 Teresa Fuertes
Teresa Fuertes Irene Salgado
Irene Salgado Virginia G. de Yébenes
Virginia G. de Yébenes- 1B Lymphocyte Biology Lab, Centro Nacional de Investigaciones Cardiovasculares, Madrid, Spain
- 2Department of Immunology, Ophthalmology and ENT, Universidad Complutense de Madrid School of Medicine, Madrid, Spain
- 3Inmunología Linfocitaria Lab, Hospital 12 de Octubre Health Research Institute (imas12), Madrid, Spain
Germinal centers (GCs) are complex multicellular structures in which antigen-specific B cells undergo the molecular remodeling that enables the generation of high-affinity antibodies and the differentiation programs that lead to the generation of plasma–antibody-secreting cells and memory B cells. These reactions are tightly controlled by a variety of mechanisms, including the post-transcriptional control of gene expression by microRNAs (miRNAs). Through the development of animal models with B cell-specific modified miRNA expression, we have contributed to the understanding of the role of miRNAs in the regulation of GC responses and in B cell neoplasia. Here, we review recent advances in the understanding of the role of miRNAs in the regulation of B cell and T follicular helper physiology during the GC response and in the diseases associated to GC response dysregulation.
Introduction
The germinal center (GC) response is a key B lymphocyte maturation and differentiation program essential for the generation of competent protective immunity. The GC response is initiated in mature B lymphocytes after antigen encounter and leads to the generation of memory B cells and plasma antibody-secreting cells that produce antibodies with high antigen affinity and with different immunoglobulin (Ig) isotypes, conferring the Ig molecule with the ability to orchestrate different immune effector responses (1, 2). At the molecular level, these reactions are initiated by the activity of activation induced deaminase (AID), an enzyme that deaminates cytosines in the Ig genes, triggering somatic hypermutation (SHM) and class switch recombination (CSR), processes respectively responsible for the changes in affinity and isotype in the Ig genes. At the cellular level, initiation of the GC reaction requires the cognate interaction of antigen-activated B-lymphocytes with a specialized subset of GC T CD4 cells, the follicular T helper (Tfh) cells. Tfh-GC B cell interactions are dependent on a number of molecule interactions that signal for full B and Tfh cell differentiation together with cellular localization in follicles. These interactions include T-cell receptor recognition of B cell peptide-MHC complexes as well as CD40 and ICOS ligand co-receptor interactions (3). Developing Tfh and B GC cells are influenced by changing cytokine, chemokine and cellular environments through the induction of specific transcriptional programs (4). Gene transcription in Tfh and B cells is regulated by key GC transcription factors such as BCL6, as well as by RNA-binding proteins and microRNAs (miRNAs) (3, 5). miRNAs are small non-coding RNA molecules that drive post-transcriptional negative regulation of gene expression by promoting the degradation or translational blockade of partially complementary target mRNAs. Mature miRNAs are 21-24-nucleotide RNA molecules processed from longer RNA precursors in two consecutive cleavage steps mediated by the RNase III enzymes Drosha and Dicer (6). Ablation of miRNAs in miRNA-processing-enzyme deletion knockout models has demonstrated that miRNAs play essential roles in diverse developmental, cellular, and physiological processes (7, 8). miRNAs fine-tune cellular gene expression networks and have emerged as essential regulators of GC differentiation responses.
miRNAs in Physiological GC Regulation
Studies of global miRNA depletion in GC B and T cell-specific models showed that miRNAs are essential for proper GC formation (9, 10). Dicer-mediated miRNA depletion after AID expression in early activated GC B cells impaired the production of high-affinity class-switched antibodies and the generation of memory B and long-lived plasma cells after T cell-dependent immunization due to defects in B cell proliferation and survival (9). Likewise, DGCR8-Drosha complex-mediated miRNA depletion in CD4 T cells showed that CD4 T cell-expressed miRNAs are essential for the differentiation of Tfh cells and the induction of GC B cells during T cell-dependent immunizations (10). Interestingly, miRNAs are not only required to regulate Tfh and GC B cell function in a cell intrinsic manner, but are also important contact-independent mediators of T-B cellular communication (Figure 1). This communication occurs through the transfer to B cells of a restricted set of T cell-derived miRNAs in extracellular vesicles and modulates the efficiency of GC generation and antibody secretion in response to immunization (11).
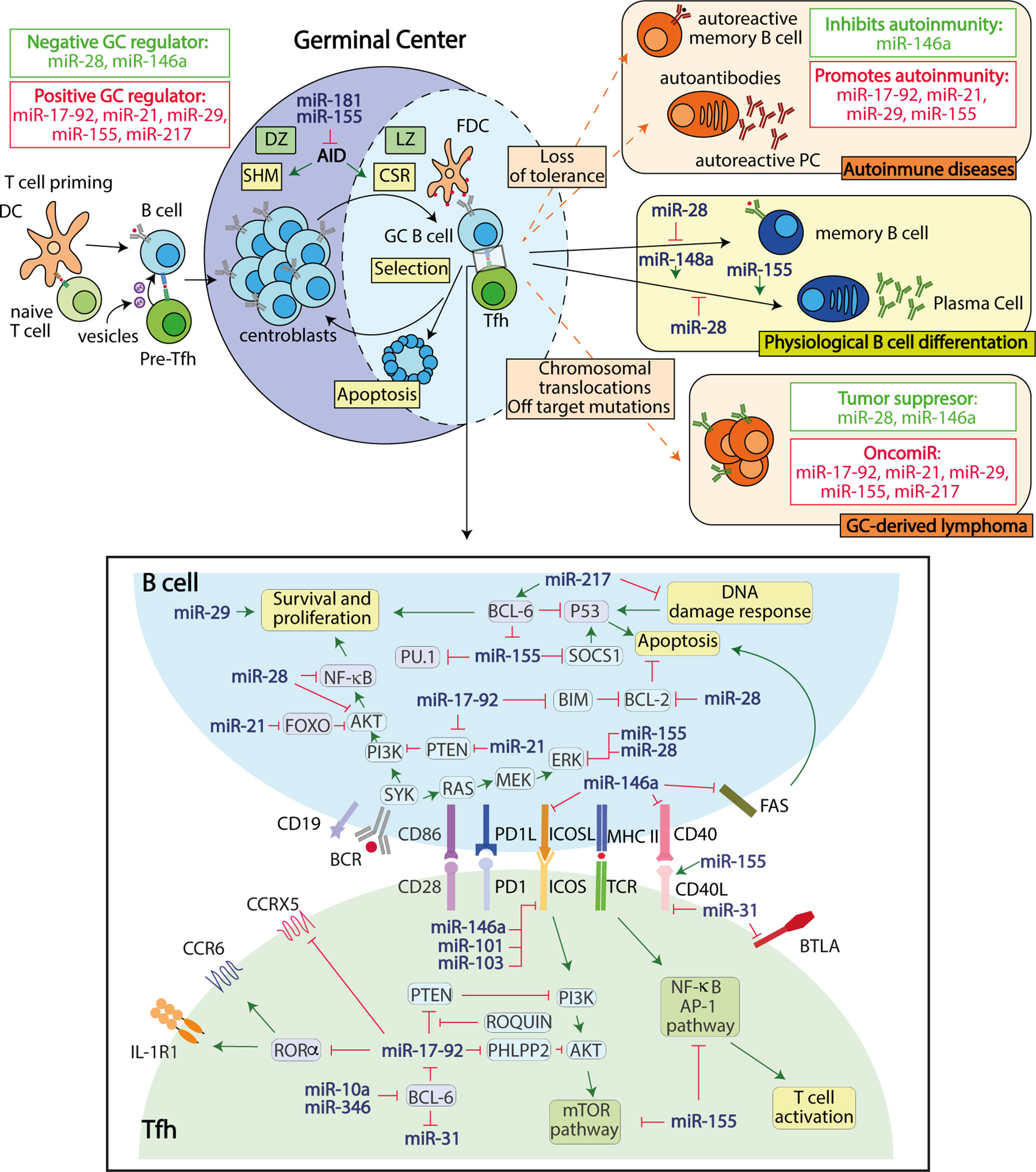
Figure 1 miRNAs regulate gene expression in B and Tfh germinal center cells. Regulated miRNA expression is required to regulate B-Tfh cell interactions and ensure proper GC responses. GC-derived dysfunctions caused by miRNA alterations can lead to the development of autoimmunity and/or B cell neoplasia through the disruption of post-transcriptional control mechanisms required for the maintenance of GC homeostasis.
