 Andrew Kent
Andrew Kent Natalie V. Longino
Natalie V. Longino Allison Christians
Allison Christians Eduardo Davila
Eduardo Davila
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 03 May 2021
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.658611
This article is part of the Research Topic Engineered Immune Cells in Cancer Immunotherapy (EICCI) View all 47 articles
T cell-based immunotherapies including genetically engineered T cells, adoptive transfer of tumor-infiltrating lymphocytes, and immune checkpoint blockade highlight the impressive anti-tumor effects of T cells. These successes have provided new hope to many cancer patients with otherwise poor prognoses. However, only a fraction of patients demonstrates durable responses to these forms of therapies and many develop significant immune-mediated toxicity. These heterogeneous clinical responses suggest that underlying nuances in T cell genetics, phenotypes, and activation states likely modulate the therapeutic impact of these approaches. To better characterize known genetic variations that may impact T cell function, we 1) review the function of early T cell receptor-specific signaling mediators, 2) offer a synopsis of known mutations and genetic alterations within the associated molecules, 3) discuss the link between these mutations and human disease and 4) review therapeutic strategies under development or in clinical testing that target each of these molecules for enhancing anti-tumor T cell activity. Finally, we discuss novel engineering approaches that could be designed based on our understanding of the function of these molecules in health and disease.
T cell activation, differentiation, and effector functions are tightly controlled by highly specialized and interconnected signaling pathways. Major early mediators of T cell activation include: (1) activation via the α and β or γ and δ chains of the T cell receptor (TCR) molecules (1); (2) CD3 signal-transduction molecules (2); (3) CD4 and CD8 co-receptors that help stabilize TCR-peptide-MHC interactions (3, 4); (4) early signaling mediators such as LCK, FYN, and ZAP70 (5, 6); and (5) the LAT-signalosome that leads to activation of a myriad of downstream signaling intermediates and pathways (Figure 1) (7). These proximal signaling prote13ins interact with myriad of intermediate molecules to ultimately initiate various multiple cellular processes including differentiation and effector function (7). We review the molecules involved in early TCR signaling and the receptors in T cells and consider how mutations or alterations in these molecules contribute to human disease, particularly immunity to cancer. We highlight therapeutic strategies designed to utilize this fundamental knowledge of molecular function for cancer treatment, with an emphasis on novel strategies that are showing early clinical potential. A discussion on T cell based immune therapy would be remiss without mentioning immune checkpoint inhibitors (ICIs) including monoclonal antibodies targeting PD-1, PD-L1 and CTLA4, as well as chimeric antigen receptor (CAR) T cells and tumor-infiltrating lymphocyte (TIL) therapy. These agents have revolutionized cancer therapy, but due to the wealth of literature on these topics, we will not address them directly in this review except as they relate to specific molecules discussed in each section below.
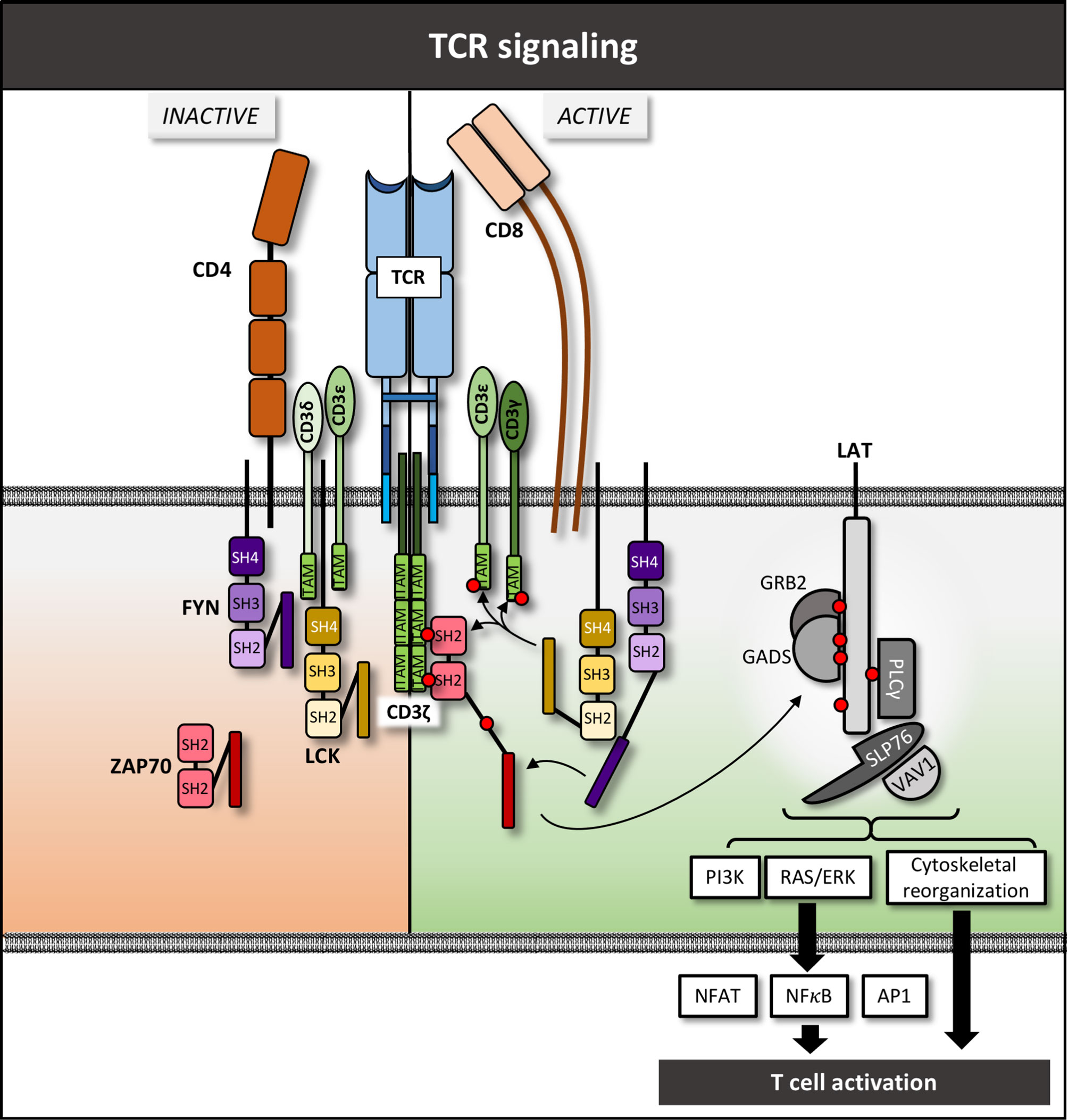
Figure 1 TCR complex and downstream signaling pathway – schematic overview. Red circles denote phosphate groups.
Unique from all other cell types, T cells express antigen-specific TCRs. The TCR complex is a heterodimer composed of either α/β or γ/δ chains, defining two major ”flavors” of T cells. In humans, 95% of T cells are α/β heterodimers while only 5% are γ/δ homodimers (Figure 2). Within these heterodimers, each chain is comprised of extracellular variable and constant domains, a short linker peptide, and a transmembrane domain (8). The variable domains undergo somatic rearrangement during T cell development and contact the antigenic peptide presented by major-histocompatibility complex (pMHC). The interaction forms the physical basis by which T cells can recognize a myriad of targets (9, 10). The linker and transmembrane domains allow association with additional molecules such as the CD3 chains as well as CD4 and CD8 in their respective T cell types.
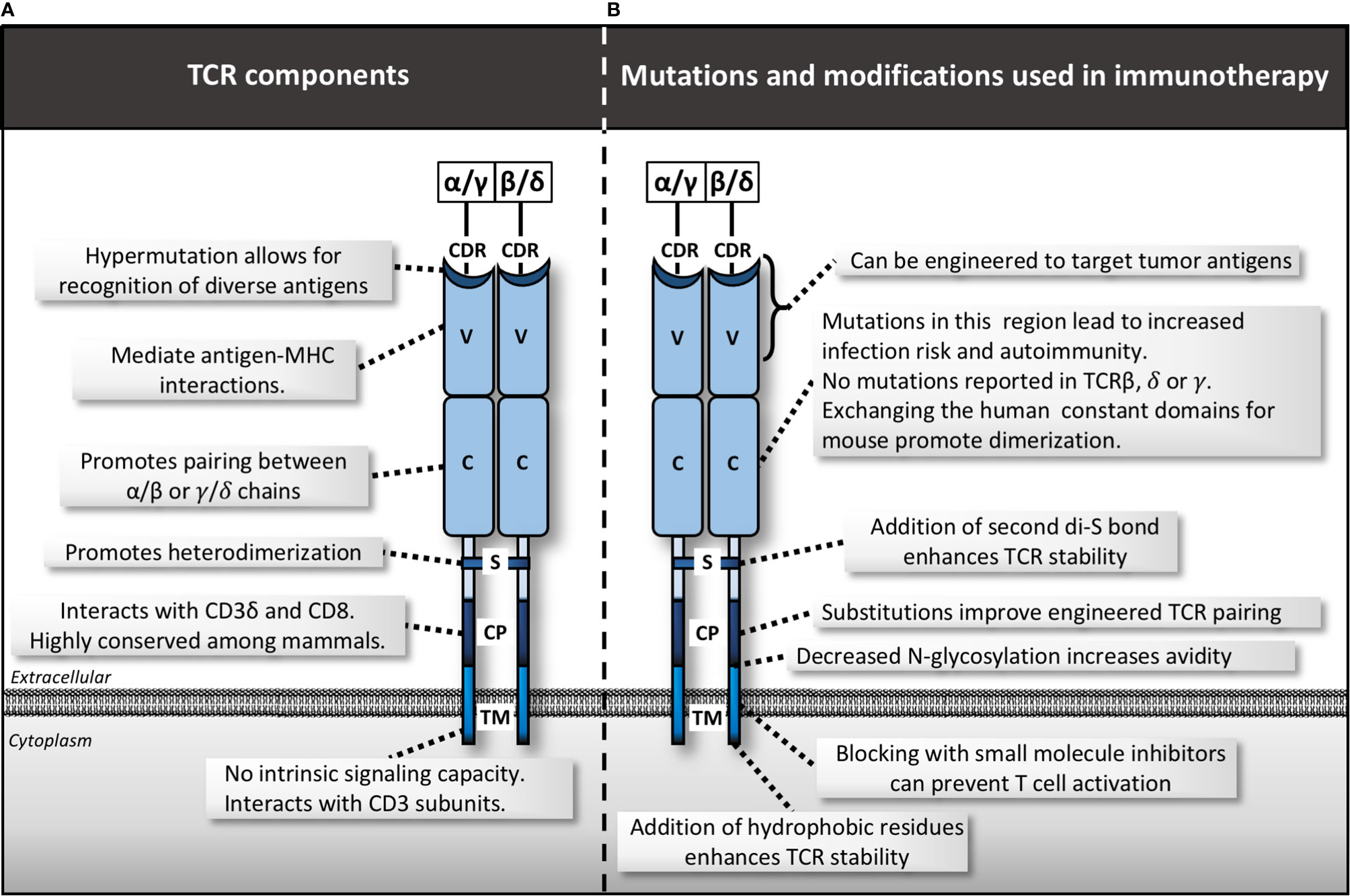
Figure 2 TCR complex structure, mutations and manipulations for immunotherapy. (A) Various domains within the T cell receptor (TCR) including the complementarity-determining region (CDR), variable domain (V), constant domain (C), disulfide bond (S), membrane-proximal connecting peptides (CP) and transmembrane (TM). (B) Mutations and modifications used in immunotherapy by region.
As briefly stated above, the variable domain of the TCR genes undergoes somatic recombination early in T cell development, thereby forming the basis for TCR diversity (11). This highly controlled process creates an array of T cells each expressing a unique TCR that can bind varied pMHC complexes. It is estimated that the human T cell repertoire can target on the order of 10 (12) unique specifities (13). Upon recognition of a unique pMHC expressed on target cells, various extracellular and intracellular accessory molecules are recruited to mediate a massive transcriptional shift towards T cell effector functions that allow effective killing of infected, malignant, or altered-self targets (14). Genetic variations in these TCR genes between individuals affect the clonality and diversity of that individual’s T cell repertoire and can lead to disease. Numerous studies have linked intratumoral and peripheral blood TCR clonality and diversity with cancer prognosis and response to various treatment modalities. In cervical cancer fewer TCR clonotypes in sentinel lymph nodes correlated with worse outcomes (15), and in colorectal cancer, patients with metastatic disease harbored less TCR diversity in tumor draining lymph nodes (12). Comparing healthy individuals to a variety of cancer patients, Simnica et al. found that as people age, their TCR diversity diminishes and that cancer patients have reduced TCR diversity relative to healthy age-matched controls (16). Additional studies in melanoma and pancreatic cancer have shown that increased TCR diversity correlates with improved outcomes after immune checkpoint inhibition (ICI) (17–19), suggesting this treatment modality is reliant on an individual’s T cell repertoire and ability to recognize tumor antigens for its beneficial effect. These data highlight the ability of T cells to recognize and fight cancer and suggest that therapeutic efficacy relies on a diverse TCR pool.
While most anti-tumor studies have manipulated α/β T cells, γ/δ T cells have unique features that could be exploited for cancer treatment. In pre-clinical studies, mice lacking γ/δ T cells have increased incidence of various cancer types (20–23). This anti-tumor effect is mediated via γ/δ T cell recognition of stress-associated molecules often upregulated in cancer such as heat-shock proteins, non-classical MHC molecules, and various phospho-antigens, a process encapsulated under the term the “lymphoid stress-surveillance response.” (24) Furthermore, unique from α/β T cells, γ/δ T cells have potent activation responses prior to expansion, and express various NK cell receptors such as NKG2D that further enhance their ability to recognize altered or damaged self-cells (24). The proportion of γ/δ T cells infiltrating tumors is predictive of favorable prognosis (25). Among the various tumor types studied, melanoma tumors harbored the highest proportion of γ/δ T cells and the presence of these cells correlated with a lower risk of metastasis (26). Interestingly, γ/δ T cells are often over-represented in the heterogeneous adoptively transferred cell populations of successful tumor infiltrating lymphocyte (TIL) therapies, again suggesting potent anti-tumor activity (27). However, in vivo γ/δ T cell stimulating approaches using IL-2 and bisphosphonates has had underwhelming results (28, 29). Further studies are needed to define optimal ex vivo expansion strategies of γ/δ T cells, and to piece apart the differential benefit of γ/δ and α/β T cell subsets in adoptive transfer approaches.
Aside from the extracellular variable region of the TCR, subtle changes in the extracellular constant and transmembrane (TM) domains can affect the ability of the entire TCR complex to assemble and function (Figure 2). The only in-human mutation linked to the TCR α constant (TRAC) domain, is a G to A substitution at the C-terminus of exon 3. This mutation results in a complete lack of α/β T cells, implicating a key role in TRAC in regulating development of this cell type (30). No identified human diseases so far have been linked to mutations in the TM domain, or the TCR β, γ, or δ constant domains (TRBC, TRGC, or TRDC), suggesting they are all highly evolutionarily conserved. Indeed, numerous mutational and structural studies have confirmed the essential functions of both the TM and constant domains in assembly of the TCR complex and signal transduction largely via CD3 subunit recruitment and activation (31). More recent studies have only just begun to piece apart the nuanced mechanisms of these interactions (32–34).
New strategies are emerging to manipulate the TM and constant domains of the TCR for therapeutic benefit. For example, the TCR constant domain harbors a disulfide bond that promotes heterodimerization. Adding a second disulfide bond within this region has been shown to enhance TCR stability, signaling and T cell mediated tumor killing in cancer models (35). Adding additional hydrophobic residues to the TM domain enhanced surface expression and T cell avidity, leading to increased anti-tumor T cell activity in vitro (36). In mouse and human models, removal of conserved N-glycosylation sites in the TCR variable and constant domains improves T cell avidity and tumor cell recognition (37). Other studies have improved engineered-TCR technology via manipulation of constant domains. Exchanging the human constant-region for the murine equivalent (38), or just a 9 amino acid fragment thereof (39), prevents native-non-native heterodimerization. This results in improved pairing of TCR subunits with the desired specificity, enhanced CD3/TCR stability, and increased anti-tumor activity. Introduction of additional cysteine residues in the constant region achieved similar results (40). Finally, new approaches to target transmembrane domains with novel peptides, such as core peptide (CP) targeting of the TM domain of the TCR molecules, has shown early promise in various diseases such as autoimmune disease (41). Similar strategies could be extrapolated to anti-cancer applications. These insights highlight that modulation of anti-tumor immunity should be approached cautiously to maximize the effect on the malignant cells, while preventing detrimental side effects on the host.
Signaling through the TCR requires interaction with several CD3 subunits, as the TCR chains themselves do not contain intracellular signaling domains (42). There are four CD3 types: CD3ε and CD3δ form a heterodimer that binds to TCRα, CD3ε and CD3γ form another heterodimer that binds to the TCRβ chain, and two CD3ζ chains form a homodimer that associates with both TCR α and β chains (Figure 3) (31). In the case of the γ/δ TCR, two CD3ε/γ homodimers are involved in lieu of CD3δ (43). Despite extensive study, the exact geometry and binding sites between these CD3 molecules and the TCR have not been fully elucidated, but likely involve a combination of residues in the constant regions as well as ionizable and hydrophobic residues in the transmembrane regions (44). Signaling itself is mediated through phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) within the CD3 and ζ chains (45).
