 Michael Pisano1,2Yan Cheng1Fumou Sun1Binod Dhakal1Anita D’Souza1
Michael Pisano1,2Yan Cheng1Fumou Sun1Binod Dhakal1Anita D’Souza1 Saurabh Chhabra1Jennifer M. Knight3
Saurabh Chhabra1Jennifer M. Knight3 Sridhar Rao4,5
Sridhar Rao4,5 Fenghuang Zhan6
Fenghuang Zhan6 Parameswaran Hari1
Parameswaran Hari1 Siegfried Janz1*
Siegfried Janz1*- 1Division of Hematology and Oncology, Department of Medicine, Medical College of Wisconsin, Milwaukee, WI, United States
- 2Interdisciplinary Graduate Program in Immunology, University of Iowa, Iowa City, IA, United States
- 3Departments of Psychiatry, Medicine, and Microbiology & Immunology, Medical College of Wisconsin, Milwaukee, WI, United States
- 4Division of Hematology, Oncology and Marrow Transplant, Department of Pediatrics, Medical College of Wisconsin, Milwaukee, WI, United States
- 5Blood Research Institute, Versiti Wisconsin, Milwaukee, WI, United States
- 6Myeloma Center, Department of Internal Medicine and Winthrop P. Rockefeller Cancer Institute, University of Arkansas for Medical Sciences, Little Rock, AR, United States
Mouse models of human cancer provide an important research tool for elucidating the natural history of neoplastic growth and developing new treatment and prevention approaches. This is particularly true for multiple myeloma (MM), a common and largely incurable neoplasm of post-germinal center, immunoglobulin-producing B lymphocytes, called plasma cells, that reside in the hematopoietic bone marrow (BM) and cause osteolytic lesions and kidney failure among other forms of end-organ damage. The most widely used mouse models used to aid drug and immunotherapy development rely on in vivo propagation of human myeloma cells in immunodeficient hosts (xenografting) or myeloma-like mouse plasma cells in immunocompetent hosts (autografting). Both strategies have made and continue to make valuable contributions to preclinical myeloma, including immune research, yet are ill-suited for studies on tumor development (oncogenesis). Genetically engineered mouse models (GEMMs), such as the widely known Vκ*MYC, may overcome this shortcoming because plasma cell tumors (PCTs) develop de novo (spontaneously) in a highly predictable fashion and accurately recapitulate many hallmarks of human myeloma. Moreover, PCTs arise in an intact organism able to mount a complete innate and adaptive immune response and tumor development reproduces the natural course of human myelomagenesis, beginning with monoclonal gammopathy of undetermined significance (MGUS), progressing to smoldering myeloma (SMM), and eventually transitioning to frank neoplasia. Here we review the utility of transplantation-based and transgenic mouse models of human MM for research on immunopathology and -therapy of plasma cell malignancies, discuss strengths and weaknesses of different experimental approaches, and outline opportunities for closing knowledge gaps, improving the outcome of patients with myeloma, and working towards a cure.
Introduction
Multiple myeloma (MM) is a neoplasm of terminally differentiated, post-germinal center, immunoglobulin (Ig)-producing B-lymphocytes, called plasma cells, that reside in the hematopoietic bone marrow (BM). Quintessential disease manifestations include a serum M-spike (monoclonal Ig, paraprotein) and signs of end-organ damage known as CRAB symptoms: hypercalcemia, renal insufficiency, anemia, and lytic bone lesions (1). The most recent estimate of the US National Cancer Institute SEER (Surveillance, Epidemiology, and End Results) Program predicts slightly more than 32 thousand cases of newly diagnosed myeloma (NDMM) and nearly 13 thousand disease-specific deaths in 2020. This renders MM the second most common and one of the deadliest blood cancers in the United States. Owing to newly developed myeloma agents, particularly proteasome inhibitors (PIs), immunomodulatory drugs (IMiDs) and monoclonal antibodies (mAbs), the outcome for patients with MM has significantly improved in recent years (2), making it possible, at long last, to cure a tangible number of patients (3). However, in the great majority of cases, following a period of successful therapy, myeloma relapses as a drug-refractory, aggressive disease that leaves few, if any, therapeutic options. The root causes of progression to relapsed and/or therapy-refractory myeloma (RRMM) include tumor cell-intrinsic changes such as point mutations in drug response genes (4), copy number alterations that may abrogate tumor suppressor pathways (5), epigenomic aberrations modifying gene expression (6), and increased cancer stemness, which may impact lineage fidelity and tumor dormancy to name but two changes (7). An equally important yet tumor cell-extrinsic driver of RRMM pathophysiology is the tumor microenvironment (TME), which provides myeloma-promoting interactions with resident BM cells, including specimens of the innate and adaptive immune system (8). Enhanced understanding of immune regulation of the BM microenvironment (BMME) has not only shed light on pathways of myeloma progression but also greatly advanced myeloma treatment over the past decade (9).
Mouse models of human myeloma have provided preclinical research tools for elucidating the role of the immune system in the natural history of plasma cell neoplasia and in assessing candidate immunotherapies for myeloma (10, 11). Numerous experimental model systems are available, however, none perfectly replicate human myeloma (Figure 1). The most widely used models rely on in vivo propagation of human myeloma cells in immunodeficient hosts (human-in-mouse xenografting) (12–14) or myeloma-like plasma cells from C57BL/6 (B6) (15) or BALB/c (C) mice (16) in genetically compatible (syngeneic) immunocompetent hosts (mouse-in-mouse autografting). Both strategies have made and continue to make important contributions to myeloma research (17, 18), but are hampered by the reality that in vivo transfer of malignant cells (tumor transplantation) is not suitable for studying tumor development (oncogenesis). In other words, xeno- and autografting of neoplastic plasma cells bypasses the natural course of human myelomagenesis that begins with monoclonal gammopathy of undetermined significance (MGUS) (19), progresses to smoldering myeloma (SMM) (20), and eventually transitions to frank neoplasia (NDMM). Laboratory mice, in which plasma cell tumors (PCT) arise de novo (spontaneously) in a fully immunocompetent microenvironment, may remedy this shortcoming yet are undermined by other limitations, including complex breeding schemes, cost and time. Here we review the contribution of mouse models to advances in immunopathology and -therapy of human myeloma, discuss strengths and weaknesses of different experimental approaches, and outline opportunities for closing knowledge gaps.
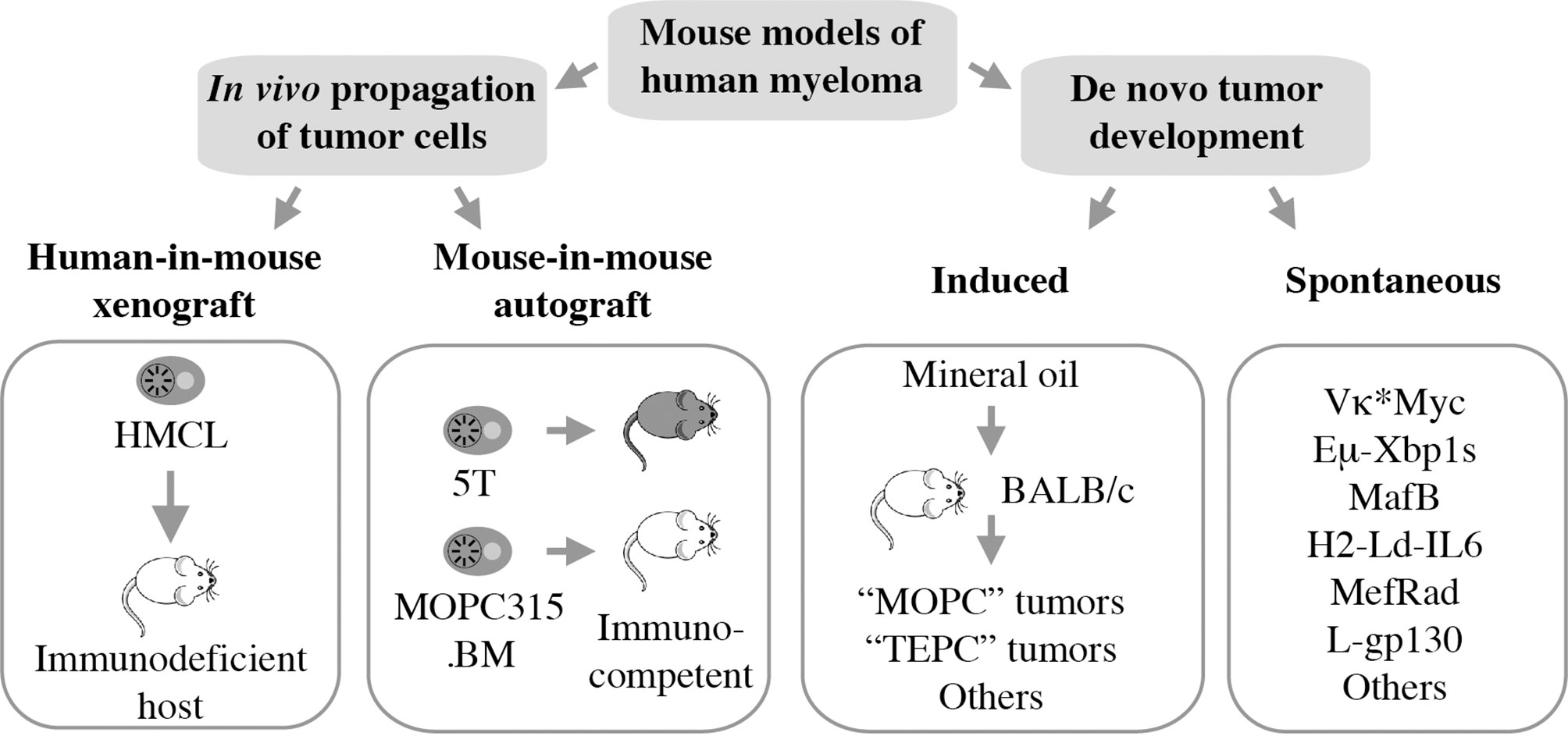
Figure 1 Mouse models of human myeloma. Xeno- and autografting relies on in vivo propagation of fully transformed tumor cells. Models of tumor development include peritoneal plasmacytomas that can be readily induced in genetically susceptible BALB/c mice and myeloma-like tumors that arise spontaneously in a variety of transgenic mice.
Immunopathology And -Therapy Of Multiple Myeloma
Immune Editing and Immune Dysfunction in Myeloma
As mentioned above, frank myeloma is invariably preceded by the precursor conditions MGUS (19) and SMM (21). MGUS is, in most cases, asymptomatic (22) and is usually detected years before frank MM manifests. MGUS progresses to active myeloma at the slow and constant rate of approximately 1% per year (21, 23). Consistent with the more advanced stage of tumor development, the progression rate of SMM is higher: 10% per annum in the first 5 years and 3% thereafter (24). Notably, BM plasma cells of individuals with MGUS exhibit a gene expression profile that is highly similar to that of myeloma (25) and MGUS plasma cells contain many of the cytogenetic changes (chromosomal translocations, gains and deletions) typically seen in myeloma cells (26–28). These findings have long raised suspicion that MGUS may in fact be a malignancy that is suppressed by a strong, extrinsic force, such as a cognate cytotoxic immune response (immune surveillance). Because most cases of MGUS do not progress to MM, this surveillance mechanism must be effective and enduring, essentially covering the entire lifespan of most individuals harboring an aberrant plasma cell clone of this sort. A large body of recent work expertly reviewed elsewhere (29, 30) strongly suggests that the breakdown of immunologic surveillance is at the heart of the MGUS-to-MM transition. According to this theory, known as cancer immunoediting (Figure 2A), the immune system is initially highly successful in eliminating abnormal plasma cells (Elimination stage), but then switches to an impasse that permits a limited number of these cells to survive in a quiescent or dormant state (Equilibrium) that may last for many years in individuals with MGUS (34). For reasons that are not yet clear, the equilibrium is eventually disrupted in a subset of patients, allowing the aberrant cell clone to evade immune control (Escape) and fuel the malignant growth that underlies active myeloma. In sync with that scenario, immune suppression caused by regulatory T cells, myeloid derived suppressor cells (MDSC) and dysfunctional effector T lymphocytes, is a hallmark of NDMM (Figure 2B). Growth and survival support of myeloma cells by innate immune cells, including conventional and plasmacytoid dendritic cells (35, 36) and eosinophils (37), is the flip side of the same coin. Of practical relevance for patient care is the knowledge that immune dysfunction in patients with myeloma may lead to increased risk of infections (38) and lack of a vigorous vaccination response (39) – something to be mindful about in the midst of SARS-CoV-2 (40).
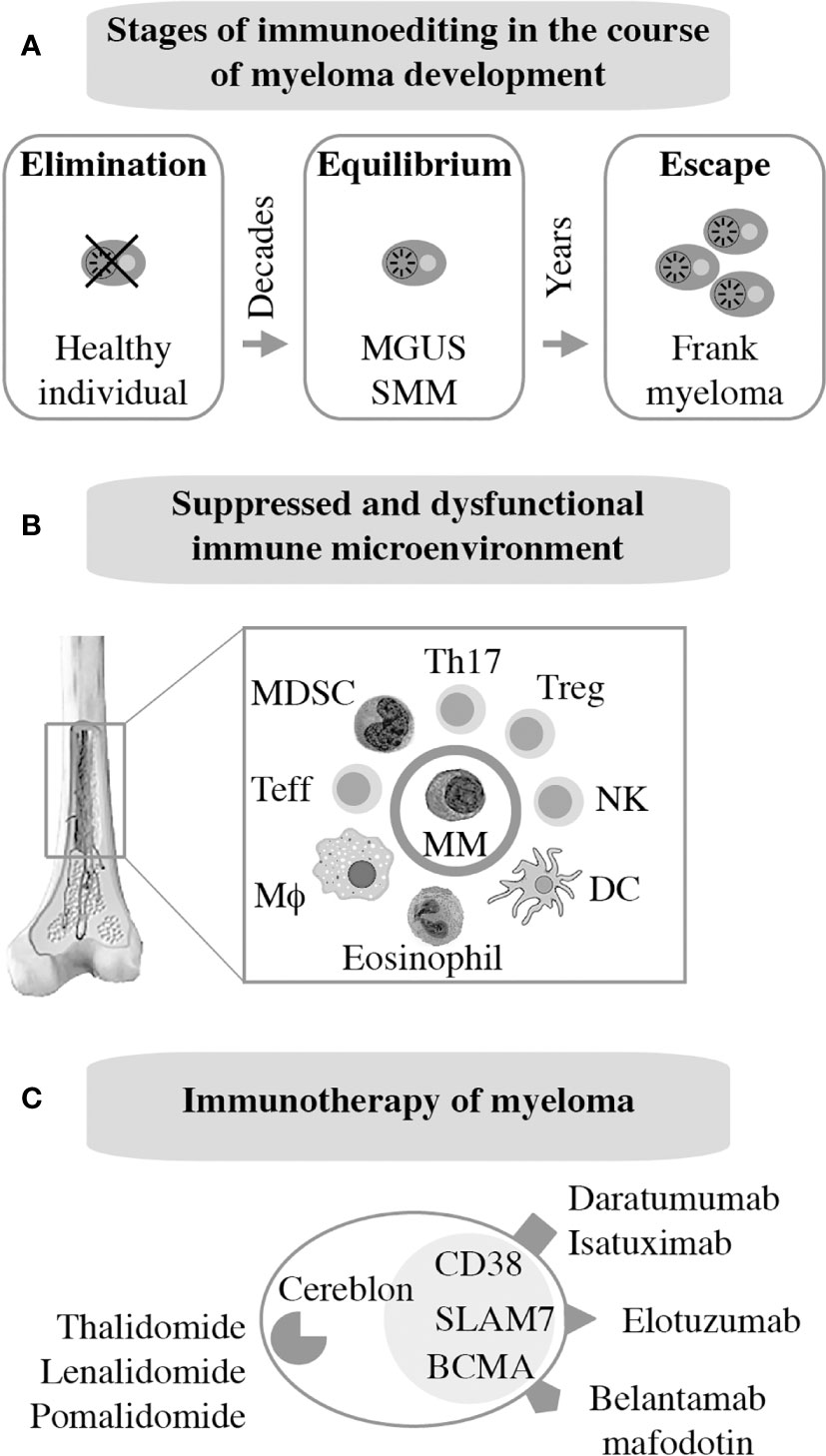
Figure 2 Immunopathology and -therapy of myeloma. Evidence indicates that myeloma development is promoted, in part, by the gradual breakdown of immunosurveillance (A). Consequently, patients with myeloma have a suppressed and dysfunctional immune microenvironment (B). 2 subsets of Tregs that discriminate MGUS from MM (31); 2 subsets of terminal effector T cells (TTE) are involved in MGUS to MM transition (32); Attrition of BM-resident T cells due to loss of “stem-like” TCF1/7hi T cells may underlie loss of immune surveillance in myeloma (33). Enhanced understanding of this microenvironment has been key for the development of immunotherapies of myeloma (C).