miRNAs in the Regulation of B Cells in the GC
The most extensively studied GC B cell miRNA is miR-155, whose expression is upregulated after mature B cell activation and in GC B cells (12–15). Infection of miR-155-deficient mice with pathogenic bacteria showed that miR-155 expression is required to control pathogen-induced disease (16). Characterization of the response to T cell-dependent immunizations in miR-155-/- loss-of-function and miR-155KI gain-of-function mouse models revealed that miR-155 expression is required for efficient adaptive immune responses, including the generation of GC B cells and the secretion of antigen-specific antibodies (12, 16). miR-155 is a positive regulator of the GC response, and deficiency in miR-155 expression leads to reduced cytokine production, IgG1 secretion, impaired affinity maturation, and plasmablast B cell generation in a B cell autonomous manner (12, 17, 18). miR-155 controls affinity-based selection, at least in part, by protecting light zone (LZ) GC c-MYC+ B cells from apoptosis (19).
Transcriptome studies showed that miR-155 regulates the expression of numerous mRNAs in B cells (17, 18), although the functional consequences of miR-155-dependent mRNA regulation in GC B cells has been characterized for only a few miR-155 targets. The transcription factor PU.1 is a direct miR-155 target implicated in miR-155 mediated effects on CSR (17). PU.1 is encoded by Sfpi1, and the consequences of disrupting miR-155–Sfpi1 mRNA interaction in vivo were determined by generating knock-in mice with a mutation in the miR-155 recognition site in the Sfpi1 mRNA 3’UTR. miR-155-mediated PU.1 post-transcriptional regulation was shown to be required for efficient terminal plasma B cell differentiation and antigen-specific immunoglobulin (Ig) secretion through the downregulation of Pax5 expression and genes involved in adhesion and B-T cell interactions (20).
The other well characterized miR-155 target in GC B cells is activation-induced deaminase (AID), the enzyme responsible for the molecular remodeling of Igs in the GC. Knock-in mice with a disruption of the miR-155 recognition site in the Aicda mRNA 3’UTR demonstrated that miR-155 expression in GC B cells is needed to limit AID expression, allow proper affinity maturation, and restrict oncogenic AID-mediated MYC-IgH chromosomal translocations (21, 22). GC tolerance of DNA damage is multilayered and temporally regulated (23), and miR-155 expression is in turn limited by the expression of BCL6 (24, 25), an important transcriptional regulator and proto-oncogene that inhibits the DNA damage response in GC B cells (26). In addition, miR-155 negatively regulates the expression of Socs1, a P53 activator important for the DNA damage response (27). miR-155 thus plays a dual role in modulating the accumulation of DNA double-strand breaks (DSB) associated with the GC reaction, regulating P53 activity by controlling the expression levels of Aicda and Socs1.
AID expression is directly regulated in B cells by yet other miRNAs in B cells. miR-361 is another BCL6-downregulated miRNA that targets Aicda, presumably in light-zone GC B cells (25). miR-181b, which is highly expressed in mature resting B cells and whose expression diminishes upon B cell activation, targets Aicda directly through the binding of several partly complementary sequences found in its mRNA 3’UTR (28). Thus, AID levels are controlled by different miRNAs at different stages of B cell activation.
Another miRNA that positively regulates the GC response upon its induction during B cell activation and in GC B cells is miR-217. Using gain- and loss-of-function mouse models, we showed that miR-217 promotes the generation of GC B cells and increases the generation of class-switched antibodies and the frequency of somatic hypermutation in B cells. We found that miR-217 regulates a DNA damage response and repair gene network that stabilizes BCL6 expression in GC B cells (29). Thus, miR-217 downregulates a network of genes that sense and repair genotoxic events on DNA, which in turn can increase GC B cell tolerance to DNA damage in the context of AID activity, very much like BCL6. Notably, we found that miR-217 protects BCL6 from previously described genotoxic stress-induced degradation (23), suggesting that both molecules form part of the same network that renders GC cells permissive to genomic instability and prone to malignant transformation.
Positive regulation of terminal post-GC plasma B cell differentiation has been suggested to be regulated by other miRNAs. A likely candidate is miR-148a, the most abundant miRNA in human and murine plasma cells, which has been shown to promote plasma cell differentiation and survival in vitro. Importantly, miR-148a expression was shown to downregulate the expression of the GC transcription factors Mitf and Bach2, which block premature plasma cell maturation and favor cell death (30). Definition of the role of miR-148a as a regulator of GC-dependent plasma cell differentiation in vivo would require the development of gain- or loss-of-function miR-148a B cell-specific mouse models.
GC miRNAs can also act as regulators that restrict the GC response, the best-characterized negative regulators of GC responses being miR-28 and miR-146a. miR-28 is a GC-specific miRNA (14, 15) whose expression is lost in numerous mature B-cell neoplasms (31–33). By combining gain- and loss-of-function approaches, we showed that miR-28 negatively regulates CSR and immunization-triggered GC and post-GC plasma and memory B cell generation. Combined transcriptome and proteome analysis upon inducible re-expression of miR-28 in B cells revealed that miR-28 expression induces the coordinated downregulation of the key BCR signaling gene network regulating B-cell proliferation and cell death (33), thus supporting the notion that miR-28 limits the strength of BCR signaling and regulates proliferation and survival of GC B cells.
miR-146a is expressed in B cells upon stimulation and within GC B cells (15), and loss of miR-146a causes a B cell-intrinsic increase in the GC response to immunization (34), spontaneous GC generation in aged mice, and increased production of anti-double–stranded DNA (dsDNA) auto-antibodies (35). miR-146a was shown to limit B cell GC functional responses by downregulating B cell expression of signaling pathway components involved in GC B Tfh cellular interactions, such as ICOSL (34) and CD40 (35).
Other miRNAs have also been suggested to negatively regulate terminal post-GC plasma and memory B cell differentiation. miR-125b, a miRNA highly expressed in dividing centroblasts in GC B cells (36), has been shown to inhibit plasma cell generation and antibody secretion in vitro (37, 38). Importantly, direct mRNA targeting by miR-125b was shown to downregulate the expression of BLIMP-1 and IRF-4 transcription factors, which are essential for plasma cell differentiation (36–39). Prdm1, the gene encoding BLIMP-1, is a direct target of other highly expressed GC B cell miRNAs, including miR-9, miR-30a, and let-7 family miRNAs (40–43). Interestingly, the expression of miR-30a and miR-125b is regulated epigenetically in B cells and can be modulated using histone deacetylase inhibitors to inhibit BLIMP-1 expression in the context of antibody responses and GC-derived diseases (44–46). Memory B cell generation is associated to changes in chromatin accessibility and miRNA expression, and miR-181 has been recently identified as a major gene expression regulator during memory B cell differentiation (47).
