 Hengqi B. Zheng
Hengqi B. Zheng M. Teresa de la Morena
M. Teresa de la Morena David L. Suskind1,2
David L. Suskind1,2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 26 May 2021
Sec. Primary Immunodeficiencies
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.675186
This article is part of the Research Topic Genetic Studies of Immune Dysregulation: Exploration of the Knowns and Unknowns with Genomics View all 7 articles
Very Early Onset Inflammatory Bowel Disease (VEO-IBD) represents a cohort of inflammatory bowel disease (IBD) patients diagnosed before 6 years of age. Unlike IBD diagnosed at older ages, VEO-IBD can be associated with underlying primary immunodeficiencies. VEO-IBD has been linked to monogenic variations in over 70 genes involved in multiple pathways of immunity. As sequencing technologies and platforms evolve and become readily available, an increasing number of genes linked to VEO-IBD have emerged. Although monogenic defects are rare in VEO-IBD, diagnosis of these variants can often dictate specific treatment. In this mini-review, we set out to describe monogenic variants previously characterized in multiple patients in the literature that contribute to VEO-IBD, diagnostic tools, unique treatment modalities for specific genetic diagnoses, and future directions in the field of VEO-IBD. Although this mini-review is by no means comprehensive of all the novel monogenic variants linked to VEO-IBD, we hope to provide relevant information that is readily accessible to clinicians and educators.
Inflammatory bowel disease (IBD) which includes ulcerative colitis, Crohn disease, and indeterminate IBD, are autoimmune diseases of the gastrointestinal tract. While the etiopathogenesis of IBD is not fully elucidated, IBD is believed to be a result of an exaggerated host inflammatory response to the resident intestinal microbiome. Both genetic and environmental factors have been implicated in the development of IBD (1). IBD diagnosed during childhood or adolescence comprises a fourth of all diagnosed IBD (2). Very Early Onset Inflammatory Bowel Disease (VEO-IBD) represents a subgroup of pediatric IBD diagnosed before the age of 6 years (3) with further subclassification into infantile-onset IBD when diagnosed before 2 years of age and neonatal-onset if diagnosed by 28 days of age (3). Genome-wide association studies (GWAS) reveal the involvement of over 260 loci in IBD. While the associated genotypic mutations are known, the mechanism by which most mutations contribute to disease phenotype development remains to be understood (1, 4). In older children and adults, IBD is more likely polygenic (1). In contrast, VEO-IBD can be rarely associated with monogenic mutations which is often linked to inborn errors of immunity (5). Depending on the patient population, parental consanguinity can be associated with higher risk for genetic mutations in infantile and neonatal forms of IBD. Parental history of consanguinity should prompt a heightened level of suspicion for underlying monogenic variation of VEO-IBD (6, 7). Over 70 genes have been causatively linked to VEO-IBD (3) and additional genetic candidates are being discovered as sequencing technology become faster and provide wider genomic coverage.
The incidence of pediatric IBD is between 2 to 12 cases annually per 100,000 children in industrialized countries (8–10). Over the past decade, the incidence of pediatric IBD and VEO-IBD has continued to rise (8). VEO-IBD represents the fastest growing age group of IBD in some countries. A recent Canadian multi-province population study showed a rising incidence of VEO-IBD (6 months to 5 years) by about 7% per year (10). Thus far, the etiology behind the rise in incidence in pediatric IBD and VEO-IBD is unclear and further research efforts are needed to alleviate disease and healthcare burden.
There is a growing number of causative monogenic variants of VEO-IBD. These monogenic variants are clinically important and dictate divergent pathways in treatment of VEO-IBD. Monogenic etiologies may underlie 15-20% of VEO-IBD patients (3, 11–14). To date, the monogenic variants can be classified into 5 distinct groups: (1) Epithelial barrier defects; (2) Phagocytic defects; (3) T and B cell defects; (4) T regulatory cells and signaling; and (5) Hyper- and auto-inflammatory conditions (see Table 1) (3, 11–13).
1. Epithelial barrier defects can result in inflammation of the skin and intestines early on in life. The IKBKG gene encodes the regulatory protein NEMO on the X chromosome which is a subunit of the of NF-kB kinase (IKK) complex needed for activation of the NF-kB family of transcription factors and X-linked ectodermal dysplasia and antibody deficiencies occurs in the absence of a functional NEMO protein (15). ADAM17 loss-of-function mutation leads to a lack of disintegrin and metalloproteinase 17 which converts TNF-alpha from membrane-bound form to soluble TNF-alpha (16). Clinical manifestations of ADAM17 deficiency can lead to erythematous psoriasiform rash, dermatitis, and diarrhea (16). TTC7A gene encodes for a regulatory protein which with EFR3 homolog B to regulate phosphatidylinositol kinase to maintain plasma membrane homeostasis (17). TTC7A deficiency can present in the neonatal period with intestinal atresia, apoptotic enterocolitis, and severe combined immunodeficiency (SCID) (17). Dystrophic epidermolysis bullosa and Crohn’s disease can occur with mutations in the COL7A1 which encodes for collagen VII (18). Kindler syndrome results from mutations in the FERMT1 gene whose gene product are adhesion proteins binding cytoplasmic domains and clinically presents with skin blistering, atrophy and cancer along with ulcerative colitis (19–21). Loeys-Dietz Syndrome results from transforming growth factor beta receptors (TGFBR) 1 and 2 mutations leading to cytokine signaling defects manifesting as severe IBD, skeletal abnormalities, craniofacial abnormalities, and vascular injury (22). Aberrations in the SLC9A3 gene which encodes for intestinal chloride/bicarbonate exchanger, leads to congenital chloride diarrhea, and gain of function GUCY2 which is an intestinal receptor for bacterial enterotoxins, leads to familial diarrhea disorders (23, 24).
