
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 30 March 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.660194
This article is part of the Research TopicFunction and Dysfunction of Complement Factor HView all 13 articles
 Felix Poppelaars1
Felix Poppelaars1 Elena Goicoechea de Jorge2
Elena Goicoechea de Jorge2 Ilse Jongerius3,4
Ilse Jongerius3,4 Antje J. Baeumner5
Antje J. Baeumner5 Mark-Steven Steiner6
Mark-Steven Steiner6 Mihály Józsi7,8
Mihály Józsi7,8 Erik J. M. Toonen9
Erik J. M. Toonen9 Diana Pauly10,11* and the SciFiMed consortium
Diana Pauly10,11* and the SciFiMed consortiumInflammation is a common denominator of diseases. The complement system, an intrinsic part of the innate immune system, is a key driver of inflammation in numerous disorders. Recently, a family of proteins has been suggested to be of vital importance in conditions characterized by complement dysregulation: the human Factor H (FH) family. This group of proteins consists of FH, Factor H-like protein 1 and five Factor H-related proteins. The FH family has been linked to infectious, vascular, eye, kidney and autoimmune diseases. In contrast to FH, the functions of the other highly homologous proteins are largely unknown and, hence, their role in the different disease-specific pathogenic mechanisms remains elusive. In this perspective review, we address the major challenges ahead in this emerging area, including 1) the controversies about the functional roles of the FH protein family, 2) the discrepancies in quantification of the FH protein family, 3) the unmet needs for validated tools and 4) limitations of animal models. Next, we also discuss the opportunities that exist for the immunology community. A strong multidisciplinary approach is required to solve these obstacles and is only possible through interdisciplinary collaboration between biologists, chemists, geneticists and physicians. We position this review in light of our own perspective, as principal investigators of the SciFiMed Consortium, a consortium aiming to create a comprehensive analytical system for the quantitative and functional assessment of the entire FH protein family.
“When you can measure what you are speaking about, and express it in numbers, you know something about it”
– William Thomson, 1st Baron Kelvin
The complement system forms a major arm of innate immunity and is of importance to fight invading pathogens (1). It consists of over 50 proteins that activate each other in a fixed order via three distinct pathways; the classical (CP), lectin (LP) and alternative pathway (AP), which all lead to cleavage of C3 and C5. This results in labeling of pathogens with C3b, attraction of immune cells via the anaphylatoxins C3a and C5a, and formation of the membrane attack complex [reviewed in (2)]. While the complement system is traditionally seen as a plasma system, recent studies also describe its importance locally, perhaps even inside cells (3). In health, the complement system is tightly regulated to prevent unwanted activation, inflammation and tissue damage. It has long been known that complement dysregulation contributes to various inflammatory and autoimmune diseases (4–6). A number of membrane-bound and fluid phase regulators ensure that the complement system is well-controlled (reviewed in (7)). Here, we will focus on the main regulator of the alternative pathway, namely Factor H (FH). FH can distinguish between self and non-self, and prevents complement activation both on cellular surfaces and in the circulation (8). More specifically, FH can function as a co-factor for Factor I (FI)-mediated proteolysis of C3b into iC3b, a molecule that cannot further propagate pathway activation. FH can also compete with Factor B (FB) to inhibit formation of the C3(H2O)B fluid phase tickover complex. In addition, FH promotes the decay of existing C3bBb-complexes (i.e., the C3-convertase), as well as the C4bC2aC3b and C3bBbC3b-complexes (i.e., the C5 convertases). FH is composed of 20 repetitive units, called complement control protein (CCP) domains, in a “beads on a string” configuration. The CCPs are ~65 amino acids in length and contain two invariant disulfide bonds. The FH N-terminal (CCPs 1–4) is important for decay accelerating activity and co-factor activity, while the internal region (CCPs 6–8) and the C-terminal (CCPs 19–20) are needed for host/ligand recognition and thus also for complement regulation on host surfaces (9–11). The human gene for FH is located on chromosome 1 within the Regulators of Complement Activation (RCA) gene cluster. The RCA gene cluster contains more than sixty genes and includes a ~700 Kb region in which FH as well as the Factor H-Related (FHR) proteins are encoded (described below). The complement FHR genes (CFHR) contain several repeating regions believed to have resulted from large genomic duplication events leading to the production of FHR proteins with partly similar domains to FH (12).
FH like-1 (FHL-1) is an alternatively-spliced version of FH and shares the first 7 CCP domains of FH before terminating with a unique four amino acid C-terminal tail. FHL-1 contains the C3b binding and regulatory domains of FH and thereby retains the regulatory function of FH. Also, since FHL-1 contains the CCP domains 6–7 of FH, it is assumed that FHL-1 shares some of the FH ligands and the ability to regulate complement on certain surfaces (13–15). Indeed, FHL-1 has been shown to bind similar ligands as FH such as C-reactive protein (CRP), pentraxin 3, heparin and malondialdehyde epitopes (16–18). Nevertheless, clear differences exist between these two proteins such as the extra binding domains in FH (CCP 8–20), the distinctive three-dimensional conformation of both proteins, and the unique C-terminus of FHL-1. This suggests that FH and FHL-1 also bind to distinct ligands expressed in certain tissues. Moreover, it has been implied that FHL-1 has a local and tissue specific role instead of a systemic function like FH (13).
Humans also have five FHR proteins; FHR-1, FHR-2, FHR-3, FHR-4 and FHR-5, whose functions are poorly characterized (described in more detail in (8)). Yet, their importance is shown by the causal link between genetic alterations in CFHR and various diseases (i.e., IgA nephropathy (IgAN) (19–23), age-related macular degeneration (AMD) (24–28), invasive meningococcal disease (29–31), atypical hemolytic uremic syndrome (aHUS) (32) and C3 glomerulopathy (C3G) (33). All FHR proteins share a high degree of similarity with FH in their N-terminus (varying between 36 and 94%) and their C-terminus (varying between 36 and 100%) (34). Notably, the N-terminus of the FHR proteins resembles CCPs 6–8 of FH, while the C-terminus is similar to CCPs 19–20 of FH. FHR-5 is an exception to this, since FHR-5 shares homology to CCPs 6–7 as well as CCPs 10–14 and CCPs 19–20 of FH. The homology of the FHR proteins to the surface recognition domains of FH enables these proteins to bind similar ligands on surfaces including heparin and C3 activation fragments such as C3b or C3d (32). However, since all FHR proteins lack the domains of FH responsible for the regulatory activity, the FHR proteins will, unlike FH, most likely not provide protection to these surfaces against complement attack. The current belief is, therefore, that the FHR proteins antagonize the ability of FH to regulate complement activation (35). Furthermore, some FHR proteins can form dimers. FHR-1, FHR-2, and FHR-5 contain a dimerization motif in their N-terminal domains, while in FHR-3 and FHR-4 this motif is missing. This would enable FHR-1, FHR-2, and FHR-5 to form both homodimers and heterodimers. Accordingly, structural and sequence analyses suggested that, in addition to homodimers, FHR-1 can form heterodimers with FHR-2 and FHR-5, while FHR-2/FHR-5 heterodimers would only be formed in sera partially or totally deficient in FHR-1 (36). However, recently, another study suggested that only four dimers occur in circulation: homodimers of FHR-1, FHR-2, and FHR-5, as well as FHR-1/FHR-2 heterodimers (37). Further studies are therefore needed to confirm the exact nature of the dimer composition, as well as the precise function of these dimers.
In hindsight, the earliest publication about the FH family was in 1965, when Nilsson and Muller-Eberhard initially isolated FH from human serum and identified this novel protein as β1H globulin (38). Yet, it wasn’t until 1976 that two groups independently of each other discovered the C3b inhibitory activity of FH as well as its regulatory activity on the C3-convertase (39–41). In 1983, regulation of C5 convertases by FH was first described (42). Finally, in 1988, the genetic code of FH and its amino acid sequence were identified (43). This discovery was essential to uncover the structure of FH as discussed above. At around the same time of this breakthrough, Schwaeble et al. demonstrated the expression of an additional smaller truncated form of FH in the human liver, which we now know as FHL-1 (44). In 1989, the same group demonstrated that FHL-1 had FI-cofactor activity (45). In the end, in 1991, FH and FHL-1 were shown to be derived from the same gene by a process of alternative splicing (46, 47).
One of the earliest mentions on any of the FHR proteins was in a paper describing the isolation of murine FHR proteins by Vik et al. in 1990 (48). Due to the extensive number of large genomic duplications between the exons of CFH and the CFHR genes, determining the genomic positions of the human CFHR genes was challenging and was performed throughout the early to mid-1990’s. In 1991 and 1992, mRNA transcripts encoding for FHR-1, FHR-2 and FHR-3 were revealed (47, 49–51). Expression of FHRs on protein level were characterized and described soon after (44, 50, 52). The position of CFHR2 was the first of the FHR proteins to be determined when it was identified within the region between CFH and Factor XIII (53). In the next years, the other three CFHR genes were mapped within the RCA cluster between CFH and CFHR2 (54). However, due to the high sequence resemblances between these genes, the determination of their exact positioning was not possible. The last CFHR gene discovered was for FHR-5, which was first described at protein level in 2001 in studies of immune-complex-mediated kidney diseases (55, 56). Finally, the genetic location of CFHR5 was determined using fluorescence in situ hybridization (FISH), radiation hybrid mapping and BLAST alignment (57). Ultimately, it wasn’t until 2002 that the genomic segment containing the CFH and CFHR gene family was confirmed and to have the gene positions from centromere to telomere: CFH, CFHR3, CFHR1, CFHR4, CFHR2, CFHR5 (a schematic overview of the genomic organization of the CFH gene family is provided in (35)). Furthermore, nowadays other forms of the FH protein family have also been described, namely the alternatively spliced forms of CFHR4 named FHR-4A and FHR-4B leading to a total of 8 proteins that are encoded by the human CFH and CFHR gene family (excluding the different glycosylation variants as well as the homo- and heterodimers) (an outline of the protein structure of the FH family is provided in (8, 35, 58).
In recent years, numerous conditions have been associated with mutations or polymorphisms in the CFH gene family [an overview is provided in (32, 35, 59)]. These findings support the notion that complement dysregulation due to alterations in the FH family are a unifying pathogenic feature of various pathologies. Deciphering the pathogenic mechanism by which this protein family leads to disease is crucial for establishing the right diagnosis and therapeutic interventions. Despite the association of genetic variants in the CFH gene family with diseases, little is known regarding the biological processes leading to inflammation and tissue injury. Understanding the molecular mechanisms behind these genetic associations is challenging and represents an area of intense research. Notably, the disease-associated genetic variants in the CFHR1-5 are particularly difficult to interpret due to the lack of knowledge regarding the biological role of the FHRs. We and others have shown the existence of genotype-phenotype correlations between gene variants in the CFH-CFHR1-5 and complement-mediated diseases demonstrating that, although the same genes associate with various diseases, the molecular mechanisms behind these associations are specific of each condition (15, 59–61). In this context, the studies performed with FH, the best-known member of the family, were the first ones to illustrate such genotype–phenotype correlations. Mutations causing plasma FH deficiencies were amongst the first CFH alterations described. When these CFH mutations are present in homozygosis or compound heterozygosis they lead to complete FH deficiency, which cause massive complement activation in fluid phase, and are commonly associated with C3G, a heterogeneous histopathological entity characterized by glomerular C3 accumulation (62). However, when the null CFH alleles are in heterozygosis they only lead to partial FH deficiencies, and are equally associated with C3G as well as other diseases such as aHUS, AMD and IgAN. In this scenario, the combination with other genetic, acquired and/or environmental risk factors that are specific for each disease determines the final phenotype outcome. Interestingly, missense mutations within the C-terminus of FH are prototypical of aHUS and cause an inappropriate regulation of complement on endothelial surfaces leading to tissue damage, but without altering complement regulation in the fluid phase (63–66).
In addition to CFH, strong associations between genetic modifications in CFHR1-5 and pathologic outcome have also emerged (32). Amongst the disease-associated CFHR1-5 variants, genomic rearrangements leading to deletions, duplications, or hybrid genes are the most remarkable and informative ones (59). Perhaps, the most extensively studied is the deletion of CFHR3 and CFHR1, which has a variable allele frequency between 0–55% in various ethnic groups (67). Moreover, this common gene variant is associated with protection against the development of AMD and IgAN, while it increases the susceptibility for aHUS (due to anti-FH autoantibodies) and systemic lupus erythematosus (SLE) (21, 24, 68, 69). These different disease associations highlight the relevance of the context in defining the effect of the FHR-1 and FHR-3 deficiency, illustrating those situations where the promotion of complement activation by the FHR proteins may be detrimental (i.e., Bruch´s membrane and mesangium) or where it may be beneficial (i.e., apoptotic cells). Another captivating type of a genomic rearrangement of the CFHRs is the duplication of the dimerization domains in FHR-1, FHR-2 or FHR-5. These CFHR variations are exclusively associated with C3G (33, 70–73). In this case, the resulting proteins are gain-of-function mutants that present an increased avidity for their ligands (36). Hence, these mutant proteins are postulated to out–compete the binding of FH to C3b deposited on surfaces and impair complement regulation more efficiently than the corresponding wild-type proteins (32, 36).