Overall, these studies have identified a set of miRNAs that are required to promote or limit the GC reaction through post-transcriptional gene expression regulation in B cells (Figure 1 and Table 1).
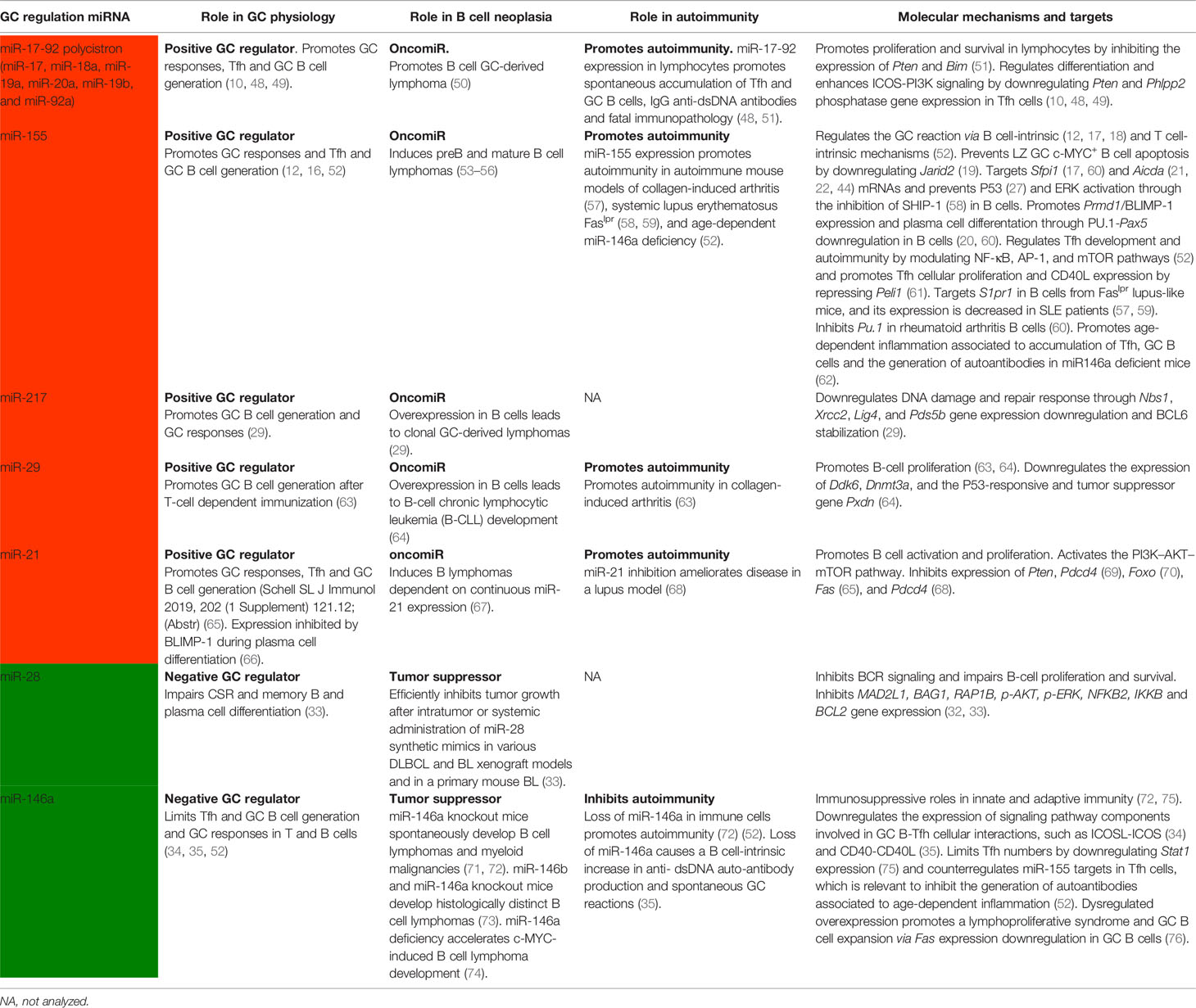
Table 1 Identified roles of miRNAs in the regulation of physiological GC responses and in GC-derived dysfunctions.
miRNAs in the Regulation of Follicular Helper T Cells
The induction of the GC reaction is critically dependent on the colocalization of B cells with Tfh cells and interaction between the two. This GC B-Tfh cell interaction and the resulting intracellular signaling are also controlled by miRNAs expressed in Tfh cells. Remarkably, miR-146a downregulates the inducible costimulatory Icos expression in Tfh cells (34), and thus the expression of the two interacting molecules (ICOS and ICOSL) of this costimulatory pathway are negatively controlled in both cell subsets by the same miRNA. ICOS directly controls the migration of CD4+ T cells from the T cell-B cell border into the B cell follicles of peripheral lymphoid organs (77). Importantly, ICOS signaling in T cells was shown to be important for miR-146a mediated Tfh and GC regulation (34). Icos expression is also negatively regulated by two other miRNAs whose expression is downregulated during Tfh differentiation, miR-101 and miR-103 (78, 79). Thus, ICOS co-stimulatory receptor expression is redundantly regulated by miRNAs in Tfh cells presumably to limit or end the GC reaction.
GC B-Tfh derived ICOS signaling is mediated by the PI3K/AKT pathway (80) and inhibited by PTEN phosphatase activity in Tfh cells (81). This key signaling pathway for Tfh activation and differentiation (3) is additionally regulated at different levels in Tfh cells by miRNAs from the miR-17-92 cluster. miR-17-92 cluster expression is induced early in T cell activation (48) and is repressed by BCL6, the critical transcriptional factor that regulates Tfh differentiation (82). T cell-specific miR-17-92 gain- and loss-of function mouse models showed that the microRNAs of the cluster are critical promoters of Tfh and GC B cell differentiation and antigen-specific antibody generation during both T-cell dependent immune responses and chronic viral infection (10, 48, 49). miR-17-92 cluster miRNAs regulate the ICOS-PI3K signaling pathway in Tfh cells through the simultaneous targeting of different pathway inhibitory components. miR-17-92 inhibits PTEN phosphatase expression upstream of AKT (10, 48, 51) as well as the downstream AKT phosphatase PHLPP2 (48). This pathway is additionally regulated in Tfh cells by Roquin, an RNA-binding protein that recognizes specific stem-loop structures in the 3’UTRs of target mRNAs and which interferes with miR-17-92 binding to an overlapping binding site in the Pten mRNA 3’UTR (83). Important miR-17-92 targets mediating other aspects of the Tfh differentiation program include the transcription factor RORα; responsible for the induction of IL-1R1 and CCR6 expression in Tfh cells (10), and CXCR5, a hallmark Tfh molecule that influences Tfh cell localization to follicles in which the ligand CXCL13 is expressed (82).
BCL6 represses the expression of a significant fraction of the miRNAs expressed in mouse CD4+ T cells (82); however, the functional contribution of this repression to the Tfh cell transcriptional program has been characterized for few miRNAs outside the miR-17-92 cluster. BCL6 represses miR-31 expression in human Tfh cells through direct binding to its promoter (84). miR-31 inhibits the expression of CD40L, SAP, and BTLA, which are crucial for Tfh cell helper activity and cross-talk with B cells (85–87). Accordingly, Tfh cells forced to express miR-31 display decreased B-helper activity (84). Although BCL6 controls Tfh activity in humans and mice, the role of miR-31 is restricted to human Tfh cell differentiation, reflecting a species specificity on the action of some miRNAs.
