 Anna Johansson-Percival1,2*
Anna Johansson-Percival1,2* Ruth Ganss1,2
Ruth Ganss1,2- 1Cancer Microenvironment Laboratory, Harry Perkins Institute of Medical Research, QEII Medical Centre, Nedlands, WA, Australia
- 2Centre for Medical Research, The University of Western Australia, Crawley, WA, Australia
Improving the effectiveness of anti-cancer immunotherapy remains a major clinical challenge. Cytotoxic T cell infiltration is crucial for immune-mediated tumor rejection, however, the suppressive tumor microenvironment impedes their recruitment, activation, maturation and function. Nevertheless, solid tumors can harbor specialized lymph node vasculature and immune cell clusters that are organized into tertiary lymphoid structures (TLS). These TLS support naïve T cell infiltration and intratumoral priming. In many human cancers, their presence is a positive prognostic factor, and importantly, predictive for responsiveness to immune checkpoint blockade. Thus, therapeutic induction of TLS is an attractive concept to boost anti-cancer immunotherapy. However, our understanding of how cancer-associated TLS could be initiated is rudimentary. Exciting new reagents which induce TLS in preclinical cancer models provide mechanistic insights into the exquisite stromal orchestration of TLS formation, a process often associated with a more functional or “normalized” tumor vasculature and fueled by LIGHT/LTα/LTβ, TNFα and CC/CXC chemokine signaling. These emerging insights provide innovative opportunities to induce and shape TLS in the tumor microenvironment to improve immunotherapies.
Introduction
Unprecedented success of immune checkpoint blockade (ICB) in melanoma patients has sparked considerable interest in immunotherapies (1). Treatment with immune modulatory antibodies has also highlighted the critical importance of an immune “hot” tumor environment for therapeutic responsiveness (2). Considerable efforts are now being directed into increasing responsiveness to ICB in all cancer patients.
The tumor microenvironment including stromal innate immune cells, fibroblasts and the vasculature has become a major target for new therapies aiming to increase intratumoral T cell numbers and their activation status prior to ICB (3, 4). Spontaneous and/or therapeutic increase of T cell numbers into tumors can result in the formation of TLS (3, 5). These TLS have the ability to effectively prime naïve T cells entering through high endothelia venules (HEV) (6). Notably, the presence of TLS predicts and improves efficacy of immunotherapy in mice and humans (7).
In this review, we delineate common features of peripheral lymph nodes (LNs), inflammation- and cancer-associated TLS, and discuss the relationship between the presence of TLS, lymphocyte priming and response to immunotherapy. We further elaborate on potential drivers for intratumoral TLS formation and how TLS could be exploited therapeutically, in particular for non-responsive, immune “cold” cancers.
The Beginning: Development of Lymphoid Tissue
The immune system is comprised of organs and cell types that protect the host from foreign pathogens and disease. The highly specialized adaptive immune system consists of T and B lymphocytes that form in the bone marrow and later reside in secondary lymphoid organs (SLOs). SLOs are strategically placed to facilitate immune surveillance and priming of naïve T cells and also include LNs (8). The structural framework of LNs are fibroblastic reticular cells (FRCs) which mediate cross-talk between various immune cell populations throughout the LN. In addition, follicular dendritic cells (FDCs) that reside within B cell zones maximize interactions between antigens, antigen presenting cells and naïve lymphocytes (9). Embedded in the paracortical region of LNs are HEVs, highly specialized post capillary venules that serve as entry portals for naïve and central memory lymphocytes from the blood; this migration process is mediated by interactions of L-selectin expressed on lymphocytes and peripheral node addressins (PNAds) on HEVs (10). TLS are lymphoid aggregates similar to SLOs which develop in non-lymphoid tissue, for instance at sites of chronic inflammation (11). TLS vary in composition and maturity but share with SLOs separated B and T cell zones, stromal cells, and HEVs.
One proposed mechanism for the initiation of LN development is upregulation of chemokine (C-X-C motif) ligand 13 (Cxcl13) by lymphotoxin beta receptor (LTβR) expressing mesenchymal precursors known as lymphoid tissue organizer (LTo) cells (12). Cxcl13 subsequently attracts hematopoietic precursors or lymphoid tissue inducer (LTi) cells resulting in the first cluster of LTi cells and the initiation of LN development (12). Mature LTi express lymphotoxin alpha 1 beta 2 (LTα1ß2) which binds LTβR in activated LTo, resulting in further LTo maturation and expression of intercellular adhesion molecule 1 (ICAM1), vascular cell adhesion molecule 1 (VCAM1), chemokine (C-C motif) ligand 19 (Ccl19) and 21 (Ccl21), and Cxcl13 which recruit more LTi and promote interactions between LTi and LTo (8, 9). Mouse LTo may give rise to stromal lineages such as FRCs, FDCs, lymphatic endothelium and vascular endothelium within adult LNs (13).
Emerging evidence also highlights a crucial role of vascular endothelium in the development of LNs. In adult LNs, endothelial cells (ECs) and lymphatic endothelial cells (LECs) express LTβR; EC-specific deletion of LTβR by crossing vascular endothelial cadherin (VE-Cad)-Cre and LTβRfl/fl mice results in compromised LN development with a reduced HEV network demonstrating the importance of EC-specific LTβR for HEV development and lymphocyte trafficking (14). Moreover, EC and to a lesser extent LEC-specific deletion of NFκB-inducing kinase (NIK), one of the major pathways downstream of LTβR signaling, results in an almost complete loss of peripheral LNs (15). In the remaining LN anlagen of these mice, CD4+ LTi cells are drastically reduced coinciding with very low VCAM1, ICAM1, Cxcl13 and Ccl19 expression levels suggesting that failure of LTi to engage with ECs during LN development prevents LTo activation. Furthermore, forced retention of LTi following treatment of pregnant mice with the drug FTY720 which sequesters lymphocytes in LNs, results in formation of mature ectopic LNs in the inguinal fat pad of the progeny (15). These findings imply that the numbers of LTi retained by EC/LECs may be an additional determinant of LN development, alongside interactions between LTi and mesenchymal LTo (16).
TLS Form Under Inflammatory Conditions in Mice
Although the initial events of LN development are not fully resolved, LTβR signaling is crucial for subsequent LN maturation, and also plays a major role in TLS formation during chronic inflammation in mice (Figure 1). For instance, in apolipoprotein E (ApoE)-/- mice, LTβR expressing aortic smooth muscle cells (SMC) over time become activated and produce TLS inducing cytokines such as Cxcl13, Ccl21 and LTβ (17). This leads to the formation of mature aortic TLS containing B cell follicles and germinal centers (GCs), T cells and HEVs. Importantly, TLS assembly can be prevented by blocking LTβR signaling in vivo (17).
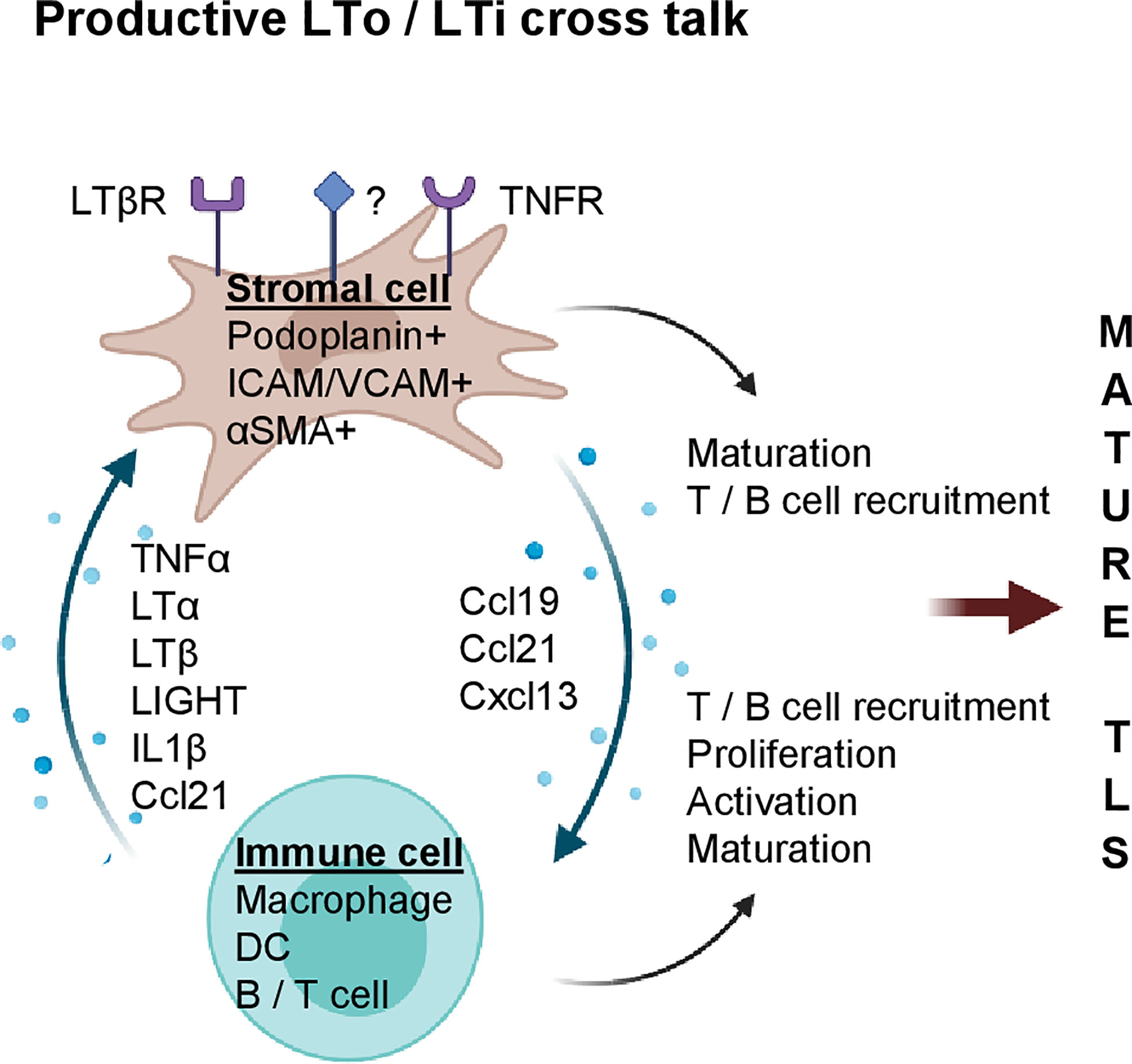
Figure 1 Stromal and immune cell cross talk mediate TLS formation during chronic inflammation. Potential cytokines/chemokines involved in immune (LTi) and stromal cell (LTo) cross-talk. Stromal cells express cytokine receptors such as LTβR and TNFR (and potentially others, marked with “?”); upon activation, LN inducing chemokines such as Ccl19, Ccl21 and Cxcl13 are secreted by stromal cells which increase immune cell density and foster their own maturation. Activated stroma and immune cells coordinate formation of LN aggregates which can mature into clusters containing T cells, B cells, FDCs and MECA79+ HEVs (mature TLS). Created with BioRender.com.
