 Jose Manuel Sánchez-Maldonado1,2,3Rafael Cáliz1,3,4
Jose Manuel Sánchez-Maldonado1,2,3Rafael Cáliz1,3,4 Miguel Ángel López-Nevot3,5Antonio José Cabrera-Serrano1,2,3Ana Moñiz-Díez1,2,3
Miguel Ángel López-Nevot3,5Antonio José Cabrera-Serrano1,2,3Ana Moñiz-Díez1,2,3 Helena Canhão6,7Rob Ter Horst8
Helena Canhão6,7Rob Ter Horst8 Luca Quartuccio9Signe B. Sorensen10,11Bente Glintborg12,13Merete L. Hetland12,13Ileana Filipescu14Eva Pérez-Pampin15Pablo Conesa-Zamora16Jerzy Swierkot17Alfons A. den Broeder18Salvatore De Vita9Eva Rabing Brix Petersen19
Luca Quartuccio9Signe B. Sorensen10,11Bente Glintborg12,13Merete L. Hetland12,13Ileana Filipescu14Eva Pérez-Pampin15Pablo Conesa-Zamora16Jerzy Swierkot17Alfons A. den Broeder18Salvatore De Vita9Eva Rabing Brix Petersen19 Yang Li8,20Miguel A. Ferrer3Alejandro Escudero21
Yang Li8,20Miguel A. Ferrer3Alejandro Escudero21 Mihai G. Netea8,22
Mihai G. Netea8,22 Marieke J. H. Coenen23Vibeke Andersen9,10,24
Marieke J. H. Coenen23Vibeke Andersen9,10,24 João E. Fonseca25,26Manuel Jurado1,2,3
João E. Fonseca25,26Manuel Jurado1,2,3 Katarzyna Bogunia-Kubik27
Katarzyna Bogunia-Kubik27 Eduardo Collantes21
Eduardo Collantes21 Juan Sainz1,2,3,28*
Juan Sainz1,2,3,28*- 1Genomic Oncology Area, Centre for Genomics and Oncological Research (GENYO), Parque tecnológico de la Salud (PTS) Granada, Granada, Spain
- 2Hematology Department, Virgen de las Nieves University Hospital, Granada, Spain
- 3Instituto de Investigación Biosanitaria (IBs) Granada, Granada, Spain
- 4Department of Rheumatology, Virgen de las Nieves University Hospital, Granada, Spain
- 5Immunology Department, Virgen de las Nieves University Hospital, Granada, Spain
- 6EpiDoC Unit, CEDOC, NOVA Medical School and National School of Public Health, Universidade Nova de Lisboa, Lisbon, Portugal
- 7Comprehensive Health Research Center (CHRC), NOVA Medical School, Lisbon, Portugal
- 8Department of Internal Medicine and Radboud Center for Infectious Diseases, Radboud University Nijmegen Medical Center, Nijmegen, Netherlands
- 9Department of Medical Area, Clinic of Rheumatology, University of Udine, Udine, Italy
- 10Molecular Diagnostic and Clinical Research Unit, IRS-Center Sonderjylland, University Hospital of Southern Jutland, Aabenraa, Denmark
- 11Institute of Molecular Medicine, Faculty of Health Sciences, University of Southern Denmark, Odense, Denmark
- 12The Danish Rheumatologic Biobank and Copenhagen Center for Arthritis Research (DANBIO) Registry, The Danish Rheumatologic Biobank and Copenhagen Center for Arthritis Research (COPECARE), Center for Rheumatology and Spine Diseases, Centre of Head and Orthopaedics, Rigshospitalet, Glostrup, Denmark
- 13Department of Clinical Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
- 14Rheumatology Department, University of Medicine and Pharmacy “Iuliu Hatieganu”, Cluj-Napoca, Romania
- 15Rheumatology Unit, University Hospital of Santiago de Compostela, Santiago de Compostela, Spain
- 16Clinical Analysis Department, Santa Lucía University Hospital, Cartagena, Spain
- 17Department of Rheumatology and Internal Medicine, Wroclaw Medical University, Wroclaw, Poland
- 18Radboud Institute for Health Sciences, Department of Rheumatology, Radboud University Medical Center, Nijmegen, Netherlands
- 19Department of Biochemistry and Immunology, University Hospital of Southern Jutland, Aabenraa, Denmark
- 20Centre for Individualised Infection Medicine (CiiM) & Centre for Experimental and Clinical Infection Research (TWINCORE), Helmholtz-Centre for Infection Research (HZI) and The Hannover Medical School (MHH), Hannover, Germany
- 21Rheumatology Department, Reina Sofía Hospital/Instituto Maimónides de Investigación Biomédica de Córdoba (IMIBIC)/University of Córdoba, Córdoba, Spain
- 22Department for Immunology & Metabolism, Life and Medical Sciences Institute (LIMES), University of Bonn, Bonn, Germany
- 23Radboud Institute for Health Sciences, Department of Human Genetics, Radboud University Medical Center, Nijmegen, Netherlands
- 24Institute of Regional Research, Faculty of Health Sciences, University of Southern Denmark, Odense, Denmark
- 25Rheumatology and Metabolic Bone Diseases Department, Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte (CHLN), Lisbon, Portugal
- 26Rheumatology Research Unit, Instituto de Medicina Molecular, Faculty of Medicine, University of Lisbon, Lisbon Academic Medical Center, Lisbon, Portugal
- 27Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, Wrocław, Poland
- 28Department of Biochemistry and Molecular Biology I, University of Granada, Granada, Spain
We aimed to validate the association of 28 GWAS-identified genetic variants for response to TNF inhibitors (TNFi) in a discovery cohort of 1361 rheumatoid arthritis (RA) patients monitored in routine care and ascertained through the REPAIR consortium and DANBIO registry. We genotyped selected markers and evaluated their association with response to TNFi after 6 months of treatment according to the change in disease activity score 28 (ΔDAS28). Next, we confirmed the most interesting results through meta-analysis of our data with those from the DREAM cohort that included 706 RA patients treated with TNFi. The meta-analysis of the discovery cohort and DREAM registry including 2067 RA patients revealed an overall association of the LINC02549rs7767069 SNP with a lower improvement in DAS28 that remained significant after correction for multiple testing (per-allele ORMeta=0.83, PMeta=0.000077; PHet=0.61). In addition, we found that each copy of the LRRC55rs717117G allele was significantly associated with lower improvement in DAS28 in rheumatoid factor (RF)-positive patients (per-allele ORMeta=0.67, P=0.00058; PHet=0.06) whereas an opposite but not significant effect was detected in RF-negative subjects (per-allele ORMeta=1.38, P=0.10; PHet=0.45; PInteraction=0.00028). Interestingly, although the identified associations did not survive multiple testing correction, the meta-analysis also showed overall and RF-specific associations for the MAFBrs6071980 and CNTN5rs1813443 SNPs with decreased changes in DAS28 (per-allele ORMeta_rs6071980 = 0.85, P=0.0059; PHet=0.63 and ORMeta_rs1813443_RF+=0.81, P=0.0059; PHet=0.69 and ORMeta_rs1813443_RF-=1.00, P=0.99; PHet=0.12; PInteraction=0.032). Mechanistically, we found that subjects carrying the LINC02549rs7767069T allele had significantly increased numbers of CD45RO+CD45RA+ T cells (P=0.000025) whereas carriers of the LINC02549rs7767069T/T genotype showed significantly increased levels of soluble scavengers CD5 and CD6 in serum (P=0.00037 and P=0.00041). In addition, carriers of the LRRC55rs717117G allele showed decreased production of IL6 after stimulation of PBMCs with B burgdorferi and E coli bacteria (P=0.00046 and P=0.00044), which suggested a reduced IL6-mediated anti-inflammatory effect of this marker to worsen the response to TNFi. In conclusion, this study confirmed the influence of the LINC02549 and LRRC55 loci to determine the response to TNFi in RA patients and suggested a weak effect of the MAFB and CNTN5 loci that need to be further investigated.
Introduction
Rheumatoid Arthritis (RA) is a complex and chronic disease marked by symptoms of inflammation and pain in the joints that eventually lead to joint destruction, loss of function and disability. These symptoms of inflammation are mostly driven by certain central cytokines that modulate both cellular and humoral immune responses in the synovial fluid and synovium of patients (1). Although RA remains as a chronic and incurable autoimmune disease that occurs in as much as 0.5-1% of the general population (2), the introduction of biological agents to target deregulated cytokines has substantially improved the signs and symptoms of the disease (3). Among these cytokines, tumor necrosis factor alpha (TNFα) has attracted most attention as it has been found to be deregulated in patients with autoimmune diseases including RA (4). It has been reported, for instance, that TNFα activates macrophages, synoviocytes, chondrocytes, and osteoclasts in a dose-dependent manner (5) and that high levels of circulating TNFα correlate with disease activity and disease progression (6).
The increasing number of biological agents approved by the FDA and the increased prevalence of the disease all around the world (7) have placed a substantial economic burden for health care systems. Although the introduction of biosimilars in clinical practice reduced the cost of these treatments in many countries (8), there is still an unmet need to optimize biologic therapies, avoiding unnecessary adverse effects risks and reducing costs (9).
The interplay between genetics and drug response has been the subject of intense investigations during last decades. Response to biologics has been shown to vary between individuals and that a large proportion of patients show no clinical improvement (10). Given the high cost of these drugs and the potential impairment of non-responding patients, the identification of genetic biomarkers associated with drug response to specific biological agents would help to know which patients might benefit from a particular treatment. However, to date, only a few genome-wide association studies (GWAS) (11–17) or well powered candidate gene association studies have been conducted (18–26). We are far from being able to optimize drug dosing or prioritize drug combinations based on genetic findings. In fact, attempts to validate the association of most of the genetic markers identified in these association studies have failed (27), which confirms the limited application of genetic findings in a clinical setting. Considering that the validation of previous GWAS findings is an essential step to tailor treatments for RA and to approach personalized medicine, we aimed to validate the association of GWAS-identified variants for response to TNF inhibitors (TNFi) in a two-stage nested case-control association study including a cohort of 1361 anti-TNF naïve RA patients ascertained through the REPAIR consortium and DANBIO registry and an independent replication cohort of 706 RA patients treated with TNFi from the DREAM registry. We also investigated whether the effect of selected markers on the response to TNFi could be modified by rheumatoid factor (RF) status, and whether genetic variants could influence immune responses and affect the serological concentration of 108 plasmatic inflammatory proteins, 7 serum steroid hormones or counts of 91 blood-derived immune cell populations.
