 Emilio G. Vozza
Emilio G. Vozza Michelle E. Mulcahy†
Michelle E. Mulcahy† Rachel M. McLoughlin
Rachel M. McLoughlin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 16 June 2021
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.667387
This article is part of the Research Topic Fighting an Elusive Enemy: Staphylococcus aureus and its Antibiotic Resistance, Immune-Evasion and Toxic Mechanisms View all 20 articles
The success of Staphylococcus aureus as a human commensal and an opportunistic pathogen relies on its ability to adapt to several niches within the host. The innate immune response plays a key role in protecting the host against S. aureus infection; however, S. aureus adeptness at evading the innate immune system is indisputably evident. The “Trojan horse” theory has been postulated to describe a mechanism by which S. aureus takes advantage of phagocytes as a survival niche within the host to facilitate dissemination of S. aureus to secondary sites during systemic infection. Several studies have determined that S. aureus can parasitize both professional and non-professional phagocytes by manipulating the host autophagy pathway in order to create an intracellular survival niche. Neutrophils represent a critical cell type in S. aureus infection as demonstrated by the increased risk of infection among patients with congenital neutrophil disorders. However, S. aureus has been repeatedly shown to survive intracellularly within neutrophils with evidence now supporting a pathogenic role of host autophagy. By manipulating this pathway, S. aureus can also alter the apoptotic fate of the neutrophil and potentially skew other important signalling pathways for its own gain. Understanding these critical host-pathogen interactions could lead to the development of new host directed therapeutics for the treatment of S. aureus infection by removing its intracellular niche and restoring host bactericidal functions. This review discusses the current findings surrounding intracellular survival of S. aureus within neutrophils, the pathogenic role autophagy plays in this process and considers the therapeutic potential for targeting this immune evasion mechanism.
Staphylococcus aureus has evolved with the human immune system in order to survive as a commensal organism as well as a causative agent of disease. S. aureus is one of the most frequent causes of bloodstream infection worldwide, with a high hospital mortality rate ranging from 15-40% and a significant risk of reoccurrence in susceptible individuals (1, 2). Complications of S. aureus bacteraemia include secondary or metastatic infection, sepsis and septic shock (3). Metastatic infection by S. aureus is defined as infection at a secondary anatomical site that is distinct from the primary site of infection. Metastatic infections that occur during S. aureus bacteraemia include infective endocarditis, septic arthritis and vertebral osteomyelitis with reported prevalence ranges from 5.7-75.3% (4). Intracellular survival of S. aureus within host phagocytes has been highlighted as a mechanism employed by S. aureus to achieve metastatic infection and host dissemination (5–7).
In 2011, Thwaites et al. presented the idea that polymorphonuclear neutrophils (PMN) may represent a “privileged site” for S. aureus and may act as a Trojan horse for intracellular survival and dissemination to secondary sites in the host following an initial focus of infection (8). Evidence of this already existed in murine PMN (9); however, it has since been proven that S. aureus can survive intracellularly within PMN in murine and zebrafish in vivo models of infection as well as in primary human neutrophils (7, 10–12). Whilst much remains to be understood about how S. aureus achieves this, a key mechanism for intracellular survival within PMN has been identified. The autophagy pathway, a homeostatic cellular process of nutrient recycling within the cell, is co-opted by S. aureus as an intracellular survival niche (13). This review discusses what we now know about how S. aureus survives within PMN, since the initial opinion of Thwaites et al. that PMN act as a Trojan horse for S. aureus. We highlight the recent advances in uncovering the autophagy pathway as a conduit for intracellular survival. Furthermore, we speculate how this mechanism, that is subverted by S. aureus, could in-turn potentially be subverted by us as a novel approach to the treatment of S. aureus disease.
Historically, S. aureus has been classed as an extracellular pathogen, with uptake and phagocytosis simply considered a consequence of the host immune response. However, the concept of S. aureus as a facultative intracellular pathogen, capable of invading, surviving and proliferating within non-professional and professional phagocytes has been consistently shown (6, 14–16), with intracellular survival now accepted as an active mechanism of immune evasion employed by this bacterium. Several in vitro studies have shown that intracellular survival of S. aureus can be achieved in a wide array of cell types (Table 1). However, the exact mechanism and life cycle of intracellular S. aureus is not fully understood and most likely varies in a strain and host cell dependant manner. Commonalities among many in vitro studies do highlight three potential key mechanisms; 1) The survival of S. aureus within acidified phagosomes potentially inducing bacterial stress responses that delay or perturb phagolysosome formation, 2) The formation of quasi dormant single colony variants (SCVs) and persisters resistant to killing and sensing by the host, 3) The induction of host autophagy and survival within host autophagosomes. However, it is likely that some overlap and redundancy exists among these mechanisms to enable intracellular survival.
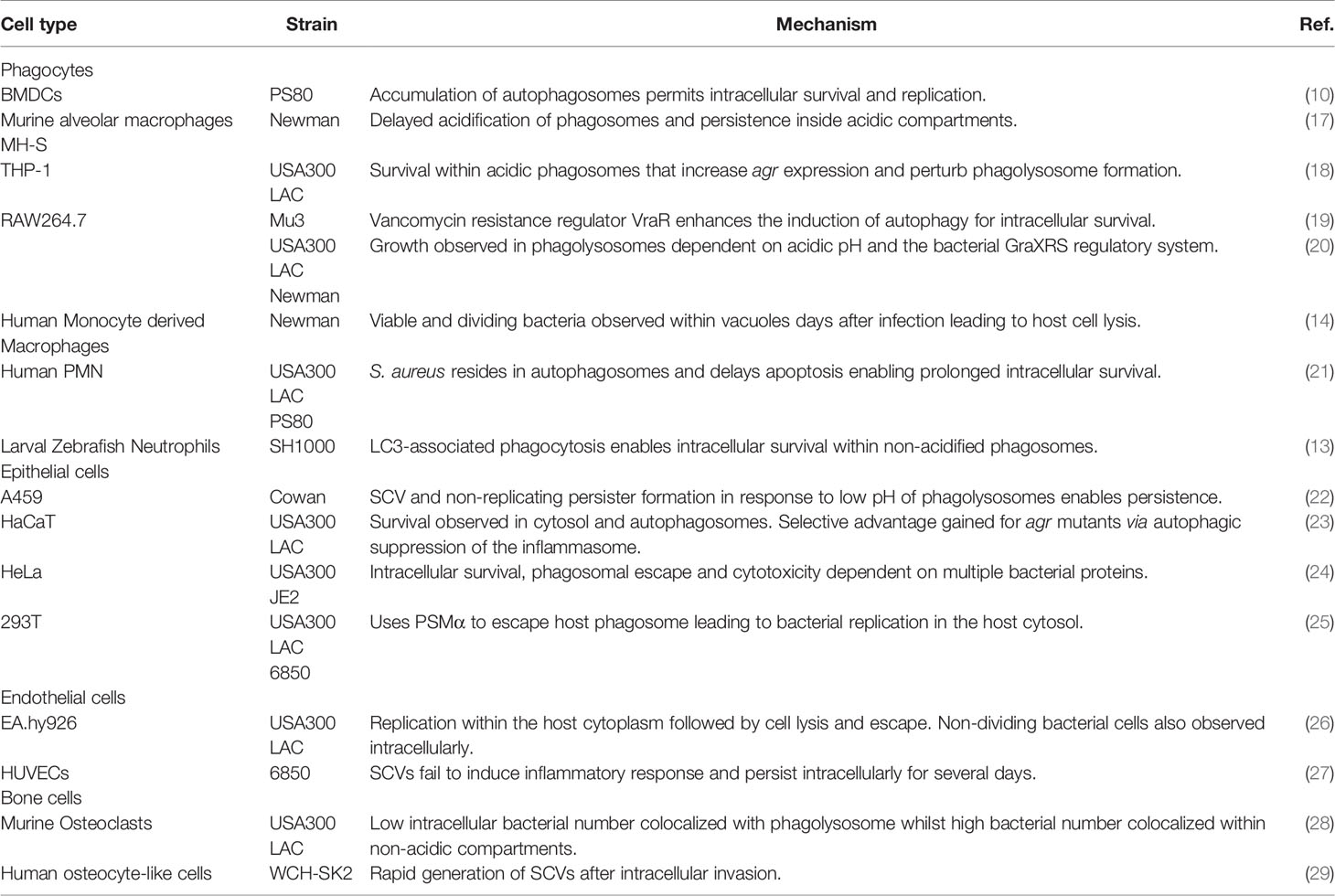
Table 1 Intracellular survival of S. aureus has been shown across a range of cell types and involving multiple mechanisms.
Impressively, several studies have reported the ability of S. aureus to survive intracellularly within dendritic cells, macrophages and neutrophils, phagocytic cells critical in the defence against and killing of invading pathogens (7, 10, 12, 13). S. aureus survival within macrophages has now been well-documented in studies using macrophage cell lines and primary cells. S. aureus was shown to survive over 24h in murine macrophage cell lines (17, 30) and up to 3-4 days in human monocyte-derived macrophages (14). Furthermore, live S. aureus has been observed to perturb phagolysosomal formation and reside within acidified phagosomes of THP-1 macrophages (18). When treated with phagosomal acidification inhibitors, viable intracellular S. aureus was dramatically reduced indicating S. aureus can manipulate host phagosomes for its own gain. In one particular study, alpha toxin (hla), produced by intracellular S. aureus, was shown to prevent mitochondrial recruitment to host phagosomes impairing ROS mediated killing within human monocytes and BMDMs (31), implicating perturbation of host bactericidal mechanisms in maturing phagosomes as the intracellular survival strategy of S. aureus whilst also implicating hla. In vivo, Kupffer cells localised to the liver were found to be the primary reservoir for S. aureus in a murine model of systemic infection (7). However, the complete removal of Kupffer cells using clodronate liposomes led to a reduction in S. aureus sequestration in the liver, persistent bacteraemia and 100% mortality indicating their importance in controlling infection. Regardless of their importance, intracellularly viable S. aureus was found to replicate within these Kupffer cells inside LAMP1-decorated acidic compartments with several compartments failing to generate superoxide. This model indicates that S. aureus survives within perturbed phagolysosomes of Kupffer cells as it’s intracellular niche before overwhelming and lysing the host cell. After cell lysis, extracellular S. aureus was then rapidly phagocytosed by infiltrating PMN. A follow up to this study showed that upon release from Kupffer cells, S. aureus disseminated to the peritoneal cavity and resided within GATA6+ macrophages for up to 2 days (32). This ultimately led to further dissemination to peritoneal organs which was reduced in mice lacking GATA6+ large peritoneal macrophages.
Two important virulence regulators have been heavily implicated in the intracellular survival of S. aureus within phagocytes; the accessory gene regulator (agr) and to a lesser extent, the staphylococcal accessory element (Sae) (10, 33–36). Both regulators govern the vast majority of cytolytic toxins produced by S. aureus, and when absent, lead to significant reduction in phagosomal escape as well as reduced intracellular burden. Additionally, the agr locus has also been shown to activate the Sae locus which may highlight a synergistic or overlapping relationship between the two regulators and their associated factors (37). An initial study of intracellular survival within human monocyte derived macrophages (hMDMs) highlighted the importance of the agr locus and the stress response and virulence regulator SigB, as the absence of either regulator led to significant reduction in intracellular burden and cell death (14). More recently, the agr and Sae locus have been further implicated in intracellular survival as mutant strains of either regulator showed significant reductions in phagosomal escape within THP-1 cells and hMDMs, with double mutants showing the greatest reduction in cytotoxicity and phagosomal retention (35). These results were attributed to a group of cytolytic peptides regulated by the agr locus and the Sae locus; Phenol Soluble Modulin (PSM) α, Leucocidin A/B (lukAB) and Panton–Valentine leucocidin (PVL). The importance of the agr locus in intracellular survival has been demonstrated using the Nebraska transposon mutant library and the CA-MRSA strain JE2. Single mutants for agrA, agrB, agrC and PSM transporter pmtC showed the greatest reduction in phagosomal escape within Hela cells which potentially mimics the mechanism found in phagocytes (24). Grosz et al., showed that phagosomal escape of the MRSA strains LAC USA300 and MW2 USA400 as well as the MSSA strain 6850 was exclusively dependent on PSMα (25). This was determined using mutant strains of PSMα which failed to escape host phagosomes in professional and non-professional phagocytes with PSMα complementation restoring phagosomal escape. Surewaard et al., showed that PSMs are functionally inhibited by serum lipoproteins with human serum, which protect human PMN from PSM induced cell lysis (38). This implies that in the body, serum proteins render PSMs redundant in the extracellular environment which the authors theorize implicate the role of PSMs to be found intracellularly. Indeed, the authors showed that PSMα expression was induced between 45 minutes to 2 hours post phagocytosis within human PMN implicating PSMs in the intracellular environment.
