 Stuart I. Mannering
Stuart I. Mannering Alan F. Rubin
Alan F. Rubin Ruike Wang
Ruike Wang Pushpak Bhattacharjee
Pushpak Bhattacharjee
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 30 April 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.667870
This article is part of the Research Topic Autoimmune Diabetes: Molecular Mechanisms and Neoantigens View all 13 articles
In 2016 Delong et al. discovered a new type of neoepitope formed by the fusion of two unrelated peptide fragments. Remarkably these neoepitopes, called hybrid insulin peptides, or HIPs, are recognized by pathogenic CD4+ T cells in the NOD mouse and human pancreatic islet-infiltrating T cells in people with type 1 diabetes. Current data implicates CD4+ T-cell responses to HIPs in the immune pathogenesis of human T1D. Because of their role in the immune pathogenesis of human T1D it is important to identify new HIPs that are recognized by CD4+ T cells in people at risk of, or with, T1D. A detailed knowledge of T1D-associated HIPs will allow HIPs to be used in assays to monitor changes in T cell mediated beta-cell autoimmunity. They will also provide new targets for antigen-specific therapies for T1D. However, because HIPs are formed by the fusion of two unrelated peptides there are an enormous number of potential HIPs which makes it technically challenging to identify them. Here we review the discovery of HIPs, how they form and discuss approaches to identifying new HIPs relevant to the immune pathogenesis of human type 1 diabetes.
Type 1 diabetes (T1D) is an autoimmune disease caused the T-cell mediated destruction of the pancreatic beta cells (1). How and why the immune system turns against otherwise healthy tissue, to cause an autoimmune disease, is currently not known. However, for T1D it is clear that T-cells infiltrate the islet of Langerhans within the pancreas and destroy the insulin producing beta cells (2).
Genetic studies have revealed that risk of developing T1D is associated with class II HLA (3). The strongest genetic risk for developing T1D is associated with the HLA class II haplotypes HLA-DR3-DQ2 (Odds ratio (OR)>3.6) and HLA-DR4-DQ8 (OR>11.37) (3). Within these haplotypes the HLA-DQ alleles confer the majority of the risk (4). Intriguingly, people who are HLA-DR3-DQ2; DR4-DQ8 heterozygous are at greatest risk (OR>16.6) of developing type 1 diabetes (3). HLA-DQ2/8 heterozygous antigen presenting cells express four types of HLA-DQαβ heterodimers. The ‘regular’ HLA-DQ2cis and –DQ8cis (α and β chains encoded on the same chromosome) and the ‘trans’ dimers (α and β chains encoded on different chromosomes) (5). Collectively, this work suggests that CD4+ T-cell responses against beta cell derived antigens play a central role in the pathogenesis of T1D, the identity of the antigens targeted by pathogenic CD4+ T cells has not been fully resolved.
Insulin and its precursor, proinsulin, have long been considered to be central to the autoimmune pathogenesis of T1D (1, 6). Proinsulin is the major protein product of beta cells accounting for ~10% of the cell’s protein (7). Mature insulin forms when the central ‘C-peptide’ of proinsulin is excised in the beta-cell granules leaving mature insulin and ‘free’ C-peptide (8). A genetic susceptibility locus maps to a variable number of tandem repeats (VNTR) upstream of the insulin gene and regulates its expression in the thymus, which in turn affects central tolerance to (pro)insulin (9). This locus is strongly associated with risk of developing type 1 diabetes (OR>2.5), second only to the HLA (10). In 2015, Pathiraja et al. (11) were the first to show that proinsulin specific CD4+ T cells infiltrate human islets in type 1 diabetes. Human islet-infiltrating CD4+ T cells recognize several epitopes derived from the C-peptide presented by HLA-DQ8, or DQ8trans (11).
