 Stefanie Krug
Stefanie Krug Sadiya Parveen
Sadiya Parveen William R. Bishai*
William R. Bishai*- Department of Medicine, Division of Infectious Diseases, Johns Hopkins University School of Medicine, Baltimore, MD, United States
Following infection with Mycobacterium tuberculosis, the causative agent of tuberculosis (TB), most human hosts are able to contain the infection and avoid progression to active TB disease through expression of a balanced, homeostatic immune response. Proinflammatory mechanisms aiming to kill, slow and sequester the pathogen are key to a successful host response. However, an excessive or inappropriate pro-inflammatory response may lead to granuloma enlargement and tissue damage, which may prolong the TB treatment duration and permanently diminish the lung function of TB survivors. The host also expresses certain anti-inflammatory mediators which may play either beneficial or detrimental roles depending on the timing of their deployment. The balance between the timing and expression levels of pro- and anti-inflammatory responses plays an important role in the fate of infection. Interestingly, M. tuberculosis appears to manipulate both sides of the human immune response to remodel the host environment for its own benefit. Consequently, therapies which modulate either end of this spectrum of immune responses at the appropriate time may have the potential to improve the treatment of TB or to reduce the formation of permanent lung damage after microbiological cure. Here, we highlight host-directed TB therapies targeting pro- or anti-inflammatory processes that have been evaluated in pre-clinical models. The repurposing of already available drugs known to modulate these responses may improve the future of TB therapy.
Introduction
Tuberculosis (TB) is a devastating communicable disease caused by Mycobacterium tuberculosis (M.tb) that is responsible for approximately 10 million infections and 1.4 million human deaths every year (1). Global TB control is complicated by long treatment durations and emerging drug resistance (1). Interestingly, most people infected with M.tb develop lifelong latent TB without ever experiencing signs and symptoms of disease. Successful containment is the result of a multifaceted immune response that restricts bacterial expansion but may fail to completely eliminate the pathogen (2). When sterilization is not achieved, the host may nevertheless successfully contain the infection by forming granulomas. However, in individuals who progress to active TB, granulomatous containment breaks down, resulting in lesion expansion, necrosis and liquefaction accompanied by bacterial proliferation and lung damage (2). This granulomatous inflammation during active TB may permanently diminish lung function even after completion of TB therapy (3).
The host utilizes both anti- and pro-inflammatory mechanisms in an effort to contain the infection: during latent M.tb infection, the immune response is successfully balanced but during active disease, this homeostatic balance is lost and disease progression occurs. Anti-inflammatory responses, mediated by regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), M2-polarized macrophages and cytokines such as interleukin (IL)-10, are observed during active TB and may antagonize the bactericidal effects of the immune system (4). Despite the presence of these immuno-tolerizing cells, host pro-inflammatory responses during active TB are often inappropriately expressed at high levels, either spatially or temporally, resulting in lung damage. Consequently, host-directed therapies (HDTs) that modify these non-productive immunologic responses may offer potential benefit as adjunctive agents alongside antimicrobial TB therapy (5). In this mini-review, we highlight FDA-approved drugs as well as select agents in development that have immunomodulatory activity and are under study as HDTs for TB in pre-clinical models and/or human clinical trials.
Improving TB Therapy by Modulating Pro-Inflammatory Responses
In immunocompetent patients with active TB, pro-inflammatory immune responses are often robust but fail to contain bacterial proliferation, leading to tissue damage and nonproductive inflammation. Nearly half of all active TB patients suffer from persistent or even progressive pulmonary dysfunction and face an increased risk of chronic lung disease even after microbiologically successful cure (3, 6–9). Post-TB lung defects (PTLD) include obstructive or restrictive lung disease, both of which may lead to chronic dyspnea, cough, reduced exercise tolerance, and a heightened risk for infections (3). In addition to shortening the duration of therapy, a parallel goal for TB HDTs is to avoid the development of irreversible lung damage from nonproductive inflammatory responses and to concomitantly improve the quality of life of TB survivors (3, 10). In this section, we discuss several classes of HDTs that may reduce nonproductive inflammation and PTLD (Figure 1, left; Table 1, top).
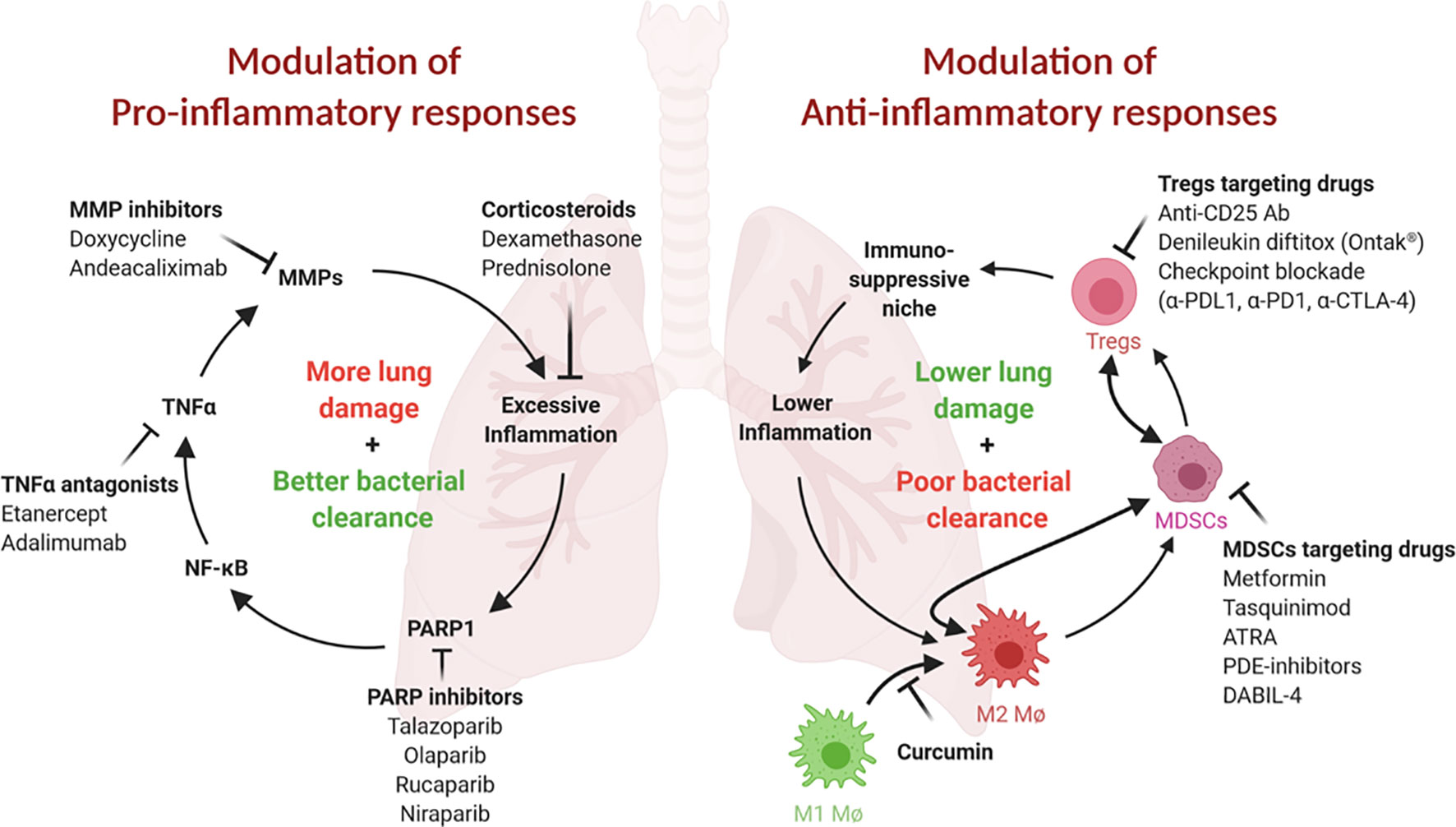
Figure 1 Both pro- and ani-inflammatory responses play critical roles in TB pathogenesis. (Left) Proinflammatory responses and tissue remodeling in TB are important for bacterial clearance but may lead to excessive inflammation and persisting lung damage. Adjunct modulation of lung remodeling (for example, via TNFα or MMP inhibition) or inflammation (for example, by corticosteroids) may improve the outcome of TB therapy. Inhibition of PARP1, an essential NF-κB, TNFα and MMP cofactor and driver of lung inflammation, may be similarly beneficial. (Right) Anti-inflammatory responses safeguard against tissue damage but may result in less than desirable bacterial clearance. These responses are often mediated by immunosuppressive cell populations, such as MDSCs, Tregs and M2 macrophages. Inhibition or elimination of these cell types may be achieved using the inhibitors shown. This figure was created using BioRender.
