
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 13 August 2021
Sec. B Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.670280
This article is part of the Research TopicMouse Models of B Cell MalignanciesView all 14 articles
 Joanna C. Dawes1,2*
Joanna C. Dawes1,2* Anthony G. Uren1,2*
Anthony G. Uren1,2*Cancer genome sequencing has identified dozens of mutations with a putative role in lymphomagenesis and leukemogenesis. Validation of driver mutations responsible for B cell neoplasms is complicated by the volume of mutations worthy of investigation and by the complex ways that multiple mutations arising from different stages of B cell development can cooperate. Forward and reverse genetic strategies in mice can provide complementary validation of human driver genes and in some cases comparative genomics of these models with human tumors has directed the identification of new drivers in human malignancies. We review a collection of forward genetic screens performed using insertional mutagenesis, chemical mutagenesis and exome sequencing and discuss how the high coverage of subclonal mutations in insertional mutagenesis screens can identify cooperating mutations at rates not possible using human tumor genomes. We also compare a set of independently conducted screens from Pax5 mutant mice that converge upon a common set of mutations observed in human acute lymphoblastic leukemia (ALL). We also discuss reverse genetic models and screens that use CRISPR-Cas, ORFs and shRNAs to provide high throughput in vivo proof of oncogenic function, with an emphasis on models using adoptive transfer of ex vivo cultured cells. Finally, we summarize mouse models that offer temporal regulation of candidate genes in an in vivo setting to demonstrate the potential of their encoded proteins as therapeutic targets.
B cell neoplasms can be categorized by their cell of origin, each subtype being representative of a discrete stage in differentiation with characteristic phenotypes and genetic lesions (Table 1, Figure 1) [reviewed in (1, 2)]. Accurate modelling of these diseases in mice requires alteration of various biological processes at different stages of B cell development including differentiation, migration, rearrangement of immunoglobulin genes, T helper interactions, positive selection for antigen and negative selection against autoreactivity. Like most tumors, B cell malignancies accumulate amplifications, deletions, rearrangements, deregulation of methylation and non-synonymous point mutations. Additionally, B cells are characterized by remodeling of the immunoglobulin loci by recombinase activating gene (RAG) mediated V(D)J recombination of immunoglobulin variable regions and by activation induced cytidine deaminase (AID) mediated class switch recombination. Errors of these processes mean immunoglobulin loci are the most frequent translocation partners of B cell malignancies, often placing oncogenes under the control of the highly expressed immunoglobulin promoters and enhancers and causing deregulated and constitutive expression (3–6) (Figure 2). Somatic hypermutation by AID is also a known source of oncogenic mutations in B neoplasms as evidenced by the mutation fingerprint of commonly mutated genes, and aberrant somatic hypermutation of non-immunoglobulin genes at hotspots located throughout the genome also contributes to lymphomagenesis (4, 7–9).

Table 1 Major subtypes of B cell malignancies.
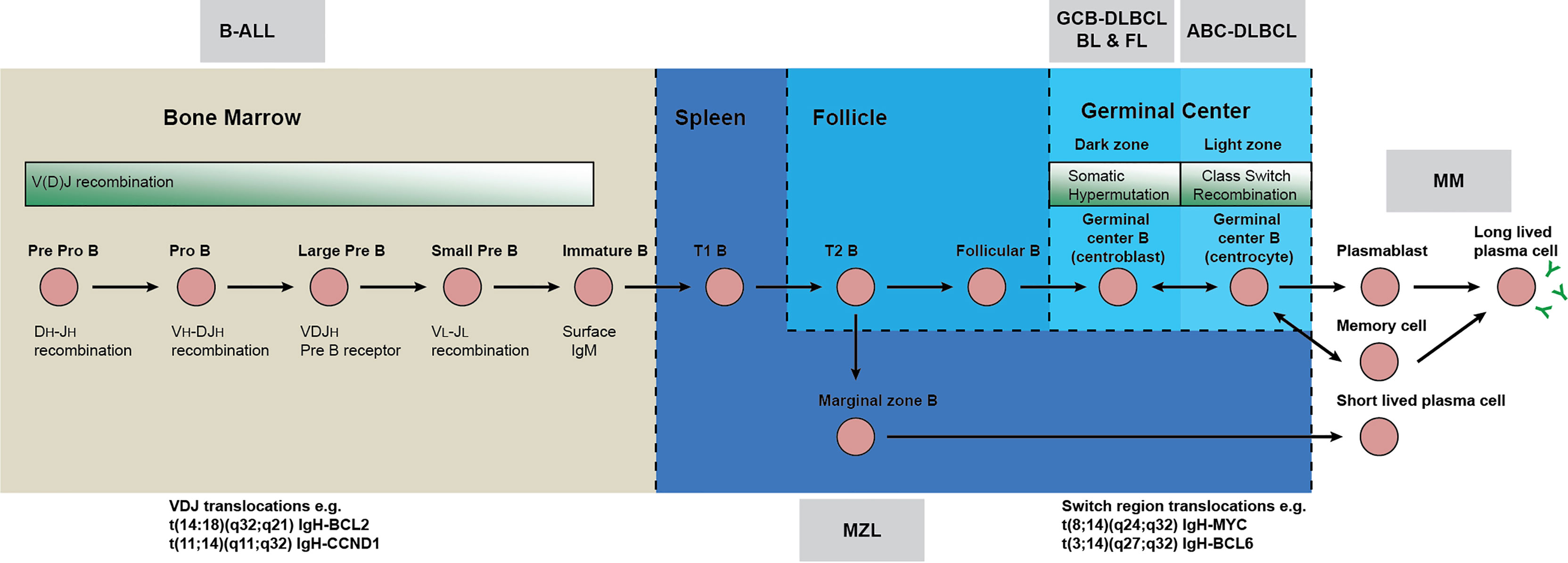
Figure 1 Neoplastic mechanisms throughout B cell development. Early B-cell development in adult bone marrow is characterized by generation of the B-cell receptor (BCR). Immunoglobulin heavy chain (IgH) gene rearrangements (VH, DH and JH), together with the VL-JL rearrangements of light chain (IgL) gene segments generate a B-cell repertoire of antibodies that recognize a variety of antigens. If the BCR does not form correctly, or is autoreactive, the cell undergoes apoptosis or receptor editing. Immature B-cells with functional BCRs, migrate to secondary lymphoid organs like the spleen and lymph nodes where they form follicles during the T-cell dependent immune response. Within the follicle, B-cells undergo two further remodeling steps in the germinal centers (GC): somatic hypermutation (SHM) and class switch recombination (CSR). SHM introduces point mutations, deletions or duplications into the variable region of the immunoglobulin genes. CSR replaces the IgH chain constant region, altering the effector response but not the antigen binding domain. GC B-cells that have undergone CSR and have high affinity IgG BCRs are selected for and differentiate into plasma cells and long-lived plasma cells, the effectors of adaptive immunity. The assignment of translocations to either V(D)J recombination in pro/pre B cells in the marrow or class switch recombination in the germinal center is based on the location and sequences of junctions present in each translocation. For instance, V(D)J derived translocations typically give rise to BCL2 or CCND1 translocations to the Ig variable regions whereas class switching frequently results in translocations between BCL6 or MYC and Ig constant regions.

Figure 2 Aberrent V(D)J & class switch recombination creates oncogenic translocations. Immunoglobulin variable region rearrangement and class switch recombination are error prone processes. (A) The immunoglobulin heavy chain locus is divided into different repetitive elements including variable segments (VH purple), diversity segments (DH green), joining segments (JH blue) and constant regions (yellow). Vertical bars depict both functional gene segments (typical numbers are indicated), non-functional pseudogenes (numbers not indicated) and repetitive elements (adapted from imgt.org). (B) Expression of recombinase activating genes (RAG) during pre pro, pro and pre B stages creates breaks between V(D)J segments, which are resolved by excision of intervening DNA. Activation induced cytidine deaminase (AID) deaminates cytosine residues of single stranded DNA which are exposed during transcription. The resulting mismatch is processed by error prone DNA repair mechanisms (including endonuclease G) resulting in excision of the default IgM/IgD constant region and fusion of the joining segments with constant regions of other isotypes. (C) Errors in resolving breaks initiated by RAG and AID can resolve as translocations with non-immunoglobulin loci. Break points adjacent to the heavy chain JH or DH segments indicate that translocations occur at the pre pro B cell stage in the bone marrow (IgH/BCL2 or IgH/CRLF2). Similar translocations also originate from immunoglobulin light chain loci. Translocations may also result from class-switch recombination in the light zone of the germinal center, evidenced by breakpoints in the immunoglobulin constant region (IgH/Myc). Breakpoints adjacent to successfully rearranged V(D)J segments with somatically mutated variable portions indicate that an AID mediated translocation has occurred later in development in the germinal center dark zone.
Speculation on the developmental stage that each malignancy is derived from is based on a combination of genetic lesions and phenotypic characteristics [reviewed in (1, 2)]. The cell of origin suggested by translocation breakpoints does not always match the cell of origin suggested by somatic hypermutations and/or markers expressed on the cell surface. Follicular lymphoma (FL) and diffuse large B cell lymphoma (DLBCL) appear to have a mature B cell phenotype. Nonetheless, they share a common translocation IgH/BCL2 t(14;18)(q32;q21) which occurs early in B cell development during V(D)J recombination mediated by RAG in the primary lymphoid organs (10). This translocation places the anti-apoptotic BCL2 gene under control of the immunoglobulin heavy chain (IgH) enhancer and plays a causal role in germinal center derived malignancies. Though this translocation is commonly an initiating event in development of FL, it alone is not sufficient to initiate lymphomagenesis and similar translocations can be detected in the peripheral blood and lymph nodes of healthy individuals (11). Similarly, whilst chronic lymphocytic leukemia (CLL) is also considered a mature B cell malignancy, the propensity for cells to become malignant appears to arise as early as the hematopoietic stem cell (HSC) stage and driver lesions are detectable in the hematopoietic progenitors of some patients (12–14). CLL is also characterized by distinct subtypes that either have unmutated variable regions or hypermutated post germinal center variable regions. Mantle cell lymphoma (MCL) has a similar bifurcation between mutated/unmutated variable regions.
Further complicating matters, some B cell neoplasms result from the transformation of non-malignant monoclonal outgrowths or from other neoplasm subtypes. DLBCL can arise from the transformation of CLL, small lymphocytic leukemia (SLL) or FL (15) or less commonly from mucosa associated lymphoid tissue (MALT) lymphoma (16, 17) or Hodgkin lymphoma (HL) (18). CLL can itself develop from the precursor condition monoclonal B cell lymphocytosis (19) and a high proportion of multiple myeloma (MM) arise from monoclonal gammopathy of undetermined significance (20, 21).
Recent genomic studies of B cell malignancies implicate a handful of well-characterized, commonly mutated or translocated genes as well as a long tail of genes that have limited or no experimental evidence to support a role in disease. Since it is unlikely that all these mutations are driving oncogenesis there is need for validation methods such as functional genomic screens, forward genetic screens and comparative genomics to scrutinize driver mutations and better prioritize candidates for further study. The diverse mechanisms by which mutations contribute to disease means that many can only be tested within the context of specific developmental stages, predisposing mutations and optimal antigen receptor stimulation. For secondary/later stage mutations, oncogenic function and selection may only occur within a specific context of germline variants and/or early stage initiating mutations. This exponentially expanding set of parameters for validation experiments therefore requires prioritization by analysis of mutation profiles and higher throughput testing methods.
In some respects, sequencing human tumors can be viewed as a special case of forward genetics, i.e. surveying genetic diversity under selection in order to identify which genetic changes drive a biological process. Forward genetic screens can also be targeted to the somatic cells of mice using either chemical or insertional mutagens. In the context of mouse cancer models, reverse genetics is the introduction of variation known to be associated with cancer in order to study the function of that variation. This variation can be introduced to the germline or targeted to specific populations of somatic cells.
The benefit of using mouse models to study hematologic malignancies is that the major genes and biological processes driving hematopoiesis and immunity are sufficiently conserved such that human malignancies can be recapitulated by the introduction of equivalent mutations. Mice also allow the complexity of immune system biology to be recapitulated in vivo in ways not yet possible with cultured cells or organoids.
This review covers mouse models used to rapidly screen for and validate large numbers of candidate genes driving B cell malignancies and is aimed at readers seeking to understand various techniques for targeting different developmental stages and disease subtypes. We provide a history of forward and reverse genetic approaches in mice that balance the trade-offs between fidelity, precision, and throughput. In the first half we discuss how forward genetics in mice can independently validate the oncogenic function of rarely mutated genes from human studies, and in some cases is a tool leading to discovery of human cancer genes. In the second half we cover reverse genetic screens and validation experiments where throughput is increased through the use of transplantation of primary cells transduced with ORF, shRNA and CRISPR-Cas viral constructs. Throughout we also emphasize strategies to direct models toward specific malignancy subtypes by a combination of germline lesions, targeted cell populations and temporal control of gene expression.
Many driver genes are difficult to identify because they are subject to deregulation by non-coding mutations, copy number changes or epigenetic modification. Comparative genomic analyses of human tumors with the findings from forward genetic screens in mouse models can narrow down the identity of these driver genes. Mice with germline mutations that sensitize them to a specific disease subtypes are subjected to mutagenesis of their somatic cells and the resultant tumors are sequenced. We discuss here the combined use of genetically modified mice, insertional mutagens and exome sequencing as tools to perform forward genetic screens in B cell neoplasms (summarized in Table 2). Observing which mutations are present in both mouse and human cohorts, or which mutation combinations are selected to co-occur more or less frequently than expected by chance can prioritize validation experiments.

Table 2 Forward genetic screens in mouse models of B cell malignancies.
Somatic insertional mutagenesis screens are one of the most efficient tools for performing forward genetic screens in mouse models of cancer. When insertional mutagens are integrated randomly throughout the genome of somatic cells, they can deregulate and disrupt genes in a manner analogous to chromosomal rearrangements, deletions, non-coding mutations and truncating coding mutations. Mouse tissues are typically mutagenized by replicative retroviruses or germline copies of transposons mobilized in different tissues. Under optimal conditions a subset of these mutagenized cells eventually give rise to malignancies.
The primary benefit of analyzing integration mutations is the ease with which they can be mapped to the genome by amplifying sequences that flank the integration using various ligation mediated PCR methods. In analyzing cohorts of tumors in these screens, loci found to have insertion sites in independent tumors, more frequently than expected by chance, are defined as common insertion sites or common integration sites (CISs). Selection of mutations at these CIS loci indicates that they cause changes in expression levels or cause disruption/truncation of cancer drivers. There can be substantial phenotypic and genetic variability between malignancies of a single cohort and driving specificity toward B cells rather than T or myeloid lineages has been achieved through a combination of limiting mutagenesis to B lineages, screening mice with a predisposition toward B malignancy subtypes and curating uniform subsets of B lineage tumors from mixed cohorts.
Slow transforming retroviruses have an extensive history of use as insertional mutagens in the hematopoietic compartment [reviewed in (62)]. When newborn mice are infected with slow transforming retroviruses, they fail to mount an immune response and consequently develop a viremia that lasts the lifetime of the animal. Successive rounds of reinfection lead to an accumulation of insertion mutations in cells of the hematopoietic compartment, where the high rate of proliferation during early postnatal development makes them the preferred host cells for virus propagation. In some strains the spontaneous activation of endogenous ecotropic retroviruses gives rise to disease by a similar process. Over time, mutations providing a selective advantage will lead to the clonal expansion of cells with multiple oncogenic insertion mutations.
These viruses induce a range of hematologic malignancies. B cell tumor cohorts have been generated by choosing specific combinations of virus strain and host mouse strain. Sequences within the virus LTRs are responsible for cell type-specific expression and alterations can skew tumors toward B cell subtypes (63, 64). Akv is an endogenous, ecotropic murine leukemia virus isolated from the AKR strain which causes mature B cell lymphomas with an FL and DLBCL phenotype (65). Mutation of the Akv LTR enhancer sequences causes plasmacytoma like disease (66) and mutation of splice sites broadens the diversity of B malignancy subtypes (32). Somatic reactivation of endogenous ecotropic proviruses can also give rise to B lineage malignancies in inbred mouse strains (SJL/J mice, CWD/LeAgl, SEA/GnJ, SL/Hk) (67, 68), recombinant inbred strains (AKXD) (69) and the NFS.V+ congenic mice (bearing ecotropic MuLV loci from AKXD or C58/Lw) (70, 71). When integration sites from B malignancies of these models were cloned, many were found recurrently at sites that were known or have subsequently been found to be drivers of B cell malignancies (27, 30–39, 41, 42) (Table 2).
Retroviral integrations tend to increase the expression of oncogenes although occasionally intragenic integrations cause loss of function of tumor suppressors. One of the more innovative strategies to increase the proportion of tumor suppressors identified is the screening of mice on a Blm hypomorphic background (40). Blm encodes a recQ DNA helicase the loss of which creates genomic instability. This encourages DNA repair by homologous recombination, which in turn leads to increased sister chromatid exchange and large stretches of loss of heterozygosity. When integration of a virus disrupts a tumor suppressor gene, any subsequent deletions or loss of heterozygosity that removes the wild type copy will increase selection of tumor suppressor mutations.
Enforced expression of B cell lymphoma oncogenes such as MYC, NMYC or BCL2, in the B cell compartment or loss of tumor suppressors can also skew MuLV driven malignancies toward B cell lymphoma and leukemias (22–26). Recent large scale analyses of the common integration sites of two BCL2 transgenic strains infected with Moloney MuLV (MoMuLV) shows a statistically significant bias toward verified drivers of human B lymphoma and leukemia (Pou2f2, Pax5, Ikzf3, Ebf1) in addition to dozens of other candidate loci (29).
The use of DNA transposons as somatic insertional mutagens in mice has led to dozens of tissue and cell type specific screens [reviewed in (72–74)]. The Sleeping Beauty (SB) transposon isolated from salmonid fish and the piggyBac (PB) transposon isolated from the cabbage looper moth have both been adapted for use in mammalian cells. When mice bearing a concatemer of a transposon as a germline transgene are crossed to mice that express the cognate transposase, this causes mobilization of the transposon in somatic cells. By controlling expression of the transposase and the cargo of the transposon (typically a promoter trap and/or gene trap) this technique generates a wider spectrum of tumor types than slow transforming retroviruses, although this versatility is tempered by several caveats. Transposons have a tendency to hop in cis which can complicate analysis of integrations near the initial concatemer. Furthermore, remobilization of the transposon during tumor development can cause local hopping around the region of tumor initiating mutations and there is typically a higher level of background non-CIS mutations that provide no selective advantage to tumor cells. The screens discussed in detail below have successfully targeted transposon mutagenesis to the B cell compartment using a combination of predisposing mutations combined with tissue specific transposase expression.
