 Domenick E. Kennedy
Domenick E. Kennedy Marcus R. Clark
Marcus R. Clark
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 31 March 2021
Sec. B Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.659151
This article is part of the Research Topic Germinal Centers in Lymphoid and Non-Lymphoid Tissues: Adaptive and Evolving Structures View all 14 articles
Protective high affinity antibody responses emerge through an orchestrated developmental process that occurs in germinal centers (GCs). While GCs have been appreciated since 1930, a wealth of recent progress provides new insights into the molecular and cellular dynamics governing humoral immunity. In this review, we highlight advances that demonstrate that fundamental GC B cell function, selection, proliferation and SHM occur within distinct cell states. The resulting new model provides new opportunities to understand the evolution of immunity in infectious, autoimmune and neoplastic diseases.
Since the histologic identification in 1930, almost a century of investigation has revealed the central importance of germinal centers (GCs) in humoral immunity (1). Fundamental to GC function is the orchestration of the molecular programs of immunoglobulin gene somatic hypermutation (SHM), selection for antibody affinity and specificity, and proliferative expansion of selected cells. Within the GC, these processes are coordinated with remarkable rapidity such that a B cell transits through these processes in four to six hours allowing for numerous rounds of selection and immune amplification during the course of a typical acute GC reaction of 14 to 21 days (2, 3). From the GC circuit both plasma cells (PCs) and memory B cells (MBCs) are produced ensuring both acute and durable antigen-specific immunity.
The importance of GCs in infection and vaccine responses has been demonstrated in numerous studies (3–5). However, the molecular nature of the GC incurs risk. One risk is that, through stochastic SHM, autoreactive and potentially pathogenic antibodies arise. However, elegant studies have demonstrated that GCs strongly select against autoreactivity (6). Indeed, selection against autoreactivity appears to have primacy over selection for affinity. The mechanisms by which GCs purge the antigen-selected repertoire of autoreactivity remain unclear.
The other risk of ongoing SHM and proliferation is neoplastic transformation. The GC B cell molecular program establishes a state that promotes survival in the presence of increased genomic stress (7, 8). This both enables SHM and increases the risk of lymphoma (9, 10). Indeed, GC B cells are precursors for large B cell lymphoma, follicular lymphoma, and Burkitt lymphoma (11, 12). Transformation has been linked to off-target AID activity (13). Studies in humans and mice have revealed multiple genetic, epigenetic and signaling mechanisms that individually mitigate the risk of neoplastic transformation inherit to the GC response. Recent studies are providing insights into how these mechanisms are coordinated with those that drive affinity maturation (14–16).
In this review, we will discuss the current understanding of how SHM, selection and proliferation are coordinated within the GC. We will first discuss the cellular architecture of the GC and then what is known about underlying molecular programs. Finally, we will propose a model that segregates fundamental GC molecular functions into separate cell states and niches in a way that enables effective adaptive immunity and mitigates the risks of neoplastic transformation.
The initial histological description of GCs noted characteristic dark and light zones (DZ and LZ respectively). Live cell imaging, and other complementary approaches, have provided remarkable insights into how GCs form and polarize into these two zones that perform very different functions. However, as described below, it has become clear that simple division of cells into DZ and LZ populations obscures underlying molecular GC functions.
Activation of naïve B cells in primary follicles induces migration to interfollicular areas and conjugates with antigen-specific T cells. B cells then present antigens to T cells and receive help through CD40-CD40L signals that promote B cell survival (5, 17, 18). Based on intravital microscopy experiments, these B-T cell conjugates can be found within a day after immunization (19, 20). NF-κB signaling is critical downstream of BCR activation to form GCs (21). However, while GCs do not form in mice with impaired NF-κB signaling, responding B cells are still able to migrate to the B-T cell border and present antigen to T cells. Instead, NF-κB signaling regulates expression of the transcription factors (TFs) IRF4 and BCL6, which are critical for both the GC and PC developmental programs (18, 20, 22, 23).
The formation of B-T cell conjugates is followed by T cells entering the B cell follicle on day 3. Subsequently, on day 4, B cells re-enter the B cell follicle and proliferate to form early GCs (18–20). Contrary to these distinct cellular events, the molecular regulation underlying the early events after antigen encounter is still being defined. Recent studies suggest there are multiple pre-GC B cell states leading up to commitment to the GC B cell program (24). Some of these are controlled by the transcriptional repressor BCL6 (20, 25). BCL6 is upregulated in the outer follicle after antigen encounter and is important for forming B-T cell conjugates prior to GC commitment (20). Within early and mature GCs, BCL6 controls B cell positioning within the B cell follicle (20, 23, 26–28). It also enhances GC B cell proliferation by making them more tolerant of DNA damage (7, 8). Ultimately, BCL6 drives the GC B cell program and prevents PC differentiation through inhibition of Prdm1 (BLIMP) (29).
Recent studies further reinforce the importance of BCL6 in GC B cell commitment. By modulating the amount of Bcl6 expression in transgenic mice, one study found that Bcl6hi B cells responding to immunization were more likely to commit to the GC program (30). In complementary studies, Zhang et al. found that while T-B cell conjugates are important to generate GC B cell precursors, increasing the time of T-B cell conjugates or CD40 signaling reduces progression to a Bcl6hi state and favors plasmablast (PB) differentiation in the extrafollicular region (24).
