 Dipranjan Laha
Dipranjan Laha Robert Grant
Robert Grant Prachi Mishra
Prachi Mishra Naris Nilubol
Naris Nilubol- Surgical Oncology Program, Center for Cancer Research, National Cancer Institute, NIH, Bethesda, MD, United States
The tumor microenvironment (TME) is an intricate system within solid neoplasms. In this review, we aim to provide an updated insight into the TME with a focus on the effects of tumor necrosis factor-α (TNF-α) on its various components and the use of TNF-α to improve the efficiency of drug delivery. The TME comprises the supporting structure of the tumor, such as its extracellular matrix and vasculature. In addition to cancer cells and cancer stem cells, the TME contains various other cell types, including pericytes, tumor-associated fibroblasts, smooth muscle cells, and immune cells. These cells produce signaling molecules such as growth factors, cytokines, hormones, and extracellular matrix proteins. This review summarizes the intricate balance between pro-oncogenic and tumor-suppressive functions that various non-tumor cells within the TME exert. We focused on the interaction between tumor cells and immune cells in the TME that plays an essential role in regulating the immune response, tumorigenesis, invasion, and metastasis. The multifunctional cytokine, TNF-α, plays essential roles in diverse cellular events within the TME. The uses of TNF-α in cancer treatment and to facilitate cancer drug delivery are discussed. The effects of TNF-α on tumor neovasculature and tumor interstitial fluid pressure that improve treatment efficacy are summarized.
Introduction
The tumor microenvironment (TME) is a complex biological ecosystem of solid tumors encompassing all the cells and structures found in healthy organ tissue (1). These include, but are not limited to, blood vessels, immune infiltrates, fibroblasts, and the extracellular matrix. Tumor cells, immune cells, fibroblasts, myofibroblasts, and microvascular structures such as vascular endothelial cells and pericytes found within the TME play various critical roles in regulating tumor initiation and progression (1–3). These cell types can control tumor growth through their normal regulatory functions. However, the dysregulation of these cells can promote tumor growth and metastasis. Recent studies demonstrate that the relationships between cancer cells and their surrounding microenvironments affect cancer cell survival, growth, proliferation, epithelial-mesenchymal transition (EMT), and metastasis (4). Thus, the modulation of the TME as a target for clinical applications is an area of interest in cancer treatments. Communication between neoplastic cells and the TME is conducted mainly through standard mechanisms observed in communication between healthy cells in normal organ tissue: through intercellular junctions and receptor-signal pathways encased in a three-dimensional extracellular matrix (ECM). Glycoproteins, proteoglycans, cytokines, and growth factors provide structural support and information exchange (5). In both normal tissue and solid cancers, TNF-α has diverse regulatory roles in the TME depending on the type of cells.
TNF-α is, to a large degree, produced by macrophages but also by other immune cell types, including lymphoid cells, mast cells, and by non-immune cells such as endothelial cells, fibroblasts, and smooth muscle cells (6–9). The members of the TNF-α family exert their effects through two distinct receptors, TNFRSF1A (TNFR1) and TNFRSF1B (TNFR2). TNFR1 is ubiquitously expressed and found in all cell types. Among many diverse effects induced by TNF-α, the major role of TNFR1 is the initiation of apoptosis through its death domain, which is not found on TNFR2 (9). Seemingly contradictory to its apoptotic signaling function, activation of TNFR1 also can induce cell survival mechanisms (9). The determination of the final downstream activity of this receptor is based on the concentration of TNF-α in the microenvironment as well as the effects of other involved cytokines. TNFR2 is mostly found on immune cells, where its pathway activation by TNF-α assists in regulating the immune response and inflammation. TNFR2 activation on immune cells within the TME and cancer cells themselves can promote tumor growth and progression (10). Increased TNFR2 expression found on regulatory T cells within the TME can suppress the immune response and prevent activation of cytotoxic T cells, which decreases the ability of the immune response to suppress the tumor (10, 11). Myeloid-derived suppressor (MDSC) are a group of immature cells that can differentiate into several different immune cell types, but in their immature state, are potently immunosuppressive (10). TNF-α can suppress the MDSC differentiation and induce accumulation of MDSC, which enhances their immunosuppressive effects in the TME through TNFR2 signaling (10, 12, 13).
Through its roles in apoptosis, angiogenesis, and immune cell recruitment and regulation, as well as its function in assisting with the construction of the ECM, this review summarizes many roles of TNF-α and its relation to the various components of the TME. Many cell-signaling mechanisms are involved in these functions, and we attempt to explain the roles of these pathways in relation to this versatile cytokine. Through the understanding of these pathways, scientists and clinicians are finding ways to exploit them as therapeutic targets. For example, inhibition of endogenous TNF-α is a standard of care for chronic inflammatory diseases such as ulcerative colitis, Crohn’s disease, rheumatoid arthritis, and several other diseases. In addition, TNF-α was previously used with good efficacy in patients with limb soft tissue sarcomas (STS) and in-transit melanoma by targeting the hap-hazard neovascular growth within the TME of these lesions (14–16). In this review, it is evident that there are many potential applications to manipulate TNF-α pathways, specifically in its role in the TME for cancer therapy.
TNF-α Pathway at Glance
Research dating back more than twenty years has shown that the TNF-α superfamily comprises at least 19 members that signal through 29 receptors (17). It is a pleiotropic cytokine, binds two receptors – TNFR1(receptor type 1: CD120a; p55/60) and TNRFR2 (TNF receptor type 2; CD120b; p75/80) – and is produced by many different types of cells (17). Unlike the TNFR2 expression which is limited to immune cells and a few other cell types, TNFR1 expression is present in various cell types (18–20). TNF-α binds to these receptors resulting in several diverse effects, cell proliferation, survival, and apoptosis (21–23). Abnormal production of TNF-α and TNF receptor signaling has been associated with the pathogenesis of several inflammatory diseases including rheumatoid arthritis, Crohn’s disease, atherosclerosis, psoriasis, and cancer (24). TNF-α has both tumor-promoting and tumor-suppressing roles in the TME. It is well reported that the tumor parenchyma and the TME continuously produce endogenous TNF-α, which induces tumor angiogenesis and promotes progression. The innate immune cells of the TME secrete various cytokines such as TNF-α and interleukin-6 (IL-6), which can promote cancer cell survival (25) and induce the expression of vascular endothelial cell adhesion molecules (CAM) that facilitate extravasation of leukocytes (26). TNF-α mediated matrix metalloproteinase (MMP) production in tumor cells or the TME also promotes tumor expansion (27, 28).
TNF-α plays an important role in tumor metastasis. It increases the expression of angiogenic factors such as basic fibroblast growth factor (bFGF), interleukin-8 (IL-8), and vascular endothelial growth factor (VEGF) in endothelial cells of the TME. TNF-α induced the expression of adhesion molecules such as intracellular adhesion molecule (ICAM)-1, E-selectin, and VCAM-1 in liver sinusoidal endothelial cells and induced tumor metastasis (29). So far, several FDA-approved TNF-α inhibitors, such as infliximab, etanercept, and adalimumab have been used to treat various human illnesses (30). We have summarized the multiple roles of TNF-α in different solid cancers based on preclinical studies (Table 1).
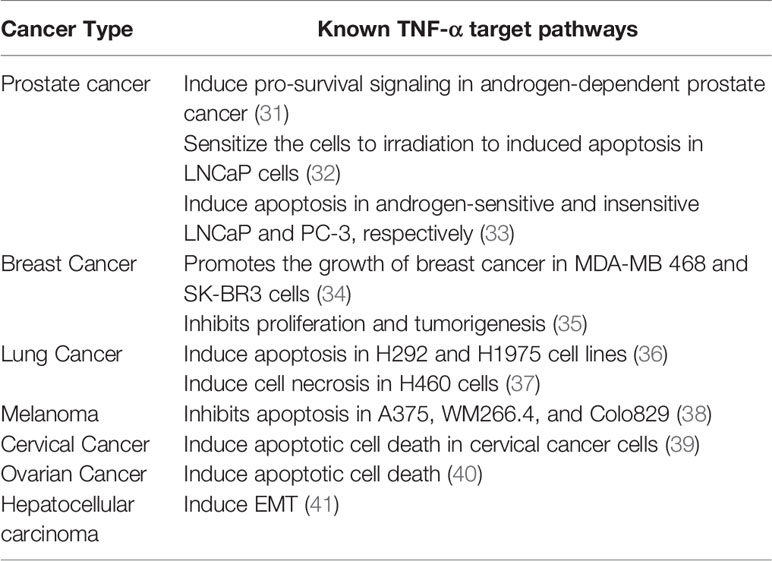
Table 1 The roles of TNF-α in different cancer types.
TNF-α Mediated Signaling Pathways in the TME
Two different forms of TNF-α have been identified: (i) soluble TNF-α (sTNF-α) and (ii) transmembrane TNF-α (mTNF-α) (42). The mTNF-α is the precursor of sTNF-α. TNF-α converting enzyme (TACE) can cleave mTNF-α, releasing sTNF-α. Previous findings reported dual roles of TNF-α based on the exposure time and cytokine levels reached within the TME (33). Soluble TNF-α mostly binds with TNFR1 and controls the inflammatory immune response, whereas mTNF-α mostly binds with TNFR2 and controls cell proliferation, survival, and other biological activities. The interaction between mTNF-α and TNFR2 is dependent on responses to different signaling pathways. In colorectal cancer, TNFR2 modulates the expression of Ki67, influences fibroblast associated protein and α-smooth muscle actin, and increases cellular proliferation and migration (43). Anti-TNFR2 antibodies suppress tumor-associated regulatory T cells (Tregs) and inhibit ovarian cancer cell proliferation (44). Ligand binding to TNFR2 leads to the activation of NF-κB and several kinase pathways, including JNK, p38, MAPK, ERK, and PI3K (45). Apart from NF-κB and kinase pathways, other processes and signaling pathways, such as EMT and TGF-β receptor-mediated signaling, are also critically regulated through TNF-α signaling (described in detail below). We have summarized different oncogenic signaling pathways such as β-catenin, STAT3, PI3K/PTEN/AKT/mTOR, p53, which are directly or indirectly regulated by TNF-α in Table 2.
