 Elodie Renaude1,2
Elodie Renaude1,2 Marie Kroemer1,3Christophe Borg1,2,4Paul Peixoto1,5
Marie Kroemer1,3Christophe Borg1,2,4Paul Peixoto1,5 Eric Hervouet1,5,6
Eric Hervouet1,5,6 Romain Loyon1†
Romain Loyon1† Olivier Adotévi1,2,4*†
Olivier Adotévi1,2,4*†- 1University of Bourgogne Franche-Comté, INSERM, EFS BFC, UMR1098, Interactions Hôte-Greffon-Tumeur/Ingénierie Cellulaire et Génique, Besançon, France
- 2Centre Hospitalier Universitaire de Besançon, Centre d’Investigation Clinique, INSERM CIC 1431, Besançon, France
- 3Department of Pharmacy, University Hospital of Besançon, Besançon, France
- 4Department of Medical Oncology, University Hospital of Besançon, Besançon, France
- 5EPIGENEXP Platform, University of Bourgogne Franche-Comté, Besançon, France
- 6DImaCell Platform, University of Bourgogne Franche-Comté, Besançon, France
Evidences highlight the role of various CD4+ helper T cells (CD4+ Th) subpopulations in orchestrating the immune responses against cancers. Epigenetics takes an important part in the regulation of CD4+ Th polarization and plasticity. In this review, we described the epigenetic factors that govern CD4+ T cells differentiation and recruitment in the tumor microenvironment and their subsequent involvement in the antitumor immunity. Finally, we discussed how to manipulate tumor reactive CD4+ Th responses by epigenetic drugs to improve anticancer immunotherapy.
Introduction
Distinct Roles of CD4+ Helper T Cells in Cancer
Tumor reactive CD4+ T cells are a heterogeneous subset that have different effector functions depending on the type of cytokines they produce (1, 2).
T helper 1 (Th1): Among CD4+ subsets, Th1 cells produce IFN-γ, IL-2, tumor necrosis factor α (TNFα) and express the T-box transcription factor (T-bet). Th1 cells play a well-defined role in antitumor protection by orchestrating cell-mediated immunity against cancer cells. Notably, these cells show the capacity to enhance tumor specific CD8+ T-cell generation, function, memory and survival (3–5). Additionally, the secretion of IFN-γ by Th1 cells can also promote CXCL9 and CXCL10 expression in the tumor microenvironment (TME) and therefore ensure the recruitment of CXCR3 expressing CD8+ T cells at tumor site (6, 7). Moreover, the production of IFN-γ by Th1 cells may enhance MHC class I and II expression at the surface of tumor cells, thus increasing tumor-derived peptide presentation. IFN- γ can also influence the polarization of macrophages toward proinflammatory M1 phenotype (8, 9). Emerging functions of Th1 cells also indicate their involvement in tumor angiogenesis inhibition, promoting cancer cell senescence, highly sensitive neoepitopes recognition, and protecting effector cytotoxic T lymphocytes (CTL) from exhaustion (4, 10). As a result, the presence of IFN-γ producing Th1 cells within the tumor microenvironment is associated with a good prognosis in several human cancers (11).
T helper 2 (Th2): Th2 cells produce IL-4, IL-5 and IL-13 and their differentiation is governed by the transcription factor GATA3 (12). Ambivalent roles have been described for these cells in the context of cancer. Indeed, their presence in the TME can be either beneficial or detrimental for patient survival (1, 2). In a mouse model wherein ovalbumin (OVA) is used as a specific tumor antigen, it has been shown that OVA-specific Th2 cells elicited a long-lasting antitumor response in mice. Indeed, IL-4 produced by memory anti-OVA Th2 cells directly stimulated natural killer (NK) cells cytotoxic activity against tumor (13–15). Moreover, the blockage of TGF-β signaling in CD4+ T cells of mice promoted a Th2 cell-differentiation program and reduced tumor growth. Indeed, IL4 produced by TGF-β receptor 2 (TGFBR2)-deficient CD4+ T cells in a mouse model of breast cancer could reprogram the tumor vasculature and triggered cancer cell hypoxia and death. These results support the antitumor role of Th2 cells (16). However, in human cancers, Th2 cells infiltration in the TME is commonly associated with poor clinical outcome (17, 18).
T helper 17 (Th17): Th17 cells are characterized by the master transcription factor retinoic acid receptor–related orphan receptor γt (RORγt). Th17 cells produce IL-17, IL-21, and IL-22. Their presence in the TME is either associated with a good or a poor prognosis (17, 19). In colorectal cancer, hepatocellular carcinoma, gastric and pancreatic cancer, Th17 cells polarization in TME was associated with an unfavorable prognosis whereas its presence in prostate, epithelial ovarian cancer or uterine cervical cancer was associated with a better clinical outcome (20–24). The pro-tumoral roles of Th17 cells may be attributed to IL-17 proangiogenic functions. Indeed, IL-17 elicited VEGF production in colorectal cancer (25). Moreover, IL-17 signaling in tumor cells and tumor-associated stromal cells can lead to IL-6 production and induce STAT3 activation. STAT3 acts as an oncogenic factor by up-regulating pro-survival genes expression as well as metalloproteinases expression, thus favoring tumor invasion and metastases formation (26). IL-17 produced by Th17 cells may also influence the TME by promoting the recruitment of myeloid-derived suppressor cells (MDSCs) (27, 28).
Regulatory T cells (Treg): Treg cells express among others, the master transcription factor forkhead box P3 (FOXP3) and the high-affinity heterotrimeric IL-2 receptor (CD25). The presence of Treg cells in the tumor microenvironment is generally associated with poor clinical outcome in human cancers (11, 17, 19). These observations can be explained by their production of immunosuppressive cytokines (IL-10, TGF-β, IL-35) in the environment that can inhibit antitumor immunity (29–31). Treg cells have other mechanisms of humoral immunosuppression. For instance, Treg cells are highly dependent of IL-2 and can reduce its availability for effector T cells via their constitutive expression of CD25. Moreover, ATP can be converted into adenosine by CD39 and CD73 expressed on Treg cells. Adenosine is an immunomodulatory metabolite that can provide immunosuppressive signals to effector T cells and antigen-presenting cells (APC) via engagement of adenosine A2A receptor (A2AR). The secretion of granzyme and perforin by Treg cells can also damage effector T cells. In addition, Treg cells display immunosuppressive functions by a contact-dependent mechanism through the expression of immunosuppressive receptors such as LAG-3 (lymphocyte-activation gene 3) or CTLA-4 (cytotoxic T-lymphocyte-associated protein 4). CTLA-4 binds to CD80 and CD86 on APC thus transmitting suppressive signals to these cells. Moreover, the co-stimulatory signals induced upon the binding of CD80 or CD86 to CD28 is prevented by CTLA-4, thus restraining the activation of effector B and T lymphocytes. Additionally, the interaction of LAG-3 with MHC class II on APC can suppresses dendritic cell maturation and immunostimulatory capacity (32–35). However, other studies in colorectal cancer associated Treg cells infiltration with a better prognosis (36, 37).
T follicular helper (Tfh): Tfh cells express the transcription factor B cell lymphoma 6 (BCL-6) and secrete CXCL13 and IL-21. Tfh help is essential for B lymphocytes activation, antibody production and memory formation. Tfh cells support the development of adaptive antitumor humoral responses and the formation of tertiary lymphoid structures (TLS) (38). Thus, Tfh cells infiltration in the TME is associated with a good prognosis in breast cancer and colorectal cancer (39, 40).
Epigenetic Modifications in T Cells
Epigenetic modifications such as DNA methylation and post-translational histone modifications can regulate gene expression by modulating chromatin accessibility to transcription factors. Epigenetics is a reversible process as there are enzymes catalyzing the apposition of the post translational modifications such as histone methyltransferases (HMT) and histone acetylases (HAT) (epigenetic writers) and enzyme responsible for the demethylation and deacetylation of histones (HDAC) which are referred to as epigenetic eraser (Figure 1). Histone acetylation is associated with a permissive chromatin state whereas histone deacetylation generates a close chromatin state which is transcriptionally inactive. Histone methylation can be either favorable or unfavorable to transcription depending on the number and the position of the methyl groups on the histone tail. Trimethylation of the lysine 9 of histone 3 (H3K9me3) or H3K27me3 can be recognized by the heterochromatin protein HP1 which is responsible for chromatin silencing. On the contrary, H3K4me3 is positively correlated with gene transcription as it is recognized by NURF (nucleosome remodeling factor). The NURF complex is associated with a permissive chromatin and is enriched at promoter transcription starting sites (41, 42). The combination of post-translational histone modifications is called the histone code and overall defines chromatin accessibility to transcription factors (43) (Figure 1). DNA methylation on cytosines occurs preferentially at CpG islands which are composed of a high GC base density and located at promoter or distal cis-regulatory elements, like enhancers. DNA methylation is catalyzed by DNA methyltransferases (DNMT) and is associated with gene silencing (44). At the post transcriptional level, noncoding RNA (ncRNA) can target complementary mRNA epigenetic and impair mRNA translation. Indeed, the double strand complex is then recognized by the RNA-induced silencing complex (RISC) and can result in the cleavage of the targeted mRNA or the inhibition of its translation (45).
