 Tamsin Cargill
Tamsin Cargill Emma L. Culver
Emma L. Culver- 1Peter Medawar Building for Pathogen Research, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom
- 2Oxford Liver Unit, John Radcliffe Hospital, Oxford, United Kingdom
B cells form a branch of the adaptive immune system, essential for the body’s immune defense against pathogens. B cell dysfunction has been implicated in the pathogenesis of immune mediated liver diseases including autoimmune hepatitis, IgG4-related hepatobiliary disease, primary biliary cholangitis and primary sclerosing cholangitis. B cells may initiate and maintain immune related liver diseases in several ways including the production of autoantibodies and the activation of T cells via antigen presentation or cytokine production. Here we comprehensively review current knowledge on B cell mechanisms in immune mediated liver diseases, exploring disease pathogenesis, B cell therapies, and novel treatment targets. We identify key areas where future research should focus to enable the development of targeted B cell therapies.
Introduction
B cells form a branch of the adaptive immune system that confer long-lived targeted responses to pathogens and other non–self-proteins (1). Developing in the bone marrow and spleen, B cells mature from lymphoid progenitors to mature B cells which express B cell receptors, each with its own unique antigen affinity. On meeting its cognate antigen, naïve B cells proliferate within B cell follicles producing short lived antigen secreting cells and germinal center (GC) B cells. GC B cells undergo rounds of proliferation and somatic hypermutation within GCs, resulting in populations of cells including long-lived memory B cells, plasmablasts (PB) and plasma cells (PC), which are able to secrete high affinity antibodies (1). Antibody subclass is directed by class switch recombination, which enables the production of several immunoglobulin (Ig) subtypes IgM, IgD, IgG (types 1-4), IgA and IgE [reviewed in (2)]. B cells can affect immune control directly via neutralizing antibodies, or indirectly via communication with the complement cascade and other effector immune cells such as macrophages and T cells through cytokine production or antigen presentation [reviewed in (3)]. B cells can also have regulatory functions, through production of anti-inflammatory cytokines [reviewed in (4)].
B cell dysfunction has been implicated in the pathogenesis of a number of immune mediated liver diseases including autoimmune hepatitis (AIH), IgG4-related hepatobiliary disease (IgG4-HBD), primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC). Here we review current knowledge on B cell mechanisms in immune mediated liver diseases, exploring disease pathogenesis, B cell therapies, and novel treatment targets. The role of B cells in biliary atresia and drug-induced liver injury has been explored elsewhere (5) and will not be covered in this review.
Autoimmune Hepatitis
Autoimmune hepatitis (AIH) is a chronic inflammatory liver disease characterized by raised serum total immunoglobulin G (IgG), the presence of circulating autoantibodies and liver histology showing interface hepatitis with a lymphoplasmacytic infiltrate (6). Although no single disease trigger has been identified, multiple genetic, epigenetic and environmental factors are associated with AIH development [reviewed in (7, 8)]. Corticosteroids induce disease remission and immunosuppressives such as thiopurines and mycophenolate are used to maintain the therapeutic response. Biochemical and histological disease relapse is frequent on withdrawal of immunosuppression.
Whilst early studies supported T cell dysregulation as being central to AIH pathogenesis [reviewed in (8)], a role for B cells became evident when B cell depletion with Rituximab was shown to induce clinical improvement in AIH patients refractory to conventional therapy (9, 10) and in murine models of AIH (11). Several pathogenic mechanisms of B cells in AIH have been proposed including the generation of auto-reactive antibodies, B cell overactivation, excess immunoglobulin production and the recruitment of T cells through cytokine production and antigen presentation.
Auto-Antibodies in AIH
Specific autoreactive antibodies can be detected in AIH patients, including anti-nuclear (ANA), anti-smooth muscle (SMA), anti-liver-kidney microsomal type 1 or 3 (anti-LKM-1, anti-LKM-3), anti-liver cytosol type 1 (anti-LC1), anti-soluble liver antigen (anti-SLA) or anti-asialoglycoprotein receptor (ASGPR) [reviewed in (12)]. Total IgG levels and titers of autoantibodies are part of the diagnostic criteria for AIH and can correlate with liver biochemical and histological markers of disease activity (6, 13). Autoantibody detection can also be used to sub-classify AIH into type 1 and type 2, the latter affecting younger individuals with increased disease severity (Table 1).
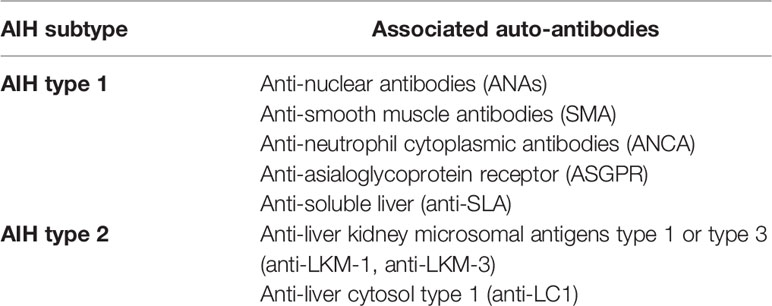
Table 1 Autoantibodies in AIH.
The events leading to autoantibody production in AIH are not clear, but some evidence suggests they might form in response to infection with hepatitis C virus or human herpes virus 6, where viral and human epitopes share sequence homology (14, 15). Although autoantibody detection has a predominantly diagnostic role, there is evidence that anti-LKM-1 and anti-LC1 are directly pathogenic. Anti-LKM-1 production is triggered by expression of cytochrome P450 2D6 (CYP2D6) on the surface of hepatocytes in AIH. It may be directly cytotoxic or activate auto-reactive T cells that target CYP2D6 expressing hepatocytes (14).
Activation of B Cells and IgG Production
Activated B cells may be a determinant of disease activity in AIH. In new onset AIH there are increased numbers of activated B cells and PC compared to healthy controls (16). PCs positively correlate with serum IgG levels, suggesting they might be the source of excess IgG production, characteristic of active disease (16).
Several factors might regulate B cell overactivation in AIH. Circulating follicular helper T (Tfh) cells necessary for B cell differentiation and maturation and their hallmark cytokine IL-21, which drives B cell activation, PC differentiation, and immunoglobulin production, are increased in AIH and correlate with serum IgG and hepatic inflammation (16, 17). Blockade of IL-21 suppresses Tfh cell generation and can prevent AIH development in murine models. B cell activating factor (BAFF), necessary for B cell survival and differentiation, is also increased in the serum of individuals with AIH (18, 19). BAFF levels positively correlate with liver transaminases and histological liver inflammation, but not with serum IgG titer (19).
Finally, T cells themselves might be the drivers of B cell activation in AIH. Sequencing of B and T cell receptors in the blood and liver of individuals with AIH showed skewing of T rather than B cell receptor profiles (20). T cell expression of activation markers PD1 and CD38 and the magnitude of ex-vivo cytokine responses towards autoantigenic peptides correlates with AIH disease activity (20–22). Autoreactive T cell responses specific to autoantigen SLA have been investigated by flow cytometry and single cell RNA sequencing (23), showing SLA CD4 T cells transcriptionally upregulate expression of genes associated with B cell help and inflammation. These cells are similar in profile to so called T peripheral helper (Tph) cells that have been identified as drivers of B cell inflammation in other autoimmune conditions such as Rheumatoid Arthritis and IgG4-related disease (24). The population of PD1 positive activated Tph cells was expanded amongst all CD4 T cells in AIH, produced IL-21 and interferon gamma (IFNγ) on ex-vivo restimulation and drove B cell differentiation in co-cultures (23).
Together these data suggest overactivation of the B cell axis in AIH is associated with IgG production and liver inflammation.
Antigen Presentation in AIH
Antigen presentation by B cells to T cells may play an important role in AIH pathogenesis. In patients with new onset AIH, there is increased expression of CD86 on B cells (16), suggesting they are primed for T cell co-stimulation. In a murine model of AIH, B cell depletion was associated with a reduced cytotoxic and proliferative capacity of intrahepatic T cells, implying in this model at least, B cells are necessary for T cell function (11). Ex-vivo proliferation assays of T cells from these AIH mice showed that CD19+ B cells as compared to CD19- lymphocytes were effective antigen presenting cells to CD4+ T cells (11). There is also evidence that B and T cells might share epitopes for the same autoantigens, as LKM1 specific immune responses have overlap between B and T cell epitope sequences (21). Together these observations suggest the B and T cell arms of the adaptive immune response respond to the same autoantigens in AIH, and B cells are necessary to support T cell function through antigen presentation.
Cytokines Regulating Inflammation and Recruitment in AIH
Cytokine and chemokines provide signals that orchestrate the immune response. In a murine model of AIH, hepatic B cells expressed higher levels of proinflammatory cytokines including interferon gamma (IFNγ) than B cells from healthy murine counterparts. These proinflammatory cytokines might attract T cells, which are found co-located with B cells within inflammatory lesions in the liver (11). B cells can also produce C-X-C motif chemokine ligand 10 (CXCL10 or IP-10), which correlates with liver transaminase levels in individuals with AIH (25). After B cell depletion in AIH there is a decrease in proinflammatory cytokines as well as CXCL10 (10, 11), supporting that B cells are an important source of proinflammatory signals during active disease.
The role of B cells in anti-inflammatory cytokine production has also been investigated. In a mouse model of AIH, B cell depletion prior to AIH onset led a more severe AIH phenotype with increased liver inflammation (26). Adoptive transfer of IL-10 dependent B regulatory cells, ameliorated inflammation by inhibition of T cell responses in this model (26). This was in contrast to a different mouse model of AIH, where B cell depletion prevented AIH development (11). These results suggest different B cell subtypes might have different roles in AIH pathogenesis, and that the balance between pro- and anti-inflammatory cytokines might be key.
The Gut Microbiome in AIH
The liver is anatomically and physiologically linked to the gut microbiota via the enterohepatic circulation (27). The gut microbiota is essential to maintain immune homeostasis of the gut-liver axis and is a major modulator of autoimmunity (27, 28). Shifts in microbiota composition activates a mucosal immune response, causing an imbalance of homeostasis, translocation of bacteria and migration of immune cells into the liver (28). Microbial antigens are recognized by the immune system via presentation on major histocompatibility complex (MHC) class II molecules to CD4+ T cells. Intestinal microbial exposure triggers expansion of B cell populations and antibody production in the liver, with proliferating antibody secreting plasmablasts derived from gut associated lymphoid tissues present in animal and human liver disease (29).
Animal models of AIH provided the first evidence that AIH was associated with reduced genetic diversity of the gut microbiome (30). A recent study in humans confirmed these findings, showing individuals with steroid-naïve AIH have a distinct microbial composition compared with healthy volunteers, with lower intraindividual diversity (31). Expansion of potential pathobiont genus Veillonella was associated with active disease status and therefore may be directly involved in AIH pathogenesis (31). Further studies are required to understand whether B cells or other immune mediators are directly involved in the response to the Veillonella genus.
IgG4-Related Hepatobiliary Disease
IgG4-related hepatobiliary disease (IgG4-HBD) is part of the chronic multi-system fibroinflammatory condition IgG4-related disease (IgG4-RD). IgG4-HBD is characterized clinically by masses and/or biliary strictures, elevated serum immunoglobulin G4 (IgG4) subclass levels, and organ infiltration with IgG4-positive plasma cells and CD4 T helper cells, a storiform pattern of fibrosis and obliterative phlebitis (32). IgG4-HBD has an elderly male preponderance and is associated with other autoimmune conditions such as thyroiditis and coeliac disease. The disease is steroid responsive, but many patients experience disease relapse.
B cells play several potential roles in disease pathogenesis including the production of autoreactive antibodies, presentation of antigen to T cells, cytokine/chemokine production and directly contributing to fibrosis. B cell depletion with rituximab has shown efficacy in treating active IgG4-RD as both an induction and maintenance agent, and is commissioned in the UK as a third-line therapy (33). Postulated mechanisms of B cell involvement in IgG4-HBD are summarized in Figure 1.