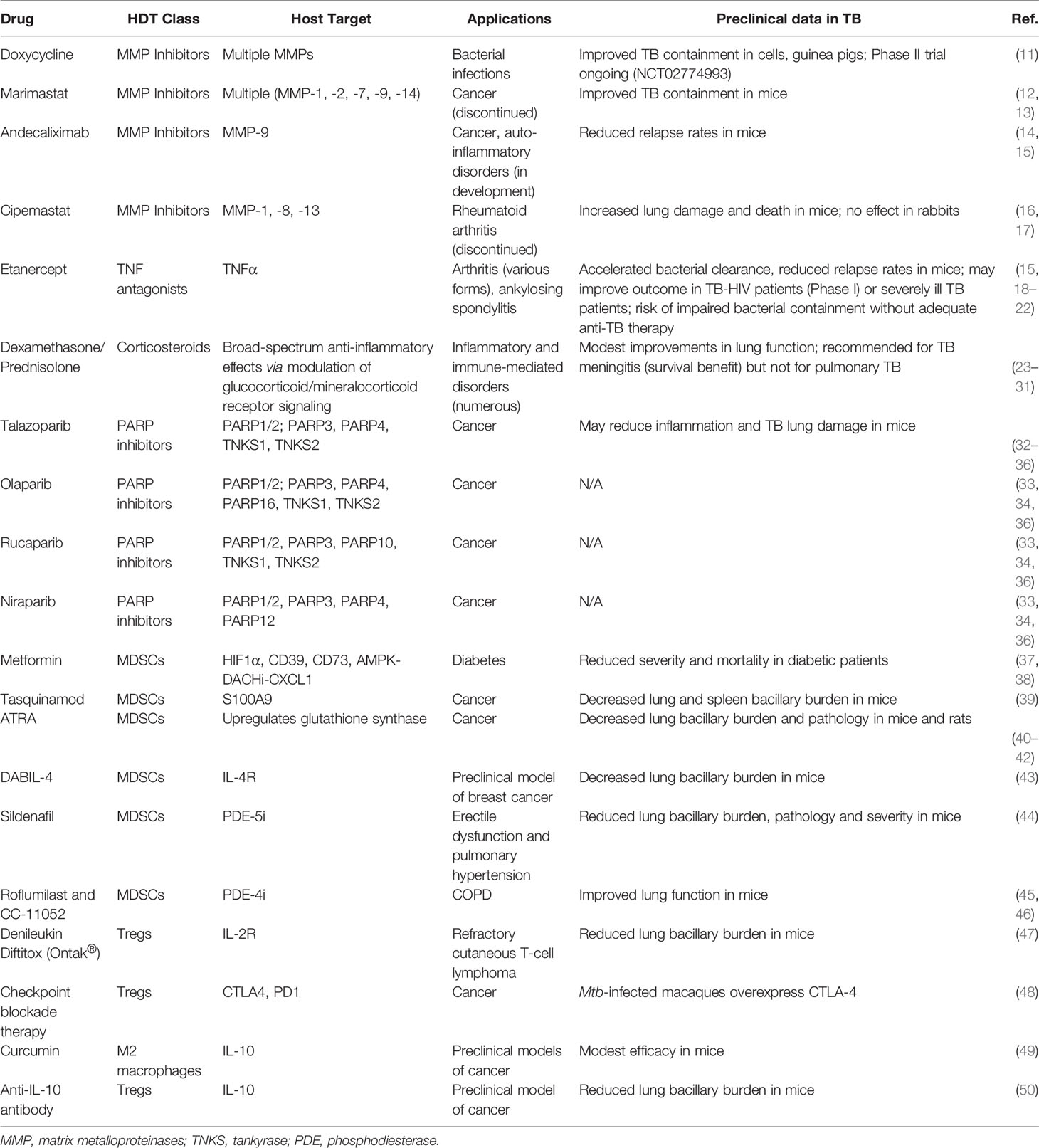
Table 1 Immune-modulatory drugs that may improve TB therapy.
MMP Inhibitors
Tissue-degrading matrix metalloproteinases (MMPs), in particular MMP-1, -3 and -9, are major drivers of TB-associated lung damage (51–55). While extracellular matrix remodeling is important for immune cell migration and granuloma formation, MMP levels in TB patients remain elevated even after treatment completion and thus may drive progressive lung dysfunction (55, 56). Consequently, adjunctive MMP inhibition has been studied as an HDT to improve TB outcome. Doxycycline, a well-known antibacterial agent, also is known to have MMP inhibitory properties, making it the only currently FDA-approved MMP inhibitor. Doxycycline has been shown to not only inhibit TB-induced MMP activation but also to contain mycobacterial growth in cells and guinea pigs (11). Results from a phase II pilot study (NCT02774993) that evaluated the efficacy of adjuvant doxycycline as a novel HDT for pulmonary TB are pending and may offer insights into the safety and efficacy of this approach.
Excess MMP activity is observed in a number of human degenerative diseases and hence several targeted MMP inhibitors have been developed and evaluated in human studies. While adverse effects hindered early MMP inhibitors, there is newfound optimism that this may be overcome with a newer generation of inhibitors (57). For example, the broad-spectrum MMP inhibitor marimastat (BB-2516) reduced granuloma formation and bacterial growth in vitro and increased the efficacy of TB antibiotics in mice but its clinical development was discontinued due to its side effects (12, 13). However, the humanized monoclonal MMP-9 antibody andecaliximab is in late-stage development for cancer and auto-inflammatory disorders (14) and might improve TB outcome since the addition of an anti-MMP-9 antibody has been shown to reduce TB relapse rates in mice (15). In contrast, the MMP-1 inhibitor cipemastat increased immunopathology and death in M.tb-infected C3HeB/FeJ mice and failed to prevent M.tb-mediated cavity-generation in a rabbit model (16, 17). Nonetheless, the next generation of MMP inhibitors with improved selectivity, specificity and safety is a promising class of drugs that warrants consideration for HDT activity in TB.
TNF Antagonists
An alternative to direct MMP inhibition is to modulate the factors that promote MMP expression and TB inflammation, such as tumor necrosis factor α (TNFα) and the transcription factor NF-κB (55). TNFα is an important driver of TB lung damage by enhancing granuloma progression, cavitation, and MMP expression, and its expression levels are inversely correlated with the resolution of lung lesions during TB therapy (58–61). Correspondingly, HIV-positive TB patients generally have less lung damage than HIV-negative TB patients, and TB-immune reconstitution inflammatory syndrome (TB-IRIS) following antiretroviral therapy is associated with increased lung damage and reduced lung function (8, 62–64). TNFα also contributes to restrictive and obstructive airflow deficits by promoting fibrogenesis (18, 65, 66). Adjuvant administration of the TNFα antagonist etanercept accelerated bacterial clearance and reduced relapse rates in mice, and a promising phase I trial showed that etanercept may improve lung involvement and treatment responses in TB-HIV patients (15, 19, 20). There have also been case reports of TNFα inhibitors being used successfully to improve the clinical course of patients with advanced drug-susceptible TB who were doing poorly (21). In contrast, however, TNFα inhibitors are well-known to impair bacterial containment when used without accompanying multidrug anti-TB therapy (18, 22). While TNFα antagonists have the potential to improve TB therapy when used as adjunctive agents, there have been concerns about their expense, their need to be given parenterally, and the potential for disease worsening if administered without adequate anti-TB chemotherapy, and due to these concerns advanced clinical trials to test them as adjunctive HDTs for TB have not been performed (18, 60, 67).
Corticosteroids
Corticosteroids are another class of anti-inflammatory drugs that have garnered attention as potential TB-HDTs (68). In pulmonary TB, adjunctive corticosteroids, including the broadly immunosuppressive agents dexamethasone and prednisolone, have been studied for their ability to reduce post-treatment morbidity. Indeed, while some studies have demonstrated modest improvements in clinical outcomes, such as preservation of lung vital capacity, major improvements in the prevention of lung disability have not been shown (23–28). Thus, corticosteroids are not recommended in current TB treatment guidelines for the management of pulmonary TB (29, 30). Corticosteroids have also been evaluated in the management of tuberculous pericarditis, but they do not appear to change outcomes and are currently not recommended in that setting (69). In contrast, well-controlled studies have demonstrated a clear-cut survival benefit for use of corticosteroids in TB meningitis, and hence corticosteroids are considered mandatory in the treatment of that form of TB (31).
