 Domenico Somma
Domenico Somma Fatma O. Kok1
Fatma O. Kok1 Christine A. Wells
Christine A. Wells Ruaidhrí J. Carmody
Ruaidhrí J. Carmody
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 13 October 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.669906
Since its discovery over 30 years ago the NF-ĸB family of transcription factors has gained the status of master regulator of the immune response. Much of what we understand of the role of NF-ĸB in immune development, homeostasis and inflammation comes from studies of mice null for specific NF-ĸB subunit encoding genes. The role of inflammation in diseases that affect a majority of individuals with health problems globally further establishes NF-ĸB as an important pathogenic factor. More recently, genomic sequencing has revealed loss of function mutations in the NFKB1 gene as the most common monogenic cause of common variable immunodeficiencies in Europeans. NFKB1 encodes the p105 subunit of NF-ĸB which is processed to generate the NF-ĸB p50 subunit. NFKB1 is the most highly expressed transcription factor in macrophages, key cellular drivers of inflammation and immunity. Although a key role for NFKB1 in the control of the immune system is apparent from Nfkb1-/- mouse studies, we know relatively little of the role of NFKB1 in regulating human macrophage responses. In this study we use the THP1 monocyte cell line and CRISPR/Cas9 gene editing to generate a model of NFKB1-/- human macrophages. Transcriptomic analysis reveals that activated NFKB1-/- macrophages are more pro-inflammatory than wild type controls and express elevated levels of TNF, IL6, and IL1B, but also have reduced expression of co-stimulatory factors important for the activation of T cells and adaptive immune responses such as CD70, CD83 and CD209. NFKB1-/- THP1 macrophages recapitulate key observations in individuals with NFKB1 haploinsufficiency including decreased IL10 expression. These data supporting their utility as an in vitro model for understanding the role of NFKB1 in human monocytes and macrophages and indicate that of loss of function NFKB1 mutations in these cells is an important component in the associated pathology.
Common variable immunodeficiency (CVID) is the most common primary immuno-deficiency in humans and is a clinically and genetically heterozygous disorder characterised by increased susceptibility to infection, hypogammaglobulinaemia and poor responses to vaccination. Recent advances in genome sequencing have led to the identification of genetic determinants of CVID in a significant number of clinical cases. These analyses have identified loss of function variants in the gene encoding the NF-ĸB p105 subunit (NFKB1) as the most common monogenic cause of CVID in Europeans (1–3). In these studies loss of function variants of NFKB1 had the highest probability of disease association and explained the largest number of CVID cases. CVID resulting from NFKB1 loss of function is characterised by lymphadenopathy, splenomegaly, and autoimmunity, as well as an increased incidence of cancers including solid tumours and hematologic malignancies. In addition to loss of function mutations, monogenic heterogeneous mutations affecting the nuclear localisation and stability of NF-ĸB1 can also lead to immune-pathologies including autoinflammatory diseases such as Behçet disease (4).
The NF-ĸB family of transcription factors are critical regulators of immune homeostasis and the inflammatory response. NF-ĸB is activated by virtually all immunoreceptors, including cytokine-, antigen- and Toll-like- receptors, and co-ordinates the expression of more than 500 genes encoding pro-inflammatory factors such as cytokines and chemokines, as well as genes controlling cell survival, proliferation, and differentiation. There are five NF-ĸB subunits, RelA, c-Rel, p50, RelB and p52, which form homo-and heterodimers to regulate gene transcription. The NF-ĸB p50 subunit is generated from the limited proteasomal processing of the p105 precursor protein which also functions as an inhibitor of NF-ĸB activation. p50 lacks a transactivation domain and so may act as a transcriptional repressor as well as activator depending on whether it forms homodimers or heterodimers with other NF-ĸB subunits (5, 6).
Studies of individuals with loss of function NFKB1 variants reveal significant effects on immune cell development and function. These analyses show that NFKB1 haploinsufficiency leads to reduced invariant natural killer cell numbers as well as reduced CD4 and CD8 T cells at early stages of development, while leading to an accumulation of effector memory T cells. B cell homeostasis is similarly disrupted leading to an overall B cell deficiency including reduced memory and class switched B cells (3). B cells with NFKB1 loss of function mutations also show impaired proliferation following B cell receptor activation or stimulation with Toll-like receptor (TLR) ligand and display reduced IgG production ex vivo (3). In contrast, stimulation of NFKB1 deficient whole blood with TLR ligand results in elevated expression of the pro-inflammatory cytokines IL-1β and TNFα (7). A recently identified homozygous mutation in NFKB1 (G960R) led to perturbations in both T- and B-cell maturation and function, reduced CD4+ memory T cell numbers, while T cell responses to mitogens and cytokine secretion were reduced. These studies also revealed skewed T-cell receptor repertoire, and reduced output of naïve T cells in the NFKB1 variant patient (8).
NFKB1 haplo-insufficiency phenotypes have some overlap with those of the Nfkb1-/- mouse. Similar to NFKB1 loss of function variants in humans, Nfkb1-/- mice are more susceptible to bacterial infection and have significantly reduced peripheral B cell numbers. In mice, Nfkb1 is important for mature B cell survival but does not appear to have a significant role in B cell development. However, when Nfkb1 is deleted in combination with Rel or Nfkb2 B cell development is abnormal (9–12). Nfkb1-/- B cells also proliferate poorly in response to TLR stimulation but, in contrast to NFKB1 haplo-insufficient B cells, proliferate normally in response to B cell receptor activation (13). In contrast to the apparent role for NFKB1 in T cell maturation in humans, in mouse Nfkb1 appears dispensable for normal T cell maturation but is required for the development of Th2 responses involving IL-4, IL-5 and IL-13 (14, 15). These differences suggested that further study of NF-ĸB1 function in human cells is warranted to more completely understand its role in the regulation of immune responses.
NFKB1 is the most abundantly expressed transcription factor in macrophages (16), key initiators of chronic inflammatory disease. In addition to regulating TLR-induced pro-inflammatory gene expression in activated macrophages, NFKB1 also plays a key role in shaping macrophage polarisation and innate immune memory responses (17). Innate immune memory refers to the adaptation of innate immune cells to re-activation with the same or different stimulus and can result in enhanced or reduced inflammatory responses (18). Nfkb1-/- macrophages are defective in LPS tolerance, a form of innate immune memory characterized by reduced expression of pro-inflammatory cytokines such as Tnf (19). Loss of this anti-inflammatory function in Nfkb1-/- mice results in chronic low-grade inflammation leading to accelerated aging and a shortened life span (20). Conversely, pro-inflammatory Nfkb1-/- macrophages adopt an anti-tumour phenotype that reduces the tumour burden in mouse models of colitis associated cancer (21). Despite the recognized physiological roles of NFKB1 in macrophages, the consequences of NFKB1 deficiency in human macrophages is not known. Here we describe an in vitro model cell system for the study of NFKB1 function in human monocytes and macrophages using CRISPR/Cas9 gene editing to generate a NFKB1-/- THP1 human monocyte cell line. Deletion of NFKB1 does not alter the activation of the NF-ĸB pathway in response to TLR stimulation, however transcriptomic analysis reveals a critical role for NFKB1 in regulating key pro-inflammatory genes including co-stimulatory factors of T cell mediated adaptive immune responses.
To explore the role of p105/p50 in the regulation of human macrophage responses we employed CRISPR/Cas9 gene editing approaches to generate a NFKB1-/- THP1 human monocyte cell line. THP1 is a human monocytic cell line that can be differentiated into macrophages by phorbol myristate acetate (PMA) and is an established in vitro model for the study of human monocyte and macrophage responses (22). Targeted CRISPR/Cas9 guided double strand breaks generated a 5 base pair deletion in exon 5 of the NFKB1 gene which created a premature stop codon (Figures 1A–D). The absence of p105/p50 protein was confirmed by western blot analysis (Figure 1E).
