 Christina Lamers
Christina Lamers Carla Johanna Plüss
Carla Johanna Plüss Daniel Ricklin
Daniel Ricklin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 April 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.662164
The β2-integrin receptor family has a broad spectrum of physiological functions ranging from leukocyte adhesion, cell migration, activation, and communication to the phagocytic uptake of cells and particles. Among the members of this family, complement receptor 3 (CR3; CD11b/CD18, Mac-1, αMβ2) is particularly promiscuous in its functional profile and ligand selectivity. There are close to 100 reported structurally unrelated ligands for CR3, and while many ligands appear to cluster at the αMI domain, molecular details about binding modes remain largely elusive. The versatility of CR3 is reflected in its functional portfolio, which includes prominent roles in the removal of invaders and cell debris, induction of tolerance and synaptic pruning, and involvement in the pathogenesis of numerous autoimmune and chronic inflammatory pathologies. While CR3 is an interesting therapeutic target for immune modulation due to these known pathophysiological associations, drug development efforts are limited by concerns of potential interference with host defense functions and, most importantly, an insufficient molecular understanding of the interplay between ligand binding and functional impact. Here, we provide a systematic summary of the various interaction partners of CR3 with a focus on binding mechanisms and functional implications. We also discuss the roles of CR3 as an immune receptor in health and disease, as an activation marker in research and diagnostics, and as a therapeutic target.
Integrins are a group of diverse cell surface receptors that play key roles in cell adhesion, communication, activation, migration, and cellular uptake. They provide major molecular links between extracellular matrix components, adhesion molecules, and plasma proteins. As a phylogenetically ancient family of large glycoproteins with origins that can be traced back 750 million years, integrins coevolved with the development of specialized cells, tissue, and metabolic and host defense systems such as the complement system (1). Integrins transmit signals bidirectionally across the plasma membrane and participate in a wide range of processes, such as inflammation, tissue homeostasis, and angiogenesis. Dysregulation of integrin activity has been associated with various clinical conditions, including autoimmune, thrombotic, and vascular diseases and cancer metastasis. Extensive efforts have therefore been directed towards the development of integrin antagonists (2–4), but few have yielded clinically approved drugs. In general, integrins are considered poor drug targets due to their molecular complexity, functional versatility and, in some cases, ligand promiscuity of the integrin receptors.
Few members of the integrin family illustrate the complexity of integrin-mediated interactions and functions as impressively as complement receptor 3 (CR3; CD11b/CD18, Mac-1, αMβ2). This gives CR3 immense translational potential as a diagnostic marker and pharmacological target, which is reflected in a ligand repertoire approaching 100 molecules of natural and synthetic origin. CR3 is involved in leukocyte adhesion and migration, phagocytic elimination of pathogens, induction of both inflammatory and tolerogenic responses, and modulation of parallel or downstream host defense pathways (Figure 1). Despite numerous studies on CR3, aspects of its roles in health and disease and how it can be influenced on a molecular level remain elusive. Before unraveling the interactome and functional spectrum of CR3, we will provide a brief summary of the general molecular features of integrin receptors with a focus on the β2 family.
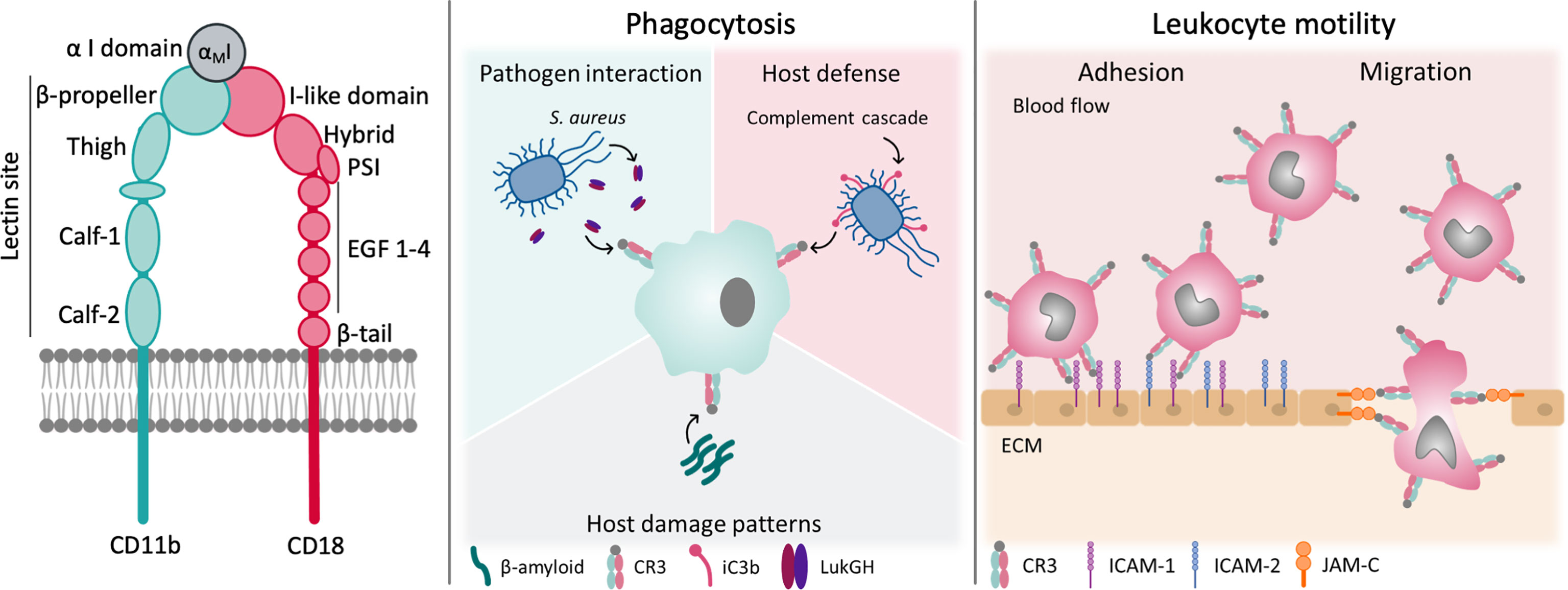
Figure 1 Structure and function of CR3. CR3 exerts a broad variety of functions in host-defense and leukocyte motility. Some of them are shown.
Integrins are heterodimers that consist of two non-covalently associated α and β subunits, which are both type I transmembrane glycoproteins. The human integrin family encompasses 24 members, assembled from 18 different α and 8 different β subunits. In general, each subunit is defined by a large multidomain extracellular section, a transmembrane domain, and a typically short intracellular tail, the latter of which is important for integrin regulation and linkage to the actin cytoskeleton inside the cell (5). Whereas all α subunits share a β-propeller, a thigh, and two calf domains in their ectodomain, half of them, including CD11b of CR3, also have a globular I domain that is inserted in the β-propeller (6). These I domains contain a metal ion-dependent adhesion site (MIDAS), which coordinates divalent cations (e.g., Mg2+), and are the major binding area for integrin ligands (7). All β subunits contain an I-like domain with a similar fold to the α-I domains alongside a plexin-semaphorin-integrin (PSI), four epidermal growth factors (EGF), and a β-tail domain (Figure 1). These I-like domains contain a MIDAS, which is flanked by two adjacent metal coordination sites (termed AMIDAS and LIMBS/SyMBS) that bind Ca2+ and exert modulatory functions (5). The headpiece of integrins is formed by the β-propeller and I-domain of the α-subunit in contact with the I-like domain of β-subunit (5). In integrins that lack the I domain, the I-like domain makes extensive contacts with the β-propeller, and provides an interface for ligand binding (2).
Integrin receptors derive their unparalleled functional versatility, including adhesion, “inside-out” and “outside-in” signaling, and/or ligand and particle uptake (8), from the unique composition of a compact headpiece that serves as an interaction platform, two flexible legs, and a transmembrane link to the cytoskeleton. The activity state of integrin receptors is defined by their three distinct conformations (Figure 2). In the activated state, both subunits are fully extended to expose over 4000 Å2 of solvent accessible surface (2). In the inactive state, the receptor is bent, and the globular head is kept close to the membrane surface, shielding the surface from solvent. In the intermediate state, the receptor is extended, but the cytoplasmatic tails are not separated, which seems to be induced by ligand binding to the bent conformation (5).
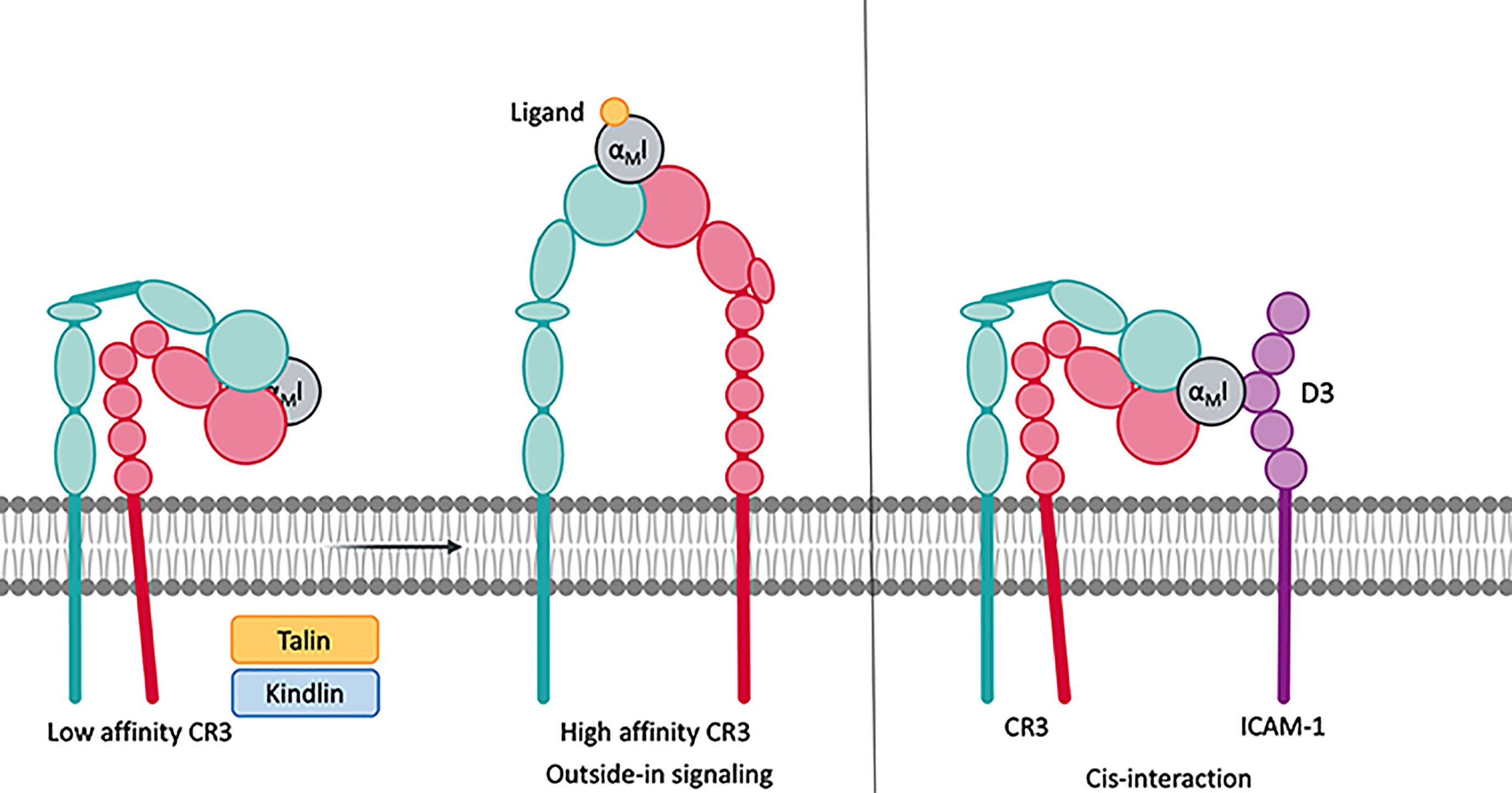
Figure 2 Integrins remain in a low affinity, bent-closed state. Cytoplasmic factors such as talin and kindlin connect the cytoplasmic tail of the integrins to the cytoskeleton. This leads to an extension of the extracellular domains with an open, high affinity, ligand accessible headpiece. CR3 is able to bind to protein on the same surface, which is termed cis-ligation/cis-interaction.
In the absence of stimuli, the integrins generally remain in the inactive, bent-close state, wherein the transmembrane domains of the two subunits are associated (9). Upon cell-stimulatory signals, such as cell surface receptors [e.g., toll-like receptor (TLR) or T-cell receptors], talin and kindlin are recruited to the cytoplasmatic tail of the integrins to connect them to the cytoskeleton (10). The resulting tensile force separates the intracellular tails and extends the extracellular domains to assume the active state (5, 11). This mode of signal transduction is referred to as “inside-out” signaling and may be further influenced by proteins that interact with the cytoplasmatic tail of the β-subunit. Mn2+ can induce integrin activation without separation of the cytoplasmatic tails, though bent and extended conformations with an open and closed headpiece coexist (8, 11). During leukocyte extravasation, P-selectin ligation induces the extended conformation with the closed headpiece, thereby enabling leukocyte rolling. Upon ICAM-1 ligation to integrins, this leukocyte rolling is decelerated. Finally, chemokine signaling via its GPCR receptors triggers leukocyte arrest, which opens the headpiece (12, 13).
The switch from the bent to extended conformation has profound consequences for ligand binding, which improves by several orders of magnitude (e.g., 4000-fold for cRGD binding to α5β1) (14). Affinity enhancements are often driven by a large reduction in ligand dissociation (15). This indicates that the extension of the receptor improves the accessibility of the headpiece and also induces conformational changes in the ligand binding domains (6). Indeed, this so-called “switchblade model” suggests a two-step activation process, during which extension of the legs is followed by a rearrangement of the binding area on the headpiece. For I domains, the extension leads to the downward axial displacement of the C-terminal helix to enhance ligand access and affects the position of the three loops that confine the MIDAS region.
While it is well established that ligand binding induces “outside-in” signal transduction, the underlying processes are less understood. Upon binding of extracellular ligands, integrin receptors can form clusters on the cell surface that extend from tenths of angstroms (microcluster) to > 200 nM (macrocluster) (16) and, by affecting binding avidity, enhance cell adhesion (17). Integrin clustering is observed on platelets (18), leukocytes (13), and, as patterned arrays, on primary neutrophils (12). Similar to “inside-out” signaling, conformational changes appear to play a critical role in “outside-in” signal transduction. Ligand-induced conformational propagation and receptor clustering trigger numerous intracellular signaling cascades after assembly of focal signaling complexes at the cytoplasmic face of the cell membrane, which may include kinases and adaptors. Nearly 60 proteins have been identified as constituents of this adhesome (19).
The family of β2 integrins, comprising four members that all contain an α-I domain, are all found on leukocytes, yet each has a distinct expression pattern (20, 21). Whereas CD11a/CD18 (lymphocyte function-associate antigen 1 or LFA-1; αLβ2) is found on all leukocytes, its expression is more prominent on lymphocytes. CD11b/CD18 (CR3) is the predominant integrin on neutrophils and is common on other myeloid cells, including macrophages, monocytes, eosinophils. It is also found on natural killer (NK) cells, mast cells, and B and T lymphocytes. While CD11c/CD18 (CR4, p150,95, αXβ2) can be detected on NK, B, and T cells, it is predominantly expressed on myeloid dendritic cells, macrophages, and dendritic cells of the splenic white pulp and marginal zone. Finally, CD11d/CD18 (αDβ2) is detected on most circulating monocytes and neutrophils, NK cells, and a small fraction of circulating T cells (22).
Despite their leukocyte-centered and partially overlapping expression profiles, the β2 integrin family has distinct ligand binding patterns. LFA-1 primarily binds to intercellular adhesion molecules (ICAM-1 to ICAM-5) and is critical for leukocyte trafficking by enabling firm adhesion to the endothelial layer and subsequent extravasation (6). Moreover, LFA-1 is an essential component of the immunological synapse between T cells and antigen-presenting cells (APC). LFA-1 also modulates the differentiation, survival, and activity of various lymphocyte subpopulations. Whereas CR3 and CR4 are also involved in leukocyte adhesion and migration, they are versatile in their interactions and functions, including phagocytosis of opsonized particles, podosome formation, and effector molecule enhancement (e.g., FcγR, uPAR, CD14). Among the most intriguing aspects of CR3 is its involvement in the removal of superfluous synapses during synaptic pruning (23). Comparatively, little is known about the functional spectrum of CD11d/CD18, which binds ICAM-3, VCAM-1, and matrix proteins (24). It must be noted that some of the functional aspects of β2 integrins have only been investigated in animal models and await confirmation in humans.
The tight involvement of β2 integrins in host defense and immune modulation (25–28) renders them a potential Achilles’ heel in the susceptibility for infectious, inflammatory, and/or autoimmune diseases. As the most prominent example of this, the autosomal recessive disorder leukocyte adhesion deficiency (LAD) presents with immunodeficiency and recurrent infections due to genetic mutations that encode the β2-subunit (25). Furthermore, β2 integrins are associated with the pathogenesis of leukocyte-mediated inflammation (during atherosclerosis or reperfusion injury), autoimmune disorders [e.g., systemic lupus erythematosus (SLE)], and dry eye disease. CR3 is an especially important regulator to balance the induction of tolerance, as it may inhibit the release of cytokines but may also generate an inflammation response. However, the regulation of this dual effect is not yet understood. It may be a combination of ligands, co-receptor ligation, or the pre-activation of the cell, and the cell types (e.g., macrophage vs dendritic cells) (28).
Several antagonistic compounds that target β2 integrins, such as small molecules and antibodies, have been in development as therapeutics (2–4, 26). However, few have reached the clinic. Therefore, it is critical to understand the functional and ligand binding spectrum of β2 integrins to increase access to therapeutic intervention.
When assessing the interactome of CR3, this β2 integrin family member is unique due to its distinct selectivity profile and its breadth of reported ligands. Whereas most integrins show a preference for RGD and/or LDV motifs (29, 30), these tripeptides do not seem to play a major role in CR3 recognition (31). CD11b has more than 100 reported ligands from soluble mediators, counterreceptors, and ECM components to microbial recognition and evasion molecules. The globular head of CR3, and in particular the αMI domain (7), is regarded as the main binding site, with some ligands that also engage with the β-propeller (2) of the α subunit or sites on the β2 domain. A lectin domain located on the C-terminus of the α subunit has been proposed but is poorly characterized. Finally, the glycosylation pattern of CR3 might account for binding to other receptors such as DC-SIGN (32), which renders CR3 a ligand itself. The quantity and promiscuity of ligands might be explained by the complexity of molecular interactions of CR3, which include multiple binding sites on the heterodimer and within the αMI domain and the glycosylation of CR3.
For most of the reported ligands, binding to CR3 was demonstrated in interaction assays using CR3-expressing cells, the purified heterodimer, or recombinant αMI. The recombinant αMI is expressed as either a wildtype variant or with a I316G mutation that yields a high affinity variant, which has been replicated in a mouse knock-in model (33, 34). The binding site has been mapped through competitive experiments with anti-CD11b antibodies and known CR3 ligands (Figure 3), although allosteric effects impede the interpretation of these results (35). As an aside, abolished ligand binding in the presence of EDTA is often interpreted as a MIDAS-mediated interaction with αMI, though the function of the β2 subunit is equally dependent on divalent cations. Additionally, EDTA may affect the potential binding of lectins to CR3 glycans. Otherwise, the exact binding mode has not been well investigated; a specific interaction to the αMI domain was confirmed with biophysical methods, though for less than 20% of reported ligands. Even with recent advances in integrin structure determination, structural insight into ligand binding remains scarce, with only a few available crystal structures of the αMI domain in complex with ligands (19). Despite these challenges, the conformational change between open and closed I-domains has been elucidated (36). Advances in cryo-EM (37, 38) and small angle x-ray scattering (39) have provided insight into the global conformation and interaction of CR3. Furthermore, the functional impact of CR3 ligation has been investigated in vivo for a few ligands (Supplementary Tables 1–9). However, the results from rodent models should be interpreted cautiously as little is reported about differences in CR3 across species. For example, mouse and human CR3 protein sequences share 78% similarity (40, 41), but the LukGH ligand shows specificity for human CR3 and no affinity to mouse CR3 (41, 42). In light of the imprecise description and insufficient validation of CR3 ligation (Supplementary Tables 1–9), some aspects about the interactome of this receptor must be regarded with care. Clearly, more investigations are warranted to fully understand CR3 ligand interactions, signaling, and functional consequences.
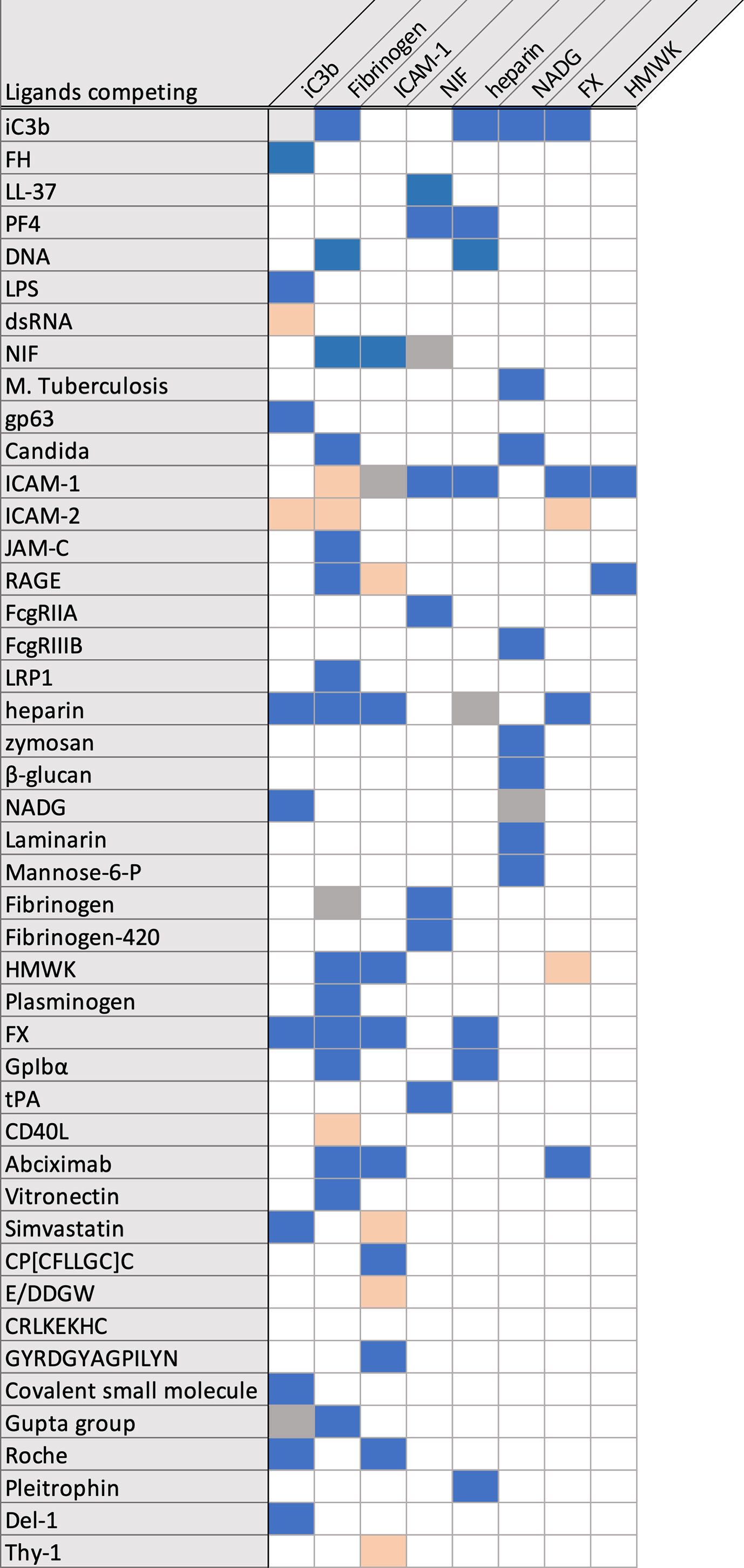
Figure 3 Overview over reported competing/not competing CR3 ligands. Competing ligands are marked in blue, not competing ligands are marked in salmon.
The αMI domain, first crystallized in 1995 (43), coordinates the divalent metal ion with amino acids S142, S144, and D140 (α1 loop) of the MIDAS domain, and D242 (α5 loop) and T209 (α3-α4 loop). The sixth coordination site of the metal ion is free and can be filled by a carboxylic acid (Asp, Glu), either intrinsic partner, or by an extrinsic ligand (6, 43). The identification of multiple, overlapping binding sites in αMI (Figure 4) led to the introduction of a mosaic model of interaction areas (44). The βD-α5 loop and α5-helix (K245-A261) within the alphaMI domain are exclusive to CD11b in comparison to CD11a, that have been associated with the recognition of several CR3 ligands (45–47). Several groups have tried to identify a common binding motif for ligand interactions with the αMI domain, which led to the proposal of two opposite minimal binding sequences. Vorup-Jensen et al. postulated that a single carboxyl group, which coordinates with the divalent cation of the MIDAS, may define the relevant structural feature for ligand binding, as shown for simvastatin (48). While no specific pattern has been postulated, there are likely more amino acid contacts surrounding the MIDAS. However, this minimal binding feature would not apply to ligands that do not bind the MIDAS (e.g., CD40L, Figure 4). Alternatively, Podolnikova et al. proposed a minimal binding sequence of a basic amino acid surrounded by lipophilic ones such as HyBHy, HyHyBHy, HyBHyHy, and HyHyBHyHy (where Hy represents any hydrophobic residue and B is arginine or lysine) (49).