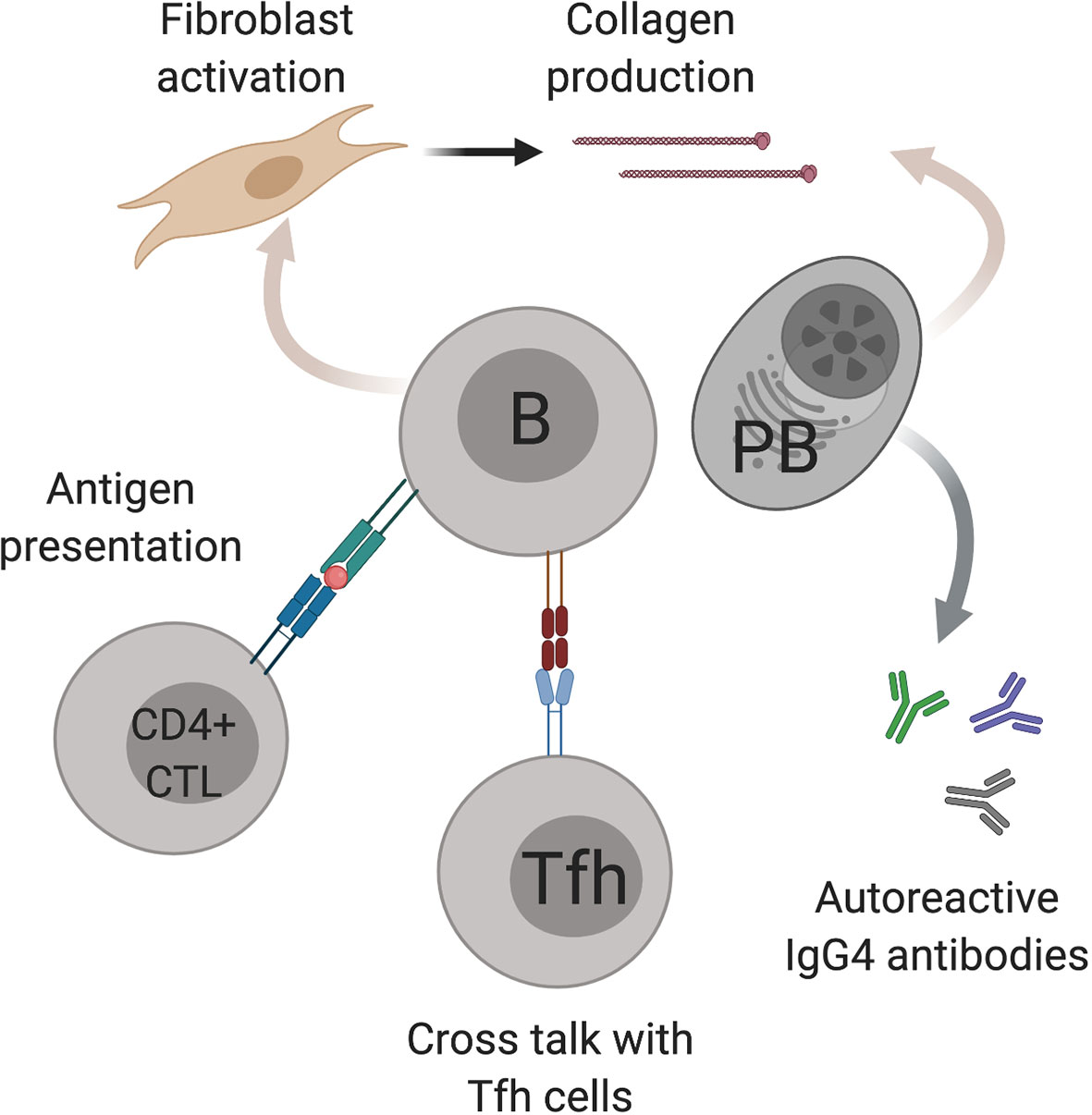
Figure 1 Postulated mechanisms of B cell involvement in IgG4-hepato biliary disease. B cells (B) have several possible mechanisms in IgG4-hepato biliary disease pathogenesis including antigen presentation to cytotoxic CD4 T cells (CD4+ CTL), cross talk with T follicular helper cells (Tfh) and fibroblast activation. IgG4 positive plasmablasts (PB) also contribute to fibrosis as well as producing IgG4 antibodies. Image created using BioRender.com.
IgG4 Antibodies in IgG4-HBD
Abundance of circulating and tissue-infiltrating IgG4 antibodies are evident in IgG4-HBD. Elevated serum IgG4 levels are a useful adjunct in disease diagnosis and if elevated they are helpful in disease monitoring and in relapse prediction. Circulating IgG4+ PB also show utility in disease monitoring in those with active disease (34).
IgG4 responses comprise approximately 4% of IgG subclass circulating antibodies in health (35). They have a unique structure and function and can undergo exchange of their antigen binding fragments (Fab) in a process known as Fab arm exchange (36). The resulting heterodimeric antibodies are poor complement binders that are unable to induce the formation of large immune complexes (37, 38). Circulating memory B cells have lower surface expression of complement receptor 2 (CR2, CD21) on IgG4 compared to IgG1 B cells, suggesting IgG4 memory B cells themselves are less responsive to complement signals (39). Hypocomplementemia occurs in IgG4-RD involving the renal, pancreatic and biliary systems, but its role in pathogenesis is not yet clear.
IgG4 B cells are produced in response to repetitive antigenic stimulation (40), developing for example after administration of therapeutic monoclonal antibodies (41). IgG4 responses may be formed as a protective mechanism to dampen immune reactions towards antigens encountered repeatedly [reviewed in (42, 43)]. IgG4 B cells also localize to tissue sites of inflammation (44), supporting that their recruitment may be a local mechanism to contain immune responses. Both type 2 helper T cells (Th2) and their signature interleukins (IL) -4, -10 and -13, and Tfh cells producing IL-21 are associated with IgG4 production (45–52). However, in IgG4-HBD the exact mechanisms triggering and sustaining the predominance of the IgG4 subclass remain elusive.
Autoantibodies and B Cell Clonality in IgG4-HBD
Excess IgG4 antibodies may be autoreactive to an as yet unidentified antigen. Autoantibodies against a range of different antigens such as galectin-3, prohibin, annexin A11 and laminin 511-E8 have been identified in patients with IgG4-RD (53, 54). However, all are non-specific ubiquitous proteins, expressed with variable frequencies across organs in patients with systemic disease. Indeed, polyclonal expansion of IgG4 autoantibodies has been demonstrated in response to multiple common environmental antigens in IgG4-HBD (55), indicating the predominance of IgG4 responses might be a more generalized phenomenon.
Next generation sequencing data of both circulating IgG4 B cells and PBs shows oligoclonal expansion of B cell receptor clones in IgG4-HBD (56–58). Circulating PB that re-expand on disease relapse have a different clonality to those at disease presentation (58). This suggests alternative epitopes might drive subsequent rounds of inflammation, although it does not rule out a common driving autoantigen. Further studies are required to understand in vivo, whether IgG4 B cells are pathogenic themselves or whether other immunological conditions drive and sustain B cell proliferation and IgG4 antibody production.
Antigen Presentation to Pathogenic T Cells in IgG4-HBD
Expanded B cells and PBs may present antigen to pathogenic subsets of T cells in IgG4-HBD. Expansion of clonally restricted CD4+ SLAMF7+ cytotoxic T cells (CTLs) cells that interact with B cells and produce pro-fibrotic cytokines have been demonstrated in the circulation and inflamed tissue of individuals with IgG4-RD. Depletion of B cells with rituximab leads to profound clinical and radiological improvement in disease activity, paralleled by a decline in PB and CD4 CTLs, implying B cells are necessary to sustain the expanded CTL population (59, 60).
Antigen-experienced B cells can regulate Tfh cell differentiation by priming naïve CD4 T cells and polarizing them towards IL-21-producing Tfh cells that enhance immunoglobulin production in cocultured B cells (61). Tfh are activated and expanded in IgG4-HBD, and play a crucial role in B cell differentiation and GC formation in involved organs (51, 52, 62–66). Circulating activated Tfh cells have been observed to positively correlate with PB number, IgG4 class switch promoting cytokines such as IL-4, serum IgG4 levels, disease activity and organ involvement in IgG4-HBD (51, 52, 62–66). It is plausible that bidirectional signaling between Tfh and B cells might sustain a cycle of IgG4 humoral responses in IgG4-HBD, although this has yet to be shown definitively in vivo.
Fibrogenic B Cells in IgG4-HBD
Storiform fibrosis is an integral histopathological feature of lesions in IgG4-HBD. Indirect evidence that B cells play a role in fibrosis came from histological sections showing B cells in close proximity to fibrotic ducts in IgG4-HBD (67). Further evidence shows in vitro B cells derived from individuals with IgG4-RD are able to activate primary pancreatic human fibroblasts to produce collagen (67). PBs rather than naïve or memory B cells were most successful in collagen production when cultured in vitro with fibroblasts. Transcriptomic analysis showed genes associated with fibroblast proliferation, including lysyl oxidase homolog 2 (LOXL2), were upregulated and demonstrated direct expression of collagen by PBs themselves (67). B cell depletion with Rituximab reduces clinical disease activity, the Enhanced Liver Fibrosis score (ELF score), a surrogate marker of fibrosis (68), and the number and size of tissue myofibroblasts in IgG4-RD. These observations support a role for B cell depletion in treating those with fibrotic disease and the potential to reverse fibrosis in the longer term.
The Gut Microbiome in IgG4-HBD
A single microbiome study in IgG4-RD evaluating stool microbiome by metagenomics showed IgG4 patients to have a decreased alpha diversity compared with healthy controls, and expansion of potentially pathogenic and pro inflammatory species such as Th17-inducing strain of E. lenta (69). There was potential microbiome-driven skewing of the immune cell population to favor both fibrotic and pre-inflammatory pathways in this disease, with microbial similarities seen to systemic sclerosis (in terms of fibrosis, perhaps sharing the CTLs that shape B cell proliferation and antibody production) and rheumatoid arthritis (in terms of inflammation) but differing to the pathogens observed in inflammatory bowel disease (69). Further studies to understand the relationship between the gut microbiome and B cells in IgG4-HBD as well as its response to B cell depletion therapy will further our understanding of its role in disease pathogenesis.
Primary Biliary Cholangitis
Primary biliary cholangitis is a chronic cholestatic disease characterized by inflammation of the small intrahepatic bile ducts, resulting in fibrosis and cirrhosis (70). PBC has a female preponderance and is frequently associated with concurrent autoimmune disorders such as Sjögren’s syndrome. Immunosuppressive agents are not effective in PBC treatment, but ursodeoxycholic acid (UDCA) is able to induce disease remission in a proportion of patients.
In PBC both environmental and genetic risk factors contribute to a loss of immune tolerance towards biliary epithelial cells (71). Although T cells have been implicated in disease pathogenesis, B cells are also thought to play an important role. B cells and PCs can be detected in the inflammatory infiltrate surrounding bile ducts in PBC (72). Circulating CD19+ B cells are increased in PBC, and positively correlate with biochemical indices of disease activity including alkaline phosphatase (ALP) levels (73). Genetic studies have identified several risk loci coding for proteins involved in B cell development and function including CD80, CXCR5, POU2AF1, SPI, IKZF3, ARID3A and CD40L (74–79). Current evidence supports a role for B cells in PBC pathogenesis though autoantibody production, cytokine secretion and reduced regulatory functions.
Autoantibodies and B Cell Clonality in PBC
Antimitochondrial antibodies (AMA) are detectable in the majority of individuals with PBC and, being highly sensitive and specific, are useful for disease diagnosis (70). Pyruvate dehydrogenase complex - E2 subunit (PDC-E2), a major target of AMA, is aberrantly expressed on biliary epithelial cells in individuals with PBC (80).
Several lines of evidence support that AMAs might be pathogenic in PBC. The frequency of B cells specific to mitochondrial antigens were found to be correlated with increasing stage of PBC in the early inflammatory phase (81). Antibody secreting PBs and PCs isolated from the livers of individuals with PBC were able to produce AMA ex-vivo (82) and PCs surrounding inflamed bile ducts on liver biopsies were observed to correlate with AMA titers (72). AMA have also been shown to stimulate macrophages to produce pro-inflammatory cytokines (83) that can directly drive inflammation in PBC.
The association of PBC with AMA and their ability to inhibit energy generation by the pyruvate dehydrogenase complex led to the hypothesis that fatigue in PBC might be ameliorated by B cell depletion. However a phase II randomized controlled trial of treatment with the B cell depleting agent Rituximab in PBC conferred no significant improvement in fatigue (84), despite being associated with reduced AMA titers (84, 85). Further, a minority of PBC patients do not have detectable AMAs, suggesting they might not be pathogenic in of themselves. CD4 T cells with autoreactivity to PDC-E2 are present even in AMA negative persons with PBC, indicating T rather than B cell responses towards PDC-E2 might drive immunopathogenesis (86).
Alternative auto-antibodies can be useful to diagnose individuals with AMA negative PBC (70). The presence of the antinuclear antibodies anti-Sp100 anti-gp210 are associated with a more severe clinical course (87, 88). Other autoantibodies include kelch-like 12 (KLHL12) and hexokinase 1 (HK1) are specific to PBC (89, 90). Studies investigating the clonality of the B cell repertoire in PBC have reported an oligoclonal expansion within the B cell repertoire and have identified disease associated clones shared between different PBC donors (91, 92). This suggests the selection of common autoantigens might drive the altered B cell clonality in PBC, but it is unclear why loss of tolerance towards self-antigen occurs.
As well as specific autoantibodies, a polyclonal IgM response is commonly observed in PBC which is mirrored by infiltrating IgM positive PBs in the portal tracts on liver histology (93–95). T cell expression of CD40-ligand (CD40L) can interact with CD40 on B cells, promoting B cell activation and differentiation. In PBC, methylation of the CD40L promotor is reduced, resulting in increased CD40L expression on CD4 T cells and higher serum IgM levels (96). CD40L has also been identified as a central upstream regulator in PBC by GWAS and microarray analysis (79), supporting that T and B cell communication via the CD40L-CD40 axis has an important role in pathogenesis.