PARP Inhibitors
Poly(ADP-ribose) Polymerase (PARP) inhibitors (PARP-Is) are a new class of anticancer drugs introduced in the last decade, and four such agents are already FDA-approved. The PARP family of enzymes, comprised of at least 17 members, regulates wide-ranging cellular functions via the post-translational modification of mono- or poly(ADP-ribosyl)ation (70–74). PARP1, the founding member of the PARP family, is a eukaryotic master regulator particularly important for inflammatory processes and stress responses and accounts for at least 85% of cellular poly-ADP-ribose (PAR) formation (75). Importantly, PARP1 amplifies and sustains chronic inflammation by inducing inflammatory mediators that further stimulate its own activation (75, 76). Consequently, PARP1 contributes to disorders such as endotoxic shock, sepsis, asthma, COPD and ARDS, and PARP-Is have been shown to reduce inflammation and disease severity in numerous inflammatory conditions (75–78). PARP1 is an essential NF-κB, TNFα and MMP cofactor, and PARP-Is protect against tissue degradation by inhibiting multiple MMPs (71, 79–86). Therefore, PARP-Is have been proposed as HDTs for reducing TB-induced inflammation and lung disease (32). There are currently four FDA-approved PARP-Is for cancer therapies, talazoparib (Talzenna, Pfizer), olaparib (Lynparza, AstraZeneca), rucaparib (Rubraca, Clovis Oncology) and niraparib (Zejula, GlaxoSmithKline), with many more in various phases of development looking to expand their application in cancer therapy and beyond (33–35). Since it has been shown that PARP1 inhibition can ameliorate numerous inflammatory conditions, including rheumatoid arthritis, asthma, atherosclerosis and allergy-, toxicity- and injury-induced inflammation, the addition of a PARP inhibitor might similarly improve TB therapy by reducing inflammation and lung damage (75, 79, 87).
Improving TB Therapy by Modulating Anti-Inflammatory Responses
An important theme in TB pathogenesis research in recent decades has been the observation that M.tb carries virulence traits that subvert normal host immune responses and lead to pathogen survival and/or proliferation. One such mechanism is the recruitment of immunosuppressive or tolerizing cells to the site of infection, resulting in blunted bactericidal responses and the expression of elevated levels of IL-10 which further promotes anti-inflammatory responses (88). Indeed, it has recently been shown that the microbial polypeptide ESAT6 is one mediator that promotes the differentiation of M1 macrophages into anti-inflammatory M2 macrophages (89). Other tolerizing, immunosuppressive cells that are recruited to the site of infection include MDSCs, Tregs and M2-polarized macrophages. In this section, we highlight the major cell types involved in these anti-inflammatory responses and discuss drugs that target them and may be candidate TB HDTs (Figure 1, right; Table 1, bottom).
Myeloid-Derived Suppressor Cells (MDSCs)
MDSCs represent an immunosuppressive cell population increasingly recognized as an important driver of TB pathogenesis. MDSCs are comprised of two distinct subsets: polymorphonuclear MDSCs (PMN-MDSCs) and mononuclear MDSCs (M-MDSCs). In mice, PMN-MDSCs are defined as CD11b+ Ly6G+ Ly6Clow and M-MDSCs as CD11b+ Ly6G- Ly6CHigh. In humans, MDSCs are identified as CD11b+ CD33+ HLA-DRlow∕neg cells (90, 91), and these are further subdivided into PMN-MDSCs by the markers CD14− CD66b+ CD15+, and M-MDSCs as CD14+ (92–95). While the role of MDSCs in suppressing inflammation has been extensively studied in cancer, it is becoming increasingly evident that MDSCs play an important role in the establishment of chronic infections including TB. Clinical studies have revealed that levels of MDSCs are high in the blood and sputum of active TB patients at the time of diagnosis and that they decline in response to successful chemotherapy (96–98). This association suggests that MDSCs may play an important role in the pathogenesis of active TB pathology and its dysfunctional inflammatory processes. Further evidence comes from murine studies where the relative abundance of MDSCs has also been found to correlate with the TB susceptibility of a given mouse strain. Relatively high levels of MDSCs are observed in susceptible mouse strains, such as 129S2 and C3HeB/FeJ, while lower MDSC levels are found in relatively resistant strains, such as BALB/c and C57BL/6 (99). Multiple HDTs have been tested in both pre-clinical and clinical settings that (1) inhibit the recruitment, expansion or function of MDSCs; or (2) specifically or non-specifically deplete their population.
Metformin. The widely used diabetes drug metformin inhibits the frequency and recruitment of MDSCs in cancer by modulating the expression and activity of HIF-1α, CD39, and CD73 and the AMPK-DACH1-CXCL1 axis (100, 101). A widely cited study in 2014 revealed that metformin reduced disease severity and inflammation in mice and was retrospectively associated with a lower degree of disease severity in diabetic patients with active TB who happened to be taking metformin during TB treatment (37). Another retrospective study showed that metformin therapy reduces the elevated TB mortality observed in diabetics (38). In spite of these observations, long-term chemotherapy studies in mice have failed to demonstrate a significant beneficial effect of adjunctive metformin together with standard TB chemotherapy (102). Clearly, prospective human studies are needed, and the NIH has recently funded a prospective Phase 2A study of metformin in patients with TB (103).
Tasquinimod is an experimental quinoline-3-carboxamide drug that has been studied in human prostate cancer (104). It has been shown to slow tumor growth in murine cancer models and to reduce MDSC tumor infiltration (105). It is believed to act by binding to and inhibiting the activity of the S100A9 protein; S100A9 together with S100A8 are known to modulate myeloid cell activity though TLR4 binding (104, 106). Because of its anti-MDSC properties, tasquinimod has been tested in murine TB models, and it has been shown not only to deplete MDSCs but also to decrease the relative bacterial burden in both lungs and spleens of infected animals (39).
All-trans Retinoic Acid (ATRA, tretinoin, a vitamin A derivative) is an FDA-approved drug which has been tested extensively in cancer models and has been shown to deplete MDSCs and slow tumor growth. While its precise mechanism of action is unknown, ATRA upregulates glutathione synthase (GSS), neutralizes high levels of reactive oxygen species (ROS) and induces differentiation of myeloid cells away from the MDSC phenotype (107). Importantly, however, ATRA has pleotropic effects on numerous cell types so in instances where it was found to be effective, one cannot be certain that its efficacy was through MDSC inhibition. Multiple groups have tested the effects of ATRA in murine TB models both as a monotherapy and in combination with standard TB therapy. In M. tb.-infected mice and rats, ATRA has been shown to reduce relative bacterial burden and lung pathology in a manner that correlates with MDSC depletion. The drug also exhibits anti-mycobacterial activity in vitro (96, 108).
In addition to the non-specific depletion of MDSCs, our group has recently tested the diphtheria toxin-related IL-4 fusion protein, DABIL-4, as a targeting agent against MDSCs which are known to express the IL-4 receptor, CD124. In an acute murine model of TB, DABIL-4 administration depleted IL-4R+ MDSCs, IL-4R+ M2 macrophages and IL-4R+ lymphocytes. Depletion of these cell populations coincided with a significant reduction in the lung bacillary burden at day 21 post infection (43). We have also tested DABIL-4 in a murine breast cancer model and demonstrated that targeted depletion of MDSCs results in slower tumor growth and reduced splenomegaly and metastasis (109).
Phosphodiesterase inhibitors. Sildenafil, an FDA-approved type 5 phosphodiesterase-selective inhibitor (PDE-5i), is used in human patients for the treatment of erectile dysfunction and pulmonary hypertension. The drug downregulates arginase-1 and nitric oxide synthase-2 (NOS2) in a cGMP-dependent fashion, thereby hampering the immunosuppressive potential of MDSCs (110). Maiga et al. showed that the combination of sildenafil and cilostazol (an FDA-approved PDE-3 inhibitor) reduced pathology, disease severity and bacterial burden in murine TB; however, monotherapy with sildenafil alone showed no statistically significant benefit in the same mouse model (44, 111). PDE-4 inhibitors, such as roflumilast and CC-11052, a Celgene PDE4 inhibitor in development, have also shown promising activity against TB in animal models (45, 46). A clinical trial evaluating CC-11052 as an adjunctive HDT alongside standard therapy has been conducted (NCT02968927), and preliminary results suggest that use of CC-11052 was associated with improvements in lung function (112).