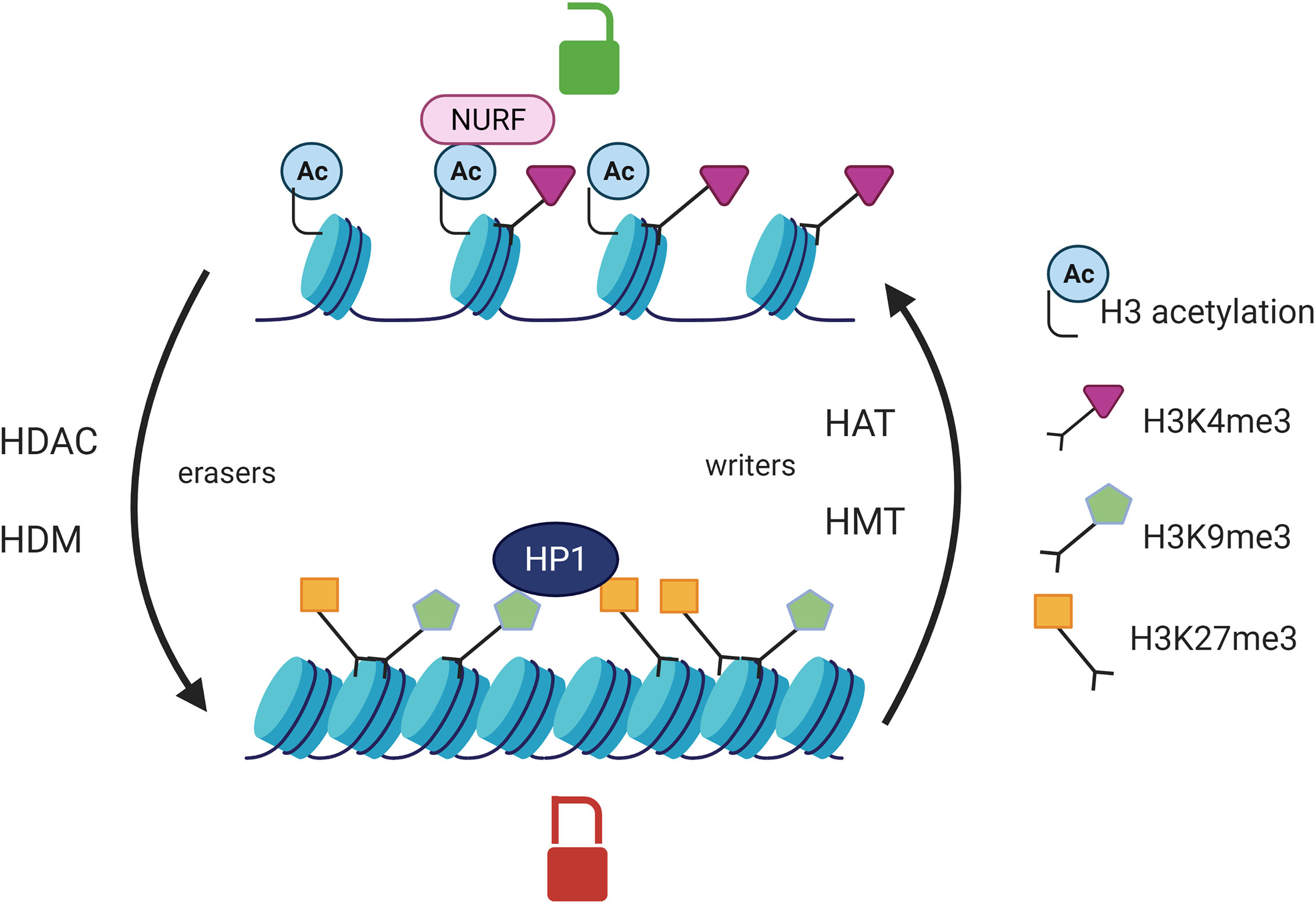
Figure 1 Post translational histone modifications and their effects on transcription. Histone acetylation catalyzed by HAT is associated with an open chromatin whereas HDAC erase this mark and are responsible for chromatin compaction. HMT add methyl groups on the lysines of histone H3 protein whereas HDM catalyze the opposite reaction (eraser). The effect of histone methylation on transcription is variable. HP1 (heterochromatin protein 1) can recognize H3K9me3 or H3K27me3 and is responsible for chromatin silencing. NURF (nucleosome remodeling factor) recognizes the histone mark H3K4me3 and is associated with a chromatin permissive to transcription. HAT, histone acetyltransferases; HDAC, Histone deacetylases; HMT, Histone methyltransferases; HDM, histone demethylases.
Epigenetics in CD4+ T Cells Differentiation and Plasticity
Epigenetics Regulates CD4+ T Cells Differentiation
The cytokines present in the microenvironment during TCR activation drive a cascade of signalization and activate transcription factors, which in turn modulate epigenetic enzymes leading to the CD4+ T cell polarization. Epigenetic modifications trigger the chromatin accessibility to lineage specific master transcription factors (T-bet, GATA3, RORγt, FOXP3) responsible of CD4+ T cells commitment (Figure 2). There is an increase of DNA demethylation and permissive histone modifications at lineage specific genes loci as well as a decrease of repressive histone marks which allow the expression of specific transcription factors and effector cytokines (46, 47). Indeed, the CD4+ T cell profile is dependent of the cytokines produced in the environment by innate immune cells following danger signals (48, 49).
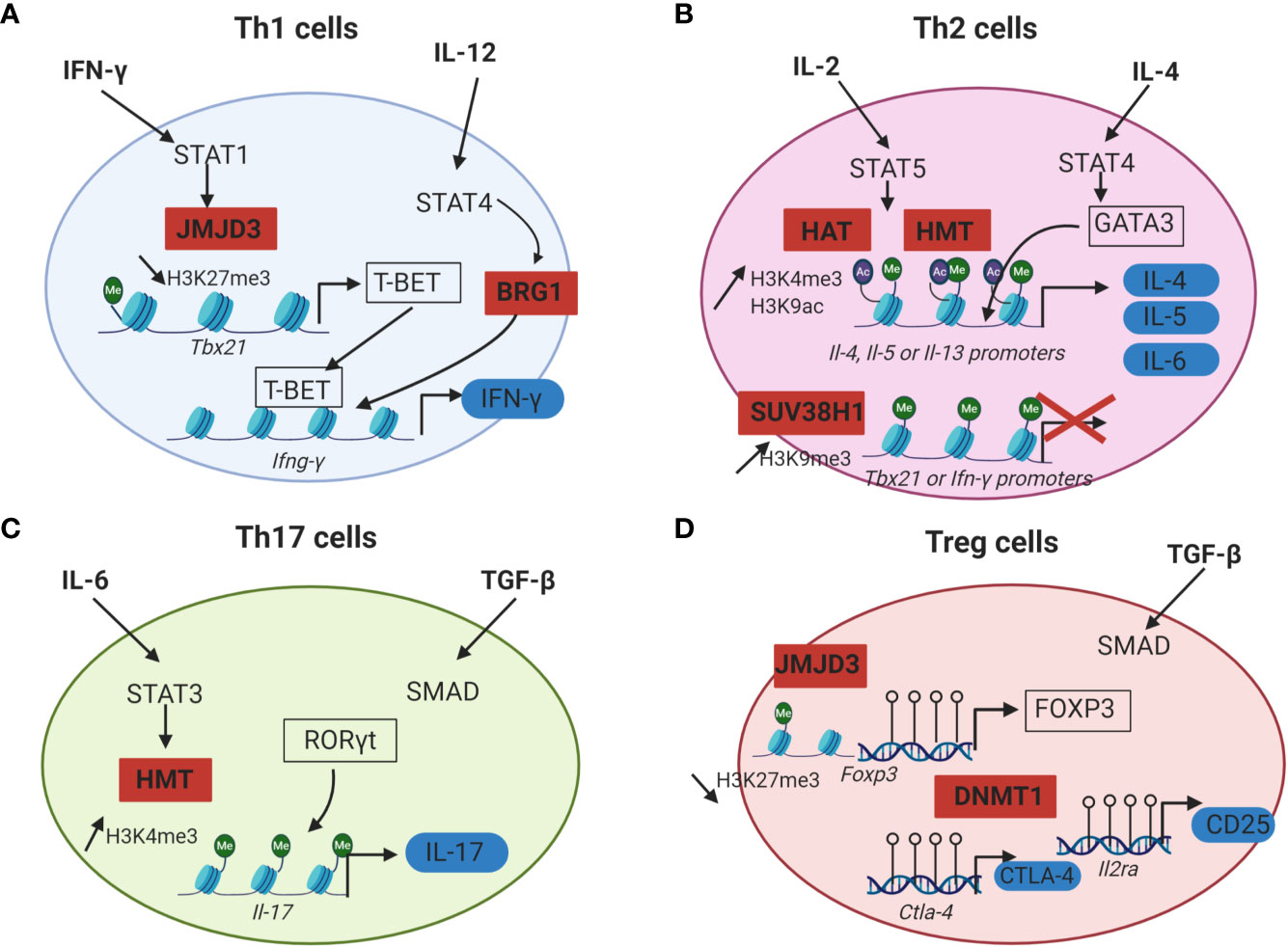
Figure 2 Epigenetic mechanisms that regulate CD4+ T cells differentiation. For each CD4+ T cell subset, cytokines initiating Th cells differentiation are in bold, lineage specific transcription factors are framed in black and effector proteins are surrounded in blue. Epigenetic enzymes regulating CD4+ T cells differentiation are in red squares. (A) IFN-γ and IL-12 drive Th1 cells differentiation by inducing STAT1 and STAT4 activation. STAT1 recruits JMJD3 (H3K27me3 histone demethylases) at the promoter of Tbx21. This promotes the loss of the repressive mark H3K27me3 and allows T-BET expression. STAT4 induces the recruitment of a chromatin remodeling complex (BRG1) at the locus of Ifn-γ which is responsible for a chromatin permissive to transcription. T-BET can then bind to the Ifn-γ promoter and induce its transcription. (B) In Th2 cells, IL-2 signals through STAT5 and triggers the recruitment of HAT and HMT responsible for the increase of the permissive marks H3K9ac and H3K4me3 at Il-4, Il-5 and Il-13 gene loci. IL-4 plays a role in inducing STAT6 activation, which upregulates the expression of the master regulator GATA3. GATA3 can then bind to the promoters of Il-4, Il-5 and Il-13 and induce the transcription of these effector cytokines. In Th2 cells, Th1 associated genes are silenced by the HMT SUV39H1 which increases the repressive mark H3K9me3 at the loci of Tbx21 and Ifn-γ and allows Th2 lineage stability. (C) During Th17 differentiation, IL-6 induces STAT3 activation and binding to the Il-17 promoter. STAT3 then recruits histone modification enzymes at the Il-17 locus, resulting in an increase of the permissive marks H3K4me3 and chromatin accessibility. RORγt can thus bind to the Il-17 promoter and induce IL-17 expression. (D) Treg cells differentiation from naïve CD4+ T cells requires TGF-β. The expression of the master transcription factor FOXP3 as well as the immune checkpoint CTLA-4 and the IL-2RA (CD25) is dependent of DNA demethylation at the loci of these genes by DNMT1. Empty circles on DNA: unmethylated CpG island.
Th1 cells differentiation is dependent of IFN-γ and IL-12. IFN-γ signaling in the activated CD4+ T cells triggers the activation of STAT1 signalization pathway and thus promotes T-bet expression. T-bet further increases the IFN-γ production and IL-12 receptor expression in the differentiating cells. IL-12 signaling induces STAT4 activation which promotes IFN-γ production by an epigenetic mechanism. Indeed, STAT4 bound to the Ifn-γ promoter in Th1 cells and recruits a chromatin remodeling complex called Brahma related gene 1 (BRG1), that remodels the nucleosomes and enhances the transcription of IFN-γ (Figure 2A) (50). Moreover, chromatin immunoprecipitation (ChIP) assays revealed that the level of permissive histone H4 acetylation marks at the Ifn-γ promoter of activated CD4+ T cells cultured under Th1 polarizing conditions (IL-12 and anti-IL-4) were higher compared to Th2 cells or undifferentiated Th cells. ChIP assays also indicated that differentiation into Th1 cells was accompanied by a decrease of HDAC-Sin3A recruitment at the Ifn-γ locus. By contrast, in undifferentiated CD4+ T cells, the level of HDAC-Sin3A was enhanced at this locus, thus preventing the formation of stable H4 acetylation marks. The transduction of T-bet into undifferentiated CD4+ T cells was sufficient to reduce the level of HDAC-Sin3A at the Ifn-γ locus and was associated with a gain of H4 acetylation across this locus as well as IFN-γ production. Taken together, these results indicated that the loss of HDAC-Sin3A at the Ifn-γ locus in effector Th1 cells was actively regulated by T-bet thus allowing the expression of IFN-γ (51). T-bet not only induces Th1 differentiation, but also prevents the differentiation toward the other subsets of CD4+ T cells by repressing their master transcription factors (GATA3 and RORγt) (52, 53).