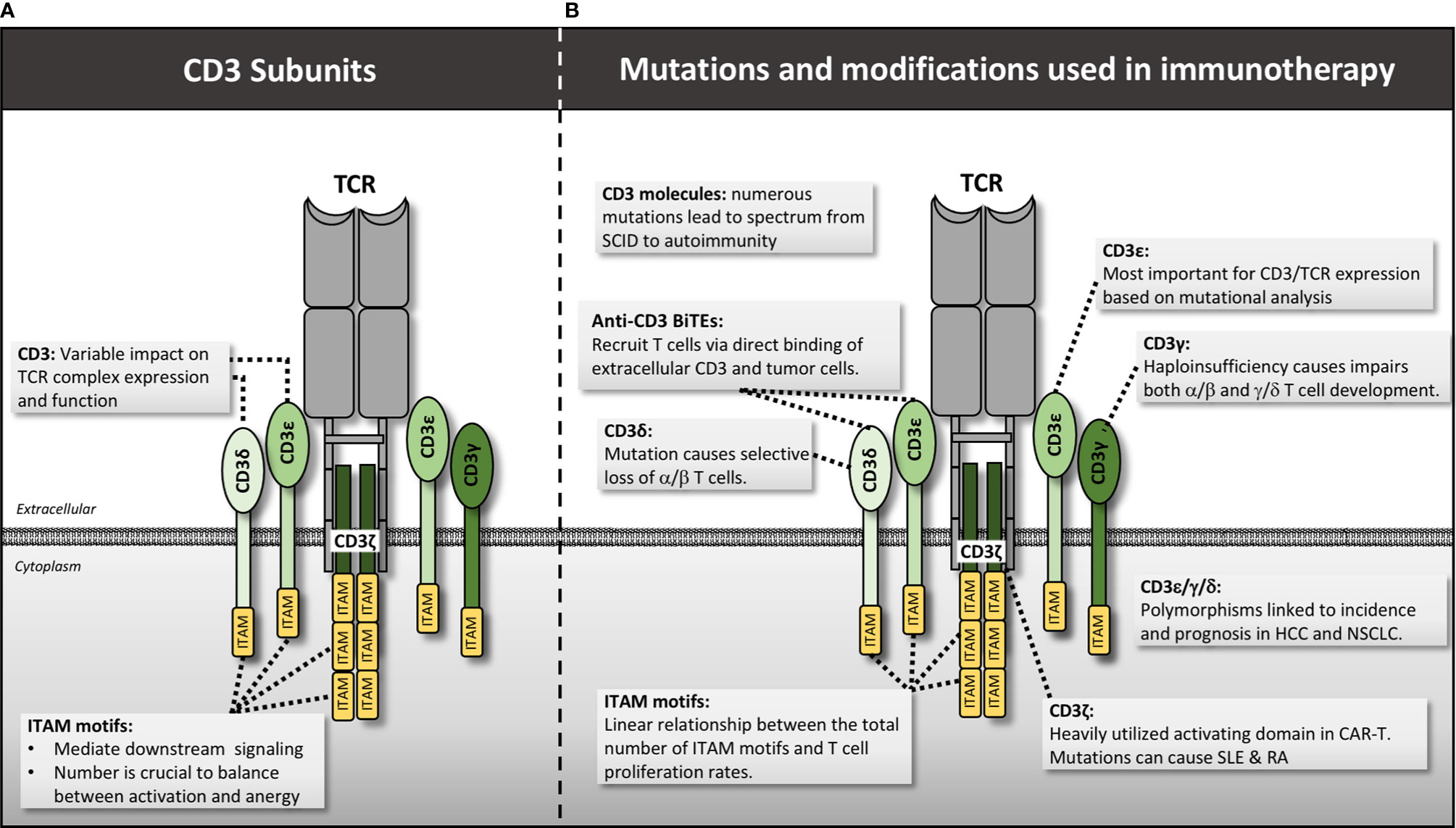
Figure 3 CD3 structure, mutations and manipulations for immunotherapy. (A) CD3 molecules interact with the TCR in CD3δ/ϵ and CD3ϵ/γ heterodimers while CD3ζ forms a single homodimer. Each CD3 molecule contains one or more ITAM motif that is essential for downstream signaling. (B) Various mutations and genetic engineering approaches alter or manipulate CD3 function thereby impacting T cell activity. HCC, hepatocellular carcinoma; NSCLC, non-small cell lung cancer.
Mutations in the CD3 molecules are associated with an array of human diseases ranging from severe-combined immunodeficiency (SCID) (46, 47) to autoimmune disorders (48). Regarding immunodeficiency, frameshift, nonsense, and splice variant mutations disrupt the ability of the CD3 molecules to be expressed or, if successfully translated, to bind to the TCR molecules to form a functional TCR complex (49). Without functional TCR signaling, T cells fail selection in the thymus, resulting in a complete lack of T cells and severe disease (46, 47, 50–52). More mild immunodeficiencies result in patients where TCR/CD3 is expressed but at much lower levels than normal, impairing T cell activation and function (53–55). The CD3 molecule affected determines the type of deficiency. For example, the CD3δ mutations C202T (that results in a premature stop codon at residue 68), and G to A substitution at position +5 of intron 2, were both found to cause a selective lack of α/β T cells but preserve the γ/δ T cell pool because the CD3δ molecule is not included within the γ/δ TCR complex (56–58). In contrast, CD3γ haploinsufficiency had a larger effect on the γ/δ T cell pool, suggesting this molecule is more important in γ/δ T cell development in humans (59).
At the opposite end of the spectrum are CD3 mutations that pre-dispose patients to autoimmunity. Of all the CD3 subtypes, mutations in CD3ζ and CD3γ harbor the strongest link to autoimmune and inflammatory conditions. Numerous SNPs and splice variants, affecting primarily the 3’UTR, intron 1, and exon 7 of CD3ζ have been associated with pathogenesis in systemic lupus erythematosus (SLE) (48, 60–64) and rheumatoid arthritis (RA) (65). A single SNP in intron 1 (rs858554) has a strong association with both diseases, as well as immune thrombocytopenia (ITP), suggesting a common underlying mechanism (48, 66) possibly via down-regulation of CD3ζ (67). Aside from germline mutations themselves, hypermethylation of the CD3ζ gene has been associated with severe SLE phenotypes and correlates with reduced CD3ζ expression (60). One might predict down-regulation of CD3ζ would decrease T cell activation. However, decreased CD3ζ expression in SLE patients’ T cells was found to result in aberrant recruitment of FcRγ to the TCR complex in lieu of CD3ζ (68). Aside from mutations in CD3ζ, CD3γ mutations can also result in a range of phenotypes including mild immunodeficiency to autoimmunity. For example, Recio et al. found a common A to T mutation in nucleotide 205 of exon 3 in multiple patients from two different families in Turkey (69). This mutation results in a premature stop codon at residue 69 of the CD3γ protein. Despite sharing the exact same mutation, the patients from the first family had severe SCID and died in infancy, while the second patient was largely asymptomatic despite having similarly decreased levels of TCR/CD3 expression. A second study identified a mutation in another Turkish family at position -1 of exon 3 resulting in a premature stop codon at the protein level. This mutation disrupted CD3γ expression, resulting in a variety of autoimmune diseases (70). Thus CD3γ deficiency results in disparate phenotypes that may be augmented by the larger genetic and environmental context of each individual. To elucidate this further, Rowe et al. studied the function and clonality of T cells from CD3γ deficient patients predisposed to autoimmunity. They found that these individuals harbor decreased Treg function, and enrichment of hydrophobic residues at positions 6 and 7 of the CDR3 chain within the variable domain of the TCR, a feature previously linked to auto-reactivity (71). Another detailed analysis of T cell subsets in CD3γ-deficient patients revealed impairment specifically in CD8 T cell development, but not CD4 development (72). This shift in the balance of T cell subsets could be responsible for the concurrent immunodeficiency and autoimmunity observed. In general, the heterogeneity of phenotypes resulting from CD3 mutations, particularly CD3γ, suggests complex underlying biology that is not fully encapsulated within our current mechanistic models. It will be interesting to apply this foundational knowledge of CD3-chain function to the improved design of engineered TCR’s and CAR’s, and their application in cancer treatment.
The number of ITAM motifs in the signaling domain appears to be key to regulating signal strength and thereby T cell activity. For example, Holst et al. conducted preclinical mouse studies using T cells with different numbers of ITAMs (73). They reconstituted Rag-/- mice with T cells in which almost every possible permutation of ITAM expression on all CD3 chains (a total of 25 recombinant conditions) was tested for its effect on T cell development and function. Their main conclusions include a quantitative linear relationship between the cumulative number of ITAM motifs in the CD3 complex and T cell proliferation rates. Despite decreased proliferation rates with fewer ITAM’s, they observed that fewer than 7 total ITAMs results in severe autoimmune disease (73), thought due to failed negative selection in the thymus of highly self-reactive T cell clones. An important qualitative difference was observed in mice harboring 6 total ITAMs: autoimmune disease occurred in those with mutated CDζa, the proximal ITAM motif, and CDζc, the distal ITAM motif, but not with mutated intermediate CDζb. No autoimmune disease was observed in mice with wildtype CDζ that lacked ITAMs from CD3δ, γ, and ε. It is not known if these phenotypes recapitulate human biology but does imply that individual ITAM’s perform specific roles and that the total number has important implications for T cell function. These types of detailed studies of CD3-subtype roles within the TCR complex will lead to better predictors of disease outcomes and means to genetically target and intervene for clinical benefit.
Notably, a few germline alterations in the CD3 subunits have been linked to cancer. An insertion/deletion polymorphism in the CD3γ promoter was linked with increased hepatocellular carcinoma incidence (74), and another (rs3181259T>C) in CD3δ was linked to recurrence in non-small cell lung cancer (NSCLC) (75). SNP rs967591G>A in CD3ε correlated with lower CD3ε expression and shorter survival in NSCLC suggesting a functional consequence could be impaired TCR signaling (76). It is surprising that such little data exists linking CD3 dysfunction with cancer given its essential role in T cell function and thereby adaptive immunosurveillance. Other disease factors in the heterogeneity of human cancers may cloud identification of a CD3 mutation signature in studies based on genetic analysis alone.
A variety of therapies have been designed to exploit CD3 subtype functions for cancer treatment. Non-specific stimulation of T cell pools with anti-CD3 antibodies has been used to enhance anti-tumor T cell responses. In TIL therapy, ex vivo culture with activating anti-CD3 antibodies along with IL-2 is the preferred method for activation and expansion (77). Compared to α/β T cell activation, CD3 conformational changes do not play as prominent a role in conventional γ/δ T cell activation. Nevertheless, binding of anti-CD3 antibodies (78) or Fab fragments (79) to γ/δ T cells enhanced tumor killing in vitro, suggesting exploiting the CD3 signaling pathway could augment novel anti-tumor properties of this unique cell type.
In CAR-T engineering, derivatives of CD3ζ chains are the favored intracellular signaling moieties incorporated into most CARs (80). Next generation CARs also include intracellular signaling components of costimulatory molecules such as 41BB or CD28 fused to the CD3ζ ITAM domains to further enhance CAR T activation (81). Engineered CAR proteins have been shown to interact with and signal via endogenously expressed TCR components (82). Therefore, fine-tuning the intracellular and transmembrane components may result in varied and potentially desirable enhancements of CAR T function. For instance the number and type of ITAM’s impacts the risk of autoimmune disease development in mice (73). With this knowledge in mind, Feucht et al. selectively mutated 1 or 2 ITAM’s of the CD3ζ within a CD19-CD28-CD3ζ CAR and tested the resultant impact on T cell function (83). CAR’s with mutations (X) in the second and third ITAMs (denoted 1XX), were the most efficacious and induced long-term remission in a pre-B acute lymphoblastic leukemia mouse model. Based on the identification of aberrant FcRγ recruitment in autoimmune disease when CD3ζ was mutationally defective (84), one might predict that incorporation of the intracellular portion of FcRγ into CARs in lieu of CD3ζ domains would result in increased CAR T activation. However, FcRγ ITAM domains showed no benefit over CD3ζ when utilized in CAR technology (85, 86), suggesting this synthetic biology does not fully mimic mutation-driven phenotypes observed in nature. Other strategies to enhance CAR T signaling could be inspired by the hyper-activated T cell states observed in autoimmune diseases caused by mutations in other CD3 subunits, and by experimenting with ITAM number and CD3 subunit of origin. However, consideration into the potential risk of chronic activation would have to be investigated.
Another approach that exploits CD3 activity in T cell signaling is to use bi-specific T cell engagers (BITEs). These constructs are comprised of one antibody moiety binding to an antigen of choice and an opposing antibody moiety binding CD3 subunits on T cells (87). The BiTE could be thought of as a soluble CAR, bridging a T cell and target tumor cell. However, in contrast to CARs, BiTEs are still reliant on endogenous CD3 expression in the T cells they recruit. BiTEs have been shown to induce responses in polyclonal populations of CD4 and CD8 T cells (88), and do not have to be custom made for each patient. Currently most bind to the extracellular component of CD3ε (88). Based on our above appreciation for the heterogenous involvement of various CD3 subunits in T cell function and disease, targeting other CD3 subunits could be used to fine-tune desired T cell recruitment with BiTEs. BiTE efficacy is in part limited by specific tumor cell phenotypes, such as the expression of sialophorin which limits T cell to tumor cell adhesion (89), and by similar side effects of CRS and neurotoxicity observed in CAR Therapy (90, 91). One study also identified a polymorphism in the CD3ζ chain (SNP rs2949655) that correlated with reduced cytotoxicity in response to BiTE treatment (92), demonstrating how CD3 mutational profiling could be used to help guide personalized treatment approaches targeting this pathway. Further improving our understanding of CD3 subunit function and signaling will help elucidate additional strategies for therapeutic intervention.
The co-receptors CD4 and CD8 are essential to T cell development and formation of a functional TCR-MHC synapse (93). They are considered co-receptors because they stabilize MHC-antigen-TCR complexes and contribute to the functions of CD4+ helper T cells and CD8+ cytotoxic T cells without direct antigen binding (94, 95). CD4 is comprised of four extracellular Ig domains that bind to MHC-II. CD8 is a dimer comprised of either a CD8α homodimer or a CD8α and CD8β heterodimer (96, 97). These extracellular domains are attached to a long extracellular stalk domain that, by means of differential sialylation/glycosylation, helps regulate CD8-MHC-I binding affinity (98). CD8αα is found on γδT cells, intestinal and dermal intra-epithelial T cells, as well as NK cells (99). CD8αβ, on the other hand, is primarily expressed on conventional cytotoxic CD8 T cells (100). Crystal structures have shown CD4 binds at membrane-proximal α2 and β2 domains of the MHC-II molecules at residues conserved between the different MHC-II types (101), while CD8 binds mainly at the α3 domain of MHC-I (102). MHC-II/CD4 or MHC-I/CD8 binding occurs at much lower affinity than MHC/TCR, presumably to help calibrate appropriate T cell selection during thymic development and avoid autoimmunity (100). Both CD4 and CD8 have transmembrane domains that contribute to the formation of their respective TCR complexes, as well as intracellular domains that associate with LCK to facilitate intracellular signaling.
The only known polymorphisms in CD8α (p.Gly111Ser) associated with human disease affect functional expression of the CD8α molecule, resulting in compete lack of CD8 T cells (103). No other mutations or even polymorphisms in the CD4 molecule or CD8β have thus far been linked to human disease, likely because each has such refined and essential functional requirements for T cell function. In mice, studies have shown that missense or non-functional mutations of CD4 and CD8 result in lack of either CD4 or CD8 T cell subtypes due to failure of thymic selection (104, 105).
The use of CD4 and CD8 as markers for specific T cell subsets and their functions has profound implications to tumor immunology and immunotherapy. Within the tumor microenvironment, increased T cell infiltration, specifically of CD8 cytotoxic T cells, and high CD8/FoxP3 ratios correlate with better overall survival in multiple cancer types (106). Th1 phenotypes of TILs correlate with improved outcomes (107). In adoptive cell therapy, it was thought that the CD8 component of TILs was the most important due to the known cytotoxic capability of CD8 T cells, with CD4 T cells playing a merely supportive role (108, 109). Indeed, in melanoma patients a higher frequency of CD8 T cells amongst the infused cells correlated with better responses (110). However, CD4 T cells have been shown to exert anti-tumor effects through largely unknown and likely multivalent mechanisms (111). Additionally, recent evidence has shown CD4 T cells can acquire cytotoxic capabilities in the presence of IL-2 and mediate direct tumor cell killing (112). For CAR T cell therapy, a combination of CD4 and CD8 T cells in a defined 1:1 ratio appears to be most efficacious (113). Current TIL therapy regimens use ex vivo IL-2 expansion, but do not select for T cell subsets prior to re-infusion (77). More detailed studies of CD4 and CD8 TIL subsets and means to expand and enhance their function ex vivo, as well as the optimal balance of CD4 and CD8 cell types in adoptive cell therapies, are required.
Various approaches to target CD4 have been applied or are in development for immunotherapy. CARs targeting CD4 have shown promise in pre-clinical models of peripheral T cell lymphomas (PTCLs) (114). Anti-CD4 antibodies have been used in patients with PTCLs as well as cutaneous T cell lymphomas with some early clinical benefit (115–118). Anti-CD4 antibodies have also shown benefit in non-hematologic malignancies via depletion of anti-inflammatory CD4 T cell subsets including Tregs, thereby allowing for enhanced proliferation of anti-tumor CD8 cytotoxic T cells (119). This approach is further bolstered with the addition of immune-checkpoint blockade (120) and is now being applied in early clinical trials (121).