Regulatory T-Cells (Tregs)
Tregs comprise an immunosuppressive CD4+ T-cell population which express CD25 and FoxP3. CD8+ Tregs also exist but their role in TB has not been extensively studied. Classic CD4+ CD25+ FoxP3+ Tregs are anti-inflammatory cells which keep effector T-cell function in check while promoting MDSC recruitment and maturation to further facilitate immunosuppression. Their presence in active TB is believed to inhibit anti-bacterial immune responses and to contribute to disease progression (113). Consistent with this, elevated Treg levels have been described in the blood and pleural fluid in pulmonary TB patients compared with healthy controls, and Treg levels were observed to decline to healthy control levels after successful TB chemotherapy (114).
Treg-depleting immunotherapies. The administration of anti-CD25 monoclonal antibodies in various cancer models has not only depleted Tregs but also slowed tumor progression (115). Anti-CD25 antibodies have been tested in the mouse TB model and were found to reduce relative bacillary loads in the lung and spleen and to improve lung pathology (116). Denileukin diftitox (Ontak®), a diphtheria toxin-related IL-2 fusion protein that was previously approved by the FDA for the treatment of refractory cutaneous T-cell lymphoma, is known to have potent Treg-depleting activity and has also been tested in murine TB models (47, 117). Ontak® monotherapy not only decreased Treg and MDSC frequencies in lungs and spleens but also significantly reduced relative bacterial CFU counts in a short-term TB mouse model. Additionally, the fusion protein toxin when combined with standard TB therapy significantly accelerated bacterial clearance in mice (47, 117).
Checkpoint Blockade Immunotherapy
Checkpoint blockade therapies, such as anti-PD-1 and anti-CTLA4 antibodies, have revolutionized the field of immunotherapy and have become an essential part of standard care for various human malignancies (118). In M.tb-infected macaques, Tregs have been shown to express CTLA-4, suggesting that anti-CTLA-4-directed checkpoint inhibitors may offer a potential HDT TB treatment (48). However, several groups have reported TB reactivation in cancer patients treated with checkpoint blockade therapy (119–121). While this does not necessarily indicate that checkpoint inhibitors given as adjuvants alongside appropriate anti-TB chemotherapy will fail to accelerate TB cure, more studies will be needed reach a conclusion regarding the efficacy of checkpoint blockade therapy as HDT for TB.
Anti-IL-10 Therapies
IL-10 is a key anti-inflammatory cytokine secreted by CD4+ T cells, macrophages and MDSCs that suppresses T-cell function, blunts inflammatory responses, and promotes TB disease progression (50). IL-10 has been implicated in the M2-polarization of macrophages and this may further contribute to anti-inflammatory responses. An abundance of M2-polarized macrophages has been described in human lung granulomas (122), although it remains unclear if these M2 macrophages are causal in granuloma formation or rather a secondary consequence. IL-10 inhibitors would be expected to inhibit the direct anti-inflammatory effects of IL-10 and also prevent conversion of M1 macrophages into M2 macrophages. Indeed, IL-10 inhibitors have been tested both in cancer models and also in models of TB. Curcumin (diferuloylmethane), one of the active compounds found in turmeric, has been shown to modulate IL-10 levels and the frequency of M2 macrophages (123). Preparations of curcumin have been shown to drive a therapeutic benefit in a murine metastatic breast cancer model (124). In the context of TB, curcumin has also been shown to control the growth of M.tb in THP-1 macrophages and in primary alveolar macrophages derived from healthy human controls (125). More recently, a nanoparticle preparation of curcumin was tested in a murine TB model where it showed modest activity as monotherapy and more potent activity in combination with isoniazid (49). Direct inhibition of IL-10 with an anti-IL-10-receptor antibody in a murine TB model was shown to reduce bacterial CFU counts although it had little impact on the lung pathology (50).
Discussion
Host-directed therapies have the potential to improve the treatment of TB by modulating either pro- or anti-inflammatory immune mechanisms. Interference with certain pro-inflammatory mechanisms offers the potential to reduce lung damage, increase antibiotic efficacy and shorten treatment duration. On the other hand, modulation of certain immunosuppressive immune responses may enhance the innate bactericidal activity of the immune system and thus accelerate bacterial clearance. Repurposing drugs that are safe and approved for human use is an approach that may fast-track the clinical development of new host-directed TB treatment regimens. Here, we reviewed HDTs of interest for TB that target pro- or anti-inflammatory immune mechanisms (Figure 1; Table 1). On the proinflammatory side, we highlighted MMP inhibition, TNFα antagonists, corticosteroids and PARP inhibition to reduce TB-associated lung damage and inflammation. However, immune modulation in TB should be approached with caution as disrupting the intricate host-pathogen relationship can also increase the risk for disease progression or exacerbate inflammation. It is important that the dosing, frequency and timing of TB-HDTs are carefully optimized to minimize potentially harmful effects. Moreover, HDTs should be primarily evaluated as treatment adjuvants to be utilized alongside fully active traditional anti-TB chemotherapy. A related concern is that of drug-drug interactions and the potential for one agent to reduce the circulating concentration of another.
Even though TB-associated persistent lung dysfunction is a common disability in TB survivors, there are currently no guidelines for the diagnosis or management of PTLDs, and it is unclear to what extent they contribute to the economic burden of TB (8, 126). Reducing TB-associated lung dysfunction has the potential to greatly improve the quality of life after TB by reducing morbidity and loss of income. While pulmonary function testing in early TB carries some risk of TB transmission, it has been safely implemented in numerous clinical trials. We therefore recommend that more consideration should be given to the routine assessment of lung function in TB clinical trials. In addition to HDTs, non-pharmacological interventions, such as pulmonary rehabilitation, may improve lung function after completion of TB therapy and should be considered in the management of TB patients on a case-by-case basis (127). Importantly, we hope to increase awareness that the fight against TB does not end with microbiological cure.
Author Contributions
SK and SP contributed equally to this mini-review. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully acknowledge the support of NIH grants AI 152688, 155602, 130595 and HL 140812.
References
1. World Health Organization. Global tuberculosis report 2019. Geneva: World Health Organization (2019). Licence: CC BY-NC-SA 3.0 IGO.
