 Violetta S. Gogoleva1,2*
Violetta S. Gogoleva1,2* Kamar-Sulu N. Atretkhany1
Kamar-Sulu N. Atretkhany1 Arina P. Dygay2,3,4
Arina P. Dygay2,3,4 Taisiya R. Yurakova2,3,4
Taisiya R. Yurakova2,3,4 Marina S. Drutskaya1,2
Marina S. Drutskaya1,2 Sergei A. Nedospasov1,2,4
Sergei A. Nedospasov1,2,4- 1Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow, Russia
- 2Department of Immunobiology and Biomedicine, Sirius University of Science and Technology, Sirius, Russia
- 3Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow, Russia
- 4Department of Immunology, Faculty of Biology, Lomonosov Moscow State University, Moscow, Russia
TNF is a multifunctional cytokine with its key functions attributed to inflammation, secondary lymphoid tissue organogenesis and immune regulation. However, it is also a physiological regulator of hematopoiesis and is involved in development and homeostatic maintenance of various organs and tissues. Somewhat unexpectedly, the most important practical application of TNF biology in medicine is anti-TNF therapy in several autoimmune diseases. With increased number of patients undergoing treatment with TNF inhibitors and concerns regarding possible adverse effects of systemic cytokine blockade, the interest in using humanized mouse models to study the efficacy and safety of TNF-targeting biologics in vivo is justified. This Perspective discusses the main functions of TNF and its two receptors, TNFR1 and TNFR2, in steady state, as well as in emergency hematopoiesis. It also provides a comparative overview of existing mouse lines with humanization of TNF/TNFR system. These genetically engineered mice allow us to study TNF signaling cascades in the hematopoietic compartment in the context of various experimental disease models and for evaluating the effects of various human TNF inhibitors on hematopoiesis and other physiological processes.
Introduction to Hematopoiesis
Hematopoiesis is the process of blood cell development that in vertebrates is initiated early during embryogenesis and may be divided into 3 phases or so-called distinct waves of hematopoiesis.
The first (or primitive) wave takes place in the yolk sac starting in mice at embryonic day 7.5 (E7.5) and generates unipotent blood cell types (1). The second (or pro-definitive) wave occurs in the yolk sac, embryo proper and allantois of the mouse embryo and gives rise to multipotent progenitors (2). The third wave of hematopoiesis represents definitive hematopoiesis and is dependent on the activity of hematopoietic stem cells (HSCs), which are the basic units of the adult hematopoietic system. HSCs generated in the embryonic aorta-gonad-mesonephros region first colonize the fetal liver (E10.5) and then shortly before birth (E16) migrate to the bone marrow (BM), where the majority of HSCs reside to sustain steady state hematopoiesis (3).
HSCs are multipotent, self-renewing cells capable of differentiating into all mature blood cell lineages over the lifespan of the animal. Lineage choice may be directed both intrinsically and extrinsically via activation of transcription factors or extrinsically by cytokines (4). The majority of HSCs are quiescent under steady-state conditions, and few HSCs cycle to sustain hematopoiesis (5). In order to maintain hematopoietic homeostasis and to prevent development of malignancies, the self-renewal and differentiation capacities of HSCs are tightly regulated. This is, at least partly, achieved by the specialized network of interactions between distinct cell types (6, 7) and secreted factors (8, 9) in the BM niche that maintains HSC activity in steady-state conditions.
However, in the case of systemic infections and pathological conditions, such as myeloablation after chemo- or radiotherapy, some HSCs may respond and exit their quiescent state. These ‘activated’ HSCs contribute to the pool of hematopoietic progenitor cells, which will undergo further differentiation in order to replenish the population of immune cells being in high demand at the sites of inflammation in the process of so-called emergency hematopoiesis. This is possible because hematopoietic stem and progenitor cells (HSPCs) express Toll-like receptors (10) and cytokine receptors (11) and, thus, can respond to inflammatory signals. Activation of TLR signaling in HSPCs not only drives myeloid cell differentiation (10), but also leads to production of cytokines, which regulate myeloid differentiation and HSPC proliferation (12). HSPCs may respond to cytokines released during inflammation either systemically or locally by cells in the hematopoietic microenvironment or BM niche. Indeed, it was shown that HSPCs express various cytokine receptors, including IL-1R (13), IL-6Rα, as well as both TNF receptors, TNFR1 and TNFR2 (12).
As mentioned above, proinflammatory cytokines are critical components of inflammation-induced myelopoiesis. However, inflammatory signals may also be implicated in the maintenance of homeostatic hematopoiesis. During embryonic development proinflammatory cytokines control HSPC specification in the pro-definitive wave of hematopoiesis (14). Moreover, proinflammatory cytokines may regulate adult hematopoiesis and maintain the balance between HSC dormancy and lineage commitment (15). This question is important because systemic and long-term anti-cytokine therapy is being applied to treat an increasing number of conditions, including autoimmune disorders. One of the proinflammatory cytokines implicated in hematopoiesis is TNF, which we discuss in the context of humanized mouse models in this Perspective.
TNF in Steady-State Hematopoiesis
TNF is a pleiotropic cytokine involved in inflammation, development of secondary lymphoid organs and immune regulation. TNF is produced as a transmembrane protein and can be proteolytically cleaved into a soluble form. TNF exerts its functions via two distinct receptors – TNFR1 and TNFR2 (16). Both receptors also may interact with soluble (LTα3) and membrane-bound (LTα2β1) lymphotoxins, respectively (17). Interestingly, TNF may play a role both in embryonic and in adult hematopoiesis (Figure 1).
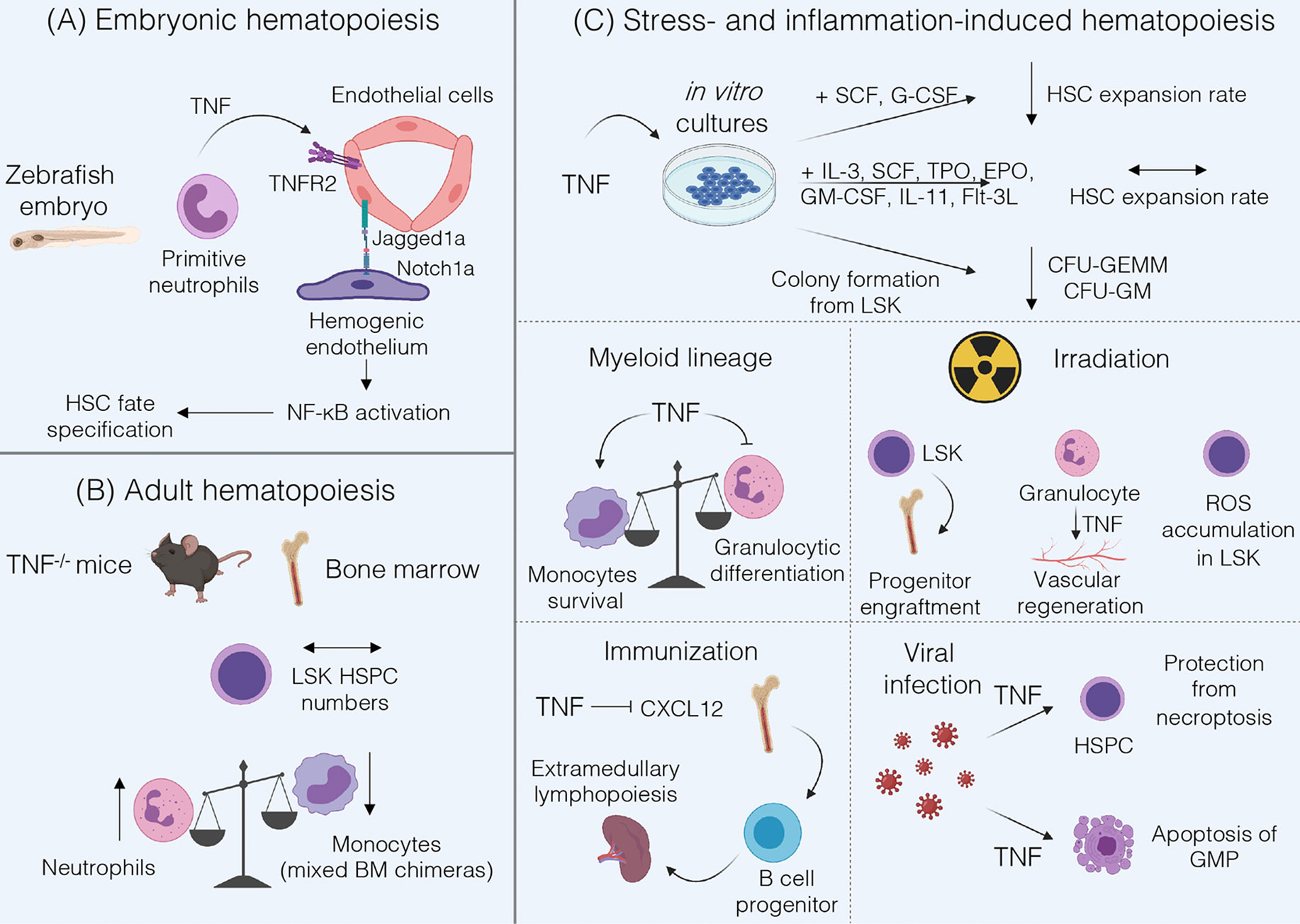
Figure 1 Summary of TNF functions in hematopoiesis. (A) During fetal hematopoiesis in zebrafish TNF/TNFR2 signaling is required to establish HSC fate via activation of Notch and NF-kB signaling (18). (B) Bone marrow of adult TNF-deficient mice is characterized by normal LSK HSPC numbers and by an increase in Gr-1+ neutrophils (19). Mixed Tnf-/- and Tnf+/+ BM chimeras underrepresent TNF-deficient monocytes (20). (C) TNF may inhibit HSC expansion when cultured with SCF and G-CSF, but not in cytokine-rich medium (21). Addition of TNF to LSK cultures inhibits formation of CFU-GEMM and CFU-GM (22). TNF promotes monocytes survival (20) and inhibits proliferation and differentiation of granulocyte progenitors (19, 23, 24). Under inflammatory conditions induced by irradiation TNF may be beneficial for progenitor engraftment (25) but stromal cell-derived TNF induces ROS accumulation in HSPCs (26), and granulocyte-derived TNF is involved in vascular regeneration (27). Following immunization, TNF may suppress CXCL12-dependent retention of B cell progenitors in the bone marrow leading to their migration (28). In the case of viral infections TNF protects HSPCs from necroptosis, enhances myelopoiesis and induces apoptosis of GMP (21).