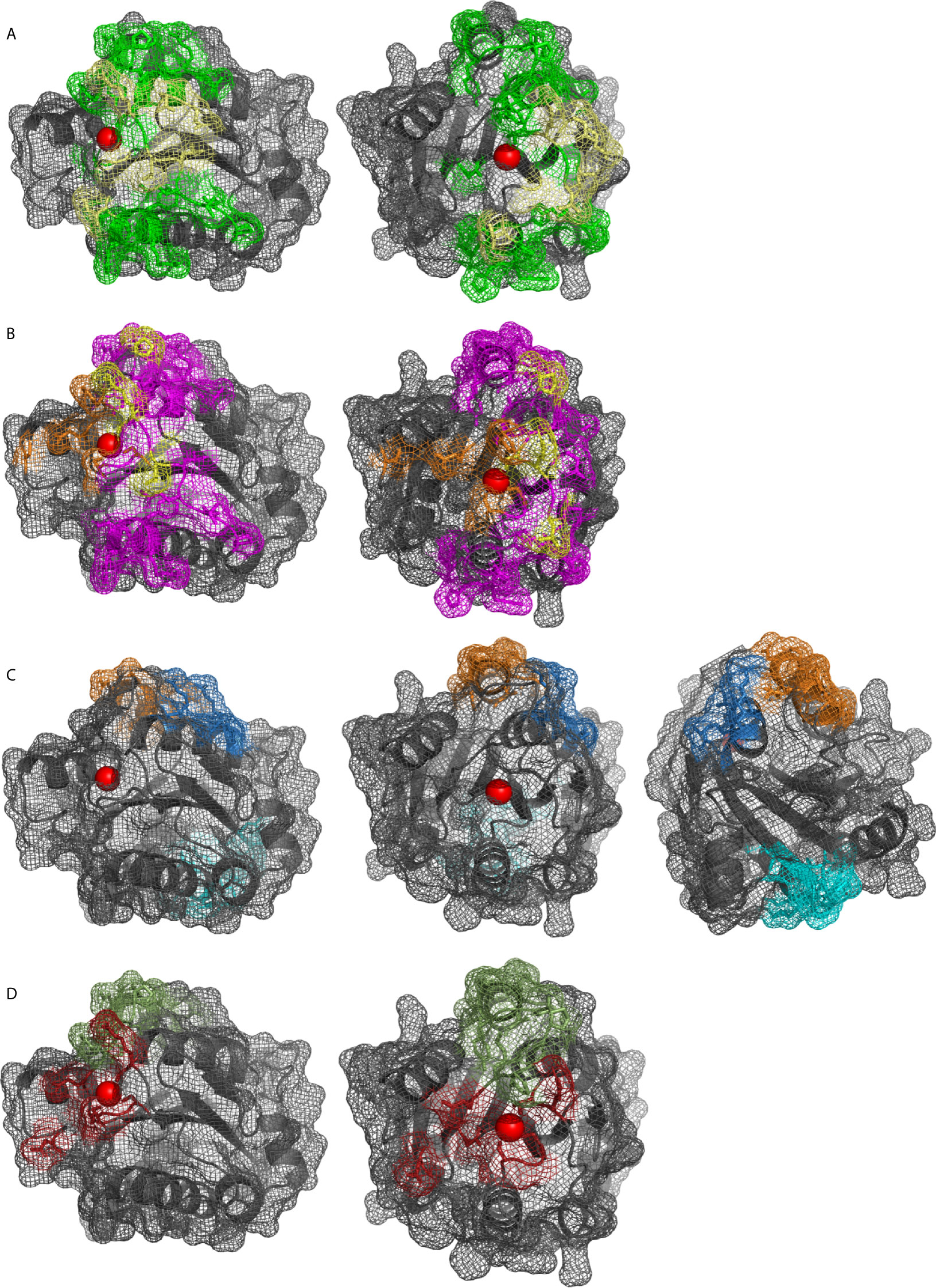
Figure 4 αMI domain (pdb: 1IDO) as cartoon and surface shown as mesh, Mg2+ in red. Binding sites of ligands reported by mutational, competition, structural or docking studies: (A) iC3b (green), C3d (yellow), (B) NIF (pink), LukGH (orange), (yellow for overlapping), (C) glucosamine (blue), FCgRII (orange), and CD40L and LRP1 (cyan), (D) GPIbα (ruby), fibrinogen (green).
A binding site was identified on the α-subunit of CR3 and termed the “lectin domain”, which interacts with carbohydrate structures and is unique among integrins. This site is distinct from the binding areas at the globular head and was mapped C-terminally to the αMI domain, which comprises the broad stretch of amino acids 400–1092 on the leg of CD11b (Figure 1) (50, 51). Ligation of the lectin site is suggested to induce a primed state of CR3 (52), which induces phosphorylation of the CD18 cytoplasmatic tail by protein kinase C (53), without increasing CR3 surface-expression. This process leads to a magnesium-dependent conformational change of the αMI domain and exposes the CBRM1/5 activation epitope. However, the epitope of the monoclonal antibody (mAb) 24 remains unexposed, which would indicate high affinity binding for ICAM-1 (54).
The simultaneous recognition of ligands via the lectin and αMI domains induces distinct responses. For example, co-ligation of CR3 by microbial β-glucan (lectin site ligand) and iC3b (αMI ligand) triggers phagocytosis and degranulation, while ligation of iC3b on erythrocytes or tumor cells in the absence of lectin site-ligation induced no cytotoxicity (55). Furthermore, β-glucans by themselves can induce ROS production and respiratory burst, whereas CR3 co-ligation by β-glucan and fibrinogen is required to release neutrophil extracellular traps (NET) (56).
The binding specificity of the lectin domain seems rather broad and includes polysaccharides containing mannose, N-acetyl-D-glucosamine (NADG), and glucose (51, 57). Ligands that bind CR3 at least partially via the lectin domain are microbial cell wall polysaccharides, such as zymosan, β-glucan,and laminarin (57) as well as glycans found in bacterial pili and fungal hyphae. In contrast, dextran and α-mannan do not bind.
Cis-ligation is the simultaneous interaction of proteins on the same cell surface, as opposed to trans-ligation, which involves another cell. In addition to trans-ligation, CR3 has been shown to interact with several membrane proteins via cis-ligation, which is typically mediated by the lectin and/or the αMI domains (see above). Depending on the ligation site, cis-ligation may result in either amplification of the response or in negative regulation. For example, cis-ligation of the lectin site leads to the formation of multi-protein complexes, which may substantially enhance effector function, such as phagocytosis. Conversely, cis-ligation by αMI domain interactions seems to trap the integrin in a bent, and thus inactive, conformation and impair receptor functions. Protein-complexes with increased activity after cis-ligation with CR3 include FCγRIII (58, 59), uPAR (60), tPA (61), and SLAMF7 (62). Negative regulation upon cis-ligation was reported for ICAM-1 (13), IL-13Rα1 (63, 64), FcγRIIa (65), and CD22 (66).
As major leukocyte receptors, β2 integrins in general and CR3 in particular profoundly shape the cellular immune response through numerous interactions. The repertoire of CR3 ligands associated with leukocyte function are structurally diverse but can still be clustered based on their function (Table 1 and Supplementary Table 1). CR3 is a major receptor for the phagocytosis of opsonized particles. As such, CR3 plays an important role in host defense and the removal of cell debris and apoptotic cells. At the same time, pathogens use this integrin for host/cell invasion and immune evasion. Moreover, CR3 is involved in leukocyte migration, especially during inflammation, with several ligands reported as counterreceptors for this purpose. Finally, the crosstalk of CR3 with proteins of the hemostatic systems plays a pivotal role in thrombosis and links hemostasis to inflammation.
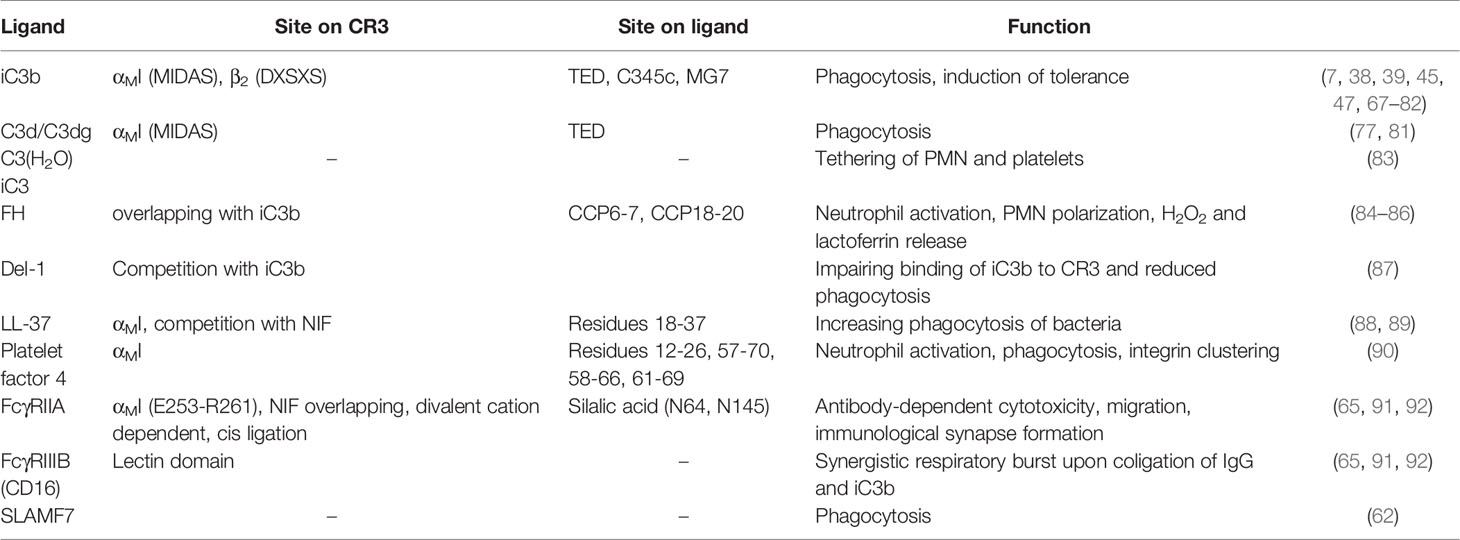
Table 1 Host defense mechanisms.
CR3 was shown to regulate neutrophil apoptosis, which leads to an enhanced accumulation of CD11b-/- neutrophils (93). Mature neutrophils are terminally differentiated cells with a short circulation half-life. They are cleared from circulation by spontaneous apoptosis and subsequent phagocytosis by resident macrophages in the spleen, bone marrow, and Kupffer cells in the liver (94). A hallmark of inflammatory resolution is the engulfment of apoptotic neutrophils by macrophages (efferocytosis). Inflammatory stimuli (e.g., G-CSF, TNFα, IL-6, LTB4, C5a or LPS) and endothelial transmigration (95) via adhesion to ICAM-1 or fibrinogen (96) prolong the life span of neutrophils. Conversely, high levels of TNFα accelerate apoptosis (97). Complement-opsonized targets induce apoptosis of polymorphonuclear leukocytes (PMNs) via CR3 (98, 99), while pro-survival signals can be triggered by CR3 clustering-dependent binding to fibrinogen and plasminogen (95, 96, 100). This clustering is induced by ligands that bind both sub-domains of CR3, but not those that solely bind the αMI domain (e.g., NIF or P1) (101). On a molecular level, the pro-survival signal via activation of Akt is dependent on specific ligands rather than integrin activation. This effect was shown with αMI mutants locked in the active state, which failed to induce the pro-survival signal (101).
CR3 is a major regulator in host defense and tissue homeostasis due to its central role in the removal of invaders, damaged cells, and protein aggregates by mediating phagocytosis (67, 68). Phagocytosis via CR3 largely depends on prior opsonization of surfaces by the complement system and can result in pro-inflammatory signaling, induce tolerance with reduced IL-12 levels, and upregulate tolerogenic IL-10 and TGFβ (102). The complement cascade is initiated upon recognition of antibody clusters or non-self-signatures on targeted cells, and this initiation cleaves the plasma protein C3 to generate an anaphylatoxin (C3a) and the opsonic C3b fragment that is deposited at the site of activation. C3b can form C3 convertase complexes that activate more C3. This fuels an amplification loop that culminates in the cleavage of C5 with subsequent formation of the inflammatory mediator C5a and lytic membrane attack complexes. At the same time, complement regulators degrade C3b into the fragments iC3b and C3dg, which do not participate in amplification, though do mediate immune functions (103).
Among these fragments, iC3b is considered the main ligand recognized by CR3 that leads to phagocytosis (67, 68), and the interaction was shown to be dependent on divalent cations (69). To determine the CR3-iC3b binding interaction, investigations identified multiple discontinuous sites on CR3 (70, 104). On the αMI domain, this included areas surrounding the MIDAS (i.e., βA-α1, α3-α4, βD-α5, and βE-α6 loops) (7, 71, 72). The fourth blade of the β-propeller in CD11b (73) and a conserved amino acid sequence (DXSXS) in CD18 (72, 74, 75) also participate in the interaction. Consistent with these results, transgenic cells that express CR3 and lack αMI can still bind to iC3b, albeit with lower affinity (76).
On host cells that express complement receptor 1 (CR1, CD35) as cofactor, iC3b is further degraded by factor I to surface-tethered C3dg and C3d and soluble C3c. While C3b and C3c have weak affinities for CR3 and CR4, comparable to the affinity of a sole acidic side chain that interacts with the MIDAS, C3d and C3dg display strong affinity for CR3 and weak for CR4 (77). While the distinct selectivities of the related complement receptors was unexpected, they show that differential patterns of complement opsonization/regulation and CR3/CR4 expression may fine-tune an immune response.
In addition to its cleavage by convertases, C3 can also assume an activated state through low-level hydrolysis in circulation or upon contact with various surfaces. The resulting C3(H2O) is structurally similar to C3b and can be degraded to iC3, which largely corresponds to iC3b. Both C3(H2O) and iC3 have been detected on activated platelets and facilitate the tethering of platelets to PMNs (83).
Factor H (FH), a soluble complement regulator, was reported to adhere to PMNs in a divalent cation and CR3-dependent manner, which leads to activation and polarization of adhered PMN with increased release of H2O2 and lactorferrin (84). FH domains CCP6-7 and CCP18-20 were identified as interaction sites for CR3, and the observed competition with iC3b suggested overlapping binding sites on the receptor (85). FH, and CFHR1 that contains domains homologous to the binding areas of FH and may therefore act as CR3 ligand, can bind simultaneously to neutrophils and Candida albicans. Consequently, this enhances neutrophil adherence and pathogen killing (86).
Developmental endothelial locus-1 (Del-1) is a matrix glycoprotein expressed and secreted by endothelial cells. It has been shown to behave as an antagonist for CR3-mediated phagocytosis through competition with iC3b (87). Del-1 is also able to inhibit the LFA-1:ICAM-1 interaction, which reduces inflammatory cell recruitment and IL-17 induction (105).
Upon recognition of danger signals, leukocytes secrete certain host defense molecules that also bind to CR3. LL-37, an endogenous antimicrobial peptide, is secreted by a wide variety of cells (e.g., neutrophils, NK, mast cells, B and T cells, and epithelial cells) and binds to the αMI domain of CR3. LL-37 also has a high affinity for bacterial cells, which increases phagocytosis (88, 89). Platelet factor 4 (PF4, also known as CXCL4) is a cationic protein secreted from α-granules of activated platelets to activate neutrophils, augment phagocytosis, and induce integrin clustering in a CR3-dependent manner (90). Direct binding of PF4 to isolated αMI domains has been observed with biolayer inerferometry (90).
Members of the Fc receptor family bind immunoglobulins with distinct specificities and functional consequences, which include phagocytic uptake of antibody-coated cells and particles. Simultaneous phagocytosis of iC3b-opsonized and IgG-labeled particles leads to a synergistic respiratory burst (106). Additionally, CR3 is needed for optimal phagocytosis via FcγR (107, 108). The interplay between CR3 and FcγR is considered critical for immunological defense mechanisms. For example, the ligation of FcγRIIa is necessary for antibody-dependent cellular cytotoxicity (ADCC) and critical for FcγRIIa-mediated cell-spreading, migration (91, 109), and immunological synapse formation (110). Co-ligation of CR3 and FcγRIII leads to an association of FcγRII with the actin cytoskeleton and subsequent phosphorylation that induces a respiratory burst (92). A soluble form of FcγRIII (sCD16) is shed from cell surfaces and triggers cell activation by interacting with CR3 and CR4, which leads to “outside-in” signaling and cytokine production (111). For FcγRIIa (CD32a), cis-ligation to the CR3 αMI domain inhibits neutrophil recruitment, which is absent in the SLE-associated SNP rs1143679 (R77H) (65).
The direct interaction of FcγRIII with CR3 was elucidated by resonance energy transfer studies and microscopy (112). In competition experiments with NADG, the relevant binding site was mapped to the lectin site of CR3. In contrast, the interaction with FcγRIIa was insensitive to NADG, and binding studies with recombinant αMI confirmed an overlapping binding site to neutrophil inhibitory factor (NIF) (91). A detailed investigation of the binding mode showed that sialic acids on FcγRIIa glycans (attached to N64 and N145) are responsible for the αMI interaction, as treatment with neuraminidase diminished binding (65).
SLAMF7 (CD319) is a robust marker of malignant plasma cells in multiple myeloma and was shown to be responsible for the phagocytosis of cells after disruption of the tumor surveillance checkpoint CD47-SIRPα. The colocalization of SLAMF7 with CR3 in a cis interaction was shown with co-immunoprecipitation studies and confocal microscopy. Furthermore, phagocytosis was inhibited with anti-CD11b mAbs and CD11b knock-out (62).
Although CR3-dependent phagocytosis is typically mediated by complement and/or antibody opsonization, several pathogens are directly recognized by CR3, which facilitates phagocytosis and induces an antimicrobial response. However, some intracellular pathogens exploit CR3 as an effective entry port to invade cells. Such pathogen interactions with complement integrin receptors have been reviewed recently (113), so in the following sections, we focus on the molecular determinant and involved ligands (Tables 2, 3 and Supplementary Tables 2, 3) that enable pathogen recognition by CR3.
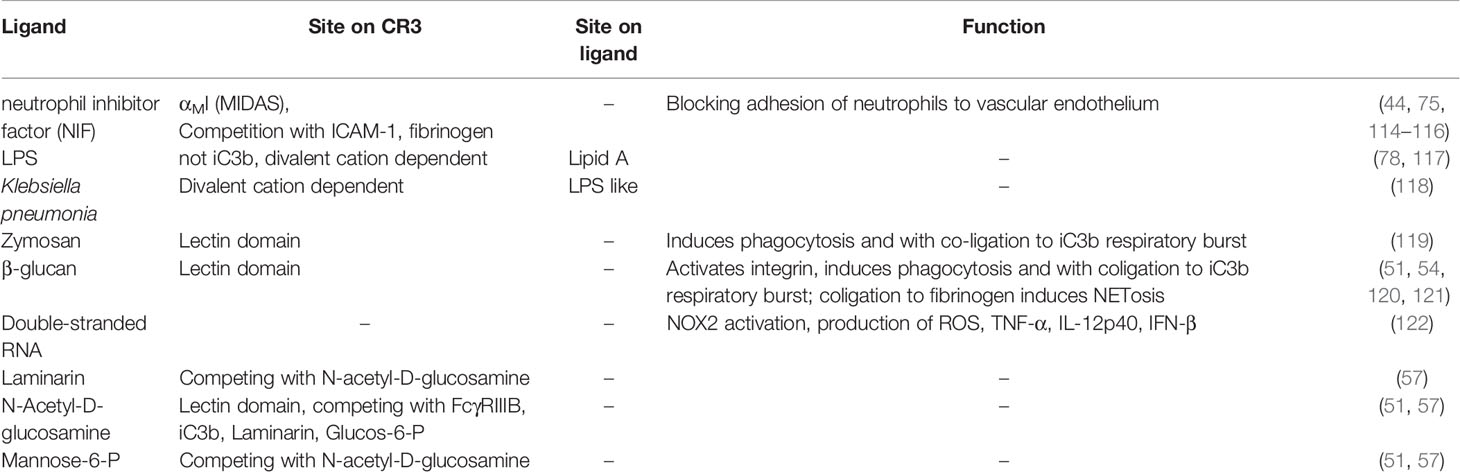
Table 2 Defense against pathogens.
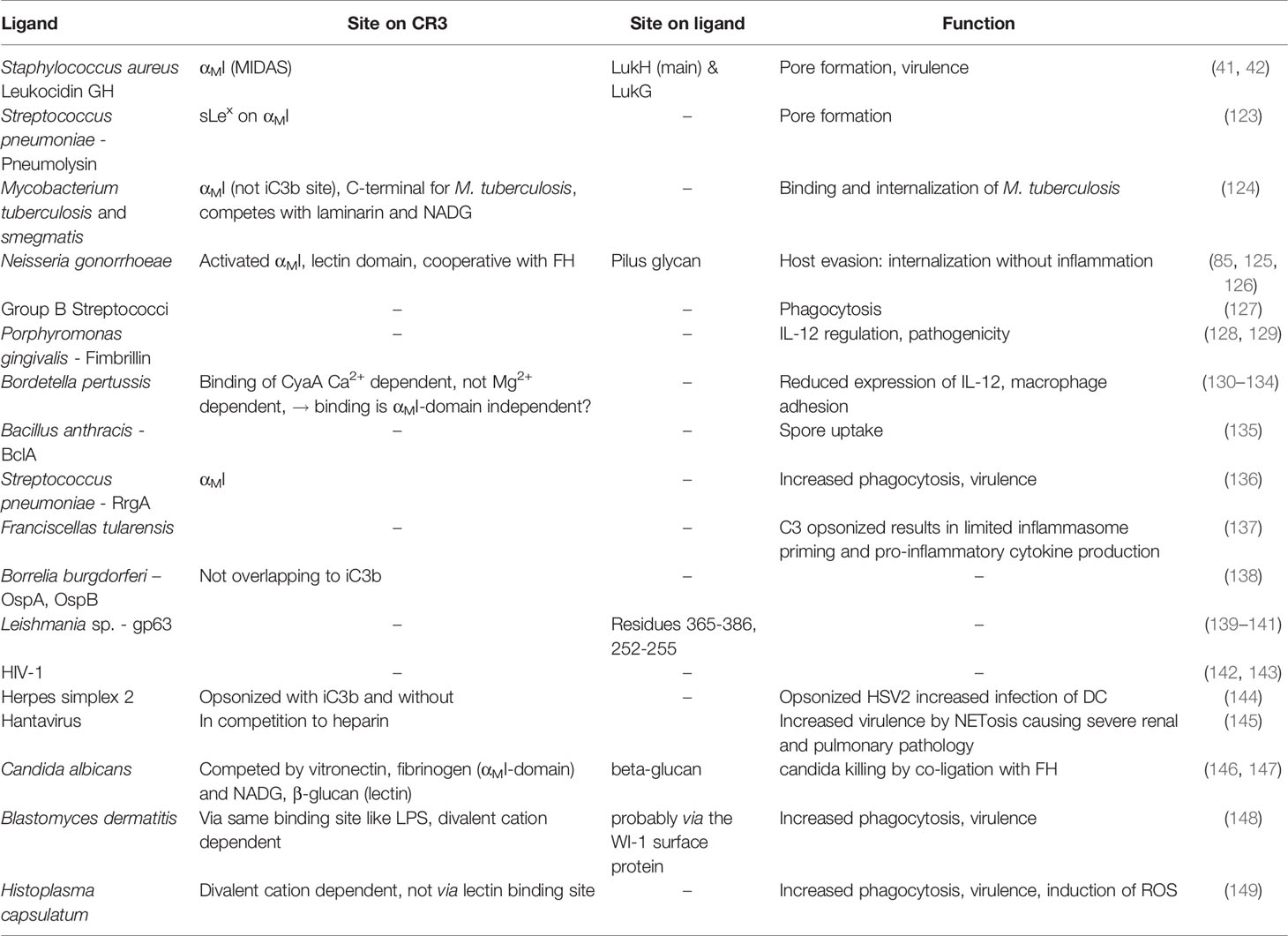
Table 3 Pathogen evasion.
Neutrophil inhibitory factor (NIF), a glycoprotein of hookworms, blocks adhesion of neutrophils to the vascular endothelium (150) by binding CR3 with high affinity (low nM range) and long target residence (t½ ~ 8 h) (114). Interestingly, the CR3-NIF interaction is independent of neutrophil activation (114). The binding site is located within the αMI domain (115) with several reported contact areas (75, 114). NIF binding is cation-dependent (70) and competes with fibrinogen and ICAM-1 (116), but not with FX (44). Reports on competitive binding for iC3b are contradictory (44, 116), which may be attributed to different experimental setups (i.e., recombinant αMI vs the full heterodimer). A homolog-scanning mutagenesis approach highlighted the importance of contact areas that surround MIDAS and identified the αXI domain of CR4 as a potent receptor for NIF (116).