LTβR binds two ligands, the developmentally important LN-inducing cytokine LTα1ß2 and tumor necrosis factor superfamily (TNFSF) 14 or LIGHT. Increased LIGHT expression coincides with TLS formation in the pancreas of aged non-obese diabetic (NOD) mice; in vivo inhibition of LTβR prevents TLS formation and diabetes (18). TLS in mouse pancreatic islets can also be induced by overexpressing C-X-C chemokine receptor type 5 (Cxcr5), the receptor for Cxcl13 (19), Cxcl12, Ccl19 or Ccl21 (20) under the control of the rat insulin gene promotor. Interestingly, LTβR or LTα1β2 blockade prevents TLS formation in chemokine overexpressing mice (19, 20), implying that LTα1β2 and/or LIGHT are bona fide TLS inducers under inflammatory conditions. However, mechanisms leading to inflammation-associated TLS formation are complex and can involve a network of multiple immune and stromal cell types, and - besides LTα1β2 – other cytokines such as tumor necrosis factor alpha (TNFα), IL6, IL13, IL17, IL22 and IL23 (21–25).
In mouse inflammatory lesions, stromal cells can function as LTo by upregulation of the FRC markers podoplanin, Ccl19, Ccl21 and Cxcl13 which in turn stimulate lymphocyte recruitment to sites of inflammation (26, 27). For instance, in patients with primary Sjögren’s syndrome (pSS) and a mouse model of salivary gland inflammation, IL13 production by activated fibroblast activation protein (FAP)+ podoplanin+ fibroblasts, termed “immunofibroblasts”, is the earliest detectable event during TLS neogenesis which precedes lymphocyte recruitment into tissue and subsequent IL22/LTα1β2 secretion (24). As demonstrated in mice deficient for IL13 or its receptor IL4R, “immunofibroblast” activation is dependent on IL13/IL4R signaling and precedes their expansion which is subsequently regulated by lymphocyte-derived IL22 (28). Furthermore, genetic deletion of FAP+ fibroblasts abolishes TLS formation highlighting the LTo role of fibroblasts during TLS formation (24).
During ear inflammation in mice, induction of podoplanin+ stromal cells is dependent on myeloid cells, since depletion of CD11+ Gr1+ cells using monoclonal antibodies significantly reduces podoplanin+ cells (26). This suggests that circulating monocytes can acquire a postnatal role as LTi. Indeed, myeloid cells have been implicated in the development of TLS in various experimental systems. For instance, global overexpression of TNFα in mice by expressing a stabilized TNFα mRNA (TNFΔARE) leads to the development of TLS in the intestine in a process which is dependent on F4/80+ myeloid cells (21). Mechanistically, F4/80+CD11b+ myeloid cells in the LN anlagen are the major source of TNFα and inducers of stromal maturation and expression of LTo chemokines such as Cxcl13, Ccl19 and Ccl21. The potency of these myeloid cells was further demonstrated by surgical transplantation of LN anlagen from TNF/RORc(γt)-/- mice under the kidney capsule of RORC(γt)-/- mice that lack classical LTi; this leads to LN development in the majority of mice thus demonstrating that TNFα producing myeloid cells have the capacity to induce LN formation (21). In atherosclerosis, M1-polarized macrophages act as LTi cells and produce high levels of LN-inducing cytokines such as TNFα and LTα (29). In vitro stimulation of vascular SMCs (vSMC) with M1 macrophage conditioned media induces an LTo profile and triggers the formation of TLS in vivo following vSMC injection (29). VSMC activation is dependent on TNFR signaling as blockade of TNFR1/2 in vivo abolishes the LTo phenotype and prevents TLS formation. Similarly, adipose tissue-associated TLS formation is dependent on myeloid derived TNFα and stromal expression of TNFR, but independent of LTβR signaling (27).
The effects of DCs on lymph angiogenesis and TLS induction have also been studied in multiple models (30–35). For instance, in a mouse model of atopic dermatitis, CD11c+ DCs accumulate around newly formed HEVs; inhibition of LTβR signaling or depletion of CD11c+ cells inhibits HEV formation (33). Similarly, following influenza virus infection in mice, lung CD11c+ DCs express TLS-inducing cytokines such as LTβ, Cxcl13, Ccl19 and Ccl21 which correlates with formation of mature TLS; in vivo depletion of CD11c+ cells or inhibition of LTβR signaling perturbs TLS formation (34). Moreover, in plaques arising in ApoE-/- mice, LTβ producing CD11c+ CD68+ Ly6Clo monocytes reside in close proximity to vSMCs and induce Cxcl13 and Ccl21 secretion, indicating a potential role of DCs as LTi (17). Overall, multiple models of chronic inflammation show that stromal cells can gain LTo function whilst inflammatory myeloid cells play a crucial role as LTi. Moreover, in the process of TLS formation, TNFα and LTβ serve important non-redundant roles.
Spontaneous TLS Formation in Human Cancer
Tumors are described as “wounds that never heal” (36), and indeed rely on continuous stromal remodeling, inflammation and angiogenesis to support the rapidly growing cancer. The abnormal angiogenic tumor vasculature often lacks adhesion molecules such as ICAM/VCAM which prevents efficient lymphocyte-EC binding (37, 38). However, despite this “anergic” tumor vasculature, the tumor microenvironment (TME) can support naïve T cell infiltration, and spontaneous intratumoral TLS formation has been observed in a subset of patients across cancer types (7).
Although the precise mechanism of spontaneous TLS formation in human cancers is unknown, the presence of intratumoral TLS structures is often associated with a favorable clinical outcome and extended disease-free survival (7, 39–47). In hepatocellular carcinoma (HCC) for instance, the presence of intratumoral TLS reduces the risk for early relapse following tumor resection (43). In addition, mature TLS harboring GCs rather than poorly defined lymphocyte aggregates have the lowest recurrence risk (43). In human breast cancer, the presence of HEVs correlates with overall T and B cell infiltration, and improved prognosis (44, 45). Moreover, flow cytometry and gene expression analysis of CD4+ T cell subsets revealed that highly infiltrated breast cancers also harbor TLS, and express markers such as Cxcl13, ICOS, IFNγ and TBX21/T-bet, commonly associated with follicular T helper (Tfh) and Th1 profiles (39, 40). In multiple human cancers such as lung, breast, pancreatic, gastric cancers and melanoma, TLShigh tumors harbor more activated, cytotoxic or naïve CD8+ T cells together with CD4+ T cells which are skewed to a Th1 and/or Th17 phenotype when compared to TLSlow tumors (41, 42, 44–47).
The presence of intratumoral TLS can be determined by analyzing chemokine gene-expression signatures which were first described in colorectal cancer (48) and subsequently validated for other types of cancer such as HCC, breast cancer and melanoma (43, 49, 50). The ability to assess TLS status prior to therapy is of clinical significance and may offer an opportunity to improve immunotherapy (49).
However, the predictive value of TLS for patient outcome is complex, and other parameters besides presence or absence of lymphocyte aggregates seem to be important. In colorectal cancer, for instance, TLS structures with high densities of M2 macrophages and T helper cells expressing GATA3, a master regulator of Th2 differentiation, contribute to immune suppression and thus correlate with relapse rather than improved prognosis (51). In HCC, TLS in the tumor margin are associated with an increased risk of recurrence (52). Moreover, TLS which arise in HCC patients, or mice with persistent and high NFκB activation in hepatocytes, promote tumor progression rather than anti-tumor immunity (52). Similarly, early human hepatic lesions can harbor immature TLS characterized by the expression of immune suppressive cytokines and T cell exhaustion markers such as IL10RA, TGFβ1, TIM-3 and PD-L2 (53). In other cancer types, for instance breast, colorectal and pancreatic cancers, TLS are often found in peri-tumoral locations, and are associated with more advanced disease (41, 54, 55). Overall, these studies indicate that intratumoral location and TLS maturity are crucial parameters for productive anti-tumor immunity and improved patient outcome (7, 56).
TLS as Intratumoral Priming Sites for Adaptive Immunity
It is commonly accepted that naïve lymphocytes do not enter peripheral tissues or tumors, but circulate through lymphoid organs to encounter cognate antigen for activation. However, there is emerging evidence that HEV+ TLS may activate effector T cells intratumorally thus bypassing the need for tumor-antigen presentation in draining LNs (57). For instance, LIGHT accelerates development of diabetes in NOD mice even after surgical removal of pancreatic draining LNs implying that naïve T cells are primed within TLS in pancreatic islets (18). In B16 melanoma-bearing mice, adoptively transferred naïve anti-tumor T cells differentiate into effector cells, reduce tumor growth and improve survival even when lymphocyte egress from LNs is blocked by FTY720 (6, 58). This suggests that HEV+ mouse melanomas can facilitate naïve T cell infiltration, and support subsequent priming and differentiation (6).
Naïve T cell activation in TLS relies on the presence of antigen presenting cells such as B cells and DCs. Indeed, in lung (42, 59), breast (60) and renal cancers (61), a high density of TLS-associated mature DCs correlates with the degree of Th1 effector T cell infiltration and improved prognosis. Interestingly, DCs are also involved in HEV function. In peripheral LNs, for instance, DCs maintain HEV maturity and thus naïve T cell infiltration through LTβR signaling (30). In human breast cancer, DCs produce high levels of LTβ and the density of mature DC-LAMP+ DCs strongly correlates with the frequency of HEVs (60). Collectively, this indicates that DCs maintain HEV maturity and facilitate T cell egress and priming in both LNs and TLS.
B cells are an integral part of mature TLS and potent antigen presenting cells. In some cancers, B cells have been shown to foster tumor development by secreting factors which contribute to a pro-tumorigenic immune environment (62). However, mature B cells in TLS produce antibodies within GCs which correlates with a higher degree of T cell infiltration and disease free survival (63–65). Improved prognosis in human breast cancer is associated with CD4+ Tfh cells which produce an abundance of Cxcl13 and support B cell differentiation, TLS formation and GC maturation (39, 66). In pancreatic adenocarcinoma (PDAC), the presence of B cells within mature TLS correlates with improved prognosis in patients, or increased immune response to vaccination in mice (67). Furthermore, initial evidence in human melanoma suggested a potential link between antibody producing B cells and ICB responsiveness (68, 69). This has now been confirmed in a series of studies which performed in-depth molecular analyses in ICB responder and non-responder tumor tissues (70–72). For instance in human sarcoma, ICB responders are characterized by B cell-rich intratumoral TLS and an immune gene signature related to T cell infiltration and activation, immune checkpoints and expression of Cxcl13 (70). In human melanoma, B cell-enriched TLS confer improved survival and responsiveness to ICB, and also contain naïve and/or memory T cells and an immune signature indicative of enhanced B-T cell interactions and antigen presentation (71, 72). In contrast, T cells in TLS negative melanomas expressed elevated TIM3 and PD-1 levels which may indicate a dysfunctional state (72). Furthermore, RNA-seq analysis of B cell receptors (BCRs) in melanomas showed greater BCR diversity and B cell maturity in ICB responders versus non-responders supporting an active role for B cells in anti-tumor immunity (71). In summary, these studies demonstrate a major role of TLS-associated B cells in antigen presentation, T cell polarization and activation thus placing B cells at the center of TLS function (62, 70–75).
