 Manuel Ruiz-Pablos
Manuel Ruiz-Pablos Bruno Paiva
Bruno Paiva Rosario Montero-Mateo3
Rosario Montero-Mateo3 Nicolas Garcia
Nicolas Garcia Aintzane Zabaleta
Aintzane Zabaleta- 1Faculty of Medicine of the European University of Madrid, Madrid, Spain
- 2Clinica Universidad de Navarra, Centro de Investigación Medica Aplicada (CIMA), IdiSNA, Instituto de Investigación Sanitaria de Navarra, Pamplona, Spain
- 3Department of Pediatrics, Hospital Clínico San Carlos, Madrid, Spain
Myalgic encephalomyelitis or chronic fatigue syndrome (ME/CFS) affects approximately 1% of the general population. It is a chronic, disabling, multi-system disease for which there is no effective treatment. This is probably related to the limited knowledge about its origin. Here, we summarized the current knowledge about the pathogenesis of ME/CFS and revisit the immunopathobiology of Epstein-Barr virus (EBV) infection. Given the similarities between EBV-associated autoimmune diseases and cancer in terms of poor T cell surveillance of cells with EBV latency, expanded EBV-infected cells in peripheral blood and increased antibodies against EBV, we hypothesize that there could be a common etiology generated by cells with EBV latency that escape immune surveillance. Albeit inconclusive, multiple studies in patients with ME/CFS have suggested an altered cellular immunity and augmented Th2 response that could result from mechanisms of evasion to some pathogens such as EBV, which has been identified as a risk factor in a subset of ME/CFS patients. Namely, cells with latency may evade the immune system in individuals with genetic predisposition to develop ME/CFS and in consequence, there could be poor CD4 T cell immunity to mitogens and other specific antigens, as it has been described in some individuals. Ultimately, we hypothesize that within ME/CFS there is a subgroup of patients with DRB1 and DQB1 alleles that could confer greater susceptibility to EBV, where immune evasion mechanisms generated by cells with latency induce immunodeficiency. Accordingly, we propose new endeavors to investigate if anti-EBV therapies could be effective in selected ME/CFS patients.
Introduction
Myalgic encephalomyelitis or chronic fatigue syndrome (ME/CFS) is a life-limiting, multi-system disease for which there is no effective treatment (1). It is characterized by unexplained disabling fatigue and a combination of unspecific symptoms that last for at least 6 months (2, 3). At least nine disease definitions have been developed. Its prevalence ranges in between 0.1% and 2.5% of the general population (4), depending on the diagnostic criteria being applied (Supplemental Table 1). The annual incidence of cases with ME/CFS in the United Kingdom is of 14.8 per 100,000 people (5).
Pathogenesis of ME/CFS
The cause of this syndrome is unknown. However, there is a growing body of evidence supporting the role of dysfunction in immune, neuro-endocrine, and autonomic systems, and several biologically based theories are currently being investigated. Hormonal alterations have been identified in some individuals with ME/CFS, whereby hypocortisolism might produce fatigue-like symptoms (1, 6–8). Lipid and energy metabolism dysfunction are also thought to contribute to the etiology of ME/CFS. Genetic predisposition or common environmental exposure to infectious or toxic agents, may be associated with some family histories with high prevalence of ME/CFS (1, 9–14).
Infectious triggering a chronic inflammatory response has long been a hypothesized risk factor for the development of ME/CFS due to the large number of individuals with a history of infection prior to the onset of symptoms. Clinical manifestations of the disease such as chronic fatigue and flu-like symptoms, may be explained by the presence immunological alterations leading to reduced cytotoxic activity and altered metabolism of natural killer (NK) cells and T lymphocytes, reduced T cell responses to mitogens and other specific antigens, or the presence of chronic low-grade systemic inflammation with increased levels of proinflammatory cytokines and oxidative stress (1, 2, 15–18).
Another hypothesis is autoimmunity due to the presence of autoantibodies against nuclear, membrane and neurotransmitter receptor structures in some patients (1, 19). This hypothesis has prompted several research groups to seek for an association between the expression of certain HLA-II alleles and the development of ME/CFS. Accordingly, HLA-DQA1*01, HLA-DQB1*06 (20), DQB1*0303 (21), HLA-DQ3, HALA-DR4 and HLA-DR5 (22) have been associated with increased risk of ME/CFS, but the robustness of the data supporting this association is limited (20, 23). Thus, the possibility that some individuals with genetic predisposition may develop ME/CFS after stimulus (e.g., infection) and subsequent autoimmunity, remains to be demonstrated (12, 20, 22). In such cases, the diagnostic criteria could take into account the pathogen involved in the disease onset, which could potentially improve patient’ stratification and management.
Interestingly, ME/CFS was first described in reference to a post Epstein-Barr fatigue. Infection with viruses such as Epstein-Barr (EBV), but also with human herpesvirus (HHV) -6, cytomegalovirus (CMV), human parvovirus B19 and enteroviruses (24–30), as well as bacterial and parasite infections (31), have been suggested as risk factors. However, infection prior to its onset is not true of all ME/CFS patients and its etiological significance remains uncertain.
Here, we revisited the immunopathobiology of EBV infection before summarizing contradictory data about a possible association between chronic EBV infection and ME/CFS in some patients. Consistent with this hypothesis, we finalized this mini review describing possible therapeutic options against EBV.
Immunopathobiology of the Epstein-Barr Virus
EBV belongs to the family of γ-herpesvirus (32). Amongst other cell types, it infects B cells of adjacent lymphoid tissues before establishing a lifelong latent infection in memory B cells (I/0 latencies). Indeed, the virus maintains a latent state as an episome without expressing viral genes, allowing B cells with latency 0 and some B cells with latency I to escape immune surveillance (Figure 1A) (33–36).
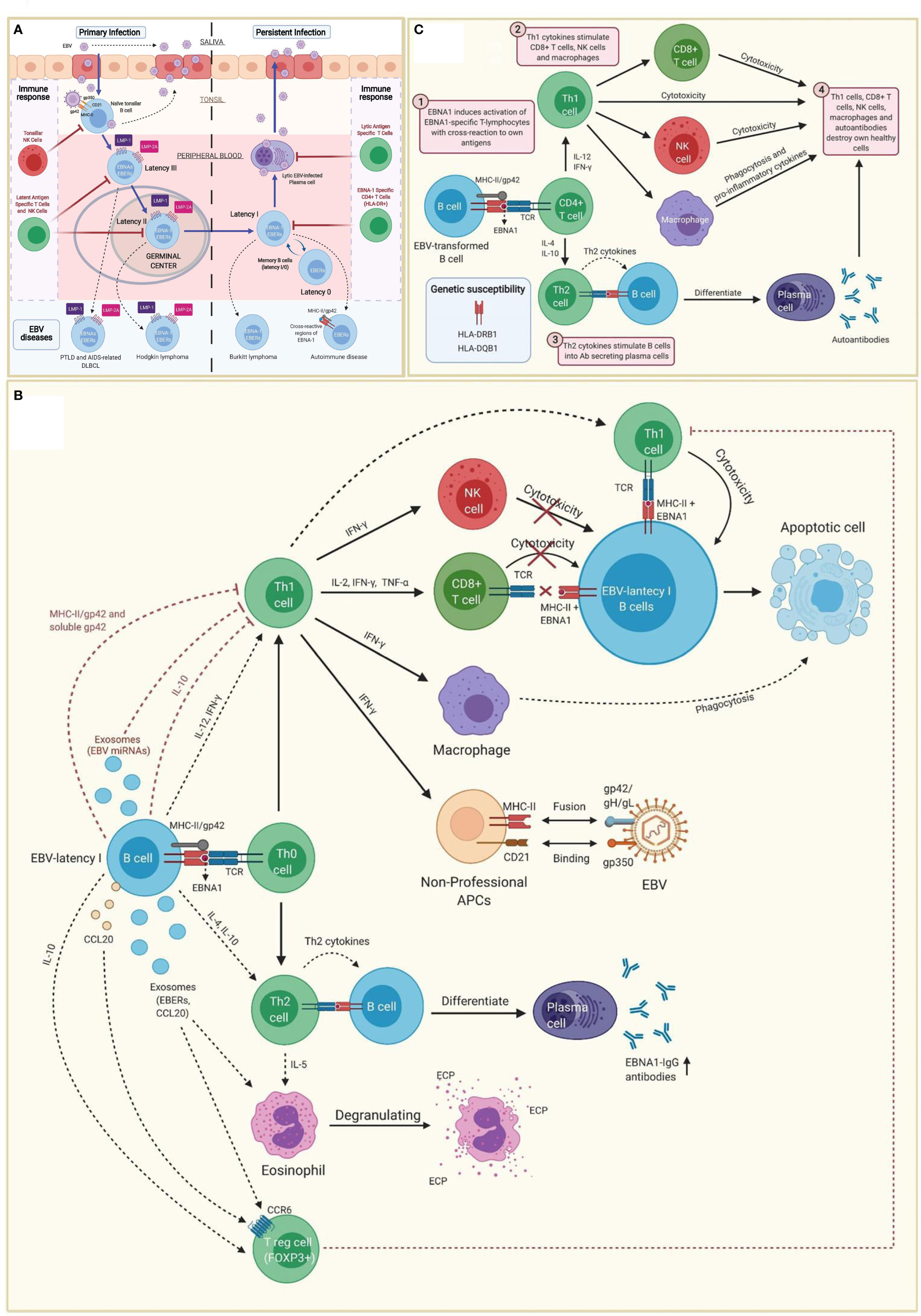
Figure 1 Immunopathobiology of Epstein-Barr virus (EBV) infection. (A) EBV is transmitted to the host through saliva from a carrier individual, and infects pharyngeal epithelial cells followed by naïve tonsillar B cells through interactions between gp350 and gp42 glycoproteins of the viral envelope with CD21 and MHC class II molecules (MHC-II), respectively. Lytic infection produces new viral particles that infect more epithelial cells. Subsequently, these EBV-infected B cells enter into a latency phase in the periphery, where they express a specific set of viral genes, including LMP1, LMP2A, EBNAs and EBER (latency III). These latent III B cells progress through the germinal center reaction into latency II and emerge as memory B cells with I/0 latencies that establish a lifelong latent infection. The immune response of the healthy host is sufficient to maintain control of the EBV infection. NK cells in tonsil produce high levels of IFN-γ that withhold the transformation of B cells by EBV during earlier stages of the infection. Both, type III and type II latent B cells are controlled by NK and T cells specific to latent proteins. By contrast, memory B cells with type I latency are only