2. Huynh KK, Joshi SA, Brown EJ. A delicate dance: host response to mycobacteria. Curr Opin Immunol (2011) 23(4):464–72. doi: 10.1016/j.coi.2011.06.002
3. Ravimohan S, Kornfeld H, Weissman D, Bisson GP. Tuberculosis and lung damage: from epidemiology to pathophysiology. Eur Respir Rev (2018) 27(147):170077. doi: 10.1183/16000617.0077-2017
4. Cicchese JM, Evans S, Hult C, Joslyn LR, Wessler T, Millar JA, et al. Dynamic balance of pro- and anti-inflammatory signals controls disease and limits pathology. Immunol Rev (2018) 285(1):147–67. doi: 10.1111/imr.12671
5. Zitko J. Doležal M: Old Drugs and New Targets as an Outlook for the Treatment of Tuberculosis. Curr Medicinal Chem (2018) 25(38):5142–67. doi: 10.2174/0929867324666170920154325
6. Chushkin MI, Ots ON. Impaired pulmonary function after treatment for tuberculosis: the end of the disease? Jornal brasileiro pneumologia publicacao oficial da Sociedade Bras Pneumologia e Tisilogia (2017) 43(1):38–43. doi: 10.1590/s1806-37562016000000053
7. Manji M, Shayo G, Mamuya S, Mpembeni R, Jusabani A, Mugusi F. Lung functions among patients with pulmonary tuberculosis in Dar es Salaam - a cross-sectional study. BMC Pulm Med (2016) 16(1):58–8. doi: 10.1186/s12890-016-0213-5
8. Meghji J, Lesosky M, Joekes E, Banda P, Rylance J, Gordon S, et al. Patient outcomes associated with post-tuberculosis lung damage in Malawi: a prospective cohort study. Thorax (2020) 75(3):269–78. doi: 10.1136/thoraxjnl-2019-213808
9. Malherbe ST, Shenai S, Ronacher K, Loxton AG, Dolganov G, Kriel M, et al. Persisting positron emission tomography lesion activity and Mycobacterium tuberculosis mRNA after tuberculosis cure. Nat Med (2016) 22(10):1094–100. doi: 10.1038/nm.4177
10. Plit ML, Anderson R, Van Rensburg CE, Page-Shipp L, Blott JA, Fresen JL, et al. Influence of antimicrobial chemotherapy on spirometric parameters and pro-inflammatory indices in severe pulmonary tuberculosis. Eur Respir J (1998) 12(2):351–6. doi: 10.1183/09031936.98.12020351
11. Walker NF, Clark SO, Oni T, Andreu N, Tezera L, Singh S, et al. Doxycycline and HIV infection suppress tuberculosis-induced matrix metalloproteinases. Am J Respir Crit Care Med (2012) 185(9):989–97. doi: 10.1164/rccm.201110-1769OC
12. Parasa VR, Muvva JR, Rose JF, Braian C, Brighenti S, Lerm M. Inhibition of Tissue Matrix Metalloproteinases Interferes with Mycobacterium tuberculosis-Induced Granuloma Formation and Reduces Bacterial Load in a Human Lung Tissue Model. Front Microbiol (2017) 8:2370. doi: 10.3389/fmicb.2017.02370
13. Xu Y, Wang L, Zimmerman MD, Chen KY, Huang L, Fu DJ, et al. Matrix metalloproteinase inhibitors enhance the efficacy of frontline drugs against Mycobacterium tuberculosis. PloS Pathog (2018) 14(4):e1006974. doi: 10.1371/journal.ppat.1006974
14. Shah MA, Yanez Ruiz EP, Bodoky G, Starodub A, Cunningham D, Yip D, et al. A phase III, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of andecaliximab combined with mFOLFOX6 as first-line treatment in patients with advanced gastric or gastroesophageal junction adenocarcinoma (GAMMA-1). J Clin Oncol (2019) 37(4_suppl):4–4. doi: 10.1200/JCO.2019.37.4_suppl.4
15. Ordonez AA, Pokkali S, Kim S, Carr B, Klunk MH, Tong L, et al. Adjunct antibody administration with standard treatment reduces relapse rates in a murine tuberculosis model of necrotic granulomas. PloS One (2018) 13(5):e0197474. doi: 10.1371/journal.pone.0197474
16. Ordonez AA, Pokkali S, Sanchez-Bautista J, Klunk MH, Urbanowski ME, Kubler A, et al. Matrix Metalloproteinase Inhibition in a Murine Model of Cavitary Tuberculosis Paradoxically Worsens Pathology. J Infect Dis (2019) 219(4):633–6. doi: 10.1093/infdis/jiy373
17. Urbanowski ME, Ihms EA, Bigelow K, Kubler A, Elkington PT, Bishai WR. Repetitive Aerosol Exposure Promotes Cavitary Tuberculosis and Enables Screening for Targeted Inhibitors of Extensive Lung Destruction. J Infect Dis (2018) 218(1):53–63. doi: 10.1093/infdis/jiy127
18. Tsenova L, O’Brien P, Holloway J, Peixoto B, Soteropoulos P, Fallows D, et al. Etanercept Exacerbates Inflammation and Pathology in a Rabbit Model of Active Pulmonary Tuberculosis. J Interferon Cytokine Res (2014) 34(9):716–26. doi: 10.1089/jir.2013.0123
19. Skerry C, Harper J, Klunk M, Bishai WR, Jain SK. Adjunctive TNF inhibition with standard treatment enhances bacterial clearance in a murine model of necrotic TB granulomas. PloS One (2012) 7(6):e39680. doi: 10.1371/journal.pone.0039680
20. Wallis RS, Kyambadde P, Johnson JL, Horter L, Kittle R, Pohle M, et al. A study of the safety, immunology, virology, and microbiology of adjunctive etanercept in HIV-1-associated tuberculosis. AIDS (London England) (2004) 18(2):257–64. doi: 10.1097/00002030-200401230-00015
21. Wallis RS, van Vuuren C, Potgieter S. Adalimumab treatment of life-threatening tuberculosis. Clin Infect Dis an Off Publ Infect Dis Soc America (2009) 48(10):1429–32. doi: 10.1086/598504
22. Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. New Engl J Med (2001) 345(15):1098–104. doi: 10.1056/NEJMoa011110
23. Malik SK, Martin CJ. Tuberculosis, corticosteroid therapy, and pulmonary function. Am Rev Respir Dis (1969) 100(1):13–8. doi: 10.1164/arrd.1969.100.1.13
24. Angel JH, Chu LS, Lyons HA. Corticotropin in the treatment of tuberculosis. A controlled study. Arch Internal Med (1961) 108:353–69. doi: 10.1001/archinte.1961.03620090025004
25. Marcus H, Yoo OH, Akyol T, Williams MH Jr. A randomized study of the effects of corticosteroid therapy on healing of pulmonary tuberculosis as judged by clinical, roentgenographic, and physiologic measurements. Am Rev Respir Dis (1963) 88:55–64. doi: 10.1164/arrd.1963.88.1.55
26. Dooley DP, Carpenter JL, Rademacher S. Adjunctive corticosteroid therapy for tuberculosis: a critical reappraisal of the literature. Clin Infect Dis an Off Publ Infect Dis Soc America (1997) 25(4):872–87. doi: 10.1086/515543
27. Critchley JA, Young F, Orton L, Garner P. Corticosteroids for prevention of mortality in people with tuberculosis: a systematic review and meta-analysis. Lancet Infect Dis (2013) 13(3):223–37. doi: 10.1016/S1473-3099(12)70321-3
28. Wallis RS. Corticosteroid Effects on Sputum Culture in Pulmonary Tuberculosis: A Meta-Regression Analysis. Open Forum Infect Dis (2014) 1(1):ofu020. doi: 10.1093/ofid/ofu020
29. Nahid P, Dorman SE, Alipanah N, Barry PM, Brozek JL, Cattamanchi A, et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America Clinical Practice Guidelines: Treatment of Drug-Susceptible Tuberculosis. Clin Infect Dis an Off Publ Infect Dis Soc America (2016) 63(7):e147–95. doi: 10.1093/cid/ciw376
30. Nahid P, Mase SR, Migliori GB, Sotgiu G, Bothamley GH, Brozek JL, et al. Treatment of Drug-Resistant Tuberculosis. An Official ATS/CDC/ERS/IDSA Clinical Practice Guideline. Am J Respir Crit Care Med (2019) 200(10):e93–e142. doi: 10.1164/rccm.201909-1874ST
31. Thwaites GE, Nguyen DB, Nguyen HD, Hoang TQ, Do TT, Nguyen TC, et al. Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. New Engl J Med (2004) 351(17):1741–51. doi: 10.1056/NEJMp048227
32. Krug S, Kumar P, Kang BG, Dawson TM, Dawson VL, Bishai WR. Pyrazinamide inhibits the host enzyme PARP1 to reduce inflammation and accelerate bacterial clearance. In: Tuberculosis: Science Aimed at Ending the Epidemic (eSymposia). Keystone, CO, USA: Virtual Keystone Symposia (2020).
33. Berger NA, Besson VC, Boulares AH, Burkle A, Chiarugi A, Clark RS, et al. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br J Pharmacol (2018) 175(2):192–222. doi: 10.1111/bph.13748
34. Curtin NJ, Szabo C. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol aspects Med (2013) 34(6):1217–56. doi: 10.1016/j.mam.2013.01.006
36. Thorsell A-G, Ekblad T, Karlberg T, Löw M, Pinto AF, Trésaugues L, et al. Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors. J Medicinal Chem (2017) 60(4):1262–71. doi: 10.1021/acs.jmedchem.6b00990
37. Singhal A, Jie L, Kumar P, Hong GS, Leow MK-S, Paleja B, et al. Metformin as adjunct antituberculosis therapy. Sci Trans Med (2014) 6(263):263ra159–263ra159. doi: 10.1126/scitranslmed.3009885
38. Degner NR, Wang JY, Golub JE, Karakousis PC. Metformin Use Reverses the Increased Mortality Associated With Diabetes Mellitus During Tuberculosis Treatment. Clin Infect Dis (2018) 66(2):198–205. doi: 10.1093/cid/cix819
39. Gupta S, Krug S, Pokkali S, Leanderson T, Isaacs JT, Srikrishna G, et al. Pharmacologic Exhaustion of Suppressor Cells with Tasquinimod Enhances Bacterial Clearance during Tuberculosis. Am J Respir Crit Care Med (2019) 199(3):386–9. doi: 10.1164/rccm.201805-0820LE
40. Coleman MM, Basdeo SA, Coleman AM, Cheallaigh CN, Peral de Castro C, McLaughlin AM, et al. All-trans Retinoic Acid Augments Autophagy during Intracellular Bacterial Infection. Am J Respir Cell Mol Biol (2018) 59(5):548–56. doi: 10.1165/rcmb.2017-0382OC
41. Wheelwright M, Kim EW, Inkeles MS, De Leon A, Pellegrini M, Krutzik SR, et al. All-trans retinoic acid-triggered antimicrobial activity against Mycobacterium tuberculosis is dependent on NPC2. J Immunol (Baltimore Md 1950) (2014) 192(5):2280–90. doi: 10.4049/jimmunol.1301686
42. Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res (2003) 63(15):4441–9.
