
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 07 September 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.667097
This article is part of the Research TopicNew Biomarkers for the Diagnosis and Treatment of Systemic Lupus ErythematosusView all 16 articles
 Kun Xiang1,2†
Kun Xiang1,2† Peng Wang3†
Peng Wang3† Zhiwei Xu4†Yu-Qian Hu1,2Yi-Sheng He1,2Yue Chen1,2Ya-Ting Feng1,2
Zhiwei Xu4†Yu-Qian Hu1,2Yi-Sheng He1,2Yue Chen1,2Ya-Ting Feng1,2 Kang-Jia Yin1,2Ji-Xiang Huang1,2Jie Wang1,2Zheng-Dong Wu1,2Xiao-Ke Yang5De-Guang Wang6
Kang-Jia Yin1,2Ji-Xiang Huang1,2Jie Wang1,2Zheng-Dong Wu1,2Xiao-Ke Yang5De-Guang Wang6 Dong-Qing Ye1,2*
Dong-Qing Ye1,2* Hai-Feng Pan1,2*
Hai-Feng Pan1,2*The observational association between gut microbiome and systemic lupus erythematosus (SLE) has been well documented. However, whether the association is causal remains unclear. The present study used publicly available genome-wide association study (GWAS) summary data to perform two-sample Mendelian randomization (MR), aiming to examine the causal links between gut microbiome and SLE. Two sets of MR analyses were conducted. A group of single nucleotide polymorphisms (SNPs) that less than the genome-wide statistical significance threshold (5 × 10-8) served as instrumental variables. To obtain a comprehensive conclusion, the other group where SNPs were smaller than the locus-wide significance level (1 × 10-5) were selected as instrumental variables. Based on the locus-wide significance level, the results indicated that there were causal effects of gut microbiome components on SLE risk. The inverse variance weighted (IVW) method suggested that Bacilli and Lactobacillales were positively correlated with the risk of SLE and Bacillales, Coprobacter and Lachnospira were negatively correlated with SLE risk. The results of weighted median method supported that Bacilli, Lactobacillales, and Eggerthella were risk factors for SLE and Bacillales and Coprobacter served as protective factors for SLE. The estimates of MR Egger suggested that genetically predicted Ruminiclostridium6 was negatively associated with SLE. Based on the genome-wide statistical significance threshold, the results showed that Actinobacteria might reduce the SLE risk. However, Mendelian randomization pleiotropy residual sum and outlier (MR-PRESSO) detected significant horizontal pleiotropy between the instrumental variables of Ruminiclostridium6 and outcome. This study support that there are beneficial or detrimental causal effects of gut microbiome components on SLE risk.
Systemic lupus erythematosus (SLE) is an autoimmune connective tissue disease involving multiple organs, and it presents with a range of clinical symptoms, including skin rash, pericarditis, nephritis, and neurological and hematological involvement. Loss of tolerance to autoantigens is one of the hallmarks of SLE. Genetic, hormonal, and environmental factors interact in susceptible individuals, resulting in autoantibodies deposition and abnormal production of proinflammatory cytokines (1). In addition, ultraviolet light and infections induce DNA damage and apoptosis which increase exposure to autoantigens are potential triggers for SLE as well (2). The current treatment strategy is mainly the use of non-selective immunosuppressive agents. Long-term use of immunosuppressants weakens the immunity and results in severe infections (3). Therefore, it is imperative to explore the etiology of SLE to facilitate the development of treatment strategies with low damage or even no side effects.
Recently, the causal link between the gut microbiome composition and SLE risk has attracted widespread attention. The intestinal microbiota plays a critical role in the maturation of the host immune response and provide protection against pathogen overgrowth (4). A study demonstrated that the gut microbiome was related to the dynamics of human immune cells, suggesting that the gut microbiome drove the modulation of the immune system (5). The dysbiosis of gut microbiome affected immune responses, which contributed to the occurrence of autoimmune diseases (6). One possible explanation was that the presence of commensal gut microbiome influenced the autoimmune responses to nuclear antigens (7). Several studies indicated that SLE patients had dysbiosis of gut microbiome and decreased species richness (8, 9). Furthermore, the decrease in species diversity was particularly significant in patients with high SLE activity index (10), suggesting that intestinal flora might be involved in the immune pathogenesis of autoimmune diseases. Nevertheless, it remains unclear as to whether there is a causal relationship between gut microbiome and SLE.
Mendelian randomization (MR) is an approach integrating summary data of genome-wide association study (GWAS), and hence, the impact of confounding factors (e.g., environment) is minimized. MR is a common method to infer whether there are causal relationships between exposure and complex outcomes. Genetic variants that are significantly related to exposure are selected as instrumental variables to infer the causality (11). The instrumental variables that affect the exposure will affect the results proportionally if the exposure is causal. In the current study, the two-sample MR was conducted to examine if there is a causal relationship between gut microbiome composition and SLE risk.
Single-nucleotide polymorphisms (SNPs) related to human gut microbiome composition were selected as instrumental variables from a GWAS with 18,473 individuals, including 122,110 variant sites (12). It was a multi-ethnic large-scale GWAS that recruited 25 population-based cohorts from the United States, Canada, Israel, the Netherlands, Belgium, Sweden, South Korea, Germany, Denmark, Finland, and the UK to explore the association between autosomal human genetic variants and the gut microbiome. Effect estimates of the SNPs related to SLE risk were extracted from a large SLE GWAS, which involved 7,219 cases and 15,991 controls of European ancestry (13).
To ensure the authenticity and accuracy of the conclusions on the causal link between gut microbiome and SLE risk, the following quality control steps were used to select optimal instrument variables. First, SNPs significantly related to gut microbiome were selected as instrumental variables. Two thresholds were used to select the instrumental variable. A set of SNPs less than the genome-wide statistical significance threshold (5 × 10-8) served as instrumental variables. In order to obtain more comprehensive results, the other group where SNPs are smaller than the locus-wide significance level (1 × 10-5) was selected as instrumental variables. Second, the minor allele frequency (MAF) threshold of variants of interest was 0.01. Third, one of the principles of the MR approach is that there is no linkage disequilibrium (LD) among the included instrumental variables, since the presence of strong LD might result in biased results. In the current study, the clumping process (R2 < 0.001 and clumping distance = 10,000kb) were conducted to assess the LD between the included SNPs. Fourth, an important step of MR is to ensure that the effects of the SNPs on the exposure correspond to the same allele as the effects on the outcome. In accordance with the principle, palindromic SNPs would not be included in the instrumental variables. Fifth, when SNPs related to exposure were absent in the outcome GWAS, the proxy SNPs significantly associated with the variants of interest were selected (r2 > 0.8).