Escherichia coli binds to macrophages even in the absence of complement components. This might be possible due to its surface glycoprotein, lipopolysaccharide (LPS), which was identified as a binding partner to β2 integrins. The lipid A part of LPS appears to play a critical role in this binding interaction, since polymyxin B sulfate can block binding (117). The binding site on CD11b was shown to be distinct from iC3b binding, but this was not further characterized (78). The acylpolygalactosides of Klebsiella, which consist of a poly(1, 3)galactose chain, glucosamine disaccharide, and lipid βOH myristates, are similar in structure to LPS. These structures mediate Ca2+- and Mg2+-dependent binding to monocytes, with involvement from both CD14 and CR3 (118).
Zymosan A, a preparation of cell wall glucans from Saccharomyces cerevisiae, is a known activator of the complement system, but also directly activates macrophages and induces phagocytosis (119, 151), with β-glucan as the responsible component. β-glucans are high-molecular-weight polysaccharides comprised of β-D-glucose and are typically found in the cell walls of bacteria, fungi, and plants [laminarin (57)]. β-glucans can activate macrophages that mediate phagocytosis and respiratory burst (51) via CR3 (119). They have shown remarkable activity as an immune-modulatory compound in animal models and clinical trials (152). Imprime PGG (see section 3.4) is a β-glucan derivative, which has been investigated in clinical trials in combination with therapeutic antibodies for oncotherapy (153–156).
β-glucans do not bind to the canonical binding sites, such as the αMI domain or the β2 globular head, but rather to the lectin domain (50, 54). Monosaccharides such as mannose, galactose, fucose, and glucose can only compete with β-glucan at very high concentrations (>200 mM), while NADG, α- and β-methylmannoside, α- and β-methylglucoside, and mannose-6-P are stronger competitors (51, 57). Interestingly, distinct responses between soluble β-glucans (sbglu) and immobilized β-glucans have been reported (52). Binding of sbglu to CR3 yields similar responses as induced “inside-out” signaling (i.e., increased CR3 expression, conformational change to an extended state, increased affinity to fibrinogen), albeit to a lesser extent. Remarkably, the extension induced by sbglu results in an intermediate conformation that leads to cellular activation, which can be detected by the phosphorylation of proteins associated with transcriptional regulation, mRNA processing, and alternative splicing. Another study suggested that sbglu binds CR3 indirectly via opsonization with iC3b (120). One possible explanation for those conflicting results might be the use of different species between studies, since dectin-1 is the predominant receptor for β-glucan phagocytosis in mice, while CR3 is solely responsible for β-glucan signaling in humans (121).
Double stranded RNA (dsRNA), as a result of viral infection, is bound and internalized by CR3 via an unspecified molecular mechanism, which leads to increased NOX2 activity, ROS production, and elevated levels of proinflammatory cytokines, such as TNF-α and IFN-β, as shown in a mouse model (122).
Leukocidin is a pore-forming toxin from Staphylococcus aureus that lyses phagocytic cells, such as neutrophils, monocytes, and macrophages. CD11b and CD18 are both targets of this bi-component cytotoxin, which is composed of LukG and LukH subunits (41) that were shown via crystal structure to form a heterotrimer with CR3. The heterotrimer oligomerizes into a β-barrel pore, which is inserted into the cell membrane through bending of the β2 integrin (42). LukGH establishes extensive polar contacts and salt bridges to the αMI domain, including the MIDAS, as well as lipophilic interactions (42). Cytolysins are able to form pores in a similar way as leucocidin. For example, pneumolysin from Staphylococcus pneumoniae interacts with CR3 with high affinity (KD ~ 7 nM) via the sialyl-LewisX of the αMI domain (123).
Mycobacterium tuberculosis, a bacterium that colonizes intracellularly in mononuclear phagocytes, is another pathogen that uses CR3. Binding and internalization of M. tuberculosis is mediated by iC3b-opsonization and through direct binding to CR3 (157, 158). The direct binding was mapped to the αMI domain, distinct from the iC3b binding site, and to the C-terminus, which is likely the lectin domain (124). Mycobacterium strains with capsular polysaccharides composed of glucose, arabinose, and mannose (e.g., M. tuberculosis) involve the lectin domain in binding, whereas strains with low densities of phosphatidylinositol mannoside (e.g., M. smegmatis) are primarily opsonized by iC3b and phagocytized (159).
Neisseria gonorrhoeae exploits CR3 for internalization without inducing inflammation via several mechanisms (160). iC3b-opsonized gonococci and the outer membrane proteins of porins and pili are all known ligands of the αMI domain (125, 161). Additionally, a cooperative mechanism of FH-mediated bridging between gonococci and CR3 has been postulated (85). The C-terminal CCP18-20 binds to CR3 when the FH domains CCP6-20 assume the appropriate spatial orientation, while CCP6-10 and CCP18-20 are able to adhere to gonococci (85). In addition, glycans displayed on the pili contribute to binding CR3 in its closed conformation, thereby activating it (126), which indicates an involvement of the lectin domain.
Additional pathogens that bind to CR3, mostly via membrane-bound glycoproteins, and can increase virulence are group B streptococci (127), Porphyromonas gingivalis (128, 129), Bordetella pertussis (130–133), Bacillus anthracis (162), Streptococcus pneumoniae (136), Francisella tularensis (137), Borellia burgdorferi (138), and the parasite Leishmania (139, 163). CR3-mediated binding induces phagocytosis, which often results in decreased IL-12 levels (129, 134) and inhibition of inflammasome activation (137), which leads to pathogen evasion from immune surveillance. For most of these pathogens, binding to CR3 was reported without knowledge of the exact binding site. In the case of Leishmania, its glycoprotein gp63 was shown to interact with the αMI domain (140, 141) and compete with iC3b binding, which indicates an overlapping binding site.
Some viruses hijack CR3 for host cell entry and immune evasion. For example, non-opsonized and iC3b-opsonized HIV-1 both use CR3 for internalization by monocytes and immature dendritic cells for effective transfer to CD4+ T cells (142). Complement-mediated opsonization of HIV-1 leads to enhanced infection due to decreased anti-viral and anti-inflammatory responses (143). Similarly, opsonized herpes simplex virus 2 is internalized via CR3, which leads to increased infection of dendritic cells (144). Other viruses, such as hantavirus, use CR3 as an entry receptor but induce strong NETosis (145), thereby harming the host.
Large granular lymphocytes adhere to the hyphae (polymeric β-glucan structures) of C. albicans via CR3 and inhibit their growth (146, 164). The molecular binding site on CR3 was mapped to the αMI domain, although the lectin domain also seems to play a role, since N-acetyl-D-glucosamine, D-mannose, and β-glucan compete with binding. Other fungi, such as Blastomyces dermatitis (148) and Histoplasma capsulatum (149), also adhere to CR3.
Damage-associated molecular patterns (DAMPs) typically refer to changes in cell-surface expression profiles upon host cell damage or apoptosis, cellular debris, and aggregated proteins. Such cells and debris are typically removed by phagocytes to maintain homeostasis. Similar to its direct recognition of pathogens, CR3 can directly bind to various DAMP ligands (Table 4 and Supplementary Table 4) and thereby contribute to homeostasis.
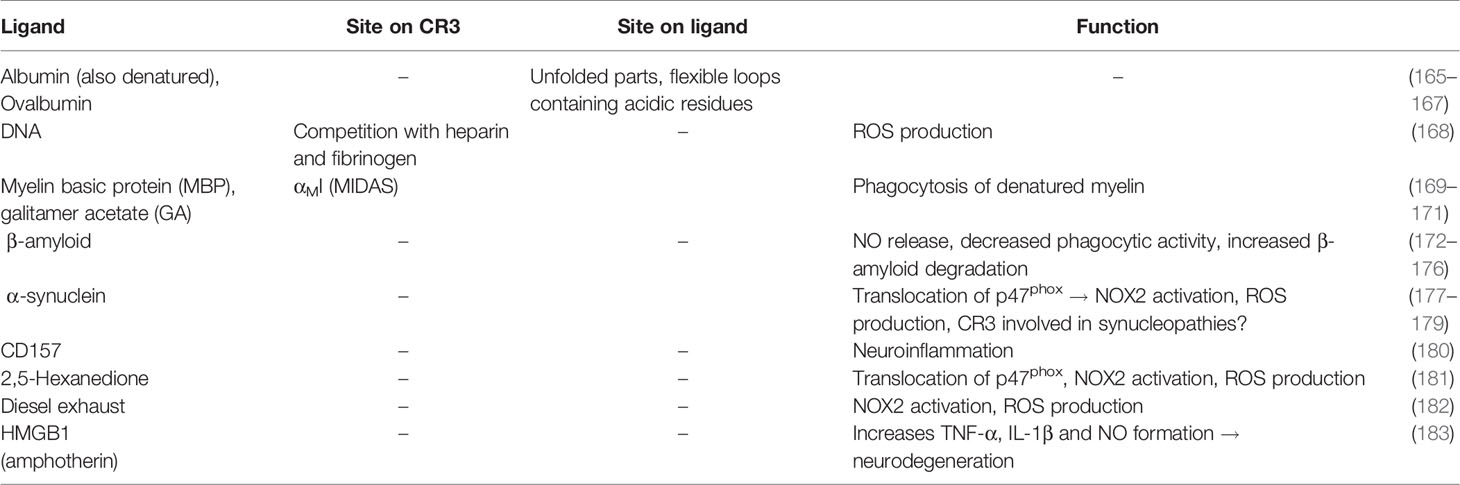
Table 4 Recognition of host damage patterns.
Synthetic DNA oligodeoxynucleotides are bound and internalized by CR3, which induces production of ROS in TNF-α/fMLP-stimulated PMNs (168). Furthermore, CR3 can recognize many denatured proteins. For example, denatured BSA and ovalbumin bind CR3 on neutrophils (165). Albumin immobilized on polystyrene and perfluorinated surfaces is a ligand for CR3, which indicates that BSA is not a suitable blocking agent for in vitro assays with leukocytes and CR3 (166, 167).
Myelin basic protein (MBP) is a potential autoantigen in multiple sclerosis (MS), and a mouse model of MS showed a correlation between MBP binding to CR3 and experimental autoimmune encephalitis (EAE) (169, 170, 184). CR3-deficient mice are protected from developing symptoms in an EAE model (184). Mimicking unfolded MBP, the mixture of basic peptides known as glatiramer acetate (GA), is an active ingredient of an MS therapeutic that competes with MBP binding by also binding the CR3 αMI domain (171). Finally, direct binding of MBP to CR3 is dependent on divalent cations (Ca2+, Mg2+, and Mn2+) (171).
Aggregated β-amyloid is associated with Alzheimer’s disease (172) as well as increased CR3 expression. In mouse models, β-amyloid binding to CR3 increased NO release, which led to neurotoxicity, but also the induction of phagocytosis and the degradation of β-amyloid (173–175). These data indicate a complex or dual role of CR3 in pathogenesis. Similarly, in the neurodegenerative Parkinson’s disease (PD), α-synuclein aggregates are associated with the induction of neurotoxicity by activation of microglia that leads to NOX2 (NADPH oxidase) activation and ROS production via Src-Erk- and Rho-dependent pathways (177, 178), where CD11b-/- microglia are better protected (179). Another risk factor for PD, CD157, a member of the NADase/ADP-ribosyl cyclase family, can functionally associate with CR3 to potentially drive neuroinflammation (180). Similarly to α-synuclein, 2,5-hexanedione (181) and diesel exhaust particles (182) induce dopaminergic neurodegeneration via activated rodent microglia, CR3-dependent NOX2 activation, and ROS production via the Src-Erk pathway. When found extracellularly, the intracellular, chromatin-binding protein high mobility group box protein 1 (HMGB1, amphoterin), causes increases in TNFα, IL-1β, and NO secretion. In this process, the NFκB pathway activates NADPH oxidase, which induces progressive neurodegeneration in rodents. The direct interaction between HMGB1 and CR3 was shown by co-immunoprecipitation experiments (183), but the involved domains and mechanism of action remains unknown.
A major role of β2 integrins is the mediation of leukocyte adhesion to endothelial or interstitial matrix with subsequent leukocyte migration and tissue inflitration. This process is essential for directing immune cells to sites of inflammation and is facilitated by β2 integrins that employ a set of ligands to control leukocyte motility (Table 5 and Supplementary Table 5).
Table 5 Leukocyte adhesion and extravasation.
ICAMs belong to the immunoglobulin superfamily and serve as primary counterreceptors in this process. They differ in expression pattern and cellular distribution, with ICAM-1 expressed at low levels on leukocytes, endothelial cells, keratinocytes, and fibroblasts and up-regulated upon inflammatory stimuli, such as cytokines. ICAM-2 is constitutively expressed by platelets, leukocytes, and endothelial cells, while ICAM-3 is highly expressed on leukocytes but is absent from endothelial cells. ICAM-4 and ICAM-5 are expressed exclusively on erythrocytes and on a subset of neurons in the telencephalon, respectively (192). Whereas CD11a binds ICAM-1-5, the complement integrins CD11b and CD11c both recognize ICAM-1, -2, and -4. CD11d appears to primarily bind ICAM-3 (207).
ICAM-1 (CD54) plays a major role in the leukocyte adhesion cascade during extravasation into inflamed tissue. Circulating neutrophils start rolling on endothelial cell upon interaction with P-selectin and arrest under flow after activation by IL-8, which leads to CR3 activation by “inside-out” signaling. CR3 binds to the third Ig-like domain of ICAM-1, while LFA-1 binds the first Ig-like domain (185), which enables simultaneous binding of both integrin receptors. The binding of CR3 to ICAM-1 seems to be dependent on the level of ICAM-1 glycosylation, as CR3 binds with higher avidity to ICAM-1 with low glycosylation (186). The αMI domain is the major binding site for ICAM-1 (7, 72, 187, 188), but the conserved DXSXS motif of the β2-subunit may also be important (74). Recent studies indicate that ICAM-1 can bind to CR3 on the same cell (cis-ligation), with the integrin in a bent conformation but with an active headpiece. The cis-ligation between ICAM-1 and CR3 renders the bent conformation more stable (13) and was therefore proposed as negative regulation mechanism.
ICAM-2 (CD102) has been shown to be a ligand of CR3 (190) through interaction with the αMI domain via its first Ig-like domain (191). A peptide derived from the Ig-like D1 domain competes with ICAM-2 binding to CR3 using a binding site distinct from iC3b, FX, and fibrinogen. Binding of the peptide activates CR3 and increases ICAM-1-mediated neutrophil adhesion as well as CR3´s binding to fibrinogen and iC3b (191).
ICAM-4 is expressed on erythrocytes and binds isolated CD11a and CD11b in a divalent cation-dependent manner (192, 208), where CR3 is the stronger binding partner. ICAM-4 mediates binding via its Ig-like domains D1 and D2 (192). However, the binding site on CR3 has not been investigated in detail.
The junctional adhesion molecule (JAM) receptor family is also part of the type-I Ig superfamily and is found on platelets (193, 209), T-cells, and NK-cells as well as at the desmosomes of endothelial cells and intestinal epithelial cells (194). JAM receptors are believed to play a role in neutrophil-platelet interaction and neutrophil transepithelial migration. JAM-C binds to CR3 via a binding site that overlaps with that of fibrinogen (193).
Other counterreceptors involved in CR3-mediated leukocyte recruitment are basigin (CD147), extracellular matrix metalloproteinase inducer (EMMPRIN) (195), and the receptor for advanced glycation end products (RAGE), the latter of which is a multiligand receptor expressed on inflamed vascular cells. In a mouse model, neutrophil extravasation into the peritoneum was primarily mediated by CR3 and ICAM-1, with RAGE accounting for 25% of total binding (196). In diabetic mice, RAGE-mediated extravasation increased to 50% (196), which highlights the relevance of this receptor interaction in diabetes. Similarly, Thy-1 (CD90), expressed on endothelial cells, may serve as a CR3-counterreceptor, as shown for psoriasis (197).
SIRPα (MFR), another member of the Ig superfamily, is involved in macrophage fusion, which accompanies chronic inflammatory conditions. The αMI domain mediates the interaction of CR3 with this counterreceptor (210) and exhibits binding to the soluble Ig1-2-3 ectodomains of SIRPα (199). Interestingly, the inactive form of αMI can still bind SIRPα, albeit to a lesser extent (199). Basic amino acids flanked by lipophilic residues were identified as binding motifs within SIRPα (49).
CD40L is a member of the tumor necrosis factor (TNF) superfamily and regulates B- and T-cell function by interacting with CD40. CD40L is expressed on macrophages, endothelial cells, and smooth muscle cells and plays a role in chronic inflammatory diseases, such as atherosclerosis. It stabilizes arterial thrombi by ligation to GpIIb/IIIa (211). However, CD40L also mediates atherogenesis independently of CD40 via CR3 (212). CD40L binds the αMI domain at a binding site that is distinct from fibrinogen. While the corresponding site on CD40L has not been investigated, CR3 binding does not compete with CD40 or GPIIb/IIIa binding (200). Blocking the CD40L:CR3 interaction by an anti-CD40L antibody or a cyclic peptide derived from the αMI binding site attenuated atherosclerosis in mice, which resulted in less inflamed, smaller, and more stable atherosclerotic lesions without affecting bleeding time and thrombus formation (200).
Human leukocyte elastase (HLE) and azurophilic granule proteins [such as myloperoxidase (201)] also bind to CR3. HLE binding can be blocked by a serine protease inhibitor, which indicates active site involvement in binding to CR3. Azurodicin, a homolog protein lacking protease activity, binds with even higher affinity, though the exact molecular meachanism and binding site on CR3 was not investigated (202). A complex of pro-MMP-9 and CR3 is formed intracellularly in granules and translocated to the cell surface during cell activation (203). The binding sequence was mapped to the catalytic domain of pro-MMP-9, and binding of pro-MMP-2 and MMP-8 to the αMI domain has also been reported (204). Studies show that CR3 is cleaved by those proteases, which is a critical process for neutrophil detachment during chemotaxis (213).
Pleiotrophin, a cationic cytokine and growth factor, is expressed in injured tissue during regeneration and has affinity for heparin/glycosaminoglycans and CR3. Pleiotrophin is involved in the adherence and migration of neutrophils and leads to MAP kinase activation upon CR3 binding (205).
Dynorphin A, a member of the class of endogenous opioid receptor peptides, can induce phagocytosis and ROS production by ligation to CR3. Several CR3-binding motifs that involve basic and hydrophobic residues were identified by sequence analysis, with reported interaction with the αMI domain (206).
DC-SIGN is a C-type lectin expressed on dendritic cells, where it serves as an adhesion receptor for endothelial cells and PMNs. DC-SIGN binds to the sialyl-LewisX motif of CR3, which is expressed solely on PMNs (32).
The extracellular matrix (ECM) is composed of various proteins, such as collagen, elastin, fibronectin, laminin, thrombospondin, and glycosaminoglycans (214), and is involved in neutrophil recruitment to inflammation sites (215). Several ECM components have been identified as CR3 ligands (Table 6 and Supplementary Table 6), and binding of thrombospondin, vitronectin (217), fibrinogen, and fibronectin activates PMN and induces H2O2 secretion (216). CR3 interactions have also been confirmed for other ECM components, such as collagen IV (215), undulin, laminin (240), and lumican (243). Mindin appears to play a dual role in mediating leukocyte migration and serving as an opsonin (244, 261). It also induces phagocytosis via the Syk pathway and activates NFκB (245). Similarly, CCN1 and CCN2 binding to CR3 leads to NFκB activation (246–248). The oxidation of polyunsaturated fatty acids forms 2-(ω-carboxyethyl)pyrrole (CEP), which can then modify ECM proteins. CEP is associated with inflammation and mediates the expression of pro-inflammatory cytokines in macrophages. It also increases adhesion and migration via binding to CR3 and CR4 (249).
Table 6 Leukocyte migration on extracellular matrix.
Fibrinogen is the zymogen form of fibrin, which is primarily known as a central component of hemostasis. However, fibrinogen also ligates CR3 and CR4 and plays important roles in inflammatory processes. Early observations showed that PMNs bind to fibrin and can migrate on a fibrinogen matrix (262, 263), with CR3 identified as the responsible receptor (218). The γ-chain of fibrinogen, which also interacts with the platelet integrin GpIIb/IIIa via its RGD motif, can bind CR3 (218) independently of RGD (219–222). A cyclic peptide, derived from the fibrinogen γ-chain (CNRLTIGC) (222), also binds to CR3 (250). Interestingly, the binding motifs within the fibrinogen structure are buried in soluble fibrinogen, but become accessible to CR3 upon fibrinogen binding to surfaces or proteolytic cleavage (46, 223).
Fibrinogen binding to CR3 is likely the best-characterized ligand interaction of this versatile integrin receptor (188, 218, 219). A study that associated sequence homologies in the I domains of β2 integrins with fibrinogen binding identified the βD-α5 loop (K245-R261) as the binding site within the αMI domain (47, 221, 224). This loop is not part of the MIDAS, so fibrinogen binding occurs cation-independently. Competition experiments with FX and iC3b identified partial overlap in the binding regions (224). The detrimental influence of mutations within the β2 I-like domain on fibrinogen binding (47) may be caused by regulatory roles rather than direct binding (264), since a αMI-less CR3 mutant does not bind fibrinogen (76). Alternative splicing produces two forms of fibrinogen called Fg-340 and Fg-420, with greater abundance found for Fg-340. Both forms function similarly in regard to clotting, crosslinking of FXIII, and fibrinolysis. Interestingly, Fg-420 features an extended domain, which has 40% sequence homology to the γ-chain. This results in a threefold higher affinity of CR3 for Fg-420 relative to Fg-340 (225).
The CR3 interaction with fibrinogen is detrimental for inflammatory responses and is distinct from the role of fibrinogen in hemostasis. Deletion of the CR3-binding motif diminishes adhesion of primary neutrophils and macrophages (226), thereby reducing renal pathology in mice with sickle cell anemia due to the decreased production of IL-6, IL-1, and TNFα (227). The CR3-fibrinogen interaction is also responsible for phagocyte accumulation at sites of biomaterial implantation (265).
Plasminogen is the zymogen form of plasmin, which is responsible for dissolving fibrin clots. It interacts with the ECM after plasma leakage into tissue and can bind to leukocytes. Besides VLA-4, CR3 was identified as the responsible receptor, with binding mediated by the αMI domain (250). Angiostatin, which consists of the kringle domains 1-3 or 1-4 of plasminogen, is expressed during inflammation and wound healing. It competes with fibrinogen and ICAM-1 binding to CR3, which inhibits the extravasation of neutrophils and reduces inflammatory responses (251). Lipoprotein(a), a risk factor for coronary heart disease, has pro-inflammatory effects and is highly homologous to the kringle domain 4 of plasminogen, which enables binding to CR3. Preincubation with homocysteine, another risk factor for coronary heart disease, leads to enhanced binding of lipoprotein(a) to CR3 and, consequently, increased cardiovascular risk (252).
The urokinase receptor (uPAR, CD87) is a GPI-tethered glycoprotein binding urokinase (urokinase-type plasminogen activator, uPA), a serine protease that activates plasminogen to plasmin. uPAR directs plasminogen activator activity to specific areas on the cell surface and plays a central role in thrombolysis, ECM degradation, and leukocyte migration. The association between CR3 and uPAR (59, 253), and its impact on leukocyte recruitment, was reported in the 1990’s (58, 254). The interaction is enhanced by the addition of Zn2+, an effect that is mediated by uPAR (266). The uPAR binding site has been mapped to the W4 blade of the β-propeller of the CD11b subunit (255). However, CR3 is postulated to build complexes with GPI-anchored proteins via the lectin domain (60), thereby acting as transmembrane signaling adaptor. Ligation of uPAR and CR3 leads to CR3 activation (priming), similar to ligation of FcyRIIIB and CR3 (58, 59). The complex of urokinase, uPAR, and CR3 enhances CR3-binding to fibrinogen, with no effect on ICAM-1 binding. The enhancement is initiated by the urokinase:uPAR:CR3 complex and is mediated by FAK phosphorylation via the MAP kinase pathway (256). Furthermore, uPA is attracted to this complex, which both enables the cleavage of ECM components to facilitate neutrophil migration and negatively modulates the stability of the uPAR:CR3 complex (59). A phage–display-derived peptide against uPAR (M25, STYHHLSLGYMYTLN) can disrupt the uPAR:CR3 complex, which attenuates binding to fibrinogen (255).
Like uPA, tissue plasminogen activator (tPA) is a serine protease on endothelial cells that catalyzes the conversion of plasminogen to plasmin. In analogy to urokinase, tPA binds to CR3 and enhances recognition of fibrin, which can be blocked by NIF (257). tPA also forms a complex with Annexin A2, which is a Ca2+ and phospholipid-binding protein found intra- and extracellularly. It has no transmembrane domain and associates transiently with the membrane, without the ability to transmit signals into the cell. Annexin A2 was identified as receptor for tPA and co-immunoprecipitates with CD11b. This complex of Annexin A2, tPA, and CD11b initiates an “outside-in” signal that activates integrin-linked kinase (ILK) and, subsequently, the NFκB pathway. This signaling is dependent on CD11b, as shown in control experiments with a CD11b-neutralizing antibody and CD11b knock out mice (61). Neutrophil antigen BI (NB1/CD177), a GPI-anchored protein in the Ly6/urokinase plasminogen activator receptor family, is also reported to bind CR3, which leads to neutrophil activation and release of ROS (258, 267).