Bcl6 gene expression is also regulated by miRNAs in CD4+ T cells, and this regulation influences the generation of Tfh cells from T cell precursors. miR-10a, a miRNA highly expressed in mouse regulatory T cells (Treg), has been proposed to attenuate the conversion of inducible Tregs to Tfh cells through Bcl6 repression in mice (88). miR-346 has been suggested to repress BCL6 gene expression in human Tfh cells (62).
Another key miRNA regulator of both CG B and Tfh cells is the positive GC regulator miR-155. Immunization of T cell-specific miR-155-deficient Cd4-Cre miR155fl/fl mice revealed impaired in GC B and Tfh cell generation and antigen-specific antibody production (52), revealing that miR-155 expression regulates Tfh development during immunization responses through T cell intrinsic mechanisms. The same study showed that miR-155 regulates different Tfh-cell targets important for Tfh development and autoimmunity in the NF-κB, AP-1 and mTOR pathways. Interestingly, miR-155 promotes Tfh cell accumulation during chronic, low-grade inflammation by counteracting the effect of miR-146a in Tfh cells (52). A later study showed that miR-155 promotes Tfh cell proliferation and CD40L expression by repressing expression of Peli1, a ubiquitin ligase that promotes the degradation of the NF-κB family transcription factor c-REL (61). These data suggest that miR-155 contributes to increased Tfh-mediated GC B activation through increased CD40L–CD40 interaction, which is known to be a limiting step in B cell clonal expansion, GC formation, isotype switching, affinity maturation, and the generation of long-lived plasma cells (89, 90).
Thus, miRNAs regulate Tfh cellular differentiation and interaction with B cells in the GC at multiple levels and through multilayer regulatory molecular circuits (Figure 1).
miRNAs in GC-Derived B Cell Neoplasia and Autoimmune Diseases
Defects in GC regulation lead to immune diseases such as autoimmunity and mature B-cell neoplasia. These diseases are ultimately caused by the dysregulation of two distinct GC checkpoints; a breakdown of immune tolerance in autoimmunity and a surpassing of the DNA damage-tolerance threshold associated with Ig remodeling during CSR and SHM in B cell neoplasia. However, both diseases share a contribution from some of the mechanisms that promote GC dysfunction, including lymphoproliferative aberrant GC persistence, abnormal cellular components, and abnormal cellular signaling (91–93).
Recent studies addressing the contribution of miRNAs to these two GC-derived diseases revealed that dysregulated miRNA expression in GC B or Tfh cells can trigger B cell neoplasia or autoimmunity (Figure 1). Interestingly, several miRNAs that positively regulate the GC response also promote autoimmunity and B cell neoplasia (Table 1). For instance, miR-155, which promotes GC responses through T and B cell intrinsic mechanisms (12, 16, 52), also promotes autoimmune diseases characterized by switched auto-antibodies (52, 57–60, 94) and B cell neoplasia (53–56), likely through multilayer mechanisms that can lead to aberrant GC persistence due to increased proliferation, reduced cell death and altered cellular signaling of Tfh and GC B cells (Table 1). Similarly, the miR-17-92 polycistron, which promotes GC responses by enhancing Tfh and B lymphocyte proliferation and survival by inhibiting the expression of Pten and Bim (10, 48, 49, 51), when overexpressed in different mouse models promotes the generation of spontaneous GCs, IgG anti-double–stranded DNA (dsDNA) autoantibodies linked to fatal immunopathology (48, 51), and B cell GC-derived lymphoma (50). Accordingly, miR-155 and miR-17-92 are upregulated in mature B cell neoplasia and GC-derived autoimmune diseases (95–97), suggesting their involvement in the enhancement of GC-derived human diseases. Other miRNAs that positively regulate GC responses and have been found to promote both autoimmunity and B-cell derived neoplasia include miR-29 and miR-21 (Table 1). Further studies are required to establish whether the switched autoantibodies generated in the context of positive GC miRNA regulator overexpression are derived from GC-derived plasma cells or are also generated from extrafollicular plasma cells.
Several miRNAs that negatively regulate the GC reaction have the opposite effect on B cell neoplasia and autoimmunity development, limiting the generation of GC-derived diseases (Table 1). miR-28 and miR-146a, well-characterized negative regulators of GC responses (33–35, 52), have both been found to exert tumor suppressor activity in B cell lymphoma development by limiting cell proliferation, promoting cell death and regulating cell signaling (33, 71, 73, 74) (Table 1). However, protection against autoimmune diseases has only been explored for miR-146a, which inhibits autoimmunity, anti-dsDNA auto-antibody production (72), and spontaneous GC reactions (52) counterregulating miR-155 targets in Tfh cells (62) and through B cell-intrinsic mechanisms, likely by targeting CD40 signaling pathway components (35). Nevertheless, GC B cell miR-146a expression needs to be tightly regulated because forced overexpression promotes a lymphoproliferative syndrome via Fas downregulation (76). Thus, both superabundant or insufficient miR-146a expression are harmful for GC homeostasis.
Overall, these studies show that regulated miRNA expression is required to ensure proper GC responses and that GC-derived dysfunctions caused by miRNA alterations frequently lead to the development of both autoimmunity and B cell neoplasia through the disruption of post-transcriptional control mechanisms required for the maintenance of GC homeostasis, regulated cell signaling, cell death and proliferation. Further studies are needed to characterize with more detail the molecular mechanisms leading to both neoplastic transformation and autoimmunity caused by miRNA-dependent GC gene expression dysregulation.
Conclusions and Perspective
Studies by many groups in the field have shown that miRNAs play a key role in GC-response regulation and are required to prevent GC-derived autoimmunity and B cell neoplasia. The description of the role of dysregulated miRNAs in mature B cell oncogenic transformation and GC-derived autoimmunity has led to the clinical use of miRNAs as disease biomarkers with prognostic and predictive value and to the identification of targets for miRNA-based therapy (97, 98). The mechanisms leading to dysregulated miRNA expression in GC cells are poorly understood, and their characterization will likely provide new opportunities for therapeutic intervention. Strategies to restore or inhibit dysregulated miRNA expression have already established the therapeutic potential of miRNA modulation in in vivo models of GC-derived B cell neoplasia and autoimmunity (33, 44, 56, 68, 99–105). Moreover, synthetic miRNA mimics or anti-miR molecules are suitable for the generation of miRNA-based drugs that can be coupled to different types of nanocarriers and conjugates for effective delivery [reviewed in (106)].
The unique molecular features of miRNAs make them attractive tools for the development of miRNA-based therapies, and miRNA-based drugs are currently being tested in clinical trials for several diseases, including different types of cancer [reviewed in (97)]. This emerging and promising field faces a number of challenges regarding the clinical translation of miRNA-based therapies for B cell neoplasia and autoimmunity. Major challenges include i) the development of cell-type specific miRNA-based drug targeting approaches to improve specificity and reduce toxicity derived from miRNA delivery to healthy cells and ii) the development of models of human mature B cell neoplasia and GC-derived autoimmunity that faithfully recapitulate human disease to improve pre-clinical testing. The rapid pace of research in the field ensures the continuing excitement and expectations in building the path from basic science to translational miRNA-mediated GC regulation.
Author Contributions
TF, IS and VY contributed substantially to the content and structure of this review. All authors contributed to the article and approved the submitted version.
Funding
Our work is supported by the Spanish Ministerio de Ciencia e Innovación (PID2019-107551RB-I00). TF is supported by a PhD fellowship from the Spanish Ministerio de Ciencia, Innovacion y Universidades (BES-2017-079759), VY by funding from the Universidad Complutense de Madrid, and IS by a student research grant from the Ministerio de Educación y Formación Profesional.
Conflict of Interest
A European Patent Application titled ‘miRNA compositions for the treatment of mature B cell neoplasms’ EP16722679, EP17382740 was filed on March 3 2017.