The efficacy of anti-cancer effector T cells is intimately linked to the presence or absence of CD4+CD25+FoxP3+ regulatory T cells (T regs), and interestingly, T reg depletion induces TLS. For instance, in a mouse model of chemically induced fibrosarcoma genetic T reg deletion triggers intratumoral HEV formation, T cell recruitment and tumor control (76, 77). Similarly, T-reg depletion in a model of autochthonous lung adenocarcinoma induces TLS, increases T cell proliferation and DC activation with ensuing tumor control (78).
Overall, current evidence strongly supports a role of intratumoral TLS as priming sites for anti-tumor immunity and prognostic indicators for ICB efficacy. Spontaneous formation of mature and functional TLS in cancer is highly orchestrated and context-dependent; insights into this process will provide exciting opportunities for innovative drug development.
From Concept to Treatment: Therapeutic Induction of TLS
Experimental TLS induction in animal models provides an important opportunity to study the complex interplay between immune cell populations which foster adaptive anti-cancer immunity. Therapeutic TLS induction in cancer patients holds the promise to advance immunotherapy. Numerous attempts have been made to induce TLS in mouse models, so far with mixed outcomes. For instance, both Ccl21 and LTβR play important roles during peripheral LN development. Early work in a mouse melanoma model indeed found that a recombinant antibody targeting LTα to melanoma cells induced intratumoral HEVs, B and T cell zones, and improved survival (79). In contrast, Ccl21 overexpressing melanoma cells promoted infiltration of suppressive immune cells and cytokines which collectively stimulate tumor growth (80). Thus, to harness TLS therapeutically better mechanistic insights into intratumoral TLS formation are urgently needed.
More recent attempts to induce TLS in mouse tumors have employed sophisticated technologies such as artificial scaffolds, gene engineering, and vaccination strategies. Given the crucial role of LTo cells in the recruitment of LTi during LN development (8, 12), a role of stromal cells as TLS inducers has been widely explored (48, 81–83). For instance, LTα overexpression in a stromal cell line derived from thymus induces lymphoid-like organoids in mice when co-implanted with DCs in a collagenous scaffold (81). Moreover, a collagen sponge with a cocktail of LN-inducing cytokines when implanted under the kidney capsule also initiates formation of artificial LN-like TLS (artTLS) with distinct B/T cell zones, FDC/FRCs and HEVs. Intriguingly, implantation of these sponges into immunodeficient mice generates antibody producing cells following immunization (82), further supporting a role of TLS in adaptive immunity. Similarly, a LN-derived stromal cell line which expresses high levels of the FRC marker podoplanin and chemokines such as Ccl19, Ccl21, Cxcl10 and Cxcl13 - reminiscent of the chemokine gene signature first identified in human colorectal cancer (48) - when implanted subcutaneously in mice also generates TLS (83). Within these TLS, resident T cells were successfully activated into effector T cells by tumor-lysate-pulsed DCs which suppressed the growth of adjacent MC38 colon cancer cells (83).
In gene engineering studies, DCs were generated to produce high levels of T-bet/Tbx21, a transcription factor that drives the development and functionality of immune cells, particularly by producing the key Th1 cytokine IFNγ. T-bet overexpressing DCs also produce high levels of pro-inflammatory cytokines such as TNFα, IL12p40 and IL-36γ, and induce TLS in a mouse colon cancer model; even in the absence of peripheral LNs intratumoral DC-Tbet therapy prolongs survival (84). In contrast, tumor growth control is abolished in IL36R-deficient mice indicating a crucial role of T-bet/IL-36γ in therapeutic TLS induction (84). This is supported by findings in human colon cancer where IL-36γ is highly expressed in M1 macrophages and cells of the vasculature, including vSMCs and HEVs, and correlates with spontaneous TLS formation (85).
In human papilloma virus (HPV) 16-positive cervical cancer, intramuscular vaccination targeting HPV16 E6/E7 antigens induces intratumoral TLS which contain antigen-experienced effector memory T cells (86). Moreover, TLS-rich tumor stroma harbors a typical Th1 gene signature with increased levels of Cxcr3, TBX21, IFNγ and IFNβ.
In human PDAC, T cell infiltration and activation is positively linked to survival in some patients (87, 88), and TLS can be induced following an allogeneic granulocyte-macrophage colony stimulating factor secreting vaccine (GVAX) when given in combination with T reg-depleting cyclophosphamide (89). TLS display a distinct Th17 gene signature, a high T effector to T reg ratio, and serve as a prognostic tool to segregate long term from short term survivors (89). Although this clinical trial provides rare evidence for therapeutic TLS induction in humans, PDAC can harbor spontaneous intratumoral TLS which are linked to better prognosis (41). Interestingly, spontaneous TLS in PDAC are associated with a more mature vascular network that expresses the vascular adhesion molecule VE-Cadherin and is covered by αSMA+ pericytes, a mural cell type which wraps around and supports the endothelium (41), suggesting a possible link between TLS formation and stabilized tumor vessels.
A Potential Link Between Vascular Normalization and TLS Induction
T cell infiltration into solid cancers is controlled by the vasculature which co-evolves with an immune-suppressive microenvironment and plays an active part in limiting T cell influx (37, 90–93). In contrast, activating tumor blood vessels to express adhesion molecules such as ICAM and VCAM enables productive endothelial-T cell interactions and fosters effector T cell transmigration (3, 92, 94–97). Moreover, tumor vessel normalization which improves vascular morphology and function lowers hypoxia and indirectly changes the tumor microenvironment to support Th1-driven anti-tumor immunity (98–100). Therefore, compounds which normalize tumor blood vessels and attract T cells may have the capacity to induce intratumoral TLS. Indeed, a fusion compound of the cytokine LIGHT conjugated to a homing peptide (vascular targeting peptide or VTP) which delivers LIGHT specifically to angiogenic tumor vessels is such a reagent (95). LTβR and Herpes virus entry mediator (HVEM) are major LIGHT receptors, expressed in stroma and immune cells, respectively, and thus link LIGHT to LN neogenesis and immune regulation (101–106). Treatment of neuroendocrine pancreatic cancer (PNET) in mice with low dose LIGHT-VTP normalizes blood vessels and induces intratumoral TLS with distinct B and T cell zones and high expression of the T cell attractant Ccl21 in vascular cells as well as macrophages (Figures 2A, B) (3, 95). Importantly, the capacity to induce TLS correlates with the degree of vessel normalization and is abolished with high dose LIGHT-VTP which induces vessel death, demonstrating a causal link between vessel normalization and TLS formation (3). Other treatment regimens which are known to normalize tumor vessels in PNET such as low dose anti-vascular endothelial growth factors (VEGF) or anti-angiopoietin-2/anti-VEGF therapies facilitate lymphocyte infiltration but do not induce TLS as monotherapies (107, 108). Similarly, cytokine fusion compounds which deliver for instance TNFα or IFNγ to tumor vessels in PNET induce vessel normalization and/or vessel wall inflammation without TLS formation demonstrating the unique opportunities of targeting LIGHT into the tumor microenvironment (97, 109). Furthermore, intratumoral treatment of melanoma-bearing mice with low dose stimulator of interferon genes (STING) agonist (ADU S-100) normalizes angiogenic blood vessels and upregulates TLS-inducing factors such as Ccl19, Ccl21, LTαβ and LIGHT (110). This induces unstructured HEV-containing lymphocyte aggregates resembling TLS which contain T cells and CD11c+ DCs. STING activation enables recruitment of pre-primed peripheral T cells and expansion of unique T cell clonotypes in the TME thus further supporting the benefits of reagents with dual capacity to induce vessel normalization and intratumoral priming. Nevertheless, the anti-tumor effects of LIGHT-VTP or STING monotherapies are modest, and the clinical relevance of these reagents lies in increasing the potency of current immunotherapies (3, 110).