LDL receptor-related protein (LRP) is a large endocytic receptor on macrophages, which is involved in macrophage migration via interaction with β2 integrins (259). LRP-1 (CD91) binds to a complex of CR3, tPA, fibrin, and PAI-1, which is subsequently internalized. In turn, this leads to detachment of the macrophages and their migration on the ECM (257). Essential amino acids for this complex formation reside on a site opposite the MIDAS, though binding is not limited to the αMI domain (260). A soluble form of LRP1 (sLRP1), which is found in elevated concentrations (~ 10 nM) in plasma during inflammation, competes with CR3 binding to fibrinogen, which might confer a regulatory mechanism for resolving inflammation (260).
Fibronectin (Fn), a known ligand of β1 integrins such as α5β1, also binds to CR3 via the αMI domain. Interestingly, binding of Fn to CR3 does not increase migration, like binding to α5β1, and in fact, the opposite effect is observed. Considering the correlation with the density of CR3 surface expression, this effect likely occurs in response to inflammatory signaling, when CR3 is shuttled from intracellular storage vesicles to the cell surface, thereby increasing the receptor density by approximately a factor of 10 (239).
Any breach of the body’s physical barriers requires rapid, coordinated defense system responses to stop blood loss, initiate wound repair, and combat microbial intruders. CR3 interactions with ligands of the contact and hemostatic system (Table 7 and Supplementary Table 7) can contribute successful defense reactions, but may also play a role in thrombo-inflammatory disorders. Firm adhesion and trans-platelet migration of leukocytes on vascular thrombus is dependent on CR3 when GPIbα is a counter-ligand (268). GPIbα and CR3 binding induces a bidirectional signal that yields proinflammatory and prothrombotic responses. By blocking CR3:GPIbα-mediated leukocyte–platelet interactions with antibodies, leukocyte accumulation after arterial injury could be reduced (286). In addition, responses to tissue injury in models of vasculitis (287), glomerulonephritis (288), and EAE (289) have proven to be dependent on CR3:GPIbα binding. By blocking this interaction, NET formation could be prevented (290). The importance of this interaction was also demonstrated using CR3-deficient mice, which were protected in a thrombotic glomerulonephritis model (288) and exhibited impaired thrombus formation without affecting the coagulation time, platelet count, and activation (269). Furthermore, CR3 clustering from GPIbα binding induced phosphorylation of PKCδ and downregulated Foxp1, thereby reducing tissue factor (TF) expression (269). The CR3:GPIbα binding is promoted by the αMI domain (228), with a binding site that is distinct from that of fibrinogen, ICAM-1, and JAM-C (286). The leucine-rich C-terminal flanking region of GPIbα (GPIbαN) was identified as the interaction site (228), which binds with a glutamic acid to the αMI MIDAS, similarly to the internal ligand of the α7-helix (229). Further interactions are formed towards a groove on the surface of the αMI domain created by F246 and R206 (229).

Table 7 Leukocyte interaction with homeostasis and thrombi.
Heparin (HMW and LMW), an anticoagulant in pharmacotherapy, adheres to neutrophils in an interaction mediated by β2 integrins (273). The structural similarity of heparin to β-glucans suggests binding via the lectin site. However, the αMI domain appears to mediate heparin binding (273), and heparin competes with CR3-binding of FX, ICAM-1, and iC3b, even at lower concentrations than needed for anticoagulation (weak prolongation of the activated partial thromboplastin time (aPPT), 0.1–1 U/mL) (274). Interestingly, desulfated heparin loses its affinity for CR3, which indicates that sulfate groups are key to the interaction (273). A similar influence of sulfation was reported for fucosylated proteoglycans (57), such as fucoidan (271). Accordingly, glucosamine, heparin, and related mucopolysaccharides of the sulfated glucosamine glycan type (except for chondroitin sulfate) bind to CR3 and can mediate the binding of proteins decorated with glycans to CR3, as seen with CD44v3 (272).
Glucosamine has been described as an inhibitor of the CR3:GPIbα interaction through direct binding to the αMI domain (269). Glucosamine decreased thrombus formation in an in vivo model of carotid artery photochemical injury without prolonging bleeding time, which demonstrated its anti-thrombotic potential (269).
Activated monocytes induce coagulation processes through CR3-mediated binding of FX in a calcium-dependent and saturable manner (275, 291). Although the binding site has been postulated to lie outside the αMI domain (76, 188), FX binding can be blocked by iC3b (219). An FX-derived peptide also directly competes with ICAM-1 (35). Interestingly, while FX is a ligand for CR3, activated FXa is not able to bind (276).
High-molecular-weight kininogen (HMWK) is an abundant plasma protein that acts as an initiator of the contact activation pathway during coagulation. It circulates as a complex with plasma prekallikrein (PPK) and is activated upon binding to negatively charged surfaces, where it facilitates the activation of FXII and prevents thrombin binding. When cleaved to HMWKa by plasma kallikrein (PK), it liberates the potent vasodilator, bradykinin. HMWK can bind to neutrophils with high affinity (KD 9–18 nM) and is dependent on Zn2+ (278). Of note, HMWKa can still bind to CR3 (KD ~ 60 nM) (279). HMWK domain 3 and the C-terminus of domain 5 were identified as binding sites (270, 280, 281), which seems to overlap with fibrinogen (230) and ICAM-1 (279, 280), but not FX, and indicates involvement of the αMI domain. HMWK-binding to CR3 enhances the formation of CR3:GPIbα complexes by 2-fold, likely by bridging the two receptors via domain 5 (to CR3) and domain 3 (to GPIbα) (270). The CR3-HMWK interaction leads to an increased release of neutrophil elastase (NE) (278). Meanwhile, HMWKa binding to mononuclear cells leads to the release of cytokines (TNF-α, IL-1β, IL-6) and chemokines (IL-8 and MCP-1) (282).
Besides promoting coagulation, CR3 also interacts with anticoagulant factors. Thrombomodulin is a multi-domain anticoagulant with anti-inflammatory properties, where domain 3 is a ligand for CR3, while domains 1 and 2 are important for its anticoagulant and anti-inflammatory effects (283). Furthermore, thrombomodulin interferes with the binding of anti-neutrophil cytoplasmic autoantibodies, which inhibits NET formation (284). The endothelial protein C-receptor (EPCR) plays a crucial role in the protein C anticoagulant pathway by promoting protein C activation. Its soluble form binds CR3 on activated neutrophils. Further investigation is needed to identify the molecular binding site on EPCR or CR3 (285) and to elucidate the functional implications of ligation.
There are close to 100 reported ligands of CR3, but not all can be associated with a functional cluster, despite their importance in physiological/pathophysiological processes (Table 8 and Supplementary Table 8). Intracellular proteins located in the endoplasmic reticulum (ER) interact with CR3, which supports the translation of CR3 and its trafficking to the cell surface. Protein disulfide isomerase (PDI) contributes to the proper folding of proteins by catalyzing disulfide bond formation in the ER, but is also expressed on cell surfaces. In the case of CR3, disulfide bonds stabilize the open activated conformation (296), and PDI is involved in neutrophil adhesion during vascular inflammation (292). B-cell receptor-associated protein 31 (BAP31) is an ER-associated transmembrane protein that is involved in regulating cellular anterograde transport. Its reported binding to CR3 might play a role in protein trafficking to the cell membrane (293).

Table 8 Involvement of CR3 in additional interactions.
CD22 is a glycoprotein on B-cells involved in the negative-feedback regulation of B-cell receptor (BCR) signaling through formation of a complex with CD11b, BCR Lyn, and SHP-1. The negative feedback is dependent on functional CD11b, as R77H polymorphism results in decreased co-localization with the CD22:Lyn : SHP-1 complex. In turn, this leads to enhanced B-cell proliferation and Ca2+ influx (66). Interestingly, the R77H mutation seems to affect glycosylation, since the negative feedback interaction of CD22 with CD11b was also diminished when CD11b was treated with neuramidase, and lectin binding was abolished when R77H CD11b was transfected into CHO cells.
CD23, a C-type lectin, is a ligand for CD11b and CD11c on resting monocytes. When bound, it induces NO formation and the secretion of pro-inflammatory cytokines (IL-1β, IL-6, and TNFα) (294).
The cytokine interleukin 13 (IL-13) partially shares the signalling pathway with IL-4 through engaging with the IL-4 receptor type II. This receptor is a heterodimer, consisting of IL-13Rα1 and IL-4Rα, which assembles after ligand binding (IL-13 binds IL-13Rα1; IL-4 binds the first IL-4Rα chain). Ligand binding to the receptor triggers the expression of pro-inflammatory genes such as 15-LO and CD36. CR3 colocalizes with and binds the IL-13Rα1 chain via cis-ligation as a negative-feedback mechanism, thereby reducing the formation of foam cells (63, 64). Potential interaction areas were identified within the W5 blade of the β-propeller, the αMI domain, and the CD11b leg from a co-evolution analysis of CR3 and IL-13Rα1 (63).
Interestingly, adhesion of monocytes to steel used for stents was shown to depend on CR3, which can lead to restenosis. The binding could be blocked by coating with the semiconductor silicon carbide (295).
CR3 has long been an interesting translational target due to its highly dynamic involvement in clinically relevant processes that include inflammation and thrombosis and immune cell adhesion, activation, and trafficking. As a result, a plethora of antibodies, inhibitors, and synthetic molecules have been developed that interact with CR3 (Table 9 and Supplementary Table 9).

Table 9 CR3 as a potential therapeutic target.
CD11b expression has been a longstanding immunological marker to identify subsets of leukocytes. However, the dynamically increased expression in various disease states also points to the utility of CR3 as a biomarker. The value of monitoring CR3 expression has been shown in acute myeloid leukemia (315, 316), childhood acute lymphoblastic leukemia (317), neonatal sepsis (318), inflammatory lung disease (319), metabolic syndrome (320), Alzheimer’s disease (321), and gastric cancer. In cancer, elevated numbers of CD11b+ cells are often considered predictors of a poor prognosis (322). Recently, CR3 expression was found to be elevated in hypoxic COVID-19 patients, but not in patients with mild symptoms. Therefore, CR3 may serve as marker for disease severity in COVID-19 patients (323).
The association of CR3 and other β2 integrins with immuno-inflammatory dysregulation in numerous diseases has sparked considerable therapeutic interest in this receptor class (2–4, 26). For example, CR3 has been suggested as an intervention target for ischemic stroke in a rodent model due to its upregulated expression and the beneficial outcome (e.g., reduced neutrophil infiltration) of receptor inhibition. However, clinical trials with a NIF-based CD11b inhibitor and the anti-CD18 antibody rovelizumab for treatment of reperfusion injury in stroke were halted due to lack of efficacy (324). There has been mounting interest in recent years in CR3 for its role in CNS development, homeostasis, and neurodegenerative diseases. However, the biology of CR3 in neurological processes is complex. In rodent models, neuroinflammation in Alzheimer’s and Parkinson’s disease is reduced in a CR3-dependent manner by phagocytotic plaque clearance (172) and Aβ level reductions (174). Conversely, binding of β-amyloid (325) and α-synuclein (178) to CR3 induces production of ROS.
The clinical introduction of β2 integrin modulators has been met with challenges after efalizumab, an anti-CD11a mAb for the treatment of psoriasis, was retracted from the market for reactivation of John Cunningham virus. The virus induced potentially life-threatening progressive multifocal leukoencephalopathy (PML) (326). In contrast, the CR3 small molecule agonist LA-1/ADH-503 had positive effects in various inflammatory- and leukocyte-driven disease models in rodents (308, 327, 328) in addition to Aβ42 levels (174). Recent investigations identified a role of CD11b activation in anti-tumor innate immunity (34) and possible therapeutic applications in pancreatic (329) and lung cancer (330). LA-1/ADH-503 is currently under clinical investigation (licensed to Gossamer Bio as GB1275) in Phase I/II trials for solid tumors (NCT04060342). Imprime PGG, a β-glucan (see section 3.1.2.1) that targets the lectin domain of CR3 primes phagocytes to exert cytotoxicity against tumor cells (152). Combination therapies of Imprime PGG with immune checkpoint inhibitors are currently under evaluation in Phase 2 trials for treating melanoma and metastatic breast cancer (NCT02981303). The efficacy and safety of CR3 agonists remains to be determined.
Despite the challenges in clinical applications, various CR3-targeting compounds have been developed that include mAbs, recombinant proteins, peptides, and small molecules. While they are not all designed as therapeutics, they are valued for their potential towards unraveling the complex biology of CR3. Indeed, several mAbs have been developed as probes to investigate binding sites, conformational states, and functional outcomes of CR3 ligation in in vitro settings (Figure 5) (331). Abciximab (7E3 fab), a therapeutic antibody against GpIIb/IIIa and platelet aggregation inhibitor, binds to CR3 and competes with ICAM-1, fibrinogen, and FX (332). However, this antibody is no longer in clinical use due to supply problems.
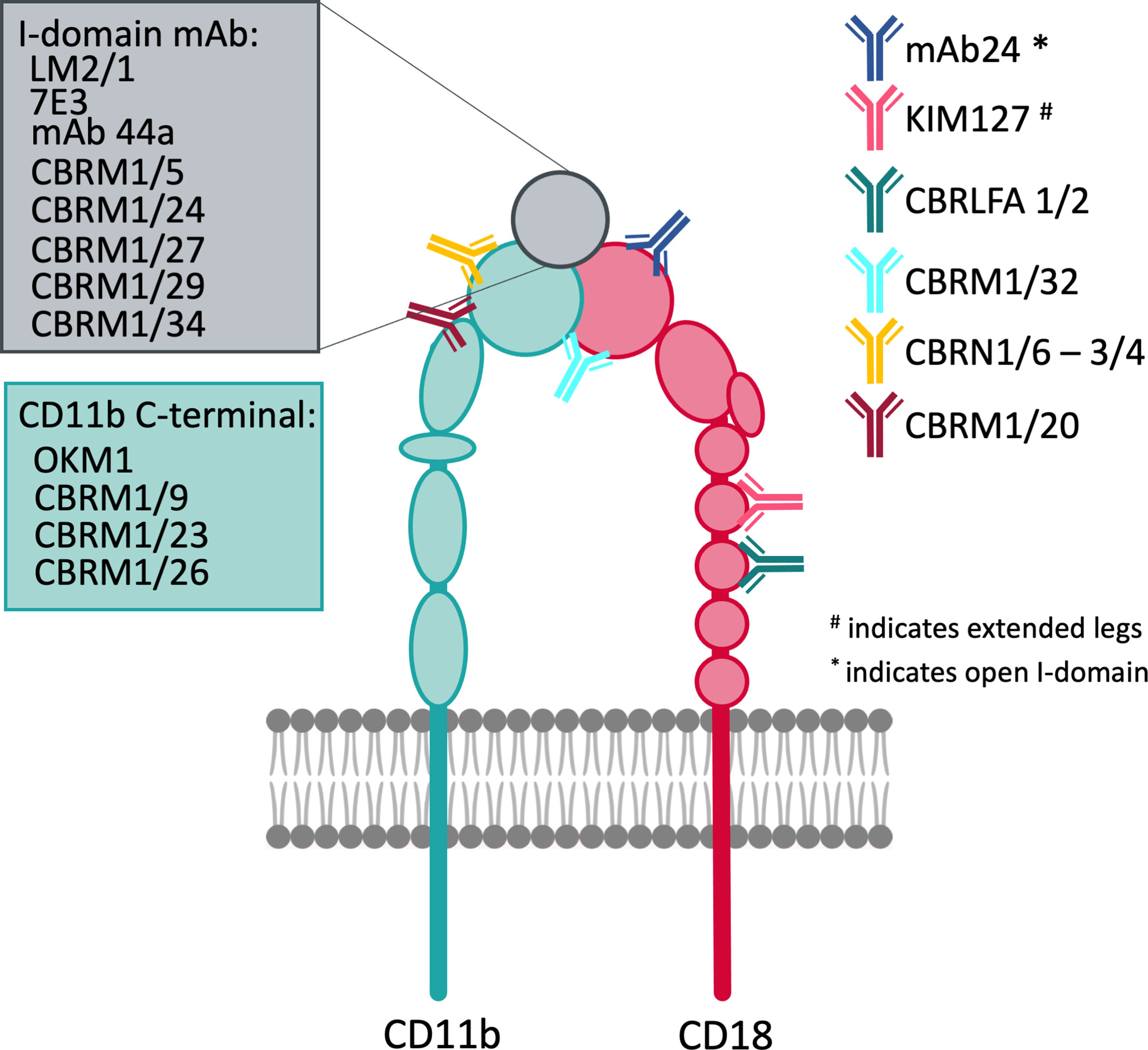
Figure 5 Overview of important antibodies developed against CD11b and CD18 mapped to their reactive regions.
Saccharides that impact leukocytes might induce effects similar to Imprime PGG when bound to the lectin site of CR3. The acidic fraction of lactose-derived oligosaccharides from human breast milk inhibits leukocyte adhesion (333) and transendothelial migration (297). This has led to the preclinical evaluation of lactosyl derivatives (Gu-4) for the treatment of severe burn-related shock and sepsis in animal models (297, 334). Moreover, hydroxyethyl starch 130/0.4 (HES), a synthetic colloid used in volume replacement therapies, binds to fMLP-activated neutrophils via CR3, which leads to “outside-in” signaling via increased activation of the PI3K/Akt pathway, with reduced migration, chemotaxis, and impaired binding to fibrinogen (298).
Attempts have been made to identify selective small-molecule CR3 antagonists. Fluorenylalkanoic and benzoic acids, which originated from Fmoc amino acid derivatives (leumidins) that inhibit neutrophil recruitment, were among the first compounds proposed as novel leukocyte adhesion inhibitors (300). Next-generation compounds replaced the carbamate moiety and increased activity 10-fold with the lead molecule (Figure 6 and Compound 1) that had an IC50 = 5 µM in neutrophil adherence assays (300). While CR3 has been suggested as the responsible target, this has not been confirmed by direct binding studies (301).

Figure 6 Small molecules developed as possible CR3 modulators.
In a screening assay with immobilized CR3 and iC3b as binding partners, two compounds (Figure 6 and Compounds 2; 3) were identified as inhibitors of the CR3:iC3b interaction, with IC50 values of 0.14 and 0.33 µM, respectively. At concentrations up to 10 µM, they showed no binding to LFA-1, αVβ3, α4β1, or α4β7. Interestingly, the binding of both compounds is not easily reversed, and covalent binding can be induced by light (302).
The Gupta group screened >92,500 commercially available compounds in a high-throughput screening assay to identify antagonists competing with fibrinogen binding to CR3, the addition of Mn2+ rendered the integrin active. From this work, 63 compounds were identified that inhibit fibrinogen binding with an activity < 1 µM, though only a subset of compounds was disclosed (Figure 6 and Compounds 4–10) (303).
Roche and Genentech patented compounds characterized as LFA-1 antagonists (Figure 6 and Compounds 11–13), which perturb the interface between the αI domain and βI-like domain. Consequently, these compounds lock the I-like domain in an active conformation, which leads to integrin extension, while the I domain is locked in its inactive state (304). In addition to being antagonists of LFA-1, the compounds also inhibit CR3 binding to iC3b (11, 3µM and 12, 0.1 µM, 13 XVA143) (304).
Simvastatin and other statins were identified as allosteric inhibitors of LFA-1, which bind to the so-called L-site on the opposite side of the MIDAS (335). In the CR3 interaction with simvastatin, a recent crystal structure shows that the carboxylic acid of the hydrolyzed lactone interacts with the MIDAS to form a salt bridge, which seems to be the only relevant contact formed. Further studies are needed to explore the specificity and impact of this interaction. In surface plasmon resonance experiments, simvastatin partially inhibited the interaction of CR3 αMI with iC3b and C3d, whereas the CR3:ICAM-1 interaction was not inhibited. At micromolar concentrations, simvastatin inhibits monocyte binding to iC3b in flow cytometry and adhesion assays. However, this interaction does not explain the plethora of pleiotropic effects of simvastatin (48).
Several phage display screening campaigns aimed at β2 integrins have been conducted, which panned against full CR3 (305), the αMI domain (204), or one of its ligands (231). Koivunen et al. panned against the full CR3 purified from blood samples with CX7C and CX9C libraries (cyclic peptides with two fixed cysteines and 7 or 9 variable positions, respectively) in Mn2+-containing buffer. They identified peptides that bind the αMI domain in a divalent cation-dependent manner, with cross reactivity to LFA-1 and CR4. The minimum consensus sequence was CLLGC, where the most potent peptide (CP[CFLLGC]C) had an IC50 of 20 µM. This peptide inhibited leukocyte adhesion to ICAM-1 and von Willebrand factor (305). A different phage display screen against the purified αMI domain with a pool of random peptide libraries (CX7-10C and X9-10) yielded E/DDGW as a minimal consensus sequence. This sequence is also found in MMPs, which were identified as CR3 ligands (see above). The DDGW peptide is an effective inhibitor of MMP-9-binding to the αMI (IC50 = 20 µM) as well as the αLI domain, though not in competition with CR3-binding to ICAM-1 (204). As an alternative target, the anti-αMI mAb 44a, known to block binding of fibrinogen, iC3b, and C1q, but not ICAM-1, was also used for phage display panning. Screening of a CX6C library displayed on M13 filamentous phage yielded the peptide (CRLKEKHC), which can dose-dependently inhibit binding of fibrinogen to CR3 (IC50 = 3.35 µM) (231).
Another approach to developing binding peptides is to derive them from the complementarity-determining regions of anti-CR3 antibodies, which block binding to ICAM-1. The most potent peptides derived from this method inhibited binding of CR3-expressing CHO cells to ICAM-1 with an IC50 = 30 µM. Generation of a focused library based on the identified sequence yielded the active peptide (GYRDGYAGPILYN) (306, 309).
A high-throughput screen was implemented to identify antagonists in competition with DDGW-displaying phages from a pool of 10,000 commercially available small molecules. This screen generated 19 lead compounds with significant and reproducible inhibition of DDGW-phage binding. Most of the compounds had a common 2-thioxothiazolidin-4-one substructure (Figure 6 and Compound 14–16). Surprisingly, although the compounds inhibited phage binding to the αMI domain, they also greatly enhanced the binding of pro-MMP-9 and fibrinogen. A binding site in a hydrophobic cavity that appears in the open conformation of the αMI domain was proposed using computational modeling methods. Compound 16 was tested in vivo and reduced inflammation response as determined by reduced neutrophil emigration (307). Similarly, a high-throughput screen was conducted by the Gupta group with a library of >13,500 small molecules to identify compounds that increased CR3 binding to fibrinogen in Ca2+/Mg2+-containing buffer. A large subset of the hits contained a central 2-thioxothiazolidin-4-one motif, which was a previously reported hit structure in an independent screen against the αMI-domain in competition with DDGW-phages (307). Compound 17 (Figure 6) had 2-fold higher binding to CD11b over CD11a, with an EC50 = 13.6 ± 5 µM. In silico docking experiments also suggested that the compounds (termed leukadherins) bind between helix α7 and α1 and the central β-sheet (310), in agreement with prior reports (307). In contrast to activating antibodies, leukadherins do not induce integrin clusterin or intracellular signaling due to a lack of a global conformational change during binding (311). Leukadherin-1 (LA-1, ADH-503) (Figure 6 and Compound 18), the most prominent compound of the class, is currently in clinical development as GB1275 (see above). It increases CR3-dependent cell adhesion while reducing chemotaxis and transendothelial migration. Consequently, it has showed efficacy in leukocyte-dependent disease models, such as acute peritonitis in mice, vascular injury in rats, and experimental nephristis in mice (312). Furthermore, LA-1 can suppress human innate inflammatory signaling (313), TLR-dependent inflammation, and autoimmunity in SLE (308), in addition to ameliorating endothelial barrier damage in critically ill patients (314). In addition, LA-1 promotes pro-inflammatory macrophage polarisation, which drives anti-tumor innate immunity (34).
Integrins are a versatile class of cell surface receptors with a broad spectrum of functions in inter-cellular communication, tissue development, maintenance, and repair. Integrins have a well established link to the ECM and play a role in leukocyte migration and function. Within this family, CR3 has particularly broad ligand promiscuity. Its ability to bind ligands spans the I-domain, adjunct areas, and distinct regions like the lectin domain. Whereas CR3 ligands were first reported 40 years ago, recent crystallographic insights and binding studies reveal a considerably complex picture. While CR3 has close to 100 ligands, binding for most of these have been demonstrated through biological methods without full investigation at the molecular level, including specific binding sites, competition with other ligands, or functional implications. Additionally, the treasure chest of CR3-mediated interactions and functions is still not exhausted. For example, recent studies showed a link between the complement system and integrins (especially LFA-1) in orchestrating metabolism and immunity (1) through regulation of the intracellular complement system (i.e. the complosome) (336), which is required for Th1 responses. Cultured cells can take up some of the required C3 for those responses in the form of hydrolyzed C3, which serves as ligand for CR3. Looking forward, further and in-depth investigations are needed to elucidate the complex roles of leukocyte integrins and their crosstalk across numerous pathways.