Weakly oncogenic mutations observed in human tumors do not always lead to tumor development in mouse models due to a lack of cooperating mutations. One of the first B cell malignancy SB screens used a mouse model of the t(12;21)(p13;q22) ETV6/RUNX1 (TEL/AML1) fusion transcript, seen frequently in B cell acute lymphoblastic leukemia (ALL). Mice expressing the ETV6/RUNX1 fusion and the SB transposase variant HSB5 from the endogenous Etv6 locus had a background of long latency hematologic malignancies similar to wild type controls i.e. the fusion itself caused only slightly higher rates of lymphomagenesis (43). When this allele was combined with a gene trap/promoter trap transposon the mice developed a spectrum of acute myeloid leukemia (AML), T cell ALL and B cell precursor ALL. By sequencing the integrations of B ALL samples, recurrent truncating mutations were observed in Ebf1, a tumor suppressor of B ALL (75, 76) which encodes a transcriptional activator of another B cell ALL tumor suppressor Pax5.
STAT5 activating mutations are observed in a small proportion of B ALL, however mice expressing a constitutively active form of STAT5B (STAT5-CA N642H) do not develop ALL (46). Combining this allele with a transposon gene/promoter trap concatemer, a Cre-inducible SB and Cd79a-Cre (switching in developing B cells) induced progenitor B cell leukemias thereby verifying STAT5B N642H as an oncogene in B ALL. Integrations of 65 mice identified 12 CIS, the most commonly mutated genes suggesting three major mechanisms for STAT5 mediated transformation: disruption of B-cell development (Sos1, Kdm2a, Ikzf1, Klf3), enhanced JAK/STAT5 signaling (Jak1) and modification of the CDKN2A tumor-suppressor pathway (Bmi1, Cdkn2a).
The exomes of CLL samples average less than one coding driver mutation per sample, making mechanisms of transformation difficult to identify. The Eµ-TCL1 transgenic model develops a disease that is phenotypically similar to CLL. The B cells of these mice were mutagenized using a conditional SB transposase switched by CD19-Cre (pan B cell) (44, 59). Transposon mobilization decreased latency of Eµ-TCL leukemias and integrations of 15 mice yielded 8 CIS loci, four of which implicate Nf-kappaB signaling (Nfkb1, Tab2, Map3K14, and Nfkbid). NF-kappaB activating mutations are rare in the coding regions of human CLL however it has been shown that CLL cells are frequently dependent on extracellular signaling pathways that activate Nf-kappaB (77) and several mouse models of CLL/SLL have constitutive activation of NF-kappaB pathways (78–80).
The Cre strains used to switch conditional SB alleles need not be entirely B cell specific in order to generate B cell malignancies. The Cnp-Cre strain expresses Cre in the nervous system and splenic germinal centers (47). Combining Cnp-Cre with an SB11 conditional allele, a transposon concatemer and a conditional oncogenic point mutation of tumor suppressor Trp53 (Trp53R270H) generated a mixture of solid tumors as well as lymphoid malignancies with an FL/DLBCL like phenotype. Integrations from 23 samples identified Bach2 as the most frequently mutated locus. Pten disrupting mutations were also identified and the authors verified Pten as a tumor suppressor in the Cnp-Cre driven model. These results are consistent with human germinal center B cell DLBCL where PTEN is mutated or deleted (81) and reduced expression correlates with AKT activation (82).
More recently the piggyBac transposon has also been adapted for somatic insertional mutagenesis. Mice expressing constitutive piggyBac transposase were crossed with a gene trap piggyBac transposon concatemer allele onto a Blmm3/m3 hypomorph background (48). Gene trap transposons are more likely to generate intragenic loss of function mutations which are in turn more likely to be selected if they occur in tumor suppressors. The use of a Blm hypomorph background to enhance loss of heterozygosity events is the same strategy used to enrich for tumor suppressors in the retroviral screen of AKXD Blmm3/m3 mice cited in the previous section (40). A spectrum of solid and hematological tumors were generated, with the majority of mice having DLBCL like B lymphoid malignancies. These had recurrent amplification of Bcl11a and Rel, this region also being amplified in human DLBCL. Integration site cloning using the semiquantitative QiSeq insert cloning protocol (83) on 43 samples identified nearly 300,000 integrations. Inactivating/disrupting mutations were observed in tumor suppressors known to be mutated in human DLBCL and there was significant overlap between the orthologs of CIS genes and the set of genes that are down regulated in human DLBCL vs non-malignant B cells. This suggests CIS genes may inform the identification of genes that are down regulated in human DLBCL by epigenetic variation and/or large-scale deletions.
Improvement in next generation sequencing platforms and ligation mediated PCR methods have increased the throughput and sensitivity of integration site cloning. Ligation mediated-PCR methods such as vectorette and splinkerette-PCR use a non-complimentary adaptor that facilitates enrichment of integrated sequences separately from the remainder of the genome (84–86). Shearing of tumor DNA prior to construction of sequencing libraries allows the abundance of each integration within a sample to be quantitated by counting the number of unique read fragment lengths mapping to each integration (83, 87–89). This approach to quantitation minimizes sequence and amplification biases particularly when compared to libraries prepared by non-random digestion at restriction enzymes recognition sites. Other studies have quantitated integrations through analyses of whole genome amplification of single cells (SBCapSeq) (90, 91) or using unique molecular identifiers (LUMI-PCR) (92).
Collectively, these recent methods allow affordable, genome wide identification of thousands of subclonal mutations with a sensitivity spanning more than two orders of magnitude. This quantitative coverage in turn allows estimation of the order in which lesions occurred (93) and cloning large numbers of subclonal mutations provides the statistical power needed to derive genetic associations between early-stage clonal mutations and late-stage subclonal mutations (29). Translating these types of analyses to human tumors is currently limited by depth of sequencing coverage. Currently whole genome sequencing of human tumors rarely exceeds 100x coverage and therefore offers less sensitive detection of subclonal mutations than recent integration site cloning studies. Nonetheless, recent studies of single cell sequencing of human tumors include thousands of cells which can therefore identify subclonal mutations within limited/targeted regions of the genome (94–97).
The scale of these datasets requires innovations in statistical methodology, both to identify selected mutations and to demonstrate associations between them. For density based estimates of mutation selection (98–100) the background integration biases of insertional mutagens can be compensated for by the use of unselected cell populations for comparison with tumor mutation distributions (29, 93, 101). Alternatively, identifying local ratios of forward/reverse strand mutations throughout the genome is analogous to using ratios of synonymous and non-synonymous coding mutations in that it does not require correction for integration site biases of the mutagen (29, 93). Strand bias analyses requires a higher density of integrations to reach statistical significance and will only identify loci with a strong bias in one direction or the other.
Analyses of concurrent mutations is complicated by the inclusion of subclonal mutations. When sequencing coverage is deep enough to include very subclonal mutations, some of the most frequently mutated loci can be found mutated in 100% of samples. Any two loci found to have subclonal mutations in all samples will appear to be 100% concomitant, even when these mutations are not found within the same subclones of each sample. For this reason, quantifying the significance of concurrent mutations requires the use of contingency table tests that exclusively use clonal mutations i.e. mutations with a high likelihood of occurring within the same cells. Alternatively it is possible to use asymmetrical contingency tables that classify all tumors on the basis of their early stage “trunk” clonal mutations and/or germline mutations at one locus and then test for the distribution of all mutations (both clonal and subclonal) at other loci (29, 102, 103). The number of genome-wide pairwise tests that can be performed on these cohorts can number in the thousands and this in turn requires stringent multiple testing correction to limit false positive associations. Nonetheless, with high coverage of subclonal mutations it is now possible to identify hundreds of significantly co-mutated loci pairs from a cohort of several hundred mice infected with MoMuLV (29).
The expense of exome and whole genome sequencing is generally reserved for the study of human tumors however, an increasing number of mouse tumor exomes have also been sequenced, albeit with cohort sizes that are far smaller than their human counterparts. In most cases this sequencing is to verify that the model accurately recapitulates human disease, however, some studies also suggest or verify candidate drivers where coding mutations in human cohorts fail to reach statistical significance. Several examples are discussed below.
Combining Cre recombinase inducible alleles of mutant Myd88 (L252P) and Rosa-26 expressed Bcl2 with germinal center specific Cγ1-Cre generated a human ABC-DLBCL like model (60). Sequencing samples from 17 mice confirmed mutation of human DLBCL drivers including Pim1, Myc, Kmt2d, Nfkbia, Stat3, Pou2f2, and Hist1h1e. In a similar study, conditional expression in germinal center B cells of Myc and constitutively activated Akt (fusing p85 to p110) gave rise to Burkitt’s lymphoma (BL) like disease with recurrent mutations in Ccnd3, which is mutated in BL and known to regulate germinal center B cell proliferation (49).
TET1 and TET2 encode methylcytosine dioxygenase enzymes that are thought to play a role in demethylation of DNA by catalyzing conversion of 5-methylcytosine to 5-hydroxymethylcytosine. TET2 is mutated in a range of hematopoietic malignancies including a subset of DLBCL where mutations can be traced back to HSC populations, suggesting TET2 mutation is an early event in lymphomagenesis (104). Mice lacking Tet2 globally or in B cell subpopulations (CD19-Cre or Vav-Cre) develop B cell lymphoma (57, 105). One study sequencing six B cell tumors matched to germline counterparts revealed thirty-four acquired mutations, albeit not recurrently mutated, presumably due to the limited cohort size. Nonetheless, more than half of these genes are also mutated in either DLBCL, CLL or both.
TET1 is not mutated in human B malignancies however it is downregulated in human DLBCL and FL which inconclusively suggests a role for TET1 in human B lymphoma. Tet1 deficient mice develop mature B cell malignancies which when sequenced were found to have missense mutations in the mouse homologues of known human B cell malignancy drivers including Gna13, Kmt2d, Myd88, Cd83, Pim1, Cd79B and Fas (106). Additional mutations were observed in linker histone genes, histone variants and histone modifying enzymes. Histone linker mutations are a feature of FL (107). These mutations corroborate a role for TET1 down regulation in human B cell malignancies.
Mouse models that accurately recapitulate human disease can also identify novel drivers. The Eμ -Myc model is a mainstay of B cell leukemia/lymphoma research but exomes from this model have only recently been sequenced (54). Sequencing of 23 lymphoma exomes revealed recurrent mutations in known drivers such as Trp53, Cdkn2a and Kras, but also the “BCL6 corepressor” gene Bcor. Inactivating mutations in Bcor accelerated Eμ-Myc driven lymphomas in transplantation assays. An independent study sequenced NUP98/PHF23 transgenic mice which primarily develop T cell lymphoma but in a minority of cases also develop progenitor B-1 type ALL. This study also identified Bcor mutations in addition to mutations of Jak1, Jak2, Jak3 and Stat5a (51, 56). Bcor loss of function mutations were subsequently found to cause B-1 progenitor ALL in mice (108).
Aside from driver detection, exome sequencing can also be a powerful tool to examine the mutagenic processes of early premalignant states under defined experimental conditions. Sungalee et al. developed an innovative model of t(14;18) translocation where human BCL2 expression from an Eμ enhancer is sporadically induced by RAG recombinase in pro/pre B cells (50). Repeat immunization demonstrated a survival advantage for hBCL2 expressing cells after multiple rounds of germinal center reentry. Exome sequencing was performed on BCL2-enriched germinal center and memory cell fractions from three chronically immunized mice and a control (empty vector) mouse. B cell subsets of the BCL2-transduced mice had between 111 and 2,565 nonsilent SNVs compared with 63 to 70 SNVs in the control mouse, despite the absence of overt morphological differences between hBCL2 and control animals. This model demonstrates a role for BCL2 in enhanced survival of cells that accumulate potentially oncogenic off target mutations of AID.
PAX5 encodes a B cell development transcription factor that activates B cell specific genes and represses genes of alternative lineages. It is one of the most frequently altered genes in B ALL and a varied spectrum of somatic rearrangements, translocations, mutations and deletions have been observed at this locus (76, 109–111). Karyotypic analysis indicates translocations are early events whereas deletion of a second allele appears to be a secondary later event. Germline point mutations that inhibit PAX5 function also increase the likelihood of B ALL and some are accompanied by somatic deletion of the other PAX5 allele or cooperating PAX5 mutations (112–115). The discovery of frequent PAX5 deletion and translocations in human ALL was rapidly followed by a large number of studies using some form of Pax5 deletion or translocation in mice to model B ALL. To date six independent studies have included exome sequencing and/or insertional mutagenesis of mouse B ALL samples generated by mutation of Pax5. The convergence of findings of these studies demonstrates the value of multimodal screening in mice to validate mutation profiles of human tumors.
Pax5 heterozygosity combined with thymectomy skews ENU and MoMuLV driven lymphomas toward a B lineage disease since thymectomy removes competing T cell malignancies (28). Array-CGH copy number analyses of these malignancies identified amplification of chromosome 15 (and consequently Myc) and focal deletions of the second allele of Pax5. Exome sequencing revealed B ALL samples from ENU treated mice had 5-fold more exonic mutations than MuLV infected mice and identified recurrent mutations in the human ALL mutated genes: Pax5, Jak3, Ptpn11, Jak1, and Nras. Pax5 mutations were almost exclusively within the DNA-binding paired domain and equivalent to mutations observed in human B ALL. Jak1 and Jak3 mutations were located in the pseudokinase domains of JAK1 and JAK3, and are known to induce cytokine-independent proliferation and activation of downstream signaling pathways sensitive to JAK inhibitors. MoMuLV CIS included an overlapping set of B ALL drivers including Myc, Stat5b, Zeb2, Jak1, Jak3, Ikzf1, Gsdmc, Ebf1 and Ptpn11.
In another study multiple Cre strains (Cd19, Mb1(Cd79a) or Mx1) were used to induce conditional expression of a E2A/PBX1 fusion, resulting in B cell precursor ALLs, which arrested at the pro B/large pre B II stages in a manner similar to human E2A/PBX1 ALL (53). Copy number analyses and exome sequencing of leukemia samples revealed that 30% harbored inactivating mutations or deletion of Pax5 that resulted in decreased expression. Combining E2A/PBX1 with Pax5 deletion increased the penetrance and shortened the latency of leukemia. Other mutated loci from E2A/PBX1 tumors included Cdkn2a, Jak1, Jak3, Ptpn11 and Kras.
The ETV6/RUNX1-SB model described in the previous section (43) was subsequently combined with heterozygous deletion of Pax5 which increased the incidence of B-cell precursor (BCP)-ALL (45). Targeted exome sequencing identified recurrent mutations in Jak3, Trp53 and Jak1, with Jak1/3 mutations again being found in the pseudokinase domain. Insertional mutagenesis identified 6 CIS, four of which (Zfp423, Cblb, Stat5b and Foxp1) have well-characterized roles in B cell maturation. Increased ZNF423 expression is associated with ETV6/RUNX1+ B ALL and Pax5 or Ebf1 mutations synergize with STAT5 in B ALL. It is worth noting this mutation profile is more likely a function of Pax5 loss since a different model of ETV6/RUNX1 with wild type Pax5 primarily identified loss of function mutations in the KDM lysine demethylase family (55).
B ALL models have also been used to study the relationship between gut microbiota and leukemia incidence (52, 55, 61). Pax5 heterozygous mice do not develop leukemia when housed in specific pathogen free (SPF) conditions however they are prone to precursor B ALL when exposed to common pathogens and sequencing of these leukemias identified recurrent Jak3 mutations (52). In an independent study it was also shown that Pax5 heterozygous mice or mice bearing a Sca1-ETV6/RUNX1 transgene also do not develop B ALL when housed in SPF conditions. Nonetheless both mouse strains had sufficiently altered immune responses to alter the gut microbiome composition when housed in a non-SPF facility and Pax5 heterozygous mice in SPF conditions treated with antibiotics developed B ALL (61). The authors hypothesize that dysbiosis caused by non-SPF housing or the absence a normal gut biome in SPF conditions with antibiotics drives disease on this background, however secondary mutations were also required. Exome sequencing of 17 B ALL cases from non-SPF conditions identified recurrent mutations affecting the JAK/STAT and RAS signaling pathways (Jak1, Jak3, and Ptpn11). Non-SPF mice treated with antibiotics also harbored recurrent mutations in Kit, Flt3, and Cbl that have not been observed in other Pax5+/– cohorts. This may suggest that selection of secondary mutations reflects differences in gut biome.
PAX5 is frequently mutated by translocation events creating fusion proteins in human tumors and some of these have been shown to cause B ALL in mice (58, 116). In one of these studies exome sequencing of five leukemic mice expressing a PAX5/ENL fusion identified mutations in Ptpn11, Kras, Pax5, and Jak3 genes (58). Of these genes PAX5, PTPN11, KRAS and its homologue NRAS were found recurrently mutated across a panel of human B ALL samples from diverse subtypes.
These six independent studies using complementary experimental designs all identify mutations in Jak3 by exome sequencing and in some cases also by insertional mutagenesis. Furthermore, four studies identified mutations in Jak1, three studies identified Ras family member mutations (albeit through different mechanisms) and four had Ptpn11 mutations. Collectively this work demonstrates how mutagenesis and exome sequencing of mouse models with defined lesions can reliably and reproducibly reveal cooperating mutations in independent cohorts, and that Pax5 heterozygosity in mice faithfully reproduces mutational profiles observed in human B ALL.
Over the past decade the expanding number of candidate mutations associated with B cell malignancies has increased the requirement for medium to high throughput methods of validation. There is an extensive literature of transgenes, knockouts and conditional alleles for B cell lymphomas [reviewed in (117–121)]. Although tissue specific switching of conditional alleles is arguably the gold standard for mouse tumor models, the obvious drawback of this approach is the time taken to generate strains and cohorts of multiallelic models. In this section we discuss higher throughput reverse genetic models including transplantation/adoptive transfer models with an emphasis on models using primary cells transduced with viral constructs. We also summarize recently developed methods for rapid generation of multiallelic strains and several approaches to temporal control of gene expression.
Early transplantation-based B cell malignancy models used cell lines derived from spontaneous malignancies injected into syngeneic hosts and/or human malignant cell lines injected as xenografts into immunocompromised mouse strains [reviewed in (122)]. More recently primary B cell malignancies have been transduced with viral constructs prior to transplantation, either to overexpress oncogenes (123–125) or to express shRNAs/sgRNAs that validate essential genes, therapeutic targets and tumor suppressors (126–134) (Table 3).