An important twist in proinsulin’s role in T1D came in 2016 when Delong et al. (12) described hybrid insulin peptides (HIPs). A hybrid insulin peptide, or HIP, is a CD4+ T-cell epitope that is formed by the posttranslational fusion of two peptide fragments [reviewed by (13)]. To date, as the name suggests, at least one of the peptide fragments derives from insulin or proinsulin. The peptide fragments that form a HIP can be derived from the same parental protein molecule(s), or from two distinct proteins. Delong and Haskins’ goal was to identify the epitopes recognized by a family of T-cell clones, known as BDC, that had been isolated from NOD mice (14). Importantly, many of the BDC lines were known to cause diabetes (15). Because of their clear pathogenic role, the BDC clones have been well studied, particularly BDC-2.5 (16, 17). However, the naturally arising agonist epitope for BDC-2.5, and many other BDC clones, remained elusive. Several mimotopes peptides were identified that stimulated BDC-2.5 (18–20), but a broadly accepted, naturally arising, target antigen was not found. Despite this, it became clear that an epitope from the beta-cell secretory granule protein, chromogranin-A (ChgA) was, at least, a partial target (21, 22). More specifically, it was clear that the WE14 fragment of chromogranin-A could stimulate the BDC-2.5 cells albeit weakly. Synthetic peptide analogues of the WE14 fragment were made with amino acid substitutions at each position and tested for their capacity to stimulate BDC-2.5 cells. This analysis showed that substitutions at the carboxyl terminus of the putative epitope impacted upon BDC-2.5 stimulation (22). Furthermore, a membrane extract from a beta-cell tumor was a more potent stimulator of BDC-2.5 than synthetic WE14 peptide (22) - clearly something was missing.
Delong, Haskins and co-workers resolved the enigma of the natural agonist for BDC-2.5 (and several other BDC lines) when they made the intuitive leap that CD4+ T-cell epitopes could form by fusion of two peptide fragments. They showed, by mass spectrometry and functional T-cell assays, that the missing half of the epitope was a fragment of the C-peptide from proinsulin (LQTLAL). The optimal agonist for BDC-2.5 (and BDC-9.46 and BDC-10.1) was a peptide formed by the fusion of a fragment of C-peptide with a fragment derived from WE14 (LQTLAL-WSRMD). A similar HIP (LQTLAL-NAARD) formed by the same C-peptide fragment (LQTLAL) fused to a fragment of IAPP2 (NAARD) was also identified. These new types of peptides were called ‘hybrid insulin peptides’ or HIP. Titration experiments, using synthetic peptides, revealed that a Cpeptide-ChgA HIP was an extremely potent (at nM concentrations) stimulator of BDC-2.5 CD4+ T cells, making it a very plausible candidate for the natural agonist for BDC-2.5. Solving the mystery of the ligand for BDC-2.5 was interesting, but it was not clear if HIP specific CD4+ T cells arose in people with T1D and if they played any role the pathogenesis of T1D. We addressed this question by testing a panel of 16 putative human HIPs, designed based on the HIPs identified in the NOD mouse, against of human islet-infiltrating CD4+ T-cell clones. From these experiments we identified two clones that responded to a Cpeptide-IAPP2 HIP (12). Independently, Sally Kent’s group screened the same panel of 16 HIPs and identified an islet-infiltrating CD4+ T-cell line that responded to a Cpept-NYP HIP (12). Subsequently, further evidence for a pathogenic role of CD4+ T-cell responses to HIPs has accumulated. For example, further screening of this panel of 16 HIPs against human islet-infiltrating T-cells lines revealed responses to Cpept-IAPP1, Cpept-InsA and Cpept-IAPP2 HIPs (23) (See Table 1). Analysis of HIP specific responses in the PBMC of people with recent onset T1D revealed responses to several HIPs that were less frequently detected in control subjects who did not have T1D (25).