The inhibition of Th1 associated genes in Th2 cells is mediated by epigenetics. In Th2 cells, the expression of IFN- γ is inhibited by the deposition of the repressive histone mark H3K27me3 to the Ifn-γ locus by EZH2 (54). Moreover, the silencing of Th1 gene loci was reported to be associated with an increase of the repressive histone modification H3K9me3 at these loci. The HMT SUV39H1 is responsible for H3K9 methylation. The deposition of H3K9me3 then recruits the heterochromatin protein 1α (HP1α) and promotes transcriptional silencing with the formation of heterochromatin at the promoter of Th1 specific genes, thus allowing Th2 lineage stability (55).
Regarding the initiation of Th2 differentiation, IL-2 and IL-4 are essential for Th2 cells generation from naive CD4+ T cells. IL-2 signals through STAT5 whereas IL-4 plays a role in inducing STAT6 activation, which upregulates the expression of the master regulator GATA3 (56). ChIP analysis of Th2-primed cells revealed that STAT5A could bind the Il-4 gene but failed to do so in cells primed under Th1 conditions or with anti-IL-2. Upon activation and differentiation of human Th2 cells from naïve CD4+ T cells, the permissive marks H3K9 acetylation and H3K4me3 were increased at Il-4, Il-5 and Il-13 gene loci. This chromatin remodeling occurring at most of the Th2 cytokine gene loci revealed the important part played by epigenetics in Th2 cell differentiation (Figure 2B) (57).
Epigenetics also plays a key role in the differentiation of Th17 cells [for a review: Renaude et al. (58)]. TGF- β, IL-6, IL-21 and IL-23 are required for Th17 differentiation from naive T cells. Briefly, TGF-β activates the Smad signaling pathway whereas IL-6 induces STAT3 activation (59). STAT3 binding to the Il-17 promoter correlates with an increase of the permissive marks H3K4me3 at the Il-17 locus (60) (Figure 2C). The deposition of permissive or repressive histone marks at Th17 specific gene loci regulates the chromatin accessibly to these genes and is therefore essential for the expression of Th17 specific cytokines IL-17 and IL-21 (61).
The comparison of the DNA methylation landscape between conventional CD4+ T cells and Treg cells revealed that DNA of Treg cells is globally hypomethylated. This observation correlated with the expression of genes vital for Treg cell function, such as Foxp3, Ctla4 and Il2ra (62, 63) (Figure 2D). Moreover, conditional deletion of Dnmt1 in CD4+ T cells of mice resulted in a decrease of the number and the immunosuppressive function of peripheral Treg cells, leading to lethal autoimmunity (64). Additionally, JMJD3, a histone H3K27 demethylase, was found to skew CD4+ T cell differentiation in vivo. The specific ablation of JMJD3 in the T cells of mice promoted Th2 and Th17 differentiation and inhibited Th1 and Treg cells polarization. Mechanistically, ChIP assays revealed that JMJD3 KO T cells were associated with a decrease of the permissive mark H3K4me3 at the Foxp3 promoter as well as an increase of the repressive mark H3K27me3 at the Ifn-γ promoter (65).
Tfh cells differentiation is also regulated by an epigenetic mechanism. Indeed, in a model of acute viral infection in mice, an increased level of the histone mark H3K27me3 was observed in Tfh cells compared to Th1 cells. The ablation of EZH2 (H3K27 HMT) in virus-specific CD4+ T cells resulted in an impaired differentiation of Tfh cells. EZH2 was found to regulate the chromatin accessibility of Tfh lineage associated genes such as Bcl-6 and Il-21 (66, 67). These data support evidence for the implication of epigenetics in the formation of CD4+ helper T cells differentiation.
Epigenetic Regulation of CD4+ T Cells Plasticity
CD4+ T cells are characterized by plastic capacities and can transdifferentiate into other subsets and this phenomenon is mediated by epigenetics. First evidence of CD4+ T cell plasticity was brought by the existence of cells expressing simultaneously genes that were specific of other CD4+ T cell lineages. For example, the co-expression of cytokines specific of Th17 and Th2 cells, IL-17 and IL-4, was identified in human circulating CD4+ T cells and revealed the potential plasticity of Th17/Th2 cells (68). Moreover, Th17 cells were found to convert into Th1 cells with the production of IFN-γ. In some cases, IL-17 expression was maintained in these cells whereas Th1 arising from Th17 cells could also totally extinguished IL-17 expression, indicating a progressive process and a terminal differentiation toward the Th1 phenotype. The conversion of Th17 cells into Treg cells was also observed in mouse and human. These cells expressed the lineage transcription factor FOXP3 as well as IL-17 and showed immunosuppressive capacities with the expression of IL-10 (69, 70).
Among the CD4+ helper subsets, Th17 cells elicit the greatest plastic capacities and can eventually convert into the other subsets under specific conditions. This particularity can be attributed to the fact that the expression of the cytokines and transcription factors defining Th17 cell lineage is unstable. Indeed, ChIP sequencing assays performed on the different CD4+ T cell subsets have revealed the existence of a “poised” chromatin state in Th17 cells. This was characterized by the presence of both repressive and permissive histone marks at other lineage specific loci and was associated with an absence of DNA methylation. These results indicate that the expression of other lineage specific genes is not strongly repressed in Th17 cells (71, 72). Moreover, the expression of the Th1 cell-like phenotype induced by the in vitro culture of Th17 cells with IL-12 was correlated with a decrease of the permissive marks H3K4me and histone acetylation at the Il-17 locus and an increase in these marks at the Ifn-γ locus These modifications resulted in an increased production of IFN-γ by these cells and a reduction of IL-17 secretion (73). The implication of epigenetics in CD4+ T cell plasticity was confirmed by Li et al., (65), as Jmjd3 deficiency in mice restrains the plasticity of the conversion of Th2, Th17 or Treg cells to Th1 cells (65).
The activity of epigenetic enzymes can also be modulated by the metabolism and can therefore affect CD4+ T cell plasticity. As an example, the metabolite 2-hydroxyglutarate is an inhibitor of ten-eleven translocation (TET) enzymes, which are implicated in DNA demethylation. The reduction of 2-hydroxyglutarate level in Th17 cells after aminooxy-acetic acid treatment led to the diminution of Foxp3 promoter methylation, therefore stabilizing FOXP3 expression and driving the Th17/Treg plasticity (74, 75). Epigenetic enzymes activity is dependent of the availability of metabolites like S-adenosylmethionine (SAM), a donor of the methyl group needed for DNA and histone methyltransferases activity and can therefore be affected by changes in their availability. An excess of SAM in the microenvironment may increase gene silencing by enhancing DNA methylation and affect CD4+ T cell plasticity (76).
Epigenetics in CD4+ T Cells Memory
Epigenetics is also involved in the maturation process of CD4+ T cells and the generation of the immune memory. Durek et al., (77), studied the epigenetic landscape of naive, central memory, effector memory, and terminally differentiated CD4+ T cells from the blood (77). They performed extensive epigenetic profiling: DNA methylation analysis, chromatin accessibility studies and ChIP sequencing for H3K4me1, H3K4me3, H3K9me3, H3K27ac, H3K27me3, and H3K36me3 as well as sequencing of coding and non-coding RNA of these cells. Their results showed a global loss of DNA methylation upon transition from the naive to the memory stages. Progressive changes in the transcriptomes and in the DNA accessibility profiles suggested that the differentiation of CD4+ T memory subsets support a linear model. Moreover, the authors identified important factors that drive or maintain the CD4+ memory phenotype. Among these factors, they identified many non-coding RNAs (ncRNAs), which were differentially expressed in naive and memory T cells. The transcription factor FOXP1 might act as an important regulator for the naive to memory transition as its expression was higher in naive CD4+ T cells compared to the other subsets of memory T cells. Foxp1 expression was repressed by DNA methylation in memory T cells.
Involvement of Epigenetic in the Control of Tumor Reactive CD4+ T Cells
It has been demonstrated that epigenetic modulation of CD4+ T cells polarization at tumor site might influence cancer patients’ outcome. Since CD4+ T cells polarization is epigenetically regulated, characterization of immune cells infiltration at tumor site may be assessed by epigenetic analysis. Among Treg cells, two subsets have been defined based on their developmental origin. Thymus-derived natural Tregs (nTregs) show a stable expression of FOXP3 associated with DNA demethylation, whereas peripherally induced Tregs (iTregs) cells do not express FOXP3 constitutively. In mouse tumor models and primary human tumors, analysis of DNA methylation patterns at the Foxp3 locus as well as functional studies revealed that Treg cells infiltrating in the TME mainly corresponded to nTreg and not iTreg cells (78). The functionality of conventional helper T cells as well as Treg cells in the TME can be modulated by epigenetics. Genome wide DNA methylation landscape of tumor infiltrating and blood CD4+ T cells from glioblastoma patients revealed differentially methylation pattern. The methylation changes were associated with transcriptomic changes for 341 genes in CD4+ tumor infiltrating T cells compared to blood. This study revealed that the TME may induce epigenetic alterations in tumor infiltrating CD4+ T cells (79). Depletion of tumor-associated macrophages (TAMs) in pancreatic cancer could also reprogram the epigenetic profile of tumor infiltrating CD8+ and CD4+ T cells. In a model of pancreatic cancer in mice, TAMs were depleted using trabectedin and the cytokine and epigenetic profile of T cells were assessed. Tumor infiltrating CD4+ T cells displayed a regulatory phenotype with high IL-10 expression and low IFN-γ production. On the contrary, in trabectedin-treated mice, the permissive histone mark H3K4me3 was decreased and the repressive mark H3K27me3 was increased at the Il-10 promoter resulting in low IL-10 expression. This study highlighted the role of TAMs in modulating the epigenetic profile of tumor infiltrating CD4+ T cells towards a pro-tumoral phenotype (80). In melanoma patients, EZH2 expression was increased in Treg cells infiltrating the tumor compared to Treg cells from the peripheral blood. Genetic deletion of Ezh2 in Treg cells from tumor-bearing mice reduced FOXP3 expression and modulated their functionality. Indeed, Ezh2-deficient tumor infiltrating Treg cells showed an increased production of the pro-inflammatory cytokines TNF-α, IFN-γ, and IL-2 and a reduced expression of IL-10. This observation was correlated with an enhanced recruitment and function of CD8+ and CD4+ effector T cells in the TME and led to the tumor elimination in mice (81). Epigenetics is also involved in CD4+ T cells exhaustion in the TME. The inhibitory receptor programmed cell death-1 (PD-1) was found to be epigenetically regulated in tumor-reactive lymphocytes. Indeed, upon T cell activation, the chromatin organizer special AT-rich sequence-binding protein-1 (SATB-1) inhibited PD-1 expression by recruiting a nucleosome remodeling deacetylase (NuRD) complex to Pdcd1 regulatory regions. Inhibition of STAB-1 in CD4+ and CD8+ T cells increased PD-1 expression and resulted in the loss of their effector activity more rapidly than wild-type lymphocytes. The transfer type -deficient tumor-reactive CD4+ T cells in Lewis Lung Carcinoma-bearing mice, resulted in a decreased survival rate. Therefore SATB-1 functions to prevent premature T cell exhaustion by regulating Pdcd1 expression upon T cell activation (82). Moreover, in colorectal cancer patients, other immune checkpoint inhibitors have been found to be epigenetically regulated in CD4+ and CD8+ tumor infiltrating lymphocytes. The promoter regions of T-cell immunoglobulin and mucin-domain containing-3 (Tim-3), Ctla-4, Pdcd1, Programmed cell death-ligand 1 (Pd-l1), Tox and Tox2 were highly demethylated whereas the promoter of Lag-3 was highly methylated. These results correlated with the transcriptional upregulation of Tim-3, Pd-1, Pd-l1, Ctla-4, Tox and Tox2 in CD4+ and CD8+ infiltrating colorectal cancer (83).