The role of TNF during fetal hematopoiesis was mostly studied in zebrafish. These studies revealed that TNF derived from primitive neutrophils binds to TNFR2 on endothelial cells resulting in the upregulation of Notch ligand Jagged1a, which in turn binds to Notch1a receptor on the neighboring hemogenic endothelium triggering HSC fate specification. Moreover, TNF/TNFR2 axis also activates canonical NF-κB pathway in hemogenic endothelium, which triggers a transcriptional program to establish HSC generation (18). Of note, not only TNF but also other inflammatory stimuli such as TLR4-MyD88 signaling or G-CSF may lead to NF-κB activation required for HSC development (29). Interestingly, inflammatory signaling represents a highly conserved pathway regulating the HSC development. Studies in E9.5 mouse embryos revealed that hematopoietic cluster cells and endothelial cells respond to IFNγ and to a lesser extent to TNF stimulation. The most likely source of TNF in the mouse embryo is the population of primitive F4/80+ macrophages, similar to the situation in zebrafish embryo (30). However, the precise contribution of TNF to mammalian HSC development is not completely understood. Taken together, TNF signaling may be required for HSC emergence in the developing embryo via activation of evolutionarily conserved signaling pathways, but it might be partially redundant with other inflammatory stimuli.
Many studies have been performed to examine the role of TNF in the adult hematopoiesis; however, most of these have relied on cell culture and/or bone marrow chimeras, which could indirectly affect HSPC phenotype and functions. Another challenge in understanding the role of TNF in steady-state conditions is due to its capacity to induce systemic inflammation when administered in vivo which in turn may activate stress-induced hematopoiesis thus obscuring TNF contribution to hematopoiesis. Therefore, in this section we will focus on in vivo studies using gene-deficient mice. TNF deficiency did not alter the number of Lin-Sca-1+c-kit+ (LSK) in the BM, consisting mostly of lineage-biased multipotent progenitors (19). In early studies TNFR1-deficient bone marrow was characterized by increased number of LSK (22). However, examination of purified LSKFLT3- HSPCs revealed no differences in the numbers of HSPC isolated from the adult BM of TNFR1/TNFR2 double knockout mice (31). Hence, it is likely that TNF does not affect HSPC compartment under steady-state conditions in vivo.
Regarding the differentiation of HSCs to more committed progenitors, TNFR1-deficient mice showed a decrease in pre-B cell compartment and an increase in myeloid progenitors (32). Accordingly, TNF-deficient mice demonstrated an increase in the number of Gr-1+ neutrophils both in the BM and in peripheral blood (19). Transcriptome analysis of monocytes and their BM precursors revealed an increase in TNF expression upon differentiation of Ly6Chi/Ly6Cint monocytes into Ly6Clo monocytes (20). Therefore, TNF may control granulocyte number in the blood and BM and support monocytic differentiation in vivo.
TNF in Stress- and Inflammation-Induced Hematopoiesis
Numerous studies on the role of TNF in HSPC functions relied on in vitro and in vivo colony formation assays together with the assessment of reconstitution potential, engraftment and survival abilities of multipotent progenitors upon transplantation into irradiated recipient mice. However, results obtained from these studies should be carefully interpreted since these setups may affect HSPC proliferation, survival, self-renewal and differentiation. We will discuss some experiments and their possible applicability to hematopoietic compartment.
Studies with competitive co-transplantation of TNFR1-/-TNFR2-/- CD45.2+ and wild-type (WT) CD45.1+ BM cells into lethally irradiated congenic CD45.1+CD45.2+ WT recipients showed enhanced activity of TNFR1-/-TNFR2-/- HSCs as determined by long-term reconstitution by TNFR-deficient HSCs following transplantation. Moreover, in vivo administration of TNF to WT mice led to a decrease in BM cellularity and to reduction in HSC cycling activity in a competitive transplantation assay (31). These data suggest that TNF blockade may be beneficial for post-transplantation reconstitution and also supports the idea that TNF may suppress HSC activity. In contrast, transplantation of bone marrow cells from 6 months old TNFR1-deficient mice into lethally irradiated recipients showed reduced repopulating ability of TNFR1-/- BM cells as compared to WT cells (32). However, this effect was shown on non-purified HSPCs and under long-term transplantation conditions that may affect the outcome of the experiment. TNF does not inhibit expansion of highly purified Lin-Sca-1+c-kit+Flk2-CD150+CD48- HSCs in medium supplemented with IL-3, SCF, TPO, EPO, GM-CSF, IL-11 and Flt3-L, or so-called cytokine-rich medium, regardless of TNF concentration. Under cytokine-poor conditions (medium supplemented with SCF and G-CSF only) HSC expansion rate upon addition of TNF was significantly decreased (21). Similar findings were reported by Pronk et al. when TNF was added to the culture of LSK cells (31). Importantly, studies by Yamashita et al. revealed that this effect was due to the inhibition of autophagy by TNF, which sensitized HSCs to cell death in cytokine-deprived environment (21). Taken together, HSCs appear to be resistant to TNF cytotoxicity but this resistance may be changed by the environmental stress.
Addition of exogenous TNF to bone marrow cultures of LSK resulted in reduced numbers of large-sized colonies, such as CFU-GEMMs (colony-forming unit − granulocyte, erythroid, macrophage, megakaryocyte) and CFU-GMs (granulocyte/macrophage colony forming units), in methylcellulose assay (22). LSK isolated from TNF-deficient mice gave rise to an increased number of splenic colony-forming units in lethally irradiated recipient mice (19). Experiments with TNFR1 and TNFR2 agonists demonstrated that TNFR2 is essential for TNF-mediated inhibition of colony formation of early Lin-Sca-1+ progenitors, while addition of TNFR1 agonist had no significant effect on the number of Lin-Sca-1+ proliferative clones (23). Thus, TNF may inhibit formation of CFU-GEMM and CFU-GM colonies and differentiation potential of multipotent progenitors, presumably via TNFR2.
Evaluation of TNF effects on myeloid lineage commitment revealed that addition of TNF or TNFR1 agonists resulted in a decrease in G-CSF-induced colony formation and in inhibition of G-CSF expression by BM cultures (23). This was further supported in vitro in TNF-deficient long-term bone marrow cultures, which were characterized by increased proliferative potential of granulocytic progenitors and increased numbers of CFU-GMs within sorted LSK population, as compared to WT BM cultures (19). Consistent with in vitro data, CFU-GM formation was reduced in WT-recipient mice reconstituted with TNF-deficient BM, suggesting a possible role of TNF, expressed by hematopoietic and not by stromal cells, in inducing cell death at the GMP stage (33). This is also in agreement with data showing elimination of GMPs by TNF in a dose-dependent manner (21). Next, TNF was demonstrated to block granulocytic differentiation and IL-3-dependent proliferation of granulocyte-committed cells (24). At the same time, an autonomous effect of TNF on monocytes was proposed. Specifically, TNF is required for monocyte survival in vivo (20). Thus, TNF stands at the crossroad of myeloid lineage commitment via negatively affecting granulocytic cell differentiation and driving the survival of monocytes. Interestingly, these findings suggest possibilities to control the differentiation of myeloid progenitors. For example, TNF was shown to directly upregulate a central transcription factor for myeloid lineage commitment, PU.1, in HSPC in vivo during acute inflammation (34). Moreover, in some abnormal hematopoiesis conditions, such as clonal hematopoiesis associated with aging, TNF blockade may help to overcome TET2-mutant HSPC skewing toward myeloid lineage and the formation of CFU-GMs (35).