In 2019, it was discovered that class switch recombination (CSR), the process by which B cells perform DNA rearrangements at the heavy chain locus to replace IgM and IgD, for IgA, IgG, or IgE, occurs during the GC initiation phase (31). Most studies on CSR have been performed in vitro and focused on the molecular mechanism, reviewed (32). However, strong evidence for when CSR occurs in vivo was lacking. Through a combination of imaging and molecular experiments Roco et al. found that CSR occurs in the first few days after activation and prior to GC commitment (31). Evidence for this included the observation of predominantly IgM+ GCs as well as the visualization of CSR prior to mature GC formation. This study resolved a critical question in the field placing CSR in the early events during GC initiation, and validated earlier evidence that CSR might occur prior to mature GCs (33, 34). The transcriptional states associated with CSR have now been resolved at the single cell level, further accelerating our understanding of early events in the GC reaction (35).
By day 4 after immunization, GCs precursors begin to expand and polarize to form LZ and DZ areas by day 7 (18, 36). The LZ contains more sparse populations of B cells that capture antigen from follicular dendritic cells (FDCs) and receive help from cognate T follicular helper (TFH) cells (37). B cells in the LZ are selected based on their competency to present antigen to TFH cells as well as BCR signal strength (38–40). These B cell interactions with TFH cells guide the major known GC fates which include cyclic re-entry, cell death as well as PC and MBC differentiation (37). Tingible body macrophages (TBMs), which lie within the DZ, clear dying B cells and thereby likely prevent inflammation and autoimmunity (37, 41).
A wealth of data assign both proliferation and somatic hypermutation to the DZ (3, 18, 37). However, genomic mutation, and the attendant genotoxic stress are incompatible and indeed antagonistic to proliferation. It is possible that mechanisms intrinsic to genotoxic stress, such as the sensing of DNA breaks by p53, segregate proliferation from SHM within the DZ (42). However, as discussed below, these incompatible processes occur in different cell populations each occupying a unique niche within the GC.
An extensive body of literature has revealed that the LZ and DZ perform very different functions and that this is associated with great molecular complexity. However, when LZ (CD83hiCXCR4low) and DZ(CD83loCXCR4hi) cells are isolated and characterized for RNA expression and genomic accessibility, they are remarkably similar (14, 38, 43). Taken at face value, these data suggest that primarily post-transcriptional mechanisms regulate cycling through the dark and light zones. This seems attractive as it could provide for very rapid cell fate transitions. Alternatively, it is possible that simply dividing GC B cells into two populations obscures important underlying molecular dynamics.
There are data supporting the latter possibility. In addition to driving affinity maturation, the GC selects for differentiation into both PCs and MBCs. Precursors of these populations must exist in the GC and, indeed, some have been identified (16, 44, 45). However, these populations are not observed upon simple division of the GC into two populations.
Given the rapidity of the GC cycle, it is also possible that B cells are always in transition (46). This is suggested by available single cell RNA-Seq studies, where GC B cells do not resolve into discrete cell populations (14–16). However, some cell fate decisions are discrete and are associated with definitive checkpoints. A cell either undergoes mitosis or it does not. Likewise for apoptosis. It is possible that SHM occurs along a gradient of cell states. However, we would argue that such a strategy would incur unnecessary genomic risk. Furthermore, during B lymphopoiesis genomic recombination and proliferation are segregated into very different cell populations (47, 48). As described below, this strategy is recapitulated in the GC.
The canonical DZ encompasses a gradient of CXCR4 and CD83 surface expression. Differing levels of CXCR4 raises the possibility that these cells could occupy different niches within the DZ and therefore be imbued with different functions. Remarkably, when we split the DZ into two populations (CXCR4hiCD83+ and CXCR4+CD83-), and therefore GC B cells into three populations, there were striking differences in RNA and protein expression (14). It became clear that proliferation was restricted to the CXCR4hiCD83+ population. More specifically, these cells were the site of mitosis marked by increased cell size, enrichment of cells in G2/M and high expression of the mitotic factor Cyclin B1. Previous work has indicated that GC B cells can transit the G1/S checkpoint in any compartment (49, 50). Our data indicate that mitosis occurs in a discrete cell state, which we refer to as proliferative DZ cells or DZp cells.
Conversely, CXCR4+CD83- cells have features of cells undergoing SHM. These include high expression of Aicda and genotoxic stress genes as well as induced phosphoproteomic pathways of DNA damage response. These cells were also in the G1 phase of the cell cycle where AID activity is known to occur (14, 51). We refer to this DZ subset as DZd for DZ differentiation. While immunoglobulin gene transcription, which is necessary for AID targeting (52), is induced in DZd, it was effectively repressed in DZp cells. These and other data indicate that both positive and repressive mechanisms ensure segregation of mitosis and SHM into different cell states.
Comparison across RNA expression and proteomic data sets indicated a cyclic progression in which cells selected in the LZ transit to the DZp for mitosis and then to the DZd for SHM (14). These data also revealed how the molecular programs in one cell state primes for functions in the next. Myc provides an example. The Myc locus displayed increased genome accessibility and transcription in LZ B cells compared to the DZ populations. However, phosphorylated MYC protein was observed in LZ and DZp B cells, with the downstream MYC program uniquely high in DZp cells.