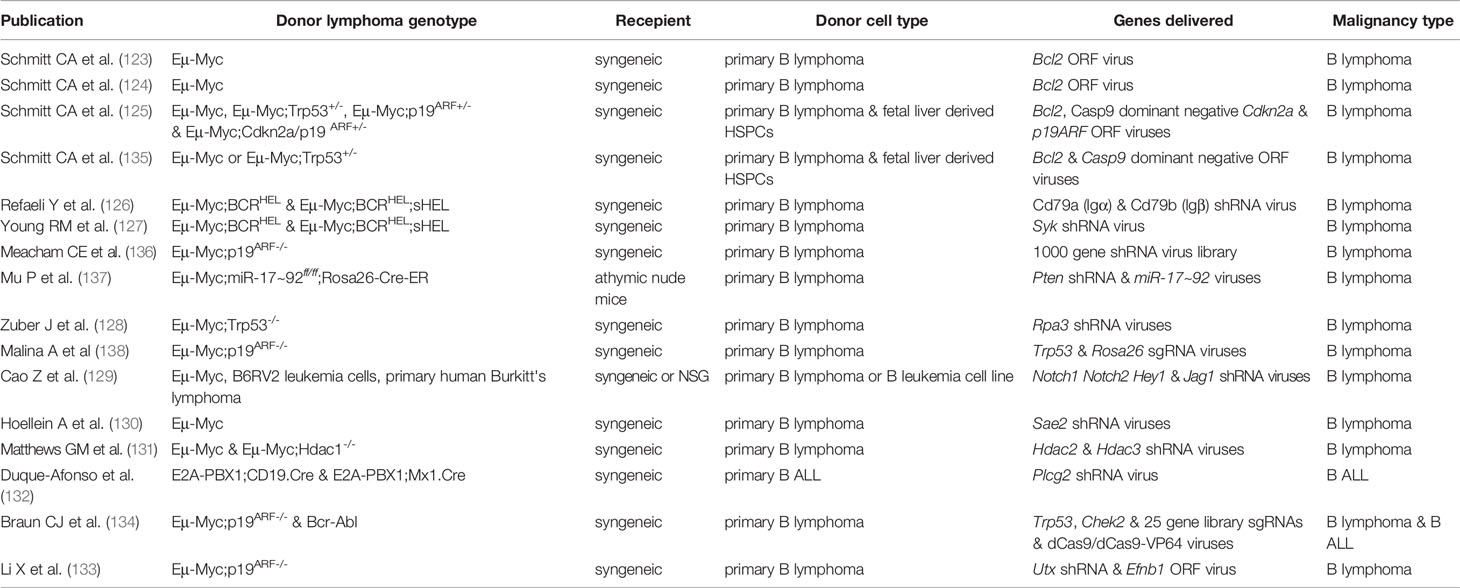
Table 3 Reverse genetic B cell malignancy transplantation assays using primary lymphomas.
Primary B lymphomas have also been used to perform in vivo shRNA screens. In one study shRNAs against 1000 candidate cancer genes were selected to identify genes that are required for B lymphoma growth in vivo and this identified regulators of actin dynamics and cell motility (136). In a more targeted study, combining an in vitro screen with in vivo verification, conditional deletion of the miR-17∼92 cluster was found to cause apoptosis in Eμ-Myc lymphomas. In vitro screening of shRNAs against targets of these microRNAs identified Pten suppression as a mediator of miR-17∼92 survival signals which was verified when shRNAs against Pten rescued in vivo lymphoma growth of miR-17∼92 deleted Eμ-Myc lymphoma cells (137).
Studying the transition of non-malignant cells into lymphomas and leukemias requires the use of untransformed primary cells. Retroviral transduction of primary cell suspensions from spleen, bone marrow and fetal liver can reveal the transforming potential of candidate genes and mutations. In early experiments both viral and cellular genes were found to facilitate colony formation in agar, clonal outgrowth of cell lines and development of malignancies from adoptive transfer into irradiated or immuno-compromised recipients [reviewed in (139, 140)]. The malignancy subtypes generated are a function of the genes being transduced and the lineage of infected cells, with B cell malignancies being successfully generated from wild type donor cells by using cellular genes (141, 142), acute transforming retrovirus oncogenes (143–147) and the fusion transcripts of oncogenic translocations (145, 148–152) (summarized in Table 4). Experiments using the mixed populations of cell types from whole bone marrow as donor cells can yield a mixture of malignancy subtypes from a single cohort. The use of purified target cells offers greater control of malignancy subtype, and protocols for the culture and transduction of purified HSCs, hematopoietic stem and precursor cells (HSPCs), B lineage precursors/progenitors and splenic B cells (188–190) have all been adapted to generate B lineage malignancies.

Table 4 Reverse genetic B cell malignancy transplantation assays using primary cells.
Combining both sensitized germline alleles with transduction of retroviruses also influences the cell type and developmental stage of the disease being modelled. One of the most frequently used strain to derive B cell malignancies by adoptive transfer of transduced cells is the Eμ-Myc model of Burkitt lymphoma (191). When bone marrow derived pre B cells from Eμ-Myc transgenic mice were transformed by transduction with v-H-ras or v-raf, the resulting clones forming subcutaneous tumors in nude mice (153). Later protocols using transplanted Eμ-Myc fetal liver cells (typically HSCs and HSPCs) yielded B cell lymphomas that could be accelerated by loss of tumor suppressors in the germline (e.g. Trp53, Cdkn2a, p19ARF) and by retroviral expression of cooperating ORFs (125, 135, 157, 162). The use of fetal liver cells is particularly useful for the generation of lymphomas where one or more germline alleles prevent normal development after E17. This approach has been used to demonstrate that deletion of the essential mediators of apoptosis Apaf1 and Casp9 does not affect reconstitution by fetal liver HSCs (192) or alter the phenotype or latency of lymphomas derived from reconstitution by Eμ-Myc fetal liver HSCs (193).
Viral constructs capable of robust expression of short RNAs have facilitated rapid verification of putative tumor suppressors and oncogenic microRNAs. In an early study a series of Trp53 shRNAs introduced into Eμ-Myc HSCs were found to accelerate lymphomagenesis in a manner that correlated with the strength of knockdown (155) and similar results were obtained for the mir-17–19b microRNA (160) and for shRNAs against Puma (Bbc3, a proapoptotic downstream target of Trp53) (156) and Bcor (a tumor suppressor of Burkitt lymphoma) (54). The Eμ-Myc model has also been used to screen pools of shRNAs against a set of 1000 known and putative cancer genes for tumor suppressor activity. Infecting fetal liver HSCs and transplanting them into recipients identified Sfrp1, Numb, Mek1, Ang2 as tumor suppressors, as well as identifying known components of the DNA damage response machinery including Rad17 (166). A more focused shRNA screen of known targets of Trp53 was conducted using fetal liver HSCs of mice that were either Eμ-Myc Puma-/- or Cdkn1a(p21)-/-Puma-/- in order to identify targets of p53 that were responsible for its tumor suppressor role independently of Cdkn1a or Puma (184). This identified a spectrum of shRNAs against DNA damage repair genes including Mlh1, Msh2, Rnf144b, Cav1 and Ddit4 which were capable of accelerating Eμ-Myc fetal liver HSC transplant lymphomas.
The most common strains used in these transplantation experiments are wild type mice, Eμ-Myc transgenics, Trp53 mutants and Cdkn2a/p19ARF mutants however many other strains have also been used to probe the cooperative or epistatic relationships of transduced genes with germline alleles (108, 158, 159, 161, 162, 165, 174–176, 180, 187) (summarized in Table 4). HSCs transduced with Myc retroviruses develop into aggressive pre B cell lymphomas when transplanted (159), and point mutants of Myc typically found in Burkitt’s lymphoma increased penetrance and accelerated latency. Replacing wild type donor cells with Trp53-/+, Trp53-/- or Bim-/- donor HSCs accelerates latency of wild type Myc more than Myc mutants, suggesting these mutations reduce Myc induced apoptosis mediated by Trp53/Bim and reduce selection for Trp53 inactivation. Thus, epistatic effects and mutation redundancy can be rapidly investigated using germline alleles that predispose animals to disease.
The “Philadelphia chromosome” translocation, t(9;22)(q34;q11), that expresses the BCR/ABL1 translocation product is observed in both human CML and B ALL. Retroviral transduction of hematopoietic cells with the BCR/ABL1 fusion transcript and transplantation into syngeneic recipients is one of the most extensively studied transplantation models in the literature. BCR/ABL1 transduced bone marrow gives rise to various lineages of malignancies including, B lineage, myeloid and mixed lineage leukemias. Disease subtype and latency can be altered by varying donor cell lineage, genotype, culture protocols and the use of different BCR/ABL1 isoforms. B lineage disease is particularly promoted by culture conditions that enhance pro/pre B differentiation and the use of the p185(p190) isoform (152, 170). Myeloid disease can be promoted by the p210 isoform and by pre-treating donor mice with 5-fluorouracil which depletes bone marrow of cycling cells and prompts cycling of quiescent HSCs (152, 170).
Mutations that determine which cell lineages can form different malignancy subtypes have been extensively studied using this model. Transplantation of purified HSCs transduced with BCR/ABL1 retrovirus typically gives rise to CML, however loss of both Cdkn2a/p19ARF or only p19ARF in donor cells gives rise to B ALL (165). This is consistent with the observation of concomitant deletion of the CDKN2A/p14ARF locus in human B ALL but not in CML. p19ARF loss also enables B ALL to develop rapidly from BCR/ABL1 transduced common lymphoid progenitors, pro B cells and pre B cells. Furthermore, p19ARF null pro B cell derived disease was phenotypically different from wild type pro B cell derived disease which had substantially lower penetrance and delayed kinetics. When compared to p19ARF null pro B cell derived disease, p19ARF null HSC derived B ALL had greater colony forming potential in methylcellulose, greater resistance to dexamethasone and reduced resistance to the kinase inhibitor imatinib mesylate (Gleevec) (162, 165). Germline truncations of p19ARF have also identified a specific region responsible for Trp53 induction and suppression of BCR/ABL1 leukemias (180).
These differences of BCR/ABL1 driven disease in different lineages might suggest a separate cell of origin for B cell and myeloid disease, however using purified HSCs as donor cells suggests both CML and B ALL can arise from a long-term HSC cell of origin (170). In this model disease subtype is primarily influenced by the use of different isoforms of BCR/ABL1, with the longer p210 isoform giving rise to CML whereas a shorter isoform p185 causes B ALL. Despite this common cell of origin, the cancer-propagating “cancer stem cell” populations responsible for maintaining these diseases appear to be distinct. The CML disease was maintained by a long-term HSC population whereas B ALL was maintained by a differentiated pro B population.
The interactions of BCR/ABL1 with other donor cell lesions has also identified additional essential mediators of disease. Stat5a and Stat5b are necessary for establishment and maintenance of malignancies derived from BCR/ABL1 infected bone marrow (163, 167) and N-terminal truncation of Stat5a and Stat5b skews BCR/ABL1 p210 transplants toward a B cell lineage but BCR/ABL1 p185 transplants are unaffected (154). BCR/ABL p210 fails to induce disease when using Cdk6−/− bone marrow, possibly due to defects in HSC cycling in these mice (175). BCR/ABL1 transduced bone marrow lacking three Src kinases (Lyn-/-;Hck-/-;Fgr-/-) resulted in delayed onset of B ALL compared with wild type donors however this delay was not seen for the onset of CML derived using 5-fluorouracil treated donors (158). This demonstrates a B lineage specific requirement of Src kinases for BRC/ABL1 driven malignancies, and is consistent with synergy of pan Src kinase inhibitor CGP76030 with imatinib mesylate in treating BCR/ABL1-induced B ALL but not CML.
Collectively the above literature of BCR/ABL1 malignancy models demonstrate how the genotype and developmental stage of transduced donor cells can alter disease subtype, clonogenicity and treatment response. These studies also demonstrate the value of transplantation models in defining the tumor initiating cell types of genetically similar (if not identical) but phenotypically distinct diseases.
One disadvantage of using HSCs or mixed populations of bone marrow as donor cells is their potential to develop a wide spectrum of malignancy lineages from undefined developmental stages. Culture systems for in vitro expansion of B cell progenitors (189, 194, 195) have been further refined to use B220+Kit+ fetal liver cells cultured with IL-7 on ST-2 stromal cells (168) and more recently stroma free pro B cell culture conditions have also been developed using IL-7, SCF and FLT3 ligand (196). These systems allow expansion and transduction of progenitor/precursor B cells for use in transplantation studies. In one study, pro B cells were transduced with combinations of Bcl2, Myc and Ccnd1, and transplanted into severe combined immunodeficient (SCID) mice. The recipients develop an immature B cell lymphoma/leukemia which infiltrated the lymph nodes, spleen, thymus and bone marrow. Ex vivo cultured pro B cells transduced with Bcl2 and Myc were also transduced with a cDNA library and selected for continued growth in the absence of IL-7 and ST-2 stromal cells. CCND3 and NRAS both rendered these cultures independent of IL-7 and ST-2 cells and accelerated Myc/Bcl2 driven disease when transplanted into SCID mice.
Long term proliferative pre B cell lines derived from fetal liver have also been used to identify and characterize genes that cooperate with Myc in an in vivo setting (195). Tetracycline inducible vectors expressing both Myc and Pim1 have demonstrated an interdependent role of these genes in allowing in vivo expansion of pre B cell lines, and this expansion is dependent upon continued expression of both genes (169). In a similar study, doxycycline inducible Myc and Bcl2l1 (Bcl-xL) pre BI cells were differentiated to immature B1 cells in vitro, and timed CpG stimulation generated either pre BII-like or mature B1-cell lines and IgM-secreting B1 cells. Introduction of these pre BI cells into Rag1-/- mice gives rise to plasmablast and plasma cell hyperplasia that was reversible when doxycycline was removed (179).
This model was also used to screen a library of cDNAs in pre B cell lines to identify genes that cooperate with Myc (178). Cells were selected in vitro in the absence of stromal cells and IL7 and in vivo by transplantation into Rag1-/- immunocompromised recipients. These screens identified multiple Myc cooperating genes including Exosc1, Rpl18a, Rpl35a Ndufs7, Cacybp, and Ptprcap. Overexpression of both a full length and truncated form of Exosc1 (a component of the RNA exosome) were validated as cooperating with Myc and the authors proposed the mechanism is a function of global inhibition of mRNA degradation.
Later stages of B cell development can also be reproduced in vitro. Propagating naïve splenic B cells on a layer of 3T3 fibroblasts expressing both CD40L and BAFF causes them to proliferate into germinal center B cell like cells called in-vitro-induced germinal center B (iGB) cells (190). When iGB cells were cultured with IL-4 they differentiated toward memory B cell precursors and IL-21 steered differentiation to a long-lived plasma cell phenotype. Retroviral transduction of these iGB cells with Myc and Bcl2 and transplantation into sublethally irradiated syngeneic recipients led to development of aggressive DLBCL like disease (172).
It is worth noting that in vivo library based screens of B cell biology need not be strictly limited to readouts of malignancy. Various models of B cell development and positive/negative selection of B cells are also relevant to malignancy. The IgMb-macroself mouse strain ubiquitously expresses a superantigen that causes deletion of immature B cells through reaction of surface IgM with a “self” superantigen. Transduction of bone marrow HSPCs with retroviruses encoding a miRNA expression library, and reconstitution of macroself recipients led to selection of cells expressing miR-148a, which was then verified to prevent deletion of self-reactive B cells (197).
The heavy chain of the B1-8hi transgenic strain has an increased affinity to the 4-Hydroxy-3-nitrophenylacetyl (NP) hapten after immunization and this model has been used to screen for modifiers of germinal center formation. Splenic B cells from B1-8hi transgenic mice were stimulated with anti-CD180 antibody, transduced with a library of shRNAs and transplanted into wild-type C57BL/6 mice (198). Recipients were then immunized with NP-chicken gamma globulin and alum to stimulate germinal center formation by the transplanted cells. Comparing the ratio of shRNA transduced B cells from germinal centers to non-germinal center cells revealed selection against Zdhhc2 shRNA, thereby demonstrating a role for this gene in productive germinal center formation. Zdhhc2 shRNA also inhibited development of iGB cells ex vivo. Although these two studies do not model tumor development, they demonstrate how specific stages of B cell development can be probed using adoptive transfer of primary B cells to perform reverse genetic screens.
Various site-specific nucleases such as zinc finger nucleases, transcription activator-like effector nucleases (TALENs) and Cas endonucleases have been adapted to targeted editing of mammalian genomes (199). Since the proof of concept of CRISPR-Cas editing in mammalian cells (200, 201) this system has gained popularity primarily because the target site specificity is determined by a short guide RNA (sgRNA) sequence that includes a ~20 nucleotide segment that is complementary to the target site sequence. This property makes CRISPR-Cas targeting constructs easier to use than zinc finger nuclease and TALEN constructs which express large open reading frames and require longer sequences of protein encoded DNA binding motifs to determine target site specificity. There can also be gene/locus specific differences between the technologies, with TALENs demonstrating greater activity in heterochromatin and Cas9 greater activity in euchromatin (202).
The short targeting sequence of CRISPR-Cas endonucleases facilitates functional genomics experiments with a similar throughput to shRNA vectors, with the additional benefit of editing target alleles permanently rather than changing gene expression by lowering mRNA levels. Like shRNAs, sgRNAs can vary in their efficiency and off target effects. Double stranded DNA breaks created by SpCas9 are primarily repaired by nonhomologous end joining (NHEJ) which is an error prone process. Consequently, some earlier versions of CRISPR-Cas editing have been shown to create imprecise lesions. Depending on the delivery system used, a subset of target sites may generate deletions of thousands of bases, complex rearrangements and crossover events (203–207). Nonetheless, technical innovations including novel Cas variants and fusions, improved sgRNA design, optimized delivery methods and allele replacement by homologous recombination have reduced off target effects, increased efficiency, improved specificity and even permitted mutation of single bases (208–225).
CRISPR-Cas genome editing has been adapted to modelling a variety of tumor types either through loss of function mutations, chromosomal engineering or gain of expression [reviewed in (226, 227)]. CRISPR-Cas models of B cell malignancy are rare although there is now an extensive literature on conditions that increase the efficiency of editing primary B cells or HSCs. Some researchers use cells from mice expressing Cas9 and transduce these with viruses expressing sgRNAs (228–233). In one of these studies, activated B cells from mice expressing a Cre inducible Cas9 IRES GFP from the Rosa26 locus were transduced with sgRNA viruses, achieving up to 80% knockout efficiency in cultured primary B cells (231). Another study transduced lipopolysaccharide (LPS) stimulated B cells expressing Cas9 from the Rosa26 locus with virus expressing sgRNA and a puromycin resistance marker, followed by selection with puromycin to enrich for edited cells (230). Others have used Cas9 expressing Lineage- Sca-1+ Kit+ (LSK) HSCs from bone marrow, transduced these with lentivirus sgRNA constructs, yielding edited cells of multiple hematopoietic lineages in reconstituted recipients including B cells (232). B cell specificity has also been achieved via the transduction of LSK HSCs bearing a Cre switchable Cas9 under control of a CD19-Cre transgene (233). The knockout efficiency of target genes in CD34+ HSPCs from human cord blood can also be improved by changing the order of delivery and the sgRNA structure, and these cells remain transplantable into immunocompromised mice after transduction (234).