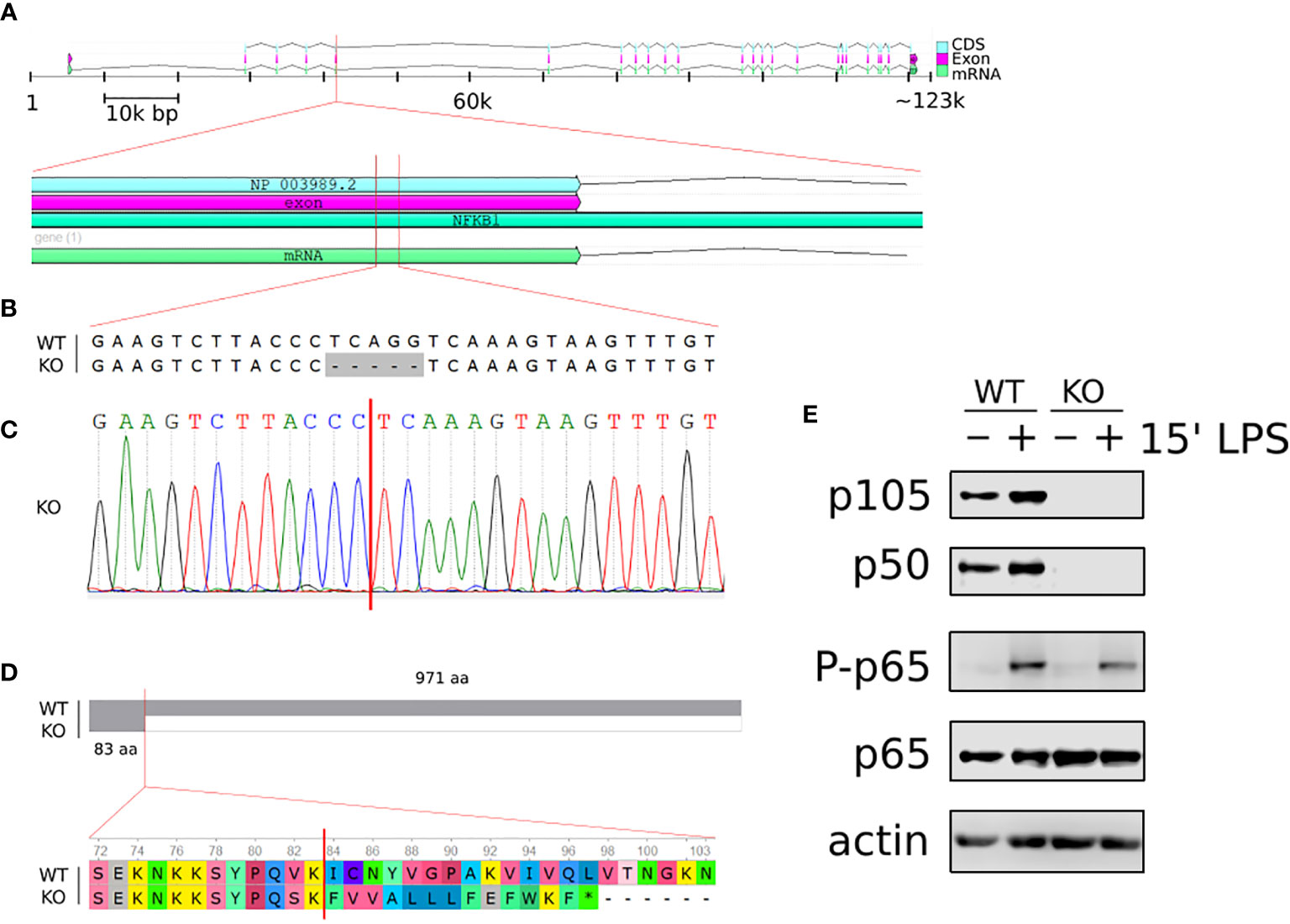
Figure 1 Generation of NFKB1-/- THP1 cells. (A) Schematic overview of homo sapiens NFKB1 gene organisation on chromosome 4; cDNA sequence (CDS), exon and mRNA are indicated. (B) Alignment of NFKB1 exon 5 DNA sequences of wild type (WT) and NFKB1-/- (KO) cells showing CRISPR/Cas9 generated deletion. (C) Sanger sequencing chromatogram of exon 5 in NFKB1-/- cells containing a frameshift mutation. (D) Alignment of NFKB1 open reading frames in WT and NFKB1-/- cells, grey area indicates sequence identify. (E) PMA-induced macrophages from WT and NFKB1-/- THP1 cells were stimulated with LPS (10 ng/mL) for 15 min, and p105/p50 (NFKB1 protein products) analysed by western blot. Antibodies specific for p65 phosphorylation at S536 (p-p65) were used to measure NF-κB pathway activation, anti- p65 and anti-actin antibodies were used as loading controls.
PMA-induced differentiation of NFKB1-/- THP1 cells to macrophage was indistinguishable to that observed for wild type (WT) THP1 cells (data not shown) confirming that NFKB1 is not required for macrophage differentiation as previously reported in Nfkb1-/- mice (19). To investigate the functional response of NFKB1-/- THP1 derived macrophages to inflammatory stimuli we next stimulated cells with the Toll-like receptor (TLR) 4 ligand lipopolysaccharide (LPS). First, we assessed the impact of NFKB1 knockout on the activation of the NF-ĸB pathway as measured by phosphorylation of the p65 subunit of NF-ĸB at S536 by the IKK complex, an event strongly linked to NF-ĸB activation (23). This revealed similar levels of p65 S536 phosphorylation in LPS stimulated NFKB1-/- cells when compared to WT controls (Figure 1E), in line with data from Nfkb1-/- mice demonstrating that NFKB1 is not required for IKK activation in the NF-ĸB pathway. Collectively, these data show that deletion of NFKB1 in THP1 does not affect PMA induced differentiation to macrophages, or LPS-induced activation of the NF-ĸB pathway, thereby demonstrating their suitability for investigating role of NFKB1 in LPS-stimulated cells.
To determine the role of NFKB1 in regulating inflammatory gene expression we stimulated PMA induced NFKB1-/- and WT THP1 macrophages with LPS for 3 hours prior to analysis by RNA-seq alongside untreated controls. Unsupervised principal component analysis revealed distinct transcriptional profiles for WT and NFKB1-/- cells in both unstimulated and LPS-stimulated conditions (Figure 2A). This analysis showed that the main variance between experimental groups was LPS stimulation and NFKB1 deletion (Figure 2A).
