 Arjuna Singanayagam
Arjuna Singanayagam Evangelos Triantafyllou
Evangelos Triantafyllou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 02 April 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.661182
This article is part of the Research Topic Macrophage Plasticity in Sterile and Pathogen-Induced Inflammation View all 18 articles
Chronic liver injury results in immune-driven progressive fibrosis, with risk of cirrhosis development and impact on morbidity and mortality. Persistent liver cell damage and death causes immune cell activation and inflammation. Patients with advanced cirrhosis additionally experience pathological bacterial translocation, exposure to microbial products and chronic engagement of the immune system. Bacterial infections have a high incidence in cirrhosis, with spontaneous bacterial peritonitis being the most common, while the subsequent systemic inflammation, organ failure and immune dysregulation increase the mortality risk. Tissue-resident and recruited macrophages play a central part in the development of inflammation and fibrosis progression. In the liver, adipose tissue, peritoneum and intestines, diverse macrophage populations exhibit great phenotypic and functional plasticity determined by their ontogeny, epigenetic programming and local microenvironment. These changes can, at different times, promote or ameliorate disease states and therefore represent potential targets for macrophage-directed therapies. In this review, we discuss the evidence for macrophage phenotypic and functional alterations in tissue compartments during the development and progression of chronic liver failure in different aetiologies and highlight the potential of macrophage modulation as a therapeutic strategy for liver disease.
Liver disease is a global health burden with recent estimates suggesting that 844 million people worldwide have chronic liver disease (CLD), with a mortality rate of 2 million deaths per year: approximately, 1 million due to complications of cirrhosis and 1 million due to viral hepatitis and hepatocellular carcinoma (HCC) (1, 2). Chronic liver injury, most commonly caused by alcohol, infection or liver fat accumulation associated with features of the metabolic syndrome, triggers the activation of liver-resident and infiltrating immune cells, resulting in inflammation, progressive fibrosis, disrupted architecture, vascular changes and aberrant regeneration, which are defining characteristics of liver cirrhosis (2). Management of patients with cirrhosis is limited to treating the underlying cause and, where appropriate, liver transplantation. However, the latter is difficult to access in much of the world where donor organ supply is insufficient to meet demands (2). Therefore, developing new effective therapies for CLD patients would likely have a considerable benefit on morbidity and mortality. This review focuses on the diverse and dynamic role of macrophages in CLD and how their modulation might offer novel therapeutics.
Cirrhosis comprises two consecutive but potentially reversible stages: compensated (asymptomatic) and decompensated cirrhosis (3, 4). The term acute decompensation (AD) of cirrhosis defines the acute development of one or more major complication(s) in patients (5). These complications include ascites, hepatic encephalopathy and variceal haemorrhage. The first AD episode marks the transition from the compensated to the decompensated stage. During AD, patients are extremely prone to develop bacterial infections, to the point that bacterial infections have been considered as the fourth major complication of the disease (5). Cirrhosis features a gradually dysfunctional immune response that encompasses systemic inflammation, which can exacerbate clinical manifestations of cirrhosis (e.g., hemodynamic derangement and kidney injury) and, as disease progresses, immunodeficiency and impaired antimicrobial functions that are associated with high infection risk (Figure 1). The term cirrhosis-associated immune dysfunction (CAID) defines the dynamic spectrum of the immunological perturbations that develop in these patients [reviewed in (6–8)].
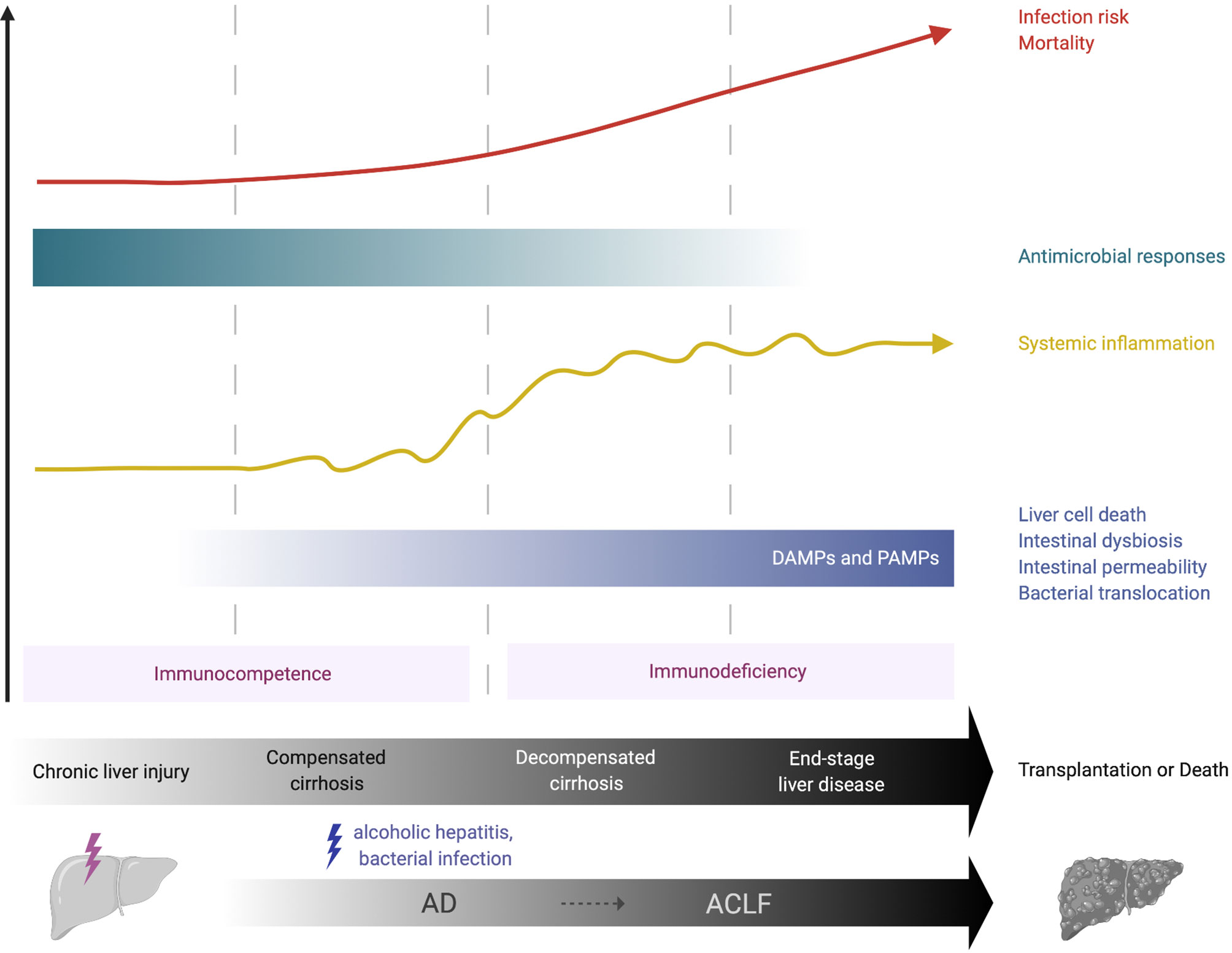
Figure 1 Immunological features of chronic liver failure. Over the course of compensated or decompensated cirrhosis, an acute precipitating event such as alcoholic hepatitis or bacterial infection may challenge liver homeostasis and lead to acute-on-chronic liver failure (ACLF). In compensated cirrhosis, liver-derived DAMPs, for example from necrotic hepatocytes, may activate the local immune system and initiate systemic inflammation. In decompensated cirrhosis, increasing alterations in intestinal homeostasis (e.g., bacterial overgrowth, increased permeability) allow the pathological translocation of bacteria, bacterial products (e.g., LPS) and PAMPs into the liver, thus providing further chronic stimulation of innate immune cells and propagation of local and systemic inflammation. This may be crucial in the transition from the immunocompetent state (pro-inflammatory immune response) to a more hyporesponsive, immunodeficient state that is observed in advanced cirrhosis. The latter increases the risk of life-threatening infections and substantially contributes to the high morbidity and mortality observed in these patients. ACLF, acute-on-chronic liver failure; AD, acute decompensation; DAMP, damage-associated molecular pattern; PAMP, pathogen-associated molecular pattern.
Over the course of compensated or decompensated cirrhosis, an acute precipitating event, most commonly bacterial infections and acute alcoholic hepatitis, challenge liver homeostasis and may lead to a syndrome called acute-on-chronic liver failure (ACLF) (9, 10). ACLF is defined by acute decompensation of cirrhosis, hepatic and/or extra-hepatic organ failure with high short-term (28-day) mortality (3, 4). Inadequate immune responses to the precipitating event are key to its pathogenesis, with high grade of systemic/local inflammation and immunodeficiency, that lead to further organ failure. This is profound in the setting of ACLF, which resembles the immunopathology of sepsis, with an initial systemic inflammatory response (cytokine storm) followed by a compensatory anti-inflammatory response that can impair immune defence against infections (3, 4).
Patients with cirrhosis develop a range of complications, with infections being one of the most clinically important issues, associated with high morbidity and mortality. Bacterial infections have been shown to occur frequently (32-34%) in patients with advanced cirrhosis; in 30–50%, infection is the cause of hospital admission, and a further 15–35% develop nosocomial infections (as compared to 5-7% of general population) (11). Infections can further trigger hepatic decompensation and are well-known precipitants for encephalopathy, hepatorenal syndrome (HRS) and development of ACLF (5). Most recent data derive from the CANONIC and PREDICT studies, that were observational prospective investigations in large cohorts of non-selected patients hospitalised with AD. Among 407 patients with AD-ACLF, the incidence of infections at hospital admission and during a 28-day follow-up period was 65% (12). The corresponding incidence of infections in 1,071 patients with AD-no-ACLF was 53% (9, 13). Globally, the most common infections in cirrhotic patients are spontaneous bacterial peritonitis (SBP) (27%), urinary tract infections (22%), pneumonia (19%), spontaneous bacteraemia (8%), skin and soft tissue infections (8%) (14). The majority of infections identified in cirrhotic patients are caused by Gram-negative bacteria (e.g., Escherichia coli, Klebsiella Pneumoniae) of intestinal origin; Gram-positive bacterial infections are less frequent (e.g., Staphylococcus aureus) (14, 15). The epidemiology of infections in cirrhosis continuously change, with recent studies demonstrating increasing prevalence of multidrug resistant organisms (29% to 38% increase from 2011 to 2017-2018) (14, 15).
Innate immune cells (mainly neutrophils, monocytes and macrophages) are primed to detect tissue damaging or infectious insults, and therefore are key orchestrators of inflammatory responses. During progression of CLD these cells initiate and drive both liver and systemic inflammation by recognising/responding to damage-associated molecular patterns (DAMPs) released from injured/activated liver cells and/or pathogen-associated molecular patterns (PAMPs) (16). Fibrosis occurs following chronic liver injury from an insult (toxic, metabolic, or infectious) which can perpetuate inflammation. Fibrogenesis is the common pathological mechanism that causes cirrhosis; it is a complex and dynamic process that involves an array of activated resident or recruited inflammatory cells (e.g., platelets, macrophages), hepatic stellate cells (HSCs), hepatocytes, other extracellular matrix (ECM) producing cells and extracellular signals [e.g., cytokines, chemokines, adipokines or reactive oxygen species (ROS)] (17–19). The activation of HSCs - transdifferentiation of quiescent cells into proliferative, fibrogenic myofibroblasts - is a well-established driver of fibrosis [reviewed in (17–19)]. This is a cardinal process in which quiescent HSCs responding to hepatic injury downregulate the expression of GFAP, peroxisome proliferator-activated (PPAR)-γ and vitamin A and become activated HSCs (18). Following stimulation with cytokines and fibrogenic signals, of which transforming growth factor (TGF)-β is the most potent, HSCs upregulate α-smooth muscle actin expression, increase transcription of collagen type I via SMAD-3 amongst other routes, and traffic to sites of injury where they secrete ECM resulting in fibrous scar formation (20). With fibrosis progression, cirrhosis may ultimately develop.
Advanced cirrhosis is closely associated with pathological bacterial translocation (BT), the increased rate of translocation of bacteria or bacterial products (e.g. lipopolysaccharide [LPS], bacterial DNA and peptidoglycans) from the gut to mesenteric lymph nodes and other tissues (21). SBP is considered to be the hallmark manifestation of pathological BT and is a multi-faceted process. This involves small bowel dysmotility, small intestinal bacterial overgrowth (SIBO), increased intestinal permeability and dysbiosis. The exact mechanisms of enhanced gut permeability are not yet fully understood; loosening of the intestinal epithelial tight junctions and reductions in luminal antimicrobial peptides, such as secretory component and mucins, may have a role (21, 22). Dysbiosis sees a decrease in the quantity of beneficial autochthonous bacteria (e.g., Ruminococcacaea and Lachnospiracaea) and an increase in potentially pathogenic taxa (e.g., Enterobacteriaceae and Bacteroidaceae), termed the cirrhosis dysbiosis ratio (23). Why such intestinal changes occur in cirrhosis is unclear but alcohol, portal hypertension and impaired local immune responses have been implicated (24). Impaired hepatic immune responses and portosystemic collaterals in advanced cirrhosis provide further opportunity for translocated products to pass from the intestinal lumen to the systemic circulation, chronically stimulating innate immune cells and enhancing systemic inflammation. This increase in bacterial products may be crucial in the switch from the initially pro-inflammatory immune response described in early cirrhosis to a more hyporesponsive, immunodeficient state observed in decompensated cirrhosis (Figure 1).
Circulating monocytes, a key component of the mononuclear phagocyte immune system, play pivotal roles in defence against infections and contribute to the systemic inflammation in chronic liver failure. In addition, they augment the local macrophage pool via their recruitment to inflammatory sites after a sterile/tissue-damaging or infectious insult to the liver (10, 16, 25). Human monocytes are divided into three major subsets: classical (CD14+CD16−), intermediate (CD14+CD16+) and non-classical (CD14dimCD16+). Each of these subsets can be distinguished from each other by their differential expression of surface markers and their distinct functions [for an up-to-date review see (26)]. Classical monocytes are comparable to the inflammatory Ly6C+/high CCR2high murine subset whilst non-classical monocytes resemble the patrolling Ly6C-/low CX3CR1high subset. The former cells rapidly infiltrate tissues in response to injury, have the capacity to differentiate into dendritic-like cells, and are primed for phagocytosis and innate immune sensing. Ly6Clow monocytes crawl along endothelial cells, coordinating the clearance of apoptotic cells and cellular debris (27). Similarly, non-classical human monocytes are suited to adhesion and Fc gamma-mediated phagocytosis. Whilst intermediate monocytes do not demonstrate such crawling behaviour, they are a heterogenous population adapted for antigen presentation, cytokine production and apoptosis regulation (26, 28–30). Interestingly, a study using deuterium labelling in humans has suggested that intermediate and non-classical monocytes emerge sequentially from the pool of classical monocytes (31).