Editing can also be made more efficient by delivering Cas9 as a protein, as an mRNA or as a ribonuclear protein of Cas9 precomplexed with sgRNA [reviewed in (217)]. Activating culture conditions also play a role in efficient transduction of B cells. Human peripheral blood B cells can be expanded by IL4 and CD40L cross linking and then electroporated. Electroporation with CD19 targeting sgRNAs in combination with either Cas9 RNA or Cas9 protein, can achieve knockout of CD19 in up to 70% of cells (235). Mouse splenic B cells activated using LPS (as a TLR4/CD180 agonist), or human peripheral blood B cells activated by anti-CD180 antibody can be edited by electroporated Cas9/sgRNA ribonuclear protein combined with adeno associated virus templates (236) or transfection of Cas9/sgRNA ribonuclear protein with ssDNA template (237). Human peripheral B cells cultured in IL-2, IL-10, IL-15, multimerized CD40 ligand and CpG oligodeoxynucleotide are efficiently edited by electroporation with CRISPR/Cas9 ribonuclear protein and adeno associated virus template constructs (238).
Although CRISPR-Cas has not been used to study lymphoma and leukemia as often as shRNAs, the technology has proven utility in validation of tumor suppressor candidates. One of the earliest proof of concept experiments used a vector expressing both Cas9 and an sgRNA against Trp53. When established Eμ-Myc p19ARF-/- lymphomas were transduced and transplanted into recipients, lymphoma growth in vivo was accelerated (138). Another model used a vector constitutively expressing Cas9 with a doxycycline inducible vector expressing Trp53 sgRNA in Eμ-Myc HSCs. This yielded rapid 100% penetrant disease where both Trp53 alleles could be found modified by a range of insertions, indels, deletions and large deletions (173). A similar model was subsequently developed using a doxycycline inducible Cas9 transgene in donor cells (183).
Three of the forward genetic studies mentioned previously in this review used similar Eμ-Myc fetal liver HSC transplant models to verify the tumor suppressors Bcor (using a vector expressing both Cas9 and sgRNA) (54, 108) and Rfx7 and Phip (combining a sgRNA vector with a Cas9 transgene) (48). Eμ-Myc HSCs have also been used to perform CRISPR-Cas9 functional genomic screens. In one study a library of 75 sgRNAs was designed against rarely mutated genes in Burkitt’s lymphoma and cloned into a Cas9 expressing vector. Pools of up to 5 sgRNAs were then transduced into Eμ-Myc fetal liver cells. Transduced cells were transplanted into irradiated recipients and when lymphomas developed, sgRNAs were quantitated by PCR and sequencing; genomic editing was then measured by T7 endonuclease I mismatch assays. Of the candidate sgRNAs identified by the screen Phip and Sp3 were functionally validated as tumor suppressors using the same assay (177).
Cas/sgRNA targeting has also been expanded to the regulation of transcription by fusing nuclease deficient SpCas9 to transcriptional domains that either activate transcription (referred to as CRISPRa) or inhibit transcription (referred to as CRISPRi) (239). CRISPRi constructs have modified in vivo growth of primary Eμ-Myc;p19ARF-/- lymphoma and have been used to conduct an in vivo screen in Bcr-Abl driven B ALL (134). From a 25 gene library of sgRNAs, Chk2 was identified as a modifier of temozolomide resistance.
CRISPR-Cas has also been used to engineer translocation events in somatic cells for mouse models of cancer. One of the earliest examples was induction of the EML4/ALK translocations in a lung cancer model (240) and since then CRISPR-Cas induced translocations have also been adapted to hematological malignancies, with a handful of models generating a subset of B lineage malignancies alongside other subtypes. One small study using mouse donor cells introduced SpCas9 and guide RNAs that translocate Npm1 to Alk in mouse fetal liver HSCs. Transplanting these into recipients lead to two cases of T lineage ALK+ anaplastic large-cell lymphoma and one case of anaplastic ALK+ large B-cell lymphoma (186). Two studies discussed in the next section have also used CRISPR-Cas editing to cause malignancies by introducing these same translocations in human HSCs (182, 185).
Human cord-blood derived HSCs have been used in a manner similar to mouse derived HSCs to generate adoptive transfer models of various hematologic malignancies. The t(11;19)(q23;p13.3) translocation produces the MLL/ENL (KMT2A/MLLT1) fusion and is found in acute leukemias of B, T and myeloid lineages. The t(9;11)(p21;q23) translocation produces the MLL/AF9 (KMT2A/MLLT3) fusion and is primarily observed in childhood AML but also in a subset of ALL in children below 1 year of age. Lineage-depleted human umbilical cord blood infected with MLL/ENL virus gave rise to B precursor ALL when injected into sublethally irradiated NOD/LtSz-scid/scid mice (164). An MLL/AF9 virus in the same model gives rise to a phenotypically similar B ALL and less frequently an AML like disease.
The lineage restriction of tumor initiating and tumor propagating cells was also investigated. The use of myeloid promoting suspension culture protocols skewed both transgenes toward a myeloid leukemia when injected. Transplantation of limiting cell numbers of primary tumors into secondary recipients indicated self-renewing leukemia cells were rare. Southern blots of retroviral vector integration and immunoglobulin heavy chain rearrangement suggested that leukemia initiating stem cells with germline immunoglobulin can differentiate into tumor propagating B lineage cell type having undergone immunoglobulin rearrangement. Lineage switching between B and myeloid lineages was also observed in culture.
More recently both translocations have been recreated in human HSCs using CRISPR-Cas. One model used CRISPR-Cas to induce the MLL/AF9 (KMT2A/MLLT3) translocation in cultured human cord blood HSCs. Transplanting these cells into sublethally irradiated NSG mice gave rise to B ALL, acute myeloid leukemia or mixed phenotype acute leukemia, as well as mice with a mixture of AML/ALL (185). Another study induced CRISPR-Cas mediated translocations between MLL(KMT2A) and ENL(MLLT1) in human HSCs. When these cells were introduced to NSG mice, myeloid disease was most commonly observed however secondary outgrowths of B ALL were also identified (182).
Other B cell malignancies have also been modelled. Transducing human CD133+ HSCs with MYCT58A mutant and BCL2 from a lentiviral vector gives rise to a “double hit” lymphoma like disease when transplanted into NOD.Cg-PrkdcscidIL2rgtm1Wjl/SzJ (NSG) mice (171). NSG mice with a humanized lymphoid compartment can be generated by injecting human fetal liver CD34+ hematopoietic progenitor cells into 1 Gy irradiated NSG recipients and these mice can serve as a model for virus induced primary effusion lymphoma caused by coinfection with Kaposi sarcoma-associated herpesvirus and Epstein-Barr virus (181).
Historically, developing models using new germline alleles is time consuming. Traditional transgenesis by pronuclear injection of fertilized zygotes leads to unpredictable integration sites of unkown copy number. Gene targetting by homologous recombination in ES cells requires constructs with long homology arms and extensive screening and characterization of clones prior to blastocyst injection. Founder animals must then be crossed to verify germline transmission and to combine multiple alleles in sufficient animals to form experimental cohorts.
Higher throughput strategies to generating these models and cohorts have now been developed using various combinations of CRISPR-Cas, recombinase mediated cassette exchange and routine derivation of new ES cell lines from mice bearing multiple germline alleles [reviewed in (241)]. Superficially, generating null alleles by zygote injection of CRISPR-Cas components is less laborious and time consuming than generating constructs for allele replacement, targeting of ES cells and blastocyst injection. In practice however, the methods used to generate mouse strains have diversified and now vary considerably in the time taken to generate targeting constructs and/or the reliability and efficiency of allele generation.
One of the main advantages of CRISPR-Cas is that the targeting of multiple alleles simultaneously can disrupt both copies of a gene and/or disrupt multiple genes a single founder animal. Simultaneous CRISPR-Cas mutation of both germline alleles of B lymphoma tumor suppressors Tet1 and Tet2 have been achieved as inactivating mutations (242) and even conditional loss of function alleles (243), potentially saving generations of breeding time in order to establish mutant cohorts. The efficiency of generating floxed alleles by injection of zygotes has been improved by the use of Cas9-avidin fusion with biotin tagged replacement template DNA (223). As mentioned previously some versions of CRISPR-Cas editing can generate imprecise lesions and off target effects (203–207). As such the speed gained from generating multiple CRISPR-Cas edited alleles simultaneously may be somewhat offset by the need for backcrossing and the need to repeat experiments using mice derived from independent founders. Where greater precision of allele generation is required (e.g. null alleles are lethal) the use of newly rederived ES cell lines from mouse strains carrying four or more alleles of interest offers an alternative for rapid generation of multiallelic models (244, 245).
CRISPR-Cas also potentially allows novel approaches to increase the throughput of gene editing of many alleles simultaneously (246). Multi sgRNA alleles can be generated by placing a lox71 site downstream of a U6 promoter and then downstream of this a concatemers of alternating sgRNA sequences with loxKR3 lox sites. LoxKR3 sites cannot combine with each other but can recombine with lox71. As such expression of Cre recombinase will lead to a single recombination event where only one of the sgRNA sequences chosen at random will be expressed in each cell. This allows Cre inducible sgRNA mosaicism that can be employed to study a range of genes in a single animal, or using germline switching the generation of many sgRNA strains from a single progenitor allele.
Studies cited in previous sections drive specific disease subtypes through combinations of germline alleles, viral vectors and the cell types being cultured and transplanted. Additional control can also be achieved though vectors or alleles that are regulated by Cre recombinase or ligands such as doxycycline (for tetracycline regulatable promoters) and 4-hydroxytamoxifen (to regulate nuclear localization of estrogen receptor fusions) (247). Early regulatable models of lymphoma include mouse strains expressing human MYC from tetracycline/doxycycline regulatable promoters (248, 249) which develop T, B and myeloid lineage malignancies that regress upon doxycycline induced repression of MYC expression. A similar doxycycline repressible BCL2 transgene when combined with the Eμ-Myc transgene gave rise to B lymphoblastic leukemia. These leukemias undergo remission upon repression of BCL2 expression (250). Studies such as these demonstrate the continued requirement of cancer initiating oncogenes in tumor maintenance and thereby the potential of these proteins as therapeutic targets.
Similar regulation of shRNAs has also been achieved using tetracycline/doxycycline repressible germline alleles. Constitutive knockdown of the Pax5 tumor suppressor by shRNA in combination with constitutively activated STAT5b causes a B ALL like disease (251). Repression of Pax5 shRNA expression by doxycycline treatment removes a block in differentiation and when these established leukemias are transplanted into Rag1-/- recipients, doxycycline treatment causes remission of disease (251). An analogous model of tumor suppressor restoration is a mouse strain that fuses the carboxy terminus of the endogenous Trp53 open reading frame to the estrogen receptor ligand binding domain. This fusion renders mice functionally Trp53 null until treated with 4-hydroxytamoxifen (252). Restoring Trp53 function in established Eμ-Myc lymphomas demonstrates the therapeutic potential for Trp53 restoration (253), which is analogous to the stabilization of Trp53 by treatment with Mdm2/Mdmx inhibitors.
Constructs that express open reading frames or shRNAs from tetracycline/doxycycline regulatable promoters have been transduced into B cell lines or hematopoietic stem cells used for transplant studies (128, 254–258). Placing the expressed sequences of a retrovirus between tandem loxP sequences allows controlled expression of an invertible open reading frame cassette that is limited by lineage specific Cre expression (259). Viral vectors expressing doxycycline regulatable Cas9 (260) or sgRNAs (173) have also been developed. Aside from the previously mentioned strains expressing Cas9 under control of Cre recombinase (228, 233), several mouse strains have also been generated that place Cas9 under the control of tetracycline regulatable promoters (183, 261, 262). In one of these studies, the Col1a1 locus was targetted in ES cells with a construct expressing multiple sgRNAs constitutively and a tetracycline/doxycyline regulatable Cas9 expression cassette. The resulting mice were crossed to mice with a reverse tetracycline transactivator allowing doxycycline controlled biallelic mutation of multiple genes in somatic cells using only two alleles (262).
CRISPRi gene regulation has similarly been placed under doxycycline control; Sp-dCas9 (nuclease deficient Cas9) is fused with the VP64 transactivation domain (i.e. four copies of the herpesvirus VP16 domain), the RELA p65 transactivation domain and the reverse tetracycline activation domain (263). This fusion has relatively low activity until cells are treated with doxycycline at which point the coexpression of sgRNA sequences causes activated expression of genes determined by the sgRNA target sequence.
Human populations are highly polymorphic and this, in combination with the mutator phenotype generated by many tumours, can make it difficult to subtract the background noise of unselected passenger mutations from the evidence for driver mutations. The growing volume of human cancer genome sequencing data has created an exponentially expanding number of potential validation experiments. This in turn creates a prioritization bottleneck deciding which combinations of mutations should be tested and the developmental stages in which they should be modeled.
Forward genetics in mice can aid this prioritization by cross species comparative genomics i.e. identifying the overlap of mutation spectra identified between human and mouse cohorts. There is a tradeoff between the extent to which forward genetic screens recapitulate human disease and the throughput of mutation detection. Exome sequencing arguably better mimics human tumor exomes, however insertional mutagenesis screens better represent the effects of translocation/copy number/non-coding mutations and are relatively lower cost and higher yield in terms of the number of driver mutations identified per mouse. This cost benefit analysis becomes relevant when considering the statistical limits of proving selection of non-coding point mutations from WGS of human cohorts. Insertional mutagenesis models also have greater statistical power to identify cooperating mutations, especially when sequencing coverage is sufficient to identify subclonal mutations.
Forward genetic screens in mice also have limitations. In some cases, the expected homologs of human drivers are not identified because functionally equivalent mutations that are specific to mice are identified instead. Another limitation of both forward and reverse genetic models is that the time frame for development of malignancies in mice is far shorter than observed in human lifespans. Consequently, predisposing mutations must be introduced and this in turn increases the chance of multiple clones arising independently in a single animal. Furthermore, Cre expressing strains used in some studies (such as Cd19 and Aicda) may replace the endogenous gene thereby altering the dosage of crucial B cell development genes.
Since the emphasis of this review is experimental approaches with higher throughput to identify and validate large numbers of candidate genes, for the most part we have not covered reverse genetic models that exclusively employ cohorts generated by germline alleles, which is a vast literature in and of itself. Rapid model prototyping using transplantation of virus transduced cells reduces the commitment of resources for initial testing of candidate genes. This allows more genes to be tested before robust germline models are designed and, in a handful of instances, allows for high throughput screens of dozens or even hundreds of candidate genes within a single cohort. This approach does, however, lack the precision of inducible targeted germline alleles and the dosage of transduced constructs can be highly variable and potentially reach expression levels not observed in human cells. Many cancer genes have physiological roles that are highly sensitive to dosage, and non-physiological expression levels from vectors may block or drive differentiation, sensitize cells to apoptosis or alter proliferation in a manner not typically observed as a result of somatic mutations.
Despite the number of labs working with B cell transplantation models, very few have published functional genomic screens where selection takes place in vivo. In vivo functional genomic screens are arguably a uniquely powerful application of adoptive transfer protocols, however in writing this review we only identified a limited number of studies using libraries of ORFs, shRNAs or gRNAs in B cell malignancy models. This would suggest that there is unrealized potential for more screens and/or that there are technical barriers to the reproducible identification of constructs selected in parallel. Functional genomic screens are a tradeoff between increasing the number of constructs screened whilst controlling for the stochasticity of selection of each construct relative to others in the library. B cell survival, migration, selection, differentiation and expansion are highly stochastic processes that may obscure selection in complex libraries. One approach to controlling for this stochasticity employed by several researchers is screening smaller pools of limited numbers of constructs to make selection of individual constructs easier to observe. Another approach to increasing confidence in the set of candidates identified in a screen is performing parallel screens using both shRNAs and sgRNAs to find genes that are selected by methodologies (264).
In aggregate, the studies discussed in this review suggest several lines of research that are ripe for further innovation. The literature of transplantation-based models is dominated by the use of Eμ-Myc fetal liver HSCs as donor cells (often in combination with mutation of Trp53 or the Cdkn2a locus). Transplantation models could easily be diversified through the use of different germline lesions or culturing and transplanting later stages of B cell development. Many of the studies we discuss have successfully cultured and transduced primary B cells with high efficiency, though currently only a handful have used transplantation of transduced B cells as the basis of lymphoma models.
Another promising direction for future development is the use of humanized models of human HSCs transplanted into immunodeficient mice. Immunodeficient host strains vary considerably in their engraftment potential, and although most are missing later stages of B cell development, they can recapitulate earlier stages of lymphoid development and have formed the basis of useful models for lymphoblastic leukemias. Immunodeficient mouse strains have various defects in the hematopoietic microenvironment and/or stromal cell proteins that are incompatible with their human targets and this can inhibit engraftment. For instance, IL2 receptor common gamma chain null mice (including NSG mice) lack lymphoid tissue inducer cells and innate lymphoid cells and do not express HLA molecules on thymic epithelia thereby preventing MHC restriction of T cells. Some models also develop graft-versus-host disease. There now is a growing literature of newer humanized models that can better recapitulate all aspects of human hematopoiesis (265–268). Newer strains compensate for these deficiencies or enhance engraftment by transgenic expression of human proteins (including SCF, GM-CSF, IL3, SIRPA, HLA, B2M, TPO and TSLP). Other deficiencies can be partly compensated by co-engraftment of human HSCs with human mesenchymal stem/stromal cells. Although this review has mostly emphasized the use of models dissecting the biology and genetics of B cell malignancies, there is likely unrealized potential for transplant models in preclinical studies of treatment efficacy and/or identification of novel therapeutic targets.
Modelling the antigenic context of B cell malignancies is also rarely addressed in the literature. The role of antigenic stimulation or tolerization by commensal flora is frequently overlooked and SPF or germ free conditions may not accurately represent the microbiome of human cancer patients, particularly where disease is influenced by a combination of normal gut flora and infectious agents, as is the case with H.pylori infection in MALT lymphoma (269).
Identification of human cancer drivers and the cell types that are most responsible for tumor initiation and propagation are critical foundations for the discovery of novel targeted therapies. The “post-genomic era” with seemingly limitless genomic data to interrogate has brought its own challenges of how to prioritize the use of this data. Modeling large numbers of mutations over the many stages of B cell development is a vast undertaking, particularly when differentiating between genes responsible for tumor initiation and maintenance and when testing potential targets across diverse genetic contexts of a single disease subtype. As such even the highest throughput experimental approaches discussed here can benefit from the parallel use of complementary methods.
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
JD and AU were supported by MRC programme grant MC_A652_5PZ20.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Andrej Alendar, Heather Verkade and Kira Behrens for critical reading of the manuscript.