Other studies revealed that addition of TNF to the LSK cultures negatively regulated both long-term and short-term reconstituting activity after transplantation of LSK into lethally irradiated recipient mice (36). However, TNF produced by BM microenvironment is required for long-term engraftment and survival of purified LSK in allogeneic and syngeneic recipients (37). This is in line with the fact that the engraftment of Lin- BM cells from TNFR1- and TNFRs-deficient donors in wild-type recipients was defective suggesting a stimulatory role of TNF in successful progenitor engraftment. Interestingly, homing of the engrafted progenitors to the BM was primarily mediated by TNFR1 (25). On the other hand, total body irradiation and inflammation within BM was associated with elevated levels of TNF. Subsequently, TNF induced ROS accumulation in LSK leading to impairment of their reconstitution ability. Addition of TNFR1 antagonistic peptide to LSK cultures inhibited ROS accumulation suggesting that TNFR1 blockade prior to transplantation may lead to improved reconstitution capability (26). Altogether, more precise in vivo studies with defined protocols of total body irradiation and transplantation are needed to identify the role of TNF in the engraftment and survival of HSPCs. For example, TNF may contribute to regeneration of BM niche after HSC transplantation, since TNF-deficient mice displayed reduced number of BM endothelial cells upon myeloablation with a single injection of 5-fluorouracil. Furthermore, Gr1+CD115- granulocyte-derived TNF promoted vascular regeneration following transplantation (27). In the context of hematological malignancies, such as myeloproliferation, inflammation may be implicated in the disruption of BM microenvironment. Flt3ITD/ITD mice, harboring the most common somatic mutation in patients with acute myeloid leukemia, were shown to upregulate TNF production by endothelial cells in the BM niche, which subsequently may lead to suppression of HSC activity. Treatment of Flt3+/+ and Flt3ITD/ITD mice with Etanercept resulted in partial rescue of LSKCD150+CD48- engraftment capacities (38).
As a consequence of inflammation, TNF may mobilize B cell progenitors from BM to peripheral tissues by suppressing their CXCL12-induced retention in the BM. This mobilization establishes extramedullary lymphopoiesis possibly needed for resolution of inflammation (28). Contribution of TNF to emergency myelopoiesis was clearly demonstrated by Yamashita et al. (21). In a model of poly(I:C)-induced inflammation, TNF, on one hand, induced NF-kB activity, which protected HSPCs from inflammation-induced necroptosis and, on the other hand, promoted myelopoiesis and induced apoptosis of GMPs (21). This effect may be protective in pathogenesis of some viral infections, in which case GMPs may act as the latent reservoirs for viruses in the BM and should be eliminated (39).
Defects in hematopoiesis may lead to the development of hematologic disorders. Although the exact role of TNF in different hematological diseases remains incompletely understood, elevated levels of TNF were found in patients suffering from myeloid leukemia (40) and myelodysplastic syndromes (41, 42), Fanconi anemia (43), Hodgkin’s disease (44) and Non-Hodgkin lymphoma (45).
Moreover, since TNF inhibitors are widely used to treat autoimmune disorders, it is important to address possible side effects of anti-TNF therapy. Indeed, hematological complications were reported in patients on TNF blockade (Table 1). For example, a case report was published showing pancytopenia after treatment with Infliximab (47). Furthermore, side effects of Etanercept [which also neutralizes both LTα3 and LTα2β1 (17)] treatment on hematopoietic cells have been reported including aplastic anemia (52), thrombocytopenia (55), and bone marrow aplasia with pancytopenia (64). Interestingly, a common phenomenon in patients receiving anti-TNF therapy is the development of neutropenia (81). Apart from that, some lymphoproliferative disorders were reported. For example, treatment of rheumatoid arthritis and inflammatory bowel disease (IBD) patients with Etanercept or Infliximab, respectively, led to formation of cutaneous and systemic T-cell lymphomas (75). A case of Hodgkin-type lymphoproliferative lesions was reported for an IBD patient treated with Infliximab for a 6-month period (79).

Table 1 Hematological complications from systemic TNF blockade.
Altogether, TNF may play a crucial role in inflammation-induced hematopoiesis and may be implicated in the pathogenesis of some hematologic disorders. Anti-TNF therapy may lead to rare but severe side effects affecting hematopoietic compartment and resulting in the development of hematological complications and even malignancies. The exact mechanisms of these side effects are not well understood and should be addressed in the future using humanized mouse models.
Humanized Mice as the Tools to Study Hematopoiesis and to Evaluate the Consequences of Systemic Cytokine Ablation
To identify the effects and to evaluate the efficacy and safety of clinically available or novel human TNF (hTNF) inhibitors proper animal models should be generated and validated. Importantly, in spite of a conservative nature of TNF family of cytokines and their corresponding genes, most hTNF inhibitors do not block mouse TNF (82). Therefore, various panels of humanized mice that express hTNF and/or TNFRs are required to facilitate the research (Table 2). Such preclinical models were first generated in 1991 by G. Kollias group, when the first mice with overexpression of human TNF were reported (83).
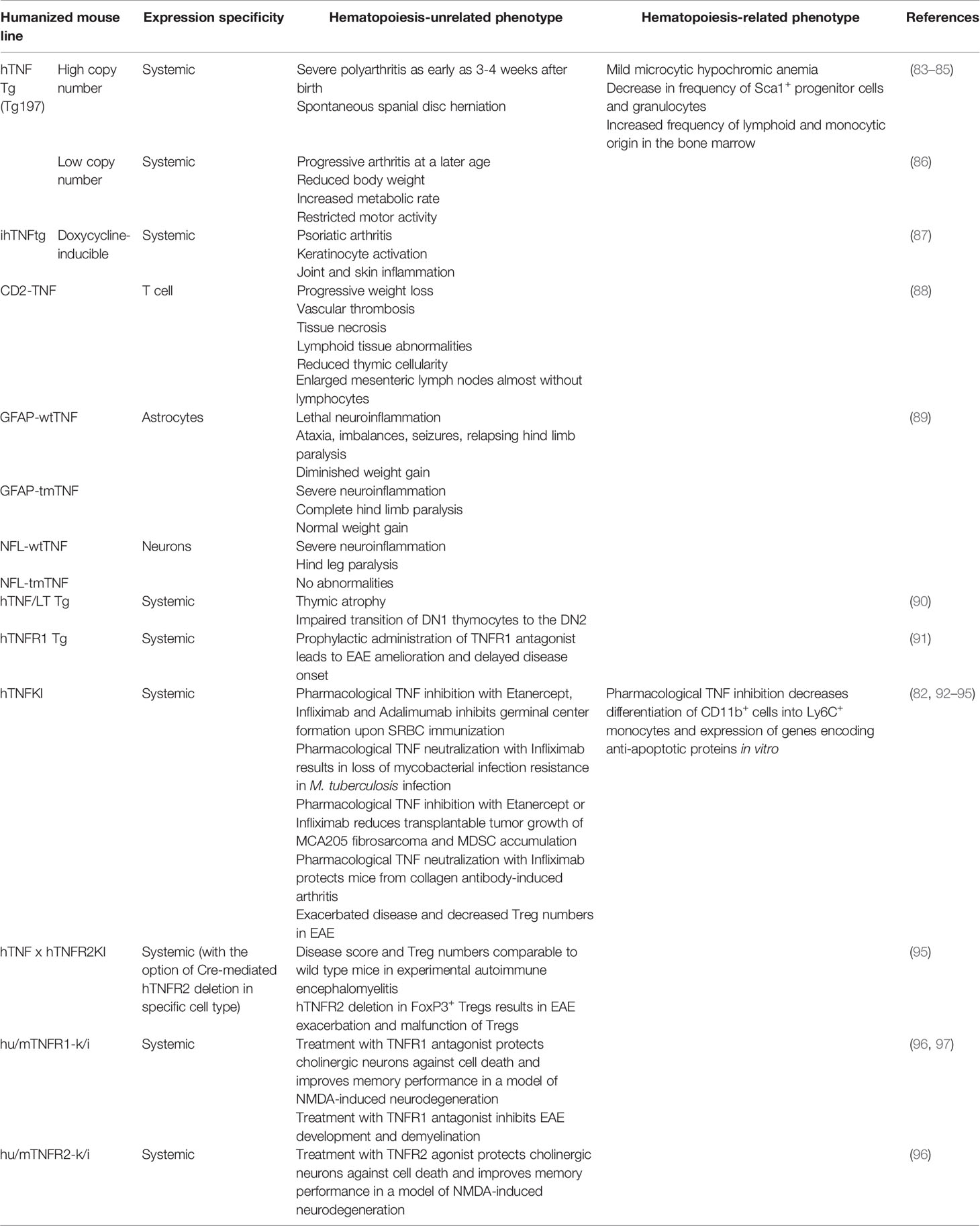
Table 2 Humanization of TNF/LT system in mice as a tool to study human hematopoiesis.
Mice With the Overexpression of Human TNF
The very first TNF humanized model was a transgenic mouse with the overexpression of TNF due to intentional dysregulation of TNF mRNA half-life (83). In general, overexpression of cytokines in animal models is a powerful tool to study molecular mechanisms associated with increased cytokine production (98). It is well established that dysregulated TNF production is detrimental in various autoimmune diseases including rheumatoid arthritis, psoriasis and IBD (99). Mice with human TNF overexpression (hTNF Tg mice with a high transgene copy number and dysregulated control) start to develop severe polyarthritis as early as 3-4 weeks after birth with similar characteristics observed in rheumatoid arthritis patients. Administration of antibodies to hTNF Tg mice led to the suppression of arthritis (83). Moreover, TNF overexpression in hTNF Tg mice led to increased incidence of spontaneous spinal disc herniation, which is involved in the development of acute radicular pain (84). These hTNF Tg mice displayed a decrease in hemoglobin associated with mild microcytic hypochromic anemia at the age of 9 weeks. Furthermore, TNF overexpression was associated with a decrease in the frequency of Sca-1+ progenitor cells and granulocytes with concurrent increase in the frequency of cells of both lymphoid and monocytic origin in the bone marrow (85). In summary, constitutive TNF overexpression is associated with the development of spontaneous autoimmune conditions. hTNF Tg mice served as an excellent model for associated disorders, such as progressive rheumatoid arthritis, although physiological relevance of using these mice in order to delineate the in vivo effects of systemic TNF inhibition on other functions was limited. To overcome the limitations of non-physiological levels of systemic TNF overproduction, other mouse models, such as tissue-specific, inducible and low copy number hTNF transgenic mice were developed.