Acknowledgments
We thank Simon Bartlett for English editing.
References
1. Victora GD, Nussenzweig MC. Germinal Centers. Annu Rev Immunol (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
2. Mesin L, Ersching J, Victora GD. Germinal Center B Cell Dynamics. Immunity (2016) 45:471–82. doi: 10.1016/j.immuni.2016.09.001
3. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular Helper T Cells. Annu Rev Immunol (2016) 34:335–68. doi: 10.1146/annurev-immunol-041015-055605
4. Stebegg M, Kumar SD, Silva-Cayetano A, Fonseca VR, Linterman MA, Graca L. Regulation of the Germinal Center Response. Front Immunol (2018) 9:2469. doi: 10.3389/fimmu.2018.02469
5. Song S, Matthias PD. The Transcriptional Regulation of Germinal Center Formation. Front Immunol (2018) 9:2026. doi: 10.3389/fimmu.2018.02026
6. Gebert LFR, MacRae IJ. Regulation of Microrna Function in Animals. Nat Rev Mol Cell Biol (2019) 20:21–37. doi: 10.1038/s41580-018-0045-7
7. Guo WT, Wang Y. Dgcr8 Knockout Approaches to Understand Microrna Functions in Vitro and in Vivo. Cell Mol Life Sci (2019) 76:1697–711. doi: 10.1007/s00018-019-03020-9
8. Park CY, Choi YS, McManus MT. Analysis of Microrna Knockouts in Mice. Hum Mol Genet (2010) 19:R169–75. doi: 10.1093/hmg/ddq367
9. Xu S, Guo K, Zeng Q, Huo J, Lam KP. The Rnase III Enzyme Dicer is Essential for Germinal Center B-Cell Formation. Blood (2012) 119:767–76. doi: 10.1182/blood-2011-05-355412
10. Baumjohann D, Kageyama R, Clingan JM, Morar MM, Patel S, de Kouchkovsky D, et al. The Microrna Cluster Mir-17 Approximately 92 Promotes TFH Cell Differentiation and Represses Subset-Inappropriate Gene Expression. Nat Immunol (2013) 14:840–8. doi: 10.1038/ni.2642
11. Fernandez-Messina L, Rodriguez-Galan A, de Yebenes VG, Gutierrez-Vazquez C, Tenreiro S, Seabra MC, et al. Transfer of Extracellular Vesicle-Microrna Controls Germinal Center Reaction and Antibody Production. EMBO Rep (2020) 21:e48925. doi: 10.15252/embr.201948925
12. Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, et al. Regulation of the Germinal Center Response by Microrna-155. Science (2007) 316:604–8. doi: 10.1126/science.1141229
13. Tam W. Identification and Characterization of Human BIC, a Gene on Chromosome 21 That Encodes a Noncoding RNA. Gene (2001) 274:157–67. doi: 10.1016/S0378-1119(01)00612-6
14. Basso K, Sumazin P, Morozov P, Schneider C, Maute RL, Kitagawa Y, et al. Identification of the Human Mature B Cell Mirnome. Immunity (2009) 30:744–52. doi: 10.1016/j.immuni.2009.03.017
15. Kuchen S, Resch W, Yamane A, Kuo N, Li Z, Chakraborty T, et al. Regulation of Microrna Expression and Abundance During Lymphopoiesis. Immunity (2010) 32:828–39. doi: 10.1016/j.immuni.2010.05.009
16. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of Bic/Microrna-155 for Normal Immune Function. Science (2007) 316:608–11. doi: 10.1126/science.1139253
17. Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, et al. Microrna-155 Regulates the Generation of Immunoglobulin Class-Switched Plasma Cells. Immunity (2007) 27:847–59. doi: 10.1016/j.immuni.2007.10.009
18. Arbore G, Henley T, Biggins L, Andrews S, Vigorito E, Turner M, et al. Microrna-155 is Essential for the Optimal Proliferation and Survival of Plasmablast B Cells. Life Sci Alliance (2019) 2(3):e201800244. doi: 10.26508/lsa.201800244
19. Nakagawa R, Leyland R, Meyer-Hermann M, Lu D, Turner M, Arbore G, et al. Microrna-155 Controls Affinity-Based Selection by Protecting C-MYC+ B Cells From Apoptosis. J Clin Invest (2016) 126:377–88. doi: 10.1172/JCI82914
20. Lu D, Nakagawa R, Lazzaro S, Staudacher P, Abreu-Goodger C, Henley T, et al. The Mir-155-PU.1 Axis Acts on Pax5 to Enable Efficient Terminal B Cell Differentiation. J Exp Med (2014) 211:2183–98. doi: 10.1084/jem.20140338
21. Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, et al. Microrna-155 is a Negative Regulator of Activation-Induced Cytidine Deaminase. Immunity (2008) 28:621–9. doi: 10.1016/j.immuni.2008.03.015
22. Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, Robbiani DF, et al. Microrna-155 Suppresses Activation-Induced Cytidine Deaminase-Mediated Myc-Igh Translocation. Immunity (2008) 28:630–8. doi: 10.1016/j.immuni.2008.04.002
23. Phan RT, Saito M, Kitagawa Y, Means AR, Dalla-Favera R. Genotoxic Stress Regulates Expression of the Proto-Oncogene Bcl6 in Germinal Center B Cells. Nat Immunol (2007) 8:1132–9. doi: 10.1038/ni1508
24. Phan RT, Dalla-Favera R. The BCL6 Proto-Oncogene Suppresses P53 Expression in Germinal-Centre B Cells. Nature (2004) 432:635–9. doi: 10.1038/nature03147
25. Basso K, Schneider C, Shen Q, Holmes AB, Setty M, Leslie C, et al. BCL6 Positively Regulates AID and Germinal Center Gene Expression Via Repression of Mir-155. J Exp Med (2012) 209:2455–65. doi: 10.1084/jem.20121387
26. Basso K, Dalla-Favera R. Roles of BCL6 in Normal and Transformed Germinal Center B Cells. Immunol Rev (2012) 247:172–83. doi: 10.1111/j.1600-065X.2012.01112.x
27. Bouamar H, Jiang D, Wang L, Lin AP, Ortega M, Aguiar RC. Microrna 155 Control of P53 Activity is Context Dependent and Mediated by Aicda and Socs1. Mol Cell Biol (2015) 35:1329–40. doi: 10.1128/MCB.01446-14
28. de Yebenes VG, Belver L, Pisano DG, Gonzalez S, Villasante A, Croce C, et al. Mir-181b Negatively Regulates Activation-Induced Cytidine Deaminase in B Cells. J Exp Med (2008) 205:2199–206. doi: 10.1084/jem.20080579
29. de Yebenes VG, Bartolome-Izquierdo N, Nogales-Cadenas R, Perez-Duran P, Mur SM, Martinez N, et al. Mir-217 is an Oncogene That Enhances the Germinal Center Reaction. Blood (2014) 124:229–39. doi: 10.1182/blood-2013-12-543611
30. Porstner M, Winkelmann R, Daum P, Schmid J, Pracht K, Corte-Real J, et al. Mir-148a Promotes Plasma Cell Differentiation and Targets the Germinal Center Transcription Factors Mitf and Bach2. Eur J Immunol (2015) 45:1206–15. doi: 10.1002/eji.201444637