1. Willis TG, Dyer MJS. The Role of Immunoglobulin Translocations in the Pathogenesis of B-Cell Malignancies. Blood (2000) 96:808–22. doi: 10.1182/blood.V96.3.808
2. Husby S, Grønbæk K. Mature Lymphoid Malignancies: Origin, Stem Cells, and Chronicity. Blood Adv (2017) 1:2444–55. doi: 10.1182/bloodadvances.2017008854
3. Busslinger M, Klix N, Pfeffer P, Graninger PG, Kozmik Z. Deregulation of PAX-5 by Translocation of the Emu Enhancer of the IgH Locus Adjacent to Two Alternative PAX-5 Promoters in a Diffuse Large-Cell Lymphoma. Proc Natl Acad Sci (1996) 93:6129–34. doi: 10.1073/pnas.93.12.6129
4. Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Küppers R, et al. Hypermutation of Multiple Proto-Oncogenes in B-Cell Diffuse Large-Cell Lymphomas. Nature (2001) 412:341–6. doi: 10.1038/35085588
5. Küppers R. Mechanisms of B-Cell Lymphoma Pathogenesis. Nat Rev Cancer (2005) 5:251–62. doi: 10.1038/nrc1589
6. Basso K, Dalla-Favera R. Germinal Centres and B Cell Lymphomagenesis. Nat Rev Immunol (2015) 15:172–84. doi: 10.1038/nri3814
7. Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, et al. AID Is Required for Germinal Center-Derived Lymphomagenesis. Nat Genet (2008) 40:108–12. doi: 10.1038/ng.2007.35
8. Khodabakhshi AH, Morin RD, Fejes AP, Mungall AJ, Mungall KL, Bolger-Munro M, et al. Recurrent Targets of Aberrant Somatic Hypermutation in Lymphoma. Oncotarget (2012) 3:1308–19. doi: 10.18632/oncotarget.653
9. Pasqualucci L, Guglielmino R, Malek SN, Novak U, Compagno M, Nanjangud G, et al. Aberrant Somatic Hypermutation Targets an Extensive Set of Genes in Diffuse Large B-Cell Lymphoma. Blood (2004) 104:1528–8. doi: 10.1182/blood.V104.11.1528.1528
10. Jäger U, Böcskor S, Le T, Mitterbauer G, Bolz I, Chott A, et al. Follicular Lymphomas’ BCL-2/IgH Junctions Contain Templated Nucleotide Insertions: Novel Insights Into the Mechanism of T(14;18) Translocation. Blood (2000) 95:3520–9. doi: 10.1182/blood.V95.11.3520
11. Limpens J, Stad R, Vos C, De Vlaam C, De Jong D, Van Ommen GJB, et al. Lymphoma-Associated Translocation T(14;18) in Blood B Cells of Normal Individuals. Blood (1995) 85:2528–36. doi: 10.1182/blood.v85.9.2528.bloodjournal8592528
12. Damm F, Mylonas E, Cosson A, Yoshida K, Della Valle V, Mouly E, et al. Acquired Initiating Mutations in Early Hematopoietic Cells of CLL Patients. Cancer Discovery (2014) 4:1088–101. doi: 10.1158/2159-8290.CD-14-0104
13. Gahn B, Schäfer C, Neef J, Troff C, Feuring-Buske M, Hiddemann W, et al. Detection of Trisomy 12 and Rb-Deletion in CD34+ Cells of Patients With B-Cell Chronic Lymphocytic Leukemia. Blood (1997) 89:4275–81. doi: 10.1182/blood.V89.12.4275
14. Kikushige Y, Ishikawa F, Miyamoto T, Shima T, Urata S, Yoshimoto G, et al. Self-Renewing Hematopoietic Stem Cell Is the Primary Target in Pathogenesis of Human Chronic Lymphocytic Leukemia. Cancer Cell (2011) 20:246–59. doi: 10.1016/j.ccr.2011.06.029
15. Montgomery ND, Mathews SP. Transformation in Low-Grade B-Cell Neoplasms. Surg Pathol Clin (2016) 9:79–92. doi: 10.1016/j.path.2015.09.004
16. Chan JKC, Ng CS, Isaacson PG. Relationship Between High-Grade Lymphoma and Low-Grade B-Cell Mucosa-Associated Lymphoid Tissue Lymphoma (MALToma) of the Stomach. Am J Pathol (1990) 136:1153–64.
17. Peng H, Du M, Diss TC, Isaacson PG, Pan L. Genetic Evidence for a Clonal Link Between Low and High-Grade Components in Gastric MALT B-Cell Lymphoma. Histopathology (1997) 30:425–9. doi: 10.1046/j.1365-2559.1997.5450786.x
18. Al-Mansour M, Connors JM, Gascoyne RD, Skinnider B, Savage KJ. Transformation to Aggressive Lymphoma in Nodular Lymphocyte-Predominant Hodgkin’s Lymphoma. J Clin Oncol (2010) 28:793–9. doi: 10.1200/JCO.2009.24.9516
19. Landgren O, Albitar M, Ma W, Abbasi F, Hayes RB, Ghia P, et al. Caporaso NE. B-Cell Clones as Early Markers for Chronic Lymphocytic Leukemia. N Engl J Med (2009) 360:659–67. doi: 10.1056/NEJMoa0806122
20. Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A Monoclonal Gammopathy Precedes Multiple Myeloma in Most Patients. Blood (2009) 113:5418–22. doi: 10.1182/blood-2008-12-195008
21. Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, Hayes RB, et al. Monoclonal Gammopathy of Undetermined Significance (MGUS) Consistently Precedes Multiple Myeloma: A Prospective Study. Blood (2009) 113:5412–7. doi: 10.1182/blood-2008-12-194241
22. van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, van der Gulden H, Berns A. Identification of Cooperating Oncogenes in E Mu-Myc Transgenic Mice by Provirus Tagging. Cell (1991) 65:737–52. doi: 10.1016/0092-8674(91)90382-9
23. Haupt Y, Alexander WS, Barri G, Peter Klinken S, Adams JM. Novel Zinc Finger Gene Implicated as Myc Collaborator by Retrovirally Accelerated Lymphomagenesis in Eμ-Myc Transgenic Mice. Cell (1991) 65:753–63. doi: 10.1016/0092-8674(91)90383-A
24. Shinto Y, Morimoto M, Katsumata M, Uchida A, Aozasa K, Okamoto M, et al. Moloney Murine Leukemia Virus Infection Accelerates Lymphomagenesis in E Mu-Bcl-2 Transgenic Mice. Oncogene (1995) 11:1729–36.
25. van der Lugt NM, Domen J, Verhoeven E, Linders K, van der Gulden H, Allen J, et al. Proviral Tagging in E Mu-Myc Transgenic Mice Lacking the Pim-1 Proto-Oncogene Leads to Compensatory Activation of Pim-2. EMBO J (1995) 14:2536–44. doi: 10.1002/j.1460-2075.1995.tb07251.x
26. Sheppard RD, Samant SA, Rosenberg M, Silver LM, Cole MD. Transgenic N-Myc Mouse Model for Indolent B Cell Lymphoma: Tumor Characterization and Analysis of Genetic Alterations in Spontaneous and Retrovirally Accelerated Tumors. Oncogene (1998) 17:2073–85. doi: 10.1038/sj.onc.1202125
27. Mikkers H, Allen J, Knipscheer P, Romeijn L, Hart A, Vink E, et al. High-Throughput Retroviral Tagging to Identify Components of Specific Signaling Pathways in Cancer. Nat Genet (2002) 32:153–9. doi: 10.1038/ng950
28. Dang J, Wei L, de Ridder J, Su X, Rust AG, Roberts KG, et al. PAX5 Is a Tumor Suppressor in Mouse Mutagenesis Models of Acute Lymphoblastic Leukemia. Blood (2015) 125:3609–17. doi: 10.1182/blood-2015-02-626127
29. Webster P, Dawes JC, Dewchand H, Takacs K, Iadarola B, Bolt BJ, et al. Subclonal Mutation Selection in Mouse Lymphomagenesis Identifies Known Cancer Loci and Suggests Novel Candidates. Nat Commun (2018) 9:2649. doi: 10.1038/s41467-018-05069-9
30. Martín-Hernández J, Sørensen AB, Pedersen FS. Murine Leukemia Virus Proviral Insertions Between the N-Ras and Unr Genes in B-Cell Lymphoma DNA Affect the Expression of N-Ras Only. J Virol (2001) 75:11907–12. doi: 10.1128/JVI.75.23.11907-11912.2001
31. Ma SL, Sørensen AB, Kunder S, Sørensen KD, Quintanilla-Martinez L, Morris DW, et al. The Icsbp Locus Is a Common Proviral Insertion Site in Mature B-Cell Lymphomas/Plasmacytomas Induced by Exogenous Murine Leukemia Virus. Virology (2006) 352:306–18. doi: 10.1016/j.virol.2006.05.006
32. Sørensen AB, Lund AH, Kunder S, Quintanilla-Martinez L, Schmidt J, Wang B, et al. Impairment of Alternative Splice Sites Defining a Novel Gammaretroviral Exon Within Gag Modifies the Oncogenic Properties of Akv Murine Leukemia Virus. Retrovirology (2007) 4:46. doi: 10.1186/1742-4690-4-46
33. Liu J, Sørensen A, Wang B, Wabl M, Nielsen A, Pedersen F. Identification of Novel Bach2 Transcripts and Protein Isoforms Through Tagging Analysis of Retroviral Integrations in B-Cell Lymphomas. BMC Mol Biol (2009) 10:2. doi: 10.1186/1471-2199-10-2
34. Pyrz M, Wang B, Wabl M, Pedersen FS. A Retroviral Mutagenesis Screen Identifies Cd74 as a Common Insertion Site in Murine B-Lymphomas and Reveals the Existence of a Novel IFNgamma-Inducible Cd74 Isoform. Mol Cancer (2010) 9:86. doi: 10.1186/1476-4598-9-86
35. Hartley JW, Chattopadhyay SK, Lander MR, Taddesse-Heath L, Naghashfar Z, Morse HC, et al. Accelerated Appearance of Multiple B Cell Lymphoma Types in NFS/N Mice Congenic for Ecotropic Murine Leukemia Viruses. Lab Investig (2000) 80:159–69. doi: 10.1038/labinvest.3780020
36. Suzuki T, Shen H, Akagi K, Morse HC, Malley JD, Naiman DQ, et al. New Genes Involved in Cancer Identified by Retroviral Tagging. Nat Genet (2002) 32:166–74. doi: 10.1038/ng949
37. Tsuruyama T, Nakamura T, Jin G, Ozeki M, Yamada Y, Hiai H. Constitutive Activation of Stat5a by Retrovirus Integration in Early Pre-B Lymphomas of SL/Kh Strain Mice. Proc Natl Acad Sci (2002) 99:8253–8. doi: 10.1073/pnas.112202899
38. Jin G, Tsuruyama T, Yamada Y, Hiai H. Svi3: A Provirus Common Integration Site in C-Myc in SL/Kh Pre-B Lymphomas. Cancer Sci (2003) 94:791–5. doi: 10.1111/j.1349-7006.2003.tb01520.x
39. Shin MS, Fredrickson TN, Hartley JW, Suzuki T, Agaki K, Morse HC. High-Throughput Retroviral Tagging for Identification of Genes Involved in Initiation and Progression of Mouse Splenic Marginal Zone Lymphomas. Cancer Res (2004) 64:4419–27. doi: 10.1158/0008-5472.CAN-03-3885
40. Suzuki T, Minehata K, Akagi K, Jenkins NA, Copeland NG. Tumor Suppressor Gene Identification Using Retroviral Insertional Mutagenesis in Blm-Deficient Mice. EMBO J (2006) 25:3422–31. doi: 10.1038/sj.emboj.7601215
41. Weiser KC, Liu B, Hansen GM, Skapura D, Hentges KE, Yarlagadda S, et al. Retroviral Insertions in the VISION Database Identify Molecular Pathways in Mouse Lymphoid Leukemia and Lymphoma. Mamm Genome (2007) 18:709–22. doi: 10.1007/s00335-007-9060-2
42. Tsuruyama T, Imai Y, Takeuchi H, Hiratsuka T, Maruyama Y, Kanaya K, et al. Dual Retrovirus Integration Tagging: Identification of New Signaling Molecules Fiz1 and Hipk2 That Are Involved in the IL-7 Signaling Pathway in B Lymphoblastic Lymphomas. J Leukoc Biol (2010) 88:107–16. doi: 10.1189/jlb.1109748
43. van der Weyden L, Giotopoulos G, Rust AG, Matheson LS, van Delft FW, Kong J, et al. Modeling the Evolution of ETV6-RUNX1–induced B-Cell Precursor Acute Lymphoblastic Leukemia in Mice. Blood (2011) 118:1041–51. doi: 10.1182/blood-2011-02-338848
44. Zanesi N, Balatti V, Riordan J, Burch A, Rizzotto L, Palamarchuk A, et al. A Sleeping Beauty Screen Reveals NF-kB Activation in CLL Mouse Model. Blood (2013) 121:4355–8. doi: 10.1182/blood-2013-02-486035
45. van der Weyden L, Giotopoulos G, Wong K, Rust AG, Robles-Espinoza CD, Osaki H, et al. Somatic Drivers of B-ALL in a Model of ETV6-RUNX1; Pax5(+/-) Leukemia. BMC Cancer (2015) 15:585. doi: 10.1186/s12885-015-1586-1
46. Heltemes-Harris LM, Larson JD, Starr TK, Hubbard GK, Sarver AL, Largaespada DA, et al. Sleeping Beauty Transposon Screen Identifies Signaling Modules That Cooperate With STAT5 Activation to Induce B-Cell Acute Lymphoblastic Leukemia. Oncogene (2016) 35:3454–64. doi: 10.1038/onc.2015.405
47. Rahrmann EP, Wolf NK, Otto GM, Heltemes-Harris L, Ramsey LB, Shu J, et al. Sleeping Beauty Screen Identifies RREB1 and Other Genetic Drivers in Human B-Cell Lymphoma. Mol Cancer Res (2019) 17:567–82. doi: 10.1158/1541-7786.MCR-18-0582
48. Weber J, de la Rosa J, Grove CS, Schick M, Rad L, Baranov O, et al. PiggyBac Transposon Tools for Recessive Screening Identify B-Cell Lymphoma Drivers in Mice. Nat Commun (2019) 10:1415. doi: 10.1038/s41467-019-09180-3
49. Sander S, Calado DP, Srinivasan L, Köchert K, Zhang B, Rosolowski M, et al. Synergy Between PI3K Signaling and MYC in Burkitt Lymphomagenesis. Cancer Cell (2012) 22:167–79. doi: 10.1016/j.ccr.2012.06.012
50. Sungalee S, Mamessier E, Morgado E, Grégoire E, Brohawn PZ, Morehouse CA, et al. Germinal Center Reentries of BCL2-Overexpressing B Cells Drive Follicular Lymphoma Progression. J Clin Invest (2014) 124:5337–51. doi: 10.1172/JCI72415
51. Gough SM, Goldberg L, Pineda M, Walker RL, Zhu YJ, Bilke S, et al. Spontaneous Mutations of Bcor and Jak1/2 Genes Lead to an Aggressive Leukemia of B-1 Progenitor B Cells. Blood (2014) 124:3573–3. doi: 10.1182/blood.V124.21.3573.3573
52. Martín-Lorenzo A, Hauer J, Vicente-Dueñas C, Auer F, González-Herrero I, García-Ramírez I, et al. Infection Exposure Is a Causal Factor in B-Cell Precursor Acute Lymphoblastic Leukemia as a Result of Pax5 -Inherited Susceptibility. Cancer Discovery (2015) 5:1328–43. doi: 10.1158/2159-8290.CD-15-0892
53. Duque-Afonso J, Feng J, Scherer F, Lin C-H, Wong SHK, Wang Z, et al. Comparative Genomics Reveals Multistep Pathogenesis of E2A-PBX1 Acute Lymphoblastic Leukemia. J Clin Invest (2015) 125:3667–80. doi: 10.1172/JCI81158
54. Lefebure M, Tothill RW, Kruse E, Hawkins ED, Shortt J, Matthews GM, et al. Genomic Characterisation of Eμ-Myc Mouse Lymphomas Identifies Bcor as a Myc Co-Operative Tumour-Suppressor Gene. Nat Commun (2017) 8:14581. doi: 10.1038/ncomms14581
55. Rodríguez-Hernández G, Hauer J, Martín-Lorenzo A, Schäfer D, Bartenhagen C, García-Ramírez I, et al. Infection Exposure Promotes ETV6-RUNX1 Precursor B-Cell Leukemia via Impaired H3K4 Demethylases. Cancer Res (2017) 77:4365–77. doi: 10.1158/0008-5472.CAN-17-0701
56. Gough SM, Goldberg L, Pineda M, Walker RL, Zhu YJ, Bilke S, et al. Progenitor B-1 B-Cell Acute Lymphoblastic Leukemia Is Associated With Collaborative Mutations in 3 Critical Pathways. Blood Adv (2017) 1:1749–59. doi: 10.1182/bloodadvances.2017009837
57. Mouly E, Ghamlouch H, Della-Valle V, Scourzic L, Quivoron C, Roos-Weil D, et al. B-Cell Tumor Development in Tet2-Deficient Mice. Blood Adv (2018) 2:703–14. doi: 10.1182/bloodadvances.2017014118
58. Jamrog L, Chemin G, Fregona V, Coster L, Pasquet M, Oudinet C, et al. PAX5-ELN Oncoprotein Promotes Multistep B-Cell Acute Lymphoblastic Leukemia in Mice. Proc Natl Acad Sci (2018) 115:10357–62. doi: 10.1073/pnas.1721678115
59. Zaborsky N, Gassner FJ, Höpner JP, Schubert M, Hebenstreit D, Stark R, et al. Exome Sequencing of the TCL1 Mouse Model for CLL Reveals Genetic Heterogeneity and Dynamics During Disease Development. Leukemia (2019) 33:957–68. doi: 10.1038/s41375-018-0260-4
60. Flümann R, Rehkämper T, Nieper P, Pfeiffer P, Holzem A, Klein S, et al. An Autochthonous Mouse Model of Myd88 - and BCL2 -Driven Diffuse Large B-Cell Lymphoma Reveals Actionable Molecular Vulnerabilities. Blood Cancer Discovery (2021) 2:70–91. doi: 10.1158/2643-3230.BCD-19-0059
61. Vicente-Dueñas C, Janssen S, Oldenburg M, Auer F, González-Herrero I, Casado-García A, et al. An Intact Gut Microbiome Protects Genetically Predisposed Mice Against Leukemia. Blood (2020) 136:2003–17. doi: 10.1182/blood.2019004381
62. Kool J, Berns A. High-Throughput Insertional Mutagenesis Screens in Mice to Identify Oncogenic Networks. Nat Rev Cancer (2009) 9:389–99. doi: 10.1038/nrc2647
63. Fan H. Leukemogenesis by Moloney Murine Leukemia Virus: A Multistep Process. Trends Microbiol (1997) 5:74–82. doi: 10.1016/S0966-842X(96)10076-7
64. Hanecak R, Pattengale PK, Fan H. Addition of Substitution of Simian Virus 40 Enhancer Sequences Into the Moloney Murine Leukemia Virus (M-MuLV) Long Terminal Repeat Yields Infectious M-MuLV With Altered Biological Properties. J Virol (1988) 62:2427–36. doi: 10.1128/JVI.62.7.2427-2436.1988
65. Lovmand J, Sorensen AB, Schmidt J, Ostergaard M, Luz A, Pedersen FS. B-Cell Lymphoma Induction by Akv Murine Leukemia Viruses Harboring One or Both Copies of the Tandem Repeat in the U3 Enhancer. J Virol (1998) 72:5745–56. doi: 10.1128/JVI.72.7.5745-5756.1998
66. Sørensen KD, Kunder S, Quintanilla-Martinez L, Sørensen J, Schmidt J, Pedersen FS. Enhancer Mutations of Akv Murine Leukemia Virus Inhibit the Induction of Mature B-Cell Lymphomas and Shift Disease Specificity Towards the More Differentiated Plasma Cell Stage. Virology (2007) 362:179–91. doi: 10.1016/j.virol.2006.12.016
67. Mucenski ML, Bedigian HG, Shull MM, Copeland NG, Jenkins NA. Comparative Molecular Genetic Analysis of Lymphomas From Six Inbred Mouse Strains. J Virol (1988) 62:839–46. doi: 10.1128/jvi.62.3.839-846.1988
68. Yamada Y, Matsushiro H, Ogawa MS, Okamoto K, Nakakuki Y, Toyokuni S, et al. Genetic Predisposition to Pre-B Lymphomas in SL/Kh Strain Mice. Cancer Res (1994) 54:403–7.