These data, in conjunction with scRNA-Seq data (14, 15) indicate that expression across each stage is dynamic and that the initiation of transcriptional programs rapidly induces proteomic and functional programs as cells transit through each stage. Overall, these data help explain the rapidity of the GC cycle (2, 3, 38, 50, 53–57).
Remarkably, ATAC-Seq of the LZ, DZp and DZd revealed enhancer accessibility was also very dynamic across the three cell states (14). These differences were not only quantitative but also qualitative with characteristic TF binding motifs becoming accessible in each GC B cell state. In fact, the most enriched TF binding motifs found in each subpopulation were for TFs known to have importance within the GC. These included CTCF in the DZd, OCT2 in the DZp, and SpiB and PU.1 in the LZ (58–62). OCT2 is required for GC B cell proliferation and is dysregulated in B cell lymphomas (62). While OCT2 binding motifs were enriched in DZp accessible regions, Oct2 expression did not change between subsets. Instead, expression of Pou2af1 (binding partner of OCT2) was increased in DZp cells (14, 63–65). These data suggest that epigenetic mechanisms play important and complementary roles to TFs in regulating GC B cell fate. We propose that, by mechanisms yet to be defined, GC B cells integrate regulatory mechanisms across the whole vertical pathway of protein expression to affect rapid cell fate decisions.
Our analysis revealed several markers of the DZp population that allowed identification of distinct clusters of DZp cells within the larger DZd pool (14). These clusters did not arise solely from clonal proliferative expansion. Rather, cells appear to migrate to these DZp niches to undergo mitosis. Why would mitosis occur in a distinct niche? Subsequent analysis demonstrated that DZp cells are intimately intertwined with TBMs. Furthermore, within these cellular aggregates are GC DZp cells that are apoptotic and appear to be undergoing engulfment by TBMs. Integration of these and molecular data suggests a new model of GC compartmentalization (Figure 1).
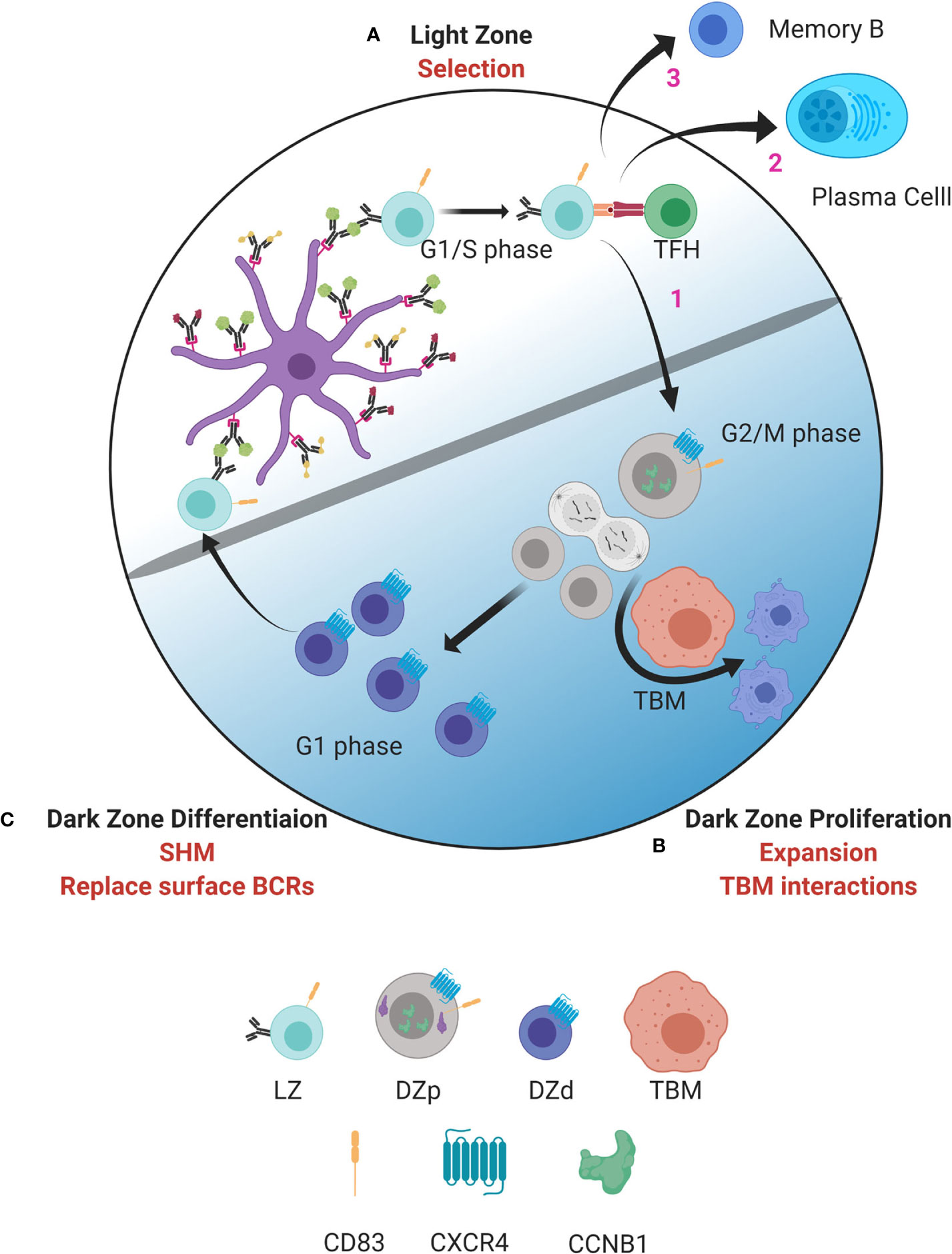
Figure 1 Model of germinal center dynamics and compartmentalization. GC B cells progress through a series of molecular states compartmentalizing key functions to distinct spatial niches. (A) GC B cell selection occurs in the LZ. B cells entering the LZ from the DZd first attempt to capture antigen deposited on follicular dendritic cells (FDC). This is followed by antigen processing and presentation to TFH cells in the context of MHC class II. Interactions between LZ B cells and TFH determine if B cells are fated to differentiate into MBCs or PBs, undergo cyclic re-entry into the DZp, or initiate apoptosis. Cells selected for cyclic re-entry migrate to the DZp (B). We propose that B cells that initiate apoptosis in the LZ are cleared by TBMs in the DZp niche. Those cells that are successfully selected in the LZ, and therefore are not cleared by TBMs, undergo mitosis. (C) After one or more rounds of cell division, GC B cells transit to the DZd compartment where differentiative functions, such as SHM and replacing old BCRs with newly mutated BCRs, are proposed to occur. B cells that successfully complete processes in the DZd migrate to the LZ to undergo selection. Figure created with BioRender.com.