Besides genetic modifications, systemic levels of FHRs may also be crucial in disease processes. In anti-neutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis, increased systemic levels of FHR-1 were found compared to healthy controls (74). Additionally, FHR-1 levels were shown to weakly correlate with lower renal function and the percentage of relapses increased with growing FHR-1 concentrations. In IgAN, two groups independently of each other reported that plasma levels of FHR-1 and the FHR-1/FH-ratio are elevated in these patients and associate with progressive disease (75, 76). In contrast, plasma FHR-5 and the FHR-5/FH-ratio were not associated with progressive disease (76). However, higher FHR-5 levels in IgAN did associate with histological disease severity. In aHUS, plasma FHR-3 levels were demonstrated to be elevated compared to controls even when taking the CFHR3 genotype into account (77). Also, the aHUS-risk CFH–CFHR3–CFHR1 haplotype was shown to be associated with increased plasma levels of FHR-3, suggesting that an imbalance between FH and FHR-3 concentration may predispose individuals to aHUS. Recently, increased systemic FHR-4 levels were shown to be strongly associated with AMD (28). This is the first time that FHR-4 has been associated with a disease. A genome-wide association study revealed that an intronic variant in CFHR4 correlated with systemic complement activation in AMD patients and associated with an increased risk of AMD development (26). A follow-up study demonstrated that the CFHR4 variant was associated with higher levels of FHR-4 (28). Moreover, circulating FHR-4 levels and the FHR-4/FH-ratio were demonstrated to be elevated in AMD compared to controls, and the protein co-localized with complement activation products in choriocapillaris beneath the retina.
In addition to autoimmune diseases, the FH family has also been known to be involved in infections (78). Pathogens evade complement attack by recruiting complement regulators such as FH onto their surface, and it is suggested that the FHR proteins have evolved as decoys to reduce the amount of FH that is acquired by the microbes (35, 79). An illustrative example of this situation was described by Caesar et al., who showed that FHR-3 competes with FH for the binding of a FH-binding protein on Neisseria meningitidis, acting as a FH antagonist, which explains why the CFH haplotype 3, characterized by low FH and high FHR-3 plasma levels, is associated with lower susceptibility to meningococcal disease (78, 80). However, in contrast, the deletion of CFHR3 and CFHR1 was found to be associated with better survival in patients with bacterial meningitis (81). Altogether, this demonstrates the complex and multifaceted roles of the FH family in infections.
Altogether, the associations of the FH family with these diseases illustrate the relevance of the delicate balance between the different family members. Notably, the ratio between the levels of the regulator FH and the FHR proteins (i.e., FHR-1, FHR-3, FHR-4 and FHR-5) seems crucial in determining the outcome. Hence, either genetic or environmental factors altering the protein levels or the functionality of these proteins will have an impact on complement regulation and will define the susceptibility for the development of pathological conditions.
In the last few decades, major strides have been made in our understanding of the FH protein family. From these findings an appreciation has emerged of the vast complexity of this group of proteins as well as of the monoclonal antibodies developed to specifically detect the different members of the Factor H protein family. Unfortunately, we still face multiple unmet challenges. Many of these involve the need for reagents and models to better understand the function of FHR proteins in health and pathology. Here, we will discuss four specific unmet challenges that need to be resolved (Figure 1).
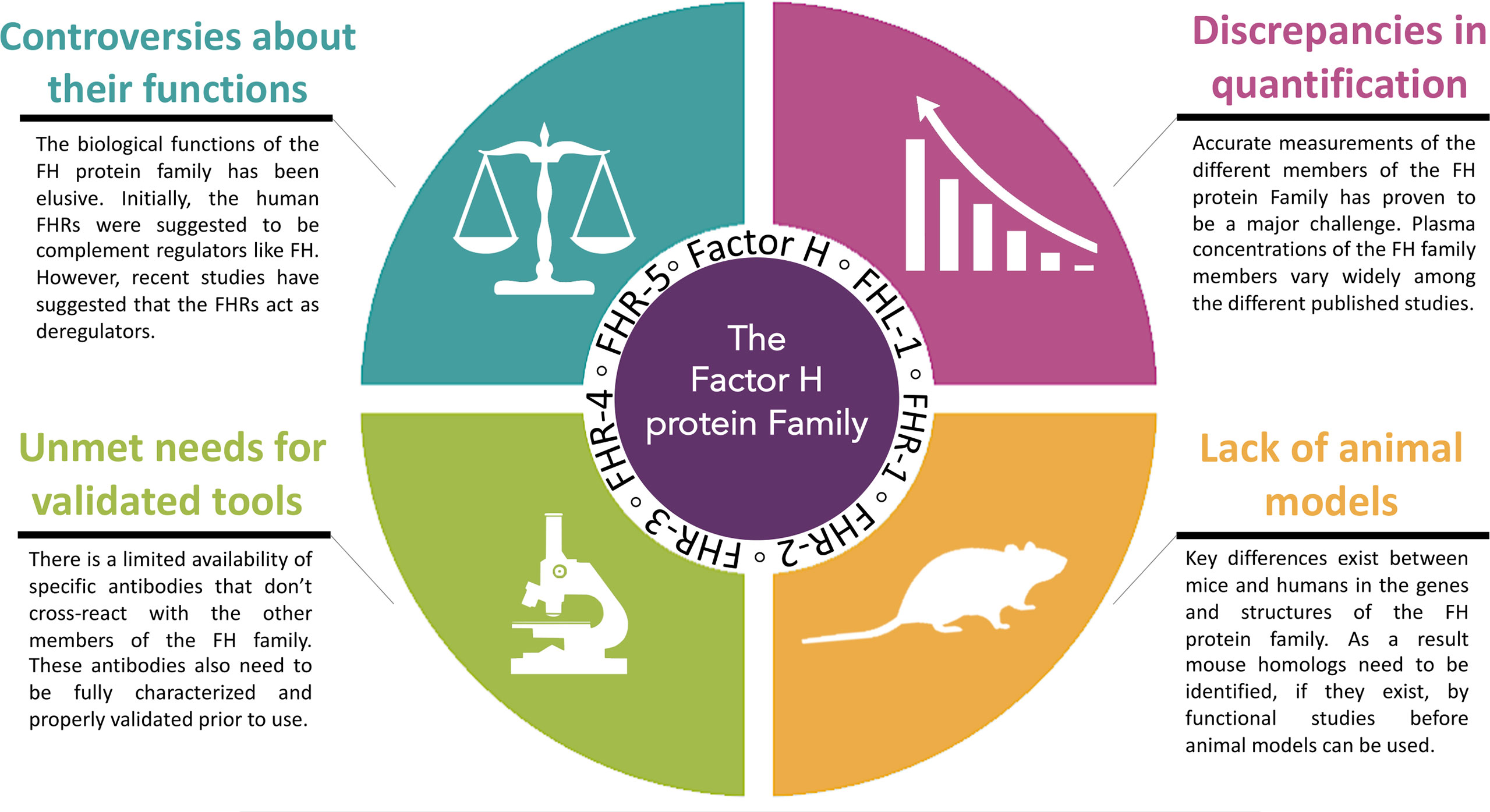
Figure 1 Four specific unmet challenges for the Factor H protein Family. The major challenges ahead in this emerging area are: 1. the controversies about the functional roles of the Factor H protein family, 2. the discrepancies in quantification of the Factor H protein family, 3. the unmet needs for validated tools and 4. limitations of animal models.
The biological function of the FH protein family has been elusive. While it is clear that FH is a potent inhibitor of the complement system, and most data point towards a similar role for FHL-1, the functions of the more recently discovered FHR proteins are less well characterized and therefore remain uncertain. Initially, it was suggested that certain family members (e. g. FHR-1, FHR-3 and FHR-4) had no specific function or, at least, no essential function within the complement system. This rationale was largely based on the high frequency of CFHR gene deletions in the general population and the high homology among these proteins, suggesting some degree of functional redundancy. Instead, genetic studies revealed that alterations in the CFHR genes were indeed associated with pathology (described above), thereby providing the first piece of evidence that FHR proteins could be key pathogenic drivers of human disease. Since the pathogenesis of these diseases involve complement dysregulation, initial functional studies primarily focused on the potential regulatory functions of the FHR proteins. As a result, considerable controversy exists over whether the FHR proteins have complement regulatory capacity or not. Results from these earlier studies indicated that the FHR proteins primarily functioned as complement regulators at specific steps of the cascade, while this concept was contested by later studies (82). Specifically, first the interaction of FHR proteins with C3 was investigated by functional studies, as an indicator of their potential complement inhibiting capacity, since the related proteins were assumed to be functional analogues of FH. Indeed, FHR-3 and FHR-4B were able to bind to the C3d region of C3b (83). Yet, when FHR-3 and FHR-4 were first studied for their effect on Factor I-mediated C3b inactivation, direct co-factor activity was very weak and only detectable at very high, and non-physiological, concentrations (i.e., 400 µg/ml). Moreover, the addition of FHR-3 and FHR-4 also enhanced the inhibitory activity of FH. A later study demonstrated a small inhibitory effect for FHR-3 in the hemolysis assays using FH-depleted serum (84). The regulatory effect of FHR-3 was shown to be based on cofactor activity, although supraphysiological concentrations were once again used. In contrast, others could not show any significant cofactor activity for FHR-4, even at high concentrations (i.e., 650 µg/ml) (85). In this study, FHR-4 did slightly enhance the inhibitory activity of FH. Later, FHR-2 was shown to bind C3b and C3d (86). While no cofactor or decay accelerating activity was found for FHR-2, it was shown to inhibit the activity of the C3bBb-convertase. For FHR-5, weak cofactor activity and fluid phase C3-convertase inhibiting activity were reported, once again at very high concentrations (87). FHR-5 was also found to inhibit both the C5-convertases of the CP and AP in an artificial, bead-based in vitro model (88). In these latter assays, the effective FHR-5 concentrations were close to serum levels measured in samples from healthy donors and patients with glomerulonephritis (37, 76, 87, 89). In conformity, FHR-5 produced by glioblastoma cells was also shown to act as a co-factor of Factor I and inhibit terminal pathway activation, although solely at high concentrations (90). Likewise, inhibition at the C5 level and/or the terminal pathway has also been reported for FHR-1, FHR-2 and FHR-3 (84, 86, 91). Fittingly, in a mouse model of a neurological autoimmune disease, injection of FHR-1 expressing neural stem cells ameliorated brain injury. Human FHR-1 was shown to protect astrocytes from complement activation by inhibiting the formation of the membrane attack complex (92). However, others have not been able to find any significant inhibiting activity of FHR-1 on the terminal pathway (36, 93–95).
Despite these initial studies, it has been very difficult to reconcile the reported regulatory activities of FHR proteins with their structures, especially considering the lack of structural homology of the related proteins with the regulatory domains of FH. In recent years, accumulating data strongly indicated a role for the FHR proteins as promoters of complement activation that stands in contrast to the regulatory function of FH and FHL-1. The study by Hebecker and Józsi was the first to challenge the paradigm, demonstrating that, by binding C3b, FHR-4 in fact enhances alternative pathway activation (85), a mechanism which was also suggested previously by Närkiö-Mäkelä et al. (96). This property of FHR-4 was recently exploited to overcome complement resistance of HER-2 positive tumor cells by applying FHR-4 based immunoconjugates (97). Studies by Tortajada et al. and Goicoechea et al. soon followed and described another mechanism, namely de-regulation by FHRs through competition with FH. FHR-1, FHR-2 and FHR-5 were shown to form homo- and hetero-oligomeric complexes, while a C3G-associated mutation in FHR-1 resulted in the duplication of the dimerization domain leading to the formation of unusually large multimeric FHR complexes that exhibited increased avidity for C3 activation fragments (36, 71). Similarly, in IgAN, elevated levels of FHR proteins were shown to be associated with enhanced complement activation, while the absence of FHR-1 and FHR-3 was shown to decrease complement activation in AMD. These functional roles are opposite to that proposed in prior studies but are entirely consistent with the FHR structures, wherein the homologies of FHR proteins with FH are in the surface ligand-binding sites. Moreover, these studies strongly suggest that FHR proteins compete with FH (and FHL-1) mediated inhibition and thereby antagonize this key regulator of the complement system. Overall, FHRs were indeed shown to enhance complement activation both directly and indirectly (i.e., via competing with FH), thus emerging as “regulators of the regulator” (34). Competition between FHRs and FH has been described for several binding ligands. FHR-1, FHR-3, FHR-4 and FHR-5 were all shown to compete with FH for binding to C3b, to variable extent. Some of these differential effects may be related to the different avidities also determined by homo- or heterodimerization of FHR-1 and FHR-5 (36, 80, 84, 91). In addition, FHR-5 has been shown to strongly inhibit the binding of FH to the pentraxins (i.e., CRP and PTX3), as well as to extracellular matrix and malondialdehyde-acetaldehyde epitopes (98, 99). Subsequently, FHR-5 enhanced AP activation on these ligands. For FHR-1, FHR-4 and FHR-5 it has been shown that, by binding C3b, they can serve as a platform for the assembly of a functionally active C3bBbP convertase, consequently enhancing activation of the AP (85, 95, 98). Furthermore, FHR-5 can also induce AP activating by the recruitment of properdin via the CCPs 1-2 (99). Interestingly, in addition to modulation of the AP, FHR-1 and FHR-4 were both shown to activate the CP as well through the binding of CRP (i.e., FHR-1 the monomeric form, and FHR-4 the native, pentameric CRP) (85, 95, 100). More recently, FHR-1 and FHR-5 were shown to compete with FH for binding to DNA and thus promote AP activation, as well as to modulate both AP and CP activation on the surface of necrotic cells via interactions with monomeric CRP and PTX3 (101).