B Cell Expansion and Activation in PBC
Studies of circulating lymphocytes in PBC have observed increases in CD19+ B cells and activated CD25+ B cells, correlating with disease stage (73, 97). Within the B cell compartment, different B cells subsets seem to be altered including decreased circulating memory B cells and increased naïve B cells and PBs (73, 98). In liver biopsy specimens from individuals with PBC, CD20+ B cells were found in lymphoid follicle like aggregations a distance from portal tracts, or more scantily as part of the lymphoplasmacytic infiltrate surrounding inflamed bile ducts. Plasma cells expressing IgM or IgG were consistently found surrounding bile ducts in a coronal distribution alongside CD4 and CD8 T cells, and thus local interactions between PCs and T cells via cytokine production or direct antigen presentation are possible (72).
Several factors have been identified that might drive B cell expansion in PBC. CXCR5+ Tfh cells, necessary for B cell differentiation, were found to be expanded in the blood and liver of individuals with PBC and ex-vivo, enhanced AMA production from autologous B cells in co-culture (73). Furthermore the Tfh associated cytokine IL-21 is increased in the serum and livers of treatment naïve PBC patients, is positively correlated with the percentage of circulating PBs and reduces with successful treatment with UDCA (73). In addition, CD19+ B cells have higher expression of the IL-21 receptor in PBC, suggesting they might be more receptive to IL-21 signaling (73). BAFF, another signal associated with B cell maturation, has been shown to be increased in PBC and positively correlates with AST and bilirubin (99). These observations support that B cell expansion and activation in PBC are driven by signals such as BAFF and IL-21 from Tfh cells.
Cytokines Regulating Inflammation and Recruitment in PBC
There is evidence that B cells produce cytokines in PBC that might contribute to disease pathogenesis.
Ex-vivo CD19+ B cells from PBC patients produced increased inflammatory cytokines interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α) compared to CD19+ cells from healthy controls (73). Circulating CXCL10, which can be produced by a variety of cells including B cells, has been observed to be increased in PBC patients and is expressed in portal regions or by hepatocytes in areas of focal necrosis (25, 100). These data support that B cells might contribute the inflammatory milieu in PBC, leading to the perpetuation of inflammation.
Other reports have investigated anti-inflammatory regulatory B cells in PBC. One study found although peripheral B regulatory cells (CD19+CD24hiCD38hi) were increased in PBC, ex-vivo these cells produce lower inhibitory signals such as IL-10 and showed less inhibitory activity towards CD4+ T cells (101). Instead, proinflammatory cytokines such as IL-6 and IL-12 were produced and CD4 T cell differentiation towards a Th1 phenotype occurred (101). However, other studies have not detected differences in circulating B regulatory cells in PBC (73) and a recent analysis of immune cells at the single cell level found a different population of regulatory B cells with a B10 phenotype (high CD24 and IgD expression) were decreased rather than increased in PBC patients (98). Together these results suggest the role of different B cell subsets in PBC pathogenesis is likely to be nuanced, and dependent on the fine tuning of pro- and anti- inflammatory cytokines.
The Gut Microbiome in PBC
Several studies support that the gut microbiome may play an important role in PBC pathogenesis. AMAs have been found to cross react with several bacterial proteins including Escherichia coli, which might initiate their production early in PBC (21, 102–104). Changes in the fecal microbiome may drive the polyclonal IgM response in PBC, which is less diverse in persons with PBC than in healthy counterparts (105–107). Specific bacterial populations in PBC are associated with alterations in bile acids, which are thought to directly contribute to biliary epithelial cell damage (108). Alterations in bile acid composition correlate with serum IgM in PBC both before and after treatment (108). Further work is required to determine causality between these associations.
Primary Sclerosing Cholangitis
Primary sclerosing cholangitis is a chronic disease characterized by immune mediated damage of the biliary tree resulting in concentric fibrosis and stricturing of the extrahepatic or intrahepatic bile ducts (109). Currently there is no effective treatment. PSC is strongly associated with Inflammatory Bowel Disease (IBD). PSC-IBD subjects have been shown to have a distinct gut microbiome as compared to individuals with IBD alone or healthy controls (110), leading to the hypothesis that loss of immunological tolerance to shared gut and liver antigens might be involved in PSC pathogenesis. The role of B cells in PSC pathogenesis is incompletely understood, but there is evidence that autoantigens towards microbiome sensing proteins and antibody secreting plasma cells might play a role.
Autoantibodies and B Cell Clonality in PSC
The association of several autoantibodies with PSC and evidence of their potential roles in pathogenesis is accumulating. Anti-neutrophil cytoplasmic antibodies (ANCA) are commonly detected in PSC, but their presence is not specific, and over 10 antigenic targets have been proposed [reviewed in (111)]. In one large European PSC cohort, 80% were positive for ANCA predominantly with a perinuclear staining pattern (p-ANCA) (112). Whether the presence of ANCA antibodies in PSC is protective or detrimental is not yet clear. ANCA specific for proteinase 3 (PR3), detected in 35% of PSC patients in one cohort, was shown to positively correlate with liver transaminase and ALP levels (113). An association between ANCA negative PSC patients and biliary cancer has also been reported (112).
Through the connection of PSC with IBD, common autoantibody targets have been investigated. Autoantibodies of the IgA subclass targeting glycoprotein 2 (GP2) were identified in patients with both PSC and IBD (114, 115). GP2 is not known to be expressed on the biliary epithelium but is present on intestinal microfold (M) cells and plays a role in antigen sensing of the gut microbiome (116, 117). This supports that the gut-liver axis might be key in PSC pathogenesis and loss of tolerance may initially occur in the gut (reviewed in (118)). The presence of anti-GP2 antibodies in individuals with PSC is associated with cholangiocarcinoma, increased mortality and reduced transplant free survival (119, 120), indicating anti-GP2 antibodies might play a pathogenic role. However observational studies report only 31–50% of PSC patients are anti-GP2 positive (120), suggesting other mechanisms are likely to be relevant.
Autoantibodies directed at the biliary epithelium itself have also been investigated. Sera from individuals with PSC has been shown to induce the production of pro-inflammatory cytokines such as IL-6 by biliary epithelial cells (121, 122). Supernatants from liver infiltrating B cells isolated from PSC patients and cultured ex-vivo have been shown to react to several proteins in an array assays, including nucleolar protein 3 (NOD3), expressed by cholangiocytes (82). B cell clonality studies also point to the involvement of as yet unidentified auto-antigens, with individuals with PSC exhibiting increased B cell clonality in the liver and gut compared to healthy tissue (123). This study also found significant overlap in expanded B cell clones shared between the gut and liver (123), further supporting the hypothesis that loss of tolerance in the gut is important.
Liver Infiltrating B Cells in PSC
A single study has addressed the phenotype of infiltrating B cells in PSC livers (123). Although B cells were significantly lower than in livers from PBC patients, PSC specimens had proportionally higher percentages of IgA or IgG positive PBs amongst antibody-secreting cells, and ex-vivo were able to secrete IgA and IgG immunoglobulins (82). This supports that PBs might be the predominant local source of autoantibody production in PSC, although further work is necessary to validate this and understand the mechanisms maintaining PB expansion.
Mdr2-/- mice which lack the biliary transport protein Mdr2, demonstrate sclerosing cholangitis and progressive liver fibrosis and have been used to model autoimmune cholestatic diseases including PSC (124). A recent study investigating the role of B cells in the pathogenesis of this model has revealed intrahepatic B cells produce IgG constitutively, the levels of which positively correlated with serum BAFF. BAFF blockade or depletion of CD20+ B cells by monoclonal antibody reduced hepatic fibrosis in this model (125), raising the possibility that similar effects could be seen with B cell depletion in PSC patients. However, as the primary deficit in this model is due to impairment in bile transport rather than alterations in the gut-liver axis, the translation of these findings to PSC may not bare out.
The Gut Microbiome in PSC
In PSC and PSC-IBD, microbiome association studies in both stool and the intestinal mucosa have identified alterations in the bacterial component of the microbiome (126). Increased relative abundance of Enterococcus, Veilonella and Streptococcus genera have been reported in multiple studies, with Klebsiella pneumoniae-harboring microbiomes from patients with PSC/UC being implicated as a driver of Th17 responses in gnotobiotic mouse models (127). Human studies to examine the relationship between the gut microbiome and B cells in PSC will be required to understand whether they are causally linked.
B Cell Responses to Immunomodulatory Therapies
Corticosteroids
Corticosteroids are steroid hormones used as first line induction therapy to control inflammation in patients with AIH, IgG4-HBD and autoimmune overlap with cholestatic liver disease, usually leading to both clinical and biochemical improvement (13, 70, 109). On reduction of steroid dose or withdrawal of medication, disease relapse occurs frequently (>80% in AIH and 35-60% in IgG4-HBD) (128, 129). Prednisolone therapy has been shown to decrease activated T cells, inhibit the differentiation of B lymphocytes into PC, reduce IL-10 and IL-21 cytokine levels, and the expression of BLIMP-1 and Bcl6 that regulate PC differentiation in mouse models of autoimmune disease (130). Intra-hepatic B cells and T regulatory cell proliferation is suppressed by prednisolone in adults with AIH (131). Circulating PBs and activated PD1+ Tfh cells are decreased and clonal B cell expansion is reduced within 12 weeks of prednisolone therapy in adults with IgG4-HPB disease (57, 132, 133).
Ursodeoxycholic Acid
Ursodeoxycholic acid (UDCA) is a secondary bile acid used to treat cholestatic liver diseases. UDCA main mechanisms of actions include a) protection of cholangiocytes against cytotoxicity of hydrophobic bile acids, b) stimulation of hepatobiliary secretion and c) protection of hepatocytes against bile acid induced apoptosis. In patients with PBC UDCA improves liver biochemistry, may delay disease progression to severe fibrosis/cirrhosis, and prolongs transplant free survival. In patients with PSC it improves liver biochemistry and surrogate markers of prognosis (70, 109). In patients with IgG4-HBD it may also improve liver biochemistry but has no known impact on disease progression or survival. Reductions in CD19+ B cells and B cell clonal expansion have been observed after UDCA treatment in patients with PBC (92), although it is not known if this has a direct or indirect relationship to UDCA treatment, and whether the observed alterations in the B cell compartment contribute to disease remission. Similar studies in PSC and IgG4-HBD in response to UDCA have not been done.
Rituximab
Rituximab is a monoclonal antibody which targets the humoral immune response by inducing B lymphocyte depletion and decreased production of autoantibodies. It has been used to treat individuals intolerant or unresponsive to standard therapy therapies in AIH and IgG4-HBD (28, 114) and in a clinical trial for symptomatic fatigue in PBC (84).
A prospective open label trial (134) and several retrospective observational studies of between 10 and 60 participants support rituximab as an effective agent to treat active IgG4-RD [Table 2 (68, 134–141)]. Maintenance treatment with further rituximab infusions reduces the risk of IgG4-RD relapse compared with induction therapy alone (141), but this has not been confirmed in randomized trials. Side effects including infusion reactions, hypogammaglobulinemia and infection have been reported (134–136, 139, 140). In IgG4-HBD, rituximab not only depletes CD20+ B cells, but also short-lived CD20- PBs and reduces cytotoxic CD4+ T cells (137, 138). Long-lived CD20- PCs that reside in the bone marrow are also not depleted and might represent a niche of pathogenic cells that drive disease relapse. If disease reactivation occurs expanded PBs of different clonalities are observed (59), indicating B cells with different antigen affinities might be involved in repeated rounds of inflammation. Interestingly, rituximab leads to selective decreases in serum IgG4 levels in both patients with IgG4-RD and in Rheumatoid arthritis where serum IgG4 levels at baseline are normal (134, 144, 145). This suggests short-lived PB are the main contributors to serum IgG4 levels and disease activity is reduced through depletion of these cells.
