 Ananya Sen
Ananya Sen James A. Imlay
James A. Imlay

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 April 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.667343
This article is part of the Research Topic The Role of Reactive Oxygen Species in Protective Immunity View all 13 articles
Microbes rely upon iron as a cofactor for many enzymes in their central metabolic processes. The reactive oxygen species (ROS) superoxide and hydrogen peroxide react rapidly with iron, and inside cells they can generate both enzyme and DNA damage. ROS are formed in some bacterial habitats by abiotic processes. The vulnerability of bacteria to ROS is also apparently exploited by ROS-generating host defense systems and bacterial competitors. Phagocyte-derived can toxify captured bacteria by damaging unidentified biomolecules on the cell surface; it is unclear whether phagocytic H2O2, which can penetrate into the cell interior, also plays a role in suppressing bacterial invasion. Both pathogenic and free-living microbes activate defensive strategies to defend themselves against incoming H2O2. Most bacteria sense the H2O2 via OxyR or PerR transcription factors, whereas yeast uses the Grx3/Yap1 system. In general these regulators induce enzymes that reduce cytoplasmic H2O2 concentrations, decrease the intracellular iron pools, and repair the H2O2-mediated damage. However, individual organisms have tailored these transcription factors and their regulons to suit their particular environmental niches. Some bacteria even contain both OxyR and PerR, raising the question as to why they need both systems. In lab experiments these regulators can also respond to nitric oxide and disulfide stress, although it is unclear whether the responses are physiologically relevant. The next step is to extend these studies to natural environments, so that we can better understand the circumstances in which these systems act. In particular, it is important to probe the role they may play in enabling host infection by microbial pathogens.
Life evolved 3.8 billion years ago in an anoxic world. The biochemical pathways of these primordial organisms were based upon iron-cofactored enzymes, as this transition metal is adept at both redox and ligand-exchange processes. One billion years later, the appearance of photosystem II began the release of diatomic oxygen into the atmosphere. The oxygen levels in the atmosphere remained relatively low for another billion years, because photosynthetically generated O2 was quickly consumed through its chemical reduction by environmental ferrous iron and sulfide. Only later, once these reductants had been largely titrated, did oxygen accumulate to higher concentrations (1). However, when it did, O2 created an environment that was—and remains—incompatible with extant organisms, which have inherited their iron-centric metabolic plans from their anoxic forbears.
In part the problem is that molecular oxygen oxidizes ferrous iron to insoluble ferric hydroxide precipitates, making it difficult for cells to acquire enough iron to charge their enzymes. That problem has been substantially ameliorated by the evolution of a variety of iron-import tactics (2). However, in addition, oxygen is kinetically active as a univalent oxidant (3). It disrupts the metabolism of anaerobic organisms by oxidizing their low-potential metal cofactors, thereby inactivating key enzymes, and by adducting the radical-based enzymes that play specialized roles in metabolism. Contemporary anaerobes have not solved this problem: In order to optimize their anaerobic growth, they continue to rely upon enzymes that are directly disrupted by oxygen, and so these microbes are constrained to anoxic niches (4).
In contrast, organisms that committed to life in oxic environments were able to dispense with low-potential catalytic strategies, and they employ enzymes that molecular oxygen does not damage at an important rate. Yet aerobes have residual problems with oxygen. Molecular oxygen is a passable univalent oxidant, and inside cells it adventitiously steals electrons from the cofactors of redox-active enzymes (5, 6). The transfer of a single electron results in the formation of superoxide (Figure 1A); the transfer of two electrons results in the formation of hydrogen peroxide. Both these species are more-potent univalent oxidants than molecular oxygen itself, and if left unchecked they can oxidize the exposed iron cofactors of enzymes that are found throughout metabolism. Further, a secondary reaction between hydrogen peroxide and cellular iron pools creates hydroxyl radicals (Figure 1), which are extremely potent species that can directly oxidize all cellular biomolecules (7). This vulnerability to partially reduced oxygen species (ROS) is universal among contemporary organisms. This review specifically aims to describe the strategies that are used by organisms to defray the toxicity of hydrogen peroxide—and to highlight the circumstances in which these defenses may not be adequate.
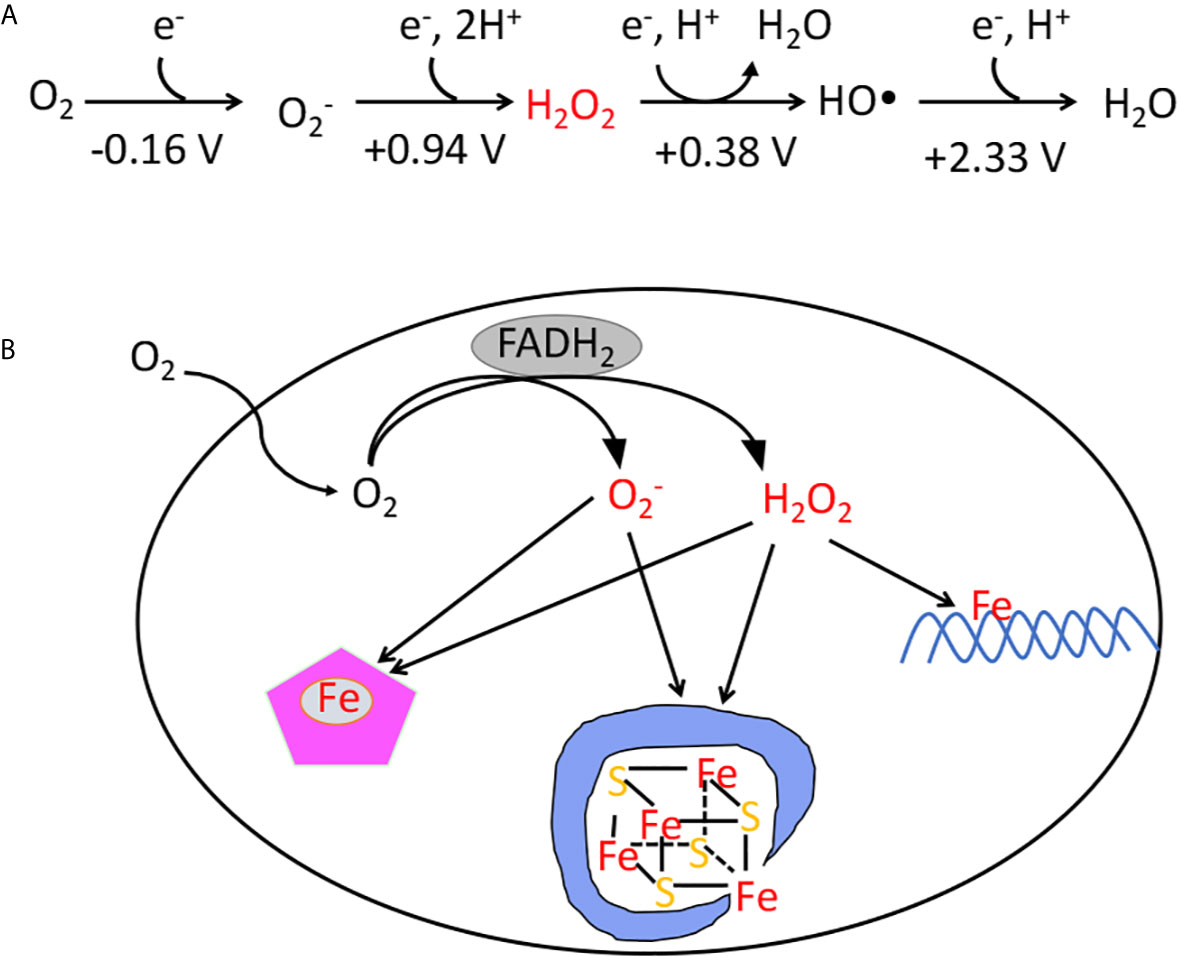
Figure 1 (A) The reduction potential of oxygen and reactive oxygen species. The standard reduction potentials (pH 7) indicate that unlike O2, superoxide, hydrogen peroxide, and hydroxyl radicals are potent univalent oxidants. The standard concentration of oxygen is regarded as 1 M. (B) The classes of damage caused by intracellular and H2O2. The transfer of electrons from redox enzymes to oxygen generates superoxide and hydrogen peroxide. Both species can oxidize the solvent-exposed iron centers of mononuclear iron enzymes and [4Fe-4S] dehydratases. Additionally, H2O2 directly reacts with the intracellular iron pool, which is loosely associated with biomolecules, including DNA. The reaction generates hydroxyl radicals, which can damage DNA.
The significance of hydrogen peroxide (H2O2) was first suggested by the discovery in 1900 of an enzyme devoted to degrading it: catalase (8). The discoverer, Oscar Loew, noted that it is found in virtually all tissues, and he made the inference that H2O2 was likely a by-product of metabolism that, if not removed, must be toxic to cells. Seventy years later Joe McCord and Irwin Fridovich chanced upon an enzyme that degrades superoxide () (9). Subsequent work has extended the cohort of scavenging enzymes to include peroxidases and superoxide reductases, and it has confirmed that virtually no organism lacks the ability to degrade H2O2 and .
The model bacterium Escherichia coli contains two superoxide dismutases in its cytoplasm and one in its periplasm. Its cytoplasm also features both an NADH peroxidase (AhpCF) and two catalases (10) (Figure 2). Interestingly, some of these enzymes take advantage of the fact that iron can react with and H2O2: The original superoxide dismutase was likely an iron-dependent SOD, and most catalases use heme to degrade H2O2. Looking more broadly, similar scavenging systems are distributed through all biological kingdoms and in most cellular compartments, including the mitochondria, peroxisomes, and cytoplasm of eukarya.
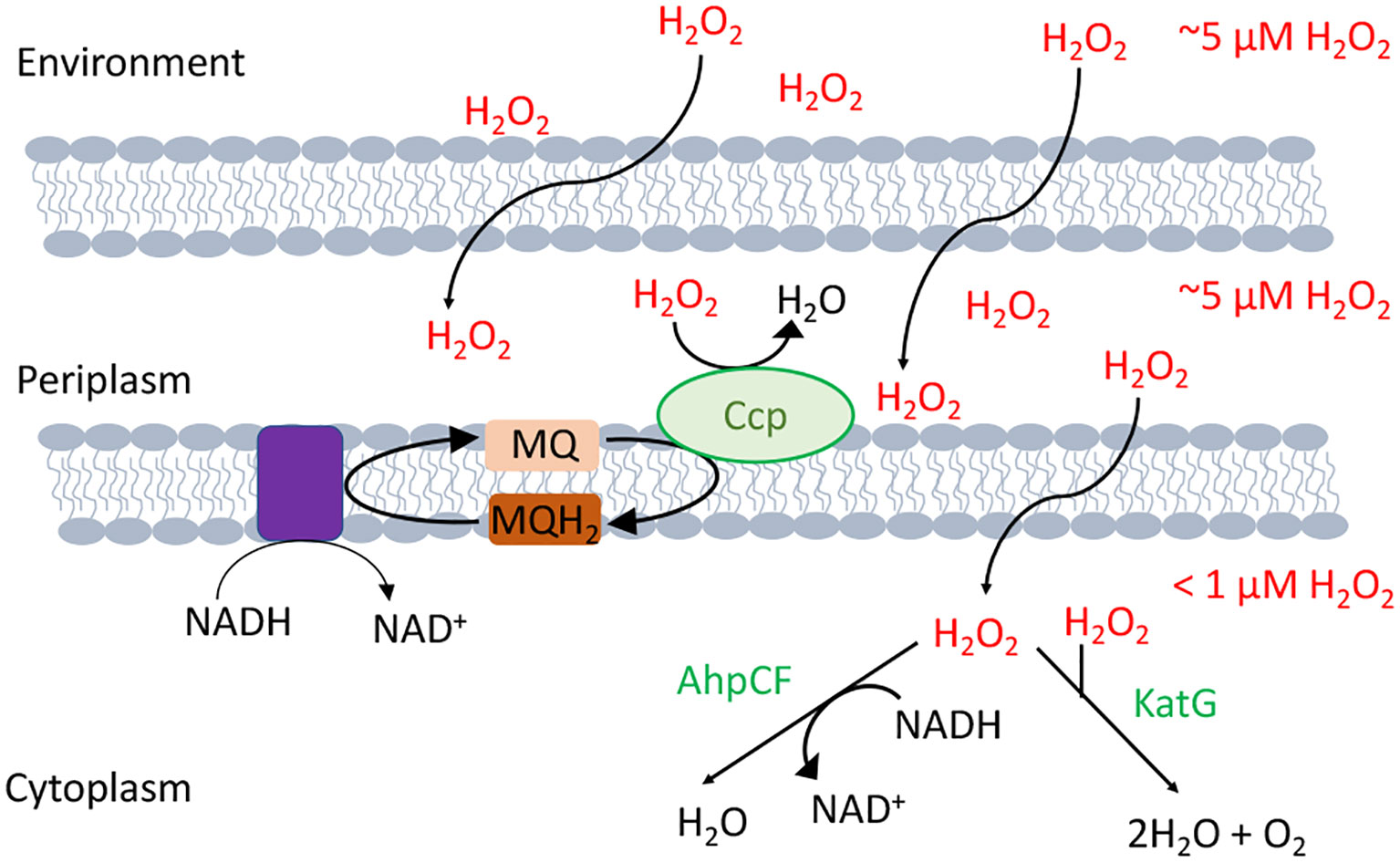
Figure 2 H2O2-scavenging enzymes in E. coli. Environmental H2O2 gradually diffuses into the cytoplasm, where it is degraded by NADH peroxidase (AhpCF) and catalase (KatG). Both are induced by OxyR. Cytoplasmic H2O2 is therefore substantially lower in concentration than is extracellular H2O2. Under hypoxic conditions OxyR also induces the periplasmic cytochrome c peroxidase (Ccp), which allows the respiratory chain to employ H2O2 as a terminal oxidant. Because H2O2 rapidly crosses through OM porins, and Ccp activity is moderate, the periplasmic H2O2 concentration is likely equivalent to that outside the cell.
The importance of these enzymes was revealed by genetic studies of E. coli: Mutants that lack cytoplasmic SODs, or that lack Ahp and catalases, were found to be unable to grow under oxic conditions in a standard glucose medium (10, 11). The observation confirmed Loew’s postulate, proving both that these species are generated internally and that if not scavenged they will cripple cellular metabolism. Direct observation of production is not possible, but mutants that cannot degrade H2O2 release it into growth media at a rate that connotes an internal production rate of 10 µM/sec (12). Measurements show that as little as 0.5 µM intracellular H2O2 is sufficient to poison select biosynthetic pathways (13–15). The titers and kinetics of the scavenging enzymes are sufficient to suppress internal concentrations to about 50 nM—low enough to enable metabolism to operate without bottlenecks (12).
The orbital structure of O2 restricts it to accepting a single electron at a time, and its reduction potential (- 0.16 V) is too modest for it to directly oxidize most biomolecules (3). However, O2 can accept electrons from electron donors such as metal centers, flavins, and quinones. These are all prominent electron carriers in the E. coli respiratory chain, yet the rate at which cells produce endogenous H2O2 did not substantially diminish in mutants that lacked the respiratory enzymes, suggesting that in this bacterium and H2O2 are primarily produced by the accidental autooxidation of non-respiratory flavoproteins (5, 6) (Figure 1). These proteins are found throughout metabolism, and many, including glutathione reductase, lipoamide dehydrogenase, and glutamate synthase, have been shown to release ROS in vitro (16–18). The small size of O2 prevents its exclusion from most active sites, and its collision with reduced flavins triggers the consecutive transfer of one or two electrons, generating or H2O2, respectively (19). The rate is naturally proportionate to collision frequency, meaning that ROS formation is more rapid in highly oxic environments. This math presumably underlies the observation that most organisms cannot tolerate oxygen levels that substantially exceed those of their natural habitat.
The phenotypes of SOD mutants and of catalase/peroxidase mutants enabled investigators to track down the specific injuries that these ROS create. The mutants can grow at wild-type rates under anoxic conditions, but they require supplementation with aromatic and branched-chain amino acids if they are to grow in oxic media (11, 13–15, 20). They are also unable to use any carbon source, such as acetate, that required a fully functional TCA cycle (21, 22). Further scrutiny identified particular enzymes whose damage resulted in these defects.
The aromatic biosynthesis defect derives from the ability of H2O2 to oxidize the Fe(II) cofactor of the first enzyme of the pathway, DAHP synthase (15). The resulting Fe(III) atom dissociates. The role of the iron atom is both to bind substrate and to stabilize an oxyanion intermediate in the catalytic cycle; therefore, the resultant apoenzyme is completely inactive and the pathway fails. Other mononuclear Fe(II) enzymes such as ribulose-5-phosphate 3-epimerase, peptide deformylase, threonine dehydrogenase, and cytosine deaminase are similarly damaged (13, 14). Notably, the reaction between Fe(II) and H2O2 also generates a hydroxyl radical. If the catalytic Fe(II) atom is coordinated by a cysteine side chain, the hydroxyl radical reacts immediately with this sacrificial residue, creating a sulfenic acid (Figure 3) (13). Cellular thioredoxins and glutaredoxins can reduce this moiety back to a native cysteine residue, thereby allowing Fe(II) to bind and the activity to be restored (13). In contrast, when H2O2 oxidizes the Fe(II) of enzymes that lack such a residue, the nascent hydroxyl radical oxidizes other active-site ligands, creating lesions that are irreversible. These reactions are one source of the protein carbonylation that can be detected in H2O2-stressed cells (13).