43. Parveen S, Lun S, Urbanowski ME, Cardin M, Murphy JR, Bishai WR. Effective host-directed therapy for tuberculosis by targeted depletion of myeloid-derived suppressor cells using a diphtheria toxin-based fusion protein. bioRxiv (2020) 2020.2012.2010.420224. doi: 10.1101/2020.12.10.420224
44. Maiga M, Ammerman NC, Maiga MC, Tounkara A, Siddiqui S, Polis M, et al. Adjuvant host-directed therapy with types 3 and 5 but not type 4 phosphodiesterase inhibitors shortens the duration of tuberculosis treatment. J Infect Dis (2013) 208(3):512–9. doi: 10.1093/infdis/jit187
45. Maiga MC, Ahidjo BA, Maiga M, Bishai WR. Roflumilast, a Type 4 Phosphodiesterase Inhibitor, Shows Promising Adjunctive, Host-Directed Therapeutic Activity in a Mouse Model of Tuberculosis. Antimicrob Agents Chemother (2015) 59(12):7888–90. doi: 10.1128/AAC.02145-15
46. Subbian S, Tsenova L, Holloway J, Peixoto B, O’Brien P, Dartois V, et al. Adjunctive Phosphodiesterase-4 Inhibitor Therapy Improves Antibiotic Response to Pulmonary Tuberculosis in a Rabbit Model. EBioMedicine (2016) 4:104–14. doi: 10.1016/j.ebiom.2016.01.015
47. Gupta S, Cheung L, Pokkali S, Winglee K, Guo H, Murphy JR, et al. Suppressor Cell-Depleting Immunotherapy With Denileukin Diftitox is an Effective Host-Directed Therapy for Tuberculosis. J Infect Dis (2017) 215(12):1883–7. doi: 10.1093/infdis/jix208
48. Green AM, Mattila JT, Bigbee CL, Bongers KS, Lin PL, Flynn JL. CD4(+) regulatory T cells in a cynomolgus macaque model of Mycobacterium tuberculosis infection. J Infect Dis (2010) 202(4):533–41. doi: 10.1086/654896
49. Tousif S, Singh DK, Mukherjee S, Ahmad S, Arya R, Nanda R, et al. Nanoparticle-Formulated Curcumin Prevents Posttherapeutic Disease Reactivation and Reinfection with Mycobacterium tuberculosis following Isoniazid Therapy. Front Immunol (2017) 8:739. doi: 10.3389/fimmu.2017.00739
50. Beamer GL, Flaherty DK, Assogba BD, Stromberg P, Gonzalez-Juarrero M, de Waal Malefyt R, et al. Interleukin-10 promotes Mycobacterium tuberculosis disease progression in CBA/J mice. J Immunol (Baltimore Md 1950) (2008) 181(8):5545–50. doi: 10.4049/jimmunol.181.8.5545
51. Elkington P, Shiomi T, Breen R, Nuttall RK, Ugarte-Gil CA, Walker NF, et al. MMP-1 drives immunopathology in human tuberculosis and transgenic mice. J Clin Invest (2011) 121(5):1827–33. doi: 10.1172/JCI45666
52. Ong CW, Elkington PT, Brilha S, Ugarte-Gil C, Tome-Esteban MT, Tezera LB, et al. Neutrophil-Derived MMP-8 Drives AMPK-Dependent Matrix Destruction in Human Pulmonary Tuberculosis. PloS Pathog (2015) 11(5):e1004917. doi: 10.1371/journal.ppat.1004917
53. Arpino V, Brock M, Gill SE. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol J Int Soc Matrix Biol (2015) 44-46:247–54. doi: 10.1016/j.matbio.2015.03.005
54. Kübler A, Luna B, Larsson C, Ammerman NC, Andrade BB, Orandle M, et al. Mycobacterium tuberculosis dysregulates MMP/TIMP balance to drive rapid cavitation and unrestrained bacterial proliferation. J Pathol (2015) 235(3):431–44. doi: 10.1002/path.4432
55. Sabir N, Hussain T, Mangi MH, Zhao D, Zhou X. Matrix metalloproteinases: Expression, regulation and role in the immunopathology of tuberculosis. Cell Proliferation (2019) 52(4):e12649. doi: 10.1111/cpr.12649
56. Ugarte-Gil CA, Elkington P, Gilman RH, Coronel J, Tezera LB, Bernabe-Ortiz A, et al. Induced sputum MMP-1, -3 & -8 concentrations during treatment of tuberculosis. PloS One (2013) 8(4):e61333. doi: 10.1371/journal.pone.0061333
57. Fields GB. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells (2019) 8(9):984. doi: 10.3390/cells8090984
58. Elkington PT, Green JA, Emerson JE, Lopez-Pascua LD, Boyle JJ, O’Kane CM, et al. Synergistic up-regulation of epithelial cell matrix metalloproteinase-9 secretion in tuberculosis. Am J Respir Cell Mol Biol (2007) 37(4):431–7. doi: 10.1165/rcmb.2007-0011OC
59. Elkington PTG, Emerson JE, Lopez-Pascua LDC, O’Kane CM, Horncastle DE, Boyle JJ, et al. Mycobacterium tuberculosis Up-Regulates Matrix Metalloproteinase-1 Secretion from Human Airway Epithelial Cells via a p38 MAPK Switch. J Immunol (2005) 175(8):5333–40. doi: 10.4049/jimmunol.175.8.5333
60. Mootoo A, Stylianou E, Arias MA, Reljic R. TNF-alpha in tuberculosis: a cytokine with a split personality. Inflammation Allergy Drug Targets (2009) 8(1):53–62. doi: 10.2174/187152809787582543
61. Su W-L, Perng W-C, Huang C-H, Yang C-Y, Wu C-P, Chen J-H. Association of Reduced Tumor Necrosis Factor Alpha, Gamma Interferon, and Interleukin-1β (IL-1β) but Increased IL-10 Expression with Improved Chest Radiography in Patients with Pulmonary Tuberculosis. Clin Vaccine Immunol (2010) 17(2):223–31. doi: 10.1128/CVI.00381-09
62. Ravimohan S, Tamuhla N, Kung S-J, Nfanyana K, Steenhoff AP, Gross R, et al. Matrix Metalloproteinases in Tuberculosis-Immune Reconstitution Inflammatory Syndrome and Impaired Lung Function Among Advanced HIV/TB Co-infected Patients Initiating Antiretroviral Therapy. EBioMedicine (2016) 3:100–7. doi: 10.1016/j.ebiom.2015.11.040
63. Chamie G, Luetkemeyer A, Walusimbi-Nanteza M, Okwera A, Whalen CC, Mugerwa RD, et al. Significant variation in presentation of pulmonary tuberculosis across a high resolution of CD4 strata. Int J Tuberc Lung Dis (2010) 14(10):1295–302.