Immunotherapy of Myeloma
The past decade has witnessed tremendous progress in immunotherapeutic approaches to myeloma. Authoritative and up-to-date reviews are available (29, 41). Current FDA-approved interventions include monoclonal antibodies targeting CD38 [daratumumab (42), isatuximab (43)] or SLAMF7 [elotuzumab (44)] on the surface of tumor cells. An antibody-drug conjugate (ADC) that binds to BCMA [belantamab mafodotin (45)] has also been approved just recently. Additional BCMA-targeted therapies, in particular chimeric antigen receptor (CAR) T cells and bispecific T cell engagers (BITEs), are in advanced stages of clinical development. Immune modulation using small-drug inhibitors of cereblon, a component of an E2 ubiquitin ligase complex, is also approved for treatment of myeloma and is widely used for maintenance therapy internationally. Immunomodulatory drugs of this sort, dubbed IMIDs, include thalidomide (46), lenalidomide (47, 48), and pomalidomide (49). Next-generation cereblon-targeting agents that promise to overcome acquired resistance to IMIDs are in clinical trial (50). Figure 2C provides an overview of myeloma immunotherapies in clinical use. Not shown are proteasome inhibitors (PIs), a class of targeted myeloma agents that, in the past, have not been associated with immune-mediated myeloma-inhibiting effects. However, recent work demonstrates that modulating the immune microenvironment of myeloma, by virtue of inducing immunogenic cell death (51), primes a cytotoxic immune response to tumor cells, thereby contributing to disease control in a proteasome-independent manner.
Xenograft Models Of Myeloma
Propagating Human Myeloma Cell Lines (HMCLs) in NSG and NRG Hosts
Many advances in the field of myeloma biology, genetics, and therapy would not have been possible without preclinical investigations that relied on immunocompromised mice for hosting human myeloma cells. Human-in-mouse xenografting has a long and distinguished history in cancer, including myeloma research, beginning in the early 1960s with the discovery of the T lymphocyte-deficient nude mouse. This marked the inception of a developmental pipeline of mice that feature increasing levels of immunodeficiency. SCID (severe combined immunodeficiency) and Rag mice lack both T and B lymphocytes. The underlying genetic defects are loss-of-function mutations in Prkdc (protein kinase, DNA activated, catalytic polypeptide) and Rag1 or Rag 2 (recombination activating gene 1 or 2), respectively. Transfer of the SCID and Rag mutations on the genetic background of NOD (non-obese diabetic) led to NOD-SCID and NOD-Rag mice, which exhibit a NK (natural killer) defect on top of T and B deficiency. In addition to diminished NK cell function, NOD leads to lack of circulating complement due to deletion of the C5-encoding Hc gene and proclivity to Type 1 diabetes mellitus due to autoimmune insulinitis (52). Further modification of NOD-SCID and NOD-Rag mice by crossing in IL2Rγ (interleukin-2 receptor subunit gamma) deficiency resulted in strains NSG and NRG, which are devoid of T, B and NK cells. Lack of IL2Rγ (a.k.a. CD132 or common gamma chain, γc) abrogates NK cells and compromises at least 6 interleukin signaling pathways: IL-2, 4, 7, 9, 15 and 21 (Figure 3B). NSG and NRG mice, which are commercially available and widely used in myeloma research, readily permit engraftment of human myeloma cells upon subcutaneous (SC), intravenous (IV), intraperitoneal (IP), or intratibial injection (53, 54). Strengths and limitations of myeloma xenografting have been reviewed in great depth elsewhere (55). Care must be taken to select the most appropriate HMCL for a given research question, as significant differences between cell lines exist (56, 57). 3D in vitro culture of myeloma cells using a variety of artificial scaffolds is an emerging technology that competes with xenografting and holds promise for drug testing for personalized myeloma therapy (58).
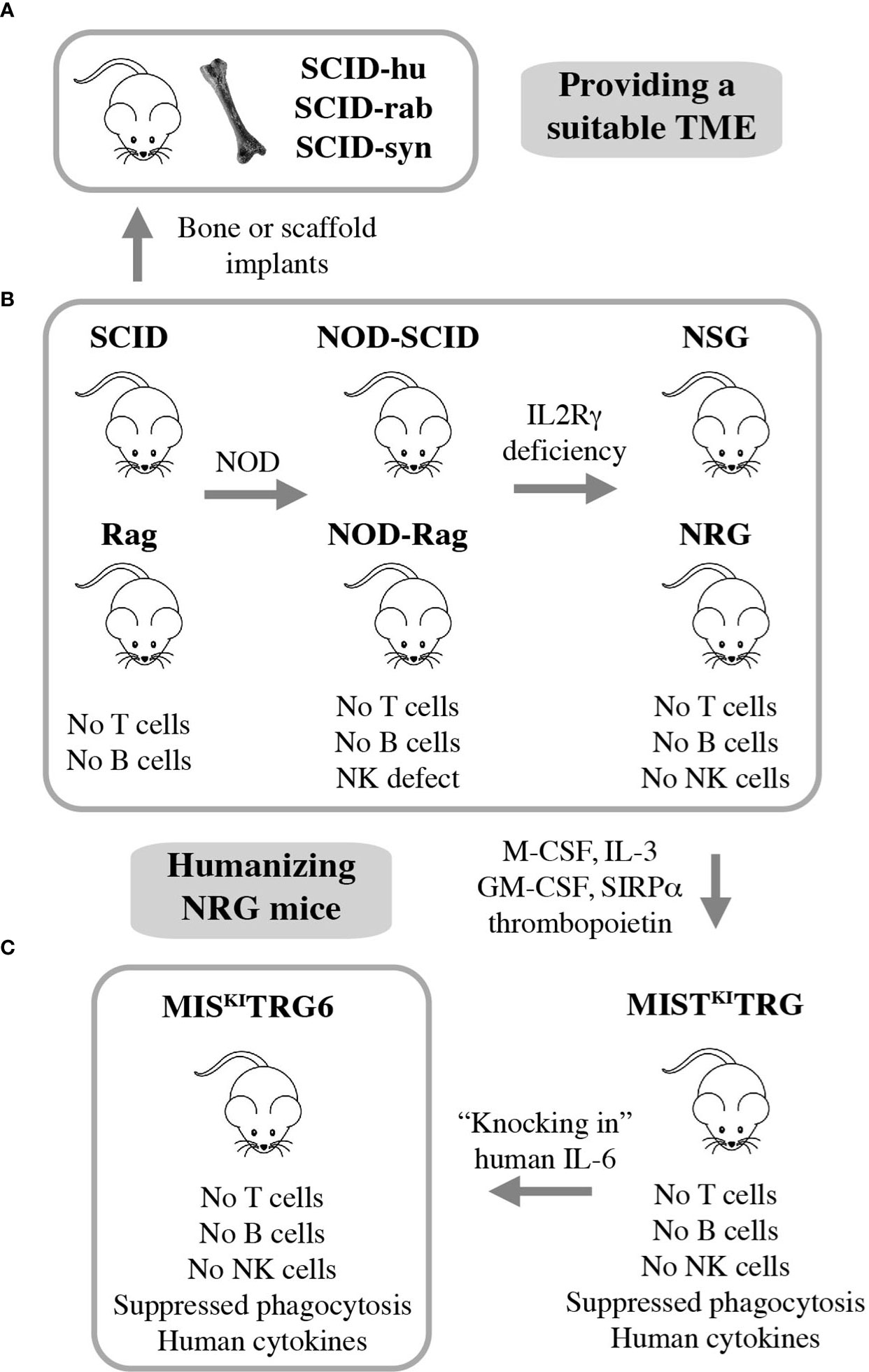
Figure 3 Xenografting human myeloma in immunodeficient mice. NSG and NRG mice are widely employed for preclinical studies using human myeloma cell lines (HMCLs) but are limited in terms of hosting primary, patient-derived myeloma cells (B). Implantation of bone chips or artificial scaffolds in SCID mice can overcome this limitation (A) but is faced with practical limitations and the inability to support MGUS and SMM plasma cells. Additional incremental steps in humanizing NSG and NRG mice may solve this problem. A promising step in this direction is the recent development of IL-6 transgenic MISKITRG mice, which can support homing and survival of plasma cells not only from patients with frank and smoldering MM but also individuals with MGUS (C).
Engrafting Patient-Derived Myeloma Cells in Implanted Bone Chips
Major limitations of myeloma xenografting described above include the reality that tumor growth occurs mostly outside the BM and primary patient-derived tumors do not grow at all. The latter is a fundamental flaw caused by the stringent dependency of myeloma cells on support from the human BMME. HMCLs do not exhibit this dependency because they are derived from malignant plasma cells that circulated in the peripheral blood or occurred in body cavity effusions of patients with plasma cell leukemia, the end stage of myeloma progression. The derivation from leukemic cells is also in line with the extramedullary growth pattern (plasmacytoma) of HMCL-in-mouse xenografts mentioned above. To provide a TME that is more conducive for primary myeloma cells, investigators modified SCID mice by implanting small pieces of human or rabbit bone subcutaneously. Synthetic 3D scaffolds serving as surrogate bone provide an alternative approach. These experimental model systems have come to be known as SCID-hu (12, 59–61), SCID-rab (62), and SCID-syn (14) (Figure 3A). They are equally capable and have greatly enhanced preclinical myeloma research (10, 11) by addressing knowledge gaps in treatment and pathophysiology: SCID-hu (63–65), SCID-rab (66–68), and SCID-syn (69, 70). However, more widespread use of these models is hampered by ethical and practical limitations, such as reliable provision of fetal human bone and difficulties in administering human myeloma cells to small implants. The fact that fetal BM does not equate with adult BM in terms of cellular composition and immune milieu, and that rabbit or synthetic bone does not fully recapitulate the myeloma-supporting properties of human bone adds an additional biological limitation. This backdrop helps explain why the use of SCID-hu, SCID-rab, and SCID-syn has been declining in recent years and why researchers have been looking for alternative strategies to propagate myeloma in laboratory mice. The most promising development to date is the humanization of NRG mice, as described below.
Maintaining MGUS/SMM Plasma Cells in Humanized NRG Mice
Gene targeting in embryonic stem cells is a convenient research tool for substituting mouse genes with human counterparts and, thereby, humanizing laboratory mice. This approach has generated highly immunodeficient mice in which two major obstacles to engraftment of human cells have been largely overcome: innate immune rejection via phagocytosis and lack of activity of certain cytokines and growth factors across the human-mouse species barrier (71). Strain MIS(KI)TRG is an excellent example of recent developments. It features, on the genetic background of NRG, the expression of 5 human “knock in” genes encoding M-CSF (macrophage colony-stimulating factor a.k.a. colony stimulating factor 1 or CSF1), IL-3 (interleukin-3), GM-CSF (granulocyte-macrophage colony-stimulating factor a.k.a. colony stimulating factor 2 or CSF2), SIRPα (signal regulatory protein α), and thrombopoietin (72). MIS(KI)TRG mice exhibit improved engraftment of hematopoietic stem and progenitor cells and lend themselves to hosting PDX (patient-derived xenograft) tumors from many human cancer types. However, these mice were still unsuitable for stable engraftment of primary myeloma cells. Cognizant of the critical role of IL-6 for growth and survival of neoplastic plasma cells (73), Madhav Dhodapkar and his associates added a human IL-6 allele to strain MIS(KI)TRG, thus generating IL-6 transgenic MIS(KI)TRG, or MIS(KI)TRG6 mice (Figure 3C) (74). The introduction of human IL-6 resulted in a remarkable breakthrough for preclinical myeloma research, because for the first time it allowed engraftment and propagation of primary MM cells in a reliable and reproducible fashion. What is more, MIS(KI)TRG6 supported engraftment of SMM and MGUS plasma cells upon transfer of CD3-depleted BM mononuclear cells to recipient bone. The finding that, unlike NDMM cells, RRMM cells actively homed to and expanded in other sites of the skeleton resembled the more advanced tumor progression stage of relapsed compared to new myeloma. Finally, while RRMM remained confined to bone, tumor samples from patients with plasma cell leukemia demonstrated the kind of systemic, extramedullary dissemination pattern that one might expect from a leukemic cell clone.
Utility of Myeloma Xenografting in Immunotherapy Research
Of the three principal model systems of myeloma xenografting described above, the HMCL-in-mouse approach, has probably had the greatest impact on immunotherapy research in myeloma. While MIS(KI)TRG6 mice have not yet been used to that end, the implantation-based SCID models have largely been supplanted by HMCL-in-mouse xenografts, as mentioned above. Indeed, HMCL xenografting appears to lend itself readily to the complex requirements of experimental immunotherapy. Often this involves adoptive immune cell transfer to study mice and testing of new therapeutic antibodies or antibody-drug conjugates (ADCs) in mice co-treated with established myeloma drugs or candidate small-drug inhibitors. HMCL xenografts are often used as a first choice when the efficacy of cytotoxic T cells, NK cells, or engineered killer cells to remove myeloma in an intact organism in vivo is to be evaluated. To highlight but a few examples of their utility, HMCL xenografts have majorly contributed to the development of CAR-T treatments for BCMA (75) and newly emerging CAR-T targets such as CD229 (SLAMF3, LY9) (76). HMCL xenografts have been successfully employed to assess a monoclonal antibody to transferrin receptor 1, a newly emerging molecular target expressed on the surface of myeloma cells (77, 78). Similarly, HMCL xenografts have been used to evaluate AMG 701, a half-life extended BITE that binds to BCMA on myeloma cells and CD3 on T cells (79), and to demonstrate that the therapeutic efficacy of daratumumab in myeloma may be enhanced when CD38 in NK cells has been deleted (80). This body of work illustrates the rapid evolution of the myeloma immunotherapy landscape and that HMCL xenografting is poised to add further value to the field as we go forward.