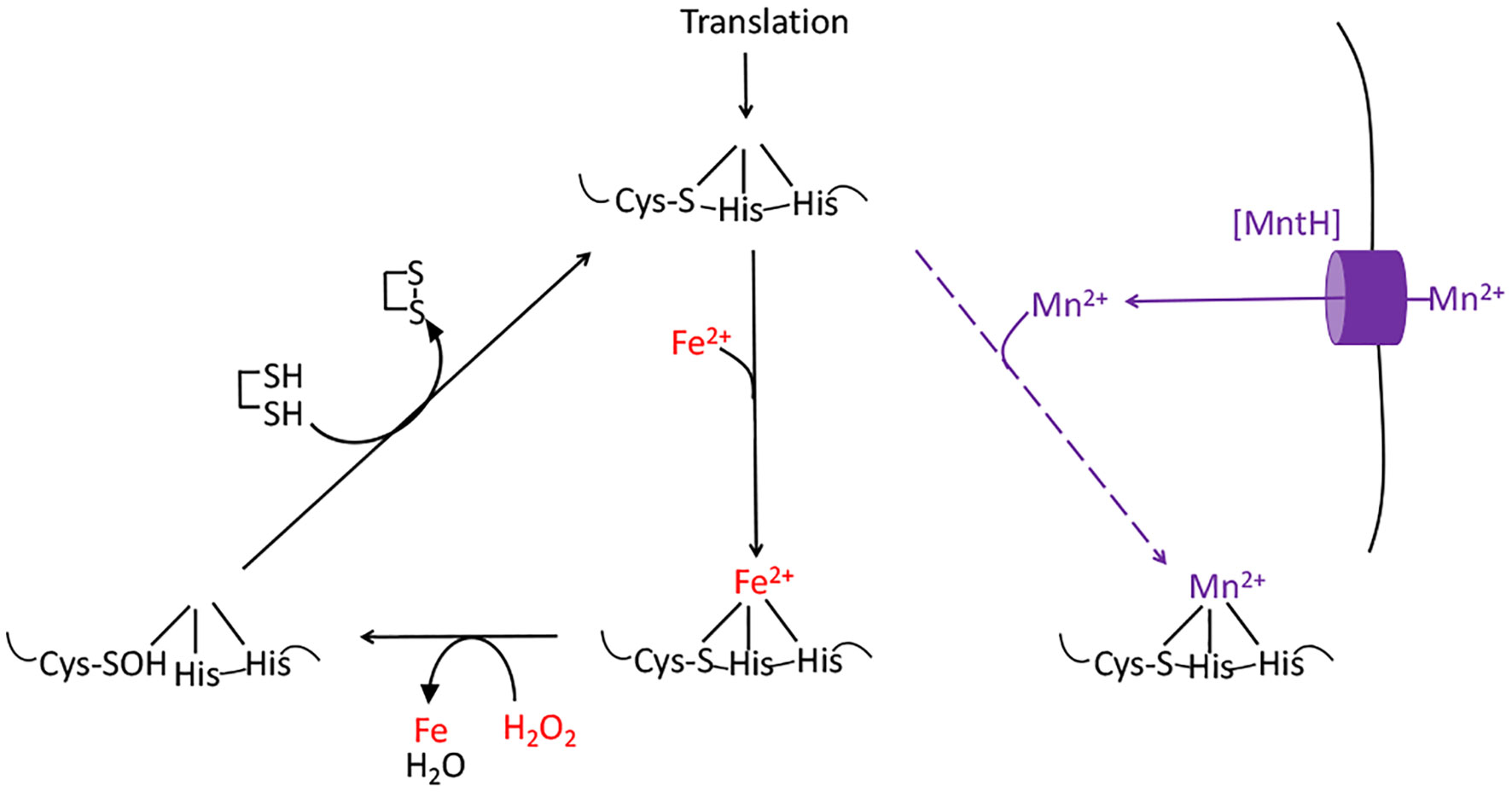
Figure 3 The damage caused to mononuclear iron proteins by hydrogen peroxide. H2O2 directly oxidizes the solvent-exposed Fe(II) cofactor, which then dissociates. The ferryl (FeO2+) species that is formed in this reaction can directly oxidize the polypeptide ligands to the iron atom, irreversibly inactivating the enzyme. However, if a cysteine residue coordinates the iron, it will quench the ferryl radical (as shown). The enzyme activity can then be restored by reduction of the cysteine sulfenate residue, probably by thioredoxins. OxyR induces the MntH manganese importer, allowing the proteins to be metallated with Mn(II), which provides activity and does not react with H2O2.
The branched-chain biosynthetic defects as well as the TCA-cycle defects are caused by the oxidation of the [4Fe-4S] clusters of dehydratase enzymes (20, 22–26). A solvent-exposed iron atom of these clusters binds substrate directly, activating it for deprotonation and subsequently completing the dehydration by abstracting a hydroxide anion. But the same exposed iron atom can be oxidized by hydrogen peroxide to an unstable [4Fe-4S]3+ state (Figure 4); this valence is unstable, and the cluster quickly disintegrates into a [3Fe-4S]+ form that lacks the catalytic, solvent-exposed iron atom (24). Interestingly, the hydroxyl radical that is formed during this process pulls a second electron from the iron-sulfur cluster; thus, a hydroxide anion, rather than a hydroxyl radical, is released into the active site, and polypeptide oxidation is avoided. Cells continuously repair these damaged clusters, so that the steady-state activity of these enzymes reflects the balance between the oxidation and repair rates (21, 27, 28).
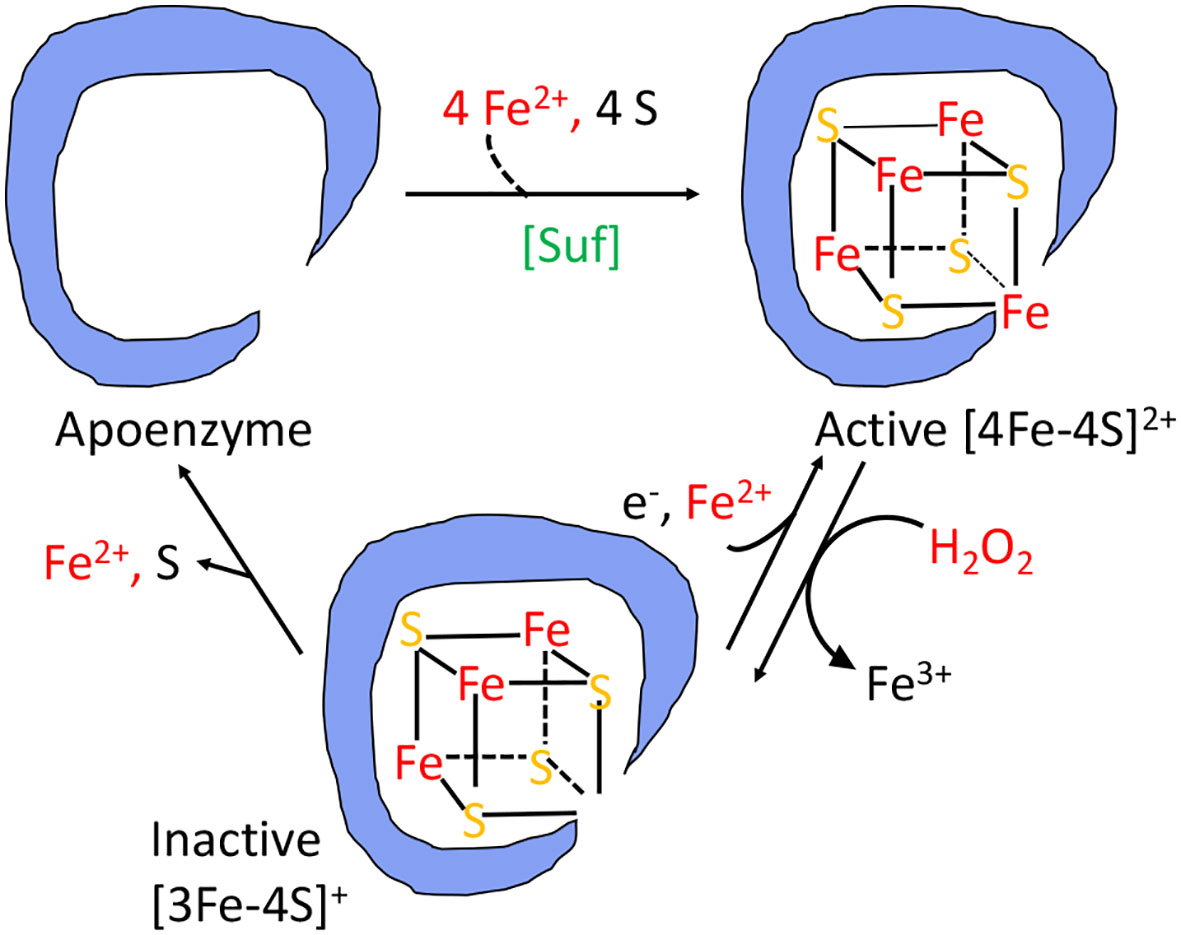
Figure 4 The damage caused to [4Fe-4S]2+ cluster enzymes by hydrogen peroxide. The catalytic Fe atom of the dehydratase enzyme reacts with H2O2 and dissociates, leaving behind an inactive [3Fe-4S]+ cluster. That cluster can be reactivated by reduction and remetallation. In some dehydratases the cluster completely disintegrates to form an apoenzyme. OxyR induces the Suf system to rebuild a functional holoenzyme.
In addition to the metabolic defects described above, H2O2 can also react with the cytoplasmic pool of loose iron that is used to metallate nascent iron-dependent enzymes (Figure 1B). Iron is sticky, and this pool is thought to adhere to a wide variety of biomolecules, including the surface of nucleic acids (29). DNA thereby acts as a locus of hydroxyl radical production, and so DNA damage is a universal consequence of H2O2 stress (30–33). Some oxidative base lesions are mutagenic; others comprise replication blocks. All organisms therefore wield enzymes devoted to the excision or recombinational repair of oxidative lesions. Even though the level of endogenous H2O2 is well-controlled in scavenger-proficient E. coli, the rate of DNA oxidation remains high enough that mutants lacking these repair pathways cannot grow in oxic environments (34, 35).
Thus, organisms that dwell in oxic habitats can do so only because they have acquired an array of both scavenging and repair functions. The level of these enzymes is high and their synthesis is costly; accordingly, their titers have been calibrated to barely withstand the amount of stress commensurate with the oxygen level of the native environment (36, 37). This arrangement is successful under routine growth conditions. However, we shall see that it becomes inadequate if special circumstances elevate the production of ROS.
Both environmental photochemistry and chemical redox reactions generate (38), but the steady-state level of formed in this way is unlikely to be high enough to pose a risk for cells. Notably, the known targets of are iron enzymes that are cytoplasmic, and is a charged species that cannot cross membranes to get at them (39, 40). However, both microbes and higher organisms have evolved mechanisms by which they can use to poison unwanted competitors.
Mammals, plants, and amoebae have all weaponized an NADPH oxidase to kill bacteria (41, 42). Mammalian phagocytes engulf microbial invaders and spray them with superoxide that is formed by an inducible NADPH oxidase (Figure 5). The importance of this enzyme is reflected by the observation that humans and mice that lack it are vulnerable to infections (43, 44). However, it is still unclear how the phagocytic ROS production inhibits microbial growth. The acidic environment inside the phagosome can partially protonate , resulting in a neutral species that in principle can penetrate captive bacteria; however, Salmonella enterica mutants lacking the periplasmic superoxide dismutase are hypersensitive, suggesting that does not gain access into the cytosol and instead acts on a target on the cell surface or in the periplasm (45, 46). Because protonated HO2 is a better oxidant than , it is possible that the acidity of the phagosome expands the range of biomolecules that superoxide can damage. The target has not yet been identified. A key difficulty is that in vitro systems have been unable to match the micromolar doses of superoxide (46, 47) that are sustained in the phagosome.
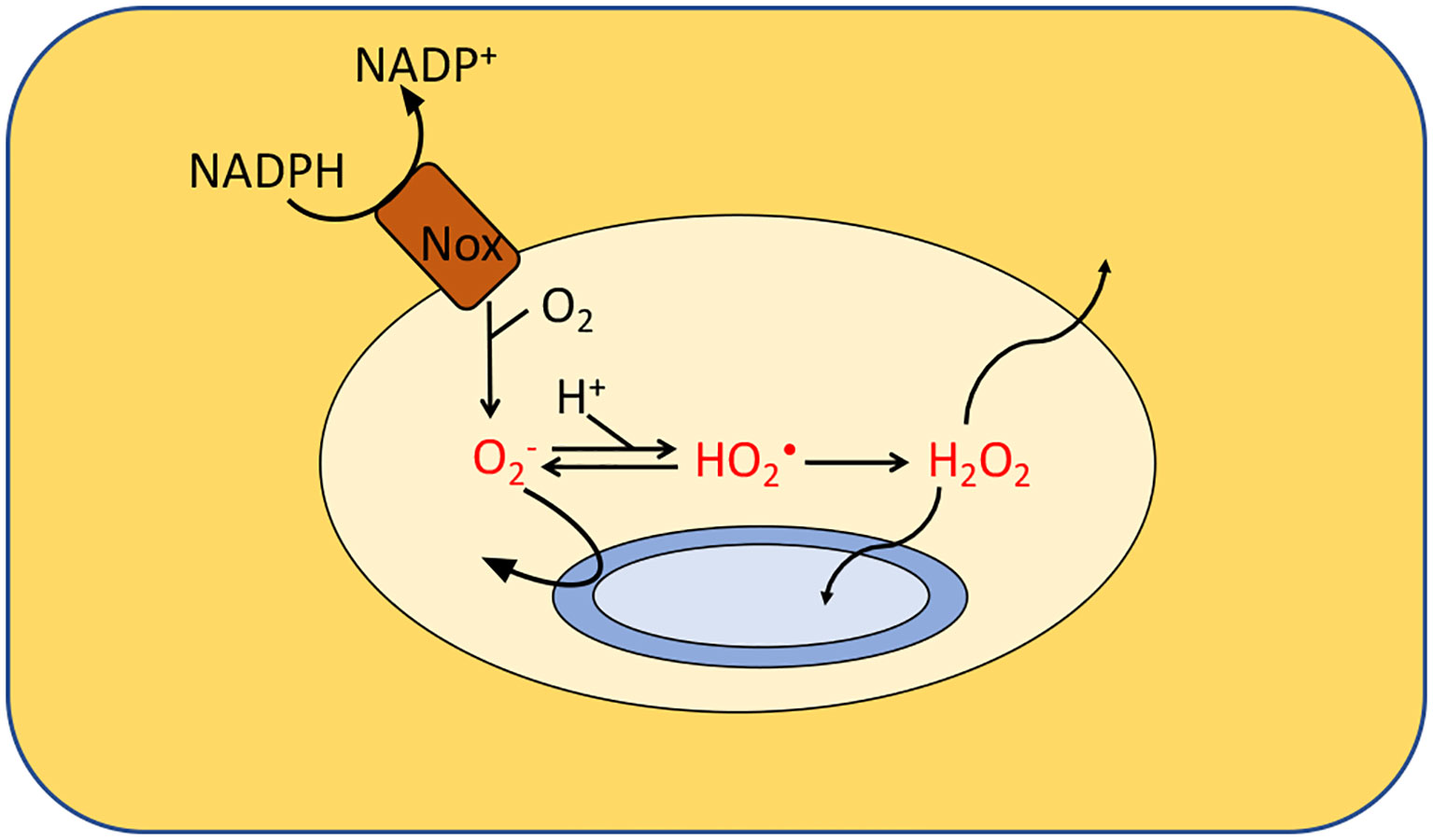
Figure 5 Formation of ROS by phagosomes. NADPH oxidase (Nox) generates superoxide which cannot penetrate the cytoplasmic membranes of the engulfed bacteria. It is believed that either superoxide or its protonated form injures extracytoplasmic targets. Additionally, membrane-permeable H2O2 is generated through dismutation. Calculations suggested that the levels of , HO2• and H2O2 in isolated macrophages range from 10–50 μM, 0.1–4 μM and 1–4 μM, respectively, depending upon phagosomal pH. Modeling predicts a similar H2O2 concentration inside neutrophils. The H2O2 levels would rise, however, if it accumulates it the surrounding tissue.
Bacteria also have found a way to impose stress upon competitors—and in this case the is aimed at the cytoplasm. A wide range of bacteria (and plants) secrete redox-cycling antibiotics (48–51). These are primarily soluble quinones and phenazines; they penetrate target cells, oxidize their redox enzymes, and transfer the electrons to oxygen. Enteric bacteria protect themselves from these compounds by activating the SoxRS regulon (52, 53). Its components elevate the titer of cytoplasmic SOD, pump out the drugs, and modify the cell envelope to diminish their entry (54).
Over the past dozen years microbiologists have examined the possibility that other bacterial stresses might also owe their potency, in part, to oxidative stress. The clinical antibiotics ampicillin, kanamycin, norfloxacin and trimethoprim have been particular foci of these studies, but similar hypotheses have been ventured for metal overload, nanoparticles, solvent stress, toxin/antitoxin systems, and many others (55). Key observations have been that the stressed cells accumulate oxidized forms of cell-penetrating dyes, which are thought to be oxidized by hydroxyl radicals; that cell death is slowed by the administration of thiol compounds, which might scavenge ROS, and by iron chelators that would block hydroxyl-radical formation; and that toxicity is diminished in mutants whose TCA cycle is blocked, ostensibly diminishing the rate of respiration and any associated ROS formation (56). However, this interpretation has been challenged (55, 57–59). Most of these protectants also have the capacity to slow metabolism, and it is well known that the efficacy of antibiotic action depends upon a robust growth rate. Moreover, tests of antibiotic action did not detect oxidative damage to ROS-sensitive enzymes or to DNA; the rate of H2O2 formation (measured in scavenging mutants) was not accelerated; and the E. coli response to H2O2 stress (below) was not triggered. Finally, no clear model has emerged to explain how such diverse stresses could create toxic levels of ROS. More work must be done to resolve these contradictory observations.