2. Phagocytic defects can lead to alterations in pathogen recognition and clearance. Chronic granulomatous disease (CGD) is a clinical manifestation of defective phagocytic activity in pathogen killing and clearance. Components of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex [gp91-phox (CYBB), p22-phox (CYBA), p47-phox (NCF1), p67-phox (NCF2), p40-phox (NCF4)] are defective, most commonly in granulocytes (25). Clinical manifestations of CGD include wide phenotypic variations with severe bacterial and fungal infections throughout the body, cutaneous and deep organ abscesses, and intestinal inflammation appearing similar to fistulizing Crohn’s disease with microscopic granulomatous colitis (26). Intestinal obstruction and strictures along with perianal abscesses and fistulae can be present as non-infectious manifestations of CGD (27, 28). Leukocyte adhesion defect 1 (LAD1) results from a mutation in ITGB2 gene encoding CD18, a subunit of the beta2 integrin leading to impairment of leukocyte migration (29). LAD1 is associated with an IBD phenotype, severe infection, delayed umbilical cord separation, and a high reactive leukocytosis consistent with immobile peripheral granulocytes (29).
3. T and B cell defects within the adaptive immune system can present with intestinal inflammation. Mutations in genes that affect the development or function of T and B cells [DCLRE1C(ARTEMIS), ZAP70, RAG1/2, IL2RG, ILRA7, LIG4, JAK3, ADA] can result in intestinal inflammation along with recurrent infections from severe combined immunodeficiency (SCID) (30–32). Omenn syndrome is an autosomal recessive type of SCID with residual T cell function manifesting severe eczema, hair and cartilage hypoplasia, and intestinal inflammation with defects in IL7R (33, 34). Cytotoxic T-lymphocyte antigen 4 (CTLA4) encodes for a protein receptor which functions as a negative regulator of T regulatory cells (35). CTLA4 protein deficiency leads to a lack of self-tolerance and hyperinflammatory pathway manifesting in enteropathy, cytopenia, type 1 diabetes, and respiratory symptoms (35). Mutations in the ICOS gene encoding for costimulatory receptor for inducible T cell activation can present with common variable immunodeficiency (CVID) resulting in recurrent bacterial and viral infections, splenomegaly, and colitis (36, 37). Bruton’s or X-linked agammaglobulinemia with a defect in the Bruton’s tyrosine kinase (Btk) gene leads to sinusitis, acute otitis media, and colitis (38). Wiskott-Aldrich Syndrome (WAS) occurs with a mutated WAS gene leading to a reduced or absent WAS protein expression which leads to actin filament defect in hematopoietic cells (39). WAS patients can present with the classic findings of thrombocytopenia, eczema, eosinophilia, and immune deficiencies along with colitis (39).
4. T regulatory (Treg) cells and the regulatory pathway play a critical role in the maintenance of intestinal homeostasis. Forkhead box protein 3 (FOXP3) is a critical transcription factor for CD4+ Foxp3+ Tregs and pathogenic variants in FOXP3 leads to immunodysregulation, polyendocrinopathy, enteropathy X-linked (IPEX) syndrome and prototype of a group of disorders called PIRD (Primary Immune Regulatory Disorders) (40, 41). Over 63 FOXP3 mutations have been reported though severity of disease is independent of level of protein expression. Classic IPEX syndrome presents as the clinical triad of profuse diarrhea (autoimmune enteropathy), type-1 diabetes (endocrinopathy), and dermatitis with a propensity for skin and respiratory infections (42). In addition to classical presentation, variable clinical manifestations of IPEX patients show no apparent genotype-phenotype correlation and disease course or outcome (43). Other inborn errors of immunity can manifest in an IPEX-like manner with normal FOXP3 expression, associated immune dysregulation, eczema, and enteropathy (44). Gain of function mutations in signal transducer and activator of transcription1 (STAT1) and 3 (STAT3) can lead to enteropathy, severe viral and bacterial infections, and endocrinopathy (44). LPS-responsive beige-like anchor (LRBA) mutation leads absent or dysfunctional protein needed for Treg function and can present with enteropathy, cytopenias, lymphadenopathy, and hepatosplenomegaly (44). IL2RA mutations leads to CD25 protein deficiency and impaired Treg function, resulting in enteropathy, eczema, recurrent viral infections, and autoimmune anemia (45). Defects in the IL-10 pathway were some of the first monogenic variants discovered to be associated with VEO-IBD especially with presentation during the neonatal period (46, 47). IL-10 is a regulatory cytokine which limits secretion of proinflammatory cytokines and acts on multiple innate immune cells such as macrophages, monocytes, and Natural Killer (NK) cells along with adaptive immune cells (48). Aberrations in the IL-10 signaling pathway leaves pro-inflammatory pathways unopposed and can manifest as severe colitis, perianal fistulas, folliculitis, and occasional arthritis (46–48). The IL-10 hetrotetrameric receptor consists of two alpha subunits (IL-10R1) and two beta subunits (IL-10R2) and results in downstream activation of signal transducer and activator of transcription (STAT-3) (48). Mutations in the genes IL-10RA and IL-10RB which encode for IL-10R1 and IL-10R2 are defects leading to infant onset-IBD (46, 47). IL-10R deficiency patients have been reported to develop large B-cell lymphoma through a mechanism that is yet to be fully elucidated but thought to be due to an impaired immune surveillance (49, 50).