controlled by activated EBNA-1 specific CD4 T cells. EBV-infected plasma cells can periodically enter in lytic phase, but are controlled by CD4 and CD8 T cells with specificity for EBV lytic proteins. The programs of the viral latent cycle are expressed in various EBV-associated diseases. Latency I is found in Burkitt lymphoma, latency II in Hodgkin lymphoma and latency III in post-transplantation lymphoproliferative disease (PTLD) and AIDS-associated diffuse large B-cell lymphoma (DLBCL). EBV-latency I B cells escaping the surveillance of EBNA-1-specific CD4 T cells could lead to autoimmunity by presenting EBNA-1 in MHC-II/gp42, which may cause cross-reaction with own antigens. (B) EBNA1 is presented to CD4 T cells on MHC class II molecules in EBV-infected B cells. Both, MHC-II bound gp42 and soluble gp42 facilitate immune evasion by preventing activation and recognition of T cell receptors (TCR) in CD4 T cells. In addition, some EBV miRNAs could directly reduce CD4 T cell cytotoxicity through the intercellular exosomal pathway or inhibit MHC class II-mediated antigen processing and presentation in the host cell. By contrast, activation of Th1 CD4 T cells, would favor co-stimulation of CD8 T cells, NK cells and macrophages. Increased levels of IFN-γ in response to EBV infection can induce expression of MHC class II molecules in other cells such as epithelial cells, endothelial cells, pancreatic beta cells, fibroblasts, keratinocytes and glial cells, allowing them to act as non-professional antigen-presenting cells that can become infected by EBV through gp42/MHC-II interaction. If these cells also express low levels of CD21 (thymocytes, a subset of peripheral T lymphocytes, follicular dendritic cells, astrocytes and some epithelial cells), they may further facilitate EBV entry by interacting gp350 with CD21. IFN-γ released by NK cells may withhold the transformation of B cells by EBV during the early stages of infection, but it fails to inhibit the proliferation of fully transformed EBV-infected B cells (latency). CD8 T cells do not recognize EBNA1 in EBV-transformed cells, since it is presented in MHC class II molecules. Only EBNA1-specific CD4 T cells have cytotoxic activity against EBNA1-expressing B cells, causing them to enter apoptosis and become phagocyted by macrophages. However, EBV transformed B cells release IL-10, TGF-β, CCL20 and exosomes (containing EBERs and CCL20), which attract T regulatory (Treg) cells to the site of infection inhibiting antigen-stimulated CD4 effector T cells. IL-10 released by EBV transformed B cells during EBNA1 presentation, favors a Th2 (over Th1) immune response that also inhibits CD8 T cells and NK cells. These EBNA1 specific Th2 cells further induce antibody secretion by plasma cells. EBERs released on exosomes can activate other cells such as eosinophils that degranulate and release the cationic eosinophilic protein (ECP). (C) EBNA1 is one of the main candidates in the generation of autoantibodies and EBNA1 specific self-reactive cytotoxic Th1 cells in genetically predisposed individuals. EBNA-1 is subjected to citrullination and presented in MHC class II molecules after macroautophagy in EBV-infected B cells. As gp42 binds peripherally to the β1 domain of the β chain of HLA-DR -DQ, it may confer greater susceptibility or resistance to this interaction, depending on the host DRB1* and DQB1* allele. Post-translational modifications, such as citrullination, may form neoantigens that can generate autoimmunity when recognized by CD4 T cells. If polarized into a Th2 phenotype, CD4 T cells will stimulate B cell differentiation into plasma cells that could secrete autoantibodies. Antigen-specific CD4 T cells with a Th1 cytokine pattern may have cytotoxic activity, apart from co-stimulating CD8 T cells, NK cells and macrophages. Together with autoantibodies produced by plasma cells, all these cell types would participate in the destruction of healthy cells and the development of autoimmunity.
Efficient B cell infection by EBV requires up to 5 envelope glycoproteins, whereby gp42 ultimately promotes entry of the virus into B cells through the interaction with host MHC class II molecules (37, 38). Gp42 binds to β1 domain of the β chain of HLA-DR -DQ, or –DP and blocks TCR-HLA-DR interactions impairing antigen presentation (37–39). Hence, gp42-expressing B cells show reduced ability to activate CD4 T cells (37). Furthermore, gp42 can be presented as a membrane protein bound to MHC-II molecules or in a soluble form (s-gp42). Both have been detected during the lytic phase in Burkitt lymphoma (latency I), suggesting that soluble gp42 is generated during EBV lytic infection and inhibits the presentation of HLA-II restricted antigens to T cells (40). Noteworthy, EBNA-1 specific cytotoxic CD4 T cells are decreased in patients with post-transplant lymphoproliferative disorders (41), in some pediatric forms of Burkitt lymphoma (42, 43), in EBV-positive lymphoma (44), in lymphomas associated with HIV infection (45), and in lymphoma infiltrating the central nervous system (45). By contrast, the immune response in healthy individuals is sufficient to control EBV infection (35, 46).
Genetic Predisposition to EBV Infection
The diversity of human leukocyte antigen (HLA) molecules results from selective pressure during co-evolution with pathogens (47, 48). A characteristic of HLA diversity is the long-term persistence of allelic lineages, which causes trans-species polymorphisms to be shared among closely related species (48). In humans, there are 13 allelic lineages of DRB1 (48) and, according to the phylogenetic relationship between the different DRB genes of primates (hominoids, New World and Old World monkeys) described by Bontrop et al, the DRB1*04, *03 and *02 lineages are the oldest, with the DRB1*04 lineage being the most ancestral (49). Since EBV is the only human-adapted member of the genus Lymphocryptovirus, transferred to a hominid ancestor (50), it could be hypothesized that immune evasion mechanisms of the EBV have more effectively evolved among older allelic lineages of DRB1. Such an hypothesis could help explaining why individuals with haplotypes DR2-DQ6, DR3-DQ2 or DR4-DQ8 are less resistant to EBV infection and are at greater risk of developing EBV-related disorders (Supplemental Table 2) (34).
Other genetic predisposition factors to consider are glutamic acid 46 (E46) and arginine 72 (R72) from HLA class II molecules (51). R72 is fully preserved in the HLA-DR, HLA-DQ and -DP sequences (52); by contrast, E46 is conserved in all HLA-DR,-DP alleles and only in a small subset of HLA-DQ alleles β * 02 (β * 0201, β * 0202 and β * 0203) (53). This suggests that individuals with HLA-DQ alleles β * 02 may have higher susceptibility to infection in tissues with cells expressing only HLA-DQ, as well as a higher rate of EBV infection in those cells expressing different HLA class II isotypes along with HLA-DQ β * 02 (53).
EBV-Associated Diseases
EBV is present in more than 90% of the population and the decreased ability of the immune system to control/eliminate EBV infection may be responsible for causing EBV-associated diseases (35, 54) such as rheumatoid arthritis (34, 55, 56), systemic lupus erythematosus (34, 57, 58), Sjögren’s syndrome (34), multiple sclerosis (59–61), myasthenia gravis (62), diabetes mellitus type 1 (53), fulminant type diabetes (63), celiac disease (64), autoimmune thyroiditis (65, 66), Hodgkin and non-Hodgkin lymphoma (35, 67) (Supplemental Table 2).
