicate that pro-inflammatory or anti-inflammatory microglia have divergent effects, which have significant implications for understanding microglia function via intracellular and extracellular signal-regulated pathways. Besides, this review provides the first comprehensive assessment of cellular and molecular mechanisms and pathways responsible for regulating microglia, including an in-depth analysis of signaling pathways strongly associated with microglia following ICH. Notwithstanding the relatively limited number of reliable clinical trials and the lack of molecular genetic studies on the phenotypic change of microglia, this work offers valuable insights into a novel therapeutic strategy for ICH that targets microglia. Further research on interventions associated with microglial physiology is an essential next step in confirming a framework for assessing the feasibility of the novel therapy mentioned above.
Data Availability Statement
The data that support the findings of this study are openly available in pubmed at https://pubmed.ncbi.nlm.nih.gov/. Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from National Natural Science Foundation of China (81771294) and the National undergraduate innovation training project (2020105330289).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Eldahshan W, Fagan SC, Ergul A. Inflammation Within the Neurovascular Unit: Focus on Microglia for Stroke Injury and Recovery. Pharmacol Res (2019) 147:104349. doi: 10.1016/j.phrs.2019.104349
2. Bian Z, Gong Y, Huang T, Lee CZW, Bian L, Bai Z, et al. Deciphering Human Macrophage Development At Single-Cell Resolution. Nature (2020) 582(7813):571–6. doi: 10.1038/s41586-020-2316-7
3. Prinz M, Jung S, Priller J. Microglia Biology: One Century of Evolving Concepts. Cell (2019) 179(2):292–311. doi: 10.1016/j.cell.2019.08.053
4. Wan S, Cheng Y, Jin H, Guo D, Hua Y, Keep RF, et al. Microglia Activation and Polarization After Intracerebral Hemorrhage in Mice: The Role of Protease-Activated Receptor-1. Transl Stroke Res (2016) 7(6):478–87. doi: 10.1007/s12975-016-0472-8
5. Wolf SA, Boddeke HW, Kettenmann H. Microglia in Physiology and Disease. Annu Rev Physiol (2017) 79:619–43. doi: 10.1146/annurev-physiol-022516-034406
6. Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep (2018) 22(3):832–47. doi: 10.1016/j.celrep.2017.12.066
7. Yang Z, Zhao T, Zou Y, Zhang JH, Feng H. Curcumin Inhibits Microglia Inflammation and Confers Neuroprotection in Intracerebral Hemorrhage. Immunol Lett (2014) 160(1):89–95. doi: 10.1016/j.imlet.2014.03.005
8. Wang M, Hua Y, Keep RF, Wan S, Novakovic N, Xi G. Complement Inhibition Attenuates Early Erythrolysis in the Hematoma and Brain Injury in Aged Rats. Stroke (2019) 50(7):1859–68. doi: 10.1161/strokeaha.119.025170
9. Vinukonda G, Liao Y, Hu F, Ivanova L, Purohit D, Finkel DA, et al. Human Cord Blood-Derived Unrestricted Somatic Stem Cell Infusion Improves Neurobehavioral Outcome in a Rabbit Model of Intraventricular Hemorrhage. Stem Cells Transl Med (2019) 8(11):1157–69. doi: 10.1002/sctm.19-0082