The number of circulating monocytes increases in relation to the severity of cirrhosis (from Child-Pugh A to Child-Pugh C and AD) (32, 33). Of note, an expansion of intermediate monocytes has been described in the circulation and liver of patients with cirrhosis progression (32, 34). Dysregulation of circulating monocytes and their contribution to systemic immune paresis and the pathophysiology of cirrhosis have been well-documented [reviewed in (10, 35)]. The most consistent finding with regard to monocyte phenotype in chronic liver failure is reduced HLA-DR expression. This reduction, that likely compromises their antigen presentation capacity and the development of adaptive immunity, is considered a hallmark of “endotoxin tolerance”, the immunosuppressed state in which monocytes are refractory to further LPS stimulation or other microbial stimuli. Monocyte HLA-DR expression levels diminish progressively in relation to cirrhosis severity and are associated with adverse clinical outcomes (33, 34, 36–38). Furthermore, monocyte production of pro-inflammatory cytokines in response to microbial cues (e.g., LPS) is key to innate defences against infection as such cytokines can enhance their antimicrobial functions. Monocyte inflammatory (e.g., TNF-α, IL-6) cytokine secretion appears to diminish with advancing chronic liver disease particularly in AD and late stage ACLF (32–34, 36–39). Similarly, plasma derived from AD, but not compensated, cirrhosis suppresses LPS-stimulated TNF-α by healthy monocytes (40).
A role for the anti-inflammatory cytokine IL-10 in driving monocytes towards this “endotoxin tolerant state” has been proposed but this mechanism may involve a variety of pathogen-derived signals or cell-secreted cytokines and proteins (34–37, 41). Recently, the reduced HLA-DR expression on monocytic cells in advanced stages of cirrhosis (AD/ACLF) has been attributed to the expansion of monocytic (CD11b+CD15-CD14+HLA-DR-) myeloid-derived suppressor cells (M-MDSCs) in the circulation (38). M-MDSCs have great pathological significance in chronic liver failure as they exert highly immune-suppressive functions: they decrease T cell proliferation, produce low TNF-α and IL-6 levels after toll-like receptor (TLR) stimulation and have reduced bacterial (E. coli) phagocytosis capacity (38).
The TAM (TYRO3/AXL/MERTK) family of receptors, which are critical regulators of innate immune responses and promote clearance of apoptotic cells (termed efferocytosis), are shown to contribute to systemic immune paresis in liver failure (33, 42, 43). Brenig et al. recently identified a distinct immune-regulatory population of (CD14+HLA-DR+) AXL-expressing monocytes that is expanded in parallel with progression of cirrhosis prior to the AD/ACLF stage and correlates with development of infection and one-year mortality (33). This subset displayed attenuated TNF-α and IL-6 secretion following TLR stimulation, reduced T cell activation but had preserved bacterial phagocytosis and enhanced efferocytosis capacity (33). In patients with AD/ACLF, the emergence of a MERTK-expressing monocyte population is described to decrease innate immune responses to LPS and to associate with disease severity and adverse outcomes (34, 42). Proof-of-concept in vitro inhibition of either AXL or MERTK was shown to enhance monocyte inflammatory responses in ACLF samples. Therefore, TAM receptor targeting may be a new therapeutic strategy to restore monocyte function in cirrhosis. However, this needs further clinical evaluation (35).
Macrophages are the most abundant hepatic immune cells with pivotal roles: from maintaining liver homeostasis and immune tolerance in the face of continuous exposure to harmless gut-derived antigens from food and commensal microbes or their products, to rapidly identifying pathogens and orchestrating immune responses for their elimination (16). In addition, they are functionally important for clearing cellular debris and metabolic waste, and regulating iron and cholesterol homeostasis (16). During steady state, the predominant macrophage population comprises resident Kupffer cells (KCs) which are capable of self-renewal and have a specific transcriptional program defined by their unique niche (44–46). KCs reside within sinusoidal blood vessels, in continuous contact with liver sinusoidal endothelial cells (LSECs), while they always extend a substantial fraction of their cell body into the perisinusoidal space of Disse where they can closely interact with HSCs and hepatocytes (47). This position of KCs is functionally important during liver homeostasis and injury. In the setting of inflammation, following a toxic, metabolic or infectious insult, the liver macrophage pool can further consist of recruited monocyte-derived macrophages (MoMFs) (10, 16). In mice, resident KCs can be identified by their specific (CD11bintF4/80high) MARCO+CLEC4F+TIM4+ expression profile while MoMFs are (CD11bhighF4/80int) CX3CR1+ cells (45, 46, 48, 49). An additional population of liver capsular macrophages (LCMs) has been described to occupy the hepatic capsule. LCMs are CX3CR1highTIM4- MoMFs, replenished at steady state by blood monocytes, that exert key immune surveillance and antimicrobial functions and therefore defend the liver from infections traversing the peritoneal cavity (50).
In humans, liver single cell RNA sequencing (scRNA-seq) studies have identified CD68+ CD163+MARCO+TIMD4+ resident KCs, CD68+MARCO- recruited MoMFs and infiltrating CD14+ monocytes (51–53). Unbiased cross-species comparison suggests a highly conserved transcriptional signature among human and mouse KCs (53, 54). The dichotomous concept of M1/M2 polarisation cannot be reliably applied to hepatic macrophages, as with most tissue macrophages, since they simultaneously express M1/M2 markers and exhibit great plasticity which is dependent on their ontogeny, local and systemic environmental mediators and epigenetic programming. Liver macrophage ontogeny and heterogeneity is extensively covered elsewhere (16, 44, 55, 56).
Tissue damage and hepatocyte injury generate coordinated wound healing responses that aim to restore healthy hepatic parenchyma. Release of DAMPs, ROS and changes in hepatocyte gene expression are triggered, promoting an immune response, clearance of apoptotic or dead cells, hepatocyte proliferation, matrix remodelling, and angiogenesis (57). Such changes result in increased expression of profibrogenic TGF-β, hedgehog signalling ligands and CXCL10 amongst others (58–61). If the insult abates, then resolution responses, including macrophage efferocytosis, restore tissue homeostasis. However, chronic injury eventually leads to dysregulated tissue repair and development of liver fibrosis. This is a dynamic process in which tissue damage and inflammation result in activation of HSCs, their epigenetic reprogramming and trans-differentiation into activated myofibroblast-like cells, cytokine and chemokine release, thus contributing to a profibrogenic microenvironment with subsequent ECM accumulation (mainly type I collagen) that limits parenchymal regeneration (19, 62). Early fibrosis can regress with injury removal and the activity of matrix degradation and remodelling by several matrix metalloproteinases (MMPs), myofibroblast senescence and HSC apoptosis (63, 64). MMPs are counteracted by tissue inhibitors of MMPs (TIMPs) and the balance of activity favouring TIMPs is associated with advancing fibrosis (64, 65).
Chronic liver injury eventually leads to progressive liver fibrosis and the development of cirrhosis with formation of fibrous septa, structural and vascular changes and regenerative nodules (58). Macrophage adaption during liver injury can depend on the nature of the insult (e.g., toxic, metabolic or infectious). In alcohol-induced liver damage, the number of hepatic macrophages increases via MoMF recruitment (66). Both KCs and MoMFs exhibit substantial plasticity with phenotypic and functional alterations depending on microenvironmental signals. Alcohol sensitizes KCs to TLR4-signalling and incites oxidative stress, which promotes an M1-like phenotype and LPS-induced cytokine production, particularly TNF-α, IL-6, IL-1β and CCL2 (67). In murine models of alcohol steatohepatitis, MoMFs are recruited to the liver in a NOTCH1-mediated mechanism with subsequent M1-like macrophage activation (68, 69). This process is compounded by alcohol-induced intestinal gut microbial dysbiosis, reduced gut epithelial integrity and subsequent heightened translocation of gut-derived microbial products including LPS (70). Chronic excess alcohol also results in hepatocyte fat accumulation (termed steatosis) and increased hepatocyte programmed cell death via apoptosis, necrosis, pyroptosis and ferroptosis, thus promoting further inflammation and injury (71). There are protective mechanisms at play including liver macrophage autophagy (an anti-inflammatory homeostatic intracellular pathway directing damaged organelles or cytosolic macromolecules to lysosomes for degradation), in which IRF-1 degradation, mitophagy (clearance of damaged mitochondria) and downregulation of inflammasome-dependent and independent pathways occur (72–74).
The multifactorial pathophysiology of alcoholic steatohepatitis overlaps significantly with non-alcoholic fatty liver disease (NAFLD) and its inflammatory and often progressive subtype non-alcoholic steatohepatitis (NASH). Macrophage activation occurs in response to endotoxins and translocated bacteria due to increased intestinal permeability, factors released from damaged or lipoapoptotic hepatocytes, as well as alterations in the gut microbiota and nutritional components (75). Collectively, these lead to a chronic inflammatory state resulting in disease progression. In obesity-associated NAFLD, release of free fatty acids from white adipose tissue promotes hepatocyte triglyceride synthesis and storage, and lipotoxicity with production of TNF-α, IL-6, IL-1β, IL-17A and macrophage-recruiting chemokines (e.g., CCL2, CCL5 and CXCL10). Macrophages in this condition have been studied in both human and disease models. Increased numbers of CD68+ macrophages are found in biopsies of young patients with more severe NAFLD (76), while in children with NAFLD numerous activated macrophages are located in the spaces between damaged hepatocytes (77). The portal infiltration of CCR2+ macrophages (78) appears to be an early event in human NAFLD, occurring already at the stage of steatosis before inflammation or fibrosis develops, but predicting progressive disease (79). In line with these data, lipogranuloma and macrophage alignment around steatotic hepatocytes have been observed (termed hepatic crown-like structures) (80).
Similarly, macrophage infiltration has been demonstrated in murine dietary models [e.g., high-fat diet (HFD), methionine–choline-deficient (MCD) diet] which have different strengths and limitations [reviewed in (75, 81)]. Undoubtedly, liver macrophages play a major role in the pathogenesis of NAFLD/NASH (75). Experimental evidence for this has come from studies in which depleting macrophages using genetic (LysM, myeloid-specific) models and clodronate liposomes protected mice from steatosis, liver damage and inflammation (82–85). Moreover, genetic deficiency and pharmacological inhibition of CCR2 decrease monocyte recruitment into the liver and ameliorate NASH in mice (78, 86). The level of macrophage heterogeneity in NASH, however, is greater than we initially thought. Recent scRNA-seq murine studies have shed more light into this, consistently demonstrating that resident KCs are lost during NASH progression and recruited monocytes enter the liver where they respond to niche-specific and inflammatory cues to become monocyte-derived KCs (MoKCs) or temporary MoMFs (49, 87–89). The distinct roles in disease pathology, functions and interactions of these macrophage subpopulations require more exploration, as further discussed below. For an overview of the immunometabolic interplay of hepatic macrophages and the adipose tissue-fatty liver crosstalk in NAFLD/NASH see also references (75, 90–92).
In both a mouse model of hepatitis B virus (HBV)-induced liver inflammation and in patients with viral-related chronic liver failure, Tan-Garcia et al. describe an intrahepatic infiltration of a population of pro-inflammatory CD14+HLA-DRhighCD206+ myeloid cells (93). Likewise, Ohtsuki et al. observed an increase in the CD11b+F4/80+CD206+ intrahepatic macrophage population (labelled as “M2”) in HCV-infected transgenic mice, also producing more TNF-α and IL-6 following ex vivo LPS stimulation, when compared with M1 (CD11b+F4/80+CD11c+) (94). Chronic alcohol feeding of wild-type mice resulted in an increased frequency of CD206+ CD163+ M2 macrophages but also increased expression of M1 genes (TNF-α, MCP1 and IL-1β) and M2 genes (Arg1, Mrc1 and IL-10), with increased expression of Kruppel-like factor 4 (KLF4) promoting an M2 phenotype (95). Gut bacterial translocation appears important in viral chronic liver injury; interestingly, HBV-infected mice treated with oral antibiotics showed depleted hepatic CD14+CD206+ populations (93). Corroborating this, enhanced gut bacterial translocation has been noted in patients from HBV and HCV infection (96). This highlights the shared mechanisms in alcohol, NAFLD and viral-related chronic liver injury, the simplicity of the M1/M2 dichotomy and high degree of macrophage plasticity dependent on the milieu of inflammatory mediators, chemokines and gut-derived microbial products.
The role of macrophages in liver fibrosis is complex. Irrespective of the aetiology, there are common fundamental molecular mechanisms that lead to fibrosis. Mechanistically, persistent or repetitive liver injury results in hepatocyte damage and release of DAMPs which stimulate KC activation. Additionally, KC activation also occurs with the action of PAMPs derived from pathological BT across a dysfunctional gut barrier in cirrhosis, ROS, hypoxia-inducible factor-1α (HIF-1α), increased hepatic concentrations of triglycerides, cholesterol and succinate, and extracellular vesicles (16). Activated KCs produce pro-inflammatory cytokines that contribute to injury (e.g., TNF-α, IL-6 and IL-1β). They also generate, in conjunction with hepatocytes or HSCs, chemokines (e.g., CCL2, CCL5) promoting recruitment of pro-inflammatory, pro-fibrogenic MoMFs (97). Chemokine-driven recruitment may involve different mechanisms; CCL2 in addition to hepatic PC3-secreted microprotein (PSMP) promotes hepatic CCR2+ inflammatory monocyte infiltration and induction of HSCs via their CCR2 receptor (98). In NAFLD, murine and human liver biopsies demonstrate CCR2+ MoMF accumulation in the portal tracts; KCs do not express CCR2. The marker has diagnostic value with an increase in CCR2+ cells observed as fibrosis advances and CCR2+ MoMF infiltration throughout the parenchyma in end-stage cirrhosis (78). In vitro, viral hepatitis C exposed KCs secrete CCL5 that induces CCR5+ HSC activation through ERK phosphorylation (99). Where there are defined areas of injury, MoMFs form ring-like structures and where damage is more widely distributed, they convene in periportal regions (100, 101). Murine Ly6Chigh monocytes differentiate into Ly6Clow/+ macrophages that secrete proinflammatory cytokines and generate ROS. These inflammatory macrophages activate HSCs, generate TNF-α and IL-1β, that promotes HSC survival, and IL-6 that leads to HSC proliferation and production of TIMP1 (102). Other HSC activation routes occur via paracrine mechanisms involving JAK2 signalling pathways, NADPH complexes (NOX) and hepatocyte mitochondria-derived DAMPs (16, 103–106). Moreover, these MoMFs release profibrotic mediators such TGF-β, platelet-derived growth factor (PDGF), connective tissue growth factor (CTGF) and TIMPs, which promote myofibroblast ECM production (16, 58).