Autograft Models Of Myeloma
5TMM
The 5T mouse model of human multiple myeloma, or 5TMM for short, is a versatile research tool for fundamental and applied studies on plasma cell malignancies. The model is based on the genetic proclivity of inbred C57BL/KaLwRij mice (closely related to the commonly used C57BL/6) (81) to spontaneously develop a benign monoclonal gammopathy (serum paraprotein) or MGUS-like condition (82) that can progress to frank myeloma (15, 83). Using serial in vivo propagation of bone marrow cells from independent C57BL/KaLwRij donors containing different paraproteins, a number of transplantable myeloma-like plasma cell tumors were generated (Figure 4A). Two of these, dubbed 5T2 and 5T33, were fully established and generously shared with qualified investigators in Europe, the United States, and elsewhere. 5TMM tumors cause osteolytic lesions (83) and recapitulate other features of human myeloma bone disease (84, 85). 5TMM tumors grow in a fully immunocompetent BMME and are easily tracked in vivo using radiological methods including X-ray and PET imaging (86). Unlike 5T2, which can only be maintained by passaging from mouse to mouse, two continuous cell lines were derived from 5T33: 5TGM1 (87, 88) and 5T33vt (89). The genetic differences between these cell lines and 5T2 have been recently determined using NGS (90). This revealed an additional strength of 5TMM in regard to modeling human myeloma; i.e., the significant overlap of patterns of somatic mutations across the human-mouse species barrier, particularly with respect to copy number changes of genes involved in gain of 1q and deletion of 13q in human myeloma (90). The availability of cell lines, 5TGM1 and 5T33vt, has greatly enhanced the flexibility and impact of the 5TMM model. For example, the cells can be easily modified by virtue of enforced up or down regulation of genes of interest or the introduction of reporter genes for whole-body fluorescence or bioluminescence imaging of tumor growth in a quantitative, objective manner (91). The cell lines have also facilitated the examination of myeloma-immune interactions in vitro and in vivo. For example, an early study using 5TGM1 demonstrated that myeloma cells inhibit the differentiation of BM-derived dendritic cells (DCs) and interfere with their function to induce cytotoxic and humeral immune responses (92). This is relevant for ongoing efforts in human myeloma to use DCs for vaccination approaches aimed at eliciting a robust T cell-dependent cytotoxic immu response (93).
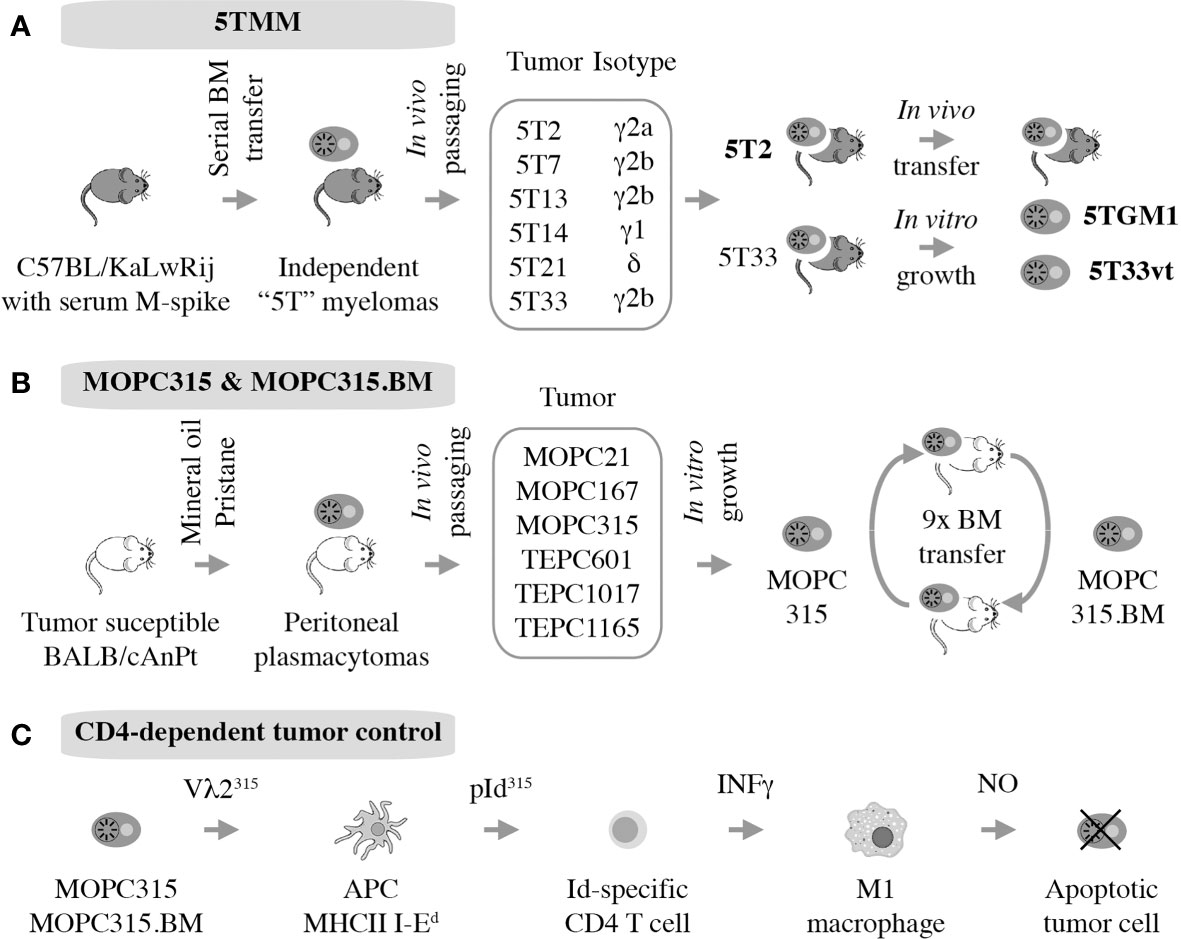
Figure 4 Autografting mouse myeloma in immunocompetent mice. Two models have been established. 5TMM is on a genetic background that is highly similar to B6 and includes two continuous cell lines, 5TGM1 and 5T33vt, that are widely used (A). MOPC315 is a peritoneal plasmacytoma on the genetic background of C that has given rise to a BM-seeking subline, MOPC315.BM, that holds great value for myeloma immunology research. (B) Decades of research by Bogen and colleagues have shown indirect CD4+ T cells mediated killing via interactions with cytotoxic macrophages, further demonstrating the utility of MOPC315.BM as an immunological research tool (C).
5TMM as Research Tool for Immune Regulation of Myeloma
The broad utility of 5TMM for studies on the immunopathology and -therapy of myeloma was recognized soon after the first tumors were established. Initial investigations described the role of the paraprotein idiotype (Id) in immune regulation (94) and immune therapy (95) of plasma cell neoplasia. Follow-up studies relying on 5TGM1 demonstrated that the idiotype is a myeloma-specific antigen that can induce an Id-specific cytotoxic T cell (CTL), T helper 1 (Th1) and T helper 2 (Th2) response (96). CTLs and Th1s were found to suppress myeloma growth, whereas Id-specific Th2 cells promoted it (96) – a preclinical clue in support of the contention that modulation of the Th1:Th2 axis might have therapeutic benefits in myeloma. The finding that 5T-bearing mice exhibit an increase in the ratio of regulatory T cells (Tregs) to T effector cells (97) proved relevant for human myeloma when it became clear that patients with new disease contain elevated numbers of Tregs in the peripheral blood (98). With respect to myeloma immunotherapy, the 5T33 model made conceptual contributions to developing DC-based MM vaccines for idiotype protein (99). This included the design of more effective adjuvants based on CpG and IFN-α (100) and the realization that myeloma cell lysates provide a more powerful DC vaccine than idiotype protein and adjuvant, alone (101). These advances were confirmed in a clinical study a few years ago showing that a patient-derived DC-MM cell fusion (hybridoma) vaccine improved the therapeutic response in a quarter of myeloma patients post-ASCT from partial to (nearly) complete (102). 5TMM has also been used to examine immunomodulatory myeloma treatments at the preclinical level; e.g., investigators demonstrated that CD4 T cells were vital for lenalidomide’s activity, while NK, B or CD8 T cells were not (103). Activation of innate-like invariant natural killer T (iNKT) cells, a cell type that has not yet been extensively examined in human myeloma, led to significantly increased survival of 5T33-bearing mice (104). The 5T33 model has also contributed early on to the CAR-T therapy field by showing that treatment using NKG2D-targeted CAR-T cells prolonged survival of tumor-bearing mice and induced a tumor-specific memory response (105). Furthermore, 5T33 not only demonstrated efficacy of immune checkpoint inhibitor (CPI) therapy using antibody to programmed death receptor-1 (PD-1) or its ligand (PDL-1), but also showed that CD8+ T cells in tumor-bearing mice post-ASCT significantly upregulated PD-1 (106, 107). In summary, the practical limitation to in vivo studies using 5TMM requiring the genetic background of C57BL/KaLwRij (108) is a small inconvenience compared to the potential contribution of this model to aiding immunotherapy development for patients with myeloma.
MOPC315.BM
MOPC315 is an IgA-producing peritoneal plasmacytoma (PCT) that arose half a century ago (109) in a BALB/c (C) mouse treated with intraperitoneal injections of mineral oil (110). MOPC315 has been used for decades in studies on immune regulation of malignant plasma cell growth (111, 112) although it is not representative of human MM, which grows in the hematopoietic BM and depends on the BMME for survival. In a major step forward, this shortcoming was remedied by the development of a subline of MOPC315, dubbed MOPC315.BM, generated by serial IV autografting of BM-derived plasma cells for nine generations. In the course of in vivo propagation, tumor cell variants with exquisite affinity to the BM increased oncogenic potency (~1 month median survival of tumor-bearing mice), and capacity for BM homing and bone destruction were preferentially selected (Figure 4B). Transfection with a luciferase reporter further increased the utility the cell line (16). MOPC315.BM is now increasingly used in preclinical myeloma research. For example, it provided the foundation for recent studies on the involvement of IL-34 and notch signaling in the pathophysiology of the focal and systemic bone loss in mice that mimics human myeloma bone disease (MBD) (113, 114). Additionally, MOPC315.BM has been employed to demonstrate that eosinophils and megakaryocytes support malignant plasma cell growth in the BM (115) and that oncolytic myxoma virus, in conjunction with ASCT, may be an effective treatment for PCT-bearing mice (116). Importantly, together with its parental line, MOPC315.BM has made major contributions to our appreciation of CD4 T cell responses in immunosurveillance and -therapy of myeloma (117), briefly summarized below.
Lesson on CD4-Dependent Control of Malignant Plasma Cell Growth
Strong evidence supports the significance of Id-specific CD4 T cells in clearance of MOPC315 plasma cells in vivo (118), confirming matching results in the 5TMM model described above. In a remarkable continuity of investigations that span more than 25 years, Bjarne Bogen and his colleagues have unequivocally shown that tumor cell-produced monoclonal Ig gives rise to a tumor-specific antigen (TSA) in the MOPC315 model system. The antigen is processed by professional antigen-presenting cells (APCs) that include tumor-infiltrating macrophages in the subcutaneous MOPC315 model and BM macrophages in the medullary MOPC315.BM model (119). With help of MHC class II-encoded I-Ed surface protein, APCs present the antigen to CD4 Th1 cells as a λ2 light-chain V region-derived idiotypic (Id) peptide; i.e., a neoepitope. This results in Th1-dependent production of IFN-γ, which activates bystander macrophages and promotes their polarization to the tumoricidal M1 phenotype. In turn, M1 macrophages upregulate inducible nitric oxide synthetase (iNOS) to produce and release nitric oxide (NO) into the extracellular milieu. NO then kills neighboring tumor cells using a mechanism that involves reactive nitrogen species (e.g., peroxynitrite) and activates the intrinsic pathway of programmed cell death (apoptosis). Thus, in the MOPC315 model system, CD4+ T cells kill tumor cells indirectly with the assistance of cytotoxic macrophages (Figure 4C). MOPC315.BM has provided additional insights into myeloma immunology. Examples include the interaction of myeloid-derived suppressor cells and T cells in vivo (120), the development of allogeneic T cell treatments for myeloma that may circumvent GvHD (121, 122), and the evaluation of novel DNA vaccines for immunotherapeutic purposes (123). Similar to the inconvenience of the genetic background of 5TMM, MOPC315.BM is on the genetic background of BALB/c (C), which is not as widely used in cancer immunology as B6. However, this is a small price to pay considering the value MOPC315.BM can add to the immune revolution in myeloma treatment (124).
Spontaneous Plasma Cell Tumors In Laboratory Mice
Inducible, Inflammation-Dependent Peritoneal Plasmacytoma
MOPC315 is but one representative of a large panel of peritoneal plasmacytomas that has been developed single handedly in the 1960s and 1970s by Dr. Michael Potter at the US National Cancer Institute, Bethesda, Maryland. He discovered that IP treatment of C mice using certain mineral oils (110) or a chemically defined component thereof, called pristane (2,6,10,14-tetramethylpentadecane) (125), induced development of MOPC (mineral oil induced plasmacytoma) and TEPC (tetramethylpentadecane induced plasmacytoma) tumors, respectively. Plasmacytomas induced in this fashion were crucial for basic research breakthroughs in antibody structure (126) and monoclonal antibody (hybridoma) technology, which began with MOPC21 (127). Unlike most inbred strains of mice, C is highly susceptible to plasmacytoma (128) due to a complex genetic trait that includes hypomorphic (weak efficiency) alleles of genes that encode the cell cycle inhibitor p16INK4a (129) and the FKBP12 rapamycin-associated protein Frap (130). Virtually all peritoneal PCTs harbor a Myc-activating chromosomal translocation (131) that takes the form of a balanced T(12;15)(Igh-Myc) in the majority (~85%) of cases. Plasmacytoma induction requires maintenance of mice in a non-SPF (specific pathogen-free) environment rich in antigenic stimuli including gut flora-derived antigens (132). Consistent with that, C mice raised under SPF or germ-free conditions exhibit a dramatically reduced tumor incidence (133) or fail to develop plasmacytoma altogether (134). Peritoneal plasmacytoma is the premier mouse model of inflammation-induced extramedullary myeloma has been used for decades to learn about immune regulation of malignant plasma cell growth (111, 112). However, BALB/c plasmacytomas are not widely used in myeloma research today because more accurate, transgenic mouse models of the disease have become available. These will be described in the following section.
Transgenic Mouse Models of Human Myeloma and Related Plasma Cell Neoplasms
Genetic modification of the mouse germline has been employed by several independent research groups to generate transgenic strains of mice that are prone to spontaneous plasma cell tumors (PCT) that replicate important features of human MM. Mice of this sort exhibit a predictable progression pattern from MGUS- and SMM-like precursor conditions to frank plasma cell neoplasia. This pattern is key for preclinical assessments of myeloma preventions, a hot topic in clinical myeloma research (135). PCT-prone mice feature a fully intact innate and adaptive immune system that is likely to adapt to tumor development much the same way as the human immune system adapts to myeloma (Figure 2A). Hence, trialing newly developed immunotherapeutics in mice that are genetically susceptible to PCT is poised to yield more complete and higher-quality information than one might get from mice that are immunocompetent but not undergoing tumorigenesis (136). The same argument can be made for the preclinical testing of complex treatment regimens that combine HSC transplantation and established myeloma drugs (PIs, IMiDs) with novel immunotherapies and small-compound inhibitors. Evaluating these types of treatment in PCT-susceptible mice may more closely mimic the response of myeloma patients exposed to triplet and quadruplet drug regimens (137). Figure 5 presents a developmental timeline of genetically engineered mouse models (GEMMs) of human myeloma and related malignancies. Table 1 provides details on tumor incidence and phenotype, genetic drivers of tumor development, and genetic background of mice. An exhaustive discussion of individual models is beyond the scope of this review. To that end, the reader is referred to the primary literature and outstanding recent reviews from Tassone (55), Vlummens (138) and their associates. A general rule that may be gleaned from the table is that single-transgenic models exhibit delayed tumor onset and a relatively low tumor incidence. To accelerate plasma cell neoplasia, investigators have taken advantage of oncogene collaboration in double-transgenic mice, such as IL6Myc (139) and Bcl-XLiMyc (140), that exhibit short tumor onset and full penetrance of the malignant phenotype (100% tumor incidence). Inducible transgenes such as L-gp130 (141) and models based on adoptive transfer of genetically modified B cells (142–144) serve the same purpose; i.e., faster and more consistent tumor development. Importantly, Vκ*MYC, developed by Marta Chesi and Leif Bergsagel at Mayo and generously shared with investigators in many countries, is the only model at this juncture for which robust immunology work is available. This is one of several reasons why Vκ*MYC is widely considered in the myeloma community as the gold standard of mouse models. Advances in immunosurveillance and immunotherapy of myeloma made possible by Vκ*MYC will be briefly discussed below.