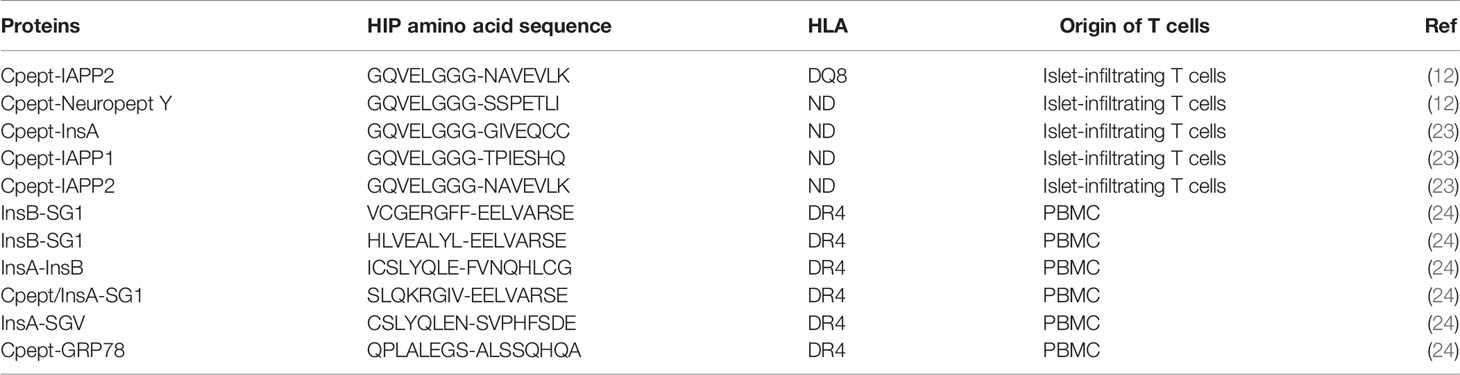
Table 1 Summary of currently known HIPs.
Protease-mediated peptide splicing, or transpeptidation, has been described in bacteria (26), plants (27), in vitro (28) and in humans. In humans, proteasome-mediated protein splicing generates epitopes recognized by tumor specific CD8+ T cells (29, 30). In all cases energy released during proteolysis is ‘recycled’ to drive the formation of a peptide bond (30). Under most circumstances this ‘reverse proteolysis’ is very inefficient, but high protein concentrations in a confined environment favor protease mediated peptide fusion (31). Beta-cell granules are extremely densely packed with insulin, C-peptide and several other proteins including chromogranins and amyloid proteins (32). Furthermore, the beta-cell granules are the site of very active proteolysis. Many proteins found in the beta cell granules, including proinsulin (PI), chromogranins (Chg) and islet amyloid polypeptide (IAPP), are cleaved by the granule proteases to generate an array of bio-active peptides (32). However, Wan et al. (33) reported that analysis of mass spectrometry data from beta-cell crinosomes revealed the presence of HIPs suggesting that HIPs form in the crinosomes. More recently, Reed et al. (34) reported that when the appropriate peptides are digested by the lysosomal protease, cathepsin L, HIPs that activate the lines BDC-2.5 and BDC-6.9 are generated. This led to the suggestion that HIPs form when senescent insulin granules fuse with lysosomes to form crinophagic granules. The high concentration of insulin granule proteins and cathepsin-L favors the formation of HIPs. Crinophagic granules can be taken up by antigen presenting cells allowing the HIPs to be presented to CD4+ T cells (35). Nonetheless, the precise location(s) of HIP formation remain unclear. C-peptide can be cleaved at sites other than the dibasic residues at the B-C and C-A chain junctions (36). This indicates that other proteases may mediate the formation of HIPs. It also remains possible that transpeptidation and HIP formation occurs in both granules and crinosomes.