10. Chang CF, Wan J, Li Q, Renfroe SC, Heller NM, Wang J. Alternative Activation-Skewed Microglia/Macrophages Promote Hematoma Resolution in Experimental Intracerebral Hemorrhage. Neurobiol Dis (2017) 103:54–69. doi: 10.1016/j.nbd.2017.03.016
11. Jing C, Bian L, Wang M, Keep RF, Xi G, Hua Y. Enhancement of Hematoma Clearance With Cd47 Blocking Antibody in Experimental Intracerebral Hemorrhage. Stroke (2019) 50(6):1539–47. doi: 10.1161/STROKEAHA.118.024578
12. Sekerdag E, Solaroglu I, Gursoy-Ozdemir Y. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Curr Neuropharmacol (2018) 16(9):1396–415. doi: 10.2174/1570159x16666180302115544
13. Su EJ, Cao C, Fredriksson L, Nilsson I, Stefanitsch C, Stevenson TK, et al. Microglial-Mediated PDGF-CC Activation Increases Cerebrovascular Permeability During Ischemic Stroke. Acta Neuropathol (2017) 134(4):585–604. doi: 10.1007/s00401-017-1749-z
14. Chen AQ, Fang Z, Chen XL, Yang S, Zhou YF, Mao L, et al. Microglia-Derived TNF-alpha Mediates Endothelial Necroptosis Aggravating Blood Brain-Barrier Disruption After Ischemic Stroke. Cell Death Dis (2019) 10(7):487. doi: 10.1038/s41419-019-1716-9
15. Geraghty JR, Davis JL, Testai FD. Neuroinflammation and Microvascular Dysfunction After Experimental Subarachnoid Hemorrhage: Emerging Components of Early Brain Injury Related to Outcome. Neurocrit Care (2019) 31(2):373–89. doi: 10.1007/s12028-019-00710-x
16. Zlokovic BV. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron (2008) 57(2):178–201. doi: 10.1016/j.neuron.2008.01.003
17. Ronaldson PT, Davis TP. Regulation of Blood-Brain Barrier Integrity by Microglia in Health and Disease: A Therapeutic Opportunity. J Cereb Blood Flow Metab (2020) 40(1_suppl):S6–s24. doi: 10.1177/0271678x20951995
18. Zhao X, Grotta J, Gonzales N, Aronowski J. Hematoma Resolution as a Therapeutic Target: The Role of Microglia/Macrophages. Stroke (2009) 40(3 Suppl):S92–4. doi: 10.1161/strokeaha.108.533158
19. Chang CF, Goods BA, Askenase MH, Hammond MD, Renfroe SC, Steinschneider AF, et al. Erythrocyte Efferocytosis Modulates Macrophages Towards Recovery After Intracerebral Hemorrhage. J Clin Invest (2018) 128(2):607–24. doi: 10.1172/jci95612
20. Wang G, Wang L, Sun XG, Tang J. Haematoma Scavenging in Intracerebral Haemorrhage: From Mechanisms to the Clinic. J Cell Mol Med (2018) 22(2):768–77. doi: 10.1111/jcmm.13441
21. Deng S, Sherchan P, Jin P, Huang L, Travis Z, Zhang JH, et al. Recombinant CCL17 Enhances Hematoma Resolution and Activation of CCR4/ERK/Nrf2/CD163 Signaling Pathway After Intracerebral Hemorrhage in Mice. Neurotherapeutics (2020) 17(4):1940–53. doi: 10.1007/s13311-020-00908-4
22. Zhuang J, Peng Y, Gu C, Chen H, Lin Z, Zhou H, et al. Wogonin Accelerates Hematoma Clearance and Improves Neurological Outcome Via the PPAR-γ Pathway After Intracerebral Hemorrhage. Transl Stroke Res (2020) 16. doi: 10.1007/s12975-020-00842-9
23. Tschoe C, Bushnell CD, Duncan PW, Alexander-Miller MA, Wolfe SQ. Neuroinflammation After Intracerebral Hemorrhage and Potential Therapeutic Targets. J Stroke (2020) 22(1):29–46. doi: 10.5853/jos.2019.02236
24. Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS Immune Privilege: Hiding in Plain Sight. Immunol Rev (2006) 213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x