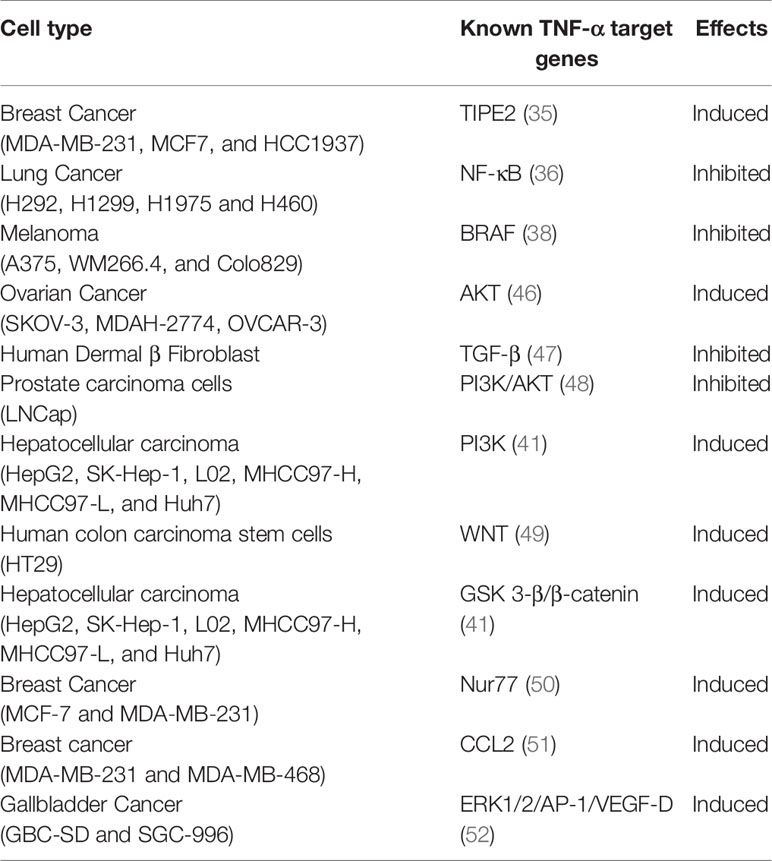
Table 2 The roles of TNF-α in different transcription factors, cytokines, and signaling pathways by cancer type.
Role of TNF-α in TGF-β Receptor Mediated Signaling Pathways
The multifunctional cytokine transforming growth factor-beta (TGF-β) regulates cell growth, extracellular matrix protein synthesis, and immune cell functions (53). In normal and premalignant cells, TGF-β acts as a tumor suppressor through the induction of apoptosis. However, when cancer cells have mutations or lose tumor suppressor genes, cells become resistant TGF-β mediated growth arrest. The crucial role of the TGF-β signaling pathway in the TME has been demonstrated in several studies. Several molecules regulate the TGF-β pathway, among them, TNF-α is of significant importance. However, it is not clear whether TNF-α directly or indirectly interacts with the TGF-β pathway. Understanding the molecular mechanisms of the antagonistic activities of TNF-α against TGF-β is critical. Studies have demonstrated that TNF-α inhibits TGF-β and ECM production, such as type I collagen and elastin in cancer cells and fibroblasts (47, 54, 55). TGF-β inhibits cancer growth through the activation of tumor angiogenesis and regulates prominent compounds involved with cancer-associated fibroblasts (CAF) in TME (56). Fibroblasts have been shown to facilitate cancer progression by supporting tumor growth, extracellular matrix remodeling, angiogenesis and by mediating tumor-promoted inflammation (57). Recent research has clarified the relationship between TGF-β regulation mediated by TNF-α and CAF. In brief, TNF-α triggers the downregulation of TGF-β receptor II leading to desensitization of human dermal fibroblasts toward TGF-β. Additionally, TNF-α impaired the response of the cells to TGF-β by regulating the turnover of TRII (47). Normal fibroblasts acquire characteristics of CAFs when stimulated with TNF-α. The most widely used marker for CAFs is α-smooth muscle actin (α-SMA). TGF-β induces α-smooth muscle actin expression in fibroblasts (58).
Role of TNF-α in NF-κB Pathways
The TME contains various types of cells, including tumor-associated macrophage (TAMs), dendritic cells, myeloid-derived suppressor cells, neutrophils, mast cells, natural killer T (NKT) cells, cancer-associated fibroblast (CAFs) and endothelial cells (59). The nuclear factor kappa-light chain enhancer of activated B cells (NF-κB) functions in these cell types and modulates inflammation, tumorigenesis, and metastasis. NF-κB activates several inflammatory genes in response to cytokines like TNF-α and IL-1, as well as to bacterial endotoxin and physical stress. Whiteside reported NF-κB activation in myeloid cells enhances inflammation in the TME (60). When cells are stimulated by an extracellular signal such as TNF-α, NF-κB is activated and enters the nucleus to bind to target genes and promotes cell death or increases cell survival in a context-dependent manner (61). In lung carcinoma cells, deoxynivalenol induces dependent proteolytic cleavage of TNFR1 through the activation of ERK and p38 MAPK, and subsequently inhibits the TNF-α-induced NF-κB signaling pathway (62). Tang et al. reported that in oral squamous cell carcinoma, TNF-α activates the NF-κB pathway, which promotes invasion and metastasis (63). In addition, NF-κB is also an important player in modulating the tumor-associated macrophages (TAM). The NF- κB pathway regulates anti and pro-inflammatory signaling pathways in the TME through tumor-associated macrophage (TAM) regulation.
Role of TNF-α in the Receptor Tyrosine Kinase Pathway
Receptor tyrosine kinases (RTK) act as a mediator between the extracellular and intracellular compartments by transferring signals from the TME into the tumor cells (64). So far, 58 different RTKs have been discovered in humans and classified into 20 different subfamilies based on their structural features, and activation of these enzymes is well defined to regulates various cellular processes (64). Mutations in RTKs and their associated downstream signaling pathways have oncogenic roles in many solid cancers, hence the development of targeted therapy specifically for these RTKs (64). However, many cancer types often acquire treatment resistance to various RTK inhibitors such as VEGFR inhibitors (bevacizumab), EGFR inhibitors (gefitinib), and FGFR inhibitors (AZD4547) (65). The mechanism associated with RTK inhibitors to disrupt neoplastic cellular growth are (i) immunogenic modulation of the TME and (ii) immune subset conditioning (66). RTK inhibitors induce immunogenic modulation via tumor cell sensitivity to immune cells-mediated lysis through an alternation in tumor cell phenotype and cause immune subset conditioning by activating immune cells and suppressing the immune suppressor cells in the TME (66).
TNF-α regulates multiple RTK pathways, including mitogen-activated protein kinases (MAPK) (i) extracellular-signal-regulated kinases (ERKs); (ii) cJun NH2-terminal kinases (JNK); and (iii) p38 MAP kinases pathways. Downregulation of MAP2K isoforms MKK4 and MKK7 in mice model prevents TNF-α mediated JNK activation (67, 68).
Apart from MAPK pathways, TNF-α signaling also controls vascular endothelial growth factor receptor (VEGFR), an RTK, which influences angiogenesis in the TME (69). This happens through TNFR1 signaling inflammatory macrophages to upregulate expression of vascular endothelial growth factor receptor 3, which causes increased production of vascular endothelial growth factor-C, and in turn, induces angiogenesis (69). The result was validated in vivo in mice with loss of function of TNFR1 (Tnfr1(-/-)) which reduced lymphangiogenesis and lymphatic metastasis (69). Tumor-mediated TNF-α and VEGF production is also associated with integrin receptor alpha V and beta 3 and beta 5 (αvβ3/αvβ5) mediated neovascularization, which shows an active interaction between tumor cells and endothelial cells through TNF-α (70). Another study demonstrated that a weakness of Akt/NF-κB signaling from TNF-α-mediated cross-talk signaling via EGFR causes the collateral sensitivity to TNF-α in a gefitinib resistant cell line (71). For instance, the over-expressions of EGFR and platelet-derived growth factor receptor α/β have been explored in tumor growth and progression (72). Sasi et al. confirmed that blocking of TNFR2/p75 with short-hairpin RNA in a Lewis Lung Carcinoma cell line induced apoptosis and decreased expression of the angiogenic growth factors VEGF, HGF, and PLGF (73). The VEGF inhibitor bevacizumab regulates a process called vessel normalization during angiogenesis through the upregulation tumor-infiltrating lymphocytes such as CD4+ and CD8+T cells in the TME (73). Below we more thoroughly discuss how TNF-α regulates T cells in the TME.