CD4+ T cells plasticity in the TME can be modulated by an epigenetic mechanism. Indeed, tumor associated macrophages (TAM) residing at tumor site can release exosomes containing miRNA. Microarray analysis of these exosomes revealed the presence of miR-29a-3p and miR-21-5p. In vitro, the treatment of CD4+ T cells by these miRNAs suppressed STAT3 and induced Th17 toward Treg commitment. These miRNAs can therefore lead to the development of Treg in the TME and thus engender an immunosuppressive context that facilitates cancer progression and metastasis (84). The plasticity of Th17 clones generated from human tumor-infiltrating lymphocytes after in vitro stimulation with OKT3 and irradiated allogeneic peripheral blood mononuclear cells was also demonstrated. Following expansion, the level of IL-17 production by these cells dropped whereas the expression of TNF-α, IFN-γ, IL-10 and TGF-β increased. Real time PCR analysis revealed that T-bet and Foxp3 expression gradually increased in Th17 clones with the expansions. The level of DNA methylation at the Foxp3 promoter in expanded Th17 cells decreased significantly with increasing stimulation and expansion cycles. These results indicated that the expansion of Th17 clones from tumor infiltrating lymphocytes promoted their conversion into mixed phenotypes that expressed FOXP3 and produced IFN-γ. These phenotypic changes resulted from the epigenetic reprogramming of lineage-specific genes (70). The expression of chemokines responsible for the attraction of CD4+ T cells at the tumor site can be modulated by epigenetics. Indeed, EZH2 (H3K27 HMT) as well as DNMT1 were found to repress the expression of the Th1-attracting chemokines CXCL9 and CXCL10 in primary ovarian cancer model. Th1 cells express the chemokine receptors CXCR3 and can thus be recruited by CXCL9 and CXCL10 enriched environments (56). The increase of the repressive mark H3K27me3 and DNA methylation at the promoter of Cxcl9 and Cxcl10 inhibited their expression and prevented Th1 cell infiltration in the TME. Inhibition of EZH2 and DNMT1 by chemical inhibitors restored the expression of CXCL9 and CXCL10 and increased effector T cell tumor infiltration (85).
Th1 cells recruitment in the TME can also be induced by the expression of endogenous retroviruses (ERV) by tumor cells. ERV originate from ancient retroviruses whose sequences are now permanently integrated in the genome. Their expression is normally repressed by DNA methylation. Increased ERV expression by DNA methyltransferases treatment can trigger the activation of viral defense pathways. Indeed, ERV can be recognized by Toll-like receptors (TLR) on the surface of innate immune cells and activate viral defense pathway. This induces IFN-γ production and triggers lymphocytes infiltration that may further favor antitumoral responses (86, 87).
How Manipulation of CD4+ T Cells by Epigenetic Therapies Can Be Used as a Strategy to Improve Anticancer Immunotherapy in Solid Tumors?
Evidence support the rational to stimulate CD4+ T cell responses for anticancer immunotherapies (88–90). Considering the important role played by epigenetics in CD4+ T cells differentiation and plasticity, strategies of epigenetic reprogramming gained growing interest to promote appropriate antitumor CD4+ helper T cells in the TME (Figure 3).
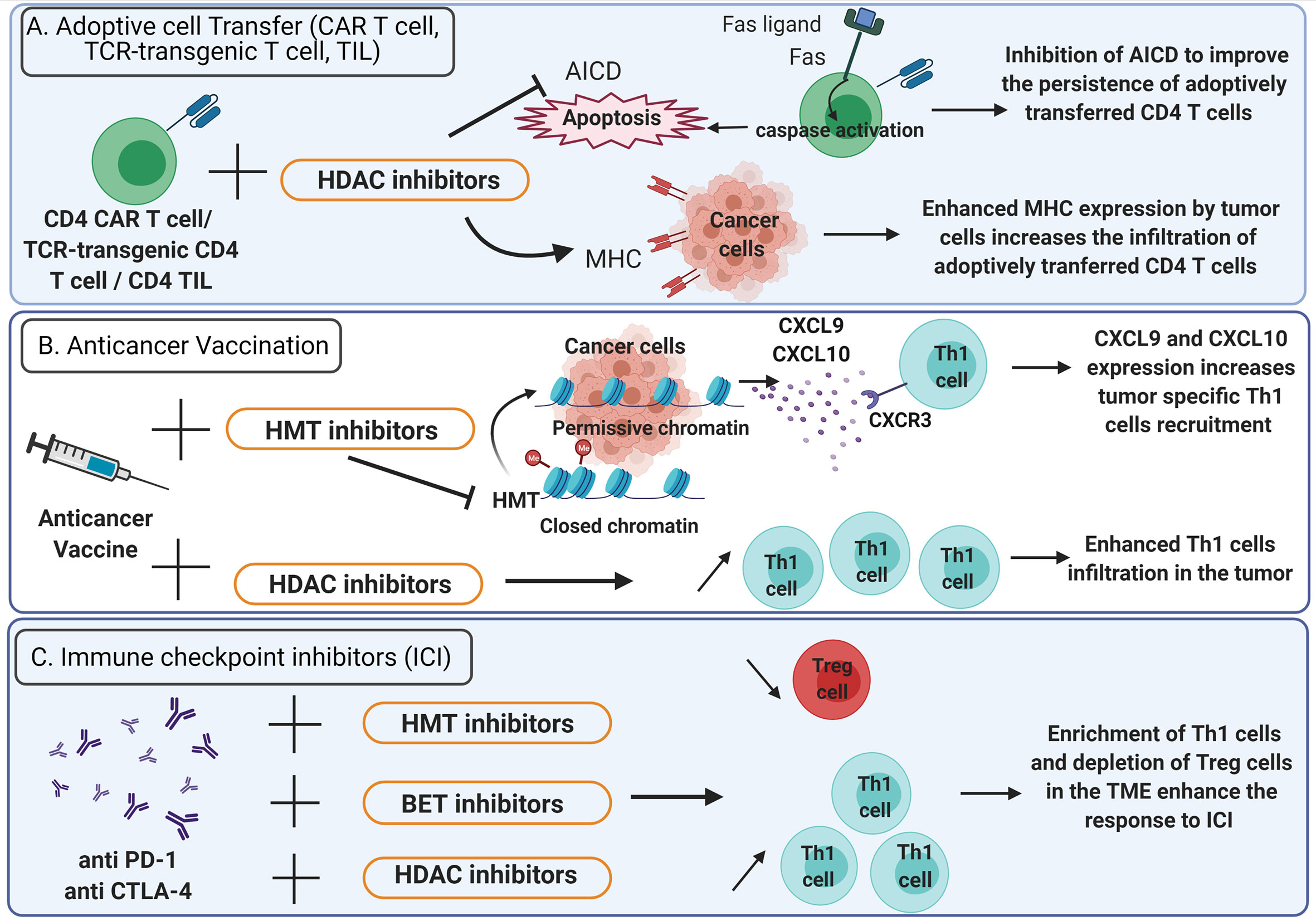
Figure 3 Epigenetic therapies potentiate the efficacy of anticancer immunotherapy. (A) Adoptive cell transfer. To improve the persistence of adoptively transferred CD4+ CAR T cells, TCR-transgenic CD4+ T cells or CD4+ TIL in the TME, activation induced cell death (AICD) can be inhibited using HDAC inhibitors (bar-headed line). AICD is mediated by Fas-Fas ligand interactions and triggers the activation of caspases that lead to apoptosis (solid arrows). HDAC inhibitors also enhance MHC expression on tumor cells thus increasing the infiltration of adoptively transferred CD4+ T cells. (B) Anticancer vaccination: HDAC inhibitors favor Th1 cells enrichment in the TME. HMT inhibitors promote the expression of CXCL9 and CXCL10 by tumor cells and enhance the recruitment of Th1 cells expressing CXCR3. (C) Immune checkpoint inhibitors: the efficacy of anti-PD-1 and anti-CTLA-4 is enhanced by HDAC, HMT and BET inhibitors which favor a TME enriched in Th1 cells and depleted in Treg cells. TME, Tumor microenvironment; TIL, Tumor infiltrating lymphocytes; CAR T cells, chimeric antigen receptor T cells; HDAC, Histone deacetylase; HMT, histone methyltransferase; BET, bromodomain and extra-terminal domain family protein.
Adoptive transfer of tumor specific CD4+ lymphocytes has proved its efficacy in the treatment of cancer (91–93). However, it is uncertain whether CD4+ T cell polarization status, acquired during in vitro stimulation, can sustain in the TME after T cell infusion. Thus, an epigenetic treatment could be administered to preserve CD4+ T cells polarization in the TME. It has been shown that epigenetic modulators can improve the efficacy of adoptive CD4+ T cell therapies by increasing MHC expression on tumor cells (Figure 3A). Several HDAC inhibitors (HDACi) such as chidamide, entinostat, vorinostat and CXD101 have been found to upregulate MHC class II expression in various cancer cell lines (94). Moreover, the expression of a constitutively active STAT5 variant (CASTAT5) by CD4+ chimeric antigen receptor (CAR) T cells targeting the B cell antigen CD19 could improve the polyfunctionality, expansion and persistence of these cells in the TME by an epigenetic mechanism. Indeed, CASTAT5-transduced CD4+ T cells displayed a genome-wide transcriptional and epigenetic remodeling. Assay for transposase-accessible chromatin using sequencing (ATAC-seq) performed on CASTAT5 CD4+ T cells identified an increased chromatin accessibility at the gene loci of Il-4, Il-13, Il-9, and GzmB but not at the Ifn- γ locus. These results were concordant with gene transcription and protein expression profiles and indicated that persistent STAT5 activation reprogrammed the epigenetic landscape of CD4+ T cells to drive polyfunctionality. In B cell lymphoma-bearing mice, the adoptive transfer of CD19 CAR T cells resulted in only transient tumor regression whereas the adoptive transfer of CASTAT5 CD19 CAR T cells was curative to nearly all mice (95). In adoptive cell transfer, the induction of CD4+ T cells persistence at tumor site represents a critical issue (96). For example, Fas-Fas ligand dependent activation-induced cell death (AICD) can be prevented by HDACi. Indeed, in a mouse model, the use of HDACi inhibits AICD of CD4+ T cells in the TME (97) (Figure 3A). Recently, it has been demonstrated that a low dose decitabine priming could enhance the persistence and the antitumor activity of CD4+ CAR T cells. Indeed, the treatment of CD4+ and CD8+ CAR T cells with the DNA methyltransferase inhibitor decitabine could upregulate the expression of memory and proliferation associated genes and downregulate the expression of T cell exhaustion related genes: Lag-3 and Ctla-4. In vitro, after co-culture with Raji tumor cells, the proliferation of CD4+ CAR T cells treated with decitabine was enhanced as well as the expression of IL-2, IFN-γ, TNF-α, Perforin, Granzyme A and Granzyme B. Upon activation, CD4+ CAR T cells treated with decitabine showed a significative increase of the expression of the chemokines CXCL8, CXCL10, CXCL1, CCL3 and CCL1. In vivo, decitabine pre-treated CD4+ and CD8+ CAR T cells targeting the CD19 antigen resulted in complete tumor regression in mice and persisted longer than untreated CAR T cells (98).