25. Wang J. Preclinical and Clinical Research on Inflammation After Intracerebral Hemorrhage. Prog Neurobiol (2010) 92(4):463–77. doi: 10.1016/j.pneurobio.2010.08.001
26. Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW. Inflammation in Intracerebral Hemorrhage: From Mechanisms to Clinical Translation. Prog Neurobiol (2014) 115:25–44. doi: 10.1016/j.pneurobio.2013.11.003
27. Zhang Z, Zhang Z, Lu H, Yang Q, Wu H, Wang J. Microglial Polarization and Inflammatory Mediators After Intracerebral Hemorrhage. Mol Neurobiol (2017) 54(3):1874–86. doi: 10.1007/s12035-016-9785-6
28. Ransohoff RM. A Polarizing Question: do M1 and M2 Microglia Exist? Nat Neurosci (2016) 19(8):987–91. doi: 10.1038/nn.4338
29. Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O’Keeffe S, Phatnani HP, et al. A Neurodegeneration-Specific Gene-Expression Signature of Acutely Isolated Microglia From an Amyotrophic Lateral Sclerosis Mouse Model. Cell Rep (2013) 4(2):385–401. doi: 10.1016/j.celrep.2013.06.018
30. Martinez FO, Gordon S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep (2014) 6:13. doi: 10.12703/p6-13
31. Ajami B, Samusik N, Wieghofer P, Ho PP, Crotti A, Bjornson Z, et al. Single-Cell Mass Cytometry Reveals Distinct Populations of Brain Myeloid Cells in Mouse Neuroinflammation and Neurodegeneration Models. Nat Neurosci (2018) 21(4):541–51. doi: 10.1038/s41593-018-0100-x
32. Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar, et al. Spatial and Temporal Heterogeneity of Mouse and Human Microglia At Single-Cell Resolution. Nature (2019) 566(7744):388–92. doi: 10.1038/s41586-019-0924-x
33. Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, Huffnagle GB, et al. Macrophage M1/M2 Polarization Dynamically Adapts to Changes in Cytokine Microenvironments in Cryptococcus Neoformans Infection. mBio (2013) 4(3):e00264–13. doi: 10.1128/mBio.00264-13
34. Desale SE, Chinnathambi S. Role of Dietary Fatty Acids in Microglial Polarization in Alzheimer’s Disease. J Neuroinflamm (2020) 17(1):93. doi: 10.1186/s12974-020-01742-3
35. Lassmann H. Pathology of Inflammatory Diseases of the Nervous System: Human Disease Versus Animal Models. Glia (2020) 68(4):830–44. doi: 10.1002/glia.23726
36. García-Revilla J, Alonso-Bellido IM, Burguillos MA, Herrera AJ, Espinosa-Oliva AM, Ruiz R, et al. Reformulating Pro-Oxidant Microglia in Neurodegeneration. J Clin Med (2019) 8(10):32. doi: 10.3390/jcm8101719
37. Rangaraju S, Dammer EB, Raza SA, Rathakrishnan P, Xiao H, Gao T, et al. Identification and Therapeutic Modulation of a Pro-Inflammatory Subset of Disease-Associated-Microglia in Alzheimer’s Disease. Mol Neurodegener (2018) 13(1):24. doi: 10.1186/s13024-018-0254-8
38. Arcuri C, Mecca C, Bianchi R, Giambanco I, Donato R. The Pathophysiological Role of Microglia in Dynamic Surveillance, Phagocytosis and Structural Remodeling of the Developing Cns. Front Mol Neurosci (2017) 10:191. doi: 10.3389/fnmol.2017.00191
39. Chen S, Peng J, Sherchan P, Ma Y, Xiang S, Yan F, et al. TREM2 Activation Attenuates Neuroinflammation and Neuronal Apoptosis Via PI3K/Akt Pathway After Intracerebral Hemorrhage in Mice. J Neuroinflamm (2020) 17(1):168. doi: 10.1186/s12974-020-01853-x