64. Meintjes G, Lawn SD, Scano F, Maartens G, French MA, Worodria W, et al. Tuberculosis-associated immune reconstitution inflammatory syndrome: case definitions for use in resource-limited settings. Lancet Infect Dis (2008) 8(8):516–23. doi: 10.1016/S1473-3099(08)70184-1
65. Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med (2011) 208(7):1339–50. doi: 10.1084/jem.20110551
66. Barlo NP, van Moorsel CH, Korthagen NM, Heron M, Rijkers GT, Ruven HJ, et al. Genetic variability in the IL1RN gene and the balance between interleukin (IL)-1 receptor agonist and IL-1β in idiopathic pulmonary fibrosis. Clin Exp Immunol (2011) 166(3):346–51. doi: 10.1111/j.1365-2249.2011.04468.x
67. Arbués A, Brees D, Chibout SD, Fox T, Kammüller M, Portevin D. TNF-α antagonists differentially induce TGF-β1-dependent resuscitation of dormant-like Mycobacterium tuberculosis. PloS Pathog (2020) 16(2):e1008312. doi: 10.1371/journal.ppat.1008312
68. Young C, Walzl G, Du Plessis N. Therapeutic host-directed strategies to improve outcome in tuberculosis. Mucosal Immunol (2020) 13(2):190–204. doi: 10.1038/s41385-019-0226-5
69. Mayosi BM, Ntsekhe M, Bosch J, Pandie S, Jung H, Gumedze F, et al. Prednisolone and Mycobacterium indicus pranii in tuberculous pericarditis. New Engl J Med (2014) 371(12):1121–30.
70. David KK, Andrabi SA, Dawson TM, Dawson VL. Parthanatos, a messenger of death. Front Biosci (Landmark edition) (2009) 14:1116–28. doi: 10.2741/3297
71. Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol (2012) 13(7):411–24. doi: 10.1038/nrm3376
72. Langelier MF, Zandarashvili L, Aguiar PM, Black BE, Pascal JM. NAD(+) analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nat Commun (2018) 9(1):844. doi: 10.1038/s41467-018-03234-8
73. Swindall AF, Stanley JA, Yang ES. PARP-1: Friend or Foe of DNA Damage and Repair in Tumorigenesis? Cancers (Basel) (2013) 5(3):943–58. doi: 10.3390/cancers5030943
74. Amé J-C, Spenlehauer C, de Murcia G. The PARP superfamily. BioEssays (2004) 26(8):882–93. doi: 10.1002/bies.20085
75. Ba X, Garg NJ. Signaling mechanism of poly(ADP-ribose) polymerase-1 (PARP-1) in inflammatory diseases. Am J Pathol (2011) 178(3):946–55. doi: 10.1016/j.ajpath.2010.12.004
76. Bai P, Virag L. Role of poly(ADP-ribose) polymerases in the regulation of inflammatory processes. FEBS Lett (2012) 586(21):3771–7. doi: 10.1016/j.febslet.2012.09.026
77. Ke Y, Wang C, Zhang J, Zhong X, Wang R, Zeng X, et al. The Role of PARPs in Inflammation-and Metabolic-Related Diseases: Molecular Mechanisms and Beyond. Cells (2019) 8(9):1047. doi: 10.3390/cells8091047
78. Giansanti V, Dona F, Tillhon M, Scovassi AI. PARP inhibitors: new tools to protect from inflammation. Biochem Pharmacol (2010) 80(12):1869–77. doi: 10.1016/j.bcp.2010.04.022
79. Rosado MM, Bennici E, Novelli F, Pioli C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology (2013) 139(4):428–37. doi: 10.1111/imm.12099
80. Liu L, Ke Y, Jiang X, He F, Pan L, Xu L, et al. Lipopolysaccharide activates ERK-PARP-1-RelA pathway and promotes nuclear factor-kappaB transcription in murine macrophages. Hum Immunol (2012) 73(5):439–47. doi: 10.1016/j.humimm.2012.02.002
81. Liaudet L, Pacher P, Mabley JG, Virag L, Soriano FG, Hasko G, et al. Activation of poly(ADP-Ribose) polymerase-1 is a central mechanism of lipopolysaccharide-induced acute lung inflammation. Am J Respir Crit Care Med (2002) 165(3):372–7. doi: 10.1164/ajrccm.165.3.2106050
82. Vuong B, Hogan-Cann AD, Alano CC, Stevenson M, Chan WY, Anderson CM, et al. NF-kappaB transcriptional activation by TNFalpha requires phospholipase C, extracellular signal-regulated kinase 2 and poly(ADP-ribose) polymerase-1. J Neuroinflamm (2015) 12:229. doi: 10.1186/s12974-015-0448-8
83. Mishra M, Kowluru RA. Role of PARP-1 as a novel transcriptional regulator of MMP-9 in diabetic retinopathy. Biochim Biophys Acta Mol basis Dis (2017) 1863(7):1761–9. doi: 10.1016/j.bbadis.2017.04.024
84. Nicolescu AC, Holt A, Kandasamy AD, Pacher P, Schulz R. Inhibition of matrix metalloproteinase-2 by PARP inhibitors. Biochem Biophys Res Commun (2009) 387(4):646–50. doi: 10.1016/j.bbrc.2009.07.080
85. Chen T, Wang W, Li J-R, Xu H-Z, Peng Y-C, Fan L-F, et al. PARP inhibition attenuates early brain injury through NF-κB/MMP-9 pathway in a rat model of subarachnoid hemorrhage. Brain Res (2016) 1644:32–8. doi: 10.1016/j.brainres.2016.05.005
86. Ghorai A, Sarma A, Chowdhury P, Ghosh U. PARP-1 depletion in combination with carbon ion exposure significantly reduces MMPs activity and overall increases TIMPs expression in cultured HeLa cells. Radiat Oncol (2016) 11(1):126. doi: 10.1186/s13014-016-0703-x
87. Gupte AN, Paradkar M, Selvaraju S, Thiruvengadam K, Shivakumar SVBY, Sekar K, et al. Assessment of lung function in successfully treated tuberculosis reveals high burden of ventilatory defects and COPD. PloS One (2019) 14(5):e0217289. doi: 10.1371/journal.pone.0217289
88. de Martino M, Lodi L, Galli L, Chiappini E. Immune Response to Mycobacterium tuberculosis: A Narrative Review. Front Pediatr (2019) 7:350. doi: 10.3389/fped.2019.00350
89. Refai A, Gritli S, Barbouche MR, Essafi M. Mycobacterium tuberculosis Virulent Factor ESAT-6 Drives Macrophage Differentiation Toward the Pro-inflammatory M1 Phenotype and Subsequently Switches It to the Anti-inflammatory M2 Phenotype. Front Cell Infect Microbiol (2018) 8:327. doi: 10.3389/fcimb.2018.00327
90. Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol (Baltimore Md 1950) (2001) 166(1):678–89. doi: 10.4049/jimmunol.166.1.678
91. Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res (2007) 13(2 Pt 2):721s–6s. doi: 10.1158/1078-0432.CCR-06-2197
92. Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res (2005) 65(8):3044–8. doi: 10.1158/0008-5472.CAN-04-4505
93. Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol Off J Am Soc Clin Oncol (2007) 25(18):2546–53. doi: 10.1200/JCO.2006.08.5829
94. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
95. Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med (2015) 66:97–110. doi: 10.1146/annurev-med-051013-052304
96. Knaul JK, Jörg S, Oberbeck-Mueller D, Heinemann E, Scheuermann L, Brinkmann V, et al. Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am J Respir Crit Care Med (2014) 190(9):1053–66. doi: 10.1164/rccm.201405-0828OC
97. du Plessis N, Loebenberg L, Kriel M, von Groote-Bidlingmaier F, Ribechini E, Loxton AG, et al. Increased frequency of myeloid-derived suppressor cells during active tuberculosis and after recent mycobacterium tuberculosis infection suppresses T-cell function. Am J Respir Crit Care Med (2013) 188(6):724–32. doi: 10.1164/rccm.201302-0249OC
98. Tsiganov EN, Verbina EM, Radaeva TV, Sosunov VV, Kosmiadi GA, Nikitina IY, et al. Gr-1dimCD11b+ immature myeloid-derived suppressor cells but not neutrophils are markers of lethal tuberculosis infection in mice. J Immunol (Baltimore Md 1950) (2014) 192(10):4718–27. doi: 10.4049/jimmunol.1301365
99. du Plessis N, Kotze LA, Leukes V, Walzl G. Translational Potential of Therapeutics Targeting Regulatory Myeloid Cells in Tuberculosis. Front Cell Infect Microbiol (2018) 8:332. doi: 10.3389/fcimb.2018.00332
100. Li L, Wang L, Li J, Fan Z, Yang L, Zhang Z, et al. Metformin-Induced Reduction of CD39 and CD73 Blocks Myeloid-Derived Suppressor Cell Activity in Patients with Ovarian Cancer. Cancer Res (2018) 78(7):1779–91. doi: 10.1158/0008-5472.CAN-17-2460
101. Qin G, Lian J, Huang L, Zhao Q, Liu S, Zhang Z, et al. Metformin blocks myeloid-derived suppressor cell accumulation through AMPK-DACH1-CXCL1 axis. Oncoimmunology (2018) 7(7):e1442167. doi: 10.1080/2162402X.2018.1442167
102. Dutta NK, Pinn ML, Karakousis PC. Metformin Adjunctive Therapy Does Not Improve the Sterilizing Activity of the First-Line Antitubercular Regimen in Mice. Antimicrob Agents Chemother (2017) 61(8):e00652-17. doi: 10.1128/AAC.00652-17
103. Kornfeld H, Singhal A, Wallis R. Trial of Metformin for TB/HIV Host-directed Therapy. National Institute of Health (NIH) (2019).