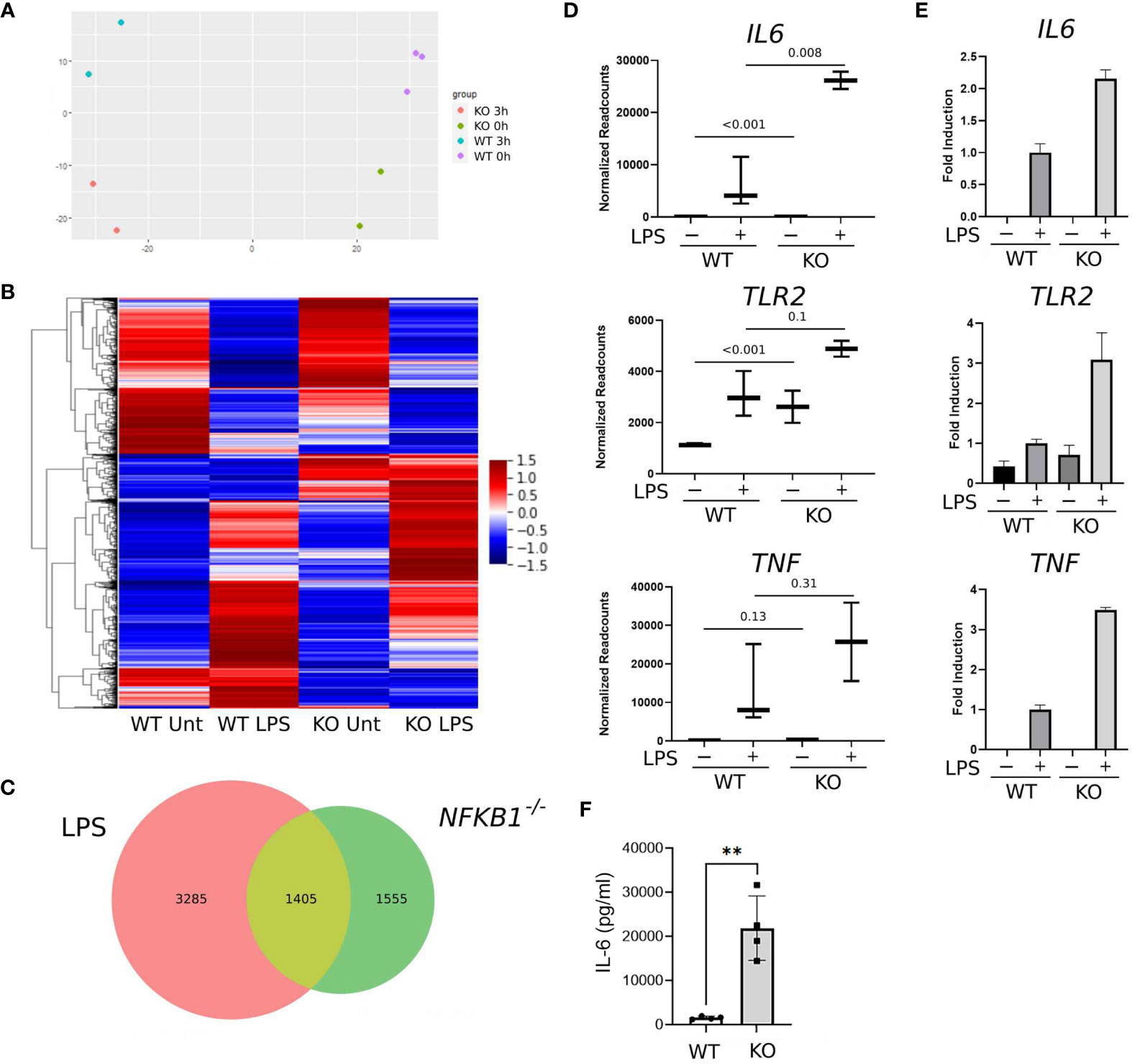
Figure 2 NFKB1 deletion alters the transcriptional profile of THP1 macrophages. (A) Principal component analysis plot showing sample similarity of RNAseq analysis of control (WT) and NFKB1-/- (KO) cells untreated (Unt) or treated with LPS. (B) Heatmap showing differentially expressed genes, Z-score normalized. Relative expression is shown as the average of replicates ranging from maximum to minimum values. (C) Venn diagram showing the overlap of LPS-inducible genes and genes differentially expressed in NFKB1-/- cells relative to WT cells in unstimulated and LPS stimulated conditions (p-value and q-value < 0.05). (D) Selected gene expression data plotted as normalized RNAseq readcounts. Data presented as mean ± SD of biological replicates, p values adjusted for Benjamini-Hochberg are indicated. (E) Patterns of expression for selected genes confirmed in an independent experiment using QPCR. Data presented as mean ± SEM of technical replicates. (F) PMA-induced macrophages from WT and NFKB1-/- THP1 cells were stimulated with LPS, and the supernatant was analysed by IL-6 ELISA. (n = 4, data presented as mean ± SD of four independent experiments). **p < 0.003 using T-test analysis.
Overall, our analysis identified 4,690 genes differentially expressed in LPS treated THP1 cells when compared to untreated controls (Figure 2C). Comparison of WT and NFKB1-/- cells identified 2,960 genes differentially expressed in NFKB1-/- cells relative to WT controls. Of this NFKB1 signature gene set, 1,405 genes were also found to be LPS regulated, indicating that 30% of genes induced by LPS are controlled by NFKB1 and 47% of NFKB1-regulated genes are induced by LPS (Figures 2B, C). Of note, the direction of regulation of genes by NFKB1 was consistent across experimental conditions and there is little overlap between genes up-regulated and genes down-regulated in NFKB1-/- cells either in steady state or LPS-stimulated conditions (Supplemental Figure S2). We validated the RNA-seq data by performing an independent QPCR analysis of selected genes including IL6, TLR2, and TNF which are known to be regulated by NFKB1 (Figures 2D, E). The increased expression of IL6 in NFKB1-/- cells relative to WT controls was further validated by ELISA analysis of cell supernatants (Figure 2F). Together these data demonstrate that NFKB1 may both positively and negatively regulate the expression of hundreds of genes, both in the steady state and upon LPS stimulation, consistent with previously described dimer-specific roles of the NF-ĸB p50 subunit (5).
To better understand which signalling pathways are regulated by NFKB1 we used the manually curated Reactome database to identify over-represented signalling pathways. We divided the genes in two subsets; one composed of NFKB1-dependent LPS-induced genes and the other of genes downregulated in NFKB1-/- cells (Supplemental Figure S2B). Reactome analysis of these two gene sets revealed that NFKB1 controls a macro “Signalling by Interleukins” pathway incorporating a number of key immuno-receptor initiated pathways (Figure 3, Supplemental Tables S1 and S2; Supplemental Figure S3). These pathways include the IL-10 signalling pathway (Supplemental Figure S4), due to the reduced expression of IL10 and IL10RA in LPS-stimulated NFKB1-/- cells compared to WT controls (Figure 4A). In contrast, expression of the inflammatory cytokine genes IL6, IL1B and IL1A is increased in NFKB1-/- cells relative to WT controls (Figure 4). In addition, reduced expression of IL4R in LPS-stimulated NFKB1-/- cells relative to WT controls also implicates NFKB1 in the regulation of IL-4 and IL-13 activated responses (Supplemental Figure S5). Similarly, reduced expression of IL2RA and IL7R in LPS-stimulated NFKB1-/- cells relative to WT controls implicates NFKB1 in the regulation of IL-2 and IL-7 activated responses (Figure 4). Increased expression of components of the interferon response, including IFNB1, IFNL1, and IFNAR2, in LPS-stimulated NFKB1-/- cells relative to WT controls also identifies NFKB1 as a regulator of interferon pathways (Figure 4).
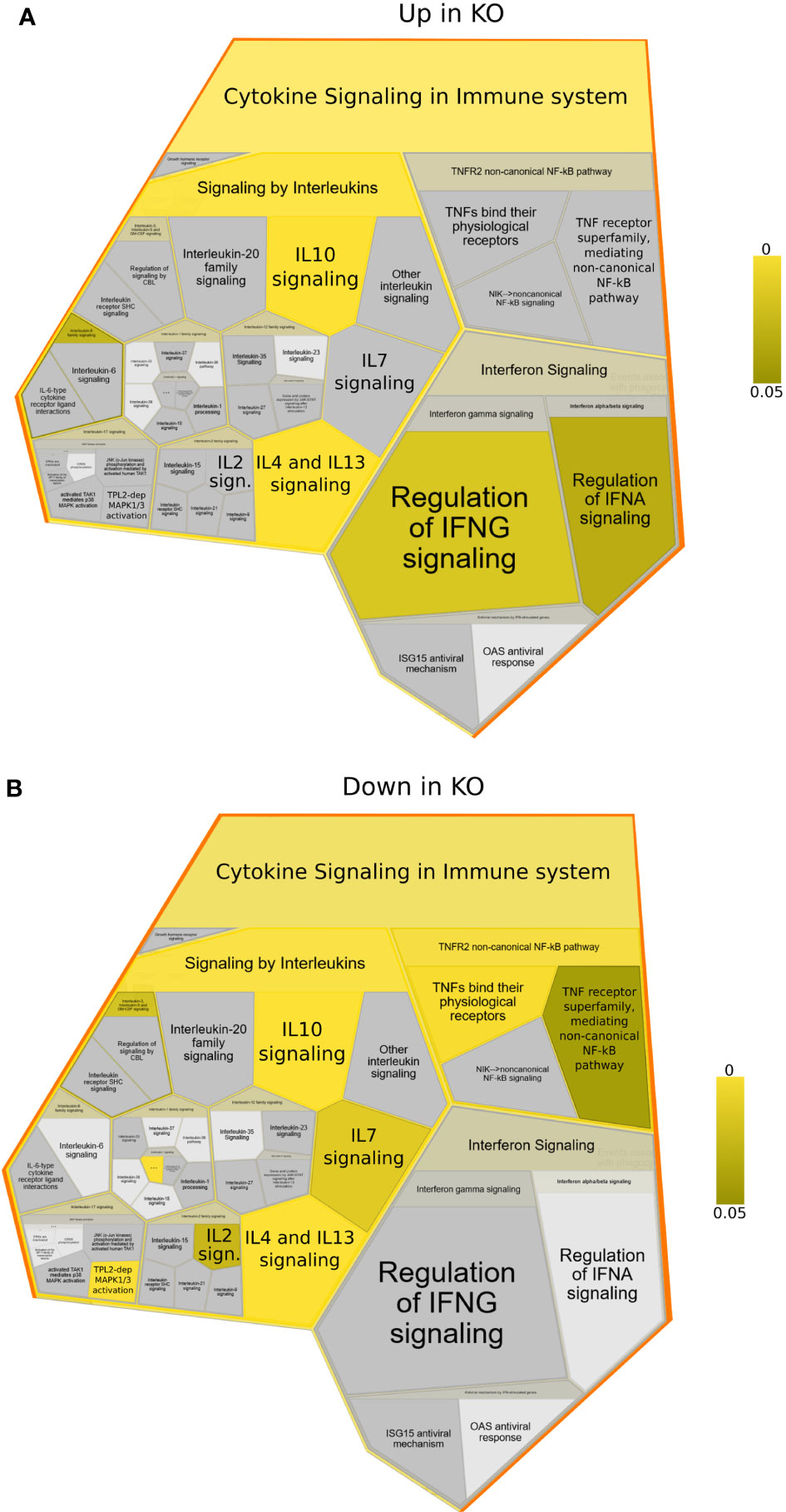
Figure 3 Reactome foam plots identifying enriched pathways using genes (A) upregulated or (B) downregulated in NFKB1-/- (KO) macrophages (either untreated or LPS treated) compared to wild type controls. Yellow bar indicates p values.