40. Brown GC, St George-Hyslop PH. Deciphering Microglial Diversity in Alzheimer’s Disease. Science (2017) 356(6343):1123–4. doi: 10.1126/science.aan7893
41. Prinz M, Erny D, Hagemeyer N. Ontogeny and Homeostasis of CNS Myeloid Cells. Nat Immunol (2017) 18(4):385–92. doi: 10.1038/ni.3703
42. Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, et al. Single Cell RNA Sequencing of Human Microglia Uncovers a Subset Associated With Alzheimer’s Disease. Nat Commun (2020) 11(1):6129. doi: 10.1038/s41467-020-19737-2
43. Ochocka N, Segit P, Walentynowicz KA, Wojnicki K, Cyranowski S, Swatler J, et al. Single-Cell RNA Sequencing Reveals Functional Heterogeneity of Glioma-Associated Brain Macrophages. Nat Commun (2021) 12(1):1151. doi: 10.1038/s41467-021-21407-w
44. Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, et al. An Environment-Dependent Transcriptional Network Specifies Human Microglia Identity. Science (2017) 356(6344):11. doi: 10.1126/science.aal3222
45. Vainchtein ID, Molofsky AV. Astrocytes and Microglia: in Sickness and in Health. Trends Neurosci (2020) 43(3):144–54. doi: 10.1016/j.tins.2020.01.003
46. Lan X, Han X, Li Q, Yang QW, Wang J. Modulators of Microglial Activation and Polarization After Intracerebral Haemorrhage. Nat Rev Neurol (2017) 13(7):420–33. doi: 10.1038/nrneurol.2017.69
47. Wu CH, Shyue SK, Hung TH, Wen S, Lin CC, Chang CF, et al. Genetic Deletion or Pharmacological Inhibition of Soluble Epoxide Hydrolase Reduces Brain Damage and Attenuates Neuroinflammation After Intracerebral Hemorrhage. J Neuroinflamm (2017) 14(1):230. doi: 10.1186/s12974-017-1005-4
48. Fang Y, Tian Y, Huang Q, Wan Y, Xu L, Wang W, et al. Deficiency of TREK-1 Potassium Channel Exacerbates Blood-Brain Barrier Damage and Neuroinflammation After Intracerebral Hemorrhage in Mice. J Neuroinflamm (2019) 16(1):96. doi: 10.1186/s12974-019-1485-5
49. Zhou K, Zhong Q, Wang YC, Xiong XY, Meng ZY, Zhao T, et al. Regulatory T Cells Ameliorate Intracerebral Hemorrhage-Induced Inflammatory Injury by Modulating Microglia/Macrophage Polarization Through the IL-10/GSK3beta/PTEN Axis. J Cereb Blood Flow Metab (2017) 37(3):967–79. doi: 10.1177/0271678x16648712
50. Taylor RA, Chang CF, Goods BA, Hammond MD, Mac Grory B, Ai Y, et al. TGF-Beta1 Modulates Microglial Phenotype and Promotes Recovery After Intracerebral Hemorrhage. J Clin Invest (2017) 127(1):280–92. doi: 10.1172/jci88647
51. Yang Z, Jiang X, Zhang J, Huang X, Zhang X, Wang J, et al. Let-7a Promotes Microglia M2 Polarization by Targeting CKIP-1 Following ICH. Immunol Lett (2018) 202:1–7. doi: 10.1016/j.imlet.2018.07.007
52. Zhang XD, Fan QY, Qiu Z, Chen S. MiR-7 Alleviates Secondary Inflammatory Response of Microglia Caused by Cerebral Hemorrhage Through Inhibiting TLR4 Expression. Eur Rev Med Pharmacol Sci (2018) 22(17):5597–604. doi: 10.26355/eurrev_201809_15824
53. Liu DL, Zhao LX, Zhang S, Du JR. Peroxiredoxin 1-Mediated Activation of TLR4/NF-kappaB Pathway Contributes to Neuroinflammatory Injury in Intracerebral Hemorrhage. Int Immunopharmacol (2016) 41:82–9. doi: 10.1016/j.intimp.2016.10.025