Figure 5 Transgenic mouse models of human plasma cell myeloma and extramedullary plasmacytoma. Shown is a timeline of model development that begins with Eµ-v-abl developed by Susan Cory’s group at WEHI and published in 1990. The H2-Ld-IL6 model of human plasmacytoma published in 2002 gave rise to the double-transgenic MycIL6 and BCL2IL6 models that take advantage of oncogene collaboration to accelerate neoplastic plasma cell development. Similarly, Vκ*MYC, the premier model of myeloma immunology research, was recently accelerated by breeding in a mutated Ras gene, leading to the highly promising VQ model published in 2020.
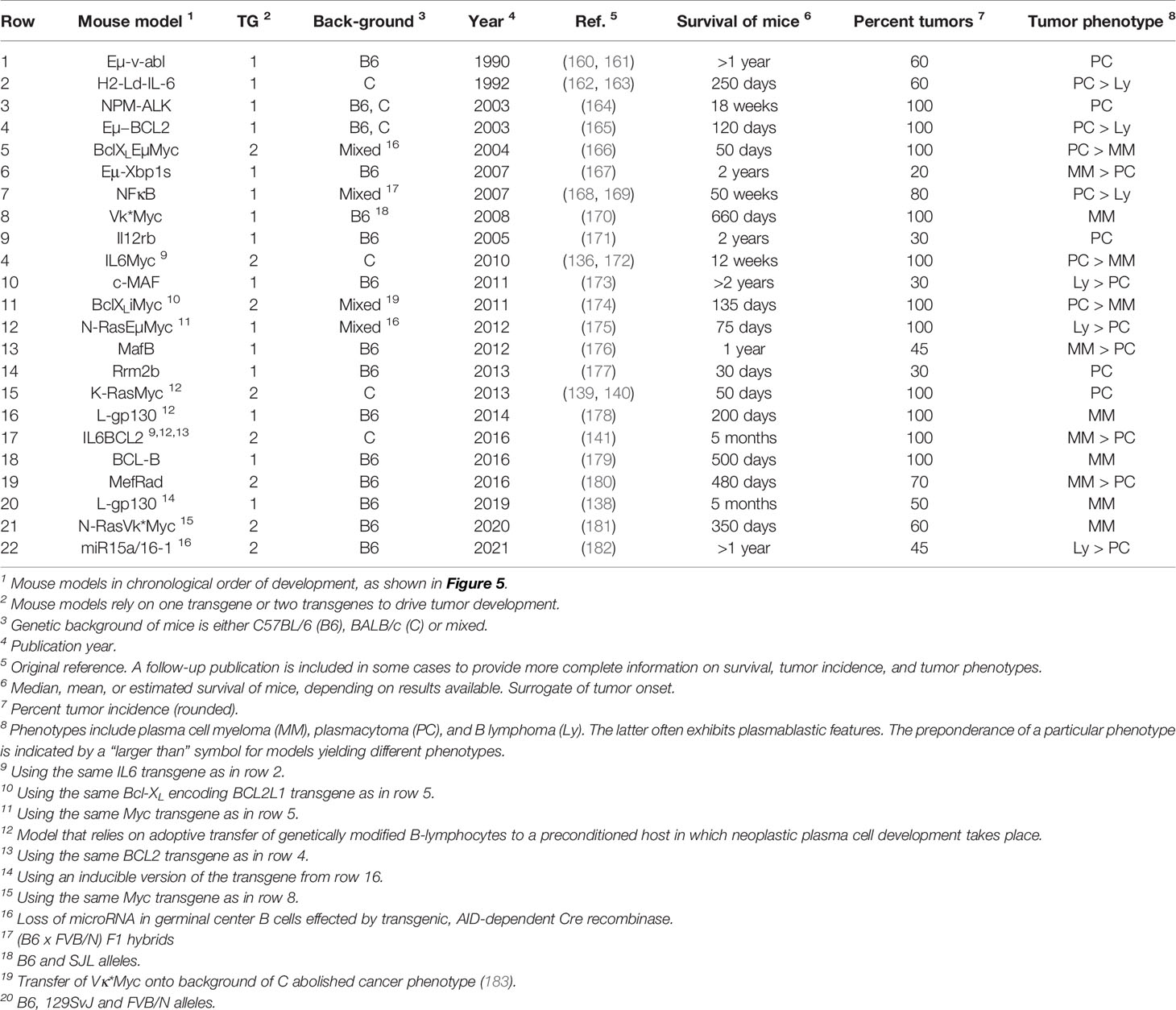
Table 1 Transgenic mice prone to spontaneous plasma cell tumors recapitulating hallmarks of human plasma cell neoplasms including multiple myeloma.
Advances in Myeloma Immunology Made Possible by Vκ*MYC
The realization that Vκ*MYC-dependent myeloma causes changes in immune regulation in mice comparable to changes seen in patients with myeloma (145) laid the foundation for mechanistic studies describing role of specific pathways of immunity to Vκ*MYC-driven tumor development. The first investigation along this line revealed the importance of CD226 for immune surveillance of myeloma. Lack of CD226 reduced the anti-myeloma response of NK and CD8 T cells, resulting in quicker tumor progression and decreased overall survival of Vκ*MYC mice (146). Another insight afforded by Vκ*MYC concerned the intriguing link between microbial gut flora and IL-17-driven tumor progression. The underlying mechanism is complex but involves an increase in Th17 cells and activation of eosinophils. Not coincidentally, therapeutic control of these changes using antibodies to IL-17 and IL-5 delayed tumor progression (147). Vκ*MYC also provided definitive genetic evidence on the involvement of the pro-inflammatory cytokine IL-18 in myeloma progression (148). This was attributable to IL-18-dependent generation of myeloid-derived suppressor cells (MDSCs), an important driver of the dysfunctional immune environment in human myeloma (Figure 2B). Another study demonstrated that tumor progression and dissemination in Vκ*MYC is not under exclusive control of the TME. Insead, it is regulated, in part, by properties of tumor cells such as expression of CD138 (149) (Figure 6A). Vκ*MYC has also impacted the field of myeloma immunotherapy in more ways than one. One study showed that treatment of mice using IAP (inhibitor of apoptosis) antagonists activated an acute inflammatory response that led to enhanced tumor phagocytosis by macrophages. Interestingly, co-treatment using antibody to PD1 led to an additional increase in survival of mice (153). By implicating the upregulation of TIGIT (T-cell immunoglobulin and ITIM domain) on CD8 T cells, Vk*MYC has also contributed to our understanding of T cell exhaustion in myeloma. Checkpoint blockade of TIGIT prolonged survival of mice and reduced levels of immunosuppressive IL-10 produced by dendritic cells (154–156). Finally, studies using Vk*MYC demonstrated that: blocking type 1 interferon signaling may inhibit Treg expansion in myeloma (157), antibody to CD137 holds promise as a consolidation treatment in myeloma (158), and HSC transplantion may facilitate both a robust anti-MM CD8 T cell response and a myeloma-specific T cell memory (159) (Figure 6B). The impressive body of work summarized above strongly suggests that Vk*MYC provides a valuable blueprint for immunological studies using other mouse myeloma models included in Table 1.
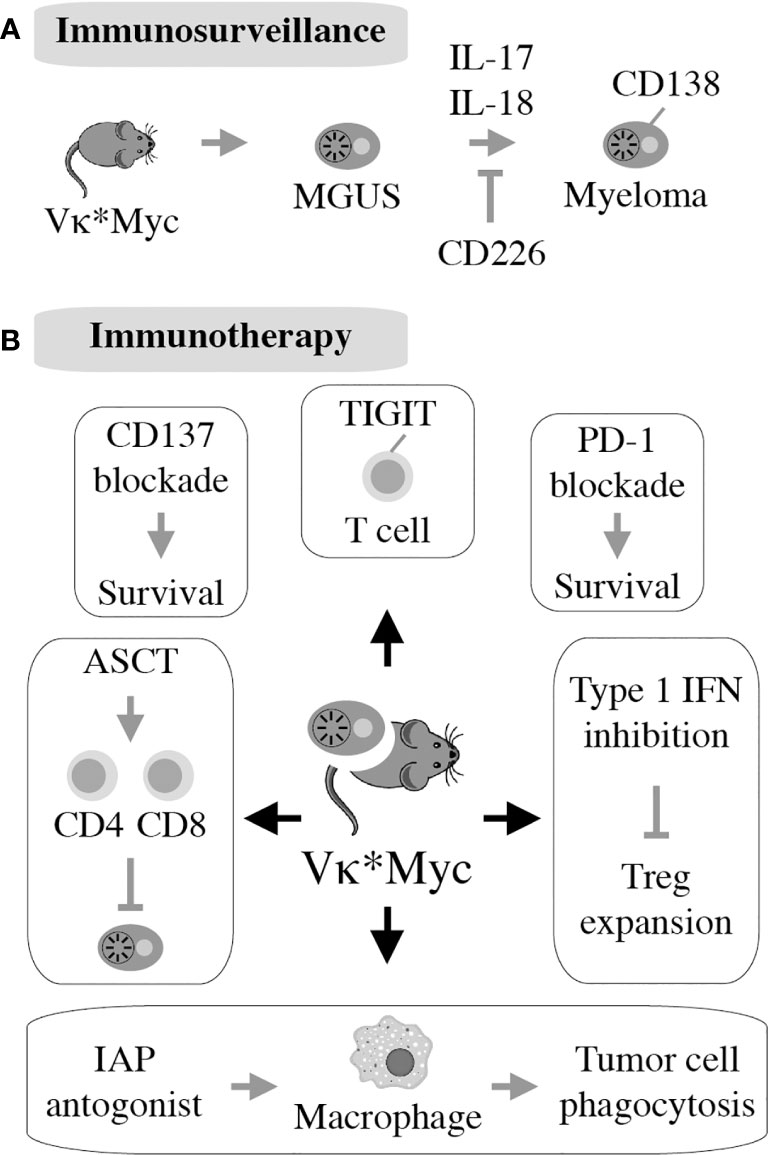
Figure 6 First described in 2008, the Vk*MYC model takes advantage of AID-activated MYC to induce myeloma on the B6 background. All 122 mice in the original study had monoclonal plasma cell expansion in the BM resembling human MM. Eighty percent of mice had measurable M-spike by 50 weeks of age. Additionally, aged Vk*MYC mice displayed many hallmarks of human MM, including bone loss and protein deposition in the kidneys (150). This allows for thorough studies of the MGUS to MM transition in this model (A). Vk*MYC mice accurately predict clinical efficacy of myeloma drugs (151) and provide a good model for experimental oncolytic immunotherapy (152) (B).
Research Gaps And Future Directions
Following in the Footsteps of Vκ*MYC
One line of future investigation should be aimed at determining whether the immune changes seen in tumor bearing Vk*MYC mice also occur in other strains of mice prone to spontaneous PCT (Table 1). Independent confirmation would lend support to the contention that the observed changes represent bona fide biological sequelae of neoplastic plasma cell development rather than a special feature of this particular model. Uncovering significant differences in immune cell compartments or pathways of immunity between different mouse models may also be of value because it may help investigators match a particular type of human myeloma in terms of progression stage (e.g., NDMM vs RRMM), outcome risk (e.g., standard vs high risk), or molecular subgroup (184) with the most appropriate mouse counterpart. Since human myeloma exhibits a great deal of diversity in cytogenetic, gene expression, epigenetic, immunologic, and other features (185), MM should be represented by a collection of models that mirror that diversity. However, this goal has not yet been achieved as the models listed in Table 1 represent but a narrow snapshot of the human myeloma landscape. With the exception of the most recently developed model that relies on a Cre recombinase effected loss of the microRNA-encoding tumor suppressor locus, miR-15a/16-1, malignant development is driven in these models by a limited set of oncogenes on the uniform, homogeneous genetic background of inbred mice (186). The overrepresentation of Myc, IL-6, and Bcl-2 family genes, particularly among the more thoroughly investigated models, underscores the narrowness and redundancy of the present situation.
Be this as it may, Vk*MYC and related models stand ready both to shed light on long-standing questions in myelomagenesis, such as the role of antigen and germinal center reentry of tumor precursors (187) and to revisit difficult issues in myeloma immunotherapy, such as the benefits of immune checkpoint inhibition (CPI) (188), which remain unclear at this juncture (189). Since Vk*MYC mice undergoing IAP inhibition responded to CPI with increased survival (153), in-depth analysis of that response may provide clues for how to incorporate CPI in human myeloma treatment protocols. Vk*MYC and other transgenics may also assist in validating novel immunotherapies that are emerging from exploratory studies using transplantation-based mouse models. Two recent advances that relied on 5TMM and HMCL, respectively, concerned the combination of vaccination and epigenetic therapies (190) and a neat strategy for enhancing the efficacy of daratumumab by genetic engineering of NK cells (191). Considering the increased interest of the clinical myeloma community in tumor prevention (192), Vk*MYC and related models may also afford opportunities for the preclinical evaluation of candidate interventions to block the progression of high-risk MGUS and SMM to frank myeloma.
Toward a Robust PDX Model of MM
Despite the breakthrough accomplishments of the MIS(KI)TRG6 mouse described above, this xenograft model of human myeloma is still limited compared to well-established PDX (patient derived xenograft) models of solid cancer (193) and emerging PDX models of lymphoma (194). Biological limitations of MIS(KI)TRG6 and the parental strain, MIS(KI)TRG, include proclivity to anemia and quick exhaustion of human grafts after cell or tissue transfer (195). There are also some thorny non-biological limitations, including intellectual property rights that have prevented the wider distribution of the MIS(KI)TRG6 thus far. Hence, additional work is warranted to improve upon this model and develop humanized laboratory mice that lend themselves to the preclinical evaluation of myeloma immunotherapy and precision medicine approaches. To that end, a fundamental conceptual consideration is the recognition that increasing levels of immunodeficiency result in better engraftment of tumor cells (Figure 3) but diminished opportunities to assess the impact of the immune system on myeloma biology and treatment responses. One way to address this conundrum is a non-genetic form of humanizing mice by means of adoptive transfer of human hematopoietic stem and progenitor, T, NK, and other blood cells. All of these cells are easily obtained from patients with myeloma, particularly those undergoing SCT, and can be engrafted in mice together with BM-derived malignant plasma cells. Disadvantages of this approach include the small experimental window in the adoptively transferred mouse (on the order of a few days) and the high risk of GvHD that may distort study results (196).
A parallel way forward is to continue with the genetic humanization of laboratory mice. The aim is to generate humanized mouse PDX myeloma models that will be as promising for immunotherapy research as the new generation of mouse PDX carcinoma models is (197, 198). Molecular targets of humanization include components of the HSC niche (e.g., c-kit and Flt3) and biological pathways of myeloid and NK reconstitution (e.g., c-kit ligand and GM-CSF). Additional targets include the major histocompatibility complex (e.g., deletion of mouse beta-2 microglobulin) and, importantly, immune checkpoints such as CTLA-4, the CD47 “don’t eat me” signal to macrophages, BTLA (CD272), TIM3, GITR, OX40 and others (199, 200). The long list of checkpoint genes underscores the elusiveness of humanizing the immune response of mice completely. The development of specialized, partially humanized mouse models dedicated to specific aspects of immunotherapy is therefore a viable compromise and a step in the right direction. A good example along this line is the generation of mice that contain a humanized form of cereblon (201), the molecular target of IMIDs. It renders the mice responsive to drugs of this sort, which are not active in normal mice. Three additional examples of humanized mouse models relevant for myeloma research are NOG-hIL-6 (202), B6-hCD3E (170) and B6-hTIGIT (203), which facilitate preclinical studies on MDSCs, BITEs and CPI, respectively.