Not all hepatic macrophages in the setting of chronic liver injury are profibrotic. MoMFs can also exert anti-fibrogenic functions, depending on their differentiation status and the disease stage. This has been shown in murine studies were the deletion of infiltrating MoMFs during fibrogenesis resulted in reduced HSC activation and ECM deposition whilst deletion of MoMFs during the regression stage impaired ECM degradation, thereby exacerbating fibrosis (107, 108). In humans, a distinct population of TREM2+CD9+ scar-associated macrophages (SAMs) have been observed to occupy fibrotic niches in livers of cirrhotic patients. SAMs are derived from recruitment and differentiation of circulating monocytes, and are expanded early in the disease course (53, 108). Originating from initially pro-inflammatory TGF-β and PDGF-producing infiltrating macrophages, SAMs evolve after a phenotypic switch in the restoration phase of tissue inflammation and function to resolve fibrosis, mainly through the expression of MMPs, including MMP-13, and mediated by macrophage migration inhibitory factor (MIF) (109, 110). TREM2+CD9+ macrophages have also been reported in NAFLD/NASH models, however, Trem2 and Cd9 are not restricted to non-KCs in mice (49, 89, 91, 111). The multiple mechanisms of fibrosis progression and resolution and the co-existence of various macrophage subsets with potentially distinct functions present several opportunities for drug therapies to reverse or ameliorate fibrosis in chronic liver diseases.
NAFLD is nowadays one of the most common CLD that is strongly associated with obesity and the metabolic syndrome, and similar to these conditions, its incidence and prevalence are worryingly increasing (91, 92). This disease is underpinned by a pathophysiology that links multiple processes including chronic low-grade inflammation, lipotoxicity, dysregulated bile acid metabolism and altered gut microbiome with enhanced BT. Adipose tissue macrophages (ATMs) are spread throughout the tissue; although their origin under homeostatic conditions is not clear, recent murine fate mapping studies have revealed that resident ATMs can derive from both yolk-sac and bone marrow (monocyte) progenitors (91, 112). How similar these two resident ATM subsets are, it remains to be further investigated. In obese adipose tissue, the size and number of adipocytes increase, to compensate for excess lipid availability. However this containment mechanism may fail, and lead to tissue dysfunction, dyslipidemia and insulin resistance (91, 92). Dying adipocytes can release various triggers (e.g., toxic lipids, adipokines) or DAMPs which create a complex local microenvironment that causes ATM activation. In response, the number of ATMs is markedly increased, with many of them surrounding necrotic adipocytes within crown-like structures, in both mice and humans, an optimal location enabling them to engulf cell debris (91, 92).
Notably, ATMs have been associated with the severity and progression of NAFLD (113). In both mice and humans, the recruitment of macrophages in adipose tissue compartment has been linked with development of insulin resistance and steatohepatitis while ablation of ATMs or surgical removal of adipose tissue in mice normalized insulin sensitivity and partially reversed liver inflammation (92). ATMs secrete inflammatory cytokines such as TNF-α, IL-1β, IL-6 and monocyte-chemoattractant protein-1 (MCP-1) which promote insulin resistance, lipolysis and hepatic lipid flux and accumulation (114). Chemokine axes such as CCL2-CCR2 and CCL5-CCR5 drive further recruitment to adipose tissue, and in part provides the rationale for their dual CCR2-CCR5 antagonism as a therapeutic approach to resolve inflammation and prevent fibrosis progression in NAFLD (75). The phenotype of ATMs does not conform to the dichotomous M1/2 polarisation as once thought, but instead has a unique blend termed a metabolically activated type with distinctive transcriptional profiles that can be influenced by the local adipose tissue microenvironment. Pro-inflammatory macrophage programming may be primed by LPS (perhaps more so with pathological BT), IFN-γ, HIF-1α and saturated fatty acids. Independent of LPS, fatty acids may be taken up via the macrophage scavenger receptor 1 (MSR1) with subsequent JNK signalling and inflammation (115). On the contrary, HIF-2α, PPAR-γ and unsaturated FAs exert an anti-inflammatory effect (91, 92, 116). Few groups have recently examined the nature of ATMs in obese adipose tissue by scRNA-seq (112, 117, 118). It has now become evident that ATMs display significant heterogeneity, with various subsets (clusters) identified that may contribute differently to the disease pathology; their specific functions need to be studied in the coming years (91).
The development of ascites, the pathological accumulation of fluid within the peritoneal cavity, is a defining feature of decompensated cirrhosis. Pathogens and bacterial products are readily absorbed by the peritoneal cavity and invoke an inflammatory reaction. This can give rise to SBP which occurs in 10% of hospitalised patients and once established can worsen prognosis including the development of multi-organ failure and increased mortality. SBP is defined by an elevated ascites polymorphonuclear cell count of >250/mm3 with or without the positive culture of microbes. In cirrhosis, the pathological BT theory implies a higher exposure to pathogenic material in the peritoneum even in steady state.
Under normal conditions, peritoneal macrophages (PMs) comprise 50-90% of the peritoneal leucocytes and are primarily responsible for clearing debris and pathogens. In both sterile and pathogen-associated injury, interferon-γ primed macrophages orchestrate the immune response, highly expressing MHC class II molecules, generating cytokines and chemokines and exerting strong antimicrobial effector mechanisms (119, 120). Similar to the dichotomy of KC and MoMF in the liver, murine PMs can be subdivided into self-renewing resident macrophages and MoMFs which slowly replace the resident population and acquire a differentiated phenotype (121). Resident PMs have a unique profile including high expression of genes encoding phagocytic receptors (e.g., Vsig4, Timd4, and Marco) (122).
In humans, two studies from the same group, one evaluating ascites of patients with cirrhosis and the other abdominal washouts from women undergoing gynaecological abdominal surgery, identified that PMs are distributed in three subpopulations: a) classical-like CD14++CD16-, b) an intermediate CD14++CD16+, and c) a large granular CD14highCD16high subset. The latter has no corresponding blood monocyte subpopulation (123, 124). The expression of CD14 (LPS related receptor) and CD16 (phagocytic Fcγ receptor) is increased in PMs, compared to circulating monocytes, suggesting that PMs are primed for pathogen defence. Moreover, in patients with cirrhosis and ascites, the CD14highCD16high peritoneal macrophage subset represented approximately 30% of total CD14+ cells, compared with approximately 15% in women undergoing gynaecological surgery, perhaps reflective of the enhanced BT in cirrhosis.
Human PMs were also found to have higher expression of other phagocytic markers (e.g., CD11c), cytokine receptors (e.g., CD116 and CD119), antigen-presentation markers (e.g., HLA-DR) and co-stimulatory molecules (e.g., CD40 and CD80) when compared with blood monocytes. The CD14highCD16high population was felt to represent an M2-polarised human resident peritoneal macrophage population, as suggested by high GATA-6 expression (similar to mice) (121, 122), enhanced expression of mannose receptor (MR) CD206 and HLA-DR, and higher frequency of IL-10 positive cells. Despite this, they seem to have an immunologically-primed status with increased phosphorylation of ERK1/2, PKB and c-Jun, sensitivity to LPS, robust oxidative burst activity, increased expression of cytosolic dsDNA sensor absent in melanoma 2 (AIM2) and AIM2 ligand-induced mature IL-1β and IL-18 production suggesting they are readied for antimicrobial defence (124–127). Immune priming may vary according to cirrhosis aetiology, with alcohol-related cirrhosis displaying a more pro-inflammatory profile, compared to viral hepatitis C, with higher ascitic IL-12 and lower IL-10 levels (128). Genetic polymorphisms in the TRAF6 gene coding for the adaptor protein in TLR signalling cascades, confers a less pro-inflammatory state of PMs in cirrhosis and increased risk of SBP (129).
Irvine et al. went further to segregate human PMs by investigating the expression of CCR2 and complement receptor for immunoglobulin (CRIg) which is encoded by the VSIG4 gene (130, 131). The investigators found a distinct human peritoneal macrophage population that was CRIghighCCR2low. Compared with CRIglow, the CRIghigh macrophages were larger, more granular, had higher expression of CD14, CD16, HLA-DR, CD169 and CD163, similar to the aforementioned CD14highCD16high resident subset. CRIghigh macrophages were characterized by increased gene transcription of efferocytosis receptors (e.g., MERTK and TIMD4), and showed enhanced phagocytic and microbicidal functions. Of note, high proportions of CRIghigh macrophages were associated with reduced liver disease severity (MELD score) and lower proportions were observed during clinical events (e.g., SBP, death) (130).
Stengel and Quickert et al., also identified a distinct subset of CD206+CCR2- large peritoneal macrophages (LPM) (in addition to being CD16+, CD163+, CRIg+ and MerTK+) in patients with cirrhosis which were transcriptionally and functionally distinct. When activated, this subset of CD206+ LPMs were more likely to produce pro-inflammatory cytokines, showed resistance to endotoxin tolerance and cleavage of surface CD206. Concentrations of the CD206 soluble form (sCD206) in the ascitic fluid were elevated in SBP, compared to non-infected ascites, reflective of PM activation; high sCD206 ascites levels were predictive of higher 90-day mortality (132). This is in contrast to the historically perceived anti-inflammatory nature of CD206+ macrophages, illustrating their plasticity (133).
Considering the entire human peritoneal macrophage population, the co-expression of M1 and M2 markers points to a constitutive plasticity, responsive to sterile and pathogenic stimuli and programmed to subsequently restore tissue homeostasis (134). The ascitic microenvironment may be crucial in altering the macrophage phenotype and function to suit the insult and promote recruitment of circulating myeloid cells. Hypoxia promotes peritoneal macrophage generation of VEGF and ADM via enhanced HIF-1α transcription (135, 136). In the setting of ACLF where cardiorespiratory organ failure might result in hypoxia, the production of vasodilator mediators in such circumstances serves as a mechanism to further worsen circulatory dysfunction. In the setting of pathological BT, high ascites bacterial DNA concentrations, even in the absence of a clinical diagnosis of SBP, are associated with increased proinflammatory cytokine production, increased inducible nitric oxide synthetase (iNOS) expression and subsequent nitric oxide (NO) overproduction, reduced CD14+ peritoneal macrophage HLA-DR expression and poor clinical outcomes (e.g. death and hospital readmission) (137–139).
Infection (SBP), bacteria or bacterial products (e.g., LPS), result in production of inflammatory cytokines such as TNF-α, IL-6, calprotectin and macrophage inflammatory protein type 1 beta (MIP-1β), which promote neutrophil and monocyte recruitment (140, 141). Counter-regulatory mechanisms, to prevent over-exuberant inflammation, result in generation of ascitic IL-10, resistin and reduction in peritoneal macrophage surface expression of CD14, CD16, HLA-DR, CD86 and CD206, which reverse on antibiotic treatment. Reduction in macrophage LPS-receptor CD14 in SBP was associated with impaired phagocytosis (142, 143). Interestingly, Wang et al. using the thermal and CCl4-induced acute liver injury murine models and intravital imaging demonstrated that peritoneal cavity (GATA6+) resident macrophages have the ability to migrate via non-vascular routes to the injured liver (144). In the damaged sites, GATA6+ macrophages could be triggered by DAMPs, such as ATP, to exhibit a more alternatively activated state and mediate cell debris clearance, thus aiding tissue repair and revascularisation. Future work will examine if these cells are present in humans and to what extent they modify other liver inflammatory diseases.
Beyond the liver, peritoneum and adipose tissue, there is little known regarding compartmental tissue macrophage phenotype and function during chronic liver failure. In health, intestinal macrophages represent a small proportion of the lamina propria immune cells, which are CD33+CD14- and exhibit anergy, hyporesponsive to LPS (145, 146). In cirrhosis, substantial evidence points to a failure of the gut barrier, permitting increased intestinal permeability and BT with increased circulating loads of bacterial DNA and other microbial derivatives, and an increased risk of infection (147). One study identified an activated CD14+TREM1+ intestinal macrophage phenotype in cirrhosis, with high expression of iNOS even in early compensated disease and secretion of NO (148). Such activated macrophages are known to be responsive to microbial challenge, with pro-inflammatory cytokine output (e.g. IL-23, TNF-α and IL-6), and may contribute to the observed intestinal defects (149). Immune defects permitting BT were also observed in mesenteric lymph nodes from patients with decompensated cirrhosis, where circulation-derived subcapsular sinus and medullary cord macrophages expressed the immune-suppressive marker MERTK, compared to non-cirrhotic controls (42). In patients with cirrhosis and portal hypertension, the number and phagocytic activity of splenic macrophages in the red pulp and marginal zone is enhanced, resulting in hypersplenism and cytopenias (150). Enhanced phagocytosis may be explained by the upregulation of micro-RNA miR-615-3p and its action on a ligand-dependent nuclear receptor corepressor (LCoR)-PPARγ axis (151). Finally, there is evidence of spleen-liver organ crosstalk, with splenectomy reversing the M1-dominant hepatic macrophage phenotype in fibrotic livers, ameliorating collagen deposition and regressing fibrosis (152). This is attributed to the disruption of splenic macrophage promotion of hepatic macrophage CCL2 expression, subsequent circulating monocyte liver recruitment and adoption of an M1 phenotype causing further injury and fibrosis (153).
Macrophages play a significant role in the development and progression of chronic liver failure. Two scavenger receptors that are highly, but not exclusively, expressed by blood monocytes and macrophages are CD163 and CD206, also known as the haemoglobin-haptoglobin receptor and mannose receptor (MR), respectively (154, 155). The soluble forms of CD163 (sCD163) and MR (sMR), that are present in plasma and body fluids, have been thoroughly investigated as macrophage-related biomarkers in liver diseases (154, 155). Over recent years, numerous studies on sCD163 and sMR have detected increased levels in relation to severity and prognosis in both acute and chronic liver diseases (154, 155). For instance, this has been demonstrated in NAFLD/NASH, viral hepatitis (e.g., HBV, HCV), autoimmune hepatitis, acute liver failure (ALF) and alcohol-related liver disease (132, 156–163). In patients with cirrhosis, sCD163 and sMR significantly correlate with severity (e.g. MELD and Child-Pugh scores) as well as the degree of portal hypertension (164–167); high plasma concentrations of sCD163 associate with variceal bleeding in cirrhotic patients and predict mortality in alcoholic hepatitis (165, 168). Bruns and colleagues recently measured sMR levels in ascites fluid from cirrhotic patients with SBP and found that its concentrations serve as a marker of peritoneal macrophage activation, inflammation and predict 90-day survival (132).
Overall, the highest sCD163 and sMR levels are detected in patients with the most severe forms of liver injury such as ALF, alcoholic hepatitis and ACLF (154, 155). In ACLF, sCD163 and sMR are independently associated with disease severity and prognosis while supplementation of these macrophage biomarkers to standard clinical scores (e.g., CLIF-C ACLF, CLIF-C AD) improved their prognostic performance (158). This accumulating clinical evidence shows robust association of macrophage-related markers with inflammation in liver pathologies and emphasizes the role of macrophages and infiltrating monocytes in liver disease development and progression. Future work will establish the incorporation of these biomarkers in current clinical practice scoring tools to improve prognosis.