Aside from using CD4 and CD8 as cell-specific markers, genetic manipulation of these molecules offers therapeutic potential. In our lab, a synthetic construct that fused the CD8α extracellular domain to a MyD88 intracellular domain, normally downstream of innate immune receptors, resulted in enhanced anti-tumor CD8 T cell function in mouse models (122). Another group demonstrated increased MHC-I binding affinity when sialylation of core 1 O-glycans on the CD8 stalk region was reduced either through neuraminidase treatment or mutation of ST3Gal-I sialyltransferase mutation (123). Through a phage display approach, Wang et al. found the substitutions Gln40Tyr and Thr45Trp in CD4 resulted in almost 500 fold increase in MHC-II binding affinity (101). Future studies are need to determine if other alterations in CD8 or CD4 extracellular domains could be used to further stabilize the TCR’s interaction with low affinity tumor antigens and thereby improve anti-tumor CD8 and CD4 effects respectively. Furthermore, docking topology of self-reactive TCR-MHC-II complexes in autoimmune T cell types is different than that in non-self-reactive T cells, and is likely influenced by CD4 molecule binding (13). One could envision that modulating CD4-TCR-MHC topologies could in a similar manner enhance T cell responses to rare self-like tumor-associated antigens (TAA). In conclusion, the CD4 and CD8 molecules are well-established markers for T cell subsets, their potential as therapeutic targets show early promise, and alteration in their function and/or binding activity warrants continued exploration.
Besides the molecules within the TCR signaling complex, many additional proximal signaling molecules also contribute to overall T cell function and hold potential as therapeutic targets. The precise events that occur following TCR activation remain controversial, but current data suggests that early proximal signaling events are largely mediated by the Src family kinases LCK and FYN (124). Upon antigenic stimulation of a T cell, pre-activated LCK is recruited first to the TCR, initiating phosphorylation of ITAMs within the CD3 intracellular domains (Figure 4). Subsequently, additional LCK bound to the CD8 and CD4 co-receptors localizes to the immune synapse, enhancing TCR-pMHC binding and enabling ZAP70 recruitment and activation (125–128). Additionally, LCK deficient mice have severely impaired T cell development (129–132) and LCK contributes to a T cells’ ability to titrate its activation level based upon the affinity of the TCR-pMHC interaction. This graded signaling response is largely regulated via distinct patterns of ITAM phosphorylation executed predominantly by LCK (133). LCK activity also modulates T cell differentiation (134, 135), CD28 costimulatory signaling (136), and even cell death (137). Given its critical role in a variety of T cell functions, it is not surprising that LCK activity is tightly regulated to maintain immune homeostasis. Consequently, genetic variations and mutations that alter the function of LCK have profound implications for the development of cancer and immune-based therapies.
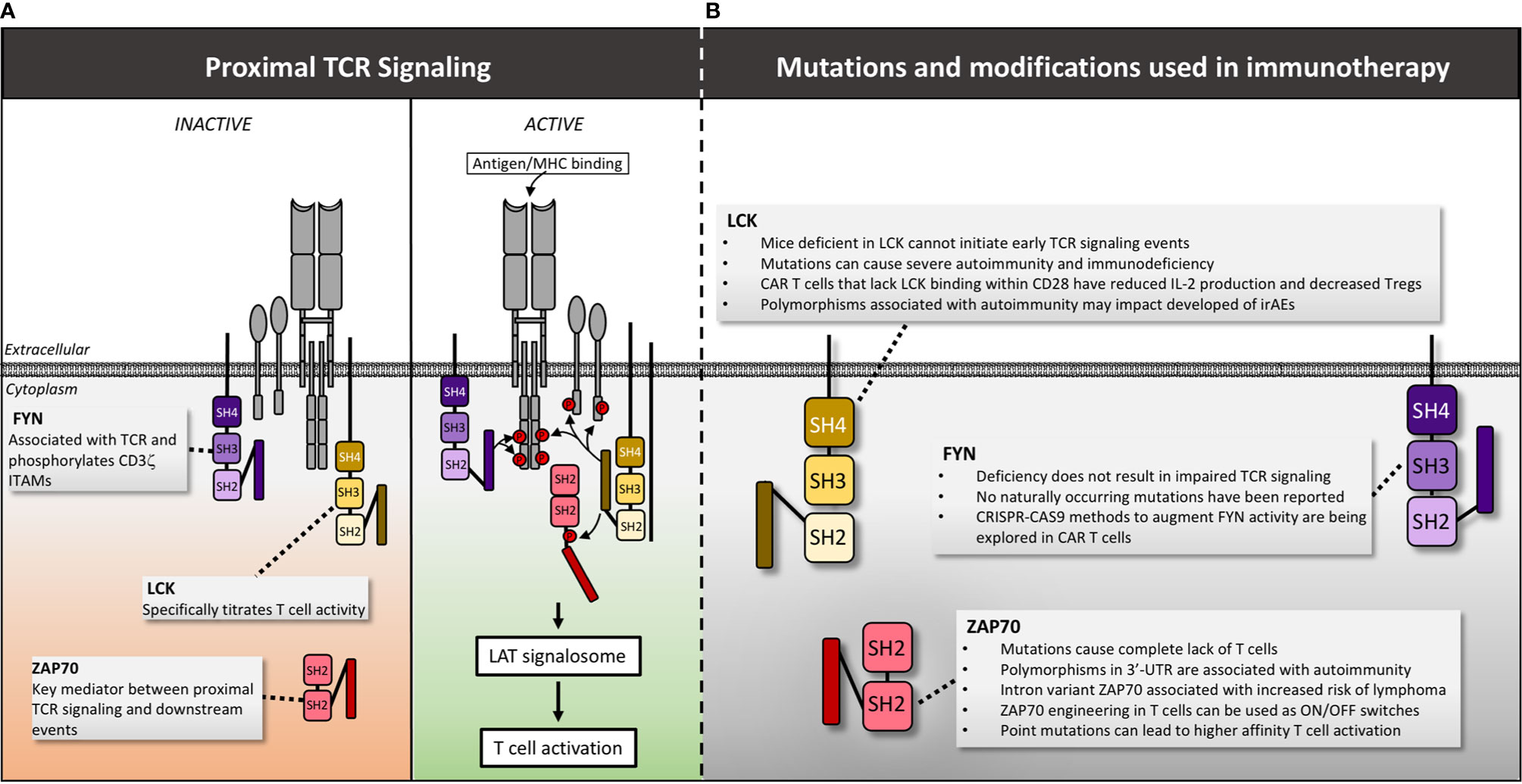
Figure 4 LCK, FYN and Zap70 structure, mutations and manipulations for immunotherapy. (A) Following antigen/MHC binding, LCK is recruited first to the TCR, initiating phosphorylation of ITAMs within the CD3 intracellular domains. Subsequently FYN is recruited and assists in phosphorylation of CD3ζ ITAMs. LCK then phosphorylates the linker sequence of ZAP70 causing a conformational change that actives the kinase activity of ZAP70 permitting additional downstream signaling. (B) The impact of mutations and genetic manipulation of LCK, FYN and ZAP70 are detailed. Red circles denote phosphate groups.
A homozygous missense mutation in a hydrophobic region of the catalytic domain (c.T1022C) has been identified in children presenting with severe recurrent infections, autoimmune manifestations and panniculitis. This mutation is associated with reduced CD4 and CD8 expression, impaired TCR activation, decreased Treg levels and expansion of an oligoclonal γ/δ T cell population (138). Additional splice mutation variants of LCK have been reported and correlate with impaired LCK function and immune dysregulation. Patients with these variants present with a range of clinical syndromes including epidermodysplasia verruciformis and recurrent bacterial infections (mutation: c.188-2A>G) (139), common variable immunodeficiency (CVID, mutation: lack of exon 7) (140), and severe combined immunodeficiency (SCID, mutation: lack of exon 7) (141). Mutations that delete a C-terminal regulatory tyrosine within LCK increase LCK activity resulting in sustained T cell activation and oncogenesis in mice (142), as well as increased IL-2 production independent of antigenic signaling in humans (143). In addition, a chromosomal translocation t(1;7)(p34;q34) has been identified in patients with T cell acute lymphoblastic leukemia (ALL) (144). This translocation also increases LCK activity and the degree of elevated activity is correlated with breakpoint location and disease severity (144). These findings are intriguing from a T cell engineering perspective given that modulating LCK function, even via changes in single residues, has significant impact on T cell function. However, based upon the stark clinical phenotypes observed among patients, it is clear that extreme caution is warranted when considering genetic manipulation of this crucial protein.
Understanding LCK activity and genetically altering this gene still holds promise for several therapeutic approaches in oncology. In particular, LCK modulation is an important area of study for CAR T cell treatment of solid tumors. For example, CAR T cells have been engineered with a deleted LCK-binding motif within CD28 (ΔCD28). In mice, these second generation ΔCD28/CD3ζ (145) and third generation ΔCD28-4-1BBζ CAR T cells (146) show reduced IL-2 production and improved tumor control in the presence of Tregs. Clinically, a patient with malignant pleural mesothelioma was treated with anti-FAP ΔCD28/CD3ζ CAR T cells and experienced stable disease for 1 year, suggesting that ΔCD28/CD3ζ CAR T cells may have contributed in controlling his disease (147). However, this finding is currently anecdotal and other studies suggest that the effect of ΔCD28/CD3ζ on CAR T cell function may depend upon the immunosuppressive mechanism within the tumor microenvironment (TME) (148). For instance, tumor models with high TGFβ within tumor tissue require an intact LCK motif within the CD28/CD3ζ CAR receptor in order to overcome TGFβ-mediated suppression (148). Therefore, patient stratification by TME immune profiles could identify patients who would benefit from ΔCD28/CD3ζ CAR T cells. Synthetic LCK modulation in CAR T cells has other therapeutic implications as well given that LCK deficient cells are resistant to activation induced cell death (149) and LCK is involved in PD-1 induced inhibition (150).
It is also important to consider the impact of LCK mutations in the context of immune checkpoint inhibition (ICI). For example, the LCK SNP rs10914542 G allele impairs TCR activation (151), suggesting that patients harboring this allele may be less likely to mount a robust response following ICI therapy. Conversely, a single amino acid variant in LCK (p.G85W of exon 4) was associated with autoimmune diseases including Sjogren syndrome, SLE and RA, suggesting that patients with this variant could be at increased risk of developing autoimmune complications in the setting of ICI (152). All together, these data suggest that artificial regulation of LCK or screening for LCK variants could inform immune-based therapeutic strategies for cancer.
FYN is another Src family tyrosine kinase involved in proximal TCR signaling, however, the precise roles of this protein are less understood than LCK. FYN also associates with the TCR and is involved in phosphorylation of CD3ζ ITAMs. However, LCK deficiency causes a much more dramatic phenotype than FYN deficiency, suggesting that FYN is not required for T cell activation (126). FYN interacts with many additional binding partners including PI3K (153, 154), lymphocyte-specific scaffold protein adhesion and degranulation-promoting adaptor protein (ADAP) (155), phosphoprotein associated with glycolipid-enriched membranes (PAG) (156), signaling lymphocyte activation molecule (SLAM) and others (157). These interactions collectively enable a diverse breadth of functions ranging from T cell activation to anergy (158). It is therefore reasonable to hypothesize that alterations in FYN function could modulate T cell activity in a variety of ways that could be exploited for therapeutic purposes.
To date, no naturally occurring mutations in FYN have been linked to disease, suggesting that either existing genetic variants are relatively benign, or they cause lethality. Despite this ambiguity, modulating FYN activity remains an active area of interest in T cell engineering. For example, in a recent review by Thakar et al., the authors propose that inhibition of the FYN-ADAP pathway using CRISPR-CAS9 could provide a unique means of selectively downregulating cytokine production by CAR T cells without impairing cytotoxicity (159). This approach could be used to reduce the severity of cytokine release syndrome, a dangerous complication of CAR T cell therapy (159). Inhibiting FYN activity may also enhance T cell migration. A study by Schaeuble et al. reported that inhibition of FYN with the small molecule SU6656 promoted enhanced CCR7-driven migratory function of nonactivated T cells in vitro (160). CCR7 is expressed by both central memory T cells (TCM) and T memory stem cells (TSCM), both of which are promising substrates for both CAR T cell (161, 162), and transgenic T cell therapy (163). Therefore, inhibiting FYN activity in genetically engineered T cells could enhance their migratory capacity prior to activation within either a tumor-draining lymph node or the TME.
Alternatively, activating some functions of FYN could improve cellular based immunotherapy. One study found that deletion of FYN in a mouse model promotes differentiation of CD4+ T cells towards a Treg phenotype and away from a Th17 phenotype (164). While speculative, this suggests that activating Fyn could promote a Th17 phenotype. Some data suggest that Th17 CD4+ T cells have superior anti-tumor function and improved persistence as compared to Th1 cells in adoptive cell therapy settings (165–167). Therefore, FYN modulation in CD4+ T cell engineering approaches may not only reduce Treg induction but could also promote a Th17 phenotype. Increasing FYN activity has other potential beneficial implications. The SH2 domain of FYN binds Tim-3, promoting T cell activation and increasing cytokine production (168), a surprising finding as Tim-3 is classically associated with T cell exhaustion. Increased FYN-Tim-3 binding via genetic modification could shift the balance of Tim-3 activity towards T cell activation and away from exhaustion (168). In contrast to the T cell activating function of FYN, FYN association with PAG and c-cbl can disrupt canonical TCR signaling and promote T cell anergy under certain conditions (156, 169–173). These findings indicate that FYN has highly varied and even paradoxical effects on T cell activity depending upon its binding partners. Importantly, the binding site for these various partners are not all known. Therefore, further characterization of the specific binding locations could enable genetic alterations to precisely tailor FYN activity and improve active immunotherapeutic approaches. Regarding passive immunotherapeutic strategies such as ICI, FYN-activating signatures have been associated with lupus nephritis, an autoimmune condition (174), loosely suggesting that increased FYN activity might increase the risk of autoimmune complications following ICI. However, no polymorphisms or genetic variants have been associated with autoimmunity thus it is unlikely that mutational profiling of FYN would predict development of immune related Adverse Events (irAE). Overall, further characterization of FYN function, binding sites and binding partners is warranted and may provide opportunities for various T cell engineering strategies in the future.
Once LCK and FYN phosphorylate ITAMs on the CD3 molecules, the next step in TCR signal transduction involves binding of ζ-chain-associated protein kinase of 70 kDa (ZAP70) (45). Distinct from the Src family kinases described above, ZAP70 along with SYK are the two prominent members of the Syk family of kinases (175). ZAP70 is comprised of an auto-inhibited kinase domain and two amino-terminal SH2 domains that bind doubly-phosphorylated ITAMs (176) Upon ITAM binding, ZAP70 undergoes a conformational change that results in additional phosphorylation of residues in the second linker sequence by LCK. This relieves inhibition of the kinase domain and results in downstream signal propagation (177).
Various ZAP70 mutations have been shown to cause a severe form of immunodeficiency characterized by complete lack of functional T cells (178). Most of these mutations affect the kinase domain (179–182), or lead to loss or destabilization of the protein transcript altogether (183–185). Syc can take the place of ZAP70 in T cell signaling when the latter is impaired, allowing for some CD4 cells to survive thymic selection, but these Syc+ZAP70- T cells are defective in IL-2 production and proliferation and provide aberrant help to B cells for antibody class switching (182, 185). In contrast, polymorphisms in the ZAP70 coding region or 3’-UTR have conversely been associated with autoimmune disorders including psoriasis and type 1 diabetes (rs17695937) (186, 187), inflammatory bowel disease (IBD, rs13420683) (188), and RA (rs2278699) (189). Finally, an intron variant of ZAP70 (rs7425883) is associated with a decreased risk of developing non-Hodgkin lymphoma (190), and aberrant elevated expression of ZAP70 in B cell CLL cells correlates with enhanced BCR signaling in the leukemic cells and poorer prognosis (191, 192). Mouse studies recapitulating some of these autoimmune or immunodeficient phenotypes have demonstrated point-mutations in the second SH2 domain, interdomain B, or paired mutations in the kinase region lead to aberrant thymic selection more permissive of higher affinity self-reactive clones, and quantitative differences in TCR signaling (193–195). Of particular interest, the tyrosine residues at positions 292, 315, and 492 have been shown to play a negative regulatory role when phosphorylated, and their mutation to phenyalanine (which prevents phosphorylation), allowed for T cell hyperactivation (196). These findings suggest that ZAP70 could be used not only as a prognostic marker of disease but also a therapeutic target.