TNF-α and Epithelial-Mesenchymal Transition Regulatory Molecules
The epithelial to mesenchymal transition (EMT) is a process whereby epithelial cells lose epithelial features and acquire properties of mesenchymal cells. The EMT is classified into three main types depending upon the biological context. Type I EMT is observed during embryonic development, Type II occurs during wound healing and tissue regeneration, and Type III occurs during cancer progression. Previous findings demonstrated that TNF-α had been implicated as a major factor in EMT through cancer initiation and progression in the TME (6, 41, 74). Wang et al. showed that TNF-α induces EMT in human HCT116 cells and promotes colorectal cancer invasion and metastasis (75). The zinc finger protein SNAI1 plays a crucial role in TNF-α induced EMT. TNF-α treatment increased the expression of SNAI1 but not SLUG, ZEB1or Twist. Overexpression of SNAI1induced a switch from E-cadherin to N-cadherin expression in HCT116 cells, which is a characteristic of EMT. Recent findings from Li et al. showed that TNF-α mediated NF-κB activation upregulates EMT regulatory gene TWIST1 expression in breast cancer cells (76). Mikesh et al. discovered the expression of molecular markers of mesenchymal phenotype in melanoma. Melanoma cells were treated with TNF-α in a 3-dimensional culture system, and the changes in the expression of E-cadherin, N-cadherin, vimentin, and fibronectin were assessed. Melanoma cells treated with TNF-α reduced the epithelial marker E-cadherin and induced mesenchymal markers N-cadherin, vimentin, fibronectin (77). The role of TNF-α in regulating the EMT in hepatocellular carcinoma cells (SMMC-7721) was studied by Chen et al. TNF-α is elevated in the supernatants of M2-tumor-associated macrophages (M2-TAMs), promoting the EMT of SMMC-7721 cells in vitro (78).
Extracellular Matrix and Tumor Microenvironment
The TME comprises various cell types embedded in an altered extracellular matrix (ECM). The ECM is a major structural component of the TME and is mainly produced by cancer-associated fibroblasts (CAF) (79). It is largely composed of fibrous proteins, glycoproteins, proteoglycans and polysaccharides (79). The ECM in solid tumors differs significantly from normal organs. During cancer progression, cancer cells cross the ECM and basement membrane. MMPs are a large family of calcium-dependent, zinc-containing endopeptidases which are proteolytic enzymes capable of degrading the macromolecules of the ECM (80). Cancer cells secrete many members of the MMP family that facilitate the cellular migration into the ECM and thereby causing local invasion and can lead to metastasis (80). This process is largely regulated by TNF-α. Dilshara et al. reported that mangiferin inhibits TNF-α induced MMP9 expression and cellular invasion by suppressing the NF-κB activity in human LNCaP prostate carcinoma cells (81). An isoquinoline derivative compound, berberine inhibits TNF-α induced MMP9 and cell invasion through the inhibition of AP-1 activity in MDA-MB-231human breast cancer cells (82).
Several studies demonstrated that the biologic phenotype of cancers not only depends upon the activities of cancer cells but also tumor-infiltrating immune cells in the TME. TNF-α is produced by macrophages and other immune cells, including dendritic cells, B cells, activated natural killer cells, and activated T cells (83, 84). We summarized the effects of TNF- α on various non-cancerous cells in the TME below (Table 3).

Table 3 The effect of TNF-α on various cell types in the TME.
Macrophages
The main function of a macrophage is to engulf and digest foreign substances, cellular debris, and other components of the TME. Once macrophages are recruited into the TME, they are polarized into M1 or M2 TAMs depending on the varying concentrations of cytokines in the TME. A high density of TAMs can be found in several cancers such as pancreatic (85), breast (86), ovarian (87), and esophageal (88) and is associated with adverse prognostic features (86, 89, 90). However, the story of TAMs is more sophisticated than simply the number of cells in the TME. There are two types of mature macrophages: (i) classically activated macrophages (M1) and (ii) alternatively activated macrophages (M2). M1 and M2 macrophages play an important role in immune regulation in the TME. TAMs are a significant producer of TNF-α within the TME and interestingly are also highly responsive to TNF-α. The M1 macrophage can be stimulated to secrete a high level of TNF-α, resulting in high concentrations of superoxide, free oxygen, and nitrogen radicals (91) which promotes cell death in TME. M2 TNF-α secretion has been shown to promote EMT and induce “stemness” in an in vitro hepatocellular carcinoma model (78). Porta et al. reported that the p50 subunit of NF-κB plays an important role in macrophage polarization both in vitro and in vivo. They concluded that the p50 homodimer inhibited the NF-κB signaling pathway and induced macrophages to display an M2 phenotype with reduced expression of TNF-α and inducible nitric oxide synthases (iNOS) (92). M2 macrophages produce anti-inflammatory cytokines such as IL-10, IL-13, and TGF-β that may facilitate tumor development in TME. Experimental therapies to date have focused on depletion of M2 macrophages in the TME, specifically in glioblastoma through the inhibition of the colony-stimulating factor-1 receptor, which skewed macrophage polarization in the TME away from M2 and toward M1 in vivo (93). Data from another group using multiple different tumor models in vitro and in vivo showed higher TNFR activation shifts the balance toward the M1 phenotype and partially inhibited gene expression, specifically characteristic of M2 TAMs (94).
Neutrophils
Like macrophages, two types of neutrophil populations have been identified within the TME. First described by Fridlender et al. in mesothelioma in vitro and xenograft model, the tumor-associated neutrophils (TAN) are polarized into a subpopulation of anti-neoplastic (N1) or, through induction by TGF-β within the TME, pro-neoplastic (N2) neutrophils (95). While TNF-α has not been directly implicated in this polarization, its role in TGF-β pathway and the other pathways in which it modulates neutrophil activity described below highlights the importance of neutrophil-neoplasm interaction. In normal vasculature in vivo, TNF-α increases neutrophil recruitment and endothelial adhesion via cytoskeletal remodeling (96, 97). TNF-α also has a “priming” effect on neutrophils, causing them to be more responsive to stimuli (98). This priming causes a respiratory burst and the generation of intracellular reactive oxygen species (ROS) and reactive nitrogen species (RNS) for neutrophils to interact with pathogens and to modulate local inflammation. In in vitro model of pancreatic ductal adenocarcinoma, neutrophils promoted EMT and metastasis in co-culture (99). In these experiments, cancer cells caused neutrophils to secrete large amounts of TNF-α and TGF-β in a co-culture model, indicating that the cytokines were responsible for regulating the EMT and the metastasis (99). A recent study demonstrated higher levels of circulating TNF-α in patients with breast cancer. The neutrophils from these patients exhibit more cytotoxicity against breast cancer cell lines ex-vivo than that of the neutrophils from patients without breast cancer (100). This work also showed that neutrophils from patients with and without breast cancer exposed to TNF-α ex-vivo exhibited enhanced cytotoxicity, with even further cytotoxicity seen in patients with breast cancer (100). TNF-α and TANs interact in many ways: 1) TANs are recruited into tumors partially from the influence of TNF-α 2) T cells attract and prime TANs with TNF-α 3) N1 TANs can attract cytotoxic T cells by TNF-α secretion 4) TANs can activate dendritic cells with TNF-α and assist CD4+ T cells with anti-neoplastic memory (101). In summary, neutrophil secretion of and reaction to TNF-α causes many interactions in the TME may be exploited as potential targets in cancer therapy.
T Cells
A heterogeneous population of tumor‐infiltrating lymphocytes, CD8+ cytotoxic T cells, CD4+ helper T cells, and regulatory T cells (Tregs) are present in the TME. As with both TAMs and TANs, these tumor-infiltrating lymphocytes either suppress or enhance tumor growth. Previous studies demonstrated that T cell responses are regulated by TNFR activation and mediated cross-talk between T cells and other cell types. The TNFR superfamily (TNFRSF) OX40, 4-1BB, CD27, and DR3 are associated with TNFR associated factors (TRAF). In detail, TRAFs can bind with the α subunit of NF-κB and I Kappa B Kinase Beta (IkB kinase-β) to assist in the activation of both canonical and non-canonical NF-κB signaling pathways. OX40, 4-1BB, and CD27 mediated activation of signaling pathways in CD4+ and CD8+ T cells increase the expression of the anti-apoptotic molecule BCL-2, which correlates with the promotion of T cell survival (102). Prior work has demonstrated that the treatment with recombinant human TNF-α in a B16F10 melanoma mouse model of lung metastasis increased tumor burden and metastatic foci and was associated with increased numbers of pulmonary regulatory CD4+/Foxp3+ T cells. TNF-α activates TNFR2 on Tregs which helps expand the immunosuppressive role of the immune cell population by inducing CD8+ and CD4+ T cells. But the accumulation of Tregs can be prevented through dysregulation of TNF-α or TNFR2 which creates less tolerogenic environment and prevents B16F10 tumor metastasis and growth (103). Hu et al. reported that TNFR2 progranulin induced the proliferation of suppressive mouse CD4+/Foxp3+ regulatory T cells (104). Therefore, the targeting of T cell-associated mechanisms has been considered a major strategy for cancer immunotherapy.
Dendritic Cells
The antigen-presenting cells (APC), dendritic cells (DC) can take up, process, and present different types of antigens, including tumor antigens, to naïve T cells. This antigen presentation can induce the creation of tumor-specific cytotoxic T cells (105). DC can also downregulate the immune response or induce immune tolerance in the TME through exposure to different stimuli. For example, when exposed to thymic stromal lymphoprotein from tumor or thymus (105), or TNF-α (106), DC express OX40 ligand, which is a member of the TNF superfamily (TNFSF4). The T cell membrane OX40 binds to OX40L on DC and induces a phenotypic subtype of CD4+ T cells, promoting tumor growth (105). DC induced CD4+ T cells to polarize to this subtype which highly secreted IL-13, which promoted breast cancer progression in vivo (107). Conversely, a subset of inflammatory DC which secrete TNF-α and produce nitric oxide are known to be tumor suppressive (108, 109). In vivo adjunctive use of this inflammatory DC subtype was shown to potentiate the effect of adoptive cell transfer, allowing for a more potent treatment strategy for cancer immunotherapy (108).