Tissue infiltrating B cells with EBV latency in a genetically predisposed individual, would trigger the activation of virus-specific IFN-γ producing Th1 CD4 T cells. High IFN-γ levels would induce the upregulation of MHC class II molecules (Figure 1B) on the surface of different cell types (e.g., epithelial cells, endothelial cells, pancreatic beta cells, fibroblasts, keratinocytes, glial cells, etc.), allowing them to function like non-professional antigen-presenting cells. Likewise, MHC class II upregulation would improve the infection capacity of EBV through the interaction with gp42 (68, 69). Co-expression of MHC-II molecules and CD21 (e.g., thymocytes, a subset of peripheral T lymphocytes, follicular dendritic cells, astrocytes and some epithelial cells) would further increase the risk of EBV infection through the interaction between gp350 and CD21 (69). These mechanisms have been hypothesized in patients with autoimmune thyroiditis with EBV transformed B cell infiltrate in the thyroid tissue (65, 66). The same principle could potentially apply to the intestinal mucosa in celiac disease (70), to the pancreatic islets in type 1 diabetes (53), to the central nervous system and multiple sclerosis (60, 71), to the exocrine glands and the Sjögren’s syndrome (34, 72), to the thymus and myasthenia gravis (62), as well as to the synovial joints and rheumatoid arthritis (38, 73, 74). Can the same principle be applied to a subgroup of patients with ME/CFS?
A Hypothetical Association Between EBV and ME/CFS
As mentioned above, EBV infection has been identified as a risk factor in a subgroup of ME/CFS patients (3, 28, 75). There are studies showing a statistically significant elevation of anti-EBV-dUTPase antibodies (75, 76), a defective EBV-specific B and T cell response (77), a high rate of active EBV infection (28), serologic evidence of EBV reactivation with elevated IgM antibodies against late VCA antigen (78–80), and a positive up-regulation of EBV-induced gene 2 (EBI2) mRNA in peripheral blood mononuclear cells (PBMC) (3) from a subgroup of patients with ME/CFS. However, serological observations related to EBV were not always confirmed and, accordingly, the association between EBV infection and ME/CFS is not established (81–85). Furthermore, the presence of an active EBV infection in a subgroup of ME/CFS patients has been actively debated, because most studies revealed no increase in EBV viral load in ME/CFS patients. If a third state of virus, defined as abortive/lytic/leaky replication (86) could explain the presence of certain EBV proteins (BRRF1 and BLLF3) with the potential capacity to contribute to the symptomatology of ME/CFS (82, 87), is an hypothesis that remains unconfirmed.
Multiple studies in patients with ME/CFS have demonstrated decreased cytotoxic activity of NK cells, increased IL-10 levels and augmented Th2 response. Also an expansion of Tregs and impaired T cell response to mitogens and other specific antigens (1, 2, 12, 16, 88–91). Based on the mechanisms of immune-escape developed by latency I (EBNA-1) cells, established after primary EBV infection, a cause-effect association between EBV infection and the disease onset in a subgroup of ME/CFS patients, could be hypothesized. Accordingly, IL-10 released by both EBV-transformed B cells and Tregs would favor a Th2 type immune response (43), and gp42-mediated disruption of TCR-MHC-II interaction would further decrease CD4 T cell activation, leading to poor CD4 T cell immunity to mitogens and other specific antigens, which have been described in some patients with ME/CFS (2). All these immune evasion mechanisms triggered by latent cells induce an immunodeficiency that allows EBV-transformed B cells, especially EBV latent I cells, to escape from immune surveillance. This could potentially help explaining the reduced EBNA-1-specific CD4 T-cell response, increased EBV latent cells (77), and increased EBV abortive lytic replication (EBV dUTPase in exosomes) (75), observed in a subgroup of patients with ME/CFS. If the infiltration and proliferation of EBV-transformed B cells in the intestinal mucosa is related to the chronic inflammation that is observed in some ME/CFS patients (15, 92–94), remains unknown. However, the results described above and the hypotheses we generated accordingly, must be interpreted with caution because there are numerous other studies that failed to reproduce the findings of decreased cytotoxic activity of NK cells, increased IL-10 levels and augmented Th2 response (83, 89, 90, 95–97). These discrepancies may be due to disease heterogeneity, different degrees of ME/CFS severity and duration, and different analytical methods (15, 28, 89, 97–99).
If the hypotheses described above were to be true, the subgroup of ME/CFS patients with EBV infection could have increased risk of developing autoimmune diseases (19, 100, 101), latent viral reactivations (e.g., herpesviruses, Parvovirus B19) and cancer (83, 102–107), namely EBV-associated lymphoma (108). Additionally, HLA-II alleles associated with increased EBV susceptibility may also explain the higher prevalence of cancer and autoimmune disorders (Figure 1C), such as rheumatoid arthritis and type 1 diabetes, in first-degree relatives of patients with ME/CFS (15, 19, 100, 109, 110). Notwithstanding, the high prevalence of EBV among the general population hinders the identification of this putative subset of patients who developed ME/CFS following infection. This is in agreement with EBV serological assessments performed in patients and healthy individuals, from where no conclusive results have been observed (85). Similarly, most studies reported no increase in EBV viral load in patients with ME/CFS (84, 111). If this is due to the fact that the possible trigger of some EBV-associated diseases are the EBV-latent cells rather than viral load (34, 35, 43, 63, 66, 67, 112–114), remains unknown in ME/CFS. Thus, to our knowledge, there is scarce evidence based in prospective cohort studies, describing rates and associations with post-infective fatigue syndrome (i.e., ME/CFS) following proven acute EBV.
Therapeutic Options Against EBV: Is There a Role in ME/CFS?
Limited knowledge about the origin of ME/CFS has hampered the development of effective treatment. Current strategies include administering nutritional supplements to overcome deficiencies and symptomatic treatment with analgesics, steroids or antidepressants (Supplemental Table 3) (1, 115). Thus, if a putative association between EBV infection and the onset of ME/CFS exists, the development of biomarkers that could identify patients in whom this may occur, would create a window of opportunity for tailored treatment against EBV.
B-cell depletion using several infusions of rituximab over 12 months was not associated with clinical improvement in patients with ME/CFS (116). Similarly, results of trials using antivirals have been inconclusive and, in some cases, contradictory (115). Accordingly, anecdotal observations of the resolution of symptoms in some ME/CFS patients with elevated levels of anti-EBV antibodies, after treatment with rituximab (100, 117) or antivirals (e.g., valaciclovir and valgancinclovir) (118, 119), have not been confirmed in large series. If B-cell depletion agents and antivirals were poised to show greater efficacy if used in a putative subgroup of patients in whom acute EBV triggered ME/CFS, remains obviously unknown. Furthermore there are no effective treatments to eradicate EBV latency in patients with EBV-associated disorders (120–122). Antiviral agents do not eradicate latent cells (120, 121), while rituximab does not remove all EBV-infected cells nor restores cellular immunity against EBV. Moreover, it induces further immunosuppression by targeting CD20 positive infected and uninfected healthy B cells (120, 122, 123). The combination of antiviral agents or intravenous immunoglobulins with rituximab can be used to treat EBV-associated disorders (121), but merely prolongs time until next relapse, since the combination of both therapies does not completely eliminate EBV infection in genetically predisposed hosts.
Other treatments such as EBV-specific adoptive T cell immunotherapy could potentially yield some benefit (124), since EBNA-1 is expressed during the lytic cycle, as well as by almost all EBV-infected cells, with the exception of those with latency 0 (125–127). However, EBNA-1 is poorly immunogenic and is not always presented in MHC class II molecules of B cells with EBV latency (46); thus, these cells (especially those with latency I) could escape EBNA-1-specific T cell immunotherapy. Another alternative proposed by Dalton et al. is the use of low doses of DNA demethylation agents (e.g., decitabine and 5-azacitidine) during a short period (Figure 2A) for the treatment of latency I EBV-associated lymphoma, which induces the transformation of latent I B-cells into latent II and III, thereby allowing EBV-specific T cells to recognize them due to increased expression of immunogenic viral antigens (i.e., LMP1, EBNA2, EBNA3A and EBNA3C) (128). Such an effect could even persist after treatment interruption (128). Furthermore, DNA demethylation agents may restore the expression of HLA-II molecules in EBV-transformed lymphoblastoid cell lines (129, 130) and induce apoptosis in EBV-transformed epithelial cells (Figure 2B) (131, 132). Therefore, if our hypothesis about the presence of an acquired immunodeficiency after EBV infection in genetically predisposed individuals with EBV-associated diseases holds true, it could be speculated that low-dose DNA demethylation agents for short-term treatment followed by EBV-specific T cell immunotherapy (128) and antiviral agents (because of the increased lytic infection by the use of DNA demethylation agents) could be beneficial. Such an approach may also be potentially useful in cases where ME/CFS develops from other viruses such as the HHV-6, CMV and human parvovirus B19 (24–29), since these DNA viruses use CpG DNA methylation as an immune evasion mechanism. Namely, the viral genome is largely methylated during the latency phase thereby preventing viral expression and genome replication; instead, it is restored to a non-methylated state during the lytic phase during latency, which is restored to a non-methylated state during the lytic phase (133–136).