To minimize the impact of bias on the results, the MR method must conform to three important assumptions. First, instrumental variables are independent of confounders that influence exposure and outcome. Second, the variants of interest used in the analysis should be significantly associated with exposure. F statistic is generally performed to assess the strength of the relevance between instrumental variables and exposure. The formula of F statistic is F = R2(n-k-1)/k(1-R2). R2 represents the exposure variance explained by the selected SNPs, n is the sample size, and k represent the number of included instrumental variables. If F is less than 10, there is a weak association between instrumental variables and exposure. Third, instrumental variables affect outcomes only through exposure, which means that there is no horizontal pleiotropy effect between instrumental variables and outcome.
In the current study, high-efficiency methods including inverse variance weighted (IVW), MR-Egger, weighted median, and weighted mode were used to infer whether there was causal effect of human gut microbiome composition on SLE risk. IVW is essentially a meta-analysis method, which converts to a weighted regression of the outcome effects of instrumental variables on the exposure effects to obtain an overall estimate of the impact of gut microbiome on the risk of SLE, where the intercept is limited to zero (14). When there is no horizontal pleiotropy, IVW can avoid the impact of confounding factors to obtain unbiased estimates. MR-Egger may be strongly influenced by outlying genetic variables, leading to inaccurate estimates. However, even if all selected instrumental variables are invalid, the MR-Egger method can still provide unbiased estimates. The weighted median can provide consistent estimates of the causal effects, even if as many as 50% of the information in the analysis comes from variations of interest are invalid instrumental variables. The weighted median method has some important advantages over the MR-Egger since it improves the accuracy of the results. When most instrumental variables with similar causal estimates are valid, the weighted mode approach is still valid even if the other instrumental variables do not meet the requirements of MR method for causal inference (15).
The MR-Egger regression was conducted to assess whether the included SNPs had potential horizontal pleiotropic effects. MR-Egger regression is a method, which has the property that both detect and adjust for pleiotropy in the MR analysis, and get a causal effect estimate (16) and examine whether the results are driven by the directional horizontal pleiotropy (17). Given the lower accuracy and statistical power of MR-Egger regression, Mendelian randomization pleiotropy residual sum and outlier (MR-PRESSO) was performed to detect any outliers reflecting likely pleiotropic biases and correct horizontal pleiotropy. Furthermore, Cochran’s Q statistic was used to quantify the heterogeneity among the selected SNPs. To determine whether there were potential strong influence SNPs, the leave-one-out sensitivity analysis was performed to verify the reliability and stability of the causal effect estimates. Statistical analyses were performed using R software (version 4.0.2, TwoSampleMR package).
Initially, 14,587 (locus-wide significance level, P < 1 × 10-5) and 456 (genome-wide statistical significance threshold, P < 5 × 10-8) SNPs were identified as instrumental variables from a large-scale GWAS. It contained 211 bacterial traits, including five biological classifications: phylum, class, order, family, and genus. After removing SNPs that had LD effects and independence from SLE, 2,105 (P < 1 × 10-5) and 13 (P < 5 × 10-8) SNPs were selected as instrumental variables. The main information of SNPs including effect allele, other allele, beta, SE, and P value were collected systematically for further analysis.
The results of IVW analyses demonstrated that Bacilli (odds ratio (OR) = 1.40, 95% confidence interval (CI), 1.02–1.93, P = 0.037) and Lactobacillales (OR = 1.40, 95% CI, 1.01–1.95, P = 0.045) were positively correlated with the risk of SLE and Bacillales (OR = 0.85, 95% CI, 0.74–0.98, P = 0.022), Coprobacter (OR = 0.78, 95% CI, 0.64–0.95, P = 0.014), and Lachnospira (OR = 0.60, 95% CI, 0.38–0.94, P = 0.027) were negatively correlated with SLE risk (Table 1). The MR estimates of weighted median indicated that Bacilli (OR = 1.59, 95% CI, 1.06–2.39, P = 0.027), Lactobacillales (OR = 1.73, 95% CI, 1.13–2.64, P = 0.011), and Eggerthella (OR = 1.41, 95% CI, 1.05–1.90, P = 0.022) were risk factors for SLE, and Bacillales (OR = 0.81, 95% CI, 0.67–0.96, P = 0.018) and Coprobacter (OR = 0.77, 95% CI, 0.59–0.99, P = 0.043) served as protective factors for SLE (Table 1). The estimates of MR Egger suggested that genetically predicted Ruminiclostridium6 were negatively associated with SLE (OR = 0.35, 95% CI, 0.15–0.83, P = 0.040). The detailed statistical results of the 211 intestinal floras were shown in Supplementary Table S1.