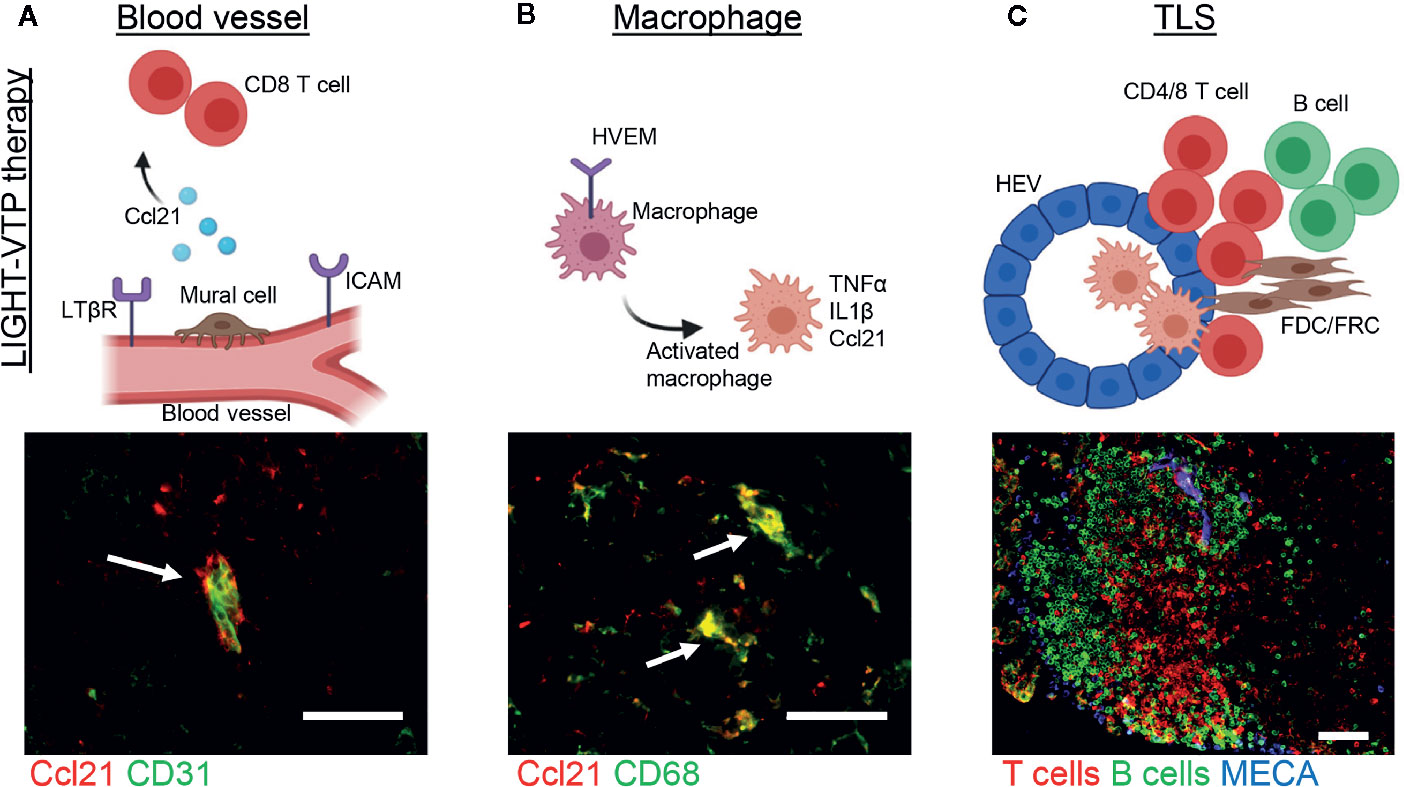
Figure 2 Induction of cancer-associated TLS during LIGHT-VTP therapy. (A, B) Treatment of transgenic PNET-bearing mice with bi-weekly i.v. injections of 20 ng LIGHT-VTP specifically targets abnormal angiogenic blood vessels and induces chemokines important for TLS formation (e.g. Ccl21) in (A) vascular cells (co-staining of CD31+ endothelium in green and Ccl21 in red, overlay in yellow marked by arrow) which attract CD8+ T cells, and (B) tumor-resident CD68+ macrophages which are recruited to the vascular niche (co-staining of CD68 in green and Ccl21 in red, overlay in yellow marked by arrows) and re-programmed to secrete other cytokines such as TNFα and IL1β which in turn attract T/B cells to form TLS (3). (C) Adoptive transfer of LIGHT-stimulated macrophages into PNET-bearing mice leads to CD68+ macrophage accumulation in the TME and subsequent formation of mature TLS 8 days after transfer. TLS with organized T cell (red) and B cell (green) zones as well as MECA79+ HEVs (blue) are depicted. Scale bars 50μm. Images are unpublished microscopic photographs similar to work published in (3). Created with BioRender.com.
TLS and Immunotherapy
Immunotherapies which boost the host’s intrinsic immunity such as anti-cancer vaccines and ICBs have dramatically changed clinical oncology. However, based on the increasing number of drug combination trials, ICB therapies will be predictably more effective in combination with other therapies such as TLS induction (7, 111).
The presence of spontaneously arising B cell-rich TLS within cancers has recently been shown to predict the response to ICB in patients with melanoma, soft-tissue sarcoma and renal cell carcinoma (see above) (70–72). In addition, a retrospective analysis of human lung cancer samples identified PD-1hi expressing CD8+ T cells within TLS to predict response to PD-1 blockade (112). These proliferating PD-1hi T cells were highly tumor-reactive, secreted Cxcl13, and are thus potential drivers of TLS formation (112). Similarly, non-small cell lung carcinoma biopsies from PD-1 blockade responders are enriched in TLS and mature B cells (113). Furthermore, patients with desmoplastic melanoma, a subtype of melanoma with dense fibroblastic stroma and high frequency of TLS, respond particularly well to PD-1 blockade compared to other advanced forms of melanoma (114). Although the correlation of TLS frequency and patient responsiveness in retrospective studies might be biased, collectively these studies support the notion that TLS induction prior to ICB is beneficial and will improve response rates to immunotherapy.
Strong evidence for beneficial TME-immune stimulating combination therapies also comes from animal studies. For instance, experimental induction of TLS with LIGHT-VTP therapy renders PNET and Lewis lung carcinoma (LLC) sensitive to ICB targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and PD-1. The combined treatment induces intratumoral activation of cytotoxic T cells with ensuing survival benefits which can be further improved when combined with anti-cancer vaccination. Notably, neither vaccination, ICB or a combination thereof match the survival outcome achieved with LIGHT-VTP combination treatment (3). In mouse breast cancer, PNET, and glioblastoma (GBM), VEGF inhibition renders tumors susceptible to anti-PD-L1 therapy. The combination treatment of anti-VEGF and anti-PD-L1 activates intratumoral DCs and T cells and reaches maximal efficacy when combined with agonistic LTRβ antibodies; this triple treatment induces HEV+ immune clusters even in highly therapy-resistant GBM (115). In the same GBM tumor model, LIGHT-VTP treatment in combination with anti-VEGF and anti-PD-L1 is even more effective than agonistic LTβR antibodies, and generates an abundance of intratumoral HEV+ TLS and granzyme B+ (GrzB) CD8+ effector T cells (116). This highlights the importance of LTβR signaling for TLS combination immune therapies but also the potential involvement of other pathways since LIGHT activates cells within the tumor microenvironment through multiple receptors including LTβR and HVEM.
Overall, there is already strong evidence that intratumoral TLS are an important prognostic tool for immunotherapies (70–72). However, beyond risk stratification, inducing TLS in combination with ICB generates a synergism which is likely to promote lymphocyte infiltration, intratumoral activation and immune rejection, in particular in immune-deserted or “cold” tumors. Given the significant toxicities of ICB as observed in recent combination trials of Nivolumab and Ipilimumab (117, 118), the presence of TLS may be helpful to select patients who will benefit most from ICB. In addition, TLS/ICB combination therapies could contribute to more effective anti-tumor responses with lower ICB doses. In this context, a preliminary study of low dose Nivolumab and Ipilimumab combined with IL-2 and hyperthermia treatment shows similar overall response rates when compared to high dose ICB with significantly lower overall toxicity (119). This indicates an exciting possibility to lower ICB doses when used in combination with other immune stimulating reagents.
Searching for the Instigator(s) in Cancer-Associated TLS
Much like LN neogenesis, formation of cancer-associated TLS presumably involves a network of stromal and immune cells linked by multiple cytokines/chemokines. However, mechanistic insights into this process are rudimentary. Since these interactions are precisely orchestrated in a 3D environment in vitro studies are challenging. Nevertheless, some cell types and cytokines/chemokines by virtue of their crucial role in experimental systems and presence in human TLS+ cancer tissue deserve further consideration (Figure 3).
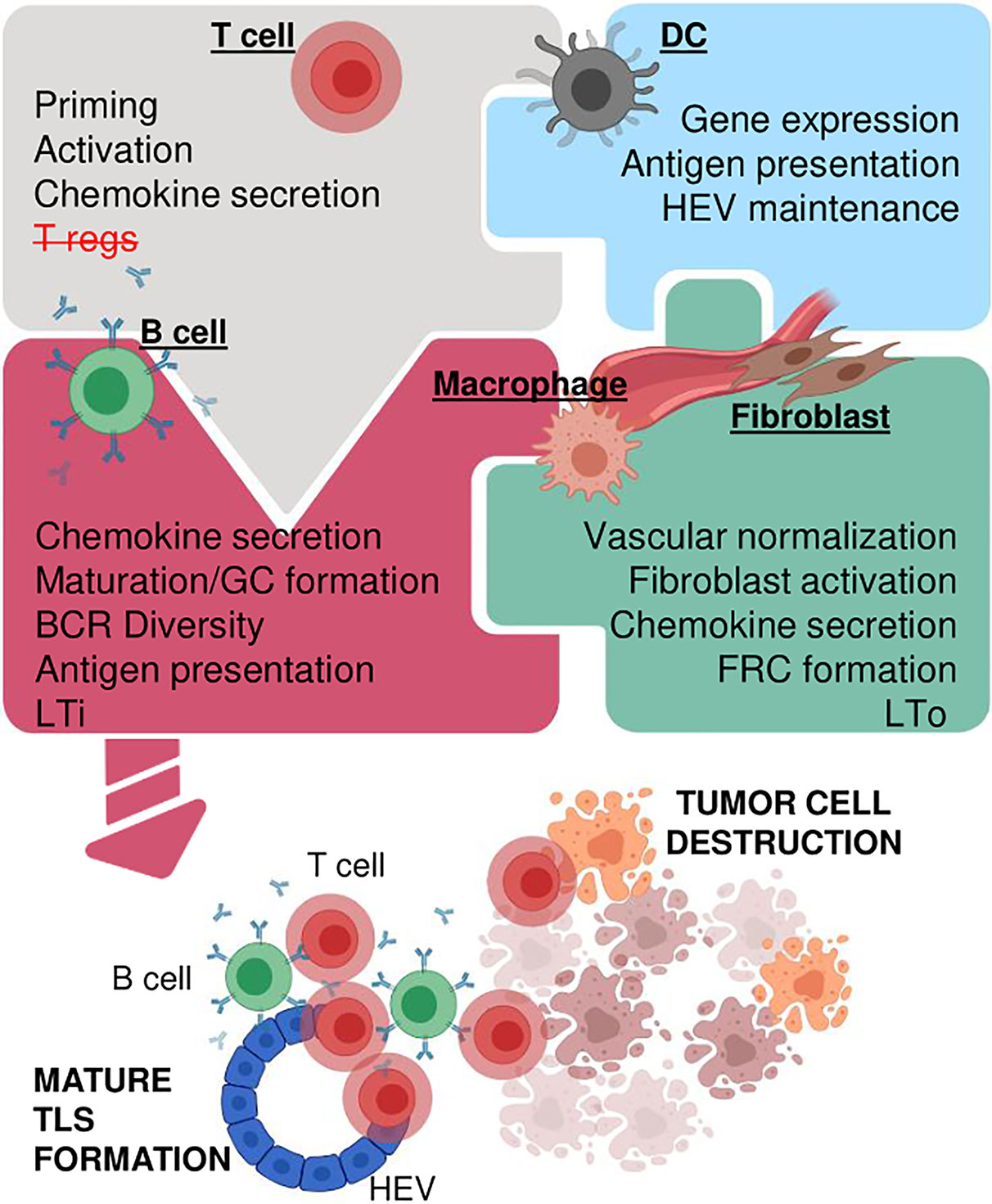
Figure 3 Concepts for creating functional TLS in cancer. Multiple immune and stromal cell types orchestrate TLS formation potentially involving activated T cells and/or depletion of T regs (upper left), DCs which express TLS supporting cytokines/chemokines, maintain HEVs and promote antigen presentation (upper right), mature B cells which produce antibodies, enhance antigen presentation, act as LTi cells (lower left), and stromal cells such as fibroblasts, macrophages and vascular cells which can act as LTo, secrete chemotactic cytokines/chemokines and/or provide structural support (lower right). Some or all cell types may be necessary in an intricate network of simultaneous or consecutive interactions to generate mature TLS which in turn enhance immunotherapy and tumor destruction. Created with BioRender.com.