Approaches to investigate tissue-restricted overexpression of human TNF in mice were also pioneered by G. Kollias group (88). For example, hTNF overexpression by T cells led to severe systemic effects, for example, CD2-hTNF Tg mice developed lethal progressive weight loss but no arthritis. hTNF overexpression by T cells also resulted in vascular thrombosis, tissue necrosis and lymphoid tissue abnormalities. Particularly, mice were characterized by reduced thymic cellularity and enlarged mesenteric lymph nodes that contained almost no lymphocytes. Overexpression of hTNF in astrocytes or neurons resulted in severe neurologic disease characterized by ataxia, seizures and relapsing hind limb paralysis (89). To delineate the contribution of soluble versus transmembrane TNF, mice that overexpress transmembrane human TNF in astrocytes (GFAP-tmTNF) or neurons (NFL-tmTNF) were engineered. Surprisingly, only astrocyte-specific overexpression of tmTNF was sufficient to trigger the development of neurologic disease (89). Thus, astrocytes appeared to be the source of pathogenic hTNF in a model of neuroinflammation.
The next step in the generation of hTNF transgenic mice was a low transgene copy number model characterized by low circulating levels of hTNF (86). In this case mice developed progressive arthritis at an older age, however, increased TNF production was also associated with reduced body weight, increased metabolic rate and restricted motor activity. This is similar to symptoms of rheumatoid cachexia in humans; therefore, these mice also represent a useful model to study conditions associated with elevated TNF production (100).
To overcome limitations of constitutive TNF overexpression, mice with reversible, doxycycline-inducible hTNF overexpression were generated. Two weeks after doxycycline administration, hTNF Tg mice developed psoriatic arthritis characterized by keratinocyte activation, joint and skin inflammation (87). Interestingly, signs of inflammation in this model were observed exclusively in the digits and, to a lesser extent, in the skin and ankles, unlike in mice with systemic TNF overexpression.
Another approach to generate mice with TNF overexpression was used by Liepinsh et al. (90). In this study mice with a large human genomic segment comprising hTNF and its two closest homologues, lymphotoxin α and β, were generated. Natural genomic context allowed hTNF/hLT genes to be expressed in response to physiological stimuli under the control of intrinsic regulatory elements. These mice demonstrated thymic atrophy and affected thymic T cell development with impaired thymocytes differentiation. Taken together, TNF humanized mouse models that partially mimic inflammatory conditions in patients with autoimmune disorders, such as rheumatoid arthritis and psoriasis, were generated and evaluated. Furthermore, to address the efficacy and possible side effects of TNF/TNFRs inhibitors in other disease models, humanized mice with regulated and cell type-specific hTNF expression were also established.
Mice With Humanization of TNF and TNFR2
Humanized TNF knock-in (hTNFKI) mice, in which case the mouse Tnf gene was substituted by its human ortholog, were generated using embryonic stem cell technology. They were used as a platform to study the effects of hTNF blockade in various disease models, including infectious, autoimmune, toxicity and transplantable tumor models (92, 93, 101, 102). Also, the efficacy of a novel myeloid-specific TNF inhibitor MYSTI in blocking hTNF and its effects in mouse models of LPS/D-galactosamine-induced hepatoxicity and collagen antibody-induced arthritis were demonstrated using these hTNFKI mice (93, 102). Additionally, these engineered mice allowed investigators to compare clinically available hTNF inhibitors such as Infliximab, Etanercept and Adalimumab (82). Furthermore, TNF ablation by pharmacological neutralization in hTNFKI mice led to the loss of the resistance to mycobacterial infection and to increased bacterial burden in the lungs (92). hTNF inhibition decreased tumor growth and MDSC accumulation in transplantable MCA 205 fibrosarcoma model, indicating a pro-tumorigenic function of TNF (94). Overall, hTNFKI mice are a useful tool to assess multiple effects of human TNF inhibition in various disease models, including adverse effects of TNF neutralization on hematopoietic compartment. To delineate the role of TNF inhibition with clinically approved blockers in myeloid cell differentiation, we isolated BM cells from hTNFKI mice, cultured them in the medium supplemented with GM-CSF and IL-4 with the addition of Infliximab and analyzed immature myeloid cell differentiation after 5 days of culturing (Figure 2A). We observed that TNF inhibition with Infliximab shifted differentiation of immature myeloid cells in vitro. Thus, TNF neutralization led to an increase in the frequency of Ly6G+Ly6Clow granulocytes and to a decrease in the frequency of Ly6G-Ly6Chigh monocytes (Figure 2B). Since TNF is important for survival of monocytes (20), we hypothesized that decreased frequency of monocytes upon anti-TNF treatment was due to induction of apoptosis. To verify that, we analyzed expression of genes encoding anti-apoptotic proteins in purified Ly6G-Ly6Chigh monocytes and found down-regulation of Bcl2, Bcl2a1a and Bcl2l1 upon treatment with Infliximab (Figure 2C). Altogether, TNF blockade with Infliximab in BM cultures from hTNFKI mice inhibited the differentiation of immature myeloid cells into monocytes probably due to the induction of apoptosis.
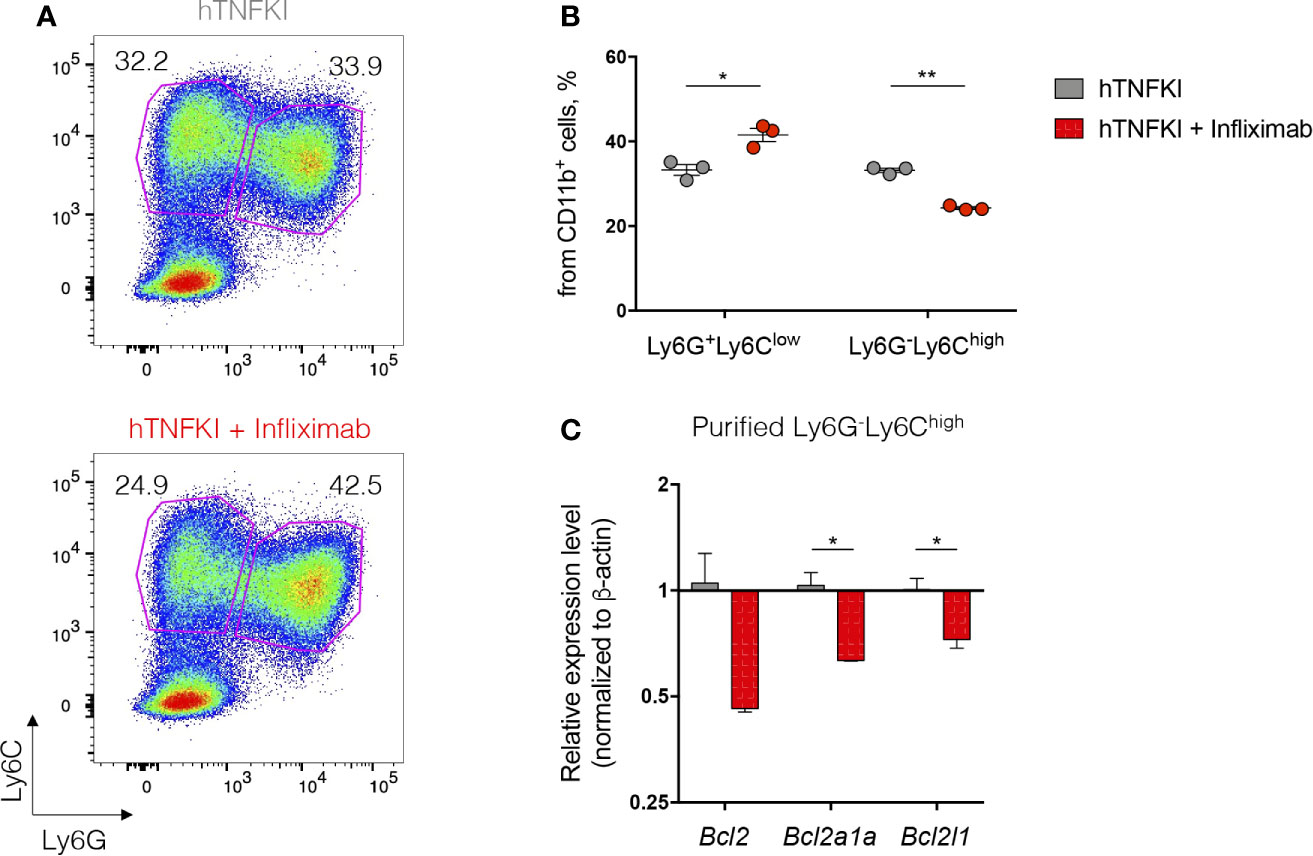
Figure 2 TNF inhibition affects immature myeloid cell development in vitro. Bone marrow cells were isolated from femurs of hTNFKI mice and cultured for 5 days in RPMI 1640 medium supplemented with L-Glutamine (2 mM), penicillin (100 U/ml), streptomycin (100 μg/ml), HEPES (10 mM), β-mercaptoethanol (50 μM), 10% FBS, GM-CSF (20 ng/ml) and IL-4 (10 ng/ml). Infliximab was added in the final concentration of 100 ng/ml. After 5 days in culture cells were stained with Fixable Viability Dye, CD11b (M1/70), Ly6C (HK1.4), Ly6G (RB6-8C5) and acquired with BD FACSCanto II flow cytometer. Data were analyzed using FlowJo software. (A) Representative FACS plots of Ly6G+Ly6Clow and Ly6G-Ly6Chigh cells gated on VD-CD11b+ cells. (B) Frequencies of Ly6G+Ly6Clow and Ly6G-Ly6Chigh cells gated on VD-CD11b+ cells. (C) Ly6G-Ly6Chigh cells were purified using Myeloid-Derived Suppressor Cell Isolation Kit (Miltenyi Biotec) according to the manufacturer’s protocol. RNA was isolated from purified cells using TRIzol Reagent (Invitrogen) according to the manufacturer’s instructions. RNA (1 μg) was treated with DNase I and reverse transcribed to cDNA with M-MuLV reverse transcriptase (RevertAid first strand cDNA synthesis kit, Thermo Scientific). Real-time quantitative PCR was performed using qPCRmix-HS SYBR+LowROX (Evrogen) and the following primer set: Actb, Forward: CTCCTGAGCGCAAGTACTCTGTG, Reverse: TAAAACGCAGCTCAGTAACAGTCC, Bcl2, Forward: GAGTTCGGTGGGGTCATGTG, Reverse: TATAGTTCCACAAAGGCATCCCAG, Bcl2a1a, Forward: GGCAGAATGGAGGTTGGGAAG, Reverse: ATTCTCGTGGGAGCCAAGGT, Bcl2l1, Forward: AGAGAGGCAGGCGATGAGTT, Reverse: TCCACAAAAGTGTCCCAGCC. Reactions were run using the following program on the Applied Biosystems 7500: 95°C for 10 min, 40 cycles of 95°C for 15 sec, 61°C for 30 sec and 72°C for 20 sec. Each point in a diagram represents a single mouse; mean ± SEM. *P < 0,05; **P < 0,01. Two-tailed unpaired Student’s t-test was used.