There is a major unmet need for effective therapies in chronic liver failure and this is of high clinical relevance considering the escalating disease prevalence. Macrophages are central to the pathogenesis of liver diseases as they are involved in the initiation, progression and regression of tissue injury. Macrophages play a key role in liver homeostasis and are among the first responders to an infectious or tissue-damaging insult exerting dual, inflammatory and anti-inflammatory/restorative, functions in the liver. Therefore, hepatic macrophages are an attractive target for developing new therapeutic approaches. Most macrophage-based strategies have been investigated in animal models while some have been evaluated in clinical trials [reviewed in (10, 16, 56, 92)].
Macrophage-targeted approaches can be categorized into those that: (i) inhibit macrophage activation, (ii) inhibit the recruitment of monocytes and monocyte-derived macrophages, (iii) reprogram macrophages via anti-inflammatory polarization mediators and signalling pathways, drug delivery nanosystems, metabolic rewiring, epigenetic mechanisms or immune checkpoint blockade (Figure 2). Cell-based therapies using autologous macrophage infusions have also been tested in patients with liver cirrhosis (169); the rationale and potential of utilizing macrophages as agents for cell therapy are reviewed in (57, 170). Here, we summarize the various approaches that have been explored for therapeutic targeting of macrophages in liver diseases.
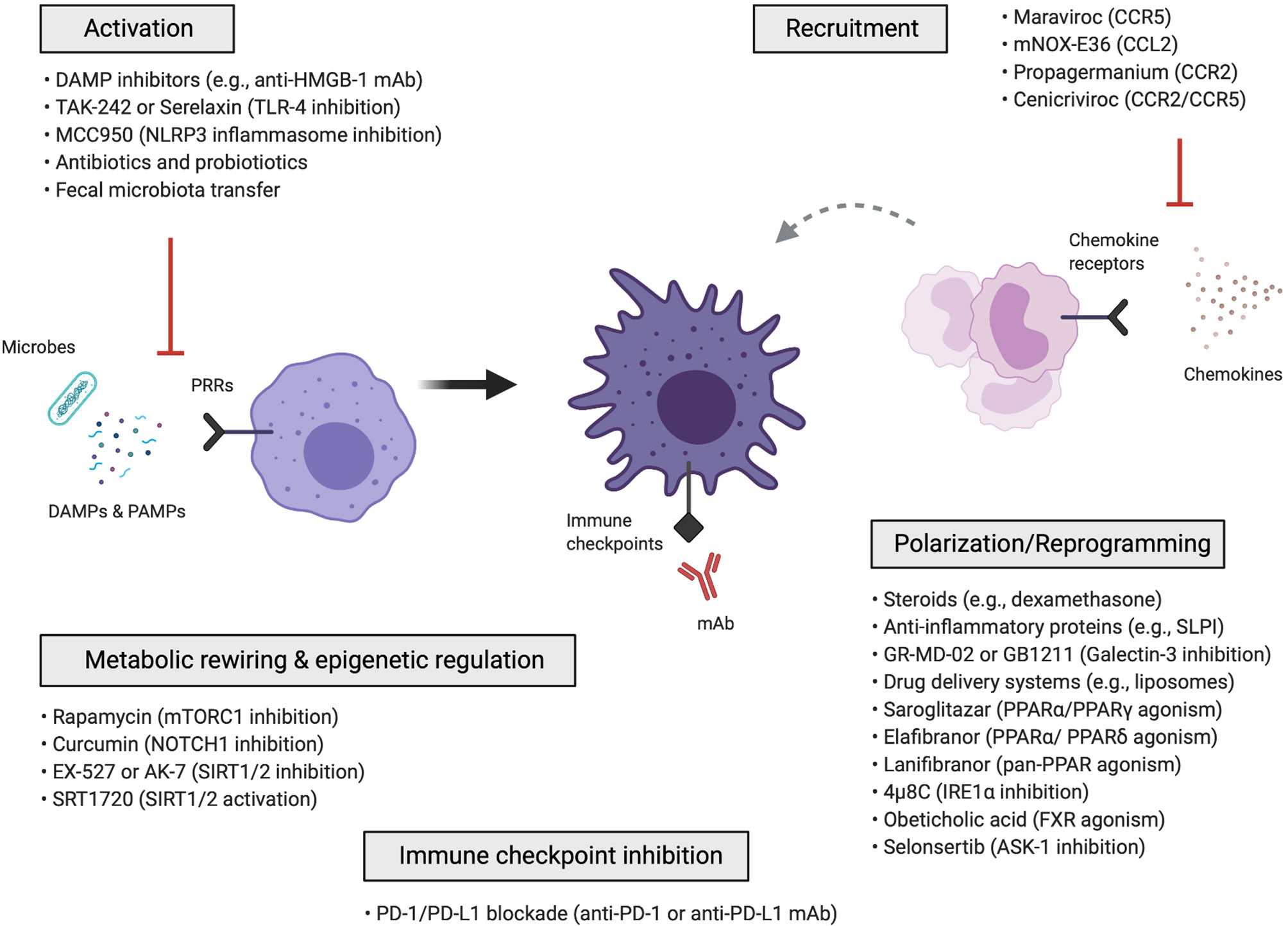
Figure 2 Macrophage-targeted therapeutic approaches for the treatment of liver disease. Schematic overview of the different macrophage-directed therapeutic approaches that can be summarized into those that: (i) inhibit the activation of macrophages, (ii) inhibit the recruitment of monocytes and monocyte-derived macrophages, (iii) reprogram macrophages through anti-inflammatory and polarization mediators, signalling pathways, drug delivery nanosystems, iv) metabolically rewire or epigenetically regulate macrophages, v) modulate macrophages via immune checkpoint inhibition. ASK-1, apoptosis signal-regulating kinase 1; CCL, C-C chemokine ligand; CCR, C-C chemokine receptor; DAMP, damage-associated molecular pattern; FXR, farnesoid X receptor; IRE1α, inositol-requiring enzyme 1α; mAb, monoclonal antibody; PAMP, pathogen-associated molecular pattern; PD-1, Programmed-cell-death-1; PD-L1, Programmed-cell-death-ligand-1; PPAR, peroxisome proliferator-activated receptor; PRR, pattern recognition receptor; SIRT1/2, sirtuin-1 and sirtuin-2; SLPI, secretory leukocyte protease inhibitor; TLR, toll-like receptor.
Changes in gut microbiota composition, increased intestinal permeability and pathological BT into the liver are characteristics of progressive chronic liver disease (e.g, cirrhosis) causing an increase in hepatic levels of endotoxin (e.g., LPS) (8, 35). These gut-derived PAMPs together with liver-derived DAMPs (e.g., HMGB-1, histones) can potentially activate macrophages via pattern recognition receptor (PRR) recognition, triggering inflammatory cascades whose activation can be modulated by several approaches. Among those PPRs, the importance of TLR4 is well documented; indeed, TLR4 inhibition (TAK-242 or Serelaxin) has been shown to ameliorate injury and inflammation in rodent models of liver fibrosis, cirrhosis and ACLF (171–174). In the same context, NLRP3 inflammasome inhibition (MCC950) reduces liver inflammation and fibrosis in experimental NASH (175). Finally, PAMP-mediated macrophage activation can be prevented with restoration of normal gut microbiome using broad-spectrum antibiotics, probiotics and fecal microbiota transfer [further discussed in (8, 16, 176)].
Tissue-infiltrating monocytes and monocyte-derived macrophages can amplify and perpetuate liver inflammation. Their recruitment into the liver is driven by the chemoattractant properties of several chemokines secreted from activated liver cells (e.g., KCs, LSECs) which interact with chemokine receptors expressed on immune cells. The CCL2/CCR2, CCL5/CCR5 and CCL1/CCR8 are very common chemoattractant axes in liver diseases (16, 56). Chemokine signalling inhibition therefore represents an interesting therapeutic strategy to reduce monocyte recruitment and has proven efficacious in experimental disease models. This can be achieved using monoclonal antibodies against chemokines or chemokine receptors, receptor antagonists, aptamer molecules or small-molecule inhibitors (16, 56).
Interference with chemokine pathways to restrict the influx of inflammatory monocytes is one of the most advanced treatments in NASH-related liver fibrosis. Cenicriviroc (CVC; a dual CCR2/CCR5 inhibitor) efficiently blocks CCL2 mediated monocyte recruitment and has been shown to exert anti-inflammatory and anti-fibrotic effects in various experimental liver disease models (78, 177, 178). These results led to human studies evaluating CVC in NASH patients with fibrosis. Following one year of CVC treatment, a significant number of NASH patients showed good response to the treatment and significant improvement in the histological stage of fibrosis (179). These positive effects were maintained in responders in the second year of CVC treatment (180) and a phase 3 clinical trial is assessing its efficacy and safety (NCT03028740). Other molecules inhibiting cell recruitment include propagermanium (CCR2 inhibitor) (181), mNOX-E36 (RNA-aptamer molecule that inhibits CCL2) (182), maraviroc (CCL5/RANTES inhibitor) (183) and small-molecule antagonist against the G protein-coupled receptor 84 (GPR84) (184) which ameliorate disease in experimental NASH.
Recent scRNA-seq studies have described the macrophage heterogeneity in murine NASH, consistently demonstrating that resident KCs (ResKCs) are lost during NASH progression and recruited monocytes enter the liver and become monocyte-derived KCs (MoKCs) or temporary MoMFs (49, 87–89). The distinct roles in disease pathology, functions and relationship between these subsets remain to be explored. For instance, what triggers ResKC death and why MoMFs are protected from this? Furthermore, it’s not clear whether the strategy to reduce monocyte recruitment in NASH via CCR2/CCL2 inhibition could affect both MoKCs and recruited MoMFs, or the latter cells only. Of note, NASH-related liver fibrosis was increased in Ccr2 KO mice (89). Could the timing of CCR2 blockade influence macrophage composition and also alter tissue remodelling? Differences between murine and human macrophage subset functions are likely. Further exploration and better understanding of macrophage heterogeneity in NASH will inform us how to specifically target these critical immune cell subpopulations.
Hepatic macrophages exhibit great functional plasticity. Therefore, a key therapeutic approach is to induce a switch from an inflammatory to an anti-inflammatory/restorative type, aimed to promote resolution of inflammation and accelerate tissue regeneration. Such macrophage reprogramming can be achieved using anti-inflammatory mediators including steroids (e.g., dexamethasone), prostaglandin E2 (PGE2), macrophage colony-stimulating-factor-1 (CSF1) and secretory leukocyte protease inhibitor (SLPI) (10, 43, 185). Galectin-3, mainly expressed in macrophages, is shown to exert inflammatory functions and profibrogenic effects on hepatic stellate cells (186). Despite showing promising results in rodent models, the galectin-3 inhibitor belapectin (GR-MD-02) did not alleviate fibrosis in a phase 2 trial in patients with NASH with cirrhosis and portal hypertension (187, 188). A study evaluating the efficacy and safety of another galectin-3 inhibitor, GB1211, in NASH patients is currently ongoing (NCT03809052). Other targets explored aiming to prevent macrophage activation and promote M2-like polarization include inositol-requiring enzyme 1α (IRE1α) (189), farnesoid X receptor (FXR) (190), apoptosis signal-regulating kinase 1 (ASK-1) (191) and obeticholic acid which is a strong FXR agonist with promising results from an early phase clinical trial in NASH (192).
Due to the anatomical location (liver sinusoids) and high scavenging capacity (e.g., mannose receptor) of KCs, the systemic administration of different drug delivery nanosystems (e.g., polymers, liposomes and microbubbles) leads to their accumulation in the liver (193, 194). This highlights their potential for developing new hepatic macrophage-targeted therapies. For instance, the administration of dexamethasone-loaded liposomes reduced liver inflammation in murine models of acute hepatitis and CCL4-induced chronic toxic liver injury, and this was associated with reduced number of hepatic T cells and induction of an M2-like/restorative macrophage phenotype (195). While such drug carrier materials target hepatic myeloid cells, liver fibrosis also affects their targeting efficiency, supporting the need to adapt nanomedicine-based approaches in chronic liver disease (194, 196).
Another strategy to induce anti-inflammatory macrophage polarization and consequently also ameliorate disease progression is by promoting signalling through the PPAR pathways. PPARs are nuclear transcription factors with multiple functions in NASH pathology, affecting inflammation, lipid and glucose metabolism (197). Recent work has shown that in vivo treatment with saroglitazar, a PPARα/PPARγ agonist, is associated with reduced inflammation and regression of fibrosis in experimental NASH models (198). In a human study, elafibranor, a PPARα/PPARδ agonist, was shown to attenuate liver inflammation without fibrosis worsening in NASH patients (199). Furthermore, in vitro treatment with lanifibranor, a pan-PPAR agonist, reduces the expression of inflammatory genes in murine macrophages and patient-derived circulating monocytes with palmitic acid and increases the expression of lipid metabolism related genes. The anti-inflammatory actions of lanifibranor can be induced through PPARδ agonism as demonstrated by evaluating the effects of individual PPAR agonists (200). Moreover, lanifibranor treatment inhibits hepatic MoMF accumulation, one of the key events preceding fibrosis (78). Lanifibranor is currently investigated in a phase 2 trial in NASH patients (NCT03008070).
Recently, LC3-associated phagocytosis (LAP), a non-canonical form of autophagy that shifts monocyte/macrophage phenotype to an anti-inflammatory type, was reported to be a protective mechanism against fibrosis and systemic inflammation in cirrhosis (201). LAP is enhanced in peripheral and hepatic monocytes from patients with liver fibrosis or those who progress to cirrhosis. Pharmacological inhibition of LAP in patient-derived monocytes or LAP genetic disruption in mice exacerbated inflammation and fibrosis after CCl4-induced liver injury whereas enhancing LAP reduced inflammation and liver fibrosis (201). Moreover, activation of LAP is lost in monocytes from ACLF patients and can be restored by targeting this pathway (201). This suggests that sustaining LAP may open new therapeutic perspectives for patients with chronic liver diseases.
An additional approach could be the metabolic rewiring of hepatic macrophages to modulate their polarization and regulate their function for liver disease treatment (92). Pharmacological promotion of autophagy by targeting a key metabolic regulator, the mammalian target of rapamycin complex 1 (mTORC1), improved high fat diet-induced steatohepatitis in mice by altering lipid metabolism, macrophage polarization, the inflammatory responses and autophagy (202). Interestingly, mice with macrophage-selective mTORC1 ablation displayed an M2-like phenotype, reduced liver inflammation and improved insulin sensitivity (202). Another study has revealed a crucial role for the NOTCH1 pathway in inducing M1-like activation of hepatic macrophages by promoting mitochondrial oxidative phosphorylation and ROS as well as M1-related gene expression (68). Conditional NOTCH1 deficiency in myeloid cells attenuated hepatic macrophage M1-like activation and inflammation in murine alcoholic steatohepatitis and markedly reduced lethality following endotoxin-mediated fulminant hepatitis (68).