Another important mechanism employed by S. aureus to establish intracellular survival is the formation of SCVs which are a slow growing bacterial phenotype that exhibit an altered and reduced metabolism compared to WT colonies (39). SCVs also show enhanced antimicrobial resistance and have long been associated with chronic and recurrent infection (40). The formation of SCVs represents an important immune evasion mechanism in sustaining long term intracellular survival that is particularly evident in non-professional phagocytic cell types. However, SCVs can form within phagocytic cell types but their formation has been shown to reduce their intracellular survival within phagocytes compared to WT colonies. For instance, THP-1 macrophages have been shown to clear SCVs more efficiently than wild type counterparts suggesting SCV formation is disadvantageous in phagocytes (41). SCVs can persist up to a week post infection within human osteoblasts and the endothelial HUVEC cell line in contrast to human macrophages which become undetectable 3 days post infection (42). SCVs show a marked reduction in agr expression, due to their altered metabolic phenotype, and show a greater reliance on SigB as mutants of this virulence factor show a significant decrease in persistence (39, 43, 44). Tuchscherr et al., showed that SigB is crucial in the formation of SCVs and intracellular persistence within human osteoblasts (45). Loss of agr and SarA expression led to increased persistence within osteoblasts whilst significantly reducing cytotoxicity against human and murine PMNs. However, each of these virulence regulators were shown to be important in vivo using both an acute and chronic murine model of infection (4 days vs 14 weeks post infection). Whilst SigB was crucial for establishment of SCVs and their associated long-term persistence, phagosomal escape was dependent on the agr and SarA loci. Whilst the agr locus is critical in subverting phagocytes, likely through subverting bactericidal mechanisms such as phagolysosomal formation, it’s absence in SCVs enables long term survival in non-professional phagocytes which potentially renders SCVs susceptible to killing by phagocytes.
Intracellular survival of S. aureus is a remarkable feat in which S. aureus can thrive in phagocytic and non-phagocytic cells by perturbing phagolysosomes and inducing SCV formation, respectively. However, a third mechanism has emerged in recent years involving host macroautophagy (herein called autophagy) that can occur within professional and non-professional phagocytes in an agr dependent or independent fashion depending on the cell type. This mechanism has been shown to affect both SCVs and WT colonies implicating the manipulation of autophagy as a critical and key step in establishing intracellular survival across various cell types and strains. Autophagy is a conserved eukaryotic homeostatic process in which damaged cellular organelles are recycled during cellular stress or starvation in order to create a supply of nutrients for cell survival (Figure 1) (52). As well as its role in homeostasis, autophagy also plays a part in the innate immune response to infection by selectively engulfing invading bacteria in a specialized autophagic process of pathogen clearance called Xenophagy (53). The xenophagic response involves the formation of an autophagosome to engulf intracellular bacteria and traffic them towards lysosomal degradation and is an important process in controlling intracellular replication of pathogens including Listeria monocytogenes, Salmonella enterica serovar Typhimurium and Mycobacterium tuberculosis (54–56). However, these pathogens as well as many other pathogens such as Escherichia coli have been reported to manipulate or subvert the autophagic pathway for survival (57–61). Moreover, many studies have described divergent mechanisms for S. aureus intracellular survival and replication using the autophagic pathway in both non-professional and professional phagocytes, with several autophagy inhibitors such as VPS34-IN1 and 3-methyladenine shown to reduce intracellular survival of S. aureus across a range of cell types (10, 21, 62, 63).
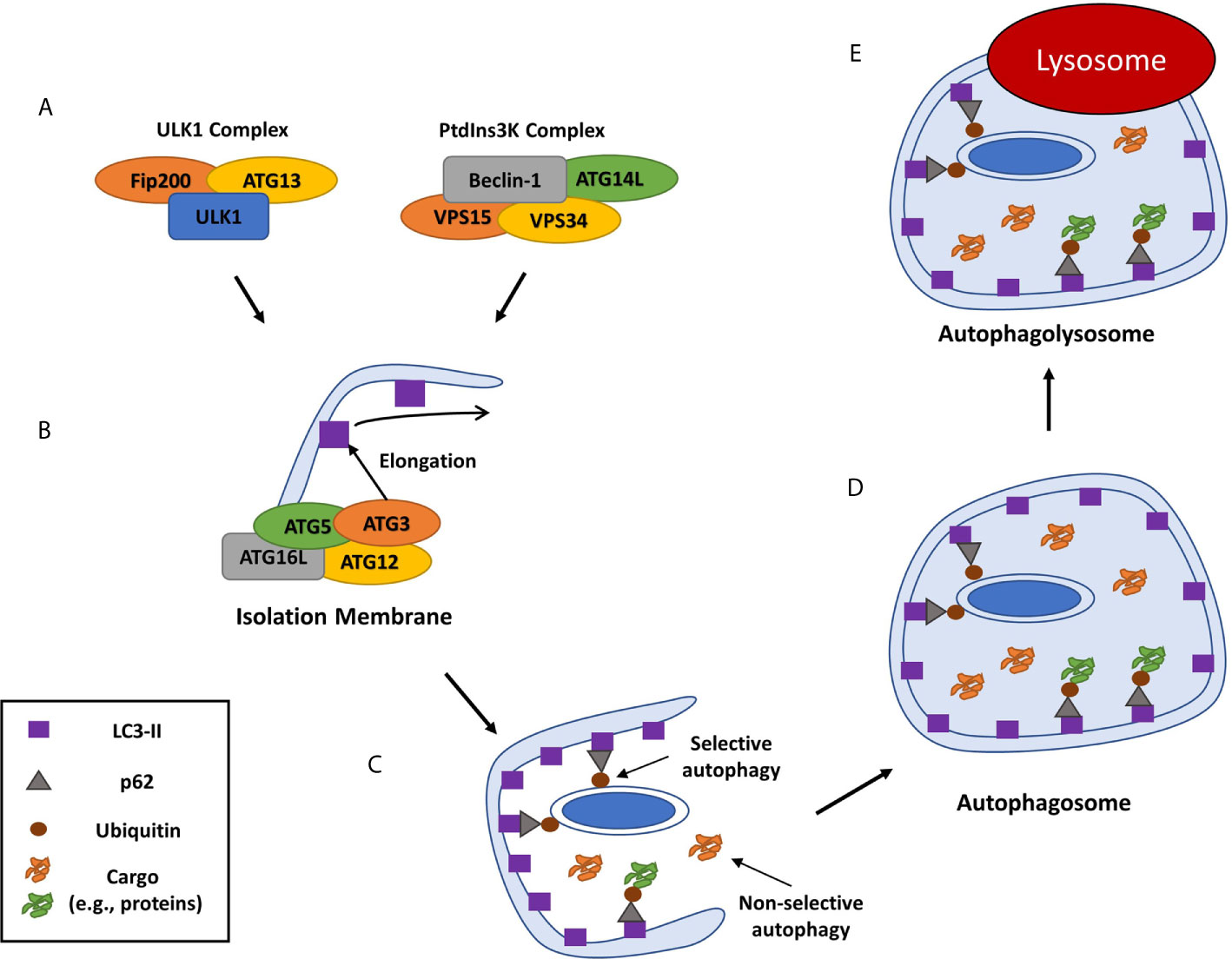
Figure 1 Overview of mammalian autophagy. Autophagy is induced through the recruitment of numerous autophagy related proteins (ATGs) to a specific cytoplasmic location. This is followed by the formation of the isolation membrane that eventually elongates and closes around organelles or cell debris to be digested to form a double-membraned phagosome, or autophagosome (46). (A) The autophagic cascade is initiated by core molecular machinery including the Ulk1 complex and an activation complex comprising of Beclin-1, the class III PI3-kinase, Vacuolar protein sorting 34 (VPS34), ATG14L and VPS15 (47). (B) This complex facilitates the de novo formation of an isolation membrane that then elongates to form a double membraned phagophore, by recruiting several ATGs, including ATG3, ATG16L and the ATG-12-ATG5 complex, which then convert the ATG8 protein LC3-I to its lipidated, membrane-bound form LC3-II (48). (C) As well as non-selective autophagy of cytoplasmic components, cargo can be specifically recognized and marked for autophagy typically by ubiquitination. Ubiquitinated cargo is then recognized by cargo receptor molecules including SQSTM1/p62, CALCOCO2 and OPTN and trafficked to the phagophore through an interaction between the cargo receptor and LC3 (49–51). (D) Continued LC3-II conjugation to the phagophore causes it to elongate and form a cargo containing autophagosome which is trafficked to the host lysosome. (E) The autophagosome matures and fuses with a lysosome, the autophagic cargo is degraded and the nutrients are released back into the cytosol to be re-used by the cell.
In a study by Schnaith et al., S. aureus survived and replicated in autophagosomes within HeLa cells (33). S. aureus was rapidly taken up by Rab7-positive endophagosomes and became enveloped in double-membraned autophagosomes within 3h of infection. S. aureus-harbouring phagosomes were mostly LC3-decorated and underwent reduced acidification. Activation of autophagy with rapamycin treatment led to a higher bacterial load within cells whereas conversely, autophagy inhibition using wortmannin reduced the bacterial burden, indicating that S. aureus replication depends on an activated autophagic pathway. Intracellular survival was followed by eventual escape into the cytoplasm coupled with caspase-independent, ATG5-dependent host cell death with high levels of vacuolization, indicative of autophagic cell death. Recently, the induction of autophagy in HeLa cells by S. aureus was compared to Salmonella infection, which is known to evade the autophagy pathway to facilitate intracellular survival (62, 64). While both bacteria showed an ability to survive intracellularly, S. aureus escaped from lysosomal compartments with less lysosomal membrane damage compared to Salmonella, suggesting that S. aureus may actively and specifically subvert the xenophagy defence pathway. Blocking autophagy mediated by the Ulk1 complex made cells more resistant to S. aureus infection compared to Salmonella infection which showed no change. Furthermore, Ulk1 inhibition reduced the ability of S. aureus to survive intracellularly, suggesting that an autophagy-dependent niche is essential for intracellular survival. This study also highlighted the importance of the agr locus as accumulation of LC3-II and p62 aggregates was shown to be partially dependent on a functional agr locus. In another study, changes to the host central carbon metabolism during S. aureus intracellular survival was evident in HeLa cells infected with MRSA strain USA300 (65). S. aureus infection led to depletion of glucose and amino acid pools as well as increased levels of glutaminolysis and activation of starvation-induced autophagy. Interestingly, although autophagy was activated by S. aureus infection, no bacteria were found within autophagosomes. This study highlights an alternative or additive mechanism of autophagy-mediated intracellular survival whereby S. aureus is manipulating the autophagy pathway in order to aid it in scavenging nutrients within the cellular space. In a study by Neumann et al., the autophagic response was induced by S. aureus infection of murine fibroblasts (66). S. aureus was encapsulated in LC3-positive phagosomes and multilamellar membranes were identified using TEM indicative of autophagosomes. In this model, intracellular S. aureus was ubiquitinated shortly after invasion and then co-localized with the trafficking proteins p62, ubiquitin and LC3 as well as trafficking proteins OPTN and CALCOCO2 indicating direct targeting of S. aureus by host autophagy. No acidification of phagosomes was reported, and S. aureus was eventually able to escape to the cytosol where bacterial replication was evident. In CHO cells, exposure to S. aureus also led to intracellular survival within non-acidic, LC3-labelled compartments and eventual escape into the cytosol (67). In a separate study using CHO cells, S. aureus induced the formation of LC3-decorated tubules from S. aureus-harbouring phagosomes (68). These tubules were observed to facilitate efficient bacterial replication.
In professional phagocytes, S. aureus can evade killing by murine bone marrow-derived dendritic cells and macrophages by manipulating the autophagy pathway in a strain-specific mechanism that appears to be dependent upon expression of the agr locus (10). Increased cytolytic protein production was associated with a disruption in autophagic flux, leading to autophagosomal accumulation and eventual cell lysis. The ability to survive by manipulating the autophagic pathway correlated with persistence in an in vivo model of systemic infection. Induction of host autophagy by S. aureus has also been reported in RAW264.7 macrophages. S. aureus expressing the vancomycin resistance-associated sensor/regulator, VraSR, were shown to enhance gene expression of beclin-1 and ATG5 compared to an isogenic VraSR mutant. The VraSR mutant also showed increased turnover of LC3-II and p62 indicating reduced disruption of autophagic flux as well as significantly reduced intracellular burden compared to the WT and VraSR complemented mutant strains (19). In bovine macrophages, S. aureus was capable of inducing host autophagy which was marked by significant increases in LC3-II and increased visualisation of host autophagosomes versus uninfected cells using TEM imaging and confocal microscopy (63). Importantly, p62 levels were elevated yet p62 degradation, a consequence of autophagosomal degradation and autolysosomal formation, was arrested indicating a disruption in autophagic flux. Furthermore, treatment with the autophagy inhibitor 3MA led to significant decreases in intracellular S. aureus. Overall, the manipulation of the autophagy pathway plays a key role in the intracellular survival of S. aureus across multiple cell types enabling enhanced survival and replication within the host cell.