Role of TNF-α in Cancer Stem Cells
The cancer stem cells (CSC) is a subpopulation within a primary cancer that have the potential to differentiate into a more mature phenotype and exhibit properties of self-renewal and immortality. CSC were first described in acute myeloid leukemia (AML) from the identification of a subset of specific surface proteins, which were found to regenerate AML in vivo (110). Since this initial description, they have been described in many different solid cancers in addition to hematologic malignancies and are implicated in chemotherapy resistance and progression to metastasis (111). Accumulating evidence suggests that TNF-α has a role in CSC regulation. TNF-α increased the breast CSC population through NF-κB/HIF1α/Slug (112). Zhao et al. reported that TNF-α treatment in a colon cancer cell-line (NCM460) derived spheroids induced NF-κB and Wnt/β-catenin pathways which can accelerate malignant transformation in intestinal stem cells (113). Additionally, using an osteosarcoma cell line in vitro, Yao et al. showed TNF-α exposure upregulated a specific microRNA (miR-155) and found that miR-155 produced a stem cell-like phenotype which promoted cancer progression (114). Suggested by these studies, the role of TNF-α in CSC may contribute to tumorigenesis and progression. However, other TNF-α-related tumor-suppressive effects may counter the effects of TNF-α on CSC.
TNF-α Mediated MicroRNA Regulation
MicroRNAs (miRNA) are short non-coding RNAs that regulate gene expression at the post-transcriptional level by binding to the 3’-untranslated region of their targeted mRNA resulting in the suppression of protein production. miRNAs are dysregulated in several cancer types. Thus, understanding the role of miRNAs in the TME is crucial (115). The remodeling of miRNAs in the TME has a role in tumor growth, metastasis, and resistance to treatment. Several miRNAs are released from neoplastic cells into the TME and regulate the functions of endothelial cells, immune cells, and fibroblasts. Previous studies from several groups reported that miR-145, miR-15a, miR-29a, miR-181A, miR-19a, miR-130a, miR-21, miR-765 are regulated by TNF-α in several cancers produced by both cancer cells and TME-related immune cells. Eleven miRNAs were shown to be differentially expressed between cancer-associated fibroblasts and healthy tissue, and 114 upregulated and 85 downregulated miRNAs have been identified in gastric cancer mesenchymal stem cells (GC-MSCs) (116). Zeng et al. showed that the tumor suppressor miR-145 is downregulated in triple-negative breast cancer cell lines MDA-MB-231, and when treated with TNF-α, this miRNA is overexpressed and induces apoptosis (117). Co-immunoprecipitation data revealed that miR-145 facilitates the formation of RIP1-FADD -caspase 8-mediated apoptotic complex with TNF-α treatment (118). Huang et al. showed that miR-19a is associated with lymph node metastasis and mediates TNF-α induced EMT in colorectal cancer (119). Furthermore, miR-19a is upregulated by TNF-α and miR-19a is required for TNF-α induced EMT and metastases in CRC cells (120). TNF-α also has a role in nuclear translocation of NF-κB followed by induction miR-130 and expression and downregulation of TNF-α (121). Higher levels of TNF-α have been observed in B-cell chronic lymphocytic leukemia (CLL) (122). Some miRNAs can regulate the TNF/TNFR gene superfamily in CLL (122). Therefore, the identification of the cross-talk between TNF-α and miRNAs could show promising effects for chemotherapeutic agents to control the TME.
Clinical Uses of TNF-α and Its Importance in Drug Delivery
Carswell et al. discovered in 1975 TNF in the serum of bacilli Calmette-Guérin infected mice inoculated with endotoxin and found that “the substance” induced in vivo hemorrhagic necrosis in sarcoma. The substance was named “tumor necrosis factor” (123). Since then, the clinical utility of TNF-α is limited due to its severe systemic toxicity. To mitigate systemic toxicities, the application of TNF-α in cancer treatments became significantly more sophisticated. Its combined use with chemotherapeutic agents in isolated limb perfusion (ILP) has shown good results in tumor response and limb salvage in patients with soft tissue sarcomas (15) and regression in locoregional metastatic melanoma of a limb (14). Because ILP uses arterial and venous canulation with a closed extracorporeal circuit, high doses of TNF- α and chemotherapy can be used in the limb with minimal systemic toxicity. However, the ILP system is cumbersome and does not address its limitation to use TNF-α systemically to improve the efficacy of cancer treatments. The work by our group has shown that when attached to gold nanoparticles (CYT-6091), recombinant human TNF-α (rhTNF) can be given systemically without dose-limiting toxicities up to 3 times that of intravenously-administered unbound rhTNF in phase I clinical trial (124). Although we demonstrated the safety of gold nanomedicine carrying TNF-α in humans, the anti-cancer efficacy was limited, with only a partial response in 27 evaluable participants with advanced solid cancers (124). To prove the concept that this cytotoxic agent was needed to improve the treatment efficacy, paclitaxel analog was added to the gold nanomedicine carrying rhTNF (CYT-21625) and the treatment efficacy, drug delivery efficiency, and systemic toxicities were assessed in multiple mouse models with pancreatic neuroendocrine tumors (pNET) and metastatic aggressive thyroid cancers. We demonstrated significantly improved treatment efficacy across all mouse models treated with CYT-21625 compared to mice treated with intravenous paclitaxel or CYT-6091, with no detectable systemic toxicities or histologic evidence of normal tissue damage or vascular leakage (125). The principle behind its efficacy is related to its effect on tumor vasculature and overcoming high intratumoral interstitial fluid pressure (IFP), allowing for more efficient drug delivery and resulting in markedly increased intratumoral concentrations of paclitaxel (126).
TNF-α has a dose-dependent effect on vascular endothelial cells, inducing angiogenesis at low levels and inhibiting or disrupting it at high levels (127). Neoplasms, just as do healthy tissues, require a blood supply to provide nutrients and allow for growth. In contrast with normal healthy vasculature, neovascular growth in cancers is imperfect, exhibiting a non-continuous endothelium and a sporadically present or absent basement membrane, which increases vessel permeability (16). This increased permeability, along with high cell density, poor venous and lymphatic outflow, and an abnormal ECM contribute to high intratumoral hydrostatic and osmotic IFP (128, 129). Elevated IFP is a barrier to efficient drug delivery in solid tumors as drug cannot effectively travel against a pressure gradient to achieve local therapeutic levels. TNF-α mitigates this barrier by disrupting tumor vasculature, reducing tumor IFP, and allowing chemotherapeutic agents to diffuse into the TME. It disrupts the vasculature by inducing endothelial cell apoptosis, specifically in the tumor while having minimal effect on native healthy vessels due to the differential expression of TNF-R1 in the cancer neovascular network. Increased levels of TNF-R1 in neovascular endothelium has been shown in several tumor types, including thyroid cancer and pNET (125).
Vascular disruption leading to the reduction in intratumoral IFP is the key concept behind the therapeutic uses of TNF-α to improve drug delivery efficiency in cancer treatments. Its most common clinical application is in ILP for locoregionally metastatic melanoma or soft tissue sarcomas of the limb. In this procedure, the limb is isolated with a tourniquet and perfused with oxygenated blood from an extracorporeal circuit, similar in concept to extracorporeal membrane oxygenation for cardiopulmonary failure. Within the perfusion circuit, hyperthermic high-dose chemotherapy (typically the alkylating agent melphalan) is infused with TNF-α. TNF-α serves two functions – inducing hemorrhagic necrosis of the tumor by disrupting the vasculature and allowing the melphalan to locally accumulate in higher levels that would otherwise cause severe systemic toxicities. The limb isolation minimizes the systemic toxicity of both the chemotherapeutic agent and TNF-α –as TNF-α is an acute phase reactant and inflammatory cytokine and can lead to distributive shock – and allows for a several-fold increase in concentration. ILP with melphalan and TNF-α was initially shown to be effective in a phase II trial, which resulted in 21 of 23 complete responses and two partial responses with no patient experiencing treatment failure (130). This cumbersome treatment strategy is only feasible in limb lesions and is not an option for primary or metastatic cancers in solid organs.
Recent work by our group has employed the concept of TNF-α as a facilitator of systemic drug delivery with a novel use of gold nanomedicine. Gold nanoparticles passively target solid tumors by the enhanced permeability and retention effect, preferentially accumulating in their tissue through a combination of highly permeable vasculature and the larger size of the particles compared to molecules dissolved in plasma (131, 132). Gold nanomedicine has a harder time passing through the tight junctions of normal vascular endothelium in non-neoplastic tissue and selectively extravasates into the TME, where it accumulates (132). In addition, the gold nanoparticles carrying rhTNF actively targets cancer neovasculature by binding to the differentially expressed TNF receptors on tumor neovascular endothelium (125). As is the case for ILP, this modality of TNF-α delivery reduces its toxic systemic effects through both active and passive tumor-specific targeting and allows for higher concentrations to be delivered. To improve treatment efficacy in cancers, we demonstrated a significantly lower tumor burden across multiple in vivo models using combined rhTNF and paclitaxel analog bound nanomedicine over both rhTNF nanomedicine alone and IV paclitaxel alone in anaplastic thyroid cancer and pNET with no apparent systemic toxicity, further indicating increased efficacy (125). In addition, radiographic imaging studies and histology showed the gold nanomedicine carrying rhTNF only and rhTNF with paclitaxel analog preferentially and specifically targeted tumor tissue and induced vascular leakage only in tumor tissue (125). The transgenic mouse model with pNET showed selective extravasation of MRI contrast in the pancreatic area, corresponding with 18F-FDG-avid lesions, from mice treated with rhTNF bound nanoparticles but not in mice treated with paclitaxel alone and vehicle control. We observed no evidence of extravasation in normal tissue, indicating that the TNF-α induced tumor-specific vascular damage (125). To demonstrate the applicability in a broader range of cancer, a pilot study showed 100% survival in vivo in mice with pancreatic ductal adenocarcinoma treated with gold nanomedicine carrying rhTNF-followed by intravenous (IV) paclitaxel compared to 50% survival in the IV paclitaxel only group and 0% in the control group at 42 days (126).