All authors contributed to the conception, writing, and editing of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Swiss National Science Foundation (31003A_176104 to DR) and the University of Basel (to CL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Dr. Kaycie Butler (ButlerSciComm) for her valuable assistance with language editing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.662164/full#supplementary-material
1. Merle N, Singh P, Rahman J, Kemper C. Integrins Meet Complement: The Evolutionary Tip of an Iceberg Orchestrating Metabolism and Immunity. Br J Pharmacol (2020), 1–17. doi: 10.1111/bph.15168
2. Shimaoka M, Springer TA. Therapeutic Antagonists and Conformational Regulation of Integrin Function. Nat Rev Drug Discovery (2003) 2:703–16. doi: 10.1038/nrd1174
3. Cox D, Brennan M, Moran N. Integrins as Therapeutic Targets: Lessons and Opportunities. Nat Rev Drug Discovery (2010) 9:804–20. doi: 10.1038/nrd3266
4. Ley K, Rivera-Nieves J, Sandborn WJ, Shattil S. Integrin-Based Therapeutics: Biological Basis, Clinical Use and New Drugs. Nat Rev Drug Discovery (2016) 15:173–83. doi: 10.1038/nrd.2015.10
5. Bachmann M, Kukkurainen S, Hytönen VP, Wehrle-Haller B. Cell Adhesion by Integrins. Physiol Rev (2019) 99:1655–99. doi: 10.1152/physrev.00036.2018
6. Arnaout MA, Mahalingam B, Xiong J-P. Integrin STRUCTURE, Allostery, AND Bidirectional Signaling. Annu Rev Cell Dev Biol (2005) 21:381–410. doi: 10.1146/annurev.cellbio.21.090704.151217
7. Michishita M, Videm V, Amin Arnaout M. A Novel Divalent Cation-Binding Site in the a Domain of the β2 Integrin CR3 (CD11b/CD18) is Essential for Ligand Binding. Cell (1993) 72:857–67. doi: 10.1016/0092-8674(93)90575-B
8. Torres-Gomez A, Cabañas C, Lafuente EM. Phagocytic Integrins: Activation and Signaling. Front Immunol (2020) 11:738. doi: 10.3389/fimmu.2020.00738
9. Fan Z, Ley K. Leukocyte Arrest: Biomechanics and Molecular Mechanisms of β2 Integrin Activation. Biorheology (2015) 52:353–77. doi: 10.3233/BIR-15085
10. Jaumouillé V, Cartagena-Rivera AX, Waterman CM. Coupling of β2 Integrins to Actin by a Mechanosensitive Molecular Clutch Drives Complement Receptor-Mediated Phagocytosis. Nat Cell Biol (2019) 21:1357–69. doi: 10.1038/s41556-019-0414-2
11. Kim M, Carman CV, Springer TA. Bidirectional Transmembrane Signaling by Cytoplasmic Domain Separation in Integrins. Sci (80 ) (2003) 301:1720–5. doi: 10.1126/science.1084174
12. Fan Z, Kiosses WB, Sun H, Orecchioni M, Ghosheh Y, Zajonc DM, et al. High-Affinity Bent β2-Integrin Molecules in Arresting Neutrophils Face Each Other Through Binding to ICAMs in Cis. Cell Rep (2019) 26:119–130.e5. doi: 10.1016/j.celrep.2018.12.038
13. Fan Z, McArdle S, Marki A, Mikulski Z, Gutierrez E, Engelhardt B, et al. Neutrophil Recruitment Limited by High-Affinity Bent β 2 Integrin Binding Ligand in Cis. Nat Commun (2016) 7:12658. doi: 10.1038/ncomms12658
14. Li J, Su Y, Xia W, Qin Y, Humphries MJ, Vestweber D, et al. Conformational Equilibria and Intrinsic Affinities Define Integrin Activation. EMBO J (2017) 36:629–45. doi: 10.15252/embj.201695803
15. Zwartz G, Chigaev A, Foutz T, Larson RS, Posner R, Sklar LA. Relationship Between Molecular and Cellular Dissociation Rates for VLA-4/VCAM-1 Interaction in the Absence of Shear Stress. Biophys J (2004) 86:1243–52. doi: 10.1016/S0006-3495(04)74198-3
16. Kim M, Carman CV, Yang W, Salas A, Springer TA. The Primacy of Affinity Over Clustering in Regulation of Adhesiveness of the Integrin αlβ2. J Cell Biol (2004) 167:1241–53. doi: 10.1083/jcb.200404160
17. Ye F, Petrich BG, Anekal P, Lefort CT, Kasirer-friede A, Sanford J, et al. The Mechanism of Kindlin-mediated Activation of Integrin α IIb β 3. Curr Biol (2013) 23:2288–95. doi: 10.1016/j.cub.2013.09.050.The
18. Gaertner F, Ahmad Z, Rosenberger G, Fan S, Nicolai L, Busch B, et al. Migrating Platelets are Mechano-scavengers That Collect and Bundle Bacteria. Cell (2017) 171:1368–82. doi: 10.1016/j.cell.2017.11.001
19. Zheng Y, Leftheris K. Insights Into Protein–Ligand Interactions in Integrin Complexes: Advances in Structure Determinations. J Med Chem (2020) 63:5675–96. doi: 10.1021/acs.jmedchem.9b01869
20. Tan S. The Leucocyte β2 (Cd18) Integrins: The Structure, Functional Regulation and Signalling Properties. Biosci Rep (2012) 32:241–69. doi: 10.1042/BSR20110101
21. Schittenhelm L, Hilkens CM, Morrison VL. β2 Integrins as Regulators of Dendritic Cell, Monocyte, and Macrophage Function. Front Immunol (2017) 8:1866. doi: 10.3389/fimmu.2017.01866
22. Arnaout MA. Biology and Structure of Leukocyte β2 Integrins and Their Role in Inflammation. F1000Research (2016) 5:2433. doi: 10.12688/f1000research.9415.1
23. Tenner AJ, Stevens B, Woodruff TM. New Tricks for an Ancient System: Physiological and Pathological Roles of Complement in the CNS. Mol Immunol (2018) 102:3–13. doi: 10.1016/j.molimm.2018.06.264
24. Yakubenko VP, Yadav SP, Ugarova TP. Integrin α Dβ 2, an Adhesion Receptor Up-Regulated on Macrophage Foam Cells, Exhibits Multiligand-Binding Properties. Blood (2006) 107:1643–50. doi: 10.1182/blood-2005-06-2509
25. Fagerholm SC, Guenther C, Asens ML, Savinko T, Uotila LM. Beta2-Integins and Interacting Proteins in Leukocyte Trafficking, Immune Supression, and Immunodeficiency Disease. Front Immunol (2019) 10:254. doi: 10.3389/fimmu.2019.00254
26. Bednarczyk M, Stege H, Grabbe S, Bros M. β2 Integrins—Multi-Functional Leukocyte Receptors in Health and Disease. Int J Mol Sci (2020) 21:1404. doi: 10.3390/ijms21041402
27. Evans R, Patzak I, Svensson L, De Filippo K, Jones K, McDowall A, et al. Integrins in Immunity. J Cell Sci (2009) 122:215–25. doi: 10.1242/jcs.019117
28. Rosetti F, Mayadas TN. The Many Faces of Mac-1 in Autoimmune Disease. Immunol Rev (2016) 269:175–93. doi: 10.1111/imr.12373
29. Pierschbacher M, Rouslahti E. Cell Attachment Activity of Fibronectin can be Duplicated by Small Synthetic Fragments of the Molecule. Nature (1984) 309:30–3. doi: 10.1038/309030a0
30. Komoriya A, Green LJ, Mervic M, Yamada SS, Yamada KM, Humphries MJ. The Minimal Essential Sequence for a Major Cell Type-Specific Adhesion Site (CS1) Within the Alternatively Spliced Type III Connecting Segment Domain of Fibronectin is Leucine-Aspartic Acid-Valine. J Biol Chem (1991) 266:15075–9. doi: 10.1016/S0021-9258(18)98588-1
31. Taniguchi-Sidle A, Isenman DE. Mutagenesis of the Arg-Gly-Asp Triplet in Human Complement Component C3 Does Not Abolish Binding of iC3b to the Leukocyte Integrin Complement Receptor Type III (Cr3, Cd11b/Cd18). J Biol Chem (1992) 267:635–43. doi: 10.1016/S0021-9258(18)48541-9
32. Van Gisbergen KPJM, Sanchez-Hernandez M, Geijtenbeek TBH, Van Kooyk Y. Neutrophils Mediate Immune Modulation of Dendritic Cells Through Glycosylation-Dependent Interactions Between Mac-1 and DC-SIGN. J Exp Med (2005) 201:1281–92. doi: 10.1084/jem.20041276
33. Martinez L, Li X, Ramos-Echazabal G, Faridi H, Zigmond ZM, Santos Falcon N, et al. A Genetic Model of Constitutively Active Integrin Cd11b/Cd18. J Immunol (2020) 205:2545–53. doi: 10.4049/jimmunol.1901402
34. Schmid MC, Khan SQ, Kaneda MM, Pathria P, Shepard R, Louis TL, et al. Integrin CD11b Activation Drives Anti-Tumor Innate Immunity. Nat Commun (2018) 9:1–14. doi: 10.1038/s41467-018-07387-4
35. Mesri M, Plescia J, Altieri DC. Dual Regulation of Ligand Binding by CD11b I Domain. J Biol Chem (1998) 273:744–8. doi: 10.1074/jbc.273.2.744
36. Vorup-Jensen T, Jensen RK. Structural Immunology of Complement Receptors 3 and 4. Front Immunol (2018) 9:2716. doi: 10.3389/fimmu.2018.02716
37. Adair BD, Xiong JP, Alonso JL, Hyman BT, Arnaout MA. Em Structure of the Ectodomain of Integrin CD11b/CD18 and Localization of Its Ligand-Binding Site Relative to the Plasma Membrane. PloS One (2013) 8:e57951. doi: 10.1371/journal.pone.0057951
38. Xu S, Wang J, Wang J-H, Springer TA. Distinct Recognition of Complement iC3b by Integrins α X β 2 and α M β 2. Proc Natl Acad Sci (2017) 114:3403–8. doi: 10.1073/pnas.1620881114
39. Jensen R, Bajic G, Sen M, Springer T, Vorup-Jensen T, Andersen G. Complement Receptor 3 Forms a Compact High Affinity Complex With Ic3b. bioRxiv (2020). doi: 10.1101/2020.04.15.043133
40. Boguslawski KM, McKeown AN, Day CJ, Lacey KA, Tam K, Vozhilla N, et al. Exploiting Species Specificity to Understand the Tropism of a Human-Specific Toxin. Sci Adv (2020) 6:eaax7515. doi: 10.1126/sciadv.aax7515
41. Dumont AL, Yoong P, Day CJ, Alonzo F, Mcdonald WH, Jennings MP, et al. Staphylococcus Aureus LukAB Cytotoxin Kills Human Neutrophils by Targeting the CD11b Subunit of the Integrin Mac-1. Proc Natl Acad Sci (2013) 110:10794–9. doi: 10.1073/pnas.1305121110
42. Trstenjak N, Milić D, Graewert MA, Rouha H, Svergun D, Djinović-Carugo K, et al. Molecular Mechanism of Leukocidin GH–integrin CD11b/CD18 Recognition and Species Specificity. Proc Natl Acad Sci USA (2020) 117:317–27. doi: 10.1073/pnas.1913690116
43. Lee J-O, Rieu P, Amin Arnaout M, Liddington R. Crystal Structure of the A Domain From the a Subunit of Integrin Cr3 (Cd11b/Cd18). Cell (1995) 80:631–8. doi: 10.1016/0092-8674(95)90517-0
44. Ustinov VA, Plow EF. Delineation of the Key Amino Acids Involved in Neutrophil Inhibitory Factor Binding to the I-domain Supports a Mosaic Model for the Capacity of Integrin αmβ2 to Recognize Multiple Ligands. J Biol Chem (2002) 277:18769–76. doi: 10.1074/jbc.M110242200
45. Zhang L, Plow EF. Amino Acid Sequences Within the α Subunit of Integrin αmβ2 (Mac-1) Critical for Specific Recognition of C3bi. Biochemistry (1999) 38:8064–71. doi: 10.1021/bi990141h
46. Ugarova TP, Yakubenko V. Recognition of Fibrinogen by Leukocyte Integrins. Ann N Y Acad Sci (2001) 936:368–85. doi: 10.1111/j.1749-6632.2001.tb03523.x
47. Yakubenko VP, Lishko VK, Lam SCT, Ugarova TP. A Molecular Basis for Integrin αmβ2 Ligand Binding Promiscuity. J Biol Chem (2002) 277:48635–42. doi: 10.1074/jbc.M208877200
48. Jensen MR, Bajic G, Zhang X, Laustsen AK, Koldsø H, Skeby KK, et al. Structural Basis for Simvastatin Competitive Antagonism of Complement Receptor 3. J Biol Chem (2016) 291:16963–76. doi: 10.1074/jbc.M116.732222
49. Podolnikova NP, Podolnikov AV, Haas TA, Lishko VK, Ugarova TP. Ligand Recognition Specificity of Leukocyte Integrin α M β 2 (Mac-1, CD11b/CD18) and Its Functional Consequences. Biochemistry (2015) 54:1408–20. doi: 10.1021/bi5013782
50. Xia Y, Ross GD. Generation of Recombinant Fragments of CD11b Expressing the Functional Beta-Glucan-Binding Lectin Site of CR3 (Cd11b/Cd18). J Immunol (1999) 162:7285–93.
51. Thornton BP, Vetvicka V, Pitman M, Goldman RC, Ross GD. Analysis of the Sugar Specificity and Molecular Location of the beta-Glucan-Binding Lectin Site of Complement Receptor Type 3 (Cd11b/Cd18). J Immunol (1996) 156:1235–46.
52. O’Brien XM, Heflin KE, Lavigne LM, Yu K, Kim M, Salomon AR, et al. Lectin Site Ligation of CR3 Induces Conformational Changes and Signaling. J Biol Chem (2012) 287:3337–48. doi: 10.1074/jbc.M111.298307
53. Rabb H, Michishita M, Sharma CP, Brown D, Arnaout MA. Cytoplasmic Tails of Human Complement Receptor Type 3 (CR3, CD11b/CD18) Regulate Ligand Avidity and the Internalization of Occupied Receptors. J Immunol (1993) 151:990–1002.
54. Větvička V, Thornton BP, Ross GD. Soluble β-Glucan Polysaccharide Binding to the Lectin Site of Neutrophil or Natural Killer Cell Complement Receptor Type 3 (CD11b/CD18) Generates a Primed State of the Receptor Capable of Mediating Cytotoxicity of iC3b-opsonized Target Cells. J Clin Invest (1996) 98:50–61. doi: 10.1172/JCI118777
55. Ross GD. Role of the Lectin Domain of Mac-1/CR3 (CD11b/CD18) in Regulating Intercellular Adhesion. Immunol Res (2002) 25:219–27. doi: 10.1385/IR:25:3:219
56. Bannerji R, Kitada S, Flinn IW, Pearson M, Young D, Reed JC, et al. Apoptotic-Regulatory and Complement-Protecting Protein Expression in Chronic Lymphocytic Leukemia: Relationship to In Vivo Rituximab Resistance. J Clin Oncol (2003) 21:1466–71. doi: 10.1200/JCO.2003.06.012
57. Zen K, Liu Y, Cairo D, Parkos CA. Cd11b/Cd18-Dependent Interactions of Neutrophils With Intestinal Epithelium Are Mediated by Fucosylated Proteoglycans. J Immunol (2002) 169:5270–8. doi: 10.4049/jimmunol.169.9.5270
58. Sitrin RG, Todd RF, Petty HR, Brock TG, Shollenberger SB, Albrecht E, et al. The Urokinase Receptor (CD87) Facilitates CD11b/CD18-mediated Adhesion of Human Monocytes. J Clin Invest (1996) 97:1942–51. doi: 10.1172/JCI118626
59. Simon BDI, Rao NK, Xu H, Wei Y, Majdic O, Ronne E, et al. Mac-L (CD11b/CD18) and the Urokinase Receptor (Cd87) Form a Functional Unit on Monocytic Cells. Blood (1996) 88:3185–94. doi: 10.1182/blood.V88.8.3185.bloodjournal8883185
60. Xia Y, Borland G, Huang J, Mizukami IF, Petty HR, Todd RF, et al. Function of the Lectin Domain of Mac-1/Complement Receptor Type 3 (CD11b/CD18) in Regulating Neutrophil Adhesion. J Immunol (2002) 169:6417–26. doi: 10.4049/jimmunol.169.11.6417
61. Lin L, Wu C, Hu K. Tissue Plasminogen Activator Activates NF-κb Through a Pathway Involving Annexin A2/CD11b and Integrin-Linked Kinase. J Am Soc Nephrol (2012) 23:1329–38. doi: 10.1681/ASN.2011111123
62. Chen J, Zhong MC, Guo H, Davidson D, Mishel S, Lu Y, et al. SLAMF7 is Critical for Phagocytosis of Haematopoietic Tumour Cells Via Mac-1 Integrin. Nature (2017) 544:493–7. doi: 10.1038/nature22076
63. Cao C, Zhao J, Doughty EK, Migliorini M, Strickland DK, Kann MG, et al. Mac-1 Regulates IL-13 Activity in Macrophages by Directly Interacting With IL-13Rα1. J Biol Chem (2015) 290:21642–51. doi: 10.1074/jbc.M115.645796
64. Yakubenko VP, Hsi LC, Cathcart MK, Bhattacharjee A. From Macrophage interleukin-13 Receptor to Foam Cell Formation: Mechanisms for αmβ2 Integrin Interference. J Biol Chem (2013) 288:2778–88. doi: 10.1074/jbc.M112.381343
65. Saggu G, Okubo K, Chen Y, Vattepu R, Tsuboi N, Rosetti F, et al. Cis Interaction Between Sialylated Fcγriia and the αi-Domain of Mac-1 Limits Antibody-Mediated Neutrophil Recruitment. Nat Commun (2018) 9:5058. doi: 10.1038/s41467-018-07506-1
66. Ding C, Ma Y, Chen X, Liu M, Cai Y, Hu X, et al. Integrin CD11b Negatively Regulates BCR Signalling to Maintain Autoreactive B Cell Tolerance. Nat Commun (2013) 4:2813. doi: 10.1038/ncomms3813
67. Ross G. Identification of a C3bi-specific Membrane Complement Receptor That is Expressed on Lymphocytes, Monocytes, Neutrophils, and Erythrocytes. J Exp Med (1982) 155:96–110. doi: 10.1084/jem.155.1.96
68. Arnaout MA, Todd RF, Dana N, Melamed J, Schlossman SF, Colten HR. Inhibition of Phagocytosis of Complement C3- or Immunoglobulin G-coated Particles and of C3bi Binding by Monoclonal Antibodies to a Monocyte-Granulocyte Membrane Glycoprotein (Mo1). J Clin Invest (1983) 72:171–9. doi: 10.1172/JCI110955
69. Ross G. Generation of Three Different Fragments of Bound C3 With Purified Factor I or Serum. II. Location of Binding Sites in the C3 Fragments for Factors B and H, Complement Receptors , and Bovine Conglutinin. J Exp Med (1983) 158:334–52. doi: 10.1084/jem.158.2.334
70. Zhang L, Plow EF. Overlapping, But Not Identical, Sites are Involved in the Recognition of C3bi, Neutrophil Inhibitory Factor, and Adhesive Ligands by the α(M)β2 Integrin. J Biol Chem (1996) 271:18211–6. doi: 10.1074/jbc.271.30.18211
71. Kamata T, Wright R, Takada Y. Critical Threonine and Aspartic Acid Residues Within the I Domains of Beta2 Integrins for Interaction With Intercellular Adhesion Molecule 1 (Icam-1) and C3bi. J Biol Chem (1995) 270:12531–5. doi: 10.1074/jbc.270.21.12531
72. McGuire SL, Bajt ML. Distinct Ligand Binding Sites in the I Domain of Integrin αmβ2 That Differentially Affect a Divalent Cation-Dependent Conformation. J Biol Chem (1995) 270:25866–71. doi: 10.1074/jbc.270.43.25866
73. Li Y, Zhang L. The Fourth Blade Within the β-Propeller Is Involved Specifically in C3bi Recognition by Integrin αmβ2. J Biol Chem (2003) 278:34395–402. doi: 10.1074/jbc.M304190200
74. Bajt ML, Godmann T, McGuire SL. Beta 2 (CD18) Mutations Abolish Ligand Recognition by I Domain Integrins Lfa-1 (Alpha L Beta 2, CD11a/CD18) and MAC-1 (Alpha M Beta 2, Cd11b/Cd18). J Biol Chem (1995) 270:94–8. doi: 10.1074/jbc.270.1.94
75. Goodman TG, Lynn Bajt M. Identifying the Putative Metal Ion-dependent Adhesion Site in the 2 (CD18) Subunit Required for L 2 and M 2 Ligand Interactions. J Biol Chem (1996) 271:23729–36. doi: 10.1074/jbc.271.39.23729
76. Yalamanchili P, Lu C, Oxvig C, Springer TA. Folding and Function of I Domain-Deleted Mac-1 and Lymphocyte Function-Associated Antigen-1. J Biol Chem (2000) 275:21877–82. doi: 10.1074/jbc.M908868199
77. Bajic G, Yatime L, Sim RB, Vorup-Jensen T, Andersen GR. Structural Insight on the Recognition of Surface-Bound Opsonins by the Integrin I Domain of Complement Receptor 3. Proc Natl Acad Sci U S A (2013) 110:16426–31. doi: 10.1073/pnas.1311261110
78. Wright SD, Levin SM, Jong MTC, Chad Z, Kabbash LG. CR3 (CD11b/CD18) Expresses One Binding Site for Arg-Gly-Asp-containing Peptides and a Second Site for Bacterial Lipopolysaccharide. J Exp Med (1989) 169:175–83. doi: 10.1084/jem.169.1.175
79. Ueda T, Rieu P, Brayer J, Arnaout MA. Identification of the Complement Ic3b Binding Site in the Beta 2 Integrin Cr3 (Cd11b/Cd18). Proc Natl Acad Sci (1994) 91:10680–4. doi: 10.1073/pnas.91.22.10680
80. Diamond MS, Garcia-Aguilar J, Bickford JK, Corbi AL, Springer TA. The I Domain is a Major Recognition Site on the Leukocyte Integrin Mac-1 (CD11b/CD18) for Four Distinct Adhesion Ligands. J Cell Biol (1993) 120:1031–43. doi: 10.1083/jcb.120.4.1031
81. Lin Z, Schmidt CQ, Koutsogiannaki S, Ricci P, Risitano AM, Lambris JD, et al. Complement C3dg-mediated Erythrophagocytosis: Implications for Paroxysmal Nocturnal Hemoglobinuria. Blood (2015) 126:891–4. doi: 10.1182/blood-2015-02-625871
82. Taniguchi-Sidle A, Isenman DE. Interactions of Human Complement Component C3 With Factor B and With Complement Receptors Type 1 (Cr1, CD35) and Type 3 (Cr3, CD11b/CD18) Involve an Acidic Sequence At the N-terminus of C3 Alpha’-Chain. J Immunol (1994) 153:5285–302.
83. Hamad OA, Mitroulis I, Fromell K, Kozarcanin H, Chavakis T, Ricklin D, et al. Contact Activation of C3 Enables Tethering Between Activated Platelets and Polymorphonuclear Leukocytes Via Cd11b/Cd18. Thromb Haemost (2015) 114:1207–17. doi: 10.1160/TH15-02-0162
84. DiScipio RG, Daffern PJ, Schraufstätter IU, Sriramarao P, DiScipio RG, Daffern PJ, et al. Human Polymorphonuclear Leukocytes Adhere to Complement Factor H Through an Interaction That Involves alphaMbeta2 (Cd11b/Cd18). J Immunol (1998) 160:4057–66.
85. Agarwal S, Ram S, Ngampasutadol J, Gulati S, Zipfel PF, Rice PA. Factor H Facilitates Adherence of Neisseria Gonorrhoeae to Complement Receptor 3 on Eukaryotic Cells. J Immunol (2010) 185:4344–53. doi: 10.4049/jimmunol.0904191
86. Losse J, Zipfel PF, Józsi M. Factor H and Factor H-Related Protein 1 Bind to Human Neutrophils Via Complement Receptor 3, Mediate Attachment to Candida Albicans, and Enhance Neutrophil Antimicrobial Activity. J Immunol (2010) 184:912–21. doi: 10.4049/jimmunol.0901702
87. Mitroulis I, Kang YY, Gahmberg CG, Siegert G, Hajishengallis G, Chavakis T, et al. Developmental Endothelial Locus-1 Attenuates Complement-Dependent Phagocytosis Through Inhibition of Mac-1-Integrin. Thromb Haemost (2014) 111:1004–6. doi: 10.1160/TH13-09-0794
88. Lishko VK, Moreno B, Podolnikova NP, Ugarova TP. Identification of Human Cathelicidin Peptide Ll-37 as a Ligand for Macrophage Integrin αmβ2 (Mac-1, CD11b/CD18) That Promotes Phagocytosis by Opsonizing Bacteria. FASEB J (2016) 2016:39–55. doi: 10.2147/RRBC.S107070
89. Zhang X, Bajic G, Andersen GR, Christiansen SH, Vorup-Jensen T. The Cationic Peptide LL-37 Binds Mac-1 (Cd11b/Cd18) With a Low Dissociation Rate and Promotes Phagocytosis. Biochim Biophys Acta (2016) 1864:471–8. doi: 10.1016/j.bbapap.2016.02.013
90. Lishko VK, Yakubenko VP, Ugarova TP, Podolnikova NP. Leukocyte Integrin Mac-1 (CD11b/CD18, Alphambeta2, CR3) Acts as a Functional Receptor for Platelet Factor 4. J Biol Chem (2018) 293:6869–82. doi: 10.1074/jbc.RA117.000515
91. Xiong Y, Cao C, Makarova A, Hyman B, Zhang L. Mac-1 Promotes Fcγriia-Dependent Cell Spreading and Migration on Immune Complexes. Biochemistry (2006) 45:8721–31. doi: 10.1021/bi060529u
92. Zhou M, Brown EJ. Cr3 (Mac-1, alphaMbeta2 , CD11b / CD18 ) and FcgRIII Cooperate in Generation of a Neutrophil Respiratory Burst: Requirement for FcgRII and Tyrosine Phosphorylation. J Cell Biol (1994) 125:1407–16. doi: 10.1083/jcb.125.6.1407
93. Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, Von Andrian UH, et al. A Novel Role for the β2 Integrin CD11b/CD18 in Neutrophil Apoptosis: A Homeostatic Mechanism in Inflammation. Immunity (1996) 5:653–66. doi: 10.1016/S1074-7613(00)80278-2
94. Pillay J, Den Braber I, Vrisekoop N, Kwast LM, De Boer RJ, Borghans JAM, et al. In Vivo Labeling With 2H2O Reveals a Human Neutrophil Lifespan of 5.4 Days. Blood (2010) 116:625–7. doi: 10.1182/blood-2010-01-259028
95. Watson RWG, Rotstein OD, Nathens AB, Parodo J, Marshall JC. Neutrophil Apoptosis is Modulated by Endothelial Transmigration and Adhesion Molecule Engagement. J Immunol (1997) 158:945–53.