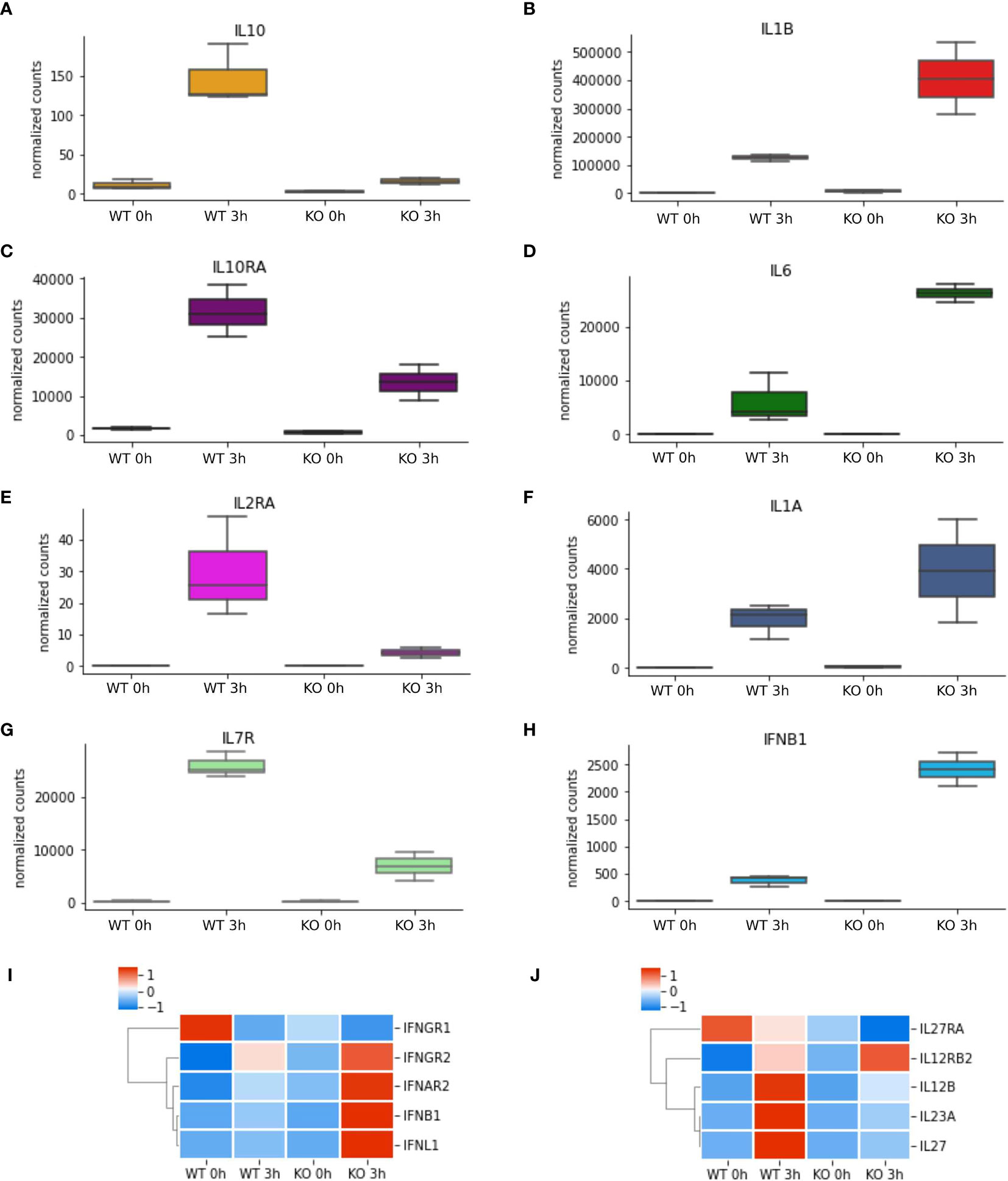
Figure 4 NFKB1-/- macrophages have an inflammatory phenotype. (A–H) RNAseq data of selected differentially expressed cytokine and cytokine receptor genes identified by Reactome pathway analysis. Heatmaps of differentially expressed (I) interferon cytokines and receptors and (J) IL-12-family of cytokines and receptors from RNAseq analysis, Z-score normalized.
Another macro pathway identified by this analysis as significantly modified in NFKB1-/- cells is “Chemokine receptors bind chemokines” (Supplemental Figure S6 and Supplemental Tables S1, S2). Here, LPS-induced increased the expression of the chemokines CXCL1, CCL1, CXCL3, CXCL8, CXCL16 and CCL20, and reduced the expression of the chemokine receptors CCR2, CXCR2, CXCR4, CCR7, and of chemokines CCL5, CCL19 and CCL22 in NFKB1-/- cells relative to WT controls and identifies NFKB1 as a regulator of chemokine signalling.
Reactome is a manually curated peer-reviewed database covering almost 11,000 human genes but excludes ~38% of the genes differentially expressed in our analysis. To get further insight into our full dataset we next used Enrichr to query the BioPlanet database which integrates several pathway databases (24). This pathway analysis confirmed the findings of the Reactome analysis such as the involvement of IL-4, IL-2, IL-1 and IL-10 pathways, but also highlighted some additional pathways such as those activated by the IL-12 family cytokines (IL12B, IL23A, IL27, IL27RA and IL12RB2) (Figure 5), as well as components of EGF, VEGF, TGF cytokine signalling (Supplemental Figure S7 and Supplemental Table S3) as altered in NFKB1-/- cells. Together this data demonstrates that deletion of NFKB1 alters not only the expression of key inflammatory cytokines such as IL1B, IL6, IL10 and IFNB1, but also the expression of cytokine and chemokine receptors.