The concept of “innate immune memory” has arisen over recent years which may open another window of opportunity for new therapies in chronic liver diseases (203). This idea stems from studies demonstrating that epigenetic mechanisms regulate macrophage function by imprinting them with a “memory response” towards future stimuli. Therefore, macrophages can mount a qualitatively different response, either exaggerated (“trained”) or impaired (“tolerant”), upon exposure to repeated challenge (204, 205). In the context of acute and excessive inflammation, tolerance can act as a protective mechanism to dampen the host’s inflammatory responses to prevent tissue damage. For instance, in response to LPS or pathogen exposure, monocytes and macrophages modify their histone acetylation and methylation traits, dictating gene expression patterns upon subsequent stimulation (206–208). Some of these mechanisms are implicated in rewiring of intracellular metabolic activities affecting the balance between glycolysis and fatty acid oxidation (209, 210). For example, inhibitors of histone deacetylases sirtuin-1 and sirtuin-2 (SIRT1/2) have shown the capacity to reverse immune paresis in experimental murine sepsis (211, 212). Whether such a molecular memory imprinting of anti-inflammatory monocytes and macrophages could be achieved in chronic liver failure is worth exploration.
Immune checkpoints constitute a complex array of receptors (e.g., PD-1, CTLA-4) and their ligands (e.g., PD-L1/PD-L2, CD80/C86) expressed on both innate and adaptive immune cells, which exert key regulatory roles during homeostasis and inflammatory pathologies, mainly in chronic infection, sepsis and cancer. Immune checkpoint inhibition has become an emerging therapeutic strategy in various liver diseases (203, 213). For instance, we have demonstrated that peripheral CD4+ T cells from ALF patients have increased CTLA-4 expression and reduced proliferative response to stimulation that can be enhanced via CTLA-4 inhibition in vitro (214). Furthermore, defects in adaptive and humoral immunity can be partially rescued by in vitro PD-1 blockade in patients with alcohol-related liver disease and viral hepatitis B infection (215, 216). Importantly, the therapeutic reversal of immune exhaustion using immune checkpoint monoclonal antibodies (mAb), alone or in combination with other drugs, has been shown to be effective and with good safety profiles in hepatocellular carcinoma (217–219).
Most checkpoint pathways have been first described as regulators of T cell immunity, but it is now clear that their effects are not limited to T cells only. For instance, accumulating evidence has revealed an crucial role for PD-1/PD-L1 signalling on altering myeloid cell function in sepsis and cancer (220–222). PD-1 and PD-L1 can be induced on monocytes and macrophages through TLR ligands (e.g., LPS) and cytokines (e.g., TNF-α, IL-6 and IL-10). Interestingly, PD-1 and PD-L1 monocyte expression is associated with increased mortality in septic patients while PD-1/PD-L1 blockade restores innate responses in experimental sepsis (223–226).
Many features of sepsis resemble those observed in acute (e.g., acetaminophen overdose) or chronic (e.g., decompensated cirrhosis, alcohol-related ACLF) liver failure patients who often acquire bacterial infections (10, 203, 213). We recently explored the PD-1/PD-L1 pathway in acetaminophen-induced ALF (227). During resolution of murine liver injury, impaired hepatic bacterial clearance and increased PD-1 and PD-L1 expression of KCs and lymphocyte subsets were detected. Compared to wild-type, PD-1 deficient or anti-PD-1 mAb treated mice with liver injury showed improved KC bacterial clearance, reduced bacterial load and protection from sepsis (227). We also found up-regulated PD-1 and PD-L1 expression of peripheral monocytes and lymphocytes in patients with ALF and increased plasma soluble PD-L1 levels predicting mortality and development of sepsis (227). Similarly, another study demonstrated overexpression of PD-L1 in peripheral monocytes and liver macrophages in cirrhotic patients who displayed impaired hepatic bacterial uptake (228). Interestingly, monocyte PD-L1 was associated with disease severity and infection risk in cirrhosis while anti-PD-L1 mAb treatment restored the phagocytic capacity of macrophages and reduced bacterial dissemination in mice with chronic liver injury (228). These studies indicate that immune checkpoint blockade may be an effective and safe strategy for restoration of defective antibacterial responses in chronic liver failure patients, where conventional treatment options are currently very limited.
In conclusion, intensive research over recent years has significantly improved our knowledge on macrophage diversity and plasticity in the context of liver diseases. Technical advances in experimental models and tools, such as single-cell approaches, have enabled us to dissect the key cellular and molecular functions of tissue macrophages, uncovering critical pathophysiological changes involved in different disease states such as liver fibrosis. This forms the basis for future studies that will further deepen our understanding of liver disease pathogenesis, thus enabling the identification of novel immunotherapeutic approaches for patients across the spectrum of chronic liver disease.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
ET has received funding support from Imperial College London (Imperial College Research Fellowship award) and funding support from the Rosetrees Trust (CF2\100002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Imperial College London and the Rosetrees Trust for ongoing support. Figures were created with BioRender.com.
1. Marcellin P, Kutala BK. Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int (2018) 38:2–6. doi: 10.1111/liv.13682
2. Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol (2019) 70:151–71. doi: 10.1016/j.jhep.2018.09.014
3. Moreau R, Jalan R, Gines P, Pavesi M, Angeli P, Cordoba J, et al. Acute-on-Chronic Liver Failure Is a Distinct Syndrome That Develops in Patients With Acute Decompensation of Cirrhosis. Gastroenterology (2013) 144:1426–37.e9. doi: 10.1053/j.gastro.2013.02.042
4. Arroyo V, Moreau R, Jalan R. Acute-on-Chronic Liver Failure. N Engl J Med (2020) 382:2137–45. doi: 10.1056/NEJMra1914900
5. Arroyo V, Angeli P, Moreau R, Jalan R, Clària J, Trebicka J, et al. The systemic inflammation hypothesis: Towards a new paradigm of acute decompensation and multiorgan failure in cirrhosis. J Hepatol (2021) 74 (3):670–85. doi: 10.1016/j.jhep.2020.11.048
6. Albillos A, Lario M, Álvarez-Mon M. Cirrhosis-associated immune dysfunction: Distinctive features and clinical relevance. J Hepatol (2014) 61(6):1385–96. doi: 10.1016/j.jhep.2014.08.010
7. Martin-Mateos R, Alvarez-Mon M, Albillos A. Dysfunctional immune response in acute-on-chronic liver failure: It takes two to tango. Front Immunol (2019) 10:973. doi: 10.3389/fimmu.2019.00973
8. Irvine KM, Ratnasekera I, Powell EE, Hume DA. Causes and Consequences of Innate Immune Dysfunction in Cirrhosis. Front Immunol (2019) 10:293:1–14. doi: 10.3389/fimmu.2019.00293
9. Trebicka J, Fernandez J, Papp M, Caraceni P, Laleman W, Gambino C, et al. PREDICT identifies precipitating events associated with the clinical course of acutely decompensated cirrhosis. J Hepatol (2020) 20:33772–7. doi: 10.1016/j.jhep.2020.11.019
10. Triantafyllou E, Woollard KJ, McPhail MJW, Antoniades CG, Possamai LA. The Role of Monocytes and Macrophages in Acute and Acute-on-Chronic Liver Failure. Front Immunol (2018) 9:2948. doi: 10.3389/fimmu.2018.02948
11. Arvaniti V, D’Amico G, Fede G, Manousou P, Tsochatzis E, Pleguezuelo M, et al. Infections in patients with cirrhosis increase mortality four-fold and should be used in determining prognosis. Gastroenterology (2010) 139:1246–56.e5. doi: 10.1053/j.gastro.2010.06.019
12. Fernández J, Acevedo J, Wiest R, Gustot T, Amoros A, Deulofeu C, et al. Bacterial and fungal infections in acute-on-chronic liver failure: prevalence, characteristics and impact on prognosis. Gut (2018) 67:1870–80. doi: 10.1136/gutjnl-2017-314240
13. Trebicka J, Fernandez J, Papp M, Caraceni P, Laleman W, Gambino C, et al. The PREDICT study uncovers three clinical courses of acutely decompensated cirrhosis that have distinct pathophysiology. J Hepatol (2020) 73:842–54. doi: 10.1016/j.jhep.2020.06.013
14. Piano S, Singh V, Caraceni P, Maiwall R, Alessandria C, Fernandez J, et al. Epidemiology and Effects of Bacterial Infections in Patients With Cirrhosis Worldwide. Gastroenterology (2019) 156(5):1368–80. doi: 10.1053/j.gastro.2018.12.005
15. Fernández J, Prado V, Trebicka J, Amoros A, Gustot T, Wiest R, et al. Multidrug-resistant bacterial infections in patients with decompensated cirrhosis and with acute-on-chronic liver failure in Europe. J Hepatol (2019) 70(3):398–411. doi: 10.1016/j.jhep.2018.10.027
16. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol (2021) 18:45–56. doi: 10.1038/s41423-020-00558-8
17. Nathwani R, Mullish B, Kockerling D, Forlano R, Manousou P, Dhar A. A review of liver fibrosis and emerging therapies. EMJ (2019) 4(4):105–16.
18. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol (2020) 18(3):151–66. doi: 10.1038/s41575-020-00372-7
19. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol (2017) 14:397–411. doi: 10.1038/nrgastro.2017.38
20. Xu F, Liu C, Zhou D, Zhang L. TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J Histochem Cytochem (2016) 64:157–67. doi: 10.1369/0022155415627681
21. Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol (2014) 60:197–209. doi: 10.1016/j.jhep.2013.07.044
22. Assimakopoulos SF, Tsamandas AC, Tsiaoussis GI, Karatza E, Triantos C, Vagianos CE, et al. Altered intestinal tight junctions’ expression in patients with liver cirrhosis: A pathogenetic mechanism of intestinal hyperpermeability. Eur J Clin Invest (2012) 42:439–46. doi: 10.1111/j.1365-2362.2011.02609.x
23. Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol (2014) 60:940–7. doi: 10.1016/j.jhep.2013.12.019
24. Fukui H. Leaky Gut and Gut-Liver Axis in Liver Cirrhosis: Clinical Studies Update. Gut Liver (2020) 1–10. doi: 10.5009/gnl20032
25. Weston CJ, Zimmermann HW, Adams DH. The Role of Myeloid-Derived Cells in the Progression of Liver Disease. Front Immunol (2019) 10:893. doi: 10.3389/fimmu.2019.00893
26. Kapellos TS, Bonaguro L, Gemünd I, Reusch N, Saglam A, Hinkley ER, et al. Human monocyte subsets and phenotypes in major chronic inflammatory diseases. Front Immunol (2019) 10:2035. doi: 10.3389/fimmu.2019.02035
27. Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-dependent Ly6Clow monocytes monitor endothelial cells and orchestrate their disposal. Cell (2013) 153:362–75. doi: 10.1016/j.cell.2013.03.010
28. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol (2005) 5:953–64. doi: 10.1038/nri1733
29. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Sci (80- ) (2017) 356:1–12. doi: 10.1126/science.aah4573
30. Guilliams M, Mildner A, Yona S. Developmental and Functional Heterogeneity of Monocytes. Immunity (2018) 49:595–613. doi: 10.1016/j.immuni.2018.10.005
31. Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med (2017) 214:1913–23. doi: 10.1084/jem.20170355
32. Zimmermann HW, Seidler S, Nattermann J, Gassler N, Hellerbrand C, Zernecke A, et al. Functional Contribution of Elevated Circulating and Hepatic Non-Classical CD14+CD16+ Monocytes to Inflammation and Human Liver Fibrosis. PloS One (2010) 5:e11049. doi: 10.1371/journal.pone.0011049
33. Brenig R, Pop OT, Triantafyllou E, Geng A, Singanayagam A, Perez-Shibayama C, et al. Expression of AXL receptor tyrosine kinase relates to monocyte dysfunction and severity of cirrhosis. Life Sci Alliance (2020) 3:e201900465. doi: 10.26508/lsa.201900465
34. Korf H, Du Plessis J, Van Pelt J, De Groote S, Cassiman D, Verbeke L, et al. Inhibition of glutamine synthetase in monocytes from patients with acute-on-chronic liver failure resuscitates their antibacterial and inflammatory capacity. Gut (2019) 68:1872–83. doi: 10.1136/gutjnl-2018-316888
35. Bernsmeier C, van der Merwe S, Périanin A. Innate immune cells in cirrhosis. J Hepatol (2020) 73:186–201. doi: 10.1016/j.jhep.2020.03.027
36. Berry PA, Antoniades CG, Carey I, McPhail MJWW, Hussain MJ, Davies ET, et al. Severity of the compensatory anti-inflammatory response determined by monocyte HLA-DR expression may assist outcome prediction in cirrhosis. Intensive Care Med (2011) 37:453–60. doi: 10.1007/s00134-010-2099-7
37. Peter J, Frey O, Stallmach A, Bruns T. Attenuated antigen-specific T cell responses in cirrhosis are accompanied by elevated serum interleukin-10 levels and down-regulation of HLA-DR on monocytes. BMC Gastroenterol (2013) 13:37. doi: 10.1186/1471-230X-13-37
38. Bernsmeier C, Triantafyllou E, Brenig R, Lebosse FJ, Singanayagam A, Patel VC, et al. CD14 + CD15 – HLA-DR – myeloid-derived suppressor cells impair antimicrobial responses in patients with acute-on-chronic liver failure. Gut (2018) 67:1155–67. doi: 10.1136/gutjnl-2017-314184
39. Albillos A, de la Hera A, Reyes E, Monserrat J, Muñoz L, Nieto M, et al. Tumour necrosis factor-alpha expression by activated monocytes and altered T-cell homeostasis in ascitic alcoholic cirrhosis: amelioration with norfloxacin. J Hepatol (2004) 40:624–31. doi: 10.1016/j.jhep.2003.12.010