96. Rubel C, Gómez S, Fernández GC, Isturiz MA, Caamaño J, Palermo MS. Fibrinogen-CD11b/CD18 Interaction Activates the NF-κb Pathway and Delays Apoptosis in Human Neutrophils. Eur J Immunol (2003) 33:1429–38. doi: 10.1002/eji.200323512
97. Takeda Y, Watanabe H, Yonehara S, Yamashita T, Salto S, Sendo F. Rapid Acceleration of Neutrophil Apoptosis by Tumor Necrosis Factor-α. Int Immunol (1993) 5:691–4. doi: 10.1093/intimm/5.6.691
98. Watson RW, Redmond HP, Wang JH, Condron C, Bouchier-Hayes D. Neutrophils Undergo Apoptosis Following Ingestion of Escherichia Coli. J Immunol (1996) 156:3986–92.
99. Zhang B, Hirahashi J, Cullere X, Mayadas TN. Elucidation of Molecular Events Leading to Neutrophil Apoptosis Following Phagocytosis. Cross-talk Between Caspase 8, Reactive Oxygen Species, and MAPK/ERK Activation. J Biol Chem (2003) 278:28443–54. doi: 10.1074/jbc.M210727200
100. Whitlock BB, Gardai S, Fadok V, Bratton D, Henson PM. Differential Roles for α(M)β2 Integrin Clustering or Activation in the Control of Apoptosis Via Regulation of Akt and ERK Survival Mechanisms. J Cell Biol (2000) 151:1305–20. doi: 10.1083/jcb.151.6.1305
101. Pluskota E, Soloviev DA, Szpak D, Weber C, Plow EF. Neutrophil Apoptosis: Selective Regulation by Different Ligands of Integrin α M β 2. J Immunol (2008) 181:3609–19. doi: 10.4049/jimmunol.181.5.3609
102. Sohn JH, Bora PS, Suk HJ, Molina H, Kaplan HJ, Bora NS. Tolerance is Dependent on Complement C3 Fragment iC3b Binding to Antigen-Presenting Cells. Nat Med (2003) 9:206–12. doi: 10.1038/nm814
103. Ricklin D, Reis ES, Lambris JD. Complement in Disease: A Defence System Turning Offensive. Nat Rev Nephrol (2016) 12:383–401. doi: 10.1038/nrneph.2016.70
104. Wright SD, Rao PE, Van Voorhis WC, Craigmyle LS, Iida K, Talle MA, et al. Identification of the C3bi Receptor of Human Monocytes and Macrophages by Using Monoclonal Antibodies. Proc Natl Acad Sci (1983) 80:5699–703. doi: 10.1073/pnas.80.18.5699
105. Eskan MA, Jotwani R, Abe T, Chmelar J, Lim J-H, Liang S, et al. The Leukocyte Integrin Antagonist Del-1 Inhibits IL-17-Mediated Inflammatory Bone Loss. Nat Immunol (2012) 13:465–73. doi: 10.1038/ni.2260
106. Ehlenberger AG, Nussenzweig V. The Role of Membrane Recepotrs for C3b and C3d in Phagocytosis. J Exp Med (1977) 145:357–71. doi: 10.1084/jem.145.2.357
107. Annenkov A, Ortlepp S, Hogg N. The β2 Integrin Mac-1 But Not P150,95 Associates With Fcγriia. Eur J Immunol (1996) 26:207–12. doi: 10.1002/eji.1830260132
108. Zhou M, Todd RF, van de Winkel JGJ, Petty HR. Cocapping of the Leukoadhesin Molecules Complement Receptor Type 3 and Lymphocyte Function-Associated Antigen-1 With Fc Gamma Receptor III on Human Neutrophils. Possible Role of Lectin-Like Interactions. J Immunol (1993) 150:3030–41. doi: 10.4049/jimmunol.1300235
109. Majima T, Ohashi Y, Nagatomi R, Iizuka A, Konno T. Defective Mononuclear Cell Antibody-Dependent Cellular Cytotoxicity (ADCC) in Patients With Leukocyte Adhesion Deficiency Emphasizing on Different Cd11/Cd18 Requirement of RcgRI Versus FcgRII in ADCC. Cell Immunol (1993) 148:385–96. doi: 10.1006/cimm.1993.1120
110. Van Spriel AB, Leusen JHW, Van Egmond M, Dijkman HBPM, Assmann KJM, Mayadas TN, et al. Mac-1 ( CD11b / CD18 ) is Essential for Fc Receptor – Mediated Neutrophil Cytotoxicity and Immunologic Synapse Formation. Blood (2001) 97:2478–86. doi: 10.1182/blood.V97.8.2478
111. Galon J, Gauchat J, Mazieres N, Spagnoli R, Storkus W, Lötze M, et al. Soluble Fcgamma Receptor Type Iii (Fcgammariii, CD16) Triggers Cell Activation Through Interaction With Complement Receptors. J Immunol (1996) 157:1184–92.
112. Poo H, Krauss JC, Mayo-Bond L, Todd RF, Petty HR. Interaction of Fcγ Receptor Type IIIB With Complement Receptor Type 3 in Fibroblast Transfectants: Evidence From Lateral Diffusion and Resonance Energy Transfer Studies. J Mol Biol (1995) 247:597–603. doi: 10.1016/S0022-2836(05)80141-X
113. Lukácsi S, Mácsik-Valent B, Nagy-Baló Z, Kovács KG, Kliment K, Bajtay Z, et al. Utilization of Complement Receptors in Immune Cell–Microbe Interaction. FEBS Lett (2020) 594:2695–713. doi: 10.1002/1873-3468.13743
114. Rieu P, Ueda T, Haruta I, Sharma CP, Arnaout MA. The A-domain of β2 Integrin CR3 (CD11b/CD18) is a Receptor for the Hookworm-Derived Neutrophil Adhesion Inhibitor Nif. J Cell Biol (1994) 127:2081–91. doi: 10.1083/jcb.127.6.2081
115. Muchowski PJ, Chang ER, Soule HR, Moyle M, Zhang L, Plow EF. Functional Interaction Between the Integrin Antagonist Neutrophil Inhibitory Factor and the I Domain of CD11b/CD18. J Biol Chem (1994) 269:26419–23. doi: 10.1016/S0021-9258(18)47210-9
116. Zhang L, Plow EF. Identification and Reconstruction of the Binding Site Within aMb2 for a Specific and High Affinity Ligand, Nif. J Biol Chem (1997) 272:17558–64. doi: 10.1074/jbc.272.28.17558
117. Wright SD, Jong MTC. Adhesion-Promoting Receptors on Human Macrophages Recognize Escherichia Colt by Binding to Lipopolysaccharide. J Exp Med (1986) 164:1876–88. doi: 10.1084/jem.164.6.1876
118. Hmama Z, Mey A, Normier G, Binz H, Revillard JP. CD14 and CD11b Mediate Serum-Independent Binding to Human Monocytes of an Acylpolygalactoside Isolated From Klebsiella Pneumoniae. Infect Immun (1994) 62:1520–7. doi: 10.1128/iai.62.5.1520-1527.1994
119. Ross GD, Cain JA, Lachmann PJ. Membrane Complement Receptor Type Three ( CR3 ) has Lectin-Like Properties Analogous to Bovine Conglutinin as Functions as a Receptor for Zymosan and Rabbit Erythrocytes as Well as a Receptor for Ic3b. J Immunol (1985) 134:3307–15.
120. Bose N, Chan ASH, Guerrero F, Maristany CM, Qiu X, Walsh RM, et al. Binding of Soluble Yeast β-Glucan to Human Neutrophils and Monocytes is Complement-Dependent. Front Immunol (2013) 4:230. doi: 10.3389/fimmu.2013.00230
121. van Bruggen R, Drewniak A, Jansen M, van Houdt M, Roos D, Chapel H, et al. Complement Receptor 3, Not Dectin-1, is the Major Receptor on Human Neutrophils for β-Glucan-Bearing Particles. Mol Immunol (2009) 47:575–81. doi: 10.1016/j.molimm.2009.09.018
122. Zhou H, Liao J, Aloor J, Nie H, Wilson BC, Fessler MB, et al. Cd11b/Cd18 (Mac-1) Is a Novel Surface Receptor for Extracellular Double-Stranded RNA to Mediate Cellular Inflammatory Responses. J Immunol (2013) 190:115–25. doi: 10.4049/jimmunol.1202136
123. Shewell LK, Day CJ, Jen FEC, Haselhorst T, Atack JM, Reijneveld JF, et al. All Major Cholesterol-Dependent Cytolysins Use Glycans as Cellular Receptors. Sci Adv (2020) 6:1–13. doi: 10.1126/sciadv.aaz4926
124. Cywes C, Godenir NL, Hoppe HC, Scholle RR, Steyn LM, Kirsch RE, et al. Nonopsonic Binding of Mycobacterium Tuberculosis to Human Complement Receptor Type 3 Expressed in Chinese Hamster Ovary Cells. Infect Immun (1996) 64:5373–83. doi: 10.1128/iai.64.12.5373-5383.1996
125. Edwards JL, Brown EJ, Uk-Nham S, Cannon JG, Blake MS, Apicella MA. A Co-Operative Interaction Between Neisseria Gonorrhoeae and Complement Receptor 3 Mediates Infection of Primary Cervical Epithelial Cells. Cell Microbiol (2002) 4:571–84. doi: 10.1046/j.1462-5822.2002.t01-1-00215.x
126. Jennings MP, Jen FEC, Roddam LF, Apicella MA, Edwards JL. Neisseria Gonorrhoeae Pilin Glycan Contributes to CR3 Activation During Challenge of Primary Cervical Epithelial Cells. Cell Microbiol (2011) 13:885–96. doi: 10.1111/j.1462-5822.2011.01586.x
127. Antal JM, Cunningham JV, Goodrum KJ. Opsonin-Independent Phagocytosis of Group B Streptococci: Role of Complement Receptor Type Three. Infect Immun (1992) 60:1114–21. doi: 10.1128/iai.60.3.1114-1121.1992
128. Takeshita A, Murakami Y, Yamashita Y, Ishida M, Fujisawa S, Kitano S, et al. Porphyromonas Gingivalis Fimbriae Use β2 Integrin (CD11/CD18) on Mouse Peritoneal Macrophages as a Cellular Receptor, and the CD18 β Chain Plays a Functional Role in Fimbrial Signaling. Infect Immun (1998) 66:4056–60. doi: 10.1128/iai.66.9.4056-4060.1998
129. Hajishengallis G. “Subversion of Innate Immunity by Periodontopathic Bacteria Via Exploitation of Complement Receptor-3”. In: Current Topics in Complement Ii. Advances in Experimental Medicine and Biology. (2008) Adv Exp Med Biol p. 203–19.
130. Relman D, Tuomanen E, Falkow S, Golenbock DT, Saukkonen K, Wright SD. Recognition of a Bacterial Adhesin by an Integrin: Macrophage Cr3 (αmβ2, CD11b CD18) Binds Filamentous Hemagglutinin of Bordetella Pertussis. Cell (1990) 61:1375–82. doi: 10.1016/0092-8674(90)90701-F
131. Wong WS, SIMON DI, ROSOFF PM, RAO NK, CHAPMAN HA. Mechanisms of Pertussis Toxin-Induced Myelomonocytic Cell Adhesion: Role of Mac-1(CD11b/CD18) and Urokinase Receptor (Cd87). Immunology (1996) 88:90–7. doi: 10.1046/j.1365-2567.1996.d01-646.x
132. Ishibashi Y, Claus S, Relman D. Bordetella Pertussis Filamentous Hemagglutinin Interacts With a Leukocyte Signal Transduction Complex and Stimulates Bacterial Adherence to Monocyte Cr3 (Cd11b/Cd18). J Exp Med (1994) 180:1225–33. doi: 10.1016/B978-1-4557-4801-3.00232-0
133. Guermonprez P, Khelef N, Blouin E, Rieu P, Ricciardi-Castagnoli P, Guiso N, et al. The Adenylate Cyclase Toxin of Bordetella Pertussis Binds to Target Cells Via the α M β 2 Integrin (Cd11b/Cd18). J Exp Med (2001) 193:1035–44. doi: 10.1084/jem.193.9.1035
134. McGuirk P, Mills KHG. Direct Anti-Inflammatory Effect of a Bacterial Virulence Factor: IL-10-dependent Suppression of IL-12 Production by Filamentous Hemagglutinin From Bordetella Peptussis. Eur J Immunol (2000) 30:415–22. doi: 10.1002/1521-4141(200002)30:2<415::AID-IMMU415>3.0.CO;2-X
135. Oliva C, Turnbough CL, Kearney JF. Cd14-Mac-1 Interactions in Bacillus Anthracis Spore Internalization by Macrophages. Proc Natl Acad Sci (2009) 106:13957–62. doi: 10.1073/pnas.0902392106
136. Orrskog S, Rounioja S, Spadafina T, Gallotta M, Norman M, Hentrich K, et al. Pilus Adhesin Rrga Interacts With Complement Receptor 3 , Thereby. MBio (2013) 4:1–12. doi: 10.1128/mBio.00535-12.Editor
137. Hoang KV, Rajaram MVS, Curry HM, Gavrilin MA, Wewers MD, Schlesinger LS. Complement Receptor 3-Mediated Inhibition of Inflammasome Priming by Ras Gtpase-Activating Protein During Francisella Tularensis Phagocytosis by Human Mononuclear Phagocytes. Front Immunol (2018) 9:561. doi: 10.3389/fimmu.2018.00561
138. Garcia RC, Murgia R, Cinco M. Complement Receptor 3 Binds the Borrelia Burgdorferi Outer Surface Proteins OspA and OspB in an iC3b-independent Manner. Infect Immun (2005) 73:6138–42. doi: 10.1128/IAI.73.9.6138-6142.2005
139. Blackwell JM. Receptors and Recognition Mechanisms of Leishmania Species. Trans R Soc Trop Med Hyg (1985) 19:606–12. doi: 10.1016/0035-9203(85)90166-X
140. Russell DG. Complement Receptor Type 3 (CR3) Binds to an Arg-Gly-Asp-containing Region of the Major Surface Glycoprotein, gp63, of Leishmania Promastigotes. J Exp Med (1988) 168:279–92. doi: 10.1084/jem.168.1.279
141. Soteriadou KP, Remoundos MS, Katsikas MC, Tzinia AK, Tsikaris V, Sakarellos C, et al. The Ser-Arg-Tyr-Asp Region of the Major Surface Glycoprotein of Leishmania Mimics the Arg-Gly-Asp-Ser Cell Attachment Region of Fibronectin. J Biol Chem (1992) 267:13980–5. doi: 10.1016/S0021-9258(19)49666-X
142. Tjomsland V, Ellegård R, Che K, Hinkula J, Lifson JD, Larsson M. Complement Opsonization of HIV-1 Enhances the Uptake by Dendritic Cells and Involves the Endocytic Lectin and Integrin Receptor Families. PloS One (2011) 6:e23542. doi: 10.1371/journal.pone.0023542
143. Ellegård R, Crisci E, Burgener A, Sjöwall C, Birse K, Westmacott G, et al. Complement Opsonization of HIV-1 Results in Decreased Antiviral and Inflammatory Responses in Immature Dendritic Cells Via CR3. J Immunol (2014) 193:4590–601. doi: 10.4049/jimmunol.1401781
144. Crisci E, Ellegård R, Nyström S, Rondahl E, Serrander L, Bergström T, et al. Complement Opsonization Promotes Herpes Simplex Virus 2 Infection of Human Dendritic Cells. J Virol (2016) 90:4939–50. doi: 10.1128/jvi.00224-16
145. Raftery MJ, Lalwani P, Krautkrämer E, Peters T, Scharffetter-Kochanek K, Krüger R, et al. β2 Integrin Mediates Hantavirus-Induced Release of Neutrophil Extracellular Traps. J Exp Med (2014) 211:1485–97. doi: 10.1084/jem.20131092
146. Forsyth CB, Mathews HL. Lymphocytes Utilize CD11b/CD18 for Adhesion to Candida Albicans. Cell Immunol (1996) 170:91–100. doi: 10.1006/cimm.1996.0138
147. Hostetter MK. Adhesins and Ligands Involved in the Interaction of Candida Spp. With Epithelial and Endothelial Surfaces. Clin Microbiol Rev (1994) 7:29–42. doi: 10.1128/CMR.7.1.29
148. Newman SL, Chaturvedi S, Klein BS. The WI-1 Antigen of Blastomyces Dermatitidis Yeasts Mediates Binding to Human Macrophage CD11b/CD18 (CR3) and CD14. J Immunol (1995) 154:753–61.
149. Bullock WE, Wright SD. Role or the Adherence-Promoting Receptors, CR3, Lfa-1, and p150,95, in Binding of Histoplasma Capsulatum by Human Macrophages. J Exp Med (1987) 165:195–210. doi: 10.1084/jem.165.1.195
150. Moyle M, Foster DL, McGrath DE, Brown SM, Laroche Y, De Meutter J, et al. A Hookworm Glycoprotein That Inhibits Neutrophil Function is a Ligand of the Integrin Cd11b/Cd18. J Biol Chem (1994) 269:10008–15. doi: 10.1016/S0021-9258(17)36982-X
151. Czop JK, Austen KF. A Beta-Glucan Inhibitable Receptor on Human Monocytes: its Identity With the Phagocytic Receptor for Particulate Activators of the Alternative Complement Pathway. J Immunol (1985) 134:2588–93.
152. Vetvicka V, Vannucci L, Sima P, Richter J. Beta Glucan: Supplement or Drug? From Laboratory to Clinical Trials. Molecules (2019) 24:1251. doi: 10.3390/molecules24071251
153. Biothera Study of Imprime PGG and Pembrolizumab in Advanced Melanoma and Triple Negative Breast Cancer. Nct02981303. (2019).
154. Biothera. Pembrolizumab + Imprime PGG for Metastatic non-Small Cell Lung Cancer After Progression on First-Line Therapy: Big Ten Cancer Research Consortium Btcrc-LUN15-017. Nct03003468. (2019).
155. Biothera Efficacy/Safety of Imprime Pgg With Cetuximab & Paclitaxel/Carboplatin Therapy in Pts With Untreated Advanced Non-Small Cell Lung Cancer. Nct00874848. (2016).
156. Biothera Efficacy and Safety Study of Imprime Pgg With Cetuximab in Subjects With Stage Iv KRAS-Mutated Colorectal Cancer. Nct00912327. (2012).
157. Schlesinger LS, Bellinger-Kawahara CG, Payne NR, Horwitz MA. Phagocytosis of Mycobacterium Tuberculosis is Mediated by Human Monocyte Complement Receptors and Complement Component C3. J Immunol (1990) 144:2771–80.
158. Hirsch CS, Ellner JJ, Russell DG, Rich EA. Complement Receptor-Mediated Uptake and Tumor Necrosis Factor-Alpha-Mediated Growth Inhibition of Mycobacterium Tuberculosis by Human Alveolar Macrophages. J Immunol (1994) 152:743–53.
159. Cywes C, Hoppe HC, Daffé M, Ehlers MRW. Nonopsonic Binding of Mycobacterium Tuberculosis to Complement Receptor Type 3 Is Mediated by Capsular Polysaccharides and Is Strain Dependent. Infect Immun (1997) 65:4258–66. doi: 10.1128/IAI.65.10.4258-4266.1997
160. Edwards JL, Brown EJ, Ault KA, Apicella MA. The Role of Complement Receptor 3 (CR3) in Neisseria Gonorrhoeae Infection of Human Cervical Epithelia. Cell Microbiol (2001) 3:611–22. doi: 10.1046/j.1462-5822.2001.00140.x
161. Edwards JL, Apicella MA. I-Domain-Containing Integrins Serve as Pilus Receptors for Neisseria Gonorrhoeae Adherence to Human Epithelial Cells. Cell Microbiol (2005) 7:1197–211. doi: 10.1111/j.1462-5822.2005.00547.x
162. Oliva CR, Swiecki MK, Griguer CE, Lisanby MW, Bullard DC, Turnbough CL, et al. The Integrin Mac-1 (CR3) Mediates Internalization and Directs Bacillus Anthracis Spores Into Professional Phagocytes. Proc Natl Acad Sci (2008) 105:1261–6. doi: 10.1073/pnas.0709321105
163. Blackwell JM, Ezekowitz AB, Roberts MB, Channon JY, Sim RB, Gordon S. Macrophage Complement and Lectin-Like Receptors Bind Leishmania in the Absence of Serum. J Exp Med (1985) 162:324–31. doi: 10.1084/jem.162.1.324
164. Forsyth CB, Mathews HL. Lymphocyte Adhesion to Candida Albicans. Infect Immun (2002) 70:517–27. doi: 10.1128/IAI.70.2.517-527.2002
165. Davis GE. The Mac-l and Pl50,95 Beta2 Integrins Bind Denatured Proteins to Mediate Leukocyte Ceil-Substrate Adhesion. Exp Cell Res (1992) 200:242–52. doi: 10.1016/0014-4827(92)90170-D
166. Godek ML, Michel R, Chamberlain LM, Castner DG, Grainger DW. Adsorbed Serum Albumin is Permissive to Macrophage Attachment to Perfluorocarbon Polymer Surfaces in Culture. J BioMed Mater Res Part A (2009) 88:503–19. doi: 10.1002/jbm.a.31886
167. Brevig T, Holst B, Ademovic Z, Rozlosnik N, Røhrmann JH, Larsen NB, et al. The Recognition of Adsorbed and Denatured Proteins of Different Topographies by β 2 Integrins and Effects on Leukocyte Adhesion and Activation. Biomaterials (2005) 26:3039–53. doi: 10.1016/j.biomaterials.2004.09.006
168. Benimetskaya L, Loike JD, Khaled Z, Loike G, Silverstein SC, Cao L, et al. Mac-1 (CD1lb/CD18) is an Oligodeoxynucleotide-Binding Protein. Nat Med (1997) 3:414–20. doi: 10.1038/nm0497-414
169. Rotshenker S. Microglia and Macrophage Activation and the Regulation of Complement-Receptor-3 (CR3/MAC-1)-Mediated Myelin Phagocytosis in Injury and Disease. J Mol Neurosci (2003) 21:65–72. doi: 10.1385/JMN:21:1:65
170. DeJong B, Smith M. A Role for Complement in Phagocytosis of Myelin. Neurochem Res (1997) 22:491–8. doi: 10.1023/a:1027372129989
171. Stapulionis R, Pinto Oliveira CL, Gjelstrup MC, Pedersen JS, Hokland ME, Hoffmann SV, et al. Structural Insight Into the Function of Myelin Basic Protein as a Ligand for Integrin αmβ2. J Immunol (2008) 180:3946–56. doi: 10.4049/jimmunol.180.6.3946
172. Fu H, Liu B, Frost JL, Hong S, Jin M, Ostaszewski B, et al. Complement Component C3 and Complement Receptor Type 3 Contribute to the Phagocytosis and Clearance of Fibrillar Aβ by Microglia. Glia (2012) 60:993–1003. doi: 10.1002/glia.22331
173. Goodwin JL, Kehrli ME, Uemura E. Integrin Mac-1 and β-Amyloid in Microglial Release of Nitric Oxide. Brain Res (1997) 768:279–86. doi: 10.1016/S0006-8993(97)00653-7
174. Czirr E, Castello NA, Mosher KI, Castellano JM, Hinkson IV, Lucin KM, et al. Microglial Complement Receptor 3 Regulates Brain Aβ Levels Through Secreted Proteolytic Activity. J Exp Med (2017) 214:1081–92. doi: 10.1084/jem.20162011
175. Choucair-Jaafar N, Laporte V, Levy R, Poindron P, Lombard Y, Gies JP. Complement Receptor 3 (CD11b/CD18) is Implicated in the Elimination of β-Amyloid Peptides. Fundam Clin Pharmacol (2011) 25:115–22. doi: 10.1111/j.1472-8206.2010.00811.x
176. Zabel M, Schrag M, Crofton A, Tung S, Beaufond P, Van Ornam J, et al. A Shift in Microglial β-Amyloid Binding in Alzheimer’s Disease is Associated With Cerebral Amyloid Angiopathy. Brain Pathol (2013) 23:390–401. doi: 10.1111/bpa.12005
177. Hou L, Wang K, Zhang C, Sun F, Che Y, Zhao X, et al. Complement Receptor 3 Mediates Nadph Oxidase Activation and Dopaminergic Neurodegeneration Through a Src-Erk-dependent Pathway. Redox Biol (2018) 14:250–60. doi: 10.1016/j.redox.2017.09.017
178. Hou L, Bao X, Zang C, Yang H, Sun F, Che Y, et al. Integrin CD11b Mediates α-Synuclein-Induced Activation of NADPH Oxidase Through a Rho-dependent Pathway. Redox Biol (2018) 14:600–8. doi: 10.1016/j.redox.2017.11.010
179. Zhang W, Dallas, Shannonhang D, Guo J-P, Pang H, Wilson B, et al. Microglial PHOX and Mac-1 are Essential to the Enhanced Dopaminergic Neurodegeneration Elicited by A30P and A53T Mutant Alpha-Synuclein. Glia (2007) 55:1178–88. doi: 10.1002/glia
180. Malinovskaya NA, Salmina AB, Prokopenko SV. The coexpression of CD157/CD11b/CD18 in an experimental model of Parkinson’s disease. Neurochem. J. (2015) 9:279–283. doi: 10.1134/S181971241504011X
181. Zhang C, Hou L, Yang J, Che Y, Sun F, Li H, et al. 2,5-Hexanedione Induces Dopaminergic Neurodegeneration Through Integrin αmβ2/Nadph Oxidase Axis-Mediated Microglial Activation Article. Cell Death Dis (2018) 9:60. doi: 10.1038/s41419-017-0091-7
182. Levesque S, Taetzsch T, Lull ME, Johnson JA, McGraw C, Block ML. The Role of MAC1 in Diesel Exhaust Particle-Induced Microglial Activation and Loss of Dopaminergic Neuron Function. J Neurochem (2013) 125:756–65. doi: 10.1111/jnc.12231
183. Gao H-M, Zhou H, Zhang F, Wilson BC, Kam W, Hong J-S. Hmgb1 Acts on Microglia Mac1 to Mediate Chronic Neuroinflammation That Drives Progressive Neurodegeneration. J Neurosci (2011) 31:1081–92. doi: 10.1523/JNEUROSCI.3732-10.2011
184. Bullard DC, Hu X, Schoeb TR, Axtell RC, Raman C, Barnum SR. Critical Requirement of CD11b (Mac-1) on T Cells and Accessory Cells for Development of Experimental Autoimmune Encephalomyelitis. J Immunol (2005) 175:6327–33. doi: 10.4049/jimmunol.175.10.6327
185. Smith CW, Marlin SD, Rothlein R, Toman C, Anderson DC. Cooperative Interactions of LFA-1 and Mac-1 With Intracellular Adhesion Molecule-1 in Facilitating Adherence and Transendothelial Migration of Human Neutrophils In Vitro. J Clin Invest (1989) 83:2008–17. doi: 10.1172/JCI114111
186. Diamond MS, Staunton DE, Marlin SD, Springer TA. Binding of the Integrin Mac-1 (CD11b/CD18) to the Third Immunoglobulin-Like Domain of ICAM-1 (CD54) and its Regulation by Glycosylation. Cell (1991) 65:961–71. doi: 10.1016/0092-8674(91)90548-D
187. Diamond MS, Springer TA. A Subpopulation of Mac-1 (Cd11b/Cd18) Molecules Mediates Neutrophil Adhesion to ICAM-1 and Fibrinogen. J Cell Biol (1993) 120:545–56. doi: 10.1083/jcb.120.2.545
188. Zhou L, Lee DHS, Plescia J, Lau CY, Altieri DC. Differential Ligand Binding Specificities of Recombinant Cd11b/Cd18 Integrin I-Domain. J Biol Chem (1994) 269:17075–9. doi: 10.1016/S0021-9258(17)32522-X
189. McCleverty CJ, Liddington RC. Engineered Allosteric Mutants of the Integrin Alphambeta2 I Domain: Structural and Functional Studies. Biochem J (2003) 372:121–7. doi: 10.1042/bj20021273
190. Xie J, Li R, Kotovuori P, Vermot-Desroches C, Wijdenes J, Arnaout MA, et al. Intercellular Adhesion Molecule-2 (Cd102) Binds to the Leukocyte Integrin CD11b/CD18 Through the A Domain. J Immunol (1995) 155:3619–28.