Identification of ‘regular’ T-cell epitopes has been scientific challenge, but over the past decades significant progress has been made (37). Progress has been facilitated by advances in peptide synthesis techniques, T-cell cloning (38), TCR expression, recombinant DNA techniques and mass spectrometry. Mapping of ‘regular’ T-cell epitopes usually reduces the target epitope from a cell, or microbe, to a protein and from there an ~8-14 amino acid peptide. However, in the case of HIPs the process is more complex, because, theoretically at least, any protein fragment can fuse with any other protein fragment. HIPs can form from fragments of the same proteins, or from two different proteins, and the fragments can fuse in either order. These possible combinations give rise to an enormous array of HIPs that are, theoretically at least, possible. For example, from a protein of 100 amino acids we calculate that there are 9,756 possible HIPs (Figure 1). This only included possible HIPs formed by fragmentation and fusion of a single 100 amino acid protein. Once other candidate proteins are considered as potential HIP fragment donors the number of possible HIPs becomes extremely large. Below we discuss the different approaches to identifying and validating novel HIPs at scale.

Figure 1 Calculating the number of HIPs that could be generated from a 100 amino acid (AA) long protein. A hypothetical 100 amino acid long peptide is indicated by the red box. The first few of all possible 10 amino acid fragments, derived from this protein are shown in red. Potential HIPs, in both orientations of the composite fragments are shown. Assumptions made in calculating the number of potential HIPs that may form from a 100 amino acid protein are shown.
Mass spectrometry has been invaluable for the identification of HIPs. The advantage of mass spectrometry is that a large number of sequences can be identified rapidly. Candidate T cell epitopes have been identified by elution of peptides from HLA/MHC on the surface of antigen-presenting cells. However, this approach is not feasible with HIPs which are present at very low concentrations in beta cells. Furthermore, since HIPs aren’t directly encoded new algorithms have been developed to facilitate the identification of HIPs, or CD8+ T-cell epitopes formed by proteosome mediated peptide splicing (39, 40). Nonetheless, a rigorous mass spectrometry work flow for the identification of HIPs from beta-cell extracts has been developed (41).
One of the disadvantages of mass spectrometry is large numbers of cells are usually required to isolate sufficient material for analysis. This makes the analysis of primary beta cells difficult. Delong et al. solved this problem by using beta cell tumors from RIP-TAg mice. More recently they have adapted their work-flow to allow for smaller numbers of cells (41) and developed internal controls to allow quantifiable confidence when matching synthetic and biologically derived peptides (42). Mass spectrometry analysis provides good evidence for the presence of a peptide, but it does not give any insights into the capacity of T cells to respond to a particular peptide for their relationship between a CD4+ T-cell response to an epitope and the disease pathogenesis. For this reason, validation of candidate HIPs with functional T-cell assays, as described above, is essential.
Arribas-Layton et al. (24) used a tetramer-based approach to identify new HIPs. They started with a database of 7,654 candidate HIPs formed by fusion of fragments of proinsulin with a variety of beta cell granule proteins. This list of peptides was stratified by predicted binding to HLA-DR4 (DRB1*04:01). The 50 peptides with the strongest predicted binding affinity to HLA-DR4 were then screened in competitive HLA binding assays and 30 confirmed HLA-DR4 binding peptides were chosen. These 30 HIPs were used to make HLA-DR4/HIP tetramers. Finally, PBMC from individuals with T1D were stimulated with pools of HIP peptides and cytokines for two weeks, then the expanded cells were stained with DR4/HIP tetramers. This work led to the identification of six new, HLA-DR4 restricted HIPs (Table 1).
The work summarized above highlights the power of combining functional T-cell responses with proteomic analysis. Observations using T-cell responses support the proteomics data and, in turn, the proteomics supports the immunology. This work brings us to the current question: is it possible to develop a high throughput and efficient protocol for identifying HIPs that are relevant to the immune pathogenesis of human T1D? If so, what would such a protocol look like? The primary challenge, as outlined above, is the enormous number of potential HIPs to be screened. Proteomic approaches use fractionation of tissue lysates and screening with disease-relevant T cells. While this approach worked for Delong et al. (12), who had the BDC lines and RIP-Tag beta cells, it will be challenging to apply this to human studies because sufficiently large numbers of beta cells are not readily available and CD4+ T cells of clear clinical relevance are not available. CD4+ T cells that infiltrate human islets (11, 43) are available, but screening large numbers of candidate HIPs against large numbers of islet-infiltrating T cells remains a challenging task.