H2O2 is chemically formed in habitats though abiotic reactions between sulfur and oxygen at oxic/anoxic surfaces and the photochemical reduction of oxygen by chromophores (60–63). Levels can reach 1 µM in the ocean (64, 65). It can also be produced as part of the plant wound response, during the inflammatory response of mammalian hosts—and, notably, as a primary metabolic by-product of many lactic acid bacteria (66–70). The latter organisms often lack respiratory chains and use the two-electron reduction of molecular oxygen to recycle reduced NADH, thereby indirectly improving the ATP yield of what is otherwise a fermentative process. It seems likely that the excreted H2O2 may suppress the growth of competitors. The ability of lactic-acid bacteria to tolerate their own H2O2 is impressive: They can achieve high densities in lab cultures in which millimolar H2O2 has accumulated. This tolerance appears to be due to the absence of oxidant-sensitive dehydratases and mononuclear Fe(II) enzymes. This adaptation is not without a price: These bacteria are unable to synthesize many amino acids, and they lack a TCA cycle and the improved energy yield that comes with it.
Unlike superoxide, H2O2 is an uncharged, albeit polar, molecule that can cross cell membranes. Because this process is relatively slow, when bacteria venture into environments containing extracellular H2O2, the high activities of intracellular catalases and peroxidases succeed at lowering internal H2O2 concentrations below that of the external environment (12). The transmembrane gradient for E. coli has been estimated to be 5- to 10-fold between the external world and the cytoplasm. Indeed, although 0.5 µM internal H2O2 is sufficient to impair the growth of this bacterium in lab cultures, external concentrations of up to 5 µM seem to be tolerated without an overt growth defect (60). Thus the limited permeability of membranes to H2O2 is essential to the efficacy of scavenging enzymes and to the ability of bacteria to grow in many habitats.
Of great interest to biologists is the role that phagocyte-derived H2O2 may play in suppressing microbial infection (Figure 5). The that is produced by host NADPH oxidase will dismutate, either spontaneously or via enzymic catalysis, to generate H2O2. The ability of H2O2 to cross membranes likely enables it to enter phagocytosed bacteria—but it also allows it to diffuse across the phagosomal membrane, into the producing cell, and potentially out into extracellular environments. Modeling suggests that this effusion sharply limits the amount of H2O2 inside the macrophage phagosome, despite the rapid rate at which it is formed. Estimates are that the steady-state level falls well below 10 µM (46). Such doses may be enough to induce stress responses in the captive bacteria, but they are unlikely to be lethal. The major caveat to this analysis is that it presumes that the environment acts as a one-way sink for the H2O2. However, if H2O2 accumulates within inflamed tissue, the H2O2 flow is bidirectional, and the level that accumulates may in principle be far higher. Clearly, this question cries out for direct measurements of H2O2 in vivo.
The rate of H2O2 production in neutrophils is substantially higher than that in macrophages—but the fact that H2O2 is a substrate of myeloperoxidase has once again been projected to cap the level at which it can accumulate (47). In sum, although at first blush it seems a no-brainer that H2O2 would contribute mightily to the killing actions of these cells, that point is not yet resolved.
When external levels of H2O2 exceed a few micromolar, its flux into microbes threatens to elevate its internal concentration to toxic levels, despite the action of scavenging enzymes. To cope, virtually all microbes possess inducible stress responses that are focused upon H2O2. The paradigmatic system is the OxyR response of E. coli (71). OxyR is a H2O2-activated transcription factor. It is not activated merely by movement of E. coli into oxic environments, and oxyR mutants are capable of normal aerobic growth. However, OxyR is activated when exogenous H2O2 accumulates to ~ 0.2 μM in the cytoplasm, which is achieved by about 3 µM external H2O2 (60, 72). Null mutants cannot grow at these levels of environmental H2O2.
The OxyR protein contains a sensory cysteine residue (C199) that is directly oxidized by H2O2, generating a sulfenic acid (-SOH) (73). As a result, the residue moves from the hydrophobic pocket in which it is buried and swings toward the C208 residue, which then condenses to form a disulfide bond (Figure 6). This bond locks OxyR into its activated conformer, and its DNA binding ability differs from that of the reduced protein. In E. coli the reduced form has little transcriptional impact upon most genes, but the oxidized form recruits RNA polymerase and thereby activates the expression of genes that possess an OxyR binding site. In many other bacteria, reduced OxyR acts as a repressor, and upon its oxidation it releases the DNA, stimulating gene expression (74, 75). In still other bacteria gene expression is both repressed by the reduced form and activated by the oxidized form. The conformational change is manifested by an alteration of its DNA footprint (76).
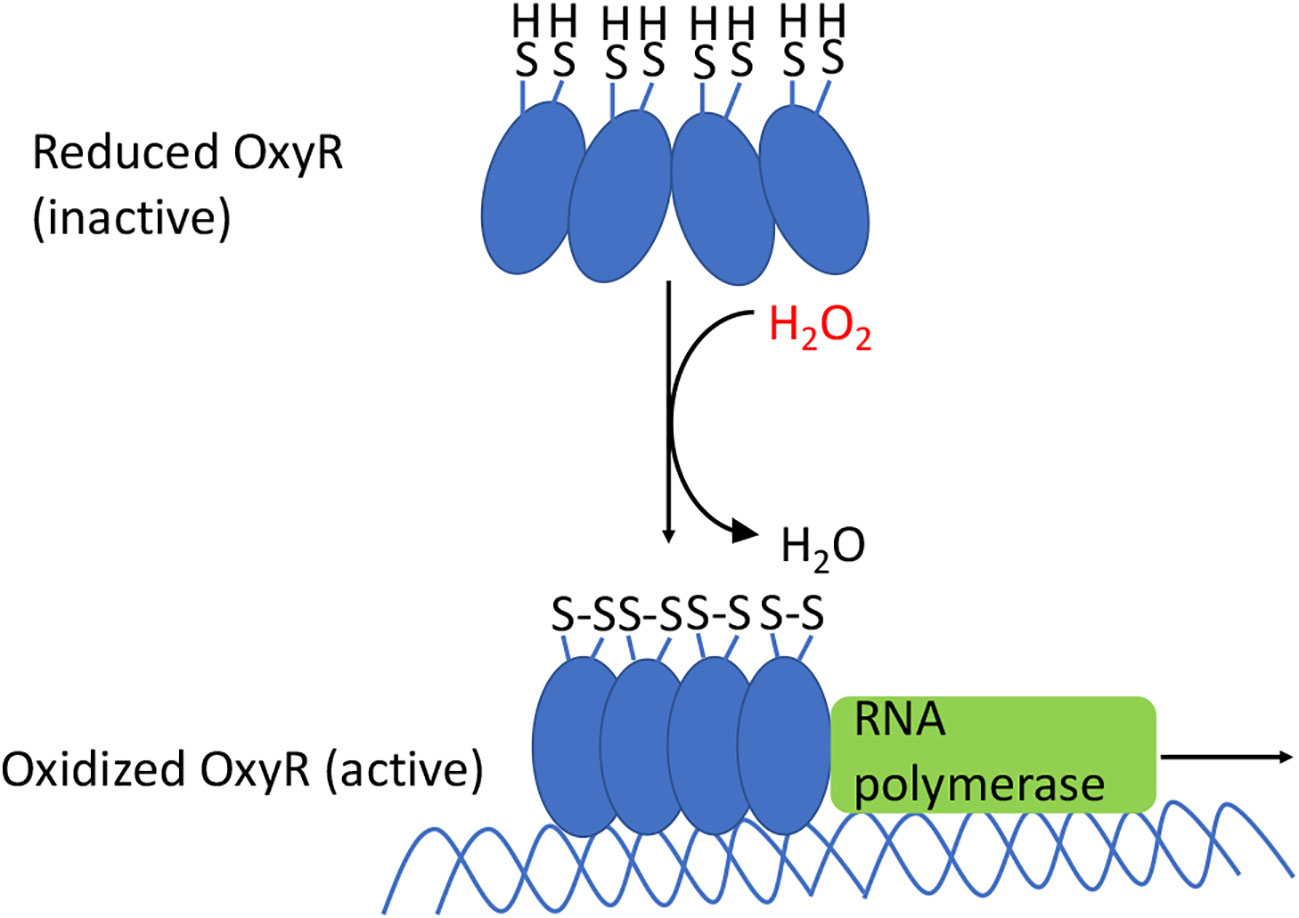
Figure 6 OxyR activation in E. coli. The oxidation of the sensory C199 cysteine by H2O2 leads to the formation of a disulfide bond between C199 and C208. The resulting conformational change causes OxyR to bind as a tetramer to the promoter regions, which recruits RNA polymerase, and results in the transcription of genes in the OxyR regulon. In many other bacteria the reduced form also binds DNA, albeit in an elongated conformation that represses transcription; oxidation again converts it to a transcriptional activator.
Free cysteine reacts very slowly with H2O2 (2 M-1 s-1), and typical cysteine residues of proteins do, too (77). Yet in OxyR the sensing cysteine residue reacts with a rate constant of 105 allowing it to detect micromolar H2O2 in seconds (72, 78). In that respect the hyperreactive cysteine of OxyR resembles the catalytic cysteine residue of thiol-based peroxidases, including AhpC of E. coli. In the latter enzyme an adjacent cationic residue facilitates the deprotonation of cysteine, which provides an order-of-magnitude improvement in its reactivity with H2O2 (79). A plausible explanation for the remaining enhancement is that the nucleophilic cysteine is arranged in a large hydrophobic cleft. Surrounding hydrogen bonds could polarize the dioxygen bond, making it vulnerable to attack, and one of the residues can protonate the hydroxide leaving group, pulling the reaction forward. As a result, thiol-based peroxidases have a rate constant of 107M-1 s-1, which is appropriate for their physiological role (80, 81). A similar physical arrangement may explain the high rate constant of OxyR as well.
It follows that the activation of OxyR is an excellent marker of H2O2 stress. This effect can be tracked by monitoring the expression of OxyR-controlled genes—or by visualizing the intrinsic fluorescence of HyPer, an engineered chimera of OxyR and yellow-fluorescent protein (82). HyPer is as responsive to H2O2 as is OxyR itself, and its oxidation status can be visualized either by microscopy or flow cytometry. Importantly, two-wavelength analysis can correct for variable HyPer content in different samples, thereby avoiding loading artifacts that can arise when redox-active dyes are employed as ROS sensors.
When E. coli is stressed by an influx of H2O2, activated OxyR stimulates the transcription of over two dozen genes (83). These mainly fall into three categories: proteins that reduce the H2O2 concentration, proteins that shrink the iron pool, and proteins that deal with the damage that H2O2 produces (Table 1). The peroxidase AhpCF and the catalase KatG are each induced more than 10-fold in order to scavenge H2O2 (Figure 2). Why two enzymes? AhpCF is an efficient scavenger when the H2O2 concentration is less than 10 μM and the cell is well-fed (10). However, AhpCF requires NADH as a reductant, and so its activity becomes limited when catabolic substrates are scarce. In contrast, catalases do not require reductants, and they can degrade H2O2 faster than Ahp. However, catalases are problematic when the H2O2 concentrations are low, since their two-step catalytic cycle can stall with the heme in its intermediate ferryl radical form. This species is a potent oxidant, and unless it is quenched by a reductant, it can abstract electrons from the surrounding polypeptide and inactivate the enzyme (81). The KatG catalase of E. coli—and of many other bacteria—has a channel that apparently enables small-molecule reductants to approach the active site and quench the high-valence heme (84); for this reason the enzyme is denoted a catalase/peroxidase, though the peroxidase activity by itself is too slow to comprise an efficient scavenging mechanism (85).
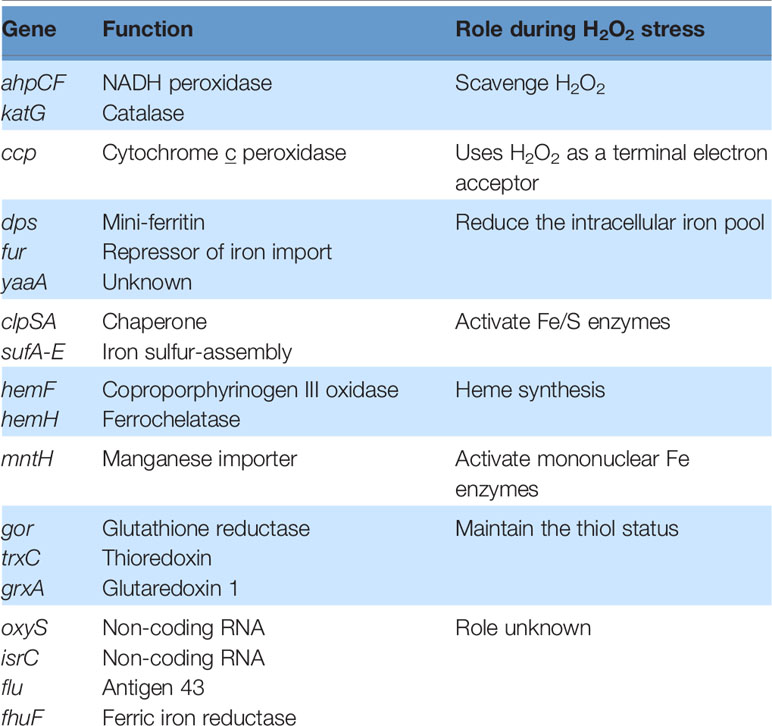
Table 1 Genes induced by OxyR during hydrogen peroxide stress in E. coli.
When H2O2 levels are high, the existential threat to bacteria is that DNA oxidation will prove lethal. This damage is driven by the intracellular iron pool (86), and so the OxyR system employs several mechanisms to diminish it (Figure 7). Dps, a dodecameric mini-ferritin, is induced to sequester unincorporated iron in its hollow core (87–89). This action requires that loose Fe(II) be oxidized to Fe(III), and for this reaction Dps apparently uses H2O2 as a co-substrate. One benefit is that Dps will stop storing iron when H2O2 concentrations drop. The iron-uptake repressor Fur is also induced (90). In unstressed cells Fur:Fe(II) complexes signal that the cell has sufficient iron, and this form occludes the promoters of genes that encode iron import systems (2). When H2O2 is present, the oxidation of Fe(II) by H2O2 can deactivate Fur, potentially leading to the disastrous import of more iron. The induction by OxyR of higher levels of Fur seems to partially correct this problem (91). Finally, YaaA is a protein whose biochemical action is not understood but which demonstrably shrinks the iron pool (92). Mutants that lack any of these proteins—Dps, Fur, or YaaA—exhibit high levels of intracellular iron and suffer rapid DNA damage during protracted H2O2 stress.
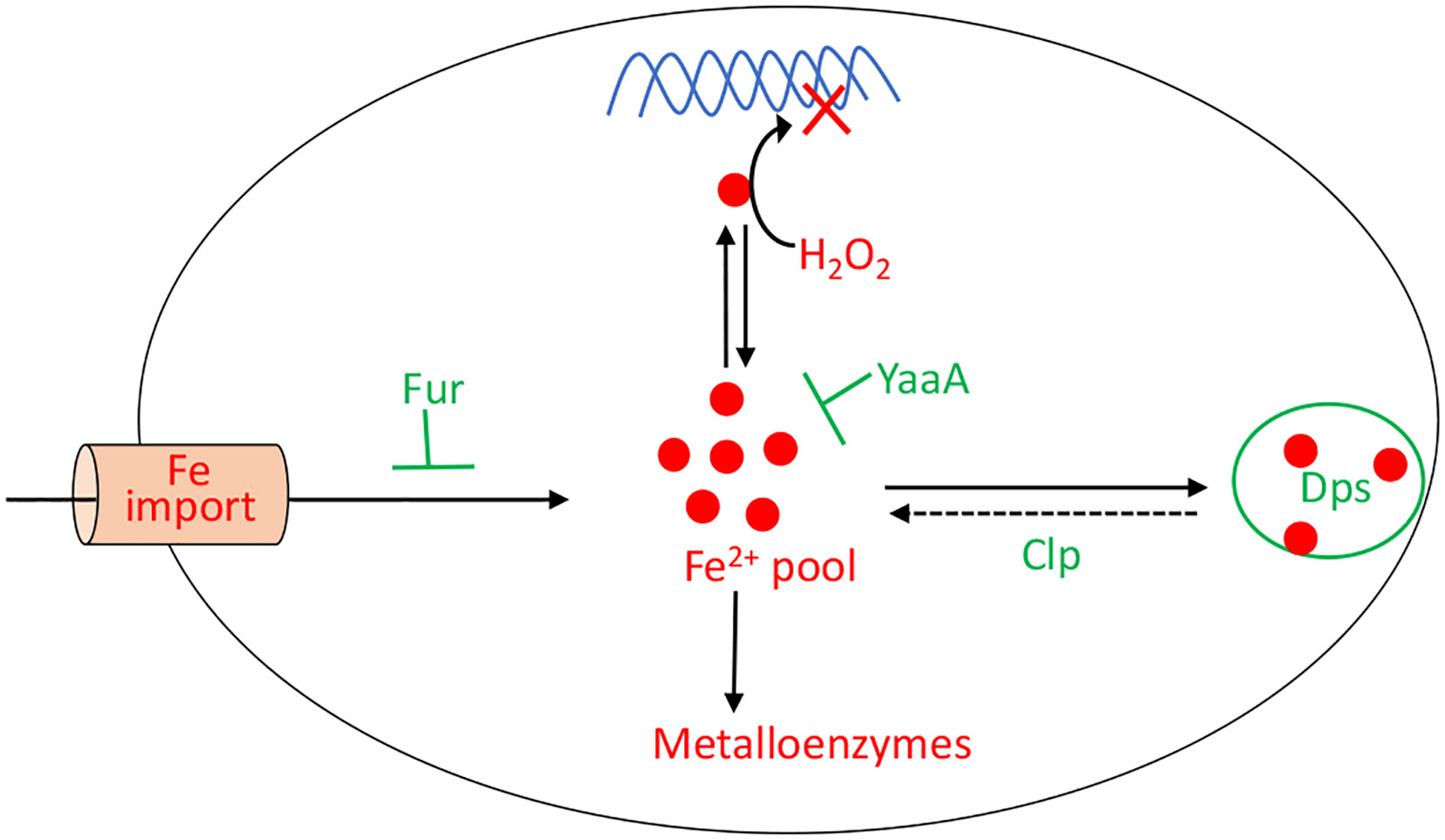
Figure 7 OxyR control of the intracellular iron pool. In order to minimize DNA damage, OxyR decreases the intracellular iron pool by inducing Dps, YaaA, and Fur. The Clp system maintains a small residual iron pool to enable synthesis of iron-dependent enzymes. The H2O2-responsive PerR regulon in Bacillus subtilis also controls Fur and MrgA, which is a Dps homolog.