Phagocytes are critical in the defence against S. aureus, yet amazingly S. aureus has developed the ability to co-opt these cells for its own survival with PMN representing the most unlikely target for intracellular survival. PMN are pivotal in the innate immune response against S. aureus infection. Neutrophils are recruited from the systemic circulation to the site of infection in response to several chemotactic factors such as chemokines and pathogen associated molecular patterns (PAMPS) where they then recognise invading bacteria via opsonised and non-opsonised mechanisms (Figure 2). S. aureus is engulfed and ultimately killed by several methods utilized by PMN including degranulation, the production of reactive oxygen species and the secretion of antimicrobial peptides (74). Additionally, extracellular S. aureus can also be killed through the production of NETs (75). The importance of PMN in S. aureus infection is most evident in individuals with deficiencies in PMN activity. Individuals with chronic granulomatous disease, a deficiency in NADPH oxidase, suffer from recurrent S. aureus infections (76, 77). In addition, frequent S. aureus infections are a consequence of severe congenital neutropenia, Chediak-Higashi syndrome and leukocyte adhesion deficiency 1 (78, 79). Furthermore, evidence from murine in vivo models have shown that neutrophil depletion leads to increased bacterial burden within the liver after intravenous injection as well as increased mortality after intratracheal injection (11, 80). Such observations highlight the importance of PMN in controlling S. aureus infection and the consequence of their absence which results in overwhelming disease.
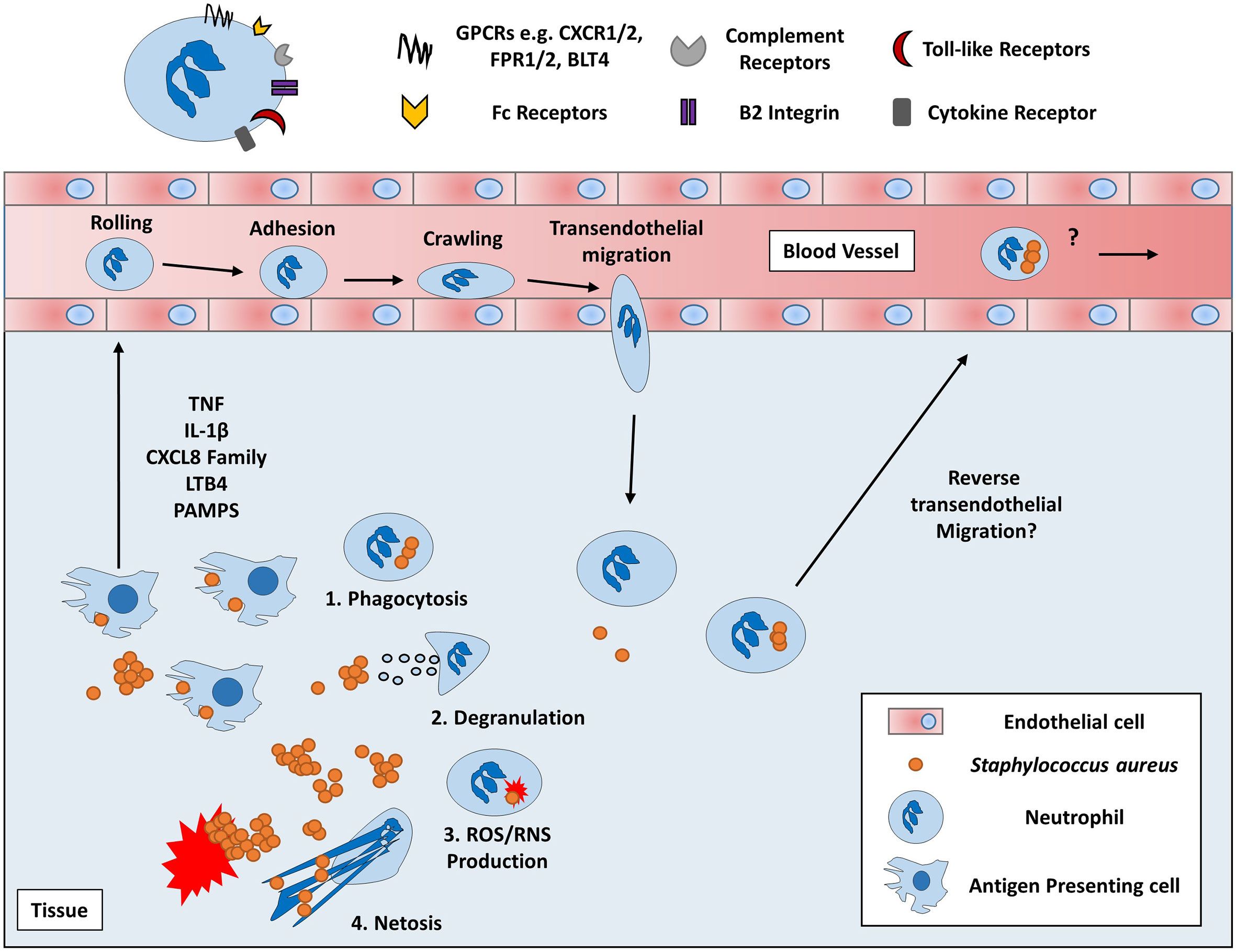
Figure 2 Overview of Neutrophil chemotaxis and effector functions. Neutrophils are circulatory immune cells which respond to host or pathogen derived chemoattractants using various receptors such as chemokine receptors (e.g. CXCR2) and pathogen recognition receptors (e.g. FPR2) (69, 70). Proinflammatory cytokines, chemokines, and lipid mediators, such as TNF, CXCL8 and leukotriene B4 (LTB4) respectively, are produced by local immune and non-immune cells which recruit PMN from the peripheral blood (71, 72). Initially, PMN slow their passage through the blood via weak interactions with the endothelium causing PMN to roll along the surface of the blood vessel. PMN then begin to adhere to the endothelium through stronger B2 integrin interactions via I-CAM1 molecules expressed on the surface of endothelial cells (73). Ultimately, PMN crawl across the endothelium toward the chemotactic gradient until they traverse through the blood vessel to the site of infection via transendothelial migration. Once at the site of infection, PMN begin to exert their various effector functions which include 1) phagocytosis via complement and Fc receptors to eliminate S. aureus via several intracellular mechanisms 2) releasing primary, secondary and tertiary granules containing antimicrobial peptides such as cathepsins, defensins and gelatinases into the extracellular space or phagosomal compartment, 3) Generating reactive oxygen or nitrogen species via NADPH oxidase, myeloperoxidase and nitric oxide synthase and 4) the release of uncondensed chromatin imbued with histones and antimicrobial peptides in a process of cell death known as NETosis (74, 75). Despite these effective killing mechanisms, S. aureus can survive intracellularly within PMN and could potentially induce rTEM in PMN to enter the peripheral blood and disseminate to secondary sites (6, 8, 73).
Although optimal PMN function is vital to protect the host against overwhelming bacterial burden during S. aureus infection, high levels of PMN recruitment can be detrimental. Limiting PMN migration during a murine model of lethal S. aureus infection led to a decrease in mortality and a reduced bacterial burden at the infection site, whereas repletion with PMN reversed this protective effect (9). Heightened CXC-chemokine driven PMN recruitment has been shown to exacerbate pathogenesis in a murine model of wound infection (81). Furthermore, IFNγ-/- mice displayed a decrease in bacterial burden at the site of infection due to reduced CXC chemokine production and subsequent PMN recruitment (82). An IFNγ-dependent increase in PMN recruitment to the site of infection provided a PMN-rich environment in which S. aureus could survive more readily. More recently, during intraperitoneal infection in mice, S. aureus survived at significantly higher levels within PMN than in macrophages or dendritic cells and were found predominantly within PMN disseminated from the peritoneal cavity to the bloodstream (10). These studies allude to intracellular survival in PMN as a possible active bacterial virulence mechanism during infection which has now been demonstrated both in vitro and in vivo. S. aureus intracellular survival was observed in PMN after gentamicin treatment during murine intraperitoneal infection (9). Surviving bacteria were viable and PMN harbouring S. aureus could cause infection when administered to naïve mice. Moreover, S. aureus survival within murine PMN after gentamicin treatment was dependent on the expression of the SarA global regulator, suggesting that intracellular survival is an active immune evasion strategy. Excessive levels of both CXCL8 and LTB4 has been shown to repel PMN migration whilst recent studies have also observed reverse transendothelial migration (rTEM) of PMN from damaged tissues (73, 83). rTEM PMN have been identified as ICAM1hiCXCR1lo expressing cells which have been detected in the peripheral blood of patients with chronic systemic inflammation (84). As such, this subpopulation of rTEM PMN could represent a key target of S. aureus to escape the primary site of infection and create secondary sites of infection.
Though the intracellular environment of neutrophils represents a harsh and dangerous space for bacteria to persist, S. aureus has amassed a large arsenal of factors to permit its intracellular survival. Neutrophils generate large amounts of reactive oxygen species (ROS) and reactive nitrogen species to effectively kill both extracellular and intracellular pathogens. To combat this, S. aureus has developed several protective enzymes to overcome free radicals, namely staphyloxanthin, catalase and superoxide dismutase (85, 86). These enzymes can effectively shield the pathogen enabling persistence during infection. Recently, a bacterial factor known as Staphylococcal peroxidase inhibitor (SPIN) was characterized and shown to inhibit myeloperoxidase (MPO), a key enzyme in neutrophils used to generate the bactericidal molecule hypochlorous acid (HOCL) (87). SPIN was shown to be heavily upregulated after phagocytosis by human PMNs leading to increased survival and resistance to neutrophil-derived ROS. This factor was shown to be regulated by the two-component regulatory system, Sae, a staphylococcal regulator previously implicated in intracellular survival of S. aureus (35).
Intracellular S. aureus recovered from PMN have been shown to upregulate RNAIII expression within the first hour post phagocytosis whilst extracellular S. aureus shows substantially less RNAIII expression (34). This is likely due to the confined space of intracellular compartments as confinement induced quorum sensing has been shown to increase agr activity due to the rapid accumulation of the autoinducing peptide (AIP) responsible for agr activation (88). Strains with a greater capacity to survive intracellularly within phagocytes show increased RNAIII expression whilst also showing greater cytolytic toxin production as measured by a vesicle lysis test (10). Whilst the agr locus is known to enhance PMN lysis, it was also been shown to enhance intracellular survival within PMN potentially in a similar fashion as observed in other phagocytes (21, 34).
As the manipulation of autophagy by intracellular S. aureus has been shown in both macrophages and dendritic cells, it can easily be theorized that autophagy too plays a central role in the manipulation of PMN for intracellular survival. Recently it has been shown using a larval zebrafish model of S. aureus infection, that S. aureus survives within spacious LC3-positive phagosomes within both PMN and macrophages (13). However, the LC3 signal decreased over time for macrophages and not for PMN, suggesting a disruption in autophagic flux in PMN. Survival was dependent on a functioning NADPH oxidase, as Cyba/p22phox knock-down in larvae, as well as chemical depletion of ROS production using DPI, led to a near complete abolition of LC3-S. aureus association. This dependence on ROS production for LC3-S. aureus co-localization meant that survival was attributed to a mechanism called LC3-associated phagocytosis (LAP). LAP uses a portion of the autophagy machinery to conjugate LC3 to single-membraned phagosomes rather than double-membraned autophagosomes indicative of the canonical autophagy pathway (89). Loss of this pathway through an ATG5/ATG16L1 double knockdown led to an almost complete loss of LC3-S. aureus vacuoles and larvae were significantly more resistant to infection, suggesting that blocking LAP results in a loss of the S. aureus survival niche. Although p62, which is a marker for selective autophagy, also colocalized with S. aureus in this model, co-localization was downstream of LAP activation and was actually attributed to a host-protective mechanism in response to S. aureus infection, as loss of p62 led to increased host susceptibility to infection (66). Furthermore, p62 deficiency had no effect on the formation of LC3-positive phagosomes. This implies that S. aureus does not subvert canonical autophagy in zebrafish PMN but rather, takes advantage of single membraned LAPosomes to avoid killing. In contrast, S. aureus has been shown to survive within primary human PMN by subverting the autophagy pathway (21). S. aureus intracellular survival coincided with a disruption in autophagic flux as determined by the accumulation of the autophagic markers p62 and LC3-II which under normal autophagic flux undergo degradation upon fusion of the autophagosome and host lysosome. Replicating S. aureus was also observed within autophagosomes using TEM microscopy of infected human PMN. Furthermore, blockade of autophagosomal formation, with the autophagy inhibitors bafilomycin A1 and VPS34-IN1, led to a significant reduction in intracellular burden thus stressing the importance of the autophagy pathway in the intracellular survival of S. aureus within PMN.