Due to its upstream role in TCR signaling, ZAP70 has been gaining interest in the world of cancer-immunotherapy. Of greatest potential, engineered ZAP70 constructs have been designed to function as on/off switches to control T cell responses. In one approach, a larger analog of the kinase inhibitor PP1 was used to selectively inhibit an engineered ZAP70 with altered inhibitor binding affinity, resulting in impaired catalytic activity (197). Interestingly, Treg function was not affected by this approach, suggesting unique non-catalytic activity of ZAP70 is functioning in Tregs (198), and that therapeutic application of this design would not affect tolerance and protective roles of adaptive immune cell types. This analog sensitive ZAP70 could be employed to turn off unwarranted activation of adoptively transferred T cell therapies. Other investigators have developed a tetracycline-inducible ZAP70 gene promoter allowing for selective turning on of ZAP70 transcription (199). This has so far only been used in studies designed to assess the role of ZAP70 in thymic selection, and clinical application may be impaired by the need for tetracycline infusion and difficulty regulating expression levels once turned on. More recently, a dual small-molecule gated ZAP70 has been created via fusion of the analog-sensitive ZAP70 to the ligand binding domain of the estrogen receptor (200), allowing for both on and off signals. In vitro, this new ZAP70 construct could be controlled on a minute-by-minute timeframe and regulated calcium flux and CD69 expression levels. However, cytokine production was impaired in the “on” configuration, thus more refinement is required before clinical application (200). Future studies could examine the role of specific ZAP70 point mutants, such as those identified above that result in autoimmune phenotypes, in regulating adoptive T cell therapy efficacy. Additionally, the differential role of Syk and ZAP70 with regards to TCR signaling is still an area of inquiry with some conflicting evidence regarding the potency of the two molecules for T cell activation (182, 185, 201, 202). Although early studies of CAR design have favored CD3ζ and ZAP70 dependent designs, recruiting the Syk tyrosine kinase in lieu of ZAP70 may have some benefit in specific contexts. Finally, CXCR3-mediated T cell chemotaxis was shown to be dependent on ZAP70 and was impaired by TCR signaling (203). Modulating ZAP70 crosstalk between these two important pathways could affect the ability of T cells to infiltrate the tumor microenvironment and maintain TCR signals and resultant activation states. Collectively, this early evidence demonstrates artificially controlling ZAP70, or variants of ZAP70 with different signaling thresholds, could be powerful tools for cancer immunotherapy.
LAT serves as a major junction point in TCR signaling, forming a nexus between the early antigen-recognition machinery and a multitude of downstream pathways (Figures 1 and 5) (204). Phosphorylation of LAT at multiple intracellular tyrosine residues by ZAP70 is a key link between TCR antigen recognition and the transcriptional paradigm shift of T cell activation (205). Due to its assembly of numerous signaling molecules, LAT has been referred to as the central platform for the “LAT signalosome.” (206) LAT is comprised of minimal extracellular and transmembrane domains, and an extensive cytoplasmic region with numerous phosphorylation and protein binding sites (207).
Mutational mapping has allowed identification of tyrosine-phosphorylation residues required for LAT to associate with individual signaling partners. For example, mutation of tyrosine 132 in human T cells results in defective binding to PLCγ-1 (208), tyrosine 171 was essential for PI3K activation (209), tyrosines 110 and 226 are required for ERK activation (208), 171 and 191 required for Gads binding, and 171, 191, and 226 together are required for Grb2 binding (Figure 5) (210). Aside from tyrosine residues, study of the human Jurkat T cell line identified 11 serine residues in the cytoplasmic domain that may be key to signal propagation. Cells expressing LAT with S->A mutations at serines 38, 40, 106, 164, and 180 exhibited decreased PLCγ-1 and SLP-76 binding, reduced IL-2 production, but increased ZAP70 phosphorylation (211). The impact of each individual serine residue has yet to be elucidated. A recent study identified as many as 90 putative binding partners for LAT, suggesting the myriad of established roles for residues in this molecule may yet underestimate the importance of LAT in T cell activation (212).
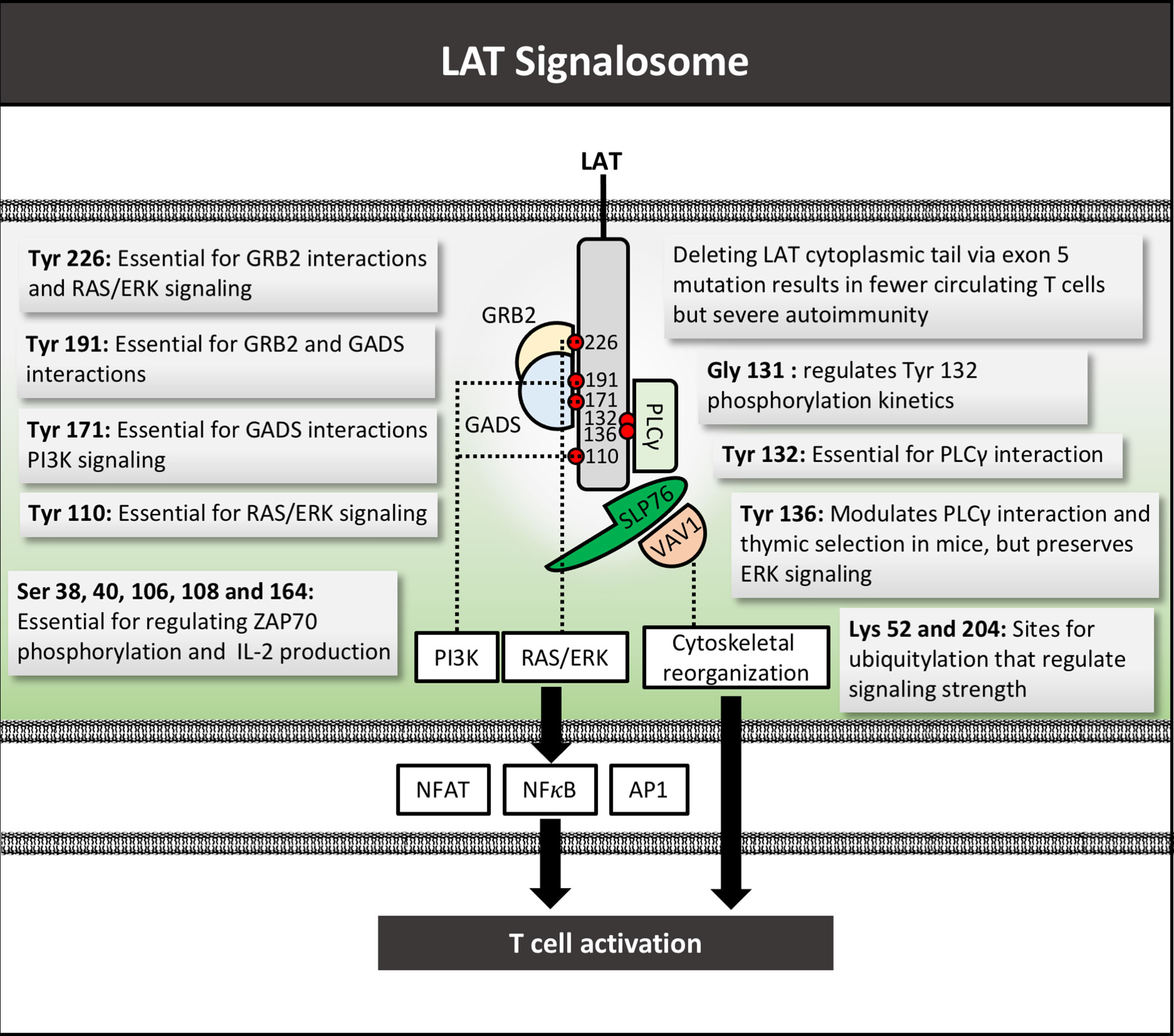
Figure 5 LAT signalosome structure and function. LAT signalosome schematic structure with function of key tyrosine residues depicted by their respective numbers, as well as mutations and respective phenotypes. Several key LAT signaling partners are depicted. Red circles denote phosphate groups. Tyr, Tyrosine; Gly, glycine; Ser, serine; Lys, Lysine.
Studies in mice have allowed mechanistic elucidation of the key function of LAT and its structure in T cell development and mature T cell functions (213–215). Completely blocking LAT expression or function in mice led to impairment in T cell development. Mutation of the distal 4 tyrosine residues of LAT to phenylalanine mimicked the phenotype of LAT-/- mice, in which T cell development is completely blocked at the double-negative 3 (DN3) stage (215). Selectively knocking-out LAT expression after the DN3 stage led to impaired development of single positive T cells in the thymus and periphery (216). Conversely, mutations or deletions of LAT in mature post-thymic T cells altered but did not completely impair T cell functions, and even led to autoimmunity and aberrant lymphoproliferation, implicating LAT in not only T cell activation but also regulation (217, 218). In one study, a mutation in tyrosine 136 and impaired PLCγ binding led to MHC-independent constitutive Th2 T cell activation and T cell dependent B cell hyperproliferation and antibody production, ultimately resulting in systemic autoimmune disease (219). Others showed that the same mutation resulted in disruption of thymic positive and negative selection (220). Similarly, a third study showed that mutation of tyrosine 136 blocked PLCγ dependent functions, but allowed for continued ERK signaling, first causing impaired T cell development but later causing a lymphoproliferative disorder (221). The mechanism of negative regulation via LAT could be mediated via association with Grb2, a known inhibitory signal mediator (222), or through binding and inhibiting the active form of LCK (223). Together these results implicate LAT in both T cell thymic selection as well as subsequent immune regulation, and that complex interactions between LAT and its multiple binding partners maintains a balance between T cell activation and inhibition.
In humans, mutations or polymorphisms in LAT recapitulate the range of mouse phenotypes ranging from autoimmunity to immunodeficiency (224). Higher expression levels of LAT (as assessed by qRT-PCR and flow cytometry) were correlated with severity of aplastic anemia, however it was unclear how this over-expression was regulated or if this was a cause or consequence of disease (225). Loss of the cytoplasmic tail of LAT due to a mutation in exon 5 resulted in immunodeficiency characterized by a decrease in circulating T cells, but simultaneous severe autoimmunity (226). T cells in these patients were still able to induce calcium influx and NF-κB activation, but had aberrant ERK signaling (226). The heterogeneity of disease resulting from LAT-deficiency has led some to distinguish LAT-dependent pathology due to immune hyper-activation from true autoimmunity (227). The mouse studies above do suggest LAT also plays a role in regulating thymic selection and can lead to true autoimmune T cells. A similar role in humans has not been entirely ruled out. These studies underscore the fine balance between pro and anti-inflammatory processes that evolved to maintain homeostasis within the immune system, and that LAT is a key regulator of this balance in T cells.
As of now, no immunotherapies have modulated LAT to enhance anti-cancer T cell responses. Based on our above understanding, it is possible that changes in LAT would lead to too many off target effects of T cell-based therapies, or persistent non-specific inflammation that would be detrimental to the host. However, careful alterations could also increase T cell activation in response to weak or rare neo-antigens or help adoptively transferred T cells maintain activation states in solid tumors. These varied effects could be achieved by selectively mutating key tyrosine or serine residues within the cytoplasmic tail. Additional modulations could include replacing lysine with arginine residues, which was shown to decrease ubiquitylation and enhance T cell signaling (228), or mutating residues neighboring key tyrosines to alter phosphorylation kinetics (229). Furthermore, the use of LAT instead of CD3ζ as the intracellular signaling component of CARs could be considered. Extensive pre-clinical testing would be required to ensure any increased T cell activation caused by LAT modulation would not result in intolerable or dangerous side effects and means to kill or turn off such cells should be incorporated as a safety mechanism.
LAT serves as a major hub after which TCR signaling networks with many other pathways in T cells, such as those downstream of chemokine and cytokine receptors and costimulatory molecules (204). As a consequence, many highly complex, and tightly regulated interactions take place including activation of canonical MAPK, NF-κB, Ca++-mediated signaling pathways that ultimately regulate the nuanced transcriptional profiles characteristic of each T cell phenotype (Figure 5) (217, 230). Abnormalities in any one of the many proteins involved in these processes can lead to aberrant T cell phenotypes. Some of the best described interactions between LAT occur with ITK, PLC-γ1, Grb2, SLP-76, VAV1, and FYB1 (230). Most of these are activating, but the interaction with Grb2 is generally inhibitory to T cell activation, as briefly mentioned above (222). As an example of the spectrum of diseases caused by these downstream proteins, gain of function of ITK is common in T cell lymphomas (231), loss-of-function mutations are associated with lymphoproliferative diseases (232), full ITK deficiency has been linked to idiopathic CD4+ T cell lymphopenia (233), and SNPs in ITK have been associated with asthma (234). Polymorphisms in FYB as well as low levels of LAT-PLC-γ1 interactions have also been implicated in susceptibility to asthma, perhaps due to skewing of T cell polarization towards an aberrant Th2 phenotype (235, 236). VAV1 has strong links to oncogenesis in various tissues (237–239), with mutations causing disruptions in multiple pathways including Ca++ signaling (240, 241). It is important to note that many of these downstream mediators are involved in signaling in other immune cell types besides T cells, and receptor driven pathways other than the TCR pathway even within T cells. For example, PLC-γ1 is involved in FcεRI-mediated mast cell degranulation, thus its association with asthma maybe more due to effects on mast cells as opposed to T cells (242). FYB is also expressed in many other immune cell types of both the innate and adaptive system with diverse functions, making cell-type or pathway specific conclusions difficult (243). Any targeting of these molecules to alter T cell function must take into account that not only T cells or the TCR pathway may be affected.
Given the complexity of this stage of T cell activation, no therapies targeting these downstream networks have yet been approved in cancer immunotherapy. However, a few groups have begun to explore distal TCR signaling modulation to benefit immunotherapy. For instance, knocking out ADAP in adoptively transferred T cells reduced PD-1 expression and increased anti-tumor efficacy in mice (244). Other pre-clinical studies have shown that Cis-mediated inhibition of CD8 T cells functions by down-regulating PLC-γ, and targeting this pathway could increase potency of CARs and other adoptive therapies (245). Outside the context of immunotherapy, directly targeting some of these molecules could also have direct anti-neoplastic effects. Much more cell-type specific, and pathway specific studies are needed before targeting these downstream mediators can be used safely and effectively in treatment of human cancers.
The TCR proximal signaling pathway that regulate T cell function comprise a complex cascade of interactions involving numerous extracellular and intracellular proteins with unique functions. Perturbations such as polymorphisms and mutations in each contribute to a myriad of human diseases ranging from immunodeficiency to autoimmunity, and some even have significant contributions to various malignancies. Understanding the nuanced role of each molecule has allowed for the design of immunotherapies to take advantage of the significant involvement of TCR signaling and cytokine activation of T cells in cancer. The genetic alterations described throughout this review are summarized in Table 1.
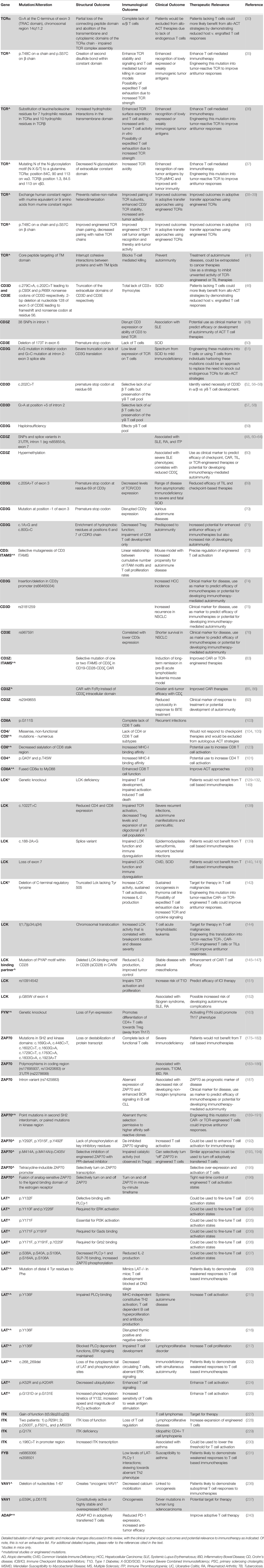
Table 1 Mutations and molecular alterations in the TCR pathways.
AK and NL wrote the manuscript and prepared the figures. AC prepared the table and provided editing and input into the manuscript. ED oversaw the entire project providing guidance as well as detailed review and editing of all aspects of the paper. All authors contributed to the article and approved the submitted version.
VA Merit Award I01BX004935; NCI R01 CA207913; NCI T32 CA23673402; University of Colorado Comprehensive Cancer Center
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the National Cancer Institute, the Department of Veterans Affairs, and the University of Colorado Comprehensive Cancer Center for the support of this work.