5. Autoimmune and hyperinflammatory systemic conditions can manifest with intestinal inflammation. X-linked inhibitor of apoptosis (XIAP) deficiency or X-linked lymphoproliferative syndrome type 2 (XLP-2) can result from various mutations within domains of BIR2 (51, 52). XIAP interacts with the serine/threonine-protein kinase 2 (RIPK2) which further mediates nuclear factor kB (NF-kB) for immune cell activation, inhibits caspase activity, and functions in the nucleotide-binding oligomerization domain-containing 1/2 (NOD1/2) intracellular pattern recognition receptor signaling pathway for maintaining intestinal homeostasis with the microbiota through the innate immune system (53, 54). Clinically, XIAP patients have recurrent splenomegaly, hemophagocytic lymphohistiocytosis (HLH) triggered by Epstein-Barr virus (EBV), and 30% with fistulizing IBD (54, 55). Mutations in NLRC4 have been reported in patients with infantile enterocolitis and autoinflammation due to constitutive inflammasome activation and production of both IL1B and IL18 (56, 57). Mevalonate kinase deficiency results with mutations in the MVK gene which leads to an accumulation of substrate mevalonate (58). Clinical symptoms manifest with a pro-inflammatory cytokine response mediated by IL-1b with episodic fevers, peritonitis, arthritis, and bloody enterocolitis (58). Tripartite motif-containing 22 (TRIM22) encodes for ubiquitin ligase which plays a role in differentiation of lymphocytes and found in macrophages and interacts with the NOD2 pathway (59). Through whole exome sequencing methods, TRIM22 mutations were recently recognized in as a causative variant in VEO-IBD with perianal disease and fistulas (59).
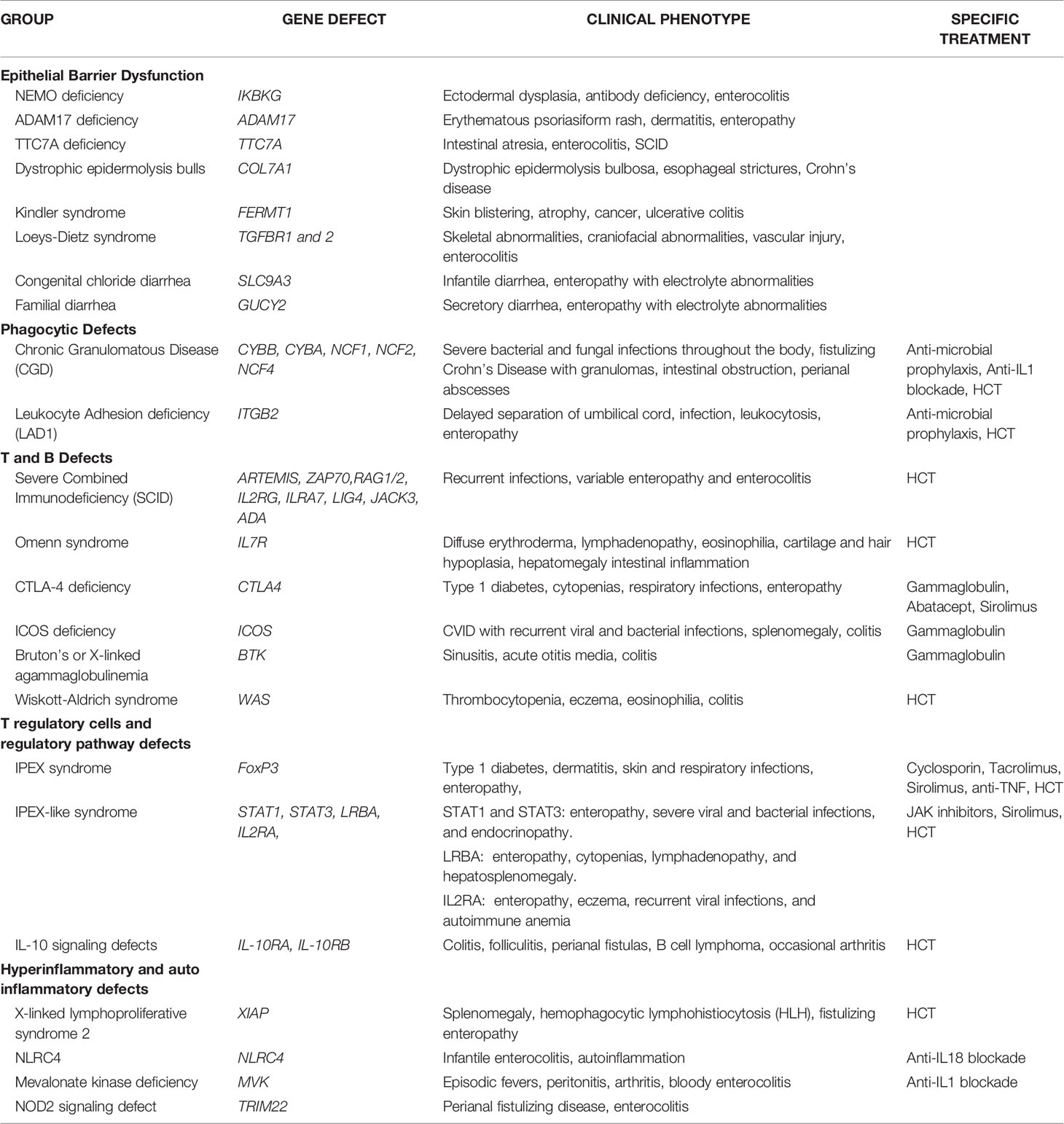
Table 1 Genetic and clinical manifestations in VEO-IBD.
VEO-IBD with underlying genetic mutation can present a diagnostic challenge. Significant time can lapse from symptom onset to final diagnosis. A high index of suspicion is required for patients presenting early in life. An expedited evaluation is required for these patients including comprehensive exam, laboratory, endoscopic, histologic, and genetic workups. A multi-disciplinary approach is required for a comprehensive evaluation of these patients including input from gastroenterology, immunology, genetics, and nutrition. A thorough evaluation is crucial especially in neonatal- and infantile-onset IBD as monogenic etiologies underlie up to 20% of VEO-IBD patients (12, 13, 60). Basic laboratory studies including complete blood count (CBC) with differential, inflammatory markers (CRP, ESR), comprehensive metabolic panel, and stool studies are often the first line of workup. Immunological panels to evaluate T cell population and memory subsets, B cell subsets, and NK cells by flow cytometry along with immunoglobulin levels and response to vaccinations can detect classic defects in adaptive immunity. A neutrophil oxidative burst assay using dihydrorhodamine identifies patients with chronic granulomatous disease. Flow cytometric based assays can also be done with intracellular protein expression looking for deficiencies in FOXP3 leading to IPEX and XIAP especially in male patients (61, 62). If available, cytokine and chemokine panels (IL-1, IL-2, IL-6, IL-10, IL-18, TNF-alpha, IFN-gamma) can be used to identify specific pathway defects, target treatment, and detect complications such as HLH development in XIAP (63, 64).