In conclusion, while complement inhibiting activity for some of the FHR proteins was described, the reported inhibitory activities were typically weak. More recent studies suggest that FHR proteins represent pattern recognition molecules that promote rather than constrain complement activation (35). To resolve the controversy, these functions need to be studied further, using physiological concentrations, and confirmed by independent research groups. The reported discrepancies may be related to the various sources of recombinant proteins used in the studies. In addition, the proteins may display different, context-dependent activities. Besides the function of the FH-family in the regulation of the complement system, non-canonical functions such as regulating cellular responses were described for FH, FHR-1 and FHR-3 (discussed in detail elsewhere (102)). Losse et al. reported that FHR-1 can bind to neutrophils via complement receptor 3 (CD11b/CD18), thereby resulting in enhanced antimicrobial activity (103). Recently, FHR-1 was shown to activate the NLRP3 inflammasome via the EMR2 receptor on monocytes and by binding to necrotic cells (74). Furthermore, these mentioned controversies raise an additional important question: How does the FH-family regulate inflammation, and what are their ligands and are there FHR protein receptors? Future studies need to define specific and shared ligands among members of this protein family, as well as conditions under which physiological or pathological functions are displayed, or competition occurs.
Accurate analysis of the FH protein family is of utmost importance for further deciphering of its function and role in complement-mediated diseases. Accurate information on physiological levels and the composition of the FH family members is also vital for functional studies, since supraphysiological levels can give misleading results. Yet, precise analysis of FH and other complement system components has proven to be challenging and systemic levels for FH family members vary widely among different studies (Table 1). These inconsistencies in levels are not only due to differences in sample type (plasma vs serum or different anticoagulants), storage (room temperature, 4°C vs -20°C or -80°C) and pre-analytical sample handling between studies, but also most likely caused by the use of different techniques (ELISA vs mass spectrometry), protocols and reagents (113). Another important explanation for the discrepancies in reported levels could be differences in characteristics of the blood donors. Age and gender have previously been demonstrated to significantly impact complement levels and functionality in the healthy population (114). Nevertheless, the impact of age and gender on the FH protein family has not been extensively studied. In healthy children, FHR-1, FHR-4A and FHR-5 levels were shown to be slightly lower in children compared to adults, but only FHR-5 levels were significantly associated with age (115). In addition, no gender differences were found. Levels of FH, FHR-2, and FHR-3 were similar to those found in adults (115). However, when corrected for genetic factors, an age-dependent increase of plasma levels of FH was seen for individuals aged 1 to 88 years (105). Furthermore, even when laboratories use the same technique, for instance ELISA, varying methods, reagents, calibrators and antibodies are used. Moreover, when antibodies are used, it is not always known whether these antibodies are truly specific for the target antigen or if cross-reactivity with other proteins may occur. Given that the FH protein family has a high degree of similarity in amino acid sequence, it is very well possible that antibodies against FH also cross-react with other FH family members. As a result, large discrepancies in levels for FH family members are observed between testing laboratories thereby hampering correct interpretation and hindering the comparison of results between studies. These inconsistencies in levels indicate the urgent need for well-characterized and standardized assays (116). Yet, validated and standardized assays for quantitative and functional analysis are not (widely) available for FH and its related proteins. Here, epitope mapping can be extremely valuable to predict (potential) cross-reactivity with other FH family members. The epitope location of a monoclonal antibody (mAb) can be determined using fragments consisting of CCP domains as previously described (111). It is important to note that in contrary to the FHR proteins, the functions of FH are well known and several functional assays for FH exist, some of which currently are being used in the clinic. FH-mediated decay-accelerating activity can be measured in both ELISA as well as surface plasmon resonance assays (117–119). The co-factor activity of FH can be determined in both fluid-phase as well as on the cell surface (120, 121). In addition, the full function of FH can be addressed on cell surfaces using cell-based assays (13, 117, 119).
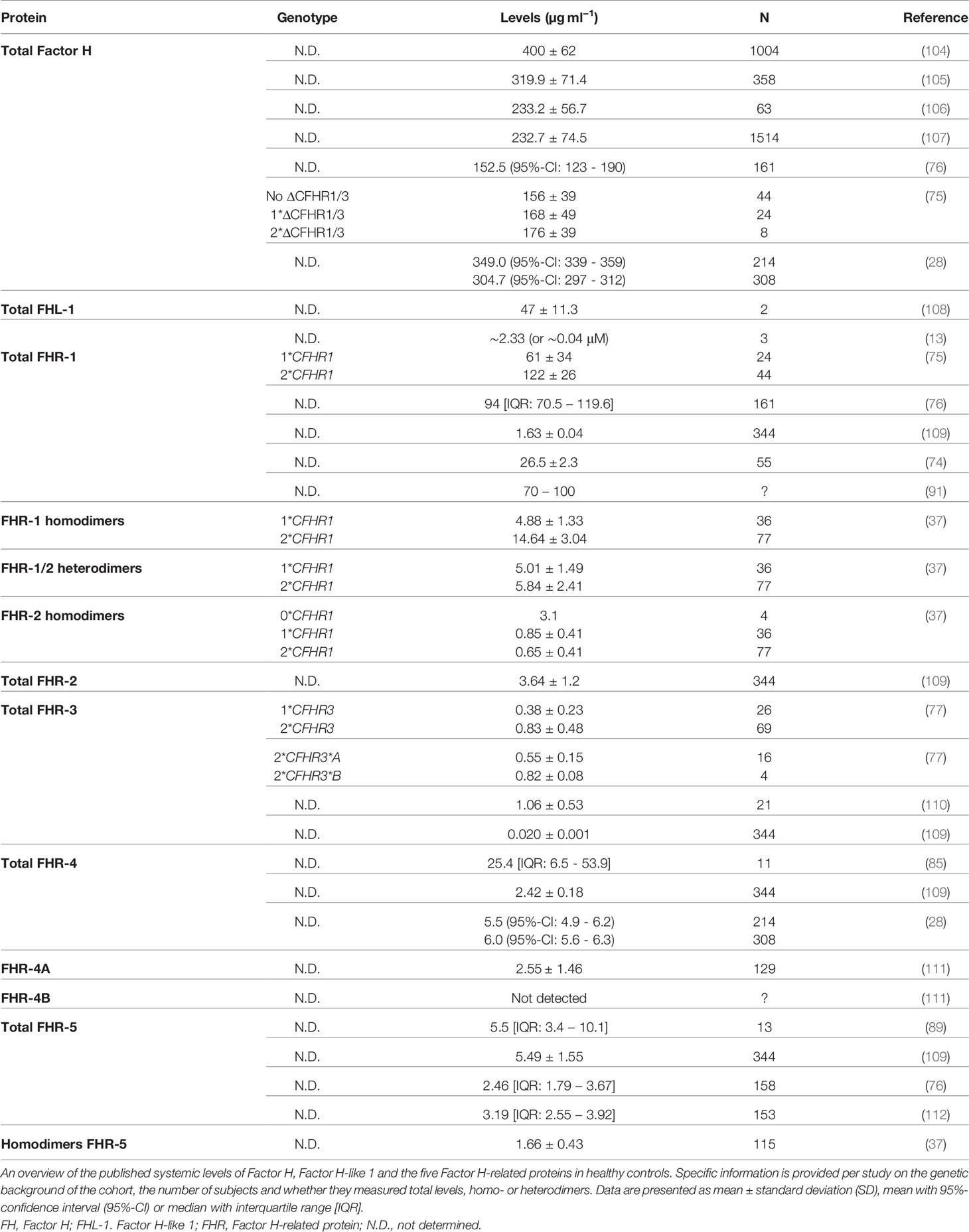
Table 1 Previously published systemic levels of the Factor H protein family.
In order to provide insight into the magnitude of the discrepancies in quantification of the FH protein family, we determined systemic FH levels in samples from healthy volunteers using seven commercially available ELISA’s (Table 2). Levels of FH were evaluated in samples that were collected, stored and handled exactly the same way. Next to those 10 samples, we also included 2 samples obtained from the Complement EQA Group. Both samples consist of a pool of serum samples derived from 5 healthy individuals. The Complement EQA Group committee aims to standardize complement analysis by providing calibrator materials and collects, evaluates and compares levels of complement components from different testing facilities (see also below). It should be noted that within the FH protein family, only quantitative analysis of serum levels of FH is included in the standardization activities of the Complement EQA Group. In 2009, this group was formally recognized and became part of the IUIS (International Union of Immunological Societies) Quality Assessment and Standardization Committee (https://iuis.org/committees/qas/) (122). We chose plasma-EDTA samples, since coagulation enzymes can also cleave complement components with subsequent generation of activation products (122). EDTA blocks the in vitro activation of the complement system by Mg2+ and Ca2+ chelation. Citrate-based anticoagulants are less useful (123). Moreover, heparin-plasma should not be used since multiple members from the FH family are heparin-binding proteins, hence heparin could interfere with the measurements. All assays were performed in parallel by the same operator according to the manufacturer’s protocols. In general, all %CV were ≤15%, indicating low variation.

Table 2 Human Factor H ELISA’s included in the comparison analysis.
First, human purified FH protein dissolved in PBS was measured in all seven assays (Cat# A137, Complement Technology Inc., TX, USA). None of the assays was able to ‘pinpoint’ this exact concentration (not corrected for the extinction coefficient). The assays from LSBio and USCN were not even able to detect purified FH protein in PBS (Figure 2A). Next, a FH depleted sample was measured as a negative control in all assays (Cat# A337, Complement Technology Inc., TX, USA). As expected, most assays did not detect FH in this sample. However, a FH concentration of 377 µg/ml was measured in the FH depleted sample using the USCN assay (Figure 2B). Subsequently, we assessed systemic FH levels in plasma-EDTA samples derived from 10 healthy controls. Results show large and significant differences in FH levels between the seven assays used (P<0.0001). No FH was detected with the LSBio assay (Figure 2C). In spite of these differences in absolute FH levels between the assays, moderate to high correlations were observed between the Abcam, Hycult, Quidel, R&Dsystems and Sanquin assays regarding the FH levels (Figure 2D). No correlation was observed between the USCN and the other assay. For the LSBio assay, no correlation could be calculated as no FH levels were detected with this assay. The FH levels in the serum pool samples obtained from the Complement EQA Group were comparable with the levels measured in the healthy control panel. Again, no FH was detected using the LSBio assay (Figure 2E). Lastly, calibrators were exchanged, except for the Abcam and Quidel ELISAs, as not enough calibrator was provided to be included in each assay as sample. Results show that the calibrators from Hycult, R&Dsystems and Sanquin were exchangeable, and yielded quantifiable and reliable levels. The calibrators from LSBio and USCN were not recognized in all other assays. In turn, the LSBio assay was not able to recognize/measure any of the calibrators except its ‘own’ calibrator. The USCN assay was able to recognize the calibrators from Hycult and Sanquin (Figure 2F). Overall, the assays from Abcam, Hycult, R&Dsystems and Sanquin were perfectly able to detect FH protein in the positive control and did not detect FH in the negative control. Additionally, the correlations between these assays were moderate to high. In contrast, the results obtained with the LSBio and the USCN assay suggest that these assays are not able to assess FH levels in samples in a reliable manner. Nevertheless, even in the reliable assays, FH levels obtained in the same sample set vary greatly. Given these discrepancies, we can conclude that the absolute FH levels determined with these assays, are probably not correct. Considering the lack of quantification, it is suggested to provide calibrators with these assays as arbitrary units (AU) only. In this instance, these assays would then be able to detect differences in FH levels between experimental groups (e.g., healthy vs disease), as long as the same assay is used for all analyzed groups. In the end, the fact that absolute FH levels vary greatly between assays may not be surprising as calibrators from different sources were used in their calibrations.