Altogether, two mechanisms utilising the autophagic machinery of PMN have thus far been identified to facilitate the intracellular survival of S. aureus (Figure 3). One mechanism relies on LAP to persist within LC3 decorated phagosomes as its primary niche, enabling the bacterium to avoid killing whilst actively replicating leading to eventual host cell lysis. The other mechanism relies upon canonical autophagy whereby intracellular S. aureus reside within autophagosomes as their primary niche in which they grow and divide presumably before overwhelming the host cell for dissemination. Remarkably, S. aureus is not only capable of subverting both canonical and non-canonical autophagy pathways for its own gain but may also act to disrupt the apoptotic pathway, presumably with the goal of protecting its intracellular niche.
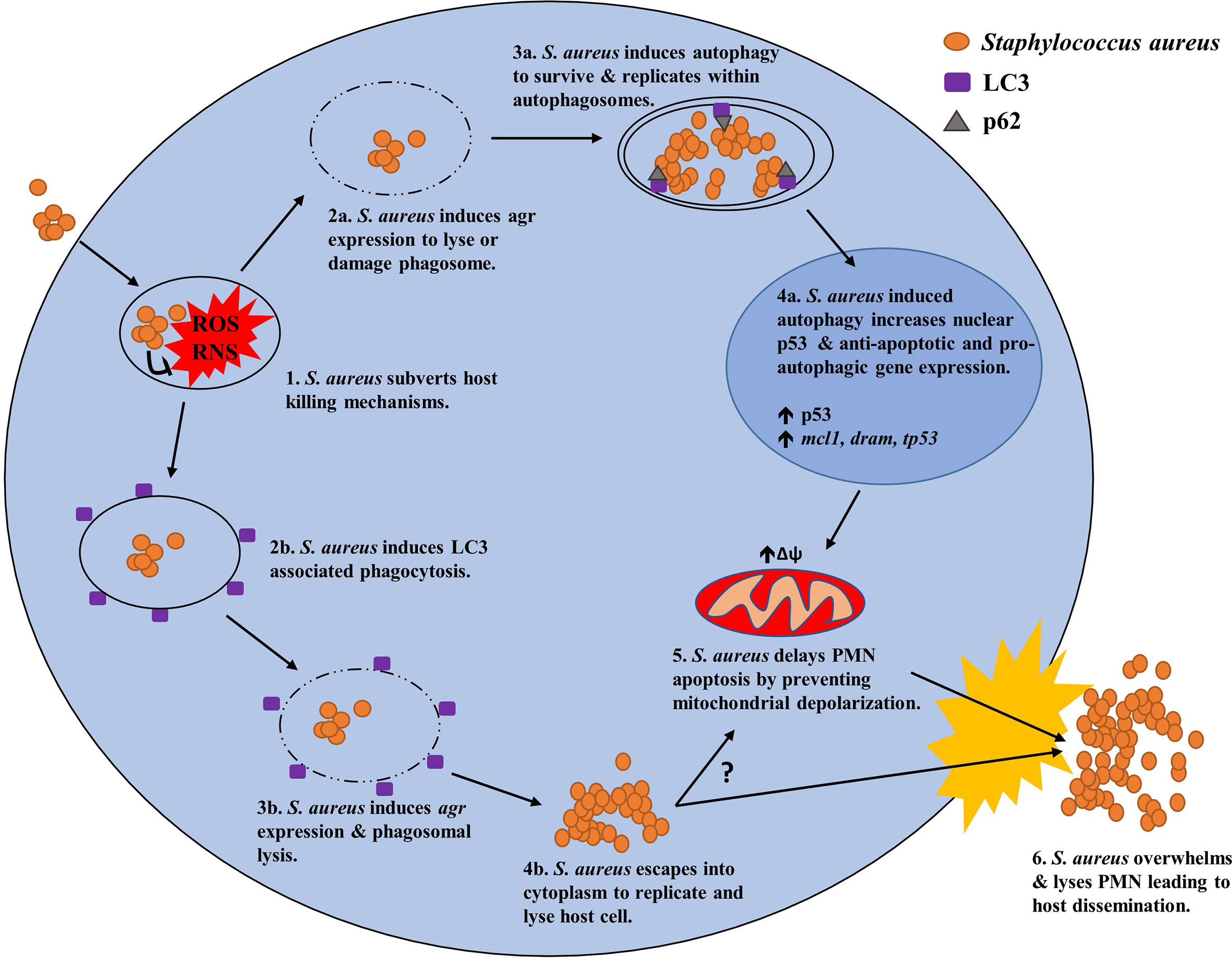
Figure 3 Potential mechanisms of S. aureus to manipulate host autophagy in PMN for intracellular survival. (1.) Upon phagocytosis by PMN, S. aureus subverts host killing mechanisms such as ROS generation and persists intracellularly by manipulating canonical or non-canonical (LAP) autophagy. (2a.) Using canonical autophagy, S. aureus lyses or damages the host phagosome using an agr regulated toxin. (3a.) Phagosomal lysis/damage induces host autophagy enabling its uptake into an autophagosome as its primary intracellular niche. (4a.) Induction of the canonical autophagy pathway leads to increased levels of nuclear p53 and anti-apoptotic and pro-autophagic gene expression of mcl1 and dram respectively. (5.) Increases in anti-apoptotic factors prevents mitochondrial depolarization and subsequent induction of the apoptotic pathway resulting in prolonged PMN survival. (6.) Eventually, S. aureus exhausts its intracellular niche and overwhelms the host cell resulting in cell lysis and host dissemination. (2b.) Alternatively, S. aureus can utilize non-canonical autophagy via LAP were the bacterium resides within LC3 decorated phagosomes as its primary niche. (3b.) S. aureus uses an agr regulated toxin to avoid bacterial killing before lysing the LC3 decorated phagosome. (4b.) S. aureus then escapes into the cytosol where it then replicates. (5.) It is unknown whether this mechanism can lead to increased PMN survival however a link between the two may exist. (6.) Ultimately S. aureus overwhelms the host cell leading to cell lysis and host dissemination.
Overall PMN represent an unlikely intracellular reservoir as mature PMN are terminally differentiated with a short circulating half-life (~7-19 hours in peripheral blood) controlled by constitutive apoptosis (90–92). However, several intracellular pathogens have been shown to delay apoptosis through the modulation of pro- and anti-apoptotic gene expression or cytokine production increasing PMN life span and thus protecting their intracellular niche (93–96). Ample evidence now exists that S. aureus does indeed survive within PMN enabling it to disseminate to secondary sites of infection. As such, any delay, even briefly, in PMN apoptosis would give S. aureus a survival advantage for long enough to potentially proliferate and disseminate, supporting the “Trojan horse” theory of S. aureus immune evasion.
Constitutive PMN apoptosis is primarily controlled by the Bcl-2 family of pro- and anti-apoptotic proteins. In PMN, the most prominent Bcl-2 family members include anti-apoptotic proteins Mcl-1 and A1, and pro-apoptotic factors such as Bid, Bim, Bad and Bax (97). Once apoptosis is initiated, PMN undergo several changes including rounding up of the cell, DNA fragmentation and phosphatidylserine exposure on the surface of the cell to facilitate macrophage efferocytosis. Under homeostatic conditions, circulating aged apoptotic PMN migrate to the liver, spleen and bone marrow were they are removed by Kupffer cells and other tissue resident macrophages (98, 99). Studies examining changes in primary human PMN during S. aureus infection in vitro reported that PMN rapidly display regular markers of apoptosis such as increased surface exposure of phosphatidylserine and loss of mitochondrial membrane potential (100). However, they also exhibited signs of a dysregulated apoptosis phenotype including sustained levels of PCNA, a lack of caspase-3 activation, increased surface expression of CD47 and inhibited macrophage efferocytosis. In other studies, the staphylococcal cell wall-associated factor lipoteichoic acid caused a delay in PMN apoptosis in vitro via TLR-2-mediated activation of NF-kB (101). The apoptotic fate of PMN during S. aureus infection was shown to be dependent on multiplicity of infection (MOI); low bacteria-PMN ratios could inhibit apoptosis whereas high ratios could induce it (102). The inhibition of PMN apoptosis in response to low MOI was shown to be cytokine dependent as blocking antibodies for IL-6 and TNF restored the apoptotic phenotype to that of uninfected PMN. The researchers speculated that high MOI of S. aureus leads to excessive TNF production that overrides the possibly anti-inflammatory effect of IL-6 seen in lowly infected PMN. These conflicting accounts of the changes to PMN lifespan during S. aureus infection, suggest that the apoptotic fate of S. aureus harbouring PMN may be context dependent.
The ability of S. aureus to delay apoptosis is a remarkable feat which suggests a pathogenic advantage is gained by the increased lifespan of infected PMN, presumably through the facilitation of intracellular survival. S. aureus has been shown to delay PMN apoptosis exemplified by a decrease in mitochondrial membrane permeabilization, a decrease in caspase-3 cleavage and lower levels of DNA degradation in S. aureus-harbouring PMN (21). Additionally, increases in the gene and protein expression of the anti-apoptotic factors Mcl-1 and A1/Bfl-1 was observed but no changes were found in the pro-apoptotic Bcl-2 member Bax. Crucially, this delay in the activation of the apoptosis pathway was dependent on an active autophagy pathway, as inhibition of autophagy activation with VPS34-IN1 led to an increase in DNA degradation and a reduction in expression of Mcl-1 (21). This close association between autophagy and apoptosis may be because these pathways share common effector molecules. For example, Mcl-1 is an anti-apoptotic homologue of the Bcl-2 family, but also plays a role in regulating autophagy by interacting directly with the critical autophagy protein beclin-1 (103). In neuronal cells, deletion of Mcl-1 leads to the induction of host autophagy implying Mcl-1 supresses autophagy likely through its binding of Beclin-1 (104). However, human neutrophils treated with tumour culture supernatants show increased survival and autophagy rates which is marked by the retainment of Mcl-1 implying a more complex relationship may exist (105). Mcl-1 is critical for PMN survival as PMN lack many Bcl-2 members and thus rely heavily on Mcl-1 for its anti-apoptotic function (106). However, Mcl-1 has a uniquely extremely high turnover rate in PMN compared to other members of the Bcl-2 family and so plays a key role in controlling PMN apoptotic cell death (106). In the context of intracellular S. aureus infection, much remains unclear as to the relationship between Mcl-1 and autophagy. However, during intracellular survival of S. aureus within PMN, inhibition of autophagy with VPS34-IN1 leads to significant reductions of Mcl-1 at the gene and protein level suggesting the induction of host autophagy leads to Mcl-1 mediated survival during S. aureus infection (21). Reduced gene expression of Mcl-1 under inhibition of autophagy was also accompanied by a significant reduction of the antiapoptotic genes bcl2 and bcl2a1.
While S. aureus can expertly delay host apoptosis in PMN, several studies have also shown that it can induce apoptosis among other forms of cell death through the production of various virulence factors. For instance, PVL has been shown to induce apoptosis in PMN through a bax-independent process involving cytochrome c release and mitochondrial associated caspase-8 and -9 activation (107). Additionally, PVL production in PMN can lead to the induction of necroptosis, a regulated form of necrotic cell death, and pyronecrosis, a variant of the highly inflammatory cell death process pyroptosis (108, 109). PVL, among many other toxins, have been shown to exacerbate lung damage through the induction of necroptosis (109) and PVL positive strains have a strong association with necrotising pneumonia (110). Although inducing necroptosis appears to be counterintuitive to the mechanism of intracellular survival, this may represent an alternative mechanism of immune evasion which could benefit extracellular S. aureus by overwhelming and eradicating local immune cells.
No specific host factor has yet been singled out as integral to autophagy-mediated S. aureus intracellular survival in PMN. However, as this autophagy dependent mechanism has been shown to delay apoptosis in PMN, any host factors that possess dual functions in both autophagy and apoptosis should be considered as potential candidates co-opted by S. aureus to facilitate intracellular survival. The antiapoptotic protein Mcl-1 has the potential to be a host-specific factor involved in autophagy-mediated intracellular survival of S. aureus. As discussed, S. aureus intracellular survival in PMN leads to the upregulation of Mcl-1 and this correlates with an anti-apoptotic phenotype in PMN (21). This anti-apoptotic phenotype occurs alongside a block in autophagic flux, creating an intracellular niche for S. aureus. Consequently, disrupted host autophagy and elevated Mcl-1 levels could result in the delayed apoptotic phenotype thus creating a suitable intracellular niche for S. aureus to reside.