As well described, selection for antigen receptor affinity occurs within the LZ with coordinated collaboration between GC LZ B cells, FDCs and TFH cells (Figure 1A). Interactions between LZ B cells and TFH cells are critical to determine GC B cell fate. Our data suggest that those LZ B cells that have been successively selected, and also those that have not, migrate to the DZp niche (Figure 1B). Those fated for apoptosis are then eliminated by TBMs, which are the principal macrophage cell population within the GC (37, 66). The mechanisms by which TBMs identify dying B cells is not fully understood. However, it is known that FDCs secrete a molecule called MFGE8, which labels apoptotic cells for phagocytosis (67, 68). This labeling occurs by binding phosphatidylserine that is externalized on the surface of cells undergoing apoptosis (69). Recently, a molecule called decay accelerating factor (DAF) has also been proposed to regulate GC B cell phagocytosis (70). Such signals likely help TBMs distinguish between healthy B cells and B cells undergoing apoptosis. We propose that B cells that have been successfully selected in the LZ, and are not cleared by TBMs, can then undergo mitosis.
Why would clearance of apoptotic cells be coordinated with mitosis? Apoptosis is the usual pathway of GC cell death (37). However, when apoptotic cells attempt division it can result in mitotic catastrophe and necrosis (71). Therefore, by positioning TBMs within the DZp niche, necrosis, and the attendant inflammation, would be prevented. Many factors affect the G1/S checkpoint and the decision to initiate proliferation (72). Our data suggest that the main quality check for cell cycle progression is at G2/M.
One of the main controllers of proliferation within the DZp is Myc. B cells selected for cyclic re-entry typically divide on average twice in the DZ (49, 50). The number of divisions is dependent on the strength of signal received from TFH cells. This, in turn, correlates with Myc protein levels implicating Myc in controlling the extent of proliferation (73). In this model, selection in the LZ determines the magnitude of Myc expression. Proliferation then continues until Myc levels are sufficiently diluted by cell division (73–75).
Upon completing one or more rounds of mitosis, GC B cells transition to the DZd stage (Figure 1C) where cell cycle exit is coordinated with induction of immunoglobulin gene transcription, which is required for AID targeting. Furthermore, dissolution of the nuclear membrane, as occurs when cells undergo mitosis, facilitates AID entering the nucleus (51). These mechanisms are predicted to restrict AID-mediated SHM to DZd cells. Indeed, these cells bear features of genotoxic stress and DNA repair associated with SHM (14). Our findings are consistent with studies suggesting that SHM occur in G1 phase cells (51, 76–79). Therefore, proliferation and SHM appear to occur in sequential and mutually exclusive cell states, DZp and DZd respectively, that ensure cells have exited cell cycle before initiating SHM. In this way, the attendant risks of gene mutation are mitigated.
B cells undergoing SHM-associated DNA damage and repair undergo an additional checkpoint prior to transit to the LZ. Upon completion of SHM, GC B cells replace old surface BCRs with the newly mutated BCR (80). B cells that generated nonfunctional BCRs due to SHM are fated for apoptosis prior to LZ entry. Thus, there are two levels of BCR selection per GC cycle, structural competency followed by relative affinity. We would postulate that those cells that express an incompetent BCR in the DZd might undergo retrograde GC cycling to be cleared by TBMs, the only known macrophage resident in the DZ (37, 66).
In our model, cells transit from the DZd to LZ without intervening proliferation. Therefore, only mutations that immediately arise on the DNA coding strand would be selected. Mutations on the non-coding strand must also be selected. However for these mutations to be selected, we predict that must arise during the preceding GC cycle and previous transit through the DZd. This would allow these single stranded mutations to be “fixed” by DNA replication in the DZp and therefore become “visible” for subsequent selection in the LZ. Alternatively, it is possible that a minor fraction of DZd cells migrate back to the DZp for proliferation and fixing of non-coding strand mutations.