Earlier biochemical studies indicated that hTNF can efficiently bind to TNFR1, but not to TNFR2 (103, 104). Indeed, inefficiency of hTNF interaction with murine TNFR2 led to disease exacerbation and decrease in Treg numbers in the periphery and CNS in a mouse model of multiple sclerosis (EAE), in which case TNFR2 signaling is protective (95). Therefore, it was desirable to generate a mouse with humanization of TNFR2 to provide efficient TNF-TNFR2 signaling. Mice were genetically designed to include two LoxP sites into human TNFR2 locus, which allowed conditional Cre-mediated deletion of TNFR2 extracellular part in the desired cell type. These doubly humanized hTNF x hTNFR2KI mice showed EAE disease severity and Treg numbers comparable to wild-type mice, confirming the restoration of protective TNF/TNFR2 signaling. Cre-mediated genetic deletion of TNFR2 gene in Treg cells resulted in EAE exacerbation and malfunction of Treg cells. Intrinsic TNFR2 signaling was important for the maintenance of suppressive functions of Tregs by sustaining expression of Treg signature molecules, such as FoxP3, CD25, CTLA-4 and GITR (95). Further, hTNFR2 agonists applied to Treg cells from doubly humanized mice induced increased Treg cells proliferation (95).
Dong et al. also generated useful TNF receptor humanized mouse models, namely hTNFR1 knock-in and hTNFR2 knock-in mice, and demonstrated decreased neuroinflammation in response to TNFR1 antagonist ATROSAB or TNFR2 agonist EHD2-scTNFR2 (TNF hexamer oligomerized using the CH2 domain of IgE) in a model of NMDA-induced neurodegeneration (96). Additionally, ATROSAB administration inhibited development of EAE, decreased CNS infiltration and demyelination in hTNFR1KI mice (97). Yet another transgenic hTNFR1 mouse strain was used for studying the efficacy of VHH (antigen binding fragment of heavy chain only camelid antibody)-based nanobody against human TNFR1 (TNFR one silencer, TROS) in EAE, in which case a prophylactic administration of TROS resulted in disease amelioration (91).
In summary, there is a growing panel of useful humanized mouse models for evaluation of biologics that affect TNF/TNFR1/TNFR2 systems, including their effects on hematopoiesis. Furthermore, restoration of affected signaling by humanization of both TNF and its receptor, TNFR2, makes it possible to comparatively evaluate not only anti-TNF drugs, but also hTNFR2 agonists and antagonists. Taken together, humanized mouse models will allow investigators to study efficacy of various TNF/TNFRs-targeting biologics and assess possible side effects on other systems for further clinical translation.
Concluding Remarks
For many years TNF was mainly considered a proinflammatory cytokine with its role in host defense, but also with detrimental effects on autoimmunity. However, basic studies on TNF biology, as well as reported side effects in patients receiving anti-TNF therapy, highlighted its homeostatic functions in many physiological processes, including hematopoiesis. Regulation of TNF/TNFRs expression in various tissues modulates the cross-talk between immune and non-immune cells, which subsequently determines the outcome of TNF action.
Future investigation of pathological versus regulatory functions of TNF and deciphering its systemic and local effects in tissues may help to improve current therapeutic approaches. Therefore, mouse models with humanized TNF/TNFRs system represent a powerful tool to study side effects of anti-TNF therapy on hematopoiesis.
Author Contributions
VG, KA, MD, and SAN designed research. VG and KA. performed research and analyzed data. VG, KA, AD, TY, MSD, and SN discussed the concept and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Reverse genetics studies were supported by the grant 075-15-2019-1660 from the Ministry of Science and Higher Education of the Russian Federation, myelopoiesis studies were supported by RFBR grant 19-34-51030.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are greatly indebted to Dr. J.R. Keller (NCI Frederick) for useful comments on the manuscript. BioRender (https://biorender.com/) was used to make the figure.
References
1. Yamane T. Mouse Yolk Sac Hematopoiesis. Front Cell Dev Biol (2018) 6:80. doi: 10.3389/fcell.2018.00080
2. Dzierzak E, Bigas A. Blood Development: Hematopoietic Stem Cell Dependence and Independence. Cell Stem Cell (2018) 22(5):639–51. doi: 10.1016/j.stem.2018.04.015
3. Muller AM, Medvinsky A, Strouboulis J, Grosveld F, Dzierzak E. Development of Hematopoietic Stem Cell Activity in the Mouse Embryo. Immunity (1994) 1(4):291–301. doi: 10.1016/1074-7613(94)90081-7
4. Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC, Schroeder T. Hematopoietic Cytokines can Instruct Lineage Choice. Science (2009) 325(5937):217–8. doi: 10.1126/science.1171461
5. Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic Stem Cells Reversibly Switch From Dormancy to Self-Renewal During Homeostasis and Repair. Cell (2008) 135(6):1118–29. doi: 10.1016/j.cell.2008.10.048
6. Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar Niches Maintain Haematopoietic Stem Cell Quiescence. Nature (2013) 502(7473):637–43. doi: 10.1038/nature12612
7. Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, et al. Distinct Bone Marrow Blood Vessels Differentially Regulate Haematopoiesis. Nature (2016) 532(7599):323–8. doi: 10.1038/nature17624
8. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity (2006) 25(6):977–88. doi: 10.1016/j.immuni.2006.10.016
9. Ogawa M, Matsuzaki Y, Nishikawa S, Hayashi S, Kunisada T, Sudo T, et al. Expression and Function of C-Kit in Hemopoietic Progenitor Cells. J Exp Med (1991) 174(1):63–71. doi: 10.1084/jem.174.1.63
10. Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-Like Receptors on Hematopoietic Progenitor Cells Stimulate Innate Immune System Replenishment. Immunity (2006) 24(6):801–12. doi: 10.1016/j.immuni.2006.04.008
11. Takizawa H, Boettcher S, Manz MG. Demand-Adapted Regulation of Early Hematopoiesis in Infection and Inflammation. Blood (2012) 119(13):2991–3002. doi: 10.1182/blood-2011-12-380113
12. Zhao JL, Ma C, O’Connell RM, Mehta A, DiLoreto R, Heath JR, et al. Conversion of Danger Signals Into Cytokine Signals by Hematopoietic Stem and Progenitor Cells for Regulation of Stress-Induced Hematopoiesis. Cell Stem Cell (2014) 14(4):445–59. doi: 10.1016/j.stem.2014.01.007
13. Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic Interleukin-1 Exposure Drives Haematopoietic Stem Cells Towards Precocious Myeloid Differentiation At the Expense of Self-Renewal. Nat Cell Biol (2016) 18(6):607–18. doi: 10.1038/ncb3346
14. Sawamiphak S, Kontarakis Z, Stainier DY. Interferon Gamma Signaling Positively Regulates Hematopoietic Stem Cell Emergence. Dev Cell (2014) 31(5):640–53. doi: 10.1016/j.devcel.2014.11.007
15. Pietras EM. Inflammation: A Key Regulator of Hematopoietic Stem Cell Fate in Health and Disease. Blood (2017) 130(15):1693–8. doi: 10.1182/blood-2017-06-780882
16. Atretkhany KN, Gogoleva VS, Drutskaya MS, Nedospasov SA. Distinct Modes of TNF Signaling Through its Two Receptors in Health and Disease. J Leukoc Biol (2020) 107(6):893–905. doi: 10.1002/JLB.2MR0120-510R