54. Bai YY, Niu JZ. miR222 Regulates Brain Injury and Inflammation Following Intracerebral Hemorrhage by Targeting ITGB8. Mol Med Rep (2020) 21(3):1145–53. doi: 10.3892/mmr.2019.10903
55. Zhang Y, Han B, He Y, Li D, Ma X, Liu Q, et al. MicroRNA-132 Attenuates Neurobehavioral and Neuropathological Changes Associated With Intracerebral Hemorrhage in Mice. Neurochem Int (2017) 107:182–90. doi: 10.1016/j.neuint.2016.11.011
56. Wang Z, Yuan B, Fu F, Huang S, Yang Z. Hemoglobin Enhances miRNA-144 Expression and Autophagic Activation Mediated Inflammation of Microglia Via mTOR Pathway. Sci Rep (2017) 7(1):11861. doi: 10.1038/s41598-017-12067-2
57. Tan X, Yang Y, Xu J, Zhang P, Deng R, Mao Y, et al. Luteolin Exerts Neuroprotection Via Modulation of the P62/Keap1/Nrf2 Pathway in Intracerebral Hemorrhage. Front Pharmacol (2019) 10:1551. doi: 10.3389/fphar.2019.01551
58. Duan XC, Wang W, Feng DX, Yin J, Zuo G, Chen DD, et al. Roles of Autophagy and Endoplasmic Reticulum Stress in Intracerebral Hemorrhage-Induced Secondary Brain Injury In Rats. CNS Neurosci Ther (2017) 23(7):554–66. doi: 10.1111/cns.12703
59. Xiao H, Chen H, Jiang R, Zhang L, Wang L, Gan H, et al. NLRP6 Contributes to Inflammation and Brain Injury Following Intracerebral Haemorrhage by Activating Autophagy. J Mol Med (Berl) (2020) 98(9):1319–31. doi: 10.1007/s00109-020-01962-3
60. Shi H, Wang J, Wang J, Huang Z, Yang Z. Il-17A Induces Autophagy and Promotes Microglial Neuroinflammation Through ATG5 and ATG7 in Intracerebral Hemorrhage. J Neuroimmunol (2018) 323:143–51. doi: 10.1016/j.jneuroim.2017.07.015
61. Ohnishi M, Katsuki H, Fujimoto S, Takagi M, Kume T, Akaike A. Involvement of Thrombin and Mitogen-Activated Protein Kinase Pathways in Hemorrhagic Brain Injury. Exp Neurol (2007) 206(1):43–52. doi: 10.1016/j.expneurol.2007.03.030
62. Zhao L, Chen S, Sherchan P, Ding Y, Zhao W, Guo Z, et al. Recombinant CTRP9 Administration Attenuates Neuroinflammation Via Activating Adiponectin Receptor 1 After Intracerebral Hemorrhage in Mice. J Neuroinflamm (2018) 15(1):215. doi: 10.1186/s12974-018-1256-8
63. Chen S, Zhao L, Sherchan P, Ding Y, Yu J, Nowrangi D, et al. Activation of Melanocortin Receptor 4 With RO27-3225 Attenuates Neuroinflammation Through AMPK/JNK/p38 MAPK Pathway After Intracerebral Hemorrhage in Mice. J Neuroinflamm (2018) 15(1):106. doi: 10.1186/s12974-018-1140-6
64. Yang Z, Yu A, Liu Y, Shen H, Lin C, Lin L, et al. Regulatory T Cells Inhibit Microglia Activation and Protect Against Inflammatory Injury in Intracerebral Hemorrhage. Int Immunopharmacol (2014) 22(2):522–5. doi: 10.1016/j.intimp.2014.06.037
65. Wang M, Cheng L, Chen ZL, Mungur R, Xu SH, Wu J, et al. Hyperbaric Oxygen Preconditioning Attenuates Brain Injury After Intracerebral Hemorrhage by Regulating Microglia Polarization in Rats. CNS Neurosci Ther (2019) 25(10):1126–33. doi: 10.1111/cns.13208
66. Sansing LH, Harris TH, Welsh FA, Kasner SE, Hunter CA, Kariko K. Toll-Like Receptor 4 Contributes to Poor Outcome After Intracerebral Hemorrhage. Ann Neurol (2011) 70(4):646–56. doi: 10.1002/ana.22528