69. Gilbert DJ, Neumann PE, Taylor BA, Jenkins NA, Copeland NG. Susceptibility of AKXD Recombinant Inbred Mouse Strains to Lymphomas. J Virol (1993) 67:2083–90. doi: 10.1128/jvi.67.4.2083-2090.1993
70. Fredrickson TN, Morse HC, Rowe WP. Spontaneous Tumors of NFS Mice Congenic for Ecotropic Murine Leukemia Virus Induction Loci2. JNCI J Natl Cancer Inst (1984) 73:521–4. doi: 10.1093/jnci/73.2.521
71. Fredrickson TN, Morse HC, Yetter RA, Rowe WP, Hartley JW, Pattengale PK. Multiparameter Analyses of Spontaneous Nonthymic Lymphomas Occurring in NFS/N Mice Congenic for Ecotropic Murine Leukemia Viruses. Am J Pathol (1985) 121:349–60.
72. Beckmann PJ, Largaespada DA. Transposon Insertion Mutagenesis in Mice for Modeling Human Cancers: Critical Insights Gained and New Opportunities. Int J Mol Sci (2020) 21:1172. doi: 10.3390/ijms21031172
73. Kawakami K, Largaespada DA, Ivics Z. Transposons As Tools for Functional Genomics in Vertebrate Models. Trends Genet (2017) 33:784–801. doi: 10.1016/j.tig.2017.07.006
74. O’Donnell KA. Advances in Functional Genetic Screening With Transposons and CRISPR/Cas9 to Illuminate Cancer Biology. Curr Opin Genet Dev (2018) 49:85–94. doi: 10.1016/j.gde.2018.03.006
75. Kuiper RP, Schoenmakers EFPM, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, et al. High-Resolution Genomic Profiling of Childhood ALL Reveals Novel Recurrent Genetic Lesions Affecting Pathways Involved in Lymphocyte Differentiation and Cell Cycle Progression. Leukemia (2007) 21:1258–66. doi: 10.1038/sj.leu.2404691
76. Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-Wide Analysis of Genetic Alterations in Acute Lymphoblastic Leukaemia. Nature (2007) 446:758–64. doi: 10.1038/nature05690
77. Mansouri L, Papakonstantinou N, Ntoufa S, Stamatopoulos K, Rosenquist R. NF-κb Activation in Chronic Lymphocytic Leukemia: A Point of Convergence of External Triggers and Intrinsic Lesions. Semin Cancer Biol (2016) 39:40–8. doi: 10.1016/j.semcancer.2016.07.005
78. Zapata JM, Krajewska M, Morse HC, Choi Y, Reed JC. TNF Receptor-Associated Factor (TRAF) Domain and Bcl-2 Cooperate to Induce Small B Cell Lymphoma/Chronic Lymphocytic Leukemia in Transgenic Mice. Proc Natl Acad Sci (2004) 101:16600–5. doi: 10.1073/pnas.0407541101
79. Pérez-Chacón G, Llobet D, Pardo C, Pindado J, Choi Y, Reed JC, et al. TNFR-Associated Factor 2 Deficiency in B Lymphocytes Predisposes to Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma in Mice. J Immunol (2012) 189:1053–61. doi: 10.4049/jimmunol.1200814
80. Pérez-Chacón G, Zapata JM. Mouse Models of Chronic Lymphocytic Leukemia. In: Oppezzo P (Ed.) Chronic Lymphocytic Leukemia. London, United Kingdom: IntechOpen Limited (2012)
81. Lenz G, Wright GW, Emre NCT, Kohlhammer H, Dave SS, Davis RE, et al. Molecular Subtypes of Diffuse Large B-Cell Lymphoma Arise by Distinct Genetic Pathways. Proc Natl Acad Sci (2008) 105:13520–5. doi: 10.1073/pnas.0804295105
82. Pfeifer M, Lenz G. PI3K/AKT Addiction in Subsets of Diffuse Large B-Cell Lymphoma. Cell Cycle (2013) 12:3347–8. doi: 10.4161/cc.26575
83. Bronner IF, Otto TD, Zhang M, Udenze K, Wang C, Quail MA, et al. Quantitative Insertion-Site Sequencing (QIseq) for High Throughput Phenotyping of Transposon Mutants. Genome Res (2016) 26:980–9. doi: 10.1101/gr.200279.115
84. Arnold C, Hodgson IJ. Vectorette PCR: A Novel Approach to Genomic Walking. Genome Res (1991) 1:39–42. doi: 10.1101/gr.1.1.39
85. Devon RS, Porteous DJ, Brookes AJ. Splinkerettes—improved Vectorettes for Greater Efficiency in PCR Walking. Nucleic Acids Res (1995) 23:1644–5. doi: 10.1093/nar/23.9.1644
86. Hui EKWW, Wang PC, Lo SJ. Strategies for Cloning Unknown Cellular Flanking DNA Sequences From Foreign Integrants. Cell Mol Life Sci (1998) 54:1403–11. doi: 10.1007/s000180050262
87. Koudijs MJ, Klijn C, van der Weyden L, Kool J, ten Hoeve J, Sie D, et al. High-Throughput Semiquantitative Analysis of Insertional Mutations in Heterogeneous Tumors. Genome Res (2011) 21:2181–9. doi: 10.1101/gr.112763.110
88. Sherman E, Nobles C, Berry CC, Six E, Wu Y, Dryga A, et al. INSPIIRED: A Pipeline for Quantitative Analysis of Sites of New DNA Integration in Cellular Genomes. Mol Ther Methods Clin Dev (2017) 4:39–49. doi: 10.1016/j.omtm.2016.11.002
89. Berry CC, Gillet NA, Melamed A, Gormley N, Bangham CRM, Bushman FD. Estimating Abundances of Retroviral Insertion Sites From DNA Fragment Length Data. Bioinformatics (2012) 28:755–62. doi: 10.1093/bioinformatics/bts004
90. Mann KM, Newberg JY, Black MA, Jones DJ, Amaya-Manzanares F, Guzman-Rojas L, et al. Analyzing Tumor Heterogeneity and Driver Genes in Single Myeloid Leukemia Cells With SBCapSeq. Nat Biotechnol (2016) 34:962–72. doi: 10.1038/nbt.3637
91. Mann M, Mann KM, Guzman-Rojas L, Amaya-Manzanares F, Jones DJ, Newberg JY, et al. SBCapSeq Protocol: A Method for Selective Cloning of Sleeping Beauty Transposon Insertions Using Liquid Capture Hybridization and Ion Torrent Semiconductor Sequencing. Protoc Exch (2016). doi: 10.1038/protex.2016.053
92. Dawes JC, Webster P, Iadarola B, Garcia-Diaz C, Dore M, Bolt BJ, et al. LUMI-PCR: An Illumina Platform Ligation-Mediated PCR Protocol for Integration Site Cloning, Provides Molecular Quantitation of Integration Sites. Mob DNA (2020) 11:7. doi: 10.1186/s13100-020-0201-4
93. Huser CA, Gilroy KL, de Ridder J, Kilbey A, Borland G, Mackay N, et al. Insertional Mutagenesis and Deep Profiling Reveals Gene Hierarchies and a Myc/p53-Dependent Bottleneck in Lymphomagenesis. PloS Genet (2014) 10:e1004167. doi: 10.1371/journal.pgen.1004167
94. van Galen P, Hovestadt V, Wadsworth MH II, Hughes TK, Griffin GK, Battaglia S, et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell (2019) 176:1265–1281.e24. doi: 10.1016/j.cell.2019.01.031
95. Wu J, Xiao Y, Sun J, Sun H, Chen H, Zhu Y, et al. A Single-Cell Survey of Cellular Hierarchy in Acute Myeloid Leukemia. J Hematol Oncol (2020) 13:128. doi: 10.1186/s13045-020-00941-y
96. Petti AA, Williams SR, Miller CA, Fiddes IT, Srivatsan SN, Chen DY, et al. A General Approach for Detecting Expressed Mutations in AML Cells Using Single Cell RNA-Sequencing. Nat Commun (2019) 10:3660. doi: 10.1038/s41467-019-11591-1
97. Miles LA, Bowman RL, Merlinsky TR, Csete IS, Ooi AT, Durruthy-Durruthy R, et al. Single-Cell Mutation Analysis of Clonal Evolution in Myeloid Malignancies. Nature (2020) 587:477–82. doi: 10.1038/s41586-020-2864-x
98. de Ridder J, Uren A, Kool J, Reinders M, Wessels L. Detecting Statistically Significant Common Insertion Sites in Retroviral Insertional Mutagenesis Screens. PloS Comput Biol (2006) 2:e166. doi: 10.1371/journal.pcbi.0020166
99. Sarver AL, Erdman J, Starr T, Largaespada DA, Silverstein KAT. TAPDANCE: An Automated Tool to Identify and Annotate Transposon Insertion CISs and Associations Between CISs From Next Generation Sequence Data. BMC Bioinf (2012) 13:134. doi: 10.1186/1471-2105-13-154
100. de Jong J, de Ridder J, van der Weyden L, Sun N, van Uitert M, Berns A, et al. Computational Identification of Insertional Mutagenesis Targets for Cancer Gene Discovery. Nucleic Acids Res (2011) 39:e105–5. doi: 10.1093/nar/gkr447
101. de Jong J, Akhtar W, Badhai J, Rust AG, Rad R, Hilkens J, et al. Chromatin Landscapes of Retroviral and Transposon Integration Profiles. PloS Genet (2014) 10:e1004250. doi: 10.1371/journal.pgen.1004250
102. Uren AG, Kool J, Matentzoglu K, de Ridder J, Mattison J, van Uitert M, et al. Large-Scale Mutagenesis in P19arf- and P53-Deficient Mice Identifies Cancer Genes and Their Collaborative Networks. Cell (2008) 133:727–41. doi: 10.1016/j.cell.2008.03.021
103. Kool J, Uren AG, Martins CP, Sie D, de Ridder J, Turner G, et al. Insertional Mutagenesis in Mice Deficient for p15Ink4b, p16Ink4a, p21Cip1, and p27Kip1 Reveals Cancer Gene Interactions and Correlations With Tumor Phenotypes. Cancer Res (2010) 70:520–31. doi: 10.1158/0008-5472.CAN-09-2736
104. Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event During Human Lymphomagenesis. Cancer Cell (2011) 20:25–38. doi: 10.1016/j.ccr.2011.06.003
105. Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Béguelin W, Fontán L, et al. TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-Cell Lymphomagenesis. Cancer Discovery (2018) 8:1632–53. doi: 10.1158/2159-8290.CD-18-0657
106. Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, Yu Y, et al. TET1 Is a Tumor Suppressor of Hematopoietic Malignancy. Nat Immunol (2015) 16:653–62. doi: 10.1038/ni.3148
107. Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones SSS, et al. Mutations in Linker Histone Genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A Underlying the Pathogenesis of Follicular Lymphoma. Blood (2014) 123:1487–98. doi: 10.1182/blood-2013-05-500264
108. Yin M, Chung YJ, Lindsley RC, Walker RL, Zhu YJ, Ebert BL, et al. Engineered Bcor Mutations Lead to Acute Leukemia of Progenitor B-1 Lymphocyte Origin in a Sensitized Background. Blood (2019) 133:2610–4. doi: 10.1182/blood.2018864173
109. Familiades J, Bousquet M, Lafage-Pochitaloff M, Béné M-C, Beldjord K, De Vos J, et al. PAX5 Mutations Occur Frequently in Adult B-Cell Progenitor Acute Lymphoblastic Leukemia and PAX5 Haploinsufficiency Is Associated With BCR-ABL1 and TCF3-PBX1 Fusion Genes: A GRAALL Study. Leukemia (2009) 23:1989–98. doi: 10.1038/leu.2009.135
110. Coyaud E, Struski S, Prade N, Familiades J, Eichner R, Quelen C, et al. Wide Diversity of PAX5 Alterations in B-ALL: A Groupe Francophone De Cytogénétique Hématologique Study. Blood (2010) 115:3089–97. doi: 10.1182/blood-2009-07-234229
111. Bastian L, Schroeder MP, Eckert C, Schlee C, Tanchez JO, Kämpf S, et al. PAX5 Biallelic Genomic Alterations Define a Novel Subgroup of B-Cell Precursor Acute Lymphoblastic Leukemia. Leukemia (2019) 33:1895–909. doi: 10.1038/s41375-019-0430-z
112. Shah S, Schrader KA, Waanders E, Timms AE, Vijai J, Miething C, et al. A Recurrent Germline PAX5 Mutation Confers Susceptibility to Pre-B Cell Acute Lymphoblastic Leukemia. Nat Genet (2013) 45:1226–31. doi: 10.1038/ng.2754
113. Auer F, Rüschendorf F, Gombert M, Husemann P, Ginzel S, Izraeli S, et al. Inherited Susceptibility to Pre B-ALL Caused by Germline Transmission of PAX5 C.547G<A. Leukemia (2014) 28:1136–8. doi: 10.1038/leu.2013.363
114. Yazdanparast S, Khatami SR, Galehdari H, Jaseb K. One Missense Mutation in Exon 2 of the PAX5 Gene in Iran. Genet Mol Res (2015) 14:17768–75. doi: 10.4238/2015.December.22.1
115. Duployez N, Jamrog LA, Fregona V, Hamelle C, Fenwarth L, Lejeune S, et al. Germline PAX5 Mutation Predisposes to Familial B-Cell Precursor Acute Lymphoblastic Leukemia. Blood (2021) 137:1424–8. doi: 10.1182/blood.2020005756
116. Smeenk L, Fischer M, Jurado S, Jaritz M, Azaryan A, Werner B, et al. Molecular Role of the PAX 5- ETV 6 Oncoprotein in Promoting B-Cell Acute Lymphoblastic Leukemia. EMBO J (2017) 36:718–35. doi: 10.15252/embj.201695495
117. Tarantul V. Transgenic Mice as an In Vivo Model of Lymphomagenesis. Int Rev Cytol (2004) 236:123–80. doi: 10.1016/S0074-7696(04)36004-3
118. You MJ. Mouse Models of Lymphoma and Lymphoid Leukemia. In: Neoplastic Hematopathology. Totowa, NJ: Humana Press. (2009) p. 583–96. doi: 10.1007/978-1-60761-384-8_36
119. Kohnken R, Porcu P, Mishra A. Overview of the Use of Murine Models in Leukemia and Lymphoma Research. Front Oncol (2017) 7:22. doi: 10.3389/fonc.2017.00022
120. Mossadegh-Keller N, Brisou G, Beyou A, Nadel B, Roulland S. Human B Lymphomas Reveal Their Secrets Through Genetic Mouse Models. Front Immunol (2021) 12:683597. doi: 10.3389/fimmu.2021.683597
121. Pasqualucci L, Meyer SN, Koul S. Mouse Models in the Study of Germinal Center Derived B-Cell Malignancies. Front Immunol (2021) 12:710711. doi: 10.3389/fimmu.2021.710711
122. Donnou S, Galand C, Touitou V, Sautès-Fridman C, Fabry Z, Fisson S. Murine Models of B-Cell Lymphomas: Promising Tools for Designing Cancer Therapies. Adv Hematol (2012) 2012:1–13. doi: 10.1155/2012/701704
123. Schmitt CA, Rosenthal CT, Lowe SW. Genetic Analysis of Chemoresistance in Primary Murine Lymphomas. Nat Med (2000) 6:1029–35. doi: 10.1038/79542
124. Schmitt CA, Lowe SW. Bcl-2 Mediates Chemoresistance in Matched Pairs of Primary Eμ-Myc Lymphomas In Vivo. Blood Cells Mol Dis (2001) 27:206–16. doi: 10.1006/bcmd.2000.0372
125. Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A Senescence Program Controlled by P53 and P16ink4a Contributes to the Outcome of Cancer Therapy. Cell (2002) 109:335–46. doi: 10.1016/s0092-8674(02)00734-1
126. Refaeli Y, Young RM, Turner BC, Duda J, Field KA, Bishop JM. The B Cell Antigen Receptor and Overexpression of MYC Can Cooperate in the Genesis of B Cell Lymphomas. PloS Biol (2008) 6:e152. doi: 10.1371/journal.pbio.0060152
127. Young RM, Hardy IR, Clarke RL, Lundy N, Pine P, Turner BC, et al. Mouse Models of Non-Hodgkin Lymphoma Reveal Syk as an Important Therapeutic Target. Blood (2009) 113:2508–16. doi: 10.1182/blood-2008-05-158618
128. Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, et al. Toolkit for Evaluating Genes Required for Proliferation and Survival Using Tetracycline-Regulated RNAi. Nat Biotechnol (2011) 29:79–83. doi: 10.1038/nbt.1720
129. Cao Z, Ding B-S, Guo P, Lee SB, Butler JM, Casey SC, et al. Angiocrine Factors Deployed by Tumor Vascular Niche Induce B Cell Lymphoma Invasiveness and Chemoresistance. Cancer Cell (2014) 25:350–65. doi: 10.1016/j.ccr.2014.02.005
130. Hoellein A, Fallahi M, Schoeffmann S, Steidle S, Schaub FX, Rudelius M, et al. Myc-Induced SUMOylation Is a Therapeutic Vulnerability for B-Cell Lymphoma. Blood (2014) 124:2081–90. doi: 10.1182/blood-2014-06-584524
131. Matthews GM, Mehdipour P, Cluse LA, Falkenberg KJ, Wang E, Roth M, et al. Functional-Genetic Dissection of HDAC Dependencies in Mouse Lymphoid and Myeloid Malignancies. Blood (2015) 126:2392–403. doi: 10.1182/blood-2015-03-632984
132. Duque-Afonso J, Lin C-H, Han K, Wei MC, Feng J, Kurzer JH, et al. E2A-PBX1 Remodels Oncogenic Signaling Networks in B-Cell Precursor Acute Lymphoid Leukemia. Cancer Res (2016) 76:6937–49. doi: 10.1158/0008-5472.CAN-16-1899
133. Li X, Zhang Y, Zheng L, Liu M, Chen CD, Jiang H. UTX Is an Escape From X-Inactivation Tumor-Suppressor in B Cell Lymphoma. Nat Commun (2018) 9:2720. doi: 10.1038/s41467-018-05084-w
134. Braun CJ, Bruno PM, Horlbeck MA, Gilbert LA, Weissman JS, Hemann MT. Versatile In Vivo Regulation of Tumor Phenotypes by Dcas9-Mediated Transcriptional Perturbation. Proc Natl Acad Sci USA (2016) 113:E3892–900. doi: 10.1073/pnas.1600582113
135. Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM, Lowe SW. Dissecting P53 Tumor Suppressor Functions In Vivo. Cancer Cell (2002) 1:289–98. doi: 10.1016/S1535-6108(02)00047-8
136. Meacham CE, Ho EE, Dubrovsky E, Gertler FB, Hemann MT. In Vivo RNAi Screening Identifies Regulators of Actin Dynamics as Key Determinants of Lymphoma Progression. Nat Genet (2009) 41:1133–7. doi: 10.1038/ng.451
137. Mu P, Han Y-C, Betel D, Yao E, Squatrito M, Ogrodowski P, et al. Genetic Dissection of the miR-17 92 Cluster of microRNAs in Myc-Induced B-Cell Lymphomas. Genes Dev (2009) 23:2806–11. doi: 10.1101/gad.1872909
138. Malina A, Mills JR, Cencic R, Yan Y, Fraser J, Schippers LM, et al. Repurposing CRISPR/Cas9 for In Situ Functional Assays. Genes Dev (2013) 27:2602–14. doi: 10.1101/gad.227132.113
139. Eva A, Pierce JH, Aaronson SA. Interactions of Retroviral and Cellular Transforming Genes With Hematopoietic Cells. Ann N Y Acad Sci (1987) 511:148–70. doi: 10.1111/j.1749-6632.1987.tb36245.x
140. Pierce JH, Eva A, Aaronson SA. Interactions of Oncogenes With Haematopoietic Cells. Clin Haematol (1986) 15:573–96. doi: 10.1016/S0308-2261(18)30003-1
141. Kitayama H, Tsujimura T, Matsumura I, Oritani K, Ikeda H, Ishikawa J, et al. Neoplastic Transformation of Normal Hematopoietic Cells by Constitutively Activating Mutations of C-Kit Receptor Tyrosine Kinase. Blood (1996) 88:995–1004. doi: 10.1182/blood.V88.3.995.995
142. Hawley TS, Fong AZC, Griesser H, Lyman SD, Hawley RG. Leukemic Predisposition of Mice Transplanted With Gene-Modified Hematopoietic Precursors Expressing Flt3 Ligand. Blood (1998) 92:2003–11. doi: 10.1182/blood.V92.6.2003.418k11_2003_2011
143. Schwartz RC, Stanton LW, Riley SC, Marcu KB, Witte ON. Synergism of V-Myc and V-Ha-Ras in the In Vitro Neoplastic Progression of Murine Lymphoid Cells. Mol Cell Biol (1986) 6:3221–31. doi: 10.1128/mcb.6.9.3221-3231.1986
144. Heard JM, Roussel MF, Rettenmier CW, Sherr CJ. Multilineage Hematopoietic Disorders Induced by Transplantation of Bone Marrow Cells Expressing the V-Fms Oncogene. Cell (1987) 51:663–73. doi: 10.1016/0092-8674(87)90135-8
145. Kelliher MA, McLaughlin J, Witte ON, Rosenberg N. Induction of a Chronic Myelogenous Leukemia-Like Syndrome in Mice With V-Abl and BCR/ABL. Proc Natl Acad Sci (1990) 87:6649–53. doi: 10.1073/pnas.87.22.9072c
146. Hawley RG, Fong AZC, Ngan BY, Hawley TS. Hematopoietic Transforming Potential of Activated Ras in Chimeric Mice. Oncogene (1995) 11:1113–23.