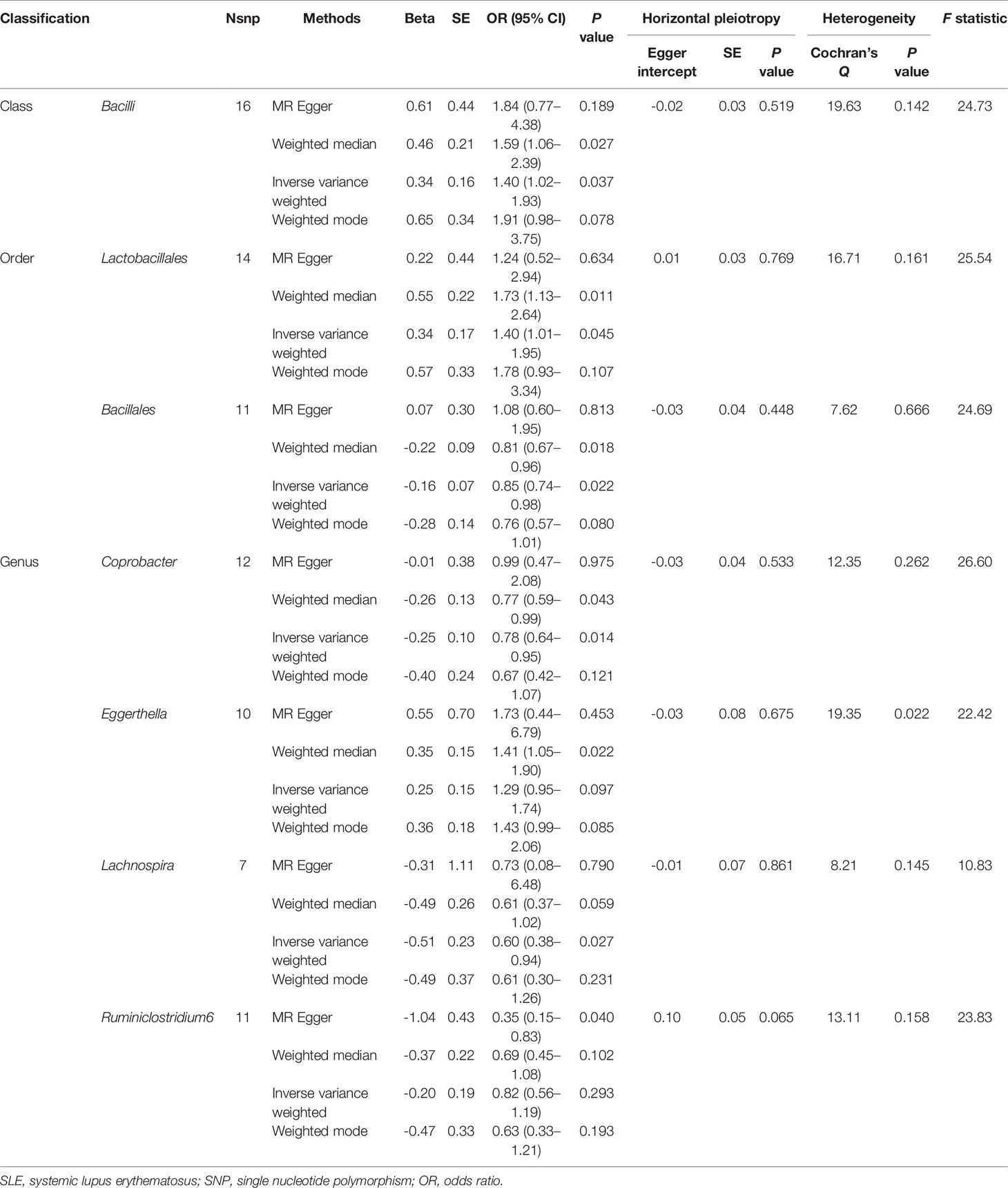
Table 1 MR results of causal links between gut microbiome and SLE risk (P < 1 × 10-5).
The horizontal pleiotropy between instrumental variables and outcome was assessed by MR-Egger regression, and the results showed that there was no evidence of horizontal pleiotropy (Table 1). No outliers were found in the analysis of Bacilli (P = 0.191), Lactobacillales (P = 0.213), Bacillales (P = 0.403), Coprobacter (P = 0.365), and Lachnospira (P = 0.301) by MR-PRESSO. MR-PRESSO suggested that there was significant horizontal pleiotropy between the instrumental variables of Eggerthella and outcome (P = 0.041), and rs1784446 was identified as outlier. However, the results did not change significantly after removing the SNP (OR = 1.42, 95% CI, 1.06–1.90, P = 0.020). In the analysis of Ruminiclostridium6, MR-PRESSO found there was significant horizontal pleiotropy (P = 0.045) and rs61060922 was identified as a pleiotropic SNP. After