67. Fang H, Chen J, Lin S, Wang P, Wang Y, Xiong X, et al. CD36-Mediated Hematoma Absorption Following Intracerebral Hemorrhage: Negative Regulation by TLR4 Signaling. J Immunol (2014) 192(12):5984–92. doi: 10.4049/jimmunol.1400054
68. Yang Z, Liu B, Zhong L, Shen H, Lin C, Lin L, et al. Toll-Like receptor-4-mediated Autophagy Contributes to Microglial Activation and Inflammatory Injury in Mouse Models of Intracerebral Haemorrhage. Neuropathol Appl Neurobiol (2015) 41(4):e95–106. doi: 10.1111/nan.12177
69. Xu J, Chen Z, Yu F, Liu H, Ma C, Xie D, et al. Il-4/STAT6 Signaling Facilitates Innate Hematoma Resolution and Neurological Recovery After Hemorrhagic Stroke in Mice. Proc Natl Acad Sci U S A (2020) 117(51):32679–90. doi: 10.1073/pnas.2018497117
70. Shi SX, Li YJ, Shi K, Wood K, Ducruet AF, Liu Q. Il (Interleukin)-15 Bridges Astrocyte-Microglia Crosstalk and Exacerbates Brain Injury Following Intracerebral Hemorrhage. Stroke (2020) 51(3):967–74. doi: 10.1161/strokeaha.119.028638
71. Yu A, Duan H, Zhang T, Pan Y, Kou Z, Zhang X, et al. Il-17A Promotes Microglial Activation and Neuroinflammation in Mouse Models of Intracerebral Haemorrhage. Mol Immunol (2016) 73:151–7. doi: 10.1016/j.molimm.2016.04.003
72. Chen Z, Xu N, Dai X, Zhao C, Wu X, Shankar S, et al. Interleukin-33 Reduces Neuronal Damage and White Matter Injury Via Selective Microglia M2 Polarization After Intracerebral Hemorrhage in Rats. Brain Res Bull (2019) 150:127–35. doi: 10.1016/j.brainresbull.2019.05.016
73. Yin M, Chen Z, Ouyang Y, Zhang H, Wan Z, Wang H, et al. Thrombin-Induced, TNFR-dependent miR-181c Downregulation Promotes MLL1 and NF-κb Target Gene Expression in Human Microglia. J Neuroinflamm (2017) 14(1):132. doi: 10.1186/s12974-017-0887-5
74. Ryu J, Pyo H, Jou I, Joe E. Thrombin Induces NO Release From Cultured Rat Microglia Via Protein Kinase C, Mitogen-Activated Protein Kinase, and NF-kappa B. J Biol Chem (2000) 275(39):29955–9. doi: 10.1074/jbc.M001220200
75. Yin M, Chen Z, Ouyang Y, Zhang H, Wan Z, Wang H, et al. Thrombin-Induced, TNFR-dependent miR-181c Downregulation Promotes MLL1 and NF-kappaB Target Gene Expression in Human Microglia. J Neuroinflamm (2017) 14(1):132. doi: 10.1186/s12974-017-0887-5
76. Zheng J, Liu Z, Li W, Tang J, Zhang D, Tang X. Lithium Posttreatment Confers Neuroprotection Through Glycogen Synthase kinase-3beta Inhibition in Intracerebral Hemorrhage Rats. J Neurosurg (2017) 127(4):716–24. doi: 10.3171/2016.7.Jns152995
77. Li R, Liu Z, Wu X, Yu Z, Zhao S, Tang X. Lithium Chloride Promoted Hematoma Resolution After Intracerebral Hemorrhage Through GSK-3beta-mediated Pathways-Dependent Microglia Phagocytosis and M2-phenotype Differentiation, Angiogenesis and Neurogenesis in a Rat Model. Brain Res Bull (2019) 152:117–27. doi: 10.1016/j.brainresbull.2019.07.019
78. Zhao S, Liu Z, Yu Z, Wu X, Li R, Tang X. BIO Alleviates Inflammation Through Inhibition of GSK-3beta in a Rat Model of Intracerebral Hemorrhage. J Neurosurg (2020) 133(2):383–402. doi: 10.3171/2019.4.Jns183501
79. Li M, Xia M, Chen W, Wang J, Yin Y, Guo C, et al. Lithium Treatment Mitigates White Matter Injury After Intracerebral Hemorrhage Through Brain-Derived Neurotrophic Factor Signaling in Mice. Transl Res (2020) 217:61–74. doi: 10.1016/j.trsl.2019.12.006