Non-Hematopoietic Stromal Cells: Blood Vessels and Fibroblasts
Tumor vasculature and TLS formation are intimately linked (3, 4, 110). For instance, LIGHT-VTP in mouse PNET increases the expression of Ccl21 in the vascular bed and in CD68+ tumor-resident macrophages associated with TLS (Figures 2A, B) (3). Moreover, a 3D scaffold environment and slow interstitial flow are essential for Ccl21 expression by LN-derived FRCs both in vitro and in vivo; without lymph flow Ccl21 expression is not detectable suggesting that fluid flow dynamics may regulate Ccl21 expression (120). It is therefore interesting to speculate that modulation of blood flow dynamics and transport of cytokines/chemokines during tumor blood vessel normalization may regulate Ccl21 expression levels in the vascular bed, and thus TLS formation in vivo.
Cancer associated fibroblasts (CAFs) form a large part of the tumor microenvironment, reduce fluid flow by increasing tumor stiffness, and support tumor-promoting inflammation (121). Thus, modulation of CAFs can enhance anti-cancer immunotherapy (121, 122) and potentially support TLS formation. More recently, a crucial role for CAFs as LTo and effector CD8+ T cells/B cells as LTi was delineated in an intraperitoneal melanoma model of spontaneous TLS formation (4). Therein, effector T cells recruit FAP- podoplanin+ fibroblasts to HEVs where they differentiate into Cxcl13 secreting FRCs via TNFR signaling, similar to previous models of chronic inflammation (26, 27). This in turn promotes recruitment and proliferation of LTα1β2 secreting B cells which further stimulate TLS formation in a positive feedback loop (4). In human and mouse lung cancer, Ccl19 producing fibroblastic stromal cells (FSC) correlate with increased CD8+ T cell infiltration and tumor growth control. Although TLS formation was not examined in this study, Ccl19-expressing FSCs reside in peri-vascular niches within LLC tumors and T cell recruitment is impaired upon Ccl19 gene deletion suggesting an early role of FSCs in forming immune-stimulating stromal niches (123). Collectively these studies support the notion that vascular cells and fibroblasts are important mediators of TLS neogenesis in cancer (4, 38, 94).
Hematopoietic Stromal Cells: Macrophages
Monocytes/macrophages are a major component of tumor stroma (124). In a hypoxic tumor environment, macrophages are immunosuppressive and support tumor growth. However, their phenotype is highly dynamic and macrophage “re-education” can support immunotherapy (125). In the context of TLS neogenesis, M1 macrophages can produce chemokines similar to those detected in TLS+ human cancers, including Ccl21 and TNFα (3, 126). Furthermore, ex vivo LIGHT-stimulated macrophages in contrast to control macrophages when adoptively transferred into tumor-bearing mice are necessary and sufficient to induce intratumoral TLS in a T cell-dependent manner (Figure 2C) (3). In addition to Ccl21, these LIGHT-stimulated macrophages also express high levels of TNFα (Figure 2B) which is a key driver of inflammation-induced TLS formation in mice (21, 27). It is therefore possible that LIGHT-stimulated macrophages drive TLS formation via the TNFα/TNFR signaling pathway which has so far not been investigated. Whilst the importance of macrophages during TLS formation in cancer is understudied, robust data in inflammatory disease support their importance in TLS neogenesis (21, 27, 29), warranting further investigations in cancer.
Hematopoietic Stromal Cells: DCs
LTα/LTβ producing CD11c+ DCs play a critical role in regulating lymphocyte trafficking and maintaining HEV phenotype and function in adult mouse LNs (30, 32), and are involved in TLS formation during chronic inflammation (30–35). In human tumors, DCs are a major source of LTβ and their density correlates with HEV formation and favorable clinical outcome in breast cancer (60). Similarly, in primary human lung and ovarian cancers the number of mature DCs correlates with the degree of CD8+ T cell infiltration, anti-tumor cytotoxicity and survival (42, 127). Furthermore, immune-stimulating and vascular normalization therapies in mice increase intratumoral CD11c+ DCs coinciding with the formation of lymphocyte aggregates and HEVs (110, 115). Treatment of B16 melanoma with low-dose STING agonist, for instance polarizes DCs to produce TLS-inducing cytokines such as LTα, IL36β and TNFα (110), implicating mature DCs in TLS neogenesis. Overall, mechanistic tumor data are still sparse; plasticity of myeloid cells as well as shared marker expression in myeloid cell and DC populations complicate interpretation of the data. Further analysis of stromal innate immune cells such as monocytes/macrophage and DCs as initiators of cancer-associated TLS is therefore warranted.
Conclusions
Although immunotherapy has shown unprecedented success in some cancer patients and tumor types, the challenge ahead lies in improving the outcome for non-responsive patients. TLS as prognostic markers for improved patient outcomes have long been recognized (7). However, only recently have mature TLS been shown to predict ICB success in patients (70–72). It is imperative to now develop strategies to increase TLS frequency and/or maturation in cancers where they naturally occur. This may be achieved by providing further innate immune stimulation as demonstrated for instance with STING agonist treatment (110). Induction of de novo TLS formation holds great therapeutic potential to overcome intrinsic immune inhibitory mechanisms within the TME and render non-responsive, immune “cold” tumors susceptible for ICB. However, the orchestration of mature immune-supportive TLS formation in cancer is complex and involves multiple cellular compartments and cytokines/chemokines; this process may also be tumor type-dependent. Emerging mechanistic insight from mouse tumors demonstrate potential LTi roles for anti-tumor effectors such as T and surprisingly B cells which requires re-definition of the role of B cells in TLS and cancer (4). Therapeutic vessel normalization which enables lymphocyte infiltration into tumors may also promote access of these LTi into the TME for more effective TLS priming (3, 110). Furthermore, intratumoral stromal cell types such a monocytes/macrophages and fibroblasts are strong candidates for LTo cells which when reprogrammed in permissive tumor “niches” can drive TLS formation (3, 4). In this context, TNFR in addition to LTβR signaling may prove crucial for tumor-associated TLS formation as opposed to primarily LTβR driven processes as seen during peripheral LN development. Overall, improving existing TLS function or priming de novo TLS formation in cancer to maximize ICB efficacy holds the potential to induce more durable anti-tumor immune responses in a higher percentage of cancer patients and warrants urgent investigation.
Author Contributions
AJ-P designed the figures. AJ-P and RG planned, constructed and wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Health and Medical Research Council of Australia (APP1157240, APP2001120), Cancer Council Western Australia, Cancer Research Institute Clinic and Laboratory Integration Program (CLIP), Worldwide Cancer Research and a Woodside Energy Fellowship (to RG). The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
2. Gajewski TF, Corrales L, Williams J, Horton B, Sivan A, Spranger S. Cancer Immunotherapy Targets Based on Understanding the T Cell-Inflamed Versus non-T Cell-Inflamed Tumor Microenvironment. Adv Exp Med Biol (2017) 1036:19–31. doi: 10.1007/978-3-319-67577-0_2
3. Johansson-Percival A, He B, Li ZJ, Kjellen A, Russell K, Li J, et al. De Novo Induction of Intratumoral Lymphoid Structures and Vessel Normalization Enhances Immunotherapy in Resistant Tumors. Nat Immunol (2017) 18(11):1207–17. doi: 10.1038/ni.3836
4. Rodriguez A, Peske JD, Woods AN, Leick KM, Mauldin IS, Young SJ, et al. Immune Mechanisms Orchestrate Tertiary Lymphoid Structures in Tumors Via Cancer-Associated Fibroblasts. Immunity (2020). doi: 10.2139/ssrn.3575119
5. Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, et al. Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell (2016) 30(3):500. doi: 10.1016/j.ccell.2016.08.011
6. Thompson ED, Enriquez HL, Fu YX, Engelhard VH. Tumor Masses Support Naive T Cell Infiltration, Activation, and Differentiation Into Effectors. J Exp Med (2010) 207(8):1791–804. doi: 10.1084/jem.20092454
7. Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary Lymphoid Structures in the Era of Cancer Immunotherapy. Nat Rev Cancer (2019) 19(6):307–25. doi: 10.1038/s41568-019-0144-6
8. van de Pavert SA, Mebius RE. New Insights Into the Development of Lymphoid Tissues. Nat Rev Immunol (2010) 10(9):664–74. doi: 10.1038/nri2832
9. Fletcher AL, Acton SE, Knoblich K. Lymph Node Fibroblastic Reticular Cells in Health and Disease. Nat Rev Immunol (2015) 15(6):350–61. doi: 10.1038/nri3846
10. Gallatin WM, Weissman IL, Butcher EC. A Cell-Surface Molecule Involved in Organ-Specific Homing of Lymphocytes. Nature (1983) 304(5921):30–4. doi: 10.1038/304030a0
11. Sautes-Fridman C, Lawand M, Giraldo NA, Kaplon H, Germain C, Fridman WH, et al. Tertiary Lymphoid Structures in Cancers: Prognostic Value, Regulation, and Manipulation for Therapeutic Intervention. Front Immunol (2016) 7:407. doi: 10.3389/fimmu.2016.00407
12. van de Pavert SA, Olivier BJ, Goverse G, Vondenhoff MF, Greuter M, Beke P, et al. Chemokine CXCL13 is Essential for Lymph Node Initiation and is Induced by Retinoic Acid and Neuronal Stimulation. Nat Immunol (2009) 10(11):1193–9. doi: 10.1038/ni.1789