40. O’Brien AJ, Fullerton JN, Massey KA, Auld G, Sewell G, James S, et al. Immunosuppression in acutely decompensated cirrhosis is mediated by prostaglandin E2. Nat Med (2014) 20(5):518–33. doi: 10.1038/nm.3516
41. Wasmuth HE, Kunz D, Yagmur E, Timmer-Stranghöner A, Vidacek D, Siewert E, et al. Patients with acute on chronic liver failure display ‘sepsis-like’ immune paralysis. J Hepatol (2005) 42:195–201. doi: 10.1016/j.jhep.2004.10.019
42. Bernsmeier C, Pop OT, Singanayagam A, Triantafyllou E, Patel VC, Weston CJ, et al. Patients With Acute-on-Chronic Liver Failure Have Increased Numbers of Regulatory Immune Cells Expressing the Receptor Tyrosine Kinase MERTK. Gastroenterology (2015) 148:603–15.e14. doi: 10.1053/j.gastro.2014.11.045
43. Triantafyllou E, Pop OT, Possamai LA, Wilhelm A, Liaskou E, Singanayagam A, et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut (2018) 67:333–47. doi: 10.1136/gutjnl-2016-313615
44. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol (2017) 17(5):306–21. doi: 10.1038/nri.2017.11
45. Beattie L, Sawtell A, Mann J, Frame TCM, Teal B, de Labastida Rivera F, et al. Bone marrow-derived and resident liver macrophages display unique transcriptomic signatures but similar biological functions. J Hepatol (2016) 65:758–68. doi: 10.1016/j.jhep.2016.05.037
46. Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating Monocyte-Derived Macrophages and Resident Kupffer Cells Display Different Ontogeny and Functions in Acute Liver Injury. J Immunol (2014) 193:344–53. doi: 10.4049/jimmunol.1400574
47. Bonnardel J, T’Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L, et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity (2019) 51:638–54.e9. doi: 10.1016/j.immuni.2019.08.017
48. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun (2016) 7:10321. doi: 10.1038/ncomms10321
49. Remmerie A, Martens L, Thoné T, Castoldi A, Seurinck R, Pavie B, et al. Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct from Kupffer Cells in the Fatty Liver. Immunity (2020) 53:641–57.e14. doi: 10.1016/j.immuni.2020.08.004
50. Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, et al. A Liver Capsular Network of Monocyte-Derived Macrophages Restricts Hepatic Dissemination of Intraperitoneal Bacteria by Neutrophil Recruitment. Immunity (2017) 47:374–88.e6. doi: 10.1016/j.immuni.2017.07.018
51. MacParland SA, Liu JC, Ma X-Z, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun (2018) 9:4383. doi: 10.1038/s41467-018-06318-7
52. Aizarani N, Saviano A, Sagar, Mailly L, Durand S, JS H, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature (2019) 572(7768):199–204. doi: 10.1038/s41586-019-1373-2
53. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature (2019) 575:512–8. doi: 10.1038/s41586-019-1631-3
54. Ramachandran P, Matchett KP, Dobie R, Wilson-Kanamori JR, Henderson NC. Single-cell technologies in hepatology: new insights into liver biology and disease pathogenesis. Nat Rev Gastroenterol Hepatol (2020) 17:457–72. doi: 10.1038/s41575-020-0304-x
55. Guillot A, Tacke F. Liver Macrophages: Old Dogmas and New Insights. Hepatol Commun (2019) 3:730–43. doi: 10.1002/hep4.1356
56. van der Heide D, Weiskirchen R, Bansal R. Therapeutic Targeting of Hepatic Macrophages for the Treatment of Liver Diseases. Front Immunol (2019) 10:2852. doi: 10.3389/fimmu.2019.02852
57. Campana L, Iredale J. Regression of Liver Fibrosis. Semin Liver Dis (2017) 37:1–10. doi: 10.1055/s-0036-1597816
58. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest (2017) 127:55–64. doi: 10.1172/JCI88881
59. Fabregat I, Moreno-Càceres J, Sánchez A, Dooley S, Dewidar B, Giannelli G, et al. TGF-β signalling and liver disease. FEBS J (2016) 283:2219–32. doi: 10.1111/febs.13665
60. Machado MV, Diehl AM. Hedgehog signalling in liver pathophysiology. J Hepatol (2018) 68:550–62. doi: 10.1016/j.jhep.2017.10.017
61. Guo X, Cen Y, Wang J, Jiang H. CXCL10-induced IL-9 promotes liver fibrosis via Raf/MEK/ERK signaling pathway. BioMed Pharmacother (2018) 105:282–9. doi: 10.1016/j.biopha.2018.05.128
62. Barcena-Varela M, Paish H, Alvarez L, Uriarte I, Latasa MU, Santamaria E, et al. Epigenetic mechanisms and metabolic reprogramming in fibrogenesis: Dual targeting of G9a and DNMT1 for the inhibition of liver fibrosis. Gut (2020) 70(2)388–400. doi: 10.1136/gutjnl-2019-320205
63. Kim K-H, Chen C-C, Monzon RI, Lau LF. Matricellular Protein CCN1 Promotes Regression of Liver Fibrosis through Induction of Cellular Senescence in Hepatic Myofibroblasts. Mol Cell Biol (2013) 33:2078–90. doi: 10.1128/mcb.00049-13
64. Parola M, Pinzani M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med (2019) 65:37–55. doi: 10.1016/j.mam.2018.09.002
65. Hemmann S, Graf J, Roderfeld M, Roeb E. Expression of MMPs and TIMPs in liver fibrosis - a systematic review with special emphasis on anti-fibrotic strategies. J Hepatol (2007) 46:955–75. doi: 10.1016/j.jhep.2007.02.003
66. Wang M, You Q, Lor K, Chen F, Gao B, Ju C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J Leukoc Biol (2014) 96:657–65. doi: 10.1189/jlb.6a0114-004rr
67. Mandrekar P, Szabo G. Signalling pathways in alcohol-induced liver inflammation. J Hepatol (2009) 50:1258–66. doi: 10.1016/j.jhep.2009.03.007
68. Xu J, Chi F, Guo T, Punj V, Paul Lee WN, French SW, et al. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J Clin Invest (2015) 125:1579–90. doi: 10.1172/JCI76468
69. Gao B, Ahmad MF, Nagy LE, Tsukamoto H. Inflammatory pathways in alcoholic steatohepatitis. J Hepatol (2019) 70:249–59. doi: 10.1016/j.jhep.2018.10.023
70. Sarin SK, Pande A, Schnabl B. Microbiome as a therapeutic target in alcohol-related liver disease. J Hepatol (2019) 70:260–72. doi: 10.1016/j.jhep.2018.10.019
71. Miyata T, Nagy LE. Programmed cell death in alcohol-associated liver disease. Clin Mol Hepatol (2020) 26:618–25. doi: 10.3350/cmh.2020.0142
72. Williams JA, Ding WX. Role of autophagy in alcohol and drug-induced liver injury. Food Chem Toxicol (2020) 136:111075. doi: 10.1016/j.fct.2019.111075
73. Liang S, Zhong Z, Kim SY, Uchiyama R, Roh YS, Matsushita H, et al. Murine macrophage autophagy protects against alcohol-induced liver injury by degrading interferon regulatory factor 1 (IRF1) and removing damaged mitochondria. J Biol Chem (2019) 294:12359–69. doi: 10.1074/jbc.RA119.007409
74. Ilyas G, Cingolani F, Zhao E, Tanaka K, Czaja MJ. Decreased Macrophage Autophagy Promotes Liver Injury and Inflammation from Alcohol. Alcohol Clin Exp Res (2019) 43:1403–13. doi: 10.1111/acer.14041
75. Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol (2019) 16:145–59. doi: 10.1038/s41575-018-0082-x
76. Park J-W, Jeong G, Kim SJ, Kim MK, Park SM. Predictors reflecting the pathological severity of non-alcoholic fatty liver disease: Comprehensive study of clinical and immunohistochemical findings in younger Asian patients. J Gastroenterol Hepatol (2007) 22:491–7. doi: 10.1111/j.1440-1746.2006.04758.x
77. Lotowska JM, Sobaniec-Lotowska ME, Lebensztejn DM. The role of Kupffer cells in the morphogenesis of nonalcoholic steatohepatitis-ultrastructural findings. the first report in pediatric patients. Scand J Gastroenterol (2013)48(3):352–7. doi: 10.3109/00365521.2012.746390
78. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology (2018) 67:1270–83. doi: 10.1002/hep.29544
79. Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, Horsfall L, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology (2014) 59:1393–405. doi: 10.1002/hep.26937
80. Itoh M, Kato H, Suganami T, Konuma K, Marumoto Y, Terai S, et al. Hepatic Crown-Like Structure: A Unique Histological Feature in Non-Alcoholic Steatohepatitis in Mice and Humans. PloS One (2013) 8:e82163. doi: 10.1371/journal.pone.0082163
81. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
82. Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, et al. Depletion of Liver Kupffer Cells Prevents the Development of Diet-Induced Hepatic Steatosis and Insulin Resistance. Diabetes (2010) 59:347–57. doi: 10.2337/db09-0016
83. Neyrinck AM, Cani PD, Dewulf EM, De Backer F, Bindels LB, Delzenne NM. Critical role of Kupffer cells in the management of diet-induced diabetes and obesity. Biochem Biophys Res Commun (2009) 385:351–6. doi: 10.1016/j.bbrc.2009.05.070
84. Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JEM, Van Rooijen N, et al. Kupffer cells promote hepatic steatosis via interleukin-1β-dependent suppression of peroxisome proliferator-activated receptor α activity. Hepatology (2010) 51(2):511–22. doi: 10.1002/hep.23337
85. Lanthier N, Molendi-Coste O, Cani PD, Rooijen N, Horsmans Y, Leclercq IA. Kupffer cell depletion prevents but has no therapeutic effect on metabolic and inflammatory changes induced by a high-fat diet. FASEB J (2011) 25(12):4301–11. doi: 10.1096/fj.11-189472
86. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest (2006) 116:115–24. doi: 10.1172/JCI24335
87. Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity (2020) 52:1057–74.e7. doi: 10.1016/j.immuni.2020.04.001
88. Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gélineau A, et al. Impaired Kupffer Cell Self-Renewal Alters the Liver Response to Lipid Overload during Non-alcoholic Steatohepatitis. Immunity (2020) 53:627–40.e5. doi: 10.1016/j.immuni.2020.06.003
89. Daemen S, Gainullina A, Kalugotla G, He L, Chan MM, Beals JW, et al. Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH. Cell Rep (2021) 34(2):10826. doi: 10.1016/j.celrep.2020.108626
90. Oates JR, McKell MC, Moreno-Fernandez ME, Damen MSMA, Deepe GS, Qualls JE, et al. Macrophage Function in the Pathogenesis of Non-alcoholic Fatty Liver Disease: The Mac Attack. Front Immunol (2019) 10:2893. doi: 10.3389/fimmu.2019.02893
91. Remmerie A, Martens L, Scott CL. Macrophage Subsets in Obesity, Aligning the Liver and Adipose Tissue. Front Endocrinol (Lausanne) (2020) 11:259. doi: 10.3389/fendo.2020.00259
92. Korf H, Boesch M, Meelberghs L, van der Merwe S. Macrophages as Key Players during Adipose Tissue-Liver Crosstalk in Nonalcoholic Fatty Liver Disease. Semin Liver Dis (2019) 39:291–300. doi: 10.1055/s-0039-1687851
93. Tan-Garcia A, Wai LE, Zheng D, Ceccarello E, Jo J, Banu N, et al. Intrahepatic CD206+ macrophages contribute to inflammation in advanced viral-related liver disease. J Hepatol (2017) 67:490–500. doi: 10.1016/j.jhep.2017.04.023
94. Ohtsuki T, Kimura K, Tokunaga Y, Tsukiyama-Kohara K, Tateno C, Hayashi Y, et al. M2 Macrophages Play Critical Roles in Progression of Inflammatory Liver Disease in Hepatitis C Virus Transgenic Mice. J Virol (2016) 90:300–7. doi: 10.1128/jvi.02293-15
95. Saha B, Bala S, Hosseini N, Kodys K, Szabo G. Krüppel-like factor 4 is a transcriptional regulator of M1/M2 macrophage polarization in alcoholic liver disease. J Leukoc Biol (2015) 97:963–73. doi: 10.1189/jlb.4a1014-485r
96. Sandler NG, Koh C, Roque A, Eccleston JL, Siegel RB, Demino M, et al. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterology (2011) 141:1220–30.e3. doi: 10.1053/j.gastro.2011.06.063
97. Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell Mol Immunol (2016) 13:316–27. doi: 10.1038/cmi.2015.104
98. She S, Wu X, Zheng D, Pei X, Ma J, Sun Y, et al. PSMP/MSMP promotes hepatic fibrosis through CCR2 and represents a novel therapeutic target. J Hepatol (2020) 72:506–18. doi: 10.1016/j.jhep.2019.09.033
99. Sasaki R, Devhare PB, Steele R, Ray R, Ray RB. Hepatitis C virus–induced CCL5 secretion from macrophages activates hepatic stellate cells. Hepatology (2017) 66:746–57. doi: 10.1002/hep.29170
100. Dal-Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CHY, Petri B, et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med (2015) 212:447–56. doi: 10.1084/jem.20141539
101. Heymann F, Hammerich L, Storch D, Bartneck M, Huss S, Rüsseler V, et al. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C-C motif chemokine receptor 8 in mice. Hepatology (2012) 55:898–909. doi: 10.1002/hep.24764
102. Nieto N. Oxidative-stress and IL-6 mediate the fibrogenic effects of rodent Kupffer cells on stellate cells. Hepatology (2006) 44:1487–501. doi: 10.1002/hep.21427
103. Akcora BÖ, Dathathri E, Ortiz-Perez A, Gabriël AV, Storm G, Prakash J, et al. TG101348, a selective JAK2 antagonist, ameliorates hepatic fibrogenesis in vivo. FASEB J (2019) 33:9466–75. doi: 10.1096/fj.201900215RR
104. An P, Wei LL, Zhao S, Sverdlov DY, Vaid KA, Miyamoto M, et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat Commun (2020) 11(2362):1–15. doi: 10.1038/s41467-020-16092-0
105. Cai B, Dongiovanni P, Corey KE, Wang X, Shmarakov IO, Zheng Z, et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab (2020) 31:406–21.e7. doi: 10.1016/j.cmet.2019.11.013
106. Paik YH, Iwaisako K, Seki E, Inokuchi S, Schnabl B, Österreicher CH, et al. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91phox mediate hepatic fibrosis in mice. Hepatology (2011) 53:1730–41. doi: 10.1002/hep.24281
107. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest (2005) 115:56–65. doi: 10.1172/JCI200522675
108. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci (2012) 109:E3186–95. doi: 10.1073/pnas.1119964109
109. Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, et al. Scar-Associated Macrophages Are a Major Source of Hepatic Matrix Metalloproteinase-13 and Facilitate the Resolution of Murine Hepatic Fibrosis. J Immunol (2007) 178:5288–95. doi: 10.4049/jimmunol.178.8.5288