The transcription factor p53 plays a regulatory role in apoptosis and is activated as part of the host response to cellular stress. Additionally, p53 can be both pro-autophagic and anti-autophagic depending on its cellular location (111, 112). Under homeostatic conditions, p53 is retained in the nucleus where it can exert pro-autophagic effects. Translocation from the nucleus to the cytoplasm leads to pro-apoptotic effects and usually occurs during cellular stress (113, 114). During S. aureus intracellular survival in primary human PMN, p53 was primarily located in the nucleus, suggesting it played a pro-autophagic role in this model (21). Damage-Regulated Autophagy Monitor (DRAM) is a direct translational product of p53 and has been shown to play a role in both autophagy activation and apoptosis (115, 116). Gene and protein expression of p53 and DRAM were increased during autophagy-mediated S. aureus intracellular survival in PMN (21). Furthermore, transcription of both were reduced after autophagy inhibition, suggesting that the autophagy pathway and the p53/DRAM pathway are directly linked during S. aureus intracellular survival in PMN. Additionally, intracellular survival of S. aureus was reduced after inhibition of p53 using the p53 inhibitor pifithrin-α. This was accompanied by a subsequent reduction in DRAM gene transcription. These results suggest that an active p53/DRAM pathway is necessary for efficient autophagy-mediated S. aureus intracellular survival. Under HIV infection, CD4+ T cells show enhanced p53 dependent gene expression of DRAM which leads to the induction of host autophagy (117). However, in this model, normal autophagic flux was detected, and increased DRAM expression was associated with enhanced lysosomal membrane permeabilization and cell death. In the intracellular survival of S. aureus, PMN could potentially be protected from this p53 dependent DRAM induced cell lysis by Mcl-1. However, PMN could still show DRAM induced autophagy which the bacterium could manipulate for its own survival. Over expression of DRAM in a zebrafish model led to hyperactivation of host autophagy leading to enhanced mtb clearance through the enhancement of host xenophagy (118). Whilst mtb is susceptible to xenophagy, S. aureus manipulates this pathway and so enhanced autophagy would be beneficial to the pathogen, potentially highlighting the importance of DRAM in positively inducing autophagy mediated intracellular survival of S. aureus in PMN.
Although the p53/DRAM pathway has been highlighted as a potential target of S. aureus to enhance host autophagy, it is likely that the highly adept S. aureus can manipulate other pathways to achieve the same outcome. Extracellular conserved chromatin-binding nuclear protein high mobility group box 1 (HMGB1) acts as a DAMP in infection and has been shown to enhance pathology in an S. aureus pneumonia model associated with a massive influx of PMN (119). However, HMGB1 was shown not to affect the influx of PMN, nor did it affect bacterial clearance yet when blocked, with anti-HMGB1, did lead to significant reductions in IL-1β production. Conversely, PMSα, a staphylococcal toxin associated with intracellular survival, has been shown to attenuate HMGB1 binding to TLR4 leading to suppression of the pro-inflammatory cytokines TNF and IL-6 in THP-1 macrophages (120). Together these results implicate the involvement and manipulation of HMGB1 in S. aureus infection which has yet to be investigated in the context of intracellular survival. Interestingly, intracellular HMGB1 can also facilitate activation of the autophagy pathway and thus should be considered in autophagy mediated intracellular survival of S. aureus. Intracellular HMGB1 contributes to the protection of mice from endotoxemia and bacterial infection by positively mediating the xenophagy pathway in peritoneal macrophages and was found to be a crucial regulator of autophagosome formation during bacterial challenge in vivo (121). LC3 processing was reduced in HMGB1-low peritoneal macrophages from mice after L. monocytogenes infection, indicating that HMGB1 is crucial for activation of bacteria-induced autophagy. Loss of HMGB1 severely reduces bacterial clearance in this model since autophagy is needed to kill L. monocytogenes. It is tempting to speculate that intracellular HMGB1 may also induce autophagy after S. aureus exposure in PMN. HMGB1 is released in bronchiolar lavage fluid and contributes to lung injury in a murine model of S. aureus pneumonia (119). This may be due to the enhanced autophagic response elicited by platelet derived HMGB1 which has been shown to enhance autophagosome formation in PMN from patients with acute myocardial infractions (122). This study also showed that HMGB1 was able to prolong PMN survival whilst preventing mitochondrial potential depletion which, in combination with the induction of autophagy, is a strikingly similar phenotype to that seen in S. aureus infection of PMN.
S. aureus clearly changes the intracellular landscape of the host cell to benefit its own survival by co-opting autophagosomes and host factor gene expression to reshape the autophagic and apoptotic fate of PMN for its own benefit. However, bacterial factors produced by S. aureus to manipulate PMN in this context represent a major gap in our current understanding of the intracellular life cycle of this bacterium. Even within the broader context of autophagy mediated S. aureus intracellular survival in professional and non-professional phagocytes, little is known. It is likely, given the pleiotropic nature and redundancy of several staphylococcal toxins, such as the PSMs, that multiple factors are involved but to date, only a few factors have been identified within a handful of cell types. For instance, several reports have highlighted the involvement of the agr regulated toxin, hla, in the induction of host autophagy. Treatment with hla alone was capable of inducing LC3 decorated vacuoles within CHO cells (67). Vacuoles containing hla mutants failed to recruit LC3 to these vacuoles in contrast to the hla expressing strain. In a follow up study, the same authors found that hla could induce LC3 decorated filaments in a Rab7 and Rab1b dependent manner which is in inhibited by cAMP (68). Studies in HACAT cells have also suggested that hla is capable of inducing autophagy possibly due to a drop in ATP/AMP ratio in response to membrane perforation (123). It is reasonable to suggest that hla expression may also induce autophagy to the same effect in neutrophils which could provide S. aureus a protective niche in which to thrive. Besides its effect on the autophagy pathway, hla has been shown to inhibit macrophage efferocytosis potentially to protect its intracellular niche within PMN from phagocytosis and killing by host macrophages (124). Regardless of the effect hla has on inducing host autophagy, it is unclear what causes the failure of host autophagosomes to mature into autophagolysosomes. However, one study showed that S. aureus was capable of disrupting autophagic flux through the expression of Immunodominant surface antigen B (IsaB) within Hela cells, THP-1 macrophages and in vivo using a skin infection model (125). Deletion of IsaB led to increased autophagic flux which was effectively reduced by recombinant IsaB thus implicating its role in the manipulation of host autophagy for intracellular survival. The role IsaB plays in subverting PMN has not yet been investigated but could potentially act as a crucial factor in achieving the abhorrent autophagic and apoptotic phenotype seen within PMN under S. aureus infection. Further research is needed to determine the bacterial factors involved in manipulating host autophagy by intracellular S. aureus across all cell types but particularly within PMN.
The manipulation of host autophagy by S. aureus for intracellular survival opens the possibility for targeting the autophagy pathway as an additive or alternative treatment for S. aureus infection, particularly in cases where antibiotic treatment is not successful. Recently, high throughput studies have begun to assess potential host directed therapies and host targets that can be used against intracellular S. aureus (126, 127). One such study used a shRNA screen in HeLa cells to assess host factors involved in S. aureus infection which highlighted several potential host targets that could lead to increased host cell viability including the pro-autophagic gene ATG10, though this target was not further assessed. However, as this review has highlighted, intracellular survival of S. aureus likely varies among strains, target host cell type and is likely dependent on bacterial/host gene expression. The complexity of these factors, and their relationship to one another, will therefore present a difficult challenge in finding a reliable target for host directed therapy. Nevertheless, drugs targeting host cells and pathways can lead to improved disease outcome in S. aureus infection, several of which that target the autophagy pathway.
The anti-malarial drug chloroquine, which can inhibit autolysosome formation, has been shown to enhance the killing of intracellular S. aureus using the antibiotic, levofloxacin, when co-administered in THP-1 macrophages (128). Although the effectiveness of chloroquine has not been attributed to its anti-autophagic ability, its clinically approved derivative, hydroxychloroquine, has been shown to modulate autophagy within a subset of patients as part of ongoing preclinical studies for anti-tumour treatments (129). Therefore, it can be speculated that this enhanced killing can be attributed to the effect’s chloroquine exerts on host autophagy and thus should be investigated in the context of autophagy mediated intracellular survival of S. aureus within PMN. Another autophagy inhibitor, Dorsomorphin, has been used in several in vitro and in vivo studies to inhibit AMPK, a key inducer of host autophagy (130). Recently, Dorsomorphin has been shown in vitro to reduce intracellular burden of S. aureus and increase cell viability in both HeLa and HUVEC cells which could potentially yield similar results in PMN through the obstruction of host autophagy (65). However, it should be noted that dorsomorphin can have off target effects which may likely hinder its potential use in a clinical setting (130). Lastly, the autophagy inhibitor, 3-MA, has recently been shown to protect against S. aureus infection in a BALB/c lung infection model (131). In this study, Raw264.7 macrophages showed increased bacterial killing of S. aureus when treated with 3MA which was associated with the reduction of the autophagic markers LC3-II and Beclin-1. In vivo, 3MA treated mice showed comparable effects to vancomycin treatment which showed reduced lung CFU and increased numbers of CD11b+F4/80+ macrophages versus control mice. The authors reasoned that S. aureus manipulates autophagy within host macrophages for its own gain and so when treated with the autophagy inhibitor, 3MA, efficient killing of the bacterium by host macrophages was restored. However, given the widespread inhibition 3MA would have across all cell types, it could be theorized that autophagy inhibition also restored neutrophil killing leading to overall improvement in infection outcome. In all, targeting host autophagy has yielded some encouraging results in the treatment of S. aureus infection in combination with antibiotics as well as a standalone treatment.
It is possible that existing clinically approved autophagy inhibitors could be re-purposed to treat S. aureus chronic infection. However, inhibiting or blocking host autophagy could have unwanted side effects and so, would need to be approached with caution. Maurer et al., generated a breed of mice with a partial knockdown of the critical LC3 processing protein ATG16L1 leading to a reduced autophagic phenotype across all tissues and cell types examined (132). ATG16L1 hypomorphic (ATG16L1HM) mice infected with S. aureus were shown to possess a significantly enhanced susceptibility to hla leading to increased mortality (133). This increased susceptibility of ATG16L1HM mice was not a result of the altered autophagy phenotype in macrophages or dendritic cells but rather due to the autophagy reduced endothelial cells. This was accredited to an inability of the ATG16L1HM mice endothelial cells to mediate hla tolerance through the autophagy pathway which under WT conditions would limit hla toxicity. Interestingly, ATG16L1HM mice were shown to have significantly enhanced survival against hla mutants compared to WT mice indicating the fine balance autophagy plays in S. aureus infection. As such, this study demonstrates the risk of inhibiting host autophagy and highlights the need for greater understanding of the relationship of S. aureus and the autophagy pathway as well as any bacterial factors involved. Of course, blanket autophagy inhibition may also inadvertently block the clearance of another pathogen. For example, treatment of cystic fibrosis patients with the antibiotic azithromycin led to opportunistic mycobacterial infections; azithromycin blocked autolysosome formation and therefore the degradation of mycobacteria within autophagosomes. It is therefore critical that these considerations are taken into account when considering clinical inhibition of autophagy. However, despite these potential downfalls, inhibition of host autophagy remains an alluring option to combat S. aureus infection.
S. aureus is a formidable foe which has developed a vast array of immune evasion mechanisms that enable its survival and persistence. Of these mechanisms, S. aureus intracellular survival represents one of the most advantageous tactics to subvert the host immune system. Such a mechanism offers both stealth and protection from host bacterial sensors, killing and even from antibiotics due to poor membrane permeability and the formation of intracellular SCVs (5, 134, 135). Intracellular survival is clearly of great benefit to S. aureus as it has been shown in numerous cell types with various mechanisms employed in both a strain and cell dependent manner. Of these mechanisms, the manipulation of host autophagy appears to play a central role in both short- and long-term intracellular survival with many professional and non-professional phagocytes shown to facilitate intracellular S. aureus in an autophagy dependent manner. Among these cell types, PMN represent a highly advantageous target of S. aureus creating an unlikely intracellular niche to shield the bacterium from extracellular threats whilst providing safe passage to secondary sites for further infection. Despite the hazardous intracellular conditions found within PMN, S. aureus can survive intracellularly and can masterfully manipulate the host autophagy pathway, a process designed to remove intracellular pathogens. In doing so, S. aureus can subvert the constitutively active apoptosis pathway in PMN to prevent destruction of its intracellular niche whilst also protecting itself from the degradative process of apoptosis and subsequent macrophage efferocytosis.
Whilst several host and bacterial factors have been identified in this context, more research is required with a focus needed in the context of host directed therapies that represent a massively important field of research currently underrepresented. Future studies should aim to give greater insight into the molecular pathways engaged within PMN by S. aureus as well as any bacterial factors involved whilst also focusing on increased in vivo studies and clinical observations of novel host directed therapies. Nevertheless, intracellular survival and host autophagy is a crucial factor in S. aureus infection which has the potential to yield powerful next generation therapeutics capable of disarming the pathogen of one of its most important immune evasion tactics.
MM and RM conceived the idea of the manuscript. EV and MM wrote the manuscript. RM edited and added invaluable insights to the manuscript. All authors contributed to the article and approved the submitted version.