Conclusion
TNF-α plays a critical role in tumor signaling pathways and immune cell manipulation within the TME. Since Carswell discovered the cytokine in 1975, our understanding of its role in cancers and chronic inflammatory diseases has improved, resulting in the development of treatments that specifically target systemic and TME-related immune cellular response. However, the clinical application such as TNF receptor blockade is only limited to the treatment of chronic inflammatory diseases. Although preclinical data of TNF-α treatment in cancers to improve drug delivery is promising, the treatment efficacy in cancers is not known due to the lack of phase II clinical trials. Because TNF-α induces diverse effects in TME, both oncogenic and tumor-suppressive effects, further studies are warranted to fully understand and selectively induce the anti-tumor effect to improve treatment efficacy in patients with TNF-α sensitive cancers.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
The preparation of this manuscript was supported by the NIH intramural research grant (# ZIA BC 011286).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
The handling editor declared a shared affiliation with the authors at the time of the review.
References
2. Chew V, Toh HC, Abastado JP. Immune Microenvironment in Tumor Progression: Characteristics and Challenges for Therapy. J Oncol (2012) 2012:608406. doi: 10.1155/2012/608406
3. Gajewski TF, Schreiber H, Fu YX. Innate and Adaptive Immune Cells in the Tumor Microenvironment. Nat Immunol (2013) 14:1014–22. doi: 10.1038/ni.2703
4. Hirata E, Ishibashi K, Kohsaka S, Shinjo K, Kojima S, Kondo Y, et al. The Brain Microenvironment Induces Dnmt1 Suppression and Indolence of Metastatic Cancer Cells. iScience (2020) 23:101480. doi: 10.1016/j.isci.2020.101480
5. Schwager SC, Taufalele PV, Reinhart-King CA. Cell-Cell Mechanical Communication in Cancer. Cell Mol Bioeng (2019) 12:1–14. doi: 10.1007/s12195-018-00564-x
6. Bates RC, Mercurio AM. Tumor Necrosis Factor-Alpha Stimulates the Epithelial-to-Mesenchymal Transition of Human Colonic Organoids. Mol Biol Cell (2003) 14:1790–800. doi: 10.1091/mbc.e02-09-0583
7. Ranta V, Orpana A, Carpen O, Turpeinen U, Ylikorkala O, Viinikka L. Human Vascular Endothelial Cells Produce Tumor Necrosis Factor-Alpha in Response to Proinflammatory Cytokine Stimulation. Crit Care Med (1999) 27:2184–7. doi: 10.1097/00003246-199910000-00019
8. Epstein Shochet G, Brook E, Israeli-Shani L, Edelstein E, Shitrit D. Fibroblast Paracrine TNF-alpha Signaling Elevates Integrin A5 Expression in Idiopathic Pulmonary Fibrosis (IPF). Respir Res (2017) 18:122. doi: 10.1186/s12931-017-0606-x
9. van Horssen R, Ten Hagen TL, Eggermont AM. TNF-Alpha in Cancer Treatment: Molecular Insights, Antitumor Effects, and Clinical Utility. Oncologist (2006) 11:397–408. doi: 10.1634/theoncologist.11-4-397
10. Sheng Y, Li F, Qin Z. Tnf Receptor 2 Makes Tumor Necrosis Factor a Friend of Tumors. Front Immunol (2018) 9:1170. doi: 10.3389/fimmu.2018.01170
11. Vanamee ES, Faustman DL. Tnfr2: A Novel Target for Cancer Immunotherapy. Trends Mol Med (2017) 23:1037–46. doi: 10.1016/j.molmed.2017.09.007
12. Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. Tumor Necrosis Factor-Alpha Blocks Differentiation and Enhances Suppressive Activity of Immature Myeloid Cells During Chronic Inflammation. Immunity (2013) 38:541–54. doi: 10.1016/j.immuni.2013.02.007
13. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF Signaling Drives Myeloid-Derived Suppressor Cell Accumulation. J Clin Invest (2012) 122:4094–104. doi: 10.1172/JCI64115
14. Deroose JP, Eggermont AM, van Geel AN, de Wilt JH, Burger JW, Verhoef C. 20 Years Experience of TNF-based Isolated Limb Perfusion for in-Transit Melanoma Metastases: TNF Dose Matters. Ann Surg Oncol (2012) 19:627–35. doi: 10.1245/s10434-011-2030-7
15. Neuwirth MG, Song Y, Sinnamon AJ, Fraker DL, Zager JS, Karakousis GC. Isolated Limb Perfusion and Infusion for Extremity Soft Tissue Sarcoma: A Contemporary Systematic Review and Meta-Analysis. Ann Surg Oncol (2017) 24:3803–10. doi: 10.1245/s10434-017-6109-7
16. Carmeliet P, Jain RK. Angiogenesis in Cancer and Other Diseases. Nature (2000) 407:249–57. doi: 10.1038/35025220
17. Aggarwal BB. Signalling Pathways of the TNF Superfamily: A Double-Edged Sword. Nat Rev Immunol (2003) 3:745–56. doi: 10.1038/nri1184
18. Speeckaert MM, Speeckaert R, Laute M, Vanholder R, Delanghe JR. Tumor Necrosis Factor Receptors: Biology and Therapeutic Potential in Kidney Diseases. Am J Nephrol (2012) 36:261–70. doi: 10.1159/000342333
19. Clouthier DL, Watts TH. Tnfrs and Control of Chronic Lcmv Infection: Implications for Therapy. Trends Immunol (2015) 36:697–708. doi: 10.1016/j.it.2015.09.005
20. Liu S, Rong L, Deng J, Zhao X, Liu X, Xu X, et al. TNFR2 Expression on non-Bone Marrow-Derived Cells is Crucial for Lipopolysaccharide-Induced Septic Shock and Downregulation of Soluble TNFR2 Level in Serum. Cell Mol Immunol (2011) 8:164–71. doi: 10.1038/cmi.2010.79
21. Pronk CJ, Veiby OP, Bryder D, Jacobsen SE. Tumor Necrosis Factor Restricts Hematopoietic Stem Cell Activity in Mice: Involvement of Two Distinct Receptors. J Exp Med (2011) 208:1563–70. doi: 10.1084/jem.20110752
22. Ghali O, Chauveau C, Hardouin P, Broux O, Devedjian JC. TNF-Alpha’s Effects on Proliferation and Apoptosis in Human Mesenchymal Stem Cells Depend on RUNX2 Expression. J Bone Miner Res (2010) 25:1616–26. doi: 10.1002/jbmr.52
23. Chau BN, Chen TT, Wan YY, DeGregori J, Wang JY. Tumor Necrosis Factor Alpha-Induced Apoptosis Requires p73 and c-ABL Activation Downstream of RB Degradation. Mol Cell Biol (2004) 24:4438–47. doi: 10.1128/MCB.24.10.4438-4447.2004
24. Farrugia M, Baron B. The Role of TNF-alpha in Rheumatoid Arthritis: A Focus on Regulatory T Cells. J Clin Transl Res (2016) 2:84–90. doi: 10.18053/jctres.02.201603.005
25. Wu TC. The Role of Vascular Cell Adhesion Molecule-1 in Tumor Immune Evasion. Cancer Res (2007) 67:6003–6. doi: 10.1158/0008-5472.CAN-07-1543
26. Khatib AM, Auguste P, Fallavollita L, Wang N, Samani A, Kontogiannea M, et al. Characterization of the Host Proinflammatory Response to Tumor Cells During the Initial Stages of Liver Metastasis. Am J Pathol (2005) 167:749–59. doi: 10.1016/S0002-9440(10)62048-2
27. Hagemann T, Robinson SC, Schulz M, Trumper L, Balkwill FR, Binder C. Enhanced Invasiveness of Breast Cancer Cell Lines Upon Co-Cultivation With Macrophages is Due to TNF-alpha Dependent Up-Regulation of Matrix Metalloproteases. Carcinogenesis (2004) 25:1543–9. doi: 10.1093/carcin/bgh146
28. Stuelten CH, DaCosta Byfield S, Arany PR, Karpova TS, Stetler-Stevenson WG, Roberts AB. Breast Cancer Cells Induce Stromal Fibroblasts to Express MMP-9 Via Secretion of TNF-alpha and TGF-Beta. J Cell Sci (2005) 118:2143–53. doi: 10.1242/jcs.02334
29. Wagener FA, Feldman E, de Witte T, Abraham NG. Heme Induces the Expression of Adhesion Molecules ICAM-1, Vcam-1, and E Selectin in Vascular Endothelial Cells. Proc Soc Exp Biol Med (1997) 216:456–63. doi: 10.3181/00379727-216-44197
30. Aggarwal BB, Sethi G, Baladandayuthapani V, Krishnan S, Shishodia S. Targeting Cell Signaling Pathways for Drug Discovery: An Old Lock Needs a New Key. J Cell Biochem (2007) 102:580–92. doi: 10.1002/jcb.21500
31. Srinivasan S, Kumar R, Koduru S, Chandramouli A, Damodaran C. Inhibiting TNF-mediated Signaling: A Novel Therapeutic Paradigm for Androgen Independent Prostate Cancer. Apoptosis (2010) 15:153–61. doi: 10.1007/s10495-009-0416-9
32. Kimura K, Bowen C, Spiegel S, Gelmann EP. Tumor Necrosis Factor-Alpha Sensitizes Prostate Cancer Cells to Gamma-Irradiation-Induced Apoptosis. Cancer Res (1999) 59:1606–14.