1. Krangel MS. Mechanics of T Cell Receptor Gene Rearrangement. Curr Opin Immunol (2009) 21:133–9. doi: 10.1016/j.coi.2009.03.009
2. Smith-Garvin JE, Koretzky GA, Jordan MS. T Cell Activation. Annu Rev Immunol (2009) 27:591–619. doi: 10.1146/annurev.immunol.021908.132706
3. Laidlaw BJ, Craft JE, Kaech SM. The Multifaceted Role of CD4(+) T Cells in CD8(+) T Cell Memory. Nat Rev Immunol (2016) 16:102–11. doi: 10.1038/nri.2015.10
4. Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, Kedl RM, et al. T Cell Responses: Naive to Memory and Everything in Between. Adv Physiol Educ (2013) 37:273–83. doi: 10.1152/advan.00066.2013
5. Walk SF, March ME, Ravichandran KS. Roles of Lck, Syk and ZAP-70 Tyrosine Kinases in TCR-mediated Phosphorylation of the Adapter Protein Shc. Eur J Immunol (1998) 28:2265–75. doi: 10.1002/(SICI)1521-4141(199808)28:08<2265::AID-IMMU2265>3.0.CO;2-P
6. Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T Cell Receptor-Proximal Signals are Sustained in Peripheral Microclusters and Terminated in the Central Supramolecular Activation Cluster. Immunity (2006) 25:117–27. doi: 10.1016/j.immuni.2006.04.010
7. Gorentla BK, Zhong XP. T Cell Receptor Signal Transduction in T Lymphocytes. J Clin Cell Immunol (2012) 2012:5. doi: 10.4172/2155-9899.S12-005
8. Wucherpfennig KW, Gagnon E, Call MJ, Huseby ES, Call ME. Structural Biology of the T-cell Receptor: Insights Into Receptor Assembly, Ligand Recognition, and Initiation of Signaling. Cold Spring Harb Perspect Biol (2010) 2:a005140. doi: 10.1101/cshperspect.a005140
9. Legut M, Cole DK, Sewell AK. The Promise of Gammadelta T Cells and the Gammadelta T Cell Receptor for Cancer Immunotherapy. Cell Mol Immunol (2015) 12:656–68. doi: 10.1038/cmi.2015.28
10. Wang JH, Reinherz EL. The Structural Basis of Alphabeta T-lineage Immune Recognition: TCR Docking Topologies, Mechanotransduction, and Co-Receptor Function. Immunol Rev (2012) 250:102–19. doi: 10.1111/j.1600-065X.2012.01161.x
11. Takaba H, Takayanagi H. The Mechanisms of T Cell Selection in the Thymus. Trends Immunol (2017) 38:805–16. doi: 10.1016/j.it.2017.07.010
12. Matsuda T, Miyauchi E, Hsu YW, Nagayama S, Kiyotani K, Zewde M, et al. TCR Sequencing Analysis of Cancer Tissues and Tumor Draining Lymph Nodes in Colorectal Cancer Patients. Oncoimmunology (2019) 8:e1588085. doi: 10.1080/2162402X.2019.1588085
13. Wucherpfennig KW, Call MJ, Deng L, Mariuzza R. Structural Alterations in peptide-MHC Recognition by Self-Reactive T Cell Receptors. Curr Opin Immunol (2009) 21:590–5. doi: 10.1016/j.coi.2009.07.008
14. Golubovskaya V, Wu L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers (Basel) (2016) 8. doi: 10.3390/cancers8030036
15. Cui JH, Lin KR, Yuan SH, Jin YB, Chen XP, Su XK, et al. Tcr Repertoire as a Novel Indicator for Immune Monitoring and Prognosis Assessment of Patients With Cervical Cancer. Front Immunol (2018) 9:2729. doi: 10.3389/fimmu.2018.02729
16. Simnica D, Akyuz N, Schliffke S, Mohme M, L VW, Mahrle T, et al. T Cell Receptor Next-Generation Sequencing Reveals Cancer-Associated Repertoire Metrics and Reconstitution After Chemotherapy in Patients With Hematological and Solid Tumors. Oncoimmunology (2019) 8:e1644110. doi: 10.1080/2162402X.2019.1644110
17. Hogan SA, Courtier A, Cheng PF, Jaberg-Bentele NF, Goldinger SM, Manuel M, et al. Peripheral Blood Tcr Repertoire Profiling May Facilitate Patient Stratification for Immunotherapy Against Melanoma. Cancer Immunol Res (2019) 7:77–85. doi: 10.1158/2326-6066.CIR-18-0136
18. Arakawa A, Vollmer S, Tietze J, Galinski A, Heppt MV, Burdek M, et al. Clonality of CD4(+) Blood T Cells Predicts Longer Survival With CTLA4 or PD-1 Checkpoint Inhibition in Advanced Melanoma. Front Immunol (2019) 10:1336. doi: 10.3389/fimmu.2019.01336
19. Hopkins AC, Yarchoan M, Durham JN, Yusko EC, Rytlewski JA, Robins HS, et al. T Cell Receptor Repertoire Features Associated With Survival in Immunotherapy-Treated Pancreatic Ductal Adenocarcinoma. JCI Insight (2018) 3. doi: 10.1172/jci.insight.122092
20. Girardi M, Oppenheim DE, Steele CR, Lewis JM, Glusac E, Filler R, et al. Regulation of Cutaneous Malignancy by Gammadelta T Cells. Science (2001) 294:605–9. doi: 10.1126/science.1063916
21. Tie G, Yan J, Khair L, Messina JA, Deng A, Kang J, et al. Hypercholesterolemia Increases Colorectal Cancer Incidence by Reducing Production of NKT and Gammadelta T Cells From Hematopoietic Stem Cells. Cancer Res (2017) 77:2351–62. doi: 10.1158/0008-5472.CAN-16-1916
22. Street SE, Hayakawa Y, Zhan Y, Lew AM, MacGregor D, Jamieson AM, et al. Innate Immune Surveillance of Spontaneous B Cell Lymphomas by Natural Killer Cells and Gammadelta T Cells. J Exp Med (2004) 199:879–84. doi: 10.1084/jem.20031981
23. Liu Z, Eltoum IE, Guo B, Beck BH, Cloud GA, Lopez RD. Protective Immunosurveillance and Therapeutic Antitumor Activity of Gammadelta T Cells Demonstrated in a Mouse Model of Prostate Cancer. J Immunol (2008) 180:6044–53. doi: 10.4049/jimmunol.180.9.6044
24. Hayday AC. Gammadelta T Cells and the Lymphoid Stress-Surveillance Response. Immunity (2009) 31:184–96. doi: 10.1016/j.immuni.2009.08.006
25. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The Prognostic Landscape of Genes and Infiltrating Immune Cells Across Human Cancers. Nat Med (2015) 21:938–45. doi: 10.1038/nm.3909
26. Cordova A, Toia F, La Mendola C, Orlando V, Meraviglia S, Rinaldi G, et al. Characterization of Human Gammadelta T Lymphocytes Infiltrating Primary Malignant Melanomas. PloS One (2012) 7:e49878. doi: 10.1371/journal.pone.0049878
27. Donia M, Ellebaek E, Andersen MH, Straten PT, Svane IM. Analysis of Vdelta1 T Cells in Clinical Grade Melanoma-Infiltrating Lymphocytes. Oncoimmunology (2012) 1:1297–304. doi: 10.4161/onci.21659
28. Nicol AJ, Tokuyama H, Mattarollo SR, Hagi T, Suzuki K, Yokokawa K, et al. Clinical Evaluation of Autologous Gamma Delta T Cell-Based Immunotherapy for Metastatic Solid Tumours. Br J Cancer (2011) 105:778–86. doi: 10.1038/bjc.2011.293
29. Pauza CD, Liou ML, Lahusen T, Xiao L, Lapidus RG, Cairo C, et al. Gamma Delta T Cell Therapy for Cancer: it Is Good to be Local. Front Immunol (2018) 9:1305. doi: 10.3389/fimmu.2018.01305
30. Morgan NV, Goddard S, Cardno TS, McDonald D, Rahman F, Barge D, et al. Mutation in the TCRalpha Subunit Constant Gene (TRAC) Leads to a Human Immunodeficiency Disorder Characterized by a Lack of Tcralphabeta+ T Cells. J Clin Invest (2011) 121:695–702. doi: 10.1172/JCI41931
31. Kuhns MS, Davis MM, Garcia KC. Deconstructing the Form and Function of the TCR/CD3 Complex. Immunity (2006) 24:133–9. doi: 10.1016/j.immuni.2006.01.006
32. Brazin KN, Mallis RJ, Boeszoermenyi A, Feng Y, Yoshizawa A, Reche PA, et al. The T Cell Antigen Receptor Alpha Transmembrane Domain Coordinates Triggering Through Regulation of Bilayer Immersion and CD3 Subunit Associations. Immunity (2018) 49:829–841 e826. doi: 10.1016/j.immuni.2018.09.007
33. Natarajan A, Nadarajah V, Felsovalyi K, Wang W, Jeyachandran VR, Wasson RA, et al. Structural Model of the Extracellular Assembly of the TCR-CD3 Complex. Cell Rep (2016) 14:2833–45. doi: 10.1016/j.celrep.2016.02.081
34. He Y, Rangarajan S, Kerzic M, Luo M, Chen Y, Wang Q, et al. Identification of the Docking Site for CD3 on the T Cell Receptor Beta Chain by Solution Nmr. J Biol Chem (2015) 290:19796–805. doi: 10.1074/jbc.M115.663799
35. Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA, et al. Enhanced Antitumor Activity of T Cells Engineered to Express T-cell Receptors With a Second Disulfide Bond. Cancer Res (2007) 67:3898–903. doi: 10.1158/0008-5472.CAN-06-3986
36. Haga-Friedman A, Horovitz-Fried M, Cohen CJ. Incorporation of Transmembrane Hydrophobic Mutations in the TCR Enhance its Surface Expression and T Cell Functional Avidity. J Immunol (2012) 188:5538–46. doi: 10.4049/jimmunol.1103020
37. Kuball J, Hauptrock B, Malina V, Antunes E, Voss RH, Wolfl M, et al. Increasing Functional Avidity of TCR-Redirected T Cells by Removing Defined N-glycosylation Sites in the TCR Constant Domain. J Exp Med (2009) 206:463–75. doi: 10.1084/jem.20082487
38. Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced Antitumor Activity of Murine-Human Hybrid T-cell Receptor (TCR) in Human Lymphocytes is Associated With Improved Pairing and TCR/CD3 Stability. Cancer Res (2006) 66:8878–86. doi: 10.1158/0008-5472.CAN-06-1450
39. Sommermeyer D, Uckert W. Minimal Amino Acid Exchange in Human TCR Constant Regions Fosters Improved Function of TCR Gene-Modified T Cells. J Immunol (2010) 184:6223–31. doi: 10.4049/jimmunol.0902055
40. Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C, et al. Facilitating Matched Pairing and Expression of TCR Chains Introduced Into Human T Cells. Blood (2007) 109:2331–8. doi: 10.1182/blood-2006-05-023069
41. Manolios N, Ali M, Bender V. T-Cell Antigen Receptor (TCR) Transmembrane Peptides: A New Paradigm for the Treatment of Autoimmune Diseases. Cell Adh Migr (2010) 4:273–83. doi: 10.4161/cam.4.2.11909
42. Courtney AH, Lo WL, Weiss A. Tcr Signaling: Mechanisms of Initiation and Propagation. Trends Biochem Sci (2018) 43:108–23. doi: 10.1016/j.tibs.2017.11.008
43. Hayes SM, Love PE. Stoichiometry of the Murine Gammadelta T Cell Receptor. J Exp Med (2006) 203:47–52. doi: 10.1084/jem.20051886
44. Dong, Zheng L, Lin J, Zhang B, Zhu Y, Li N, et al. Structural Basis of Assembly of the Human T Cell receptor-CD3 Complex. Nature (2019) 573:546–52. doi: 10.1038/s41586-019-1537-0
45. Gaud G, Lesourne R, Love PE. Regulatory Mechanisms in T Cell Receptor Signalling. Nat Rev Immunol (2018) 18:485–97. doi: 10.1038/s41577-018-0020-8
46. de Saint Basile G, Geissmann F, Flori E, Uring-Lambert B, Soudais C, Cavazzana-Calvo M, et al. Severe Combined Immunodeficiency Caused by Deficiency in Either the Delta or the Epsilon Subunit of CD3. J Clin Invest (2004) 114:1512–7. doi: 10.1172/JCI22588
47. Tasher D, Dalal I. The Genetic Basis of Severe Combined Immunodeficiency and its Variants. Appl Clin Genet (2012) 5:67–80. doi: 10.2147/TACG.S18693
48. Martins M, Williams AH, Comeau M, Marion M, Ziegler JT, Freedman BI, et al. Genetic Association of CD247 (CD3zeta) With SLE in a Large-Scale Multiethnic Study. Genes Immun (2015) 16:142–50. doi: 10.1038/gene.2014.73
49. Fischer A, de Saint Basile G, Le Deist F. CD3 Deficiencies. Curr Opin Allergy Clin Immunol (2005) 5:491–5. doi: 10.1097/01.all.0000191886.12645.79
50. Firtina S, Ng YY, Ng OH, Nepesov S, Yesilbas O, Kilercik M, et al. A Novel Pathogenic Frameshift Variant of CD3E Gene in Two T-B+ Nk+ SCID Patients From Turkey. Immunogenetics (2017) 69:653–9. doi: 10.1007/s00251-017-1005-7
51. Arnaiz-Villena A, Timon M, Corell A, Perez-Aciego P, Martin-Villa JM, Regueiro JR, et al. Brief Report: Primary Immunodeficiency Caused by Mutations in the Gene Encoding the CD3-gamma Subunit of the T-lymphocyte Receptor. N Engl J Med (1992) 327:529–33. doi: 10.1056/NEJM199208203270805
52. Yu GP, Nadeau KC, Berk DR, de Saint Basile G, Lambert N, Knapnougel P, et al. Genotype, Phenotype, and Outcomes of Nine Patients With T-B+NK+ Scid. Pediatr Transplant (2011) 15:733–41. doi: 10.1111/j.1399-3046.2011.01563.x
53. Soudais C, de Villartay JP, Le Deist F, Fischer A, Lisowska-Grospierre B. Independent Mutations of the Human CD3-epsilon Gene Resulting in a T Cell Receptor/CD3 Complex Immunodeficiency. Nat Genet (1993) 3:77–81. doi: 10.1038/ng0193-77
54. Thoenes G, Soudais C, le Deist F, Griscelli C, Fischer A, Lisowska-Grospierre B, et al. Structural Analysis of Low TCR-CD3 Complex Expression in T Cells of an Immunodeficient Patient. J Biol Chem (1992) 267:487–93. doi: 10.1016/S0021-9258(18)48521-3
55. Le Deist F, Thoenes G, Corado J, Lisowska-Grospierre B, Fischer A. Immunodeficiency With Low Expression of the T Cell Receptor/CD3 Complex. Effect on T Lymphocyte Activation. Eur J Immunol (1991) 21:1641–7. doi: 10.1002/eji.1830210709
56. Dadi HK, Simon AJ, Roifman CM. Effect of CD3delta Deficiency on Maturation of Alpha/Beta and Gamma/Delta T-cell Lineages in Severe Combined Immunodeficiency. N Engl J Med (2003) 349:1821–8. doi: 10.1056/NEJMoa031178
57. Gil J, Busto EM, Garcillan B, Chean C, Garcia-Rodriguez MC, Diaz-Alderete A, et al. A Leaky Mutation in CD3D Differentially Affects Alphabeta and Gammadelta T Cells and Leads to a Talphabeta-Tgammadelta+B+NK+ Human SCID. J Clin Invest (2011) 121:3872–6. doi: 10.1172/JCI44254
58. Garcillan B, Mazariegos MS, Fisch P, Res PC, Munoz-Ruiz M, Gil J, et al. Enrichment of the Rare CD4(+) Gammadelta T-cell Subset in Patients With Atypical CD3delta Deficiency. J Allergy Clin Immunol (2014) 133:1205–8. doi: 10.1016/j.jaci.2013.10.002
59. Munoz-Ruiz M, Perez-Flores V, Garcillan B, Guardo AC, Mazariegos MS, Takada H, et al. Human CD3gamma, But Not CD3delta, Haploinsufficiency Differentially Impairs Gammadelta Versus Alphabeta Surface TCR Expression. BMC Immunol (2013) 14:3. doi: 10.1186/1471-2172-14-3
60. Hong KM, Kim HK, Park SY, Poojan S, Kim MK, Sung J, et al. CD3Z Hypermethylation is Associated With Severe Clinical Manifestations in Systemic Lupus Erythematosus and Reduces CD3zeta-chain Expression in T Cells. Rheumatol (Oxford) (2017) 56:467–76. doi: 10.1093/rheumatology/kew405
61. Gorman CL, Russell AI, Zhang Z, Cunninghame Graham D, Cope AP, Vyse TJ, et al. Polymorphisms in the CD3Z Gene Influence TCRzeta Expression in Systemic Lupus Erythematosus Patients and Healthy Controls. J Immunol (2008) 180:1060–70. doi: 10.4049/jimmunol.180.2.1060
62. Tsuzaka K, Fukuhara I, Setoyama Y, Yoshimoto K, Suzuki K, Abe T, et al. TCR Zeta mRNA With an Alternatively Spliced 3’-Untranslated Region Detected in Systemic Lupus Erythematosus Patients Leads to the Down-Regulation of TCR Zeta and TCR/CD3 Complex. J Immunol (2003) 171:2496–503. doi: 10.4049/jimmunol.171.5.2496
63. Takeuchi T, Tsuzaka K, Pang M, Amano K, Koide J, Abe T, et al. TCR Zeta Chain Lacking Exon 7 in Two Patients With Systemic Lupus Erythematosus. Int Immunol (1998) 10:911–21. doi: 10.1093/intimm/10.7.911
64. Tsuzaka K, Takeuchi T, Onoda N, Pang M, Abe T. Mutations in T Cell Receptor Zeta Chain mRNA of Peripheral T Cells From Systemic Lupus Erythematosus Patients. J Autoimmun (1998) 11:381–5. doi: 10.1006/jaut.1998.0223
65. Teruel M, McKinney C, Balsa A, Pascual-Salcedo D, Rodriguez-Rodriguez L, Ortiz AM, et al. Association of CD247 Polymorphisms With Rheumatoid Arthritis: A Replication Study and a Meta-Analysis. PloS One (2013) 8:e68295. doi: 10.1371/journal.pone.0068295
66. Li P, Wang X, Zhao MQ, Li LJ, Zhang C, Li BZ, et al. Tcr-CD3zeta Gene Polymorphisms and Expression Profile in Rheumatoid Arthritis. Autoimmunity (2016) 49:466–71. doi: 10.1080/08916934.2016.1174855
67. Katsuyama T, Tsokos GC, Moulton VR. Aberrant T Cell Signaling and Subsets in Systemic Lupus Erythematosus. Front Immunol (2018) 9:1088. doi: 10.3389/fimmu.2018.01088
68. Enyedy EJ, Nambiar MP, Liossis SN, Dennis G, Kammer GM, Tsokos GC, et al. Fc Epsilon Receptor Type I Gamma Chain Replaces the Deficient T Cell Receptor Zeta Chain in T Cells of Patients With Systemic Lupus Erythematosus. Arthritis Rheum (2001) 44:1114–21. doi: 10.1002/1529-0131(200105)44:5<1114::AID-ANR192>3.0.CO;2-B
69. Recio MJ, Nambiar MP, Liossis SN, Dennis G, Kammer GM, Tsokos GC, et al. Differential Biological Role of CD3 Chains Revealed by Human Immunodeficiencies. J Immunol (2007) 178:2556–64. doi: 10.4049/jimmunol.178.4.2556
70. Gokturk B, Keles S, Kirac M, Artac H, Tokgoz H, Guner SN, et al. CD3G Gene Defects in Familial Autoimmune Thyroiditis. Scand J Immunol (2014) 80:354–61. doi: 10.1111/sji.12200
71. Rowe JH, Delmonte OM, Keles S, Stadinski BD, Dobbs AK, Henderson LA, et al. Patients With CD3G Mutations Reveal a Role for Human CD3gamma in Treg Diversity and Suppressive Function. Blood (2018) 131:2335–44. doi: 10.1182/blood-2018-02-835561
72. Arnaiz-Villena A, Timon M, Rodriguez-Gallego C, Iglesias-Casarrubios P, Pacheco A, Regueiro JR, et al. T Lymphocyte Signalling Defects and Immunodeficiency Due to the Lack of CD3 Gamma. Immunodeficiency (1993) 4:121–9.