17. Kucka K, Lang I, Zhang T, Siegmund D, Medler J, Wajant H. Membrane lymphotoxin-alpha2beta is a Novel Tumor Necrosis Factor (TNF) Receptor 2 (TNFR2) Agonist. Cell Death Dis (2021) 12(4):360. doi: 10.1038/s41419-021-03633-8
18. Espín-Palazón R, Stachura DL, Campbell CA, García-Moreno D, Del Cid N, Kim AD, et al. Proinflammatory Signaling Regulates Hematopoietic Stem Cell Emergence. Cell (2014) 159(5):1070–85. doi: 10.1016/j.cell.2014.10.031
19. Drutskaya MS, Ortiz M, Liepinsh DJ, Kuprash DV, Nedospasov SA, Keller JR. Inhibitory Effects of Tumor Necrosis Factor on Hematopoiesis Seen In Vitro are Translated to Increased Numbers of Both Committed and Multipotent Progenitors in TNF-deficient Mice. Exp Hematol (2005) 33(11):1348–56. doi: 10.1016/j.exphem.2005.08.001
20. Wolf Y, Shemer A, Polonsky M, Gross M, Mildner A, Yona S, et al. Autonomous TNF is Critical for In Vivo Monocyte Survival in Steady State and Inflammation. J Exp Med (2017) 214(4):905–17. doi: 10.1084/jem.20160499
21. Yamashita M, Passegue E. TNF-Alpha Coordinates Hematopoietic Stem Cell Survival and Myeloid Regeneration. Cell Stem Cell (2019) 25(3):357–72 e357. doi: 10.1016/j.stem.2019.05.019
22. Zhang Y, Harada A, Bluethmann H, Wang JB, Nakao S, Mukaida N, et al. Tumor Necrosis Factor (TNF) is a Physiologic Regulator of Hematopoietic Progenitor Cells: Increase of Early Hematopoietic Progenitor Cells in TNF Receptor p55-deficient Mice In Vivo and Potent Inhibition of Progenitor Cell Proliferation by TNF Alpha In Vitro. Blood (1995) 86(8):2930–7. doi: 10.1182/blood.V86.8.2930.bloodjournal8682930
23. Jacobsen FW, Rothe M, Rusten L, Goeddel DV, Smeland EB, Veiby OP, et al. Role of the 75-kDa Tumor Necrosis Factor Receptor: Inhibition of Early Hematopoiesis. Proc Natl Acad Sci USA (1994) 91(22):10695–9. doi: 10.1073/pnas.91.22.10695
24. Caux C, Favre C, Saeland S, Duvert V, Durand I, Mannoni P, et al. Potentiation of Early Hematopoiesis by Tumor Necrosis Factor-Alpha is Followed by Inhibition of Granulopoietic Differentiation and Proliferation. Blood (1991) 78(3):635–44. doi: 10.1182/blood.V78.3.635.635
25. Pearl-Yafe M, Mizrahi K, Stein J, Yolcu ES, Kaplan O, Shirwan H, et al. Tumor Necrosis Factor Receptors Support Murine Hematopoietic Progenitor Function in the Early Stages of Engraftment. Stem Cells (2010) 28(7):1270–80. doi: 10.1002/stem.448
26. Ishida T, Suzuki S, Lai CY, Yamazaki S, Kakuta S, Iwakura Y, et al. Pre-Transplantation Blockade of TNF-alpha-Mediated Oxygen Species Accumulation Protects Hematopoietic Stem Cells. Stem Cells (2017) 35(4):989–1002. doi: 10.1002/stem.2524
27. Bowers E, Slaughter A, Frenette PS, Kuick R, Pello OM, Lucas D. Granulocyte-Derived TNFalpha Promotes Vascular and Hematopoietic Regeneration in the Bone Marrow. Nat Med (2018) 24(1):95–102. doi: 10.1038/nm.4448
28. Ueda Y, Yang K, Foster SJ, Kondo M, Kelsoe G. Inflammation Controls B Lymphopoiesis by Regulating Chemokine CXCL12 Expression. J Exp Med (2004) 199(1):47–58. doi: 10.1084/jem.20031104
29. He Q, Zhang C, Wang L, Zhang P, Ma D, Lv J, et al. Inflammatory Signaling Regulates Hematopoietic Stem and Progenitor Cell Emergence in Vertebrates. Blood (2015) 125(7):1098–106. doi: 10.1182/blood-2014-09-601542
30. Li Y, Esain V, Teng L, Xu J, Kwan W, Frost IM, et al. Inflammatory Signaling Regulates Embryonic Hematopoietic Stem and Progenitor Cell Production. Genes Dev (2014) 28(23):2597–612. doi: 10.1101/gad.253302.114
31. Pronk CJ, Veiby OP, Bryder D, Jacobsen SE. Tumor Necrosis Factor Restricts Hematopoietic Stem Cell Activity in Mice: Involvement of Two Distinct Receptors. J Exp Med (2011) 208(8):1563–70. doi: 10.1084/jem.20110752
32. Rebel VI, Hartnett S, Hill GR, Lazo-Kallanian SB, Ferrara JL, Sieff CA. Essential Role for the p55 Tumor Necrosis Factor Receptor in Regulating Hematopoiesis At a Stem Cell Level. J Exp Med (1999) 190(10):1493–504. doi: 10.1084/jem.190.10.1493
33. Drize NI, Drutskaya MS, Gerasimova LP, Manakova TE, Chertkov IL, Turetskaya RL, et al. Changes in the Hemopoietic System of Mice Deficient for Tumor Necrosis Factor or Lymphotoxin-Alpha. Bull Exp Biol Med (2000) 130(7):676–8. doi: 10.1007/BF02682103
34. Etzrodt M, Ahmed N, Hoppe PS, Loeffler D, Skylaki S, Hilsenbeck O, et al. Inflammatory Signals Directly Instruct PU.1 in HSCs Via TNF. Blood (2019) 133(8):816–9. doi: 10.1182/blood-2018-02-832998
35. Abegunde SO, Buckstein R, Wells RA, Rauh MJ. An Inflammatory Environment Containing TNFalpha Favors Tet2-mutant Clonal Hematopoiesis. Exp Hematol (2018) 59:60–5. doi: 10.1016/j.exphem.2017.11.002
36. Bryder D, Ramsfjell V, Dybedal I, Theilgaard-Mönch K, Högerkorp CM, Adolfsson J, et al. Self-Renewal of Multipotent Long-Term Repopulating Hematopoietic Stem Cells is Negatively Regulated by Fas and Tumor Necrosis Factor Receptor Activation. J Exp Med (2001) 194(7):941–52. doi: 10.1084/jem.194.7.941
37. Rezzoug F, Huang Y, Tanner MK, Wysoczynski M, Schanie CL, Chilton PM, et al. TNF-Alpha is Critical to Facilitate Hemopoietic Stem Cell Engraftment and Function. J Immunol (2008) 180(1):49–57. doi: 10.4049/jimmunol.180.1.49
38. Mead AJ, Neo WH, Barkas N, Matsuoka S, Giustacchini A, Facchini R, et al. Niche-Mediated Depletion of the Normal Hematopoietic Stem Cell Reservoir by Flt3-ITD-induced Myeloproliferation. J Exp Med (2017) 214(7):2005–21. doi: 10.1084/jem.20161418
39. Kondo K, Kaneshima H, Mocarski ES. Human Cytomegalovirus Latent Infection of Granulocyte-Macrophage Progenitors. Proc Natl Acad Sci USA (1994) 91(25):11879–83. doi: 10.1073/pnas.91.25.11879
40. Sanchez-Correa B, Bergua JM, Campos C, Gayoso I, Arcos MJ, Bañas H, et al. Cytokine Profiles in Acute Myeloid Leukemia Patients At Diagnosis: Survival is Inversely Correlated With IL-6 and Directly Correlated With IL-10 Levels. Cytokine (2013) 61(3):885–91. doi: 10.1016/j.cyto.2012.12.023
41. Gersuk GM, Beckham C, Loken MR, Kiener P, Anderson JE, Farrand A, et al. A Role for Tumour Necrosis Factor-Alpha, Fas and Fas-Ligand in Marrow Failure Associated With Myelodysplastic Syndrome. Br J Haematol (1998) 103(1):176–88. doi: 10.1046/j.1365-2141.1998.00933.x
42. Dar S, Mundle S, Andric T, Qawi H, Shetty V, Reza S, et al. Biological Characteristics of Myelodysplastic Syndrome Patients Who Demonstrated High Versus No Intramedullary Apoptosis. Eur J Haematol (1999) 62(2):90–4. doi: 10.1111/j.1600-0609.1999.tb01727.x
43. Dufour C, Corcione A, Svahn J, Haupt R, Poggi V, Béka’ssy AN, et al. TNF-Alpha and IFN-gamma are Overexpressed in the Bone Marrow of Fanconi Anemia Patients and TNF-alpha Suppresses Erythropoiesis In Vitro. Blood (2003) 102(6):2053–9. doi: 10.1182/blood-2003-01-0114
44. Warzocha K, Bienvenu J, Ribeiro P, Moullet I, Dumontet C, Neidhardt-Berard EM, et al. Plasma Levels of Tumour Necrosis Factor and its Soluble Receptors Correlate With Clinical Features and Outcome of Hodgkin’s Disease Patients. Br J Cancer (1998) 77(12):2357–62. doi: 10.1038/bjc.1998.391
45. Salles G, Bienvenu J, Bastion Y, Barbier Y, Doche C, Warzocha K, et al. Elevated Circulating Levels of TNFalpha and its p55 Soluble Receptor are Associated With an Adverse Prognosis in Lymphoma Patients. Br J Haematol (1996) 93(2):352–9. doi: 10.1046/j.1365-2141.1996.5181059.x