33. Cruceriu D, Baldasici O, Balacescu O, Berindan-Neagoe I. The Dual Role of Tumor Necrosis Factor-Alpha (TNF-Alpha) in Breast Cancer: Molecular Insights and Therapeutic Approaches. Cell Oncol (Dordr) (2020) 43:1–18. doi: 10.1007/s13402-019-00489-1
34. Cai X, Cao C, Li J, Chen F, Zhang S, Liu B, et al. Inflammatory Factor TNF-alpha Promotes the Growth of Breast Cancer Via the Positive Feedback Loop of TNFR1/NF-kappaB (and/or P38)/P-STAT3/HBXIP/TNFR1. Oncotarget (2017) 8:58338–52. doi: 10.18632/oncotarget.16873
35. Wang K, Ren Y, Liu Y, Zhang J, He JJ. Tumor Necrosis Factor (TNF)-Alpha-Induced Protein 8-Like-2 (TIPE2) Inhibits Proliferation and Tumorigenesis in Breast Cancer Cells. Oncol Res (2017) 25:55–63. doi: 10.3727/096504016X14719078133320
36. Zhu J, Xin Y, Liu X, Wang Y, Liu Y. Nimotuzumab Enhances the Sensitivity of non-Small Cell Lung Cancer Cells to Tumor Necrosis Factor-Alpha by Inhibiting the Nuclear Factor-Small Ka, CyrillicB Signaling Pathway. Exp Ther Med (2018) 15:3345–51. doi: 10.3892/etm.2018.5856
37. Lin X, Chen Q, Huang C, Xu X. Cyld Promotes TNF-Alpha-Induced Cell Necrosis Mediated by RIP-1 in Human Lung Cancer Cells. Mediators Inflammation (2016) 2016:1542786. doi: 10.1155/2016/1542786
38. Gray-Schopfer VC, Karasarides M, Hayward R, Marais R. Tumor Necrosis Factor-Alpha Blocks Apoptosis in Melanoma Cells When BRAF Signaling is Inhibited. Cancer Res (2007) 67:122–9. doi: 10.1158/0008-5472.CAN-06-1880
39. Lee K, Lee MH, Kang YW, Rhee KJ, Kim TU, Kim YS. Parkin Induces Apoptotic Cell Death in TNF-alpha-treated Cervical Cancer Cells. BMB Rep (2012) 45:526–31. doi: 10.5483/BMBRep.2012.45.9.104
40. Tian X, Xu L, Wang P. MiR-191 Inhibits TNF-alpha Induced Apoptosis of Ovarian Endometriosis and Endometrioid Carcinoma Cells by Targeting DAPK1. Int J Clin Exp Pathol (2015) 8:4933–42.
41. Zhu Y, Cheng Y, Guo Y, Chen J, Chen F, Luo R, et al. Protein Kinase D2 Contributes to TNF-alpha-induced Epithelial Mesenchymal Transition and Invasion Via the PI3K/GSK-3beta/beta-catenin Pathway in Hepatocellular Carcinoma. Oncotarget (2016) 7:5327–41. doi: 10.18632/oncotarget.6633
42. Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T. Transmembrane TNF-alpha: Structure, Function and Interaction With anti-TNF Agents. Rheumatol (Oxford) (2010) 49:1215–28. doi: 10.1093/rheumatology/keq031
43. Wang L, Yang D, Tian J, Gao A, Shen Y, Ren X, et al. Tumor Necrosis Factor Receptor 2/AKT and ERK Signaling Pathways Contribute to the Switch From Fibroblasts to CAFs by Progranulin in Microenvironment of Colorectal Cancer. Oncotarget (2017) 8:26323–33. doi: 10.18632/oncotarget.15461
44. Torrey H, Butterworth J, Mera T, Okubo Y, Wang L, Baum D, et al. Targeting TNFR2 With Antagonistic Antibodies Inhibits Proliferation of Ovarian Cancer Cells and Tumor-Associated Tregs. Sci Signal (2017) 10(462):eaaf8608. doi: 10.1126/scisignal.aaf8608
45. Sughra K, Birbach A, de Martin R, Schmid JA. Interaction of the TNFR-receptor Associated Factor TRAF1 With I-Kappa B Kinase-2 and TRAF2 Indicates a Regulatory Function for NF-Kappa B Signaling. PloS One (2010) 5:e12683. doi: 10.1371/journal.pone.0012683
46. Cui ZY, Park SJ, Jo E, Hwang IH, Lee KB, Kim SW, et al. Cordycepin Induces Apoptosis of Human Ovarian Cancer Cells by Inhibiting CCL5-mediated Akt/NF-Kappab Signaling Pathway. Cell Death Discovery (2018) 4:62. doi: 10.1038/s41420-018-0063-4
47. Yamane K, Ihn H, Asano Y, Jinnin M, Tamaki K. Antagonistic Effects of TNF-alpha on TGF-beta Signaling Through Down-Regulation of TGF-beta Receptor Type II in Human Dermal Fibroblasts. J Immunol (2003) 171:3855–62. doi: 10.4049/jimmunol.171.7.3855
48. Jayasooriya RG, Dilshara MG, Choi YH, Moon SK, Kim WJ, Kim GY. Tianeptine Sodium Salt Suppresses TNF-alpha-induced Expression of Matrix Metalloproteinase-9 in Human Carcinoma Cells Via Suppression of the PI3K/Akt-mediated NF-Kappab Pathway. Environ Toxicol Pharmacol (2014) 38:502–9. doi: 10.1016/j.etap.2014.07.023
49. Wei X, Li X, Kong F, Ma L, Sui Y, Chen D, et al. TNF-Alpha Activates Wnt Signaling Pathway to Promote the Invasion of Human Colon Cancer Stem Cells. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2018) 34:982–8.
50. Xie L, Jiang F, Zhang X, Alitongbieke G, Shi X, Meng M, et al. Honokiol Sensitizes Breast Cancer Cells to TNF-alpha Induction of Apoptosis by Inhibiting Nur77 Expression. Br J Pharmacol (2016) 173:344–56. doi: 10.1111/bph.13375
51. Mendonca P, Horton A, Bauer D, Messeha S, Soliman KFA. The Inhibitory Effects of Butein on Cell Proliferation and TNF-alpha-induced CCL2 Release in Racially Different Triple Negative Breast Cancer Cells. PloS One (2019) 14:e0215269. doi: 10.1371/journal.pone.0215269
52. Hong H, Jiang L, Lin Y, He C, Zhu G, Du Q, et al. TNF-Alpha Promotes Lymphangiogenesis and Lymphatic Metastasis of Gallbladder Cancer Through the ERK1/2/AP-1/VEGF-D Pathway. BMC Cancer (2016) 16:240. doi: 10.1186/s12885-016-2259-4
53. Letterio JJ, Roberts AB. Regulation of Immune Responses by TGF-Beta. Annu Rev Immunol (1998) 16:137–61. doi: 10.1146/annurev.immunol.16.1.137
54. Chang NS. Transforming Growth Factor-Beta Protection of Cancer Cells Against Tumor Necrosis Factor Cytotoxicity is Counteracted by Hyaluronidase (Review). Int J Mol Med (1998) 2:653–9. doi: 10.3892/ijmm.2.6.653
55. Greenwel P, Tanaka S, Penkov D, Zhang W, Olive M, Moll J, et al. Tumor Necrosis Factor Alpha Inhibits Type I Collagen Synthesis Through Repressive CCAAT/enhancer-binding Proteins. Mol Cell Biol (2000) 20:912–8. doi: 10.1128/MCB.20.3.912-918.2000
56. Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-Associated Fibroblasts: An Emerging Target of Anti-Cancer Immunotherapy. J Hematol Oncol (2019) 12:86. doi: 10.1186/s13045-019-0770-1
57. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat Rev Cancer (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
58. Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming Growth Factor-Beta 1 Induces Alpha-Smooth Muscle Actin Expression in Granulation Tissue Myofibroblasts and in Quiescent and Growing Cultured Fibroblasts. J Cell Biol (1993) 122:103–11. doi: 10.1083/jcb.122.1.103
59. Taniguchi K, Karin M. Nf-kappaB, Inflammation, Immunity and Cancer: Coming of Age. Nat Rev Immunol (2018) 18:309–24. doi: 10.1038/nri.2017.142
60. Whiteside TL. The Tumor Microenvironment and its Role in Promoting Tumor Growth. Oncogene (2008) 27:5904–12. doi: 10.1038/onc.2008.271
61. Lawrence T. The Nuclear Factor NF-kappaB Pathway in Inflammation. Cold Spring Harb Perspect Biol (2009) 1:a001651. doi: 10.1101/cshperspect.a001651
62. Hirano S, Kataoka T. Deoxynivalenol Induces Ectodomain Shedding of TNF Receptor 1 and Thereby Inhibits the TNF-alpha-induced NF-Kappab Signaling Pathway. Eur J Pharmacol (2013) 701:144–51. doi: 10.1016/j.ejphar.2013.01.019
63. Tang D, Tao D, Fang Y, Deng C, Xu Q, Zhou J. Tnf-Alpha Promotes Invasion and Metastasis Via NF-Kappa B Pathway in Oral Squamous Cell Carcinoma. Med Sci Monit Basic Res (2017) 23:141–9. doi: 10.12659/MSMBR.903910
64. Du Z, Lovly CM. Mechanisms of Receptor Tyrosine Kinase Activation in Cancer. Mol Cancer (2018) 17:58. doi: 10.1186/s12943-018-0782-4
65. Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, et al. Overcoming EGFR(T790M) and EGFR(C797S) Resistance With Mutant-Selective Allosteric Inhibitors. Nature (2016) 534:129–32. doi: 10.1038/nature17960
66. Kwilas AR, Donahue RN, Tsang KY, Hodge JW. Immune Consequences of Tyrosine Kinase Inhibitors That Synergize With Cancer Immunotherapy. Cancer Cell Microenviron (2015) 2(1):e677. doi: 10.14800/ccm.677