While the drop in iron pools helps protect DNA, it creates a problem for the synthesis or repair of iron-cofactored enzymes. Iron cofactors comprise three main types—iron-sulfur clusters, mononuclear Fe(II) groups, and heme—and the OxyR system turns on adaptations to sustain the function of enzymes that use each. E. coli typically uses Isc-based machinery to build iron-sulfur clusters (93, 94), but during H2O2 stress the secondary Suf system is induced (95, 96) (Figure 4). For unknown reasons, this system works well when iron levels are low (97), making it better than the house-keeping Isc system, which is poisoned by H2O2 (21). The induction of a manganese importer (MntH) (98) enables mononuclear enzymes to become metallated by Mn(II) rather than Fe(II) (Figure 3). Manganese is not as efficient a catalyst as iron, but it remains available even as Dps sequesters iron, and it is unreactive with H2O2 and thereby enables the mononuclear enzymes to remain functional (13). Finally, continued heme synthesis is facilitated by the induction of HemH, which encodes ferrochelatase (99). This enzyme inserts ferrous iron into porphyrins to complete heme synthesis, but it could potentially become a bottleneck when Fe(II) levels are diminished. Induction of the enzyme helps to circumvent that problem.
Thus, maintaining a balance in the intracellular iron pool during H2O2 stress is challenging. The cells need to keep the levels low enough to avoid DNA damage, but not so low that the synthesis of Fe-dependent enzymes is inhibited. As one final gambit, E. coli uses the Clp protease system to maintain this delicate balance (27). The Clp proteins help to release some iron from Dps, allowing for the repair of [4Fe-4S] clusters (Figure 7). Interestingly, genetic data raise the possibility that the Clp proteins do so not by degrading Dps but perhaps by unfolding it.
Not all the members of the OxyR regulon have been explained. Glutaredoxin 1 is induced. This dithiol compound is capable of reducing protein disulfide bonds, and it helps to deactivate OxyR once the H2O2 stress has dissipated (75). However, the reasons for the induction of glutathione reductase and thioredoxin 2, which also reduce disulfide bonds, are less clear. It would be surprising if low-micromolar H2O2 directly oxidized typical protein thiols—the rate constants for these reactions are too low (37, 77). An alternative is that these systems repair [4Fe-4S] enzymes and mononuclear Fe enzymes: Cluster reactivation requires a dithiol in vitro (21), and the reactivation of mononuclear enzymes can require the reduction of an active-site disulfide (13). An alternative—described below—is that these sulfur reducing systems are useful if OxyR moonlights as a sensor of other thiol-derivatizing stresses.
In E. coli both the reduced and oxidized forms of OxyR repress oxyR itself by binding over its promoter; this action ensures that the titers of OxyR are controlled and do not change during H2O2 stress (76). Transcriptomic data suggest that oxidized OxyR may repress several additional genes, including those that encode the periplasmic disulfide bond chaperone DsbG, the ferric iron reductase FhuF, the inner membrane protein of unknown function YbjC, and the NADPH nitroreductase NfsA (83, 99). The significance of this regulation remains unknown.
Most members of the regulon have functions that either prevent injuries or allow the cell to tolerate them. The regulon is very successful at this: Whereas one micromolar of H2O2 in the environment fully blocks the growth of an OxyR-deficient strain, a wild-type strain is able to adapt and grow in 10 micromolar or more (60). Indeed, a final member of the OxyR regulon, cytochrome c peroxidase, is a periplasm-facing membrane-bound enzyme that allows E. coli to actually exploit environmental H2O2 as a respiratory oxidant when oxygen and nitrate are unavailable (100) (Figure 2). It is likely that the enzyme plays a role at oxic/anoxic interfaces near the intestinal epithelium, where H2O2 may be formed and may diffuse into anoxic zones.
The OxyR system has been most fully studied in E. coli, where it is presumably adapted for the enteric environment. Similarly, other bacteria seem to have adapted the OxyR regulon to suit their particular niches; they exhibit differences in terms of the regulation mechanism, the number of OxyR homologs, and the identity of the genes in the regulon. Porphyromonas gingivalis, for example, encounters two types of environments: the oral cavity where the oxygen tension is high and the hemin concentrations are low, and the periodontal pockets, which contain mixed microbial communities that lower the oxygen levels and are bathed with proteins such as hemoglobin that serve as a source of hemin. Perhaps unsurprisingly, the OxyR protein in P. gingivalis senses both signals, as evidenced by the further activation of OxyR-regulated genes in a hemin-limited environment under anaerobic conditions (101). Interestingly, measurements of the OxyR regulon genes indicate that they are constitutively expressed. This observation is consistent with the idea that the OxyR protein has mutated into a locked-on form, presumably as an adaptation to their environment, which contains H2O2-generating lactic acid bacteria.
With a few exceptions (83), OxyR in E. coli predominantly acts as an activator. However, in a variety of other organisms, it acts as a repressor and/or an activator, sometimes of the same gene: Reduced OxyR is a repressor and oxidized OxyR is an activator for the catalases katG in Burkholderia pseudomallei, katB in Shewanella oneidensis, kat in Neisseria meningitidis and Neisseria gonorrhoeae, cat in Corynebacterium diphtheriae, and katA in Pseudomonas aeruginosa (102–108). In these organisms, oxyR mutants exhibit a higher basal level of catalase expression compared to wild-type cells, as measured by gene expression and protein measurements. The presence of H2O2 further increases these levels in a wild-type cell but not in an oxyR mutant, indicating both repressor and activator function. It is unclear why there is value in having OxyR act as a repressor in some organisms and as an activator in others (Table 2). Its action as both a repressor and an activator may create a step-function-like turn-on switch, as a modest amount of H2O2 stress may be inadequate to fully convert the OxyR population to an activator form, and the residual reduced enzyme may block the action of a subpopulation of oxidized protein. In organisms that are exposed to low, continuous levels of H2O2 stress, AhpCF may suffice to protect the cell from H2O2, but when H2O2 levels become high, a full commitment to catalase synthesis may be called for. Conversely, in other organisms it may be more beneficial to preemptively synthesize basal levels of catalase to guard against a sudden deluge of oxidative stress. Under those conditions, the basal expression can help with the initial stress, and the activation of OxyR can further increase the scavenging enzyme titers.
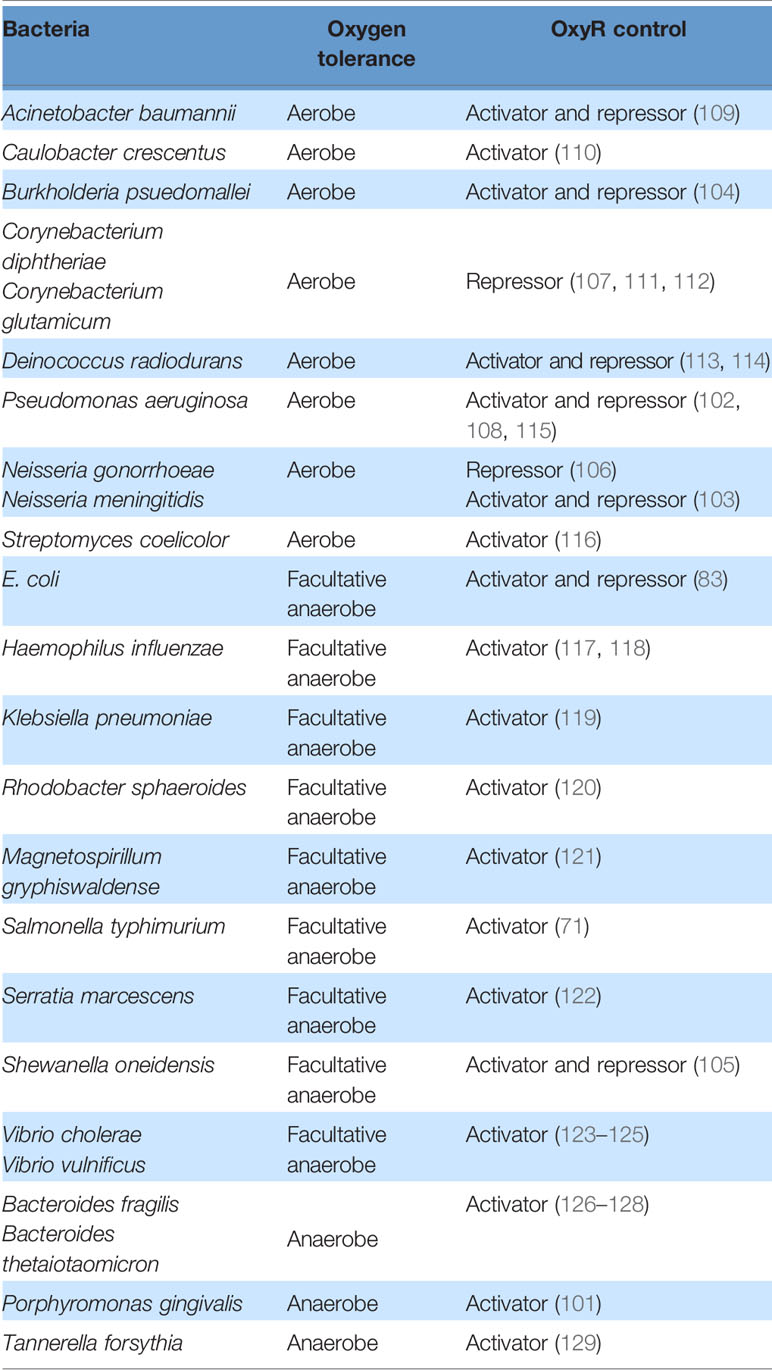
Table 2 Mechanism of OxyR control in different bacteria.
Organisms such as Vibrio cholerae, Vibrio vulnificus, and Deinococcus radiodurans each deploy two OxyR proteins. In V. vulnificus, a facultative anaerobe that occasionally encounters aeration, the two proteins are calibrated to sense different levels of H2O2 (123–125). The more sensitive OxyR (VvOxyR2) is activated by endogenous H2O2 that is formed when the cell is aerated, whereas the less sensitive OxyR (VvOxyR1) is only activated by an influx of exogenous H2O2 from the environment. Accordingly, VvOxyR2 induces a peroxidase (VvPrx2) that has a higher activity at lower H2O2 levels compared to a second peroxidase (VvPrx1) that is induced by VvOxyR1. VvPrx1 becomes necessary because high levels of H2O2 can irreversibly over-oxidize the catalytic cysteine residue of VvPrx2. It is unclear why there are two OxyR proteins in D. radiodurans. The two proteins seem to regulate different genes: OxyR1 activates katE and represses mntH and dps, and OxyR2 represses katG and the hemin transport genes (113, 114).
In some bacteria the OxyR regulon includes genes that are unrelated to iron control or H2O2 degradation. Superoxide dismutase is regulated by OxyR in P. gingivalis and Pseudomonas aeruginosa, seemingly implying that superoxide stress occurs concomitant with H2O2 stress (101, 130, 131). Perhaps the obligate anaerobe P. gingivalis, like Bacteroides thetaiotaomicron (132, 133), generates toxic doses of both superoxide and H2O2 whenever it enters oxic environments; thus, it may use H2O2 as a proxy to detect aerated habitats. In contrast, P. aeruginosa is an aerobe—but its special feature is that in competitive environments it synthesizes pyocyanin, a redox-active compound which can produce both ROS, perhaps to poison competitors (134). This behavior may necessitate the simultaneous synthesis of both superoxide and H2O2 defenses lest P. aeruginosa also poison itself. Meanwhile, DNA-binding assays and transcriptional analyses have shown that in Magnetospirillum gryphiswaldense OxyR induces the synthesis of genes involved in magnetosome production (121). Magnetotactic bacteria make internal magnetic particles as a way to orient themselves and dive into deeper water where there is less oxygen to impair these oxygen-sensitive bacteria; one infers that H2O2 is an environmental signal that triggers this defensive taxis. Together, these data indicate that OxyR has been adapted by different organisms in ways that fit their unique environmental niche.
A key question is whether OxyR plays a leading role in defending bacteria from the H2O2 that phagocytes produce as part of the cell-based immune response. Calculations predict phagosomal H2O2 levels (47, 66) that are adequate to activate OxyR, at least in E. coli (60), and local H2O2 levels could conceivably rise far higher in contained environments, such as abscesses. Infection data support this idea. OxyR is important in the colonizing ability of pathogens such as E. coli O1:K1:H7, Bacteroides fragilis, and Hemophilus influenzae: Mutants lacking oxyR were unable to colonize animal models or to induce abscesses in competition assays where they were mixed with wild-type cells (118, 126, 135). Biofilms can also shelter bacteria from external stressors—perhaps including H2O2 that is produced by an inflammatory response. The OxyR responses of Serratia marcescens, Neiserria gonorrhoeae, Klebsiella pneumoniae, and Tannerella forsythia mediate biofilm formation, a process that helps the bacteria to persist in hosts (119, 122, 129, 136). The OxyR system is required for swarming motility and the production of exotoxins by P. aeruginosa: oxyR mutants were non-motile on plates and were unable to inhibit dendritic cell proliferation (134, 137). However, while these observations demonstrate a role for OxyR in coping with host environments, they do not directly implicate the host response as the source of the H2O2 stress. Indeed, models of urinary tract infection demonstrated that oxyR mutants of E. coli were unsuccessful at colonization—but this phenotype persisted in a host that lacked its phagocytic NADPH oxidase (135). Presumably growth of the mutant was inhibited by H2O2 that was created by other environmental sources, such as competing lactic acid bacteria.
The model eukaryote Saccharomyces cerevisiae also activates a defensive response when it senses hazardous H2O2 in its environment—but although it depends upon a thiol-based sensor to do so, the sensor is unrelated to OxyR. Yap1p is the key transcription factor (138). Its amino-terminus has a bZip DNA-binding domain, but in the absence of H2O2, Yap1p shuttles between the nucleus and the cytoplasm (Figure 8). However, when H2O2 levels rise, Yap1p is indirectly activated. The cytoplasmic Gpx3 is a glutathione peroxidase whose catalytic Cys36 residue is alternatively oxidized to a sulfenic acid by H2O2 and reduced to the thiol by glutathione, with the average redox state dictated by the level of H2O2. This sulfenic acid form can react with the C598 residue of Yap1p, which is part of the cysteine-rich domain in the N-terminus, to form an interprotein disulfide intermediate. Subsequent thiol-disulfide exchange reactions lead to the formation of an intramolecular disulfide bond between Yap1 C303 and C598, causing global conformational change. As a result, the Yap1 nuclear export signal is hidden, which blocks its interaction with the nuclear exporter Crm1 (139–141). The resultant nuclear localization of Yap1p results in the activation of several genes. Thus, unlike OxyR, the cysteine residues of Yap1p do not directly react with H2O2. This difference may ensure that Yap1p stays activated even after migrating into the nucleus, where the H2O2 may not be as high as in the cytoplasm.