removing the outlier, the results changed substantially (OR = 0.48, 95% CI, 0.22–1.04, P = 0.101). The detailed information of the instrumental variables was shown in Table 2. The F statistics of the SNPs were all greater than 10, indicating that there was no weak instrumental variables bias (Table 1). Thus, the two-sample MR estimates found that Bacilli (Figure 1), Eggerthella (Supplementary Figure S1), and Lactobacillales (Supplementary Figure S2) were positively related to SLE risk, and Coprobacter (Supplementary Figure S3), Bacillales (Supplementary Figure S4), and Lachnospira (Supplementary Figure S5) played protective roles in the pathogenesis of SLE.
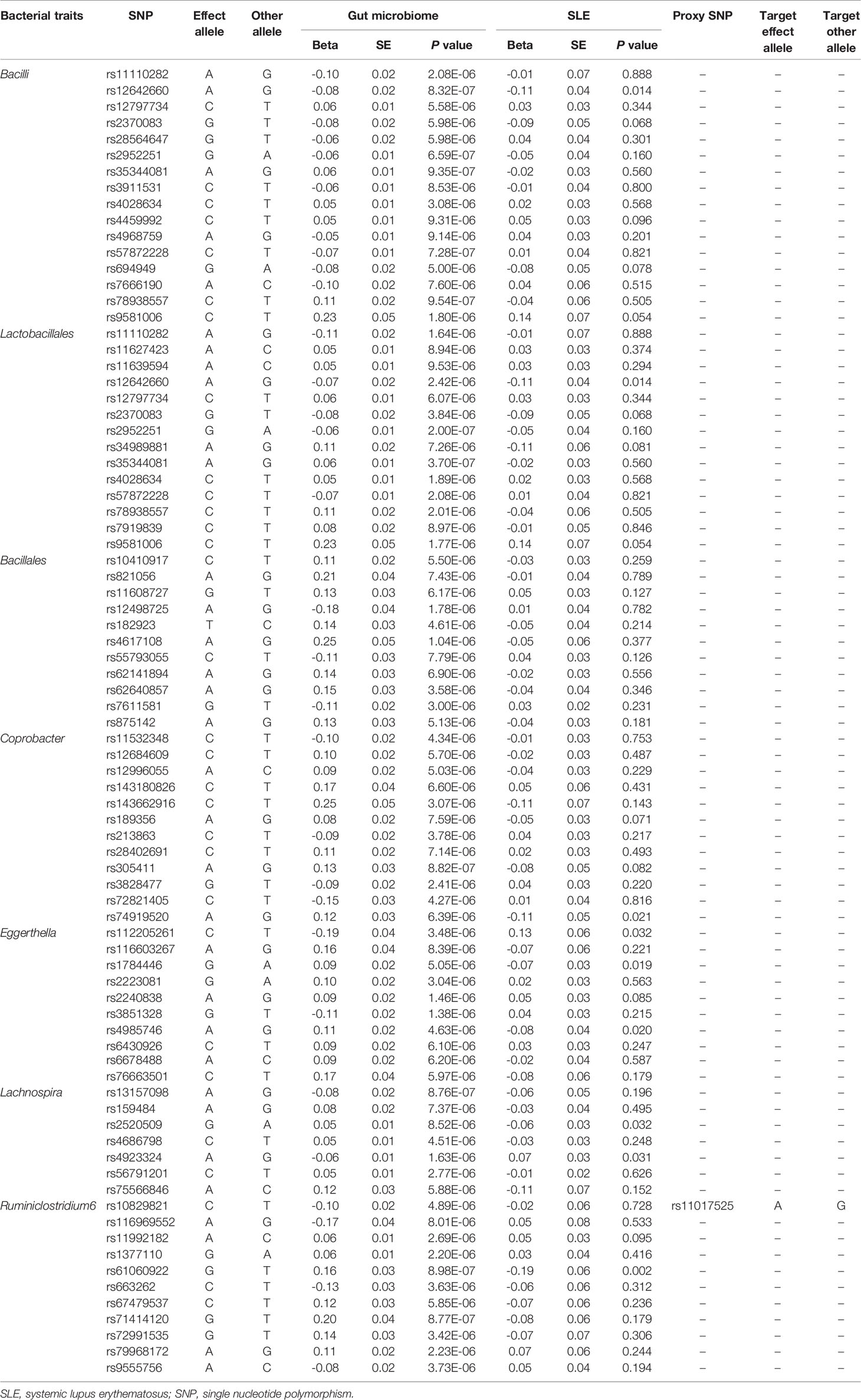
Table 2 SNPs used as instrumental variables from gut microbiome and SLE GWASs (P < 1 × 10-5).
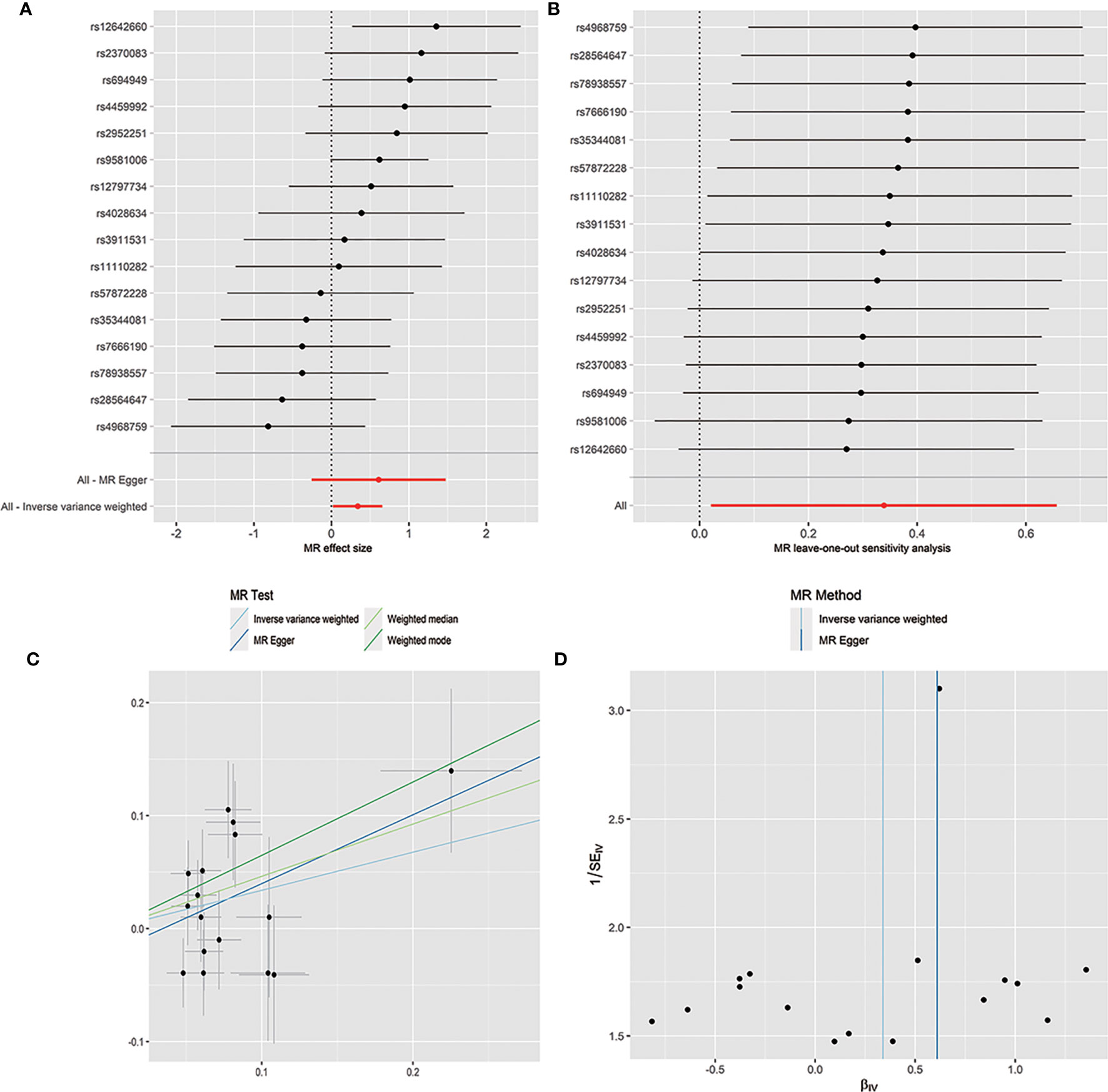
Figure 1 Forest plot (A), sensitivity analysis (B), scatter plot (C), and funnel plot (D) of the causal effect of Bacilli on SLE risk.