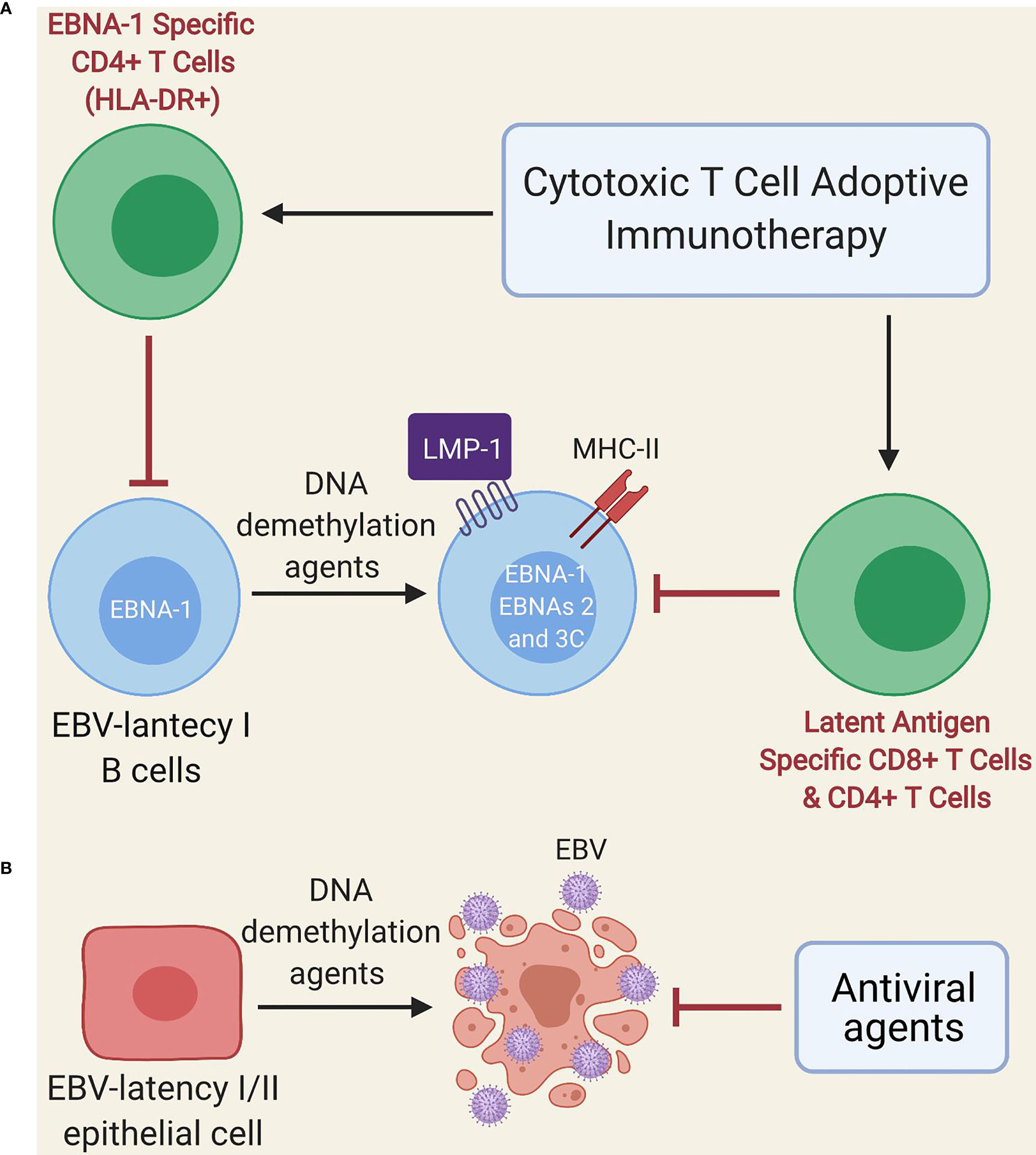
Figure 2 Schematic model of treatment with DNA demethylation agents followed by adoptive immunotherapy of EBV-specific T cells. (A) Administration of low-dose DNA demethylation agents (e.g. decitabine) restores the expression of MHC class II molecules and induces expression of LMP1, EBNA-2 and EBNA-3C in EBV-latency I B cells, improving the recognition of these cells by EBV-specific T cells (either autogenous or after adoptive immunotherapy). EBNA-1-specific CD4 T cells can only recognize latent I cells exhibiting EBNA-1 in MHC class II molecules since EBNA-1 is poorly immunogenic. (B) DNA demethylation agents (e.g. decitabine) induce lytic infection and apoptosis in EBV-transformed epithelial cells. Antiviral agents prevent viral replication.
Conclusions and Future Directions
If the link between EBV infection and ME/CFS could be demonstrated, it would warrant future research endeavors on a potential association between decreased activation of CD4 T cells and HLA class II alleles with greater predisposition to EBV infection. Such biomarkers could help to better define a hypothetical subgroup of patients with EBV-associated ME/CFS. Additional research about the efficacy of anti-EBV therapies in these patients would then be warranted. The identification of successful treatment would potentially prevent the development of this or other diseases associated with this pathogen. Interestingly, a recent prospective study investigating risk factors for developing ME/CFS in college students following infectious mononucleosis, found that those who developed ME/CFS had more physical symptoms and immune irregularities at baseline (137). If the association between the expression of certain HLA-II alleles and higher susceptibility to develop ME/CFS exists, individuals with post infectious mononucleosis fatigue syndrome could be an interesting group to study and test the hypothesis discussed in this review.
Author Contributions
All the authors listed have made a substantial, direct, and intellectual contribution to this manuscript and have approved it for publication.
Funding
Ramsay Award Program 2019 Cycle from the Solve ME/CFS Initiative.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to the Solve ME/CFS initiative and to the University of Navarra for having supported and trusted our team for research on Myalgic Encephalomyelitis or Chronic Fatigue Syndrome. All Figures were Created with BioRender.com.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.656797/full#supplementary-material
References
1. Cortes Rivera M, Mastronardi C, Silva-Aldana C, Arcos-Burgos M, Lidbury B. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Comprehensive Review. Diagnostics (2019) 9(3):91.
2. Lorusso L, Mikhaylova SV, Capelli E, Ferrari D, Ngonga GK, Ricevuti G. Immunological Aspects of Chronic Fatigue Syndrome. Autoimmun Rev Elsevier (2009) 8:287–91. doi: 10.1016/j.autrev.2008.08.003
3. Kerr JR. Epstein-Barr Virus Induced Gene-2 Upregulation Identifies a Particular Subtype of Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. Front Pediatrics Front Media SA (2019) 7:59. doi: 10.3389/fped.2019.00059
4. Bakken IJ, Tveito K, Gunnes N, Ghaderi S, Stoltenberg C, Trogstad L, et al. Two Age Peaks in the Incidence of Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: A Population-Based Registry Study From Norway 2008-2012. BMC Med (2014) 12(1):167. doi: 10.1186/s12916-014-0167-5
5. Collin SM, Bakken IJ, Nazareth I, Crawley E, White PD. Trends in the Incidence of Chronic Fatigue Syndrome and Fibromyalgia in the UK, 2001–2013: A Clinical Practice Research Datalink Study. J R Soc Med (2017) 110(6):231–44. doi: 10.1177/0141076817702530
6. Torres-Harding S, Sorenson M, Jason L, Maher K, Fletcher MA, Reynolds N, et al. The Associations Between Basal Salivary Cortisol and Illness Symptomatology in Chronic Fatigue Syndrome. J Appl Biobehav Res (2008) 13(3):157–80.