Material and Methods
Study Populations and Response to TNFi
The discovery population consisted of 1361 RA patients ascertained through the REPAIR consortium and the DANBIO registry (Table 1) (28, 29). RA patients fulfilled the 1987 revised American College of Rheumatology (ACR) (30) and/or the ACR/EULAR 2010 classification criteria (31). In order to further replicate the most interesting results, we validated the association with response to anti-TNF drugs of those SNPs showing a P<0.05 in the discovery cohort in 706 Dutch RA patients treated with TNFi from the DREAM (Dutch RhEumatoid Arthritis Monitoring) registry (Supplementary Table 1). The study followed the Declaration of Helsinki. Study participants were of European origin and gave their written informed consent to participate in the study, which was approved by the ethical review committee of participant institutions: Virgen de las Nieves University Hospital (2012/89); Santa Maria Hospital-CHLN (CE 877/121.2012); University Clinical Hospital of Santiago de Compostela (2013/156); Wroclaw Medical University (KB-625/2016); Radboud university medical center (2011/299) and by the Regional Ethics Committee of Central Denmark Region (S-20120113). A detailed description of the discovery population has been reported elsewhere (19, 20, 22, 24). All RA patients were naïve for TNFi and response to TNFi for each patient in all study populations was calculated using the change in disease activity score (DAS28CRP) between baseline and 6 months after treatment. Overall and RF-stratified linear regression analyses adjusted for age, sex and country of origin were used to determine the association between GWAS-identified SNPs and changes in DAS28. RA patients with missing values either for DAS28 (in at the time points of interest) or RF were not included in the analysis.
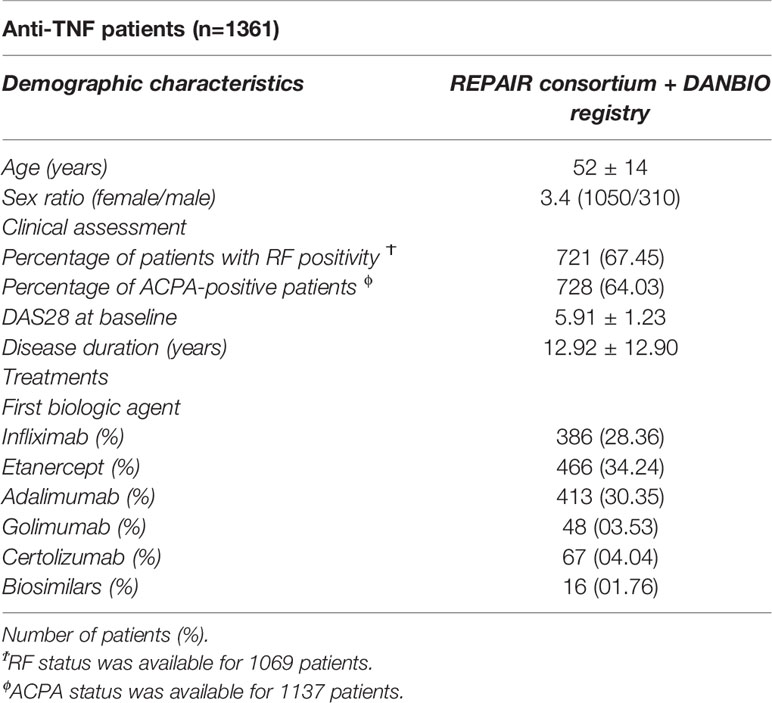
Table 1 Demographic and clinical characteristics of anti-TNF patients.
DNA Extraction, SNP Selection, Genotyping, and Quality Control
Genomic DNA from RA patients was extracted from blood samples using the QIAamp DNA Blood Mini kit (Valencia, CA, EEUU) according to manufacturer’s instructions. The single-nucleotide polymorphisms (SNPs) were selected through an extensive literature search of relevant GWAS and meta-analyses published by February 2019 using publicly available online databases. Additional criteria were potential functionality and linkage disequilibrium between the reported SNPs. Biological function was predicted according to the data publicly available in the integrated Regulome database (www.regulomedb.org), and eQTL data browsers (www.gtexportal.org/home/ and https://genenetwork.nl/bloodeqtlbrowser/). A total of 28 SNPs in 25 genes were selected for genotyping in the discovery cohort (Table 2). Genotyping of selected SNPs was performed using KASP® probes according to manufacturer’s instructions (LGC Genomics, Hoddesdon, UK). For quality control, ~5% of DNA samples were randomly included as duplicates and concordance between duplicate samples was ≥99.0%. Replication of the most interesting association was conducted in the DREAM registry (n=706) following a similar quality control genotyping strategy.
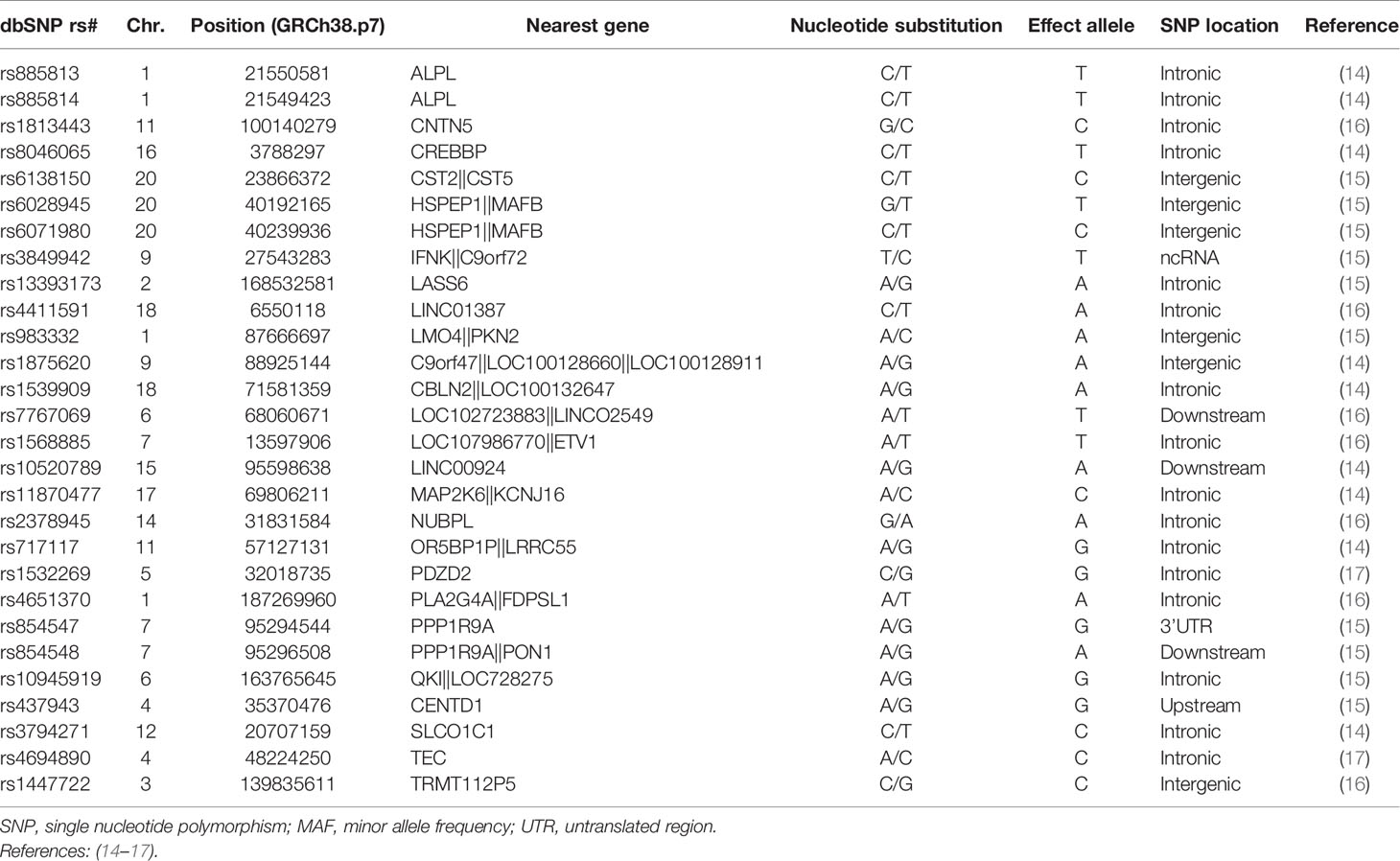
Table 2 Selection of GWAS-identified SNPs for response to anti-TNF drugs.
Hardy-Weinberg Equilibrium, Genetic Association Analysis, and Meta-Analysis
Deviation from Hardy-Weinberg Equilibrium (HWE) was tested in the control group (responders and moderate responders according to the EULAR response criteria) by chi-square (χ2), and linear regression analysis adjusted for age, sex and country of origin was used to assess the associations of the GWAS-identified polymorphisms with absolute changes in DAS28 assuming log-additive, dominant and recessive models of inheritance. Those SNPs with the lowest P-value in the discovery population according to each genetic model were advanced for replication in the DREAM cohort and meta-analysis of the discovery and replication populations using a fixed effect model was performed to validate the association observed. I2 statistic was used to assess heterogeneity between studies. Correction for multiple testing was performed using the Bonferroni method but also considering the two inheritance models tested. Given that log-additive and dominant models showed a high degree of collinearity, the significant threshold for the meta-analysis was set to 0.00089 considering log-additive/dominant and recessive inheritance models (0.05/28SNPs/2models). Overall statistical power was calculated using Quanto (v.12.4) assuming a log-additive model and a baseline risk of 30% for response to TNFi (32, 33).
Cell Isolation, Differentiation and Cytokine Quantitative Trait Loci, and Hormone Analysis in Relation to the GWAS-Identified Variants for Response To TNFi
With the aim of determining whether those SNPs associated with response to TNFi had a role in modulating immune responses, we performed in vitro stimulation experiments and measured cytokine production (IFNγ, IL1Ra, IL1β, IL6, IL8, IL10, TNFα, IL17, and IL22) after stimulation of peripheral blood mononuclear cells (PBMCs), whole blood or monocyte-derived macrophages (MDMs) from 408 healthy subjects of the 500FG cohort from the Human Functional Genomics Project (HFGP) with LPS (1 or 100 ng/ml), PHA (10μg/ml), Pam3Cys (10μg/ml), CpG (ODN M362; 10μg/ml) and B. burgdorferi and E. coli, as experimental model for cytokine production capacity. Given the sex disparities in the prevalence and course of RA and the impact of steroid hormones in modulating immune responses, we also evaluated the correlation of SNPs with serum levels of 7 steroid hormones (androstenedione, cortisol, 11-deoxy-cortisol, 17-hydroxy progesterone, progesterone, testosterone and 25 hydroxy vitamin D3) in a subset of the 500FG cohort without hormonal replacement therapy or oral contraceptives (n=280). After log transformation, cytokine or serum steroid hormone levels were correlated with the SNPs of interest using a linear regression model with age and sex as co-factors in R (http://www.r-project.org/). This analysis led to cytokine quantitative trait loci (cQTL) and hormone quantitative trait loci (hQTL). Significance thresholds were set to be 0.000463 and 0.00357 (0.05/6stimulants/9cytokines or 0.05/7hormones and 2 inheritance models) for cQTL and hQTL, respectively.