191. Li R, Xie J, Kantor C, Koistinen V, Altieri DC, Nortamo P, et al. A Peptide Derived From the Intercellular Adhesion Molecule-2 Regulates the Avidity of the Leukocyte Integrins CD11b/CD18 and CD11c/CD18. J Cell Biol (1995) 129:1143–53. doi: 10.1083/jcb.129.4.1143
192. Hermand P, Huet M, Callebaut I, Gane P, Ihanus E, Gahmberg CG, et al. Binding Sites of Leukocyte β2 Integrins (Lfa-1, Mac-1) on the Human Icam-4/Lw Blood Group Protein. J Biol Chem (2000) 275:26002–10. doi: 10.1074/jbc.M002823200
193. Santoso S, Sachs UJH, Kroll H, Linder M, Ruf A, Preissner KT, et al. The Junctional Adhesion Molecule 3 (Jam-3) on Human Platelets is a Counterreceptor for the Leukocyte Integrin Mac-1. J Exp Med (2002) 196:679–91. doi: 10.1084/jem.20020267
194. Zen K, Babbin BA, Liu Y, Whelan JB, Nusrat A, Parkos CA. Jam-C Is a Component of Desmosomes and a Ligand for CD11b/CD18-mediated Neutrophil Transepithelial Migration. Mol Biol Cell (2004) 15:3926–37. doi: 10.1091/mbc.E04
195. Heinzmann D, Noethel M, Ungern-Sternberg SV, Mitroulis I, Gawaz M, Chavakis T, et al. CD147 is a Novel Interaction Partner of Integrin αmβ2 Mediating Leukocyte and Platelet Adhesion. Biomolecules (2020) 10:7–12. doi: 10.3390/biom10040541
196. Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, et al. The Pattern Recognition Receptor (Rage) Is a Counterreceptor for Leukocyte Integrins: A Novel Pathway for Inflammatory Cell Recruitment. J Exp Med (2003) 198:15007–1515. doi: 10.1084/jem.20030800
197. Wetzel A, Wetzig T, Haustein UF, Sticherling M, Anderegg U, Simon JC, et al. Increased Neutrophil Adherence in Psoriasis: Role of the Human Endothelial Cell Receptor Thy-1 (Cd90). J Invest Dermatol (2006) 126:441–52. doi: 10.1038/sj.jid.5700072
198. Wetzel A, Chavakis T, Preissner KT, Sticherling M, Haustein U-F, Anderegg U, et al. Human Thy-1 (CD90) on Activated Endothelial Cells is a Counterreceptor for the Leukocyte Integrin Mac-1 (Cd11b/Cd18). J Immunol (2004) 172:3850–9. doi: 10.4049/jimmunol.172.6.3850
199. Podolnikova NP, Hlavackova M, Wu Y, Yakubenko VP, Faust J, Balabiyev A, et al. Interaction Between the Integrin Mac-1 and Signal Regulatory Protein α (Sirpα) Mediates Fusion in Heterologous Cells. J Biol Chem (2019) 294:7833–49. doi: 10.1074/jbc.RA118.006314
200. Wolf D, Hohmann JD, Wiedemann A, Bledzka K, Blankenbach H, Marchini T, et al. Binding of CD40L to Mac-1’s I-Domain Involves the EQLKKSKTL Motif and Mediates Leukocyte Recruitment and Atherosclerosis-But Does Not Affect Immunity and Thrombosis in Mice. Circ Res (2011) 109:1269–79. doi: 10.1161/CIRCRESAHA.111.247684
201. Johansson MW, Patarroyo M, Öberg F, Siegbahn A, Nilsson K. Myeloperoxidase Mediates Cell Adhesion Via the Amb2 Integrin (Mac-1, Cd11b/Cd18). J Cell Sci (1997) 110:1133–9.
202. Cai TQ. Human Leukocyte Elastase is an Endogenous Ligand for the Integrin Cr3 (Cd11b/Cd18, Mac-1, Alpha M Beta 2) and Modulates Polymorphonuclear Leukocyte Adhesion. J Exp Med (1996) 184:1213–23. doi: 10.1084/jem.184.4.1213
203. Stefanidakis M, Ruohtula T, Borregaard N, Gahmberg CG, Koivunen E. Intracellular and Cell Surface Localization of a Complex Between α M β 2 Integrin and Promatrix Metalloproteinase-9 Progelatinase in Neutrophils. J Immunol (2004) 172:7060–8. doi: 10.4049/jimmunol.172.11.7060
204. Stefanidakis M, Björklund M, Ihanus E, Gahmberg CG, Koivunen E. Identification of a Negatively Charged Peptide Motif Within the Catalytic Domain of Progelatinases That Mediates Binding to Leukocyte β2 Integrins. J Biol Chem (2003) 278:34674–84. doi: 10.1074/jbc.M302288200
205. Shen D, Podolnikova NP, Yakubenko VP, Ardell CL, Balabiyev A, Ugarova TP, et al. Pleiotrophin, a Multifunctional Cytokine and Growth Factor, Induces Leukocyte Responses Through the Integrin Mac-1. J Biol Chem (2017) 292:18848–61. doi: 10.1074/jbc.M116.773713
206. Podolnikova NP, Brothwell JA, Ugarova TP. The Opioid Peptide Dynorphin A Induces Leukocyte Responses Via Integrin Mac-1 (αmβ2 , Cd11b/Cd18). Mol Pain (2015) 11:33. doi: 10.1186/s12990-015-0027-0
207. Xiao X, Murk DD, Cheng CY. Intercellular Adhesion Molecules (Icams) and Spermatogenesis. Hum Reprod Update (2013) 19:167–86. doi: 10.1093/humupd/dms049
208. Bailly P, Tontti E, Hermand P, Cartron J-P, Gahmberg CG. The Red Cell LW Blood Group Protein is an Intercellular Adhesion Molecule Which Binds to CD11/CD18 Leukocyte Integrins. Eur J Immunol (1995) 25:3316–20. doi: 10.1002/eji.1830251217
209. Kummer D, Ebnet K. Junctional Adhesion Molecules (Jams): The Jam-Integrin Connection. Cells (2018) 7:25. doi: 10.3390/cells7040025
210. Podolnikova NP, Kushchayeva YS, Wu YF, Faust J, Ugarova TP. The Role of Integrins αmβ2 (Mac-1, CD11b/CD18) and αdβ2 (CD11d/CD18) in Macrophage Fusion. Am J Pathol (2016) 186:2105–16. doi: 10.1016/j.ajpath.2016.04.001
211. André P, Srinivasa Prasad KS, Denis CV, He M, Papalia JM, Hynes RO, et al. Cd40l Stabilizes Arterial Thrombi by a β3 Integrin-Dependent Mechanism. Nat Med (2002) 8:247–52. doi: 10.1038/nm0302-247
212. Zirlik A, Maier C, Gerdes N, MacFarlane L, Soosairajah J, Bavendiek U, et al. Cd40 Ligand Mediates Inflammation Independently of CD40 by Interaction With Mac-1. Circulation (2007) 115:1571–80. doi: 10.1161/CIRCULATIONAHA.106.683201
213. Zen K, Guo YL, Li LM, Bian Z, Zhang CY, Liu Y. Cleavage of the CD11b Extracellular Domain by the Leukocyte Serprocidins is Critical for Neutrophil Detachment During Chemotaxis. Blood (2011) 117:4885–94. doi: 10.1182/blood-2010-05-287722
214. Pakianathan D. Extracellular Matrix Proteins and Leukocyte Function. J Leukoc Biol (1995) 57:699–702. doi: 10.1002/jlb.57.5.699a
215. Thompson HL, Matsushima K. Human Polymorphonuclear Leucocytes Stimulated by Tumour Necrosis Factor-Alpha Show Increased Adherence to Extracellular Matrix Proteins Which is Mediated Via the CD11b/18 Complex. Clin Exp Immunol (1992) 90:280–5. doi: 10.1111/j.1365-2249.1992.tb07943.x
216. Nathan C, Srimal S, Farber C, Sanchez E, Kabbash L, Asch A, et al. Cytokine-Induced Respiratory Burst of Human Neutrophils: Dependence on Extracellular Matrix Proteins and CD11/CD18 Integrins. J Cell Biol (1989) 109:1341–9. doi: 10.1083/jcb.109.3.1341
217. Kanse SM, Matz RL, Preissner KT, Peter K. Promotion of Leukocyte Adhesion by a Novel Interaction Between Vitronectin and the Beta2 Integrin Mac-1 (alphaMbeta2, Cd11b/Cd18). Atherioscler Thromb Vasc Biol (2004) 24:2251–6. doi: 10.1161/01.ATV.0000146529.68729.8b
218. Wright SD, Weitz JI, Huang AJ, Levin SM, Silverstein SC, Loike JD. Complement Receptor Type Three (CD11b/CD18) of Human Polymorphonuclear Leukocytes Recognizes Fibrinogen. Proc Natl Acad Sci (1988) 85:7734–8. doi: 10.1073/pnas.85.20.7734
219. Altieri DC, Bader R, Mannucci PM, Edgington TS. Oligospecificity of the Cellular Adhesion Receptor Mac-1 Encompasses an Inducible Recognition Specificity for Fibrinogen. J Cell Biol (1988) 107:1893–900. doi: 10.1083/jcb.107.5.1893
220. Altieri DC, Plescia J, Plow EF. The Structural Motif Glycine 190-Valine 202 of the Fibrinogen Y Chain Interacts With CD11b/CD18 Integrin (Amb2, Mac-1) and Promotes Leukocyte Adhesion. J Biol Chem (1993) 268:1847–53. doi: 10.1016/S0021-9258(18)53932-6
221. Ugarova TP, Solovjov DA, Zhang L, Loukinov DI, Yee VC, Medved LV, et al. Identification of a Novel Recognition Sequence for Integrin aMb2 Within the Y-Chain of Fibrinogen. J Biol Chem (1998) 273:22519–27. doi: 10.1074/jbc.273.35.22519
222. Ugarova TP, Lishko VK, Podolnikova NP, Okumura N, Merkulov SM, Yakubenko VP, et al. Sequence γ377-395(P2), But Not γ190-202(P1), is the Binding Site for the αmi-Domain of Integrin αmβ2 in the αc-Domain of Fibrinogen. Biochemistry (2003) 42:9365–73. doi: 10.1021/bi034057k
223. Lishko VK, Kudryk B, Yakubenko VP, Yee VC, Ugarova TP. Regulated Unmasking of the Cryptic Binding Site for Integrin αmβ2 in the γc-Domain of Fibrinogen. Biochemistry (2002) 41:12942–51. doi: 10.1021/bi026324c
224. Yakubenko VP, Solovjov DA, Zhang L, Yee VC, Plow EF, Ugarova TP. Identification of the Binding Site for Fibrinogen Recognition Peptide γ383-395 Within the αmi-Domain of Integrin α Mβ2. J Biol Chem (2001) 276:13995–4003. doi: 10.1074/jbc.M010174200
225. Lishko VK, Yakubenko VP, Hertzberg KM, Grieninger G, Ugarova TP. The Alternatively Spliced Alpha(E)C Domain of Human fibrinogen-420 is a Novel Ligand for Leukocyte Integrins Alpha(M)Beta(2) and Alpha(X)Beta(2). Blood (2001) 98:2448–55. doi: 10.1182/blood.V98.8.2448
226. Flick MJ, Du XL, Witte DP, Jiroušková M, Soloviev DA, Busuttil SJ, et al. Leukocyte Engagement of Fibrin(Ogen) Via the Integrin Receptor αmβ2/Mac-1 is Critical for Host Inflammatory Response In Vivo. J Clin Invest (2004) 113:1596–606. doi: 10.1172/JCI20741
227. Nasimuzzaman M, Arumugam PI, Mullins ES, James JM, VandenHeuvel K, Narciso MG, et al. Elimination of the Fibrinogen Integrin α M β 2 -Binding Motif Improves Renal Pathology in Mice With Sickle Cell Anemia. Blood Adv (2019) 3:1519–32. doi: 10.1182/bloodadvances.2019032342
228. Ehlers R, Ustinov V, Chen Z, Zhang X, Rao R, Luscinskas FW, et al. Targeting Platelet-Leukocyte Interactions: Identification of the Integrin Mac-1 Binding Site for the Platelet Counter Receptor Glycoprotein Ibα. J Exp Med (2003) 198:1077–88. doi: 10.1084/jem.20022181
229. Morgan J, Saleem M, Ng R, Armstrong C, Wong SS, Caulton SG, et al. Structural Basis of the Leukocyte Integrin Mac-1 I-Domain Interactions With the Platelet Glycoprotein Ib. Blood Adv (2019) 3:1450–9. doi: 10.1182/bloodadvances.2018027011
230. Gustafson EJ, Lukasiewicz H, Wachtfogel YT, Norton KJ, Schmaier AH, Niewiarowski S, et al. High Molecular Weight Kininogen Inhibits Fibrinogen Binding to Cytoadhesins of Neutrophils and Platelets. J Cell Biol (1989) 109:377–87. doi: 10.1083/jcb.109.1.377
231. Houimel M, Mazzucchelli L. Random Phage-Epitope Library Based Identification of a Peptide Antagonist of Mac-1 β2 Integrin Ligand Binding. Matrix Biol (2012) 31:66–77. doi: 10.1016/j.matbio.2011.10.003
232. Altieri DC, Agbanyo FR, Plescia J, Ginsberg MH, Edgington TS, Plow EF. A Unique Recognition Site Mediates the Interaction of Fibrinogen With the Leukocyte Integrin Mac-1 (Cd11b/Cd18). J Biol Chem (1990) 265:12119–22. doi: 10.1016/S0021-9258(19)38316-4
233. Lishko VK, Podolnikova NP, Yakubenko VP, Yakovlev S, Medved L, Yadav SP, et al. Multiple Binding Sites in Fibrinogen for Integrin αmβ 2 (Mac-1). J Biol Chem (2004) 279:44897–906. doi: 10.1074/jbc.M408012200
234. Yakovlev S, Zhang L, Ugarova T, Medved L. Interaction of Fibrin(Ogen) With Leukocyte Receptor α Mβ2 (Mac-1): Further Characterization and Identification of a Novel Binding Region Within the Central Domain of the Fibrinogen γ-Module. Biochemistry (2005) 44:617–26. doi: 10.1021/bi048266w
235. Wang Z, Thinn AMM, Zhu J. A Pivotal Role for a Conserved Bulky Residue At the α1-Helix of the αi Integrin Domain in Ligand Binding. J Biol Chem (2017) 292:20756–68. doi: 10.1074/jbc.M117.790519
236. Vidal B, Ardite E, Suelves M, Ruiz-Bonilla V, Janué A, Flick MJ, et al. Amelioration of Duchenne Muscular Dystrophy in Mdx Mice by Elimination of Matrix-Associated Fibrin-Driven Inflammation Coupled to the αmβ2 Leukocyte Integrin Receptor. Hum Mol Genet (2012) 21:1989–2004. doi: 10.1093/hmg/dds012
237. Perez RL, Ritzenthaler JD, Roman J. Transcriptional Regulation of the Interleukin-1β Promoter Via Fibrinogen Engagement of the CD18 Integrin Receptor. J Respir Cell Mol Biol (1999) 20:1059–66. doi: 10.1165/ajrcmb.20.5.3281
238. Fan S-T, Edgington TS. Lntegrin Regulation of Leukocyte Inflammatory Functions Cd11b/Cd18 Enhancement of the Tumor Necrosis Factor-alpha Responses of Monocytes. J Immunol (1993) 150:2972–80.
239. Lishko VK, Yakubenko VP, Ugarova TP. The Interplay Between Integrins αmβ2 and α5β1 During Cell Migration to Fibronectin. Exp Cell Res (2003) 283:116–26. doi: 10.1016/S0014-4827(02)00024-1
240. Walzog B, Schuppan D, Heimpel C, Hafezi-Moghadam A, Gaehtgens P, Ley K. The Leukocyte Integrin MAc-1 (Cd11b/Cd18) Contributes to Binding of Human Granulocytes to Collagen. Exp Cell Res (1995) 218:28–38. doi: 10.1006/excr.1995.1127
241. Lahti M, Heino J, Käpylä J. Leukocyte Integrins αlβ2, αmβ2 and αxβ2 as Collagen Receptors - Receptor Activation and Recognition of GFOGER Motif. Int J Biochem Cell Biol (2013) 45:1204–11. doi: 10.1016/j.biocel.2013.03.016
242. Bohnsack JF, Akiyama SK, Damsky CH, Knapej WA, Zimmermansii GA. Human Neutrophil Adherence to Laminin in Vitro: Evidence for a Distinct Neutrophil Integrin Receptor for Laminin. J Exp Med (1990) 171:1221–37. doi: 10.1084/jem.171.4.1221
243. Lee S, Bowrin K, Hamad AR, Chakravarti S. Extracellular Matrix Lumican Deposited on the Surface of Neutrophils Promotes Migration by Binding to β2 Integrin. J Biol Chem (2009) 284:23662–9. doi: 10.1074/jbc.M109.026229
244. He YW, Li H, Zhang J, Hsu CL, Lin E, Zhang N, et al. The Extracellular Matrix Protein Mindin is a Pattern-Recognition Molecule for Microbial Pathogens. Nat Immunol (2004) 5:88–97. doi: 10.1038/ni1021
245. Liu Ys, Wang Lf, Cheng XS, Huo YN, Ouyang XM, Liang LY, et al. The Pattern-Recognition Molecule Mindin Binds Integrin Mac-1 to Promote Macrophage Phagocytosis Via Syk Activation and NF-κb p65 Translocation. J Cell Mol Med (2019) 23:3402–16. doi: 10.1111/jcmm.14236
246. Schober JM, Lau LF, Ugarova TP, Lam SCT. Identification of a Novel Integrin αmβ2 Binding Site in CCN1 (CYR61), a Matricellular Protein Expressed in Healing Wounds and Atherosclerotic Lesions. J Biol Chem (2003) 278:25808–15. doi: 10.1074/jbc.M301534200
247. Schober JM, Chen N, Grzeszkiewicz TM, Jovanovic I, Emeson EE, Ugarova TP, et al. Identification of Integrin alphaMbeta2 as an Adhesion Receptor on Peripheral Blood Monocytes for Cyr61 (CCN1) and Connective Tissue Growth Factor (CCN2): Immediate-Early Gene Products Expressed in Atherosclerotic Lesions. Blood (2002) 99:4457–65. doi: 10.1182/blood.V99.12.4457
248. Bai T, Chen C-C, Lau LF. Matricellular Protein Ccn1 Activates a Proinflammatory Genetic Program in Murine Macrophages. J Immunol (2010) 184:3223–32. doi: 10.4049/jimmunol.0902792
249. Yakubenko VP, Cui K, Ardell CL, Brown KE, West XZ, Gao D, et al. Oxidative Modifications of Extracellular Matrix Promote the Second Wave of Inflammation Via B2 Integrins. Blood (2018) 132:78–88. doi: 10.1182/blood-2017-10-810176
250. Lishko VK, Novokhatny VV, Yakubenko VP, Skomorovska-Prokvolit HV, Ugarova TP. Characterization of Plasminogen as an Adhesive Ligand for Integrins αmβ2 (Mac-1) and α5β1 (Vla-5). Blood (2004) 104:719–26. doi: 10.1182/blood-2003-09-3016
251. Chavakis T, Athanasopoulos A, Rhee JS, Orlova V, Schmidt-Wöll T, Bierhaus A, et al. Angiostatin is a Novel Anti-Inflammatory Factor by Inhibiting Leukocyte Recruitment. Blood (2005) 105:1036–43. doi: 10.1182/blood-2004-01-0166
252. Sotiriou SN, Orlova VV, Al-Fakhri N, Ihanus E, Economopoulou M, Isermann B, et al. Lipoprotein(a) in Atherosclerotic Plaques Recruits Inflammatory Cells Through Interaction With Mac-1 Integrin. FASEB J (2006) 20:559–61. doi: 10.1096/fj.05-4857fje
253. Xue W, Kindzelskii AL, Todd RF, Petty HR. Physical Association of Complement Receptor Type 3 and Urokinase-Type Plasminogen Activator Receptor in Neutrophil Membranes. J Immunol (1994) 152:4630–40.