The CD4+ T cell help is critical for the success of anticancer vaccines (99, 100). Th1 cells are essential for the induction of an effective antitumor response after vaccination (4). Thus, the administration of epigenetic therapies to modulate the CD4+ T cells polarization in the context of anticancer vaccine can be considered (Figure 3B). In preclinical models of triple-negative breast cancer 4T1 and colon carcinoma MC38, Hicks et al. (101), demonstrated that the addition of entinostat, a class I HDACi, to vaccine significantly improved the tumor control. The entinostat treatment reprogrammed the TME toward an inflamed phenotype with an enhanced expression of proinflammatory genes (IFN-γ, TNF-α) and a decrease of regulatory T cells in the TME (101) (Figure 3B). This study highlights the promising use of epigenetic therapeutics to improve the response to anticancer vaccines.
The immune checkpoint inhibitors (ICI) such as anti-CTLA-4 antibody as well as anti-PD-1, have revolutionized the treatment of several cancers but fail to control cancer progression in a significant proportion of patients (102–104). In addition, primary resistance to ICI has been observed in other types of cancer, such as pancreatic cancer and glioblastoma (105–107). The current challenge consists of using combined therapy approaches to improve ICI efficacy (108). Recent findings indicate that CD4+ helper T cells can influence the response to immunotherapy in the success of ICI (109, 110). In patients with bone metastases of castration-resistant prostate cancer, increased Th17 instead of Th1 cells were found in bone metastases after ICI therapy and this correlated with a reduced efficacy of ICI compared to Th1 cells infiltration (111). Thus, these observations support the rational to combine ICI with epigenetic therapies that induce appropriate CD4+ helper T cells polarization in the TME (Figure 3C). In a murine hepatocellular carcinoma model, the administration of the HDACi belinostat improved the response to CTLA-4 inhibition. The production of IFN-γ by tumor-reactive CD8+ T cells was enhanced and the number of splenic Treg cells was decreased in mice treated with both CTLA-4 and belinostat combination (112). Moreover, the co-administration of the HDACi trichostatin A and anti-CTLA-4 could enhance the infiltration of CD4+ T cells and was associated with better antitumor effects (97) (Figure 3C). In a mouse model of lung cancer, the use of JQ1, a bromodomain-targeted BET inhibitor, could also enhance the response to anti-PD-1 antibodies by reprogramming CD4+ T cells in the TME. Indeed, JQ1 promoted an increase of Th1 cells at tumor site as well as depletion of Treg cells and improved survival compared to mice treated with ICI alone (113). Moreover, blocking EZH2 expression by the pharmacological inhibitor CPI-1205 in MB49 tumor-bearing mice was associated with a better survival when combined with anti-CTLA-4. These results were correlated with an increased infiltration of Th1 cells and cytotoxic effector T cells in the TME as well as a depletion of Treg cells (114). In mouse and human prostate cancer organoids models, inhibition of EZH2 increased the expression of the Th1 attracting-chemokines CXCL9 and CXCL10 and derepressed endogenous double-strand RNA (ds RNA) expression. Ds RNA expression thus triggered “viral mimicry pathway” and the expression of interferon-stimulated genes via the ds RNA sensor STING. EZH2 inhibition in murine prostate cancer cell lines with DZNep and EPZ also resulted in a drastic upregulation of Th1 cytokines TNF-α, IL-2 and IL-12. Overall, EZH2 inhibition increased CD4+ and CD8+ tumor infiltration in mice and potentiate prostate cancer response to anti-PD-1 therapy (115). Additionally, the HDACi CG-745 enhanced the anti-cancer effect of anti-PD-1 therapy in syngeneic tumor mouse models by remodeling the immune microenvironment. Indeed, CG-745 increased the proliferation of helper T cells, cytotoxic T cells and NK cells, while decreasing proliferation of regulatory T cells and inhibiting myeloid-derived suppressor cells as well as M2 macrophage polarization (116). In the triple-negative 4T1 breast cancer mouse model, HDACi enhanced the in vivo response to PD-1/CTLA-4 blockade. This effect was attributed to the up-regulation of PD-L1 and HLA-DR on tumor cells as well as the decrease of the recruitment of FOXP3+ CD4+ T cells in the TME (117). By modulating CD4+ T cells polarization and recruitment at tumor site, epigenetic therapies induce a favorable immune context in the TME that improve the response to ICI (Figure 3C). The role of HDACi as an immunomodulatory agent has been extensively studied (118, 119). However, there is limited knowledge concerning the effects of HDACi on T helper cells differentiation in clinical settings (120, 121). Example of epigenetic modulators currently approved or under clinical trials are listed in Table 1.
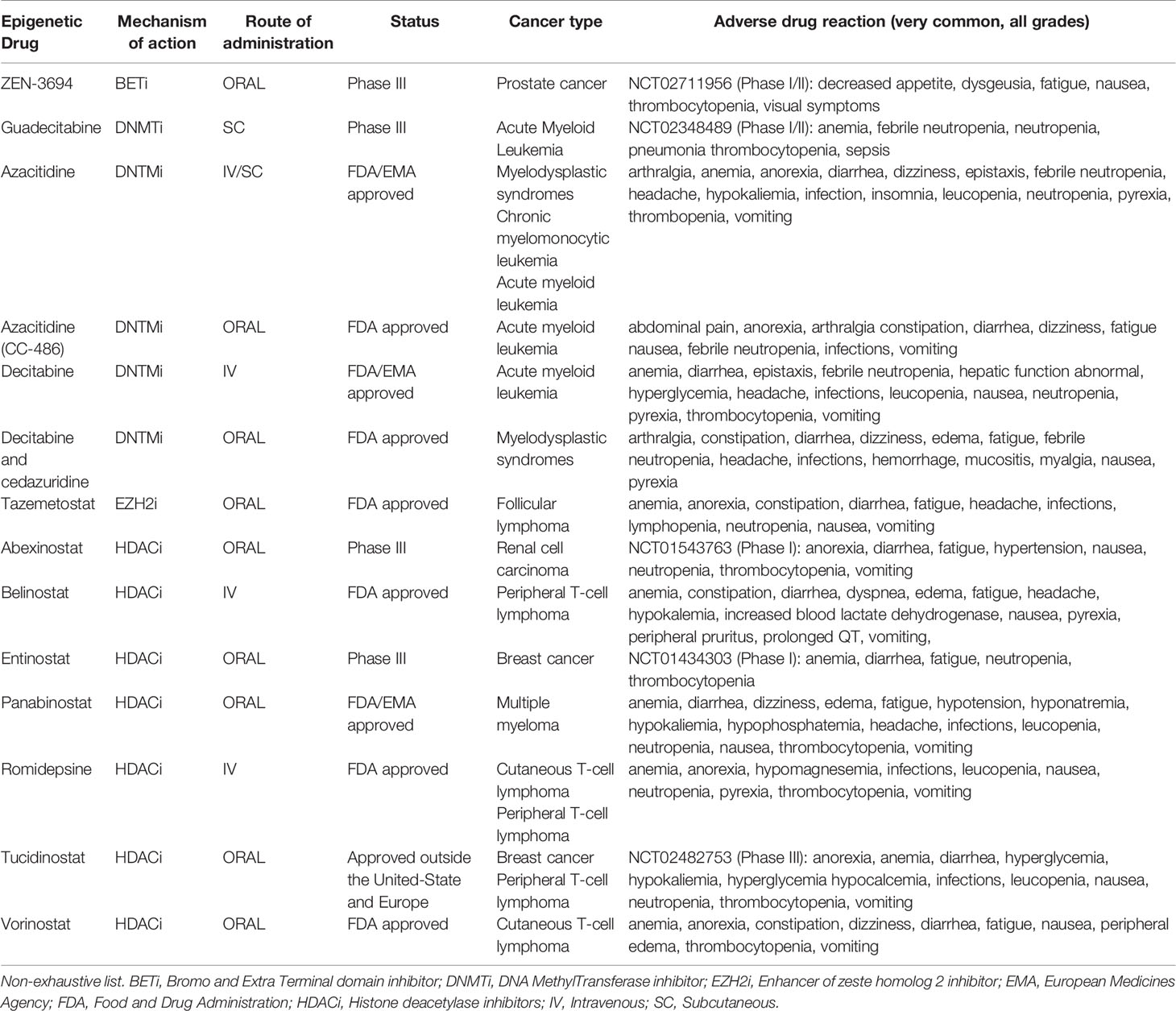
Table 1 Epigenetic modulators that may influence CD4+ T cells anticancer immunity.
Discussion
CD4+ T cells mediate antitumor immune response and are involved in the response to anticancer immunotherapies. We have seen that epigenetics plays a key role in the regulation of CD4+ T cells differentiation, plasticity and memory formation. Moreover, we described how the TME could reprogram tumor reactive CD4+ T cells epigenetic landscape and modulate their functionality and recruitment at tumor site by regulating the expression of immune inflammatory cytokines and attracting chemokines. Epigenetics also control the expression of immune checkpoints, thus regulating tumor reactive CD4+ T cells exhaustion. Additionally, the conversion of Th17 cells to Treg cells in the TME was found to be regulated by miRNAs. Since the type of CD4+ T cell infiltrating the tumor can affect differently the prognosis of patients and the response to immunotherapy, epigenetic modulators can be used to induce appropriate CD4+ helper T cells polarization and recruitment in the TME. In this review, we presented recent literature showing the growing interest of combining epigenetic treatments with immunotherapy. Epigenetic therapies can decrease the number of Treg cells in the TME or their immunosuppressive capacities, therefore improving the response to immune checkpoint blockades. Th1 cells recruitment at tumor site can be enhanced by the epigenetically induced re-expression of endogenous retroviruses or Th1-attracting chemokines (CXCL9 and CXCL10) by tumor cells. The persistence and resistance to exhaustion of adoptively transferred CD4+ T cells in the TME can be preserved by epigenetic treatments. The combination of epigenetic therapies with anticancer vaccines can also promote the expression of proinflammatory genes by CD4+ T cells, thus enhancing the anticancer immune response. Therefore, the combination of epigenetic treatment and immunotherapy provides new insights in anticancer therapy. However, the role of epigenetics on the antitumor specific immune responses remains poorly characterized. This highlights the need to investigate the role of epigenetics in CD4+ T cells differentiation and plasticity in the TME. To bring answers to these issues, our team is currently screening hundreds of drugs targeting epigenetic enzymes to understand and modulate the polarization of expanded tumor infiltrating lymphocytes.