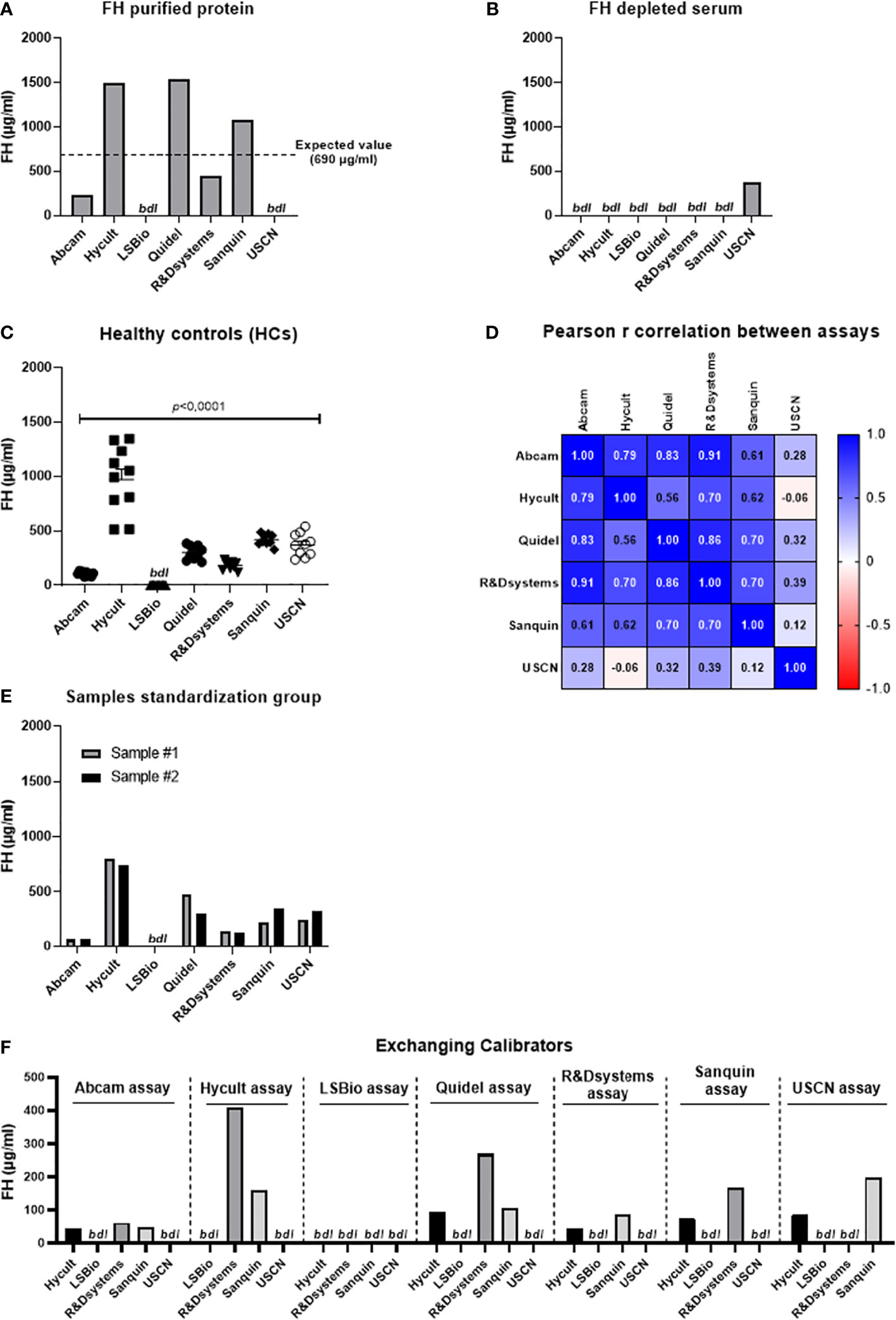
Figure 2 Assessment of Factor H levels in samples using seven different assays. (A) Assessment of Factor H (FH) purified protein in PBS (expected value is 690 µg/ml). (B) Assessment of FH levels in FH depleted serum. (C) Assessment of systemic FH levels in samples derived from healthy controls (n=10). Data are represented as mean ± SEM. Data were analyzed using the one-way ANOVA (Graphpad Prism 8.4.2, San Diego, CA, USA). A p-value <0.05 was considered significant. (D) Pearson r correlation coefficient. Pearson coefficients range from +1 to -1, with +1 representing a positive correlation of FH sample values between assays, -1 representing a negative correlation of FH sample values between assays, and 0 representing no relationship. No correlation could be calculated for the LSBio assay as no FH levels were obtained using this assay. (E) Assessment of FH levels in 2 serum pool samples obtained from the Complement EQA Group. (F) Assessment of calibrators as sample in each assay. All calibrators were exchanged between the seven assays except for the Abcam and Quidel calibrator as not enough calibrator was provided with these kits to be included in each assay as sample. FH, factor H; HCs, healthy controls; bdl, below detection limit.
For the future, we therefore recommend that a uniform protocol is used regarding sample collection, pre-analytical sample handling and storage. For the assessment itself, it is strongly recommended that standardized assays with a uniform calibrator are used. When antibodies are used for quantification, these must be characterized regarding specificity and cross-reactivity with other proteins (see also below in the next section). In this way, results can be produced that are robustly comparable between different studies.
In 2016, Nature conducted a survey to understand scientist’s view on the lack of reproducibility in research (124). When asked about the cause, a proportion pointed towards poorly-characterized tools leading to ambiguous findings, which results in an unstable knowledge foundation that is then built upon. Since this survey, different guidelines for in vitro and in vivo research have been suggested, issued and published (125, 126). Characterization of antibodies as well as validation of tools to quantify proteins is vital for every field, but particularly for the FH protein family considering the high risk of cross-reactivity due to their homology. Yet, when looking at the specific antibodies that are currently used in peer-reviewed publications, results on validation of antibodies are not always provided (Table 3). Moreover, whenever present, results on antibody validation are usually in the supplementary data, a scientific habit that perhaps should be reconsidered. Safeguarding accurate antibody validation should be a main concern for all scientists, clinical end-users, industry, journal publishers and antibody-linked research alike. Every researcher has experienced research antibodies that don’t live up to promises or expectations. Either because the antibody does not recognize the desired target, or because they recognize a different protein instead of, or as well as the desired target. Additional problems include functionality of the antibody being limited to certain applications. Too often the choice of an antibody is driven by the number of citations it has in the literature, as scientists falsely assume the antibody must have been validated previously, enabling self-perpetuating artefacts. In this regard, we offer a consensus report by the authors and hope to ignite further discussion among the community to establish recommendations for best practices to improve the reproducibility, validity and to help advance research into the FH-protein family. Obviously, these recommendations are not aimed at antibodies used for mere in vitro experiments with purified components, but rather for observational and intervention studies with a large sample size. First of all, it would be advised to (i) reference and (ii) validate the specific antibodies used for the FH protein family research field as detailed as possible (130). We further suggest a best practice protocol for the validation of all detection tools. Based on previous guidelines, we suggest the following 4-step validation protocol for antibodies against the FH protein family (131–134):
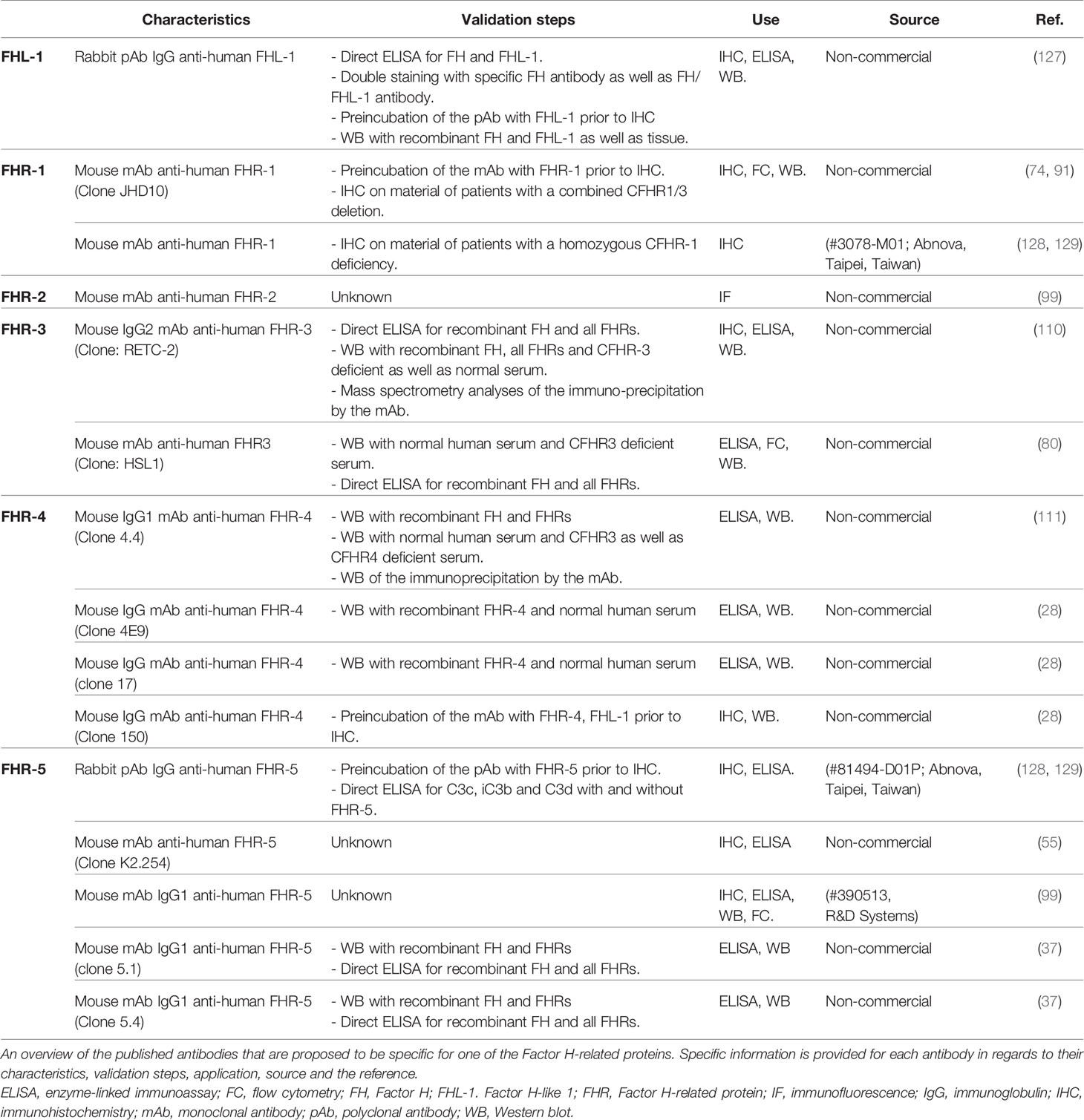
Table 3 Published antibodies that have been proposed to be specific for each of the Factor H-related proteins.
1. Show whole Western blot of target human/animals samples detected with the antibody.
Value: Knowledge regarding the respective protein size(s).
Cave: Only applicable if the antibody is reactive in Western blot.
2. Compare binding pattern of the antibody for ALL the different known FH-protein family members (e.g. ELISA, Western blot or Dot blot).
Value: Characterization of intra-protein family specificity.
Cave: Access to validation material can be restricted.
3. Test the antibody reactivity against either a.) non-depleted vs FH-protein family members depleted human serum/plasma (if available), b.) non-deficient cells vs target gene deficient cells (if available) or c.) wildtype vs knockout animals (e.g., ELISA, Western Blot or immunohistochemistry, if available).
Value: Species specificity of the antibody can be determined independent from unknown protein modifications.
Cave: Access to validation material can be restricted.
4. Characterize the specificity of an antibody using immunoprecipitation with subsequent mass spectrometry analysis from the respective target tissue.
Value: Tissue specific cross reactions of the whole antibody (even the Fc-part) will be deciphered.
Cave: Specialized collaborating laboratories are needed.
It would be preferred to perform this 4-step validation protocol in line with an already validated, antibody as a quality control (if available). A single protocol will not match all applications and researchers must ensure that the reported validation holds true for their specific use. Overall, we are convinced that validation of tools, together with transdisciplinary collaboration and transparency within the community will enable research to move forward and get one step closer towards deciphering the mode of action of the FH-protein family.
Murine models have demonstrated high value for establishing fundamental principles of complement biology (135). The murine version of FH (mFH) was first identified in 1986 and is very similar in structure and function to the human FH (136). As its human equivalent, mFH has a molecular weight of around 155 kDa, has several glycosylation sites, is positioned on chromosome 1 and is primarily produced by the liver. The plasma concentration of mFH is estimated to be similar to human FH, but an exact concentration of mFH has not been determined yet (135, 136). Clear differences exist as well between mFH and human FH. For instance, in contrast to the human CFH gene, the mCFH gene does not have an alternative splicing variant and thus no murine equivalent of FHL-1 has been identified. Overall, The mCFH gene shares 63% of sequence identity with the human CFH gene (135). Despite the differences in genes and protein structures across mouse and human, it has been observed repeatedly that essential activation and regulatory functions of this system are retained across species. In accordance, genetically deficient animals have provided a powerful tool to help understand the function of FH.