In addition to cyclic re-entry into the DZ, LZ B cell interactions with TFH govern GC exit into the PB or MBC fates (18, 81–83). While the LZ population that induces Myc contains cells destined for cyclic re-entry, this B cell pool also contains PB and MBC precursors (45, 84). PB precursors are BCL6loCD69hiIRF4+ and express high-affinity BCRs (38, 45, 85). Commitment to the PC fate is associated with stable B:TFH cell conjugates suggesting strong T cell help instructs PC differentiation (45).
In contrast MBCs develop from low-affinity B cell clones that receive low strength signals from TFH (5, 86–88). Weak T cell help also results in low Myc and mTORC1 activation, which also predisposes to differentiation into MBCs (44). Overall, these studies indicate that high affinity BCRs and strong TFH interactions predispose to PC differentiation while low affinity BCRs, and poor T cell help, leads to differentiation into MBCs.
Here we provide a three-compartment model that segregates each fundamental GC B cell function, selection, proliferation and SHM, into distinct, separate cell states. This both mitigates the risk of SHM and allows coordination of molecular processes specific to each function. Furthermore, analysis of these three populations reveal just how molecularly dynamic the GC subsets are with large differences in genomic accessibility, transcription, protein expression and protein phosphorylation across each cell state. Therefore, it is likely that many regulatory mechanisms vertically integrate across the biosynthetic pathway to both drive and maintain the integrity of the GC response. Understanding these mechanisms, and how they integrate to regulate GCs, will provide opportunities to better treat a wide breadth of infectious, autoimmune and neoplastic diseases.
DK and MC wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work is supported by the US National Institutes of Health, National Institute of Allergy and Infectious Diseases grant R01AI143778.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Röhlich K. Beitrag zur Cytologie der Keimzentren der Lymphknoten. Z Mikrosk Anat Forsch (1930) 20:287–97. doi: 10.3389/fimmu.2017.01639
2. Mayer CT, Gazumyan A, Kara EE, Gitlin AD, Golijanin J, Viant C, et al. The microanatomic segregation of selection by apoptosis in the germinal center. Science (2017) 358(6360):eaao2602. doi: 10.1126/science.aao2602
3. Mesin L, Ersching J, Victora GD. Germinal Center B Cell Dynamics. Immunity (2016) 45(3):471–82. doi: 10.1016/j.immuni.2016.09.001
4. Singh A. Eliciting B cell immunity against infectious diseases using nanovaccines. Nat Nanotechnol (2021) 16(1):16–24. doi: 10.1038/s41565-020-00790-3
5. Cyster JG, Allen CDC. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell (2019) 177(3):524–40. doi: 10.1016/j.cell.2019.03.016
6. Burnett DL, Langley DB, Schofield P, Hermes JR, Chan TD, Jackson J, et al. Germinal center antibody mutation trajectories are determined by rapid self/foreign discrimination. Science (2018) 360(6385):223–6. doi: 10.1126/science.aao3859
7. Basso K, Saito M, Sumazin P, Margolin AA, Wang K, Lim WK, et al. Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood (2010) 115(5):975–84. doi: 10.1182/blood-2009-06-227017
8. Huang C, Geng H, Boss I, Wang L, Melnick A. Cooperative transcriptional repression by BCL6 and BACH2 in germinal center B-cell differentiation. Blood (2014) 123(7):1012–20. doi: 10.1182/blood-2013-07-518605
9. Mlynarczyk C, Fontan L, Melnick A. Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol Rev (2019) 288(1):214–39. doi: 10.1111/imr.12755
10. Fernando TM, Marullo R, Pera Gresely B, Phillip JM, Yang SN, Lundell-Smith G, et al. BCL6 Evolved to Enable Stress Tolerance in Vertebrates and Is Broadly Required by Cancer Cells to Adapt to Stress. Cancer Discov (2019) 9(5):662–79. doi: 10.1158/2159-8290.CD-17-1444
11. Kuppers R, Klein U, Hansmann ML, Rajewsky K. Cellular origin of human B-cell lymphomas. N Engl J Med (1999) 341(20):1520–9. doi: 10.1056/NEJM199911113412007
12. Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell (2020) 37(4):551–68 e14. doi: 10.1016/j.ccell.2020.03.015
13. Kuppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene (2001) 20(40):5580–94. doi: 10.1038/sj.onc.1204640
14. Kennedy DE, Okoreeh MK, Maienschein-Cline M, Ai J, Veselits M, McLean KC, et al. Novel specialized cell state and spatial compartments within the germinal center. Nat Immunol (2020) 21:660–70. doi: 10.1038/s41590-020-0660-2
15. Milpied P, Cervera-Marzal I, Mollichella ML, Tesson B, Brisou G, Traverse-Glehen A, et al. Human germinal center transcriptional programs are de-synchronized in B cell lymphoma. Nat Immunol (2018) 19(9):1013–24. doi: 10.1038/s41590-018-0181-4
16. Holmes AB, Corinaldesi C, Shen Q, Kumar R, Compagno N, Wang Z, et al. Single-cell analysis of germinal-center B cells informs on lymphoma cell of origin and outcome. J Exp Med (2020) 217(10):e20200483. doi: 10.1084/jem.20200483
17. Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control systems and decision making for antibody production. Nat Immunol (2010) 11(8):681–8. doi: 10.1038/ni.1900
18. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol (2015) 15(3):137–48. doi: 10.1038/nri3804
19. Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity (2011) 34(6):947–60. doi: 10.1016/j.immuni.2011.03.024
20. Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T, et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity (2011) 34(6):961–72. doi: 10.1016/j.immuni.2011.03.025
21. Jacque E, Schweighoffer E, Visekruna A, Papoutsopoulou S, Janzen J, Zillwood R, et al. IKK-induced NF-kappaB1 p105 proteolysis is critical for B cell antibody responses to T cell-dependent antigen. J Exp Med (2014) 211(10):2085–101. doi: 10.1084/jem.20132019
22. Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity (2013) 38(5):918–29. doi: 10.1016/j.immuni.2013.04.009
23. Huang C, Gonzalez DG, Cote CM, Jiang Y, Hatzi K, Teater M, et al. The BCL6 RD2 domain governs commitment of activated B cells to form germinal centers. Cell Rep (2014) 8(5):1497–508. doi: 10.1016/j.celrep.2014.07.059
24. Zhang TT, Gonzalez DG, Cote CM, Kerfoot SM, Deng S, Cheng Y, et al. Germinal center B cell development has distinctly regulated stages completed by disengagement from T cell help. Elife (2017) 6:e19552. doi: 10.7554/eLife.19552
25. Basso K, Schneider C, Shen Q, Holmes AB, Setty M, Leslie C, et al. BCL6 positively regulates AID and germinal center gene expression via repression of miR-155. J Exp Med (2012) 209(13):2455–65. doi: 10.1084/jem.20121387
26. Cinamon G, Matloubian M, Lesneski MJ, Xu Y, Low C, Lu T, et al. Sphingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat Immunol (2004) 5(7):713–20. doi: 10.1038/ni1083
27. Gatto D, Paus D, Basten A, Mackay CR, Brink R. Guidance of B cells by the orphan G protein-coupled receptor EBI2 shapes humoral immune responses. Immunity (2009) 31(2):259–69. doi: 10.1016/j.immuni.2009.06.016
28. Pereira JP, Kelly LM, Xu Y, Cyster JG. EBI2 mediates B cell segregation between the outer and centre follicle. Nature (2009) 460(7259):1122–6. doi: 10.1038/nature08226
29. Tunyaplin C, Shaffer AL, Angelin-Duclos CD, Yu X, Staudt LM, Calame KL. Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. J Immunol (2004) 173(2):1158–65. doi: 10.4049/jimmunol.173.2.1158
30. Robinson MJ, Ding Z, Pitt C, Brodie EJ, Quast I, Tarlinton DM, et al. The Amount of BCL6 in B Cells Shortly after Antigen Engagement Determines Their Representation in Subsequent Germinal Centers. Cell Rep (2020) 30(5):1530–41 e4. doi: 10.1016/j.celrep.2020.01.009
31. Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, et al. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity (2019) 51(2):337–50 e7. doi: 10.1016/j.immuni.2019.07.001
32. Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol (2012) 12(7):517–31. doi: 10.1038/nri3216
33. Pape KA, Kouskoff V, Nemazee D, Tang HL, Cyster JG, Tze LE, et al. Visualization of the genesis and fate of isotype-switched B cells during a primary immune response. J Exp Med (2003) 197(12):1677–87. doi: 10.1084/jem.20012065
34. Cunningham AF, Gaspal F, Serre K, Mohr E, Henderson IR, Scott-Tucker A, et al. Salmonella induces a switched antibody response without germinal centers that impedes the extracellular spread of infection. J Immunol (2007) 178(10):6200–7. doi: 10.4049/jimmunol.178.10.6200
35. King HW, Orban N, Riches JC, Clear AJ, Warnes G, Teichmann SA, et al. Single-cell analysis of human B cell maturation predicts how antibody class switching shapes selection dynamics. Sci Immunol (2021) 6(56):eabe6291. doi: 10.1126/sciimmunol.abe6291
36. MacLennan IC. Germinal centers. Ann Rev Immunol (1994) 12:117–40. doi: 10.1146/annurev.iy.12.040194.001001
37. Victora GD, Nussenzweig MC. Germinal centers. Ann Rev Immunol (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
38. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell (2010) 143(4):592–605. doi: 10.1016/j.cell.2010.10.032
39. Luo W, Weisel F, Shlomchik MJ. B Cell Receptor and CD40 Signaling Are Rewired for Synergistic Induction of the c-Myc Transcription Factor in Germinal Center B Cells. Immunity (2018) 48(2):313–26 e5. doi: 10.1016/j.immuni.2018.01.008
40. Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, et al. Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science (2014) 345(6200):1058–62. doi: 10.1126/science.1257861
41. Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheumatol (2002) 46(1):191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K
42. Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell (2017) 170(6):1062–78. doi: 10.1016/j.cell.2017.08.028
43. Yoshida H, Lareau CA, Ramirez RN, Rose SA, Maier B, Wroblewska A, et al. The cis-Regulatory Atlas of the Mouse Immune System. Cell (2019) 176(4):897–912 e20. doi: 10.1016/j.cell.2018.12.036