Endoscopy with histologic evaluation via hematoxylin and eosin (H+E) stain aids in the diagnosis of VEO-IBD though lacks diagnostic specificity. The likelihood of an underlying genetic variant increases if enteritis with small bowel involvement or colitis with perianal disease is present within the VEO-IBD cohort as compared to the VEO-IBD patients with isolated colitis (65). Currently there are limited pathognomonic histological findings to suggest a monogenic cause in VEO-IBD though studies describe the increased presence of apoptosis, severe chronic architectural changes, villous blunting, and abundance of eosinophils with VEO-IBD as compared to older onset pediatric IBD (66). Some histopathological findings for specific primary immunodeficiencies should be considered when reviewing biopsies. SCID microscopic features include the presence of epithelial cell apoptosis, eosinophilic and neutrophilic infiltrations with villous atrophy (16). CGD histology may contain multiple non-caseating granulomas with minimal surrounding inflammation (13). IPEX enteropathy can endoscopically and histologically appear as villous blunting and chronic inflammation within the lamina propria (42).
Given the brevity of this mini-review, we aim to describe therapies for a few monogenic defects in VEO-IBD. In general, therapeutic options are patient specific and are influenced by underlying genetic variation or primary immunodeficiency. Interventions can include nutritional therapies, immunosuppression, as well as hematopoietic stem cell transplantation depending on the underlying genetic variations or primary immunodeficiencies (3, 11, 12, 67). In addition to nonspecific immunosuppression such as corticosteroids, some targeted treatment options exist and depend on timely diagnosis of the underlying immunologic dysfunction. Therapy for IPEX has included a range of immunosuppression regimens including yclosporin, tacrolimus, anti-TNF, and mammalian target of rapamycin (mTOR) inhibitors. Risk of infection is balanced against level of immunosuppression required to control systemic inflammation (42). Hematopoietic cell transplantation (HCT) has been performed in IPEX patients (42). In addition, anti-IL1 blockade has also been used in CGD patients with high levels of IL-1 with variable success (68, 69). Anti-microbial prophylaxis including anti-fungal and anti-bacterial medications is needed in patients with CGD and LAD1 (27, 28). HCT is the definitive therapy for phagocytic defects (70). HCT is also a treatment modality for treatment refractory XIAP and deters the risk of developing of hemophagocytic lymphohistiocytosis (HLH) (51). Mevalonate kinase deficiency (Hyper IgD syndrome) may also benefit from anti-IL1 blockade in efforts to temper the pro-inflammatory cascade (59). The diagnosis of SCID, Omenn syndrome, and WAS requires early intervention with HCT prior to the emergence of complications (39). The gastrointestinal manifestations of CTLA-4 haploinsufficiency and LRBA deficiency can be treated with gammaglobulin infusions and a number of immunomodulating agents targeting the activation of T-cells. Sirolimus, a mammalian target of rapamycin has been used successfully with caution though potential side effects profile of CMV reactivation, respiratory infections and sepsis are reported in CTLA-4 (35). Abatacept, a fusion protein containing the extracellular domain of CTLA-4, has been used successfully in patients with CTLA4 haploinsufficiency and as long-term therapy for those with LRBA deficiency (71). In severe cases, CTLA 4 may require HCT (35).
Making the correct genetic diagnosis is imperative as therapeutic interventions depends on targeting the underlying immune dysregulation. Timely identification of specific monogenic variants of VEO-IBD represents a diagnostic paradigm for patient specific precision medicine. To date, multiple sequencing techniques are offered to identify monogenic IBD and are more accessible with the advent of next-generation sequencing (NGS).
Targeted gene panels (TGP) have been developed and employed at institutions nationally. Depending on the selection and number of genes included, panels sequencing can offer high coverage of the most likely genetic culprits with a high diagnostic accuracy (72). Whole exome sequencing (WES) provides higher coverage sequencing of coding regions of the genome at higher costs depending on depth of reads. There may be some limitations in utilizing commercially available WES as some genes such as IKBKG associated with NEMO deficiency and NCF1 associated with CGD may have pseudo gene loci (60, 73, 74) Whole genome sequencing (WGS) offers the ability to capture further sequencing with the capacity to pick up copy number variations and examine noncoding areas with promoters and enhancers (72, 75, 76). However, without the availability of large research centers, WES and WGS can be difficult to come by and costly to perform.
The advent of RNA-sequencing offers a glimpse into how transcriptomics may transform our diagnostic capacity. RNA sequencing can offer target organ tissue level gene expression as certain cell types (epithelial cells, Paneth cells, M cells etc.) exist predominantly within the gastrointestinal tract. With drop-seq (77), single-cell RNA-sequencing (scRNA-seq) brings the capability to interrogate individual cellular transcripts to determine pathogenic pathways even within rare cellular subpopulations previously inaccessible using conventional techniques. A recent scRNA-seq study describes a cellular module called GIMATS (consisting of IgG plasma cells, phagocytes, activated T cells, and stromal cells) could indicate resistance to anti-TNF therapy in adult IBD patients (78). Currently both bulk and single-cell RNA-sequencing are still in the research stages of discovery but have the potential to reveal further genetic defects.
VEO-IBD represents a unique group of pediatric IBD patients diagnosed in children less than 6 years of age. These patients require a focused and multidisciplinary approach that can harmonize expertise between gastroenterology, immunology, and genetics (79). Rapid innovations in genetic molecular sequencing techniques have identified novel mutations and genetic linkage of phenotype and clinical manifestations. VEO-IBD highlights the importance and need for precision medicine in the clinical setting of IBD care.