Associated work in the lab was supported by a Science Foundation Ireland Investigator Award (15/IA/3041) and a Wellcome Investigator Award (202846/Z/16/Z) to RM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. van Hal SJ, Jensen SO, Vaska VL, Espedido BA, Paterson DL, Gosbell IB. Predictors of Mortality in Staphylococcus Aureus Bacteremia. Clin Microbiol Rev (2012) 25:362–86. doi: 10.1128/CMR.05022-11
2. Shorr AF, Tabak YP, Killian AD, Gupta V, Liu LZ, Kollef MH. Healthcare-Associated Bloodstream Infection: A Distinct Entity? Insights From a Large U.S. Database. Crit Care Med (2006) 34(10):2588–95. doi: 10.1097/01.CCM.0000239121.09533.09
3. Vos FJ, Kullberg BJ, Sturm PD, Krabbe PFM, Van Dijk APJ, Wanten GJA, et al. Metastatic Infectious Disease and Clinical Outcome in Staphylococcus Aureus and Streptococcus Species Bacteremia. Medicine (Baltimore) (2012) 91(2):86–94. doi: 10.1097/MD.0b013e31824d7ed2
4. Horino T, Hori S. Metastatic Infection During Staphylococcus Aureus Bacteremia. J Infect Chemother (2020) 26:162–9. Elsevier B.V. doi: 10.1016/j.jiac.2019.10.003
5. Garzoni C, Kelley WL. Return of the Trojan Horse: Intracellular Phenotype Switching and Immune Evasion by Staphylococcus Aureus. EMBO Mol Med (2011) 3(3):115–7. doi: 10.1002/emmm.201100123
6. Horn J, Stelzner K, Rudel T, Fraunholz M. Inside Job: Staphylococcus Aureus Host-Pathogen Interactions. Int J Med Microbiol (2018) p:607–24. Elsevier GmbH. doi: 10.1016/j.ijmm.2017.11.009
7. Surewaard BGJ, Deniset JF, Zemp FJ, Amrein M, Otto M, Conly J, et al. Identification and Treatment of the Staphylococcus Aureus Reservoir In Vivo. J Exp Med (2016) 213(7):1141–51. doi: 10.1084/jem.20160334
8. Thwaites GE, Gant V. Are Bloodstream Leukocytes Trojan Horses for the Metastasis of Staphylococcus Aureus? Nat Rev Microbiol (2011) 9:215–22. Nature Publishing Group. doi: 10.1038/nrmicro2508
9. Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. Survival of Staphylococcus Aureus Inside Neutrophils Contributes to Infection. J Immunol (2000) 164(7):3713–22. doi: 10.4049/jimmunol.164.7.3713
10. O’Keeffe KM, Wilk MM, Leech JM, Murphy AG, Laabei M, Monk IR, et al. Manipulation of Autophagy in Phagocytes Facilitates Staphylococcus Aureus Bloodstream Infection. Infect Immun (2015) 83(9):3445–57. doi: 10.1128/IAI.00358-15
11. Pollitt EJGG, Szkuta PT, Burns N, Foster SJ. Staphylococcus Aureus Infection Dynamics. PloS Pathog (2018) 14(6):e1007112. doi: 10.1371/journal.ppat.1007112
12. Prajsnar TK, Hamilton R, Garcia-Lara J, Mcvicker G, Williams A, Boots M, et al. A Privileged Intraphagocyte Niche Is Responsible for Disseminated Infection of Staphylococcus Aureus in a Zebrafish Model. Cell Microbiol (2012) 14(10):1600–19. doi: 10.1111/j.1462-5822.2012.01826.x
13. Prajsnar TK, Serba JJ, Dekker BM, Gibson JF, Masud S, Fleming A, et al. The Autophagic Response to Staphylococcus Aureus Provides An Intracellular Niche in Neutrophils. Autophagy (2021) 17(4):888–902. doi: 10.1080/15548627.2020.1739443
14. Kubica M, Guzik K, Koziel J, Zarebski M, Richter W, Gajkowska B, et al. A Potential New Pathway for Staphylococcus Aureus Dissemination: The Silent Survival of S. Aureus Phagocytosed by Human Monocyte-Derived Macrophages. PloS One (2008) 3(1):e1409. May R, editor. doi: 10.1371/journal.pone.0001409
15. Fraunholz M, Sinha B. Intracellular Staphylococcus Aureus: Live-in and Let Die. Front Cell Infect Microbiol (2012) 2:43. doi: 10.3389/fcimb.2012.00043
16. Moldovan A, Fraunholz MJ. In or Out: Phagosomal Escape of Staphylococcus Aureus. Cell Microbiol (2019) 21(3):e12997. doi: 10.1111/cmi.12997
17. Lacoma A, Cano V, Moranta D, Regueiro V, Domínguez-Villanueva D, Laabei M, et al. Investigating Intracellular Persistence of Staphylococcus Aureus Within a Murine Alveolar Macrophage Cell Line. Virulence (2017) 8(8):1761–75. doi: 10.1080/21505594.2017.1361089
18. Tranchemontagne ZR, Camire RB, O’Donnell VJ, Baugh J, Burkholder KM. Staphylococcus Aureus Strain USA300 Perturbs Acquisition of Lysosomal Enzymes and Requires Phagosomal Acidification for Survival Inside Macrophages. Infect Immun (2016) 84(1):241–53. doi: 10.1128/IAI.00704-15
19. Dai Y, Gao C, Chen L, Chang W, Yu W, Ma X, et al. Heterogeneous Vancomycin-Intermediate Staphylococcus Aureus Uses the VraSR Regulatory System to Modulate Autophagy for Increased Intracellular Survival in Macrophage-Like Cell Line RAW264.7. Front Microbiol (2019) 10(MAY):1222. doi: 10.3389/fmicb.2019.01222
20. Flannagan RS, Kuiack RC, McGavin MJ, Heinrichs DE. Staphylococcus Aureus Uses the GraXRS Regulatory System to Sense and Adapt to the Acidified Phagolysosome in Macrophages. MBio (2018) 9(4):1143. Torres VJ, editor. doi: 10.1128/mBio.01143-18
21. Mulcahy ME, O’Brien EC, O’Keeffe KM, Vozza EG, Leddy N, McLoughlin RM. Manipulation of Autophagy and Apoptosis Facilitates Intracellular Survival of Staphylococcus Aureus in Human Neutrophils. Front Immunol (2020) 11:565545/full. doi: 10.3389/fimmu.2020.565545/full
22. Leimer N, Rachmühl C, Palheiros Marques M, Bahlmann AS, Furrer A, Eichenseher F, et al. Nonstable Staphylococcus Aureus Small-Colony Variants Are Induced by Low Ph and Sensitized to Antimicrobial Therapy by Phagolysosomal Alkalinization. J Infect Dis (2016) 213(2):305–13. doi: 10.1093/infdis/jiv388
23. Soong G, Paulino F, Wachtel S, Parker D, Wickersham M, Zhang D, et al. Methicillin-Resistant Staphylococcus Aureus Adaptation to Human Keratinocytes. MBio (2015) 6(2):1–14. doi: 10.1128/mBio.00289-15
24. Blättner S, Das S, Paprotka K, Eilers U, Krischke M, Kretschmer D, et al. Staphylococcus Aureus Exploits a Non-Ribosomal Cyclic Dipeptide to Modulate Survival Within Epithelial Cells and Phagocytes. PloS Pathog (2016) 12(9):e1005857. Prince A, editor. doi: 10.1371/journal.ppat.1005857
25. Grosz M, Kolter J, Paprotka K, Winkler A-CC, Schäfer D, Chatterjee SS, et al. Cytoplasmic Replication of Staphylococcus Aureus Upon Phagosomal Escape Triggered by Phenol-Soluble Modulin α. Cell Microbiol (2014) 16(4):451–65. doi: 10.1111/cmi.12233
26. Rollin G, Tan X, Tros F, Dupuis M, Nassif X, Charbit A, et al. Intracellular Survival of Staphylococcus Aureus in Endothelial Cells: A Matter of Growth or Persistence. Front Microbiol (2017) 8(JUL):01354. doi: 10.3389/fmicb.2017.01354
27. Tuchscherr L, Heitmann V, Hussain M, Viemann D, Roth J, von Eiff C, et al. Staphylococcus Aureus Small-Colony Variants Are Adapted Phenotypes for Intracellular Persistence. J Infect Dis (2010) 202(7):1031–40. doi: 10.1086/656047
28. Krauss JL, Roper PM, Ballard A, Shih C-C, Fitzpatrick JAJ, Cassat JE, et al. Staphylococcus Aureus Infects Osteoclasts and Replicates Intracellularly. MBio (2019) 10(5):e02447–19. Whiteley M, editor. doi: 10.1128/mBio.02447-19
29. Yang D, Wijenayaka AR, Solomon LB, Pederson SM, Findlay DM, Kidd SP, et al. Novel Insights Into Staphylococcus Aureus Deep Bone Infections: The Involvement of Osteocytes. MBio (2018) 9(2):e00415–18. http://mbio.asm.org/. Smeltzer MS, Novick RP, editors. doi: 10.1128/mBio.00415-18
30. Flannagan RS, Heit B, Heinrichs DE. Intracellular Replication of Staphylococcus Aureus in Mature Phagolysosomes in Macrophages Precedes Host Cell Death, and Bacterial Escape and Dissemination. Cell Microbiol (2016) 18(4):514–35. doi: 10.1111/cmi.12527
31. Cohen TS, Boland ML, Boland BB, Takahashi V, Tovchigrechko A, Lee Y, et al. S. Aureus Evades Macrophage Killing Through NLRP3-Dependent Effects on Mitochondrial Trafficking. Cell Rep (2018) 22(9):2431–41. doi: 10.1016/j.celrep.2018.02.027
32. Jorch SK, Surewaard BGJ, Hossain M, Peiseler M, Deppermann C, Deng J, et al. Peritoneal GATA6+ Macrophages Function as a Portal for Staphylococcus Aureus Dissemination. J Clin Invest (2019) 129(11):4643–56. doi: 10.1172/JCI127286
33. Schnaith A, Kashkar H, Leggio SA, Addicks K, Krönke M, Krut O. Staphylococcus Aureus Subvert Autophagy for Induction of Caspase-Independent Host Cell Death. J Biol Chem (2007) 282(4):2695–706. doi: 10.1074/jbc.M609784200
34. Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR, Nauseef WM. Agr-Dependent Interactions of Staphylococcus Aureus USA300 With Human Polymorphonuclear Neutrophils. J Innate Immun (2010) 2(6):546–59. doi: 10.1159/000319855
35. Münzenmayer L, Geiger T, Daiber E, Schulte B, Autenrieth SE, Fraunholz M, et al. Influence of Sae-regulated and Agr-Regulated Factors on the Escape of Staphylococcus Aureus From Human Macrophages. Cell Microbiol (2016) 18(8):1172–83. doi: 10.1111/cmi.12577
36. Qazi SNA, Counil E, Morrissey J, Rees CED, Cockayne A, Winzer K, et al. Agr Expression Precedes Escape of Internalized Staphylococcus Aureus From the Host Endosome. Infect Immun (2001) 69(11):7074–82. doi: 10.1128/IAI.69.11.7074-7082.2001
37. Norvick RP, Jiang D. The Staphylococcal saeRS System Coordinates Environmental Signals Agr Quorum Sensing. Microbiology (2003) 149(10):2709–17. doi: 10.1099/mic.0.26575-0
38. Surewaard BGJ, Nijland R, Spaan AN, Kruijtzer JAW, de Haas CJC, van Strijp JAG. Inactivation of Staphylococcal Phenol Soluble Modulins by Serum Lipoprotein Particles. PloS Pathog (2012) 8(3):e1002606. doi: 10.1371/journal.ppat.1002606. Gilmore MS, editor.
39. Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M, et al. Small Colony Variants: A Pathogenic Form of Bacteria That Facilitates Persistent and Recurrent Infections. Nat Rev Microbiol (2006) 4:295–305. doi: 10.1038/nrmicro1384
40. von Eiff C, Peters G, Becker K. The Small Colony Variant (SCV) Concept—the Role of Staphylococcal SCVs in Persistent Infections. Injury (2006) 37(2):S26–33. doi: 10.1016/j.injury.2006.04.006
41. Stoneham SM, Cantillon DM, Waddell SJ, Llewelyn MJ. Spontaneously Occurring Small-Colony Variants of Staphylococcus Aureus Show Enhanced Clearance by THP-1 Macrophages. Front Microbiol (2020) 11:1300. doi: 10.3389/fmicb.2020.01300
42. Tuchscherr L, Löffler B, Proctor RA. Persistence of Staphylococcus Aureus: Multiple Metabolic Pathways Impact the Expression of Virulence Factors in Small-Colony Variants (SCVs). Front Microbiol (2020) 11:1028. doi: 10.3389/fmicb.2020.01028
43. Moisan H, Brouillette E, Jacob CL, Langlois-Bégin P, Michaud S, Malouin F. Transcription of Virulence Factors in Staphylococcus Aureus Small-Colony Variants Isolated From Cystic Fibrosis Patients Is Influenced by SigB. J Bacteriol (2006) 188(1):64–76. doi: 10.1128/JB.188.1.64-76.2006
44. Tuchscherr L, Löffler B. Staphylococcus Aureus Dynamically Adapts Global Regulators and Virulence Factor Expression in the Course From Acute to Chronic Infection. Curr Genet (2016) 62(1):15–7. doi: 10.1007/s00294-015-0503-0
45. Tuchscherr L, Bischoff M, Lattar SM, Noto Llana M, Pförtner H, Niemann S, et al. Sigma Factor SigB Is Crucial to Mediate Staphylococcus Aureus Adaptation During Chronic Infections. PloS Pathog (2015) 11(4):e1004870. Koehler TM, editor. doi: 10.1371/journal.ppat.1004870
46. He C, Klionsky DJ. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu Rev Genet (2009) 43:67–93. doi: 10.1146/annurev-genet-102808-114910
47. Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes With Mammalian Atg14 and UVRAG. Mol Biol Cell (2008) 19(12):5360–72. doi: 10.1091/mbc.e08-01-0080
48. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a Mammalian Homologue of Yeast Apg8p, Is Localized in Autophagosome Membranes After Processing. EMBO J (2000) 19(21):5720–8. doi: 10.1093/emboj/19.21.5720
49. Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu HL, et al. p62 Links the Autophagy Pathway and the Ubiqutin–Proteasome System Upon Ubiquitinated Protein Degradation. Cell Mol Biol Lett (2016) 21(1):29. doi: 10.1186/s11658-016-0031-z
50. Xie X, Li F, Wang Y, Wang Y, Lin Z, Cheng X, et al. Molecular Basis of Ubiquitin Recognition by the Autophagy Receptor CALCOCO2. Autophagy (2015) 11(10):1775–89. doi: 10.1080/15548627.2015.1082025
51. Ryan TA, Tumbarello DA. Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front Immunol (2018) 9:1024/full(MAY). doi: 10.3389/fimmu.2018.01024/full
52. Xie Z, Klionsky DJ. Autophagosome Formation: Core Machinery and Adaptations. Nat Cell Biol (2007) 9:1102–9. doi: 10.1038/ncb1007-1102
53. Gatica D, Lahiri V, Klionsky DJ. Cargo Recognition and Degradation by Selective Autophagy. Nat Cell Biol (2018) 20:233–42. doi: 10.1038/s41556-018-0037-z
54. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy Is a Defense Mechanism Inhibiting BCG and Mycobacterium Tuberculosis Survival in Infected Macrophages. Cell (2004) 119(6):753–66. doi: 10.1016/j.cell.2004.11.038
55. Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy Controls Salmonella Infection in Response to Damage to the Salmonella-containing Vacuole. J Biol Chem (2006) 281(16):11374–83. doi: 10.1074/jbc.M509157200
56. Rich KA, Burkett C, Webster P. Cytoplasmic Bacteria can be Targets for Autophagy. Cell Microbiol (2003) 5(7):455–68. doi: 10.1046/j.1462-5822.2003.00292.x
57. Chargui A, Cesaro A, Mimouna S, Fareh M, Brest P, Naquet P, et al. Subversion of Autophagy in Adherent Invasive Escherichia Coli-Infected Neutrophils Induces Inflammation and Cell Death. PloS One (2012) 7(12):e51727. doi: 10.1371/journal.pone.0051727
58. Dortet L, Mostowy S, Louaka AS, Gouin E, Nahori M-A, Wiemer EAC, et al. Recruitment of the Major Vault Protein by Inlk: A Listeria Monocytogenes Strategy to Avoid Autophagy. PloS Pathog (2011) 7(8):e1002168. doi: 10.1371/journal.ppat.1002168. Valdivia RH, editor.
59. Kim JK, Lee HM, Park KS, Shin DM, Kim TS, Kim YS, et al. MIR144* Inhibits Antimicrobial Responses Against Mycobacterium Tuberculosis in Human Monocytes and Macrophages by Targeting the Autophagy Protein DRAM2. Autophagy (2017) 13(2):423–41. doi: 10.1080/15548627.2016.1241922
60. Mitchell G, Cheng MI, Chen C, Nguyen BN, Whiteley AT, Kianian S, et al. Listeria Monocytogenes Triggers Noncanonical Autophagy Upon Phagocytosis, But Avoids Subsequent Growth-Restricting Xenophagy. Proc Natl Acad Sci (2017) 112:201716055. doi: 10.1073/pnas.1716055115
61. Feng ZZ, Jiang AJ, Mao AW, Feng Y, Wang W, Li J, et al. The Salmonella Effectors SseF and SseG Inhibit Rab1A-Mediated Autophagy to Facilitate Intracellular Bacterial Survival and Replication. J Biol Chem (2018) 293(25):9662–73. doi: 10.1074/jbc.M117.811737
62. Radhi OA, Davidson S, Scott F, Zeng RX, Jones DH, Tomkinson NCO, et al. Inhibition of the ULK1 Protein Complex Suppresses Staphylococcus-Induced Autophagy and Cell Death. J Biol Chem (2019) 294(39):14289–307. doi: 10.1074/jbc.RA119.008923
63. Cai J, Li JJ, Zhou Y, Wang J, Li JJ, Cui L, et al. Staphylococcus Aureus Facilitates its Survival in Bovine Macrophages by Blocking Autophagic Flux. J Cell Mol Med (2020) 24(6):3460–8. doi: 10.1111/jcmm.15027
64. Wu S, Shen Y, Zhang S, Xiao Y, Shi S. Salmonella Interacts With Autophagy to Offense or Defense. Front Microbiol (2020) 11:00721. doi: 10.3389/fmicb.2020.00721
65. Bravo-Santano N, Ellis JK, Mateos LM, Calle Y, Keun HC, Behrends V, et al. Intracellular Staphylococcus Aureus Modulates Host Central Carbon Metabolism to Activate Autophagy. mSphere (2018) 3(4):e00374–18. doi: 10.1128/mSphere.00374-18
66. Neumann Y, Bruns SA, Rohde M, Prajsnar TK, Foster SJ, Schmitz I. Intracellular Staphylococcus Aureus Eludes Selective Autophagy by Activating a Host Cell Kinase. Autophagy (2016) 12(11):2069–84. doi: 10.1080/15548627.2016.1226732
67. Mestre MB, Fader CM, Sola C, Colombo MI. α-Hemolysin Is Required for the Activation of the Autophagic Pathway in Staphylococcus Aureus-Infected Cells. Autophagy (2010) 6(1):110–25. doi: 10.4161/auto.6.1.10698
68. López de Armentia MM, Gauron MC, Colombo MI. Staphylococcus Aureus Alpha-Toxin Induces the Formation of Dynamic Tubules Labeled With LC3 Within Host Cells in a Rab7 and Rab1b-Dependent Manner. Front Cell Infect Microbiol (2017) 7(OCT):431. doi: 10.3389/fcimb.2017.00431
69. Rajarathnam K, Schnoor M, Richardson RM, Rajagopal S. How do Chemokines Navigate Neutrophils to the Target Site: Dissecting the Structural Mechanisms and Signaling Pathways. Cell Signal (2019) 54:69–80. Elsevier Inc. doi: 10.1016/j.cellsig.2018.11.004
70. Weiss E, Hanzelmann D, Fehlhaber B, Klos A, Von Loewenich FD, Liese J, et al. Formyl-Peptide Receptor 2 Governs Leukocyte Influx in Local Staphylococcus Aureus Infections. FASEB J (2018) 32(1):26–36. doi: 10.1096/fj.201700441r
71. Subramanian BC, Majumdar R, Parent CA. The Role of the LTB4-BLT1 Axis in Chemotactic Gradient Sensing and Directed Leukocyte Migration. Semin Immunol (2017) 33:16–29. Academic Press. doi: 10.1016/j.smim.2017.07.002
72. Kobayashi Y. The Role of Chemokines in Neutrophil Biology. Front Biosci (2008) 13:2400–7. doi: 10.2741/2853
73. Nourshargh S, Renshaw SA, Imhof BA. Reverse Migration of Neutrophils: Where, When, How, and Why? Trends Immunol (2016) 3:273–86. Elsevier Ltd. doi: 10.1016/j.it.2016.03.006
74. McGuinness W, Kobayashi S, DeLeo F. Evasion of Neutrophil Killing by Staphylococcus Aureus. Pathogens (2016) 5(1):32. doi: 10.3390/pathogens5010032
75. Pilsczek FH, Salina D, Poon KKH, Fahey C, Yipp BG, Sibley CD, et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus Aureus. J Immunol (2010) 185(12):7413–25. doi: 10.4049/jimmunol.1000675
76. Holland SM. Chronic Granulomatous Disease. Hematol Oncol Clin North Am (2013) 27:89–99. doi: 10.1016/j.hoc.2012.11.002
77. van den Berg JM, van Koppen E, Åhlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic Granulomatous Disease: The European Experience. PloS One (2009) 4(4):e5234. doi: 10.1371/journal.kpone.0005234. Alspaugh A, editor.
78. Lakshman R, Finn A. Neutrophil Disorders and Their Management. J Clin Pathol (2001) 54:7–19. doi: 10.1136/jcp.54.1.7
79. Maaloul I, Talmoudi J, Chabchoub I, Ayadi L, Kamoun TH, Boudawara T, et al. Chediak-Higashi Syndrome Presenting in Accelerated Phase: A Case Report and Literature Review. Hematol Oncol Stem Cell Ther (2016) 9(2):71–5. doi: 10.1016/j.hemonc.2015.07.002
80. Robertson CM, Perrone EE, McConnell KW, Dunne WM, Boody B, Brahmbhatt T, et al. Neutrophil Depletion Causes a Fatal Defect in Murine Pulmonary Staphylococcus Aureus Clearance. J Surg Res (2008) 150(2):278–85. doi: 10.1016/j.jss.2008.02.009
81. McLoughlin RM, Solinga RM, Rich J, Zaleski KJ, Cocchiaro JL, Risley A, et al. CD4+ T Cells and CXC Chemokines Modulate the Pathogenesis of Staphylococcus Aureus Wound Infections. Proc Natl Acad Sci U S A (2006) 103(27):10408–13. doi: 10.1073/pnas.0508961103
82. McLoughlin RM, Lee JC, Kasper DL, Tzianabos AO. IFN-γ Regulated Chemokine Production Determines the Outcome of Staphylococcus Aureus Infection. J Immunol (2008) 181(2):1323–32. doi: 10.4049/jimmunol.181.2.1323
83. Robertson AL, Holmes GR, Bojarczuk AN, Burgon J, Loynes CA, Chimen M, et al. A Zebrafish Compound Screen Reveals Modulation of Neutrophil Reverse Migration as an Anti-Inflammatory Mechanism. Sci Transl Med (2014) 6(225):225ra29–225ra29. doi: 10.1126/scitranslmed.3007672
84. Buckley CD, Ross EA, McGettrick HM, Osborne CE, Haworth O, Schmutz C, et al. Identification of a Phenotypically and Functionally Distinct Population of Long-Lived Neutrophils in a Model of Reverse Endothelial Migration. J Leukoc Biol (2006) 79(2):303–11. doi: 10.1189/jlb.0905496
85. Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, et al. Staphylococcus Aureus Golden Pigment Impairs Neutrophil Killing and Promotes Virulence Through its Antioxidant Activity. J Exp Med (2005) 202(2):209–15. doi: 10.1084/jem.20050846
86. Karavolos MH, Horsburgh M, Ingham E, Foster SJ. Role and Regulation of the Superoxide Dismutases of Staphylococcus Aureus. Microbiology (2003) 149(10):2749–58. doi: 10.1099/mic.0.26353-0
87. De Jong NWM, Ramyar KX, Guerra FE, Nijland R, Fevre C, Voyich JM, et al. Immune Evasion by a Staphylococcal Inhibitor of Myeloperoxidase. Proc Natl Acad Sci U S A (2017) 114(35):9439–44. doi: 10.1073/pnas.1707032114
88. Carnes EC, Lopez DM, Donegan NP, Cheung A, Gresham H, Timmins GS, et al. Confinement-Induced Quorum Sensing of Individual Staphylococcus Aureus Bacteria. Nat Chem Biol (2010) 6(1):41–5. doi: 10.1038/nchembio.264
89. Martinez J, Malireddi RKS, Lu Q, Cunha LD, Pelletier S, Gingras S, et al. Molecular Characterization of LC3-Associated Phagocytosis Reveals Distinct Roles for Rubicon, NOX2 and Autophagy Proteins. Nat Cell Biol (2015) 17(7):893–906. doi: 10.1038/ncb3192