Correlation Between GWAS-Identified Polymorphisms and Cell Counts of 91 Blood-Derived Immune Cell Populations and Serum/Plasmatic Proteomic Profile
We also investigated whether selected polymorphisms had an impact on blood cell counts by analyzing a set of 91 manually annotated immune cell populations and genotype data from the 500FG cohort that consisted of 408 healthy subjects (Supplementary Table 2). Cell populations were measured by 10-color flow cytometry (Navios flow cytometer, Beckman Coulter) after blood sampling (2-3 hours) and cell count analysis was performed using the Kaluza software (Beckman Coulter, v.1.3). In order to reduce inter-experimental noise and increase statistical power, cell count analysis was performed by calculating parental and grandparental percentages, which were defined as the percentage of a certain cell type within the cell-populations one or two levels higher in the hierarchical definitions of cell sub-populations (34). Detailed laboratory protocols for cell isolation, reagents, gating and flow cytometry analysis have been reported elsewhere (35) and the accession number for the raw flow cytometry data and analyzed data files are available upon request to the authors (http://hfgp.bbmri.nl). A proteomic analysis was also performed in serum and plasma samples from the 500FG cohort. Circulating proteins were measured using the commercially Olink® Inflammation panel (Olink, Sweden) that resulted in the measurement of 103 different biomarkers (Supplementary Table 3). Proteins levels were expressed on a log2-scale as normalized protein expression values, and normalized using bridging samples to correct for batch variation. Considering the number of proteins (n=103) and cell populations (n=91) tested, P-values of 0.00049 and 0.00055 were set as significant thresholds for the proteomic and cell-level variation analysis, respectively.
Results
A total of 1361 anti-TNF patients were included in the discovery population. The mean age of the RA patients was 52±14 and they showed a female/male ratio of 3.4 (1050/310). Sixty-seven percent of the RA patients were positive for RF and 64% had anti-citrullinated protein antibodies (ACPA). The median disease duration was of 12.92 years and the disease activity score 28 (DAS28CRP) calculated at patient recruitment was of 5.91 (Table 1).
Association of GWAS-Identified SNPs With Response to Anti-TNF Drugs
All SNPs were in Hardy-Weinberg equilibrium in the control group (responders according to EULAR response criteria; P>0.001) and showed a high genotyping call rate (>90%) with the exception of the LINC01387rs4411591 SNP that was excluded from the statistical analysis. The overall linear regression analysis of the discovery cohort including 1361 RA patients treated with TNFi showed that the MAFBrs6028945, MAFBrs6071980, LINC02549rs7767069, and LRRC55rs717117 SNPs had an overall significant effect on the response to TNFi at P<0.05 level (ORDominant=0.81, 95%CI 0.68-0.97, P=0.020; per-allele OR=0.83, 95%CI 0.72-0.97, P=0.020; per-allele OR=0.85, 95%CI 0.76-0.96, P=0.008; per-allele OR=0.76, 95%CI 0.60-0.97, P=0.026; Table 3). Importantly, the meta-analysis of the discovery and replication cohorts confirmed the overall association of the LINC02549rs7767069 SNP with lower DAS28 improvement that remained significant after multiple testing correction (per-allele ORMeta=0.83, 95%CI 0.76-0.91, PMeta=0.000077; PHet=0.61; Table 4). Although it did not survive multiple testing correction, the meta-analysis also showed a potentially interesting overall associations for the MAFBrs6071980 SNP with less DAS28 improvement (per-allele ORMeta_rs6071980 = 0.85, 95%CI 0.76-0.95, P=0.0059; PHet=0.63; Table 4).
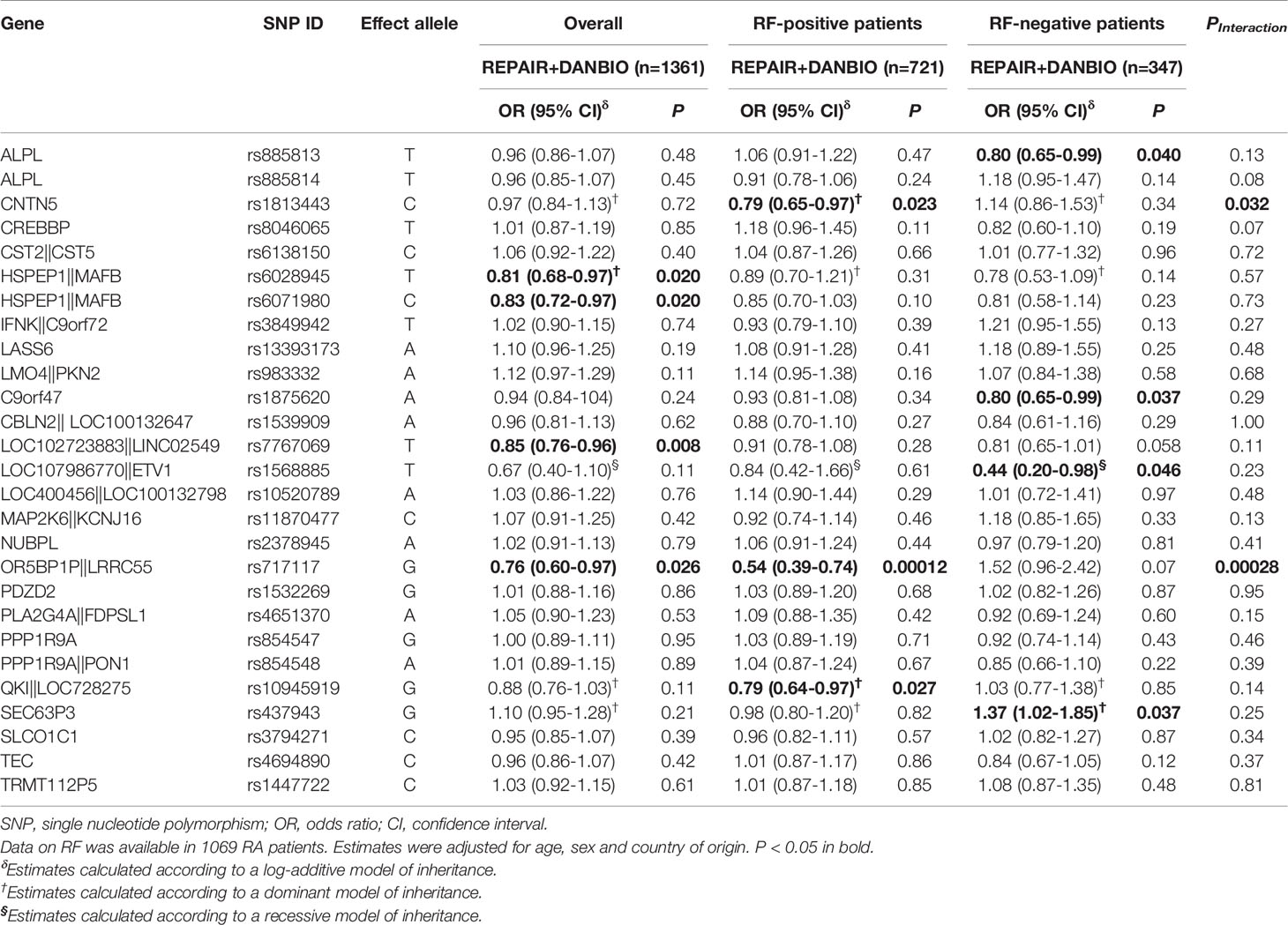
Table 3 Overall and RF-specific associations of selected polymorphisms and response to anti-TNF drugs (ΔDAS28) in the REPAIR consortium.
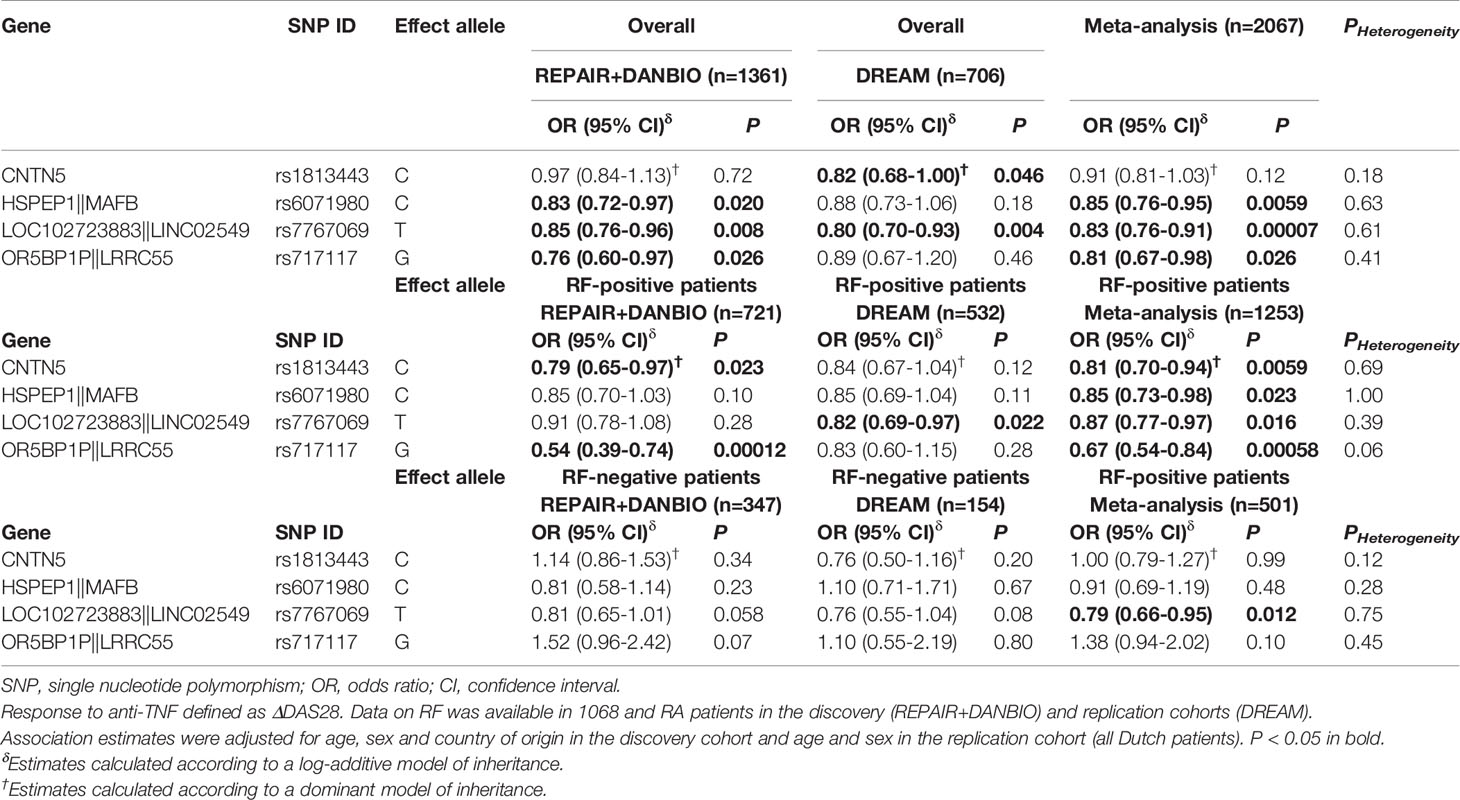
Table 4 Overall and RF-specific meta-analysis of the CNTN5rs1813443, MAFBrs607198, LINCO2549rs7767069 and LRRC55rs717117 polymorphisms and response to anti-TNF drugs.