73. Holst J, Wang H, Eder KD, Workman CJ, Boyd KL, Baquet Z, et al. Scalable Signaling Mediated by T Cell Antigen Receptor-CD3 ITAMs Ensures Effective Negative Selection and Prevents Autoimmunity. Nat Immunol (2008) 9:658–66. doi: 10.1038/ni.1611
74. Jiang L, Xu J, Ni J, Gao X, Zhu Z, Dong D, et al. A Functional Insertion/Deletion Polymorphism in the Proximal Promoter of CD3G is Associated With Susceptibility for Hepatocellular Carcinoma in Chinese Population. DNA Cell Biol (2012) 31:1480–5. doi: 10.1089/dna.2012.1706
75. Lee SY, Choi JE, Jeon HS, Choi YY, Lee WK, Lee EB, et al. A Panel of Genetic Polymorphism for the Prediction of Prognosis in Patients With Early Stage Non-Small Cell Lung Cancer After Surgical Resection. PloS One (2015) 10:e0140216. doi: 10.1371/journal.pone.0140216
76. Jeon HS, Jin G, Kang HG, Choi YY, Lee WK, Choi JE, et al. A Functional Variant At 19q13.3, rs967591G>A, is Associated With Shorter Survival of Early-Stage Lung Cancer. Clin Cancer Res (2013) 19:4185–95. doi: 10.1158/1078-0432.CCR-12-2792
77. Rohaan MW, Wilgenhof S, Haanen J. Adoptive Cellular Therapies: The Current Landscape. Virchows Arch (2019) 474:449–61. doi: 10.1007/s00428-018-2484-0
78. Dopfer EP, Hartl FA, Oberg HH, Siegers GM, Yousefi OS, Kock S, et al. The CD3 Conformational Change in the Gammadelta T Cell Receptor is Not Triggered by Antigens But can be Enforced to Enhance Tumor Killing. Cell Rep (2014) 7:1704–15. doi: 10.1016/j.celrep.2014.04.049(2014
79. Juraske C, Wipa P, Morath A, Hidalgo JV, Hartl FA, Raute K, et al. Anti-CD3 Fab Fragments Enhance Tumor Killing by Human Gammadelta T Cells Independent of Nck Recruitment to the Gammadelta T Cell Antigen Receptor. Front Immunol (2018) 9:1579. doi: 10.3389/fimmu.2018.01579
80. Abate-Daga D, Davila ML. CAR Models: Next-Generation CAR Modifications for Enhanced T-cell Function. Mol Ther Oncolytics (2016) 3:16014. doi: 10.1038/mto.2016.14
81. Rafiq S, Hackett CS, Brentjens RJ. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat Rev Clin Oncol (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
82. Bridgeman JS, Ladell K, Sheard VE, Miners K, Hawkins RE, Price DA, et al. CD3zeta-Based Chimeric Antigen Receptors Mediate T Cell Activation Via Cis- and Trans-Signalling Mechanisms: Implications for Optimization of Receptor Structure for Adoptive Cell Therapy. Clin Exp Immunol (2014) 175:258–67. doi: 10.1111/cei.12216
83. Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR Activation Potential Directs Alternative T Cell Fates and Therapeutic Potency. Nat Med (2019) 25:82–8. doi: 10.1038/s41591-018-0290-5
84. Moulton VR, Tsokos GC. Abnormalities of T Cell Signaling in Systemic Lupus Erythematosus. Arthritis Res Ther (2011) 13:207. doi: 10.1186/ar3251
85. Haynes NM, Snook MB, Trapani JA, Cerruti L, Jane SM, Smyth MJ, et al. Redirecting Mouse CTL Against Colon Carcinoma: Superior Signaling Efficacy of Single-Chain Variable Domain Chimeras Containing TCR-zeta vs Fc Epsilon RI-Gamma. J Immunol (2001) 166:182–7. doi: 10.4049/jimmunol.166.1.182
86. Ren-Heidenreich L, Mordini R, Hayman GT, Siebenlist R, LeFever A. Comparison of the TCR Zeta-Chain With the FcR Gamma-Chain in Chimeric TCR Constructs for T Cell Activation and Apoptosis. Cancer Immunol Immunother (2002) 51:417–23. doi: 10.1007/s00262-002-0301-7
87. Ellerman D. Bispecific T-cell Engagers: Towards Understanding Variables Influencing the In Vitro Potency and Tumor Selectivity and Their Modulation to Enhance Their Efficacy and Safety. Methods (2019) 154:102–17. doi: 10.1016/j.ymeth.2018.10.026
88. Slaney CY, Wang P, Darcy PK, Kershaw MH. Cars Versus Bites: A Comparison Between T Cell-Redirection Strategies for Cancer Treatment. Cancer Discovery (2018) 8:924–34. doi: 10.1158/2159-8290.CD-18-0297
89. Decker CE, Young T, Pasnikowski E, Chiu J, Song H, Wei Y, et al. Genome-Scale CRISPR Activation Screen Uncovers Tumor-Intrinsic Modulators of CD3 Bispecific Antibody Efficacy. Sci Rep (2019) 9:20068. doi: 10.1038/s41598-019-56670-x
90. Martinelli G, Boissel N, Chevallier P, Ottmann O, Gokbuget N, Topp MS, et al. Complete Hematologic and Molecular Response in Adult Patients With Relapsed/Refractory Philadelphia Chromosome-Positive B-Precursor Acute Lymphoblastic Leukemia Following Treatment With Blinatumomab: Results From a Phase Ii, Single-Arm, Multicenter Study. J Clin Oncol (2017) 35:1795–802. doi: 10.1200/JCO.2016.69.3531
91. Goebeler ME, Knop S, Viardot A, Kufer P, Topp MS, Einsele H, et al. Bispecific T-Cell Engager (Bite) Antibody Construct Blinatumomab for the Treatment of Patients With Relapsed/Refractory non-Hodgkin Lymphoma: Final Results From a Phase I Study. J Clin Oncol (2016) 34. doi: 10.1200/JCO.2014.59.1586
92. Zhang CM, Yu LY, Lv JF, Gong L, Zhou HH, Chen XP, et al. Effects of Immuno-Related Gene Polymorphisms on a Bispecific Antibody Targeting Colorectal Cancer Cell. Per Med (2018) 15:167–79. doi: 10.2217/pme-2017-0071
93. Koretzky GA. Multiple Roles of CD4 and CD8 in T Cell Activation. J Immunol (2010) 185:2643–4. doi: 10.4049/jimmunol.1090076
94. Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct Observation of Ligand Recognition by T Cells. Nature (2002) 419:845–9. doi: 10.1038/nature01076
95. Holler PD, Kranz DM. Quantitative Analysis of the Contribution of TCR/pepMHC Affinity and CD8 to T Cell Activation. Immunity (2003) 18:255–64. doi: 10.1016/s1074-7613(03)00019-0
96. Wang JH, Meijers R, Xiong Y, Liu JH, Sakihama T, Zhang R, et al. Crystal Structure of the Human CD4 N-Terminal Two-Domain Fragment Complexed to a Class II MHC Molecule. Proc Natl Acad Sci USA (2001) 98:10799–804. doi: 10.1073/pnas.191124098
97. Cole DK, Laugel B, Clement M, Price DA, Wooldridge L, Sewell AK, et al. The Molecular Determinants of CD8 Co-Receptor Function. Immunology (2012) 137:139–48. doi: 10.1111/j.1365-2567.2012.03625.x
98. Daniels MA, Devine L, Miller JD, Moser JM, Lukacher AE, Altman JD, et al. CD8 Binding to MHC Class I Molecules is Influenced by T Cell Maturation and Glycosylation. Immunity (2001) 15:1051–61. doi: 10.1016/s1074-7613(01)00252-7
99. Cheroutre H, Lambolez F. Doubting the TCR Coreceptor Function of CD8alphaalpha. Immunity (2008) 28:149–59. doi: 10.1016/j.immuni.2008.01.005
100. Li Y, Yin Y, Mariuzza RA. Structural and Biophysical Insights Into the Role of CD4 and CD8 in T Cell Activation. Front Immunol (2013) 4:206. doi: 10.3389/fimmu.2013.00206
101. Wang XX, Li Y, Yin Y, Mo M, Wang Q, Gao W, et al. Affinity Maturation of Human CD4 by Yeast Surface Display and Crystal Structure of a CD4-HLA-DR1 Complex. Proc Natl Acad Sci USA (2011) 108:15960–5. doi: 10.1073/pnas.1109438108
102. Wang R, Natarajan K, Margulies DH. Structural Basis of the CD8 Alpha Beta/MHC Class I Interaction: Focused Recognition Orients CD8 Beta to a T Cell Proximal Position. J Immunol (2009) 183:2554–64. doi: 10.4049/jimmunol.0901276
103. Mancebo E, Moreno-Pelayo MA, Mencia A, de la Calle-Martin O, Allende LM, Sivadorai P, et al. Gly111Ser Mutation in CD8A Gene Causing CD8 Immunodeficiency is Found in Spanish Gypsies. Mol Immunol (2008) 45:479–84. doi: 10.1016/j.molimm.2007.05.022
104. Rahemtulla A, Fung-Leung WP, Schilham MW, Kundig TM, Sambhara SR, Narendran A, et al. Normal Development and Function of CD8+ Cells But Markedly Decreased Helper Cell Activity in Mice Lacking CD4. Nature (1991) 353:180–4. doi: 10.1038/353180a0
105. Fung-Leung WP, Schilham MW, Rahemtulla A, Kundig TM, Vollenweider M, Potter J, et al. CD8 is Needed for Development of Cytotoxic T Cells But Not Helper T Cells. Cell (1991) 65:443–9. doi: 10.1016/0092-8674(91)90462-8
106. Gooden MJ, de Bock GH, Leffers N, Daemen T, Nijman HW. The Prognostic Influence of Tumour-Infiltrating Lymphocytes in Cancer: A Systematic Review With Meta-Analysis. Br J Cancer (2011) 105:93–103. doi: 10.1038/bjc.2011.189
107. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The Immune Contexture in Human Tumours: Impact on Clinical Outcome. Nat Rev Cancer (2012) 12:298–306. doi: 10.1038/nrc3245
108. Berg RE, Forman J. The Role of CD8 T Cells in Innate Immunity and in Antigen non-Specific Protection. Curr Opin Immunol (2006) 18:338–43. doi: 10.1016/j.coi.2006.03.010
109. Barrett DM, Grupp SA, June CH. Chimeric Antigen Receptor- and TCR-Modified T Cells Enter Main Street and Wall Street. J Immunol (2015) 195:755–61. doi: 10.4049/jimmunol.1500751
110. Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, et al. Specific Lymphocyte Subsets Predict Response to Adoptive Cell Therapy Using Expanded Autologous Tumor-Infiltrating Lymphocytes in Metastatic Melanoma Patients. Clin Cancer Res (2012) 18:6758–70. doi: 10.1158/1078-0432.CCR-12-1177
111. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient With Epithelial Cancer. Science (2014) 344:641–5. doi: 10.1126/science.1251102
112. Sledzinska A, Vila de Mucha M, Bergerhoff K, Hotblack A, Demane DF, Ghorani E, et al. Regulatory T Cells Restrain Interleukin-2- and Blimp-1-Dependent Acquisition of Cytotoxic Function by CD4(+) T Cells. Immunity (2020) 52:151–166 e156. doi: 10.1016/j.immuni.2019.12.007
113. Salmikangas P, Kinsella N, Chamberlain P. Chimeric Antigen Receptor T-Cells (Car T-Cells) for Cancer Immunotherapy - Moving Target for Industry? Pharm Res (2018) 35:152. doi: 10.1007/s11095-018-2436-z
114. Pinz K, Liu H, Golightly M, Jares A, Lan F, Zieve GW, et al. Preclinical Targeting of Human T-cell Malignancies Using CD4-specific Chimeric Antigen Receptor (CAR)-Engineered T Cells. Leukemia (2016) 30:701–7. doi: 10.1038/leu.2015.311
115. Kim YH, Duvic M, Obitz E, Gniadecki R, Iversen L, Osterborg A, et al. Clinical Efficacy of Zanolimumab (HuMax-CD4): Two Phase 2 Studies in Refractory Cutaneous T-cell Lymphoma. Blood (2007) 109:4655–62. doi: 10.1182/blood-2006-12-062877
116. Knox S, Hoppe RT, Maloney D, Gibbs I, Fowler S, Marquez C, et al. Treatment of Cutaneous T-cell Lymphoma With Chimeric anti-CD4 Monoclonal Antibody. Blood (1996) 87:893–9. doi: 10.1182/blood.V87.3.893.bloodjournal873893
117. d’Amore F, Radford J, Relander T, Jerkeman M, Tilly H, Osterborg A, et al. Phase II Trial of Zanolimumab (HuMax-CD4) in Relapsed or Refractory non-Cutaneous Peripheral T Cell Lymphoma. Br J Haematol (2010) 150:565–73. doi: 10.1111/j.1365-2141.2010.08298.x
118. Hagberg H, Pettersson M, Bjerner T, Enblad G. Treatment of a Patient With a Nodal Peripheral T-cell Lymphoma (Angioimmunoblastic T-Cell Lymphoma) With a Human Monoclonal Antibody Against the CD4 Antigen (Humax-CD4). Med Oncol (2005) 22:191–4. doi: 10.1385/MO:22:2:191
119. Aoki H, Ueha S, Shichino S, Ogiwara H, Hashimoto SI, Kakimi K, et al. Tcr Repertoire Analysis Reveals Mobilization of Novel Cd8(+) T Cell Clones Into the Cancer-Immunity Cycle Following Anti-Cd4 Antibody Administration. Front Immunol (2018) 9:3185. doi: 10.3389/fimmu.2018.03185
120. Ueha S, Yokochi S, Ishiwata Y, Ogiwara H, Chand K, Nakajima T, et al. Robust Antitumor Effects of Combined Anti-Cd4-Depleting Antibody and Anti-PD-1/PD-L1 Immune Checkpoint Antibody Treatment in Mice. Cancer Immunol Res (2015) 3:631–40. doi: 10.1158/2326-6066.CIR-14-0190
121. Shitara K, Ueha S, Shichino S, Aoki H, Ogiwara H, Nakatsura T, et al. First-in-Human Phase 1 Study of IT1208, a Defucosylated Humanized anti-CD4 Depleting Antibody, in Patients With Advanced Solid Tumors. J Immunother Cancer (2019) 7:195. doi: 10.1186/s40425-019-0677-y
122. Kaczanowska S, Ueha S, Shichino S, Aoki H, Ogiwara H, Nakatsura T, et al. A Synthetic CD8alpha:Myd88 Coreceptor Enhances CD8(+) T-Cell Responses to Weakly Immunogenic and Lowly Expressed Tumor Antigens. Cancer Res (2017) 77:7049–58. doi: 10.1158/0008-5472.CAN-17-0653
123. Moody AM, Chui D, Reche PA, Priatel JJ, Marth JD, Reinherz EL, et al. Developmentally Regulated Glycosylation of the CD8alphabeta Coreceptor Stalk Modulates Ligand Binding. Cell (2001) 07:501–12. doi: 10.1016/s0092-8674(01)00577-3(2001
124. Palacios EH, Weiss A. Function of the Src-family Kinases, Lck and Fyn, in T-cell Development and Activation. Oncogene (2004) 23:7990–8000. doi: 10.1038/sj.onc.1208074
125. Casas J, Brzostek J, Zarnitsyna VI, Hong JS, Wei Q, Hoerter JA, et al. Ligand-Engaged TCR is Triggered by Lck Not Associated With CD8 Coreceptor. Nat Commun (2014) 5:5624. doi: 10.1038/ncomms6624
126. Gascoigne NR, Casas J, Brzostek J, Rybakin V. Initiation of TCR Phosphorylation and Signal Transduction. Front Immunol (2011) 2:72. doi: 10.3389/fimmu.2011.00072
127. Jiang N, Huang J, Edwards LJ, Liu B, Zhang Y, Beal CD, et al. Two-Stage Cooperative T Cell Receptor-Peptide Major Histocompatibility complex-CD8 Trimolecular Interactions Amplify Antigen Discrimination. Immunity (2011) 34:13–23. doi: 10.1016/j.immuni.2010.12.017
128. Wu L, Wei Q, Brzostek J, Gascoigne NRJ. Signaling From T Cell Receptors (Tcrs) and Chimeric Antigen Receptors (Cars) on T Cells. Cell Mol Immunol (2020) 17:600–12. doi: 10.1038/s41423-020-0470-3
129. Goldsmith MA, Weiss A. Isolation and Characterization of a T-lymphocyte Somatic Mutant With Altered Signal Transduction by the Antigen Receptor. Proc Natl Acad Sci USA (1987) 84:6879–83. doi: 10.1073/pnas.84.19.6879
130. Straus DB, Weiss A. Genetic Evidence for the Involvement of the Lck Tyrosine Kinase in Signal Transduction Through the T Cell Antigen Receptor. Cell (1992) 70:585–93. doi: 10.1016/0092-8674(92)90428-f
131. Chiang YJ, Hodes RJ. T-Cell Development is Regulated by the Coordinated Function of Proximal and Distal Lck Promoters Active At Different Developmental Stages. Eur J Immunol (2016) 46:2401–8. doi: 10.1002/eji.201646440
132. van Oers NS, Lowin-Kropf B, Finlay D, Connolly K, Weiss A. Alpha Beta T Cell Development is Abolished in Mice Lacking Both Lck and Fyn Protein Tyrosine Kinases. Immunity (1996) 5:429–36. doi: 10.1016/s1074-7613(00)80499-9
133. Acuto O, Di Bartolo V, Michel F. Tailoring T-cell Receptor Signals by Proximal Negative Feedback Mechanisms. Nat Rev Immunol (2008) 8:699–712. doi: 10.1038/nri2397
134. Moogk D, Zhong S, Yu Z, Liadi I, Rittase W, Fang V, et al. Constitutive Lck Activity Drives Sensitivity Differences Between CD8+ Memory T Cell Subsets. J Immunol (2016) 197:644–54. doi: 10.4049/jimmunol.1600178
135. Kemp KL, Levin SD, Bryce PJ, Stein PL. Lck Mediates Th2 Differentiation Through Effects on T-bet and GATA-3. J Immunol (2010) 184:4178–84. doi: 10.4049/jimmunol.0901282
136. Dobbins J, Gagnon E, Godec J, Pyrdol J, Vignali DA, Sharpe AH, et al. Binding of the Cytoplasmic Domain of CD28 to the Plasma Membrane Inhibits Lck Recruitment and Signaling. Sci Signal (2016) 9:ra75. doi: 10.1126/scisignal.aaf0626
137. Gruber C, Henkel M, Budach W, Belka C, Jendrossek V. Involvement of Tyrosine Kinase p56/Lck in Apoptosis Induction by Anticancer Drugs. Biochem Pharmacol (2004) 67:1859–72. doi: 10.1016/j.bcp.2004.01.026
138. Hauck F, Randriamampita C, Martin E, Gerart S, Lambert N, Lim A, et al. Primary T-cell Immunodeficiency With Immunodysregulation Caused by Autosomal Recessive LCK Deficiency. J Allergy Clin Immunol (2012) 130:1144–1152 e1111. doi: 10.1016/j.jaci.2012.07.029
139. Li SL, Duo LN, Wang HJ, Dai W, Zhou EH, Xu YN, et al. Identification of LCK Mutation in a Family With Atypical Epidermodysplasia Verruciformis With T-cell Defects and Virus-Induced Squamous Cell Carcinoma. Br J Dermatol (2016) 175:1204–9. doi: 10.1111/bjd.14679
140. Sawabe T, Horiuchi T, Nakamura M, Tsukamoto H, Nakahara K, Harashima SI, et al. Defect of Lck in a Patient With Common Variable Immunodeficiency. Int J Mol Med (2001) 7:609–14. doi: 10.3892/ijmm.7.6.609
141. Goldman FD, Ballas ZK, Schutte BC, Kemp J, Hollenback C, Noraz N, et al. Defective Expression of p56lck in an Infant With Severe Combined Immunodeficiency. J Clin Invest (1998) 102:421–9. doi: 10.1172/JCI3205
142. Adler HT, Sefton BM. Generation and Characterization of Transforming Variants of the Lck Tyrosine Protein Kinase. Oncogene (1992) 7:1191–9.