The link between FH and disease is older than is frequently reported. Høgåsen et al. described more than 25 years ago that a hereditary deficiency of FH in pigs consistently led to the development of lethal renal disease, namely C3G (137). These findings were later confirmed in rodent models, demonstrating that mice deficient in FH had uncontrolled complement activation resulting in C3G (138). Interestingly, later studies revealed that aged CFH-/- mice also develop retinal abnormalities and visual dysfunction, resembling AMD (139). The broad outlines are therefore clear, dysfunction of FH leads to uncontrolled complement activation resulting in tissue injury and thus causing disease. However, it was not clear what then determines whether defects in FH cause one specific disease but not the other. Animal models have helped to attribute the different functions of FH to specific domains within the protein, and thereby reveal specific genotype–phenotype connections in FH that lead to either complement-mediated thrombotic microangiopathy (TMA) or C3G and AMD. A study by Pickering et al. uncovered that a homozygous FH deficiency in mice leads to defective function of FH in the fluid-phase triggering the development of C3G and AMD (138). In contrast, loss of the SCR 16-20 region of FH impairs the ability of FH to control complement activation on surfaces, causing spontaneous TMA (60). In conclusion, the use of animal models has helped significantly to understand the function of complement proteins and their role in disease, especially for FH.
Animal models have barely been used to study the other members of the Factor H family. As mentioned before, no murine equivalent for the human FHL-1 exists. Furthermore, the CFHR genes have arisen during evolution through duplication events of the CFH gene (12). Extensive analysis of the human CFH and CFHR gene loci using Alu/L1 repeat dating established that these duplication events occurred after the separation of rodent and primate lineages and therefore no FHR orthologues exist in mice. More specifically, the mCFHR genes differ in structure, domain composition, and sequence from the human genes (32). Like the human FHR proteins, a total of five murine CFHR genes have been suggested (mCFHR-A, mCFHR-B, mCFHR-C, mCFHR-D and mCFHR-E) and evidence for four mFHR proteins (FHR-B, FHR-C, FHR-D and FHR-E) has been derived from mRNA transcripts isolated from mouse liver (48, 140–143). However, altogether, this suggests that direct comparisons between the human and mouse FHR proteins is not informative and, therefore, any mouse FHR homologs need to be identified, if they exist, by functional studies before rodent models can be used to further study the role of the FH protein family in human health and disease. Alternatively, genetic engineering approaches could be used to create a set of humanized transgenic mice to more closely mimic the human FHR situation. Until that is achieved, the lack of animal models remains a major barrier hindering the elucidation of disease mechanisms and drug development. More importantly, the absence of appropriate animal models stresses the importance of appropriate human assays to correctly identify and study the Factor H protein family in humans.
The various disorders linked to the FH family tend to be difficult to treat and some are even incurable. An obvious therapeutic strategy for these diseases could therefore be the administration of (purified or recombinant) FH to restore complement regulation. Indeed, both in vitro and animal studies have demonstrated the therapeutic value of FH (63, 137, 144–146). In CFH-/- pigs, a single dose of 5 mg/kg porcine FH resulted in normalization of plasma C3 levels and diminished systemic complement activation for almost 3 days (137). In CFH-/- mice, both purified mouse and purified human FH led to a rapid increase of plasma C3 levels and resolution of renal C3 deposition (144, 146). However, FH supplementation as a therapy would require large amounts of biologically active protein due to high circulating levels in healthy individual, making it labor and cost intensive. Various strategies have been tested to resolve these problems. Several groups have demonstrated successful production of high yields of recombinant FH in different expression systems (such as yeast and moss) as an alternative and economically viable method (147, 148). Others have created derivates or fusion proteins from FH with enhanced pharmacokinetic and pharmacodynamic properties. Smaller constructs of FH have been created by combining the regulatory domains (N-terminus) with the surface-recognition domains (C-terminus) (121, 149, 150, 158). These FH constructs can regulate complement in vivo, and effectively reverse renal C3 deposition and restore plasma C3 levels in CFH-/- mice. However, the short half-life of these constructs remains an important limitation. FH fusion protein have also been engineered as a therapeutic approach (151–153). Most extensively studied is the CR2-FH fusion protein, that links the C3d binding domain of complement receptor 2 (CR2) to the complement inhibitory domain of FH, thus ensuring targeted regulation by FH at sites of complement activation (154). Treatment with CR2-FH was beneficial in animal models of eye, kidney and autoimmune diseases (155–157). Finally, local injection of FH (or derivates) is another approach to circumvent the need for large amounts of biologically active protein. A clinical trial investigating the safety and effectivity of recombinant FH (GEM103) administered through intravitreal injections for the treatment of geographic atrophy secondary to dry AMD is currently on-going (ClinicalTrials.gov identifier, NCT04246866).
A multidisciplinary approach is mandatory to overcome the challenges mentioned above, and is only possible through interdisciplinary collaboration between biologists, chemists, geneticists and physicians. But, where to start? As suggested by the quote of William Thomson, the authors of this paper believe that we should essentially begin with quantifying the levels and activity of the different members of the FH-family in health and disease. Detection of the FH-protein family will enable the scientific and clinical community to advance our understanding of the role of the FH-protein family in infectious, eye, kidney and autoimmune diseases, and potentially help treat these disorders.
As described, the FH-family, consists of FH, FHL-1 and the five FHR proteins which are important regulators of the complement system. Mutations and polymorphisms in the FH-family are involved in several diseases, indicating a potential crucial role of the FH-family in both health and disease. However, diagnosis and therapy of these partially incurable pathologies is to-date not related to the FH-protein family, due to a lack of fundamental knowledge of (i) the molecular mechanisms leading to disease, (ii) unknown functional, convincing principles of FH-protein family members, (iii) absent standardized diagnostics and (iv) missing suitable drug candidates. To overcome these challenges, an ardent multidisciplinary approach is required through interdisciplinary collaboration.
All authors contributed equally and the review was written together. All authors critically reviewed the manuscript prior to submission.
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 899163.
MS was employed by Microcoat Biotechnologie GmbH. ET was employed by Hycult Biotech.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This review was written on behalf of the SciFiMed Consortium. This is a European collaboration that is formed to create a comprehensive analytical system for the quantitative and functional assessment of the entire Factor H protein family. Principal investigators are (in alphabetical order): AB (Institute of Analytical Chemistry, Chemo-and Biosensors, Faculty of Chemistry and Pharmacy, University of Regensburg, Regensburg, Germany), EG (Department of Immunology, Faculty of Medicine, Complutense University and Research Institute Hospital 12 de Octubre (imas12), Madrid, Spain), IJ (Department of Immunopathology, Sanquin Research and Landsteiner Laboratory of the Academic Medical Centre, University of Amsterdam, Amsterdam, Netherlands & Department of Pediatric Immunology, Rheumatology, and Infectious Diseases, Emma Children’s Hospital, Amsterdam University Medical Centre, Amsterdam, Netherlands), MJ (MTA-ELTE Complement Research Group, Department of Immunology, ELTE Eötvös Loránd University, Budapest, Hungary), DP (Department of Ophthalmology, University Hospital Regensburg, Regensburg, Germany & Experimental Ophthalmology, University Marburg, Marburg, Germany), FP (Department of Internal Medicine, Division of Nephrology, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands), M-SS (Microcoat Biotechnologie GmbH, Bernried am Starnberger See, Germany), ET (R&D department, Hycult Biotech, Uden, The Netherlands).
aHUS, Atypical hemolytic uremic syndrome; AMD, Age-related macular degeneration; ANCA, Anti-neutrophil cytoplasmic antibody; AP, Alternative pathway; AU, Arbitrary units; BLAST, Basic local alignment search tool; C3G, C3 glomerulopathy; CCP, Complement control protein; CFH, Complement Factor H gene; CFHR, Complement Factor H-related genes; CP, Classical pathway; CRP, C-reactive protein; FB, Factor B; FH, Factor H; FHL-1, Factor H-like 1; FHR, Factor H-related protein; FI, Factor I; FISH, Fluorescence in situ hybridization; IgAN, Immunoglobulin A nephropathy; LP, Lectin pathway; mAb, Monoclonal antibody; mCFH, Murine complement Factor H gene; mCFHR, Murine complement Factor H-related gene; mFH, Murine Factor H; mFHR, Murine Factor H-related protein; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; PTX3, Pentraxin-3; RCA, Regulators of complement activation gene cluster; SLE, Systemic lupus erythematosus; TMA, Thrombotic microangiopathy.
1. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol (2015) 6:262:262. doi: 10.3389/fimmu.2015.00262
2. Reis ES, Mastellos DC, Hajishengallis G, Lambris JD. New insights into the immune functions of complement. Nat Rev Immunol (2019) 19:503–16. doi: 10.1038/s41577-019-0168-x
3. Arbore G, Kemper C, Kolev M. Intracellular complement – the complosome – in immune cell regulation. Mol Immunol (2017) 89:2–9. doi: 10.1016/j.molimm.2017.05.012
4. Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol (2017) 18:1288–98. doi: 10.1038/ni.3858
5. Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol (2016) 12:383–401. doi: 10.1038/nrneph.2016.70
6. de Boer ECW, van Mourik AG, Jongerius I. Therapeutic Lessons to be Learned From the Role of Complement Regulators as Double-Edged Sword in Health and Disease. Front Immunol (2020) 11:578069:1. doi: 10.3389/fimmu.2020.578069
7. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol (2009) 9:729–40. doi: 10.1038/nri2620
8. Sánchez-Corral P, Pouw RB, López-Trascasa M, Józsi M. Self-Damage Caused by Dysregulation of the Complement Alternative Pathway: Relevance of the Factor H Protein Family. Front Immunol (2018) 9:1607:1607. doi: 10.3389/fimmu.2018.01607
9. Zipfel PF. Complement factor H: Physiology and pathophysiology. In: . Seminars in Thrombosis and Hemostasis (Semin Thromb Hemost). (2001) p. 191–9. doi: 10.1055/s-2001-15248
10. Parente R, Clark SJ, Inforzato A, Day AJ. Complement factor H in host defense and immune evasion. Cell Mol Life Sci (2017) 74:1605–24. doi: 10.1007/s00018-016-2418-4
11. Ferreira VP, Pangburn MK, Cortés C. Complement control protein factor H: The good, the bad, and the inadequate. Mol Immunol (2010) 47:2187–97. doi: 10.1016/j.molimm.2010.05.007
12. Cantsilieris S, Nelson BJ, Huddleston J, Baker C, Harshman L, Penewit K, et al. Recurrent structural variation, clustered sites of selection, and disease risk for the complement factor H (CFH) gene family. Proc Natl Acad Sci U.S.A. (2018) 115:E4433–42. doi: 10.1073/pnas.1717600115
13. Dopler A, Guntau L, Harder MJ, Palmer A, Höchsmann B, Schrezenmeier H, et al. Self versus Nonself Discrimination by the Soluble Complement Regulators Factor H and FHL-1. J Immunol (2019) 202:2082–94. doi: 10.4049/jimmunol.1801545
14. Schwaeble W, Zwirner J, Schulz TF, Linke RP, Dierich MP, Weiss EH. Human complement factor H: expression of an additional truncated gene product of 43 kDa in human liver. Eur J Immunol (1987) 17:1485–9. doi: 10.1002/eji.1830171015