44. Inoue T, Shinnakasu R, Kawai C, Ise W, Kawakami E, Sax N, et al. Exit from germinal center to become quiescent memory B cells depends on metabolic reprograming and provision of a survival signal. J Exp Med (2021) 218(1):e20200866. doi: 10.1084/jem.20200866
45. Ise W, Fujii K, Shiroguchi K, Ito A, Kometani K, Takeda K, et al. T Follicular Helper Cell-Germinal Center B Cell Interaction Strength Regulates Entry into Plasma Cell or Recycling Germinal Center Cell Fate. Immunity (2018) 48(4):702–15 e4. doi: 10.1016/j.immuni.2018.03.027
46. Dufaud CR, McHeyzer-Williams LJ, McHeyzer-Williams MG. Deconstructing the germinal center, one cell at a time. Curr Opin Immunol (2017) 45:112–8. doi: 10.1016/j.coi.2017.03.007
47. Clark MR, Mandal M, Ochiai K, Singh H. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat Rev Immunol (2014) 14(2):69–80. doi: 10.1038/nri3570
48. Mandal M, Okoreeh MK, Kennedy DE, Maienschein-Cline M, Ai J, McLean KC, et al. CXCR4 signaling directs Igk recombination and the molecular mechanisms of late B lymphopoiesis. Nat Immunol (2019) 20(10):1393–403. doi: 10.1038/s41590-019-0468-0
49. Gitlin AD, Mayer CT, Oliveira TY, Shulman Z, Jones MJ, Koren A, et al. HUMORAL IMMUNITY. T cell help controls the speed of the cell cycle in germinal center B cells. Science (2015) 349(6248):643–6. doi: 10.1126/science.aac4919
50. Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature (2014) 509(7502):637–40. doi: 10.1038/nature13300
51. Wang Q, Kieffer-Kwon KR, Oliveira TY, Mayer CT, Yao K, Pai J, et al. The cell cycle restricts activation-induced cytidine deaminase activity to early G1. J Exp Med (2017) 214(1):49–58. doi: 10.1084/jem.20161649
52. Storb U. Why does somatic hypermutation by AID require transcription of its target genes? Adv Immunol (2014) 122:253–77. doi: 10.1016/B978-0-12-800267-4.00007-9
53. Allen CD, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science (2007) 315(5811):528–31. doi: 10.1126/science.1136736
54. Beltman JB, Allen CD, Cyster JG, de Boer RJ. B cells within germinal centers migrate preferentially from dark to light zone. Proc Natl Acad Sci U S A (2011) 108(21):8755–60. doi: 10.1073/pnas.1101554108
55. Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, et al. In vivo imaging of germinal centres reveals a dynamic open structure. Nature (2007) 446(7131):83–7. doi: 10.1038/nature05573
56. Meyer-Hermann M, Mohr E, Pelletier N, Zhang Y, Victora GD, Toellner KM. A theory of germinal center B cell selection, division, and exit. Cell Rep (2012) 2(1):162–74. doi: 10.1016/j.celrep.2012.05.010
57. Hauser AE, Junt T, Mempel TR, Sneddon MW, Kleinstein SH, Henrickson SE, et al. Definition of germinal-center B cell migration in vivo reveals predominant intrazonal circulation patterns. Immunity (2007) 26(5):655–67. doi: 10.1016/j.immuni.2007.04.008
58. Perez-Garcia A, Marina-Zarate E, Alvarez-Prado AF, Ligos JM, Galjart N, Ramiro AR. CTCF orchestrates the germinal centre transcriptional program and prevents premature plasma cell differentiation. Nat Commun (2017) 8:16067. doi: 10.1038/ncomms16067
59. Song S, Cao C, Choukrallah MA, Tang F, Christofori G, Kohler H, et al. OBF1 and Oct factors control the germinal center transcriptional program. Blood (2021). doi: 10.1182/blood.2020010175
60. Willis SN, Tellier J, Liao Y, Trezise S, Light A, O’Donnell K, et al. Environmental sensing by mature B cells is controlled by the transcription factors PU.1 and SpiB. Nat Commun (2017) 8(1):1426. doi: 10.1038/s41467-017-01605-1
61. Carotta S, Willis SN, Hasbold J, Inouye M, Pang SH, Emslie D, et al. The transcription factors IRF8 and PU.1 negatively regulate plasma cell differentiation. J Exp Med (2014) 211(11):2169–81. doi: 10.1084/jem.20140425
62. Hodson DJ, Shaffer AL, Xiao W, Wright GW, Schmitz R, Phelan JD, et al. Regulation of normal B-cell differentiation and malignant B-cell survival by OCT2. Proc Natl Acad Sci U S A (2016) 113(14):E2039–46. doi: 10.1073/pnas.1600557113
63. Strubin M, Newell JW, Matthias P. OBF-1, a novel B cell-specific coactivator that stimulates immunoglobulin promoter activity through association with octamer-binding proteins. Cell (1995) 80(3):497–506. doi: 10.1016/0092-8674(95)90500-6
64. Gstaiger M, Knoepfel L, Georgiev O, Schaffner W, Hovens CM. A B-cell coactivator of octamer-binding transcription factors. Nature (1995) 373(6512):360–2. doi: 10.1038/373360a0
65. Luo Y, Roeder RG. Cloning, functional characterization, and mechanism of action of the B-cell-specific transcriptional coactivator OCA-B. Mol Cell Biol (1995) 15(8):4115–24. doi: 10.1128/MCB.15.8.4115
66. Brink R, Phan TG. Self-Reactive B Cells in the Germinal Center Reaction. Annu Rev Immunol (2018) 36:339–57. doi: 10.1146/annurev-immunol-051116-052510
67. Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science (2004) 304(5674):1147–50. doi: 10.1126/science.1094359