67. Tournier C, Dong C, Turner TK, Jones SN, Flavell RA, Davis RJ. MKK7 is an Essential Component of the JNK Signal Transduction Pathway Activated by Proinflammatory Cytokines. Genes Dev (2001) 15:1419–26. doi: 10.1101/gad.888501
68. Sabio G, Davis RJ. TNF and MAP Kinase Signalling Pathways. Semin Immunol (2014) 26:237–45. doi: 10.1016/j.smim.2014.02.009
69. Ji H, Cao R, Yang Y, Zhang Y, Iwamoto H, Lim S, et al. TNFR1 Mediates TNF-alpha-induced Tumour Lymphangiogenesis and Metastasis by Modulating VEGF-C-VEGFR3 Signalling. Nat Commun (2014) 5:4944. doi: 10.1038/ncomms5944
70. Yujiao Gong YD, Cui J, Sun Q, Zhen Z, Gao Y, Su J, et al. Receptor Tyrosine Kinase Interaction With the Tumor Microenvironment in Malignant Progression of Human Glioblastoma, Glioma - Contemporary Diagnostic and Therapeutic Approaches, Ibrahim Omerhodžić and Kenan Arnautović, IntechOpen (2018). doi: 10.5772/intechopen/76873
71. Cobleigh MA, Tabesh B, Bitterman P, Baker J, Cronin M, Liu ML, et al. Tumor Gene Expression and Prognosis in Breast Cancer Patients With 10 or More Positive Lymph Nodes. Clin Cancer Res (2005) 11:8623–31. doi: 10.1158/1078-0432.CCR-05-0735
72. Carvalho I, Milanezi F, Martins A, Reis RM, Schmitt F. Overexpression of Platelet-Derived Growth Factor Receptor Alpha in Breast Cancer is Associated With Tumour Progression. Breast Cancer Res (2005) 7:R788–95. doi: 10.1186/bcr1304
73. Sasi SP, Yan X, Enderling H, Park D, Gilbert HY, Curry C, et al. Breaking the ‘Harmony’ of TNF-alpha Signaling for Cancer Treatment. Oncogene (2012) 31:4117–27. doi: 10.1038/onc.2011.567
74. Soria G, Ofri-Shahak M, Haas I, Yaal-Hahoshen N, Leider-Trejo L, Leibovich-Rivkin T, et al. Inflammatory Mediators in Breast Cancer: Coordinated Expression of TNFalpha & IL-1beta With CCL2 & CCL5 and Effects on Epithelial-to-Mesenchymal Transition. BMC Cancer (2011) 11:130. doi: 10.1186/1471-2407-11-130
75. Wang H, Wang HS, Zhou BH, Li CL, Zhang F, Wang XF, et al. Epithelial-Mesenchymal Transition (EMT) Induced by TNF-alpha Requires AKT/GSK-3beta-mediated Stabilization of Snail in Colorectal Cancer. PloS One (2013) 8:e56664. doi: 10.1371/journal.pone.0056664
76. Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, et al. Epithelial-Mesenchymal Transition Induced by TNF-alpha Requires NF-kappaB-mediated Transcriptional Upregulation of Twist1. Cancer Res (2012) 72:1290–300. doi: 10.1158/0008-5472.CAN-11-3123
77. Mikesh LM, Kumar M, Erdag G, Hogan KT, Molhoek KR, Mayo MW, et al. Evaluation of Molecular Markers of Mesenchymal Phenotype in Melanoma. Melanoma Res (2010) 20:485–95. doi: 10.1097/CMR.0b013e32833fafb4
78. Chen Y, Wen H, Zhou C, Su Q, Lin Y, Xie Y, et al. TNF-Alpha Derived From M2 Tumor-Associated Macrophages Promotes Epithelial-Mesenchymal Transition and Cancer Stemness Through the Wnt/beta-catenin Pathway in SMMC-7721 Hepatocellular Carcinoma Cells. Exp Cell Res (2019) 378:41–50. doi: 10.1016/j.yexcr.2019.03.005
79. Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular Matrix Structure. Adv Drug Delivery Rev (2016) 97:4–27. doi: 10.1016/j.addr.2015.11.001
80. Laronha H, Caldeira J. Structure and Function of Human Matrix Metalloproteinases. Cells (2020) 9(5):1076. doi: 10.3390/cells9051076
81. Dilshara MG, Kang CH, Choi YH, Kim GY. Mangiferin Inhibits Tumor Necrosis Factor-Alpha-Induced Matrix Metalloproteinase-9 Expression and Cellular Invasion by Suppressing Nuclear Factor-Kappab Activity. BMB (2015) Rep 48:559–64. doi: 10.5483/BMBRep.2015.48.10.003
82. Kim S, Choi JH, Kim JB, Nam SJ, Yang JH, Kim JH, et al. Berberine Suppresses TNF-alpha-induced MMP-9 and Cell Invasion Through Inhibition of AP-1 Activity in MDA-MB-231 Human Breast Cancer Cells. Molecules (2008) 13:2975–85. doi: 10.3390/molecules13122975
83. Turner M, Feldmann M. Comparison of Patterns of Expression of Tumour Necrosis Factor, Lymphotoxin and Interleukin-6 Mrna. Biochem Biophys Res Commun (1988) 153:1144–51. doi: 10.1016/S0006-291X(88)81347-0
84. Kelker HC, Oppenheim JD, Stone-Wolff D, Henriksen-DeStefano D, Aggarwal BB, Stevenson HC, et al. Characterization of Human Tumor Necrosis Factor Produced by Peripheral Blood Monocytes and its Separation From Lymphotoxin. Int J Cancer (1985) 36:69–73. doi: 10.1002/ijc.2910360112
85. Qiu SQ, Waaijer SJH, Zwager MC, de Vries EGE, van der Vegt B, Schroder CP. Tumor-Associated Macrophages in Breast Cancer: Innocent Bystander or Important Player? Cancer Treat Rev (2018) 70:178–89. doi: 10.1016/j.ctrv.2018.08.010
86. Zhao X, Qu J, Sun Y, Wang J, Liu X, Wang F, et al. Prognostic Significance of Tumor-Associated Macrophages in Breast Cancer: A Meta-Analysis of the Literature. Oncotarget (2017) 8:30576–86. doi: 10.18632/oncotarget.15736
87. Yuan X, Zhang J, Li D, Mao Y, Mo F, Du W, et al. Prognostic Significance of Tumor-Associated Macrophages in Ovarian Cancer: A Meta-Analysis. Gynecol Oncol (2017) 147:181–7. doi: 10.1016/j.ygyno.2017.07.007
88. Li J, Xie Y, Wang X, Li F, Li S, Li M, et al. Prognostic Impact of Tumor-Associated Macrophage Infiltration in Esophageal Cancer: A Meta-Analysis. Future Oncol (2019) 15:2303–17. doi: 10.2217/fon-2018-0669
89. Yu M, Guan R, Hong W, Zhou Y, Lin Y, Jin H, et al. Prognostic Value of Tumor-Associated Macrophages in Pancreatic Cancer: A Meta-Analysis. Cancer Manag Res (2019) 11:4041–58. doi: 10.2147/CMAR.S196951
90. Wu P, Wu D, Zhao L, Huang L, Chen G, Shen G, et al. Inverse Role of Distinct Subsets and Distribution of Macrophage in Lung Cancer Prognosis: A Meta-Analysis. Oncotarget (2016) 7:40451–60. doi: 10.18632/oncotarget.9625
91. Martinez FO, Helming L, Gordon S. Alternative Activation of Macrophages: An Immunologic Functional Perspective. Annu Rev Immunol (2009) 27:451–83. doi: 10.1146/annurev.immunol.021908.132532
92. Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, et al. Tolerance and M2 (Alternative) Macrophage Polarization are Related Processes Orchestrated by p50 Nuclear Factor Kappab. Proc Natl Acad Sci USA (2009) 106:14978–83. doi: 10.1073/pnas.0809784106
93. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. Csf-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat Med (2013) 19:1264–72. doi: 10.1038/nm.3337
94. Kratochvill F, Neale G, Haverkamp JM, Van de Velde LA, Smith AM, Kawauchi D, et al. Tnf Counterbalances the Emergence of M2 Tumor Macrophages. Cell Rep (2015) 12:1902–14. doi: 10.1016/j.celrep.2015.08.033
95. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-beta: “N1” Versus “N2” Tan. Cancer Cell (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
96. Silveira AAA, Dominical VM, Almeida CB, Chweih H, Ferreira WA Jr, Vicente CP, et al. TNF Induces Neutrophil Adhesion Via Formin-Dependent Cytoskeletal Reorganization and Activation of Beta-Integrin Function. J Leukoc Biol (2018) 103:87–98. doi: 10.1189/jlb.3A0916-388RR
97. Lauterbach M, O’Donnell P, Asano K, Mayadas TN. Role of TNF Priming and Adhesion Molecules in Neutrophil Recruitment to Intravascular Immune Complexes. J Leukoc Biol (2008) 83:1423–30. doi: 10.1189/jlb.0607421
98. Volk AP, Barber BM, Goss KL, Ruff JG, Heise CK, Hook JS, et al. Priming of Neutrophils and Differentiated PLB-985 Cells by Pathophysiological Concentrations of TNF-alpha is Partially Oxygen Dependent. J Innate Immun (2011) 3:298–314. doi: 10.1159/000321439
99. Lianyuan T, Gang L, Ming T, Dianrong X, Chunhui Y, Zhaolai M, et al. Tumor Associated Neutrophils Promote the Metastasis of Pancreatic Ductal Adenocarcinoma. Cancer Biol Ther (2020) 21:937–45. doi: 10.1080/15384047.2020.1807250