15. De Córdoba SR, De Jorge EG. Translational Mini-Review Series on Complement Factor H: Genetics and disease associations of human complement factor H. Clin Exp Immunol (2008) 151:1–13. doi: 10.1111/j.1365-2249.2007.03552.x
16. Kopp A, Strobel S, Tortajada A, Rodríguez de Córdoba S, Sánchez-Corral P, Prohászka Z, et al. Atypical Hemolytic Uremic Syndrome-Associated Variants and Autoantibodies Impair Binding ofand Factor H-Related Protein 1 to Pentraxin 3. J Immunol (2012) 189:1858–67. doi: 10.4049/jimmunol.1200357
17. Swinkels M, Zhang JH, Tilakaratna V, Black G, Perveen R, McHarg S, et al. C-reactive protein and pentraxin-3 binding of factor H-like protein 1 differs from complement factor H: Implications for retinal inflammation. Sci Rep (2018) 8:1643. doi: 10.1038/s41598-017-18395-7
18. Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HPN, Issa PC, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature (2011) 478:76–81. doi: 10.1038/nature10449
19. Zhu L, Zhai YL, Wang FM, Hou P, Lv JC, Xu DM, et al. Variants in complement factor H and complement factor H-related protein genes, CFHR3 and CFHR1, affect complement activation in IgA nephropathy. J Am Soc Nephrol (2015) 26:1195–204. doi: 10.1681/ASN.2014010096
20. Zhai YL, Meng SJ, Zhu L, Shi SF, Wang SX, Liu LJ, et al. Rare variants in the complement factor H-related protein 5 gene contribute to genetic susceptibility to IgA nephropathy. J Am Soc Nephrol (2016) 27:2894–905. doi: 10.1681/ASN.2015010012
21. Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet (2011) 43:321–9. doi: 10.1038/ng.787
22. Xie J, Kiryluk K, Li Y, Mladkova N, Zhu L, Hou P, et al. Fine mapping implicates a deletion of CFHR1 and CFHR3 in protection from IgA nephropathy in Han Chinese. J Am Soc Nephrol (2016) 27:3187–94. doi: 10.1681/ASN.2015111210
23. Jullien P, Laurent B, Claisse G, Masson I, Dinic M, Thibaudin D, et al. Deletion Variants of CFHR1 and CFHR3 Associate with Mesangial Immune Deposits but Not with Progression of IgA Nephropathy. J Am Soc Nephrol (2018) 29:661–9. doi: 10.1681/ASN.2017010019
24. Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet (2006) 38:1173–7. doi: 10.1038/ng1890
25. Caramoy A, Ristau T, Lechanteur YT, Ersoy L, Müller S, Gelisken F, et al. Environmental and genetic risk factors for retinal angiomatous proliferation. Acta Ophthalmol (2014) 92:745–8. doi: 10.1111/aos.12437
26. Lorés-Motta L, Paun CC, Corominas J, Pauper M, Geerlings MJ, Altay L, et al. Genome-Wide Association Study Reveals Variants in CFH and CFHR4 Associated with Systemic Complement Activation: Implications in Age-Related Macular Degeneration. Ophthalmology (2018) 125:1064–74. doi: 10.1016/j.ophtha.2017.12.023
27. den Hollander AI, Hoyng CB, Boon CJF. Complement Factor H Gene Mutations: Implications for Genetic Testing and Precision Medicine in Macular Degeneration. Ophthalmology (2019) 126:1422–3. doi: 10.1016/j.ophtha.2019.04.033
28. Cipriani V, Lorés-Motta L, He F, Fathalla D, Tilakaratna V, McHarg S, et al. Increased circulating levels of Factor H-Related Protein 4 are strongly associated with age-related macular degeneration. Nat Commun (2020) 11:778. doi: 10.1038/s41467-020-14499-3
29. Hodeib S, Herberg JA, Levin M, Sancho-Shimizu V. Human genetics of meningococcal infections. Hum Genet (2020) 139:961–80. doi: 10.1007/s00439-020-02128-4
30. Biebl A, Muendlein A, Kinz E, Drexel H, Kabesch M, Zenz W, et al. Confirmation of host genetic determinants in the CFH region and susceptibility to meningococcal disease in a central European study sample. Pediatr Infect Dis J (2015) 34:1115–7. doi: 10.1097/INF.0000000000000823
31. Davila S, Wright VJ, Khor CC, Sim KS, Binder A, Breunis WB, et al. Genome-wide association study identifies variants in the CFH region associated with host susceptibility to meningococcal disease. Nat Genet (2010) 42:772–6. doi: 10.1038/ng.640
32. Zipfel PF, Wiech T, Stea ED, Skerka C. CFHR Gene Variations Provide Insights in the Pathogenesis of the Kidney Diseases Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy. J Am Soc Nephrol (2020) 31:241–56. doi: 10.1681/ASN.2019050515
33. Chen Q, Wiesener M, Eberhardt HU, Hartmann A, Uzonyi B, Kirschfink M, et al. Complement factor H-related hybrid protein deregulates complement in dense deposit disease. J Clin Invest (2014) 124:145–55. doi: 10.1172/JCI71866
34. Cserhalmi M, Papp A, Brandus B, Uzonyi B, Józsi M. Regulation of regulators: Role of the complement factor H-related proteins. Semin Immunol (2019) 45:101341. doi: 10.1016/j.smim.2019.101341
35. Józsi M, Tortajada A, Uzonyi B, Goicoechea de Jorge E, Rodríguez de Córdoba S. Factor H-related proteins determine complement-activating surfaces. Trends Immunol (2015) 36:374–84. doi: 10.1016/j.it.2015.04.008
36. Goicoechea De Jorge E, Caesar JJE, Malik TH, Patel M, Colledge M, Johnson S, et al. Dimerization of complement factor H-related proteins modulates complement activation in vivo. Proc Natl Acad Sci U.S.A. (2013) 110:4685–90. doi: 10.1073/pnas.1219260110
37. van Beek AE, Pouw RB, Brouwer MC, van Mierlo G, Geissler J, Ooijevaar-de Heer P, et al. Factor H-related (FHR)-1 and FHR-2 form homo- and heterodimers, while FHR-5 circulates only as homodimer in human plasma. Front Immunol (2017) 8:1328. doi: 10.3389/fimmu.2017.01328
38. Nilsson UR, Mueller-Eberhard HJ. Isolation of beta if-globulin from human serum and its characterization as the fifth component of complement. J Exp Med (1965) 122:277–98. doi: 10.1084/jem.122.2.277
39. Whaley K, Ruddy S. Modulation of the alternative complement pathway by β1H globulin*. J Exp Med (1976) 144:1147–63. doi: 10.1084/jem.144.5.1147
40. Weiler JM, Daha MR, Austen KF, Fearon DT. Control of the amplification convertase of complement by the plasma protein β1H. Proc Natl Acad Sci U.S.A. (1976) 73:3268–72. doi: 10.1073/pnas.73.9.3268
41. Whaley K, Ruddy S. Modulation of C3b hemolytic activity by a plasma protein distinct from C3b inactivator. Sci (80- ) (1976) 193:1011–3. doi: 10.1126/science.948757
42. Fischer E, Kazatchkine MD. Surface-dependent modulation by H of C5 cleavage by the cell-bound alternative pathway C5 convertase of human complement. J Immunol (1983) 130(6):2821–4.
43. Ripoche J, Day AJ, Harris TJR, Sim RB. The complete amino acid sequence of human complement factor H. Biochem J (1988) 249:593–602. doi: 10.1042/bj2490593
44. Fontaine M, Demares MJ, Koistinen V, Day AJ, Davrinche C, Sim RB, et al. Truncated forms of human complement Factor H. Biochem J (1989) 258:927–30. doi: 10.1042/bj2580927
45. Misasi R, Huemer HP, Schwaeble W, Sölder E, Larcher C, Dierich MP. Human complement factor H: An additional gene product of 43kDa isolated from human plasma shows cofactor activity for the cleavage of the third component of complement. Eur J Immunol (1989) 19:1765–8. doi: 10.1002/eji.1830190936
46. Estaller C, Schwaeble W, Dierich M, Weiss EH. Human complement factor H: two factor H proteins are derived from alternatively spliced transcripts. Eur J Immunol (1991) 21:799–802. doi: 10.1002/eji.1830210337
47. Skerka C, Horstmann RD, Zipfel PF. Molecular cloning of a human serum protein structurally related to complement factor H. J Biol Chem (1991) 266:12015–20. doi: 10.1016/S0021-9258(18)99058-7
48. Vik DP, Munoz-Canoves P, Kozono H, Martin LG, Tack BF, Chaplin DD. Identification and sequence analysis of four complement factor H-related transcripts in mouse liver. J Biol Chem (1990) 265:3193–201. doi: 10.1016/S0021-9258(19)39753-4
49. Skerka C, Kuhn S, Gunther K, Lingelbach K, Zipfels PF. A Novel Short Consensus Repeat-containing Molecule Is Related to Human Complement Factor H*. (1993) 268(4):2904–8. doi: 10.1016/S0021-9258(18)53859-X
50. Skerka C, Timmann C, Horstmann RD, Zipfel PF. Two additional human serum proteins structurally related to complement factor H: Evidence for a family of factor H-related genes. J Immunol (1992) 148:3313–8.
51. Estaller C, Koistinen V, Schwaeble W, Dierich MP, Weiss EH. Cloning of the 1.4-kb mRNA species of human complement factor H reveals a novel member of the short consensus repeat family related to the carboxy terminal of the classical 150-kDa molecule. J Immunol (1991) 146:3190–6. doi: 10.5282/ubm/epub.3049
52. Skerka C, Hellwage J, Weber W, Tilkorn A, Buck F, Marti T, et al. The human factor H-related protein 4 (FHR-4). A novel short consensus repeat-containing protein is associated with human triglyceride-rich lipoproteins. J Biol Chem (1997) 272:5627–34. doi: 10.1074/jbc.272.9.5627
53. Skerka C, Zipfel PF, Moulds JAM, Taillon-Miller P, Hourcade D. The human factor H-related gene 2 (FHR2): structure and linkage to the coagulation factor XIIIb gene. Immunogenetics (1995) 42:268–74. doi: 10.1007/BF00176444
54. Díaz-Guillén MA, De Córdoba SR, Heine-Suñer D. A radiation hybrid map of complement factor H and factor H-related genes. Immunogenetics (1999) 49:549–52. doi: 10.1007/s002510050534
55. Murphy B, Georgiou T, Machet D, Hill P, McRae J. Factor H-related protein-5: A novel component of human glomerular immune deposits. Am J Kidney Dis (2002) 39:24–7. doi: 10.1053/ajkd.2002.29873
56. McRae JL, Cowan PJ, Power DA, Mitchelhill KI, Kemp BE, Morgan BP, et al. Human factor H-related protein 5 (FHR-5): A new complement-associated protein. J Biol Chem (2001) 276:6747–54. doi: 10.1074/jbc.M007495200
57. McRae JL, Murphy BF, Eyre HJ, Sutherland GR, Crawford J, Cowan PJ. Location and structure of the human FHR-5 gene. Genetica (2002) 114:157–61. doi: 10.1023/A:1015114512924
58. Józsi M, Richter H, Löschmann I, Skerka C, Buck F, Beisiegel U, et al. FHR-4A: A new factor H-related protein is encoded by the human FHR-4 gene. Eur J Hum Genet (2005) 13:321–9. doi: 10.1038/sj.ejhg.5201324
59. de Córdoba SR. Complement genetics and susceptibility to inflammatory disease. Lessons from genotype-phenotype correlations. Immunobiology (2016) 221:709–14. doi: 10.1016/j.imbio.2015.05.015
60. Pickering MC, de Jorge EG, Martinez-Barricarte R, Recalde S, Garcia-Layana A, Rose KL, et al. Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med (2007) 204:1249–56. doi: 10.1084/jem.20070301
61. Zhu L, Zhai Y-L, Wang F-M, Hou P, Lv J-C, Xu D-M, et al. and Complement Factor H-Related Protein Genes, CFHR3 and CFHR1, Affect Complement Activation in IgA Nephropathy. J Am Soc Nephrol (2015) 26:1195–204. doi: 10.1681/ASN.2014010096
62. Smith RJH, Appel GB, Blom AM, Cook HT, D’Agati VD, Fakhouri F, et al. C3 glomerulopathy — understanding a rare complement-driven renal disease. Nat Rev Nephrol (2019) 15(3):129–43. doi: 10.1038/s41581-018-0107-2
63. Sánchez-Corral P, González-Rubio C, Rodríguez De Córdoba S, López-Trascasa M. Functional analysis in serum from atypical Hemolytic Uremic Syndrome patients reveals impaired protection of host cells associated with mutations in factor H. Mol Immunol (2004) 41:81–4. doi: 10.1016/j.molimm.2004.01.003
64. Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship THJ, et al. Atypical aHUS: State of the art. Mol Immunol (2015) 67:31–42. doi: 10.1016/j.molimm.2015.03.246
65. Józsi M, Heinen S, Hartmann A, Ostrowicz CW, Hälbich S, Richter H, et al. and atypical hemolytic uremic syndrome: Mutations in the C-terminus cause structural changes and defective recognition functions. J Am Soc Nephrol (2006) 17:170–7. doi: 10.1681/ASN.2005080868
66. Manuelian T, Hellwage J, Meri S, Caprioli J, Noris M, Heinen S, et al. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J Clin Invest (2003) 111:1181–90. doi: 10.1172/JCI16651
67. Holmes LV, Strain L, Staniforth SJ, Moore I, Marchbank K, Kavanagh D, et al. Determining the Population Frequency of the CFHR3/CFHR1 Deletion at 1q32. PloS One (2013) 8:e60352. doi: 10.1371/journal.pone.0060352
68. Zhao J, Wu H, Khosravi M, Cui H, Qian X, Kelly JA, et al. Association of genetic variants in complement factor H and factor H-related genes with systemic lupus erythematosus susceptibility. PloS Genet (2011) 7. doi: 10.1371/journal.pgen.1002079
69. Zipfel PF, Edey M, Heinen S, Józsi M, Richter H, Misselwitz J, et al. Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PloS Genet (2007) 3(3):e41. doi: 10.1371/journal.pgen.0030041
70. Galebskaia L, Solovtsova I, Riumina E. Modification of proteolytic complement cascade after treatment with exogenous heparin. Vopr Med Khim. 47(1):91–7.