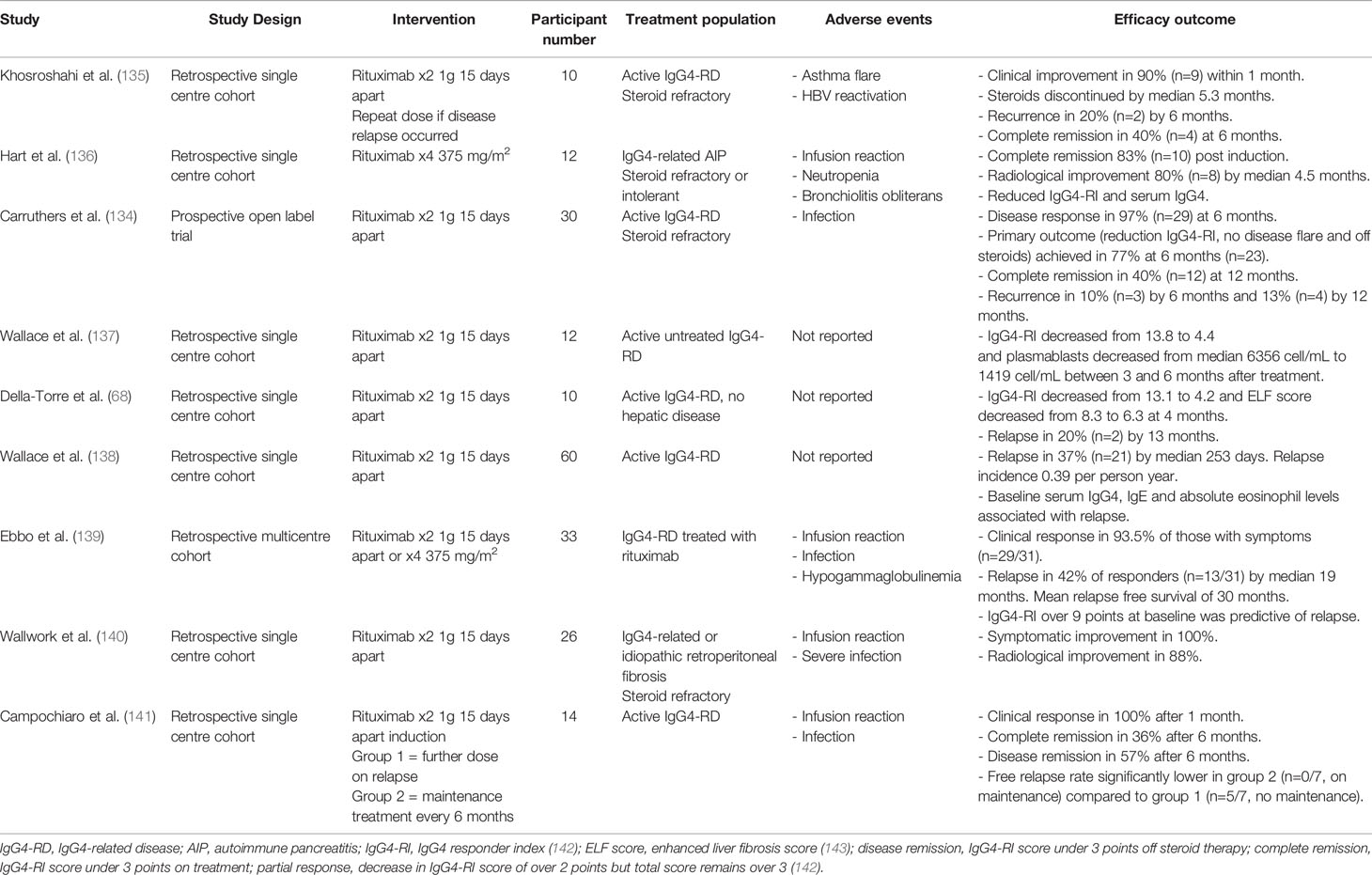
Table 2 Studies of Rituximab for treatment of IgG4-RD.
A single center open label trial of rituximab in 6 AIH patients and a retrospective cohort of 22 AIH patients in France between 2007 and 2017 demonstrated significant improvements in serum IgG and liver transaminases sustained for up to 24 months after treatment and reported no significant adverse events (9, 10). Compared to IgG4-RD, AIH involves a more cell-mediated immune process, however rituximab has been used successfully in other diseases with a similar mechanism such as multiple sclerosis, indirectly altering cell-mediated responses (146). Mouse models have shown that B lymphocyte depletion impairs CD4+ T cell activation in response to pathogen challenges, which may in part explain the effect of rituximab in AIH. In paired liver biopsies of AIH before and after rituximab therapy, inflammation grade which correlated with CD4 regulatory T cells, improved with treatment (10). This suggests B cell depletion in AIH might work therapeutically through an indirect reduction in liver infiltrating CD4 T cells.
Rituximab has been shown to reduce auto-antibody production (AMA, IgM) and biochemical values (ALP) in a small number PBC patients unresponsive to standard therapy with UDCA (85, 147). A randomized trial of rituximab in 57 early stage PBC patients on UDCA did not significantly improve fatigue (primary end point), but those in the treatment arm had significantly lower ALP levels 3 months after infusion (84). Assessment of B cell function in PBC patients after rituximab demonstrated that depletion of B cells influences the induction, maintenance, and activation of both B and T cells. Transient decreases in memory B cell and T cell frequencies and an increase in T regulatory cells are observed. This is associated with increased in mRNA levels of forkhead box P3 (FoxP3) and tissue growth factor-beta (TGF-β) and a decreased TNF-α in CD4 T cells after B cell depletion treatment (147).
Novel B Cell Therapeutic Targets
Several novel therapies are under investigation for immune-mediated liver diseases that directly or indirectly target the B cell lineage (Figure 2). These have various mechanisms of action including B cell depletion, inhibition of direct B cell signaling through cell-cell interactions and inhibition of B cell signaling through cytokine production.
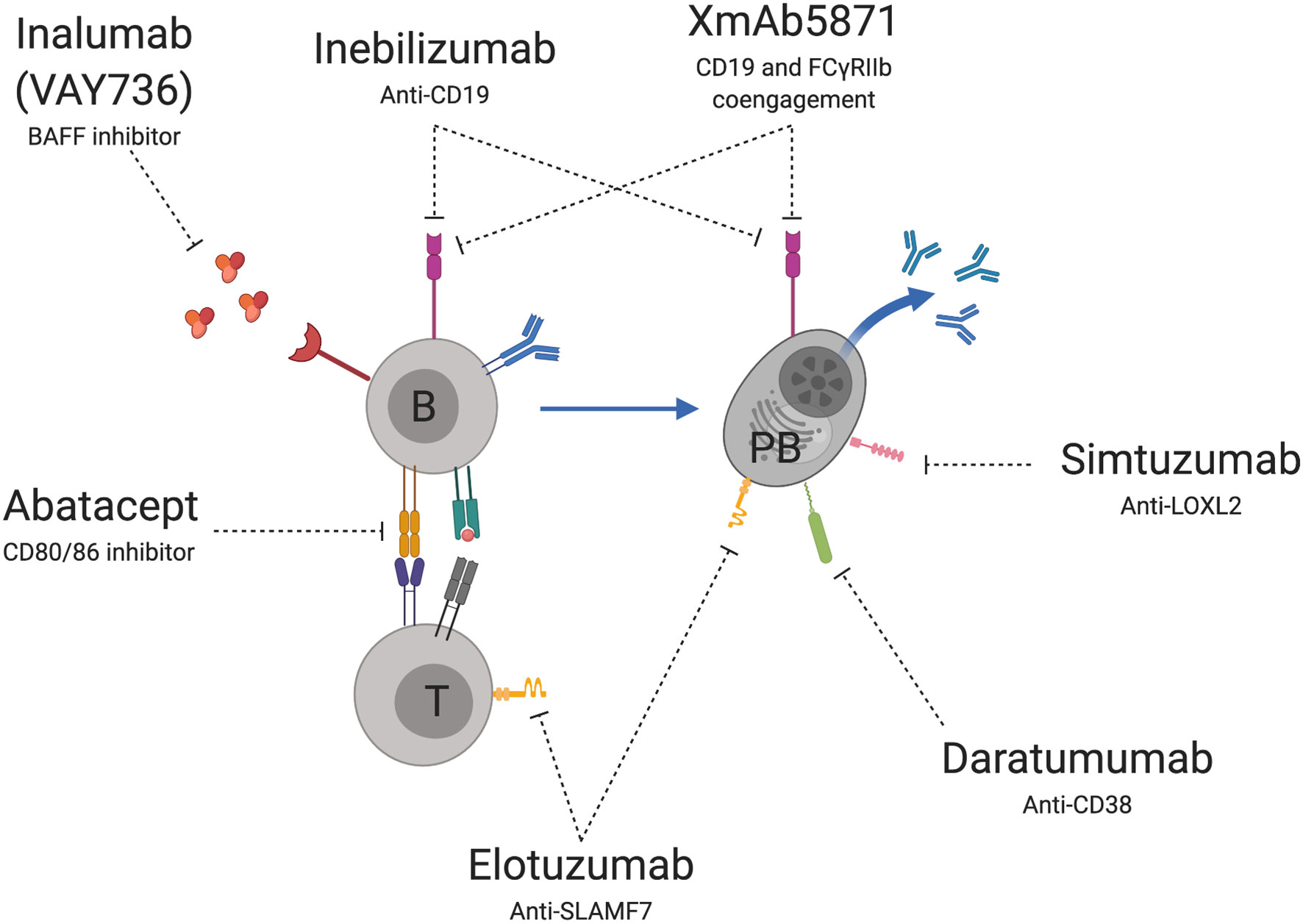
Figure 2 Novel B cell therapeutic targets for immune-mediated liver diseases. Treatments under development or evaluation include agents that deplete or disrupt B cells (B) or plasmablast (PB) signaling through CD-19, disruption of PB activation through CD-38, inhibition of PB mediated fibrosis through LOX-2 and inhibition of SLAM-F7 on PB and T cells (T). Other targets prevent B cell signaling through soluble mediators such as BAFF and receptor mediated T cell (T) co-activation through CD80/86. Image created using BioRender.com.
Agents that directly target B cells include monoclonal antibodies specific for the CD19 receptor. Treatment leads to depletion of CD19 expressing cells including B cells and PBs and has shown efficacy in treating other autoimmune disorders where B cells play a role in pathogenesis including systemic sclerosis (148), multiple sclerosis (149), and neuromyelitis optica (150). A randomized placebo-controlled trial of Inebilizumab, a monoclonal antibody specific for CD19, is currently underway in patients with active IgG4-RD (151).
Other monoclonal antibodies that directly target B cells or PBs inhibit signaling through specific surface receptor targets. These include LOXL2, which plays a role in fibrogenesis in PSC and IgG4-RD (67), SLAMF7 which is expressed highly on PBs and CTLs in IgG4-RD (59, 60) and CD38 which is expressed on expanded PBs in IgG4-RD (34, 58, 132, 152). Simtuzumab, a monoclonal antibody specific to LOXL2, has been evaluated in 234 PSC patients for its ability to reverse fibrosis. However it did not achieve a reduction in hepatic collagen content on liver biopsy or improve Ishak fibrosis stage (153). It had not been evaluated other autoimmune liver diseases. Elotuzumab, a monoclonal antibody specific to SLAMF7 and Daratumumab, a monoclonal antibody specific to CD38 are also available [reviewed in (154)] but have not been evaluated for therapy of autoimmune liver disease.
Other monoclonal antibodies alter B cell or PB surface receptor co-engagement with receptors on other cells. These include interruption of T cell activation by antigen presenting B cells through blocking CD80/86 – CD28 signaling by Abatacept, and suppression of B cell responses through co-engagement of the Fc-gamma receptor IIb with CD19 by XmAb5871 (155–157). Abatacept has shown efficacy in other B cell mediated autoimmune conditions such as Rheumatoid arthritis (158). Disappointingly, an open label trial of Abatacept in 16 PBC patients who had not responded to UDCA therapy reported that only a single patient met the primary endpoint of a reduction in ALP of over 40% from baseline and there were no significant reductions in serum IgM (159). However, clinical trials of Abatacept in IgG4-RD and AIH are ongoing and have not yet reported (160, 161). Results of XmAb5871 in autoimmune liver diseases have been more promising. XmAb5871 was evaluated in a prospective open label clinical trial in 20 individuals with active IgG4-RD (162). The primary outcome, improvement in IgG4 disease responder index by over 2 points by day 169 after therapy, was achieved in 80% of participants. During follow up, pneumonia, chronic lymphocytic leukemia and chronic inflammatory demyelinating polyneuropathy occurred certain individuals, but may not have been directly related to therapy (162, 163).
B cells and PBs are also indirectly targeted by several therapeutic agents. For example, B cell activating factor receptor (BAFF) inhibitors such as Inalumab (VAY736), prevent BAFF signaling and indirectly lead to B cell depletion. Inalumab has recently shown safety and efficacy in a phase II trial in individuals with primary Sjögren’s syndrome, a B cell mediated autoimmune disease involving exocrine glands (164) and has been used successfully to induce remission in 2 reported cases of difficult to treat AIH (165). A randomized controlled trial assessing Inalumab (VAY736) for the treatment of AIH is currently recruiting (166).
Finally, novel therapeutic agents indirectly targeting B cell signaling through cytokine production are also available. For example NI-0801 is a monoclonal directed towards CXCL10, which is produced by B cells and is increased in the serum of PBC patients (25, 100). NI-0801 has been trialed in 29 PBC patients that were unresponsive to UDCA but unfortunately was not efficacious at reducing ALP levels (167).
Currently the majority of novel B cell therapies are in early-stage clinical trials and their associated risk profiles are being elucidated. In the case of Rituximab, which is used for the treatment of several autoimmune conditions and lymphoproliferative disorders, the risks of treatment include reactivation of latent viral illness, hypogammaglobulinemia and in impaired B cell reconstitution, which can result in recurrent or reactivation of infection (168, 169). The extent to which these risks might occur with other B ell depleting or suppressing therapies in the specific context of treating autoimmune liver disease requires investigation. Despite these potential risks, overall novel B cell therapies hold great promise to improve the treatment options for autoimmune liver diseases going forward.
Current Research Gaps and Potential Developments in the Field
Current data support that B cells play a role in autoimmune liver disease pathogenesis, through autoantibody production, interaction with T cells via antigen presentation or cytokine signaling. Evidence that B cell depletion with rituximab can induce disease remission has led to support for its use for treatment refractory cases of IgG4-RD and AIH (33, 170). Several other treatments targeting B cells are under development for the treatment of immune mediated liver diseases, for example monoclonal antibodies targeting CD19 or BAFF. Some of these agents are in phase I therapeutic trials, which if successful could increase the armory of therapeutic agents for the treatment of autoimmune liver conditions (160–162, 166).