When MR analysis was performed with gut microbiome as a whole, the results of IVW (OR = 1.20, 95% CI, 0.96–1.52, P = 0.114), MR Egger (OR = 0.74, 95% CI, 0.34–1.58, P = 0.448), weighted median (OR = 1.15, 95% CI, 0.92–1.43, P = 0.221), and weighted mode (OR = 1.26, 95% CI, 0.98–1.63, P = 0.099) showed that gut microbiome was not associated with SLE risk (Table 3 and Supplementary Figure S6). The detailed information of the instrumental variables was shown in Supplementary Table S2. MR-Egger regression showed that there was no horizontal pleiotropy between instrumental variables and outcome (P = 0.213). In addition, the results of Cochrane Q statistics showed no significant heterogeneity (P = 0.052) and the F statistics was greater than 10. The results of gut microbiome classification indicated that Actinobacteria might reduce the risk of SLE (OR = 0.52, 95% CI, 0.29–0.95, P = 0.033) (Table 3). Heterogeneity and horizontal pleiotropy could not be examined due to the limited number of included SNPs.
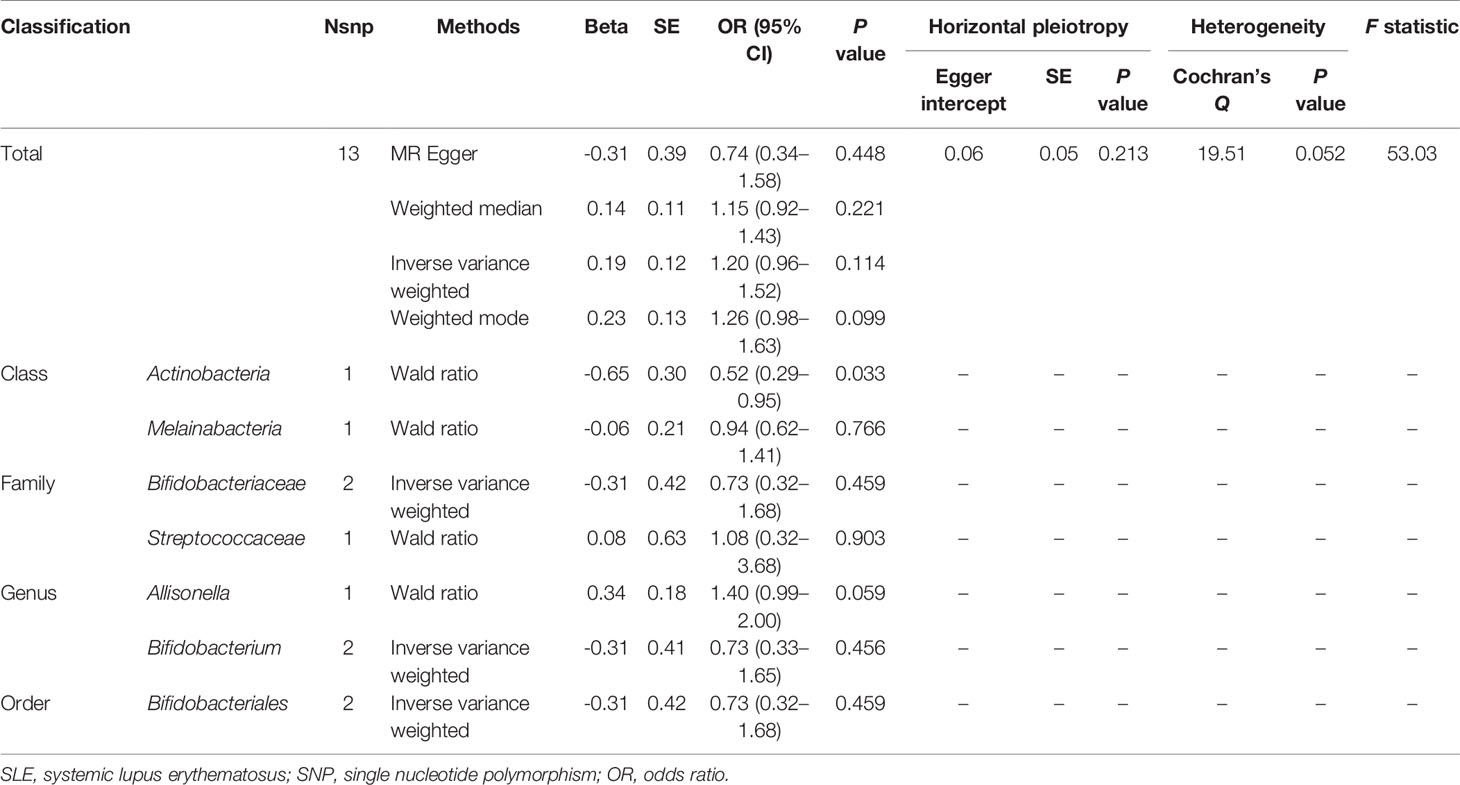
Table 3 MR results of causal links between gut microbiome and SLE risk (P < 5 × 10-8).
This two-sample MR study suggested that the levels of Bacillales, Coprobacter, Lachnospira, and Actinobacteria were negatively related to the risk of SLE, and Bacilli, Lactobacillales, and Eggerthella might be the risk factors for SLE onset. However, since there were fewer instrumental variables reaching genome-wide statistical significance threshold, the results and the precision of Actinobacteria might have been compromised.
The gastrointestinal mucosal surface of the body is abundantly colonized by trillions of symbiotic gut microbiome which participate in the modulation and maintenance of the host immune system. Therefore, the dysbiosis of gut microbiome interacts with the intestinal mucosal immune system closely (6). Several studies found that autoimmune diseases were often accompanied by gut microbiome dysbiosis or altered microbiome. The distribution of microbes from phylum to genus levels of different taxa was different between healthy subjects and early rheumatoid arthritis (RA) patients, and the difference in microbial diversity and classification indicated that gut microbes might be involved in the pathogenesis of early RA (18). Compared with healthy controls, RA patients had varying degrees of alterations in gut microbiome composition, including Bacteroides (19, 20), Prevotella (21), Verrucomicrobiae (22), and Salivarius (23). Jangi et al. (23) found an increase in Methanobrevibacter and Akkermansia in multiple sclerosis (MS) patients, and Methanobrevibacter was involved in the immunomodulatory process due to its ability to recruit inflammatory cells (24). As an autoimmune disease closely related to intestinal microbes, the occurrence of inflammatory bowel disease (IBD) was often accompanied by gut microbiome dysbiosis. A study suggested that the human intestinal microbiome had an important influence on the drug metabolism and efficacy of IBD (25). Currently, there are limited studies on the association between the candidate intestinal bacteria found in this study and complex traits. Some studies indicated that compared with healthy controls, the abundance of Bacilli was increased in encephalitis (26) and Graves’ disease patients (27). Increased Lactobacillales abundance was observed in autoimmune liver disease (28), atopic dermatitis (29), type 1 diabetes (30), and Graves’ disease (27) patients. These studies indicated that the Bacilli and Lactobacillales might have the effects of promoting inflammation. The abundance of Lachnospira was decreased in ankylosing spondylitis (31), type 1 diabetes (32), and IgE-associated allergic disease (33) patients, and the Lachnospira contributed to the alleviation of inflammation in HIV-infected patients (34), suggesting that Lachnospira might have a protective role in inflammatory conditions. These results were consistent with the present study. However, the mechanisms by which these intestinal floras exert beneficial or detrimental effects on the immune-mediated inflammatory disease remain to be further studied.