71. Tortajada A, Yébenes H, Abarrategui-Garrido C, Anter J, García-Fernández JM, Martínez-Barricarte R, et al. C3 glomerulopathy-associated CFHR1 mutation alters FHR oligomerization and complement regulation. J Clin Invest (2013) 123:2434–46. doi: 10.1172/JCI68280
72. Xiao X, Ghossein C, Tortajadam A, Zhang Y, Meyer N, Jones M, et al. Familial C3 glomerulonephritis caused by a novel CFHR5-CFHR2 fusion gene. Mol Immunol (2016) 77:89–96. doi: 10.1016/j.molimm.2016.07.007
73. Togarsimalemath SK, Sethi SK, Duggal R, Le Quintrec M, Jha P, Daniel R, et al. A novel CFHR1-CFHR5 hybrid leads to a familial dominant C3 glomerulopathy. Kidney Int (2017) 92:876–87. doi: 10.1016/j.kint.2017.04.025
74. Irmscher S, Brix SR, Zipfel SLH, Halder LD, Mutlutürk S, Wulf S, et al. Serum FHR1 binding to necrotic-type cells activates monocytic inflammasome and marks necrotic sites in vasculopathies. Nat Commun (2019) 10:2961. doi: 10.1038/s41467-019-10766-0
75. Tortajada A, Gutiérrez E, Goicoechea de Jorge E, Anter J, Segarra A, Espinosa M, et al. Elevated factor H–related protein 1 and factor H pathogenic variants decrease complement regulation in IgA nephropathy. Kidney Int (2017) 92:953–63. doi: 10.1016/j.kint.2017.03.041
76. Medjeral-Thomas NR, Lomax-Browne HJ, Beckwith H, Willicombe M, McLean AG, Brookes P, et al. Circulating complement factor H–related proteins 1 and 5 correlate with disease activity in IgA nephropathy. Kidney Int (2017) 92:942–52. doi: 10.1016/J.KINT.2017.03.043
77. Pouw RB, Delgado IG, Lera AL, de Córdoba SR, Wouters D, Kuijpers TW, et al. High complement factor H-related (FHR)-3 levels are associated with the atypical hemolytic-uremic syndrome-risk allele CFHR3*B. Front Immunol (2018) 9:848:848. doi: 10.3389/fimmu.2018.00848
78. Davila S, Wright VJ, Khor CC, Sim KS, Binder A, Breunis WB, et al. Genome-wide association study identifies variants in the CFH region associated with host susceptibility to meningococcal disease. Nat Genet (2010) 42:772–8. doi: 10.1038/ng.640
79. Józsi M. Factor H family proteins in complement evasion of microorganisms. Front Immunol (2017) 8:571. doi: 10.3389/fimmu.2017.00571
80. Caesar JJ, Lavender H, Ward PN, Exley RM, Eaton J, Chittock E, et al. Competition between antagonistic complement factors for a single protein on N. meningitidis rules Dis susceptibility (2014) 3:4008. doi: 10.7554/eLife.04008
81. Kasanmoentalib ES, Valls Serón M, Engelen-Lee JY, Tanck MW, Pouw RB, Van Mierlo G, et al. Complement factor H contributes to mortality in humans and mice with bacterial meningitis. J Neuroinflamm (2019) 16(1):279. doi: 10.1186/s12974-019-1675-1
82. Zipfel PF, Skerka C. Complement factor H and related proteins: an expanding family of complement-regulatory proteins? Immunol Today (1994) 15:121–6. doi: 10.1016/0167-5699(94)90155-4
83. Hellwage J, Jokiranta TS, Koistinen V, Vaarala O, Meri S, Zipfel PF. Functional properties of complement factor H-related proteins FHR-3 and FHR-4: Binding to the C3d region of C3b and differential regulation by heparin. FEBS Lett (1999) 462:345–52. doi: 10.1016/S0014-5793(99)01554-9
84. Fritsche LG, Lauer N, Hartmann A, Stippa S, Keilhauer CN, Oppermann M, et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Hum Mol Genet (2010) 19:4694–704. doi: 10.1093/hmg/ddq399
85. Hebecker M, Józsi M. Factor H-related protein 4 activates complement by serving as a platform for the assembly of alternative pathway C3 convertase via its interaction with C3b protein. J Biol Chem (2012) 287:19528–36. doi: 10.1074/jbc.M112.364471
86. Eberhardt HU, Buhlmann D, Hortschansky P, Chen Q, Böhm S, Kemper MJ, et al. Human factor H-related protein 2 (CFHR2) regulates complement activation. PloS One (2013) 8(11):e78617. doi: 10.1371/journal.pone.0078617
87. McRae JL, Duthy TG, Griggs KM, Ormsby RJ, Cowan PJ, Cromer BA, et al. Human Factor H-Related Protein 5 Has Cofactor Activity, Inhibits C3 Convertase Activity, Binds Heparin and C-Reactive Protein, and Associates with Lipoprotein. J Immunol (2005) 174:6250–6. doi: 10.4049/jimmunol.174.10.6250
88. Zwarthoff SA, Berends ETM, Mol S, Ruyken M, Aerts PC, Józsi M, et al. Functional characterization of alternative and classical pathway C3/C5 convertase activity and inhibition using purified models. Front Immunol (2018) 9:1691:1691. doi: 10.3389/fimmu.2018.01691
89. Vernon KA, Goicoechea De Jorge E, Hall AE, Fremeaux-Bacchi V, Aitman TJ, Cook HT, et al. Acute presentation and persistent glomerulonephritis following streptococcal infection in a patient with heterozygous complement factor H-related protein 5 deficiency. Am J Kidney Dis (2012) 60:121–5. doi: 10.1053/j.ajkd.2012.02.329
90. DeCordova S, Abdelgany A, Murugaiah V, Pathan AA, Nayak A, Walker T, et al. Secretion of functionally active complement factor H related protein 5 (FHR5) by primary tumour cells derived from Glioblastoma Multiforme patients. Immunobiology (2019) 224:625–31. doi: 10.1016/j.imbio.2019.07.006
91. Heinen S, Hartmann A, Lauer N, Wiehl U, Dahse HM, Schirmer S, et al. Factor H-related protein 1 (CFHR-1) inhibits complement C5 convertase activity and terminal complex formation. Blood (2009) 114:2439–47. doi: 10.1182/blood-2009-02-205641
92. Shi K, Wang Z, Liu Y, Gong Y, Fu Y, Li S, et al. CFHR1-Modified Neural Stem Cells Ameliorated Brain Injury in a Mouse Model of Neuromyelitis Optica Spectrum Disorders. J Immunol (2016) 197:3471–80. doi: 10.4049/jimmunol.1600135
93. Strobel S, Abarrategui-Garrido C, Fariza-Requejo E, Seeberger H, Sánchez-Corral P, Józsi M. Factor H-related protein 1 neutralizes anti-factor H autoantibodies in autoimmune hemolytic uremic syndrome. Kidney Int (2011) 80:397–404. doi: 10.1038/ki.2011.152
94. Mészáros T, Csincsi ÁI, Uzonyi B, Hebecker M, Fülöp TG, Erdei A, et al. Factor H inhibits complement activation induced by liposomal and micellar drugs and the therapeutic antibody rituximab in vitro. Nanomedicine Nanotechnology Biol Med (2016) 12:1023–31. doi: 10.1016/j.nano.2015.11.019
95. Csincsi ÁI, Szabó Z, Bánlaki Z, Uzonyi B, Cserhalmi M, Kárpáti É, et al. FHR-1 Binds to C-Reactive Protein and Enhances Rather than Inhibits Complement Activation. J Immunol (2017) 199:292–303. doi: 10.4049/jimmunol.1600483
96. Närkiö-Mäkelä M, Hellwage J, Tahkokallio O, Meri S. Complement-regulator factor H and related proteins in Otitis media with effusion. Clin Immunol (2001) 100:118–26. doi: 10.1006/clim.2001.5043
97. Seguin-Devaux C, Plesseria JM, Verschueren C, Masquelier C, Iserentant G, Fullana M, et al. FHR4-based immunoconjugates direct complement-dependent cytotoxicity and phagocytosis towards HER2-positive cancer cells. Mol Oncol (2019) 13:2531–53. doi: 10.1002/1878-0261.12554
98. Csincsi ÁI, Kopp A, Zöldi M, Bánlaki Z, Uzonyi B, Hebecker M, et al. Factor H–Related Protein 5 Interacts with Pentraxin 3 and the Extracellular Matrix and Modulates Complement Activation. J Immunol (2015) 194:4963–73. doi: 10.4049/jimmunol.1403121
99. Chen Q, Manzke M, Hartmann A, Büttner M, Amann K, Pauly D, et al. Complement factor H-related 5-hybrid proteins anchor properdin and activate complement at self-surfaces. J Am Soc Nephrol (2016) 27:1413–25. doi: 10.1681/ASN.2015020212
100. Mihlan M, Hebecker M, Dahse HM, Hälbich S, Huber-Lang M, Dahse R, et al. Human complement factor H-related protein 4 binds and recruits native pentameric C-reactive protein to necrotic cells. Mol Immunol (2009) 46:335–44. doi: 10.1016/j.molimm.2008.10.029
101. Kárpáti É, Papp A, Schneider AE, Hajnal D, Cserhalmi M, Csincsi ÁI, et al. Interaction of the Factor H Family Proteins FHR-1 and FHR-5 With DNA and Dead Cells: Implications for the Regulation of Complement Activation and Opsonization and Dead Cells: Implications for the Regulation of Complement Activation and Opsonization. Front Immunol (2020) 11:1297. doi: 10.3389/fimmu.2020.01297
102. Józsi M, Schneider AE, Kárpáti É, Sándor N. Complement factor H family proteins in their non-canonical role as modulators of cellular functions. Semin Cell Dev Biol (2019) 85:122–31. doi: 10.1016/j.semcdb.2017.12.018
103. Losse J, Zipfel PF, Józsi M. and Factor H-Related Protein 1 Bind to Human Neutrophils via Complement Receptor 3, Mediate Attachment to Candida albicans , and Enhance Neutrophil Antimicrobial Activity. J Immunol (2010) 184:912–21. doi: 10.4049/jimmunol.0901702
104. Ansari M, Mckeigue PM, Skerka C, Hayward C, Rudan I, Vitart V, et al. Genetic influences on plasma CFH and CFHR1 concentrations and their role in susceptibility to age-related macular degeneration. Hum Mol Genet (2013) 22:4857–69. doi: 10.1093/hmg/ddt336
105. Esparza-Gordillo J, Soria JM, Buil A, Almasy L, Blangero J, Fontcuberta J, et al. Genetic and environmental factors influencing the human factor H plasma levels. Immunogenetics (2004) 56:77–82. doi: 10.1007/s00251-004-0660-7
106. Hakobyan S, Harris CL, Tortajada A, Dejorge EG, Garcia-Layana A, Fernandez-Robredo P, et al. Measurement of factor H variants in plasma using variant-specific monoclonal antibodies: Application to assessing risk of age-related macular degeneration. Investig Ophthalmol Vis Sci (2008) 49:1983–90. doi: 10.1167/iovs.07-1523
107. Sofat R, Mangione PP, Gallimore JR, Hakobyan S, Hughes TR, Shah T, et al. Distribution and determinants of circulating complement factor H concentration determined by a high-throughput immunonephelometric assay. J Immunol Methods (2013) 390:63–73. doi: 10.1016/j.jim.2013.01.009
108. Friese MA, Hellwage J, Jokiranta TS, Meri S, Müller-Quernheim HJ, Peter HH, et al. Different regulation of factor H and FHL-1/reconectin by inflammatory mediators and expression of the two proteins in rheumatoid arthritis (RA). Clin Exp Immunol (2000) 121:406–15. doi: 10.1046/j.1365-2249.2000.01285.x
109. Zhang P, Zhu M, Geng-Spyropoulos M, Shardell M, Gonzalez-Freire M, Gudnason V, et al. A novel, multiplexed targeted mass spectrometry assay for quantification of complement factor H (CFH) variants and CFH-related proteins 1–5 in human plasma. Proteomics (2017) 17(6):10. doi: 10.1002/pmic.201600237
110. Schäfer N, Grosche A, Reinders J, Hauck SM, Pouw RB, Kuijpers TW, et al. Complement Regulator FHR-3 Is Elevated either Locally or Systemically in a Selection of Autoimmune Diseases. Front Immunol (2016) 7:542:542. doi: 10.3389/fimmu.2016.00542
111. Pouw RB, Brouwer MC, van Beek AE, Józsi M, Wouters D, Kuijpers TW. Complement factor H-related protein 4A is the dominant circulating splice variant of CFHR4. Front Immunol (2018) 9:729:1. doi: 10.3389/fimmu.2018.00729
112. Zhu L, Guo W YI, Su-Fang S, Li-Jun L, J-Cheng LV, Medjeral-Thomas NR, et al. Circulating complement factor H–related protein 5 levels contribute to development and progression of IgA nephropathy. Kidney Int (2018) 94:150–8. doi: 10.1016/j.kint.2018.02.023
113. Nilsson B, Ekdahl KN. Complement diagnostics: Concepts, indications, and practical guidelines. Clin Dev Immunol (2012) 2012:962702. doi: 10.1155/2012/962702
114. Gaya da Costa M, Poppelaars F, van Kooten C, Mollnes TE, Tedesco F, Würzner R, et al. Age and Sex-Associated Changes of Complement Activity and Complement Levels in a Healthy Caucasian Population. Front Immunol (2018) 9:2664:2664. doi: 10.3389/fimmu.2018.02664
115. van Beek AE, Kamp A, Kruithof S, Nieuwenhuys EJ, Wouters D, Jongerius I, et al. Reference intervals of factor H and factor H-related proteins in healthy children. Front Immunol (2018) 9:1727. doi: 10.3389/fimmu.2018.01727
116. Prohászka Z, Kirschfink M, Frazer-Abel A. Complement analysis in the era of targeted therapeutics. Mol Immunol (2018) 102:84–8. doi: 10.1016/j.molimm.2018.06.001
117. Dekkers G, Brouwer MC, Jeremiasse J, Kamp A, Biggs RM, van Mierlo G, et al. Unraveling the Effect of a Potentiating Anti–Factor H Antibody on Atypical Hemolytic Uremic Syndrome–Associated Factor H Variants. J Immunol (2020) 205:1778–86. doi: 10.4049/jimmunol.2000368
118. Pouw RB, Brouwer MC, de Gast M, van Beek AE, van den Heuvel LP, Schmidt CQ, et al. Potentiation of complement regulator factor H protects human endothelial cells from complement attack in aHUS sera. Blood Adv (2019) 3:621–32. doi: 10.1182/bloodadvances.2018025692
119. Merinero HM, García SP, García-Fernández J, Arjona E, Tortajada A, Rodríguez de Córdoba S. Complete functional characterization of disease-associated genetic variants in the complement factor H gene. Kidney Int (2018) 93:470–81. doi: 10.1016/j.kint.2017.07.015
120. Pechtl IC, Kavanagh D, Mcintosh N, Harris CL, Barlow PN. Disease-associated N-terminal complement factor H mutations perturb cofactor and decay-accelerating activities. J Biol Chem (2011) 286:11082–90. doi: 10.1074/jbc.M110.211839
121. Nichols EM, Barbour TD, Pappworth IY, Wong EKS, Palmer JM, Sheerin NS, et al. An extended mini-complement factor H molecule ameliorates experimental C3 glomerulopathy. Kidney Int (2015) 88:1314–22. doi: 10.1038/ki.2015.233
122. Prohászka Z, Nilsson B, Frazer-Abel A, Kirschfink M. Complement analysis 2016: Clinical indications, laboratory diagnostics and quality control. Immunobiology (2016) 221:1247–58. doi: 10.1016/j.imbio.2016.06.008
123. Mollnes TE, Garred P, Bergseth G. Effect of time, temperature and anticoagulants on in vitro complement activation: consequences for collection and preservation of samples to be examined for complement activation. Clin Exp Immunol (1988) 73:484–8.