A RF-stratified analysis showed a RF-specific association for the LRRC55rs717117 SNP with response to TNFi that remained statistically significant after correction for multiple testing in the discovery population. Thus, RF-positive RA patients carrying the LRRC55rs717117G allele additively decreased the drop in DAS28 (per-allele OR=0.54, 95%CI 0.39–0.74, P=0.00012) whereas RF-negative RA patients showed an opposite but not statistically significant effect (per-allele OR=1.52, 95%CI 0.96–2.42, P=0.07; PInteraction=0.00028; Table 3). Interestingly, the meta-analysis of our data with those from the DREAM registry including 2067 RA patients confirmed the RF-specific effect of this SNP to modulate the response to anti-TNF drugs (per-allele ORMeta_RF+=0.67, 95%CI 0.54-0.84, PMeta=0.00058; PHet=0.06 and per-allele ORMeta_RF-=1.38, 95%CI 0.94-2.02, P=0.10; PHet=0.45; PInteraction=0.00028; Table 4). Although it did not survive multiple testing, the meta-analysis also showed potentially interesting RF-specific association for the CNTN5rs1813443 SNP with a decreased drop in DAS28 (ORMeta_rs1813443_RF+=0.81, 95%CI 0.70-0.94, P=0.0059; PHet=0.69 and ORMeta_rs1813443_RF-=1.00, 95%CI 0.79-1.27, P=0.99; PHet=0.12; PInteraction=0.032; Table 4).
Functional Characterization of the Most Interesting Findings
Considering these results, we attempted to shed some light into the functional consequences of the overall or RF-specific effects of the LINC02549rs7767069, LRRC55rs717117, MAFBrs6071980 and CNTN5rs1813443 SNPs to modulate the response to TNFi. Interestingly, our functional experiments showed that, when considering the total number of leukocytes as reference, the LINC02549rs7767069 polymorphism significantly correlated with increased numbers of CD45RO+CD45RA+ T cells in blood (P=0.00047; Figure 1A). Subjects carrying the LINC02549rs7767069T allele (associated with poor response to TNFi in RA patients) had significantly increased numbers of CD45RO+CD45RA+ T cells (P=0.000025), which suggested that this genetic marker might influence the response to TNFi by mediating the number of this specific T cell subset in blood and, thereby contribute to inflammation. In addition, we observed that those subjects carrying two copies of the LINC02549rs7767069T allele showed significantly increased serum levels of soluble scavenger receptors CD5 and CD6 when compared with those carrying the A/T or A/A genotypes (P=0.00037 and P=0.00041; Figures 1B, C). These results also suggested a functional role of the LINC02549rs7767069 SNP in RA likely through the CD5/CD6-mediated modulation of T cells and certain subsets of B cells that control multiple processes including cellular adhesion and migration across endothelial and epithelial cells, antigen presentation by B cells and the subsequent proliferation of T cells.
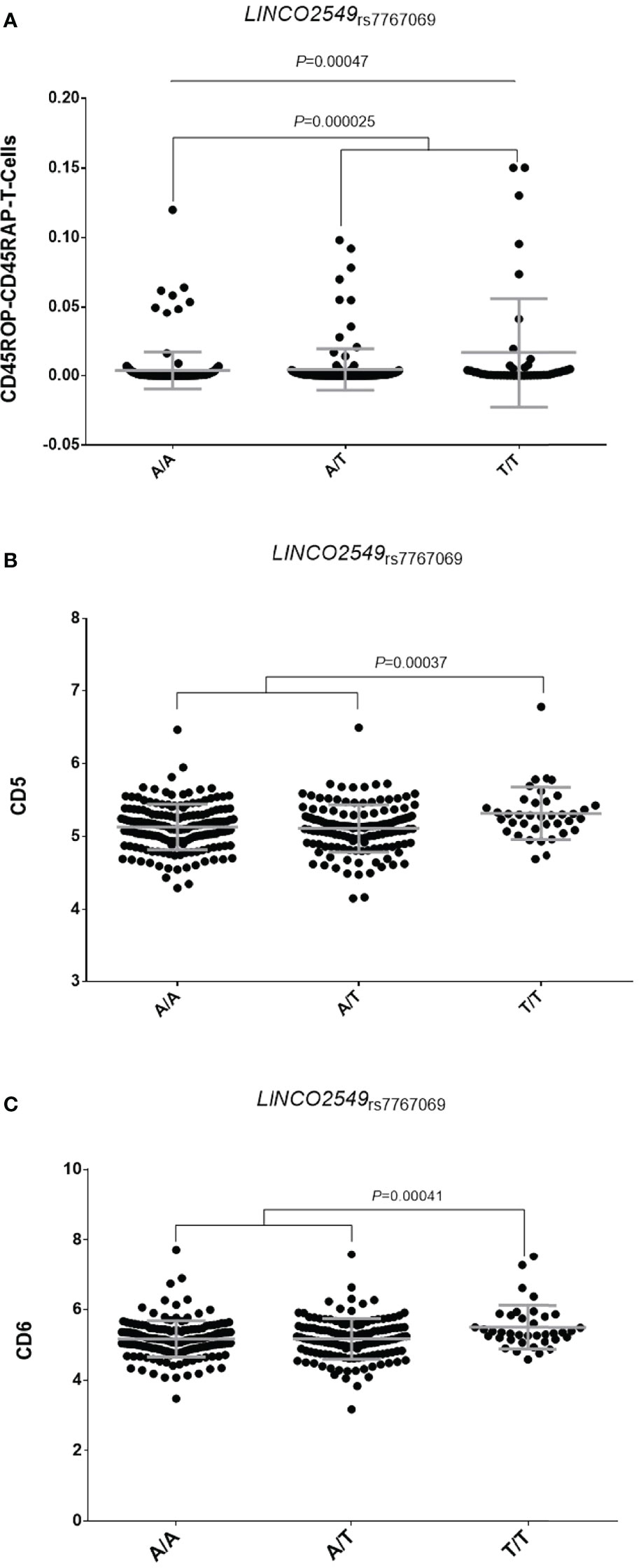
Figure 1 Correlation of the LINC02549rs7767069 polymorphism with absolute numbers of CD45RO+CD45RA+ T cells in blood (A) and serum levels of soluble scavenger receptors CD5 (B) and CD6 (C).
Furthermore, although we could not stratify our functional analyses by RF status because of the healthy nature of the blood donors, we found that carriers of the LRRC55rs717117G allele showed significantly decreased levels of IL6 production after stimulation of PBMCs with either B. burgdorferi (P=0.00046; Figure 2A) or E. coli (P=0.00044; Figure 2B), which suggested an implication of the LRRC55 locus in the modulation of the response to TNFi by regulating IL6 production and likely IL6-mediated T cell differentiation into effector Th2 cells. Functional data from Haploreg also showed that the LRRC55rs717117 variant correlates with mRNA P2RX3 expression levels, a well-known gene involved in controlling T cell proliferation. Finally, our functional experiments revealed that carriers of the MAFBrs6071980C allele showed decreased levels of Chemokine (C-C motif) ligand 23 (CCL23; P=0.0060; Figure 3A) and increased levels of serum Fibroblast growth factor 19 (FGF-19; P=0.0034; Figure 3B). Whereas FGF-19 protein modulates inflammation by mediating IL6 production, CCL23 has been implicated in monocyte recruitment during inflammation and it has been previously shown to positively correlate with drop in DAS28 after treatment with TNFi. Although the effect of the MAFB SNP to modulate either serum FGF-19 or CCL23 levels did not remain significant after correction for multiple testing, these results might indicate a weak, but still functional, effect of the MAFB locus in modulating response to anti-TNF drugs.