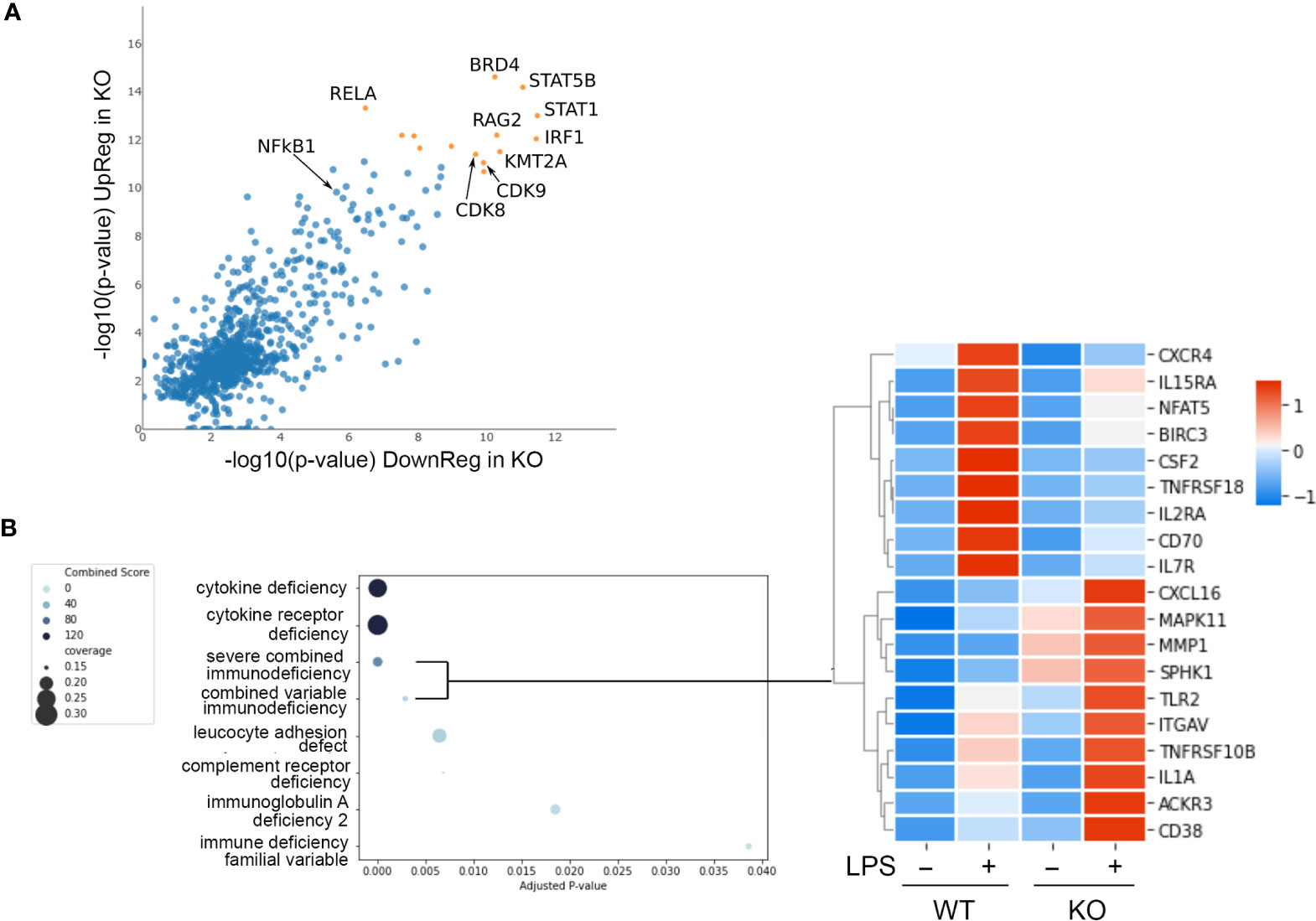
Figure 5 NFKB1 transcription factor network and disease associations. (A) Lisa analysis identifying transcription factors associated with the expression LPS dependent of genes differentially expressed in NFKB1-/- cells (p < 0.01). Top most significant factors indicated by orange symbols. (B) Identification of disease associated gene signatures in the GeneRif database with significant similarity to the transcriptional signature of LPS-stimulated NFKB1-/- cells (q value < 0.05). Heatmap on right shows selected genes associated with the severe combined and common variable immunodeficiency disease signatures, Z-score normalized.
Transcription factors form complex networks to regulate gene expression by interacting with other transcription regulators (25). To explore this aspect of NFKB1 mediated transcriptional responses we employed Lisa (epigenetic Landscape In Silico deletion Analysis) to identify potential cofactors of NFKB1. This approach uses DNase-seq and H3K27ac ChIP-seq profiles to identify the transcription factors and chromatin regulators responsible for the control of a differentially expressed gene set. This analysis confirmed the p50 and RelA NF-ĸB subunits as transcription factors associated with genes differentially expressed in LPS-stimulated NFKB1-/- cells relative to WT controls (Figure 5A). This analysis also identified the IRF1 and STAT1 transcription factors as co-regulators of these genes, supporting previous reports of a functional interaction between p50 and these factors (26, 27). In addition, this analysis also identified STAT5B, BRD4, RAG2, KMT2A and CDK8/9 as factors associated with NFKB1 regulated genes, indicating further complexity of the NF-κB1 transcriptional network. Similar findings were obtained in an independent analysis using the TRRUST database (Figure S8).
We next evaluated the potential association of the NFKB1-/- transcriptional signature (genes differentially expressed in LPS stimulated NFKB1-/- cells compared to WT controls) with disease associated gene signatures in humans. To perform this we used the GeneRif database of manually curated datasets describing gene signatures associated with human disease. This analysis revealed a significant similarity of the NFKB1-/- transcriptional signature with “Severe combined immunodeficiency” and “Common variable immunodeficiency” disease signatures (Figure 5B and Table S4). This finding suggests that loss of NFKB1 function in macrophages, and indeed other innate immune cells such as dendritic cells, is also likely to contribute the common variable immunodeficiency phenotype resulting from NFKB1 haploinsufficiency.
A key function of innate immune cell activation is co-stimulation of adaptive immune cells. To explore this aspect of NFKB1-/- macrophage responses we examined co-stimulatory gene expression in NFKB1-/- compared to WT controls (Figure 6). This revealed that expression of co-stimulatory factors such as CD70, CD83 and CD209 (Figure 6A) are decreased in LPS stimulated NFKB1-/- cells when compared to WT controls. In addition, there is also a reduction in Fc receptor gene expression (Figure 6D), as well as the CLEC family of genes (Figure 6E). These findings, along with the reduced expression of genes encoding interleukin receptors, CCR and CXCR families (Figure 6B) mentioned described above, indicate that in addition to a defective acute inflammatory response in NFKB1-/- cells, these cells are also likely to be defective the co-stimulation of T cells. Interestingly, NFKB1-/- cells have altered gene expression in steady state conditions including the anti-inflammatory CD33 (Figure 6A) which results in increased CD33 surface expression in NFKB1-/- cells as measured by flow cytometry. (Figure 6F). Conversely genes encoding inflammatory proteins such as CD38 (28) are more highly expressed in NFKB1-/- macrophage following LPS stimulation (Figure 6A) leading to increased expression of cell surface CD38 protein as confirmed by flow cytometry (Figure 6G). The increased expression of CD38 in NFKB1-/- cells has a two-phase distribution, with CD38+ high and low cells evident.
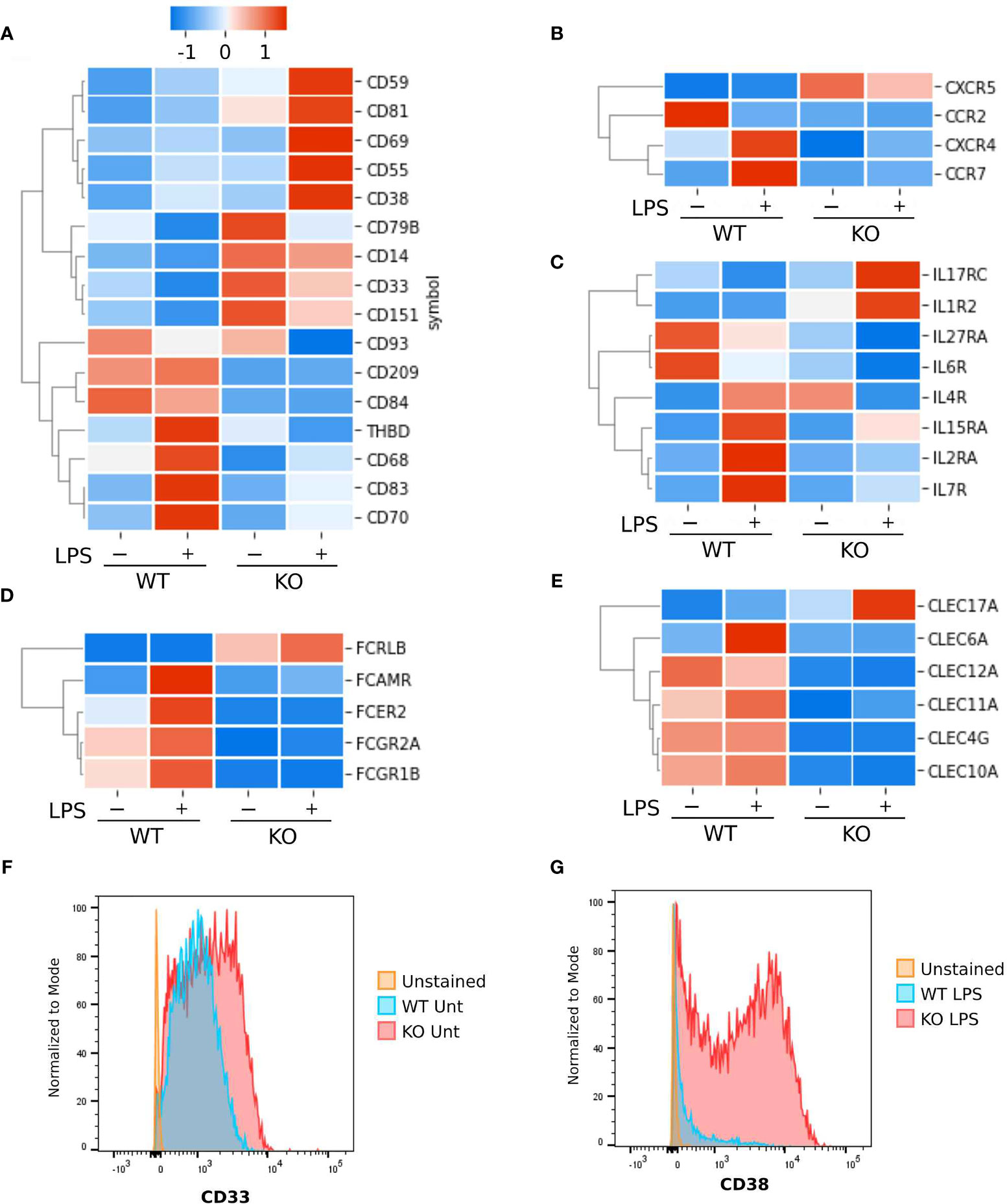
Figure 6 Surface markers differentially expressed between NFKB1-/- and wild type THP1 cells. Heatmaps Z-score normalized showing gene expressions as the average of replicates. (A) Cluster differentiation markers. (B) Chemokine and cytokines receptors. (C) Interleukin receptors. (D) Fc receptors. (E) C-type lectin domain family members. (F) Analysis by flow cytometry of CD33 and (G) CD38 cell surface protein expression in untreated (unt) or LPS stimulated wild type (WT) or NFKB1-/- (KO) THP1 cells using specific antibodies.