Author Contributions
ER, MK, and OA wrote the manuscript. RL, EH, PP, and CB edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by funding from institutional grants from INSERM, EFS and Univ. Bourgogne Franche-Comté and by the “Ligue Contre le Cancer”, the “Région Bourgogne Franche-Comté (projet d’envergure structurant C-ICI)”and european founds “Programme Interreg France-Suisse 2014-2020 (FEDER) – Projet R-TIC”.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chraa D, Naim A, Olive D, Badou A. T Lymphocyte Subsets in Cancer Immunity: Friends or Foes. J Leukoc Biol (2019) 105:243–55. doi: 10.1002/JLB.MR0318-097R1
2. Kim HJ, Cantor H. CD4 T-Cell Subsets and Tumor Immunity: The Helpful and the Not-So-Helpful. Cancer Immunol Res (2014) 2:91–8. doi: 10.1158/2326-6066.CIR-13-0216
3. Bevan MJ. Helping the CD8+ T-Cell Response. Nat Rev Immunol (2004) 4:595–602. doi: 10.1038/nri1413
4. Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4+ T Cell Help in Cancer Immunology and Immunotherapy. Nat Rev Immunol (2018) 18:635–47. doi: 10.1038/s41577-018-0044-0
5. Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, et al. CD4+ T Cell Help is Required for the Formation of a Cytolytic CD8+ T Cell Subset That Protects Against Chronic Infection and Cancer. Immunity (2019) 51:1028–42. doi: 10.1016/j.immuni.2019.10.009
6. Bos R, Sherman LA. CD4+ T-Cell Help in the Tumor Milieu is Required for Recruitment and Cytolytic Function of CD8+ T Lymphocytes. Cancer Res (2010) 70:8368–77. doi: 10.1158/0008-5472.CAN-10-1322
7. Ahrends T, Spanjaard A, Pilzecker B, Bąbała N, Bovens A, Xiao Y, et al. CD4+ T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity (2017) 47:848–61. doi: 10.1016/j.immuni.2017.10.009
8. Jabrane-Ferrat N, Faille A, Loiseau P, Poirier O, Charron D, Calvo F. Effect of Gamma Interferon on HLA Class-I and -II Transcription and Protein Expression in Human Breast Adenocarcinoma Cell Lines. Int J Cancer (1990) 45:1169–76. doi: 10.1002/ijc.2910450630
9. Wang N, Liang H, Zen K. Molecular Mechanisms That Influence the Macrophage M1-M2 Polarization Balance. Front Immunol (2014) 5:614. doi: 10.3389/fimmu.2014.00614
10. Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, et al. Tumour Ischaemia by Interferon-γ Resembles Physiological Blood Vessel Regression. Nature (2017) 545:98–102. doi: 10.1038/nature22311
11. Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The Immune Contexture in Cancer Prognosis and Treatment. Nat Rev Clin Oncol (2017) 14:717–34. doi: 10.1038/nrclinonc.2017.101
12. Zhu J, Yamane H, Cote-Sierra J, Guo L, Paul WE. GATA-3 Promotes Th2 Responses Through Three Different Mechanisms: Induction of Th2 Cytokine Production, Selective Growth of Th2 Cells and Inhibition of Th1 Cell-Specific Factors. Cell Res (2006) 16:3–10. doi: 10.1038/sj.cr.7310002
13. Kusuda T, Shigemasa K, Arihiro K, Fujii T, Nagai N, Ohama K. Relative Expression Levels of Th1 and Th2 Cytokine mRNA are Independent Prognostic Factors in Patients With Ovarian Cancer. Oncol Rep (2005) 13:1153–8. doi: 10.3892/or.13.6.1153
14. De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T Helper Type 2 Cell Infiltrate Correlates With Cancer-Associated Fibroblast Thymic Stromal Lymphopoietin Production and Reduced Survival in Pancreatic Cancer. J Exp Med (2011) 208:469–78. doi: 10.1084/jem.20101876
15. Kitajima M, Ito T, Tumes DJ, Endo Y, Onodera A, Hashimoto K, et al. Memory Type 2 Helper T Cells Induce Long-Lasting Antitumor Immunity by Activating Natural Killer Cells. Cancer Res (2011) 71:4790–8. doi: 10.1158/0008-5472.CAN-10-1572
16. Liu M, Kuo F, Capistrano KJ, Kang D, Nixon BG, Shi W, et al. Tgf-β Suppresses Type 2 Immunity to Cancer. Nature (2020) 587:115–20. doi: 10.1038/s41586-020-2836-1
17. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The Immune Contexture in Human Tumours: Impact on Clinical Outcome. Nat Rev Cancer (2012) 12:298–306. doi: 10.1038/nrc3245
18. Galon J, Bruni D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity (2020) 52:55–81. doi: 10.1016/j.immuni.2019.12.018
19. Asadzadeh Z, Mohammadi H, Safarzadeh E, Hemmatzadeh M, Mahdian-Shakib A, Jadidi-Niaragh F, et al. The Paradox of Th17 Cell Functions in Tumor Immunity. Cell Immunol (2017) 322:15–25. doi: 10.1016/j.cellimm.2017.10.015
20. Zhang J-P, Yan J, Xu J, Pang X-H, Chen M-S, Li L, et al. Increased Intratumoral IL-17-Producing Cells Correlate With Poor Survival in Hepatocellular Carcinoma Patients. J Hepatol (2009) 50:980–9. doi: 10.1016/j.jhep.2008.12.033
21. He S, Fei M, Wu Y, Zheng D, Wan D, Wang L, et al. Distribution and Clinical Significance of Th17 Cells in the Tumor Microenvironment and Peripheral Blood of Pancreatic Cancer Patients. Int J Mol Sci (2011) 12:7424–37. doi: 10.3390/ijms12117424
22. Sfanos KS, Bruno TC, Maris CH, Xu L, Thoburn CJ, DeMarzo AM, et al. Phenotypic Analysis of Prostate-Infiltrating Lymphocytes Reveals TH17 and Treg Skewing. Clin Cancer Res (2008) 14:3254–61. doi: 10.1158/1078-0432.CCR-07-5164
23. Ye Z-J, Zhou Q, Gu Y-Y, Qin S-M, Ma W-L, Xin J-B, et al. Generation and Differentiation of IL-17-Producing CD4+ T Cells in Malignant Pleural Effusion. J Immunol (2010) 185:6348–54. doi: 10.4049/jimmunol.1001728
24. Alves JJP, De Medeiros Fernandes TAA, De Araújo JMG, Cobucci RNO, Lanza DCF, Bezerra FL, et al. Th17 Response in Patients With Cervical Cancer. Oncol Lett (2018) 16:6215–27. doi: 10.3892/ol.2018.9481
25. Liu J, Duan Y, Cheng X, Chen X, Xie W, Long H, et al. IL-17 is Associated With Poor Prognosis and Promotes Angiogenesis via Stimulating VEGF Production of Cancer Cells in Colorectal Carcinoma. Biochem Biophys Res Commun (2011) 407:348–54. doi: 10.1016/j.bbrc.2011.03.021
26. Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can Promote Tumor Growth Through an IL-6-Stat3 Signaling Pathway. J Exp Med (2009) 206:1457–64. doi: 10.1084/jem.20090207
27. He D, Li H, Yusuf N, Elmets CA, Li J, Mountz JD, et al. IL-17 Promotes Tumor Development Through the Induction of Tumor Promoting Microenvironments at Tumor Sites and Myeloid-Derived Suppressor Cells. J Immunol (2010) 184:2281–8. doi: 10.4049/jimmunol.0902574
28. Song Y, Yang JM. Role of Interleukin (IL)-17 and T-Helper (Th)17 Cells in Cancer. Biochem Biophys Res Commun (2017) 493:1–8. doi: 10.1016/j.bbrc.2017.08.109
29. Jarnicki AG, Lysaght J, Todryk S, Mills KHG. Suppression of Antitumor Immunity by IL-10 and TGF-Beta-Producing T Cells Infiltrating the Growing Tumor: Influence of Tumor Environment on the Induction of CD4+ and CD8+ Regulatory T Cells. J Immunol (2006) 177:896–904. doi: 10.4049/jimmunol.177.2.896
30. Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, et al. Interleukin-35 Limits Anti-Tumor Immunity. Immunity (2016) 44:316–29. doi: 10.1016/j.immuni.2016.01.013
31. Barilla RM, Diskin B, Caso RC, Lee KB, Mohan N, Buttar C, et al. Specialized Dendritic Cells Induce Tumor-Promoting IL-10+IL-17+ FOXP3neg Regulatory CD4+ T Cells in Pancreatic Carcinoma. Nat Commun (2019) 10:1424. doi: 10.1038/s41467-019-09416-2
32. Togashi Y, Shitara K, Nishikawa H. Regulatory T Cells in Cancer Immunosuppression - Implications for Anticancer Therapy. Nat Rev Clin Oncol (2019) 16:356–71. doi: 10.1038/s41571-019-0175-7
33. Das M, Zhu C, Kuchroo VK. Tim-3 and its Role in Regulating Anti-Tumor Immunity. Immunol Rev (2017) 276:97–111. doi: 10.1111/imr.12520
34. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T Cells Inhibit Dendritic Cells by Lymphocyte Activation Gene-3 Engagement of MHC Class II. J Immunol (2008) 180:5916–26. doi: 10.4049/jimmunol.180.9.5916
35. Andrews LP, Marciscano AE, Drake CG, Vignali DAA. LAG3 (CD223) as a Cancer Immunotherapy Target. Immunol Rev (2017) 276:80–96. doi: 10.1111/imr.12519
36. Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, et al. Tumor-Infiltrating FOXP3+ T Regulatory Cells Show Strong Prognostic Significance in Colorectal Cancer. J Clin Oncol (2009) 27:186–92. doi: 10.1200/JCO.2008.18.7229
37. Frey DM, Droeser RA, Viehl CT, Zlobec I, Lugli A, Zingg U, et al. High Frequency of Tumor-Infiltrating FOXP3+ Regulatory T Cells Predicts Improved Survival in Mismatch Repair-Proficient Colorectal Cancer Patients. Int J Cancer (2010) 126:2635–43. doi: 10.1002/ijc.24989
38. Crotty S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity (2019) 50:1132–48. doi: 10.1016/j.immuni.2019.04.011
39. Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4⁺ Follicular Helper T Cell Infiltration Predicts Breast Cancer Survival. J Clin Invest (2013) 123:2873–92. doi: 10.1172/JCI67428
40. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity (2013) 39:782–95. doi: 10.1016/j.immuni.2013.10.003
41. Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, et al. Molecular Basis for Site-Specific Read-Out of Histone H3k4me3 by the BPTF PHD Finger of NURF. Nature (2006) 442:91–5. doi: 10.1038/nature04802