Conclusion
Although immunotherapy holds great promise for revolutionizing myeloma treatment (124), much remains to be learned. Currently, only a fraction of patients achieves a complete, long-lasting treatment response and a functional or definitive cure remains elusive for the great majority of patients. Accurate mouse models of myeloma are needed to close current knowledge gaps and accelerate the design and testing of new immunotherapies. The workhorses of preclinical myeloma research, HMCL-in-mouse xenografting and mouse-in-mouse autografting, will continue to be employed to that end, but we anticipate that humanized PDX models of myeloma (204) and transgenic mouse models of myeloma will become more important as we go forward. These models may elucidate the complex mechanisms underlying myeloma immunopathology and -therapy and minimize the risk of failure in challenging and expensive clinical trials. Sharing experimental model systems without strings attached, enhancing collaboration among regional core facilities and national reference centers, and establishing standards for high scientific rigor for the preclinical myeloma research will contribute to a future for patients with myeloma that is hopeful and bright.
Author Contributions
MP performed the primary literature review and prepared the table. SJ edited the text, prepared the figures and formatted the final version. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the William G. Schuett, Jr., Multiple Myeloma Research Endowment. Additional support was provided by NCI R01CA204231 (to SR), NCI R01CA236814 and DOD CA180190 (to FZ) and NCI R01 CA151354 (to SJ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
BM, bone marrow; BMME, bone marrow microenvironment; B6, C57BL/6; C, BALB/c; Ig, immunoglobulin; IMiD, immunomodulatory drug; IP, intraperitoneal; IV, intravenous; mAb, monoclonal antibody; MGUS, monoclonal gammopathy of undetermined significance; MM, multiple myeloma; NDMM, newly diagnosed multiple myeloma; PI, proteasome inhibitor; RRMM, relapsed/refractory multiple myeloma; SC, subcutaneous; SMM, smoldering multiple myeloma; TME, tumor microenvironment.
References
1. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, et al. International Myeloma Working Group Updated Criteria for the Diagnosis of Multiple Myeloma. Lancet Oncol (2014) 15:e538–48. doi: 10.1016/S1470-2045(14)70442-5
2. Rollig C, Knop S, Bornhauser M. Multiple Myeloma. Lancet (2015) 385:2197–208. doi: 10.1016/S0140-6736(14)60493-1
3. Barlogie B, Mitchell A, van Rhee F, Epstein J, Morgan GJ, Crowley J. Curing Myeloma At Last: Defining Criteria and Providing the Evidence. Blood (2014) 124:3043–51. doi: 10.1182/blood-2014-07-552059
4. Kortum KM, Mai EK, Hanafiah NH, Shi CX, Zhu YX, Bruins L, et al. Targeted Sequencing of Refractory Myeloma Reveals a High Incidence of Mutations in CRBN and Ras Pathway Genes. Blood (2016) 128:1226–33. doi: 10.1182/blood-2016-02-698092
5. Weinhold N, Ashby C, Rasche L, Chavan SS, Stein C, Stephens OW, et al. Clonal Selection and Double Hit Events Involving Tumor Suppressor Genes Underlie Relapse From Chemotherapy: Myeloma as a Model. Blood (2016) 128:1735–44. doi: 10.1182/blood-2016-06-723007
6. Agirre X, Castellano G, Pascual M, Heath S, Kulis M, Segura V, et al. Whole-Epigenome Analysis in Multiple Myeloma Reveals DNA Hypermethylation of B Cell-Specific Enhancers. Genome Res (2015) 25:478–87. doi: 10.1101/gr.180240.114
7. Nikesitch N, Ling SC. Molecular Mechanisms in Multiple Myeloma Drug Resistance. J Clin Pathol (2016) 69:97–101. doi: 10.1136/jclinpath-2015-203414
8. Gooding S, Edwards CM. New Approaches to Targeting the Bone Marrow Microenvironment in Multiple Myeloma. Curr Opin Pharmacol (2016) 28:43–9. doi: 10.1016/j.coph.2016.02.013
9. Gulla A, Anderson KC. Multiple Myeloma: The (R)Evolution of Current Therapy and a Glance Into Future. Haematologica (2020) 105(10):2358–67. doi: 10.3324/haematol.2020.247015
10. Paton-Hough J, Chantry AD, Lawson MA. A Review of Current Murine Models of Multiple Myeloma Used to Assess the Efficacy of Therapeutic Agents on Tumour Growth and Bone Disease. Bone (2015) 77:57–68. doi: 10.1016/j.bone.2015.04.004
11. Lwin ST, Edwards CM, Silbermann R. Preclinical Animal Models of Multiple Myeloma. Bonekey Rep (2016) 5:772. doi: 10.1038/bonekey.2015.142
12. Yaccoby S, Barlogie B, Epstein J. Primary Myeloma Cells Growing in SCID-hu Mice: A Model for Studying the Biology and Treatment of Myeloma and its Manifestations. Blood (1998) 92:2908–13. doi: 10.1182/blood.V92.8.2908.420a32_2908_2913
13. Miyakawa Y, Ohnishi Y, Tomisawa M, Monnai M, Kohmura K, Ueyama Y, et al. Establishment of a New Model of Human Multiple Myeloma Using NOD/SCID/gammac(null) (NOG) Mice. Biochem Biophys Res Commun (2004) 313:258–62. doi: 10.1016/j.bbrc.2003.11.120
14. Calimeri T, Battista E, Conforti F, Neri P, Di Martino MT, Rossi M, et al. A Unique Three-Dimensional SCID-polymeric Scaffold (SCID-Synth-Hu) Model for In Vivo Expansion of Human Primary Multiple Myeloma Cells. Leukemia (2011) 25:707–11. doi: 10.1038/leu.2010.300
15. Radl J, Croese JW, Zurcher C, Van den Enden-Vieveen MH, de Leeuw AM. Animal Model of Human Disease. Multiple myeloma. Am J Pathol (1988) 132:593–7.
16. Hofgaard PO, Jodal HC, Bommert K, Huard B, Caers J, Carlsen H, et al. A Novel Mouse Model for Multiple Myeloma (MOPC315.BM) That Allows Noninvasive Spatiotemporal Detection of Osteolytic Disease. PloS One (2012) 7:e51892. doi: 10.1371/journal.pone.0051892
17. Liu H, Liu Z, Du J, He J, Lin P, Amini B, et al. Thymidine Phosphorylase Exerts Complex Effects on Bone Resorption and Formation in Myeloma. Sci Transl Med 8 (2016) 8(353):353ra113. doi: 10.1126/scitranslmed.aad8949
18. Higgs JT, Lee JH, Wang H, Ramani VC, Chanda D, Hardy CY, et al. Mesenchymal Stem Cells Expressing Osteoprotegerin Variants Inhibit Osteolysis in a Murine Model of Multiple Myeloma. Blood Adv (2017) 1:2375–85. doi: 10.1182/bloodadvances.2017007310
19. Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A Monoclonal Gammopathy Precedes Multiple Myeloma in Most Patients. Blood (2009) 113:5418–22. doi: 10.1182/blood-2008-12-195008
20. Muchtar E, Kumar SK, Magen H, Gertz MA. Diagnosis and Management of Smoldering Multiple Myeloma: The Razor’s Edge Between Clonality and Cancer. Leukemia lymphoma (2018) 59:288–99. doi: 10.1080/10428194.2017.1334124
21. Kyle RA, Durie BG, Rajkumar SV, Landgren O, Blade J, Merlini G, et al. Monoclonal Gammopathy of Undetermined Significance (MGUS) and Smoldering (Asymptomatic) Multiple Myeloma: IMWG Consensus Perspectives Risk Factors for Progression and Guidelines for Monitoring and Management. Leukemia (2010) 24:1121–7. doi: 10.1038/leu.2010.60
22. Dispenzieri A. Monoclonal Gammopathies of Clinical Significance. Hematol Am Soc Hematol Educ Program (2020) 2020:380–8. doi: 10.1182/hematology.2020000122
23. Kyle RA. Monoclonal Gammopathy of Undetermined Significance and Smoldering Multiple Myeloma. Eur J Haematol Suppl (1989) 51:70–5. doi: 10.1111/j.1600-0609.1989.tb01496.x
24. Goyal G, Rajkumar SV, Lacy MQ, Gertz MA, Buadi FK, Dispenzieri A, et al. Impact of Prior Diagnosis of Monoclonal Gammopathy on Outcomes in Newly Diagnosed Multiple Myeloma. Leukemia (2019) 33:1273–7. doi: 10.1038/s41375-019-0419-7
25. Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, et al. The Molecular Classification of Multiple Myeloma. Blood (2006) 108:2020–8. doi: 10.1182/blood-2005-11-013458
26. Avet-Loiseau H, Facon T, Daviet A, Godon C, Rapp MJ, Harousseau JL, et al. 14q32 Translocations and Monosomy 13 Observed in Monoclonal Gammopathy of Undetermined Significance Delineate a Multistep Process for the Oncogenesis of Multiple Myeloma. Intergroupe Francophone du Myelome. Cancer Res (1999) 59:4546–50.
27. Kyle RA, Rajkumar SV. Monoclonal Gammopathy of Undetermined Significance. Br J haematology (2006) 134:573–89. doi: 10.1111/j.1365-2141.2006.06235.x
28. Lopez-Corral L, Gutierrez NC, Vidriales MB, Mateos MV, Rasillo A, Garcia-Sanz R, et al. The Progression From MGUS to Smoldering Myeloma and Eventually to Multiple Myeloma Involves a Clonal Expansion of Genetically Abnormal Plasma Cells. Clin Cancer Res (2011) 17:1692–700. doi: 10.1158/1078-0432.CCR-10-1066
29. Minnie SA, Hill GR. Immunotherapy of Multiple Myeloma. J Clin Invest (2020) 130:1565–75. doi: 10.1172/JCI129205
30. Nakamura K, Smyth MJ, Martinet L. Cancer Immunoediting and Immune Dysregulation in Multiple Myeloma. Blood (2020) 10;136(24):2731–40. doi: 10.1182/blood.2020006540
31. Marsh-Wakefield F, Kruzins A, McGuire HM, Yang S, Bryant C, Fazekas de St Groth B, et al. Mass Cytometry Discovers Two Discrete Subsets of CD39(-)Treg Which Discriminate Mgus From Multiple Myeloma. Front Immunol (2019) 10:1596. doi: 10.3389/fimmu.2019.01596
32. Vuckovic S, Bryant CE, Lau KHA, Yang S, Favaloro J, McGuire HM, et al. Inverse Relationship Between Oligoclonal Expanded CD69- TTE and CD69+ TTE Cells in Bone Marrow of Multiple Myeloma Patients. Blood Adv (2020) 4:4593–604. doi: 10.1182/bloodadvances.2020002237
33. Bailur JK, McCachren SS, Doxie DB, Shrestha M, Pendleton K, Nooka AK, et al. Early Alterations in Stem-Like/Resident T Cells, Innate and Myeloid Cells in the Bone Marrow in Preneoplastic Gammopathy. JCI Insight 5 (2019) 5(11). doi: 10.1172/jci.insight.127807
34. Phan TG, Croucher PI. The Dormant Cancer Cell Life Cycle. Nat Rev Cancer (2020) 20:398–411. doi: 10.1038/s41568-020-0263-0
35. Murray ME, Gavile CM, Nair JR, Koorella C, Carlson LM, Buac D, et al. CD28-Mediated Pro-Survival Signaling Induces Chemotherapeutic Resistance in Multiple Myeloma. Blood (2014) 123:3770–9. doi: 10.1182/blood-2013-10-530964
36. Ray A, Song Y, Chauhan D, Anderson KC. Blockade of Ubiquitin Receptor Rpn13 in Plasmacytoid Dendritic Cells Triggers Anti-Myeloma Immunity. Blood Cancer J (2019) 9:64. doi: 10.1038/s41408-019-0224-6
37. Wong TW, Kita H, Hanson CA, Walters DK, Arendt BK, Jelinek DF. Induction of Malignant Plasma Cell Proliferation by Eosinophils. PloS One (2013) 8:e70554. doi: 10.1371/journal.pone.0070554
38. Sorrig R, Klausen TW, Salomo M, Vangsted A, Gimsing P. Risk Factors for Infections in Newly Diagnosed Multiple Myeloma Patients: A Danish Retrospective Nationwide Cohort Study. Eur J haematology (2019) 102:182–90. doi: 10.1111/ejh.13190
39. Robertson JD, Nagesh K, Jowitt SN, Dougal M, Anderson H, Mutton K, et al. Immunogenicity of Vaccination Against Influenza, Streptococcus Pneumoniae and Haemophilus Influenzae Type B in Patients With Multiple Myeloma. Br J Cancer (2000) 82:1261–5. doi: 10.1054/bjoc.1999.1088
40. Isidori A, de Leval L, Gergis U, Musto P, Porcu P. Management of Patients With Hematologic Malignancies During the COVID-19 Pandemic: Practical Considerations and Lessons to Be Learned. Front Oncol (2020) 10:1439. doi: 10.3389/fonc.2020.01439
41. Nadeem O, Tai YT, Anderson KC. Immunotherapeutic and Targeted Approaches in Multiple Myeloma. Immunotargets Ther (2020) 9:201–15. doi: 10.2147/ITT.S240886
42. de Weers M, Tai YT, van der Veer MS, Bakker JM, Vink T, Jacobs DC, et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J Immunol (2011) 186:1840–8. doi: 10.4049/jimmunol.1003032
43. Martin T, Baz R, Benson DM, Lendvai N, Wolf J, Munster P, et al. A Phase 1b Study of Isatuximab Plus Lenalidomide and Dexamethasone for Relapsed/Refractory Multiple Myeloma. Blood (2017) 129:3294–303. doi: 10.1182/blood-2016-09-740787
44. van de Donk NW, Moreau P, Plesner T, Palumbo A, Gay F, Laubach JP, et al. Clinical Efficacy and Management of Monoclonal Antibodies Targeting CD38 and SLAMF7 in Multiple Myeloma. Blood (2016) 127:681–95. doi: 10.1182/blood-2015-10-646810
45. Trudel S, Lendvai N, Popat R, Voorhees PM, Reeves B, Libby EN, et al. Targeting B-cell Maturation Antigen With GSK2857916 Antibody-Drug Conjugate in Relapsed or Refractory Multiple Myeloma (BMA117159): A Dose Escalation and Expansion Phase 1 Trial. Lancet Oncol (2018) 19:1641–53. doi: 10.1016/S1470-2045(18)30576-X
46. Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, et al. Antitumor Activity of Thalidomide in Refractory Multiple Myeloma. New Engl J Med (1999) 341:1565–71. doi: 10.1056/NEJM199911183412102
47. Tai YT, Li XF, Catley L, Coffey R, Breitkreutz I, Bae J, et al. Immunomodulatory Drug Lenalidomide (CC-5013, IMiD3) Augments Anti-CD40 SGN-40-induced Cytotoxicity in Human Multiple Myeloma: Clinical Implications. Cancer Res (2005) 65:11712–20. doi: 10.1158/0008-5472.CAN-05-1657
48. Barlogie B, Shaughnessy J, Zangari M, Tricot G. High-Dose Therapy and Immunomodulatory Drugs in Multiple Myeloma. Semin Oncol (2002) 29:26–33. doi: 10.1016/S0093-7754(02)70058-4
49. Lacy MQ, Hayman SR, Gertz MA, Dispenzieri A, Buadi F, Kumar S, et al. Pomalidomide (CC4047) Plus Low-Dose Dexamethasone as Therapy for Relapsed Multiple Myeloma. J Clin Oncol (2009) 27:5008–14. doi: 10.1200/JCO.2009.23.6802
50. Bjorklund CC, Kang J, Amatangelo M, Polonskaia A, Katz M, Chiu H, et al. Iberdomide (Cc-220) Is a Potent Cereblon E3 Ligase Modulator With Antitumor and Immunostimulatory Activities in Lenalidomide- and Pomalidomide-Resistant Multiple Myeloma Cells With Dysregulated CRBN. Leukemia (2020) 34:1197–201. doi: 10.1038/s41375-019-0620-8
51. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat Rev (2017) 17:97–111. doi: 10.1038/nri.2016.107
52. Pearson JA, Wong FS, Wen L. The Importance of the Non Obese Diabetic (NOD) Mouse Model in Autoimmune Diabetes. J Autoimmun (2016) 66:76–88. doi: 10.1016/j.jaut.2015.08.019
53. Fryer RA, Graham TJ, Smith EM, Walker-Samuel S, Morgan GJ, Robinson SP, et al. Characterization of a Novel Mouse Model of Multiple Myeloma and its Use in Preclinical Therapeutic Assessment. PloS One (2013) 8:e57641. doi: 10.1371/journal.pone.0057641
54. Schueler J, Wider D, Klingner K, Siegers GM, May AM, Wasch R, et al. Intratibial Injection of Human Multiple Myeloma Cells in NOD/SCID Il-2Rgamma(null) Mice Mimics Human Myeloma and Serves as a Valuable Tool for the Development of Anticancer Strategies. PloS One (2013) 8:e79939. doi: 10.1371/journal.pone.0079939
55. Rossi M, Botta C, Arbitrio M, Grembiale RD, Tagliaferri P, Tassone P. Mouse Models of Multiple Myeloma: Technologic Platforms and Perspectives. Oncotarget (2018) 9:20119–33. doi: 10.18632/oncotarget.24614
56. Sarin V, Yu K, Ferguson ID, Gugliemini O, Nix MA, Hann B, et al. Evaluating the Efficacy of Multiple Myeloma Cell Lines as Models for Patient Tumors Via Transcriptomic Correlation Analysis. Leukemia (2020) 34(10):2754–65. doi: 10.1101/847368
57. Moreaux J, Klein B, Bataille R, Descamps G, Maiga S, Hose D, et al. A High-Risk Signature for Patients With Multiple Myeloma Established From the Molecular Classification of Human Myeloma Cell Lines. Haematologica (2011) 96:574–82. doi: 10.3324/haematol.2010.033456
58. Papadimitriou K, Kostopoulos IV, Tsopanidou A, Orologas-Stavrou N, Kastritis E, Tsitsilonis O, et al. Ex Vivo Models Simulating the Bone Marrow Environment and Predicting Response to Therapy in Multiple Myeloma. Cancers (Basel) (2020) 12. doi: 10.3390/cancers12082006
59. Urashima M, Chen BP, Chen S, Pinkus GS, Bronson RT, Dedera DA, et al. The Development of a Model for the Homing of Multiple Myeloma Cells to Human Bone Marrow. Blood (1997) 90:754–65. doi: 10.1182/blood.V90.2.754.754_754_765
60. Kyoizumi S, Baum CM, Kaneshima H, McCune JM, Yee EJ, Namikawa R. Implantation and Maintenance of Functional Human Bone Marrow in SCID-hu Mice. Blood (1992) 79:1704–11. doi: 10.1182/blood.V79.7.1704.bloodjournal7971704
61. Epstein J, Yaccoby S. The SCID-hu Myeloma Model. Methods Mol Med (2005) 113:183–90. doi: 10.1385/1-59259-916-8:183
62. Yata K, Yaccoby S. The SCID-rab Model: A Novel In Vivo System for Primary Human Myeloma Demonstrating Growth of CD138-expressing Malignant Cells. Leukemia (2004) 18:1891–7. doi: 10.1038/sj.leu.2403513
63. Tassone P, Neri P, Burger R, Savino R, Shammas M, Catley L, et al. Combination Therapy With Interleukin-6 Receptor Superantagonist Sant7 and Dexamethasone Induces Antitumor Effects in a Novel SCID-hu In Vivo Model of Human Multiple Myeloma. Clin Cancer Res (2005) 11:4251–8. doi: 10.1158/1078-0432.CCR-04-2611
64. Yaccoby S, Pennisi A, Li X, Dillon SR, Zhan F, Barlogie B, et al. Atacicept (Taci-Ig) Inhibits Growth of TACI(high) Primary Myeloma Cells in SCID-hu Mice and in Coculture With Osteoclasts. Leukemia (2008) 22:406–13. doi: 10.1038/sj.leu.2405048
65. van Rhee F, Szmania SM, Dillon M, van Abbema AM, Li X, Stone MK, et al. Combinatorial Efficacy of anti-CS1 Monoclonal Antibody Elotuzumab (HuLuc63) and Bortezomib Against Multiple Myeloma. Mol Cancer Ther (2009) 8:2616–24. doi: 10.1158/1535-7163.MCT-09-0483
66. Pennisi A, Li X, Ling W, Khan S, Zangari M, Yaccoby S. The Proteasome Inhibitor, Bortezomib Suppresses Primary Myeloma and Stimulates Bone Formation in Myelomatous and Nonmyelomatous Bones In Vivo. Am J Hematol (2009) 84:6–14. doi: 10.1002/ajh.21310
67. Yaccoby S, Ling W, Zhan F, Walker R, Barlogie B, Shaughnessy JD Jr. Antibody-Based Inhibition of DKK1 Suppresses Tumor-Induced Bone Resorption and Multiple Myeloma Growth In Vivo. Blood (2007) 109:2106–11. doi: 10.1182/blood-2006-09-047712
68. Li X, Ling W, Pennisi A, Wang Y, Khan S, Heidaran M, et al. Human Placenta-Derived Adherent Cells Prevent Bone Loss, Stimulate Bone Formation, and Suppress Growth of Multiple Myeloma in Bone. Stem Cells (2011) 29:263–73. doi: 10.1002/stem.572
69. Di Martino MT, Leone E, Amodio N, Foresta U, Lionetti M, Pitari MR, et al. Synthetic miR-34a Mimics as a Novel Therapeutic Agent for Multiple Myeloma: In Vitro and In Vivo Evidence. Clin Cancer Res (2012) 18:6260–70. doi: 10.1158/1078-0432.CCR-12-1708
70. Amodio N, Leotta M, Bellizzi D, Di Martino MT, D’Aquila P, Lionetti M, et al. DNA-Demethylating and Anti-Tumor Activity of Synthetic miR-29b Mimics in Multiple Myeloma. Oncotarget (2012) 3:1246–58. doi: 10.18632/oncotarget.675
71. Rongvaux A, Takizawa H, Strowig T, Willinger T, Eynon EE, Flavell RA, et al. Human Hemato-Lymphoid System Mice: Current Use and Future Potential for Medicine. Annu Rev Immunol (2013) 31:635–74. doi: 10.1146/annurev-immunol-032712-095921
72. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and Function of Human Innate Immune Cells in a Humanized Mouse Model. Nat Biotechnol (2014) 32:364–72. doi: 10.1038/nbt.2858
73. Rosean TR, Tompkins VS, Tricot G, Holman CJ, Olivier AK, Zhan F, et al. Preclinical Validation of Interleukin 6 as a Therapeutic Target in Multiple Myeloma. Immunol Res (2014) 59:188–202. doi: 10.1007/s12026-014-8528-x
74. Das R, Strowig T, Verma R, Koduru S, Hafemann A, Hopf S, et al. Microenvironment-Dependent Growth of Preneoplastic and Malignant Plasma Cells in Humanized Mice. Nat Med (2016) 22:1351–7. doi: 10.1038/nm.4202
75. Lin L, Cho SF, Xing L, Wen K, Li Y, Yu T, et al. Preclinical Evaluation of CD8+ anti-BCMA mRNA Car T Cells for Treatment of Multiple Myeloma. Leukemia (2020) 35:752–63. doi: 10.1038/s41375-020-0951-5
76. Radhakrishnan SV, Luetkens T, Scherer SD, Davis P, Vander Mause ER, Olson ML, et al. Cd229 Car T Cells Eliminate Multiple Myeloma and Tumor Propagating Cells Without Fratricide. Nat Commun (2020) 11:798. doi: 10.1038/s41467-020-14619-z
77. Daniels-Wells TR, Candelaria PV, Leoh LS, Nava M, Martinez-Maza O, Penichet ML. An IgG1 Version of the Anti-transferrin Receptor 1 Antibody Ch128.1 Shows Significant Antitumor Activity Against Different Xenograft Models of Multiple Myeloma: A Brief Communication. J Immunother (2020) 43:48–52. doi: 10.1097/CJI.0000000000000304
78. Leoh LS, Kim YK, Candelaria PV, Martinez-Maza O, Daniels-Wells TR, Penichet ML. Efficacy and Mechanism of Antitumor Activity of an Antibody Targeting Transferrin Receptor 1 in Mouse Models of Human Multiple Myeloma. J Immunol (2018) 200:3485–94. doi: 10.4049/jimmunol.1700787
79. Goldstein RL, Goyos A, Li CM, Deegen P, Bogner P, Sternjak A, et al. AMG 701 Induces Cytotoxicity of Multiple Myeloma Cells and Depletes Plasma Cells in Cynomolgus Monkeys. Blood Adv (2020) 4:4180–94. doi: 10.1182/bloodadvances.2020002565
80. Naeimi Kararoudi M, Nagai Y, Elmas E, de Souza Fernandes Pereira M, Ali SA, Imus PH, et al. CD38 Deletion of Human Primary NK Cells Eliminates Daratumumab-Induced Fratricide and Boosts Their Effector Activity. Blood (2020) 136:2416–27. doi: 10.1182/blood.2020006200
81. Amend SR, Wilson WC, Chu L, Lu L, Liu P, Serie D, et al. Whole Genome Sequence of Multiple Myeloma-Prone C57BL/KaLwRij Mouse Strain Suggests the Origin of Disease Involves Multiple Cell Types. PloS One (2015) 10:e0127828. doi: 10.1371/journal.pone.0127828
82. Radl J, De Glopper ED, Schuit HR, Zurcher C. Idiopathic Paraproteinemia. II. Transplantation of the Paraprotein-Producing Clone From Old to Young C57BL/KaLwRij Mice. J Immunol (1979) 122:609–13.
83. Croese JW, Vas Nunes CM, Radl J, van den Enden-Vieveen MH, Brondijk RJ, Boersma WJ. The 5T2 Mouse Multiple Myeloma Model: Characterization of 5T2 Cells Within the Bone Marrow. Br J Cancer (1987) 56:555–60. doi: 10.1038/bjc.1987.241
84. Radl J, Croese JW, Zurcher C, van den Enden-Vieveen MH, Brondijk RJ, Kazil M, et al. Influence of Treatment With APD-bisphosphonate on the Bone Lesions in the Mouse 5T2 Multiple Myeloma. Cancer (1985) 55:1030–40. doi: 10.1002/1097-0142(19850301)55:5<1030::AID-CNCR2820550518>3.0.CO;2-Y
85. Vanderkerken K, Goes E, De Raeve H, Radl J, Van Camp B. Follow-Up of Bone Lesions in an Experimental Multiple Myeloma Mouse Model: Description of an In Vivo Technique Using Radiography Dedicated for Mammography. Br J Cancer (1996) 73:1463–5. doi: 10.1038/bjc.1996.277
86. Soodgupta D, Hurchla MA, Jiang M, Zheleznyak A, Weilbaecher KN, Anderson CJ, et al. Very Late Antigen-4 (Alpha(4)Beta(1) Integrin) Targeted PET Imaging of Multiple Myeloma. PloS One (2013) 8:e55841. doi: 10.1371/journal.pone.0055841
87. Manning LS, Berger JD, O’Donoghue HL, Sheridan GN, Claringbold PG, Turner JH. A Model of Multiple Myeloma: Culture of 5T33 Murine Myeloma Cells and Evaluation of Tumorigenicity in the C57BL/KaLwRij Mouse. Br J Cancer (1992) 66:1088–93. doi: 10.1038/bjc.1992.415
88. Dallas SL, Garrett IR, Oyajobi BO, Dallas MR, Boyce BF, Bauss F, et al. Ibandronate Reduces Osteolytic Lesions But Not Tumor Burden in a Murine Model of Myeloma Bone Disease. Blood (1999) 93:1697–706. doi: 10.1182/blood.V93.5.1697.405a17_1697_1706
89. De Smedt E, Lui H, Maes K, De Veirman K, Menu E, Vanderkerken K, et al. The Epigenome in Multiple Myeloma: Impact on Tumor Cell Plasticity and Drug Response. Front Oncol (2018) 8:566. doi: 10.3389/fonc.2018.00566
90. Maes K, Boeckx B, Vlummens P, De Veirman K, Menu E, Vanderkerken K, et al. The Genetic Landscape of 5T Models for Multiple Myeloma. Sci Rep (2018) 8:15030. doi: 10.1038/s41598-018-33396-w
91. Alici E, Konstantinidis KV, Aints A, Dilber MS, Abedi-Valugerdi M. Visualization of 5T33 Myeloma Cells in the C57BL/KaLwRij Mouse: Establishment of a New Syngeneic Murine Model of Multiple Myeloma. Exp Hematol (2004) 32:1064–72. doi: 10.1016/j.exphem.2004.07.019
92. Wang S, Yang J, Qian J, Wezeman M, Kwak LW, Yi Q. Tumor Evasion of the Immune System: Inhibiting P38 MAPK Signaling Restores the Function of Dendritic Cells in Multiple Myeloma. Blood (2006) 107:2432–9. doi: 10.1182/blood-2005-06-2486
93. Rosenblatt J, Avigan D. Cellular Immunotherapy for Multiple Myeloma. Cancer J (2019) 25:38–44. doi: 10.1097/PPO.0000000000000356
94. Croese JW, Vissinga CS, Boersma WJ, Radl J. Immune Regulation of Mouse 5T2 Multiple Myeloma. I. Immune Response to 5t2 MM Idiotype. Neoplasma (1991) 38:457–66.
95. Croese JW, Van den Enden-Vieveen MH, Radl J. Immune Regulation of 5T2 Mouse Multiple Myeloma. II. Immunological Treatment of 5t2 MM Residual Disease. Neoplasma (1991) 38:467–74.