147. Thome KC, Radfar A, Rosenberg N. Mutation of Tp53 Contributes to the Malignant Phenotype of Abelson Virus-Transformed Lymphoid Cells. J Virol (1997) 71:8149–56. doi: 10.1128/JVI.71.11.8149-8156.1997
148. McLaughlin J, Chianese E, Witte ON. In Vitro Transformation of Immature Hematopoietic Cells by the P210 BCR/ABL Oncogene Product of the Philadelphia Chromosome. Proc Natl Acad Sci (1987) 84:6558–62. doi: 10.1073/pnas.84.18.6558
149. Daley G, Van Etten R, Baltimore D. Induction of Chronic Myelogenous Leukemia in Mice by the P210bcr/abl Gene of the Philadelphia Chromosome. Sci (80-) (1990) 247:824–30. doi: 10.1126/science.2406902
150. Elefanty AG, Hariharan IK, Cory S. Bcr-Abl, the Hallmark of Chronic Myeloid Leukaemia in Man, Induces Multiple Haemopoietic Neoplasms in Mice. EMBO J (1990) 9:1069–78. doi: 10.1002/j.1460-2075.1990.tb08212.x
151. Kuefer MU, Look AT, Pulford K, Behm FG, Pattengale PK, Mason DY, et al. Retrovirus-Mediated Gene Transfer of NPM-ALK Causes Lymphoid Malignancy in Mice. Blood (1997) 90:2901–10. doi: 10.1182/blood.V90.8.2901
152. Li S, Ilaria RL, Million RP, Daley GQ, Van Etten RA. The P190, P210, and P230 Forms of the BCR/ABL Oncogene Induce a Similar Chronic Myeloid Leukemia–like Syndrome in Mice But Have Different Lymphoid Leukemogenic Activity. J Exp Med (1999) 189:1399–412. doi: 10.1084/jem.189.9.1399
153. Alexander WS, Adams JM, Cory S. Oncogene Cooperation in Lymphocyte Transformation: Malignant Conversion of E Mu-Myc Transgenic Pre-B Cells In Vitro Is Enhanced by V-H-Ras or V-Raf But Not V-Abl. Mol Cell Biol (1989) 9:67–73. doi: 10.1128/mcb.9.1.67-73.1989
154. Sexl V, Piekorz R, Moriggl R, Rohrer J, Brown MP, Bunting KD, et al. Stat5a/b Contribute to Interleukin 7–Induced B-Cell Precursor Expansion, But Abl- Andbcr/Abl-Induced Transformation Are Independent of Stat5. Blood (2000) 96:2277–83. doi: 10.1182/blood.V96.6.2277
155. Hemann MT, Fridman JS, Zilfou JT, Hernando E, Paddison PJ, Cordon-Cardo C, et al. An Epi-Allelic Series of P53 Hypomorphs Created by Stable RNAi Produces Distinct Tumor Phenotypes In Vivo. Nat Genet (2003) 33:396–400. doi: 10.1038/ng1091
156. Hemann MT, Zilfou JT, Zhao Z, Burgess DJ, Hannon GJ, Lowe SW. Suppression of Tumorigenesis by the P53 Target PUMA. Proc Natl Acad Sci (2004) 101:9333–8. doi: 10.1073/pnas.0403286101
157. Wendel H-G, de Stanchina E, JS F, Malina A, Ray S, Kogan S, et al. Survival Signalling by Akt and Eif4e in Oncogenesis and Cancer Therapy. Nature (2004) 428:332–7. doi: 10.1038/nature02369
158. Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, et al. Requirement of Src Kinases Lyn, Hck and Fgr for BCR-ABL1-Induced B-Lymphoblastic Leukemia But Not Chronic Myeloid Leukemia. Nat Genet (2004) 36:453–61. doi: 10.1038/ng1343
159. Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, et al. Evasion of the P53 Tumour Surveillance Network by Tumour-Derived MYC Mutants. Nature (2005) 436:807–11. doi: 10.1038/nature03845
160. He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al. A microRNA Polycistron as a Potential Human Oncogene. Nature (2005) 435:828–33. doi: 10.1038/nature03552
161. Herbst A, Hemann MT, Tworkowski KA, Salghetti SE, Lowe SW, Tansey WP. A Conserved Element in Myc That Negatively Regulates its Proapoptotic Activity. EMBO Rep (2005) 6:177–83. doi: 10.1038/sj.embor.7400333
162. Williams RT, Roussel MF, Sherr CJ. Arf Gene Loss Enhances Oncogenicity and Limits Imatinib Response in Mouse Models of Bcr-Abl-Induced Acute Lymphoblastic Leukemia. Proc Natl Acad Sci (2006) 103:6688–93. doi: 10.1073/pnas.0602030103
163. Hoelbl A, Kovacic B, Kerenyi MA, Simma O, Warsch W, Cui Y, et al. Clarifying the Role of Stat5 in Lymphoid Development and Abelson-Induced Transformation. Blood (2006) 107:4898–906. doi: 10.1182/blood-2005-09-3596
164. Barabe F, Kennedy JA, Hope KJ, Dick JE. Modeling the Initiation and Progression of Human Acute Leukemia in Mice. Sci (80-) (2007) 316:600–4. doi: 10.1126/science.1139851
165. Wang P-Y, Young F, Chen C-Y, Stevens BM, Neering SJ, Rossi RM, et al. The Biologic Properties of Leukemias Arising From BCR/ABL-Mediated Transformation Vary as a Function of Developmental Origin and Activity of the P19arf Gene. Blood (2008) 112:4184–92. doi: 10.1182/blood-2008-02-142190
166. Bric A, Miething C, Bialucha CU, Scuoppo C, Zender L, Krasnitz A, et al. Functional Identification of Tumor-Suppressor Genes Through an In Vivo RNA Interference Screen in a Mouse Lymphoma Model. Cancer Cell (2009) 16:324–35. doi: 10.1016/j.ccr.2009.08.015
167. Hoelbl A, Schuster C, Kovacic B, Zhu B, Wickre M, Hoelzl MA, et al. Stat5 Is Indispensable for the Maintenance of Bcr/Abl -Positive Leukaemia. EMBO Mol Med (2010) 2:98–110. doi: 10.1002/emmm.201000062
168. Nakagawa M, Tsuzuki S, Honma K, Taguchi O, Seto M. Synergistic Effect of Bcl2, Myc and Ccnd1 Transforms Mouse Primary B Cells Into Malignant Cells. Haematologica (2011) 96:1318–26. doi: 10.3324/haematol.2011.041053
169. Bouquet C, Melchers F. Pim1 and Myc Reversibly Transform Murine Precursor B Lymphocytes But Not Mature B Lymphocytes. Eur J Immunol (2012) 42:522–32. doi: 10.1002/eji.201141987
170. Kovacic B, Hoelbl A, Litos G, Alacakaptan M, Schuster C, Fischhuber KM, et al. Diverging Fates of Cells of Origin in Acute and Chronic Leukaemia. EMBO Mol Med (2012) 4:283–97. doi: 10.1002/emmm.201100208
171. Leskov I, Pallasch CP, Drake A, Iliopoulou BP, Souza A, Shen C-HH, et al. Rapid Generation of Human B-Cell Lymphomas via Combined Expression of Myc and Bcl2 and Their Use as a Preclinical Model for Biological Therapies. Oncogene (2013) 32:1066–72. doi: 10.1038/onc.2012.117
172. Arita K, Maeda-Kasugai Y, Ohshima K, Tsuzuki S, Suguro-Katayama M, Karube K, et al. Generation of Mouse Models of Lymphoid Neoplasm Using Retroviral Gene Transduction of In Vitro-Induced Germinal Center B and T Cells. Exp Hematol (2013) 41:731–741.e9. doi: 10.1016/j.exphem.2013.04.001
173. Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O’Connor L, Milla L, et al. An Inducible Lentiviral Guide RNA Platform Enables the Identification of Tumor-Essential Genes and Tumor-Promoting Mutations In Vivo. Cell Rep (2015) 10:1422–32. doi: 10.1016/j.celrep.2015.02.002
174. Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, et al. The Histone Lysine Methyltransferase KMT2D Sustains a Gene Expression Program That Represses B Cell Lymphoma Development. Nat Med (2015) 21:1199–208. doi: 10.1038/nm.3943
175. Scheicher R, Hoelbl-Kovacic A, Bellutti F, Tigan A-S, Prchal-Murphy M, Heller G, et al. CDK6 as a Key Regulator of Hematopoietic and Leukemic Stem Cell Activation. Blood (2015) 125:90–101. doi: 10.1182/blood-2014-06-584417
176. Jiang Y, Ortega-Molina A, Geng H, Ying H-Y, Hatzi K, Parsa S, et al. CREBBP Inactivation Promotes the Development of HDAC3-Dependent Lymphomas. Cancer Discovery (2017) 7:38–53. doi: 10.1158/2159-8290.CD-16-0975
177. Katigbak A, Cencic R, Robert F, Sénécha P, Scuoppo C, Pelletier J. A CRISPR/Cas9 Functional Screen Identifies Rare Tumor Suppressors. Sci Rep (2016) 6:38968. doi: 10.1038/srep38968
178. Wolf I, Bouquet C, Melchers F. cDNA-Library Testing Identifies Transforming Genes Cooperating With C-Myc in Mouse Pre-B Cells. Eur J Immunol (2016) 46:2555–65. doi: 10.1002/eji.201646419
179. Wolf I, Bouquet C, Naumann F, Melchers F. Generation of Precursor, Immature, and Mature Murine B1-Cell Lines From C-Myc/bcl-xL-Overexpressing Pre-BI Cells. Eur J Immunol (2017) 47:911–20. doi: 10.1002/eji.201746937
180. van Oosterwijk JG, Li C, Yang X, Opferman JT, Sherr CJ. Small Mitochondrial Arf (Smarf) Protein Corrects P53-Independent Developmental Defects of Arf Tumor Suppressor-Deficient Mice. Proc Natl Acad Sci (2017) 114:7420–5. doi: 10.1073/pnas.1707292114
181. McHugh D, Caduff N, Barros MHM, Rämer PC, Raykova A, Murer A, et al. Persistent KSHV Infection Increases EBV-Associated Tumor Formation In Vivo via Enhanced EBV Lytic Gene Expression. Cell Host Microbe (2017) 22:61–73.e7. doi: 10.1016/j.chom.2017.06.009
182. Reimer J, Knöß S, Labuhn M, Charpentier EM, Göhring G, Schlegelberger B, et al. CRISPR-Cas9-Induced T(11;19)/MLL-ENL Translocations Initiate Leukemia in Human Hematopoietic Progenitor Cells In Vivo. Haematologica (2017) 102:1558–66. doi: 10.3324/haematol.2017.164046
183. Katigbak A, Robert F, Paquet M, Pelletier J. Inducible Genome Editing With Conditional CRISPR/Cas9 Mice. G3 Genes Genomes Genet (2018) 8:1627–35. doi: 10.1534/g3.117.300327
184. Janic A, Valente LJ, Wakefield MJ, Di Stefano L, Milla L, Wilcox S, et al. DNA Repair Processes Are Critical Mediators of P53-Dependent Tumor Suppression. Nat Med (2018) 24:947–53. doi: 10.1038/s41591-018-0043-5
185. Jeong J, Jager A, Domizi P, Pavel-Dinu M, Gojenola L, Iwasaki M, et al. High-Efficiency CRISPR Induction of T(9;11) Chromosomal Translocations and Acute Leukemias in Human Blood Stem Cells. Blood Adv (2019) 3:2825–35. doi: 10.1182/bloodadvances.2019000450
186. Rajan SS, Li L, Kweh MF, Kunkalla K, Amin AD, Agarwal NK, et al. CRISPR Genome Editing of Murine Hematopoietic Stem Cells to Create Npm1-Alk Causes ALK+ Lymphoma After Transplantation. Blood Adv (2019) 3:1788–94. doi: 10.1182/bloodadvances.2018025247
187. Verma D, Zanetti C, Godavarthy PS, Kumar R, Minciacchi VR, Pfeiffer J, et al. Bone Marrow Niche-Derived Extracellular Matrix-Degrading Enzymes Influence the Progression of B-Cell Acute Lymphoblastic Leukemia. Leukemia (2020) 34:1540–52. doi: 10.1038/s41375-019-0674-7
188. Williams DA, Lemischka IR, Nathan DG, Mulligan RC. Introduction of New Genetic Material Into Pluripotent Haematopoietic Stem Cells of the Mouse. Nature (1984) 310:476–80. doi: 10.1038/310476a0
189. Whitlock CA, Witte ON. Long-Term Culture of B Lymphocytes and Their Precursors From Murine Bone Marrow. Proc Natl Acad Sci (1982) 79:3608–12. doi: 10.1073/pnas.79.11.3608
190. Nojima T, Haniuda K, Moutai T, Matsudaira M, Mizokawa S, Shiratori I, et al. In-Vitro Derived Germinal Centre B Cells Differentially Generate Memory B or Plasma Cells In Vivo. Nat Commun (2011) 2:465. doi: 10.1038/ncomms1475
191. Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The C-Myc Oncogene Driven by Immunoglobulin Enhancers Induces Lymphoid Malignancy in Transgenic Mice. Nature (1985) 318:533–8. doi: 10.1038/318533a0
192. Marsden VS, O’Connor L, O’Reilly LA, Silke J, Metcalf D, Ekert PG, et al. Apoptosis Initiated by Bcl-2-Regulated Caspase Activation Independently of the Cytochrome C/Apaf-1/Caspase-9 Apoptosome. Nature (2002) 419:634–7. doi: 10.1038/nature01101
193. Scott CL, Schuler M, Marsden VS, Egle A, Pellegrini M, Nesic D, et al. Apaf-1 and Caspase-9 do Not Act as Tumor Suppressors in Myc-Induced Lymphomagenesis or Mouse Embryo Fibroblast Transformation. J Cell Biol (2004) 164:89–96. doi: 10.1083/jcb.200310041
194. Nishikawa S-I, Ogawa M, Nishikawa S, Kunisada T, Kodama H. B Lymphopoiesis on Stromal Cell Clone: Stromal Cell Clones Acting on Different Stages of B Cell Differentiation*. Eur J Immunol (1988) 18:1767–72. doi: 10.1002/eji.1830181117
195. Rolink A, Kudo A, Karasuyama H, Kikuchi Y, Melchers F. Long-Term Proliferating Early Pre B Cell Lines and Clones With the Potential to Develop to Surface Ig-Positive, Mitogen Reactive B Cells In Vitro and In Vivo. EMBO J (1991) 10:327–36. doi: 10.1002/j.1460-2075.1991.tb07953.x
196. von Muenchow L, Tsapogas P, Albertí-Servera L, Capoferri G, Doelz M, Rolink H, et al. Pro-B Cells Propagated in Stromal Cell-Free Cultures Reconstitute Functional B-Cell Compartments in Immunodeficient Mice. Eur J Immunol (2017) 47:394–405. doi: 10.1002/eji.201646638
197. Gonzalez-Martin A, Adams BD, Lai M, Shepherd J, Salvador-Bernaldez M, Salvador JM, et al. The microRNA miR-148a Functions as a Critical Regulator of B Cell Tolerance and Autoimmunity. Nat Immunol (2016) 17:433–40. doi: 10.1038/ni.3385
198. Zhao R, Zhang H, Zhang Y, Li D, Huang C, Li F. In Vivo Screen Identifies Zdhhc2 as a Critical Regulator of Germinal Center B Cell Differentiation. Front Immunol (2020) 11:1025. doi: 10.3389/fimmu.2020.01025
199. Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN, and CRISPR/Cas-Based Methods for Genome Engineering. Trends Biotechnol (2013) 31:397–405. doi: 10.1016/j.tibtech.2013.04.004
200. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Sci (80-) (2013) 339:819–23. doi: 10.1126/science.1231143
201. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-Guided Human Genome Engineering via Cas9. Sci (80-) (2013) 339:823–6. doi: 10.1126/science.1232033
202. Jain S, Shukla S, Yang C, Zhang M, Fatma Z, Lingamaneni M, et al. TALEN Outperforms Cas9 in Editing Heterochromatin Target Sites. Nat Commun (2021) 12:606. doi: 10.1038/s41467-020-20672-5
203. Canver MC, Bauer DE, Dass A, Yien YY, Chung J, Masuda T, et al. Characterization of Genomic Deletion Efficiency Mediated by Clustered Regularly Interspaced Palindromic Repeats (CRISPR)/Cas9 Nuclease System in Mammalian Cells*. J Biol Chem (2014) 289:21312–24. doi: 10.1074/jbc.M114.564625
204. Parikh BA, Beckman DL, Patel SJ, White JM, Yokoyama WM. Detailed Phenotypic and Molecular Analyses of Genetically Modified Mice Generated by CRISPR-Cas9-Mediated Editing. PloS One (2015) 10:e0116484. doi: 10.1371/journal.pone.0116484
205. Boroviak K, Fu B, Yang F, Doe B, Bradley A. Revealing Hidden Complexities of Genomic Rearrangements Generated With Cas9. Sci Rep (2017) 7:12867. doi: 10.1038/s41598-017-12740-6