46. Quartier P, Taupin P, Bourdeaut F, Lemelle I, Pillet P, Bost M, et al. Efficacy of Etanercept for the Treatment of Juvenile Idiopathic Arthritis According to the Onset Type. Arthritis Rheum (2003) 48(4):1093–101. doi: 10.1002/art.10885
47. Menon Y, Cucurull E, Espinoza LR. Pancytopenia in a Patient With Scleroderma Treated With Infliximab. Rheumatol (Oxford) (2003) 42(10):1273–4; author reply 1274. doi: 10.1093/rheumatology/keg341
48. Marchesoni A, Arreghini M, Panni B, Battafarano N, Uziel L. Life-Threatening Reversible Bone Marrow Toxicity in a Rheumatoid Arthritis Patient Switched From Leflunomide to Infliximab. Rheumatol (Oxford) (2003) 42(1):193–4. doi: 10.1093/rheumatology/key051
49. Seiderer J, Goke B, Ochsenkuhn T. Safety Aspects of Infliximab in Inflammatory Bowel Disease Patients. A Retrospective Cohort Study in 100 Patients of a German University Hospital. Digestion (2004) 70(1):3–9. doi: 10.1159/000080075
50. Feltelius N, Fored CM, Blomqvist P, Bertilsson L, Geborek P, Jacobsson LT, et al. Results From a Nationwide Postmarketing Cohort Study of Patients in Sweden Treated With Etanercept. Ann Rheum Dis (2005) 64(2):246–52. doi: 10.1136/ard.2004.023473
51. Szalay B, Acs L, Vasarhelyi B, Kovacs L, Balog A. Successful Use of Tocilizumab in a Patient With Rheumatoid Arthritis Following Severe Pancytopenia During Etanercept Therapy. J Clin Rheumatol (2011) 17(7):377–9. doi: 10.1097/RHU.0b013e318231fd99
52. Kuruvilla J, Leitch HA, Vickars LM, Galbraith PF, Li CH, Al-Saab S, et al. Aplastic Anemia Following Administration of a Tumor Necrosis Factor-Alpha Inhibitor. Eur J Haematol (2003) 71(5):396–8. doi: 10.1034/j.1600-0609.2003.00115.x
53. Feletar M, Brockbank JE, Schentag CT, Lapp V, Gladman DD. Treatment of Refractory Psoriatic Arthritis With Infliximab: A 12 Month Observational Study of 16 Patients. Ann Rheum Dis (2004) 63(2):156–61. doi: 10.1136/ard.2003.006775
54. Selby LA, Hess D, Shashidar H, de Villiers WJ, Selby LA. Crohn’s Disease, Infliximab and Idiopathic Thrombocytopenic Purpura. Inflammation Bowel Dis (2004) 10(5):698–700. doi: 10.1097/00054725-200409000-00033
55. Pathare SK, Heycock C, Hamilton J. Tnfalpha Blocker-Induced Thrombocytopenia. Rheumatol (Oxford) (2006) 45(10):1313–4. doi: 10.1093/rheumatology/kel204
56. Hamaguchi M, Kawahito Y, Ishino H, Yoshida M, Yoshikawa T. A Case Report of Tumor Necrosis Factor-Alpha Antibody-Induced Thrombocytopenia Associated With Emerging IgM Anticardiolipin Antibody in Patients With Scleroderma Overlap/Rheumatoid Arthritis. Clin Rheumatol (2007) 26(6):988–90. doi: 10.1007/s10067-006-0229-y
57. Salar A, Bessa X, Muñiz E, Monfort D, Besses C, Andreu M. Infliximab and Adalimumab-Induced Thrombocytopenia in a Woman With Colonic Crohn’s Disease. Gut (2007) 56(8):1169–70. doi: 10.1136/gut.2007.123547
58. Stinco G, Piccirillo F, Patrone P. Transient and Slight Thrombocytopaenia Induced by Etanercept During Treatment of Psoriatic Arthritis. Acta Derm Venereol (2008) 88(3):281–2. doi: 10.2340/00015555-0400
59. Brunasso AM, Massone C. Thrombocytopenia Associated With the Use of Anti-Tumor Necrosis Factor-Alpha Agents for Psoriasis. J Am Acad Dermatol (2009) 60(5):781–5. doi: 10.1016/j.jaad.2008.12.001
60. Mocciaro F, Russo G, Di Mitri R, Marino A. Infliximab-Induced Thrombocytopenia in an Elderly Patient With Ileocolonic Crohn’s Disease. Inflammation Bowel Dis (2013) 19(4):E52–53. doi: 10.1002/ibd.22989
61. Velayos Jimenez B, Cuello Garcia R, Del Olmo Martinez L, Macho Conesa A, Fernandez Salazar L. Immune Thrombocytopenic Purpura Associated With Inactive Ulcerative Colitis in Chronic Treatment With Adalimumab. Gastroenterol Hepatol (2020) 43(1):28–9. doi: 10.1016/j.gastre.2019.07.006
62. Gerloni V, Pontikaki I, Gattinara M, Fantini F. Focus on Adverse Events of Tumour Necrosis Factor Alpha Blockade in Juvenile Idiopathic Arthritis in an Open Monocentric Long-Term Prospective Study of 163 Patients. Ann Rheum Dis (2008) 67(8):1145–52. doi: 10.1136/ard.2007.069484
63. Vidal F, Fontova R, Richart C. Severe Neutropenia and Thrombocytopenia Associated With Infliximab. Ann Intern Med (2003) 139(3):W–W63. doi: 10.7326/0003-4819-139-3-200308050-00021-w4
64. Kozak N, Friedman J, Schattner A. Etanercept-Associated Transient Bone Marrow Aplasia: A Review of the Literature and Pathogenetic Mechanisms. Drugs R D (2014) 14(2):155–8. doi: 10.1007/s40268-014-0050-z
65. Favalli EG, Varenna M, Sinigaglia L. Drug-Induced Agranulocytosis During Treatment With Infliximab in Enteropathic Spondyloarthropathy. Clin Exp Rheumatol (2005) 23(2):247–50.
66. Rajakulendran S, Gadsby K, Allen D, O’Reilly S, Deighton C. Neutropenia While Receiving Anti-Tumour Necrosis Factor Treatment for Rheumatoid Arthritis. Ann Rheum Dis (2006) 65(12):1678–9. doi: 10.1136/ard.2006.056176
67. Theodoridou A, Kartsios C, Yiannaki E, Markala D, Settas L. Reversible T-large Granular Lymphocyte Expansion and Neutropenia Associated With Adalimumab Therapy. Rheumatol Int (2006) 27(2):201–2. doi: 10.1007/s00296-006-0187-3
68. Levalampi T, Korpela M, Vuolteenaho K, Moilanen E. Etanercept and Adalimumab Treatment in Patients With Rheumatoid Arthritis and Spondyloarthropathies in Clinical Practice: Adverse Events and Other Reasons Leading to Discontinuation of the Treatment. Rheumatol Int (2008) 28(3):261–9. doi: 10.1007/s00296-007-0436-0
69. Wenham C, Gadsby K, Deighton C. Three Significant Cases of Neutropenia With Etanercept. Rheumatol (Oxford) (2008) 47(3):376–7. doi: 10.1093/rheumatology/kem332
70. Ottaviani S, Cerf-Payrastre I, Kemiche F, Pertuiset E. Adalimumab-Induced Neutropenia in a Patient With Rheumatoid Arthritis. Joint Bone Spine (2009) 76(3):312–3. doi: 10.1016/j.jbspin.2008.09.017
71. Hastings R, Ding T, Butt S, Gadsby K, Zhang W, Moots RJ, et al. Neutropenia in Patients Receiving Anti-Tumor Necrosis Factor Therapy. Arthritis Care Res (Hoboken) (2010) 62(6):764–9. doi: 10.1002/acr.20037
72. Haroon M, Daly M, Harney S. Re-Challenge With Etanercept in Patients With Etanercept-Induced Neutropenia. Clin Rheumatol (2012) 31(1):151–5. doi: 10.1007/s10067-011-1822-2
73. AlAskar D, AlSardi M, Al Sulais E, Mosli M, AlAmeel T. Risk of Neutropenia in Inflammatory Bowel Disease Patients Treated With TNF Inhibitors: A Single-Center, Retrospective Cohort Study. Saudi J Gastroenterol (2020) 76(4):210–5. doi: 10.1093/ecco-jcc/jjz203.794
74. Montané E, Sallés M, Barriocanal A, Riera E, Costa J, Tena X. Antitumor Necrosis Factor-Induced Neutropenia: A Case Report With Double Positive Rechallenges. Clin Rheumatol (2007) 26(9):1527–9. doi: 10.1007/s10067-006-0415-y
75. Adams AE, Zwicker J, Curiel C, Kadin ME, Falchuk KR, Drews R, et al. Aggressive Cutaneous T-cell Lymphomas After TNFalpha Blockade. J Am Acad Dermatol (2004) 51(4):660–2. doi: 10.1016/j.jaad.2004.03.047
76. Broussais F, Kawashima M, Marotte H, Miossec P. Chronic Myeloid Leukaemia and Tuberculosis in a Patient With Rheumatoid Arthritis Treated With Infliximab. Ann Rheum Dis (2005) 64(3):509–10. doi: 10.1136/ard.2004.021378
77. Brown SL, Greene MH, Gershon SK, Edwards ET, Braun MM. Tumor Necrosis Factor Antagonist Therapy and Lymphoma Development: Twenty-Six Cases Reported to the Food and Drug Administration. Arthritis Rheum (2002) 46(12):3151–8. doi: 10.1002/art.10679