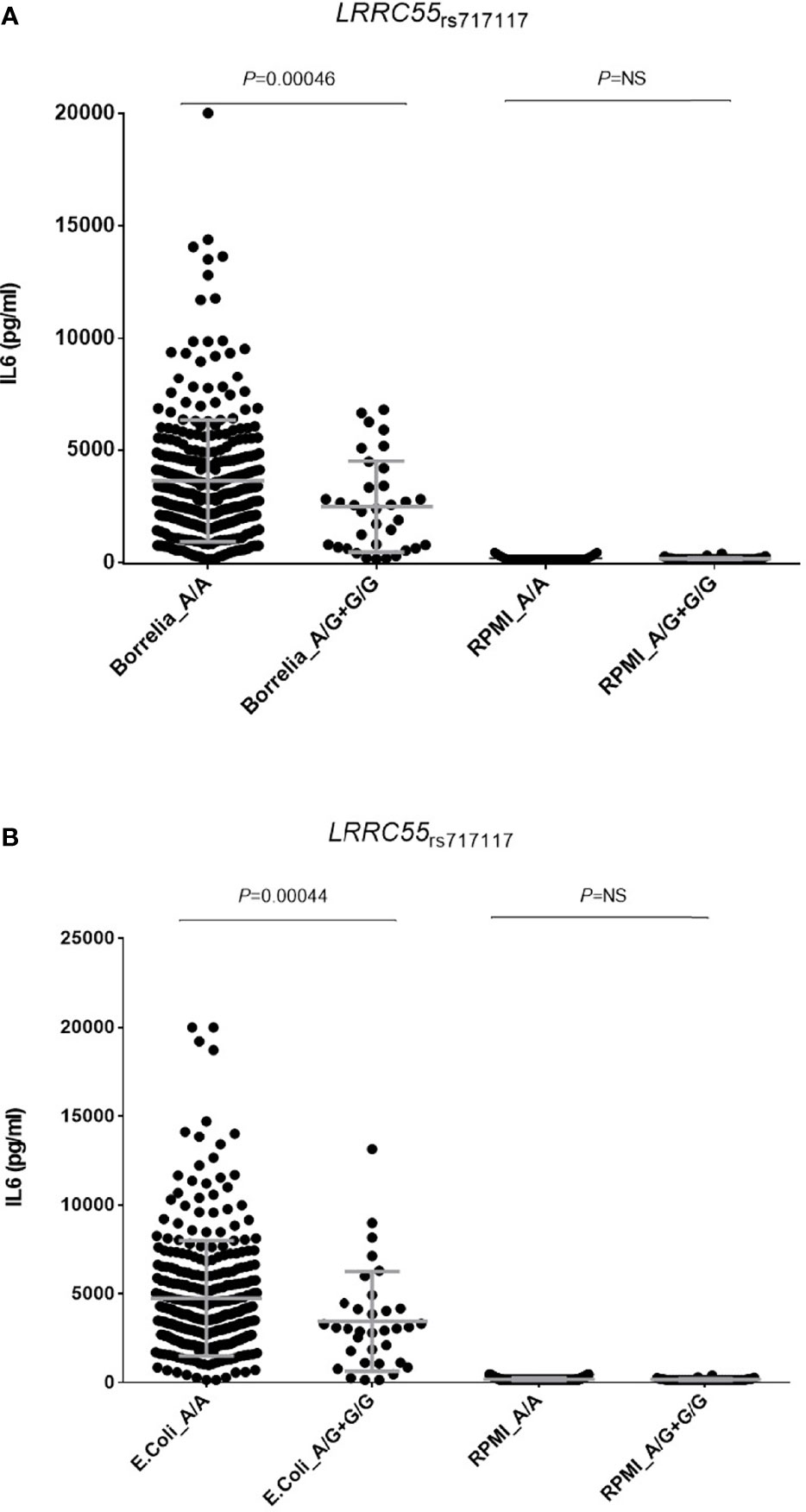
Figure 2 Correlation of the LRRC55rs717117G allele and levels of IL6 after stimulation of PBMCs either with B. burgdorferi (A) or E. coli (B).
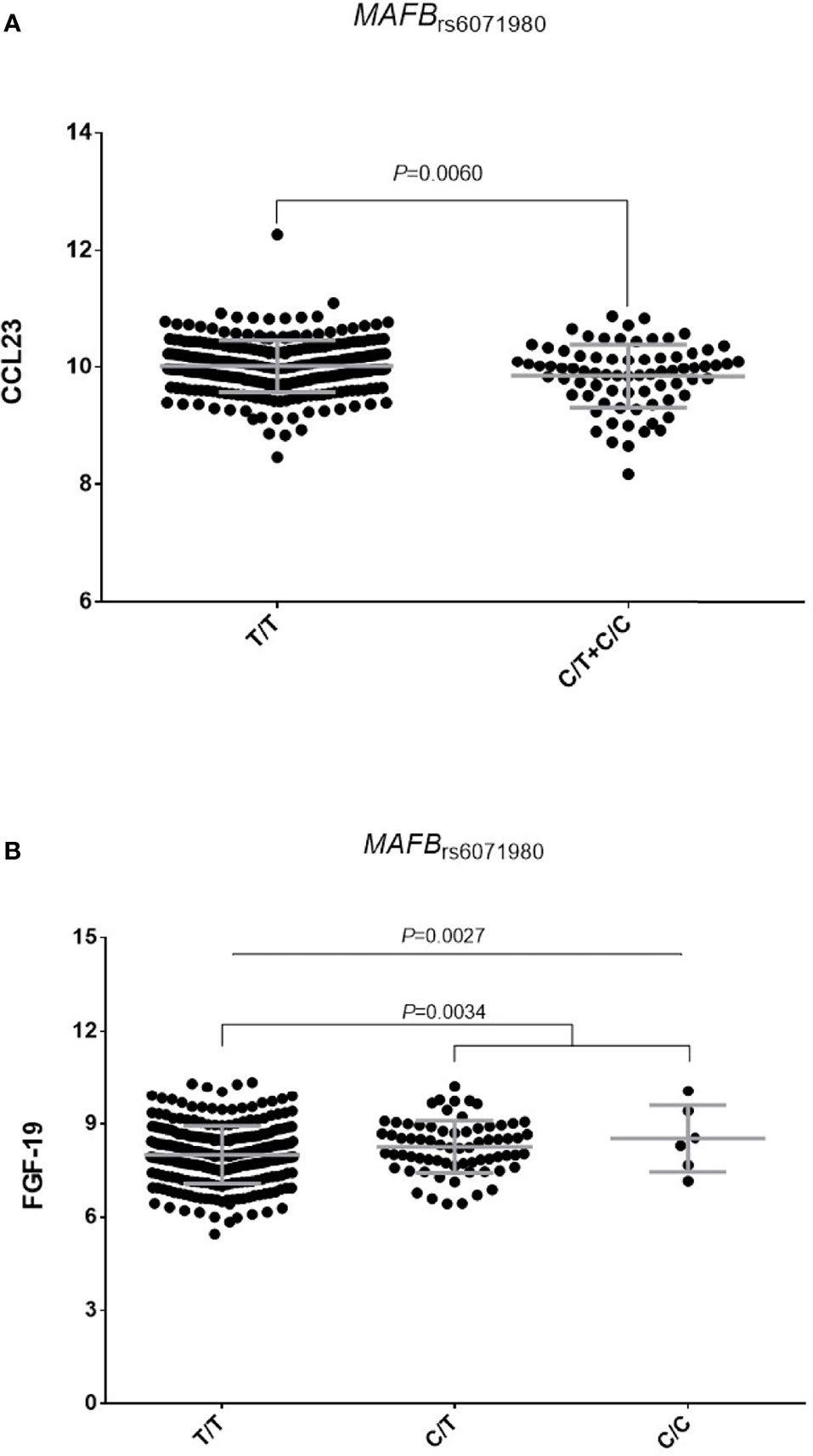
Figure 3 orrelation of the MAFBrs6071980C allele with serum levels of CCL23 (A) and FGF-19 proteins (B).
Discussion
Although treatment of RA patients using monoclonal anti-TNF drugs has been a particularly successful approach to control inflammation and to prevent joint destruction and the appearance of bone erosions, non-responsiveness is prevalent and no effective biomarkers for drug response prediction have been consistently identified (36). This comprehensive validation study aimed at confirming the association of GWAS-identified variants with response to TNFi and to shed some light into the biological mechanisms underlying the most interesting associations. For that purpose, we conducted a two-stage case control study including 2067 RA patients treated with anti-TNF drugs ascertained through the REPAIR consortium but also DANBIO and DREAM registries. The most significant result was the overall association of the LINC02549rs7767069 SNP with a poor response to anti-TNF drugs. The meta-analysis of the discovery and replication cohorts showed that each copy of the LINC02549rs7767069T allele significantly decreased the improvement in DAS28 by 17% after the treatment with a TNFi. Importantly, the association of the LINC02549rs7767069 variant with poor response to TNFi was significant in the two populations analyzed and remained significant after correction for multiple testing, which confirmed a role of the LINC02549 locus in the modulation of response to anti-TNF drugs.
LINC02549 (Long Intergenic Protein Coding RNA 2549) is an RNA gene that is affiliated with the lncRNA class, which represents a large proportion of the human transcriptome. LINC02549 maps to chromosome 6 and it is expressed in resting T cells and CD4 activated T cells. Although its function is still largely unknown, our data suggest that it might exert a role in determining the number of circulating CD45RO+CD45RA+ T cells, which are a subset of cells frequently found in the synovial fluid of both chronic arthritis (37) and RA patients (38). According to the results of Koch et al. (1990), CD45RA+ CD45RO+ T lymphocytes are mostly detected in perivascular regions, which suggest that these lymphocytes might access the RA synovial tissue via the synovial vasculature (38) and that, once there, they could play a role in promoting synovial tissue inflammation mainly by the induction of memory immune responses. Therefore, it seems to be plausible to suggest that the negative impact of the LINC02549rs7767069 SNP on the response to TNFi might be mediated by its role in modulating numbers of CD45RA+CD45RO+ T lymphocytes that could migrate to the synovial tissue and promote inflammatory responses and, thereby hamper the control of inflammation during treatment with anti-TNF drugs. In support of this hypothesis, we found that carriers of two copies of the LINC02549rs7767069T allele also showed significantly increased levels of soluble scavenger receptors CD5 and CD6 (sCD5 and sCD6) in serum that are proteins highly expressed in regulatory T cells and a specific subset of B cells (CD5+ or B1a) (39). Although the origin of these soluble scavenger receptors in RA is poorly understood, it has been suggested that they are shed in the serum by proteases from the surface of activated lymphocytes that subsequently infiltrate synovium structures (40). In fact, increased serum levels of sCD5 and sCD6 has been found in subjects diagnosed with RA (41–44) but also other autoimmune diseases such as primary Sjögren’s syndrome (42, 45, 46), systemic inflammatory response syndrome (47), multiple sclerosis (44) or dermatitis (48). Although the functional role of both soluble scavengers in autoimmune diseases is still under investigation, it is well established that sCD5 and sCD6 are regulators of T cell functions and induce autoreactivity. It is known that they are required for the initiation, differentiation and maintenance of T cell immune responses (49, 50) but also T cell migration and extravasation to the synovial tissue (51). Furthermore, it has been reported that both sCD5 and sCD6 are involved in the modulation of TCR and BCR signaling and determinate T- and B-cell survival (52) and Th17 differentiation (53, 54). Furthermore, clinical trials using humanized anti-CD6 mAbs have provided valuable information regarding the potential targeting of CD6 for the treatment of RA but also psoriasis and potentially other T cell–driven autoimmune diseases (55–57). Recent investigations have also suggested that genetic alterations within the CD6 gene associated with clinical outcome of several autoimmune diseases (44, 58) and correlated with the response to TNFi (59), which pointed to a role of these soluble scavenger receptors in modulating response to anti-TNF drugs. Considering these findings, we hypothesize that, besides its effect on modulating number of the CD45RA+CD45RO+ T lymphocytes, the LINC02549rs7767069 SNP might negatively influence the response to anti-TNF drugs by stimulating directly or indirectly the production of sCD5 and sCD6 and thereby inducing long-term T cell-mediated immune responses.