96. Hong S, Qian J, Yang J, Li H, Kwak LW, Yi Q. Roles of Idiotype-Specific T Cells in Myeloma Cell Growth and Survival: Th1 and CTL Cells are Tumoricidal While Th2 Cells Promote Tumor Growth. Cancer Res (2008) 68:8456–64. doi: 10.1158/0008-5472.CAN-08-2213
97. Laronne-Bar-On A, Zipori D, Haran-Ghera N. Increased Regulatory Versus Effector T Cell Development Is Associated With Thymus Atrophy in Mouse Models of Multiple Myeloma. J Immunol (2008) 181:3714–24. doi: 10.4049/jimmunol.181.5.3714
98. Feyler S, von Lilienfeld-Toal M, Jarmin S, Marles L, Rawstron A, Ashcroft AJ, et al. Cd4(+)Cd25(+)FoxP3(+) Regulatory T Cells are Increased Whilst CD3(+)CD4(-)CD8(-)alphabetaTCR(+) Double Negative T Cells are Decreased in the Peripheral Blood of Patients With Multiple Myeloma Which Correlates With Disease Burden. Br J Haematology (2009) 144:686–95. doi: 10.1111/j.1365-2141.2008.07530.x
99. Savelyeva N, King CA, Vitetta ES, Stevenson FK. Inhibition of a Vaccine-Induced Anti-Tumor B Cell Response by Soluble Protein Antigen in the Absence of Continuing T Cell Help. Proc Natl Acad Sci USA (2005) 102:10987–92. doi: 10.1073/pnas.0505108102
100. Hong S, Qian J, Li H, Yang J, Lu Y, Zheng Y, et al. Cpg or IFN-alpha are More Potent Adjuvants Than GM-CSF to Promote Anti-Tumor Immunity Following Idiotype Vaccine in Multiple Myeloma. Cancer Immunol Immunother (2012) 61:561–71. doi: 10.1007/s00262-011-1123-2
101. Hong S, Li H, Qian J, Yang J, Lu Y, Yi Q. Optimizing Dendritic Cell Vaccine for Immunotherapy in Multiple Myeloma: Tumour Lysates are More Potent Tumour Antigens Than Idiotype Protein to Promote Anti-Tumour Immunity. Clin Exp Immunol (2012) 170:167–77. doi: 10.1111/j.1365-2249.2012.04642.x
102. Rosenblatt J, Avivi I, Vasir B, Uhl L, Munshi NC, Katz T, et al. Vaccination With Dendritic Cell/Tumor Fusions Following Autologous Stem Cell Transplant Induces Immunologic and Clinical Responses in Multiple Myeloma Patients. Clin Cancer Res (2013) 19:3640–8. doi: 10.1158/1078-0432.CCR-13-0282
103. Zhang L, Bi E, Hong S, Qian J, Zheng C, Wang M, et al. Cd4(+) T Cells Play a Crucial Role for Lenalidomide In Vivo Anti-Tumor Activity in Murine Multiple Myeloma. Oncotarget (2015) 6:36032–40. doi: 10.18632/oncotarget.5506
104. Nur H, Fostier K, Aspeslagh S, Renmans W, Bertrand E, Leleu X, et al. Preclinical Evaluation of Invariant Natural Killer T Cells in the 5T33 Multiple Myeloma Model. PloS One (2013) 8:e65075. doi: 10.1371/journal.pone.0065075
105. Barber A, Meehan KR, Sentman CL. Treatment of Multiple Myeloma With Adoptively Transferred Chimeric NKG2D Receptor-Expressing T Cells. Gene Ther (2011) 18:509–16. doi: 10.1038/gt.2010.174
106. Hallett WH, Jing W, Drobyski WR, Johnson BD. Immunosuppressive Effects of Multiple Myeloma are Overcome by PD-L1 Blockade. Biol Blood Marrow Transplant (2011) 17:1133–45. doi: 10.1016/j.bbmt.2011.03.011
107. Kearl TJ, Jing W, Gershan JA, Johnson BD. Programmed Death receptor-1/programmed Death Receptor Ligand-1 Blockade After Transient Lymphodepletion to Treat Myeloma. J Immunol (2013) 190:5620–8. doi: 10.4049/jimmunol.1202005
108. Fowler JA, Mundy GR, Lwin ST, Edwards CM. Bone Marrow Stromal Cells Create a Permissive Microenvironment for Myeloma Development: A New Stromal Role for Wnt Inhibitor Dkk1. Cancer Res (2012) 72:2183–9. doi: 10.1158/0008-5472.CAN-11-2067
109. Potter M. Neoplastic Development in Plasma Cells. Immunol Rev (2003) 194:177–95. doi: 10.1034/j.1600-065X.2003.00061.x
110. Potter M, Boyce C. Induction of Plasma Cell Neoplasms in Strain BALB/c Mice With Mineral Oil and Mineral Oil Adjuvants. Nature (1962) 193:1086. doi: 10.1038/1931086a0
111. Mokyr MB, Prokhorova A, Rubin M, Bluestone JA. Insight Into the Mechanism of TCR-V Beta 8+/CD8+ T Cell-Mediated MOPC-315 Tumor Eradication. J Immunol (1994) 153:3123–34.
112. Wang D, Floisand Y, Myklebust CV, Burgler S, Parente-Ribes A, Hofgaard PO, et al. Autologous Bone Marrow Th Cells can Support Multiple Myeloma Cell Proliferation In Vitro and in Xenografted Mice. Leukemia (2017) 31:2114–21. doi: 10.1038/leu.2017.69
113. Baghdadi M, Ishikawa K, Nakanishi S, Murata T, Umeyama Y, Kobayashi T, et al. A Role for IL-34 in Osteolytic Disease of Multiple Myeloma. Blood Adv (2019) 3:541–51. doi: 10.1182/bloodadvances.2018020008
114. Schwarzer R, Nickel N, Godau J, Willie BM, Duda GN, Schwarzer R, et al. Notch Pathway Inhibition Controls Myeloma Bone Disease in the Murine MOPC315. BM Model Blood Cancer J (2014) 4:e217. doi: 10.1038/bcj.2014.37
115. Wong D, Winter O, Hartig C, Siebels S, Szyska M, Tiburzy B, et al. Eosinophils and Megakaryocytes Support the Early Growth of Murine MOPC315 Myeloma Cells in Their Bone Marrow Niches. PloS One (2014) 9:e109018. doi: 10.1371/journal.pone.0109018
116. Villa NY, Rahman MM, Mamola J, D’Isabella J, Goras E, Kilbourne J, et al. Autologous Transplantation Using Donor Leukocytes Loaded Ex Vivo With Oncolytic Myxoma Virus can Eliminate Residual Multiple Myeloma. Mol Ther Oncolytics (2020) 18:171–88. doi: 10.1016/j.omto.2020.06.011
117. Bogen B, Fauskanger M, Haabeth OA, Tveita A. Cd4(+) T Cells Indirectly Kill Tumor Cells Via Induction of Cytotoxic Macrophages in Mouse Models. Cancer Immunol Immunother (2019) 68:1865–73. doi: 10.1007/s00262-019-02374-0
118. Haabeth OA, Tveita A, Fauskanger M, Hennig K, Hofgaard PO, Bogen B. Idiotype-Specific CD4(+) T Cells Eradicate Disseminated Myeloma. Leukemia (2016) 30:1216–20. doi: 10.1038/leu.2015.278
119. Haabeth OAW, Hennig K, Fauskanger M, Loset GA, Bogen B, Tveita A. Cd4+ T-cell Killing of Multiple Myeloma Cells is Mediated by Resident Bone Marrow Macrophages. Blood Adv (2020) 4:2595–605. doi: 10.1182/bloodadvances.2020001434
120. Binsfeld M, Muller J, Lamour V, De Veirman K, De Raeve H, Bellahcene A, et al. Granulocytic Myeloid-Derived Suppressor Cells Promote Angiogenesis in the Context of Multiple Myeloma. Oncotarget (2016) 7:37931–43. doi: 10.18632/oncotarget.9270
121. Binsfeld M, Beguin Y, Belle L, Otjacques E, Hannon M, Briquet A, et al. Establishment of a Murine Graft-Versus-Myeloma Model Using Allogeneic Stem Cell Transplantation. PloS One (2014) 9:e113764. doi: 10.1371/journal.pone.0113764
122. Yado S, Luboshits G, Hazan O, Or R, Firer MA. Long-Term Survival Without Graft-Versus-Host-Disease Following Infusion of Allogeneic Myeloma-Specific Vbeta T Cell Families. J Immunother Cancer (2019) 7:301. doi: 10.1186/s40425-019-0776-9
123. Braathen R, Spang HCL, Hinke DM, Blazevski J, Bobic S, Fossum E, et al. A DNA Vaccine That Encodes an Antigen-Presenting Cell-Specific Heterodimeric Protein Protects Against Cancer and Influenza. Mol Ther Methods Clin Dev (2020) 17:378–92. doi: 10.1016/j.omtm.2020.01.007
124. Rasche L, Hudecek M, Einsele H. What is the Future of Immunotherapy in Multiple Myeloma? Blood (2020) 136:2491–7. doi: 10.1182/blood.2019004176
125. Anderson PN, Potter M. Induction of Plasma Cell Tumours in BALB-c Mice With 2,6,10,14-Tetramethylpentadecane (Pristane). Nature (1969) 222:994–5. doi: 10.1038/222994a0
126. Stone MJ. Monoclonal Antibodies in the Prehybridoma Era: A Brief Historical Perspective and Personal Reminiscence. Clin lymphoma (2001) 2:148–54. doi: 10.3816/CLM.2001.n.020
127. Kohler G, Milstein C. Continuous Cultures of Fused Cells Secreting Antibody of Predefined Specificity. Nature (1975) 256:495–7. doi: 10.1038/256495a0
128. Potter M, Pumphrey JG, Bailey DW. Genetics of Susceptibility to Plasmacytoma Induction. I. Balb/cAnN (C), C57BL/6N (B6), C57BL/Ka (BK), (C Times B6)F1, (C Times BK)F1, and C Times B Recombinant-Inbred Strains. J Natl Cancer Inst (1975) 54:1413–7. doi: 10.1093/jnci/54.6.1413
129. Zhang SL, DuBois W, Ramsay ES, Bliskovski V, Morse HC, Taddesse-Heath L, et al. Efficiency Alleles of the Pctr1 Modifier Locus for Plasmacytoma Susceptibility. Mol Cell Biol (2001) 21:310–8. doi: 10.1128/MCB.21.1.310-318.2001
130. Bliskovsky V, Ramsay ES, Scott J, DuBois W, Shi W, Zhang S, et al. Frap, FKBP12 Rapamycin-Associated Protein, Is a Candidate Gene for the Plasmacytoma Resistance Locus Pctr2 and Can Act as a Tumor Suppressor Gene. Proc Natl Acad Sci USA (2003) 100:14982–7. doi: 10.1073/pnas.2431627100
131. Shen-Ong GL, Keath EJ, Piccoli SP, Cole MD. Novel Myc Oncogene RNA From Abortive Immunoglobulin-Gene Recombination in Mouse Plasmacytomas. Cell (1982) 31:443–52. doi: 10.1016/0092-8674(82)90137-4
132. Niiro H, Clark EA. Regulation of B-Cell Fate by Antigen-Receptor Signals. Nat Rev (2002) 2:945–56. doi: 10.1038/nri955
133. Byrd LG, McDonald AH, Gold LG, Potter M. Specific Pathogen-Free BALB/cAn Mice are Refractory to Plasmacytoma Induction by Pristane. J Immunol (1991) 147:3632–7.
134. McIntire KR, Princler GL. Prolonged Adjuvant Stimulation in Germ-Free BALB-c Mice: Development of Plasma Cell Neoplasia. Immunology (1969) 17:481–7.
135. Lonial S, Jacobus S, Fonseca R, Weiss M, Kumar S, Orlowski RZ, et al. Randomized Trial of Lenalidomide Versus Observation in Smoldering Multiple Myeloma. J Clin Oncol (2020) 38:1126–37. doi: 10.1200/JCO.19.01740
136. Cooke RE, Koldej R, Ritchie D. Immunotherapeutics in Multiple Myeloma: How can Translational Mouse Models Help? J Oncol (2019) 2019:2186494. doi: 10.1155/2019/2186494
137. Rajkumar SV. Multiple Myeloma: Every Year a New Standard? Hematol Oncol (2019) 37 Suppl 1:62–5. doi: 10.1002/hon.2586
138. Vlummens P, De Veirman K, Menu E, De Bruyne E, Offner F, Vanderkerken K, et al. The Use of Murine Models for Studying Mechanistic Insights of Genomic Instability in Multiple Myeloma. Front Genet (2019) 10:740. doi: 10.3389/fgene.2019.00740
139. Rutsch S, Neppalli VT, Shin DM, DuBois W, Morse HC3, Goldschmidt H, et al. IL-6 and MYC Collaborate in Plasma Cell Tumor Formation in Mice. . Blood (2010) 115:1746–54. doi: 10.1182/blood-2009-08-237941
140. Boylan KL, Gosse MA, Staggs SE, Janz S, Grindle S, Kansas GS, et al. A Transgenic Mouse Model of Plasma Cell Malignancy Shows Phenotypic, Cytogenetic, and Gene Expression Heterogeneity Similar to Human Multiple Myeloma. Cancer Res (2007) 67:4069–78. doi: 10.1158/0008-5472.CAN-06-3699
141. Scherger AK, Al-Maarri M, Maurer HC, Schick M, Maurer S, Ollinger R, et al. Activated gp130 Signaling Selectively Targets B Cell Differentiation to Induce Mature Lymphoma and Plasmacytoma. JCI Insight (2019) 4(15). doi: 10.1172/jci.insight.128435
142. Hu Y, Zheng M, Gali R, Tian Z, Topal Gorgun G, Munshi NC, et al. A Novel Rapid-Onset High-Penetrance Plasmacytoma Mouse Model Driven by Deregulation of cMYC Cooperating With KRAS12V in BALB/c Mice. Blood Cancer J (2013) 3:e156. doi: 10.1038/bcj.2013.53
143. Hu Y, Song W, Cirstea D, Lu D, Munshi NC, Anderson KC. CSNK1alpha1 Mediates Malignant Plasma Cell Survival. Leukemia (2015) 29:474–82. doi: 10.1038/leu.2014.202
144. Tompkins VS, Rosean TR, Holman CJ, DeHoedt C, Olivier AK, Duncan KM, et al. Adoptive B-Cell Transfer Mouse Model of Human Myeloma. Leukemia (2016) 30:962–6. doi: 10.1038/leu.2015.197
145. Cooke RE, Gherardin NA, Harrison SJ, Quach H, Godfrey DI, Prince M, et al. Spontaneous Onset and Transplant Models of the Vk*MYC Mouse Show Immunological Sequelae Comparable to Human Multiple Myeloma. J Transl Med (2016) 14:259. doi: 10.1186/s12967-016-0994-6
146. Guillerey C, Ferrari de Andrade L, Vuckovic S, Miles K, Ngiow SF, Yong MC, et al. Immunosurveillance and Therapy of Multiple Myeloma Are CD226 Dependent. J Clin Invest (2015) 125:2904. doi: 10.1172/JCI82646
147. Calcinotto A, Brevi A, Chesi M, Ferrarese R, Garcia Perez L, Grioni M, et al. Microbiota-Driven Interleukin-17-Producing Cells and Eosinophils Synergize to Accelerate Multiple Myeloma Progression. Nat Commun (2018) 9:4832. doi: 10.1038/s41467-018-07305-8
148. Nakamura K, Kassem S, Cleynen A, Chretien ML, Guillerey C, Putz EM, et al. Dysregulated IL-18 is a Key Driver of Immunosuppression and a Possible Therapeutic Target in the Multiple Myeloma Microenvironment. Cancer Cell (2018) 33:634–48.e5. doi: 10.1016/j.ccell.2018.02.007
149. Akhmetzyanova I, McCarron MJ, Parekh S, Chesi M, Bergsagel PL, Fooksman DR. Dynamic CD138 Surface Expression Regulates Switch Between Myeloma Growth and Dissemination. Leukemia (2020) 34:245–56. doi: 10.1038/s41375-019-0519-4
150. Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, et al. AID-Dependent Activation of a MYC Transgene Induces Multiple Myeloma in a Conditional Mouse Model of Post-Germinal Center Malignancies. Cancer Cell (2008) 13:167–80. doi: 10.1016/j.ccr.2008.01.007
151. Chesi M, Matthews GM, Garbitt VM, Palmer SE, Shortt J, Lefebure M, et al. Drug Response in a Genetically Engineered Mouse Model of Multiple Myeloma is Predictive of Clinical Efficacy. Blood (2012) 120:376–85. doi: 10.1182/blood-2012-02-412783
152. Thirukkumaran CM, Shi ZQ, Nuovo GJ, Luider J, Kopciuk KA, Dong Y, et al. Oncolytic Immunotherapy and Bortezomib Synergy Improves Survival of Refractory Multiple Myeloma in a Preclinical Model. Blood Adv (2019) 3:797–812. doi: 10.1182/bloodadvances.2018025593
153. Chesi M, Mirza NN, Garbitt VM, Sharik ME, Dueck AC, Asmann YW, et al. IAP Antagonists Induce Anti-Tumor Immunity in Multiple Myeloma. Nat Med (2016) 22:1411–20. doi: 10.1038/nm.4229
154. Minnie SA, Kuns RD, Gartlan KH, Zhang P, Wilkinson AN, Samson L, et al. Myeloma Escape After Stem Cell Transplantation is a Consequence of T-cell Exhaustion and is Prevented by TIGIT Blockade. Blood (2018) 132:1675–88. doi: 10.1182/blood-2018-01-825240
155. Guillerey C, Harjunpaa H, Carrie N, Kassem S, Teo T, Miles K, et al. TIGIT Immune Checkpoint Blockade Restores CD8(+) T-Cell Immunity Against Multiple Myeloma. Blood (2018) 132:1689–94. doi: 10.1182/blood-2018-01-825265
156. Asimakopoulos F. TIGIT Checkpoint Inhibition for Myeloma. Blood (2018) 132:1629–30. doi: 10.1182/blood-2018-08-864231
157. Kawano Y, Zavidij O, Park J, Moschetta M, Kokubun K, Mouhieddine TH, et al. Blocking IFNAR1 Inhibits Multiple Myeloma-Driven Treg Expansion and Immunosuppression. J Clin Invest (2018) 128:2487–99. doi: 10.1172/JCI88169
158. Guillerey C, Nakamura K, Pichler AC, Barkauskas D, Krumeich S, Stannard K, et al. Chemotherapy Followed by Anti-CD137 mAb Immunotherapy Improves Disease Control in a Mouse Myeloma Model. JCI Insight (2019) 5(14). doi: 10.1172/jci.insight.125932
159. Vuckovic S, Minnie SA, Smith D, Gartlan KH, Watkins TS, Markey KA, et al. Bone Marrow Transplantation Generates T Cell-Dependent Control of Myeloma in Mice. J Clin Invest (2019) 129:106–21. doi: 10.1172/JCI98888
160. Rosenbaum H, Harris AW, Bath ML, McNeall J, Webb E, Adams JM, et al. An E Mu-v-abl Transgene Elicits Plasmacytomas in Concert with An Activated Myc Gene. EMBO J (1990) 9:897–905.