The recent identification of mutations in NFKB1 leading combined variable immuno-deficiencies further cements the importance of the NF-ĸB transcription factors in the control of inflammation and immunity. However, these findings also highlight that despite a number of studies using animals deficient for Nfkb1, the specific impact of loss of NFKB1 on transcriptional responses in immune cells remains largely undefined. In this study we have generated a model for the study of NFKB1 deficiency using CRISPR/Cas9 gene editing in the THP1 cell line, an established model for the study of human monocytes and macrophages. Transcriptomic analysis of TLR4 activated THP1 differentiated macrophages revealed a critical role for NFKB1 in the regulation of TLR4-induced transcriptional responses. This analysis confirms the key role of NF-κB1 in regulating TLR induced transcriptional responses in macrophages while identifying new gene targets of NFKB1 that may play a role in the pathology associated with NFKB1 haploinsufficiency.
Overall, our data shows that the expression of pro-inflammatory cytokines such as TNF, IL6, IL1B, IL10 and IFNB in NFKB1-/- THP1 macrophages show a similar pattern to that seen in studies of Nfkb1-/- mice, demonstrating that the biological functions NFKB1 are conserved across species. Moreover, our analysis of NFKB1-/- THP1 macrophages recapitulates key observations in individuals with NFKB1 haploinsufficiency including decreased IL10 expression. These data support the utility of NFKB1-/- THP1 macrophages as a model of NFKB1 deficiency in human monocytes and macrophages.
A comparison of our data with those reported from studies of Nfkb1-/- mice reveals important differences in patterns of NFKB1 regulated gene expression between mouse and human cells. This is highlighted by the expression patterns of chemokines which are differentially affected by deletion of NFKB1 in mouse and human macrophages. While studies of Nfkb1-/- mouse macrophages identified a role for Nfkb1 in promoting the expression of CCL2 and CCL17 (17), our analysis indicates that NFKB1 does not significantly regulate their expression in THP1 derived macrophages. Instead, our analysis identifies increased expression of the chemokines CXCL1, CCL1, CXCL3, CXCL8, CXCL16, and CCL20, and decreased expression of CCL5, CCL19 and CCL22 in LPS stimulated NFKB1-/- THP1 cells. While the reasons for these disparities are unclear, they may underline potential differences in chemokine regulation between human and mouse macrophages (29). What is clear however, is that NFKB1 plays an important role in coordinating the expression of chemokines and their receptors following TLR activation. Indeed, the reduced expression of CCR7 in NFKB1-/- cells reported here would suggest that cell migration to lymph nodes is likely to be impaired in individuals with NFKB1 haploinsufficiency, which may contribute to the observed pathology. The regulation by NFKB1 of genes encoding immuno-receptors in addition to chemokine receptors, such as cytokine receptors (IL6R, IL2RA, IL10RA, IL7R, IL4R, IL1R1/2), C-type lectins, Fc receptors, and co-stimulatory molecules emphasises the important role of NF-κB1 in coordinating inflammatory responses. Further understanding of the role of NF-κB1 in these processes will require in vivo experimental techniques that can assess cell extrinsic effects not possible with the in vitro approaches used in this study.
NFKB1 may influence target gene expression positively or negatively through p50 heterodimers or homodimers respectively, and also acts as an IκB protein due to the presence of C terminal ankyrin repeat domains (5). In addition, p50 may also regulate transcriptional responses through interaction with other transcription factors such as IRFs and STATs (30–32). Our analysis shows that the NFKB1 controlled transcriptome significantly overlaps with those controlled by the transcription factors IRF1, STAT5, STAT5B as well the transcriptional regulators BRD4, RAG2, KMT2A, and CDK8/9. This indicates a potential role of NFKB1 in a number of transcriptional networks and may further explain the significant impact of NFKB1 deficiency on TLR inducible responses.
Studies of Nfkb1 knockout mice have identified different roles for NF-κB1 within different immune cell types. Notably, mice heterozygous for Nfkb1 do not develop the primary immunodeficiency found in humans with NFKB1 haploinsufficiency (3), suggesting key differences in the role of NFKB1 in humans and mice. Our data demonstrate the utility of NFKB1-/- THP1 cells as an in vitro model for understanding the role of NF-κB1 in human monocytes and macrophages. THP1 cells have been extensively used for the study of monocyte and macrophage functions. Studies to date have demonstrated that the responses of THP1 cells activated with a variety of stimuli are similar to those of primary human monocytes, although the magnitude of cytokine secretion and gene expression changes may differ (22). Understanding the role of NF-κB1 in other immune cells such as T or B cells and the similarities and differences with animal models requires further study in the context of human cells. Although, generating specific mutations found in NFKB1 variant patients using similar approaches would provide a more accurate model of NFKB1 variant induced CVID that might provide further insight, this study indicates an important role for monocytes and macrophages in the immunological phenotype of NFKB1 haploinsufficiency.
THP-1 cells were cultured in RPMI containing 10% FBS with of 2 mM glutamine and 100 units/mL penicillin/streptomycin. For generation of CRISPR/Cas9 NFKB1 knock-out cells, THP1 cells were transfected with a ribonucleoprotein (RNP) complex using the Neon electroporator (ThermoFisherScientific). 2µl (20pmol) of sgRNA (: 5’-ACAAACUUACUUUGACCUGA-3’, Synthego) plus 1µl of Cas9 nuclease (20pmol) (Synthego) was added to 9.5µl Neon buffer T (ThermoFisherScientific) and incubated at room temperature for 10 minutes. THP1 cells were washed with Mg2+ and Ca2+ free phosphate buffered saline and re-suspended in Neon buffer T at a density of 2 × 107 cells/ml. 7µl of RNP complex was transfected into 10µl of cells using a 10µl Neon Transfection tip (cat# MPK2025) and settings of 1400v, 20msec, and 2xpulse. Serial dilution of cells was performed to isolate single cell clones in 96-well plates. For clone screening, genomic DNA was extracted from cells using the DNeasy Blood and Tissue Kit (QIAGEN) according to the manufacturer’s instruction. DNA was amplified by PCR using primers (F: 5’-ACCTGGCTTTTTAGCCATATCT-3’; R: 5’-TTCAGCTTAGGAGCGAAGGC-3’) and Hot-StarTaq Master Mix Kit (QIAGEN) according to the manufacturer’s instructions. Initial screens were performed by HindIII (NEB) restriction digest of PCR products purified using QIAquick PCR Purification Kit (QIAGEN), according to the manufacturer’s instructions. Gene editing of selected clones was confirmed by Sanger sequencing (GATC Biotech).