104. Williamson SC, Hartley AE, Heer R. A review of tasquinimod in the treatment of advanced prostate cancer. Drug Des Devel Ther (2013) 7:167–74. doi: 10.2147/DDDT.S31500
105. Shen L, Pili R. Tasquinimod targets suppressive myeloid cells in the tumor microenvironment. Oncoimmunology (2019) 8(10):e1072672. doi: 10.1080/2162402X.2015.1072672
106. Yoshioka Y, Mizutani T, Mizuta S, Miyamoto A, Murata S, Ano T, et al. Neutrophils and the S100A9 protein critically regulate granuloma formation. Blood Adv (2016) 1(3):184–92. doi: 10.1182/bloodadvances.2016000497
107. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res (2007) 67(22):11021–8. doi: 10.1158/0008-5472.CAN-07-2593
108. Mourik BC, Leenen PJ, de Knegt GJ, Huizinga R, van der Eerden BC, Wang J, et al. Immunotherapy Added to Antibiotic Treatment Reduces Relapse of Disease in a Mouse Model of Tuberculosis. Am J Respir Cell Mol Biol (2017) 56(2):233–41. doi: 10.1165/rcmb.2016-0185OC
109. Parveen S, Siddharth S, Cheung LS, Kumar A, Murphy JR, Sharma D, et al. IL-4 receptor targeting as an effective immunotherapy against triple-negative breast cancer. bioRxiv (2020), 2020.2008.2005.238824. doi: 10.1101/2020.08.05.238824
110. Leukes V, Walzl G, du Plessis N. Myeloid-Derived Suppressor Cells as Target of Phosphodiesterase-5 Inhibitors in Host-Directed Therapeutics for Tuberculosis. Front Immunol (2020) 11:451. doi: 10.3389/fimmu.2020.00451
111. Maiga M, Agarwal N, Ammerman NC, Gupta R, Guo H, Maiga MC, et al. Successful shortening of tuberculosis treatment using adjuvant host-directed therapy with FDA-approved phosphodiesterase inhibitors in the mouse model. PloS One (2012) 7(2):e30749. doi: 10.1371/journal.pone.0030749
112. Wallis RS, Ginindza S, Beattie T, Arjun N, Likoti M, Edward V, et al. Preliminary Results of an Experimental Medicine Trial of Adjunctive Host-Directed Therapy in Adults with Moderately or Far-Advanced Rifampin-Susceptible Pulmonary Tuberculosis. Am J Respir Crit Care Med (2019) 199:A7388. doi: 10.1164/ajrccm-conference.2019.199.1_MeetingAbstracts.A7388
113. Cardona P, Cardona P-J. Regulatory T Cells in Mycobacterium tuberculosis Infection. Front Immunol (2019) 10:2139. doi: 10.3389/fimmu.2019.02139
114. Chen X, Zhou B, Li M, Deng Q, Wu X, Le X, et al. Zhang H et al: CD4(+)CD25(+)FoxP3(+) regulatory T cells suppress Mycobacterium tuberculosis immunity in patients with active disease. Clin Immunol (2007) 123(1):50–9. doi: 10.1016/j.clim.2006.11.009
115. Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, et al. Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity (2017) 46(4):577–86. doi: 10.1016/j.immuni.2017.03.013
116. Ozeki Y, Sugawara I, Udagawa T, Aoki T, Osada-Oka M, Tateishi Y, et al. Transient role of CD4+CD25+ regulatory T cells in mycobacterial infection in mice. Int Immunol (2010) 22(3):179–89. doi: 10.1093/intimm/dxp126
117. Kumar P, Kumar A, Parveen S, Murphy JR, Bishai W. Recent advances with Treg depleting fusion protein toxins for cancer immunotherapy. Immunotherapy (2019) 11(13):1117–28. doi: 10.2217/imt-2019-0060
118. Sanseviero E, O’Brien EM, Karras JR, Shabaneh TB, Aksoy BA, Xu W, et al. Anti-CTLA-4 Activates Intratumoral NK Cells and Combined with IL15/IL15Rα Complexes Enhances Tumor Control. Cancer Immunol Res (2019) 7(8):1371–80. doi: 10.1158/2326-6066.CIR-18-0386
119. Barber DL, Sakai S, Kudchadkar RR, Fling SP, Day TA, Vergara JA, et al. Tuberculosis following PD-1 blockade for cancer immunotherapy. Sci Transl Med (2019) 11(475):eaat2702. doi: 10.1126/scitranslmed.aat2702
120. Fujita K, Terashima T, Mio T. Anti-PD1 Antibody Treatment and the Development of Acute Pulmonary Tuberculosis. J Thorac Oncol (2016) 11(12):2238–40. doi: 10.1016/j.jtho.2016.07.006
121. Reungwetwattana T, Adjei AA. Anti-PD-1 Antibody Treatment and the Development of Acute Pulmonary Tuberculosis. J Thorac Oncol (2016) 11(12):2048–50. doi: 10.1016/j.jtho.2016.10.008
122. Huang Z, Luo Q, Guo Y, Chen J, Xiong G, Peng Y, et al. Mycobacterium tuberculosis-Induced Polarization of Human Macrophage Orchestrates the Formation and Development of Tuberculous Granulomas In Vitro. PloS One (2015) 10(6):e0129744. doi: 10.1371/journal.pone.0129744
123. Abdollahi E, Momtazi AA, Johnston TP, Sahebkar A. Therapeutic effects of curcumin in inflammatory and immune-mediated diseases: A nature-made jack-of-all-trades? J Cell Physiol (2018) 233(2):830–48. doi: 10.1002/jcp.25778
124. Shiri S, Alizadeh AM, Baradaran B, Farhanghi B, Shanehbandi D, Khodayari S, et al. Dendrosomal curcumin suppresses metastatic breast cancer in mice by changing m1/m2 macrophage balance in the tumor microenvironment. Asian Pac J Cancer Prev (2015) 16(9):3917–22. doi: 10.7314/APJCP.2015.16.9.3917
125. Bai X, Oberley-Deegan RE, Bai A, Ovrutsky AR, Kinney WH, Weaver M, et al. Curcumin enhances human macrophage control of Mycobacterium tuberculosis infection. Respirology (2016) 21(5):951–7. doi: 10.1111/resp.12762
126. van Kampen SC, Wanner A, Edwards M, Harries AD, Kirenga BJ, Chakaya J, et al. International research and guidelines on post-tuberculosis chronic lung disorders: a systematic scoping review. BMJ Global Health (2018) 3(4):e000745. doi: 10.1136/bmjgh-2018-000745
Keywords: tuberculosis, PARP inhibition (PARPi), MMPs (metalloproteinases), immunotherapy, diphtheria fusion protein toxin, MDSCs, host-directed therapies
Citation: Krug S, Parveen S and Bishai WR (2021) Host-Directed Therapies: Modulating Inflammation to Treat Tuberculosis. Front. Immunol. 12:660916. doi: 10.3389/fimmu.2021.660916
Received: 29 January 2021; Accepted: 29 March 2021;
Published: 19 April 2021.
Edited by:
Suraj P. Parihar, University of Cape Town, South AfricaReviewed by:
Paulo Bettencourt, Independent Researcher, Lisbon, PortugalMohlopheni Jackson Marakalala, Africa Health Research Institute (AHRI), South Africa
Copyright © 2021 Krug, Parveen and Bishai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William R. Bishai, d2Jpc2hhaTFAamhtaS5lZHU=
†These authors have contributed equally to this work