Another interesting result that remained significant after correction for multiple testing was the RF-specific association of the LRRC55rs717117 SNP with lower changes in DAS28 after the treatment with TNFi. The meta-analysis of the discovery and replication cohorts showed that RF-positive patients carrying the LRRC55rs717117G allele have a significantly decreased drop in DAS28 after treatment with a TNFi, whereas an opposite but not statistically significant effect was observed in RF-negative RA patients. Noticeably, functional experiments showed that, after stimulation of PBMCs from healthy subjects with B. burgdorferi and E. coli bacteria, carriers of the LRRC55rs717117G allele showed significantly decreased production of IL6 when compared to those carrying the most common genotype. Although functional experiments could not be stratified by RF because of the healthy nature of blood donors, these results suggested a role of the LRRC55 locus in modulating IL6-mediated immune responses. On the other hand, functional data from Haploreg also suggested an implication of the LRRC55rs717117 variant in controlling P2RX3-mediated T cell proliferation.
LRRC55 gene maps on chromosome 11 and it encodes for the leucine-rich repeat-containing protein 55, a protein that belongs to the LRRC superfamily that include hundreds of proteins mainly expressed in brain. Several LRRC proteins have been linked to the regulation of ion channels (60) but it has been also demonstrated that LRRC proteins are also implicated in modulating immune responses against bacterial pathogens (61) and modulate cell trafficking of membrane receptors such as toll-like receptors (62). Although the interplay between LRRC55 and IL6 has not been demonstrated, our experimental data suggest that the LRRC55rs717117 SNP modulates IL6 production in response to bacteria and, therefore, might be involved in other IL6-dependent immune processes that could worsen the response to TNFi.
It is widely known that IL6 can induce both anti-inflammatory and pro-inflammatory immune responses, which depend entirely on the signalling pathway triggered. Whereas anti-inflammatory responses are mostly mediated by the classic signalling cascade (through binding to the transmembrane IL6 receptor), pro-inflammatory responses and chronic inflammation are mediated by trans-signalling (through binding to the soluble IL6 receptor) or by the interaction of IL6R with gp130 (63, 64). Considering our functional data, it is conceivable to suggest that the LRRC55rs717117 SNP might affect the response to TNFi by decreasing IL6 production and thus inhibiting the classical IL6-dependent anti-inflammatory pathway and dysregulating pro-inflammatory responses. In support of this hypothesis, several mouse models have shown that the activation of the IL6 classic signaling pathway is essential for the activation of STAT3-mediated signaling pathways which reduce inflammation and induce the regeneration of the affected tissues (65). In addition, it has been reported that IL6 is one of the earliest factors that trigger the differentiation of naive T cells into effector Th2 cells in vitro and that, when absent, aggravates the development of the inflammatory processes (64).
Finally, although the genetic association of the MAFBrs6071980 SNP with lower response to TNFi did not remain significant after correction for multiple testing, we found that the MAFBrs6071980 SNP correlated with higher levels of serum FGF-19 and decreased levels of CCL23. Given that FGF-19 is a master protein involved in the inhibition of intestinal inflammation (66, 67) and CCL23 has been positively correlated with the DAS28 score in RA patients (68), we think that it would worth to investigate more in detail the impact of this SNP on drug response in future studies. In addition, it might be interesting to further analyze the weak association of the CNTN5rs1813443 SNP with poor response to TNFi. However, given that we could not find any significant impact of this marker on immune responses, blood cell counts or serum inflammatory proteins or steroid hormones, we are prone to think that this SNP might not have a relevant role in modulating response to TNFi.
This study has both strengths and weaknesses. Among the strengths we can highlight the use of large and well-characterized RA patient populations that allowed the development of a well-powered overall association analysis but also to investigate the effect modification by RF status. Overall, we had 80% of power to detect an OR of 1.18 (α=0.00089) for a SNP with a frequency of 0.25. On the other hand, it is worth mentioning the comprehensive analysis of the functional effect of the most interesting genetic variants on modulating immune responses, which was performed using a large sample size for this kind of studies. We analysed cQTL and hQTL data but also counts of 91 blood-derived cell populations and serum levels of 103 immunological proteins. An important limitation of this study was the impossibility to adjust linear regression analyses for potential confounding factors including concomitant treatments that might influence the response to TNFi. In addition, given the healthy nature of the subjects included in the HFGP cohort, we could not control our functional experiments by RF status.
In conclusion, this study validates the overall or RF-specific association of LINC02549 and LRRC55 loci with the response to TNFi and provides new insights into the functional role of these polymorphisms in modulating immune responses and response to anti-TNF drugs.
Data Availability Statement
All data used in this project have been meticulously catalogued and archived in the BBMRI-NL data infrastructure (https://hfgp.bbmri.nl/) using the MOLGENIS open-source platform for scientific data (69). This allows flexible data querying and download, including sufficiently rich metadata and interfaces for machine processing (R statistics, REST API) and using FAIR principles to optimize Findability, Accessibility, Interoperability and Reusability (70, 71). Genetic data from the discovery and DANBIO populations can be accessed at ftp.genyo.es and data from the DREAM registry are available at https://www.synapse.org/#!Synapse:syn3280809/wiki/194735 and https://www.synapse.org/#!Synapse:syn3280809/wiki/194736.
Ethics Statement
The study was approved by the ethical review committee of participant institutions: Virgen de las Nieves University Hospital (2012/89); Santa Maria Hospital-CHLN (CE 877/121.2012); University Clinical Hospital of Santiago de Compostela (2013/156); Wroclaw Medical University (KB-625/2016); Radboud university medical center (2011/299) and by the Regional Ethics Committee of Central Denmark Region (S-20120113). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
RC and JSa designed the study and drafted the manuscript. JMSM, AM-D, and AJCS were responsible for genotyping. MALN, HC, LQ, SS, BG, MH, IF, EP-P, PC-Z, JSw, AB, SV, EP, MF, AE, MC, VA, JF, MJ, KBK, EC, and JSa coordinated the sample collection and HC, IF, and MF were involved in the records review and data acquisition. JMSM and JSa performed data quality control of genetic data and statistical analysis. MN, RTH, YL provided the functional raw data and JSa performed the analysis of functional data. All authors contributed to and approved the final version of the manuscript.
Funding
This study was supported by grants PI17/02276 and PI20/01845 from Fondo de Investigaciones Sanitarias (Madrid, Spain) and by intramural funds of GENYO and FIBAO foundation (Granada, Spain). This study was also supported by the Novo Nordisk Fonden (NNF15OC0016932, VA) and Knud og Edith Eriksens Mindefond (VA) and Gigtforeningen (A2037, A3570, VA). JS and KB-K were supported by the grant No. 2016/21/B/NZ5/01901 from the National Science Centre (Poland). MGN was supported by a Spinoza grant from the Netherlands Organization for Scientific Research. YL was supported by an ERC Starting Grant (948207) and the Radboud University Medical Centre Hypatia Grant (2018) for Scientific Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
VA has received compensation for consultancy and for being a member of an advisory board from MSD (Merck) and Janssen. BG received funding for research from AbbVie, Biogen, and Pfizer. MH received funding for research from Abbvie, Biogen, BMS, CellTrion, MSD, Novartis, Orion, Pfizer, Samsung and UCB. JF received unrestricted research grants or acted as a speaker for Abbvie, Ache, Amgen, Biogen, BMS, Janssen, Lilly, MSD, Novartis, Pfizer, Roche, UCB. AB has received congress invitations, personal fees and research fees (to the department) from Boehringer, Amgen, Abbvie, Biogen, Cellgene, Pfizer, Novartis, Galapagos, Gilead, Roche, Sanofi.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all participants who have agreed to participate in this study. Authors also thank María Dolores Casares, Ángeles Molina, Carmen Oloriz for the collection of Spanish samples and Hans Jurgen Hoffmann, Marianne Thomsen, Vibeke Østergaard Thomsen, Malene Rohr Andersen, Lise Lotte B. Laursen, Helle Jørgensen, Ram Benny Christian Dessau, Niels Steen Krogh, Ulla Vogel, Paal Skytt Andersen, Ivan Brandslund, Steffen Bank, Frederik Trier Møller, Nikolai Toft and Niels Møller Andersen for the participation in collection and purification of Danish samples. We also thank the Danish Departments of Rheumatology for their implication in the collection of clinical data from RA patients included in the DANBIO cohort and the Danish Rheumatologic Biobank. Likewise, we would like to thank Teun van Herwaarden for steroid hormone measurements in serum samples from subjects ascertained through the HFGP initiative.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.672255/full#supplementary-material