Recently, numerous human and rodent model studies have been conducted to infer the association between SLE and gut microbiome. A study found that SLE patients, especially those in the active phase, had dysbiosis in the intestinal flora (35). Luo et al. (36) found that the microbiome of active SLE patients changed compared with the non-SLE controls and the use of non-selective immunosuppressive therapies, such as dexamethasone and azathioprine, might have a broad impact on the diversity and abundance of gut microbiome. A study indicated that primary Sjögren’s syndrome (pSS) and SLE patients shared similar alterations in the composition of gut microbiome, both showing a lower bacterial abundance and Firmicutes/Bacteroidetes ratio and a higher Bacteroides species richness, which could distinguish patients from individuals in the general population (37). A rodent model indicated that there were significant differences in the composition of gut microbiome between pre-disease and diseased NZB/W F1 mice, as well as between untreated group and immunosuppressive drug treatment group (36). With the progression of diseases and drug treatment, the microbiome tended to become more diverse. The fecal microbiome of SLE mice induced the production of anti-dsDNA antibodies and stimulated inflammation, and changed the expression of SLE susceptibility genes in germfree mice (38). Consistently, Choi et al. (39) demonstrated that when transferred to sterile syngeneic C57BL/6 mice, the intestinal microbes of triple congenic lupus-prone mice stimulated autoantibodies production and modulated immune cells. Intriguingly, the horizontal transfer of intestinal flora between co-bred triple congenic lupus-prone mice and syngeneic mice could mitigate the autoimmune pathogenesis.
However, even though most studies showed that SLE patients were usually accompanied by gut microbiome dysbiosis, it might only be a clinical sign of SLE and there was no causal effect on SLE risk and gut microbiome dysbiosis. First, the use of non-selective immunosuppressive agents in SLE patients could lead to alterations in gut microbiome. Second, the intestinal flora of patients with active and inactive SLE might be different, and many studies did not take into account grouping of patients. Third, the composition of gut microbiome might be different due to the inconsistency of gender ratio and ethnicities in different studies. Fourth, although studies found that SLE patients had the phenotype of gut microbiome dysbiosis, the results of changes in specific strains were not consistent. The existence of these uncertain factors obstructed the inference of the causal link between gut microbiome and SLE risk.
The main advantage of this study was that the implementation of MR approach diminished the interference of confounding factors and reverse causality on the results, which might be more convincing than observational studies. To the best of our knowledge, the study is the first MR analysis on this topic. However, some limitations should be mentioned. First, our study was unable to determine whether overlapping participants were involved in the exposure and outcome GWAS used in the two sample MR analyses. Nevertheless, the deviation from participants overlap could be minimized by the F statistic (40). Second, since the original research lacked demographic data (e.g., gender and race), further subgroup analysis was impossible. Third, in view of the biological plausibility and the multi-stage statistical process, applying a rigorous multiple testing correction would likely have been overly conservative, which may neglect potential strains that are causally related to SLE. Therefore, we did not account for multiple testing. Fourth, since the majority of participants in the GWAS were of European ancestry, extrapolation of the results of the study to other ethnic groups might be limited.
In summary, this MR study suggests causal effects of gut microbiome on SLE. Several types of intestinal bacteria identified in this study that potentially reduced the occurrence of SLE may have the prospects for the prevention and treatment of SLE.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
H-FP and D-QY conceived the presented idea. KX, PW, and ZX performed the computations and manuscript writing. Y-QH, Y-SH, YC, Y-TF, K-JY, and J-XH were involved in acquisition of data. JW, Z-DW, X-KY, and D-GW were involved in interpretation of data. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.667097/full#supplementary-material
Supplementary Figure 1 | Forest plot (A), sensitivity analysis (B), scatter plot (C), and funnel plot (D) of the causal effect of Eggerthella on SLE risk.
Supplementary Figure 2 | Forest plot (A), sensitivity analysis (B), scatter plot (C), and funnel plot (D) of the causal effect of Lactobacillales on SLE risk.
Supplementary Figure 3 | Forest plot (A), sensitivity analysis (B), scatter plot (C), and funnel plot (D) of the causal effect of Coprobacter on SLE risk.
Supplementary Figure 4 | Forest plot (A), sensitivity analysis (B), scatter plot (C), and funnel plot (D) of the causal effect of Bacillales on SLE risk.
Supplementary Figure 5 | Forest plot (A), sensitivity analysis (B), scatter plot (C), and funnel plot (D) of the causal effect of Lachnospira on SLE risk.
Supplementary Figure 6 | Forest plot (A), sensitivity analysis (B), scatter plot (C), and funnel plot (D) of the causal effect of the whole gut microbiome on SLE risk (P < 5 × 10-8).