HZ wrote the first draft of the manuscript. DS and MM wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-Microbe Interactions Have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature (2012) 491(7422):119–24. doi: 10.1038/nature11582
2. Kelsen J, Baldassano RN. Inflammatory Bowel Disease: The Difference Between Children and Adults. Inflammation Bowel Dis (2008) 14 Suppl 2:S9–11. doi: 10.1002/ibd.20560
3. Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, Elkadri A, et al. The Diagnostic Approach to Monogenic Very Early Onset Inflammatory Bowel Disease. Gastroenterology (2014) 147(5):990–1007 e3. doi: 10.1053/j.gastro.2014.07.023
4. Yang SK, Hong M, Zhao W, Jung Y, Baek J, Tayebi N, et al. Genome-Wide Association Study of Crohn’s Disease in Koreans Revealed Three New Susceptibility Loci and Common Attributes of Genetic Susceptibility Across Ethnic Populations. Gut (2014) 63(1):80–7. doi: 10.1136/gutjnl-2013-305193
5. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification From the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
6. Fang YH, Luo YY, Yu JD, Lou JG, Chen J. Phenotypic and Genotypic Characterization of Inflammatory Bowel Disease in Children Under Six Years of Age in China. World J Gastroenterol (2018) 24(9):1035–45. doi: 10.3748/wjg.v24.i9.1035
7. Al-Hussaini A, El Mouzan M, Hasosah M, Al-Mehaidib A, AL K, Saadah OI, et al. Clinical Pattern of Early-Onset Inflammatory Bowel Disease in Saudi Arabia: A Multicenter National Study. Inflammation Bowel Dis (2016) 22(8):1961–70. doi: 10.1097/MIB.0000000000000796
8. Benchimol EI, Fortinsky KJ, Gozdyra P, Van den Heuvel M, Van Limbergen J, Griffiths AM. Epidemiology of Pediatric Inflammatory Bowel Disease: A Systematic Review of International Trends. Inflammation Bowel Dis (2011) 17(1):423–39. doi: 10.1002/ibd.21349
9. Benchimol EI, Manuel DG, To T, Griffiths AM, Rabeneck L, Guttmann A. Development and Use of Reporting Guidelines for Assessing the Quality of Validation Studies of Health Administrative Data. J Clin Epidemiol (2011) 64(8):821–9. doi: 10.1016/j.jclinepi.2010.10.006
10. Grieci T, Butter A. The Incidence of Inflammatory Bowel Disease in the Pediatric Population of Southwestern Ontario. J Pediatr Surg (2009) 44(5):977–80. doi: 10.1016/j.jpedsurg.2009.01.038
11. Conrad MA, Kelsen JR. Genomic and Immunologic Drivers of Very Early-Onset Inflammatory Bowel Disease. Pediatr Dev Pathol (2019) 22(3):183–93. doi: 10.1177/1093526619834807
12. Ouahed J, Spencer E, Kotlarz D, Shouval DS, Kowalik M, Peng K, et al. Very Early Onset Inflammatory Bowel Disease: A Clinical Approach With a Focus on the Role of Genetics and Underlying Immune Deficiencies. Inflammation Bowel Dis (2020) 26(6):820–42. doi: 10.1093/ibd/izz259
13. Kelsen JR, Sullivan KE, Rabizadeh S, Singh N, Snapper S, Elkadri A, et al. North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition Position Paper on the Evaluation and Management for Patients With Very Early-Onset Inflammatory Bowel Disease. J Pediatr Gastroenterol Nutr (2020) 70(3):389–403. doi: 10.1097/MPG.0000000000002567
14. Uhlig HH, Charbit-Henrion F, Kotlarz D, Shouval DS, Schwerd T, Strisciuglio C, et al. Clinical Genomics for the Diagnosis of Monogenic Forms of Inflammatory Bowel Disease: A Position Paper From The Paediatric IBD Porto Group of ESPGHAN. J Pediatr Gastroenterol Nutr (2020) 72(3):456–73. doi: 10.1097/MPG.0000000000003017
15. Karamchandani-Patel G, Hanson EP, Saltzman R, Kimball CE, Sorensen RU, Orange JS. Congenital Alterations of NEMO Glutamic Acid 223 Result in Hypohidrotic Ectodermal Dysplasia and Immunodeficiency With Normal Serum IgG Levels. Ann Allergy Asthma Immunol (2011) 107(1):50–6. doi: 10.1016/j.anai.2011.03.009
16. Blaydon DC, Biancheri P, Di WL, Plagnol V, Cabral RM, Brooke MA, et al. Inflammatory Skin and Bowel Disease Linked to ADAM17 Deletion. N Engl J Med (2011) 365(16):1502–8. doi: 10.1056/NEJMoa1100721
17. Avitzur Y, Guo C, Mastropaolo LA, Bahrami E, Chen H, Zhao Z, et al. Mutations in Tetratricopeptide Repeat Domain 7A Result in a Severe Form of Very Early Onset Inflammatory Bowel Disease. Gastroenterology (2014) 146(4):1028–39. doi: 10.1053/j.gastro.2014.01.015
18. Zimmer KP, Schumann H, Mecklenbeck S, Bruckner-Tuderman L. Esophageal Stenosis in Childhood: Dystrophic Epidermolysis Bullosa Without Skin Blistering Due to Collagen VII Mutations. Gastroenterology (2002) 122(1):220–5. doi: 10.1053/gast.2002.30428
19. Ussar S, Moser M, Widmaier M, Rognoni E, Harrer C, Genzel-Boroviczeny O, et al. Loss of Kindlin-1 Causes Skin Atrophy and Lethal Neonatal Intestinal Epithelial Dysfunction. PloS Genet (2008) 4(12):e1000289. doi: 10.1371/journal.pgen.1000289
20. Ussar S, Wang HV, Linder S, Fassler R, Moser M. The Kindlins: Subcellular Localization and Expression During Murine Development. Exp Cell Res (2006) 312(16):3142–51. doi: 10.1016/j.yexcr.2006.06.030