Whole cell lysates were generated from cells suspended in RIPA buffer containing 50 mM Tris–HCl (pH 7.4), 1% Nonidet P-40, 0.25% deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM NaF, 1 mM Na3VO4, 2 µg/ml aprotinin, 1 µg/ml pepstatin and 1 µg/ml leupeptin. After an additional 15 min on ice, cell extracts were centrifuged for 10 min at 14,000 g at 4°C, and supernatants. Protein concentration of lysates were determined using a Bradford assay (Bio-Rad). Denatured samples were subjected to SDS-PAGE, transferred to nitrocellulose membranes, and blocked for 1 h at room temperature using 5% fat free milk powder in PBS–Tween20. Membranes were immunoblotted with specific antibodies. p105/p50 (#12540) and anti-p65 phospho-S536 (#3031), were purchased from Cell Signalling Technologies, p65 (A301-824A) was purchased from Bethyl Laboratories, beta-actin (#A5441) was purchased from Sigma.
IL-6 ELISA (Life Technologies) was performed according to the manufacturer’s instruction using supernatants from THP1 differentiated macrophages either untreated or stimulated for 24 hours with 100 ng/ml LPS.
THP1 differentiated macrophages were stimulated 24h with LPS 100 ng/ml for 24 hours harvested after trypsin digestion, centrifuged at 400g for 5 min, washed and resuspended with FACS buffer (BD Biosciences), and transferred to FACS tubes (BD Biosciences). Fixable Viability Dye eFluor 780 (eBioscience) was added at 1:1,000 in PBS and incubated for 20 min at 4°C. Cells were then washed with FACS buffer were stained with antibodies specific for either CD33 or CD38 (Biolegend), conjugated with Alexa Fluor 700. Staining was performed in a final volume of 100 μl with antibody dilution 1:100 for 30 min on ice. Cells were washed twice with FACS buffer and resuspended in a final volume of 300 μl, filtered through a 100-μm cell strainer and analysed using a FACS Fortessa (BD Biosciences). Live macrophages were gated based on viability dye. All data generated were analysed using FlowJo software (TreeStar).
For RNA-sequencing and real-time quantitative PCR, total RNA was extracted from cells using RNeasy kits (QIAGEN), according to the manufacturer’s instructions and quantified using NanoDrop 1000 Spectrophotometer (ThermoFisher Scientific). 1 µg of isolated RNA was retro-transcribed using random primers and nanoScript reverse transcription kit (Primer Design). QPCR was performed with SYBR Green SuperMix (PerfeCTa #733-1386) using QuantiTect Primer Assays (TNF #QT00029162, IL6 #QT00083720, TLR2 #QT00236131, TBP #QT00000721) using QuantStudio 7 Flex Real-Time PCR System (ThermoFisher Scientific). All data were normalised to TBP. Gene expression changes were calculated using the 2-ΔΔCT method.
Triplicate independently generated samples for each condition were sequenced to a read depth of 20 million using an Illumina NextSeq™500 platform, generating single-end 75 bp reads fastq files. Original raw data can be found in the NCBI Gene Expression Omnibus database with the accession number GSE162015. For the analysis the University of Glasgow Galaxy (33) server (v18.09) was used and a detailed analysis pipeline is available at http://heighliner.cvr.gla.ac.uk/u/dsomma1/h/hnfkb1-wt-vs-ko. Briefly, we use FastQC (v0.72), MultiQC (v1.5.0) and Trimmomatic (v0.36.5) for FastQ files quality control check, improvement, and plots. FastQ files were mapped on human genome (Ensembl GRCh38.p13 sm primary assembly) using HISAT2 (v2.1.0). Features were assigned using Infer Experiment (v2.6.4) and featureCounts (v1.6.0.3) against human genome annotation (Ensembl GRCh38.98 filtered to contain only protein coding genes, for a total n of genes: 19976). RNAseq reads are good quality (Figure S1A), with ~90% of reads mapped uniquely (Figure S1B), and 75% assigned to the corresponding gene (Figure S1C). Two samples were excluded because PCA identify them as outliers (Figure S1D). DESeq2 (v2.11.40.2) was used to determine DE genes. Heatmaps and plots were generate using Python 3.7.4, Jupyter notebook 6.0.4, Pandas 0.23.4, Seaborn 0.10.1. Heatmap of differentially expressed genes in different replicates is shown in Figure S1E.
Genes differentially expressed between WT and NFKB1-/- cells were filtered for q value < 0.05; to be sure to identify all genes controlled by LPS. LPS-induced genes (comparing WT unstimulated vs WT LPS) were filtered for a q value < 0.1, and intersected with the lists previously generated, as indicated in Venn diagram (Figure S2B). For pathway analysis Reactome pathway database (34) was used (v71), while gene set enrichment was performed using EnrichR (35) through gseapy (v0.9.16) to query BioPlanet (24), TRRUST (36) and GeneRif (37) databases. To infer transcriptional regulators involved in gene expression Lisa (38) software website was used; top 500 genes (either upregulated or downregulated) ordered by q values were selected and LPS-dependent-p105-independent genes were used as background removal. Specifically, for Reactome, TRRUST and Lisa, genes were split into upregulated and downregulated in NFKB1-/- to dissect the contribution of each pathway and transcription factor. For BioPlanet and GeneRif analysis upregulated, and downregulated genes were used as one list to provide a broader view.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
DS, CW, and RC designed research. DS, DK, FK, and RC performed research. DK contributed new reagents. DS and CW analysed data. DS and RC wrote the paper. All authors contributed to the article and approved the submitted version.
This work was supported by the Medical Research Council (MR/M010694/1) and Biotechnology and Biological Sciences Research Council (BB/M003671/1 and BB/T007427/1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.669906/full#supplementary-material
1. Fliegauf M, Bryant VL, Frede N, Slade C, Woon ST, Lehnert K, et al. Haploinsufficiency of the NF-Kappab1 Subunit P50 in Common Variable Immunodeficiency. Am J Hum Genet (2015) 97(3):389–403. doi: 10.1016/j.ajhg.2015.07.008
2. Fliegauf M, Grimbacher B. Nuclear Factor kappaB Mutations in Human Subjects: The Devil Is in the Details. J Allergy Clin Immunol (2018) 142(4):1062–5. doi: 10.1016/j.jaci.2018.06.050
3. Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E, et al. Loss-Of-Function Nuclear Factor kappaB Subunit 1 (NFKB1) Variants are the Most Common Monogenic Cause of Common Variable Immunodeficiency in Europeans. J Allergy Clin Immunol (2018) 142(4):1285–96. doi: 10.1016/j.jaci.2018.01.039
4. Kaustio M, Haapaniemi E, Goos H, Hautala T, Park G, Syrjanen J, et al. Damaging Heterozygous Mutations in NFKB1 Lead to Diverse Immunologic Phenotypes. J Allergy Clin Immunol (2017) 140(3):782–96. doi: 10.1016/j.jaci.2016.10.054
5. Mitchell JP, Carmody RJ. NF-kappaB and the Transcriptional Control of Inflammation. Int Rev Cell Mol Biol (2018) 335:41–84. doi: 10.1016/bs.ircmb.2017.07.007
6. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-Kappab: A Blossoming of Relevance to Human Pathobiology. Cell (2017) 168(1-2):37–57. doi: 10.1016/j.cell.2016.12.012
7. Dieli-Crimi R, Martinez-Gallo M, Franco-Jarava C, Antolin M, Blasco L, Paramonov I, et al. Th1-Skewed Profile and Excessive Production of Proinflammatory Cytokines in a NFKB1-Deficient Patient With CVID and Severe Gastrointestinal Manifestations. Clin Immunol (2018) 195:49–58. doi: 10.1016/j.clim.2018.07.015