Due to the relative rarity of autoimmune liver disease, the evaluation of novel therapies with sufficient power to detect efficacy will need to be done collaboratively on a national and international scale. Inclusive trials that evaluate efficacy in a group of rare conditions with similar pathogenesis is one mechanism to recruit sufficient patients, an approach that has been taken with other immune mediated rare diseases such as the vasculitidies (171, 172). In the UK, nationally recruiting cohort studies for AIH [AIH-UK (173)], IgG4-RD [IgG4-systemic disease (174)], PBC [PBC-UK (175)] and PSC [PSC-UK (176)] provide an existing network for clinicians and scientists working collaboratively on autoimmune liver diseases, that could be harnessed for national therapeutic clinical trials.
The holistic patient experience living with autoimmune liver disease covering the spectrum of symptoms, diagnosis, treatment and management pathways should also be considered when evaluating B cell targeting therapies. Recent data from the UK-AIH national cohort study indicate quality of life is significantly impaired in AIH patients, as is particularly associated with steroid use (177). A recently published systematic review of patient reported outcomes in studies of PBC and PSC concluded that although the use of patient reported outcomes has increased over time, many are nonspecific and unvalidated (178). This underscores the importance in developing and evaluating relevant patient reported outcomes in clinical trials of novel therapeutics alongside clinical efficacy signals.
Finally, studies evaluating immunotherapies should focus on in depth evaluation of the immunological response to therapy both in the peripheral blood and if possible, at the site of disease. Recent data has shown liver fine needle aspirate (FNA) to safe in assessing the hepatic immunological milieu and has highlighted differences between immune responses in the blood and the liver (179). Studies evaluating the immunological response to B cell targeting therapies in autoimmune liver diseases should consider employing FNA to understand the local response to treatment, and in turn develop more targeted therapies towards B cells within the liver itself.
Author Contributions
TC and EC jointly wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
EC is supported by the National Institute of Health Research (NIHR) Biomedical Research Centre, based at Oxford University Hospitals Trust and Oxfordshire Health Service Research Committee (OHSRC) as part of Oxford Hospitals Charity, Oxford. TC receives funding from a Wellcome Trust Training Fellowship for Clinicians [211042/Z/18/Z]. The views expressed in this article are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
Conflict of Interest
EC is on the advisory board for Viela Bio for IgG4-realted disease.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Akkaya M, Kwak K, Pierce SK. B cell memory: building two walls of protection against pathogens. Nat Rev Immunol (2020) 20(4):229–38. doi: 10.1038/s41577-019-0244-2
2. Chi X, Li Y, Qiu X. V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: mechanism and regulation. Immunology (2020) 160(3):233–47. doi: 10.1111/imm.13176
3. Cyster JG, Allen CDC. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell (2019) 177(3):524–40. doi: 10.1016/j.cell.2019.03.016
4. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol (2012) 30:221–41. doi: 10.1146/annurev-immunol-020711-074934
5. Taylor SA, Assis DN, Mack CL. The Contribution of B Cells in Autoimmune Liver Diseases. Semin Liver Dis (2019) 39(4):422–31. doi: 10.1055/s-1688751
6. Hennes EM, Zeniya M, Czaja AJ, Parés A, Dalekos GN, Krawitt EL, et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology (2008) 48(1):169–76. doi: 10.1002/hep.22322
7. Czaja AJ. Examining pathogenic concepts of autoimmune hepatitis for cues to future investigations and interventions. World J Gastroenterol (2019) 25(45):6579–606. doi: 10.3748/wjg.v25.i45.6579
8. Webb GJ, Hirschfield GM, Krawitt EL, Gershwin ME. Cellular and Molecular Mechanisms of Autoimmune Hepatitis. Annu Rev Pathol (2018) 24(13):247–92. doi: 10.1146/annurev-pathol-020117-043534
9. Than NN, Hodson J, Schmidt-Martin D, Taubert R, Wawman RE, Botter M, et al. Efficacy of rituximab in difficult-to-manage autoimmune hepatitis: Results from the International Autoimmune Hepatitis Group. JHEP Rep (2019) 1(6):437–45. doi: 10.1016/j.jhepr.2019.10.005
10. Burak KW, Swain MG, Santodomingo-Garzon T, Santodomino-Garzon T, Lee SS, Urbanski SJ, et al. Rituximab for the treatment of patients with autoimmune hepatitis who are refractory or intolerant to standard therapy. Can J Gastroenterol = J Can Gastroenterol (2013) 27(5):273–80. doi: 10.1155/2013/512624
11. Béland K, Marceau G, Labardy A, Bourbonnais S, Alvarez F. Depletion of B cells induces remission of autoimmune hepatitis in mice through reduced antigen presentation and help to T cells. Hepatology (2015) 62(5):1511–23. doi: 10.1002/hep.27991
12. Sebode M, Weiler-Normann C, Liwinski T, Schramm C. Autoantibodies in Autoimmune Liver Disease-Clinical and Diagnostic Relevance. Front Immunol (2018) 9:609. doi: 10.3389/fimmu.2018.00609
13. Lüth S, Herkel J, Kanzler S, Frenzel C, Galle PR, Dienes HP, et al. Serologic markers compared with liver biopsy for monitoring disease activity in autoimmune hepatitis. J Clin Gastroenterol (2008) 42(8):926–30. doi: 10.1097/MCG.0b013e318154af74
14. Muratori L, Parola M, Ripalti A, Robino G, Muratori P, Bellomo G, et al. Liver/kidney microsomal antibody type 1 targets CYP2D6 on hepatocyte plasma membrane. Gut (2000) 46(4):553–61. doi: 10.1136/gut.46.4.553
15. Lapierre P, Johanet C, Alvarez F. Characterization of the B cell response of patients with anti-liver cytosol autoantibodies in type 2 autoimmune hepatitis. Eur J Immunol (2003) 33(7):1869–78. doi: 10.1002/eji.200323857
16. Ma L, Qin J, Ji H, Zhao P, Jiang Y. Tfh and plasma cells are correlated with hypergammaglobulinaemia in patients with autoimmune hepatitis. Liver Int (2014) 34(3):405–15. doi: 10.1111/liv.12245
17. Abe K, Takahashi A, Imaizumi H, Hayashi M, Okai K, Kanno Y, et al. Interleukin-21 plays a critical role in the pathogenesis and severity of type I autoimmune hepatitis. Springerplus (2016) 5(1):777. doi: 10.1186/s40064-016-2512-y
18. Nishikawa H, Enomoto H, Iwata Y, Kishino K, Shimono Y, Hasegawa K, et al. B-Cell Activating Factor Belonging to the Tumor Necrosis Factor Family and Interferon-γ-Inducible Protein-10 in Autoimmune Hepatitis. Med (Baltimore) (2016) 95(12):e3194. doi: 10.1097/MD.0000000000003194
19. Migita K, Abiru S, Maeda Y, Nakamura M, Komori A, Ito M, et al. Elevated serum BAFF levels in patients with autoimmune hepatitis. Hum Immunol (2007) 68(7):586–91. doi: 10.1016/j.humimm.2007.03.010
20. Schultheiß C, Simnica D, Willscher E, Oberle A, Fanchi L, Bonzanni N, et al. Next-generation immunosequencing reveals pathological T cell architecture in autoimmune hepatitis. Hepatology (2020). doi: 10.1002/hep.31473
21. Ma Y, Bogdanos DP, Hussain MJ, Underhill J, Bansal S, Longhi MS, et al. Polyclonal T-cell responses to cytochrome P450IID6 are associated with disease activity in autoimmune hepatitis type 2. Gastroenterology (2006) 130(3):868–82. doi: 10.1053/j.gastro.2005.12.020
22. Longhi MS, Hussain MJ, Bogdanos DP, Quaglia A, Mieli-Vergani G, Ma Y, et al. Cytochrome P450IID6-specific CD8 T cell immune responses mirror disease activity in autoimmune hepatitis type 2. Hepatology (2007) 46(2):472–84. doi: 10.1002/hep.21658
23. Renand A, Cervera-Marzal I, Gil L, Dong C, Garcia A, Kervagoret E, et al. Integrative molecular profiling of autoreactive CD4 T cells in autoimmune hepatitis. J Hepatol (2020) 73(6):1379–90.. doi: 10.1101/2020.01.06.895938
24. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature (2017) 542(7639):110–4. doi: 10.1038/nature20810
25. Nishioji K, Okanoue T, Itoh Y, Narumi S, Sakamoto M, Nakamura H, et al. Increase of chemokine interferon-inducible protein-10 (IP-10) in the serum of patients with autoimmune liver diseases and increase of its mRNA expression in hepatocytes. Clin Exp Immunol (2001) 123(2):271–9. doi: 10.1046/j.1365-2249.2001.01391.x
26. Liu X, Jiang X, Liu R, Wang L, Qian T, Zheng Y, et al. B cells expressing CD11b effectively inhibit CD4+ T-cell responses and ameliorate experimental autoimmune hepatitis in mice. Hepatology (2015) 62(5):1563–75. doi: 10.1002/hep.28001
27. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol (2018) 15(7):397–411. doi: 10.1038/s41575-0011-z
28. Wang R, Tang R, Li B, Ma X, Schnabl B, Tilg H. Gut microbiome, liver immunology, and liver diseases. Cell Mol Immunol (2021) 18(1):4–17. doi: 10.1038/s41423-020-00592-6
29. Moro-Sibilot L, Blanc P, Taillardet M, Bardel E, Couillault C, Boschetti G, et al. Mouse and Human Liver Contain Immunoglobulin A-Secreting Cells Originating From Peyer’s Patches and Directed Against Intestinal Antigens. Gastroenterology (2016) 151(2):311–23. doi: 10.1053/j.gastro.2016.04.014
30. Yuksel M, Wang Y, Tai N, Peng J, Guo J, Beland K, et al. A novel ‘humanized mouse’ model for autoimmune hepatitis and the association of gut microbiota with liver inflammation. Hepatology (2015) 62(5):1536–50. doi: 10.1002/hep.27998
31. Wei Y, Li Y, Yan L, Sun C, Miao Q, Wang Q, et al. Alterations of gut microbiome in autoimmune hepatitis. Gut (2020) Mar69(3):569–77. doi: 10.1136/gutjnl-2018-317836
32. Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol (2012) Sep25(9):1181–92. doi: 10.1038/modpathol.2012.72
33. Specialised Commissioning Team. Clinical Commissioning Policy: Rituximab for immunoglobulin G4- related disease (IgG4-RD). NHS England (2016). Available at: https://www.england.nhs.uk/wp-content/uploads/2016/12/clin-comm-pol-16057P.pdf.
34. Lanzillotta M, Della-Torre E, Milani R, Bozzolo E, Bozzalla-Cassione E, Rovati L, et al. Effects of glucocorticoids on B-cell subpopulations in patients with IgG4-related disease. Clin Exp Rheumatol (2019) 37 Suppl 118(3):159–66.
35. Morell A, Skvaril F, Steinberg AG, Van Loghem E, Terry WD. Correlations between the concentrations of the four sub-classes of IgG and Gm Allotypes in normal human sera. J Immunol (1972) 108(1):195–206.
36. Rispens T, Ooijevaar-de Heer P, Bende O, Aalberse RC. Mechanism of immunoglobulin G4 Fab-arm exchange. J Am Chem Soc (2011) 133(26):10302–11. doi: 10.1021/ja203638y
37. van der Zee JS, van Swieten P, Aalberse RC. Inhibition of complement activation by IgG4 antibodies. Clin Exp Immunol (1986) 64(2):415–22.
38. van der Zee JS, van Swieten P, Aalberse RC. Serologic aspects of IgG4 antibodies. II. IgG4 antibodies form small, nonprecipitating immune complexes due to functional monovalency. J Immunol (1986) 137(11):3566–71.
39. Lighaam LC, Vermeulen E, Bleker Td, Meijlink KJ, Aalberse RC, Barnes E, et al. Phenotypic differences between IgG4+ and IgG1+ B cells point to distinct regulation of the IgG4 response. J Allergy Clin Immunol (2014) 133(1):267–70.e1-6. doi: 10.1016/j.jaci.2013.07.044
40. Chliva C, Aggelides X, Makris M, Katoulis A, Rigopoulos D, Tiligada E. Comparable profiles of serum histamine and IgG4 levels in allergic beekeepers. Allergy (2015) 70(4):457–60. doi: 10.1111/all.12568
41. van Schouwenburg PA, Krieckaert CL, Nurmohamed M, Hart M, Rispens T, Aarden L, et al. IgG4 production against adalimumab during long term treatment of RA patients. J Clin Immunol (2012) 32(5):1000–6. doi: 10.1007/s10875-012-9705-0
42. Lighaam LC, Rispens T. The Immunobiology of Immunoglobulin G4. Semin Liver Dis (2016) 36(3):200–15. doi: 10.1055/s-0036-1584322
43. Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: an odd antibody. Clin Exp Allergy (2009) 39(4):469–77. doi: 10.1111/j.1365-2222.2009.03207.x
44. Unger P-PA, Lighaam LC, Vermeulen E, Kruithof S, Makuch M, Culver EL, et al. Divergent chemokine receptor expression and the consequence for human IgG4 B cell responses. Eur J Immunol (2020) 50(8):1113–25. doi: 10.1002/eji.201948454
45. Jeannin P, Lecoanet S, Delneste Y, Gauchat JF, Bonnefoy JY. IgE versus IgG4 production can be differentially regulated by IL-10. J Immunol (1998) 160(7):3555–61.