7. Tak LM, Cleare AJ, Ormel J, Manoharan A, Kok IC, Wessely S, et al. Meta-Analysis and Meta-Regression of Hypothalamic-Pituitary-Adrenal Axis Activity in Functional Somatic Disorders. Biol Psychol (2011) 87(2):183–94. doi: 10.1016/j.biopsycho.2011.02.002
8. Morris G, Anderson G, Maes M. Hypothalamic-Pituitary-Adrenal Hypofunction in Myalgic Encephalomyelitis (ME)/Chronic Fatigue Syndrome (CFS) as a Consequence of Activated Immune-Inflammatory and Oxidative and Nitrosative Pathways. Mol Neurobiol (2017) 54(9):6806–19. doi: 10.1007/s12035-016-0170-2
9. Underhill RA, O’gorman R. Prevalence of Chronic Fatigue Syndrome and Chronic Fatigue Within Families of CFS Patients. J Chronic Fatigue Syndr (2011) 13: (1):3–13. doi: 10.1300/J092v13n01_02
10. Walsh CM, Zainal N, Middleton SJ, Paykel ES. A Family History Study of Chronic Fatigue Syndrome. Psychiatr Genet (2001) 11(3):123–8. doi: 10.1097/00041444-200109000-00003
11. Dibble JJ, McGrath SJ, Ponting CP. Genetic Risk Factors of ME/CFS: A Critical Review. Hum Mol Genet (2020) 29(R1):R117–24. doi: 10.1093/hmg/ddaa169
12. Underhill RA. Myalgic Encephalomyelitis, Chronic Fatigue Syndrome: An Infectious Disease. Med Hypotheses (2015) 85(6):765–73. doi: 10.1016/j.mehy.2015.10.011
13. Chu L, Valencia IJ, Garvert DW, Montoya JG. Onset Patterns and Course of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front Pediatr (2019) 7(FEB). doi: 10.3389/fped.2019.00012
14. Albright F, Light K, Light A, Bateman L, Cannon-Albright LA. Evidence for a Heritable Predisposition to Chronic Fatigue Syndrome. BMC Neurol (2011) 11(1):1–6. doi: 10.1186/1471-2377-11-62
15. Mandarano AH, Maya J, Giloteaux L, Peterson DL, Maynard M, Gottschalk CG, et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients Exhibit Altered T Cell Metabolism and Cytokine Associations. J Clin Invest (2020) 130(3):1491–505. doi: 10.1172/JCI132185
16. Brenu EW, van Driel ML, Staines DR, Ashton KJ, Ramos SB, Keane J, et al. Immunological Abnormalities as Potential Biomarkers in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. J Transl Med (2011) 9(1):81. doi: 10.1186/1479-5876-9-81
17. Nguyen T, Johnston S, Clarke L, Smith P, Staines D, Marshall-Gradisnik S. Impaired Calcium Mobilization in Natural Killer Cells From Chronic Fatigue Syndrome/Myalgic Encephalomyelitis Patients Is Associated With Transient Receptor Potential Melastatin 3 Ion Channels. Clin Exp Immunol (2017) 187(2):284. doi: 10.1111/cei.12882
18. Marshall-Gradisnik S, Huth T, Chacko A, Johnston S, Smith P, Staines D. Natural Killer Cells and Single Nucleotide Polymorphisms of Specific Ion Channels and Receptor Genes in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Appl Clin Genet (2016) 9:39–47. doi: 10.2147/TACG.S99405
19. Sotzny F, Blanco J, Capelli E, Castro-Marrero J, Steiner S, Murovska M, et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome – Evidence for an Autoimmune Disease. Autoimmun Rev (2018) 17(6):601–9. doi: 10.1016/j.autrev.2018.01.009
20. Smith J, Fritz EL, Kerr JR, Cleare AJ, Wessely S, Mattey DL. Association of Chronic Fatigue Syndrome With Human Leucocyte Antigen Class II Alleles. J Clin Pathol (2005) 58(8):860–3. doi: 10.1136/jcp.2004.022681
21. Lande A, Fluge Ø, Strand EB, Flåm ST, Sosa DD, Mella O, et al. Human Leukocyte Antigen Alleles Associated With Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Sci Rep (2020) 10(1)):5267. doi: 10.1038/s41598-020-62157-x
22. Keller RH, Lane JL, Klimas N, Reiter WM, Fletcher MA, van Riel F, et al.Association between HLA Class II Antigens and the Chronic Fatigue Immune Dysfunction Syndrome. Clin Infect Dis (2021) 18 (Suppl 1):S154–6.
23. Underhill JA, Mahalingam M, Peakman M, Wessely S. Lack of Association Between HLA Genotype and Chronic Fatigue Syndrome. Eur J Immunogenet (2001) 28(3):425–8. doi: 10.1046/j.1365-2370.2001.00235.x
24. Carruthers BM, Van de Sande MI, De Meirleir KL, Klimas NG, Broderick G, Mitchell T, et al. Myalgic Encephalomyelitis: International Consensus Criteria. J Internal Med (2011) 270:327–38. doi: 10.1111/j.1365-2796.2011.02428.x
25. Dubois RE, Seeley JK, Brus I, Sakamoto K, Ballow M, Harada S, et al. Chronic Mononucleosis Syndrome. South Med J (1984) 77(11):1376–82. doi: 10.1097/00007611-198411000-00007
26. Jacobson SK, Daly JS, Thorne GM, McIntosh K. Chronic Parvovirus B19 Infection Resulting in Chronic Fatigue Syndrome: Case History and Review. Clin Infect Dis (1997) 24(6):1048–51. doi: 10.1086/513627
27. Ortega-Hernandez O-D, Shoenfeld Y. Infection, Vaccination, and Autoantibodies in Chronic Fatigue Syndrome, Cause or Coincidence? Ann N Y Acad Sci (2009) 1173(1):600–9. doi: 10.1111/j.1749-6632.2009.04799.x
28. Shikova E, Reshkova V, Kumanova А, Raleva S, Alexandrova D, Capo N, et al. Cytomegalovirus, Epstein-Barr Virus, and Human Herpesvirus-6 Infections in Patients With Myalgic Еncephalomyelitis/Chronic Fatigue Syndrome. J Med Virol (2020) 92(12):3682–8. doi: 10.1002/jmv.25744
29. Schreiner P, Harrer T, Scheibenbogen C, Lamer S, Schlosser A, Naviaux RK, et al. Human Herpesvirus-6 Reactivation, Mitochondrial Fragmentation, and the Coordination of Antiviral and Metabolic Phenotypes in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. ImmunoHorizons (2020) 4(4):201–15. doi: 10.4049/immunohorizons.2000006
30. O’Neal AJ, Hanson MR. The Enterovirus Theory of Disease Etiology in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Critical Review. Front Med (2021) 0:908.
31. Bested AC, Marshall LM. Review of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: An Evidence-Based Approach to Diagnosis and Management by Clinicians. Rev Environ Health Walter Gruyter GmbH (2015) 30:223–49. doi: 10.1515/reveh-2015-0026
32. Tzellos S, Farrell P. Epstein-Barr Virus Sequence Variation—Biology and Disease. Pathogens (2012) 1(2):156–74. doi: 10.3390/pathogens1020156
33. Khammissa RAG, Fourie J, Chandran R, Lemmer J, Feller L. Epstein-Barr Virus and Its Association With Oral Hairy Leukoplakia: A Short Review. Int J Dentistry Hindawi Publishing Corporation (2016) 2016:4941783. doi: 10.1155/2016/4941783
34. Draborg AH, Duus K, Houen G. Epstein-Barr Virus in Systemic Autoimmune Diseases. Clin Dev Immunol (2013) 2013:535738. doi: 10.1155/2013/535738
35. Hatton OL, Harris-Arnold A, Schaffert S, Krams SM, Martinez OM. The Interplay Between Epstein-Barr Virus and B Lymphocytes: Implications for Infection, Immunity, and Disease. Immunol Res (2014) 58(2–3):268–76. doi: 10.1007/s12026-014-8496-1
36. Heath E, Begue-Pastor N, Chaganti S, Croom-Carter D, Shannon-Lowe C, Kube D, et al. Epstein-Barr Virus Infection of Naïve B Cells In Vitro Frequently Selects Clones With Mutated Immunoglobulin Genotypes: Implications for Virus Biology. Sugden B, Editor. PloS Pathog (2012) 8(5):e1002697. doi: 10.1371/journal.ppat.1002697
37. Ressing ME, Van Leeuwen D, Verreck FAW, Gomez R, Heemskerk B, Toebes M, et al. Interference With T Cell Receptor-HLA-DR Interactions by Epstein-Barr Virus Gp42 Results in Reduced T Helper Cell Recognition. Proc Natl Acad Sci USA (2003) 100(20):11583–8. doi: 10.1073/pnas.2034960100
38. Trier N, Izarzugaza J, Chailyan A, Marcatili P, Houen G. Human MHC-II With Shared Epitope Motifs Are Optimal Epstein-Barr Virus Glycoprotein 42 Ligands—Relation to Rheumatoid Arthritis. Int J Mol Sci (2018) 19(1):317.