161. Vandenberg CJ, Waring P, Strasser A, Cory S. Plasmacytomagenesis in Emu-v-abl Transgenic Mice Is Accelerated When Apoptosis Is Restrained. Blood (2014) 124:1099–109. doi: 10.1182/blood-2014-04-570770
162. Suematsu S, Matsusaka T, Matsuda T, Ohno S, Miyazaki J, Yamamura K, et al. Generation of Plasmacytomas With the Chromosomal Translocation t(12;15) in Interleukin 6 Transgenic Mice. Proc Natl Acad Sci USA (1992) 89:232–5. doi: 10.1073/pnas.89.1.232
163. Kovalchuk AL, Kim JS, Park SS, Coleman AE, Ward JM, Morse HC 3rd, et al. IL-6 Transgenic Mouse Model for Extraosseous Plasmacytoma. Proc Natl Acad Sci USA (2002) 99:1509–14. doi: 10.1073/pnas.022643999
164. Chiarle R, Gong JZ, Guasparri I, Pesci A, Cai J, Liu J, et al. NPM-ALK Transgenic Mice Spontaneously Develop T-Cell Lymphomas and Plasma Cell Tumors. Blood (2003) 101:1919–27. doi: 10.1182/blood-2002-05-1343
165. Silva S, Kovalchuk AL, Kim JS, Klein G, Janz S. BCL2 Accelerates Inflammation-Induced BALB/c Plasmacytomas and Promotes Novel Tumors with Coexisting T(12;15) and T(6;15) Translocations. Cancer Res (2003) 63:8656–63.
166. Linden M, Kirchhof N, Carlson C, Van Ness B. Targeted Overexpression of Bcl-XL in B-lymphoid Cells Results in Lymphoproliferative Disease and Plasma Cell Malignancies. Blood (2004) 103:2779–86. doi: 10.1182/blood-2003-10-3399
167. Carrasco DR, Sukhdeo K, Protopopova M, Sinha R, Enos M, Carrasco DE, et al. The Differentiation and Stress Response Factor XBP-1 Drives Multiple Myeloma Pathogenesis. Cancer Cell (2007) 11:349–60. doi: 10.1016/j.ccr.2007.02.015
168. Zhang B, Wang Z, Li T, Tsitsikov EN, Ding HF. NF-kappaB2 Mutation Targets TRAF1 to Induce Lymphomagenesis. Blood (2007) 110:743–51. doi: 10.1182/blood-2006-11-058446
169. McCarthy BA, Yang L, Ding J, Ren M, King W, ElSalanty M, et al. NF-kappaB2 Mutation Targets Survival, Proliferation and Differentiation Pathways in The Pathogenesis of Plasma Cell Tumors. BMC Cancer (2012) 12:203. doi: 10.1186/1471-2407-12-203
170. Ueda O, Wada NA, Kinoshita Y, Hino H, Kakefuda M, Ito T, et al. Delta, and Gamma Humanized Mouse to Evaluate Human CD3-mediated Therapeutics. Sci Rep (2017) 7:45839. doi: 10.1038/srep45839
171. Airoldi I, Di Carlo E, Cocco C, Sorrentino C, Fais F, Cilli M, et al. Lack of Il12rb2 Signaling Predisposes to Spontaneous Autoimmunity and Malignancy. Blood (2005) 106:3846–53. doi: 10.1182/blood-2005-05-2034
172. Sun F, Cheng Y, Walsh SA, Acevedo MR, Jing X, Han SS, et al. Osteolytic Disease in IL-6 and Myc Dependent Mouse Model of Human Myeloma. Haematologica (2020) 105:e111–5. doi: 10.3324/haematol.2019.221127
173. Morito N, Yoh K, Maeda A, Nakano T, Fujita A, Kusakabe M, et al. A Novel Transgenic Mouse Model of The Human Multiple Myeloma Chromosomal Translocation t(14;16)(q32;q23). Cancer Res (2011) 71:339–48. doi: 10.1158/0008-5472.CAN-10-1057
174. Cheung WC, Kim JS, Linden M, Peng L, Van Ness B, Polakiewicz RD, et al. Novel Targeted Deregulation of c-Myc Cooperates With Bcl-X(L) to Cause Plasma Cell Neoplasms in Mice. J Clin Invest (2004) 113:1763–73. doi: 10.1172/JCI20369
175. Linden MA, Kirchhof N, Carlson CS, Van Ness BG. Targeted Overexpression of an Activated N-ras Gene Results in B-cell And Plasma Cell Lymphoproliferation and Cooperates with c-myc to Induce Fatal B-cell Neoplasia. Exp Hematol (2012) 40:216–27. doi: 10.1016/j.exphem.2011.11.006
176. Vicente-Duenas C, Romero-Camarero I, Gonzalez-Herrero I, Alonso-Escudero E, Abollo-Jimenez F, Jiang X, et al. A Novel Molecular Mechanism Involved in Multiple Myeloma Development Revealed by Targeting MafB to Haematopoietic Progenitors. EMBO J (2012) 31:3704–17. doi: 10.1038/emboj.2012.227
177. Chang L, Guo R, Huang Q, Yen Y. Chromosomal Instability Triggered by Rrm2b Loss Leads to IL-6 Secretion and Plasmacytic Neoplasms. Cell Rep (2013) 3:1389–97. doi: 10.1016/j.celrep.2013.03.040
178. Dechow T, Steidle S, Gotze KS, Rudelius M, Behnke K, Pechloff K, et al. GP130 Activation Induces Myeloma and Collaborates with MYC. J Clin Invest (2014) 124:5263–74. doi: 10.1172/JCI69094
179. Hamouda MA, Jacquel A, Robert G, Puissant A, Richez V, Cassel R, et al. BCL-B (BCL2L10) is Overexpressed in Patients Suffering From Multiple Myeloma (MM) and Drives an MM-like Disease in Transgenic Mice. J Exp Med (2016) 213:1705–22. doi: 10.1084/jem.20150983
180. Asai T, Hatlen MA, Lossos C, Ndiaye-Lobry D, Deblasio A, Murata K, et al. Generation of a Novel, Multi-Stage, Progressive, and Transplantable Model of Plasma Cell Neoplasms. Sci Rep (2016) 6:22760. doi: 10.1038/srep22760
181. Wen Z, Rajagopalan A, Flietner E, Yun G, Chesi M, Furumo Q, et al. Expression of NrasQ61R and MYC Transgene in Germinal Center B Cells Induces a Highly Malignant Multiple Myeloma in Mice. Blood (2021). doi: 10.1182/blood.2020007156
182. Sewastianik T, Straubhaar JR, Zhao JJ, Samur MK, Adler K, Tanton HE, et al. miR-15a/16-1 Deletion in Activated B Cells Promotes Plasma Cell and Mature B-cell Neoplasms. Blood (2021) 137:1905–19. doi: 10.1182/blood.2020009088
183. Affer M, Chesi M, Chen WD, Keats JJ, Demchenko YN, Tamizhmani K, et al. Promiscuous MYC Locus Rearrangements Hijack Enhancers but Mostly Super-Enhancers to Dysregulate MYC Expression in Multiple Myeloma. Leukemia (2014). doi: 10.1038/leu.2014.70
184. Corre J, Munshi NC, Avet-Loiseau H. Risk Factors in MM: Is it Time for a Revision? Blood (2020) 137(1):16–19. doi: 10.1182/blood.2019004309
185. Kumar SK, Rajkumar V, Kyle RA, van Duin M, Sonneveld P, Mateos MV, et al. Multiple Myeloma. Nat Rev Dis Primers (2017) 3:17046. doi: 10.1038/nrdp.2017.46
186. Hunter KW. Mouse Models of Cancer: Does the Strain Matter? Nat Rev Cancer (2012) 12:144–9. doi: 10.1038/nrc3206
187. Rustad EH, Yellapantula V, Leongamornlert D, Bolli N, Ledergor G, Nadeu F, et al. Timing the Initiation of Multiple Myeloma. Nat Commun (2020) 11:1917. doi: 10.1038/s41467-020-15740-9
188. Costa F, Das R, Kini Bailur J, Dhodapkar K, Dhodapkar MV. Checkpoint Inhibition in Myeloma: Opportunities and Challenges. Front Immunol (2018) 9:2204. doi: 10.3389/fimmu.2018.02204
189. Cohen AD, Raje N, Fowler JA, Mezzi K, Scott EC, Dhodapkar MV. How to Train Your T Cells: Overcoming Immune Dysfunction in Multiple Myeloma. Clin Cancer Res (2020) 26:1541–54. doi: 10.1158/1078-0432.CCR-19-2111
190. De Beck L, Melhaoui S, De Veirman K, Menu E, De Bruyne E, Vanderkerken K, et al. Epigenetic Treatment of Multiple Myeloma Mediates Tumor Intrinsic and Extrinsic Immunomodulatory Effects. Oncoimmunology (2018) 7:e1484981. doi: 10.1080/2162402X.2018.1484981
191. Ochoa MC, Perez-Ruiz E, Minute L, Onate C, Perez G, Rodriguez I, et al. Daratumumab in Combination With Urelumab to Potentiate Anti-Myeloma Activity in Lymphocyte-Deficient Mice Reconstituted With Human NK Cells. Oncoimmunology (2019) 8:1599636. doi: 10.1080/2162402X.2019.1599636
192. Lomas OC, Ghobrial IM. Clinical Controversies in the Management of Smoldering Multiple Myeloma. Am Soc Clin Oncol Educ Book (2020) 40:1–6. doi: 10.1200/EDBK_278911
193. Invrea F, Rovito R, Torchiaro E, Petti C, Isella C, Medico E. Patient-Derived Xenografts (Pdxs) as Model Systems for Human Cancer. Curr Opin Biotechnol (2020) 63:151–6. doi: 10.1016/j.copbio.2020.01.003
194. Noble JN, Mishra A. Development and Significance of Mouse Models in Lymphoma Research. Curr Hematol Malig Rep (2019) 14:119–26. doi: 10.1007/s11899-019-00504-0
195. De La Rochere P, Guil-Luna S, Decaudin D, Azar G, Sidhu SS, Piaggio E. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol (2018) 39:748–63. doi: 10.1016/j.it.2018.07.001
196. Tanaskovic O, Verga Falzacappa MV, Pelicci PG. Human Cord Blood (hCB)-CD34+ Humanized Mice Fail to Reject Human Acute Myeloid Leukemia Cells. PloS One (2019) 14:e0217345. doi: 10.1371/journal.pone.0217345
197. Tentler JJ, Lang J, Capasso A, Kim DJ, Benaim E, Lee YB, et al. Rx-5902, a Novel Beta-Catenin Modulator, Potentiates the Efficacy of Immune Checkpoint Inhibitors in Preclinical Models of Triple-Negative Breast Cancer. BMC Cancer (2020) 20:1063. doi: 10.1186/s12885-020-07500-1
198. Capasso A, Lang J, Pitts TM, Jordan KR, Lieu CH, Davis SL, et al. Characterization of Immune Responses to Anti-PD-1 Mono and Combination Immunotherapy in Hematopoietic Humanized Mice Implanted With Tumor Xenografts. J Immunother Cancer (2019) 7:37. doi: 10.1186/s40425-019-0518-z
199. Ito R, Takahashi T, Ito M. Humanized Mouse Models: Application to Human Diseases. J Cell Physiol (2018) 233:3723–8. doi: 10.1002/jcp.26045
200. Landgraf M, McGovern JA, Friedl P, Hutmacher DW. Rational Design of Mouse Models for Cancer Research. Trends Biotechnol (2018) 36:242–51. doi: 10.1016/j.tibtech.2017.12.001
201. Gemechu Y, Millrine D, Hashimoto S, Prakash J, Sanchenkova K, Metwally H, et al. Humanized Cereblon Mice Revealed Two Distinct Therapeutic Pathways of Immunomodulatory Drugs. Proc Natl Acad Sci USA (2018) 115:11802–7. doi: 10.1073/pnas.1814446115
202. Hanazawa A, Ito R, Katano I, Kawai K, Goto M, Suemizu H, et al. Generation of Human Immunosuppressive Myeloid Cell Populations in Human Interleukin-6 Transgenic Nog Mice. Front Immunol (2018) 9:152. doi: 10.3389/fimmu.2018.00152
203. Chauvin JM, Zarour HM. TIGIT in Cancer Immunotherapy. J Immunother Cancer (2020) 8(2). doi: 10.1136/jitc-2020-000957
Keywords: genetically engineered mouse models of human cancer, auto- and xenografting, immune, immunodeficient mice models, immune pathogenesis, Myeloma
Citation: Pisano M, Cheng Y, Sun F, Dhakal B, D’Souza A, Chhabra S, Knight JM, Rao S, Zhan F, Hari P and Janz S (2021) Laboratory Mice – A Driving Force in Immunopathology and Immunotherapy Studies of Human Multiple Myeloma. Front. Immunol. 12:667054. doi: 10.3389/fimmu.2021.667054
Received: 11 February 2021; Accepted: 28 April 2021;
Published: 02 June 2021.
Edited by:
Christelle Vincent-Fabert, UMR7276 Contrôle des réponses immunes B et des lymphoproliférations (CRIBL), FranceReviewed by:
Hans-Martin Jäck, University of Erlangen Nuremberg, GermanyJerome Moreaux, Université de Montpellier, France
Copyright © 2021 Pisano, Cheng, Sun, Dhakal, D’Souza, Chhabra, Knight, Rao, Zhan, Hari and Janz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Siegfried Janz, c2phbnpAbWN3LmVkdQ==