46. van de Veen W, Stanic B, Yaman G, Wawrzyniak M, Söllner S, Akdis DG, et al. IgG4 production is confined to human IL-10-producing regulatory B cells that suppress antigen-specific immune responses. J Allergy Clin Immunol (2013) 131(4):1204–12. doi: 10.1016/j.jaci.2013.01.014
47. Meiler F, Zumkehr J, Klunker S, Rückert B, Akdis CA, Akdis M. In vivo switch to IL-10-secreting T regulatory cells in high dose allergen exposure. J Exp Med (2008) 205(12):2887–98. doi: 10.1084/jem.20080193
48. Zen Y, Fujii T, Harada K, Kawano M, Yamada K, Takahira M, et al. Th2 and regulatory immune reactions are increased in immunoglobin G4-related sclerosing pancreatitis and cholangitis. Hepatology (2007) 45(6):1538–46. doi: 10.1002/hep.21697
49. Heeringa JJ, Karim AF, van Laar JAM, Verdijk RM, Paridaens D, van Hagen PM, et al. Expansion of blood IgG4+ B, TH2, and regulatory T cells in patients with IgG4-related disease. J Allergy Clin Immunol (2018) 141(5):1831–43.e10. doi: 10.1016/j.jaci.2017.07.024
50. Maehara T, Moriyama M, Nakashima H, Miyake K, Hayashida J-N, Tanaka A, et al. Interleukin-21 contributes to germinal centre formation and immunoglobulin G4 production in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz’s disease. Ann Rheum Dis (2012) 71(12):2011–9. doi: 10.1136/annrheumdis-2012-201477
51. Chen Y, Lin W, Yang H, Wang M, Zhang P, Feng R, et al. Aberrant Expansion and Function of Follicular Helper T Cell Subsets in IgG4-Related Disease. Arthritis Rheumatol (2018) 70(11):1853–65. doi: 10.1002/art.40556
52. Cargill T, Makuch M, Sadler R, Lighaam LC, Peters R, van Ham M, et al. Activated T-Follicular Helper 2 Cells Are Associated With Disease Activity in IgG4-Related Sclerosing Cholangitis and Pancreatitis. Clin Transl Gastroenterol (2019) 10(4):e00020. doi: 10.14309/ctg.0000000000000020
53. Shiokawa M, Kodama Y, Sekiguchi K, Kuwada T, Tomono T, Kuriyama K, et al. Laminin 511 is a target antigen in autoimmune pancreatitis. Sci Transl Med (2018) 10(453):eaaq0997. doi: 10.1126/scitranslmed.aaq0997
54. Perugino CA, AlSalem SB, Mattoo H, Della-Torre E, Mahajan V, Ganesh G, et al. Identification of galectin-3 as an autoantigen in patients with IgG4-related disease. J Allergy Clin Immunol (2019) 143(2):736–45.e6. doi: 10.1016/j.jaci.2018.05.011
55. Culver EL, Vermeulen E, Makuch M, van Leeuwen A, Sadler R, Cargill T, et al. Increased IgG4 responses to multiple food and animal antigens indicate a polyclonal expansion and differentiation of pre-existing B cells in IgG4-related disease. Ann Rheum Dis (2015) 74(5):944–7. doi: 10.1136/annrheumdis-2014-206405
56. de Buy Wenniger LJM, Doorenspleet ME, Klarenbeek PL, Verheij J, Baas F, Elferink RPO, et al. Immunoglobulin G4+ clones identified by next-generation sequencing dominate the B cell receptor repertoire in immunoglobulin G4 associated cholangitis. Hepatology (2013) 57(6):2390–8. doi: 10.1002/hep.26232
57. Doorenspleet ME, Hubers LM, Culver EL, de Buy Wenniger LJM, Klarenbeek PL, Chapman RW, et al. Immunoglobulin G4(+) B-cell receptor clones distinguish immunoglobulin G 4-related disease from primary sclerosing cholangitis and biliary/pancreatic malignancies. Hepatology (2016) 64(2):501–7. doi: 10.1002/hep.28568
58. Mattoo H, Mahajan VS, Della-Torre E, Sekigami Y, Carruthers M, Wallace ZS, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol (2014) 134(3):679–87. doi: 10.1016/j.jaci.2014.03.034
59. Mattoo H, Mahajan VS, Maehara T, Deshpande V, Della-Torre E, Wallace ZS, et al. Clonal expansion of CD4(+) cytotoxic T lymphocytes in patients with IgG4-related disease. J Allergy Clin Immunol (2016) 138(3):825–38. doi: 10.1016/j.jaci.2015.12.1330
60. Maehara T, Mattoo H, Ohta M, Mahajan VS, Moriyama M, Yamauchi M, et al. Lesional CD4+ IFN-γ+ cytotoxic T lymphocytes in IgG4-related dacryoadenitis and sialoadenitis. Ann Rheum Dis (2017) 76(2):377–85. doi: 10.1136/annrheumdis-2016-209139
61. de Wit J, Jorritsma T, Makuch M, Remmerswaal EBM, Klaasse Bos H, Souwer Y, et al. Human B cells promote T-cell plasticity to optimize antibody response by inducing coexpression of T(H)1/T(FH) signatures. J Allergy Clin Immunol (2015) 135(4):1053–60. doi: 10.1016/j.jaci.2014.08.012
62. Akiyama M, Suzuki K, Yamaoka K, Yasuoka H, Takeshita M, Kaneko Y, et al. Number of Circulating Follicular Helper 2 T Cells Correlates With IgG4 and Interleukin-4 Levels and Plasmablast Numbers in IgG4-Related Disease. Arthritis Rheumatol (Hoboken NJ) (2015) 67(9):2476–81. doi: 10.1002/art.39209
63. Akiyama M, Yasuoka H, Yamaoka K, Suzuki K, Kaneko Y, Kondo H, et al. Enhanced IgG4 production by follicular helper 2 T cells and the involvement of follicular helper 1 T cells in the pathogenesis of IgG4-related disease. Arthritis Res Ther (2016) 1318:167. doi: 10.1186/s13075-016-1064-4
64. Grados A, Ebbo M, Piperoglou C, Groh M, Regent A, Samson M, et al. T Cell Polarization toward TH2/TFH2 and TH17/TFH17 in Patients with IgG4-Related Disease. Front Immunol (2017) 8:235. doi: 10.3389/fimmu.2017.00235
65. Kubo S, Nakayamada S, Zhao J, Yoshikawa M, Miyazaki Y, Nawata A, et al. Correlation of T follicular helper cells and plasmablasts with the development of organ involvement in patients with IgG4-related disease. Rheumatol (Oxford) (2018) 0157(3):514–24. doi: 10.1093/rheumatology/kex455
66. Maehara T, Mattoo H, Mahajan VS, Murphy SJ, Yuen GJ, Ishiguro N, et al. The expansion in lymphoid organs of IL-4+ BATF+ T follicular helper cells is linked to IgG4 class switching in vivo. Life Sci Alliance (2018) 1(1). doi: 10.26508/lsa.201800050
67. Della-Torre E, Rigamonti E, Perugino C, Baghai-Sain S, Sun N, Kaneko N, et al. B lymphocytes directly contribute to tissue fibrosis in patients with IgG4-related disease. J Allergy Clin Immunol (2020) 145(3):968–981.e14. doi: 10.1016/j.jaci.2019.07.004
68. Della-Torre E, Feeney E, Deshpande V, Mattoo H, Mahajan V, Kulikova M, et al. B-cell depletion attenuates serological biomarkers of fibrosis and myofibroblast activation in IgG4-related disease. Ann Rheum Dis (2015) 74(12):2236–43. doi: 10.1136/annrheumdis-2014-205799
69. Plichta DR, Somani J, Pichaud M, Wallace ZS, Fernandes AD, Perugino CA, et al. Congruent microbiome signatures in fibrosis-prone autoimmune diseases: IgG4-related disease and systemic sclerosis. Genome Med (2021) 13(1):35. doi: 10.1186/s13073-021-00853-7
70. European Association for the Study of the Liver. Electronic address:ZWFzbG9mZmljZUBlYXNsb2ZmaWNlLmV1LA== European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J Hepatol (2017) 67(1):145–72. doi: 10.1016/j.jhep.2017.03.022
71. Hirschfield GM, Gershwin ME. The immunobiology and pathophysiology of primary biliary cirrhosis. Annu Rev Pathol (2013) 8:303–30. doi: 10.1146/annurev-pathol-020712-164014
72. Takahashi T, Miura T, Nakamura J, Yamada S, Miura T, Yanagi M, et al. Plasma cells and the chronic nonsuppurative destructive cholangitis of primary biliary cirrhosis. Hepatology (2012) 55(3):846–55. doi: 10.1002/hep.24757
73. Li Y, Wang W, Tang L, He X, Yan X, Zhang X, et al. Chemokine (C-X-C motif) ligand 13 promotes intrahepatic chemokine (C-X-C motif) receptor 5+ lymphocyte homing and aberrant B-cell immune responses in primary biliary cirrhosis. Hepatology (2015) 61(6):1998–2007. doi: 10.1002/hep.27725
74. Nakamura M, Nishida N, Kawashima M, Aiba Y, Tanaka A, Yasunami M, et al. Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. Am J Hum Genet (2012) 91(4):721–8. doi: 10.1016/j.ajhg.2012.08.010
75. Dong M, Li J, Tang R, Zhu P, Qiu F, Wang C, et al. Multiple genetic variants associated with primary biliary cirrhosis in a Han Chinese population. Clin Rev Allergy Immunol (2015) 48(2–3):316–21. doi: 10.1007/s12016-015-8472-0
76. Mells GF, Floyd JAB, Morley KI, Cordell HJ, Franklin CS, Shin S-Y, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet (2011) 43(4):329–32. doi: 10.1038/ng.789
77. Tanaka A, Invernizzi P, Ohira H, Kikuchi K, Nezu S, Kosoy R, et al. Replicated association of 17q12-21 with susceptibility of primary biliary cirrhosis in a Japanese cohort. Tissue Antigens (2011) 78(1):65–8. doi: 10.1111/j.1399-0039.2011.01684.x
78. Qiu F, Tang R, Zuo X, Shi X, Wei Y, Zheng X, et al. A genome-wide association study identifies six novel risk loci for primary biliary cholangitis. Nat Commun (2017) 8:14828. doi: 10.1038/ncomms14828
79. Ueno K, Aiba Y, Hitomi Y, Shimoda S, Nakamura H, Gervais O, et al. and mRNA Microarray Analysis Identified IFNG and CD40L as the Central Upstream Regulators in Primary Biliary Cholangitis. Hepatol Commun (2020) 4(5):724–38. doi: 10.1016/S0168-8278(20)30917-X
80. Tsuneyama K, Van de Water J, Leung PS, Cha S, Nakanuma Y, Kaplan M, et al. Abnormal expression of the E2 component of the pyruvate dehydrogenase complex on the luminal surface of biliary epithelium occurs before major histocompatibility complex class II and BB1/B7 expression. Hepatology (1995) 21(4):1031–7. doi: 10.1002/hep.1840210422
81. Nakamura M, Ishibashi H, Matsui M, Shimoda S, Hayashida K, Koike K, et al. Peripheral B lymphocyte repertoire to mitochondrial antigen in primary biliary cirrhosis–positive correlation between the disease activity and the frequency of circulating B lymphocytes specific for pyruvate dehydrogenase complex. Autoimmunity (1995) 21(4):253–62. doi: 10.3109/08916939509001944
82. Chung BK, Guevel BT, Reynolds GM, Gupta Udatha DBRK, Henriksen EKK, Stamataki Z, et al. Phenotyping and auto-antibody production by liver-infiltrating B cells in primary sclerosing cholangitis and primary biliary cholangitis. J Autoimmun (2017) 77:45–54. doi: 10.1016/j.jaut.2016.10.003
83. Lleo A, Bowlus CL, Yang G-X, Invernizzi P, Podda M, Van de Water J, et al. Biliary apotopes and anti-mitochondrial antibodies activate innate immune responses in primary biliary cirrhosis. Hepatology (2010) 52(3):987–98. doi: 10.1002/hep.23783
84. Khanna A, Jopson L, Howel D, Bryant A, Blamire A, Newton JL, et al. Rituximab Is Ineffective for Treatment of Fatigue in Primary Biliary Cholangitis: A Phase 2 Randomized Controlled Trial. Hepatology (2019) 70(5):1646–57. doi: 10.1002/hep.30099
85. Myers RP, Swain MG, Lee SS, Shaheen AAM. Burak KW. B-cell depletion with rituximab in patients with primary biliary cirrhosis refractory to ursodeoxycholic acid. Am J Gastroenterol (2013) 108(6):933–41. doi: 10.1038/ajg.2013.51
86. Shimoda S, Miyakawa H, Nakamura M, Ishibashi H, Kikuchi K, Kita H, et al. CD4 T-cell autoreactivity to the mitochondrial autoantigen PDC-E2 in AMA-negative primary biliary cirrhosis. J Autoimmun (2008) 31(2):110–5. doi: 10.1016/j.jaut.2008.05.003
87. Nakamura M, Kondo H, Mori T, Komori A, Matsuyama M, Ito M, et al. Anti-gp210 and anti-centromere antibodies are different risk factors for the progression of primary biliary cirrhosis. Hepatology (2007) 45(1):118–27. doi: 10.1002/hep.21472