13. Cupedo T, Jansen W, Kraal G, Mebius RE. Induction of Secondary and Tertiary Lymphoid Structures in the Skin. Immunity (2004) 21(5):655–67. doi: 10.1016/j.immuni.2004.09.006
14. Onder L, Danuser R, Scandella E, Firner S, Chai Q, Hehlgans T, et al. Endothelial Cell-Specific Lymphotoxin-Beta Receptor Signaling is Critical for Lymph Node and High Endothelial Venule Formation. J Exp Med (2013) 210(3):465–73. doi: 10.1084/jem.20121462
15. Onder L, Morbe U, Pikor N, Novkovic M, Cheng HW, Hehlgans T, et al. Lymphatic Endothelial Cells Control Initiation of Lymph Node Organogenesis. Immunity (2017) 47(1):80–92 e4. doi: 10.1016/j.immuni.2017.05.008
16. Onder L, Ludewig B. A Fresh View on Lymph Node Organogenesis. Trends Immunol (2018) 39(10):775–87. doi: 10.1016/j.it.2018.08.003
17. Grabner R, Lotzer K, Dopping S, Hildner M, Radke D, Beer M, et al. Lymphotoxin Beta Receptor Signaling Promotes Tertiary Lymphoid Organogenesis in the Aorta Adventitia of Aged ApoE-/- Mice. J Exp Med (2009) 206(1):233–48. doi: 10.1084/jem.20080752
18. Lee Y, Chin RK, Christiansen P, Sun Y, Tumanov AV, Wang J, et al. Recruitment and Activation of Naive T Cells in the Islets by Lymphotoxin Beta Receptor-Dependent Tertiary Lymphoid Structure. Immunity (2006) 25(3):499–509. doi: 10.1016/j.immuni.2006.06.016
19. Luther SA, Lopez T, Bai W, Hanahan D, Cyster JG. BLC Expression in Pancreatic Islets Causes B Cell Recruitment and Lymphotoxin-Dependent Lymphoid Neogenesis. Immunity (2000) 12(5):471–81. doi: 10.1016/S1074-7613(00)80199-5
20. Luther SA, Bidgol A, Hargreaves DC, Schmidt A, Xu Y, Paniyadi J, et al. Differing Activities of Homeostatic Chemokines CCL19, CCL21, and CXCL12 in Lymphocyte and Dendritic Cell Recruitment and Lymphoid Neogenesis. J Immunol (2002) 169(1):424–33. doi: 10.4049/jimmunol.169.1.424
21. Furtado GC, Pacer ME, Bongers G, Benezech C, He Z, Chen L, et al. Tnfalpha-Dependent Development of Lymphoid Tissue in the Absence of RORgammat(+) Lymphoid Tissue Inducer Cells. Mucosal Immunol (2014) 7(3):602–14. doi: 10.1038/mi.2013.79
22. Goya S, Matsuoka H, Mori M, Morishita H, Kida H, Kobashi Y, et al. Sustained Interleukin-6 Signalling Leads to the Development of Lymphoid Organ-Like Structures in the Lung. J Pathol (2003) 200(1):82–7. doi: 10.1002/path.1321
23. Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The Development of Inducible Bronchus-Associated Lymphoid Tissue Depends on IL-17. Nat Immunol (2011) 12(7):639–46. doi: 10.1038/ni.2053
24. Nayar S, Campos J, Smith CG, Iannizzotto V, Gardner DH, Mourcin F, et al. Immunofibroblasts are Pivotal Drivers of Tertiary Lymphoid Structure Formation and Local Pathology. Proc Natl Acad Sci USA (2019) 116(27):13490–7. doi: 10.1073/pnas.1905301116
25. Canete JD, Celis R, Yeremenko N, Sanmarti R, van Duivenvoorde L, Ramirez J, et al. Ectopic Lymphoid Neogenesis is Strongly Associated With Activation of the IL-23 Pathway in Rheumatoid Synovitis. Arthritis Res Ther (2015) 17:173. doi: 10.1186/s13075-015-0688-0
26. Peduto L, Dulauroy S, Lochner M, Spath GF, Morales MA, Cumano A, et al. Inflammation Recapitulates the Ontogeny of Lymphoid Stromal Cells. J Immunol (2009) 182(9):5789–99. doi: 10.4049/jimmunol.0803974
27. Benezech C, Luu NT, Walker JA, Kruglov AA, Loo Y, Nakamura K, et al. Inflammation-Induced Formation of Fat-Associated Lymphoid Clusters. Nat Immunol (2015) 16(8):819–28. doi: 10.1038/ni.3215
28. Barone F, Nayar S, Campos J, Cloake T, Withers DR, Toellner KM, et al. Il-22 Regulates Lymphoid Chemokine Production and Assembly of Tertiary Lymphoid Organs. Proc Natl Acad Sci USA (2015) 112(35):11024–9. doi: 10.1073/pnas.1503315112
29. Guedj K, Khallou-Laschet J, Clement M, Morvan M, Gaston AT, Fornasa G, et al. M1 Macrophages Act as LTbetaR-independent Lymphoid Tissue Inducer Cells During Atherosclerosis-Related Lymphoid Neogenesis. Cardiovasc Res (2014) 101(3):434–43. doi: 10.1093/cvr/cvt263
30. Moussion C, Girard JP. Dendritic Cells Control Lymphocyte Entry to Lymph Nodes Through High Endothelial Venules. Nature (2011) 479(7374):542–6. doi: 10.1038/nature10540
31. Marinkovic T, Garin A, Yokota Y, Fu YX, Ruddle NH, Furtado GC, et al. Interaction of Mature CD3+CD4+ T Cells With Dendritic Cells Triggers the Development of Tertiary Lymphoid Structures in the Thyroid. J Clin Invest (2006) 116(10):2622–32. doi: 10.1172/JCI28993.
32. Girard JP, Moussion C, Forster R. Hevs, Lymphatics and Homeostatic Immune Cell Trafficking in Lymph Nodes. Nat Rev Immunol (2012) 12(11):762–73. doi: 10.1038/nri3298
33. Kanameishi S, Ono S, Honda T, Kabashima K. Lymphotoxin β Receptor Signaling and CD11c-positive Dendritic Cells Form High Endothelial Venule-Like Vessels in the Skin in Murine Atopic Dermatitis Model. J Immunol (2020) 204(1 Supplement):229.2–.2.
34. GeurtsvanKessel CH, Willart MA, Bergen IM, van Rijt LS, Muskens F, Elewaut D, et al. Dendritic Cells are Crucial for Maintenance of Tertiary Lymphoid Structures in the Lung of Influenza Virus-Infected Mice. J Exp Med (2009) 206(11):2339–49. doi: 10.1084/jem.20090410
35. Halle S, Dujardin HC, Bakocevic N, Fleige H, Danzer H, Willenzon S, et al. Induced Bronchus-Associated Lymphoid Tissue Serves as a General Priming Site for T Cells and is Maintained by Dendritic Cells. J Exp Med (2009) 206(12):2593–601. doi: 10.1084/jem.20091472
36. Dvorak HF. Tumors: Wounds That do Not Heal. Similarities Between Tumor Stroma Generation and Wound Healing. N Engl J Med (1986) 315(26):1650–9. doi: 10.1056/NEJM198612253152606
37. Dirkx AE, Oude Egbrink MG, Kuijpers MJ, van der Niet ST, Heijnen VV, Bouma-ter Steege JC, et al. Tumor Angiogenesis Modulates Leukocyte-Vessel Wall Interactions In Vivo by Reducing Endothelial Adhesion Molecule Expression. Cancer Res (2003) 63(9):2322–9.
38. Johansson A, Hamzah J, Ganss R. More Than a Scaffold: Stromal Modulation of Tumor Immunity. Biochim Biophys Acta (2016) 1865(1):3–13. doi: 10.1016/j.bbcan.2015.06.001
39. Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4(+) Follicular Helper T Cell Infiltration Predicts Breast Cancer Survival. J Clin Invest (2013) 123(7):2873–92. doi: 10.1172/JCI67428
40. Gu-Trantien C, Willard-Gallo K. Tumor-Infiltrating Follicular Helper T Cells: The New Kids on the Block. Oncoimmunology (2013) 2(10):e26066. doi: 10.4161/onci.26066
41. Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K. Intratumoral Tertiary Lymphoid Organ is a Favourable Prognosticator in Patients With Pancreatic Cancer. Br J Cancer (2015) 112(11):1782–90. doi: 10.1038/bjc.2015.145
42. Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S, et al. Dendritic Cells in Tumor-Associated Tertiary Lymphoid Structures Signal a Th1 Cytotoxic Immune Contexture and License the Positive Prognostic Value of Infiltrating CD8+ T Cells. Cancer Res (2014) 74(3):705–15. doi: 10.1158/0008-5472.CAN-13-1342
43. Calderaro J, Petitprez F, Becht E, Laurent A, Hirsch TZ, Rousseau B, et al. Intra-Tumoral Tertiary Lymphoid Structures are Associated With a Low Risk of Early Recurrence of Hepatocellular Carcinoma. J Hepatol (2019) 70(1):58–65. doi: 10.1016/j.jhep.2018.09.003
44. Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie JJ, et al. Human Solid Tumors Contain High Endothelial Venules: Association With T- and B-lymphocyte Infiltration and Favorable Prognosis in Breast Cancer. Cancer Res (2011) 71(17):5678–87. doi: 10.1158/0008-5472.CAN-11-0431
45. Martinet L, Garrido I, Girard JP. Tumor High Endothelial Venules (Hevs) Predict Lymphocyte Infiltration and Favorable Prognosis in Breast Cancer. Oncoimmunology (2012) 1(5):789–90. doi: 10.4161/onci.19787
46. Martinet L, Le Guellec S, Filleron T, Lamant L, Meyer N, Rochaix P, et al. High Endothelial Venules (Hevs) in Human Melanoma Lesions: Major Gateways for Tumor-Infiltrating Lymphocytes. Oncoimmunology (2012) 1(6):829–39. doi: 10.4161/onci.20492
47. Hennequin A, Derangere V, Boidot R, Apetoh L, Vincent J, Orry D, et al. Tumor Infiltration by Tbet+ Effector T Cells and CD20+ B Cells is Associated With Survival in Gastric Cancer Patients. Oncoimmunology (2016) 5(2):e1054598. doi: 10.1080/2162402X.2015.1054598
48. Coppola D, Nebozhyn M, Khalil F, Dai H, Yeatman T, Loboda A, et al. Unique Ectopic Lymph Node-Like Structures Present in Human Primary Colorectal Carcinoma are Identified by Immune Gene Array Profiling. Am J Pathol (2011) 179(1):37–45. doi: 10.1016/j.ajpath.2011.03.007
49. Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, et al. 12-Chemokine Gene Signature Identifies Lymph Node-Like Structures in Melanoma: Potential for Patient Selection for Immunotherapy? Sci Rep (2012) 2:765. doi: 10.1038/srep00765
50. Prabhakaran S, Rizk VT, Ma Z, Cheng CH, Berglund AE, Coppola D, et al. Evaluation of Invasive Breast Cancer Samples Using a 12-Chemokine Gene Expression Score: Correlation With Clinical Outcomes. Breast Cancer Res (2017) 19(1):71. doi: 10.1186/s13058-017-0864-z
51. Yamaguchi K, Ito M, Ohmura H, Hanamura F, Nakano M, Tsuchihashi K, et al. Helper T Cell-Dominant Tertiary Lymphoid Structures are Associated With Disease Relapse of Advanced Colorectal Cancer. Oncoimmunology (2020) 9(1):1724763. doi: 10.1080/2162402X.2020.1724763
52. Finkin S, Yuan D, Stein I, Taniguchi K, Weber A, Unger K, et al. Ectopic Lymphoid Structures Function as Microniches for Tumor Progenitor Cells in Hepatocellular Carcinoma. Nat Immunol (2015) 16(12):1235–44. doi: 10.1038/ni.3290
53. Meylan M, Petitprez F, Lacroix L, Di Tommaso L, Roncalli M, Bougouin A, et al. Early Hepatic Lesions Display Immature Tertiary Lymphoid Structures and Show Elevated Expression of Immune Inhibitory and Immunosuppressive Molecules. Clin Cancer Res (2020) 26(16):4381–9. doi: 10.1158/1078-0432.CCR-19-2929