31. Iqbal J, Shen Y, Huang X, Liu Y, Wake L, Liu C, et al. Global Microrna Expression Profiling Uncovers Molecular Markers for Classification and Prognosis in Aggressive B-Cell Lymphoma. Blood (2015) 125:1137–45. doi: 10.1182/blood-2014-04-566778
32. Schneider C, Setty M, Holmes AB, Maute RL, Leslie CS, Mussolin L, et al. Microrna 28 Controls Cell Proliferation and is Down-Regulated in B-Cell Lymphomas. Proc Natl Acad Sci USA (2014) 111:8185–90. doi: 10.1073/pnas.1322466111
33. Bartolome-Izquierdo N, de Yebenes VG, Alvarez-Prado AF, Mur SM, Lopez Del Olmo JA, Roa S, et al. Mir-28 Regulates the Germinal Center Reaction and Blocks Tumor Growth in Preclinical Models of Non-Hodgkin Lymphoma. Blood (2017) 129:2408–19. doi: 10.1182/blood-2016-08-731166
34. Pratama A, Srivastava M, Williams NJ, Papa I, Lee SK, Dinh XT, et al. Microrna-146a Regulates ICOS-ICOSL Signalling to Limit Accumulation of T Follicular Helper Cells and Germinal Centres. Nat Commun (2015) 6:6436. doi: 10.1038/ncomms7436
35. Cho S, Lee HM, Yu IS, Choi YS, Huang HY, Hashemifar SS, et al. Differential Cell-Intrinsic Regulations of Germinal Center B and T Cells by Mir-146a and Mir-146b. Nat Commun (2018) 9:2757. doi: 10.1038/s41467-018-05196-3
36. Malumbres R, Sarosiek KA, Cubedo E, Ruiz JW, Jiang X, Gascoyne RD, et al. Differentiation Stage-Specific Expression of Micrornas in B Lymphocytes and Diffuse Large B-Cell Lymphomas. Blood (2009) 113:3754–64. doi: 10.1182/blood-2008-10-184077
37. Gururajan M, Haga CL, Das S, Leu CM, Hodson D, Josson S, et al. Microrna 125b Inhibition of B Cell Differentiation in Germinal Centers. Int Immunol (2010) 22:583–92. doi: 10.1093/intimm/dxq042
38. Tsai DY, Hung KH, Lin IY, Su ST, Wu SY, Chung CH, et al. Uncovering Microrna Regulatory Hubs That Modulate Plasma Cell Differentiation. Sci Rep (2015) 5:17957. doi: 10.1038/srep17957
39. Morelli E, Leone E, Cantafio ME, Di Martino MT, Amodio N, Biamonte L, et al. Selective Targeting of IRF4 by Synthetic Microrna-125b-5p Mimics Induces Anti-Multiple Myeloma Activity in Vitro and in Vivo. Leukemia (2015) 29:2173–83. doi: 10.1038/leu.2015.124
40. Nie K, Gomez M, Landgraf P, Garcia JF, Liu Y, Tan LH, et al. Microrna-Mediated Down-Regulation of PRDM1/Blimp-1 in Hodgkin/Reed-Sternberg Cells: A Potential Pathogenetic Lesion in HODGKIN Lymphomas. Am J Pathol (2008) 173:242–52. doi: 10.2353/ajpath.2008.080009
41. Nie K, Zhang T, Allawi H, Gomez M, Liu Y, Chadburn A, et al. Epigenetic Down-Regulation of the Tumor Suppressor Gene PRDM1/Blimp-1 in Diffuse Large B Cell Lymphomas: A Potential Role of the Microrna Let-7. Am J Pathol (2010) 177:1470–9. doi: 10.2353/ajpath.2010.091291
42. Zhang J, Jima DD, Jacobs C, Fischer R, Gottwein E, Huang G, et al. Patterns of Microrna Expression Characterize Stages of Human B-Cell Differentiation. Blood (2009) 113:4586–94. doi: 10.1182/blood-2008-09-178186
43. Huang X, Zhou X, Wang Z, Li F, Liu F, Zhong L, et al. CD99 Triggers Upregulation of Mir-9-Modulated PRDM1/BLIMP1 in Hodgkin/Reed-Sternberg Cells and Induces Redifferentiation. Int J Cancer (2012) 131:E382–94. doi: 10.1002/ijc.26503
44. White CA, Pone EJ, Lam T, Tat C, Hayama KL, Li G, et al. Histone Deacetylase Inhibitors Upregulate B Cell Micrornas That Silence AID and Blimp-1 Expression for Epigenetic Modulation of Antibody and Autoantibody Responses. J Immunol (2014) 193:5933–50. doi: 10.4049/jimmunol.1401702
45. Shen T, Sanchez HN, Zan H, Casali P. Genome-Wide Analysis Reveals Selective Modulation of Micrornas and Mrnas by Histone Deacetylase Inhibitor in B Cells Induced to Undergo Class-Switch DNA Recombination and Plasma Cell Differentiation. Front Immunol (2015) 6:627. doi: 10.3389/fimmu.2015.00627
46. Moroney JB, Chupp DP, Xu Z, Zan H, Casali P. Epigenetics of the Antibody and Autoantibody Response. Curr Opin Immunol (2020) 67:75–86. doi: 10.1016/j.coi.2020.09.004
47. Moroney JB, Vasudev A, Pertsemlidis A, Zan H, Casali P. Integrative Transcriptome and Chromatin Landscape Analysis Reveals Distinct Epigenetic Regulations in Human Memory B Cells. Nat Commun (2020) 11:5435. doi: 10.1038/s41467-020-19242-6
48. Kang SG, Liu WH, Lu P, Jin HY, Lim HW, Shepherd J, et al. Micrornas of the Mir-17 Approximately 92 Family are Critical Regulators of T(FH) Differentiation. Nat Immunol (2013) 14:849–57. doi: 10.1038/ni.2648
49. Wu T, Wieland A, Lee J, Hale JS, Han JH, Xu X, et al. Cutting Edge: Mir-17-92 is Required for Both CD4 Th1 and T Follicular Helper Cell Responses During Viral Infection. J Immunol (2015) 195:2515–9. doi: 10.4049/jimmunol.1500317
50. Jin HY, Oda H, Lai M, Skalsky RL, Bethel K, Shepherd J, et al. Microrna-17~92 Plays a Causative Role in Lymphomagenesis by Coordinating Multiple Oncogenic Pathways. EMBO J (2013) 32:2377–91. doi: 10.1038/emboj.2013.178
51. Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, et al. Lymphoproliferative Disease and Autoimmunity in Mice With Increased Mir-17-92 Expression in Lymphocytes. Nat Immunol (2008) 9:405–14. doi: 10.1038/ni1575
52. Hu R, Kagele DA, Huffaker TB, Runtsch MC, Alexander M, Liu J, et al. Mir-155 Promotes T Follicular Helper Cell Accumulation During Chronic, Low-Grade Inflammation. Immunity (2014) 41:605–19. doi: 10.1016/j.immuni.2014.09.015
53. Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, et al. Pre-B Cell Proliferation and Lymphoblastic Leukemia/High-Grade Lymphoma in E(Mu)-Mir155 Transgenic Mice. Proc Natl Acad Sci USA (2006) 103:7024–9. doi: 10.1073/pnas.0602266103
54. Costinean S, Sandhu SK, Pedersen IM, Tili E, Trotta R, Perrotti D, et al. Src Homology 2 Domain-Containing Inositol-5-Phosphatase and CCAAT Enhancer-Binding Protein Beta are Targeted by Mir-155 in B Cells of Emicro-Mir-155 Transgenic Mice. Blood (2009) 114:1374–82. doi: 10.1182/blood-2009-05-220814