References
1. Bluml S, Scheinecker C, Smolen JS, Redlich K. Targeting TNF Receptors in Rheumatoid Arthritis. Int Immunol (2012) 24(5):275–81. doi: 10.1093/intimm/dxs047
2. Scott DL, Wolfe F, Huizinga TW. Rheumatoid Arthritis. Lancet (2010) 376(9746):1094–108. doi: 10.1016/S0140-6736(10)60826-4
3. Pisetsky DS, Ward MM. Advances in the Treatment of Inflammatory Arthritis. Best Pract Res Clin Rheumatol (2012) 26(2):251–61. doi: 10.1016/j.berh.2012.03.001
4. Di Giovine FS, Nuki G, Duff GW. Tumour Necrosis Factor in Synovial Exudates. Ann Rheum Dis (1988) 47(9):768–72. doi: 10.1136/ard.47.9.768
5. Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor Necrosis Factor Antagonist Mechanisms of Action: A Comprehensive Review. Pharmacol Ther (2008) 117(2):244–79. doi: 10.1016/j.pharmthera.2007.10.001
6. Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor Necrosis Factor-Alpha Induces Differentiation of and Bone Resorption by Osteoclasts. J Biol Chem (2000) 275(7):4858–64. doi: 10.1074/jbc.275.7.4858
7. Safiri S, Kolahi AA, Hoy D, Smith E, Bettampadi D, Mansournia MA, et al. Global, Regional and National Burden of Rheumatoid Arthritis 1990-2017: A Systematic Analysis of the Global Burden of Disease Study 2017. Ann Rheum Dis (2019) 78(11):1463–71. doi: 10.1136/annrheumdis-2019-215920
8. Tesar T, Golias P, Kobliskova Z, Wawruch M, Kawalec P, Inotai A. Potential Cost-Savings From the Use of the Biosimilars in Slovakia. Front Public Health (2020) 8:431. doi: 10.3389/fpubh.2020.00431
9. Atzeni F, Benucci M, Salli S, Bongiovanni S, Boccassini L, Sarzi-Puttini P. Different Effects of Biological Drugs in Rheumatoid Arthritis. Autoimmun Rev (2013) 12(5):575–9. doi: 10.1016/j.autrev.2012.10.020
10. Plenge RM, Bridges SL Jr. Personalized Medicine in Rheumatoid Arthritis: Miles to Go Before We Sleep. Arthritis Rheum (2011) 63(3):590–3. doi: 10.1002/art.30126
11. Honne K, Hallgrimsdottir I, Wu C, Sebro R, Jewell NP, Sakurai T, et al. A Longitudinal Genome-Wide Association Study of Anti-Tumor Necrosis Factor Response Among Japanese Patients With Rheumatoid Arthritis. Arthritis Res Ther (2016) 18:12. doi: 10.1186/s13075-016-0920-6
12. Cui J, Stahl EA, Saevarsdottir S, Miceli C, Diogo D, Trynka G, et al. Genome-Wide Association Study and Gene Expression Analysis Identifies CD84 as a Predictor of Response to Etanercept Therapy in Rheumatoid Arthritis. PloS Genet (2013) 9(3):e1003394. doi: 10.1371/journal.pgen.1003394
13. Julia A, Fernandez-Nebro A, Blanco F, Ortiz A, Canete JD, Maymo J, et al. A Genome-Wide Association Study Identifies a New Locus Associated With the Response to Anti-TNF Therapy in Rheumatoid Arthritis. Pharmacogenom J (2016) 16(2):147–50. doi: 10.1038/tpj.2015.31
14. Krintel SB, Palermo G, Johansen JS, Germer S, Essioux L, Benayed R, et al. Investigation of Single Nucleotide Polymorphisms and Biological Pathways Associated With Response to TNFalpha Inhibitors in Patients With Rheumatoid Arthritis. Pharmacogenet Genomics (2012) 22(8):577–89. doi: 10.1097/FPC.0b013e3283544043
15. Liu C, Batliwalla F, Li W, Lee A, Roubenoff R, Beckman E, et al. Genome-Wide Association Scan Identifies Candidate Polymorphisms Associated With Differential Response to Anti-TNF Treatment in Rheumatoid Arthritis. Mol Med (2008) 14(9-10):575–81. doi: 10.2119/2008-00056.Liu
16. Umicevic Mirkov M, Cui J, Vermeulen SH, Stahl EA, Toonen EJ, Makkinje RR, et al. Genome-Wide Association Analysis of Anti-TNF Drug Response in Patients With Rheumatoid Arthritis. Ann Rheum Dis (2013) 72(8):1375–81. doi: 10.1136/annrheumdis-2012-202405
17. Plant D, Bowes J, Potter C, Hyrich KL, Morgan AW, Wilson AG, et al. Genome-Wide Association Study of Genetic Predictors of Anti-Tumor Necrosis Factor Treatment Efficacy in Rheumatoid Arthritis Identifies Associations With Polymorphisms at Seven Loci. Arthritis Rheum (2011) 63(3):645–53. doi: 10.1002/art.30130
18. Canet LM, Caliz R, Lupianez CB, Canhao H, Martinez M, Escudero A, et al. Genetic Variants Within Immune-Modulating Genes Influence the Risk of Developing Rheumatoid Arthritis and Anti-TNF Drug Response: A Two-Stage Case-Control Study. Pharmacogenet Genomics (2015) 25(9):432–43. doi: 10.1097/FPC.0000000000000155
19. Canet LM, Filipescu I, Caliz R, Lupianez CB, Canhao H, Escudero A, et al. Genetic Variants Within the TNFRSF1B Gene and Susceptibility to Rheumatoid Arthritis and Response to Anti-TNF Drugs: A Multicenter Study. Pharmacogenet Genomics (2015) 25(7):323–33. doi: 10.1097/FPC.0000000000000140
20. Canet LM, Sanchez-Maldonado JM, Caliz R, Rodriguez-Ramos A, Lupianez CB, Canhao H, et al. Polymorphisms at Phase I-Metabolizing Enzyme and Hormone Receptor Loci Influence the Response to Anti-TNF Therapy in Rheumatoid Arthritis Patients. Pharmacogenom J (2019) 19(1):83–96. doi: 10.1038/s41397-018-0057-x
21. Canhao H, Rodrigues AM, Santos MJ, Carmona-Fernandes D, Bettencourt BF, Cui J, et al. TRAF1/C5 But Not PTPRC Variants Are Potential Predictors of Rheumatoid Arthritis Response to Anti-Tumor Necrosis Factor Therapy. BioMed Res Int (2015) 2015:490295. doi: 10.1155/2015/490295
22. Manuel Sanchez-Maldonado J, Martinez-Bueno M, Canhao H, Ter Horst R, Munoz-Pena S, Moniz-Diez A, et al. NFKB2 Polymorphisms Associate With the Risk of Developing Rheumatoid Arthritis and Response to TNF Inhibitors: Results From the REPAIR Consortium. Sci Rep (2020) 10(1):4316. doi: 10.1038/s41598-020-61331-5
23. Potter C, Cordell HJ, Barton A, Daly AK, Hyrich KL, Mann DA, et al. Association Between Anti-Tumour Necrosis Factor Treatment Response and Genetic Variants Within the TLR and NF{kappa}B Signalling Pathways. Ann Rheum Dis (2010) 69(7):1315–20. doi: 10.1136/ard.2009.117309
24. Sanchez-Maldonado JM, Caliz R, Canet L, Horst RT, Bakker O, den Broeder AA, et al. Steroid Hormone-Related Polymorphisms Associate With the Development of Bone Erosions in Rheumatoid Arthritis and Help to Predict Disease Progression: Results From the REPAIR Consortium. Sci Rep (2019) 9(1):14812. doi: 10.1038/s41598-019-51255-0
25. Sode J, Vogel U, Bank S, Andersen PS, Hetland ML, Locht H, et al. Genetic Variations in Pattern Recognition Receptor Loci Are Associated With Anti-TNF Response in Patients With Rheumatoid Arthritis. PloS One (2015) 10(10):e0139781. doi: 10.1371/journal.pone.0139781
26. Sode J, Vogel U, Bank S, Andersen PS, Hetland ML, Locht H, et al. Confirmation of an IRAK3 Polymorphism as a Genetic Marker Predicting Response to Anti-TNF Treatment in Rheumatoid Arthritis. Pharmacogenom J (2018) 18(1):81–6. doi: 10.1038/tpj.2016.66
27. Suarez-Gestal M, Perez-Pampin E, Calaza M, Gomez-Reino JJ, Gonzalez A. Lack of Replication of Genetic Predictors for the Rheumatoid Arthritis Response to Anti-TNF Treatments: A Prospective Case-Only Study. Arthritis Res Ther (2010) 12(2):R72. doi: 10.1186/ar2990
28. Ibfelt EH, Jensen DV, Hetland ML. The Danish Nationwide Clinical Register for Patients With Rheumatoid Arthritis: DANBIO. Clin Epidemiol (2016) 8:737–42. doi: 10.2147/CLEP.S99490
29. Ibfelt EH, Sorensen J, Jensen DV, Dreyer L, Schiottz-Christensen B, Thygesen PH, et al. Validity and Completeness of Rheumatoid Arthritis Diagnoses in the Nationwide DANBIO Clinical Register and the Danish National Patient Registry. Clin Epidemiol (2017) 9:627–32. doi: 10.2147/CLEP.S141438
30. Arnett FC, Edworthy SM, Bloch DA, Mcshane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 Revised Criteria for the Classification of Rheumatoid Arthritis. Arthritis Rheum (1988) 31:315–24. doi: 10.1002/art.1780310302
31. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, et al. Rheumatoid Arthritis Classification Criteria: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Arthritis Rheum (2010) 62:2569–81. doi: 10.1002/art.27584
32. Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, et al. Radiographic, Clinical, and Functional Outcomes of Treatment With Adalimumab (A Human Anti-Tumor Necrosis Factor Monoclonal Antibody) in Patients With Active Rheumatoid Arthritis Receiving Concomitant Methotrexate Therapy: A Randomized, Placebo-Controlled, 52-Week Trial. Arthritis Rheum (2004) 50(5):1400–11. doi: 10.1002/art.20217
33. Weinblatt ME, Kremer JM, Bankhurst AD, Bulpitt KJ, Fleischmann RM, Fox RI, et al. A Trial of Etanercept, a Recombinant Tumor Necrosis Factor Receptor:Fc Fusion Protein, in Patients With Rheumatoid Arthritis Receiving Methotrexate. N Engl J Med (1999) 340(4):253–9. doi: 10.1056/NEJM199901283400401
34. Orru V, Steri M, Sole G, Sidore C, Virdis F, Dei M, et al. Genetic Variants Regulating Immune Cell Levels in Health and Disease. Cell (2013) 155(1):242–56. doi: 10.1016/j.cell.2013.08.041
35. Aguirre-Gamboa R, Joosten I, Urbano PCM, van der Molen RG, van Rijssen E, van Cranenbroek B, et al. Differential Effects of Environmental and Genetic Factors on T and B Cell Immune Traits. Cell Rep (2016) 17(9):2474–87. doi: 10.1016/j.celrep.2016.10.053