1. Postal M, Vivaldo JF, Fernandez-Ruiz R, Paredes JL, Appenzeller S, Niewold TB. Type I Interferon in the Pathogenesis of Systemic Lupus Erythematosus. Curr Opin Immunol (2020) 67:87–94. doi: 10.1016/j.coi.2020.10.014
2. Kim JW, Kwok SK, Choe JY, Park SH. Recent Advances in Our Understanding of the Link Between the Intestinal Microbiota and Systemic Lupus Erythematosus. Int J Mol Sci (2019) 20(19):4871. doi: 10.3390/ijms20194871
3. Tektonidou MG, Wang Z, Dasgupta A, Ward MM. Burden of Serious Infections in Adults With Systemic Lupus Erythematosus: A National Population-Based Study, 1996-2011. Arthritis Care Res (Hoboken) (2015) 67(8):1078–85. doi: 10.1002/acr.22575
4. Lynch SV, Pedersen O. The Human Intestinal Microbiome in Health and Disease. N Engl J Med (2016) 375(24):2369–79. doi: 10.1056/NEJMra1600266
5. Schluter J, Peled JU, Taylor BP, Markey KA, Smith M, Taur Y, et al. The Gut Microbiota Is Associated With Immune Cell Dynamics in Humans. Nature (2020) 588(7837):303–7. doi: 10.1038/s41586-020-2971-8
6. Xu H, Zhao H, Fan D, Liu M, Cao J, Xia Y, et al. Interactions Between Gut Microbiota and Immunomodulatory Cells in Rheumatoid Arthritis. Mediators Inflamm (2020) 2020:1430605. doi: 10.1155/2020/1430605
7. Van Praet JT, Donovan E, Vanassche I, Drennan MB, Windels F, Dendooven A, et al. Commensal Microbiota Influence Systemic Autoimmune Responses. EMBO J (2015) 34(4):466–74. doi: 10.15252/embj.201489966
8. Hevia A, Milani C, López P, Cuervo A, Arboleya S, Duranti S, et al. Intestinal Dysbiosis Associated With Systemic Lupus Erythematosus. mBio (2014) 5(5):e01548–14. doi: 10.1128/mBio.01548-14
9. He Z, Shao T, Li H, Xie Z, Wen C. Alterations of the Gut Microbiome in Chinese Patients With Systemic Lupus Erythematosus. Gut Pathog (2016) 8:64. doi: 10.1186/s13099-016-0146-9
10. Azzouz D, Omarbekova A, Heguy A, Schwudke D, Gisch N, Rovin BH, et al. Lupus Nephritis Is Linked to Disease-Activity Associated Expansions and Immunity to a Gut Commensal. Ann Rheum Dis (2019) 78(7):947–56. doi: 10.1136/annrheumdis-2018-214856
11. Dan YL, Wang P, Cheng Z, Wu Q, Wang XR, Wang DG, et al. Circulating Adiponectin Levels and Systemic Lupus Erythematosus: A Two-Sample Mendelian Randomization Study. Rheumatol (Oxford) (2020) 60(2):940–6. doi: 10.1093/rheumatology/keaa506
12. Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A, et al. Large-Scale Association Analyses Identify Host Factors Influencing Human Gut Microbiome Composition. Nat Genet (2021) 53(2):156–65. doi: 10.1038/s41588-020-00763-1
13. Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, et al. Genetic Association Analyses Implicate Aberrant Regulation of Innate and Adaptive Immunity Genes in the Pathogenesis of Systemic Lupus Erythematosus. Nat Genet (2015) 47(12):1457–64. doi: 10.1038/ng.3434
14. Choi KW, Chen CY, Stein MB, Klimentidis YC, Wang MJ, Koenen KC, et al. Assessment of Bidirectional Relationships Between Physical Activity and Depression Among Adults: A 2-Sample Mendelian Randomization Study. JAMA Psychiatry (2019) 76(4):399–408. doi: 10.1001/jamapsychiatry.2018.4175
15. Ooi BNS, Loh H, Ho PJ, Milne RL, Giles G, Gao C, et al. The Genetic Interplay Between Body Mass Index, Breast Size and Breast Cancer Risk: A Mendelian Randomization Analysis. Int J Epidemiol (2019) 48(3):781–94. doi: 10.1093/ije/dyz124
16. Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the Suitability of Summary Data for Two-Sample Mendelian Randomization Analyses Using MR-Egger Regression: The Role of the I2 Statistic. Int J Epidemiol (2016) 45(6):1961–74. doi: 10.1093/ije/dyw220
17. Burgess S, Thompson SG. Interpreting Findings From Mendelian Randomization Using the MR-Egger Method. Eur J Epidemiol (2017) 32(5):377–89. doi: 10.1007/s10654-017-0255-x
18. Jeong Y, Kim JW, You HJ, Park SJ, Lee J, Ju JH, et al. Gut Microbial Composition and Function Are Altered in Patients With Early Rheumatoid Arthritis. J Clin Med (2019) 8(5):693. doi: 10.3390/jcm8050693
19. Sun Y, Chen Q, Lin P, Xu R, He D, Ji W, et al. Characteristics of Gut Microbiota in Patients With Rheumatoid Arthritis in Shanghai, China. Front Cell Infect Microbiol (2019) 9:369. doi: 10.3389/fcimb.2019.00369
20. Rodrigues GSP, Cayres LCF, Gonçalves FP, Takaoka NNC, Lengert AH, Tansini A, et al. Detection of Increased Relative Expression Units of Bacteroides and Prevotella, and Decreased Clostridium Leptum in Stool Samples From Brazilian Rheumatoid Arthritis Patients: A Pilot Study. Microorganisms (2019) 7(10):413. doi: 10.3390/microorganisms7100413
21. Kishikawa T, Maeda Y, Nii T, Motooka D, Matsumoto Y, Matsushita M, et al. Metagenome-Wide Association Study of Gut Microbiome Revealed Novel Aetiology of Rheumatoid Arthritis in the Japanese Population. Ann Rheum Dis (2020) 79(1):103–11. doi: 10.1136/annrheumdis-2019-215743
22. Chiang HI, Li JR, Liu CC, Liu PY, Chen HH, Chen YM, et al. An Association of Gut Microbiota With Different Phenotypes in Chinese Patients With Rheumatoid Arthritis. J Clin Med (2019) 8(11):1770. doi: 10.3390/jcm8111770