55. Pedersen IM, Otero D, Kao E, Miletic AV, Hother C, Ralfkiaer E, et al. Onco-Mir-155 Targets SHIP1 to Promote Tnfalpha-Dependent Growth of B Cell Lymphomas. EMBO Mol Med (2009) 1:288–95. doi: 10.1002/emmm.200900028
56. Babar IA, Cheng CJ, Booth CJ, Liang X, Weidhaas JB, Saltzman WM, et al. Nanoparticle-Based Therapy in an in Vivo Microrna-155 (Mir-155)-Dependent Mouse Model of Lymphoma. Proc Natl Acad Sci USA (2012) 109:E1695–704. doi: 10.1073/pnas.1201516109
57. Kurowska-Stolarska M, Alivernini S, Ballantine LE, Asquith DL, Millar NL, Gilchrist DS, et al. Microrna-155 as a Proinflammatory Regulator in Clinical and Experimental Arthritis. Proc Natl Acad Sci USA (2011) 108:11193–8. doi: 10.1073/pnas.1019536108
58. Thai TH, Patterson HC, Pham DH, Kis-Toth K, Kaminski DA, Tsokos GC. Deletion of Microrna-155 Reduces Autoantibody Responses and Alleviates Lupus-Like Disease in the Fas(Lpr) Mouse. Proc Natl Acad Sci USA (2013) 110:20194–9. doi: 10.1073/pnas.1317632110
59. Xin Q, Li J, Dang J, Bian X, Shan S, Yuan J, et al. Mir-155 Deficiency Ameliorates Autoimmune Inflammation of Systemic Lupus Erythematosus by Targeting S1pr1 in Faslpr/Lpr Mice. J Immunol (2015) 194:5437–45. doi: 10.4049/jimmunol.1403028
60. Alivernini S, Kurowska-Stolarska M, Tolusso B, Benvenuto R, Elmesmari A, Canestri S, et al. Microrna-155 Influences B-Cell Function Through PU.1 in Rheumatoid Arthritis. Nat Commun (2016) 7:12970. doi: 10.1038/ncomms12970
61. Liu WH, Kang SG, Huang Z, Wu CJ, Jin HY, Maine CJ, et al. A Mir-155-Peli1-C-Rel Pathway Controls the Generation and Function of T Follicular Helper Cells. J Exp Med (2016) 213:1901–19. doi: 10.1084/jem.20160204
62. Chen J, Tian J, Tang X, Rui K, Ma J, Mao C, et al. Mir-346 Regulates CD4(+)CXCR5(+) T Cells in the Pathogenesis of Graves’ Disease. Endocrine (2015) 49:752–60. doi: 10.1007/s12020-015-0546-5
63. van Nieuwenhuijze A, Dooley J, Humblet-Baron S, Sreenivasan J, Koenders M, Schlenner SM, et al. Defective Germinal Center B-Cell Response and Reduced Arthritic Pathology in Microrna-29a-Deficient Mice. Cell Mol Life Sci (2017) 74:2095–106. doi: 10.1007/s00018-017-2456-6
64. Santanam U, Zanesi N, Efanov A, Costinean S, Palamarchuk A, Hagan JP, et al. Chronic Lymphocytic Leukemia Modeled in Mouse by Targeted Mir-29 Expression. Proc Natl Acad Sci USA (2010) 107:12210–5. doi: 10.1073/pnas.1007186107
65. Yan Y, Deng X, Ning X, Li F, Cao J. Pathogenic Mechanism of Mir-21 in Autoimmune Lymphoid Hyperplasia Syndrome. Oncol Lett (2017) 13:4734–40. doi: 10.3892/ol.2017.6039
66. Barnes NA, Stephenson S, Cocco M, Tooze RM, Doody GM. BLIMP-1 and STAT3 Counterregulate Microrna-21 During Plasma Cell Differentiation. J Immunol (2012) 189:253–60. doi: 10.4049/jimmunol.1101563
67. Medina PP, Nolde M, Slack FJ. Oncomir Addiction in an in Vivo Model of Microrna-21-Induced Pre-B-Cell Lymphoma. Nature (2010) 467:86–90. doi: 10.1038/nature09284
68. Garchow BG, Bartulos Encinas O, Leung YT, Tsao PY, Eisenberg RA, Caricchio R, et al. Silencing of Microrna-21 in Vivo Ameliorates Autoimmune Splenomegaly in Lupus Mice. EMBO Mol Med (2011) 3:605–15. doi: 10.1002/emmm.201100171
69. Gu L, Song G, Chen L, Nie Z, He B, Pan Y, et al. Inhibition of Mir-21 Induces Biological and Behavioral Alterations in Diffuse Large B-Cell Lymphoma. Acta Haematol (2013) 130:87–94. doi: 10.1159/000346441
70. Go H, Jang JY, Kim PJ, Kim YG, Nam SJ, Paik JH, et al. Microrna-21 Plays an Oncogenic Role by Targeting FOXO1 and Activating the PI3K/AKT Pathway in Diffuse Large B-Cell Lymphoma. Oncotarget (2015) 6:15035–49. doi: 10.18632/oncotarget.3729
71. Zhao JL, Rao DS, Boldin MP, Taganov KD, O’Connell RM, Baltimore D. NF-Kappab Dysregulation in Microrna-146a-Deficient Mice Drives the Development of Myeloid Malignancies. Proc Natl Acad Sci USA (2011) 108:9184–9. doi: 10.1073/pnas.1105398108
72. Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, et al. Mir-146a is a Significant Brake on Autoimmunity, Myeloproliferation, and Cancer in Mice. J Exp Med (2011) 208:1189–201. doi: 10.1084/jem.20101823
73. Mitsumura T, Ito Y, Chiba T, Matsushima T, Kurimoto R, Tanaka Y, et al. Ablation of Mir-146b in Mice Causes Hematopoietic Malignancy. Blood Adv (2018) 2:3483–91. doi: 10.1182/bloodadvances.2018017954
74. Contreras JR, Palanichamy JK, Tran TM, Fernando TR, Rodriguez-Malave NI, Goswami N, et al. Microrna-146a Modulates B-Cell Oncogenesis by Regulating Egr1. Oncotarget (2015) 6:11023–37. doi: 10.18632/oncotarget.3433
75. Wang H, Li X, Li T, Wang L, Wu X, Liu J, et al. Multiple Roles of Microrna-146a in Immune Responses and Hepatocellular Carcinoma. Oncol Lett (2019) 18:5033–42. doi: 10.3892/ol.2019.10862
76. Guo Q, Zhang J, Li J, Zou L, Zhang J, Xie Z, et al. Forced Mir-146a Expression Causes Autoimmune Lymphoproliferative Syndrome in Mice Via Downregulation of Fas in Germinal Center B Cells. Blood (2013) 121:4875–83. doi: 10.1182/blood-2012-08-452425
77. Xu H, Li X, Liu D, Li J, Zhang X, Chen X, et al. Follicular T-Helper Cell Recruitment Governed by Bystander B Cells and ICOS-Driven Motility. Nature (2013) 496:523–7. doi: 10.1038/nature12058
78. Yu D, Tan AH, Hu X, Athanasopoulos V, Simpson N, Silva DG, et al. Roquin Represses Autoimmunity by Limiting Inducible T-Cell Co-Stimulator Messenger RNA. Nature (2007) 450:299–303. doi: 10.1038/nature06253
79. Athanasopoulos V, Barker A, Yu D, Tan AH, Srivastava M, Contreras N, et al. The ROQUIN Family of Proteins Localizes to Stress Granules Via the ROQ Domain and Binds Target Mrnas. FEBS J (2010) 277:2109–27. doi: 10.1111/j.1742-4658.2010.07628.x
80. Gigoux M, Shang J, Pak Y, Xu M, Choe J, Mak TW, et al. Inducible Costimulator Promotes Helper T-Cell Differentiation Through Phosphoinositide 3-Kinase. Proc Natl Acad Sci USA (2009) 106:20371–6. doi: 10.1073/pnas.0911573106