42. Li Y, Schulz VP, Deng C, Li G, Shen Y, Tusi BK, et al. Setd1a and NURF Mediate Chromatin Dynamics and Gene Regulation During Erythroid Lineage Commitment and Differentiation. Nucleic Acids Res (2016) 44:7173–88. doi: 10.1093/nar/gkw327
43. Turner BM. Cellular Memory and the Histone Code. Cell (2002) 111:285–91. doi: 10.1016/s0092-8674(02)01080-2
44. Meier K, Recillas-Targa F. New Insights on the Role of DNA Methylation From a Global View. Front Biosci (2017) 22:644–68. doi: 10.2741/4508
45. Kung JTY, Colognori D, Lee JT. Long Noncoding Rnas: Past, Present, and Future. Genetics (2013) 193:651–69. doi: 10.1534/genetics.112.146704
46. Baguet A, Bix M. Chromatin Landscape Dynamics of the Il4-Il13 Locus During T Helper 1 and 2 Development. Proc Natl Acad Sci USA (2004) 101:11410–5. doi: 10.1073/pnas.0403334101
47. Morinobu A, Kanno Y, O’Shea JJ. Discrete Roles for Histone Acetylation in Human T Helper 1 Cell-Specific Gene Expression. J Biol Chem (2004) 279:40640–6. doi: 10.1074/jbc.M407576200
48. Miyatake S, Arai N, Arai K. Chromatin Remodeling and T Helper Subset Differentiation. IUBMB Life (2000) 49:473–8. doi: 10.1080/15216540050166990
49. Kaneko T, Hosokawa H, Yamashita M, Wang C-R, Hasegawa A, Kimura MY, et al. Chromatin Remodeling at the Th2 Cytokine Gene Loci in Human Type 2 Helper T Cells. Mol Immunol (2007) 44:2249–56. doi: 10.1016/j.molimm.2006.11.004
50. Zhang F, Boothby M. T Helper Type 1-Specific Brg1 Recruitment and Remodeling of Nucleosomes Positioned at the IFN-Gamma Promoter are Stat4 Dependent. J Exp Med (2006) 203:1493–505. doi: 10.1084/jem.20060066
51. Chang S, Collins PL, Aune TM. T-Bet Dependent Removal of Sin3A-Histone Deacetylase Complexes at the Ifng Locus Drives Th1 Differentiation. J Immunol (2008) 181:8372–81. doi: 10.4049/jimmunol.181.12.8372
52. Hwang ES, Szabo SJ, Schwartzberg PL, Glimcher LH. T Helper Cell Fate Specified by Kinase-Mediated Interaction of T-Bet With GATA-3. Science (2005) 307:430–3. doi: 10.1126/science.1103336
53. Lazarevic V, Chen X, Shim J-H, Hwang E-S, Jang E, Bolm AN, et al. Transcription Factor T-Bet Represses TH17 Differentiation by Preventing Runx1-Mediated Activation of the Rorγt Gene. Nat Immunol (2011) 12:96–104. doi: 10.1038/ni.1969
54. Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, et al. The Polycomb Protein Ezh2 Regulates Differentiation and Plasticity of CD4(+) T Helper Type 1 and Type 2 Cells. Immunity (2013) 39:819–32. doi: 10.1016/j.immuni.2013.09.012
55. Allan RS, Zueva E, Cammas F, Schreiber HA, Masson V, Belz GT, et al. An Epigenetic Silencing Pathway Controlling T Helper 2 Cell Lineage Commitment. Nature (2012) 487:249–53. doi: 10.1038/nature11173
56. Zhang Y, Zhang Y, Gu W, Sun B. TH1/TH2 Cell Differentiation and Molecular Signals. Adv Exp Med Biol (2014) 841:15–44. doi: 10.1007/978-94-017-9487-9_2
57. Cote-Sierra J, Foucras G, Guo L, Chiodetti L, Young HA, Hu-Li J, et al. Interleukin 2 Plays a Central Role in Th2 Differentiation. Proc Natl Acad Sci USA (2004) 101:3880–5. doi: 10.1073/pnas.0400339101
58. Renaude E, Kroemer M, Loyon R, Binda D, Borg C, Guittaut M, et al. The Fate of Th17 Cells is Shaped by Epigenetic Modifications and Remodeled by the Tumor Microenvironment. Int J Mol Sci (2020) 21:1673. doi: 10.3390/ijms21051673
59. Hasan M, Neumann B, Haupeltshofer S, Stahlke S, Fantini MC, Angstwurm K, et al. Activation of TGF-β-Induced Non-Smad Signaling Pathways During Th17 Differentiation. Immunol Cell Biol (2015) 93:662–72. doi: 10.1038/icb.2015.21
60. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, et al. Diverse Targets of the Transcription Factor STAT3 Contribute to T Cell Pathogenicity and Homeostasis. Immunity (2010) 32:605–15. doi: 10.1016/j.immuni.2010.05.003
61. Jiang Y, Liu Y, Lu H, Sun S-C, Jin W, Wang X, et al. Epigenetic Activation During T Helper 17 Cell Differentiation Is Mediated by Tripartite Motif Containing 28. Nat Commun (2018) 9:1424. doi: 10.1038/s41467-018-03852-2
62. Wieczorek G, Asemissen A, Model F, Turbachova I, Floess S, Liebenberg V, et al. Quantitative DNA Methylation Analysis of FOXP3 as a New Method for Counting Regulatory T Cells in Peripheral Blood and Solid Tissue. Cancer Res (2009) 69:599–608. doi: 10.1158/0008-5472.CAN-08-2361
63. Schmidl C, Klug M, Boeld TJ, Andreesen R, Hoffmann P, Edinger M, et al. Lineage-Specific DNA Methylation in T Cells Correlates With Histone Methylation and Enhancer Activity. Genome Res (2009) 19:1165–74. doi: 10.1101/gr.091470.109
64. Wang L, Liu Y, Beier UH, Han R, Bhatti TR, Akimova T, et al. Foxp3+ T-Regulatory Cells Require DNA Methyltransferase 1 Expression to Prevent Development of Lethal Autoimmunity. Blood (2013) 121:3631–9. doi: 10.1182/blood-2012-08-451765
65. Li Q, Zou J, Wang M, Ding X, Chepelev I, Zhou X, et al. Critical Role of Histone Demethylase Jmjd3 in the Regulation of CD4 + T-Cell Differentiation. Nat Commun (2014) 5:5780. doi: 10.1038/ncomms6780
66. Li F, Zeng Z, Xing S, Gullicksrud JA, Shan Q, Choi J, et al. Ezh2 Programs TFH Differentiation by Integrating Phosphorylation-Dependent Activation of Bcl6 and Polycomb-Dependent Repression of p19Arf. Nat Commun (2018) 9:5452. doi: 10.1038/s41467-018-07853-z
67. Chen X, Cao G, Wu J, Wang X, Pan Z, Gao J, et al. The Histone Methyltransferase EZH2 Primes the Early Differentiation of Follicular Helper T Cells During Acute Viral Infection. Cell Mol Immunol (2020) 17:247–60. doi: 10.1038/s41423-019-0219-z
68. Cosmi L, Maggi L, Santarlasci V, Capone M, Cardilicchia E, Frosali F, et al. Identification of a Novel Subset of Human Circulating Memory CD4(+) T Cells That Produce Both IL-17A and IL-4. J Allergy Clin Immunol (2010) 125:222–30. doi: 10.1016/j.jaci.2009.10.012
69. Obermajer N, Popp FC, Soeder Y, Haarer J, Geissler EK, Schlitt HJ, et al. Conversion of Th17 Into IL-17A(Neg) Regulatory T Cells: A Novel Mechanism in Prolonged Allograft Survival Promoted by Mesenchymal Stem Cell-Supported Minimized Immunosuppressive Therapy. J Immunol (2014) 193:4988–99. doi: 10.4049/jimmunol.1401776
70. Ye J, Su X, Hsueh EC, Zhang Y, Koenig JM, Hoft DF, et al. Human Tumor-Infiltrating Th17 Cells Have the Capacity to Differentiate Into IFN-γ+ and FOXP3+ T Cells With Potent Suppressive Function. Eur J Immunol (2011) 41:936–51. doi: 10.1002/eji.201040682
71. Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, et al. Global Mapping of H3k4me3 and H3k27me3 Reveals Specificity and Plasticity in Lineage Fate Determination of Differentiating CD4+ T Cells. Immunity (2009) 30:155–67. doi: 10.1016/j.immuni.2008.12.009
72. Kanno Y, Vahedi G, Hirahara K, Singleton K, O’Shea JJ. Transcriptional and Epigenetic Control of T Helper Cell Specification: Molecular Mechanisms Underlying Commitment and Plasticity. Annu Rev Immunol (2012) 30:707–31. doi: 10.1146/annurev-immunol-020711-075058
73. Mukasa R, Balasubramani A, Lee YK, Whitley SK, Weaver BT, Shibata Y, et al. Epigenetic Instability of Cytokine and Transcription Factor Gene Loci Underlies Plasticity of the T Helper 17 Cell Lineage. Immunity (2010) 32:616–27. doi: 10.1016/j.immuni.2010.04.016
74. Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK, et al. Metabolic Control of TH17 and Induced Treg Cell Balance by an Epigenetic Mechanism. Nature (2017) 548:228–33. doi: 10.1038/nature23475
75. Shim E-H, Livi CB, Rakheja D, Tan J, Benson D, Parekh V, et al. L-2-Hydroxyglutarate: An Epigenetic Modifier and Putative Oncometabolite in Renal Cancer. Cancer Discov (2014) 4:1290–8. doi: 10.1158/2159-8290.CD-13-0696
76. Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, et al. Histone Methylation Dynamics and Gene Regulation Occur Through the Sensing of One-Carbon Metabolism. Cell Metab (2015) 22:861–73. doi: 10.1016/j.cmet.2015.08.024
77. Durek P, Nordström K, Gasparoni G, Salhab A, Kressler C, de Almeida M, et al. Epigenomic Profiling of Human CD4+ T Cells Supports a Linear Differentiation Model and Highlights Molecular Regulators of Memory Development. Immunity (2016) 45:1148–61. doi: 10.1016/j.immuni.2016.10.022
78. Waight JD, Takai S, Marelli B, Qin G, Hance KW, Zhang D, et al. Cutting Edge: Epigenetic Regulation of FOXP3 Defines a Stable Population of CD4+ Regulatory T Cells in Tumors From Mice and Humans. J Immunol (2015) 194:878–82. doi: 10.4049/jimmunol.1402725
79. Bam M, Chintala S, Fetcko K, Williamsen BC, Siraj S, Liu S, et al. Genome Wide DNA Methylation Landscape Reveals Glioblastoma’s Influence on Epigenetic Changes in Tumor Infiltrating CD4+ T Cells. Oncotarget (2021) 12:967–81. doi: 10.18632/oncotarget.27955
80. Borgoni S, Iannello A, Cutrupi S, Allavena P, D’Incalci M, Novelli F, et al. Depletion of Tumor-Associated Macrophages Switches the Epigenetic Profile of Pancreatic Cancer Infiltrating T Cells and Restores Their Anti-Tumor Phenotype. Oncoimmunology (2018) 7:e1393596. doi: 10.1080/2162402X.2017.1393596
81. Wang D, Quiros J, Mahuron K, Pai C-C, Ranzani V, Young A, et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep (2018) 23:3262–74. doi: 10.1016/j.celrep.2018.05.050