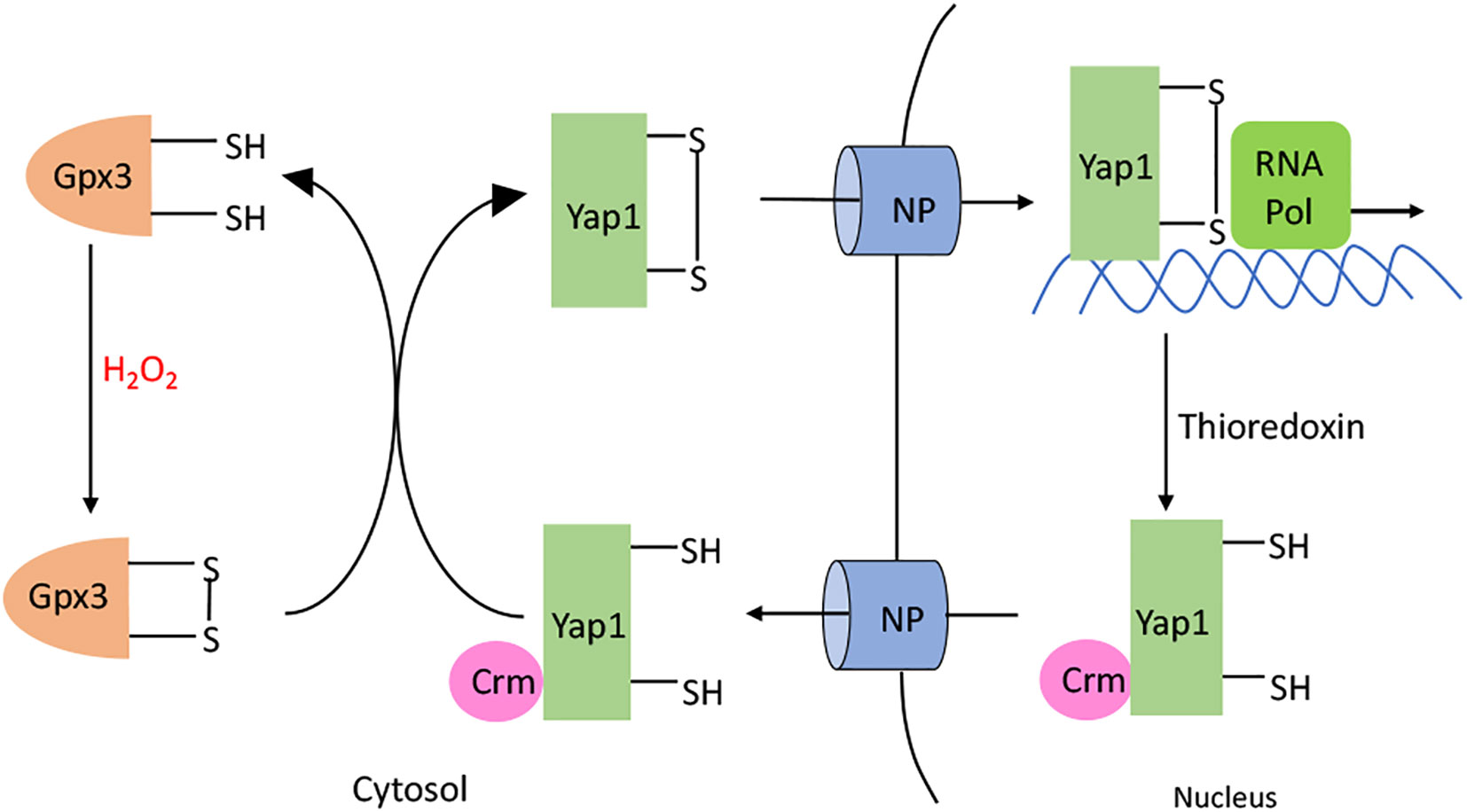
Figure 8 Yap1p activation. H2O2 oxidizes the C36 residue of glutathione peroxidase (Gpx3). The resulting sulfenate interacts with C598 in the C-terminal domain of Yap1p to form a intermolecular disulfide bond. Subsequent thiol-disulfide exchange reactions produce C303-C598 and C310-C629 disulfide bonds in Yap1p. Yap1p accumulates in the nucleus, leading to the activation of the Yap1p regulated genes. When H2O2 diminishes, Yap1p is reduced by the thioredoxin system; this change makes its nuclear export signal accessible to Crm, causing Yap1p to be transported back out of the nucleus.
In broad outline, the membership of the Yap1p regulon overlaps with that of the OxyR regulon (Table 3). These proteins scavenge H2O2, change iron levels, and influence the thiol status of the cell (146, 147). Yap1p induces several peroxidases, including Ahp1, Gpx2, and Tsa1, that scavenge cytosolic H2O2; it is currently unclear why there are three such systems. The glutathione reductase GLR1 is induced to reduce the glutathione disulfide that is formed when Gpx2 reduces H2O2. Additionally, Yap1p also drives synthesis of Ctt1, a cytosolic catalase. The mitochondrial iron exporter Mmt1 is also induced. It has been hypothesized that iron is exported in order to avoid H2O2 damage to the mitochondrial DNA (145). Alternatively, the flow of iron into the cytosol may help the repair of Fe-S clusters of H2O2-sensitive enzymes, such as LeuCD, that are localized there. Similar to OxyR, Yap1p is ultimately turned off by a thioredoxin system that consists of thioredoxins Trx1 and Trx2 and thioredoxin reductase Trr1 (148).

Table 3 Genes induced by Yap1p during hydrogen peroxide stress in yeast.
The fission yeast Schizosaccharomyces pombe features an interesting orthologue of the Yap1 system: Nuclear localization of the Pap1 transcription factor is accomplished when it receives a disulfide bond from thiol peroxidase [reviewed in (149)]. One intriguing feature of its regulon is that it includes not only familiar H2O2 defenses but also drug-resistance genes. This feature raises the possibility that H2O2 stress is frequently imposed upon S. pombe by natural antibiotics, much as the linkage of drug pumps and SOD to the SoxRS system of E. coli reveals that redox-cycling drugs are a natural source of superoxide stress.
Some bacteria rely on a thiol-independent mechanism of H2O2 sensing. Unlike OxyR and Yap1p, the transcriptional repressor PerR takes advantage of the reaction between Fe(II) and H2O2 to detect the stress. Most extensively studied in Bacillus subtilis (150–152), PerR is a Fur homolog containing a structural zinc site and a regulatory metal binding site. It was probably easy to evolve Fur to sense H2O2 stress: Fur:Fe(II) already reacts with H2O2, and Fur and PerR are sufficiently similar in primary sequence that genomic inspection cannot reliably distinguish the two.
PerR acts as a dimeric repressor when it is bound to either Mn(II) or Fe(II). Under most growth conditions, PerR has a greater binding affinity for Fe2+. However, in iron-limited medium that has been supplemented with manganese, PerR binds to Mn(II). The identity of the metal is functionally important, as Mn-bound PerR does not react with H2O2. This is probably by evolutionary design, as manganese-rich/iron-poor cells are intrinsically less vulnerable to H2O2. Because manganese supplants iron in mononuclear enzymes and a paucity of iron precludes much DNA damage, cells need not waste resources defending themselves against H2O2. In contrast, Fe-bound PerR reacts with H2O2 (Figure 9) with a rate constant of 105 M-1 s-1, which approximates that of OxyR (153); the similarity in rate constants suggests that the same level of H2O2 may be toxic in B. subtilis as in E. coli. The reaction oxidizes two of the His ligands bound to iron, forming 2-oxo-histidine; because the oxidized ligands cannot bind metal and the oxidation cannot be reversed, the repressor is permanently inactivated (154). Intriguingly, in vivo studies have shown that the majority of PerR in Staphylococcus aureus (PerRSA) is present in the oxidized form during aerobic growth, whereas this is not true of the B. subtilis PerR (PerRBS) (155). When PerRSA and PerRBS were alternately expressed in the same organism, the KatA activity and transcript levels of PerR-regulated genes were higher with PerRSA. One explanation is that PerRSA is more reactive than PerRBS and can sense lower concentrations of H2O2.
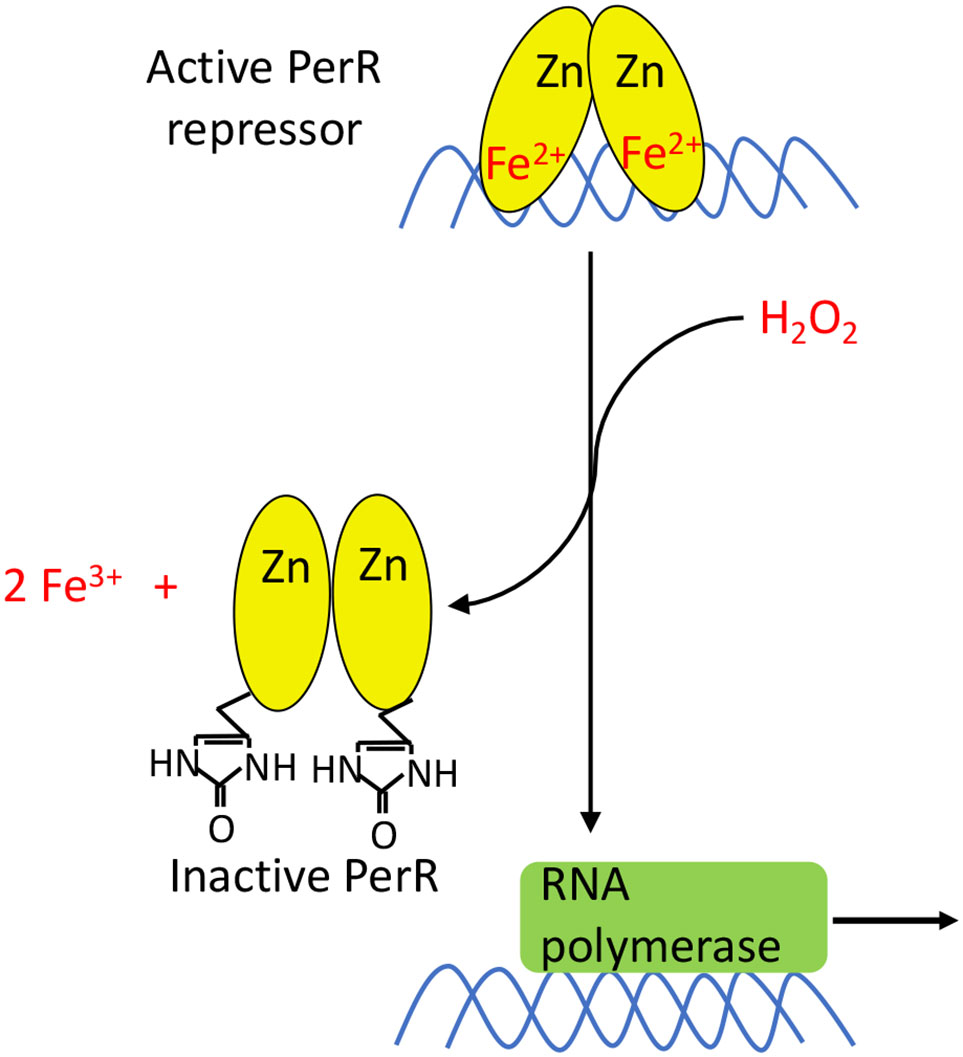
Figure 9 PerR activation. PerR is a dimeric DNA-binding protein, and it binds two metal ions per monomer. The first ion is a structural Zn2+ that is necessary for dimerization and structural integrity. The second metal ion enables DNA binding, and can either be Fe2+ or Mn2+. Only PerR bound to Fe2+ is responsive to H2O2. The oxidation of Fe2+ by H2O2 generates a localized hydroxyl/ferryl radical, which irreversibly oxidizes either of two His ligands (H37 or H91) to form 2-oxo-histidine. Metal binding is blocked, PerR dissociates from promoter sites, and the regulon is induced.
The inactivation of PerR results in the derepression of the PerR regulon, which again controls proteins that scavenge H2O2 and that lower the level of loose intracellular iron (156) (Table 4). AhpCF and KatA are induced to reduce the intracellular H2O2 levels. MrgA, which is a homolog of Dps, sequesters iron. Fur, as in E. coli, helps to reduce the intracellular iron levels by repressing iron import. The hemAXCDBL operon encodes the early steps of heme biosynthesis. Unlike E. coli, where OxyR induces the ferrochelatase HemH, B. subtilis PerR does not regulate the ferrochelatase.
Interestingly, the constitutive expression of the B. subtilis PerR regulon—in a perR mutant—causes trouble by excessively lowering the pool of intracellular iron (159). These mutants are more resistant to H2O2 but have trouble growing. It is unclear which enzyme-activity deficiency causes the poor growth. The iron deficiency was tracked to the combined repression of iron uptake by Fur, plus iron depletion due to the induction of KatA. Under these inducing conditions KatA becomes the single most abundant protein in the cell, comprising a whopping 10% of the total cell protein (159). This situation is reminiscent of OxyR-driven iron deficiency in E. coli; however, the latter is abated by induction of the Clp system (27). It seems, then, that whereas OxyR induction does not interfere with growth—and, indeed, can support it via the service of cytochrome c peroxidase—the induction of PerR is an emergency response that is incompatible with continued growth. Perhaps B. subtilis is wired to enter a period of stasis when exposed to H2O2 stress, with growth resuming only after the threat has passed, whereas OxyR allows E. coli to adjust and continue growing.
After H2O2 stress, the inactivated PerR is degraded by the protease LonA, and the repression of the PerR regulon is restored when the newly synthesized PerR binds either Mn or Fe (160).
Similar to OxyR, PerR has also been adapted by bacteria to fit their particular niches. Differences have emerged in the types of PerR, what it senses, and the genes that it controls. Most bacteria have a single PerR regulator, such as Staphylococcus aureus, Streptococcus pyogenes, Enterococcus faecalis, and Helicobacter hepaticus (161–165). On the other hand, Bacillus Licheniformis has one PerR and two PerR-like proteins, both of which can sense H2O2 by histidine oxidation (166). Other bacteria contain both OxyR and PerR, including N. gonorrhoeae, B. thetaiotaomicron, and D. radiodurans (133, 167, 168); it is not yet clear why they would require both sensing systems.
The importance of PerR in different organisms may reflect the circumstances under which these bacteria experience H2O2 stress. Low-level aeration induces the PerR regulon of the anaerobe Clostridium acetobutylicum, perhaps due to endogenous H2O2 formation (169); as oxygen levels rise, induction of the regulon is critical for cell survival. In Campylobacter jejuni, a microaerophile that lacks SoxRS and OxyR homologs, the superoxide dismutase sodB is induced in perR mutants (170). These observations indicate that PerR may not be limited to defending cells against only H2O2.
Surprisingly, it has been shown that perR mutants of S. aureus, S. pyogenes, and Group A Streptococcus have lower virulence and lower intracellular survival in infected macrophages (161, 164, 171, 172), even though the derepression of the PerR regulon might be expected to induce defenses against the oxidative stress these bacteria encounter in their hosts. It is possible that the constitutive induction of the PerR regulon causes secondary growth defects similar to what is seen in B. subtilis (159). If so, it may explain why the perR mutants of these pathogens are unable to colonize their hosts. In S. aureus and Staphylococcus epidermidis, PerR represses the expression of ferritin under low-iron conditions where PerR binds to Mn, and induces it in the presence of iron, indicating that like in B. subtilis, PerR can regulate metal homeostasis independently of oxidative stress (161, 173).
The reactive sensors of OxyR and PerR—a hyperreactive thiol and Fe(II), respectively—can be modified by reactive species other than H2O2, and this observation raises the question of whether these transcription factors profitably respond to these other stresses. The effectors that have been examined most closely are nitric oxide (NO) and disulfide stress.
cesses (174, 175), and it is deliberately generated at toxic levels by macrophages as part of the cell-based immune response (176). It is a radical species that can pair with the unpaired d-orbital electrons of iron; as a result, NO binds heme, exposed iron-sulfur clusters, and mononuclear iron, potentially inhibiting the enzymes that possess these cofactors (177–180). Many bacteria use NO-sensing transcription factors to control the synthesis of NO scavenging enzymes. In E. coli, NorR is a Fe(II)-based regulator that induces the NorVW NO reductase, while NsrR is a [2Fe-2S]-containing transcription factor whose binding by NO triggers the induction of nitric oxide dioxygenase (Hmp) (181, 182). NsrR also appears to regulate a more expansive regulon, although the roles of other members are less clear (183). The Vibrio fischeri NsrR regulates an alternative oxidase that is more resistant to inhibition by NO than are conventional respiratory oxidases; thus, this feature of the NsrR regulon allows this squid symbiont to sustain its respiration despite the NO that is generated by its host (184). NO has a second route of toxicity, too: Its reaction with superoxide, which is also produced by macrophages, forms peroxynitrite (ONOO-), a potent univalent oxidant that can penetrate into phagocytosed bacteria (185).
The Stamler group has presented evidence that OxyR also provides protection against NO stress (186, 187). Null mutants grew poorly during anaerobic respiration of nitrate, a process that might release some NO. Notably, the sensory cysteine of OxyR was nitrosylated, a modification that appeared to activate OxyR so that it induced a set of genes distinct from the conventional H2O2-driven response. The hcp operon was among those genes, and this group has proposed that Hcp contributes to the broader nitrosylation of cellular proteins, in a way that protects cells from nitrosative stress. The chemistry by which NO would chemically derivatize the OxyR thiol is not clear; NO is a radical species, so an oxidant, perhaps iron, needs to be involved to absorb the extra electron. Derivatization by Hcp is plausible; nitrosothiols readily react with activated cysteine residues, including that of OxyR, and this modification can perturb its behavior and has even been shown to initiate catalase synthesis—although it would seem to lack value in this situation.
However, other in vivo studies have elicited contradictory results. Chemostat cultures of E. coli that were grown anaerobically in the presence of 5 μM NO induced genes associated with NorR, NsrR, and Fur but not OxyR (188). Studies that have used even higher concentrations of NO sources, such as 1 mM acidified NaNO2, activated NO-detoxifying systems such as hmpA, norV, and norW, but not OxyR (189). It is possible that the identity and dose of these nitrosative stressors as well as the growth conditions contributed to the discrepancies.
The role of Yap1p in protecting yeast from nitrosative stress is also unclear. Exogenous nitrosoglutathione elicited the synthesis of superoxide dismutase and catalase, and this response depended upon Yap1p (190). However, a study that used a nitric oxide donor did not detect this effect (191). The possibility exists, then, that the effects of NO and/or nitrosothiols upon OxyR and Yap1p are adventitious. Similarly, exogenous NO can react with the Fe(II) in bacterial PerR and, by inactivating the repressor, trigger induction of its regulon (192). However, members of this regulon do not provide any obvious route to remediate this stress, and so it seems likely that effect is merely incidental to the iron-binding activity of NO.
“Disulfide stress” is a term attached to conditions that create and disseminate disulfide bonds among cellular proteins. In many studies it is imposed by exposing microbes to diamide, a manmade reagent designed to create disulfide bonds from cellular thiols (193). In some bacteria diamide elicits defensive responses that include the induction of redoxin-based disulfide-reducing systems; these regulons (194, 195) are independent of the systems that detect and suppress H2O2 stress. Diamide can activate OxyR inside E. coli, but very high doses are needed (75). One wonders, then, what the natural circumstances are that trigger “disulfide stress” responses. Further, because OxyR-controlled redoxins have not yet been assigned a role in defraying H2O2 stress, it is formally possible that the sensory thiol of OxyR serves a second purpose of detecting and defusing thiol-targeting electrophiles.