68. Kranich J, Krautler NJ, Heinen E, Polymenidou M, Bridel C, Schildknecht A, et al. Follicular dendritic cells control engulfment of apoptotic bodies by secreting Mfge8. J Exp Med (2008) 205(6):1293–302. doi: 10.1084/jem.20071019
69. Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ (2016) 23(6):962–78. doi: 10.1038/cdd.2016.11
70. Dernstedt A, Leidig J, Holm A, Kerkman PF, Mjosberg J, Ahlm C, et al. Regulation of Decay Accelerating Factor Primes Human Germinal Center B Cells for Phagocytosis. Front Immunol (2020) 11:599647. doi: 10.3389/fimmu.2020.599647
71. Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol (2011) 12(6):385–92. doi: 10.1038/nrm3115
72. Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol (2013) 14(8):518–28. doi: 10.1038/nrm3629
73. Finkin S, Hartweger H, Oliveira TY, Kara EE, Nussenzweig MC. Protein Amounts of the MYC Transcription Factor Determine Germinal Center B Cell Division Capacity. Immunity (2019) 51(2):324–36 e5. doi: 10.1016/j.immuni.2019.06.013
74. Calado DP, Sasaki Y, Godinho SA, Pellerin A, Kochert K, Sleckman BP, et al. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat Immunol (2012) 13(11):1092–100. doi: 10.1038/ni.2418
75. Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, et al. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol (2012) 13(11):1083–91. doi: 10.1038/ni.2428
76. Khair L, Guikema JE, Linehan EK, Ucher AJ, Leus NG, Ogilvie C, et al. ATM increases activation-induced cytidine deaminase activity at downstream S regions during class-switch recombination. J Immunol (2014) 192(10):4887–96. doi: 10.4049/jimmunol.1303481
77. Schrader CE, Guikema JE, Linehan EK, Selsing E, Stavnezer J. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J Immunol (2007) 179(9):6064–71. doi: 10.4049/jimmunol.179.9.6064
78. Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature (2001) 414(6864):660–5. doi: 10.1038/414660a
79. Sharbeen G, Yee CW, Smith AL, Jolly CJ. Ectopic restriction of DNA repair reveals that UNG2 excises AID-induced uracils predominantly or exclusively during G1 phase. J Exp Med (2012) 209(5):965–74. doi: 10.1084/jem.20112379
80. Stewart I, Radtke D, Phillips B, McGowan SJ, Bannard O. Germinal Center B Cells Replace Their Antigen Receptors in Dark Zones and Fail Light Zone Entry when Immunoglobulin Gene Mutations are Damaging. Immunity (2018) 49(3):477–89 e7. doi: 10.1016/j.immuni.2018.08.025
81. Akkaya M, Kwak K, Pierce SK. B cell memory: building two walls of protection against pathogens. Nat Rev Immunol (2020) 20(4):229–38. doi: 10.1038/s41577-019-0244-2
82. Merino Tejero E, Lashgari D, Garcia-Valiente R, Gao X, Crauste F, Robert PA, et al. Multiscale Modeling of Germinal Center Recapitulates the Temporal Transition From Memory B Cells to Plasma Cells Differentiation as Regulated by Antigen Affinity-Based Tfh Cell Help. Front Immunol (2020) 11:620716. doi: 10.3389/fimmu.2020.620716
83. Liu B, Lin Y, Yan J, Yao J, Liu D, Ma W, et al. Affinity-coupled CCL22 promotes positive selection in germinal centres. Nature (2021). doi: 10.1038/s41586-021-03384-8
84. Nakagawa R, Toboso-Navasa A, Schips M, Young G, Bhaw-Rosun L, Llorian-Sopena M, et al. Permissive selection followed by affinity-based proliferation of GC light zone B cells dictates cell fate and ensures clonal breadth. Proc Nat Acad Sci U S A (2021) 118(2):e2016425118. doi: 10.1073/pnas.2016425118
85. Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, et al. High affinity germinal center B cells are actively selected into the plasma cell compartment. J Exp Med (2006) 203(11):2419–24. doi: 10.1084/jem.20061254
86. Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, et al. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol (2016) 17(7):861–9. doi: 10.1038/ni.3460
87. Viant C, Weymar GHJ, Escolano A, Chen S, Hartweger H, Cipolla M, et al. Antibody Affinity Shapes the Choice between Memory and Germinal Center B Cell Fates. Cell (2020) 183(5):1298–311 e11. doi: 10.1016/j.cell.2020.09.063
Keywords: B cell, germinal center, transcription, epigenetics, T cell help, tingible body macrophage, somatic hypermutation, affinity maturation
Citation: Kennedy DE and Clark MR (2021) Compartments and Connections Within the Germinal Center. Front. Immunol. 12:659151. doi: 10.3389/fimmu.2021.659151
Received: 27 January 2021; Accepted: 15 March 2021;
Published: 31 March 2021.
Edited by:
Ann M. Haberman, Yale University, United StatesReviewed by:
Claude-Agnes Reynaud, Université Paris Descartes, FranceCopyright © 2021 Kennedy and Clark. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcus R. Clark, bWNsYXJrQHVjaGljYWdvLmVkdQ==; Domenick E. Kennedy, ZG9tZW5pY2sua2VubmVkeUBhYmJ2aWUuY29t
†Present address: Domenick E. Kennedy, Drug Discovery Science and Technology, AbbVie, North Chicago, IL, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.