88. Wesierska-Gadek J, Penner E, Battezzati PM, Selmi C, Zuin M, Hitchman E, et al. Correlation of initial autoantibody profile and clinical outcome in primary biliary cirrhosis. Hepatology (2006) 43(5):1135–44. doi: 10.1002/hep.21172
89. Norman GL, Yang C-Y, Ostendorff HP, Shums Z, Lim MJ, Wang J, et al. Anti-kelch-like 12 and anti-hexokinase 1: novel autoantibodies in primary biliary cirrhosis. Liver Int (2015) 35(2):642–51. doi: 10.1111/liv.12690
90. Hu C-J, Song G, Huang W, Liu G-Z, Deng C-W, Zeng H-P, et al. Identification of new autoantigens for primary biliary cirrhosis using human proteome microarrays. Mol Cell Proteomics (2012) 11(9):669–80. doi: 10.1074/mcp.M111.015529
91. Han F-F, Fang M-X, Zhao D-T, Dong Y-C, Yuan G-H, Gao J-E, et al. Profiling of the pattern of the human TRB/IGH-CDR3 repertoire in primary biliary cholangitis patients. Int Immunopharmacol (2020) 83:106393. doi: 10.1016/j.intimp.2020.106393
92. Tan Y-G, Wang Y-Q, Zhang M, Han Y-X, Huang C-Y, Zhang H-P, et al. Clonal Characteristics of Circulating B Lymphocyte Repertoire in Primary Biliary Cholangitis. J Immunol (2016) 197(5):1609–20. doi: 10.4049/jimmunol.1600096
93. Feizi T. Immunoglobulins in chronic liver disease. Gut (1968) 9(2):193–8. doi: 10.1136/gut.9.2.193
94. Daniels JA, Torbenson M, Anders RA, Boitnott JK. Immunostaining of plasma cells in primary biliary cirrhosis. Am J Clin Pathol (2009) 131(2):243–9. doi: 10.1309/AJCP8WHR0IEVUUOJ
95. Moreira RK, Revetta F, Koehler E, Washington MK. Diagnostic utility of IgG and IgM immunohistochemistry in autoimmune liver disease. World J Gastroenterol (2010) 16(4):453–7. doi: 10.3748/wjg.v16.i4.453
96. Lleo A, Liao J, Invernizzi P, Zhao M, Bernuzzi F, Ma L, et al. Immunoglobulin M levels inversely correlate with CD40 ligand promoter methylation in patients with primary biliary cirrhosis. Hepatology (2012) 55(1):153–60. doi: 10.1002/hep.24630
97. Cichoż-Lach H, Grywalska E, Michalak A, Kowalik A, Mielnik M, Roliński J. Deviations in Peripheral Blood Cell Populations are Associated with the Stage of Primary Biliary Cholangitis and Presence of Itching. Arch Immunol Ther Exp (Warsz) (2018) 66(6):443–52. doi: 10.1007/s00005-018-0515-9
98. Jang JS, Juran BD, Cunningham KY, Gupta VK, Son YM, Yang JD, et al. Single-cell mass cytometry on peripheral blood identifies immune cell subsets associated with primary biliary cholangitis. Sci Rep (2020) 10(1):12584. doi: 10.1038/s41598-020-69358-4
99. Migita K, Ilyassova B, Kovzel EF, Nersesov A, Abiru S, Maeda Y, et al. and APRIL levels in patients with PBC. Clin Immunol (2010) 134(2):217–25. doi: 10.1016/j.clim.2009.09.007
100. Chuang Y-H, Lian Z-X, Cheng C-M, Lan RY, Yang G-X, Moritoki Y, et al. Increased levels of chemokine receptor CXCR3 and chemokines IP-10 and MIG in patients with primary biliary cirrhosis and their first degree relatives. J Autoimmun (2005) 25(2):126–32. doi: 10.1016/j.jaut.2005.08.009
101. Chen Q, Lai L, Chi X, Lu X, Wu H, Sun J, et al. CD19+CD24hiCD38hi B Cell Dysfunction in Primary Biliary Cholangitis. Mediators Inflammation (2020) 2020:3019378. doi: 10.1155/2020/3019378
102. Fussey SP, Ali ST, Guest JR, James OF, Bassendine MF, Yeaman SJ. Reactivity of primary biliary cirrhosis sera with Escherichia coli dihydrolipoamide acetyltransferase (E2p): characterization of the main immunogenic region. Proc Natl Acad Sci U S A (1990) 87(10):3987–91. doi: 10.1073/pnas.87.10.3987
103. Bogdanos D-P, Baum H, Grasso A, Okamoto M, Butler P, Ma Y, et al. Microbial mimics are major targets of crossreactivity with human pyruvate dehydrogenase in primary biliary cirrhosis. J Hepatol (2004) 40(1):31–9. doi: 10.1016/S0168-8278(03)00501-4
104. Bogdanos D-P, Pares A, Baum H, Caballeria L, Rigopoulou EI, Ma Y, et al. Disease-specific cross-reactivity between mimicking peptides of heat shock protein of Mycobacterium gordonae and dominant epitope of E2 subunit of pyruvate dehydrogenase is common in Spanish but not British patients with primary biliary cirrhosis. J Autoimmun (2004) 22(4):353–62. doi: 10.1016/j.jaut.2004.03.002
105. Lv L-X, Fang D-Q, Shi D, Chen D-Y, Yan R, Zhu Y-X, et al. Alterations and correlations of the gut microbiome, metabolism and immunity in patients with primary biliary cirrhosis. Environ Microbiol (2016) 18(7):2272–86. doi: 10.1111/1462-2920.13401
106. Tang R, Wei Y, Li Y, Chen W, Chen H, Wang Q, et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut (2018) 67(3):534–41. doi: 10.1136/gutjnl-2016-313332
107. Furukawa M, Moriya K, Nakayama J, Inoue T, Momoda R, Kawaratani H, et al. Gut dysbiosis associated with clinical prognosis of patients with primary biliary cholangitis. Hepatol Res (2020) 50(7):840–52. doi: 10.1111/hepr.13509
108. Chen W, Wei Y, Xiong A, Li Y, Guan H, Wang Q, et al. Comprehensive Analysis of Serum and Fecal Bile Acid Profiles and Interaction with Gut Microbiota in Primary Biliary Cholangitis. Clin Rev Allergy Immunol (2020) 58(1):25–38. doi: 10.1007/s12016-019-08731-2
109. Chapman MH, Thorburn D, Hirschfield GM, Webster GGJ, Rushbrook SM, Alexander G, et al. British Society of Gastroenterology and UK-PSC guidelines for the diagnosis and management of primary sclerosing cholangitis. Gut (2019) 68(8):1356–78. doi: 10.1136/gutjnl-2018-317993
110. Kummen M, Holm K, Anmarkrud JA, Nygård S, Vesterhus M, Høivik ML, et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut (2017) 66(4):611–9. doi: 10.1136/gutjnl-2015-310500
111. Hov J-R, Boberg K-M, Karlsen T-H. Autoantibodies in primary sclerosing cholangitis. World J Gastroenterol (2008) 14(24):3781–91. doi: 10.3748/wjg.14.3781
112. Hov JR, Boberg KM, Taraldsrud E, Vesterhus M, Boyadzhieva M, Solberg IC, et al. Antineutrophil antibodies define clinical and genetic subgroups in primary sclerosing cholangitis. Liver Int (2017) 37(3):458–65. doi: 10.1111/liv.13238
113. Stinton LM, Bentow C, Mahler M, Norman GL, Eksteen B, Mason AL, et al. PR3-ANCA: a promising biomarker in primary sclerosing cholangitis (PSC). PloS One (2014) 9(11):e112877. doi: 10.1371/journal.pone.0112877
114. Papp M, Sipeki N, Tornai T, Altorjay I, Norman GL, Shums Z, et al. Rediscovery of the Anti-Pancreatic Antibodies and Evaluation of their Prognostic Value in a Prospective Clinical Cohort of Crohn’s Patients: The Importance of Specific Target Antigens [GP2 and CUZD1]. J Crohns Colitis (2015) 9(8):659–68. doi: 10.1093/ecco-jcc/jjv087
115. Kovacs G, Sipeki N, Suga B, Tornai T, Fechner K, Norman GL, et al. Significance of serological markers in the disease course of ulcerative colitis in a prospective clinical cohort of patients. PloS One (2018) 13(3):e0194166. doi: 10.1371/journal.pone.0194166
116. Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M, et al. Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature (2009) 462(7270):226–30. doi: 10.1038/nature08529
117. Terahara K, Yoshida M, Igarashi O, Nochi T, Pontes GS, Hase K, et al. Comprehensive gene expression profiling of Peyer’s patch M cells, villous M-like cells, and intestinal epithelial cells. J Immunol (2008) 180(12):7840–6. doi: 10.4049/jimmunol.180.12.7840
118. Dean G, Hanauer S, Levitsky J. The Role of the Intestine in the Pathogenesis of Primary Sclerosing Cholangitis: Evidence and Therapeutic Implications. Hepatology (2020) 72(3):1127–38. doi: 10.1002/hep.31311
119. Jendrek ST, Gotthardt D, Nitzsche T, Widmann L, Korf T, Michaels MA, et al. Anti-GP2 IgA autoantibodies are associated with poor survival and cholangiocarcinoma in primary sclerosing cholangitis. Gut (2017) 66(1):137–44. doi: 10.1136/gutjnl-2016-311739
120. Tornai T, Tornai D, Sipeki N, Tornai I, Alsulaimani R, Fechner K, et al. Loss of tolerance to gut immunity protein, glycoprotein 2 (GP2) is associated with progressive disease course in primary sclerosing cholangitis. Sci Rep (2018) 8(1):399. doi: 10.1038/s41598-017-18622-1
121. Xu B, Broome U, Ericzon B-G, Sumitran-Holgersson S. High frequency of autoantibodies in patients with primary sclerosing cholangitis that bind biliary epithelial cells and induce expression of CD44 and production of interleukin 6. Gut (2002) 51(1):120–7. doi: 10.1136/gut.51.1.120
122. Karrar A, Broomé U, Södergren T, Jaksch M, Bergquist A, Björnstedt M, et al. Biliary epithelial cell antibodies link adaptive and innate immune responses in primary sclerosing cholangitis. Gastroenterology (2007) 132(4):1504–14. doi: 10.1053/j.gastro.2007.01.039
123. Chung BK, Henriksen EKK, Jørgensen KK, Karlsen TH, Hirschfield GM, Liaskou E. Gut and Liver B Cells of Common Clonal Origin in Primary Sclerosing Cholangitis-Inflammatory Bowel Disease. Hepatol Commun (2018) 2(8):956–67. doi: 10.1002/hep4.1200
124. Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell (1993) 75(3):451–62. doi: 10.1016/0092-8674(93)90380-9
125. Thapa M, Tedesco D, Gumber S, Elrod EJ, Han J-H, Kitchens WH, et al. Blockade of BAFF Reshapes the Hepatic B Cell Receptor Repertoire and Attenuates Autoantibody Production in Cholestatic Liver Disease. J Immunol (2020) 204(12):3117–28. doi: 10.4049/jimmunol.1900391
126. Little R, Wine E, Kamath BM, Griffiths AM, Ricciuto A. Gut microbiome in primary sclerosing cholangitis: A review. World J Gastroenterol (2020) 26(21):2768–80. doi: 10.3748/wjg.v26.i21.2768
127. Nakamoto N, Sasaki N, Aoki R, Miyamoto K, Suda W, Teratani T, et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat Microbiol (2019) 4(3):492–503. doi: 10.1038/s41564-018-0333-1
128. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Autoimmune hepatitis. J Hepatol (2015) 63(4):971–1004. doi: 10.1016/j.jhep.2015.06.030
129. Okazaki K, Chari ST, Frulloni L, Lerch MM, Kamisawa T, Kawa S, et al. International consensus for the treatment of autoimmune pancreatitis. Pancreatology (2017) 17(1):1–6. doi: 10.1016/j.pan.2016.12.003
130. Yan S, Deng X, Wang Q, Sun X, Wei W. Prednisone treatment inhibits the differentiation of B lymphocytes into plasma cells in MRL/MpSlac-lpr mice. Acta Pharmacol Sin (2015) 36(11):1367–76. doi: 10.1038/aps.2015.76
131. Taubert R, Hardtke-Wolenski M, Noyan F, Wilms A, Baumann AK, Schlue J, et al. Intrahepatic regulatory T cells in autoimmune hepatitis are associated with treatment response and depleted with current therapies. J Hepatol (2014) 61(5):1106–14. doi: 10.1016/j.jhep.2014.05.034
132. Lin W, Jin L, Chen H, Wu Q, Fei Y, Zheng W, et al. B cell subsets and dysfunction of regulatory B cells in IgG4-related diseases and primary Sjögren’s syndrome: the similarities and differences. Arthritis Res Ther (2014) 16(3):R118. doi: 10.1186/ar4571