21. Kern JS, Herz C, Haan E, Moore D, Nottelmann S, von Lilien T, et al. Chronic Colitis Due to an Epithelial Barrier Defect: The Role of Kindlin-1 Isoforms. J Pathol (2007) 213(4):462–70. doi: 10.1002/path.2253
22. Naviglio S, Arrigo S, Martelossi S, Villanacci V, Tommasini A, Loganes C, et al. Severe Inflammatory Bowel Disease Associated With Congenital Alteration of Transforming Growth Factor Beta Signaling. J Crohns Colitis (2014) 8(8):770–4. doi: 10.1016/j.crohns.2014.01.013
23. Makela S, Kere J, Holmberg C, Hoglund P. SLC26A3 Mutations in Congenital Chloride Diarrhea. Hum Mutat (2002) 20(6):425–38. doi: 10.1002/humu.10139
24. Fiskerstrand T, Arshad N, Haukanes BI, Tronstad RR, Pham KD, Johansson S, et al. Familial Diarrhea Syndrome Caused by an Activating GUCY2C Mutation. N Engl J Med (2012) 366(17):1586–95. doi: 10.1056/NEJMoa1110132
25. Schappi MG, Smith VV, Goldblatt D, Lindley KJ, Milla PJ. Colitis in Chronic Granulomatous Disease. Arch Dis Child (2001) 84(2):147–51. doi: 10.1136/adc.84.2.147
26. Alimchandani M, Lai JP, Aung PP, Khangura S, Kamal N, Gallin JI, et al. Gastrointestinal Histopathology in Chronic Granulomatous Disease: A Study of 87 Patients. Am J Surg Pathol (2013) 37(9):1365–72. doi: 10.1097/PAS.0b013e318297427d
27. Anjani G, Vignesh P, Joshi V, Shandilya JK, Bhattarai D, Sharma J, et al. Recent Advances in Chronic Granulomatous Disease. Genes Dis (2020) 7(1):84–92. doi: 10.1016/j.gendis.2019.07.010
28. Henrickson SE, Jongco AM, Thomsen KF, Garabedian EK, Thomsen IP. Noninfectious Manifestations and Complications of Chronic Granulomatous Disease. J Pediatr Infect Dis Soc (2018) 7(suppl_1):S18–24. doi: 10.1093/jpids/piy014
29. van de Vijver E, Maddalena A, Sanal O, Holland SM, Uzel G, Madkaikar M, et al. Hematologically Important Mutations: Leukocyte Adhesion Deficiency (First Update). Blood Cells Mol Dis (2012) 48(1):53–61. doi: 10.1016/j.bcmd.2011.10.004
30. Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R Expression in T(-)B(+)NK(+) Severe Combined Immunodeficiency. Nat Genet (1998) 20(4):394–7. doi: 10.1038/3877
31. Dadi HK, Simon AJ, Roifman CM. Effect of CD3delta Deficiency on Maturation of Alpha/Beta and Gamma/Delta T-cell Lineages in Severe Combined Immunodeficiency. N Engl J Med (2003) 349(19):1821–8. doi: 10.1056/NEJMoa031178
32. Rohr J, Pannicke U, Doring M, Schmitt-Graeff A, Wiech E, Busch A, et al. Chronic Inflammatory Bowel Disease as Key Manifestation of Atypical ARTEMIS Deficiency. J Clin Immunol (2010) 30(2):314–20. doi: 10.1007/s10875-009-9349-x
33. Felgentreff K, Perez-Becker R, Speckmann C, Schwarz K, Kalwak K, Markelj G, et al. Clinical and Immunological Manifestations of Patients With Atypical Severe Combined Immunodeficiency. Clin Immunol (2011) 141(1):73–82. doi: 10.1016/j.clim.2011.05.007
34. Notarangelo LD. Primary Immunodeficiencies. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S182–94. doi: 10.1016/j.jaci.2009.07.053
35. Schwab C, Gabrysch A, Olbrich P, Patino V, Warnatz K, Wolff D, et al. Phenotype, Penetrance, and Treatment of 133 Cytotoxic T-lymphocyte Antigen 4-Insufficient Subjects. J Allergy Clin Immunol (2018) 142(6):1932–46.
36. Takahashi N, Matsumoto K, Saito H, Nanki T, Miyasaka N, Kobata T, et al. Impaired CD4 and CD8 Effector Function and Decreased Memory T Cell Populations in ICOS-deficient Patients. J Immunol (2009) 182(9):5515–27. doi: 10.4049/jimmunol.0803256
37. Schepp J, Chou J, Skrabl-Baumgartner A, Arkwright PD, Engelhardt KR, Hambleton S, et al. 14 Years After Discovery: Clinical Follow-Up on 15 Patients With Inducible Co-Stimulator Deficiency. Front Immunol (2017) 8:964. doi: 10.3389/fimmu.2017.00964
38. Maekawa K, Yamada M, Okura Y, Sato Y, Yamada Y, Kawamura N, et al. X-Linked Agammaglobulinemia in a 10-Year-Old Boy With a Novel Non-Invariant Splice-Site Mutation in Btk Gene. Blood Cells Mol Dis (2010) 44(4):300–4. doi: 10.1016/j.bcmd.2010.01.004
39. Catucci M, Castiello MC, Pala F, Bosticardo M, Villa A. Autoimmunity in Wiskott-Aldrich Syndrome: An Unsolved Enigma. Front Immunol (2012) 3:209. doi: 10.3389/fimmu.2012.00209
40. Barzaghi F, Passerini L, Bacchetta R. Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome: A Paradigm of Immunodeficiency With Autoimmunity. Front Immunol (2012) 3:211. doi: 10.3389/fimmu.2012.00211
41. Chan AY, Torgerson TR. Primary Immune Regulatory Disorders: A Growing Universe of Immune Dysregulation. Curr Opin Allergy Clin Immunol (2020) 20(6):582–90. doi: 10.1097/ACI.0000000000000689
42. van der Vliet HJ, Nieuwenhuis EE. IPEX as a Result of Mutations in FOXP3. Clin Dev Immunol (2007) 2007:89017. doi: 10.1155/2007/89017
43. Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, et al. Long-Term Follow-Up of IPEX Syndrome Patients After Different Therapeutic Strategies: An International Multicenter Retrospective Study. J Allergy Clin Immunol (2018) 141(3):1036–49.e5.
44. Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, et al. Clinical, Immunological, and Molecular Heterogeneity of 173 Patients With the Phenotype of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (Ipex) Syndrome. Front Immunol (2018) 9:2411. doi: 10.3389/fimmu.2018.02411
45. Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 Deficiency Causes an Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked-Like Syndrome, and Defective IL-10 Expression From CD4 Lymphocytes. J Allergy Clin Immunol (2007) 119(2):482–7. doi: 10.1016/j.jaci.2006.10.007
46. Glocker EO, Frede N, Perro M, Sebire N, Elawad M, Shah N, et al. Infant Colitis–It’s in the Genes. Lancet (2010) 376(9748):1272. doi: 10.1016/S0140-6736(10)61008-2
47. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, et al. Inflammatory Bowel Disease and Mutations Affecting the Interleukin-10 Receptor. N Engl J Med (2009) 361(21):2033–45. doi: 10.1056/NEJMoa0907206
48. Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the Interleukin-10 Receptor. Annu Rev Immunol (2001) 19:683–765. doi: 10.1146/annurev.immunol.19.1.683
49. Neven B, Mamessier E, Bruneau J, Kaltenbach S, Kotlarz D, Suarez F, et al. A Mendelian Predisposition to B-Cell Lymphoma Caused by IL-10R Deficiency. Blood (2013) 122(23):3713–22. doi: 10.1182/blood-2013-06-508267
50. Shouval DS, Ebens CL, Murchie R, McCann K, Rabah R, Klein C, et al. Large B-Cell Lymphoma in an Adolescent Patient With Interleukin-10 Receptor Deficiency and History of Infantile Inflammatory Bowel Disease. J Pediatr Gastroenterol Nutr (2016) 63(1):e15–7. doi: 10.1097/MPG.0000000000000532
51. Speckmann C, Lehmberg K, Albert MH, Damgaard RB, Fritsch M, Gyrd-Hansen M, et al. X-Linked Inhibitor of Apoptosis (XIAP) Deficiency: The Spectrum of Presenting Manifestations Beyond Hemophagocytic Lymphohistiocytosis. Clin Immunol (2013) 149(1):133–41. doi: 10.1016/j.clim.2013.07.004
52. Pedersen J, LaCasse EC, Seidelin JB, Coskun M, Nielsen OH. Inhibitors of Apoptosis (Iaps) Regulate Intestinal Immunity and Inflammatory Bowel Disease (IBD) Inflammation. Trends Mol Med (2014) 20(11):652–65. doi: 10.1016/j.molmed.2014.09.006
53. Chirieleison SM, Marsh RA, Kumar P, Rathkey JK, Dubyak GR, Abbott DW. Nucleotide-Binding Oligomerization Domain (NOD) Signaling Defects and Cell Death Susceptibility Cannot be Uncoupled in X-linked Inhibitor of Apoptosis (XIAP)-Driven Inflammatory Disease. J Biol Chem (2017) 292(23):9666–79. doi: 10.1074/jbc.M117.781500
54. Latour S, Aguilar C. XIAP Deficiency Syndrome in Humans. Semin Cell Dev Biol (2015) 39:115–23. doi: 10.1016/j.semcdb.2015.01.015
55. Aguilar C, Latour S. X-Linked Inhibitor of Apoptosis Protein Deficiency: More Than an X-Linked Lymphoproliferative Syndrome. J Clin Immunol (2015) 35(4):331–8. doi: 10.1007/s10875-015-0141-9
56. Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, et al. Mutation of NLRC4 Causes a Syndrome of Enterocolitis and Autoinflammation. Nat Genet (2014) 46(10):1135–9. doi: 10.1038/ng.3066
57. Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An Activating NLRC4 Inflammasome Mutation Causes Autoinflammation With Recurrent Macrophage Activation Syndrome. Nat Genet (2014) 46(10):1140–6. doi: 10.1038/ng.3089
58. van der Burgh R, Ter Haar NM, Boes ML, Frenkel J. Mevalonate Kinase Deficiency, a Metabolic Autoinflammatory Disease. Clin Immunol (2013) 147(3):197–206. doi: 10.1016/j.clim.2012.09.011
59. Li Q, Lee CH, Peters LA, Mastropaolo LA, Thoeni C, Elkadri A, et al. Variants in TRIM22 That Affect NOD2 Signaling Are Associated With Very-Early-Onset Inflammatory Bowel Disease. Gastroenterology (2016) 150(5):1196–207. doi: 10.1053/j.gastro.2016.01.031
60. Kammermeier J, Drury S, James CT, Dziubak R, Ocaka L, Elawad M, et al. Targeted Gene Panel Sequencing in Children With Very Early Onset Inflammatory Bowel Disease–Evaluation and Prospective Analysis. J Med Genet (2014) 51(11):748–55. doi: 10.1136/jmedgenet-2014-102624
61. Marsh RA, Villanueva J, Zhang K, Snow AL, Su HC, Madden L, et al. A Rapid Flow Cytometric Screening Test for X-Linked Lymphoproliferative Disease Due to XIAP Deficiency. Cytometry B Clin Cytom (2009) 76(5):334–44. doi: 10.1002/cyto.b.20473
62. Presicce P, Moreno-Fernandez ME, Lages CS, Orsborn KI, Chougnet CA. Association of Two Clones Allows for Optimal Detection of Human FOXP3. Cytometry A (2010) 77(6):571–9. doi: 10.1002/cyto.a.20875
63. Tatsuki M, Hatori R, Nakazawa T, Ishige T, Hara T, Kagimoto S, et al. Serological Cytokine Signature in Paediatric Patients With Inflammatory Bowel Disease Impacts Diagnosis. Sci Rep (2020) 10(1):14638. doi: 10.1038/s41598-020-71503-y
64. Wada T, Kanegane H, Ohta K, Katoh F, Imamura T, Nakazawa Y, et al. Sustained Elevation of Serum Interleukin-18 and Its Association With Hemophagocytic Lymphohistiocytosis in XIAP Deficiency. Cytokine (2014) 65(1):74–8. doi: 10.1016/j.cyto.2013.09.007
65. Charbit-Henrion F, Parlato M, Hanein S, Duclaux-Loras R, Nowak J, Begue B, et al. Diagnostic Yield of Next-Generation Sequencing in Very Early-Onset Inflammatory Bowel Diseases: A Multicentre Study. J Crohns Colitis (2018) 12(9):1104–12.