54. Figenschau SL, Fismen S, Fenton KA, Fenton C, Mortensen ES. Tertiary Lymphoid Structures are Associated With Higher Tumor Grade in Primary Operable Breast Cancer Patients. BMC Cancer (2015) 15:101. doi: 10.1186/s12885-015-1116-1
55. Bento DC, Jones E, Junaid S, Tull J, Williams GT, Godkin A, et al. High Endothelial Venules are Rare in Colorectal Cancers But Accumulate in Extra-Tumoral Areas With Disease Progression. Oncoimmunology (2015) 4(3):e974374. doi: 10.4161/2162402X.2014.974374
56. Engelhard VH, Rodriguez AB, Mauldin IS, Woods AN, Peske JD, Slingluff CL Jr. Immune Cell Infiltration and Tertiary Lymphoid Structures as Determinants of Antitumor Immunity. J Immunol (2018) 200(2):432–42. doi: 10.4049/jimmunol.1701269
57. Dieu-Nosjean MC, Giraldo NA, Kaplon H, Germain C, Fridman WH, Sautes-Fridman C. Tertiary Lymphoid Structures, Drivers of the Anti-Tumor Responses in Human Cancers. Immunol Rev (2016) 271(1):260–75. doi: 10.1111/imr.12405
58. Peske JD, Woods AB, Engelhard VH. Control of CD8 T-Cell Infiltration Into Tumors by Vasculature and Microenvironment. Adv Cancer Res (2015) 128:263–307. doi: 10.1016/bs.acr.2015.05.001
59. Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, et al. Long-Term Survival for Patients With non-Small-Cell Lung Cancer With Intratumoral Lymphoid Structures. J Clin Oncol (2008) 26(27):4410–7. doi: 10.1200/JCO.2007.15.0284
60. Martinet L, Filleron T, Le Guellec S, Rochaix P, Garrido I, Girard JP. High Endothelial Venule Blood Vessels for Tumor-Infiltrating Lymphocytes are Associated With Lymphotoxin Beta-Producing Dendritic Cells in Human Breast Cancer. J Immunol (2013) 191(4):2001–8. doi: 10.4049/jimmunol.1300872
61. Giraldo NA, Becht E, Pages F, Skliris G, Verkarre V, Vano Y, et al. Orchestration and Prognostic Significance of Immune Checkpoints in the Microenvironment of Primary and Metastatic Renal Cell Cancer. Clin Cancer Res (2015) 21(13):3031–40. doi: 10.1158/1078-0432.CCR-14-2926
62. Gunderson AJ, Coussens LM. B Cells and Their Mediators as Targets for Therapy in Solid Tumors. Exp Cell Res (2013) 319(11):1644–9. doi: 10.1016/j.yexcr.2013.03.005
63. Kroeger DR, Milne K, Nelson BH. Tumor-Infiltrating Plasma Cells Are Associated With Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin Cancer Res (2016) 22(12):3005–15. doi: 10.1158/1078-0432.CCR-15-2762
64. Garaud S, Buisseret L, Solinas C, Gu-Trantien C, de Wind A, Van den Eynden G, et al. Tumor Infiltrating B-cells Signal Functional Humoral Immune Responses in Breast Cancer. JCI Insight (2019) 4(18):e129641. doi: 10.1172/jci.insight.129641
65. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of B Cells in Tertiary Lymphoid Structures is Associated With a Protective Immunity in Patients With Lung Cancer. Am J Respir Crit Care Med (2014) 189(7):832–44. doi: 10.1164/rccm.201309-1611OC
66. Gu-Trantien C, Migliori E, Buisseret L, de Wind A, Brohee S, Garaud S, et al. CXCL13-Producing TFH Cells Link Immune Suppression and Adaptive Memory in Human Breast Cancer. JCI Insight (2017) 2(11):e91487. doi: 10.1172/jci.insight.91487
67. Castino GF, Cortese N, Capretti G, Serio S, Di Caro G, Mineri R, et al. Spatial Distribution of B Cells Predicts Prognosis in Human Pancreatic Adenocarcinoma. Oncoimmunology (2016) 5(4):e1085147. doi: 10.1080/2162402X.2015.1085147
68. Griss J, Bauer W, Wagner C, Simon M, Chen M, Grabmeier-Pfistershammer K, et al. B Cells Sustain Inflammation and Predict Response to Immune Checkpoint Blockade in Human Melanoma. Nat Commun (2019) 10(1):4186. doi: 10.1038/s41467-019-12160-2
69. Selitsky SR, Mose LE, Smith CC, Chai S, Hoadley KA, Dittmer DP, et al. Prognostic Value of B Cells in Cutaneous Melanoma. Genome Med (2019) 11(1):36. doi: 10.1186/s13073-019-0647-5
70. Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun CM, Calderaro J, et al. B Cells are Associated With Survival and Immunotherapy Response in Sarcoma. Nature (2020) 577(7791):556–60. doi: 10.1038/s41586-019-1906-8
71. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B Cells and Tertiary Lymphoid Structures Promote Immunotherapy Response. Nature (2020) 577(7791):549–55. doi: 10.1038/s41586-019-1922-8
72. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary Lymphoid Structures Improve Immunotherapy and Survival in Melanoma. Nature (2020) 577(7791):561–5. doi: 10.1038/s41586-019-1914-8
73. Carmi Y, Spitzer MH, Linde IL, Burt BM, Prestwood TR, Perlman N, et al. Allogeneic IgG Combined With Dendritic Cell Stimuli Induce Antitumour T-cell Immunity. Nature (2015) 521(7550):99–104. doi: 10.1038/nature14424
74. Bruno TC, Ruffin A, Cillo A, Liu D, Kunning S, Ferris RL, et al. Tumor Infiltrating B Cells Co-Localize With CD4 T Effector Cells Within Organized Tertiary Lymphoid Structures to Present Antigen and Educate the Anti-Tumor Immune Response in Human Primary Tumors. J Immunol (2019) 202(1 Supplement):138.15–.15.
75. Teillaud JL, Dieu-Nosjean MC. Tertiary Lymphoid Structures: An Anti-Tumor School for Adaptive Immune Cells and an Antibody Factory to Fight Cancer? Front Immunol (2017) 8:830. doi: 10.3389/fimmu.2017.00830
76. Hindley JP, Jones E, Smart K, Bridgeman H, Lauder SN, Ondondo B, et al. T-Cell Trafficking Facilitated by High Endothelial Venules is Required for Tumor Control After Regulatory T-cell Depletion. Cancer Res (2012) 72(21):5473–82. doi: 10.1158/0008-5472.CAN-12-1912
77. Colbeck EJ, Jones E, Hindley JP, Smart K, Schulz R, Browne M, et al. Treg Depletion Licenses T Cell-Driven HEV Neogenesis and Promotes Tumor Destruction. Cancer Immunol Res (2017) 5(11):1005–15. doi: 10.1158/2326-6066.CIR-17-0131
78. Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, et al. Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-Tumor T Cell Responses. Immunity (2015) 43(3):579–90. doi: 10.1016/j.immuni.2015.08.006
79. Schrama D, thor Straten P, Fischer WH, McLellan AD, Brocker EB, Reisfeld RA, et al. Targeting of Lymphotoxin-Alpha to the Tumor Elicits an Efficient Immune Response Associated With Induction of Peripheral Lymphoid-Like Tissue. Immunity (2001) 14(2):111–21. doi: 10.1016/S1074-7613(01)00094-2
80. Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of Lymphoidlike Stroma and Immune Escape by Tumors That Express the Chemokine CCL21. Science (2010) 328(5979):749–52. doi: 10.1126/science.1185837
81. Suematsu S, Watanabe T. Generation of a Synthetic Lymphoid Tissue-Like Organoid in Mice. Nat Biotechnol (2004) 22(12):1539–45. doi: 10.1038/nbt1039
82. Kobayashi Y, Watanabe T. Gel-Trapped Lymphorganogenic Chemokines Trigger Artificial Tertiary Lymphoid Organs and Mount Adaptive Immune Responses In Vivo. Front Immunol (2016) 7:316. doi: 10.3389/fimmu.2016.00316
83. Zhu G, Nemoto S, Mailloux AW, Perez-Villarroel P, Nakagawa R, Falahat R, et al. Induction of Tertiary Lymphoid Structures With Antitumor Function by a Lymph Node-Derived Stromal Cell Line. Front Immunol (2018) 9:1609. doi: 10.3389/fimmu.2018.01609
84. Weinstein AM, Chen L, Brzana EA, Patil PR, Taylor JL, Fabian KL, et al. Tbet and IL-36gamma Cooperate in Therapeutic DC-mediated Promotion of Ectopic Lymphoid Organogenesis in the Tumor Microenvironment. Oncoimmunology (2017) 6(6):e1322238. doi: 10.1080/2162402X.2017.1322238
85. Weinstein AM, Giraldo NA, Petitprez F, Julie C, Lacroix L, Peschaud F, et al. Association of IL-36gamma With Tertiary Lymphoid Structures and Inflammatory Immune Infiltrates in Human Colorectal Cancer. Cancer Immunol Immunother (2019) 68(1):109–20. doi: 10.1007/s00262-018-2259-0
86. Maldonado L, Teague JE, Morrow MP, Jotova I, Wu TC, Wang C, et al. Intramuscular Therapeutic Vaccination Targeting HPV16 Induces T Cell Responses That Localize in Mucosal Lesions. Sci Transl Med (2014) 6(221):221ra13. doi: 10.1126/scitranslmed.3007323
87. Tewari N, Zaitoun AM, Arora A, Madhusudan S, Ilyas M, Lobo DN. The Presence of Tumour-Associated Lymphocytes Confers a Good Prognosis in Pancreatic Ductal Adenocarcinoma: An Immunohistochemical Study of Tissue Microarrays. BMC Cancer (2013) 13:436. doi: 10.1186/1471-2407-13-436
88. Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of Unique Neoantigen Qualities in Long-Term Survivors of Pancreatic Cancer. Nature (2017) 551(7681):512–6. doi: 10.1038/nature24462
89. Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, et al. Immunotherapy Converts Nonimmunogenic Pancreatic Tumors Into Immunogenic Foci of Immune Regulation. Cancer Immunol Res (2014) 2(7):616–31. doi: 10.1158/2326-6066.CIR-14-0027
90. Vestweber D. How Leukocytes Cross the Vascular Endothelium. Nat Rev Immunol (2015) 15(11):692–704. doi: 10.1038/nri3908
91. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor Endothelium FasL Establishes a Selective Immune Barrier Promoting Tolerance in Tumors. Nat Med (2014) 20(6):607–15. doi: 10.1038/nm.3541
92. Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH, et al. Vascular Normalization in Rgs5-deficient Tumours Promotes Immune Destruction. Nature (2008) 453(7193):410–4. doi: 10.1038/nature06868
93. Griffioen AW, Damen CA, Blijham GH, Groenewegen G. Tumor Angiogenesis is Accompanied by a Decreased Inflammatory Response of Tumor-Associated Endothelium. Blood (1996) 88(2):667–73. doi: 10.1182/blood.V88.2.667.bloodjournal882667
94. Johansson-Percival A, He B, Ganss R. Immunomodulation of Tumor Vessels: It Takes Two to Tango. Trends Immunol (2018) 39(10):801–14. doi: 10.1016/j.it.2018.08.001
95. Johansson-Percival A, Li ZJ, Lakhiani DD, He B, Wang X, Hamzah J, et al. Intratumoral LIGHT Restores Pericyte Contractile Properties and Vessel Integrity. Cell Rep (2015) 13(12):2687–98. doi: 10.1016/j.celrep.2015.12.004
96. Ganss R, Hanahan D. Tumor Microenvironment can Restrict the Effectiveness of Activated Antitumor Lymphocytes. Cancer Res (1998) 58(20):4673–81.