133. Iwata S, Saito K, Hirata S, Tanaka Y. Phenotypic changes of lymphocyte in a patient with IgG4-related disease after corticosteroid therapy. Ann Rheum Dis (2012) 71(12):2058–9. doi: 10.1136/annrheumdis-2012-201657
134. Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis (2015) 74(6):1171–7. doi: 10.1136/annrheumdis-2014-206605
135. Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Med (Baltimore) (2012) 91(1):57–66. doi: 10.1097/MD.0b013e3182431ef6
136. Hart PA, Topazian MD, Witzig TE, Clain JE, Gleeson FC, Klebig RR, et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: the Mayo Clinic experience. Gut (2013) 62(11):1607–15. doi: 10.1136/gutjnl-2012-302886
137. Wallace ZS, Mattoo H, Carruthers M, Mahajan VS, Della Torre E, Lee H, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis (2015) 74(1):190–5. doi: 10.1136/annrheumdis-2014-205233
138. Wallace ZS, Mattoo H, Mahajan VS, Kulikova M, Lu L, Deshpande V, et al. Predictors of disease relapse in IgG4-related disease following rituximab. Rheumatol (Oxford) (2016) 55(6):1000–8. doi: 10.1093/rheumatology/kev438
139. Ebbo M, Grados A, Samson M, Groh M, Loundou A, Rigolet A, et al. Long-term efficacy and safety of rituximab in IgG4-related disease: Data from a French nationwide study of thirty-three patients. PloS One (2017) 12(9):e0183844. doi: 10.1371/journal.pone.0183844
140. Wallwork R, Wallace Z, Perugino C, Sharma A, Stone JH. Rituximab for idiopathic and IgG4-related retroperitoneal fibrosis. Med (Baltimore) (2018) 97(42):e12631. doi: 10.1097/MD.0000000000012631
141. Campochiaro C, Della-Torre E, Lanzillotta M, Bozzolo E, Baldissera E, Milani R, et al. Long-term efficacy of maintenance therapy with Rituximab for IgG4-related disease. Eur J Intern Med (2020) 74:92–8. doi: 10.1016/j.ejim.2019.12.029
142. Wallace ZS, Khosroshahi A, Carruthers MD, Perugino CA, Choi H, Campochiaro C, et al. An International Multispecialty Validation Study of the IgG4-Related Disease Responder Index. Arthritis Care Res (Hoboken) (2018) 70(11):1671–8. doi: 10.1002/acr.23543
143. Parkes J, Roderick P, Harris S, Day C, Mutimer D, Collier J, et al. Enhanced liver fibrosis test can predict clinical outcomes in patients with chronic liver disease. Gut (2010) 59(9):1245–51. doi: 10.1136/gut.2009.203166
144. Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheumatol (2010) 62(6):1755–62. doi: 10.1002/art.27435
145. Sarantopoulos A, Gkougkourelas I, Mytilinaiou M, Klonizakis P, Georgiadou A, Boura P. FRI0224 Rituximab selectively reduces IgG4 levels in rheumatoid arthritis patients. Ann Rheumatic Dis (2017) 76(Supplement 2):569. doi: 10.1136/annrheumdis-2017-eular.6690
146. Chisari CG, Sgarlata E, Arena S, Toscano S, Luca M, Patti F. Rituximab for the treatment of multiple sclerosis: a review. J Neurol (2021). doi: 10.1007/s00415-020-10362-z
147. Tsuda M, Moritoki Y, Lian Z-X, Zhang W, Yoshida K, Wakabayashi K, et al. Biochemical and immunologic effects of rituximab in patients with primary biliary cirrhosis and an incomplete response to ursodeoxycholic acid. Hepatology (2012) 55(2):512–21. doi: 10.1002/hep.24748
148. Schiopu E, Chatterjee S, Hsu V, Flor A, Cimbora D, Patra K, et al. Safety and tolerability of an anti-CD19 monoclonal antibody, MEDI-551, in subjects with systemic sclerosis: a phase I, randomized, placebo-controlled, escalating single-dose study. Arthritis Res Ther (2016) 18(1):131. doi: 10.1186/s13075-016-1021-2
149. Agius MA, Klodowska-Duda G, Maciejowski M, Potemkowski A, Li J, Patra K, et al. Safety and tolerability of inebilizumab (MEDI-551), an anti-CD19 monoclonal antibody, in patients with relapsing forms of multiple sclerosis: Results from a phase 1 randomised, placebo-controlled, escalating intravenous and subcutaneous dose study. Mult Scler (2019) 25(2):235–45. doi: 10.1177/1352458517740641
150. Cree BAC, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet (2019) 394(10206):1352–63. doi: 10.1016/S0140-6736(19)31817-3
151. Viela Bio. A Phase 3, Randomized, Double-blind, Multicenter, Placebo Controlled Study of Inebilizumab Efficacy and Safety in IgG4-Related Disease. (2020). Available at: https://www.clinicaltrials.gov/ct2/show/NCT04540497.
152. Lin W, Zhang P, Chen H, Chen Y, Yang H, Zheng W, et al. Circulating plasmablasts/plasma cells: a potential biomarker for IgG4-related disease. Arthritis Res Ther (2017) 19(1):25. doi: 10.1186/s13075-017-1231-2
153. Muir AJ, Levy C, Janssen HLA, Montano-Loza AJ, Shiffman ML, Caldwell S, et al. Simtuzumab for Primary Sclerosing Cholangitis: Phase 2 Study Results With Insights on the Natural History of the Disease. Hepatology (2019) 69(2):684–98. doi: 10.1002/hep.30237
154. Lanzillotta M, Mancuso G, Della-Torre E. Advances in the diagnosis and management of IgG4 related disease. BMJ (2020) 369:m1067. doi: 10.1136/bmj.m1067
155. Chu SY, Vostiar I, Karki S, Moore GL, Lazar GA, Pong E, et al. Inhibition of B cell receptor-mediated activation of primary human B cells by coengagement of CD19 and FcgammaRIIb with Fc-engineered antibodies. Mol Immunol (2008) 45(15):3926–33. doi: 10.1016/j.molimm.2008.06.027
156. Horton HM, Chu SY, Ortiz EC, Pong E, Cemerski S, Leung IWL, et al. Antibody-mediated coengagement of FcγRIIb and B cell receptor complex suppresses humoral immunity in systemic lupus erythematosus. J Immunol (2011) 186(7):4223–33. doi: 10.4049/jimmunol.1003412
157. Chu SY, Yeter K, Kotha R, Pong E, Miranda Y, Phung S, et al. Suppression of rheumatoid arthritis B cells by XmAb5871, an anti-CD19 antibody that coengages B cell antigen receptor complex and Fcγ receptor IIb inhibitory receptor. Arthritis Rheumatol (2014) 66(5):1153–64. doi: 10.1002/art.38334
158. Paul D, Fazeli MS, Mintzer L, Duarte L, Gupta K, Ferri L. Comparative efficacy and safety of current therapies for early rheumatoid arthritis: a systematic literature review and network meta-analysis. Clin Exp Rheumatol (2020) 38(5):1008–15.
159. Bowlus CL, Yang G-X, Liu CH, Johnson CR, Dhaliwal SS, Frank D, et al. Therapeutic trials of biologics in primary biliary cholangitis: An open label study of abatacept and review of the literature. J Autoimmun (2019) 101:26–34. doi: 10.1016/j.jaut.2019.04.005
160. Bristol-Myers Squibb. Safety and Efficacy of Abatacept in IgG4-Related Disease. (2018). Available at: https://clinicaltrials.gov/ct2/show/NCT03669861.
161. Knechtle S. Abatacept for Treatment of Recurrent or de Novo Autoimmune Hepatitis. (2020). Available at: https://clinicaltrials.gov/ct2/show/NCT04203875.
162. Xencor, Inc. Study to Evaluate the Effect of XmAb®5871 on Disease Activity in Patients With IgG4-Related Disease (RD). (2016). Available at: https://clinicaltrials.gov/ct2/show/study/NCT02725476.
163. Stone JH, Wallace ZS, Perugino CA, Fernandes A, Patel P, Foster P. Final results of an open label phase 2 study of a reversible B cell inhibitor, XmAb5871, in IgG4-related disease. Arthritis Rheumatol (2017) 69(supplement 10). doi: 10.1136/annrheumdis-2017-eular.3301
164. Dörner T, Posch MG, Li Y, Petricoul O, Cabanski M, Milojevic JM, et al. Treatment of primary Sjögren’s syndrome with ianalumab (VAY736) targeting B cells by BAFF receptor blockade coupled with enhanced, antibody-dependent cellular cytotoxicity. Ann Rheum Dis (2019) 78(5):641–7. doi: 10.1136/annrheumdis-2018-214720
165. Arvaniti P, Giannoulis G, Gabeta S, Zachou K, Koukoulis GK, Dalekos GN. Belimumab is a promising third-line treatment option for refractory autoimmune hepatitis. JHEP Rep (2020) Aug2(4):100123. doi: 10.1016/j.jhepr.2020.100123
166. Novartis Pharmaceuticals. ADCC Mediated B-Cell dEpletion and BAFF-R Blockade (AMBER). (2017). Available at: https://clinicaltrials.gov/ct2/show/NCT03217422.
167. de Graaf KL, Lapeyre G, Guilhot F, Ferlin W, Curbishley SM, Carbone M, et al. NI-0801, an anti-chemokine (C-X-C motif) ligand 10 antibody, in patients with primary biliary cholangitis and an incomplete response to ursodeoxycholic acid. Hepatol Commun (2018) 2(5):492–503. doi: 10.1002/hep4.1170
168. Sacco KA, Abraham RS. Consequences of B-cell-depleting therapy: hypogammaglobulinemia and impaired B-cell reconstitution. Immunotherapy (2018) 10(8):713–28. doi: 10.2217/imt-2017-0178
169. Kridin K, Ahmed AR. Post-rituximab immunoglobulin M (IgM) hypogammaglobulinemia. Autoimmun Rev (2020) 19(3):102466. doi: 10.1016/j.autrev.2020.102466
170. Lohse AW, Sebode M, Jørgensen MH, Ytting H, Karlsen TH, Kelly D, et al. Second-line and third-line therapy for autoimmune hepatitis: A position statement from the European Reference Network on Hepatological Diseases and the International Autoimmune Hepatitis Group. J Hepatol (2020) 73(6):1496–506. doi: 10.1016/j.jhep.2020.07.023
171. Jayne DR, Rasmussen N. Treatment of antineutrophil cytoplasm autoantibody-associated systemic vasculitis: initiatives of the European Community Systemic Vasculitis Clinical Trials Study Group. Mayo Clin Proc (1997) 72(8):737–47. doi: 10.1016/S0025-6196(11)63594-5
172. EUVAS. EUVAS. European Vascilitis Society website. (2020). Available at: https://vasculitis.org/trials/.
173. NHS Health Research Authority. The UK-AIH Cohort Research Summary. Available at: https://www.hra.nhs.uk/planning-and-improving-research/application-summaries/research-summaries/the-uk-aih-cohort.
174. IgG4-RD study group. Nuffield Department of Medicine, Oxford. IgG4-systemic disease study. Available at: https://igg4-rd.ndm.ox.ac.uk/home.
175. UK-PBC. UK-PBC - Stratified Medicine in Primary Biliary Cholangitis. Available at: http://www.uk-pbc.com/.
176. UK-PSC. UK-PSC Primary Sclerosing Cholangitis. Available at: http://www.uk-psc.com.
177. Wong LL, Fisher HF, Stocken DD, Rice S, Khanna A, Heneghan MA, et al. The Impact of Autoimmune Hepatitis and Its Treatment on Health Utility. Hepatology (2018) 68(4):1487–97. doi: 10.1002/hep.30031
178. Kim HP, Lieber SR, Rogers ME, Moon AM, Loiselle M, Walker J, et al. A Systematic Review of Patient-Reported Outcomes in Primary Biliary Cholangitis and Primary Sclerosing Cholangitis. Hepatol Commun (2020) 4(10):1502–15. doi: 10.1002/hep4.1567
Keywords: IgG4-related disease, autoimmune hepatitis, primary biliary cholangitis (PBC), primary sclerosing cholangites (PSC), B cell, Rituximab
Citation: Cargill T and Culver EL (2021) The Role of B Cells and B Cell Therapies in Immune-Mediated Liver Diseases. Front. Immunol. 12:661196. doi: 10.3389/fimmu.2021.661196
Received: 30 January 2021; Accepted: 25 March 2021;
Published: 14 April 2021.
Edited by:
Ye Htun Oo, University of Birmingham, United KingdomReviewed by:
Jochen Mattner, University of Erlangen Nuremberg, GermanyGeorg Varga, University Hospital Muenster, Germany
Copyright © 2021 Cargill and Culver. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emma L. Culver, ZW1tYS5jdWx2ZXJAbmhzLm5ldA==