90. Savill J, Dransfield I, Gregory C, Haslett C. A Blast From the Past: Clearance of Apoptotic Cells Regulates Immune Responses. Nat Rev Immunol (2002) 2:965–75. doi: 10.1038/nri957
91. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil Kinetics in Health and Disease. Trends Immunol (2010) 31:318–24. Elsevier. doi: 10.1016/j.it.2010.05.006
92. Lahoz-Beneytez J, Elemans M, Zhang Y, Ahmed R, Salam A, Block M, et al. Human Neutrophil Kinetics: Modeling of Stable Isotope Labeling Data Supports Short Blood Neutrophil Half-Lives. Blood (2016) 127(26):3431–8. doi: 10.1182/blood-2016-03-700336
93. McCracken JM, Allen LAH. Regulation of Human Neutrophil Apoptosis and Lifespan in Health and Disease. J Cell Death (2014) 7(1):15–23. doi: 10.4137/JCD.S11038
94. Laskay T, van Zandbergen G, Solbach W. Neutrophil Granulocytes as Host Cells and Transport Vehicles for Intracellular Pathogens: Apoptosis as Infection-Promoting Factor. Immunobiology (2008) 213:183–91. Urban & Fischer. doi: 10.1016/j.imbio.2007.11.010
95. Kinkead LC, Fayram DC, Allen LH. Francisella Novicida Inhibits Spontaneous Apoptosis and Extends Human Neutrophil Lifespan. J Leukoc Biol (2017) 102(3):815–28. doi: 10.1189/jlb.4MA0117-014R
96. Alberdi P, Espinosa PJ, Cabezas-Cruz A, de la Fuente J. Anaplasma Phagocytophilum Manipulates Host Cell Apoptosis by Different Mechanisms to Establish Infection. Vet Sci (2016) 3:15. doi: 10.3390/vetsci3030015
97. Rosenberg H, Oppenheim J. The 2006 Dolph Adams Award and the State of the Journal of Leukocyte Biology. J Leukoc Biol (2007) 81(2):369–71. doi: 10.1189/jlb.1106655
98. Saverymuttu SH, Peters AM, Keshavarzian A, Reavy HJ, Lavender JP. The Kinetics of 111Indium Distribution Following Injection of 111Indium Labelled Autologous Granulocytes in Man. Br J Haematol (1985) 61(4):675–85. doi: 10.1111/j.1365-2141.1985.tb02882.x
99. Martin C, Burdon PCE, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines Acting Via CXCR2 and CXCR4 Control the Release of Neutrophils From the Bone Marrow and Their Return Following Senescence. Immunity (2003) 19(4):583–93. doi: 10.1016/S1074-7613(03)00263-2
100. Greenlee-Wacker MC, Rigby KM, Kobayashi SD, Porter AR, DeLeo FR, Nauseef WM. Phagocytosis of Staphylococcus Aureus by Human Neutrophils Prevents Macrophage Efferocytosis and Induces Programmed Necrosis. J Immunol (2014) 192(10):4709–17. doi: 10.4049/jimmunol.1302692
101. Lotz S, Aga E, Wilde I, van Zandbergen G, Hartung T, Solbach W, et al. Highly Purified Lipoteichoic Acid Activates Neutrophil Granulocytes and Delays Their Spontaneous Apoptosis Via CD14 and TLR2. J Leukoc Biol (2004) 75(3):467–77. doi: 10.1189/jlb.0803360
102. Ocaña MG, Asensi V, Montes ÁH, Meana Á, Celada A, Valle-Garay E. Autoregulation Mechanism of Human Neutrophil Apoptosis During Bacterial Infection. Mol Immunol (2008) 45(7):2087–96. doi: 10.1016/j.molimm.2007.10.013
103. Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S, et al. Differential Interactions Between Beclin 1 and Bcl-2 Family Members. Autophagy (2007) 3(6):561–8. doi: 10.4161/auto.4713
104. Germain M, Slack RS. MCL-1 Regulates the Balance Between Autophagy and Apoptosis. Autophagy (2011) 7:549–51. doi: 10.4161/auto.7.5.15098
105. Li XF, Chen DP, Ouyang FZ, Chen MM, Wu Y, Kuang DM, et al. Increased Autophagy Sustains the Survival and Pro-Tumourigenic Effects of Neutrophils in Human Hepatocellular Carcinoma. J Hepatol (2015) 62(1):131–9. doi: 10.1016/j.jhep.2014.08.023
106. Moulding DA, Akgul C, Derouet M, White MR, Edwards SW. BCL-2 Family Expression in Human Neutrophils During Delayed and Accelerated Apoptosis. J Leukoc Biol (2001) 70(5):783–92. doi: 10.1189/jlb.70.5.783
107. Genestier AL, Michallet MC, Prévost G, Bellot G, Chalabreysse L, Peyrol S, et al. Staphylococcus Aureus Panton-Valentine Leukocidin Directly Targets Mitochondria and Induces Bax-independent Apoptosis of Human Neutrophils. J Clin Invest (2005) 115(11):3117–27. doi: 10.1172/JCI22684
108. Holzinger D, Gieldon L, Mysore V, Nippe N, Taxman DJ, Duncan JA, et al. Staphylococcus Aureus Panton-Valentine Leukocidin Induces an Inflammatory Response in Human Phagocytes Via the NLRP3 Inflammasome. J Leukoc Biol (2012) 92(5):1069–81. doi: 10.1189/jlb.0112014
109. Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, et al. Toxin-Induced Necroptosis Is a Major Mechanism of Staphylococcus Aureus Lung Damage. PloS Pathog (2015) 11(4):e1004820. Miller LS, editor. doi: 10.1371/journal.ppat.1004820
110. Saeed K, Gould I, Espositio S, Ahmad-Saeed N, Ahmed SS, Alp E, et al. Panton–Valentine Leukocidin-Positive Staphylococcus Aureus: A Position Statement From the International Society of Chemotherapy. Int J Antimicrob Agents (2018) 51:16–25. Elsevier B.V. doi: 10.1016/j.ijantimicag.2017.11.002
111. Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy Regulation by P53. Curr Opin Cell Biol (2010) 22:181–5. doi: 10.1016/j.ceb.2009.12.001
112. Livesey KM, Kang R, Vernon P, Buchser W, Loughran P, Watkins SC, et al. p53/HMGB1 Complexes Regulate Autophagy and Apoptosis. Cancer Res (2012) 72(8):1996–2005. doi: 10.1158/0008-5472.CAN-11-2291
113. Zhang Y, Xiong Y. A p53 Amino-Terminal Nuclear Export Signal Inhibited by DNA Damage-Induced Phosphorylation. Science (80- ) (2001) 292(5523):1910–5. doi: 10.1126/science.1058637
114. Mrakovcic M, Fröhlich LF. P53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules (2018) 8(2):14. doi: 10.3390/biom8020014
115. Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a P53-Induced Modulator of Autophagy, Is Critical for Apoptosis. Cell (2006) 126(1):121–34. doi: 10.1016/j.cell.2006.05.034
116. Crighton D, Wilkinson S, Ryan KM. DRAM Links Autophagy to p53 and Programmed Cell Death. Autophagy (2007) 3:72–4. doi: 10.4161/auto.3438
117. Laforge M, Limou S, Harper F, Casartelli N, Rodrigues V, Silvestre R, et al. Dram Triggers Lysosomal Membrane Permeabilization and Cell Death in CD4+ T Cells Infected With HIV. PloS Pathog (2013) 9(5):e1003328. doi: 10.1371/journal.ppat.1003328. Aiken C, editor.
118. Van Der Vaart M, Korbee CJ, Lamers GEM, Tengeler AC, Hosseini R, Haks MC, et al. The DNA Damage-Regulated Autophagy Modulator DRAM1 Links Mycobacterial Recognition Via TLP-MYD88 to Authophagic Defense. Cell Host Microbe (2014) 15(6):753–67. doi: 10.1016/j.chom.2014.05.005
119. Achouiti A, van der Meer AJ, Florquin S, Yang H, Tracey KJ, van ‘t Veer C, et al. High-Mobility Group Box 1 and the Receptor for Advanced Glycation End Products Contribute to Lung Injury During Staphylococcus Aureus Pneumonia. Crit Care (2013) 17(6):R296. doi: 10.1186/cc13162
120. Chu M, Zhou M, Jiang C, Chen X, Guo L, Zhang M, et al. Staphylococcus Aureus Phenol-Soluble Modulins A1-α3 Act as Novel Toll-Like Receptor (TLR) 4 Antagonists to Inhibit HMGB1/TLR4/NF-κB Signaling Pathway. Front Immunol (2018) 9(APR):862. doi: 10.3389/fimmu.2018.00862
121. Yanai H, Matsuda A, An J, Koshiba R, Nishio J, Negishi H, et al. Conditional Ablation of HMGB1 in Mice Reveals its Protective Function Against Endotoxemia and Bacterial Infection. Proc Natl Acad Sci U S A (2013) 110(51):20699–704. doi: 10.1073/pnas.1320808110
122. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated Platelets Present High Mobility Group Box 1 to Neutrophils, Inducing Autophagy and Promoting the Extrusion of Neutrophil Extracellular Traps. J Thromb Haemost (2014) 12(12):2074–88. doi: 10.1111/jth.12710
123. Kloft N, Neukirch C, Bobkiewicz W, Veerachato G, Busch T, Von Hoven G, et al. Pro-Autophagic Signal Induction by Bacterial Pore-Forming Toxins. Med Microbiol Immunol (2010) 199(4):299–309. doi: 10.1007/s00430-010-0163-0
124. Cohen TS, Jones-Nelson O, Hotz M, Cheng L, Miller LS, Suzich J, et al. S. Aureus Blocks Efferocytosis of Neutrophils by Macrophages Through the Activity of its Virulence Factor Alpha Toxin. Sci Rep (2016) 6(1):35466. doi: 10.1038/srep35466
125. Liu PF, Cheng JS, Sy CL, Huang WC, Yang HC, Gallo RL, et al. IsaB Inhibits Autophagic Flux to Promote Host Transmission of Methicillin-Resistant Staphylococcus Aureus. J Invest Dermatol (2015) 135(11):2714–22. doi: 10.1038/jid.2015.254
126. Bravo-Santano N, Stölting H, Cooper F, Bileckaja N, Majstorovic A, Ihle N, et al. Host-Directed Kinase Inhibitors Act as Novel Therapies Against Intracellular Staphylococcus Aureus. Sci Rep (2019) 9(1):4876. doi: 10.1038/s41598-019-41260-8
127. Bravo-Santano N, Capilla-Lasheras P, Mateos LM, Calle Y, Behrends V, Letek M. Identification of Novel Targets for Host-Directed Therapeutics Against Intracellular Staphylococcus Aureus. Sci Rep (2019) 9(1):15435. doi: 10.1038/s41598-019-51894-3
128. Nguyen HA, Grellet J, Paillard D, Dubois V, Quentin C, Saux MC. Factors Influencing the Intracellular Activity of Fluoroquinolones: A Study Using Levofloxacin in a Staphylococcus Aureus THP-1 Monocyte Model. J Antimicrob Chemother (2006) 57(5):883–90. doi: 10.1093/jac/dkl079
129. Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, et al. Combined MTOR and Autophagy Inhibition: Phase I Trial of Hydroxychloroquine and Temsirolimus in Patients With Advanced Solid Tumors and Melanoma. Autophagy (2014) 10(8):1391–402. doi: 10.4161/auto.29119
130. Dasgupta B, Seibel W. Compound C/Dorsomorphin: its Use and Misuse as an AMPK Inhibitor. In: Neumann D, Viollet B, (eds). Methods in Molecular Biology. New York, NY: Humana Press Inc (2018). p. 195–202.
131. Zhu Y, Li H, Ding S, Wang Y. Autophagy Inhibition Promotes Phagocytosis of Macrophage and Protects Mice From Methicillin-Resistant Staphylococcus Aureus Pneumonia. J Cell Biochem (2018) 119(6):4808–14. doi: 10.1002/jcb.26677
132. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A Key Role for Autophagy and the Autophagy Gene Atg16l1 in Mouse and Human Intestinal Paneth Cells. Nature (2008) 456(7219):259–63. doi: 10.1038/nature07416
133. Maurer K, Reyes-Robles T, Alonzo F, Durbin J, Torres VJ, Cadwell K. Autophagy Mediates Tolerance to Staphylococcus Aureus Alpha-Toxin. Cell Host Microbe (2015) 17(4):429–40. doi: 10.1016/j.chom.2015.03.001
134. Darouiche RO, Hamill RJ. Antibiotic Penetration of and Bactericidal Activity Within Endothelial Cells. Antimicrob Agents Chemother (1994) 38(5):1059–64. doi: 10.1128/AAC.38.5.1059
Keywords: Staphylococcus aureus, intracellular survival, autophagy, neutrophils, apoptosis
Citation: Vozza EG, Mulcahy ME and McLoughlin RM (2021) Making the Most of the Host; Targeting the Autophagy Pathway Facilitates Staphylococcus aureus Intracellular Survival in Neutrophils. Front. Immunol. 12:667387. doi: 10.3389/fimmu.2021.667387
Received: 12 February 2021; Accepted: 28 May 2021;
Published: 16 June 2021.
Edited by:
Amy Rasley, United States Department of Energy (DOE), United StatesReviewed by:
Roberta Olmo Pinheiro, Instituto Oswaldo Cruz (Fiocruz), BrazilCopyright © 2021 Vozza, Mulcahy and McLoughlin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rachel M. McLoughlin, cmFjaGVsLm1jbG91Z2hsaW5AdGNkLmll
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.