80. Jung KH, Chu K, Lee ST, Kim SJ, Song EC, Kim EH, et al. Blockade of AT1 Receptor Reduces Apoptosis, Inflammation, and Oxidative Stress in Normotensive Rats With Intracerebral Hemorrhage. J Pharmacol Exp Ther (2007) 322(3):1051–8. doi: 10.1124/jpet.107.120097
81. Li Y, Zhu ZY, Lu BW, Huang TT, Zhang YM, Zhou NY, et al. Rosiglitazone Ameliorates Tissue Plasminogen Activator-Induced Brain Hemorrhage After Stroke. CNS Neurosci Ther (2019) 25(12):1343–52. doi: 10.1111/cns.13260
82. Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, et al. Hematoma Resolution as a Target for Intracerebral Hemorrhage Treatment: Role for Peroxisome Proliferator-Activated Receptor Gamma in Microglia/Macrophages. Ann Neurol (2007) 61(4):352–62. doi: 10.1002/ana.21097
83. Xu R, Wang S, Li W, Liu Z, Tang J, Tang X. Activation of Peroxisome Proliferator-Activated Receptor-Gamma by a 12/15-Lipoxygenase Product of Arachidonic Acid: A Possible Neuroprotective Effect in the Brain After Experimental Intracerebral Hemorrhage. J Neurosurg (2017) 127(3):522–31. doi: 10.3171/2016.7.Jns1668
84. Wang Y, Chen Q, Tan Q, Feng Z, He Z, Tang J, et al. Simvastatin Accelerates Hematoma Resolution After Intracerebral Hemorrhage in a PPARgamma-dependent Manner. Neuropharmacology (2018) 128:244–54. doi: 10.1016/j.neuropharm.2017.10.021
85. Wang G, Li T, Duan SN, Dong L, Sun XG, Xue F. PPAR-Gamma Promotes Hematoma Clearance Through Haptoglobin-Hemoglobin-CD163 in a Rat Model of Intracerebral Hemorrhage. Behav Neurol (2018) 2018:7646104. doi: 10.1155/2018/7646104
86. Miao H, Li R, Han C, Lu X, Zhang H. Minocycline Promotes Posthemorrhagic Neurogenesis Via M2 Microglia Polarization Via Upregulation of the TrkB/BDNF Pathway in Rats. J Neurophysiol (2018) 120(3):1307–17. doi: 10.1152/jn.00234.2018
87. Yao ST, Cao F, Chen JL, Chen W, Fan RM, Li G, et al. NLRP3 is Required for Complement-Mediated Caspase-1 and IL-1beta Activation in ICH. J Mol Neurosci (2017) 61(3):385–95. doi: 10.1007/s12031-016-0874-9
88. Liang H, Sun Y, Gao A, Zhang N, Jia Y, Yang S, et al. Ac-YVAD-cmk Improves Neurological Function by Inhibiting caspase-1-mediated Inflammatory Response in the Intracerebral Hemorrhage of Rats. Int Immunopharmacol (2019) 75:105771. doi: 10.1016/j.intimp.2019.105771
89. Yang X, Sun J, Kim TJ, Kim YJ, Ko SB, Kim CK, et al. Pretreatment With Low-Dose Fimasartan Ameliorates NLRP3 Inflammasome-Mediated Neuroinflammation and Brain Injury After Intracerebral Hemorrhage. Exp Neurol (2018) 310:22–32. doi: 10.1016/j.expneurol.2018.08.013
90. Wells JE, Biernaskie J, Szymanska A, Larsen PH, Yong VW, Corbett D. Matrix Metalloproteinase (MMP)-12 Expression has a Negative Impact on Sensorimotor Function Following Intracerebral Haemorrhage in Mice. Eur J Neurosci (2005) 21(1):187–96. doi: 10.1111/j.1460-9568.2004.03829.x
91. Wasserman JK, Schlichter LC. Minocycline Protects the Blood-Brain Barrier and Reduces Edema Following Intracerebral Hemorrhage in the Rat. Exp Neurol (2007) 207(2):227–37. doi: 10.1016/j.expneurol.2007.06.025
92. Xue M, Hollenberg MD, Yong VW. Combination of Thrombin and Matrix Metalloproteinase-9 Exacerbates Neurotoxicity in Cell Culture and Intracerebral Hemorrhage in Mice. J Neurosci (2006) 26(40):10281–91. doi: 10.1523/jneurosci.2806-06.2006