39. Spriggs MK, Armitage RJ, Comeau MR, Strockbine L, Farrah T, Macduff B, et al. The Extracellular Domain of the Epstein-Barr Virus BZLF2 Protein Binds the HLA-DR Beta Chain and Inhibits Antigen Presentation. J Virol (1996) 70(8):5557–63. doi: 10.1128/jvi.70.8.5557-5563.1996
40. Ressing ME, van Leeuwen D, Verreck FAW, Keating S, Gomez R, Franken KLMC, et al. Epstein-Barr Virus Gp42 Is Posttranslationally Modified To Produce Soluble Gp42 That Mediates HLA Class II Immune Evasion. J Virol (2005) 79(2):841–52. doi: 10.1128/JVI.79.2.841-852.2005
41. Jones K, Nourse JP, Morrison L, Nguyen-Van D, Moss DJ, Burrows SR, et al. Expansion of EBNA1-Specific Effector T Cells in Posttransplantation Lymphoproliferative Disorders. Blood (2010) 116(13):2245–52. doi: 10.1182/blood-2010-03-274076
42. Moormann AM, Heller KN, Chelimo K, Embury P, Ploutz-Snyder R, Otieno JA, et al. Children With Endemic Burkitt Lymphoma Are Deficient in EBNA1-Specific IFN-γ T Cell Responses. Int J Cancer (2009) 124(7):1721–6. doi: 10.1002/ijc.24014
43. Futagbi G, Gyan B, Nunoo H, Tetteh J, Welbeck J, Renner L, et al. High Levels of IL-10 and CD4+CD25hi+ Treg Cells in Endemic Burkitt’s Lymphoma Patients. Biomedicines (2015) 3(3):224–36. doi: 10.3390/biomedicines3030224
44. Heller KN, Arrey F, Steinherz P, Portlock C, Chadburn A, Kelly K, et al. Patients With Epstein Barr Virus-Positive Lymphomas Have Decreased CD4 + T-Cell Responses to the Viral Nuclear Antigen 1. Int J Cancer (2008) 123(12):2824–31. doi: 10.1002/ijc.23845
45. Piriou E, Van Dort K, Nanlohy NM, Van Oers MHJ, Miedema F, Van Baarle D. Loss of EBNA1-Specific Memory CD4+ and CD8+ T Cells in HIV-Infected Patients Progressing to AIDS-Related Non-Hodgkin Lymphoma. Blood (2005) 106(9):3166–74. doi: 10.1182/blood-2005-01-0432
46. Leung CS, Haigh TA, Mackay LK, Rickinson AB, Taylor GS. Nuclear Location of an Endogenously Expressed Antigen, EBNA1, Restricts Access to Macroautophagy and the Range of CD4 Epitope Display. Proc Natl Acad Sci USA (2010) 107(5):2165–70. doi: 10.1073/pnas.0909448107
47. Meyer D, Vitor VR, Bitarello BD, Débora DY, Nunes K. A Genomic Perspective on HLA Evolution. Immunogenetics Springer Verlag (2018) 70:5–27. doi: 10.1007/s00251-017-1017-3
48. Yasukochi Y, Satta Y. A Human-Specific Allelic Group of the MHC DRB1 Gene in Primates. J Physiol Anthropol (2014) 33(1):14. doi: 10.1186/1880-6805-33-14
49. Bontrop RE, Otting N, De Groot NG, Doxiadis GGM. Major Histocompatibility Complex Class II Polymorphisms in Primates. Immunol Rev (1999) 167:339–50. doi: 10.1111/j.1600-065X.1999.tb01403.x
50. Ehlers B, Spieß K, Leendertz F, Peeters M, Boesch C, Gatherer D, et al. Lymphocryptovirus Phylogeny and the Origins of Epstein-Barr Virus. J Gen Virol (2010) 91(3):630–42. doi: 10.1099/vir.0.017251-0
51. McShane MP, Mullen MM, Haan KM, Jardetzky TS, Longnecker R. Mutational Analysis of the HLA Class II Interaction With Epstein-Barr Virus Glycoprotein 42. J Virol (2003) 77(13):7655–62. doi: 10.1128/JVI.77.13.7655-7662.2003
52. Mullen MM, Haan KM, Longnecker R, Jardetzky TS. Structure of the Epstein-Barr Virus Gp42 Protein Bound to the MHC Class II Receptor HLA-Dr1. Mol Cell (2002) 9(2):375–85. doi: 10.1016/S1097-2765(02)00465-3
53. Haan KM, Longnecker R. Coreceptor Restriction Within the HLA-DQ Locus for Epstein-Barr Virus Infection. Proc Natl Acad Sci USA (2000) 97(16):9252–7. doi: 10.1073/pnas.160171697
54. Hislop AD, Taylor GS. T-Cell Responses to EBV. Curr Topics Microbiol Immunol (2015) 325–53. doi: 10.1007/978-3-319-22834-1_11
55. Magnusson M, Brisslert M, Zendjanchi K, Lindh M, Bokarewa MI. Epstein–Barr Virus in Bone Marrow of Rheumatoid Arthritis Patients Predicts Response to Rituximab Treatment. Rheumatology (2010) 49(10):1911–9. doi: 10.1093/rheumatology/keq159
56. Franssila R, Hedman K. Viral Causes of Arthritis. Best Pract Research: Clin Rheumatol (2006) 20:1139–57. doi: 10.1016/j.berh.2006.08.007
57. Draborg A, Izarzugaza JMG, Houen G. How Compelling Are the Data for Epstein–Barr Virus Being a Trigger for Systemic Lupus and Other Autoimmune Diseases? Curr Opin Rheumatol (2016) 28(4):398–404. doi: 10.1097/BOR.0000000000000289
58. Draborg AH, Duus K, Houen G. Epstein-Barr Virus and Systemic Lupus Erythematosus. Clin Dev Immunol (2012) 2012 p. 370516. doi: 10.1155/2012/370516
59. Ascherio A, Munger KL, Lennette ET, Spiegelman D, Hernán MA, Olek MJ, et al. Epstein-Barr Virus Antibodies and Risk of Multiple Sclerosis: A Prospective Study. J Am Med Assoc (2001) 286(24):3083–8. doi: 10.1001/jama.286.24.3083
60. Guan Y, Jakimovski D, Ramanathan M, Weinstock-Guttman B, Zivadinov R. The Role of Epstein-Barr Virus in Multiple Sclerosis: From Molecular Pathophysiology to In Vivo Imaging. Neural Regener Res (2019) 14(3):373.
61. Xiao D, Ye X, Zhang N, Ou M, Guo C, Zhang B, et al. A Meta-Analysis of Interaction Between Epstein-Barr Virus and HLA-DRB1∗1501 on Risk of Multiple Sclerosis. Sci Rep (2015) 5(1):18083.
62. Cavalcante P, Marcuzzo S, Franzi S, Galbardi B, Maggi L, Motta T, et al. Epstein-Barr Virus in Tumor-Infiltrating B Cells of Myasthenia Gravis Thymoma: An Innocent Bystander or an Autoimmunity Mediator? Oncotarget (2017) 8(56):95432–49. doi: 10.18632/oncotarget.20731
63. Fujiya A, Ochiai H, Mizukoshi T, Kiyota A, Shibata T, Suzuki A, et al. Fulminant Type 1 Diabetes Mellitus Associated With a Reactivation of Epstein-Barr Virus That Developed in the Course of Chemotherapy of Multiple Myeloma. J Diabetes Investig (2010) 1(6):286–9. doi: 10.1111/j.2040-1124.2010.00061.x
64. Harley JB, Chen X, Pujato M, Miller D, Maddox A, Forney C, et al. Transcription Factors Operate Across Disease Loci, With EBNA2 Implicated in Autoimmunity. Nat Genet (2018) 50(5):699–707. doi: 10.1038/s41588-018-0102-3
65. Stiefel P, Aparicio R, Dolores Nieto M, Alfaro V. Infección Por El Virus De Epstein-Barr Y Enfermedad De Graves-Basedow: ¿Simple Casualidad O Algo Más? Med Clin (Barc) (2006) 126(7):278–9. doi: 10.1157/13085286
66. Janegova A, Janega P, Rychly B, Kuracinova K, Babal P. The Role of Epstein-Barr Virus Infection in the Development of Autoimmune Thyroid Diseases. Endokrynol Pol (2015) 66(2):132–6. doi: 10.5603/EP.2015.0020
67. Beltramino M, Calmet R, Gatica Valdés M. Virus De Epstein-Barr Y Su Relación Con El Desarrollo De Enfermedades Linfoproliferativas. Hematología (2005) 9(2):39–54.
68. Ghasemi F, Tessier TM, Gameiro SF, Maciver AH, Cecchini MJ, Mymryk JS. High MHC-II Expression in Epstein–Barr Virus-Associated Gastric Cancers Suggests That Tumor Cells Serve an Important Role in Antigen Presentation. Sci Rep (2020) 10(1):1–16. doi: 10.1038/s41598-020-71775-4
69. Haan KM, Kwok WW, Longnecker R, Speck P. Epstein-Barr Virus Entry Utilizing HLA-DP or HLA-DQ as a Coreceptor. J Virol (2000) 74(5):2451–4. doi: 10.1128/JVI.74.5.2451-2454.2000
70. Perfetti V, Baldanti F, Lenti MV, Vanoli A, Biagi F, Gatti M, et al. Detection of Active Epstein–Barr Virus Infection in Duodenal Mucosa of Patients With Refractory Celiac Disease. Clin Gastroenterol Hepatol (2016) 14(8):1216–20. doi: 10.1016/j.cgh.2016.03.022
71. Pender MP, Burrows SR. Epstein–Barr Virus and Multiple Sclerosis: Potential Opportunities for Immunotherapy. Clin Transl Immunol (2014) 3(10):e27. doi: 10.1038/cti.2014.25
72. Fox RI, Luppi M, Kang H, Pisa P. Reactivation of Epstein-Barr Virus in Sjögren’s Syndrome. Springer Semin Immunopathol (1991) 13(2):217–31. doi: 10.1007/BF00201470
73. Mahabadi M, Faghihiloo E, Alishiri GH, Ataee MH, Ataee RA. Detection of Epstein-Barr Virus in Synovial Fluid of Rheumatoid Arthritis Patients. Electron Physician (2016) 8(3):2181–6. doi: 10.19082/2181
74. Toussirot E, Roudier J. Pathophysiological Links Between Rheumatoid Arthritis and the Epstein-Barr Virus: An Update. Joint Bone Spine Elsevier Masson; (2007) 74:418–26. doi: 10.1016/j.jbspin.2007.05.001
75. PhD MVW, Cox B, PhD WPL, Ariza ME. Epstein-Barr Virus Dutpase Induces Neuroinflammatory Mediators: Implications for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Clin Ther (2019) 41(5):848–63.