81. Rolf J, Bell SE, Kovesdi D, Janas ML, Soond DR, Webb LM, et al. Phosphoinositide 3-Kinase Activity in T Cells Regulates the Magnitude of the Germinal Center Reaction. J Immunol (2010) 185:4042–52. doi: 10.4049/jimmunol.1001730
82. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The Transcriptional Repressor Bcl-6 Directs T Follicular Helper Cell Lineage Commitment. Immunity (2009) 31:457–68. doi: 10.1016/j.immuni.2009.07.002
83. Essig K, Hu D, Guimaraes JC, Alterauge D, Edelmann S, Raj T, et al. Roquin Suppresses the PI3K-Mtor Signaling Pathway to Inhibit T Helper Cell Differentiation and Conversion of Treg to Tfr Cells. Immunity (2017) 47:1067–82.e12. doi: 10.1016/j.immuni.2017.11.008
84. Ripamonti A, Provasi E, Lorenzo M, De Simone M, Ranzani V, Vangelisti S, et al. Repression of Mir-31 by BCL6 Stabilizes the Helper Function of Human Follicular Helper T Cells. Proc Natl Acad Sci USA (2017) 114:12797–802. doi: 10.1073/pnas.1705364114
85. Deenick EK, Ma CS. The Regulation and Role of T Follicular Helper Cells in Immunity. Immunology (2011) 134:361–7. doi: 10.1111/j.1365-2567.2011.03487.x
86. Cannons JL, Qi H, Lu KT, Dutta M, Gomez-Rodriguez J, Cheng J, et al. Optimal Germinal Center Responses Require a Multistage T Cell:B Cell Adhesion Process Involving Integrins, SLAM-Associated Protein, and CD84. Immunity (2010) 32:253–65. doi: 10.1016/j.immuni.2010.01.010
87. Kashiwakuma D, Suto A, Hiramatsu Y, Ikeda K, Takatori H, Suzuki K, et al. B and T Lymphocyte Attenuator Suppresses IL-21 Production From Follicular Th Cells and Subsequent Humoral Immune Responses. J Immunol (2010) 185:2730–6. doi: 10.4049/jimmunol.0903839
88. Takahashi H, Kanno T, Nakayamada S, Hirahara K, Sciume G, Muljo SA, et al. TGF-Beta and Retinoic Acid Induce the Microrna Mir-10a, Which Targets Bcl-6 and Constrains the Plasticity of Helper T Cells. Nat Immunol (2012) 13:587–95. doi: 10.1038/ni.2286
89. Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 Interactions At the Interface of Tolerance and Immunity. Annu Rev Immunol (2004) 22:307–28. doi: 10.1146/annurev.immunol.22.012703.104533
90. Perez-Melgosa M, Hollenbaugh D, Wilson CB. Cutting Edge: CD40 Ligand is a Limiting Factor in the Humoral Response to T Cell-Dependent Antigens. J Immunol (1999) 163:1123–7.
91. Vinuesa CG, Sanz I, Cook MC. Dysregulation of Germinal Centres in Autoimmune Disease. Nat Rev Immunol (2009) 9:845–57. doi: 10.1038/nri2637
92. Elsner RA, Shlomchik MJ. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity (2020) 53:1136–50. doi: 10.1016/j.immuni.2020.11.006
93. Mlynarczyk C, Fontan L, Melnick A. Germinal Center-Derived Lymphomas: The Darkest Side of Humoral Immunity. Immunol Rev (2019) 288:214–39. doi: 10.1111/imr.12755
94. Wen Z, Xu L, Chen X, Xu W, Yin Z, Gao X, et al. Autoantibody Induction by DNA-Containing Immune Complexes Requires HMGB1 With the TLR2/Microrna-155 Pathway. J Immunol (2013) 190:5411–22. doi: 10.4049/jimmunol.1203301
95. Kuo G, Wu CY, Yang HY. Mir-17-92 Cluster and Immunity. J Formos Med Assoc (2019) 118:2–6. doi: 10.1016/j.jfma.2018.04.013
96. Leng RX, Pan HF, Qin WZ, Chen GM, Ye DQ. Role of Microrna-155 in Autoimmunity. Cytokine Growth Factor Rev (2011) 22:141–7. doi: 10.1016/j.cytogfr.2011.05.002
97. Fuertes T, Ramiro AR, de Yebenes VG. Mirna-Based Therapies in B Cell Non-Hodgkin Lymphoma. Trends Immunol (2020) 41:932–47. doi: 10.1016/j.it.2020.08.006
98. Long H, Wang X, Chen Y, Wang L, Zhao M, Lu Q. Dysregulation of Micrornas in Autoimmune Diseases: Pathogenesis, Biomarkers and Potential Therapeutic Targets. Cancer Lett (2018) 428:90–103. doi: 10.1016/j.canlet.2018.04.016
99. Zhang Y, Roccaro AM, Rombaoa C, Flores L, Obad S, Fernandes SM, et al. LNA-Mediated Anti-Mir-155 Silencing in Low-Grade B-Cell Lymphomas. Blood (2012) 120:1678–86. doi: 10.1182/blood-2012-02-410647
100. Zhu FQ, Zeng L, Tang N, Tang YP, Zhou BP, Li FF, et al. Microrna-155 Downregulation Promotes Cell Cycle Arrest and Apoptosis in Diffuse Large B-Cell Lymphoma. Oncol Res (2016) 24:415–27. doi: 10.3727/096504016X14685034103473
101. Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C, et al. Microrna Silencing for Cancer Therapy Targeted to the Tumour Microenvironment. Nature (2015) 518:107–10. doi: 10.1038/nature13905
102. Leone E, Morelli E, Di Martino MT, Amodio N, Foresta U, Gulla A, et al. Targeting Mir-21 Inhibits in Vitro and in Vivo Multiple Myeloma Cell Growth. Clin Cancer Res (2013) 19:2096–106. doi: 10.1158/1078-0432.CCR-12-3325
103. Su YL, Wang X, Mann M, Adamus TP, Wang D, Moreira DF, et al. Myeloid Cell-Targeted Mir-146a Mimic Inhibits NF-Kappab-Driven Inflammation and Leukemia Progression in Vivo. Blood (2020) 135:167–80. doi: 10.1182/blood.2019002045
104. Zhang J, Jia G, Liu Q, Hu J, Yan M, Yang B, et al. Silencing Mir-146a Influences B Cells and Ameliorates Experimental Autoimmune Myasthenia Gravis. Immunology (2015) 144:56–67. doi: 10.1111/imm.12347
105. Casali P, Shen T, Xu Y, Qiu Z, Chupp DP, Im J, et al. Estrogen Reverses HDAC Inhibitor-Mediated Repression of Aicda and Class-Switching in Antibody and Autoantibody Responses by Downregulation of Mir-26a. Front Immunol (2020) 11:491. doi: 10.3389/fimmu.2020.00491
Keywords: microRNA, germinal center, antibodies, autoimmunity, neoplasia
Citation: Fuertes T, Salgado I and de Yébenes VG (2021) microRNA Fine-Tuning of the Germinal Center Response. Front. Immunol. 12:660450. doi: 10.3389/fimmu.2021.660450
Received: 29 January 2021; Accepted: 31 March 2021;
Published: 19 April 2021.
Edited by:
Thierry Fest, University of Rennes 1, FranceReviewed by:
Paolo Casali, University of Texas Health Science Center at San Antonio, United StatesMasaki Hikida, Akita University, Japan
Copyright © 2021 Fuertes, Salgado and de Yébenes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Virginia G. de Yébenes, dmdhcmNpYXlAdWNtLmVz