Thus far, the only condition under which disulfide stress is known to naturally occur in E. coli is during periods of rapid cystine import (196). This situation arises when sulfur-limited cells, which induce all forms of sulfur importers, encounter cystine. Gross over-import of cystine results, and disulfide-exchange reactions cause disulfide bonds to be transferred from the imported cystine to cytoplasmic proteins. OxyR is modified, and it induces its regulon. Both the thioredoxin and glutaredoxin systems that it induces act to minimize the disulfide stress. Disulfide stress can also be imposed by exposing cells to antimicrobial plant compounds such as diallyl thiosulfinate and diallyl polysulfanes; the induction of ahpC, trxA, and trxC results (197). It seems unlikely that E. coli naturally encounters these chemicals.
Similar stresses can activate Yap1p of yeast. It is directly modified by diamide—without the mediation of Gpx3—and the H2O2-sensing C303 is not involved in the resultant conformational change (198). Instead, disulfide linkages are formed between C598 and C620, C620 and C629, and C598 and C629. Glutathione reductase and thioredoxin are subsequently induced. However, broadly speaking, the activation of these systems by disulfide-generating agents—and perhaps by nitrogen species—has attracted a fair amount of attention without compelling evidence that these outcomes are not accidental.
To date, the mechanisms by which H2O2 poisons bacteria have been explored primarily in test tubes. From those studies we have learned that H2O2 stress is iron-focused, driven by reactions with enzymic iron-sulfur clusters and Fe(II) prosthetic groups and with the pool of loose iron. The high rate constants of these reactions, and the abundance of vulnerable enzymes, means that low-micromolar concentrations of H2O2 suffice to bring bacterial growth to a halt. The known defenses remediate the same injuries that have been discovered, which provides confidence that the overall picture has come into shape. In E. coli, the most-examined microbe, it appears that the endogenous levels of H2O2 in the fully aerated bacterium falls just short of the threshold for H2O2 toxicity—and for the induction of emergency responses. Those responses, then, evidently exist to shield the cell from external H2O2.
The next step is to figure out how this information translates to real-world environments. The broad distribution of H2O2 defenses, and in particular of inducible defenses, suggests that H2O2 stress is a pervasive phenomenon that extends, at least episodically, to most biological habitats. Yet the actual circumstances and severity of oxidative stress remain poorly understood. Plausible sources of stress range from the redox collision that occurs at oxic/anoxic interfaces to the oxidative burst of mammalian and plant defenses. We infer that different microbes may encounter this stress in different circumstances, as the defensive regulons have been modified to suit their specific situations. The rationale for these differences is not always clear; in particular, we do not yet understand why some organisms use OxyR as a sensor, why others use PerR, and why a third set employ both. Thus, if we are to fully understand oxidative stress, we will need to continue to expand these studies beyond E. coli and yeast, and we will need to evaluate the intensity of H2O2 stress and the role of these systems in natural habitats.
For many microbiologists, the key questions concern whether host-generated H2O2 plays a role in suppressing invasion by most bacteria—and, if it does so, by which strategies dedicated pathogens manage to circumvent this stress. It may seem intuitively obvious that phagocytic H2O2 is a potent defense, but back-of-the-envelope calculations suggest that the H2O2 levels may not rise as high as workers have sometimes assumed. Part of the difficulty here is that phagocytic action has typically been studied using as prey the same professional pathogens that have, by definition, developed ways to elude the toxicity. We endorse the idea of studying the process using the 99% of bacteria that phagocytes efficiently kill.
AS is the first author and JI is the last author. Both authors have contributed to the writing. All authors contributed to the article and approved the submitted version.
This work was supported by grant GM49640 from the National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
2. Wandersman C, Delepelaire P. Bacterial iron sources: from siderophores to hemophores. Ann Rev Microbiol (2004) 58:611–47. doi: 10.1146/annurev.micro.58.030603.123811
3. Naqui A, Chance B. Reactive oxygen intermediates in biochemistry. Ann Rev Biochem (1986) 55:137–66. doi: 10.1146/annurev.bi.55.070186.001033
4. Khademian M, Imlay JA. How microbes evolved to tolerate oxygen. Trends Microbiol (2020) 20:30262–6. doi: 10.1016/j.tim.2020.10.001
5. Korshunov S, Imlay JA. Two sources of endogenous hydrogen peroxide in Escherichia coli. Mol Microbiol (2010) 75:1389–401. doi: 10.1111/j.1365-2958.2010.07059.x
6. Seaver LC, Imlay JA. Are respiratory enzymes the primary sources of intracellular hydrogen peroxide? J Biol Chem (2004) 279:48742–50. doi: 10.1074/jbc.M408754200
7. Davies MJ. The oxidative environment and protein damage. Biochim Biophys Acta (2005) 1703:93–109. doi: 10.1016/j.bbapap.2004.08.007
8. Loew O. A new enzyme of general occurrence in organismis. Science (1900) 11:701–2. doi: 10.1126/science.11.279.701
9. McCord J, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem (1969) 244:6049–55. doi: 10.1016/S0021-9258(18)63504-5
10. Seaver LC, Imlay JA. Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. J Bacteriol (2001) 183:7173–81. doi: 10.1128/JB.183.24.7173-7181.2001
11. Carlioz A, Touati D. Isolation of superoxide dismutase mutants in Escherichia coli: is superoxide dismutase necessary for aerobic life? EMBO J (1986) 5:623–30. doi: 10.1002/j.1460-2075.1986.tb04256.x
12. Seaver LC, Imlay JA. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J Bacteriol (2001) 183:7182–9. doi: 10.1128/JB.183.24.7182-7189.2001
13. Anjem A, Imlay JA. Mononuclear iron enzymes are primary targets of hydrogen peroxide stress. J Biol Chem (2012) 287:15544–56. doi: 10.1074/jbc.M111.330365
14. Sobota JM, Imlay JA. Iron enzyme ribulose-5-phosphate 3-epimerase in Escherichia coli is rapidly damaged by hydrogen peroxide but can be protected by manganese. Proc Natl Acad Sci USA (2011) 108:5402–7. doi: 10.1073/pnas.1100410108
15. Sobota JM, Gu M, Imlay JA. Intracellular hydrogen peroxide and superoxide poison 3-deoxy-D-arabinoheptulosonate 7-phosphate synthase, the first committed enzyme in the aromatic biosynthetic pathway of Escherichia coli. J Bacteriol (2014) 196:1980–91. doi: 10.1128/JB.01573-14
16. Geary LE, Meister A. On the mechanism of glutamine-dependent reductive amination of α-ketoglutarate catalyzed by glutamate synthase. J Biol Chem (1977) 252:3501–8. doi: 10.1016/S0021-9258(17)40419-4
17. Grinblat L, Sreider CM, Stoppani AO. Superoxide anion production by lipoamide dehydrogenase redox-cycling: effect of enzyme modifiers. Biochem Int (1991) 23:83–92.
18. Massey V, Strickland S, Mayhew SG, Howell LG, Engel PC, Matthews RG, et al. The production of superoxide anion radicals in the reaction of reduced flavins and flavoproteins with molecular oxygen. Biochem Biophys Res Commun (1969) 36:891–7. doi: 10.1016/0006-291X(69)90287-3
19. Messner KR, Imlay JA. Mechanism of superoxide and hydrogen peroxide formation by fumarate reductase, succinate dehydrogenase, and aspartate oxidase. J Biol Chem (2002) 277:42563–71. doi: 10.1074/jbc.M204958200
20. Jang S, Imlay JA. Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron-sulfur enzymes. J Biol Chem (2007) 282:929–37. doi: 10.1074/jbc.M607646200
21. Jang S, Imlay JA. Hydrogen peroxide inactivates the Escherichia coli Isc iron-sulphur assembly system, and OxyR induces the Suf system to compensate. Mol Microbiol (2010) 78(6):1448–67. doi: 10.1111/j.1365-2958.2010.07418.x
22. Gardner PR, Fridovich I. Superoxide sensitivity of the Escherichia coli aconitase. J Biol Chem (1991) 266:19328–33. doi: 10.1016/S0021-9258(18)55001-8
23. Flint DH, Smyk-Randall E, Tuminello JF, Draczynska-Lusiak B, Brown OR. The inactivation of dihydroxyacid dehydratase in Escherichia coli treated with hyperbaric oxygen occurs because of the destruction of its Fe-S cluster, but the enzyme remains in the cell in a form that can be reactivated. J Biol Chem (1993) 268:25547–52. doi: 10.1016/S0021-9258(19)74426-3
24. Flint DH, Tuminello JF, Emptage MH. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J Biol Chem (1993) 268:22369–76. doi: 10.1016/S0021-9258(18)41538-4
25. Gardner PR, Fridovich I. Superoxide sensitivity of the Escherichia coli 6-phosphogluconate dehydratase. J Biol Chem (1991) 266:1478–83. doi: 10.1016/S0021-9258(18)52319-X
26. Kuo CF, Mashino T, Fridovich I. α,β-dihydroxyisovalerate dehydratase: a superoxide-sensitive enzyme. J Biol Chem (1987) 262:4724–7. doi: 10.1016/S0021-9258(18)61255-4
27. Sen A, Zhou Y, Imlay JA. During oxidative stress the Clp proteins of Escherichia coli ensure that iron pools remain sufficient to reactivate oxidized metalloenzymes. J Bacteriol (2020) 202:e00235–20. doi: 10.1128/JB.00235-20
28. Gardner PR, Fridovich I. Inactivation-reactivation of aconitase in Escherichia coli. A sensitive measure of superoxide radical. J Biol Chem (1992) 267:8757–63. doi: 10.1016/S0021-9258(19)50343-X
29. Henle ES, Han Z, Tang N, Rai P, Luo Y, Linn S. Sequence-specific DNA cleavage by Fe2+-mediated Fenton reactions has possible biological implications. J Biol Chem (1999) 274:962–71. doi: 10.1074/jbc.274.2.962
30. Farr SB, D’Ari R, Touati D. Oxygen-dependent mutagenesis in Escherichia coli lacking superoxide dismutase. Proc Natl Acad Sci USA (1986) 83:8268–72. doi: 10.1073/pnas.83.21.8268
31. Dizdaroglu M. Chemical determination of free radical-induced damage to DNA. Free Rad Biol Med (1991) 10:225–42. doi: 10.1016/0891-5849(91)90080-M
32. Hutchinson F. Chemical changes induced in DNA by ionizing radiation. Prog Nucl Acid Res (1985) 32:116–54. doi: 10.1016/S0079-6603(08)60347-5
33. Park S, You X, Imlay JA. Substantial DNA damage from submicromolar intracellular hydrogen peroxide detected in Hpx- mutants of Escherichia coli. Proc Natl Acad Sci USA (2005) 102:9317–22. doi: 10.1073/pnas.0502051102
34. Morimyo M. Anaerobic incubation enhances the colony formation of a polA recB strain of Escherichia coli K-12. J Bacteriol (1982) 152:208–14.
35. Boling M, Adler H, Masker W. Restoration of viability to an Escherichia coli mutant deficient in the 5’ to 3’ exonuclease of DNA polymerase I. J Bacteriol (1984) 160:706–10. doi: 10.1128/JB.160.2.706-710.1984
36. Gort AS, Imlay JA. Balance between endogenous superoxide stress and antioxidant defenses. J Bacteriol (1998) 180:1402–10. doi: 10.1128/JB.180.6.1402-1410.1998
37. Imlay JA. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol (2013) 11:443–54. doi: 10.1038/nrmicro3032
38. Imlay JA. Where in the world do bacteria experience oxidative stress? Environ Microbiol (2018) 21:521–30. doi: 10.1111/1462-2920.14445
39. Korshunov SS, Imlay JA. A potential role for periplasmic superoxide dismutase in blocking the penetration of external superoxide into the cytosol of phagocytosed bacteria. Mol Microbiol (2002) 43:95–106. doi: 10.1046/j.1365-2958.2002.02719.x
40. Lynch R, Fridovich I. Permeation of the erythrocyte stroma by superoxide radical. J Biol Chem (1978) 253:4697–9. doi: 10.1016/S0021-9258(17)30446-5
41. Bedard K, Lardy B, Krause KH. NOX family NADPH oxidases: Not just in mammals. Biochemie (2007) 89:1107–12. doi: 10.1016/j.biochi.2007.01.012
42. Kawahara T, Quinn MT, Lambeth JD. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. MBC Evol Biol (2007) 7:109. doi: 10.1186/1471-2148-7-109
43. Shiloh MU, MacMicking JD, Nicholson S, Brause JE, Potter S, Marino M, et al. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity (1999) 10:29–38. doi: 10.1016/S1074-7613(00)80004-7
44. Thomas DC. The phagocyte respiratory burst: Historical perspectives and recent advances. Immunol Lett (2017) 192:88–96. doi: 10.1016/j.imlet.2017.08.016
45. Craig M, Slauch J. Phagocytic superoxide specifically damages an extracytoplasmic target to inhibit or kill Salmonella. PLoS One (2009) 4(3):e4975. doi: 10.1371/journal.pone.0004975
46. Slauch JM. How does the oxidative burst of macrophages kill bacteria? Mol Microbiol (2011) 80:580–3. doi: 10.1111/j.1365-2958.2011.07612.x
47. Winterbourn CC, Hampton MB, Livesey JH, Kettle AJ. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome. Implications for microbial killing. J Biol Chem (2006) 281:39860–9. doi: 10.1074/jbc.M605898200
48. Hassan HM, Fridovich I. Intracellular production of superoxide radical and of hydrogen peroxide by redox active compounds. Arch Biochem Biophys (1979) 196:385–95. doi: 10.1016/0003-9861(79)90289-3
49. Inbaraj JJ, Chignell CF. Cytotoxic action of juglone and plumbagin: a mechanistic study using HaCaT keratinocytes. Chem Res Toxicol (2004) 17:55–62. doi: 10.1021/tx034132s
50. Paiva SRD, Figueiredo MR, Aragão TV, Kaplan MAC. Antimicrobial activity in vitro of plumbagin isolated from Plumbago species. Mem Inst Oswaldo Cruz (2003) 98:959–61. doi: 10.1590/S0074-02762003000700017
51. Turner JM, Messenger AJ. Occurrence, biochemistry, and physiology of phenazine pigment production. Adv Microb Physiol (1986) 27:211–75. doi: 10.1016/S0065-2911(08)60306-9
52. Greenberg JT, Monach P, Chou JH, Josephy PD, Demple B. Positive control of a global antioxidant defense regulon activated by superoxide-generating agents in. Escherichia coli Proc Natl Acad Sci USA (990) 87:6181–5. doi: 10.1073/pnas.87.16.6181
53. Tsaneva IR, Weiss B. soxR, a locus governing a superoxide response regulon in Escherichia coli K-12. J Bacteriol (1990) 172:4197–205. doi: 10.1128/JB.172.8.4197-4205.1990
54. Pomposiello PJ, Bennik MH, Demple B. Genome-wide transcriptional profiling of the Escherichia coli responses to superoxide stress and sodium salicylate. J Bacteriol (2001) 183:3890–902. doi: 10.1128/JB.183.13.3890-3902.2001
55. Imlay JA. Diagnosing oxidative stress in bacteria: not as easy as you might think. Curr Opin Microbiol (2015) 24:124–31. doi: 10.1016/j.mib.2015.01.004
56. Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell (2007) 130:797–810. doi: 10.1016/j.cell.2007.06.049
57. Liu Y, Imlay JA. Cell death from antibiotics without the involvement of reactive oxygen species. Science (2013) 339:1210–3. doi: 10.1126/science.1232751
58. Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K. Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science (2013) 339:1213–6. doi: 10.1126/science.1232688
59. Ezraty B, Vergnes A, Banzhaf M, Duverger Y, Huguenot A, Brochado AR, et al. Fe-S cluster biosynthesis controls uptake of aminoglycosides in a ROS-less death pathway. Science (2013) 340:1583–7. doi: 10.1126/science.1238328
60. Li X, Imlay JA. Improved measurements of scant hydrogen peroxide enable experiments that define its threshold of toxicity for Escherichia coli. Free Rad Biol Med (2018) 120:217–27. doi: 10.1016/j.freeradbiomed.2018.03.025
61. Ogino T, Maegawa S, Shigeno S, Fujikura K, Toyohara H. Highly sensitive avoidance plays a key role in sensory adaptation to deep-sea hydrothermal vent environments. PLoS One (2018) 13:e0189902. doi: 10.1371/journal.pone.0189902
62. Wilson CL, Hinman NW, Cooper WJ, Brown CF. Hydrogen peroxide cycling in surface geothermal waters of Yellowstone National Park. Environ Sci Technol (2000) 34:2655–62. doi: 10.1021/es9906397
63. Wilson CL, Hinman NW, Sheridan RP. Hydrogen peroxide formation and decay in iron-rich geothermal waters: the relative roles of abiotic and biotic mechanisms. Photochem Photobiol (2000) 71:691–9. doi: 10.1562/0031-8655(2000)071<0691:HPFADI>2.0.CO;2
64. Lesser MP. Oxidative stress in marine environments: biochemistry and physiological ecology. Annu Rev Physiol (2006) 68:253–78. doi: 10.1146/annurev.physiol.68.040104.110001
65. Mesle MM, Beam JP, Jay ZJ, Bodle B, Bogenschutz E, Inskeep WP. Hydrogen peroxide cycling in high-temperature acidic geothermal springs and potential implications for oxidative stress response. Front Mar Sci (2017) 4:130. doi: 10.3389/fmars.2017.00130
66. Imlay JA. Oxidative Stress. In: Neidhardt FC, editor. EcoSal–Escherichia coli and Salmonella: Cellular and Molecular Biology. Washington, D.C.: ASM Press (2009). Available at: http://www.ecosal.org.