76. Halpin P, Williams MV, Klimas NG, Fletcher MA, Barnes Z, Ariza ME. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Gulf War Illness Patients Exhibit Increased Humoral Responses to the Herpesviruses-Encoded Dutpase: Implications in Disease Pathophysiology. J Med Virol (2017) 89(9):1636–45. doi: 10.1002/jmv.24810
77. Loebel M, Strohschein K, Giannini C, Koelsch U, Bauer S, Doebis C, et al. Deficient EBV-Specific B- and T-Cell Response in Patients With Chronic Fatigue Syndrome. PloS One (2014) 9(1):85387. doi: 10.1371/journal.pone.0085387
78. Kawai K, Kawai A. Studies on the Relationship Between Chronic Fatigue Syndrome and Epstein-Barr Virus in Japan. Intern Med (1992) 31(3):313–8. doi: 10.2169/internalmedicine.31.313
79. Sairenji T, Yamanishi K, Tachibana Y, Bertoni G, Kurata T. Antibody Responses to Epstein-Barr Virus, Human Herpesvirus 6 and Human Herpesvirus 7 in Patients With Chronic Fatigue Syndrome. Intervirology (1995) 38(5):269–73. doi: 10.1159/000150450
80. Lerner AM, Beqaj SH, Deeter RG, Fitzgerald JT. IgM Serum Antibodies to Epstein-Barr Virus Are Uniquely Present in a Subset of Patients With the Chronic Fatigue Syndrome. In Vivo (Brooklyn) (2004) 18(2)):101–6.
81. Loebel M, Eckey M, Sotzny F, Hahn E, Bauer S, Grabowski P, et al. Serological Profiling of the EBV Immune Response in Chronic Fatigue Syndrome Using a Peptide Microarray. PloS One (2017) 12(6):e0179124. doi: 10.1371/journal.pone.0179124
82. A ME. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: The Human Herpesviruses Are Back! Biomolecules (2021) 11(2):1–17. doi: 10.3390/biom11020185
83. Rasa S, Nora-Krukle Z, Henning N, Eliassen E, Shikova E, Harrer T, et al. Chronic Viral Infections in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). J Transl Med (2018) 16(1):268. doi: 10.1186/s12967-018-1644-y
84. Cameron C, Flamand L, Juwana H, Middeldorp J, Naing Z, Rawlinson W, et al. Serological and Virological Investigation of the Role of the Herpesviruses EBV, CMV and HHV-6 in Post-Infective Fatigue Syndrome. J Med Virol (2010) 82(10):1684–8. doi: 10.1002/jmv.21873
85. Soto NE, Straus SE. Chronic Fatigue Syndrome and Herpesviruses: The Fading Evidence. Herpes (2000) 7(2):46–50.
86. Laichalk LL, Thorley-Lawson DA. Terminal Differentiation Into Plasma Cells Initiates the Replicative Cycle of Epstein-Barr Virus In Vivo. J Virol (2005) 79(2):1296–307. doi: 10.1128/JVI.79.2.1296-1307.2005
87. Hong GK, Delecluse HJ, Gruffat H, Morrison TE, Feng W-H, Sergeant A, et al. The BRRF1 Early Gene of Epstein-Barr Virus Encodes a Transcription Factor That Enhances Induction of Lytic Infection by BRLF1. J Virol (2004) 78(10):4983–92. doi: 10.1128/jvi.78.10.4983-4992.2004
88. ter Wolbeek M, van Doornen DLJ, Kavelaars A, van de Putte EM, Schedlowski M, Heijnen CJ. Longitudinal Analysis of Pro- and Anti-Inflammatory Cytokine Production in Severely Fatigued Adolescents. Brain Behav Immun (2007) 21(8):1063–74. doi: 10.1016/j.bbi.2007.04.007
89. Giloteaux L, O’Neal A, Castro-Marrero J, Levine SM, Hanson MR. Cytokine Profiling of Extracellular Vesicles Isolated From Plasma in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Pilot Study. J Transl Med (2020) 18118(1):1–17. doi: 10.1186/s12967-020-02560-0
90. Yang T, Yang Y, Wang D, Li C, Qu Y, Guo J, et al. The Clinical Value of Cytokines in Chronic Fatigue Syndrome. J Transl Med (2019) 17(1):213. doi: 10.1186/s12967-019-1948-6
91. Broderick G, Fuite J, Kreitz A, Vernon SD, Klimas N, Fletcher MA. A Formal Analysis of Cytokine Networks in Chronic Fatigue Syndrome. Brain Behav Immun (2010) 24(7):1209. doi: 10.1016/j.bbi.2010.04.012
92. Lakhan SE, Kirchgessner A. Gut Inflammation in Chronic Fatigue Syndrome. Nutr Metab (2010) 7:79. doi: 10.1186/1743-7075-7-79
93. Giloteaux L, Goodrich JK, Walters WA, Levine SM, Ley RE, Hanson MR. Reduced Diversity and Altered Composition of the Gut Microbiome in Individuals With Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Microbiome (2016) 4(1):30. doi: 10.1186/s40168-016-0171-4
94. Nagy-Szakal D, Williams BL, Mishra N, Che X, Lee B, Bateman L, et al. Fecal Metagenomic Profiles in Subgroups of Patients With Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Microbiome (2017) 5(1):44. doi: 10.1186/s40168-017-0261-y
95. Gupta S, Aggarwal S, See D, Starr A. Cytokine Production by Adherent and Non-Adherent Mononuclear Cells in Chronic Fatigue Syndrome. J Psychiatr Res (1997) 31(1):149–56. doi: 10.1016/s0022-3956(96)00063-5
96. Borish L, Schmaling K, DiClementi JD, Streib J, Negri J, Jones JF. Chronic Fatigue Syndrome: Identification of Distinct Subgroups on the Basis of Allergy and Psychologic Variables. J Allergy Clin Immunol (1998) 102(2):222–30. doi: 10.1016/s0091-6749(98)70090-9
97. Corbitt M, Eaton-Fitch N, Staines D, Cabanas H, Marshall-Gradisnik S. A Systematic Review of Cytokines in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis/Systemic Exertion Intolerance Disease (CFS/ME/SEID). BMC Neurol (2019) 19(1):207. doi: 10.1186/s12883-019-1433-0
98. Blundell S, Ray KK, Buckland M, White PD. Chronic Fatigue Syndrome and Circulating Cytokines: A Systematic Review. Brain Behav Immun (2015) 50:186–95. doi: 10.1016/j.bbi.2015.07.004
99. Jonsjö MA, Olsson GL, Wicksell RK, Alving K, Holmström L, Andreasson A. The Role of Low-Grade Inflammation in ME/CFS (Myalgic Encephalomyelitis/Chronic Fatigue Syndrome) - Associations With Symptoms. Psychoneuroendocrinology (2020) 113:104578. doi: 10.1016/j.psyneuen.2019.104578
100. Fluge Ø, Risa K, Lunde S, Alme K, Rekeland IG, Sapkota D, et al. B-Lymphocyte Depletion in Myalgic Encephalopathy/ Chronic Fatigue Syndrome. An Open-Label Phase II Study With Rituximab Maintenance Treatment. Van Der Feltz-Cornelis C, Editor. PloS One (2015) 10(7):e0129898. doi: 10.1371/journal.pone.0129898
101. Li H, Yu X, Liles C, Khan M, Vanderlinde-Wood M, Galloway A, et al. Autoimmune Basis for Postural Tachycardia Syndrome. J Am Heart Assoc (2014) 3(1):e000755. doi: 10.1161/JAHA.113.000755
102. Appay V, Zaunders JJ, Papagno L, Sutton J, Jaramillo A, Waters A, et al. Characterization of CD4 + CTLs Ex Vivo. J Immunol (2002) 168(11):5954–8. doi: 10.4049/jimmunol.168.11.5954
103. Kumar A, Perdomo MF, Kantele A, Hedman L, Hedman K, Franssila R. Granzyme B Mediated Function of Parvovirus B19-Specific CD4+ T Cells. Clin Transl Immunol (2015) 4(7):e39. doi: 10.1038/cti.2015.13
104. Alcami A, Koszinowski UH. Viral Mechanisms of Immune Evasion. Immunol Today (2000) 21:447–55. doi: 10.1016/S0167-5699(00)01699-6
105. Muraro E, Merlo A, Martorelli D, Cangemi M, Santa SD, Dolcetti R, et al. Fighting Viral Infections and Virus-Driven Tumors With Cytotoxic CD4+ T Cells. Front Immunol (2017) 8:197. doi: 10.3389/fimmu.2017.00197
106. Jason LA, Corradi K, Gress S, Williams S, Torres-Harding S. Causes of Death Among Patients With Chronic Fatigue Syndrome. Health Care Women Int (2006) 27(7):615–26. doi: 10.1080/07399330600803766
107. Lee J-S, Lacerda EM, Nacul L, Kingdon CC, Norris J, O’Boyle S, et al. Salivary DNA Loads for Human Herpesviruses 6 and 7 Are Correlated With Disease Phenotype in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front Med (2021) 0:656692/full. doi: 10.3389/fmed.2021.656692/full