The development of improved synthetic peptide and DNA technologies increases the feasibility of screening approaches. Large libraries of candidate HIPs can be generated in silico and as peptides or synthetic DNA. For example, Arribas-Layton et al. refined their pool of over 7,000 candidates HIPs by predicting and then confirming binding to HLA-DR4. This gave them a manageable number of HIPs to screen with PBMC from people with T1D using DR4/HIP tetramers. The limitation of this approach is that it focuses on a single HLA restriction and relies upon the accuracy of the HLA-binding predictions.
An ideal HIP-identification protocol would have the following features: (i) it would allow the screening of very large numbers of candidate HIPs, reducing the incentive to use HLA binding predictions to enrich for candidates with higher predicted binding capacities. (ii) it would screen full haplotypes of T1D-associated HLA alleles at one time, avoiding the need to screening for a particular restriction. (iii) it would identify epitopes based on their capacity to stimulate disease relevant T cells, such as CD4+ T cells isolated from the islets of organ donors who had T1D. The frequency of beta cell antigen specific T cells in peripheral blood is too low to allow these cells to be used in screening assays. Expansion of cells of possible relevance biases the screen towards the specificities already thought to be important. To our knowledge, an assay that meets all these criteria does not currently exist. Recent developments may make such an assay feasible. For example, the T-Scan protocol (44), which can screen large numbers of candidate CD8+ T-cell epitopes, has many of the desirable feature. Our challenge now is to develop a comparable assay that can be used to identify human CD4+ T-cell epitopes, including HIPs.
The discovery of HIP peptides has revealed a new array of targets for the autoimmune responses underlying type 1 diabetes. Discoveries made using the NOD mouse have rapidly been supported by experiments using human materials. It is now clear that HIPs do form in beta cells and can be the targets of autoimmune responses. However, it seems very likely that we have only identified a fraction of the HIPs that may play a role in T1D. Emerging approaches to identify and validate new HIPs. Once defined these epitopes will be of great interest, and utility, in T cell assays to monitor changes in beta cell autoimmunity (45) and antigen-specific therapies for T1D.
SM and PB conceived the scope and focus of the review. SM wrote the first draft. AR, RW and PB contributed to the editing and preparation of the final manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Juvenile Diabetes Research Foundation (2-SRA-2020-909-S-B) and the National Health and Medical Research Council (NHMRC GNT123586, 1138717) (SM). PB is supported by a St. Vincent’s Institute Rising Star Award. The authors acknowledge the support of the Operational Infrastructure Support Program of the Victorian Government.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors gratefully acknowledge the contribution of their colleagues in the Immunology of Diabetes Unit at St Vincent’s Institute for helpful discussions.