67. Liu X, Ramsey MM, Chen X, Koley D, Whiteley M, Bard AJ. Real-time mapping of a hydrogen peroxide concentration profile across a polymicrobial bacterial biofilm using scanning electrochemical microscopy. Proc Natl Acad Sci USA (2011) 108:2668–73. doi: 10.1073/pnas.1018391108
68. Seki M, Iida K, Saito M, Nakayama H, Yoshida S. Hydrogen peroxide production in Streptococcus pyogenes: involvement of lactase oxidase and coupling with aerobic utilization of lactate. J Bacteriol (2004) 186:2046–51. doi: 10.1128/JB.186.7.2046-2051.2004
69. Spellerberg B, Cundell DR, Sandros J, Pearce BJ, Idanpaan-Heikkila I, Rosenow C, et al. Pyruvate oxidase, as a determinant of virulence in Streptococcus pneumoniae. Mol Microbiol (1996) 19:803–13. doi: 10.1046/j.1365-2958.1996.425954.x
70. Tong H, Chen W, Merritt J, Qi F, Shi W, Dong X. Streptococcus oligofermentans inhibits Streptococcus mutans through conversion of lactic acid into inhibitory H2O2: a possible counteroffensive strategy for interspecies competition. Mol Microbiol (2007) 63:872–80. doi: 10.1111/j.1365-2958.2006.05546.x
71. Christman MF, Morgan RW, Jacobson FS, Ames BN. Positive control of a regulon for defenses against oxidative stress and some heat-shock proteins in Salmonella typhimurium. Cell (1985) 41:753–62. doi: 10.1016/S0092-8674(85)80056-8
72. Aslund F, Zheng M, Beckwith J, Storz G. Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc Natl Acad Sci USA (1999) 96:6161–5. doi: 10.1073/pnas.96.11.6161
73. Choi H, Kim S, Mukhopadhyay P, Cho S, Woo J, Storz G, et al. Structural basis of the redox switch in the OxyR transcription factor. Cell (2001) 105:103–13. doi: 10.1016/S0092-8674(01)00300-2
74. Tao K, Fujita N, Ishihama A. Involvement of the RNA polymerase alpha subunit C-terminal region in co-operative interaction and transcriptional activation with OxyR protein. Mol Microbiol (1993) 7:859–64. doi: 10.1111/j.1365-2958.1993.tb01176.x
75. Zheng M, Aslund F, Storz G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science (1998) 279:1718–21. doi: 10.1126/science.279.5357.1718
76. Toledano MB, Kullik I, Trinh F, Baird PT, Schneider TD, Storz G. Redox-dependent shift of OxyR-DNA contacts along an extended DNA-binding site: a mechanism for differential promoter selection. Cell (1994) 78:897–909. doi: 10.1016/S0092-8674(94)90702-1
77. Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Rad Biol Med (1999) 27:322–8. doi: 10.1016/S0891-5849(99)00051-9
78. Pedre B, Young D, Charlier D, Mourenza A, Rosado LA, Marcos-Pascual L, et al. Structural snapshots of OxyR reveal the peroxidatic mechanism of H2O2 sensing. Proc Natl Acad Sci USA (2018) 115:e11623–32. doi: 10.1073/pnas.1807954115
79. Nelson KJ, Parsonage D, Hall A, Karplus PA, Poole LB. Cysteine pK(a) values for the bacterial peroxiredoxin AhpC. Biochemistry (2008) 47:12860–8. doi: 10.1021/bi801718d
80. Poole LB. The catalytic mechanism of peroxiredoxins. Subcell Biochem (2007) 44:61–81. doi: 10.1007/978-1-4020-6051-9_4
81. Putnam CD, Arvai AS, Bourne Y, Tainer JA. Active and inhibited human catalase structures: ligand and NADPH binding and catalytic mechanism. J Mol Biol (2000) 296:295–309. doi: 10.1006/jmbi.1999.3458
82. Belousov VV, Fradkov AF, Lukyanov KA, Staroverov DB, Shakhbazov KS, Terskikh AV. Genetically encoded fluorescent indictor for intracellular hydrogen peroxide. Nat Methods (2006) 3:281–6. doi: 10.1128/JB.183.15.4562-4570.2001
83. Zheng M, Wang X, Templeton LJ, Smulski DR, LaRossa RA, Storz G. DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J Bacteriol (2001) 183:4562–70. doi: 10.1128/JB.183.15.4562-4570.2001
84. Diaz A, Loewen PC, Fita I, Carpena X. Thirty years of heme catalases structural biology. Arch Biochem Biophys (2012) 525:102–10. doi: 10.1016/j.abb.2011.12.011
85. Singh R, Wiseman B, Deemagarn T, Jha V, Switala J, Loewen PC. Comparative study of catalase-peroxidases (KatGs). Arch Biochem Biophys (2007) 471:207–14. oi: 10.1016/j.abb.2007.12.008
86. Imlay JA, Chin SM, Linn S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science (1988) 240:640–2. doi: 10.1126/science.2834821
87. Altuvia S, Almiron M, Huisman G, Kolter R, Storz G. The dps promoter is activated by OxyR during growth and by IHF and sigma S in stationary phase. Mol Microbiol (1994) 13:265–72. doi: 10.1111/j.1365-2958.1994.tb00421.x
88. Grant RA, Filman DJ, Finkel SE, Kolter R, Hogle JM. The crystal structure of Dps, a ferritin homolog that binds and protects DNA. Nat Struct Biol (1998) 5:294–303. doi: 10.1038/nsb0498-294
89. Ilari A, Ceci P, Ferrari D, Rossi G, Chiancone E. Iron incorporation into E. coli Dps gives rise to a ferritin-like microcrystalline core. J Biol Chem (2002) 277:37619–23. doi: 10.1074/jbc.M206186200
90. Zheng M, Doan B, Schneider TD, Storz G. OxyR and SoxRS regulation of fur. J Bacteriol (1999) 181:4639–43. doi: 10.1128/JB.181.15.4639-4643.1999
91. Varghese S, Wu A, Park S, Imlay KRC, Imlay JA. Submicromolar hydrogen peroxide disrupts the ability of Fur protein to control free-iron levels in Escherichia coli. Mol Microbiol (2007) 64:822–30. doi: 10.1111/j.1365-2958.2007.05701.x
92. Liu Y, Bauer SC, Imlay JA. The YaaA protein of the Escherichia coli OxyR regulon lessens hydrogen peroxide toxicity by diminishing the amount of intracellular unincorporated iron. J Bacteriol (2011) 193:2186–96. doi: 10.1128/JB.00001-11
93. Giel JL, Rodionov D, Liu M, Blattner FR, Kiley PJ. IscR-dependent gene expression links iron-sulphur cluster assembly to the control of O2-regulated genes in Escherichia coli. Mol Microbiol (2006) 60:1058–75. doi: 10.1111/j.1365-2958.2006.05160.x
94. Schwartz CJ, Giel JL, Patschkowski T, Luther C, Ruzicka FJ, Beinert H, et al. IscR, an Fe-S cluster-containing transcription factor, represses expression of Escherichia coli genes encoding Fe-S cluster assembly proteins. Proc Natl Acad Sci USA (2001) 98:14895–900. doi: 10.1073/pnas.251550898
95. Lee JH, Yeo WS, Roe JH. Induction of the sufA operon encoding Fe-S assembly proteins by superoxide generators and hydrogen peroxide: involvement of OxyR, IHF and an unidentified oxidant-responsive factor. Mol Microbiol (2004) 51:1745–55. doi: 10.1111/j.1365-2958.2003.03946.x
96. Yeo WS, Lee JH, Lee KC, Roe JH. IscR acts as an activator in response to oxidative stress for the suf operon encoding Fe-S assembly proteins. Mol Microbiol (2006) 61:206–18. doi: 10.1111/j.1365-2958.2006.05220.x
97. Djaman O, Outten FW, Imlay JA. Repair of oxidized iron-sulfur clusters in Escherichia coli. J Biol Chem (2004) 279:44590–9. doi: 10.1074/jbc.M406487200
98. Kehres DG, Janakiraman A, Slauch JM, Maguire ME. Regulation of Salmonella enterica serovar Typhimurium mntH transcription by H2O2, Fe2+, and Mn2+. J Bacteriol (2002) 184:3151–8. doi: 10.1128/JB.184.12.3151-3158.2002
99. Mancini S, Imlay JA. The induction of two biosynthetic enzymes helps Escherichia coli sustain heme synthesis and activate catalase during hydrogen peroxide stress. Mol Microbiol (2015) 96:744–63. doi: 10.1111/mmi.12967
100. Khademian M, Imlay JA. Escherichia coli cytochrome c peroxidase is a respiratory oxidase that enables the use of hydrogen peroxide as a terminal electron acceptor. Proc Natl Acad Sci USA (2017) 114:E6922–31. doi: 10.1073/pnas.1701587114
101. Xie H, Zheng C. OxyR activation in Porphyromonas gingivalis in response to a hemin-limited environment. Infect Immun (2012) 80(10):3471–80. doi: 10.1128/IAI.00680-12
102. Heo YJ, Chung IY, Cho WJ, Lee BY, Kim JH, Choi KH, et al. The major catalase gene (katA) of Pseudomonas aeruginosa PA14 is under both positive and negative control of the global transactivator OxyR in response to hydrogen peroxide. J Bacteriol (2010) 192:381–90. doi: 10.1128/JB.00980-09
103. Ieva R, Roncarati D, Metruccio MM, Seib KL, Scarlato V, Delany I. OxyR tightly regulates catalase expression in Neisseria meningitidis through both repression and activation mechanisms. Mol Microbiol (2008) 70:1152–65. doi: 10.1111/j.1365-2958.2008.06468.x
104. Jangiam W, Loprasert S, Smith DR, Tungpradabkul S. Burkholderia pseudomallei RpoS regulates OxyR and the katG-dpsA operon under conditions of oxidative stress. Microbiol Immunol (2010) 54:389–97. doi: 10.1111/j.1348-0421.2010.00230.x
105. Jiang Y, Dong Y, Luo Q, Li N, Wu G, Gao H. Protection from oxidative stress relies mainly on derepression of OxyR-dependent KatB and Dps in Shewanella oneidensis. J Bacteriol (2014) 196:445–58. doi: 10.1128/JB.01077-13
106. Tseng HJ, McEwan AG, Apicella MA, Jennings MP. OxyR acts as a repressor of catalase expression in Neisseria gonorrhoeae. Infect Immun (2003) 71:550–6. doi: 10.1128/iai.71.1.550-556.2003
107. Kim JS, Holmes RK. Characterization of OxyR as a negative transcriptional regulator that represses catalase production in Corynebacterium diphtheriae. PLoS One (2012) 7:e31709. doi: 10.1371/journal.pone.0031709
108. Ochsner UA, Vasil ML, Alsabbagh E, Parvatiyar K, Hassett DJ. Role of the Pseudomonas aeruginosa oxyR-recG operon in oxidative stress defense and DNA repair: OxyR-dependent regulation of the katB-ankB, ahpB, and ahpC-ahpF. J Bacteriol (2000) 182:4533–44. doi: 10.1128/JB.182.16.4533-4544.2000
109. Juttukonda LJ, Green ER, Lonergan ZR, Heffern MC, Chang CJ, Skaar EP. Acinetobacter baumannii OxyR regulates the transcriptional response to hydrogen peroxide. Infect Immun (2018) 87:e00413–18. doi: 10.1128/IAI.00413-18
110. Silva LG, Lorenzetti APR, Ribeiro RA, Alves IR, Leaden L, Galhardo RS, et al. OxyR and the hydrogen peroxide stress response in Caulobacter crescentus. Gene (2019) 700:70–84. doi: 10.1016/j.gene.2019.03.003
111. Milse J, Petri K, Ruckert C, Kalinowski J. Transcriptional response of Corynebacterium glutamicum ATCC 13032 to hydrogen peroxide stress and characterization of the OxyR regulon. J Biotechnol (2014) 190:40–54. doi: 10.1016/j.jbiotec.2014.07.452
112. Teramoto H, Inui M, Yukawa H. OxyR acts as a transcriptional repressor of hydrogen peroxide-inducible antioxidant genes in Corynebacterium glutamicum R. FEBS J (2013) 280:3298–312. doi: 10.1111/febs.12312
113. Chen H, Xu G, Zhao Y, Tian B, Lu H, Yu X, et al. A novel OxyR sensor and regulator of hydrogen peroxide stress with one cysteine residue in Deinococcus radiodurans. PLoS One (2008) 3:e1602. doi: 10.1371/journal.pone.0001602
114. Yin L, Wang L, Lu H, Xu G, Chen H, Zhan H, et al. DRA0336, another OxyR homolog, involved in the antioxidation mechanisms in Deinococcus radiodurans. J Microbiol (2010) 48:473–9. doi: 10.1007/s12275-010-0043-8
115. Wei Q, Minh PN, Dotsch A, Hildebrand F, Panmanee W, Elfarash A, et al. Global regulation of gene expression by OxyR in an important human opportunistic pathogen. Nucl Acids Res (2012) 40:4320–33. doi: 10.1093/nar/gks017
116. Hahn JS, Oh SY, Roe JH. Role of OxyR as a peroxide-sensing positive regulator in Streptomyces coelicolor A3(2). J Bacteriol (2002) 184:5214–22. doi: 10.1128/JB.184.19.5214-5222.2002
117. Maciver I, Hansen EJ. Lack of expression of the global regulator OxyR in Haemophilus influenzae has a profound effect on growth phenotype. Infect Immun (1996) 64:4618–29. doi: 10.1128/IAI.64.11.4618-4629.1996
118. Whitby PW, Morton DJ, Vanwagoner TM, Seale TW, Cole BK, Mussa HJ, et al. Haemophilus influenzae OxyR: characterization of its regulation, regulon and role in fitness. PLoS One (2012) 7:e50588. doi: 10.1371/journal.pone.0050588
119. Hennequin C, Forestier C. oxyR, a LysR-type regulator involved in Klebsiella pneumoniae mucosal and abiotic colonization. Infect Immun (2009) 77:5449–57. doi: 10.1128/IAI.00837-09
120. Remes B, Berghoff BA, Förstner KU. Role of oxygen and the OxyR protein in response to iron limitation in Rhodobacter sphaeroides. BMC Genomics (2014) 15:794. doi: 10.1186/1471-2164-15-794
121. Niu W, Zhang Y, Liu J, Wen T, Miao T, Basit A, et al. OxyR controls magnetosome formation by regulating magnetosome island (MAI) genes, iron metabolism, and redox state. Free Radic Biol Med (2020) 161:272–82. doi: 10.1016/j.freeradbiomed.2020.10.015
122. Shanks RM, Stella NA, Kalivoda EJ, Doe MR, O’Dee DM, Lathrop KL, et al. A Serratia marcescens OxyR homolog mediates surface attachment and biofilm formation. J Bacteriol (2007) 189:7262–72. doi: 10.1128/JB.00859-07
123. Kim S, Bang Y-J, Kim D, Lim JG, Oh MH, Choi SH. Distinct characteristics of OxyR2, a new OxyR-type regulator, ensuring expression of peroxiredoxin 2 detoxifying low levels of hydrogen peroxide in Vibrio vulnificus. Mol Microbiol (2014) 93:992–1009. doi: 10.1111/mmi.12712
124. Wang H, Naseer N, Chen Y, Zhu AY, Kuai X, Galagedera N, et al. OxyR2 modulates OxyR1 activity and Vibrio cholerae oxidative stress response. Infect Immun (2017) 85:e00929–16. doi: 10.1128/IAI.00929-16
125. Bang YJ, Lee ZW, Kim D, Jo I, Ha NC, Choi SH. OxyR2 functions as a three-state redox switch to tightly regulate production of Prx2, a peroxiredoxin of Vibrio vulnificus. J Biol Chem (2016) 291:16038–47. doi: 10.1074/jbc.M115.710343
126. Sund CJ, Rocha ER, Tzianabos AO, Wells WG, Gee JM, Reott MA, et al. The Bacteroides fragilis transcriptome response to oxygen and H2O2: the role of OxyR and its effect on survival and virulence. Mol Microbiol (2008) 67:129–42. doi: 10.1111/j.1365-2958.2007.06031.x
127. Rocha ER, Owens G Jr., Smith CJ. The redox-sensitive transcriptional activator OxyR regulates the peroxide response regulon in the obligate anaerobe Bacteroides fragilis. J Bacteriol (2000) 182:5059–69. doi: 10.1128/JB.182.18.5059-5069.2000
128. Rocha ER, Herren CD, Smalley DJ, Smith CJ. The complex oxidative stress response of Bacteroides fragilis: the role of OxyR in control of gene expression. Anaerobe (2003) 9:165–73. doi: 10.1016/S1075-9964(03)00118-5
129. Honma K, Mishima E, Inagaki S, Sharma A. The OxyR homologue in Tannerella forsythia regulates expression of oxidative stress responses and biofilm formation. Microbiology (2009) 155:1912–22. doi: 10.1099/mic.0.027920-0
130. Diaz PI, Slakeski N, Reynolds EC, Morona R, Rogers AH, Kolenbrander PE. Role of oxyR in the oral anaerobe Porphyromonas gingivalis. J Bacteriol (2006) 188:2454–62. doi: 10.1128/JB.188.7.2454-2462.2006
131. Vinckx T, Wei Q, Matthijs S, Noben JP, Daniels R, Cornelis P. A proteome analysis of the response of a Pseudomonas aeruginosa oxyR mutant to iron limitation. Biometals (2011) 24:523–32. doi: 10.1007/s10534-010-9403-4
132. Lu Z, Sethu R, Imlay JA. Endogenous superoxide is a key effector of the oxygen sensitivity of a model obligate anaerobe. Proc Natl Acad Sci USA (2018) 115:e3266–75. doi: 10.1073/pnas.1800120115
133. Mishra S, Imlay JA. An anaerobic bacterium, Bacteroides thetaiotaomicron, uses a consortium of enzymes to scavenge hydrogen peroxide. Mol Microbiol (2013) 90:1356–71. doi: 10.1111/mmi.12438
134. Vinckx T, Wei Q, Matthijs S, Cornelis P. The Pseudomonas aeruginosa oxidative stress regulator OxyR influences production of pyocyanin and rhamnolipids: protective role of pyocyanin. Microbiology (2010) 156:678–86. doi: 10.1099/mic.0.031971-0
135. Johnson JR, Clabots C, Rosen H. Effect of inactivation of the global oxidative stress regulator OxyR on the colonization ability of Escherichia coli O1:K1:H7 in a mouse model of ascending urinary tract infection. Infect Immun (2006) 74:461–8. doi: 10.1128/IAI.74.1.461-468.2006
136. Seib KL, Wu HJ, Srikhanta YN, Edwards JL, Falsetta ML, Hamilton AJ, et al. Characterization of the OxyR regulon of Neisseria gonorrhoeae. Mol Microbiol (2007) 63:54–68. doi: 10.1111/j.1365-2958.2006.05478.x
137. Melstrom KA Jr., Kozlowski R, Hassett DJ, Suzuki H, Bates DM, Gamelli RL, et al. Cytotoxicity of Pseudomonas secreted exotoxins requires OxyR expression. J Surg Res (2007) 143:50–7. doi: 10.1016/j.jss.2007.02.046
138. Delaunay A, Isnard A-D, Toledano MB. H2O2 sensing through oxidation of the Yap1 transcription factor. EMBO J (2000) 19:5157–66. doi: 10.1093/emboj/19.19.5157
139. Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell (2002) 111:471–81. doi: 10.1016/S0092-8674(02)01048-6
140. Wood MJ, Andrade EC, Storz G. The redox domain of the Yap1p transcription factor contains two disulfide bonds. Biochemistry (2003) 42:11982–91. doi: 10.1021/bi035003d
141. Wood MJ, Storz S, Tjandra N. Structural basis for redox regulation of Yap1 transcription factor localization. Nature (2004) 430:913–7. doi: 10.1038/nature02790
142. Wong CM, Siu KL, Jin DY. Peroxiredoxin-null yeast cells are hypersensitive to oxidative stress and are genomically unstable. J Biol Chem (2004) 279(22):23207–13. doi: 10.1074/jbc.M402095200
143. Trotter EW, Rand JD, Vickerstaff J, Grant CM. The yeast Tsa1 peroxiredoxin is a ribosome-associated antioxidant. Biochem J (2008) 412:73–80. doi: 10.1042/BJ20071634
144. Avery AM, Avery SV. Saccharomyces cerevisiae expresses three phospholipid hydroperoxide glutathione peroxidases. J Biol Chem (2001) 276:33730–5. doi: 10.1074/jbc.M105672200
145. Li L, Bertram S, Kaplan J, Jia X, Ward DM. The mitochondrial iron exporter genes MMT1 and MMT2 in yeast are transcriptionally regulated by Aft1 and Yap1. J Biol Chem (2020) 295:1716–26. doi: 10.1074/jbc.RA119.011154
146. Stephen DW, Rivers SL, Jamieson DJ. The role of the YAP1 and YAP2 genes in the regulation of the adaptive oxidative stress responses of Saccharomyces cerevisiae. Mol Microbiol (1995) 16:415–23. doi: 10.1111/j.1365-2958.1995.tb02407.x
147. Dumond H, Danielou N, Pinto M, Bolotin-Fukuhara M. A large-scale study of Yap1p-dependent genes in normal aerobic and H2O2-stress conditions. Mol Microbiol (2000) 36:830–45. doi: 10.1046/j.1365-2958.2000.01845.x
148. Carmel-Harel O, Storz G. Roles of the glutathione- and thioredoxin-dependent reduction systems in the Escherichia coli and Saccharomyces cerevisiae responses to oxidative stress. Annu Rev Microbiol (2000) 54:439–61. doi: 10.1146/annurev.micro.54.1.439
149. Boronaat S, Domenech A, Paulo E, Calvo IA, Garcia-Santamarina S, Garcia P, et al. Thiol-based H2O2 signaling in microbial systems. Red Bio (2014) 2:395–9.