254. May AE, Kanse SM, Lund LR, Gisler RH, Imhof BA, Preissner KT. Urokinase Receptor (Cd87) Regulates Leukocyte Recruitment Via β 2 Integrins In Vivo. J Exp Med (1998) 188:1029–37. doi: 10.1084/jem.188.6.1029
255. Simon DI, Wei Y, Zhang L, Rao NK, Xu H, Chen Z, et al. Identification of a Urokinase Receptor-Integrin Interaction Site. Promiscuous Regulator of Integrin Function. J Biol Chem (2000) 275:10228–34. doi: 10.1074/jbc.275.14.10228
256. Zhang H, Colman RW, Sheng N. Regulation of CD11b/CD18 (Mac-1) Adhesion to Fibrinogen by Urokinase Receptor (Upar). Inflammation Res (2003) 52:86–93. doi: 10.1007/s000110300006
257. Cao C, Lawrence DA, Li Y, Von Arnim CAF, Herz J, Su EJ, et al. Endocytic Receptor LRP Together With tPA and PAI-1 Coordinates Mac-1-dependent Macrophage Migration. EMBO J (2006) 25:1860–70. doi: 10.1038/sj.emboj.7601082
258. Jerke U, Rolle S, Dittmar G, Bayat B, Santoso S, Sporbert A, et al. Complement Receptor Mac-1 is an Adaptor for NB1 (CD177)-Mediated PR3-ANCA Neutrophil Activation. J Biol Chem (2011) 286:7070–81. doi: 10.1074/jbc.M110.171256
259. Spijkers PPEM, da Costa Martins P, Westein E, Gahmberg CG, Zwaginga JJ, Lenting PJ. LDL-Receptor – Related Protein Regulates beta2-integrin – Mediated Leukocyte Adhesion. Blood (2005) 105:170–7. doi: 10.1182/blood-2004-02-0498.Supported
260. Ranganathan S, Cao C, Catania J, Migliorini M, Zhang L, Strickland DK. Molecular Basis for the Interaction of Low Density Lipoprotein Receptor-Related Protein 1 (LRP1) With Integrin αmβ2: Identification of Binding Sites Within αmβ2 for LRP1. J Biol Chem (2011) 286:30535–41. doi: 10.1074/jbc.M111.265413
261. Jia W, Li H, He Y-W. Pattern Recognition Molecule Mindin Promotes Intranasal Clearance of Influenza Viruses. J Immunol (2008) 180:6255–61. doi: 10.4049/jimmunol.180.9.6255
262. Trezzini C, Jungi TW, Kuhnert P, Peterhans E. Fibrinogen Association With Human Monocytes: Evidence for Constitutive Expression of Fibrinogen Receptors and for Involvement of Mac-1 (Cd18, CR3) in the Binding. Biochem Biophys Res Commun (1988) 156:477–84. doi: 10.1016/S0006-291X(88)80866-0
263. Forsyth CB, Solovjov DA, Ugarova TP, Plow EF. Integrin αmβ2-Mediated Cell Migration to Fibrinogen and its Recognition Peptides. J Exp Med (2001) 193:1123–33. doi: 10.1084/jem.193.10.1123
264. Lu C, Shimaoka M, Zang Q, Takagi J, Springer TA. Locking in Alternate Conformations of the Integrin αlβ2 I Domain With Disulfide Bonds Reveals Functional Relationships Among Integrin Domains. Proc Natl Acad Sci U S A (2001) 98:2393–8. doi: 10.1073/pnas.041618598
265. Tang BL, Eaton JW. Fibrin ( Ogen ) Mediates Acute Inflammatory Responses to Biomaterials. J Exp Med (1993) 178:2147–56. doi: 10.1084/jem.178.6.2147
266. Chavakis T, May AE, Preissner KT, Kanse SM. Molecular Mechanisms of Zinc-Dependent Leukocyte Adhesion Involving the Urokinase Receptor and β2-Integrins. Blood (1999) 93:2976–83. doi: 10.1182/blood.v93.9.2976.409k21_2976_2983
267. Wang JX, Bair AM, King SL, Shnayder R, Huang YF, Shieh CC, et al. Ly6G Ligation Blocks Recruitment of Neutrophils Via a β2-Integrin- Dependent Mechanism. Blood (2012) 120:1489–98. doi: 10.1182/blood-2012-01-404046
268. Simon DI, Chen Z, Xu H, Li CQ, Dong JF, McIntire LV, et al. Platelet Glycoprotein Ibalpha is a Counterreceptor for the Leukocyte Integrin Mac-1 (Cd11b/Cd18). J Exp Med (2000) 192:193–204. doi: 10.1084/JEM.192.2.193
269. Wang Y, Gao H, Shi C, Erhardt PW, Pavlovsky A, Soloviev DA, et al. Leukocyte Integrin Mac-1 Regulates Thrombosis Via Interaction With Platelet Gpibα. Nat Commun (2017) 8:15559. doi: 10.1038/ncomms15559
270. Chavakis T, Santoso S, Clemetson KJ, Sachs UJH, Isordia-Salas I, Pixley RA, et al. High Molecular Weight Kininogen Regulates Platelet-Leukocyte Interactions by Bridging Mac-1 and Glycoprotein Ib. J Biol Chem (2003) 278:45375–81. doi: 10.1074/jbc.M304344200
271. Hidalgo A, Peired AJ, Weiss LA, Katayama Y, Frenette PS. The Integrin αmβ2 Anchors Hematopoietic Progenitors in the Bone Marrow During Enforced Mobilization. Blood (2004) 104:993–1001. doi: 10.1182/blood-2003-10-3702
272. Zen K, Liu DQ, Li LM, Chen CXJ, Guo YL, Ha B, et al. The Heparan Sulfate Proteoglycan Form of Epithelial Cd44v3 Serves as a CD11b/CD18 Counter-Receptor During Polymorphonuclear Leukocyte Transepithelial Migration. J Biol Chem (2009) 284:3768–76. doi: 10.1074/jbc.M807805200
273. Diamond MS, Alon R, Parkos CA, Quinn MT, Springer TA. Heparin is an Adhesive Ligand for the Leukocyte Integrin Mac-1 (Cd11b/Cd18). J Cell Biol (1995) 130:1473–82. doi: 10.1083/jcb.130.6.1473
274. Peter K, Schwarz M, Conradt C, Nordt T, Moser M, Kübier W, et al. Heparin Inhibits Ligand Binding to the Leukocyte Integrin Mac-1 (Cd11b/Cd18). Circulation (1999) 100:1533–9. doi: 10.1161/01.CIR.100.14.1533
275. Altieri DC, Edgington TS. The Saturable High Affinity Association of Factor X to ADP-stimulated Monocytes Defines a Novel Function of the Mac-1 Receptor. J Biol Chem (1988) 263:7007–15. doi: 10.1016/S0021-9258(18)68596-5
276. Altieri DC, Etingin OR, Fair DS, Brunck TK, Geltosky JE, Hajjar DP, et al. Structurally Homologous Ligand Binding of Integrin Mac-1 and Viral Glycoprotein C Receptors. Sci (80) (1991) 254:1200–2. doi: 10.1126/science.1957171
277. Plescia J, Altieri DC. Activation of Mac-1 (Cd11b/Cd18)-Bound Factor X by Released Cathepsin G Defines an Alternative Pathway of Leucocyte Initiation of Coagulation. Cell (1996) 879:873–9. doi: 10.1042/bj3190873
278. Gustafson EJ, Schmaier AH, Wachtfogel YT, Kaufman N, Kucich U, Colman RW. Human Neutrophils Contain and Bind High Molecular Weight Kininogen. J Clin Invest (1989) 84:28–35. doi: 10.1172/JCI114151
279. Sheng N, Fairbanks MB, Heinrikson RL, Canziani G, Chaiken IM, Mosser DM, et al. Cleaved High Molecular Weight Kininogen Binds Directly to the Integrin Cd11b/Cd18 (Mac-1) and Blocks Adhesion to Fibrinogen and ICAM-1. Blood (2000) 95:3788–95. doi: 10.1182/blood.v95.12.3788.012k47_3788_3795
280. Wachtfogel YT, DeLa Cadena RA, Kunapuli SP, Rick L, Miller M, Schultze RL, et al. High Molecular Weight Kininogen Binds to Mac-1 on Neutrophils by its Heavy Chain (Domain 3) and its Light Chain (Domain 5). J Biol Chem (1994) 269:19307–12. doi: 10.1016/S0021-9258(17)32168-3
281. Chavakis T, Kanse SM, Pixley RA, May AE, Isordia-Salas I, Colman RW, et al. Regulation of Leukocyte Recruitment by Polypeptides Derived From High Molecular Weight Kininogen. FASEB J (2001) 15:2365–76. doi: 10.1096/fj.01-0201com
282. Khan MM, Bradford HN, Isordia-Salas I, Liu Y, Wu Y, Espinola RG, et al. High-Molecular-Weight Kininogen Fragments Stimulate the Secretion of Cytokines and Chemokines Through Upar, Mac-1, and gC1qR in Monocytes. Arterioscler Thromb Vasc Biol (2006) 26:2260–6. doi: 10.1161/01.ATV.0000240290.70852.c0
283. Kawamoto E, Okamoto T, Takagi Y, Honda G, Suzuki K, Imai H, et al. LFA-1 and Mac-1 Integrins Bind to the Serine/Threonine-Rich Domain of Thrombomodulin. Biochem Biophys Res Commun (2016) 473:1005–12. doi: 10.1016/j.bbrc.2016.04.007
284. Watanabe-Kusunoki K, Nakazawa D, Kusunoki Y, Kudo T, Hattanda F, Nishio S, et al. Recombinant Thrombomodulin Ameliorates Autoimmune Vasculitis Via Immune Response Regulation and Tissue Injury Protection. J Autoimmun (2020) 108:102390. doi: 10.1016/j.jaut.2019.102390
285. Fink K, Busch HJ, Bourgeois N, Schwarz M, Wolf D, Zirlik A, et al. Mac-1 Directly Binds to the Endothelial Protein C-Receptor: A Link Between the Protein C Anticoagulant Pathway and Inflammation? PloS One (2013) 8:1–5. doi: 10.1371/journal.pone.0053103
286. Wang Y, Sakuma M, Chen Z, Ustinov V, Shi C, Croce K, et al. Leukocyte Engagement of Platelet Glycoprotein Ibα Via the Integrin Mac-1 is Critical for the Biological Response to Vascular Injury. Circulation (2005) 112:2993–3000. doi: 10.1161/CIRCULATIONAHA.105.571315
287. Hirahashi J, Mekala D, Van Ziffle J, Xiao L, Saffaripour S, Wagner DD, et al. Mac-1 Signaling Via Src-Family and Syk Kinases Results in Elastase-Dependent Thrombohemorrhagic Vasculopathy. Immunity (2006) 25:271–83. doi: 10.1016/j.immuni.2006.05.014
288. Hirahashi J, Hishikawa K, Kaname S, Tsuboi N, Wang Y, Simon DI, et al. Mac-1 (CD11b/CD18) Links Inflammation and Thrombosis After Glomerular Injury. Circulation (2009) 120:1255–65. doi: 10.1161/CIRCULATIONAHA.109.873695
289. Langer HF, Choi EY, Zhou H, Schleicher R, Chung KJ, Tang Z, et al. Platelets Contribute to the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Circ Res (2012) 110:1202–10. doi: 10.1161/CIRCRESAHA.111.256370
290. Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, et al. Mediators and Molecular Pathways Involved in the Regulation of Neutrophil Extracellular Trap Formation Mediated by Activated Platelets. J Leukoc Biol (2016) 99:153–62. doi: 10.1189/jlb.3a0415-161r
291. Altieri DC, Morrissey JH, Edgington TS. Adhesive Receptor Mac-1 Coordinates the Activation of Factor X on Stimulated Cells of Monocytic and Myeloid Differentiation: An Alternative Initiation of the Coagulation Protease Cascade. Proc Nati Acad Sci USA (1988) 85:7462–6. doi: 10.1073/pnas.85.20.7462
292. Hahm E, Li J, Kim K, Huh S, Rogelj S, Cho J. Extracellular Protein Disulfide Isomerase Regulates Ligand-Binding Activity of Amb2 Integrin and Neutrophil Recruitment During Vascular Inflammation. Blood (2013) 121:3789–800. doi: 10.1182/blood-2012
293. Zen K, Utech M, Liu Y, Soto I, Nusrat A, Parkos CA. Association of BAP31 With CD11b/CD18. Potential Role in Intracellular Trafficking of CD11b/CD18 in Neutrophils. J Biol Chem (2004) 279:44924–30. doi: 10.1074/jbc.M402115200
294. Lecoanet-Henchoz S, Gauchat JF, Aubry JP, Graber P, Life P, Paul-Eugene N, et al. Cd23 Regulates Monocyte Activation Through a Novel Interaction With the Adhesion Molecules CD11b-CD18 and CD11c-CD18. Immunity (1995) 3:119–25. doi: 10.1016/1074-7613(95)90164-7
295. Schuler P, Assefa D, Ylänne J, Basler N, Olschewski M, Ahrens I, et al. Adhesion of Monocytes to Medical Steel as Used for Vascular Stents is Mediated by the Integrin Receptor Mac-1 (Cd11b/Cd18; αm β2) and can be Inhibited by Semiconductor Coating. Cell Commun Adhes (2003) 10:17–26. doi: 10.1080/15419060302065
296. Shimaoka M, Lu C, Salas A, Xiao T, Takagi J, Springer TA. Stabilizing the Integrin αm Inserted Domain in Alternative Conformations With a Range of Engineered Disulfide Bonds. Proc Natl Acad Sci U S A (2002) 99:16737–41. doi: 10.1073/pnas.252633099
297. Yan TT, Li Q, Zhou HT, Zhao YT, Yu SQ, Xu GL, et al. Gu-4 Suppresses Affinity and Avidity Modulation of CD11b and Improves the Outcome of Mice With Endotoxemia and Sepsis. PloS One (2012) 7:e30110. doi: 10.1371/journal.pone.0030110
298. Trentini A, Murganti F, Rosta V, Cervellati C, Manfrinato MC, Spadaro S, et al. Hydroxyethyl Starch 130/0.4 Binds to Neutrophils Impairing Their Chemotaxis Through a Mac-1 Dependent Interaction. Int J Mol Sci (2019) 20:817. doi: 10.3390/ijms20040817
299. Schwarz M, Nordt T, Bode C, Peter K. The GP Iib/Iiia Inhibitor Abciximab (C7e3) Inhibits the Binding of Various Ligands to the Leukocyte Integrin Mac-1 (Cd11b/Cd18, αmβ2). Thromb Res (2002) 107:121–8. doi: 10.1016/S0049-3848(02)00207-4
300. Hamilton GS, Mewshaw RE, Bryant CM, Feng Y, Endemann G, Madden KS, et al. Fluorenylalkanoic and Benzoic Acids as Novel Inhibitors of Cell Adhesion Processes in Leukocytes. J Med Chem (1995) 38:1650–6. doi: 10.1021/jm00010a009
301. Endemann G, Feng Y, Bryant CM, Hamilton GS, Perumattam J, Mewshaw RE, et al. Novel Anti-Inflammatory Compounds Prevent Cd11b/Cd18, αmβ2 (Mac-1)-Dependent Neutrophil Adhesion Without Blocking Activation-Induced Changes in Mac-1. J Pharmacol Exp Ther (1996) 276:5–12.
302. Bansal VS, Vaidya S, Somers EP, Kanuga M, Shevell D, Weikel R, et al. Small Molecule Antagonists of Complement Receptor Type 3 Block Adhesion and Adhesion-Dependent Oxidative Burst in Human Polymorphonuclear Leukocytes. J Pharmacol Exp Ther (2003) 304:1016–24. doi: 10.1124/jpet.102.045286
303. Faridi MH, Maiguel D, Brown BT, Suyama E, Barth CJ, Hedrick M, et al. High-Throughput Screening Based Identification of Small Molecule Antagonists of Integrin CD11b/CD18 Ligand Binding. Biochem Biophys Res Commun (2010) 394:194–9. doi: 10.1016/j.bbrc.2010.02.151
304. Shimaoka M, Salas A, Yang W, Weitz-Schmidt G, Springer TA. Small Molecule Integrin Antagonists That Bind to the Beta2 Subunit I-Like Domain and Activate Signals in One Direction and Block Them in the Other. Immunity (2003) 19:391–402. doi: 10.1016/s1074-7613(03)00238-3
305. Koivunen E, Ranta TM, Annila A, Taube S, Uppala A, Jokinen M, et al. Inhibition of β2 Integrin-Mediated Leukocyte Cell Adhesion by Leucine-Leucine-Glycine Motif-Containing Peptides. J Cell Biol (2001) 153:905–15. doi: 10.1083/jcb.153.5.905
306. Feng Y, Chung D, Garrard L, McEnroe G, Lim D, Scardina J, et al. Peptides Derived From the Complementarity-Determining Regions of Anti- Mac-1 Antibodies Block Intercellular Adhesion Molecule-1 Interaction With Mac-1. J Biol Chem (1998) 273:5625–30. doi: 10.1074/jbc.273.10.5625
307. Björklund M, Aitio O, Stefanidakis M, Suojanen J, Salo T, Sorsa T, et al. Stabilization of the Activated αmβ2 Integrin by a Small Molecule Inhibits Leukocyte Migration and Recruitment. Biochemistry (2006) 45:2862–71. doi: 10.1021/bi052238b
308. Faridi MH, Khan SQ, Zhao W, Lee HW, Altintas MM, Zhang K, et al. Cd11b Activation Suppresses TLR-dependent Inflammation and Autoimmunity in Systemic Lupus Erythematosus. J Clin Invest (2017) 127:1271–83. doi: 10.1172/JCI88442
309. Yao X, Dong G, Zhu Y, Yan F, Zhang H, Ma Q, et al. Leukadherin-1-Mediated Activation of CD11b Inhibits LPS-Induced Pro-Inflammatory Response in Macrophages and Protects Mice Against Endotoxic Shock by Blocking LPS-TLR4 Interaction. Front Immunol (2019) 10:215. doi: 10.3389/fimmu.2019.00215
310. Faridi MH, Maiguel D, Barth CJ, Stoub D, Day R, Schürer S, et al. Identification of Novel Agonists of the Integrin Cd11b/Cd18. Bioorg Med Chem Lett (2009) 19:6902–6. doi: 10.1016/j.bmcl.2009.10.077
311. Faridi MH, Altintas MM, Gomez C, Duque JC, Vazquez-Padron RI, Gupta V. Small Molecule Agonists of Integrin CD11b/CD18 do Not Induce Global Conformational Changes and are Significantly Better Than Activating Antibodies in Reducing Vascular Injury. Biochim Biophys Acta (2013) 1830:3696–710. doi: 10.1016/j.bbagen.2013.02.018
312. Maiguel D, Faridi MH, Wei C, Kuwano Y, Balla KM, Hernandez D, et al. Small Molecule-Mediated Activation of the Integrin Cd11b/Cd18 Reduces Inflammatory Disease. Sci Signal (2011) 4:1–15. doi: 10.1126/scisignal.2001811
313. Roberts AL, Fürnrohr BG, Vyse TJ, Rhodes B. The Complement Receptor 3 (Cd11b/Cd18) Agonist Leukadherin-1 Suppresses Human Innate Inflammatory Signalling. Clin Exp Immunol (2016) 185:361–71. doi: 10.1111/cei.12803
314. Dickson CM, LeBlanc B, Edhi MM, Heffernan DS, Faridi MH, Gupta V, et al. Leukadherin-1 Ameliorates Endothelial Barrier Damage Mediated by Neutrophils From Critically Ill Patients. J Intensive Care (2018) 6:1–10. doi: 10.1038/s41575-019-0191-1
315. Graf M, Reif S, Kröll T, Hecht K, Nuessler V, Schmetzer H. Expression of MAC-1 (CD11b) in Acute Myeloid Leukemia (AML) is Associated With an Unfavorable Prognosis. Am J Hematol (2006) 81:227–35. doi: 10.1002/ajh.20526
316. Xu S, Li X, Zhang J, Chen J, Stover CM. Prognostic Value of CD11b Expression Level for Acute Myeloid Leukemia Patients: A Meta-Analysis. PloS One (2015) 10:1–14. doi: 10.1371/journal.pone.0135981
317. Rhein P, Mitlohner R, Basso G, Gaipa G, Dworzak MN, Kirschner-Schwabe R, et al. CD11b is a Therapy Resistance- and Minimal Residual Disease-Specific Marker in Precursor B-Cell Acute Lymphoblastic Leukemia. Blood (2010) 115:3763–71. doi: 10.1182/blood-2009-10-247585
318. Qiu X, Li J, Yang X, Tang J, Shi J, Tong Y, et al. Is Neutrophil CD11b a Special Marker for the Early Diagnosis of Sepsis in Neonates? A Systematic Review and Meta-Analysis. BMJ Open (2019) 9:e025222. doi: 10.1136/bmjopen-2018-025222
319. Duan M, Steinfort DP, Smallwood D, Hew M, Chen W, Ernst M, et al. Cd11b Immunophenotyping Identifies Inflammatory Profiles in the Mouse and Human Lungs. Mucosal Immunol (2016) 9:550–63. doi: 10.1038/mi.2015.84
320. Jialal I, Adams-Huet B, Devaraj S. Monocyte Cell Adhesion Molecule Receptors in Nascent Metabolic Syndrome. Clin Biochem (2016) 49:505–7. doi: 10.1016/j.clinbiochem.2015.12.009
321. Akiyama H, McGeer PL. Brain Microglia Constitutively Express β-2 Integrins. J Neuroimmunol (1990) 30:81–93. doi: 10.1016/0165-5728(90)90055-R
322. Okita Y, Tanaka H, Ohira M, Muguruma K, Kubo N, Watanabe M, et al. Role of Tumor-Infiltrating Cd11b+ Antigen-Presenting Cells in the Progression of Gastric Cancer. J Surg Res (2014) 186:192–200. doi: 10.1016/j.jss.2013.08.024
323. Gupta R, Gant VA, Williams B, Enver T. Increased Complement Receptor-3 Levels in Monocytes and Granulocytes Distinguish COVID-19 Patients With Pneumonia From Those With Mild Symptoms. Int J Infect Dis (2020) 99:381–5. doi: 10.1016/j.ijid.2020.08.004
324. Edwards DN, Bix GJ. The Inflammatory Response After Ischemic Stroke: Targeting β2 and β1 Integrins. Front Neurosci (2019) 13:540. doi: 10.3389/fnins.2019.00540
325. Zhang D, Hu X, Qian L, Chen SH, Zhou H, Wilson B, et al. Microglial MAC1 Receptor and PI3K are Essential in Mediating β-Amyloid Peptide-Induced Microglial Activation and Subsequent Neurotoxicity. J Neuroinflammation (2011) 8:1–14. doi: 10.1186/1742-2094-8-3
326. Mitroulis I, Alexaki V, Kourtzelis I, Ziogas A, Hajishengallis G, Chavakis T. Leukocyte Integrins: Role in Leukocyte Recruitment and as Therapeutic Targets in Inflammatory Disease. Pharmacol Ther (2015) 147:123–35. doi: 10.1016/j.pharmthera.2014.11.008.Leukocyte
327. Khan SQ, Guo L, Cimbaluk DJ, Elshabrawy H, Faridi MH, Jolly M, et al. A Small Molecule β2 Integrin Agonist Improves Chronic Kidney Allograft Survival by Reducing Leukocyte Recruitment and Accompanying Vasculopathy. Front Med (2014) 1:45. doi: 10.3389/fmed.2014.00045
328. Jagarapu J, Kelchtermans J, Rong M, Chen S, Hehre D, Hummler S, et al. Efficacy of Leukadherin-1 in the Prevention of Hyperoxia-Induced Lung Injury in Neonatal Rats. Am J Respir Cell Mol Biol (2015) 53:793–801. doi: 10.1165/rcmb.2014-0422OC
329. Panni RZ, Herndon JM, Zuo C, Hegde S, Hogg GD, Knolhoff BL, et al. Agonism of CD11b Reprograms Innate Immunity to Sensitize Pancreatic Cancer to Immunotherapies. Sci Transl Med (2019) 11:eaau9240. doi: 10.1126/scitranslmed.aau9240
330. Geraghty T, Rajagopalan A, Aslam R, Pohlman A, Venkatesh I, Zloza A, et al. Positive Allosteric Modulation of CD11b as a Novel Therapeutic Strategy Against Lung Cancer. Front Oncol (2020) 10:748. doi: 10.3389/fonc.2020.00748
331. Rosetti F, Chen Y, Sen M, Thayer E, Azcutia V, Herter JM, et al. A Lupus-Associated Mac-1 Variant has Defects in Integrin Allostery and Interaction With Ligands Under Force. Cell Rep (2015) 10:1655–64. doi: 10.1016/j.celrep.2015.02.037
332. Plescia J, Conte MS, VanMeter G, Ambrosini G, Altieri DC. Molecular Identification of the Cross-Reacting Epitope on α(M)β2 Integrin I Domain Recognized by Anti-α(Iib)β3 Monoclonal Antibody 7E3 and its Involvement in Leukocyte Adherence. J Biol Chem (1998) 273:20372–7. doi: 10.1074/jbc.273.32.20372
333. Bode L, Kunz C, Muhly-Reinholz M, Mayer K, Seeger W, Rudloff S. Inhibition of Monocyte, Lymphocyte, and Neutrophil Adhesion to Endothelial Cells by Human Milk Oligosaccharides. Thromb Haemost (2004) 92:1402–10. doi: 10.1160/TH04-01-0055
334. Zhao Z, Li Q, Hu J, Li Z, Liu J, Liu A, et al. Lactosyl Derivatives Function in a Rat Model of Severe Burn Shock by Acting as Antagonists Against CD11b of Integrin on Leukocytes. Glycoconj J (2009) 26:173–88. doi: 10.1007/s10719-008-9174-0
335. Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, et al. Statins Selectively Inhibit Leukocyte Function Antigen-1 by Binding to a Novel Regulatory Integrin Site. Nat Med (2001) 7:687–92. doi: 10.1038/89058
Keywords: complement, integrin, CR3 (CD11b/CD18), inflammation, autoimmune diseases, host defense
Citation: Lamers C, Plüss CJ and Ricklin D (2021) The Promiscuous Profile of Complement Receptor 3 in Ligand Binding, Immune Modulation, and Pathophysiology. Front. Immunol. 12:662164. doi: 10.3389/fimmu.2021.662164
Received: 31 January 2021; Accepted: 12 April 2021;
Published: 29 April 2021.
Edited by:
Mihály Józsi, Eötvös Loránd University, HungaryReviewed by:
Thomas Vorup-Jensen, Aarhus University, DenmarkCopyright © 2021 Lamers, Plüss and Ricklin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christina Lamers, Y2hyaXN0aW5hLmxhbWVyc0B1bmliYXMuY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.