143. Luo K, Sefton BM. Activated Lck Tyrosine Protein Kinase Stimulates Antigen-Independent Interleukin-2 Production in T Cells. Mol Cell Biol (1992) 12:4724–32. doi: 10.1128/mcb.12.10.4724
144. Tycko B, Smith SD, Sklar J. Chromosomal Translocations Joining LCK and TCRB Loci in Human T Cell Leukemia. J Exp Med (1991) 174:867–73. doi: 10.1084/jem.174.4.867
145. Kofler DM, Chmielewski M, Rappl G, Hombach A, Riet T, Schmidt A, et al. CD28 Costimulation Impairs the Efficacy of a Redirected T-Cell Antitumor Attack in the Presence of Regulatory T Cells Which can be Overcome by Preventing Lck Activation. Mol Ther (2011) 19:760–7. doi: 10.1038/mt.2011.9
146. Suryadevara CM, Desai R, Farber SH, Choi BD, Swartz AM, Shen SH, et al. Preventing Lck Activation in CAR T Cells Confers Treg Resistance But Requires 4-1bb Signaling for Them to Persist and Treat Solid Tumors in Nonlymphodepleted Hosts. Clin Cancer Res (2019) 25:358–68. doi: 10.1158/1078-0432.CCR-18-1211
147. Gulati P, Ruhl J, Kannan A, Pircher M, Schuberth P, Nytko KJ, et al. Aberrant Lck Signal Via CD28 Costimulation Augments Antigen-Specific Functionality and Tumor Control by Redirected T Cells With PD-1 Blockade in Humanized Mice. Clin Cancer Res (2018) 24:3981–93. doi: 10.1158/1078-0432.CCR-17-1788
148. Golumba-Nagy V, Kuehle J, Hombach AA, Abken H. CD28-Zeta CAR T Cells Resist TGF-Beta Repression Through IL-2 Signaling, Which can Be Mimicked by an Engineered Il-7 Autocrine Loop. Mol Ther (2018) 26:2218–30. doi: 10.1016/j.ymthe.2018.07.005
149. Yu XZ, Levin SD, Madrenas J, Anasetti C. Lck is Required for Activation-Induced T Cell Death After TCR Ligation With Partial Agonists. J Immunol (2004) 172:1437–43. doi: 10.4049/jimmunol.172.3.1437
150. Arulraj T, Barik D. Mathematical Modeling Identifies Lck as a Potential Mediator for PD-1 Induced Inhibition of Early TCR Signaling. PloS One (2018) 13:e0206232. doi: 10.1371/journal.pone.0206232
151. Zhu Q, Wang J, Zhang L, Bian W, Lin M, Xu X, et al. Lck rs10914542-G Allele Associates With Type 1 Diabetes in Children Via T Cell Hyporesponsiveness. Pediatr Res (2019) 86:311–5. doi: 10.1038/s41390-019-0436-2
152. Wang Y, Chen S, Chen J, Xie X, Gao S, Zhang C, et al. Germline Genetic Patterns Underlying Familial Rheumatoid Arthritis, Systemic Lupus Erythematosus and Primary Sjogren’s Syndrome Highlight T Cell-Initiated Autoimmunity. Ann Rheum Dis (2020) 79:268–75. doi: 10.1136/annrheumdis-2019-215533
153. Raab M, Cai YC, Bunnell SC, Heyeck SD, Berg LJ, Rudd CE, et al. p56Lck and p59Fyn Regulate CD28 Binding to Phosphatidylinositol 3-Kinase, Growth Factor Receptor-Bound Protein GRB-2, and T Cell-Specific Protein-Tyrosine Kinase ITK: Implications for T-cell Costimulation. Proc Natl Acad Sci USA (1995) 92:8891–5. doi: 10.1073/pnas.92.19.8891
154. Susa M, Rohner D, Bichsel S. Differences in Binding of PI 3-Kinase to the Src-Homology Domains 2 and 3 of p56 Lck and p59 Fyn Tyrosine Kinases. Biochem Biophys Res Commun (1996) 220:729–34. doi: 10.1006/bbrc.1996.0472
155. da Silva AJ, Li Z, de Vera C, Canto E, Findell P, Rudd CE, et al. Cloning of a Novel T-cell Protein FYB That Binds FYN and SH2-domain-containing Leukocyte Protein 76 and Modulates Interleukin 2 Production. Proc Natl Acad Sci USA (1997) 94:7493–8. doi: 10.1073/pnas.94.14.7493
156. Filby A, Seddon B, Kleczkowska J, Salmond R, Tomlinson P, Smida M, et al. Fyn Regulates the Duration of TCR Engagement Needed for Commitment to Effector Function. J Immunol (2007) 179:4635–44. doi: 10.4049/jimmunol.179.7.4635
157. Martin-Cofreces NB, Martin-Cofreces NB, Sancho D, Fernandez E, Vicente-Manzanares M, Gordon-Alonso M, et al. Role of Fyn in the Rearrangement of Tubulin Cytoskeleton Induced Through TCR. J Immunol (2006) 176:4201–7. doi: 10.4049/jimmunol.176.7.4201
158. Gerbec ZJ, Thakar MS, Malarkannan S. The Fyn-ADAP Axis: Cytotoxicity Versus Cytokine Production in Killer Cells. Front Immunol (2015) 6:472. doi: 10.3389/fimmu.2015.00472
159. Thakar MS, Kearl TJ, Malarkannan S. Controlling Cytokine Release Syndrome to Harness the Full Potential of CAR-Based Cellular Therapy. Front Oncol (2019) 9:1529. doi: 10.3389/fonc.2019.01529
160. Schaeuble K, Hauser MA, Singer E, Groettrup M, Legler DF. Cross-Talk Between TCR and CCR7 Signaling Sets a Temporal Threshold for Enhanced T Lymphocyte Migration. J Immunol (2011) 187:5645–52. doi: 10.4049/jimmunol.1101850
161. Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, et al. Phase 1 Studies of Central Memory-Derived CD19 Car T-cell Therapy Following Autologous HSCT in Patients With B-cell Nhl. Blood (2016) 127:2980–90. doi: 10.1182/blood-2015-12-686725
162. Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T Memory Stem Cells in Health and Disease. Nat Med (2017) 23:18–27. doi: 10.1038/nm.4241
163. He Q, Jiang X, Zhou X, Weng J. Targeting Cancers Through TCR-peptide/MHC Interactions. J Hematol Oncol (2019) 12:139. doi: 10.1186/s13045-019-0812-8
164. Ueda A, Zhou L, Stein PL. Fyn Promotes Th17 Differentiation by Regulating the Kinetics of RORgammat and Foxp3 Expression. J Immunol (2012) 188:5247–56. doi: 10.4049/jimmunol.1102241
165. Bowers JS, Nelson MH, Majchrzak K, Bailey SR, Rohrer B, Kaiser AD, et al. Th17 Cells are Refractory to Senescence and Retain Robust Antitumor Activity After Long-Term Ex Vivo Expansion. JCI Insight (2017) 2:e90772. doi: 10.1172/jci.insight.90772
166. Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, et al. Tumor-Specific Th17-polarized Cells Eradicate Large Established Melanoma. Blood (2008) 112:362–73. doi: 10.1182/blood-2007-11-120998
167. Kuen DS, Kim BS, Chung Y. Il-17-Producing Cells in Tumor Immunity: Friends or Foes? Immune Netw (2020) 20:e6. doi: 10.4110/in.2020.20.e6
168. Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, et al. Phosphotyrosine-Dependent Coupling of Tim-3 to T-cell Receptor Signaling Pathways. Mol Cell Biol (2011) 31:3963–74. doi: 10.1128/MCB.05297-11
169. Gajewski TF, Fields P, Fitch FW. Induction of the Increased Fyn Kinase Activity in Anergic T Helper Type 1 Clones Requires Calcium and Protein Synthesis and is Sensitive to Cyclosporin a. Eur J Immunol (1995) 25:1836–42. doi: 10.1002/eji.1830250707
170. Boussiotis VA, Freeman GJ, Berezovskaya A, Barber DL, Nadler LM. Maintenance of Human T Cell Anergy: Blocking of IL-2 Gene Transcription by Activated Rap1. Science (1997) 278:124–8. doi: 10.1126/science.278.5335.124
171. Watson AR, Janik DK, Lee WT. Superantigen-Induced CD4 Memory T Cell Anergy. I. Staphylococcal Enterotoxin B Induces Fyn-mediated Negative Signaling. Cell Immunol (2012) 276:16–25. doi: 10.1016/j.cellimm.2012.02.003
172. Davidson D, Schraven B, Veillette A. PAG-Associated FynT Regulates Calcium Signaling and Promotes Anergy in T Lymphocytes. Mol Cell Biol (2007) 27:1960–73. doi: 10.1128/MCB.01983-06
173. Utting O, Priatel JJ, Teh SJ, Teh HS. p59fyn (Fyn) Promotes the Survival of Anergic CD4-CD8- Alpha Beta TCR+ Cells But Negatively Regulates Their Proliferative Response to Antigen Stimulation. J Immunol (2001) 166:1540–6. doi: 10.4049/jimmunol.166.3.1540
174. Mkaddem SB, Murua A, Flament H, Titeca-Beauport D, Bounaix C, Danelli L, et al. Lyn and Fyn Function as Molecular Switches That Control Immunoreceptors to Direct Homeostasis or Inflammation. Nat Commun (2017) 8:246. doi: 10.1038/s41467-017-00294-0
175. Bradshaw JM. The Src, Syk, and Tec Family Kinases: Distinct Types of Molecular Switches. Cell Signal (2010) 22:1175–84. doi: 10.1016/j.cellsig.2010.03.001
176. Yan Q, Barros T, Visperas PR, Deindl S, Kadlecek TA, Weiss A, et al. Structural Basis for Activation of ZAP-70 by Phosphorylation of the SH2-kinase Linker. Mol Cell Biol (2013) 33:2188–201. doi: 10.1128/MCB.01637-12
177. Au-Yeung BB, Deindl S, Hsu LY, Palacios EH, Levin SE, Kuriyan J, et al. The Structure, Regulation, and Function of ZAP-70. Immunol Rev (2009) 228:41–57. doi: 10.1111/j.1600-065X.2008.00753.x
178. Elder ME. Zap-70 and Defects of T-cell Receptor Signaling. Semin Hematol (1998) 35:310–20. doi: 10.1016/S0161-5890(98)90371-6
179. Arpaia E, Shahar M, Dadi H, Cohen A, Roifman CM. Defective T Cell Receptor Signaling and CD8+ Thymic Selection in Humans Lacking zap-70 Kinase. Cell (1994) 76:947–58. doi: 10.1016/0092-8674(94)90368-9
180. Chan AC, Kadlecek TA, Elder ME, Filipovich AH, Kuo WL, Iwashima M, et al. Zap-70 Deficiency in an Autosomal Recessive Form of Severe Combined Immunodeficiency. Science (1994) 264:1599–601. doi: 10.1126/science.8202713
181. Elder ME. Severe Combined Immunodeficiency Due to a Defect in the Tyrosine Kinase ZAP-70. Pediatr Res (1996) 39:743–8. doi: 10.1203/00006450-199605000-00001
182. Toyabe S, Watanabe A, Harada W, Karasawa T, Uchiyama M. Specific Immunoglobulin E Responses in ZAP-70-deficient Patients are Mediated by Syk-dependent T-Cell Receptor Signalling. Immunology (2001) 103:164–71. doi: 10.1046/j.1365-2567.2001.01246.x
183. Gelfand EW, Weinberg K, Mazer BD, Kadlecek TA, Weiss A. Absence of ZAP-70 Prevents Signaling Through the Antigen Receptor on Peripheral Blood T Cells But Not on Thymocytes. J Exp Med (1995) 182:1057–65. doi: 10.1084/jem.182.4.1057
184. Matsuda S, Suzuki-Fujimoto T, Minowa A, Ueno H, Katamura K, Koyasu S, et al. Temperature-Sensitive ZAP70 Mutants Degrading Through a Proteasome-Independent Pathway. Restoration of a Kinase Domain Mutant by Cdc37. J Biol Chem (1999) 274:34515–8. doi: 10.1074/jbc.274.49.34515
185. Noraz N, Schwarz K, Steinberg M, Dardalhon V, Rebouissou C, Hipskind R, et al. Alternative Antigen Receptor (TCR) Signaling in T Cells Derived From ZAP-70-deficient Patients Expressing High Levels of Syk. J Biol Chem (2000) 275:15832–8. doi: 10.1074/jbc.M908568199
186. Stawczyk-Macieja M, Rebala K, Szczerkowska-Dobosz A, Wysocka J, Cybulska L, Kapinska E, et al. Evaluation of Psoriasis Genetic Risk Based on Five Susceptibility Markers in a Population From Northern Poland. PloS One (2016) 11:e0163185. doi: 10.1371/journal.pone.0163185
187. Ferjeni Z, Bouzid D, Fourati H, Stayoussef M, Abida O, Kammoun T, et al. Association of TCR/CD3, Ptpn22, CD28 and ZAP70 Gene Polymorphisms With Type 1 Diabetes Risk in Tunisian Population: Family Based Association Study. Immunol Lett (2015) 163:1–7. doi: 10.1016/j.imlet.2014.11.005
188. Bouzid D, Fourati H, Amouri A, Marques I, Abida O, Haddouk S, et al. Association of ZAP70 and PTPN6, But Not BANK1 or CLEC2D, With Inflammatory Bowel Disease in the Tunisian Population. Genet Test Mol Biomarkers (2013) 17:321–6. doi: 10.1089/gtmb.2012.0372
189. Chen SY, Liu MF, Wang CR. Genetic Polymorphism of 3’ Untranslated Region of Zeta-Chain Associated Protein Kinase 70 kDa in Southern Taiwanese Patients With Rheumatoid Arthritis. Clin Rheumatol (2016) 35:747–50. doi: 10.1007/s10067-015-3044-5
190. Cerhan JR, Fredericksen ZS, Novak AJ, Ansell SM, Kay NE, Liebow M, et al. A Two-Stage Evaluation of Genetic Variation in Immune and Inflammation Genes With Risk of non-Hodgkin Lymphoma Identifies New Susceptibility Locus in 6p21.3 Region. Cancer Epidemiol Biomarkers Prev (2012) 21:1799–806. doi: 10.1158/1055-9965.EPI-12-0696
191. Chen L, Huynh L, Apgar J, Tang L, Rassenti L, Weiss A, et al. Zap-70 Enhances IgM Signaling Independent of its Kinase Activity in Chronic Lymphocytic Leukemia. Blood (2008) 111:2685–92. doi: 10.1182/blood-2006-12-062265
192. Rassenti LZ, Huynh L, Toy TL, Chen L, Keating MJ, Gribben JG, et al. Zap-70 Compared With Immunoglobulin Heavy-Chain Gene Mutation Status as a Predictor of Disease Progression in Chronic Lymphocytic Leukemia. N Engl J Med (2004) 351:893–901. doi: 10.1056/NEJMoa040857
193. Sakaguchi S, Sakaguchi N, Yoshitomi H, Hata H, Takahashi T, Nomura T, et al. Spontaneous Development of Autoimmune Arthritis Due to Genetic Anomaly of T Cell Signal Transduction: Part 1. Semin Immunol (2006) 18:199–206. doi: 10.1016/j.smim.2006.03.007
194. Siggs OM, Miosge LA, Yates AL, Kucharska EM, Sheahan D, Brdicka T, et al. Opposing Functions of the T Cell Receptor Kinase ZAP-70 in Immunity and Tolerance Differentially Titrate in Response to Nucleotide Substitutions. Immunity (2007) 27:912–26. doi: 10.1016/j.immuni.2007.11.013