150. Chen L, Keramati L, Helmann JD. Coordinate regulation of Bacillus subtilis peroxide stress genes by hydrogen peroxide and metal ions. Proc Natl Acad Sci USA (1995) 92:8190–4. doi: 10.1073/pnas.92.18.8190
151. Helmann JD. Specificity of metal sensing: iron and manganese homeostasis in Bacillus subtilis. J Biol Chem (2014) 289:28112–20. doi: 10.1074/jbc.R114.587071
152. Herbig AF, Helmann JD. Roles of metal ions and hydrogen peroxide in modulating the interaction of the Bacillus subtilis PerR peroxide regulon repressor with operator DNA. Mol Microbiol (2001) 41:849–59. doi: 10.1046/j.1365-2958.2001.02543.x
153. Lee J-W, Helmann JD. Biochemical characterization of the structural Zn2+ site in the Bacillus subtilis peroxide sensor PerR. J Biol Chem (2006) 281:23567–78. doi: 10.1074/jbc.M603968200
154. Lee JW, Helmann JD. The PerR transcription factor senses H2O2 by metal-catalyzed histidine oxidation. Nature (2006) 440:363–7. doi: 10.1038/nature04537
155. Ji CJ, Kim JH, Won YB, Lee YE, Choi TW, Ju SY, et al. Staphylococcus aureus PerR is a hypersensitive hydrogen peroxide sensor using iron-mediated histidine oxidation. J Biol Chem (2015) 290:20374–86. doi: 10.1074/jbc.M115.664961
156. Bsat N, Herbig A, Casillas-Martinez L, Setlow P, Helmann JD. Bacillus subtilis contains multiple Fur homologues: identification of the iron uptake (Fur) and peroxide regulon (PerR) repressors. Mol Microbiol (1998) 29:189–98. doi: 10.1046/j.1365-2958.1998.00921.x
157. Helmann JD, Wu MFW, Gaballa A, Kobel PA, Morshedi MM, Fawcett P, et al. The global transcriptional response of Bacillus subtilis to peroxide stress is coordinated by three transcription factors. J Bacteriol (2003) 185:243–53. doi: 10.1128/JB.185.1.243-253.2003
158. Faulkner MJ, Helmann JD. Peroxide stress elicits adaptive changes in bacterial metal ion homeostasis. Antioxid Redox Signal (2011) 15:175–89. doi: 10.1089/ars.2010.3682
159. Faulkner MJ, Ma Z, Fuangthong M, Helmann JD. Derepression of the Bacillus subtilis PerR peroxide stress response leads to iron deficiency. J Bacteriol (2012) 194:1226–35. doi: 10.1128/JB.06566-11
160. Ahn BE, Baker TA. Oxidization without substrate unfolding triggers proteolysis of the peroxide-sensor, PerR. Proc Natl Acad Sci USA (2016) 113:E23–31. doi: 10.1073/pnas.1522687112
161. Horsburgh MJ, Clements MO, Crossley H, Ingham E, Foster SJ. PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect Immun (2001) 69:3744–54. doi: 10.1128/IAI.69.6.3744-3754.2001
162. Belzer C, van Schendel BA, Hoogenboezem T, Kusters JG, Hermans PW, van Vliet AH, et al. PerR controls peroxide- and iron-responsive expression of oxidative stress defense genes in Helicobacter hepaticus. Eur J Microbiol Immunol (Bp) (2011) 1:215–22. doi: 10.1556/EuJMI.1.2011.3.5
163. King KY, Horenstein JA, Caparon MG. Aerotolerance and peroxide resistance in peroxidase and PerR mutants of Streptococcus pyogenes. J Bacteriol (2000) 182:5290–9. doi: 10.1128/JB.182.19.5290-5299.2000
164. Ricci S, Janulczyk R, Bjorck L. The regulator PerR is involved in oxidative stress response and iron homeostasis and is necessary for full virulence of Streptococcus pyogenes. Infect Immun (2002) 70:4968–76. doi: 10.1128/IAI.70.9.4968-4976.2002
165. Verneuil N, Rince A, Sanguinetti M, Posteraro B, Fadda G, Auffray Y, et al. Contribution of a PerR-like regulator to the oxidative-stress response and virulence of Enterococcus faecalis. Microbiol (Reading) (2005) 151:3997–4004. doi: 10.1099/mic.0.28325-0
166. Kim JH, Ji CJ, Ju SY, Yang YM, Ryu SH, Kwon Y, et al. Bacillus licheniformis contains two more PerR-like proteins in addition to PerR, Fur, and Zur orthologues. PLoS One (2016) 11:e0155539. doi: 10.1371/journal.pone.0155539
167. Liu C, Wang L, Li T, Lin L, Dai S, Tian B, et al. A PerR-like protein involved in response to oxidative stress in the extreme bacterium Deinococcus radiodurans. Biochem Biophys Res Commun (2014) 450:575–80. doi: 10.1016/j.bbrc.2014.06.015
168. Wu HJ, Seib KL, Srikhanta YN, Kidd SP, Edwards JL, Maguire TL, et al. PerR controls Mn-dependent resistance to oxidative stress in Neisseria gonorrhoeae. Mol Microbiol (2006) 60:401–16. doi: 10.1111/j.1365-2958.2006.05079.x
169. Hillmann F, Fischer RJ, Saint-Prix F, Girbal L, Bahl H. PerR acts as a switch for oxygen tolerance in the strict anaerobe Clostridium acetobutylicum. Mol Microbiol (2008) 68:848–60. doi: 10.1111/j.1365-2958.2008.06192.x
170. Kim JC, Oh E, Hwang S, Ryu S, Jeon B. Non-selective regulation of peroxide and superoxide resistance genes by PerR in Campylobacter jejuni. Front Microbiol (2015) 6:126. doi: 10.3389/fmicb.2015.00126
171. Brenot A, King KY, Caparon MG. The PerR regulon in peroxide resistance and virulence of Streptococcus pyogenes. Mol Microbiol (2005) 55(1):221–34. doi: 10.1111/j.1365-2958.2004.04370.x
172. Gryllos I, Grifantini R, Colaprico A, Cary ME, Hakansson A, Carey DW, et al. PerR confers phagocytic killing resistance and allows pharyngeal colonization by group A Streptococcus. PLoS Pathog (2008) 4:e1000145. doi: 10.1371/journal.ppat.1000145
173. Morrissey JA, Cockayne A, Brummell K, Williams P. The staphylococcal ferritins are differentially regulated in response to iron and manganese and via PerR and Fur. Infect Immun (2004) 72:972–9. doi: 10.1128/IAI.72.2.972-979.2004
174. Rowley G, Hensen D, Felgate H, Arkenberg A, Appia-Ayme C, Prior K, et al. Resolving the contributions of the membrane-bound and periplasmic nitrate reductase systems to nitric oxide and nitrous oxide production in Salmonella enterica serovar Typhimurium. Biochem J (2012) 441:755–62. doi: 10.1042/BJ20110971
175. Hassan J, Bergaust LL, Molstad L, de Vries S, Bakken LR. Homeostatic control of nitric oxide (NO) at nanomolar concentrations in denitrifying bacteria—Modeling and experimental determination of NO reductase kinetics in vivo in Paracoccus denitrificans. Environ Microbiol (2016) 18:2964–78. doi: 10.1111/1462-2920.13129
176. Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol (2004) 2:820–32. doi: 10.1038/nrmicro1004
177. Gardner PR, Constantino G, Szabo C, Salzman AL. Nitric oxide sensitivity of aconitases. J Biol Chem (1997) 272:25071–6. doi: 10.1074/jbc.272.40.25071
178. Hyduke DR, Jarboe LR, Tran LM, Chou KJ, Liao JC. Integrated network analysis identifies nitic oxide response networks and dihydroxyacid dehydratase as a crucial target in Escherichia coli. Proc Natl Acad Sci USA (2007) 104:8484–9. doi: 10.1073/pnas.0610888104
179. Stevanin TM, Ioannidis N, Mills CE, Kim SO, Hughes MN, Poole RK. Flavohemoglobin Hmp affords inducible protection for Escherichia coli respiration, catalyzed by cytochromes bo‘ or bd, from nitric oxide. J Biol Chem (2000) 275:35868–75. doi: 10.1074/jbc.M002471200
180. Radi R. Reactions of nitric oxide with metalloproteins. Chem Res Toxicol (1996) 9:828–35. doi: 10.1021/tx950176s
181. Tucker NP, D’Autreaux B, Yousafzai FK, Fairhurst SA, Spiro S, Dixon R. Analysis of the nitric oxide-sensing non-heme iron center in the NorR regulatory protein. J Biol Chem (2008) 283:908–18. doi: 10.1074/jbc.M705850200
182. Bodenmiller DM, Spiro S. The yjeB (nsrR) gene of Escherichia coli encodes a nitric oxide sensitive transcriptional regulator. J Bacteriol (2006) 188:874–81. doi: 10.1128/JB.188.3.874-881.2006
183. Karlinsey JE, Bang I-S, Becker LA, Frawley ER, Porwollik S, Robbins HF, et al. The NsrR regulon in nitrosative stress resistance of Salmonella enterica serovar Typhimurium. Mol Microbiol (2012) 85:1179–93. doi: 10.1111/j.1365-2958.2012.08167.x
184. Dunn AK, Karr EA, Wang Y, Batton AR, Ruby EG, Stabb EV. The alternative oxidase (AOX) gene in Vibrio fischeri is controlled by NsrR and upregulated in response to nitric oxide. Mol Microbiol (2010) 77:44–55. doi: 10.1111/j.1365-2958.2010.07194.x
185. Ischiropoulos H, Zhu L, Beckman JS. Peroxynitrite formation from macrophage-derived nitric oxide. Arch Biochem Biophys (1992) 298:446–51. doi: 10.1016/0003-9861(92)90433-w
186. Seth D, Hausladen A, Wang YJ, Stamler JS. Endogenous protein S-nitrosylation of E. coli: regulation by OxyR. Science (2012) 336:470–3. doi: 10.1126/science.1215643
187. Kim SO, Merchant K, Nudelman R, Beyer WF Jr., Keng T, DeAngelo J, et al. OxyR: a molecular code for redox-related signaling. Cell (2002) 109:383–96. doi: 10.1016/s0092-8674(02)00723-7
188. Pullan ST, Gidley MD, Jones RA, Barrett J, Stevanin TM, Read RC, et al. Nitric oxide in chemostat-cultured Escherichia coli is sensed by Fnr and other global regulators: unaltered methionine biosynthesis indicates lack of S nitrosation. J Bacteriol (2007) 189:1845–55. doi: 10.1128/JB.01354-06
189. Mukhopadhyay P, Zheng M, Bedzyk LA, LaRossa RA, Storz G. Prominent roles of the NorR and Fur regulators in the Escherichia coli transcriptional response to reactive nitrogen species. Proc Natl Acad Sci USA (2004) 101:745–50. doi: 10.1073/pnas.0307741100
190. Lushchak OV, Inoue Y, Lushchak VI. Regulatory protein Yap1 is involved in response of yeast Saccharomyces cerevisiae to nitrosative stress. Biochem (Mosc) (2010) 75:629–64. doi: 10.1134/S0006297910050135
191. Horan S, Bourges I, Meunier B. Transcriptional response to nitrosative stress in Saccharomyces cerevisiae. Yeast (2006) 23:519–35. doi: 10.1002/yea.1372
192. Moore CM, Nakano MM, Wang T, Ye RW, Helmann JD. Response of Bacillus subtilis to nitric oxide and the nitrosating agent sodium nitroprusside. J Bacteriol (2004) 186:4655–64. doi: 10.1128/JB.186.14.4655-4664.2004
193. Kosower NS, Kosower EM, Wertheim B. Diamide, a new reagent for the intracellular oxidation of glutathione to the disulfide. Biochem Biophys Res Commun (1969) 37:593–6. doi: 10.1016/0006-291X(69)90850-X
194. Zuber P. Management of oxidative stress in Bacillus. Annu Rev Microbiol (2009) 63:575–97. doi: 10.1146/annurev.micro.091208.073241
195. Park J-H, Lee J-H, Roe J-H. SigR, a hub of multilayered rgulation of redox and antibiotic stress responses. Mol Microbiol (2019) 112:420–31. doi: 10.1111/mmi.14341
196. Korshunov S, Imlay KRC, Imlay JA. Cystine import is a valuable but risky process whose hazards Escherichia coli minimizes by inducing a cysteine exporter. Mol Microbiol (2020) 113:22–39. doi: 10.1111/mmi.14403
197. Muller A, Eller J, Albrecht F, Prochnow P, Kuhlmann K, Bandow JE, et al. Allicin induces thiol stress in bacteria through S-allylmercapto modification of protein cysteines. J Biol Chem (2016) 291:11477–90. doi: 10.1074/jbc.M115.702308
Keywords: reactive oxygen species, OxyR regulator, peroxide sensing repressor (PerR), Yap1p, nitric oxide
Citation: Sen A and Imlay JA (2021) How Microbes Defend Themselves From Incoming Hydrogen Peroxide. Front. Immunol. 12:667343. doi: 10.3389/fimmu.2021.667343
Received: 12 February 2021; Accepted: 19 March 2021;
Published: 27 April 2021.
Edited by:
Michael Walch, Université de Fribourg, SwitzerlandCopyright © 2021 Sen and Imlay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James A. Imlay, amltbGF5QGlsbGlub2lzLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.