23. Jangi S, Gandhi R, Cox LM, Li N, von Glehn F, Yan R, et al. Alterations of the Human Gut Microbiome in Multiple Sclerosis. Nat Commun (2016) 7:12015. doi: 10.1038/ncomms12015
24. Bang C, Weidenbach K, Gutsmann T, Heine H, Schmitz RA. The Intestinal Archaea Methanosphaera Stadtmanae and Methanobrevibacter Smithii Activate Human Dendritic Cells. PloS One (2014) 9(6):e99411. doi: 10.1371/journal.pone.0099411
25. Crouwel F, Buiter HJC, de Boer NK. Gut Microbiota-Driven Drug Metabolism in Inflammatory Bowel Disease. J Crohns Colitis (2020) 15(2):307–15. doi: 10.1093/ecco-jcc/jjaa143
26. Xu R, Tan C, He Y, Wu Q, Wang H, Yin J. Dysbiosis of Gut Microbiota and Short-Chain Fatty Acids in Encephalitis: A Chinese Pilot Study. Front Immunol (2020) 11:1994. doi: 10.3389/fimmu.2020.01994
27. Yan HX, An WC, Chen F, An B, Pan Y, Jin J, et al. Intestinal Microbiota Changes in Graves' Disease: A Prospective Clinical Study. Biosci Rep (2020) 40(9):BSR20191242. doi: 10.1042/BSR20191242
28. Abe K, Takahashi A, Fujita M, Imaizumi H, Hayashi M, Okai K, et al. Dysbiosis of Oral Microbiota and Its Association With Salivary Immunological Biomarkers in Autoimmune Liver Disease. PloS One (2018) 13(7):e0198757. doi: 10.1371/journal.pone.0198757
29. Jeong DY, Ryu MS, Yang HJ, Jeong SY, Zhang T, Yang HJ, et al. Pediococcus Acidilactici Intake Decreases the Clinical Severity of Atopic Dermatitis Along With Increasing Mucin Production and Improving the Gut Microbiome in Nc/Nga Mice. BioMed Pharmacother (2020) 129:110488. doi: 10.1016/j.biopha.2020.110488
30. Higuchi BS, Rodrigues N, Gonzaga MI, Paiolo JCC, Stefanutto N, Omori WP, et al. Intestinal Dysbiosis in Autoimmune Diabetes Is Correlated With Poor Glycemic Control and Increased Interleukin-6: A Pilot Study. Front Immunol (2018) 9:1689. doi: 10.3389/fimmu.2018.01689
31. Zhang L, Han R, Zhang X, Fang G, Chen J, Li J, et al. Fecal Microbiota in Patients With Ankylosing Spondylitis: Correlation With Dietary Factors and Disease Activity. Clin Chim Acta (2019) 497:189–96. doi: 10.1016/j.cca.2019.07.038
32. Leiva-Gea I, Sánchez-Alcoholado L, Martín-Tejedor B, Castellano-Castillo D, Moreno-Indias I, Urda-Cardona A, et al. Gut Microbiota Differs in Composition and Functionality Between Children With Type 1 Diabetes and MODY2 and Healthy Control Subjects: A Case-Control Study. Diabetes Care (2018) 41(11):2385–95. doi: 10.2337/dc18-0253
33. Simonyté Sjödin K, Hammarström ML, Rydén P, Sjödin A, Hernell O, Engstrand L, et al. Temporal and Long-Term Gut Microbiota Variation in Allergic Disease: A Prospective Study From Infancy to School Age. Allergy (2019) 74(1):176–85. doi: 10.1111/all.13485
34. Serrano-Villar S, Vázquez-Castellanos JF, Vallejo A, Latorre A, Sainz T, Ferrando-Martínez S, et al. The Effects of Prebiotics on Microbial Dysbiosis, Butyrate Production and Immunity in HIV-Infected Subjects. Mucosal Immunol (2017) 10(5):1279–93. doi: 10.1038/mi.2016.122
35. Li Y, Wang HF, Li X, Li HX, Zhang Q, Zhou HW, et al. Disordered Intestinal Microbes Are Associated With the Activity of Systemic Lupus Erythematosus. Clin Sci (Lond) (2019) 133(7):821–38. doi: 10.1042/CS20180841
36. Luo XM, Edwards MR, Mu Q, Yu Y, Vieson MD, Reilly CM, et al. Gut Microbiota in Human Systemic Lupus Erythematosus and a Mouse Model of Lupus. Appl Environ Microbiol (2018) 84(4):e02288–17. doi: 10.1128/AEM.02288-17
37. van der Meulen TA, Harmsen HJM, Vila AV, Kurilshikov A, Liefers SC, Zhernakova A, et al. Shared Gut, But Distinct Oral Microbiota Composition in Primary Sjögren's Syndrome and Systemic Lupus Erythematosus. J Autoimmun (2019) 97:77–87. doi: 10.1016/j.jaut.2018.10.009
38. Ma Y, Xu X, Li M, Cai J, Wei Q, Niu H. Gut Microbiota Promote the Inflammatory Response in the Pathogenesis of Systemic Lupus Erythematosus. Mol Med (2019) 25(1):35. doi: 10.1186/s10020-019-0102-5
39. Choi SC, Brown J, Gong M, Ge Y, Zadeh M, Li W, et al. Gut Microbiota Dysbiosis and Altered Tryptophan Catabolism Contribute to Autoimmunity in Lupus-Susceptible Mice. Sci Transl Med (2020) 12(551):eaax2220. doi: 10.1126/scitranslmed.aax2220
Keywords: autoimmune disease, Mendelian randomization, gut microbiome, systemic lupus erythematosus, causality
Citation: Xiang K, Wang P, Xu Z, Hu Y-Q, He Y-S, Chen Y, Feng Y-T, Yin K-J, Huang J-X, Wang J, Wu Z-D, Yang X-K, Wang D-G, Ye D-Q and Pan H-F (2021) Causal Effects of Gut Microbiome on Systemic Lupus Erythematosus: A Two-Sample Mendelian Randomization Study. Front. Immunol. 12:667097. doi: 10.3389/fimmu.2021.667097
Received: 12 February 2021; Accepted: 16 August 2021;
Published: 07 September 2021.
Edited by:
Trine N. Jorgensen, Case Western Reserve University, United StatesReviewed by:
Christopher Michael Reilly, The Edward Via College of Osteopathic Medicine (VCOM), United StatesCopyright © 2021 Xiang, Wang, Xu, Hu, He, Chen, Feng, Yin, Huang, Wang, Wu, Yang, Wang, Ye and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hai-Feng Pan, cGFuaGFpZmVuZzE5ODJAc2luYS5jb20=; Dong-Qing Ye, eWRxYWhtdUAxMjYuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.