110. Barnes MA, McMullen MR, Roychowdhury S, Madhun NZ, Niese K, Olman MA, et al. Macrophage migration inhibitory factor is required for recruitment of scar-associated macrophages during liver fibrosis. J Leukoc Biol (2015) 97:161–9. doi: 10.1189/jlb.3a0614-280r
111. Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol Cell (2019) 75:644–60.e5. doi: 10.1016/j.molcel.2019.07.028
112. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell (2019) 178:686–98.e14. doi: 10.1016/j.cell.2019.05.054
113. du Plessis J, van Pelt J, Korf H, Mathieu C, van der Schueren B, Lannoo M, et al. Association of Adipose Tissue Inflammation With Histologic Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology (2015) 149:635–48.e14. doi: 10.1053/j.gastro.2015.05.044
114. Lefere S, Tacke F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep (2019) 1(1):30–43. doi: 10.1016/j.jhepr.2019.02.004
115. Kragh Petersen S, Bilkei-Gorzo O, Govaere O, Härtlova A. Macrophages and scavenger receptors in obesity-associated non-alcoholic liver fatty disease (NAFLD). Scand J Immunol (2020) 92:1–7. doi: 10.1111/sji.12971
116. Ni Y, Ni L, Zhuge F, Xu L, Fu Z, Ota T. Adipose Tissue Macrophage Phenotypes and Characteristics: The Key to Insulin Resistance in Obesity and Metabolic Disorders. Obesity (2020) 28:225–34. doi: 10.1002/oby.22674
117. Hill DA, Lim H-W, Kim YH, Ho WY, Foong YH, Nelson VL, et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci (2018) 115:E5096–105. doi: 10.1073/pnas.1802611115
118. Silva HM, Báfica A, Rodrigues-Luiz GF, Chi J, Santos P d’Emery A, Reis BS, et al. Vasculature-associated fat macrophages readily adapt to inflammatory and metabolic challenges. J Exp Med (2019) 216:786–806. doi: 10.1084/jem.20181049
119. Heel KA, Hall JC. Peritoneal defences and peritoneum-associated lymphoid tissue. Br J Surg (1996) 83:1031–6. doi: 10.1002/bjs.1800830804
120. Capobianco A, Cottone L, Monno A, Manfredi AA, Rovere-Querini P. The peritoneum: healing, immunity, and diseases. J Pathol (2017) 243:137–47. doi: 10.1002/path.4942
121. Bain CC, Hawley CA, Garner H, Scott CL, Schridde A, Steers NJ, et al. Long-lived self-renewing bone marrow-derived macrophages displace embryo-derived cells to inhabit adult serous cavities. Nat Commun (2016) 7:ncomms11852. doi: 10.1038/ncomms11852
122. Rosas M, Davies LC, Giles PJ, Liao CTC-T, Kharfan B, Stone TC, et al. The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Sci (80- ) (2014) 344:645–8. doi: 10.1126/science.1251414
123. Ruiz-Alcaraz AJ, Tapia-Abellán A, Fernández-Fernández MD, Tristán-Manzano M, Hernández-Caselles T, Sánchez-Velasco E, et al. A novel CD14(high) CD16(high) subset of peritoneal macrophages from cirrhotic patients is associated to an increased response to LPS. Mol Immunol (2016) 72:28–36. doi: 10.1016/j.molimm.2016.02.012
124. Ruiz-Alcaraz AJ, Carmona-Martínez V, Tristán-Manzano M, Machado-Linde F, Sánchez-Ferrer ML, García-Peñarrubia P, et al. Characterization of human peritoneal monocyte/macrophage subsets in homeostasis: Phenotype, GATA6, phagocytic/oxidative activities and cytokines expression. Sci Rep (2018) 8:12794. doi: 10.1038/s41598-018-30787-x
125. Ruiz-Alcaraz AJ, Martínez-Esparza M, Caño R, Hernández-Caselles T, Recarti C, Llanos L, et al. Peritoneal macrophage priming in cirrhosis is related to ERK phosphorylation and IL-6 secretion. Eur J Clin Invest (2011) 41:8–15. doi: 10.1111/j.1365-2362.2010.02368.x
126. Ahmed AMM, Bomford A, Nouri-Aria KT, Davies T, Smith R, Williams R. Peritoneal macrophages from patients with cirrhotic ascites show impaired phagocytosis and vigorous respiratory burst. Results Immunol (2011) 1:53–9. doi: 10.1016/j.rinim.2011.08.004
127. Lozano-Ruiz B, Bachiller V, García-Martínez I, Zapater P, Gómez-Hurtado I, Moratalla A, et al. Absent in melanoma 2 triggers a heightened inflammasome response in ascitic fluid macrophages of patients with cirrhosis. J Hepatol (2015) 62:64–71. doi: 10.1016/j.jhep.2014.08.027
128. Tapia-Abellán A, Martínez-Esparza M, Ruiz-Alcaraz AJ, Hernández-Caselles T, Martínez-Pascual C, Miras-López M, et al. The peritoneal macrophage inflammatory profile in cirrhosis depends on the alcoholic or hepatitis C viral etiology and is related to ERK phosphorylation. BMC Immunol (2012) 13:42. doi: 10.1186/1471-2172-13-42
129. Mai M, Stengel S, Al-Herwi E, Peter J, Schmidt C, Rubio I, et al. Genetic variants of TRAF6 modulate peritoneal immunity and the risk of spontaneous bacterial peritonitis in cirrhosis: A combined prospective-retrospective study. Sci Rep (2017) 7:4914. doi: 10.1038/s41598-017-04895-z
130. Irvine KM, Banh X, Gadd VL, Wojcik KK, Ariffin JK, Jose S, et al. CRIg-expressing peritoneal macrophages are associated with disease severity in patients with cirrhosis and ascites. JCI Insight (2016) 1:668–78. doi: 10.1172/jci.insight.86914
131. Zeng Z, Surewaard BGJ, Wong CHY, Geoghegan JA, Jenne CN, Kubes P. CRIg Functions as a Macrophage Pattern Recognition Receptor to Directly Bind and Capture Blood-Borne Gram-Positive Bacteria. Cell Host Microbe (2016) 20:99–106. doi: 10.1016/j.chom.2016.06.002
132. Stengel S, Quickert S, Lutz P, Ibidapo-Obe O, Steube A, Köse-Vogel N, et al. Peritoneal Level of CD206 Associates With Mortality and an Inflammatory Macrophage Phenotype in Patients With Decompensated Cirrhosis and Spontaneous Bacterial Peritonitis. Gastroenterology (2020) 158:1745–61. doi: 10.1053/j.gastro.2020.01.029
133. Kruger AJ. Can Macrophages in Cirrhotic Ascites Fluid Predict Clinical Outcome in Spontaneous Bacterial Peritonitis? Gastroenterology (2020) 158:1540–3. doi: 10.1053/j.gastro.2020.02.040
134. Ruiz-Alcaraz AJ, Martínez-Banaclocha H, Marín-Sánchez P, Carmona-Martínez V, Iniesta-Albadalejo MA, Tristán-Manzano M, et al. Isolation of functional mature peritoneal macrophages from healthy humans. Immunol Cell Biol (2020) 98:114–26. doi: 10.1111/imcb.12305
135. Pérez-Ruiz M, Ros J, Morales-Ruiz M, Navasa M, Colmenero J, Ruiz-del-Arbol L, et al. Vascular endothelial growth factor production in peritoneal macrophages of cirrhotic patients: Regulation by cytokines and bacterial lipopolysaccharide. Hepatology (1999) 29:1057–63. doi: 10.1002/hep.510290416
136. Cejudo-Martín P, Ros J, Navasa M, Fernández J, Fernádez-Varo G, Ruiz-del-Arbol L, et al. Increased production of vascular endothelial growth factor in peritoneal macrophages of cirrhotic patients with spontaneous bacterial peritonitis. Hepatology (2001) 34:487–93. doi: 10.1053/jhep.2001.27093
137. Francés R, Rodríguez E, Muñoz C, Zapater P, la Sen MLD, Ndongo M, et al. Intracellular cytokine expression in peritoneal monocyte/macrophages obtained from patients with cirrhosis and presence of bacterial DNA. Eur J Gastroenterol Hepatol (2005) 17:45–51. doi: 10.1097/00042737-200501000-00010
138. Francés R, Muñoz C, Zapater P, Uceda F, Gascón I, Pascual S, et al. Bacterial DNA activates cell mediated immune response and nitric oxide overproduction in peritoneal macrophages from patients with cirrhosis and ascites. Gut (2004) 53:860–4. doi: 10.1136/gut.2003.027425
139. Fagan KJ, Rogers GB, Melino M, Arthur DM, Costello ME, Morrison M, et al. Ascites bacterial burden and immune cell profile are associated with poor clinical outcomes in the absence of overt infection. PloS One (2015) 10:1–15. doi: 10.1371/journal.pone.0120642
140. Lesińska M, Hartleb M, Gutkowski K, Nowakowska-Duława E. Procalcitonin and macrophage inflammatory protein-1 beta (MIP-1β) in serum and peritoneal fluid of patients with decompensated cirrhosis and spontaneous bacterial peritonitis. Adv Med Sci (2014) 59:52–6. doi: 10.1016/j.advms.2013.07.006
141. Hadjivasilis A, Tzanis A, Ioakim KJ, Poupoutsi I, Agouridis AP, Kouis P. The diagnostic accuracy of ascitic calprotectin for the early diagnosis of spontaneous bacterial peritonitis. Eur J Gastroenterol Hepatol (2020) 33(3):312–8. doi: 10.1097/meg.0000000000001813
142. Nieto JC, Nieto JC, Sanchez E, Sanchez E, Romero C, Romero C, et al. Impaired innate immune response of leukocytes from ascitic fluid of patients with spontaneous bacterial peritonitis. J Leukoc Biol (2015) 98:819–25. doi: 10.1189/jlb.3ab0315-106r
143. Nieto JC, Perea L, Soriano G, Zamora C, Cantó E, Medina A, et al. Ascitic fluid regulates the local innate immune response of patients with cirrhosis. J Leukoc Biol (2018) 104:833–41. doi: 10.1002/jlb.3a0218-072r
144. Wang J, Kubes P. A Reservoir of Mature Cavity Macrophages that Can Rapidly Invade Visceral Organs to Affect Tissue Repair. Cell (2016) 165:668–78. doi: 10.1016/j.cell.2016.03.009
145. Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, et al. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest (2005) 115:66–75. doi: 10.1172/JCI200519229
146. Smith PD, Smythies LE, Shen R, Greenwell-Wild T, Gliozzi M, Wahl SM. Intestinal macrophages and response to microbial encroachment. Mucosal Immunol (2011) 4:31–42. doi: 10.1038/mi.2010.66
147. Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol (2020) 72:558–77. doi: 10.1016/j.jhep.2019.10.003
148. Du Plessis J, Vanheel H, Janssen CEI, Roos L, Slavik T, Stivaktas PI, et al. Activated intestinal macrophages in patients with cirrhosis release NO and IL-6 that may disrupt intestinal barrier function. J Hepatol (2013) 58:1125–32. doi: 10.1016/j.jhep.2013.01.038
149. Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, et al. Unique CD14+ intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-γ axis. J Clin Invest (2008) 118:2269–80. doi: 10.1172/JCI34610
150. Yongxiang W, Zongfang L, Guowei L, Zongzheng J, Xi C, Tao W. Effects of splenomegaly and splenic macrophage activity in hypersplenism due to cirrhosis. Am J Med (2002) 113:428–31. doi: 10.1016/S0002-9343(02)01210-X
151. Jiang A, Zhang S, Li Z, Liang R, Ren S, Li J, et al. miR-615-3p promotes the phagocytic capacity of splenic macrophages by targeting ligand-dependent nuclear receptor corepressor in cirrhosis-related portal hypertension. Exp Biol Med (2011) 236:672–80. doi: 10.1258/ebm.2011.010349
152. Yada A, Iimuro Y, Uyama N, Uda Y, Okada T, Fujimoto J. Splenectomy attenuates murine liver fibrosis with hypersplenism stimulating hepatic accumulation of Ly-6Clo macrophages. J Hepatol (2015) 63:905–16. doi: 10.1016/j.jhep.2015.05.010
153. Li L, Wei W, Li Z, Chen H, Li Y, Jiang W, et al. The spleen promotes the secretion of CCL2 and supports an M1 dominant phenotype in hepatic macrophages during liver fibrosis. Cell Physiol Biochem (2018) 51:557–74. doi: 10.1159/000495276
154. Nielsen MC, Hvidbjerg Gantzel R, Clària J, Trebicka J, Møller HJ, Grønbæk H. Macrophage Activation Markers, CD163 and CD206, in Acute-on-Chronic Liver Failure. Cells (2020) 9:1175. doi: 10.3390/cells9051175
155. Grønbæk H, Gantzel RH, Laursen TL, Kazankov K, Møller HJ. Macrophage markers and innate immunity in cirrhosis. J Hepatol (2020) 73:1586–8. doi: 10.1016/j.jhep.2020.07.033
156. Kazankov K, Barrera F, Møller HJ, Rosso C, Bugianesi E, David E, et al. The macrophage activation marker sCD163 is associated with morphological disease stages in patients with non-alcoholic fatty liver disease. Liver Int (2016) 36:1549–57. doi: 10.1111/liv.13150
157. Kazankov K, Barrera F, Møller HJ, Bibby BM, Vilstrup H, George J, et al. Soluble CD163, a macrophage activation marker, is independently associated with fibrosis in patients with chronic viral hepatitis B and C. Hepatology (2014) 60:521–30. doi: 10.1002/hep.27129
158. Grønbæk H, Rødgaard-Hansen S, Aagaard NK, Arroyo V, Moestrup SK, Garcia E, et al. Macrophage activation markers predict mortality in patients with liver cirrhosis without or with acute-on-chronic liver failure (ACLF). J Hepatol (2016) 64:813–22. doi: 10.1016/j.jhep.2015.11.021
159. Grønbaek H, Kreutzfeldt M, Kazankov K, Jessen N, Sandahl T, Hamilton-Dutoit S, et al. Single-centre experience of the macrophage activation marker soluble (s)CD163 - associations with disease activity and treatment response in patients with autoimmune hepatitis. Aliment Pharmacol Ther (2016) 44:1062–70. doi: 10.1111/apt.13801
160. Triantafyllou E, Pop OT, Abeles RD, Khamri W, Possamai L, Hussain M, et al. P138 SERONEGATIVE ACUTE LIVER FAILURE REPRESENTS A MACROPHAGE–T CELL ACTIVATION SYNDROME. J Hepatol (2014) 60:S111–2. doi: 10.1016/S0168-8278(14)60300-7
161. Antoniades C, Triantafyllou E, Gadhok R, Abeles R, Quaglia A, Khamri W, et al. 1013 CD163 IS A MECHANISTIC BIOMARKER IN ACUTE LIVER FAILURE REFLECTING A MACROPHAGE ACTIVATION LIKE SYNDROME. J Hepatol (2013) 58:S417. doi: 10.1016/S0168-8278(13)61015-6
162. Sandahl TD, Støy SH, Laursen TL, Rødgaard-Hansen S, Møller HJ, Møller S, et al. The soluble mannose receptor (sMR) is elevated in alcoholic liver disease and associated with disease severity, portal hypertension, and mortality in cirrhosis patients. PloS One (2017) 12:e0189345. doi: 10.1371/journal.pone.0189345
163. Rainer F, Horvath A, Sandahl TD, Leber B, Schmerboeck B, Blesl A, et al. Soluble CD163 and soluble mannose receptor predict survival and decompensation in patients with liver cirrhosis, and correlate with gut permeability and bacterial translocation. Aliment Pharmacol Ther (2018) 47:657–64. doi: 10.1111/apt.14474
164. Holland-Fischer P, Grønbæk H, Sandahl TD, Moestrup SK, Riggio O, Ridola L, et al. Kupffer cells are activated in cirrhotic portal hypertension and not normalised by TIPS. Gut (2011) 60(10):1389–93. doi: 10.1136/gut.2010.234542
165. Waidmann O, Brunner F, Herrmann E, Zeuzem S, Piiper A, Kronenberger B. Macrophage activation is a prognostic parameter for variceal bleeding and overall survival in patients with liver cirrhosis. J Hepatol (2013) 58:956–61. doi: 10.1016/j.jhep.2013.01.005