195. Hsu LY, Tan YX, Xiao Z, Malissen M, Weiss A. A Hypomorphic Allele of ZAP-70 Reveals a Distinct Thymic Threshold for Autoimmune Disease Versus Autoimmune Reactivity. J Exp Med (2009) 206:2527–41. doi: 10.1084/jem.20082902
196. Wang H, Kadlecek TA, Au-Yeung BB, Goodfellow HE, Hsu LY, Freedman TS, et al. Zap-70: An Essential Kinase in T-cell Signaling. Cold Spring Harb Perspect Biol (2010) 2:a002279. doi: 10.1101/cshperspect.a002279
197. Levin SE, Zhang C, Kadlecek TA, Shokat KM, Weiss A. Inhibition of ZAP-70 Kinase Activity Via an Analog-Sensitive Allele Blocks T Cell Receptor and CD28 Superagonist Signaling. J Biol Chem (2008) 283:15419–30. doi: 10.1074/jbc.M709000200
198. Au-Yeung BB, Levin SE, Zhang C, Hsu LY, Cheng DA, Killeen N, et al. A Genetically Selective Inhibitor Demonstrates a Function for the Kinase Zap70 in Regulatory T Cells Independent of its Catalytic Activity. Nat Immunol (2010) 11:1085–92. doi: 10.1038/ni.1955
199. Saini M, Sinclair C, Marshall D, Tolaini M, Sakaguchi S, Seddon B, et al. Regulation of Zap70 Expression During Thymocyte Development Enables Temporal Separation of CD4 and CD8 Repertoire Selection At Different Signaling Thresholds. Sci Signal (2010) 3:ra23. doi: 10.1126/scisignal.2000702
200. Wong NML, Wong WW. Engineering a Dual Small Molecule Gated ZAP70 Switch in T Cells. ACS Synth Biol (2018) 7:969–77. doi: 10.1021/acssynbio.7b00394
201. Williams BL, Schreiber KL, Zhang W, Wange RL, Samelson LE, Leibson PJ, et al. Genetic Evidence for Differential Coupling of Syk Family Kinases to the T-cell Receptor: Reconstitution Studies in a ZAP-70-deficient Jurkat T-cell Line. Mol Cell Biol (1998) 18:1388–99. doi: 10.1128/mcb.18.3.1388
202. Nambiar MP, Fisher CU, Kumar A, Tsokos CG, Warke VG, Tsokos GC, et al. Forced Expression of the Fc Receptor Gamma-Chain Renders Human T Cells Hyperresponsive to TCR/CD3 Stimulation. J Immunol (2003) 170:2871–6. doi: 10.4049/jimmunol.170.6.2871
203. Dar WA, Knechtle SJ. CXCR3-Mediated T-cell Chemotaxis Involves ZAP-70 and is Regulated by Signalling Through the T-cell Receptor. Immunology (2007) 120:467–85. doi: 10.1111/j.1365-2567.2006.02534.x
204. Balagopalan L, Kortum RL, Coussens NP, Barr VA, Samelson LE. The Linker for Activation of T Cells (LAT) Signaling Hub: From Signaling Complexes to Microclusters. J Biol Chem (2015) 290:26422–9. doi: 10.1074/jbc.R115.665869
205. Chakraborty AK, Weiss A. Insights Into the Initiation of TCR Signaling. Nat Immunol (2014) 15:798–807. doi: 10.1038/ni.2940
206. Malissen B, Aguado E, Malissen M. Role of the LAT Adaptor in T-cell Development and Th2 Differentiation. Adv Immunol (2005) 87:1–25. doi: 10.1016/S0065-2776(05)87001-4
207. Zhang W, Sloan-Lancaster J, Kitchen J, Trible RP, Samelson LE. LAT: The ZAP-70 Tyrosine Kinase Substrate That Links T Cell Receptor to Cellular Activation. Cell (1998) 92:83–92. doi: 10.1016/s0092-8674(00)80901-0
208. Lin J, Weiss A. Identification of the Minimal Tyrosine Residues Required for Linker for Activation of T Cell Function. J Biol Chem (2001) 276:29588–95. doi: 10.1074/jbc.M102221200
209. Paz PE, Wang S, Clarke H, Lu X, Stokoe D, Abo A, et al. Mapping the Zap-70 Phosphorylation Sites on LAT (Linker for Activation of T Cells) Required for Recruitment and Activation of Signalling Proteins in T Cells. Biochem J (2001) 356:461–71. doi: 10.1042/0264-6021:3560461
210. Zhang W, Trible RP, Zhu M, Liu SK, McGlade CJ, Samelson LE, et al. Association of Grb2, Gads, and Phospholipase C-gamma 1 With Phosphorylated LAT Tyrosine Residues. Effect of LAT Tyrosine Mutations on T Cell Angigen Receptor-Mediated Signaling. J Biol Chem (2000) 275:23355–61. doi: 10.1074/jbc.M000404200
211. Martinez-Florensa M, Garcia-Blesa A, Yelamos J, Munoz-Suano A, Dominguez-Villar M, Valdor R, et al. Serine Residues in the LAT Adaptor are Essential for TCR-dependent Signal Transduction. J Leukoc Biol (2011) 89:63–73. doi: 10.1189/jlb.0509342
212. Roncagalli R, Hauri S, Fiore F, Liang Y, Chen Z, Sansoni A, et al. Quantitative Proteomics Analysis of Signalosome Dynamics in Primary T Cells Identifies the Surface Receptor CD6 as a Lat Adaptor-Independent TCR Signaling Hub. Nat Immunol (2014) 15:384–92. doi: 10.1038/ni.2843
213. Zhang W, Irvin BJ, Trible RP, Abraham RT, Samelson LE. Functional Analysis of LAT in TCR-mediated Signaling Pathways Using a LAT-deficient Jurkat Cell Line. Int Immunol (1999) 11:943–50. doi: 10.1093/intimm/11.6.943
214. Nunez-Cruz S, Aguado E, Richelme S, Chetaille B, Mura AM, Richelme M, et al. LAT Regulates Gammadelta T Cell Homeostasis and Differentiation. Nat Immunol (2003) 4:999–1008. doi: 10.1038/ni977
215. Sommers CL, Menon RK, Grinberg A, Zhang W, Samelson LE, Love PE. Knock-in Mutation of the Distal Four Tyrosines of Linker for Activation of T Cells Blocks Murine T Cell Development. J Exp Med (2001) 194:135–42. doi: 10.1084/jem.194.2.135
216. Shen S, Zhu M, Lau J, Chuck M, Zhang W. The Essential Role of LAT in Thymocyte Development During Transition From the Double-Positive to Single-Positive Stage. J Immunol (2009) 182:5596–604. doi: 10.4049/jimmunol.0803170
217. Brownlie R, Zamoyska R. LAT Polices T Cell Activation. Immunity (2003) 31:174–6. doi: 10.1016/j.immuni.2009.08.002(2009
218. Mingueneau M, Roncagalli R, Gregoire C, Kissenpfennig A, Miazek A, Archambaud C, et al. Loss of the LAT Adaptor Converts Antigen-Responsive T Cells Into Pathogenic Effectors That Function Independently of the T Cell Receptor. Immunity (2009) 31:197–208. doi: 10.1016/j.immuni.2009.05.013
219. Genton C, Wang Y, Izui S, Malissen B, Delsol G, Fournie GJ, et al. The Th2 Lymphoproliferation Developing in LatY136F Mutant Mice Triggers Polyclonal B Cell Activation and Systemic Autoimmunity. J Immunol (2006) 177:2285–93. doi: 10.4049/jimmunol.177.4.2285
220. Sommers CL, Lee J, Steiner KL, Gurson JM, Depersis CL, El-Khoury D, et al. Mutation of the Phospholipase C-gamma1-binding Site of LAT Affects Both Positive and Negative Thymocyte Selection. J Exp Med (2005) 201:1125–34. doi: 10.1084/jem.20041869
221. Sommers CL, Park CS, Lee J, Feng C, Fuller CL, Grinberg A, et al. A LAT Mutation That Inhibits T Cell Development Yet Induces Lymphoproliferation. Science (2002) 296:2040–3. doi: 10.1126/science.1069066
222. Yamasaki S, Nishida K, Sakuma M, Berry D, McGlade CJ, Hirano T, et al. Gads/Grb2-mediated Association With LAT is Critical for the Inhibitory Function of Gab2 in T Cells. Mol Cell Biol (2003) 23:2515–29. doi: 10.1128/mcb.23.7.2515-2529.2003
223. Kabouridis PS, Isenberg DA, Jury EC. A Negatively Charged Domain of LAT Mediates its Interaction With the Active Form of Lck. Mol Membr Biol (2011) 28:487–94. doi: 10.3109/09687688.2011.624990
224. Bacchelli C, Moretti FA, Carmo M, Adams S, Stanescu HC, Pearce K, et al. Mutations in Linker for Activation of T Cells (LAT) Lead to a Novel Form of Severe Combined Immunodeficiency. J Allergy Clin Immunol (2017) 139:634–642 e635. doi: 10.1016/j.jaci.2016.05.036
225. Sheng W, Liu C, Fu R, Wang H, Qu W, Ruan E, et al. Abnormalities of Quantities and Functions of Linker for Activations of T Cells in Severe Aplastic Anemia. Eur J Haematol (2014) 93:214–23. doi: 10.1111/ejh.12327
226. Keller B, Zaidman I, Yousefi OS, Hershkovitz D, Stein J, Unger S, et al. Early Onset Combined Immunodeficiency and Autoimmunity in Patients With Loss-of-Function Mutation in LAT. J Exp Med (2016) 213:1185–99. doi: 10.1084/jem.20151110
227. Roncagalli R, Mingueneau M, Gregoire C, Malissen M, Malissen B. LAT Signaling Pathology: An “Autoimmune” Condition Without T Cell Self-Reactivity. Trends Immunol (2010) 31:253–9. doi: 10.1016/j.it.2010.05.001
228. Balagopalan L, Ashwell BA, Bernot KM, Akpan IO, Quasba N, Barr VA, et al. Enhanced T-cell Signaling in Cells Bearing Linker for Activation of T-cell (LAT) Molecules Resistant to Ubiquitylation. Proc Natl Acad Sci USA (2011) 108:2885–90. doi: 10.1073/pnas.1007098108
229. Lo WL, Shah NH, Rubin SA, Zhang W, Horkova V, Fallahee IR, et al. Slow Phosphorylation of a Tyrosine Residue in LAT Optimizes T Cell Ligand Discrimination. Nat Immunol (2019) 20:1481–93. doi: 10.1038/s41590-019-0502-2
230. Malissen B, Gregoire C, Malissen M, Roncagalli R. Integrative Biology of T Cell Activation. Nat Immunol (2014) 15:790–7. doi: 10.1038/ni.2959(2014
231. Liang PI, Chang ST, Lin MY, Hsieh YC, Chu PY, Chen CJ, et al. Angioimmunoblastic T-cell Lymphoma in Taiwan Shows a Frequent Gain of ITK Gene. Int J Clin Exp Pathol (2014) 7:6097–107. doi: 10.1097/01.PAT.0000454436.01801.25
232. Linka RM, Risse SL, Bienemann K, Werner M, Linka Y, Krux F, et al. Loss-of-Function Mutations Within the IL-2 Inducible Kinase ITK in Patients With EBV-associated Lymphoproliferative Diseases. Leukemia (2012) 26:963–71. doi: 10.1038/leu.2011.371
233. Serwas NK, Cagdas D, Ban SA, Bienemann K, Salzer E, Tezcan I, et al. Identification of ITK Deficiency as a Novel Genetic Cause of Idiopathic CD4+ T-Cell Lymphopenia. Blood (2014) 124:655–7. doi: 10.1182/blood-2014-03-564930
234. Lee SH, Chang HS, Jang AS, Park SW, Park JS, Uh ST, et al. The Association of a Single-Nucleotide Polymorphism of the IL-2 Inducible T-cell Kinase Gene With Asthma. Ann Hum Genet (2011) 75:359–69. doi: 10.1111/j.1469-1809.2010.00637.x
235. Addobbati C, Brandao LA, Guimaraes RL, Pancotto JA, Donadi EA, Crovella S, et al. FYB Gene Polymorphisms are Associated With Susceptibility for Systemic Lupus Erythemathosus (SLE). Hum Immunol (2013) 74:1009–14. doi: 10.1016/j.humimm.2013.04.026
236. Peng X, Cui Z, Gu W, Xu W, Guo X. Low Level of LAT-PLC-gamma1 Interaction is Associated With Th2 Polarized Differentiation: A Contributing Factor to the Etiology of Asthma. Cell Immunol (2014) 290:131–7. doi: 10.1016/j.cellimm.2014.05.012
237. Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, et al. Integrated Molecular Analysis of Adult T Cell Leukemia/Lymphoma. Nat Genet (2015) 47:1304–15. doi: 10.1038/ng.3415
238. Coppola J, Bryant S, Koda T, Conway D, Barbacid M. Mechanism of Activation of the Vav Protooncogene. Cell Growth Differ (1991) 2:95–105.
239. Deckert M, Tartare-Deckert S, Couture C, Mustelin T, Altman A. Functional and Physical Interactions of Syk Family Kinases With the Vav Proto-Oncogene Product. Immunity (1996) 5:591–604. doi: 10.1016/s1074-7613(00)80273-3
240. Knyazhitsky M, Moas E, Shaginov E, Luria A, Braiman A. Vav1 Oncogenic Mutation Inhibits T Cell Receptor-Induced Calcium Mobilization Through Inhibition of Phospholipase Cgamma1 Activation. J Biol Chem (2012) 287:19725–35. doi: 10.1074/jbc.M111.309799
241. Shalom B, Farago M, Pikarsky E, Katzav S. Vav1 Mutations Identified in Human Cancers Give Rise to Different Oncogenic Phenotypes. Oncogenesis (2018) 7:80. doi: 10.1038/s41389-018-0091-1
242. Athari SS. Targeting Cell Signaling in Allergic Asthma. Signal Transduct Target Ther (2019) 4:45. doi: 10.1038/s41392-019-0079-0
243. Rudolph J, Meinke C, Voss M, Guttek K, Kliche S, Reinhold D, et al. Immune Cell-Type Specific Ablation of Adapter Protein ADAP Differentially Modulates Eae. Front Immunol (2019) 10:2343. doi: 10.3389/fimmu.2019.02343
244. Li C, Li W, Xiao J, Jiao S, Teng F, Xue S, et al. ADAP and SKAP55 Deficiency Suppresses PD-1 Expression in CD8+ Cytotoxic T Lymphocytes for Enhanced Anti-Tumor Immunotherapy. EMBO Mol Med (2015) 7:754–69. doi: 10.15252/emmm.201404578
Keywords: T cell (antigen) receptor, cellular immunotherapy, cytokines, T cell signaling, cellular engineering
Citation: Kent A, Longino NV, Christians A and Davila E (2021) Naturally Occurring Genetic Alterations in Proximal TCR Signaling and Implications for Cancer Immunotherapy. Front. Immunol. 12:658611. doi: 10.3389/fimmu.2021.658611
Received: 26 January 2021; Accepted: 06 April 2021;
Published: 03 May 2021.
Edited by:
Axel Schambach, Hannover Medical School, GermanyReviewed by:
Luis De La Cruz-Merino, Virgen Macarena University Hospital, SpainCopyright © 2021 Kent, Longino, Christians and Davila. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eduardo Davila, ZWR1YXJkby5kYXZpbGFAY3VhbnNjaHV0ei5lZHU=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.