66. Conrad MA, Carreon CK, Dawany N, Russo P, Kelsen JR. Distinct Histopathological Features at Diagnosis of Very Early Onset Inflammatory Bowel Disease. J Crohns Colitis (2019) 13(5):615–25. doi: 10.1093/ecco-jcc/jjy212
67. Kelsen JR, Baldassano RN. The Role of Monogenic Disease in Children With Very Early Onset Inflammatory Bowel Disease. Curr Opin Pediatr (2017) 29(5):566–71. doi: 10.1097/MOP.0000000000000531
68. Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A. Inflammasome Activation in NADPH Oxidase Defective Mononuclear Phagocytes From Patients With Chronic Granulomatous Disease. Blood (2010) 116(9):1570–3. doi: 10.1182/blood-2010-01-264218
69. Hahn KJ, Ho N, Yockey L, Kreuzberg S, Daub J, Rump A, et al. Treatment With Anakinra, a Recombinant Il-1 Receptor Antagonist, Unlikely to Induce Lasting Remission in Patients With CGD Colitis. Am J Gastroenterol (2015) 110(6):938–9. doi: 10.1038/ajg.2015.135
70. Kato K, Kojima Y, Kobayashi C, Mitsui K, Nakajima-Yamaguchi R, Kudo K, et al. Successful Allogeneic Hematopoietic Stem Cell Transplantation for Chronic Granulomatous Disease With Inflammatory Complications and Severe Infection. Int J Hematol (2011) 94(5):479–82. doi: 10.1007/s12185-011-0932-6
71. Kiykim A, Ogulur I, Dursun E, Charbonnier LM, Nain E, Cekic S, et al. Abatacept as a Long-Term Targeted Therapy for LRBA Deficiency. J Allergy Clin Immunol Pract (2019) 7(8):2790–800 e15. doi: 10.1016/j.jaip.2019.06.011
72. Uhlig HH, Muise AM. Clinical Genomics in Inflammatory Bowel Disease. Trends Genet (2017) 33(9):629–41. doi: 10.1016/j.tig.2017.06.008
73. Aradhya S, Bardaro T, Galgoczy P, Yamagata T, Esposito T, Patlan H, et al. Multiple Pathogenic and Benign Genomic Rearrangements Occur at a 35 Kb Duplication Involving the NEMO and LAGE2 Genes. Hum Mol Genet (2001) 10(22):2557–67. doi: 10.1093/hmg/10.22.2557
74. Heyworth PG, Noack D, Cross AR. Identification of a Novel NCF-1 (p47-phox) Pseudogene Not Containing the Signature GT Deletion: Significance for A47 Degrees Chronic Granulomatous Disease Carrier Detection. Blood (2002) 100(5):1845–51. doi: 10.1182/blood-2002-03-0861
75. Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, et al. Whole-Genome Sequencing Is More Powerful Than Whole-Exome Sequencing for Detecting Exome Variants. Proc Natl Acad Sci USA (2015) 112(17):5473–8. doi: 10.1073/pnas.1418631112
76. Hegde M, Santani A, Mao R, Ferreira-Gonzalez A, Weck KE, Voelkerding KV. Development and Validation of Clinical Whole-Exome and Whole-Genome Sequencing for Detection of Germline Variants in Inherited Disease. Arch Pathol Lab Med (2017) 141(6):798–805. doi: 10.5858/arpa.2016-0622-RA
77. Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively Parallel Digital Transcriptional Profiling of Single Cells. Nat Commun (2017) 8:14049. doi: 10.1038/ncomms14049
78. Martin JC, Chang C, Boschetti G, Ungaro R, Giri M, Grout JA, et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated With Resistance to Anti-TNF Therapy. Cell (2019) 178(6):1493–508.e20. doi: 10.1016/j.cell.2019.08.008
Keywords: very early onset IBD (VEOIBD), primary immunodeficiencies (PID), genetic testing, IPEX (immune dysregulation), next generation (deep) sequencing (NGS)
Citation: Zheng HB, de la Morena MT and Suskind DL (2021) The Growing Need to Understand Very Early Onset Inflammatory Bowel Disease. Front. Immunol. 12:675186. doi: 10.3389/fimmu.2021.675186
Received: 02 March 2021; Accepted: 04 May 2021;
Published: 26 May 2021.
Edited by:
Rajarshi Ghosh, National Institute of Allergy and Infectious Diseases (NIH), United StatesReviewed by:
Judith Kelsen, Children’s Hospital of Philadelphia, United StatesCopyright © 2021 Zheng, de la Morena and Suskind. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hengqi B. Zheng, YmV0dHkuemhlbmdAc2VhdHRsZWNoaWxkcmVucy5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.