100. Comen E, Wojnarowicz P, Seshan VE, Shah R, Coker C, Norton L, et al. TNF is a Key Cytokine Mediating Neutrophil Cytotoxic Activity in Breast Cancer Patients. NPJ Breast Cancer (2016) 2:16009. doi: 10.1038/npjbcancer.2016.9
101. Fridlender ZG, Albelda SM. Tumor-Associated Neutrophils: Friend or Foe? Carcinogenesis (2012) 33:949–55. doi: 10.1093/carcin/bgs123
102. Croft M. The Role of TNF Superfamily Members in T-cell Function and Diseases. Nat Rev Immunol (2009) 9:271–85. doi: 10.1038/nri2526
103. Chopra M, Riedel SS, Biehl M, Krieger S, von Krosigk V, Bauerlein CA, et al. Tumor Necrosis Factor Receptor 2-Dependent Homeostasis of Regulatory T Cells as a Player in TNF-induced Experimental Metastasis. Carcinogenesis (2013) 34:1296–303. doi: 10.1093/carcin/bgt038
104. Hu Y, Xiao H, Shi T, Oppenheim JJ, Chen X. Progranulin Promotes Tumour Necrosis Factor-Induced Proliferation of Suppressive Mouse CD4(+) Foxp3(+) Regulatory T Cells. Immunology (2014) 142:193–201. doi: 10.1111/imm.12241
105. Palucka K, Banchereau J. Cancer Immunotherapy Via Dendritic Cells. Nat Rev Cancer (2012) 12:265–77. doi: 10.1038/nrc3258
106. Nakae S, Suto H, Iikura M, Kakurai M, Sedgwick JD, Tsai M, et al. Mast Cells Enhance T Cell Activation: Importance of Mast Cell Costimulatory Molecules and Secreted TNF. J Immunol (2006) 176:2238–48. doi: 10.4049/jimmunol.176.4.2238
107. Aspord C, Pedroza-Gonzalez A, Gallegos M, Tindle S, Burton EC, Su D, et al. Breast Cancer Instructs Dendritic Cells to Prime Interleukin 13-Secreting CD4+ T Cells That Facilitate Tumor Development. J Exp Med (2007) 204:1037–47. doi: 10.1084/jem.20061120
108. Marigo I, Zilio S, Desantis G, Mlecnik B, Agnellini AHR, Ugel S, et al. T Cell Cancer Therapy Requires CD40-CD40L Activation of Tumor Necrosis Factor and Inducible Nitric-Oxide-Synthase-Producing Dendritic Cells. Cancer Cell (2016) 30:377–90. doi: 10.1016/j.ccell.2016.08.004
109. Fu C, Jiang A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front Immunol (2018) 9:3059. doi: 10.3389/fimmu.2018.03059
110. Bonnet D, Dick JE. Human Acute Myeloid Leukemia is Organized as a Hierarchy That Originates From a Primitive Hematopoietic Cell. Nat Med (1997) 3:730–7. doi: 10.1038/nm0797-730
111. Kusoglu A, Biray Avci C. Cancer Stem Cells: A Brief Review of the Current Status. Gene (2019) 681:80–5. doi: 10.1016/j.gene.2018.09.052
112. Storci G, Sansone P, Mari S, D’Uva G, Tavolari S, Guarnieri T, et al. Tnfalpha Up-Regulates SLUG Via the NF-kappaB/HIF1alpha Axis, Which Imparts Breast Cancer Cells With a Stem Cell-Like Phenotype. J Cell Physiol (2010) 225:682–91. doi: 10.1002/jcp.22264
113. Zhao X, Ma L, Dai L, Zuo D, Li X, Zhu H, et al. Tnfalpha Promotes the Malignant Transformation of Intestinal Stem Cells Through the NFkappaB and Wnt/betacatenin Signaling Pathways. Oncol Rep (2020) 44:577–88. doi: 10.3892/or.2020.7631
114. Yao J, Lin J, He L, Huang J, Liu Q. TNF-Alpha/miR-155 Axis Induces the Transformation of Osteosarcoma Cancer Stem Cells Independent of TP53INP1. Gene (2020) 726:144224. doi: 10.1016/j.gene.2019.144224
115. Pan Z, Tian Y, Niu G, Cao C. Role of microRNAs in Remodeling the Tumor Microenvironment (Review). Int J Oncol (2020) 56:407–16. doi: 10.3892/ijo.2019.4952
116. Wang M, Zhao C, Shi H, Zhang B, Zhang L, Zhang X, et al. Deregulated microRNAs in Gastric Cancer Tissue-Derived Mesenchymal Stem Cells: Novel Biomarkers and a Mechanism for Gastric Cancer. Br J Cancer (2014) 110:1199–210. doi: 10.1038/bjc.2014.14
117. Ding L, Gu H, Xiong X, Ao H, Cao J, Lin W, et al. Micrornas Involved in Carcinogenesis, Prognosis, Therapeutic Resistance and Applications in Human Triple-Negative Breast Cancer. Cells (2019) 8(12):1492. doi: 10.3390/cells8121492
118. Ranjan K, Pathak C. FADD Regulates NF-kappaB Activation and Promotes Ubiquitination of cFLIPL to Induce Apoptosis. Sci Rep (2016) 6:22787. doi: 10.1038/srep22787
119. Liu Y, Liu R, Yang F, Cheng R, Chen X, Cui S, et al. miR-19a Promotes Colorectal Cancer Proliferation and Migration by Targeting TIA1. Mol Cancer (2017) 16:53. doi: 10.1186/s12943-017-0625-8
120. Huang L, Wang X, Wen C, Yang X, Song M, Chen J, et al. Hsa-miR-19a is Associated With Lymph Metastasis and Mediates the TNF-alpha Induced Epithelial-to-Mesenchymal Transition in Colorectal Cancer. Sci Rep (2015) 5:13350. doi: 10.1038/srep13350
121. Zhang J, Wu H, Li P, Zhao Y, Liu M, Tang H. Nf-kappaB-modulated miR-130a Targets TNF-alpha in Cervical Cancer Cells. J Transl Med (2014) 12:155. doi: 10.1186/1479-5876-12-155
122. Balatti V, Pekarky Y, Croce CM. Role of microRNA in Chronic Lymphocytic Leukemia Onset and Progression. J Hematol Oncol (2015) 8:12. doi: 10.1186/s13045-015-0112-x
123. Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An Endotoxin-Induced Serum Factor That Causes Necrosis of Tumors. Proc Natl Acad Sci USA (1975) 72:3666–70. doi: 10.1073/pnas.72.9.3666
124. Libutti SK, Paciotti GF, Byrnes AA, Alexander HR Jr, Gannon WE, Walker M, et al. and Pharmacokinetic Studies of CYT-6091, a Novel PEGylated Colloidal gold-rhTNF Nanomedicine. Clin Cancer Res (2010) 16:6139–49. doi: 10.1158/1078-0432.CCR-10-0978
125. Nilubol N, Yuan Z, Paciotti GF, Tamarkin L, Sanchez C, Gaskins K, et al. Novel Dual-Action Targeted Nanomedicine in Mice With Metastatic Thyroid Cancer and Pancreatic Neuroendocrine Tumors. J Natl Cancer Inst (2018) 110:1019–29. doi: 10.1093/jnci/djy003
126. Libutti SK, Tamarkin L, Nilubol N. Targeting the Invincible Barrier for Drug Delivery in Solid Cancers: Interstitial Fluid Pressure. Oncotarget (2018) 9:35723–5. doi: 10.18632/oncotarget.26267
127. Fajardo LF, Kwan HH, Kowalski J, Prionas SD, Allison AC. Dual Role of Tumor Necrosis Factor-Alpha in Angiogenesis. Am J Pathol (1992) 140:539–44.
128. Cairns R, Papandreou I, Denko N. Overcoming Physiologic Barriers to Cancer Treatment by Molecularly Targeting the Tumor Microenvironment. Mol Cancer Res (2006) 4:61–70. doi: 10.1158/1541-7786.MCR-06-0002
129. Heldin CH, Rubin K, Pietras K, Ostman A. High Interstitial Fluid Pressure - an Obstacle in Cancer Therapy. Nat Rev Cancer (2004) 4:806–13. doi: 10.1038/nrc1456
130. Lienard D, Ewalenko P, Delmotte JJ, Renard N, Lejeune FJ. High-Dose Recombinant Tumor Necrosis Factor Alpha in Combination With Interferon Gamma and Melphalan in Isolation Perfusion of the Limbs for Melanoma and Sarcoma. J Clin Oncol (1992) 10:52–60. doi: 10.1200/JCO.1992.10.1.52
131. Gabizon A, Bradbury M, Prabhakar U, Zamboni W, Libutti S, Grodzinski P. Cancer Nanomedicines: Closing the Translational Gap. Lancet (2014) 384:2175–6. doi: 10.1016/S0140-6736(14)61457-4
Keywords: tumor microenvironment, tumor necrosis factor-α, transforming growth factor beta, extracellular matrix, interstitial fluid pressure
Citation: Laha D, Grant R, Mishra P and Nilubol N (2021) The Role of Tumor Necrosis Factor in Manipulating the Immunological Response of Tumor Microenvironment. Front. Immunol. 12:656908. doi: 10.3389/fimmu.2021.656908
Received: 21 January 2021; Accepted: 07 April 2021;
Published: 27 April 2021.
Edited by:
Peng Qu, National Institutes of Health (NIH), United StatesReviewed by:
Guohao Wang, National Institutes of Health (NIH), United StatesGiuseppe Murdaca, University of Genoa, Italy
Andrey Kruglov, German Rheumatism Research Center (DRFZ), Germany
Copyright © 2021 Laha, Grant, Mishra and Nilubol. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naris Nilubol, bmFyaXMubmlsdWJvbEBtYWlsLm5paC5nb3Y=