166. Grønbæk H, Sandahl TD, Mortensen C, Vilstrup H, Møller HJ, Møller S. Soluble CD163, a marker of Kupffer cell activation, is related to portal hypertension in patients with liver cirrhosis. Aliment Pharmacol Ther (2012) 36(2):173–80. doi: 10.1111/j.1365-2036.2012.05134.x
167. Rode A, Nicoll A, Møller HJ, Lim L, Angus PW, Kronborg I, et al. Hepatic macrophage activation predicts clinical decompensation in chronic liver disease. Gut (2013) 62:1231–2. doi: 10.1136/gutjnl-2012-304135
168. Saha B, Tornai D, Kodys K, Adejumo A, Lowe P, McClain C, et al. Biomarkers of Macrophage Activation and Immune Danger Signals Predict Clinical Outcomes in Alcoholic Hepatitis. Hepatology (2019) 70:1134–49. doi: 10.1002/hep.30617
169. Moroni F, Dwyer BJ, Graham C, Pass C, Bailey L, Ritchie L, et al. Safety profile of autologous macrophage therapy for liver cirrhosis. Nat Med (2019) 25:1560–5. doi: 10.1038/s41591-019-0599-8
170. Starkey Lewis PJ, Moroni F, Forbes SJ. Macrophages as a Cell-Based Therapy for Liver Disease. Semin Liver Dis (2019) 39:442–51. doi: 10.1055/s-0039-1688502
171. Shah N, Montes De Oca M, Jover-Cobos M, Tanamoto KI, Muroi M, Sugiyama KI, et al. Role of toll-like receptor 4 in mediating multiorgan dysfunction in mice with acetaminophen induced acute liver failure. Liver Transplant (2013) 19(7):751–61. doi: 10.1002/lt.23655
172. Seki E, De Minicis S, Österreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat Med (2007) 13(11):1324–32. doi: 10.1038/nm1663
173. Engelmann C, Sheikh M, Sharma S, Kondo T, Loeffler-Wirth H, Zheng YB, et al. Toll-like receptor 4 is a therapeutic target for prevention and treatment of liver failure. J Hepatol (2020) 73(1):102–12. doi: 10.1016/j.jhep.2020.01.011
174. Bennett RG, Simpson RL, Hamel FG. Serelaxin increases the antifibrotic action of rosiglitazone in a model of hepatic fibrosis. World J Gastroenterol (2017) 23(22):3999–4006. doi: 10.3748/wjg.v23.i22.3999
175. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol (2017) 66(5):1037–46. doi: 10.1016/j.jhep.2017.01.022
176. Mazagova M, Wang L, Anfora AT, Wissmueller M, Lesley SA, Miyamoto Y, et al. Commensal microbiota is hepatoprotective and prevents liver fibrosis in mice. FASEB J (2015) 29(3):1043–55. doi: 10.1096/fj.14-259515
177. Ambade A, Lowe P, Kodys K, Catalano D, Gyongyosi B, Cho Y, et al. Pharmacological Inhibition of CCR2/5 Signaling Prevents and Reverses Alcohol-Induced Liver Damage, Steatosis, and Inflammation in Mice. Hepatology (2019) 69:1105–21. doi: 10.1002/hep.30249
178. Kruger AJ, Fuchs BC, Masia R, Holmes JA, Salloum S, Sojoodi M, et al. Prolonged cenicriviroc therapy reduces hepatic fibrosis despite steatohepatitis in a diet-induced mouse model of nonalcoholic steatohepatitis. Hepatol Commun (2018) 2:529–45. doi: 10.1002/hep4.1160
179. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology (2018) 67:1754–67. doi: 10.1002/hep.29477
180. Ratziu V, Sanyal A, Harrison SA, Wong VW, Francque S, Goodman Z, et al. Cenicriviroc Treatment for Adults With Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology (2020) 72:892–905. doi: 10.1002/hep.31108
181. Tamura Y, Sugimoto M, Murayama T, Ueda Y, Kanamori H, Ono K, et al. Inhibition of CCR2 ameliorates insulin resistance and hepatic steatosis in db/db mice. Arterioscler Thromb Vasc Biol (2008) 28(12):2195–201. doi: 10.1161/ATVBAHA.108.168633
182. Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N, et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C + macrophage infiltration in mice. Hepatology (2014) 59:1060–72. doi: 10.1002/hep.26783
183. Perez-Martinez L, Perez-Matute P, Aguilera-Lizarraga J, Rubio-Mediavilla S, Narro J, Recio E, et al. Maraviroc, a CCR5 antagonist, ameliorates the development of hepatic steatosis in a mouse model of non-alcoholic fatty liver disease (NAFLD). J Antimicrob Chemother (2014) 69:1903–10. doi: 10.1093/jac/dku071
184. Puengel T, De Vos S, Hundertmark J, Kohlhepp M, Guldiken N, Pujuguet P, et al. The Medium-Chain Fatty Acid Receptor GPR84 Mediates Myeloid Cell Infiltration Promoting Steatohepatitis and Fibrosis. J Clin Med (2020) 9:1140. doi: 10.3390/jcm9041140
185. Antoniades CG, Khamri W, Abeles RD, Taams LS, Triantafyllou E, Possamai LA, et al. Secretory leukocyte protease inhibitor: A pivotal mediator of anti-inflammatory responses in acetaminophen-induced acute liver failure. Hepatology (2014) 59:1564–76. doi: 10.1002/hep.26933
186. Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, Iredale JP, et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci (2006) 103:5060–5. doi: 10.1073/pnas.0511167103
187. Iacobini C, Menini S, Ricci C, Fantauzzi CB, Scipioni A, Salvi L, et al. Galectin-3 ablation protects mice from diet-induced NASH: A major scavenging role for galectin-3 in liver. J Hepatol (2011) 54(5):975–83. doi: 10.1016/j.jhep.2010.09.020
188. Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients With Nonalcoholic Steatohepatitis With Cirrhosis and Portal Hypertension. Gastroenterology (2020) 158:1334–45.e5. doi: 10.1053/j.gastro.2019.11.296
189. Shan B, Wang X, Wu Y, Xu C, Xia Z, Dai J, et al. The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat Immunol (2017) 18(5):519–29. doi: 10.1038/ni.3709
190. McMahan RH, Wang XX, Cheng LL, Krisko T, Smith M, El KK, et al. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J Biol Chem (2013) 288(17):11761–70. doi: 10.1074/jbc.M112.446575
191. Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology (2018) 67(2):549–59. doi: 10.1002/hep.29514
192. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet (2015) 385(9972):956–65. doi: 10.1016/S0140-6736(14)61933-4
193. Ergen C, Heymann F, Al Rawashdeh W, Gremse F, Bartneck M, Panzer U, et al. Targeting distinct myeloid cell populations in vivo using polymers, liposomes and microbubbles. Biomaterials (2017) 114:106–20. doi: 10.1016/j.biomaterials.2016.11.009
194. Colino CI, Lanao JM, Gutierrez-Millan C. Targeting of Hepatic Macrophages by Therapeutic Nanoparticles. Front Immunol (2020) 11:218. doi: 10.3389/fimmu.2020.00218
195. Bartneck M, Scheyda KM, Warzecha KT, Rizzo LY, Hittatiya K, Luedde T, et al. Fluorescent cell-traceable dexamethasone-loaded liposomes for the treatment of inflammatory liver diseases. Biomaterials (2015) 37:367–82. doi: 10.1016/j.biomaterials.2014.10.030
196. Ergen C, Niemietz PM, Heymann F, Baues M, Gremse F, Pola R, et al. Liver fibrosis affects the targeting properties of drug delivery systems to macrophage subsets in vivo. Biomaterials (2019) 206:49–60. doi: 10.1016/j.biomaterials.2019.03.025
197. Boeckmans J, Natale A, Rombaut M, Buyl K, Rogiers V, De Kock J, et al. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells (2019) 9(1):37. doi: 10.3390/cells9010037
198. Jain MR, Giri SR, Bhoi B, Trivedi C, Rath A, Rathod R, et al. Dual PPARα/γ agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int (2018) 38(6):1084–94. doi: 10.1111/liv.13634
199. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology (2016) 150(5):1147–59. doi: 10.1053/j.gastro.2016.01.038
200. Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A, et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages☆. J Hepatol (2020) 73:757–70. doi: 10.1016/j.jhep.2020.04.025
201. Wan J, Weiss E, Ben Mkaddem S, Mabire M, Choinier P-M, Picq O, et al. LC3-associated phagocytosis protects against inflammation and liver fibrosis via immunoreceptor inhibitory signaling. Sci Transl Med (2020) 12:eaaw8523. doi: 10.1126/scitranslmed.aaw8523
202. Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol (2013) 58(5):993–9. doi: 10.1016/j.jhep.2013.01.011
203. Riva A, Mehta G. Regulation of Monocyte-Macrophage Responses in Cirrhosis—Role of Innate Immune Programming and Checkpoint Receptors. Front Immunol (2019) 10(167);1–7. doi: 10.3389/fimmu.2019.00167
204. Netea MG, Latz E, Mills KHG, O’Neill LAJ. Innate immune memory: A paradigm shift in understanding host defense. Nat Immunol (2015) 16:675–9. doi: 10.1038/ni.3178
205. Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, et al. Trained immunity: A program of innate immune memory in health and disease. Sci (80- ) (2016) 352:aaf1098–aaf1098. doi: 10.1126/science.aaf1098
206. Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature (2007) 447:972–8. doi: 10.1038/nature05836
207. De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The Histone H3 Lysine-27 Demethylase Jmjd3 Links Inflammation to Inhibition of Polycomb-Mediated Gene Silencing. Cell (2007) 130(6):1083–94. doi: 10.1016/j.cell.2007.08.019
208. Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, McCall CE. NAD + -dependent SIRT1 Deacetylase Participates in Epigenetic Reprogramming during Endotoxin Tolerance. J Biol Chem (2011) 286:9856–64. doi: 10.1074/jbc.M110.196790
209. Tao J, Zhang J, Ling Y, McCall CE, Liu TF. Mitochondrial sirtuin 4 resolves immune tolerance in monocytes by rebalancing glycolysis and glucose oxidation homeostasis. Front Immunol (2018) 9:419. doi: 10.3389/fimmu.2018.00419
210. Bekkering S, Blok BA, Joosten LAB, Riksen NP, Van Crevel R, Netea MG. In Vitro experimental model of trained innate immunity in human primary monocytes. Clin Vaccine Immunol (2016) 23(12):926–33. doi: 10.1128/CVI.00349-16
211. Vachharajani VT, Liu T, Brown CM, Wang X, Buechler NL, Wells JD, et al. SIRT1 inhibition during the hypoinflammatory phenotype of sepsis enhances immunity and improves outcome. J Leukoc Biol (2014) 96:785–96. doi: 10.1189/jlb.3ma0114-034rr
212. Wang X, Buechler NL, Martin A, Wells J, Yoza B, McCall CE, et al. Sirtuin-2 regulates sepsis inflammation in ob/ob mice. PloS One (2016) 11(8):e160431. doi: 10.1371/journal.pone.0160431
213. Riva A, Chokshi S. Immune checkpoint receptors: homeostatic regulators of immunity. Hepatol Int (2018) 12:223–36. doi: 10.1007/s12072-018-9867-9
214. Khamri W, Abeles RD, Hou TZ, Anderson AE, El-Masry A, Triantafyllou E, et al. Increased Expression of Cytotoxic T-Lymphocyte–Associated Protein 4 by T Cells, Induced by B7 in Sera, Reduces Adaptive Immunity in Patients With Acute Liver Failure. Gastroenterology (2017) 153:263–276.e8. doi: 10.1053/j.gastro.2017.03.023
215. Markwick LJL, Riva A, Ryan JM, Cooksley H, Palma E, Tranah TH, et al. Blockade of PD1 and TIM3 restores innate and adaptive immunity in patients with acute alcoholic hepatitis. Gastroenterology (2015) 148:590–602.e10. doi: 10.1053/j.gastro.2014.11.041
216. Salimzadeh L, Le Bert N, Dutertre CA, Gill US, Newell EW, Frey C, et al. PD-1 blockade partially recovers dysfunctional virus-specific B cells in chronic hepatitis B infection. J Clin Invest (2018) 128:4573–87. doi: 10.1172/JCI121957
217. Pinato DJ, Guerra N, Fessas P, Murphy R, Mineo T, Mauri FA, et al. Immune-based therapies for hepatocellular carcinoma. Oncogene (2020) 39:3620–37. doi: 10.1038/s41388-020-1249-9
218. Pinter M, Jain RK, Duda DG. The Current Landscape of Immune Checkpoint Blockade in Hepatocellular Carcinoma. JAMA Oncol (2020) 7(1):113–23. doi: 10.1001/jamaoncol.2020.3381
219. Flynn MJ, Sayed AA, Sharma R, Siddique A, Pinato DJ. Challenges and Opportunities in the Clinical Development of Immune Checkpoint Inhibitors for Hepatocellular Carcinoma. Hepatology (2019) 69:2258–70. doi: 10.1002/hep.30337
220. Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol (2020) 5:1–15. doi: 10.1126/sciimmunol.aay1863
221. Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, et al. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front Immunol (2019) 10:2298. doi: 10.3389/fimmu.2019.02298
222. Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv (2020) 6(38):eabd2712. doi: 10.1126/sciadv.abd2712
223. Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS. Frontline Science: Defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J Leukoc Biol (2016) 100:1239–54. doi: 10.1189/jlb.4hi0616-255r
224. Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, Hotchkiss RS. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol (2010) 88:233–40. doi: 10.1189/jlb.0110037
225. Zasada M, Lenart M, Rutkowska-Zapała M, Stec M, Durlak W, Grudzień A, et al. Analysis of PD-1 expression in the monocyte subsets from non-septic and septic preterm neonates. PloS One (2017) 12:1–13. doi: 10.1371/journal.pone.0186819
226. Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U.S.A. (2009) 106:6303–8. doi: 10.1073/pnas.0809422106
227. Triantafyllou E, Gudd CLC, Mawhin M-A, Husbyn HC, Trovato FM, Siggins MK, et al. PD-1 blockade improves Kupffer cell bacterial clearance in acute liver injury. J Clin Invest (2021) 131(4):1–15. doi: 10.1172/JCI140196
Keywords: cirrhosis, macrophages, liver injury, liver inflammation, liver fibrosis, chronic liver failure, Kupffer cells
Citation: Singanayagam A and Triantafyllou E (2021) Macrophages in Chronic Liver Failure: Diversity, Plasticity and Therapeutic Targeting. Front. Immunol. 12:661182. doi: 10.3389/fimmu.2021.661182
Received: 30 January 2021; Accepted: 18 March 2021;
Published: 02 April 2021.
Edited by:
Ioannis Kourtzelis, University of York, United KingdomReviewed by:
Antonios Chatzigeorgiou, National and Kapodistrian University of Athens, GreeceCopyright © 2021 Singanayagam and Triantafyllou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Evangelos Triantafyllou, ZS50cmlhbnRhZnlsbG91QGltcGVyaWFsLmFjLnVr
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.