93. Wasserman JK, Zhu X, Schlichter LC. Evolution of the Inflammatory Response in the Brain Following Intracerebral Hemorrhage and Effects of Delayed Minocycline Treatment. Brain Res (2007) 1180:140–54. doi: 10.1016/j.brainres.2007.08.058
94. Wang G, Li Z, Li S, Ren J, Suresh V, Xu D, et al. Minocycline Preserves the Integrity and Permeability of BBB by Altering the Activity of DKK1-Wnt Signaling in ICH Model. Neuroscience (2019) 415:135–46. doi: 10.1016/j.neuroscience.2019.06.038
95. Liang H, Guan D, Gao A, Yin Y, Jing M, Yang L, et al. Human Amniotic Epithelial Stem Cells Inhibit Microglia Activation Through Downregulation of Tumor Necrosis Factor-Alpha, interleukin-1beta and Matrix metalloproteinase-12 In Vitro and in a Rat Model of Intracerebral Hemorrhage. Cytotherapy (2014) 16(4):523–34. doi: 10.1016/j.jcyt.2013.11.007
96. Chen M, Li X, Zhang X, He X, Lai L, Liu Y, et al. The Inhibitory Effect of Mesenchymal Stem Cell on Blood-Brain Barrier Disruption Following Intracerebral Hemorrhage in Rats: Contribution of TSG-6. J Neuroinflamm (2015) 12:61. doi: 10.1186/s12974-015-0284-x
97. Wu H, Wu T, Xu X, Wang J, Wang J. Iron Toxicity in Mice With Collagenase-Induced Intracerebral Hemorrhage. J Cereb Blood Flow Metab (2011) 31(5):1243–50. doi: 10.1038/jcbfm.2010.209
98. Zhao F, Hua Y, He Y, Keep RF, Xi G. Minocycline-Induced Attenuation of Iron Overload and Brain Injury After Experimental Intracerebral Hemorrhage. Stroke (2011) 42(12):3587–93. doi: 10.1161/strokeaha.111.623926
99. Li Q, Wan J, Lan X, Han X, Wang Z, Wang J. Neuroprotection of Brain-Permeable Iron Chelator VK-28 Against Intracerebral Hemorrhage in Mice. J Cereb Blood Flow Metab (2017) 37(9):3110–23. doi: 10.1177/0271678x17709186
100. Dai S, Hua Y, Keep RF, Novakovic N, Fei Z, Xi G. Minocycline Attenuates Brain Injury and Iron Overload After Intracerebral Hemorrhage in Aged Female Rats. Neurobiol Dis (2019) 126:76–84. doi: 10.1016/j.nbd.2018.06.001
101. Apolloni S, Amadio S, Fabbrizio P, Morello G, Spampinato AG, Latagliata EC, et al. Histaminergic Transmission Slows Progression of Amyotrophic Lateral Sclerosis. J Cachexia Sarcopenia Muscle (2019) 10(4):872–93. doi: 10.1002/jcsm.12422
Keywords: intracerebral hemorrhage, microglia, phenotype switch, neuroimmunology, neuroinflammation
Citation: Liu J, Liu L, Wang X, Jiang R, Bai Q and Wang G (2021) Microglia: A Double-Edged Sword in Intracerebral Hemorrhage From Basic Mechanisms to Clinical Research. Front. Immunol. 12:675660. doi: 10.3389/fimmu.2021.675660
Received: 03 March 2021; Accepted: 19 April 2021;
Published: 06 May 2021.
Edited by:
Amanda Sierra, Achucarro Basque Center for Neuroscience, SpainReviewed by:
Adam Denes, Institute of Experimental Medicine (MTA), HungarySusanne Kooistra, University Medical Center Groningen, Netherlands
Copyright © 2021 Liu, Liu, Wang, Jiang, Bai and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaiqing Wang, d2FuZ2dxMDhAMTI2LmNvbQ==; d2FuZ2dxMDhAMTYzLmNvbQ==; orcid.org/0000-0002-8977-1383
†These authors have contributed equally to this work