78. Wolfe F, Michaud K. Lymphoma in Rheumatoid Arthritis: The Effect of Methotrexate and Anti-Tumor Necrosis Factor Therapy in 18,572 Patients. Arthritis Rheum (2004) 50(6):1740–51. doi: 10.1002/art.20311
79. Vieites B, Avila R, Biscuola M, Carvajo F. Cutaneous Hodgkin-type Lymphoproliferative Lesion Associated With Immunomodulatory Therapy for Ulcerative Colitis. J Cutan Pathol (2011) 38(5):443–7. doi: 10.1111/j.1600-0560.2010.01663.x
80. D’Haens G, Reinisch W, Colombel JF, Panes J, Ghosh S, Prantera C, et al. Five-Year Safety Data From ENCORE, a European Observational Safety Registry for Adults With Crohn’s Disease Treated With Infliximab [Remicade(R)] or Conventional Therapy. J Crohns Colitis (2017) 11(6):680–9. doi: 10.1093/ecco-jcc/jjw221
81. Bessissow T, Renard M, Hoffman I, Vermeire S, Rutgeerts P, Van Assche G. Review Article: non-Malignant Haematological Complications of Anti-Tumour Necrosis Factor Alpha Therapy. Aliment Pharmacol Ther (2012) 36(4):312–23. doi: 10.1111/j.1365-2036.2012.05189.x
82. Winsauer C, Kruglov AA, Chashchina AA, Drutskaya MS, Nedospasov SA. Cellular Sources of Pathogenic and Protective TNF and Experimental Strategies Based on Utilization of TNF Humanized Mice. Cytokine Growth Factor Rev (2014) 25(2):115–23. doi: 10.1016/j.cytogfr.2013.12.005
83. Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, et al. Transgenic Mice Expressing Human Tumour Necrosis Factor: A Predictive Genetic Model of Arthritis. EMBO J (1991) 10(13):4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x
84. Gorth DJ, Shapiro IM, Risbud MV. Transgenic Mice Overexpressing Human TNF-alpha Experience Early Onset Spontaneous Intervertebral Disc Herniation in the Absence of Overt Degeneration. Cell Death Dis (2018) 10(1):7. doi: 10.1038/s41419-018-1246-x
85. Capocasale RJ, Makropoulos DA, Achuthanandam R, Stowell N, Quinn J, Rafferty PA, et al. Myelodysplasia and Anemia of Chronic Disease in Human Tumor Necrosis Factor-Alpha Transgenic Mice. Cytometry A (2008) 73(2):148–59. doi: 10.1002/cyto.a.20512
86. Hayward MD, Jones BK, Saparov A, Hain HS, Trillat AC, Bunzel MM, et al. An Extensive Phenotypic Characterization of the hTNFalpha Transgenic Mice. BMC Physiol (2007) 7:13. doi: 10.1186/1472-6793-7-13
87. Retser E, Schied T, Skryabin BV, Vogl T, Kanczler JM, Hamann N, et al. Doxycycline-Induced Expression of Transgenic Human Tumor Necrosis Factor Alpha in Adult Mice Results in Psoriasis-Like Arthritis. Arthritis Rheum (2013) 65(9):2290–300. doi: 10.1002/art.38026
88. Probert L, Keffer J, Corbella P, Cazlaris H, Patsavoudi E, Stephens S, et al. Wasting, Ischemia, and Lymphoid Abnormalities in Mice Expressing T Cell-Targeted Human Tumor Necrosis Factor Transgenes. J Immunol (1993) 151(4):1894–906.
89. Akassoglou K, Probert L, Kontogeorgos G, Kollias G. Astrocyte-Specific But Not Neuron-Specific Transmembrane TNF Triggers Inflammation and Degeneration in the Central Nervous System of Transgenic Mice. J Immunol (1997) 158(1):438–45.
90. Liepinsh DJ, Kruglov AA, Galimov AR, Shakhov AN, Shebzukhov YV, Kuchmiy AA, et al. Accelerated Thymic Atrophy as a Result of Elevated Homeostatic Expression of the Genes Encoded by the TNF/lymphotoxin Cytokine Locus. Eur J Immunol (2009) 39(10):2906–15. doi: 10.1002/eji.200839191
91. Steeland S, Van Ryckeghem S, Van Imschoot G, De Rycke R, Toussaint W, Vanhoutte L, et al. TNFR1 Inhibition With a Nanobody Protects Against EAE Development in Mice. Sci Rep (2017) 7(1):13646. doi: 10.1038/s41598-017-13984-y
92. Olleros ML, Chavez-Galan L, Segueni N, Bourigault ML, Vesin D, Kruglov AA, et al. Control of Mycobacterial Infections in Mice Expressing Human Tumor Necrosis Factor (TNF) But Not Mouse Tnf. Infect Immun (2015) 83(9):3612–23. doi: 10.1128/IAI.00743-15
93. Drutskaya MS, Nosenko MA, Gorshkova EA, Mokhonov VV, Zvartsev RV, Polinova AI, et al. Effects of Myeloid Cell-Restricted TNF Inhibitors In Vitro and In Vivo. J Leukoc Biol (2020) 107(6):933–9. doi: 10.1002/JLB.3AB0120-532R
94. Atretkhany KS, Nosenko MA, Gogoleva VS, Zvartsev RV, Qin Z, Nedospasov SA, et al. Tnf Neutralization Results in the Delay of Transplantable Tumor Growth and Reduced Mdsc Accumulation. Front Immunol (2016) 7:147. doi: 10.3389/fimmu.2016.00147
95. Atretkhany KN, Mufazalov IA, Dunst J, Kuchmiy A, Gogoleva VS, Andruszewski D, et al. Intrinsic TNFR2 Signaling in T Regulatory Cells Provides Protection in CNS Autoimmunity. Proc Natl Acad Sci USA (2018) 115(51):13051–6. doi: 10.1073/pnas.1807499115
96. Dong Y, Fischer R, Naudé PJ, Maier O, Nyakas C, Duffey M, et al. Essential Protective Role of Tumor Necrosis Factor Receptor 2 in Neurodegeneration. Proc Natl Acad Sci USA (2016) 113(43):12304–9. doi: 10.1073/pnas.1605195113
97. Williams SK, Fairless R, Maier O, Liermann PC, Pichi K, Fischer R, et al. Anti-TNFR1 Targeting in Humanized Mice Ameliorates Disease in a Model of Multiple Sclerosis. Sci Rep (2018) 8(1):13628. doi: 10.1038/s41598-018-31957-7
98. Gorshkova EA, Zvartsev RV, Drutskaya MS, Gubernatorova EO. [Humanized Mouse Models as a Tool to Study Proinflammatory Cytokine Overexpression]. Mol Biol (Mosk) (2019) 53(5):755–73. doi: 10.1134/S0026893319050078
99. Kalliolias GD, Ivashkiv LB. TNF Biology, Pathogenic Mechanisms and Emerging Therapeutic Strategies. Nat Rev Rheumatol (2016) 12(1):49–62. doi: 10.1038/nrrheum.2015.169
100. Roubenoff R, Roubenoff RA, Cannon JG, Kehayias JJ, Zhuang H, Dawson-Hughes B, et al. Rheumatoid Cachexia: Cytokine-Driven Hypermetabolism Accompanying Reduced Body Cell Mass in Chronic Inflammation. J Clin Invest (1994) 93(6):2379–86. doi: 10.1172/JCI117244
101. Kruglov AA, Tumanov AV, Grivennikov SI, Shebzukhov YV, Kuchmiy AA, Efimov GA, et al. Modalities of Experimental TNF Blockade In Vivo: Mouse Models. Adv Exp Med Biol (2011) 691:421–31. doi: 10.1007/978-1-4419-6612-4_44
102. Efimov GA, Kruglov AA, Khlopchatnikova ZV, Rozov FN, Mokhonov VV, Rose-John S, et al. Cell-type-restricted Anti-Cytokine Therapy: TNF Inhibition From One Pathogenic Source. Proc Natl Acad Sci USA (2016) 113(11):3006–11. doi: 10.1073/pnas.1520175113
103. Ameloot P, Fiers W, De Bleser P, Ware CF, Vandenabeele P, Brouckaert P. Identification of Tumor Necrosis Factor (TNF) Amino Acids Crucial for Binding to the Murine P75 TNF Receptor and Construction of Receptor-Selective Mutants. J Biol Chem (2001) 276(40):37426–30. doi: 10.1074/jbc.M102020200
Keywords: cytokines, cytokine blockade, steady-state hematopoiesis, emergency hematopoiesis, humanized mouse models
Citation: Gogoleva VS, Atretkhany KN, Dygay AP, Yurakova TR, Drutskaya MS and Nedospasov SA (2021) Current Perspectives on the Role of TNF in Hematopoiesis Using Mice With Humanization of TNF/LT System. Front. Immunol. 12:661900. doi: 10.3389/fimmu.2021.661900
Received: 31 January 2021; Accepted: 19 April 2021;
Published: 13 May 2021.
Edited by:
Yasuyuki Saito, Kobe University, JapanReviewed by:
Masayuki Yamashita, The University of Tokyo, JapanGeorge Kalliolias, Hospital for Special Surgery, United States
Copyright © 2021 Gogoleva, Atretkhany, Dygay, Yurakova, Drutskaya and Nedospasov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Violetta S. Gogoleva, dmlvbGV0dGUuZ29nb2xldmFAZ21haWwuY29t