124. Baker M, Penny D. Is there a reproducibility crisis? Nature (2016) 533:452–4. doi: 10.1038/533452A
125. Emmerich CH, Harris CM. “Minimum Information and Quality Standards for Conducting, Reporting, and Organizing In Vitro Research,”. In: Bespalov A., Michel M., Steckler T. Cham: Springer (2019) 257:177–96. doi: 10.1007/164_2019_284
126. Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. J Physiol (2020) 598:3793–801. doi: 10.1113/JP280389
127. Clark SJ, Schmidt CQ, White AM, Hakobyan S, Morgan BP, Bishop PN. Identification of Factor H–like Protein 1 as the Predominant Complement Regulator in Bruch’s Membrane: Implications for Age-Related Macular Degeneration. J Immunol (2014) 193:4962–70. doi: 10.4049/jimmunol.1401613
128. Medjeral-Thomas NR, Moffitt H, Lomax-Browne HJ, Constantinou N, Cairns T, Cook HT, et al. Glomerular Complement Factor H–Related Protein 5 (FHR5) Is Highly Prevalent in C3 Glomerulopathy and Associated With Renal Impairment. Kidney Int Rep (2019) 4:1387–400. doi: 10.1016/j.ekir.2019.06.008
129. Medjeral-Thomas NR, Troldborg A, Constantinou N, Lomax-Browne HJ, Hansen AG, Willicombe M, et al. Progressive IgA Nephropathy Is Associated With Low Circulating Mannan-Binding Lectin–Associated Serine Protease-3 (MASP-3) and Increased Glomerular Factor H–Related Protein-5 (FHR5) Deposition. Kidney Int Rep (2018) 3:426–38. doi: 10.1016/j.ekir.2017.11.015
130. Rizner TL, Sasano H, Choi MH, Odermatt A, Adamski J. Recommendations for description and validation of antibodies for research use. J Steroid Biochem Mol Biol (2016) 156:40–2. doi: 10.1016/j.jsbmb.2015.11.021
131. Uhlen M, Bandrowski A, Carr S, Edwards A, Ellenberg J, Lundberg E, et al. A proposal for validation of antibodies. Nat Methods (2016) 13:823–7. doi: 10.1038/nmeth.3995
132. Acharya P, Quinlan A, Neumeister V. The ABCs of finding a good antibody: How to find a good antibody, validate it, and publish meaningful data. F1000Research (2017) 6:851. doi: 10.12688/f1000research.11774.1
133. Pauly D, Hanack K. How to avoid pitfalls in antibody use. F1000Research (2015) 4:691. doi: 10.12688/f1000research.6894.1
134. Voskuil JLA. The challenges with the validation of research antibodies. F1000Research (2017) 6:161. doi: 10.12688/f1000research.10851.1
135. Pouw RB, Vredevoogd DW, Kuijpers TW, Wouters D. Of mice and men: The factor H protein family and complement regulation. Mol Immunol (2015) 67:12–20. doi: 10.1016/j.molimm.2015.03.011
136. Kristensen T, Tack BF. Murine protein H is comprised of 20 repeating units, 61 amino acids in length. Proc Natl Acad Sci U.S.A. (1986) 83:3963–7. doi: 10.1073/pnas.83.11.3963
137. Høgåsen K, Jansen JH, Mollnes TE, Hovdenes J, Harboe M. Hereditary porcine membranoproliferative glomerulonephritis type II is caused by factor H deficiency. J Clin Invest (1995) 95:1054–61. doi: 10.1172/JCI117751
138. Pickering MC, Cook HT, Warren J, Bygrave AE, Moss J, Walport MJ, et al. Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet (2002) 31:424–8. doi: 10.1038/ng912
139. Coffey PJ, Gias C, McDermott CJ, Lundh P, Pickering MC, Sethi C, et al. Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc Natl Acad Sci (2007) 104:16651–6. doi: 10.1073/pnas.0705079104
140. Antonioli AH, White J, Crawford F, Renner B, Marchbank KJ, Hannan JP, et al. Modulation of the Alternative Pathway of Complement by Murine Factor H–Related Proteins. J Immunol (2018) 200:316–26. doi: 10.4049/jimmunol.1602017
141. Li X, Hao Z, Liu X, Li W. Deficiency of Mouse FHR-1 Homolog, FHR-E, Accelerates Sepsis, and Acute Kidney Injury Through Enhancing the LPS-Induced Alternative Complement Pathway. Front Immunol (2020) 11:1123:1123. doi: 10.3389/fimmu.2020.01123
142. Cserhalmi M, Csincsi ádám I, Mezei Z, Kopp A, Hebecker M, Uzonyi B, et al. The murine factor H-Related protein FHR-B promotes complement activation. Front Immunol (2017) 8:1145. doi: 10.3389/fimmu.2017.01145
143. Hellwage J, Eberle F, Babuke T, Seeberger H, Richter H, Kunert A, et al. Two factor H-related proteins from the mouse: Expression analysis and functional characterization. Immunogenetics (2006) 58:883–93. doi: 10.1007/s00251-006-0153-y
144. Fakhouri F, Goicoechea De Jorge E, Brune F, Azam P, Cook HT, Pickering MC. Treatment with human complement factor H rapidly reverses renal complement deposition in factor H-deficient mice. (2010) 78(3):279–86. doi: 10.1038/ki.2010.132
145. Józsi M, Oppermann M, Lambris JD, Zipfel PF. The C-terminus of complement factor H is essential for host cell protection. Mol Immunol (2007) 44:2697–706. doi: 10.1016/j.molimm.2006.12.001
146. Paixão-Cavalcante D, Hanson S, Botto M, Cook HT, Pickering MC. Factor H facilitates the clearance of GBM bound iC3b by controlling C3 activation in fluid phase. Mol Immunol (2009) 46:1942–50. doi: 10.1016/j.molimm.2009.03.030
147. Michelfelder S, Parsons J, Bohlender LL, Hoernstein SNW, Niederkrüger H, Busch A, et al. Moss-produced, glycosylation-optimized human Factor H for therapeutic application in complement disorders. J Am Soc Nephrol (2017) 28:1462–74. doi: 10.1681/ASN.2015070745
148. Schmidt CQ, Slingsby FC, Richards A, Barlow PN. Production of biologically active complement factor H in therapeutically useful quantities. Protein Expr Purif (2011) 76:254–63. doi: 10.1016/j.pep.2010.12.002
149. Yang Y, Denton H, Davies OR, Smith-Jackson K, Kerr H, Herbert AP, et al. An engineered complement factor H construct for treatment of C3 glomerulopathy. J Am Soc Nephrol (2018) 29:1649–61. doi: 10.1681/ASN.2017091006
150. Hebecker M, Alba-Domínguez M, Roumenina LT, Reuter S, Hyvärinen S, Dragon-Durey M-A, et al. An Engineered Construct Combining Complement Regulatory and Surface-Recognition Domains Represents a Minimal-Size Functional Factor H. J Immunol (2013) 191:912–21. doi: 10.4049/jimmunol.1300269
151. Fridkis-Hareli M, Storek M, Mazsaroff I, Risitano AM, Lundberg AS, Horvath CJ, et al. Design and development of TT30, a novel C3d-targeted C3/C5 convertase inhibitor for treatment of human complement alternative pathway-mediated diseases. Blood (2011) 118:4705–13. doi: 10.1182/blood-2011-06-359646
152. Shaughnessy E, Lambris JD, Ram S, Anna D, Blom M, Magda M, et al. Streptococcus pyogenes Gram-Positive Bacterial Pathogen Therapeutic Approach against the IgG Chimeric Proteins as a – Factor H. (2021) 199(11):3828–39. doi: 10.4049/jimmunol.1700426
153. Michelfelder S, Fischer F, Wäldin A, Hörle KV, Pohl M, Parsons J, et al. The MFHR1 fusion protein is a novel synthetic multitarget complement inhibitor with therapeutic potential. J Am Soc Nephrol (2018) 29:1141–53. doi: 10.1681/ASN.2017070738
154. Holers VM, Tomlinson S, Kulik L, Atkinson C, Rohrer B, Banda N, et al. New therapeutic and diagnostic opportunities for injured tissue-specific targeting of complement inhibitors and imaging modalities. Semin Immunol (2016) 28:260–7. doi: 10.1016/j.smim.2016.05.007
155. Rohrer B, Long Q, Coughlin B, Brooks Wilson R, Huang Y, Qiao F, et al. A targeted inhibitor of the alternative complement pathway reduces angiogenesis in a mouse model of age-related macular degeneration. Investig Ophthalmol Vis Sci (2009) 50:3056–64. doi: 10.1167/iovs.08-2222
156. Huang Y, Qiao F, Atkinson C, Holers VM, Tomlinson S. A Novel Targeted Inhibitor of the Alternative Pathway of Complement and Its Therapeutic Application in Ischemia/Reperfusion Injury. J Immunol (2008) 181:8068–76. doi: 10.4049/jimmunol.181.11.8068
157. Sekine H, Hsieh Kinser TT, Qiao F, Martinez E, Paulling E, Ruiz P, et al. The benefit of targeted and selective inhibition of the alternative complement pathway for modulating autoimmunity and renal disease in MRL/lpr mice. Arthritis Rheum (2011) 63:1076–85. doi: 10.1002/art.30222
Keywords: complement system, factor H (FH), factor H-related protein, factor H-like protein 1, challenges - development directions
Citation: Poppelaars F, Goicoechea de Jorge E, Jongerius I, Baeumner AJ, Steiner M-S, Józsi M, Toonen EJM, Pauly D and the SciFiMed consortium (2021) A Family Affair: Addressing the Challenges of Factor H and the Related Proteins. Front. Immunol. 12:660194. doi: 10.3389/fimmu.2021.660194
Received: 29 January 2021; Accepted: 08 March 2021;
Published: 30 March 2021.
Edited by:
Cees Van Kooten, Leiden University, NetherlandsReviewed by:
Michael Kirschfink, Heidelberg University, GermanyCopyright © 2021 Poppelaars, Goicoechea de Jorge, Jongerius, Baeumner, Steiner, Józsi, Toonen, Pauly and the SciFiMed consortium. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana Pauly, ZGlhbmEucGF1bHlAdWtyLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.