36. Cuppen BV, Welsing PM, Sprengers JJ, Bijlsma JW, Marijnissen AC, van Laar JM, et al. Personalized Biological Treatment for Rheumatoid Arthritis: A Systematic Review With a Focus on Clinical Applicability. Rheumatol (Oxford) (2016) 55(5):826–39. doi: 10.1093/rheumatology/kev421
37. Summers KL, O’Donnell JL, Hart DN. Co-Expression of the CD45RA and CD45RO Antigens on T Lymphocytes in Chronic Arthritis. Clin Exp Immunol (1994) 97(1):39–44. doi: 10.1111/j.1365-2249.1994.tb06576.x
38. Koch AE, Robinson PG, Radosevich JA, Pope RM. Distribution of CD45RA and CD45RO T-Lymphocyte Subsets in Rheumatoid Arthritis Synovial Tissue. J Clin Immunol (1990) 10(4):192–9. doi: 10.1007/BF00918651
39. Aruffo A, Melnick MB, Linsley PS, Seed B. The Lymphocyte Glycoprotein CD6 Contains a Repeated Domain Structure Characteristic of a New Family of Cell Surface and Secreted Proteins. J Exp Med (1991) 174(4):949–52. doi: 10.1084/jem.174.4.949
40. Calvo J, Places L, Espinosa G, Padilla O, Vila JM, Villamor N, et al. Identification of a Natural Soluble Form of Human CD5. Tissue Antigens (1999) 54(2):128–37. doi: 10.1034/j.1399-0039.1999.540203.x
41. Lydyard PM, Youinou PY, Cooke A. CD5-Positive B Cells in Rheumatoid Arthritis and Chronic Lymphocytic Leukemia. Immunol Today (1987) 8(2):37–9. doi: 10.1016/0167-5699(87)90235-0
42. Youinou P, Mackenzie L, Broker BM, Isenberg DI, Drogou-Lelong A, Gentric A, et al. The Importance of CD5-Positive B Cells in Nonorgan-Specific Autoimmune Diseases. Scand J Rheumatol Suppl (1988) 76:243–9. doi: 10.3109/03009748809102975
43. Jamin C, Magadur G, Lamour A, Mackenzie L, Lydyard P, Katsikis P, et al. Cell-Free CD5 in Patients With Rheumatic Diseases. Immunol Lett (1992) 31(1):79–83. doi: 10.1016/0165-2478(92)90014-F
44. Consuegra-Fernandez M, Lin F, Fox DA, Lozano F. Clinical and Experimental Evidence for Targeting CD6 in Immune-Based Disorders. Autoimmun Rev (2018) 17(5):493–503. doi: 10.1016/j.autrev.2017.12.004
45. Ramos-Casals M, Font J, Garcia-Carrasco M, Calvo J, Places L, Padilla O, et al. High Circulating Levels of Soluble Scavenger Receptors (Scd5 and Scd6) in Patients With Primary Sjogren’s Syndrome. Rheumatol (Oxford) (2001) 40(9):1056–9. doi: 10.1093/rheumatology/40.9.1056
46. Dauphinee MJ, Tovar Z, Ballester A, Talal N. The Expression and Function of CD3 and CD5 in Patients With Primary Sjogren’s Syndrome. Arthritis Rheum (1989) 32(4):420–9. doi: 10.1002/anr.1780320411
47. Aibar J, Martinez-Florensa M, Castro P, Carrasco E, Escoda-Ferran C, Fernandez S, et al. Pattern of Soluble CD5 and CD6 Lymphocyte Receptors in Critically Ill Patients With Septic Syndromes. J Crit Care (2015) 30(5):914–9. doi: 10.1016/j.jcrc.2015.04.120
49. Zimmerman AW, Joosten B, Torensma R, Parnes JR, van Leeuwen FN, Figdor CG. Long-Term Engagement of CD6 and ALCAM Is Essential for T-Cell Proliferation Induced by Dendritic Cells. Blood (2006) 107(8):3212–20. doi: 10.1182/blood-2005-09-3881
50. Nair P, Melarkode R, Rajkumar D, Montero E. CD6 Synergistic Co-Stimulation Promoting Proinflammatory Response Is Modulated Without Interfering With the Activated Leucocyte Cell Adhesion Molecule Interaction. Clin Exp Immunol (2010) 162(1):116–30. doi: 10.1111/j.1365-2249.2010.04235.x
51. Alonso-Ramirez R, Loisel S, Buors C, Pers JO, Montero E, Youinou P, et al. Rationale for Targeting CD6 as a Treatment for Autoimmune Diseases. Arthritis (2010) 2010:130646. doi: 10.1155/2010/130646
52. Burgueno-Bucio E, Mier-Aguilar CA, Soldevila G. The Multiple Faces of CD5. J Leukoc Biol (2019) 105(5):891–904. doi: 10.1002/JLB.MR0618-226R
53. Li Y, Singer NG, Whitbred J, Bowen MA, Fox DA, Lin F. CD6 as a Potential Target for Treating Multiple Sclerosis. Proc Natl Acad Sci USA (2017) 114(10): 2687–92. doi: 10.1073/pnas.1615253114
54. Consuegra-Fernandez M, Julia M, Martinez-Florensa M, Aranda F, Catala C, Armiger-Borras N, et al. Genetic and Experimental Evidence for the Involvement of the CD6 Lymphocyte Receptor in Psoriasis. Cell Mol Immunol (2018) 15(10):898–906. doi: 10.1038/cmi.2017.119
55. Rodriguez PC, Prada DM, Moreno E, Aira LE, Molinero C, Lopez AM, et al. The Anti-CD6 Antibody Itolizumab Provides Clinical Benefit Without Lymphopenia in Rheumatoid Arthritis Patients: Results From a 6-Month, Open-Label Phase I Clinical Trial. Clin Exp Immunol (2018) 191(2):229–39. doi: 10.1111/cei.13061
56. Rodriguez PC, Torres-Moya R, Reyes G, Molinero C, Prada D, Lopez AM, et al. A Clinical Exploratory Study With Itolizumab, an Anti-CD6 Monoclonal Antibody, in Patients With Rheumatoid Arthritis. Results Immunol (2012) 2:204–11. doi: 10.1016/j.rinim.2012.11.001
57. Krupashankar DS, Dogra S, Kura M, Saraswat A, Budamakuntla L, Sumathy TK, et al. Efficacy and Safety of Itolizumab, a Novel Anti-CD6 Monoclonal Antibody, in Patients With Moderate to Severe Chronic Plaque Psoriasis: Results of a Double-Blind, Randomized, Placebo-Controlled, Phase-III Study. J Am Acad Dermatol (2014) 71(3):484–92. doi: 10.1016/j.jaad.2014.01.897
58. Kofler DM, Farkas A, von Bergwelt-Baildon M, Hafler DA. The Link Between CD6 and Autoimmunity: Genetic and Cellular Associations. Curr Drug Targets (2016) 17(6):651–65. doi: 10.2174/1389450117666160201105934
59. Krintel SB, Essioux L, Wool A, Johansen JS, Schreiber E, Zekharya T, et al. CD6 and Syntaxin Binding Protein 6 Variants and Response to Tumor Necrosis Factor Alpha Inhibitors in Danish Patients With Rheumatoid Arthritis. PloS One (2012) 7(6):e38539. doi: 10.1371/journal.pone.0038539
60. Yan J, Aldrich RW. BK Potassium Channel Modulation by Leucine-Rich Repeat-Containing Proteins. Proc Natl Acad Sci USA (2012) 109(20):7917–22. doi: 10.1073/pnas.1205435109
61. Shi XZ, Feng XW, Sun JJ, Zhao XF, Wang JX. Leucine-Rich Repeats Containing Protein Functions in the Antibacterial Immune Reaction in Stomach of Kuruma Shrimp Marsupenaeus Japonicus. Fish Shellfish Immunol (2017) 61:130–7. doi: 10.1016/j.fsi.2016.12.029
62. Tatematsu M, Funami K, Ishii N, Seya T, Obuse C, Matsumoto M. LRRC59 Regulates Trafficking of Nucleic Acid-Sensing TLRs From the Endoplasmic Reticulum via Association With UNC93B1. J Immunol (2015) 195(10):4933–42. doi: 10.4049/jimmunol.1501305
63. Gabay C. Interleukin-6 and Chronic Inflammation. Arthritis Res Ther (2006) 8 Suppl 2:S3. doi: 10.1186/ar1917
64. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The Pro- and Anti-Inflammatory Properties of the Cytokine Interleukin-6. Biochim Biophys Acta (2011) 1813(5):878–88. doi: 10.1016/j.bbamcr.2011.01.034
65. Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, et al. TGF-Beta Suppresses Tumor Progression in Colon Cancer by Inhibition of IL-6 Trans-Signaling. Immunity (2004) 21(4):491–501. doi: 10.1016/j.immuni.2004.07.020
66. Gadaleta RM, Garcia-Irigoyen O, Cariello M, Scialpi N, Peres C, Vetrano S, et al. Corrigendum to ‘Fibroblast Growth Factor 19 Modulates Intestinal Microbiota and Inflammation in Presence of Farnesoid X Receptor’: [EBioMedicine 54 (2020) 102719]. EBioMedicine (2020) 57:102873. doi: 10.1016/j.ebiom.2020.102719
67. Gadaleta RM, Garcia-Irigoyen O, Cariello M, Scialpi N, Peres C, Vetrano S, et al. Fibroblast Growth Factor 19 Modulates Intestinal Microbiota and Inflammation in Presence of Farnesoid X Receptor. EBioMedicine (2020) 54:102719. doi: 10.1016/j.ebiom.2020.102719
68. Rioja I, Hughes FJ, Sharp CH, Warnock LC, Montgomery DS, Akil M, et al. Potential Novel Biomarkers of Disease Activity in Rheumatoid Arthritis Patients: CXCL13, CCL23, Transforming Growth Factor Alpha, Tumor Necrosis Factor Receptor Superfamily Member 9, and Macrophage Colony-Stimulating Factor. Arthritis Rheum (2008) 58(8):2257–67. doi: 10.1002/art.23667
69. Swertz MA, Dijkstra M, Adamusiak T, van der Velde JK, Kanterakis A, Roos ET, et al. The MOLGENIS Toolkit: Rapid Prototyping of Biosoftware at the Push of a Button. BMC Bioinf (2010) 11 Suppl 12:S12. doi: 10.1186/1471-2105-11-S12-S12
70. Wilkinson MD, Dumontier M, Jan Aalbersberg I, Appleton G, Axton M, Baak A, et al. Addendum: The FAIR Guiding Principles for Scientific Data Management and Stewardship. Sci Data (2019) 6(1):6. doi: 10.1038/s41597-019-0009-6
Keywords: GWAS, genetic variant, rheumatoid arthritis, drug response, TNF inhibitors
Citation: Sánchez-Maldonado JM, Cáliz R, López-Nevot MÁ, Cabrera-Serrano AJ, Moñiz-Díez A, Canhão H, Ter Horst R, Quartuccio L, Sorensen SB, Glintborg B, Hetland M L, Filipescu I, Pérez-Pampin E, Conesa-Zamora P, Swierkot J, den Broeder AA, De Vita S, Petersen ERB, Li Y, Ferrer MA, Escudero A, Netea M G, Coenen MJH, Andersen V, Fonseca JE, Jurado M, Bogunia-Kubik K, Collantes E and Sainz J (2021) Validation of GWAS-Identified Variants for Anti-TNF Drug Response in Rheumatoid Arthritis: A Meta-Analysis of Two Large Cohorts. Front. Immunol. 12:672255. doi: 10.3389/fimmu.2021.672255
Received: 25 February 2021; Accepted: 11 October 2021;
Published: 27 October 2021.
Edited by:
Pier Luigi Meroni, Istituto Auxologico Italiano (IRCCS), ItalyReviewed by:
Matteo Doglio, San Raffaele Hospital (IRCCS), ItalyAlessandra Bettiol, University of Florence, Italy
Copyright © 2021 Sánchez-Maldonado, Cáliz, López-Nevot, Cabrera-Serrano, Moñiz-Díez, Canhão, Ter Horst, Quartuccio, Sorensen, Glintborg, Hetland, Filipescu, Pérez-Pampin, Conesa-Zamora, Swierkot, den Broeder, De Vita, Petersen, Li, Ferrer, Escudero, Netea, Coenen, Andersen, Fonseca, Jurado, Bogunia-Kubik, Collantes and Sainz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Sainz, anVhbi5zYWluekBnZW55by5lcw==