1. Mannering SI, Pathiraja V, Kay TW. The Case for an Autoimmune Aetiology of Type 1 Diabetes. Clin Exp Immunol (2016) 183(1):8–15. doi: 10.1111/cei.12699
2. Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, et al. Demonstration of Islet-Autoreactive CD8 T Cells in Insulitic Lesions From Recent Onset and Long-Term Type 1 Diabetes Patients. J Exp Med (2012) 209(1):51–60. doi: 10.1084/jem.20111187
3. Noble JA. Immunogenetics of Type 1 Diabetes: A Comprehensive Review. J Autoimmun (2015) 64:101–12. doi: 10.1016/j.jaut.2015.07.014
4. Todd JA, Bell JI, McDevitt HO. Hla-DQ Beta Gene Contributes to Susceptibility and Resistance to Insulin-Dependent Diabetes Mellitus. Nature (1987) 329(6140):599–604. doi: 10.1038/329599a0
5. Nepom BS, Schwarz D, Palmer JP, Nepom GT. Transcomplementation of HLA Genes in IDDM. Hla-DQ Alpha- and Beta-Chains Produce Hybrid Molecules in DR3/4 Heterozygotes. Diabetes (1987) 36(1):114–7. doi: 10.2337/diab.36.1.114
6. Narendran P, Mannering SI, Harrison LC. Proinsulin-a Pathogenic Autoantigen in Type 1 Diabetes. Autoimmun Rev (2003) 2(4):204–10. doi: 10.1016/S1568-9972(03)00009-0
7. Weir GC, Bonner-Weir S. Islet Beta Cell Mass in Diabetes and How it Relates to Function, Birth, and Death. Ann New York Acad Sci (2013) 1281:92–105. doi: 10.1111/nyas.12031
8. Dodson G, Steiner D. The Role of Assembly in Insulin’s Biosynthesis. Curr Opin Struct Biol (1998) 8(2):189–94. doi: 10.1016/S0959-440X(98)80037-7
9. Bennett ST, Lucassen AM, Gough SC, Powell EE, Undlien DE, Pritchard LE, et al. Susceptibility to Human Type 1 Diabetes At IDDM2 is Determined by Tandem Repeat Variation At the Insulin Gene Minisatellite Locus. Nat Genet (1995) 9(3):284–92. doi: 10.1038/ng0395-284
10. Concannon P, Rich SS, Nepom GT. Genetics of Type 1a Diabetes. New Engl J Med (2009) 360(16):1646–54. doi: 10.1056/NEJMra0808284
11. Pathiraja V, Kuehlich JP, Campbell PD, Krishnamurthy B, Loudovaris T, Coates PT, et al. Proinsulin-Specific, HLA-DQ8, and HLA-DQ8-transdimer-restricted Cd4+ T Cells Infiltrate Islets in Type 1 Diabetes. Diabetes (2015) 64(1):172–82. doi: 10.2337/db14-0858
12. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T Cells in Type 1 Diabetes Recognize Epitopes Formed by Peptide Fusion. Science (2016) 351(6274):711–4. doi: 10.1126/science.aad2791
13. Wiles TA, Delong T. Hips and HIP-Reactive T Cells. Clin Exp Immunol (2019) 198(3):306–13. doi: 10.1111/cei.13335
14. Haskins K, Portas M, Bergman B, Lafferty K, Bradley B. Pancreatic Islet-Specific T-cell Clones From Nonobese Diabetic Mice. Proc Natl Acad Sci U S A (1989) 86(20):8000–4. doi: 10.1073/pnas.86.20.8000
15. Haskins K, McDuffie M. Acceleration of Diabetes in Young NOD Mice With a CD4+ Islet-Specific T Cell Clone. Science (1990) 249(4975):1433–6. doi: 10.1126/science.2205920
16. Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a Diabetogenic T Cell From Genesis Through Pathogenesis. Cell (1993) 74(6):1089–100. doi: 10.1016/0092-8674(93)90730-E
17. Ramirez L, Hamad AR. Status of Autoimmune Diabetes 20-Year After Generation of BDC-2.5-TCR Transgenic non-Obese Diabetic Mouse. World J Diabetes (2013) 4(4):88–91. doi: 10.4239/wjd.v4.i4.88
18. Haskins K. Pathogenic T-cell Clones in Autoimmune Diabetes: More Lessons From the NOD Mouse. Adv Immunol (2005) 87:123–62. doi: 10.1016/S0065-2776(05)87004-X