82. Stephen TL, Payne KK, Chaurio RA, Allegrezza MJ, Zhu H, Perez-Sanz J, et al. SATB1 Expression Governs Epigenetic Repression of PD-1 in Tumor-Reactive T Cells. Immunity (2017) 46:51–64. doi: 10.1016/j.immuni.2016.12.015
83. Sasidharan Nair V, Saleh R, Toor SM, Taha RZ, Ahmed AA, Kurer MA, et al. Epigenetic Regulation of Immune Checkpoints and T Cell Exhaustion Markers in Tumor-Infiltrating T Cells of Colorectal Cancer Patients. Epigenomics (2020) 12:1871–82. doi: 10.2217/epi-2020-0267
84. Zhou J, Li X, Wu X, Zhang T, Zhu Q, Wang X, et al. Exosomes Released From Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol Res (2018) 6:1578–92. doi: 10.1158/2326-6066.CIR-17-0479
85. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic Silencing of TH1-Type Chemokines Shapes Tumour Immunity and Immunotherapy. Nature (2015) 527:249–53. doi: 10.1038/nature15520
86. Gonzalez-Cao M, Karachaliou N, Santarpia M, Viteri S, Meyerhans A, Rosell R. Activation of Viral Defense Signaling in Cancer. Ther Adv Med Oncol (2018) 10:1-12. doi: 10.1177/1758835918793105
87. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell (2015) 162:961–73. doi: 10.1016/j.cell.2015.07.056
88. Galaine J, Borg C, Godet Y, Adotévi O. Interest of Tumor-Specific CD4 T Helper 1 Cells for Therapeutic Anticancer Vaccine. Vaccines (Basel) (2015) 3:490–502. doi: 10.3390/vaccines3030490
89. Zanetti M. Tapping CD4 T Cells for Cancer Immunotherapy: The Choice of Personalized Genomics. J Immunol (2015) 194:2049–56. doi: 10.4049/jimmunol.1402669
90. Tay RE, Richardson EK, Toh HC. Revisiting the Role of CD4+ T Cells in Cancer Immunotherapy—New Insights Into Old Paradigms. Cancer Gene Ther (2021) 28:5–17. doi: 10.1038/s41417-020-0183-x
91. Muranski P, Restifo NP. Adoptive Immunotherapy of Cancer Using CD4(+) T Cells. Curr Opin Immunol (2009) 21:200–8. doi: 10.1016/j.coi.2009.02.004
92. Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EKM, et al. Naive Tumor-Specific CD4(+) T Cells Differentiated In Vivo Eradicate Established Melanoma. J Exp Med (2010) 207:651–67. doi: 10.1084/jem.20091921
93. Wang D, Aguilar B, Starr R, Alizadeh D, Brito A, Sarkissian A, et al. Glioblastoma-Targeted CD4+ CAR T Cells Mediate Superior Antitumor Activity. JCI Insight (2018) 3. doi: 10.1172/jci.insight.99048
94. Blaszczak W, Liu G, Zhu H, Barczak W, Shrestha A, Albayrak G, et al. Immune Modulation Underpins the Anti-Cancer Activity of HDAC Inhibitors. Mol Oncol (2021). doi: 10.1002/1878-0261.12953
95. Ding Z-C, Shi H, Aboelella NS, Fesenkova K, Park E-J, Liu Z, et al. Persistent STAT5 Activation Reprograms the Epigenetic Landscape in CD4+ T Cells to Drive Polyfunctionality and Antitumor Immunity. Sci Immunol (2020) 5. doi: 10.1126/sciimmunol.aba5962
96. Lesch S, Benmebarek M-R, Cadilha BL, Stoiber S, Subklewe M, Endres S, et al. Determinants of Response and Resistance to CAR T Cell Therapy. Semin Cancer Biol (2020) 65:80–90. doi: 10.1016/j.semcancer.2019.11.004
97. Cao K, Wang G, Li W, Zhang L, Wang R, Huang Y, et al. Histone Deacetylase Inhibitors Prevent Activation-Induced Cell Death and Promote Anti-Tumor Immunity. Oncogene (2015) 34:5960–70. doi: 10.1038/onc.2015.46
98. Wang Y, Tong C, Dai H, Wu Z, Han X, Guo Y, et al. Low-Dose Decitabine Priming Endows CAR T Cells With Enhanced and Persistent Antitumour Potential via Epigenetic Reprogramming. Nat Commun (2021) 12:409. doi: 10.1038/s41467-020-20696-x
99. van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJM. Vaccines for Established Cancer: Overcoming the Challenges Posed by Immune Evasion. Nat Rev Cancer (2016) 16:219–33. doi: 10.1038/nrc.2016.16
100. Melssen M, Slingluff CL. Vaccines Targeting Helper T Cells for Cancer Immunotherapy. Curr Opin Immunol (2017) 47:85–92. doi: 10.1016/j.coi.2017.07.004
101. Hicks KC, Knudson KM, Lee KL, Hamilton DH, Hodge JW, Figg WD, et al. Cooperative Immune-Mediated Mechanisms of the HDAC Inhibitor Entinostat, an IL15 Superagonist, and a Cancer Vaccine Effectively Synergize as a Novel Cancer Therapy. Clin Cancer Res (2020) 26:704–16. doi: 10.1158/1078-0432.CCR-19-0727
102. Hirsch L, Zitvogel L, Eggermont A, Marabelle A. PD-Loma: A Cancer Entity With a Shared Sensitivity to the PD-1/PD-L1 Pathway Blockade. Br J Cancer (2019) 120:3–5. doi: 10.1038/s41416-018-0294-4
103. Ribas A, Wolchok JD. Cancer Immunotherapy Using Checkpoint Blockade. Science (2018) 359:1350–5. doi: 10.1126/science.aar4060
104. Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and Activity of Anti-PD-L1 Antibody in Patients With Advanced Cancer. N Engl J Med (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
105. Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J Immunother (2010) 33:828–33. doi: 10.1097/CJI.0b013e3181eec14c
106. Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant Anti-PD-1 Immunotherapy Promotes a Survival Benefit With Intratumoral and Systemic Immune Responses in Recurrent Glioblastoma. Nat Med (2019) 25:477–86. doi: 10.1038/s41591-018-0337-7
107. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant Nivolumab Modifies the Tumor Immune Microenvironment in Resectable Glioblastoma. Nat Med (2019) 25:470–6. doi: 10.1038/s41591-018-0339-5
108. Sharma P, Allison JP. The Future of Immune Checkpoint Therapy. Science (2015) 348:56–61. doi: 10.1126/science.aaa8172
109. Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, et al. Systemic Immunity is Required for Effective Cancer Immunotherapy. Cell (2017) 168:487–502.e15. doi: 10.1016/j.cell.2016.12.022
110. Zuazo M, Arasanz H, Fernández-Hinojal G, García-Granda MJ, Gato M, Bocanegra A, et al. Functional Systemic CD4 Immunity is Required for Clinical Responses to PD-L1/PD-1 Blockade Therapy. EMBO Mol Med (2019) 11:e10293. doi: 10.15252/emmm.201910293
111. Jiao S, Subudhi SK, Aparicio A, Ge Z, Guan B, Miura Y, et al. Differences in Tumor Microenvironment Dictate T Helper Lineage Polarization and Response to Immune Checkpoint Therapy. Cell (2019) 179:1177–1190.e13. doi: 10.1016/j.cell.2019.10.02984
112. Llopiz D, Ruiz M, Villanueva L, Iglesias T, Silva L, Egea J, et al. Enhanced Anti-Tumor Efficacy of Checkpoint Inhibitors in Combination With the Histone Deacetylase Inhibitor Belinostat in a Murine Hepatocellular Carcinoma Model. Cancer Immunol Immunother (2019) 68:379–93. doi: 10.1007/s00262-018-2283-0
113. Adeegbe DO, Liu S, Hattersley MM, Bowden M, Zhou CW, Li S, et al. BET Bromodomain Inhibition Cooperates With PD-1 Blockade to Facilitate Antitumor Response in Kras-Mutant Non-Small Cell Lung Cancer. Cancer Immunol Res (2018) 6:1234–45. doi: 10.1158/2326-6066.CIR-18-0077
114. Goswami S, Apostolou I, Zhang J, Skepner J, Anandhan S, Zhang X, et al. Modulation of EZH2 Expression in T Cells Improves Efficacy of Anti-CTLA-4 Therapy. J Clin Invest (2018) 128:3813–8. doi: 10.1172/JCI99760
115. Morel KL, Sheahan AV, Burkhart DL, Baca SC, Boufaied N, Liu Y, et al. EZH2 Inhibition Activates a Dsrna-STING-Interferon Stress Axis That Potentiates Response to PD-1 Checkpoint Blockade in Prostate Cancer. Nat Cancer (2021) 2:444–56. doi: 10.1038/s43018-021-00185-w
116. Kim Y-D, Park S-M, Ha HC, Lee AR, Won H, Cha H, et al. HDAC Inhibitor, CG-745, Enhances the Anti-Cancer Effect of Anti-PD-1 Immune Checkpoint Inhibitor by Modulation of the Immune Microenvironment. J Cancer (2020) 11:4059–72. doi: 10.7150/jca.44622
117. Terranova-Barberio M, Thomas S, Ali N, Pawlowska N, Park J, Krings G, et al. HDAC Inhibition Potentiates Immunotherapy in Triple Negative Breast Cancer. Oncotarget (2017) 8:114156–72. doi: 10.18632/oncotarget.23169
118. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase Inhibition Promotes the Generation and Function of Regulatory T Cells. Nat Med (2007) 13:1299–307. doi: 10.1038/nm1652
119. Wu Q, Nie J, Gao Y, Xu P, Sun Q, Yang J, et al. Reciprocal Regulation of Rorγt Acetylation and Function by P300 and HDAC1. Sci Rep (2015) 5:16355. doi: 10.1038/srep16355
120. Ho TCS, Chan AHY, Ganesan A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J Med Chem (2020) 63:12460–84. doi: 10.1021/acs.jmedchem.0c00830
Keywords: epigenetics, CD4+ helper T cells, plasticity, tumor microenvironment, immunotherapy
Citation: Renaude E, Kroemer M, Borg C, Peixoto P, Hervouet E, Loyon R and Adotévi O (2021) Epigenetic Reprogramming of CD4+ Helper T Cells as a Strategy to Improve Anticancer Immunotherapy. Front. Immunol. 12:669992. doi: 10.3389/fimmu.2021.669992
Received: 19 February 2021; Accepted: 15 June 2021;
Published: 28 June 2021.
Edited by:
José Manuel González-Navajas, Hospital General Universitario de Alicante, SpainReviewed by:
Hao Shi, St. Jude Children’s Research Hospital, United StatesPeng Jiang, National Institutes of Health (NIH), United States
Copyright © 2021 Renaude, Kroemer, Borg, Peixoto, Hervouet, Loyon and Adotévi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivier Adotévi, b2xpdmllci5hZG90ZXZpQHVuaXYtZmNvbXRlLmZy
†These authors have contributed equally to this work