97. Johansson A, Hamzah J, Payne CJ, Ganss R. Tumor-Targeted TNFalpha Stabilizes Tumor Vessels and Enhances Active Immunotherapy. Proc Natl Acad Sci U S A (2012) 109(20):7841–6. doi: 10.1073/pnas.1118296109
98. Ganss R, Ryschich E, Klar E, Arnold B, Hammerling GJ. Combination of T-cell Therapy and Trigger of Inflammation Induces Remodeling of the Vasculature and Tumor Eradication. Cancer Res (2002) 62(5):1462–70.
99. Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, et al. Mutual Regulation of Tumour Vessel Normalization and Immunostimulatory Reprogramming. Nature (2017) 544(7649):250–4. doi: 10.1038/nature21724
100. Zheng X, Fang Z, Liu X, Deng S, Zhou P, Wang X, et al. Increased Vessel Perfusion Predicts the Efficacy of Immune Checkpoint Blockade. J Clin Invest (2018) 128(5):2104–15. doi: 10.1172/JCI96582
101. Crowe PD, VanArsdale TL, Walter BN, Ware CF, Hession C, Ehrenfels B, et al. A Lymphotoxin-Beta-Specific Receptor. Science (1994) 264(5159):707–10. doi: 10.1126/science.8171323
102. Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes Simplex Virus-1 Entry Into Cells Mediated by a Novel Member of the TNF/NGF Receptor Family. Cell (1996) 87(3):427–36. doi: 10.1016/S0092-8674(00)81363-X
103. Lu TT, Browning JL. Role of the Lymphotoxin/LIGHT System in the Development and Maintenance of Reticular Networks and Vasculature in Lymphoid Tissues. Front Immunol (2014) 5:47. doi: 10.3389/fimmu.2014.00047
104. Browning JL, Allaire N, Ngam-Ek A, Notidis E, Hunt J, Perrin S, et al. Lymphotoxin-Beta Receptor Signaling is Required for the Homeostatic Control of HEV Differentiation and Function. Immunity (2005) 23(5):539–50. doi: 10.1016/j.immuni.2005.10.002
105. Chang YH, Hsieh SL, Chao Y, Chou YC, Lin WW. Proinflammatory Effects of LIGHT Through HVEM and LTbetaR Interactions in Cultured Human Umbilical Vein Endothelial Cells. J BioMed Sci (2005) 12(2):363–75. doi: 10.1007/s11373-005-1360-5
106. Morel Y, Schiano de Colella JM, Harrop J, Deen KC, Holmes SD, Wattam TA, et al. Reciprocal Expression of the TNF Family Receptor Herpes Virus Entry Mediator and its Ligand LIGHT on Activated T Cells: LIGHT Down-Regulates its Own Receptor. J Immunol (2000) 165(8):4397–404. doi: 10.4049/jimmunol.165.8.4397
107. Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, Kiialainen A, et al. Dual Angiopoietin-2 and VEGFA Inhibition Elicits Antitumor Immunity That is Enhanced by PD-1 Checkpoint Blockade. Sci Transl Med (2017) 9(385):eaak9670. doi: 10.1126/scitranslmed.aak9670
108. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular Normalizing Doses of Antiangiogenic Treatment Reprogram the Immunosuppressive Tumor Microenvironment and Enhance Immunotherapy. Proc Natl Acad Sci U.S.A. (2012) 109(43):17561–6. doi: 10.1073/pnas.1215397109
109. Johansson A, Hamzah J, Ganss R. Intratumoral TNFalpha Improves Immunotherapy. Oncoimmunology (2012) 1(8):1395–7. doi: 10.4161/onci.20981
110. Chelvanambi M, Fecek RJ, Taylor JL, Storkus WJ. STING Agonist-Based Treatment Promotes Vascular Normalization and Tertiary Lymphoid Structure Formation in the Therapeutic Melanoma Microenvironment. J Immunother Cancer (2021) 9(2):e001906. doi: 10.1136/jitc-2020-001906
111. Upadhaya S, Neftelino ST, Hodge JP, Oliva C, Campbell JR, Yu JX. Combinations Take Centre Stage in PD1/PDL1 Inhibitor Clinical Trials. Nat Rev Drug Discovery (2020) 20:168–9. doi: 10.1038/d41573-020-00204-y
112. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A Transcriptionally and Functionally Distinct PD-1(+) Cd8(+) T Cell Pool With Predictive Potential in non-Small-Cell Lung Cancer Treated With PD-1 Blockade. Nat Med (2018) 24(7):994–1004. doi: 10.1038/s41591-018-0057-z
113. Cottrell TR, Thompson ED, Forde PM, Stein JE, Duffield AS, Anagnostou V, et al. Pathologic Features of Response to Neoadjuvant anti-PD-1 in Resected non-Small-Cell Lung Carcinoma: A Proposal for Quantitative Immune-Related Pathologic Response Criteria (Irprc). Ann Oncol (2018) 29(8):1853–60. doi: 10.1093/annonc/mdy218
114. Eroglu Z, Zaretsky JM, Hu-Lieskovan S, Kim DW, Algazi A, Johnson DB, et al. High Response Rate to PD-1 Blockade in Desmoplastic Melanomas. Nature (2018) 553(7688):347–50. doi: 10.1038/nature25187
115. Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, et al. Combined Antiangiogenic and anti-PD-L1 Therapy Stimulates Tumor Immunity Through HEV Formation. Sci Transl Med (2017) 9(385):eaak9679. doi: 10.1126/scitranslmed.aak9679
116. He B, Jabouille A, Steri V, Johansson-Percival A, Michael IP, Kotamraju VR, et al. Vascular Targeting of LIGHT Normalizes Blood Vessels in Primary Brain Cancer and Induces Intratumoural High Endothelial Venules. J Pathol (2018) 245(2):209–21. doi: 10.1002/path.5080
117. Xing P, Zhang F, Wang G, Xu Y, Li C, Wang S, et al. Incidence Rates of Immune-Related Adverse Events and Their Correlation With Response in Advanced Solid Tumours Treated With NIVO or NIVO+IPI: A Systematic Review and Meta-Analysis. J Immunother Cancer (2019) 7(1):341. doi: 10.1186/s40425-019-0779-6
118. Callahan MK, Kluger H, Postow MA, Segal NH, Lesokhin A, Atkins MB, et al. Nivolumab Plus Ipilimumab in Patients With Advanced Melanoma: Updated Survival, Response, and Safety Data in a Phase I Dose-Escalation Study. J Clin Oncol (2018) 36(4):391–8. doi: 10.1200/JCO.2017.72.2850
119. Kleef R, Nagy R, Baierl A, Bacher V, Bojar H, McKee DL, et al. Low-Dose Ipilimumab Plus Nivolumab Combined With IL-2 and Hyperthermia in Cancer Patients With Advanced Disease: Exploratory Findings of a Case Series of 131 Stage IV Cancers - a Retrospective Study of a Single Institution. Cancer Immunol Immunother (2021) 70(5):1393–403. doi: 10.1007/s00262-020-02751-0
120. Tomei AA, Siegert S, Britschgi MR, Luther SA, Swartz MA. Fluid Flow Regulates Stromal Cell Organization and CCL21 Expression in a Tissue-Engineered Lymph Node Microenvironment. J Immunol (2009) 183(7):4273–83. doi: 10.4049/jimmunol.0900835
121. Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-Associated Fibroblasts: An Emerging Target of Anti-Cancer Immunotherapy. J Hematol Oncol (2019) 12(1):86. doi: 10.1186/s13045-019-0770-1
122. Monteran L, Erez N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front Immunol (2019) 10:1835. doi: 10.3389/fimmu.2019.01835
123. Cheng HW, Onder L, Cupovic J, Boesch M, Novkovic M, Pikor N, et al. CCL19-Producing Fibroblastic Stromal Cells Restrain Lung Carcinoma Growth by Promoting Local Antitumor T-cell Responses. J Allergy Clin Immunol (2018) 142(4):1257–71 e4. doi: 10.1016/j.jaci.2017.12.998
124. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-Associated Macrophages as Treatment Targets in Oncology. Nat Rev Clin Oncol (2017) 14(7):399–416. doi: 10.1038/nrclinonc.2016.217
125. DeNardo DG, Ruffell B. Macrophages as Regulators of Tumour Immunity and Immunotherapy. Nat Rev Immunol (2019) 19(6):369–82. doi: 10.1038/s41577-019-0127-6
126. Allavena P, Sica A, Garlanda C, Mantovani A. The Yin-Yang of Tumor-Associated Macrophages in Neoplastic Progression and Immune Surveillance. Immunol Rev (2008) 222:155–61. doi: 10.1111/j.1600-065X.2008.00607.x
Keywords: light, LTβR, tumor, TLS, ICB, vascular normalization
Citation: Johansson-Percival A and Ganss R (2021) Therapeutic Induction of Tertiary Lymphoid Structures in Cancer Through Stromal Remodeling. Front. Immunol. 12:674375. doi: 10.3389/fimmu.2021.674375
Received: 01 March 2021; Accepted: 04 May 2021;
Published: 27 May 2021.
Edited by:
Anna Dimberg, Uppsala University, SwedenReviewed by:
Tibor Bakacs, Alfred Renyi Institute of Mathematics, HungaryTiziana Schioppa, University of Brescia, Italy
Copyright © 2021 Johansson-Percival and Ganss. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Johansson-Percival, YW5uYS5hLmpvaGFuc3NvbkBwZXJraW5zLm9yZy5hdQ==