8. Mandola AB, Sharfe N, Nagdi Z, Dadi H, Vong L, Merico D, et al. Combined Immunodeficiency Caused by a Novel Homozygous NFKB1 Mutation. J Allergy Clin Immunol (2021) 147(2):727–33.e2. doi: 10.1016/j.jaci.2020.08.040
9. Grumont RJ, Rourke IJ, O'Reilly LA, Strasser A, Miyake K, Sha W, et al. B Lymphocytes Differentially Use the Rel and Nuclear Factor Kappab1 (NF-Kappab1) Transcription Factors to Regulate Cell Cycle Progression and Apoptosis in Quiescent and Mitogen-Activated Cells. J Exp Med (1998) 187(5):663–74. doi: 10.1084/jem.187.5.663
10. Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, et al. Requirement for NF-kappaB in Osteoclast and B-Cell Development. Genes Dev (1997) 11(24):3482–96. doi: 10.1101/gad.11.24.3482
11. Horwitz BH, Scott ML, Cherry SR, Bronson RT, Baltimore D. Failure of Lymphopoiesis After Adoptive Transfer of NF-kappaB-Deficient Fetal Liver Cells. Immunity (1997) 6(6):765–72. doi: 10.1016/s1074-7613(00)80451-3
12. Pohl T, Gugasyan R, Grumont RJ, Strasser A, Metcalf D, Tarlinton D, et al. The Combined Absence of NF-Kappa B1 and C-Rel Reveals That Overlapping Roles for These Transcription Factors in the B Cell Lineage are Restricted to the Activation and Function of Mature Cells. Proc Natl Acad Sci USA (2002) 99(7):4514–9. doi: 10.1073/pnas.072071599
13. Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted Disruption of the P50 Subunit of NF-Kappa B Leads to Multifocal Defects in Immune Responses. Cell (1995) 80(2):321–30. doi: 10.1016/0092-8674(95)90415-8
14. Artis D, Kane CM, Fiore J, Zaph C, Shapira S, Joyce K, et al. Dendritic Cell-Intrinsic Expression of NF-Kappa B1 Is Required to Promote Optimal Th2 Cell Differentiation. J Immunol (2005) 174(11):7154–9. doi: 10.4049/jimmunol.174.11.7154
15. Das J, Chen CH, Yang L, Cohn L, Ray P, Ray A. A Critical Role for NF-Kappa B in GATA3 Expression and TH2 Differentiation in Allergic Airway Inflammation. Nat Immunol (2001) 2(1):45–50. doi: 10.1038/83158
16. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity (2014) 40(2):274–88. doi: 10.1016/j.immuni.2014.01.006
17. Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, et al. Tolerance and M2 (Alternative) Macrophage Polarization Are Related Processes Orchestrated by P50 Nuclear Factor kappaB. Proc Natl Acad Sci USA (2009) 106(35):14978–83. doi: 10.1073/pnas.0809784106
18. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained Immunity: A Program of Innate Immune Memory in Health and Disease. Science (2016) 352(6284):aaf1098. doi: 10.1126/science.aaf1098
19. Bohuslav J, Kravchenko VV, Parry GC, Erlich JH, Gerondakis S, Mackman N, et al. Regulation of an Essential Innate Immune Response by the P50 Subunit of NF-kappaB. J Clin Invest (1998) 102(9):1645–52. doi: 10.1172/JCI3877
20. Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, et al. Chronic Inflammation Induces Telomere Dysfunction and Accelerates Ageing in Mice. Nat Commun (2014) 2:4172. doi: 10.1038/ncomms5172
21. Porta C, Ippolito A, Consonni FM, Carraro L, Celesti G, Correale C, et al. Protumor Steering of Cancer Inflammation by P50 NF-kappaB Enhances Colorectal Cancer Progression. Cancer Immunol Res (2018) 6(5):578–93. doi: 10.1158/2326-6066.CIR-17-0036
22. Chanput W, Mes JJ, Wichers HJ. THP-1 Cell Line: An In Vitro Cell Model for Immune Modulation Approach. Int Immunopharmacol (2014) 23(1):37–45. doi: 10.1016/j.intimp.2014.08.002
23. Christian F, Smith EL, Carmody RJ. The Regulation of NF-kappaB Subunits by Phosphorylation. Cells (2016) 5(1). doi: 10.3390/cells5010012
24. Huang R, Grishagin I, Wang Y, Zhao T, Greene J, Obenauer JC, et al. The NCATS BioPlanet - An Integrated Platform for Exploring the Universe of Cellular Signaling Pathways for Toxicology, Systems Biology, and Chemical Genomics. Front Pharmacol (2019) 10:445. doi: 10.3389/fphar.2019.00445
25. Griffon A, Barbier Q, Dalino J, van Helden J, Spicuglia S, Ballester B. Integrative Analysis of Public ChIP-Seq Experiments Reveals a Complex Multi-Cell Regulatory Landscape. Nucleic Acids Res (2015) 43(4):e27. doi: 10.1093/nar/gku1280
26. Piaszyk-Borychowska A, Szeles L, Csermely A, Chiang HC, Wesoly J, Lee CK, et al. Signal Integration of IFN-I and IFN-II With TLR4 Involves Sequential Recruitment of STAT1-Complexes and NFkappaB to Enhance Pro-Inflammatory Transcription. Front Immunol (2019) 10:1253. doi: 10.3389/fimmu.2019.01253
27. Sanceau J, Boyd DD, Seiki M, Bauvois B. Interferons Inhibit Tumor Necrosis Factor-Alpha-Mediated Matrix Metalloproteinase-9 Activation via Interferon Regulatory Factor-1 Binding Competition With NF-Kappa B. J Biol Chem (2002) 277(38):35766–75. doi: 10.1074/jbc.M202959200
28. Amici SA, Young NA, Narvaez-Miranda J, Jablonski KA, Arcos J, Rosas L, et al. CD38 Is Robustly Induced in Human Macrophages and Monocytes in Inflammatory Conditions. Front Immunol (2018) 9:1593. doi: 10.3389/fimmu.2018.01593
29. Hagai T, Chen X, Miragaia RJ, Rostom R, Gomes T, Kunowska N, et al. Gene Expression Variability Across Cells and Species Shapes Innate Immunity. Nature (2018) 563(7730):197–202. doi: 10.1038/s41586-018-0657-2
30. Della Chiara G, Crotti A, Liboi E, Giacca M, Poli G, Lusic M. Negative Regulation of HIV-1 Transcription by a Heterodimeric NF-Kappab1/P50 and C-Terminally Truncated STAT5 Complex. J Mol Biol (2011) 410(5):933–43. doi: 10.1016/j.jmb.2011.03.044
31. Shen CH, Stavnezer J. Interaction of Stat6 and NF-Kappab: Direct Association and Synergistic Activation of Interleukin-4-Induced Transcription. Mol Cell Biol (1998) 18(6):3395–404. doi: 10.1128/mcb.18.6.3395
32. Yu Z, Zhang W, Kone BC. Signal Transducers and Activators of Transcription 3 (STAT3) Inhibits Transcription of the Inducible Nitric Oxide Synthase Gene by Interacting With Nuclear Factor kappaB. Biochem J (2002) 367(Pt 1):97–105. doi: 10.1042/BJ20020588
33. Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2018 Update. Nucleic Acids Res (2018) 46(W1):W537–W44. doi: 10.1093/nar/gky379
34. Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res (2020) 48(D1):D498–503. doi: 10.1093/nar/gkz1031
35. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res (2016) 44(W1):W90–7. doi: 10.1093/nar/gkw377
36. Han H, Cho JW, Lee S, Yun A, Kim H, Bae D, et al. TRRUST V2: An Expanded Reference Database of Human and Mouse Transcriptional Regulatory Interactions. Nucleic Acids Res (2018) 46(D1):D380–D6. doi: 10.1093/nar/gkx1013
37. Jimeno-Yepes AJ, Sticco JC, Mork JG, Aronson AR. GeneRIF Indexing: Sentence Selection Based on Machine Learning. BMC Bioinf (2013) 14:171. doi: 10.1186/1471-2105-14-171
Keywords: NF-κB, macrophage, NFKB1, transcriptomics, toll-like receptors, THP1 cells
Citation: Somma D, Kok FO, Kerrigan D, Wells CA and Carmody RJ (2021) Defining the Role of Nuclear Factor (NF)-κB p105 Subunit in Human Macrophage by Transcriptomic Analysis of NFKB1 Knockout THP1 Cells. Front. Immunol. 12:669906. doi: 10.3389/fimmu.2021.669906
Received: 19 February 2021; Accepted: 13 September 2021;
Published: 13 October 2021.
Edited by:
Cheol-Heui Yun, Seoul National University, South KoreaReviewed by:
George Hsiao, Taipei Medical University, TaiwanCopyright © 2021 Somma, Kok, Kerrigan, Wells and Carmody. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Domenico Somma, ZG9tZW5pY28uc29tbWFAZ2xhc2dvdy5hYy51aw==; Ruaidhrí J. Carmody, cnVhaWRocmkuY2FybW9keUBnbGFzZ293LmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.