19. Dai YD, Jensen KP, Lehuen A, Masteller EL, Bluestone JA, Wilson DB, et al. A Peptide of Glutamic Acid Decarboxylase 65 can Recruit and Expand a Diabetogenic T Cell Clone, BDC-2.5, in the Pancreas. J Immunol (2005) 175(6):3621–7. doi: 10.4049/jimmunol.175.6.3621
20. Judkowski V, Pinilla C, Schroder K, Tucker L, Sarvetnick N, Wilson DB. Identification of MHC Class II-restricted Peptide Ligands, Including a Glutamic Acid Decarboxylase 65 Sequence, That Stimulate Diabetogenic T Cells From Transgenic BDC-2.5 Nonobese Diabetic Mice. J Immunol (2001) 166(2):908–17. doi: 10.4049/jimmunol.166.2.908
21. Nikoopour E, Sandrock C, Huszarik K, Krougly O, Lee-Chan E, Masteller EL, et al. Cutting Edge: vasostatin-1-derived Peptide ChgA29-42 is an Antigenic Epitope of Diabetogenic BDC-2.5 T Cells in Nonobese Diabetic Mice. J Immunol (2011) 186(7):3831–5. doi: 10.4049/jimmunol.1003617
22. Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, et al. Chromogranin A is an Autoantigen in Type 1 Diabetes. Nat Immunol (2010) 11(3):225–31. doi: 10.1038/ni.1844
23. Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, et al. Analysis of Self-Antigen Specificity of Islet-Infiltrating T Cells From Human Donors With Type 1 Diabetes. Nat Med (2016) 22(12):1482–7. doi: 10.1038/nm.4203
24. Arribas-Layton D, Guyer P, Delong T, Dang M, Chow IT, Speake C, et al. Hybrid Insulin Peptides Are Recognized by Human T Cells in the Context of DRB1*04:01. Diabetes (2020) 69(7):1492–502. doi: 10.2337/db19-0620
25. Baker RL, Rihanek M, Hohenstein AC, Nakayama M, Michels A, Gottlieb PA, et al. Hybrid Insulin Peptides Are Autoantigens in Type 1 Diabetes. Diabetes (2019) 68(9):1830–40. doi: 10.2337/db19-0128
26. Marraffini LA, Dedent AC, Schneewind O. Sortases and the Art of Anchoring Proteins to the Envelopes of Gram-Positive Bacteria. Microbiol Mol Biol Rev (2006) 70(1):192–221. doi: 10.1128/MMBR.70.1.192-221.2006
27. Carrington DM, Auffret A, Hanke DE. Polypeptide Ligation Occurs During Post-Translational Modification of Concanavalin a. Nature (1985) 313(5997):64–7. doi: 10.1038/313064a0
28. Fodor S, Zhang Z. Rearrangement of Terminal Amino Acid Residues in Peptides by Protease-Catalyzed Intramolecular Transpeptidation. Anal Biochem (2006) 356(2):282–90. doi: 10.1016/j.ab.2006.06.023
29. Hanada K, Yewdell JW, Yang JC. Immune Recognition of a Human Renal Cancer Antigen Through Post-Translational Protein Splicing. Nature (2004) 427(6971):252–6. doi: 10.1038/nature02240
30. Vigneron N, Stroobant V, Chapiro J, Ooms A, Degiovanni G, Morel S, et al. An Antigenic Peptide Produced by Peptide Splicing in the Proteasome. Science (2004) 304(5670):587–90. doi: 10.1126/science.1095522
31. Somalinga BR, Roy RP. Volume Exclusion Effect as a Driving Force for Reverse Proteolysis. Implications for Polypeptide Assemblage in a Macromolecular Crowded Milieu. J Biol Chem (2002) 277(45):43253–61. doi: 10.1074/jbc.M207974200
32. Suckale J, Solimena M. The Insulin Secretory Granule as a Signaling Hub. Trends Endocrinol Metab (2010) 21(10):599–609. doi: 10.1016/j.tem.2010.06.003
33. Wan X, Vomund AN, Peterson OJ, Chervonsky AV, Lichti CF, Unanue ER. The MHC-II Peptidome of Pancreatic Islets Identifies Key Features of Autoimmune Peptides. Nat Immunol (2020) 21(4):455–63. doi: 10.1038/s41590-020-0623-7