108. Chang CM, Warren JL, Engels EA. Chronic Fatigue Syndrome and Subsequent Risk of Cancer Among Elderly US Adults. Cancer (2012) 118(23):5929–36. doi: 10.1002/cncr.27612
109. Endicott NA. Chronic Fatigue Syndrome in Private Practice Psychiatry: Family History of Physical and Mental Health. J Psychosom Res (1999) 47(4):343–54. doi: 10.1016/s0022-3999(99)00013-6
110. Blomberg J, Gottfries C-G, Elfaitouri A, Rizwan M, Rosén A. Infection Elicited Autoimmunity and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: An Explanatory Model. Front Immunol (2018) 9(FEB):15. doi: 10.3389/fimmu.2018.00229
111. Kristiansen MS, Stabursvik J, O'Leary EC, Pedersen M, Asprusten T, Leegaard TT, et al. Clinical Symptoms and Markers of Disease Mechanisms in Adolescent Chronic Fatigue Following Epstein-Barr Virus Infection: An Exploratory Cross-Sectional Study. Brain Behav Immun (2019) 80:551–63. doi: 10.1016/j.bbi.2019.04.040
112. Draborg AH, Jacobsen S, Westergaard M, Mortensen S, Larsen JL, Houen G, et al. Reduced Response to Epstein-Barr Virus Antigens by T-Cells in Systemic Lupus Erythematosus Patients. Lupus Sci Med (2014) 1(1):15. doi: 10.1136/lupus-2014-000015
113. Pender MP, Csurhes PA, Burrows JM, Burrows SR. Defective T-Cell Control of Epstein–Barr Virus Infection in Multiple Sclerosis. Clin Transl Immunol (2017) 6(1):e126. doi: 10.1038/cti.2016.87
114. Toussirot É, Roudier J. Epstein-Barr Virus in Autoimmune Diseases. Best Pract Research: Clin Rheumatol Baillière Tindall (2008) 22:883–96. doi: 10.1016/j.berh.2008.09.007
115. Richman S, Morris MC, Broderick G, Craddock TJA, Klimas NG, Fletcher MA. Pharmaceutical Interventions in Chronic Fatigue Syndrome: A Literature-Based Commentary. Clin Ther (2019) 41(5):798–805. doi: 10.1016/j.clinthera.2019.02.011
116. Fluge Ø, Rekeland IG, Lien K, Thürmer H, Borchgrevink PC, Schäfer C, et al. B-Lymphocyte Depletion in Patients With Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Randomized, Double-Blind, Placebo-Controlled Trial. Ann Intern Med (2019) 170(9):585–93. doi: 10.7326/M18-1451
117. Fluge Ø, Bruland O, Risa K, Storstein A, Kristoffersen EK, Sapkota D, et al. Benefit From B-Lymphocyte Depletion Using the Anti-CD20 Antibody Rituximab in Chronic Fatigue Syndrome. A Double-Blind and Placebo-Controlled Study. Reindl M, Editor. PloS One (2011) 6(10):e26358. doi: 10.1371/journal.pone.0026358
118. Lerner AM, Beqaj SH, Deeter RG, Fitzgerald JT. Valacyclovir Treatment in Epstein-Barr Virus Subset Chronic Fatigue Syndrome: Thirty-Six Months Follow-Up. In Vivo (Brooklyn) (2007) 21(5):707–14.
119. Montoya JG, Kogelnik AM, Bhangoo M, Lunn MR, Flamand L, Merrihew LE, et al. Randomized Clinical Trial to Evaluate the Efficacy and Safety of Valganciclovir in a Subset of Patients With Chronic Fatigue Syndrome. J Med Virol (2013) 85(12):2101–9. doi: 10.1002/jmv.23713
120. Heslop HE. How I Treat EBV Lymphoproliferation. Blood (2009) 114(19):4002–8. doi: 10.1182/blood-2009-07-143545
121. Pender MP. Preventing and Curing Multiple Sclerosis by Controlling Epstein-Barr Virus Infection. Autoimmun Rev (2009) 8:563–8. doi: 10.1016/j.autrev.2009.01.017
122. Savoldo B, Rooney CM, Quiros-Tejeira RE, Caldwell Y, Wagner H, Lee T, et al. Cellular Immunity to Epstein-Barr Virus in Liver Transplant Recipients Treated With Rituximab for Post-Transplant Lymphoproliferative Disease. Am J Transplant (2005) 5(3):566–72. doi: 10.1111/j.1600-6143.2004.00693.x
123. Suzan F, Ammor M, Ribrag V. Fatal Reactivation of Cytomegalovirus Infection After Use of Rituximab for a Post-Transplantation Lymphoproliferative Disorder. N Engl J Med (2001) 345(13):1000–0. doi: 10.1056/NEJM200109273451315
124. Nikiforow S, Bottomly K, Miller G. CD4+ T-Cell Effectors Inhibit Epstein-Barr Virus-Induced B-Cell Proliferation. J Virol (2001) 75(8):3740–52. doi: 10.1128/JVI.75.8.3740-3752.2001
125. Schaefer BC, Strominger JL, Speck SH. A Simple Reverse Transcriptase PCR Assay to Distinguish EBNA1 Gene Transcripts Associated With Type I and II Latency From Those Arising During Induction of the Viral Lytic Cycle. J Virol (1996) 70(11):8204–8. doi: 10.1128/jvi.70.11.8204-8208.1996
126. Amyes E, Hatton C, Montamat-Sicotte D, Gudgeon N, Rickinson AB, McMichael AJ, et al. Characterization of the CD4+ T Cell Response to Epstein-Barr Virus During Primary and Persistent Infection. J Exp Med (2003) 198(6):903–11. doi: 10.1084/jem.20022058
127. Frappier L. The Epstein-Barr Virus EBNA1 Protein. Scientifica (Cairo) (2012) 2012:1–15. doi: 10.6064/2012/438204
128. Dalton T, Doubrovina E, Pankov D, Reynolds R, Scholze H, Selvakumar A, et al. Epigenetic Reprogramming Sensitizes Immunologically Silent EBV+ Lymphomas to Virus-Directed Immunotherapy. Blood (2020) 135(21):1870–81. doi: 10.1182/blood.2019004126
129. Turrini R, Merlo A, Dolcetti R, Zanovello P, Rosato A. Differential Down-Modulation of HLA Class I and II Molecule Expression on Human Tumor Cell Lines Upon In Vivo Transfer. Cancer Immunol Immunother (2011) 60(11):1639–45. doi: 10.1007/s00262-011-1086-3
130. Merlo A, Turrini R, Bobisse S, Zamarchi R, Alaggio R, Dolcetti R, et al. Virus-Specific Cytotoxic CD4 + T Cells for the Treatment of EBV-Related Tumors. J Immunol (2010) 184(10):5895–902. doi: 10.4049/jimmunol.0902850
131. Nishikawa J, Iizasa H, Yoshiyama H, Nakamura M, Saito M, Sasaki S, et al. The Role of Epigenetic Regulation in Epstein-Barr Virus-Associated Gastric Cancer. Int J Mol Sci (2017) 18(8):1606. doi: 10.3390/ijms18081606
132. Nakamura M, Nishikawa J, Saito M, Sakai K, Sasaki S, Hashimoto S, et al. Decitabine Inhibits Tumor Cell Proliferation and Up-Regulates E-Cadherin Expression in Epstein-Barr Virus-Associated Gastric Cancer. J Med Virol (2017) 89(3):508–17. doi: 10.1002/jmv.24634
133. Gallinella G. Parvovirus B19 Achievements and Challenges. ISRN Virol (2013) 2013:1–33. doi: 10.5402/2013/898730
134. Bonvicini F, Manaresi E, Di Furio F, de Falco L, Gallinella G. Parvovirus B19 DNA CpG Dinucleotide Methylation and Epigenetic Regulation of Viral Expression. PloS One (2012) 7(3):e33316. doi: 10.1371/journal.pone.0033316
135. Flower K, Thomas D, Heather J, Ramasubramanyan S, Jones S, Sinclair AJ. Epigenetic Control of Viral Life-Cycle by a DNA-Methylation Dependent Transcription Factor. PloS One (2011) 6(10):e25922. doi: 10.1371/journal.pone.0025922
136. Minarovits J. Epigenotypes of Latent Herpesvirus Genomes. Curr Topics Microbiol Immunol (2006) 310:61–80. doi: 10.1007/3-540-31181-5_5
Keywords: chronic fatigue syndrome, myalgic encephalomyelitis, EBV EBNA-1, HLA-II alleles, cancer, CD4+ CTL, autoimmunity, immunotherapy
Citation: Ruiz-Pablos M, Paiva B, Montero-Mateo R, Garcia N and Zabaleta A (2021) Epstein-Barr Virus and the Origin of Myalgic Encephalomyelitis or Chronic Fatigue Syndrome. Front. Immunol. 12:656797. doi: 10.3389/fimmu.2021.656797
Received: 21 January 2021; Accepted: 19 October 2021;
Published: 15 November 2021.
Edited by:
Linda F. Van Dyk, University of Colorado Denver, United StatesReviewed by:
Andrew R. Lloyd, Kirby Institute, AustraliaHirokazu Kanegane, Tokyo Medical and Dental University, Japan
Copyright © 2021 Ruiz-Pablos, Paiva, Montero-Mateo, Garcia and Zabaleta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuel Ruiz-Pablos, bWFucnVpcGFAZ21haWwuY29t; Aintzane Zabaleta, YXphYmFsZXRhYUB1bmF2LmVz