206. Adikusuma F, Piltz S, Corbett MA, Turvey M, McColl SR, Helbig KJ, et al. Large Deletions Induced by Cas9 Cleavage. Nature (2018) 560:E8–9. doi: 10.1038/s41586-018-0380-z
207. Kosicki M, Tomberg K, Bradley A. Repair of Double-Strand Breaks Induced by CRISPR–Cas9 Leads to Large Deletions and Complex Rearrangements. Nat Biotechnol (2018) 36:765–71. doi: 10.1038/nbt.4192
208. Ran FA, Hsu PD, Lin C-Y, Gootenberg JS, Konermann S, Trevino AE, et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell (2013) 154:1380–9. doi: 10.1016/j.cell.2013.08.021
209. Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas Nuclease Specificity Using Truncated Guide RNAs. Nat Biotechnol (2014) 32:279–84. doi: 10.1038/nbt.2808
210. Kleinstiver BP, Sousa AA, Walton RT, Tak YE, Hsu JY, Clement K, et al. Engineered CRISPR–Cas12a Variants With Increased Activities and Improved Targeting Ranges for Gene, Epigenetic and Base Editing. Nat Biotechnol (2019) 37:276–82. doi: 10.1038/s41587-018-0011-0
211. Walton RT, Christie KA, Whittaker MN, Kleinstiver BP. Unconstrained Genome Targeting With Near-PAMless Engineered CRISPR-Cas9 Variants. Sci (80-) (2020) 368:290–6. doi: 10.1126/science.aba8853
212. Hirakawa MP, Krishnakumar R, Timlin JA, Carney JP, Butler KS. Gene Editing and CRISPR in the Clinic: Current and Future Perspectives. Biosci Rep (2020) 40(4):BSR20200127. doi: 10.1042/BSR20200127
213. Manghwar H, Li B, Ding X, Hussain A, Lindsey K, Zhang X, et al. CRISPR/Cas Systems in Genome Editing: Methodologies and Tools for sgRNA Design, Off-Target Evaluation, and Strategies to Mitigate Off-Target Effects. Adv Sci (2020) 7:1902312. doi: 10.1002/advs.201902312
214. Koblan LW, Doman JL, Wilson C, Levy JM, Tay T, Newby GA, et al. Improving Cytidine and Adenine Base Editors by Expression Optimization and Ancestral Reconstruction. Nat Biotechnol (2018) 36:843–6. doi: 10.1038/nbt.4172
215. Doman JL, Raguram A, Newby GA, Liu DR. Evaluation and Minimization of Cas9-Independent Off-Target DNA Editing by Cytosine Base Editors. Nat Biotechnol (2020) 38:620–8. doi: 10.1038/s41587-020-0414-6
216. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-And-Replace Genome Editing Without Double-Strand Breaks or Donor DNA. Nature (2019) 576:149–57. doi: 10.1038/s41586-019-1711-4
217. Filippova J, Matveeva A, Zhuravlev E, Stepanov G. Guide RNA Modification as a Way to Improve CRISPR/Cas9-Based Genome-Editing Systems. Biochimie (2019) 167:49–60. doi: 10.1016/j.biochi.2019.09.003
218. Nakade S, Tsubota T, Sakane Y, Kume S, Sakamoto N, Obara M, et al. Microhomology-Mediated End-Joining-Dependent Integration of Donor DNA in Cells and Animals Using TALENs and CRISPR/Cas9. Nat Commun (2014) 5:5560. doi: 10.1038/ncomms6560
219. Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally Engineered Cas9 Nucleases With Improved Specificity. Sci (80-) (2016) 351:84–8. doi: 10.1126/science.aad5227
220. Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-Fidelity CRISPR-Cas9 Nucleases With No Detectable Genome-Wide Off-Target Effects. Nature (2016) 529:490–5. doi: 10.1038/nature16526
221. Lee K, Mackley VA, Rao A, Chong AT, Dewitt MA, Corn JE, et al. Synthetically Modified Guide RNA and Donor DNA Are a Versatile Platform for CRISPR-Cas9 Engineering. Elife (2017) 6:e25312. doi: 10.7554/eLife.25312.001
222. Yao X, Wang X, Hu X, Liu Z, Liu J, Zhou H, et al. Homology-Mediated End Joining-Based Targeted Integration Using CRISPR/Cas9. Cell Res (2017) 27:801–14. doi: 10.1038/cr.2017.76
223. Ma M, Zhuang F, Hu X, Wang B, Wen XZ, Ji JF, et al. Efficient Generation of Mice Carrying Homozygous Double-Floxp Alleles Using the Cas9-Avidin/Biotin-Donor DNA System. Cell Res (2017) 27:578–81. doi: 10.1038/cr.2017.29
224. Miura H, Quadros RM, Gurumurthy CB, Ohtsuka M. Easi-CRISPR for Creating Knock-in and Conditional Knockout Mouse Models Using Long ssDNA Donors. Nat Protoc (2018) 13:195–215. doi: 10.1038/nprot.2017.153
225. Gu B, Posfai E, Rossant J. Efficient Generation of Targeted Large Insertions by Microinjection Into Two-Cell-Stage Mouse Embryos. Nat Biotechnol (2018) 36:632–7. doi: 10.1038/nbt.4166
226. Weber J, Rad R. Engineering CRISPR Mouse Models of Cancer. Curr Opin Genet Dev (2019) 54:88–96. doi: 10.1016/j.gde.2019.04.001
227. Weber J, Braun CJ, Saur D, Rad R. In Vivo Functional Screening for Systems-Level Integrative Cancer Genomics. Nat Rev Cancer (2020) 20:573–93. doi: 10.1038/s41568-020-0275-9
228. Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, et al. CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell (2014) 159:440–55. doi: 10.1016/j.cell.2014.09.014
229. Gundry MC, Brunetti L, Lin A, Mayle AE, Kitano A, Wagner D, et al. Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. Cell Rep (2016) 17:1453–61. doi: 10.1016/j.celrep.2016.09.092
230. Chu VT, Weber T, Graf R, Sommermann T, Petsch K, Sack U, et al. Efficient Generation of Rosa26 Knock-in Mice Using CRISPR/Cas9 in C57BL/6 Zygotes. BMC Biotechnol (2016) 16:4. doi: 10.1186/s12896-016-0234-4
231. Chu VT, Graf R, Wirtz T, Weber T, Favret J, Li X, et al. Efficient CRISPR-Mediated Mutagenesis in Primary Immune Cells Using CrispRGold and a C57BL/6 Cas9 Transgenic Mouse Line. Proc Natl Acad Sci (2016) 113:12514–9. doi: 10.1073/pnas.1613884113
232. LaFleur MW, Nguyen TH, Coxe MA, Yates KB, Trombley JD, Weiss SA, et al. A CRISPR-Cas9 Delivery System for In Vivo Screening of Genes in the Immune System. Nat Commun (2019) 10:1668. doi: 10.1038/s41467-019-09656-2
233. Ten Hacken E, Yin S, Clement K, Redd RA, Hernandez-Sanchez M, Li S, et al. Interrogation of Individual CLL Loss-Of-Function Lesions By CRISPR In Vivo Editing Reveals Common and Unique Pathway Alterations. Blood (2019) 134:684–4. doi: 10.1182/blood-2019-127673
234. Yudovich D, Bäckström A, Schmiderer L, Žemaitis K, Subramaniam A, Larsson J. Combined Lentiviral- and RNA-Mediated CRISPR/Cas9 Delivery for Efficient and Traceable Gene Editing in Human Hematopoietic Stem and Progenitor Cells. Sci Rep (2020) 10:22393. doi: 10.1038/s41598-020-79724-x
235. Johnson MJ, Laoharawee K, Lahr WS, Webber BR, Moriarity BS. Engineering of Primary Human B Cells With CRISPR/Cas9 Targeted Nuclease. Sci Rep (2018) 8:12144. doi: 10.1038/s41598-018-30358-0
236. Nahmad AD, Raviv Y, Horovitz-Fried M, Sofer I, Akriv T, Nataf D, et al. Engineered B Cells Expressing an Anti-HIV Antibody Enable Memory Retention, Isotype Switching and Clonal Expansion. Nat Commun (2020) 11:5851. doi: 10.1038/s41467-020-19649-1
237. Hartweger H, McGuire AT, Horning M, Taylor JJ, Dosenovic P, Yost D, et al. HIV-Specific Humoral Immune Responses by CRISPR/Cas9-Edited B Cells. J Exp Med (2019) 216:1301–10. doi: 10.1084/jem.20190287
238. Moffett HF, Harms CK, Fitzpatrick KS, Tooley MR, Boonyaratanakornkit J, Taylor JJ. B Cells Engineered to Express Pathogen-Specific Antibodies Protect Against Infection. Sci Immunol (2019) 4:eaax0644. doi: 10.1126/sciimmunol.aax0644
239. Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell (2014) 159:647–61. doi: 10.1016/j.cell.2014.09.029
240. Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han Y-C, et al. In Vivo Engineering of Oncogenic Chromosomal Rearrangements With the CRISPR/Cas9 System. Nature (2014) 516:423–7. doi: 10.1038/nature13902
241. Huijbers IJ. Generating Genetically Modified Mice: A Decision Guide. In: . Methods Mol Biol (2017) 1642:1–19. doi: 10.1007/978-1-4939-7169-5_1
242. Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, et al. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell (2013) 153:910–8. doi: 10.1016/j.cell.2013.04.025
243. Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-Step Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell (2013) 154:1370–9. doi: 10.1016/j.cell.2013.08.022
244. Huijbers IJ, Bin Ali R, Pritchard C, Cozijnsen M, Kwon MC, Proost N, et al. Rapid Target Gene Validation in Complex Cancer Mouse Models Using Re-Derived Embryonic Stem Cells. EMBO Mol Med (2014) 6:212–25. doi: 10.1002/emmm.201303297
245. Huijbers IJ, Del Bravo J, Bin Ali R, Pritchard C, Braumuller TM, van Miltenburg MH, et al. Using the GEMM-ESC Strategy to Study Gene Function in Mouse Models. Nat Protoc (2015) 10:1755–85. doi: 10.1038/nprot.2015.114
246. Chen Y, Mao S, Liu B, Jing Z, Zang Y, Xia J, et al. Novel Mosaic Mice With Diverse Applications. bioRxiv (2020). doi: 10.1101/2020.03.21.001388
247. Zhang J, Chen L, Zhang J, Wang Y. Drug Inducible CRISPR/Cas Systems. Comput Struct Biotechnol J (2019) 17:1171–7. doi: 10.1016/j.csbj.2019.07.015
248. Felsher DW, Bishop JM. Reversible Tumorigenesis by MYC in Hematopoietic Lineages. Mol Cell (1999) 4:199–207. doi: 10.1016/S1097-2765(00)80367-6
249. Marinkovic D, Marinkovic T, Mahr B, Hess J, Wirth T. Reversible Lymphomagenesis in Conditionally C-MYC Expressing Mice. Int J Cancer (2004) 110:336–42. doi: 10.1002/ijc.20099
250. Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 Is Required for Maintenance of a Model Leukemia. Cancer Cell (2004) 6:241–9. doi: 10.1016/j.ccr.2004.07.011
251. Liu GJ, Cimmino L, Jude JG, Hu Y, Witkowski MT, McKenzie MD, et al. Pax5 Loss Imposes a Reversible Differentiation Block in B-Progenitor Acute Lymphoblastic Leukemia. Genes Dev (2014) 28:1337–50. doi: 10.1101/gad.240416.114
252. Christophorou MA, Martin-Zanca D, Soucek L, Lawlor ER, Brown-Swigart L, Verschuren EW, et al. Temporal Dissection of P53 Function In Vitro and In Vivo. Nat Genet (2005) 37:718–26. doi: 10.1038/ng1572
253. Martins CP, Brown-Swigart L, Evan GI. Modeling the Therapeutic Efficacy of P53 Restoration in Tumors. Cell (2006) 127:1323–34. doi: 10.1016/j.cell.2006.12.007
254. Scheijen B, Ngo HT, Kang H, Griffin JD. FLT3 Receptors With Internal Tandem Duplications Promote Cell Viability and Proliferation by Signaling Through Foxo Proteins. Oncogene (2004) 23:3338–49. doi: 10.1038/sj.onc.1207456
255. Klucher KM, Lopez DV, Daley GQ. Secondary Mutation Maintains the Transformed State in BaF3 Cells With Inducible BCR/ABL Expression. Blood (1998) 91:3927–34. doi: 10.1182/blood.v91.10.3927
256. Dickins RA, Hemann MT, Zilfou JT, Simpson DR, Ibarra I, Hannon GJ, et al. Probing Tumor Phenotypes Using Stable and Regulated Synthetic microRNA Precursors. Nat Genet (2005) 37:1289–95. doi: 10.1038/ng1651
257. Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, et al. An Optimized microRNA Backbone for Effective Single-Copy RNAi. Cell Rep (2013) 5:1704–13. doi: 10.1016/j.celrep.2013.11.020
258. Dugray A, Geay J, Foudi A, Bonnet M, Vainchenker W, Wendling F, et al. Rapid Generation of a Tetracycline-Inducible BCR-ABL Defective Retrovirus Using a Single Autoregulatory Retroviral Cassette. Leukemia (2001) 15:1658–62. doi: 10.1038/sj.leu.2402225
259. Csikós T, Reijmers RM, Uren AG, Spaargaren M, Pals ST. Instant Conditional Transgenesis in the Mouse Hematopoietic Compartment. J Immunol Methods (2008) 339:259–63. doi: 10.1016/j.jim.2008.08.009
260. Cao J, Wu L, Zhang S-M, Lu M, Cheung WKC, Cai W, et al. An Easy and Efficient Inducible CRISPR/Cas9 Platform With Improved Specificity for Multiple Gene Targeting. Nucleic Acids Res (2016) 44:e149. doi: 10.1093/nar/gkw660
261. Lundin A, Porritt MJ, Jaiswal H, Seeliger F, Johansson C, Bidar AW, et al. Development of an ObLiGaRe Doxycycline Inducible Cas9 System for Pre-Clinical Cancer Drug Discovery. Nat Commun (2020) 11:4903. doi: 10.1038/s41467-020-18548-9
262. Dow LE, Fisher J, O’Rourke KP, Muley A, Kastenhuber ER, Livshits G, et al. Inducible In Vivo Genome Editing With CRISPR-Cas9. Nat Biotechnol (2015) 33:390–4. doi: 10.1038/nbt.3155
263. Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, Iyer E PR, et al. Highly Efficient Cas9-Mediated Transcriptional Programming. Nat Methods (2015) 12:326–8. doi: 10.1038/nmeth.3312
264. Morgens DW, Deans RM, Li A, Bassik MC. Systematic Comparison of CRISPR/Cas9 and RNAi Screens for Essential Genes. Nat Biotechnol (2016) 34:634–6. doi: 10.1038/nbt.3567
265. De La Rochere P, Guil-Luna S, Decaudin D, Azar G, Sidhu SS, Piaggio E. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol (2018) 39:748–63. doi: 10.1016/j.it.2018.07.001
266. Saito Y, Shultz LD, Ishikawa F. Understanding Normal and Malignant Human Hematopoiesis Using Next-Generation Humanized Mice. Trends Immunol (2020) 41:706–20. doi: 10.1016/j.it.2020.06.004
267. Martinov T, McKenna KM, Tan WH, Collins EJ, Kehret AR, Linton JD, et al. Building the Next Generation of Humanized Hemato-Lymphoid System Mice. Front Immunol (2021) 12:643852. doi: 10.3389/fimmu.2021.643852
268. Stripecke R, Münz C, Schuringa JJ, Bissig K, Soper B, Meeham T, et al. Innovations, Challenges, and Minimal Information for Standardization of Humanized Mice. EMBO Mol Med (2020) 12:e8662. doi: 10.15252/emmm.201708662
Keywords: B cell leukemia, B cell lymphoma, mouse models, insertional mutagenesis, exome sequencing, reverse genetics, CRISPR-Cas, shRNA
Citation: Dawes JC and Uren AG (2021) Forward and Reverse Genetics of B Cell Malignancies: From Insertional Mutagenesis to CRISPR-Cas. Front. Immunol. 12:670280. doi: 10.3389/fimmu.2021.670280
Received: 20 February 2021; Accepted: 09 July 2021;
Published: 13 August 2021.
Edited by:
Christelle Vincent-Fabert, UMR7276 Contrôle des réponses immunes B et des lymphoproliférations (CRIBL), FranceReviewed by:
Rodney P. DeKoter, Western University, CanadaCopyright © 2021 Dawes and Uren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony G. Uren, YW50aG9ueS51cmVuQGdtYWlsLmNvbQ==; Joanna C. Dawes, ZGF3ZXNqb2FubmFAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.