34. Reed B, Crawford F, Hill RC, Jin N, White J, Krovi SH, et al. Lysosomal Cathepsin Creates Chimeric Epitopes for Diabetogenic CD4 T Cells Via Transpeptidation. J Exp Med (2020) 218(2). doi: 10.1084/jem.20192135
35. Vomund AN, Zinselmeyer BH, Hughes J, Calderon B, Valderrama C, Ferris ST, et al. Beta Cells Transfer Vesicles Containing Insulin to Phagocytes for Presentation to T Cells. Proc Natl Acad Sci (2015) 112(40):E5496–502. doi: 10.1073/pnas.1515954112
36. Verchere CB, Paoletta M, Neerman-Arbez M, Rose K, Irminger JC, Gingerich RL, et al. Des-(27-31)C-Peptide. A Novel Secretory Product of the Rat Pancreatic Beta Cell Produced by Truncation of Proinsulin Connecting Peptide in Secretory Granules. J Biol Chem (1996) 271(44):27475–81. doi: 10.1074/jbc.271.44.27475
37. James EA, Mallone R, Kent SC. Dilorenzo TP. T-Cell Epitopes and Neo-epitopes in Type 1 Diabetes: A Comprehensive Update and Reappraisal. Diabetes (2020) 69(7):1311–35. doi: 10.2337/dbi19-0022
38. Mannering SI, Dromey JA, Morris JS, Thearle DJ, Jensen KP, Harrison LC. An Efficient Method for Cloning Human Autoantigen-Specific T Cells. J Immunol Methods (2005) 298(1-2):83–92. doi: 10.1016/j.jim.2005.01.001
39. Faridi P, Li C, Ramarathinam SH, Vivian JP, Illing PT, Mifsud NA, et al. A Subset of HLA-I Peptides are Not Genomically Templated: Evidence for Cis- and Trans-Spliced Peptide Ligands. Sci Immunol (2018) 3(28). doi: 10.1126/sciimmunol.aar3947
40. Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, Sette A, et al. A Large Fraction of HLA Class I Ligands are Proteasome-Generated Spliced Peptides. Science (2016) 354(6310):354–8. doi: 10.1126/science.aaf4384
41. Wiles AT, Powell R, Michel CR, Scott Beard K, Hohenstein A, Bradley B, et al. Identification of Hybrid Insulin Peptides (Hips) in Mouse and Human Islets by Mass Spectrometry. J Proteome Res (2019) 18(3):814–25. doi: 10.1021/acs.jproteome.8b00875
42. Wiles TA, Saba LM, Delong T. Peptide-Spectrum Match Validation With Internal Standards (P-Vis): Internally-Controlled Validation of Mass Spectrometry-Based Peptide Identifications. J Proteome Res (2021) 20(1):236–49. doi: 10.1021/acs.jproteome.0c00355
43. Kent SC, Mannering SI, Michels AW, Babon JAB. Deciphering the Pathogenesis of Human Type 1 Diabetes (T1D) by Interrogating T Cells From the “Scene of the Crime”. Curr Diabetes Rep (2017) 17(10):95. doi: 10.1007/s11892-017-0915-y
44. Kula T, Dezfulian MH, Wang CI, Abdelfattah NS, Hartman ZC, Wucherpfennig KW, et al. T-Scan: A Genome-Wide Method for the Systematic Discovery of T Cell Epitopes. Cell (2019) 178(4):1016–28.e13. doi: 10.1016/j.cell.2019.07.009
Keywords: hybrid insulin peptides (HIPs), CD4+ T cell, autoimmunity, type 1 diabetes, epitope
Citation: Mannering SI, Rubin AF, Wang R and Bhattacharjee P (2021) Identifying New Hybrid Insulin Peptides (HIPs) in Type 1 Diabetes. Front. Immunol. 12:667870. doi: 10.3389/fimmu.2021.667870
Received: 15 February 2021; Accepted: 15 April 2021;
Published: 30 April 2021.
Edited by:
Arnaud Zaldumbide, Leiden University Medical Center, NetherlandsReviewed by:
Cheryl Lichti, Washington University in St. Louis, United StatesCopyright © 2021 Mannering, Rubin, Wang and Bhattacharjee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stuart I. Mannering, c21hbm5lcmluZ0BzdmkuZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.