
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 05 July 2021
Sec. Primary Immunodeficiencies
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.652487
 Ilaria Mormile1
Ilaria Mormile1 Alessandra Punziano1
Alessandra Punziano1 Carlo Alberto Riolo2
Carlo Alberto Riolo2 Francescopaolo Granata1
Francescopaolo Granata1 Michela Williams1
Michela Williams1 Amato de Paulis1,3
Amato de Paulis1,3 Giuseppe Spadaro1,3*
Giuseppe Spadaro1,3* Francesca Wanda Rossi1,3
Francesca Wanda Rossi1,3Common variable immunodeficiency (CVID) is the most common clinically significant primary immunodeficiency in adulthood, which presents a broad spectrum of clinical manifestations, often including non-infectious complications in addition to heightened susceptibility to infections. These protean manifestations may significantly complicate the differential diagnosis resulting in diagnostic delay and under-treatment with increased mortality and morbidity. Autoimmunity occurs in up to 30% of CVID patients, and it is an emerging cause of morbidity and mortality in this type of patients. 95 patients (42 males and 53 females) diagnosed with CVID, basing on ESID diagnostic criteria, were enrolled in this retrospective cohort study. Clinical phenotypes were established according to Chapel 2012: i) no other disease-related complications, ii) cytopenias (thrombocytopenia/autoimmune hemolytic anemia/neutropenia), iii) polyclonal lymphoproliferation (granuloma/lymphoid interstitial pneumonitis/persistent unexplained lymphadenopathy), and iv) unexplained persistent enteropathy. Clinical items in the analysis were age, gender, and clinical features. Laboratory data included immunoglobulin (Ig)G, IgM and IgA levels at diagnosis, flow-cytometric analysis of peripheral lymphocytes (CD3+, CD3+CD4+, CD3+CD8+, CD19+, CD4+CD25highCD127low, CD19hiCD21loCD38lo, and follicular T helper cell counts). Comparisons of continuous variables between groups were performed with unpaired t-test, when applicable. 39 patients (41%) showed autoimmune complications. Among them, there were 21 females (53.8%) and 18 males (46.2%). The most prevalent autoimmune manifestations were cytopenias (17.8%), followed by arthritis (11.5%), psoriasis (9.4%), and vitiligo (6.3%). The most common cytopenia was immune thrombocytopenia, reported in 10 out of 95 patients (10.5%), followed by autoimmune hemolytic anemia (n=3, 3.1%) and autoimmune neutropenia (n=3, 3.1%). Other autoimmune complications included thyroiditis, coeliac disease, erythema nodosum, Raynaud’s phenomenon, alopecia, recurring oral ulcers, autoimmune gastritis, and primary biliary cholangitis. There were no statistically significant differences comparing immunoglobulin levels between CVID patients with or without autoimmune manifestations. There was no statistical difference in CD3+, CD8+, CD4+CD25highCD127low T, CD19, CD19hiCD21loCD38lo, and follicular T helper cell counts in CVID patients with or without autoimmune disorders. In conclusion, autoimmune manifestations often affect patients with CVID. Early recognition and tailored treatment of these conditions are pivotal to ensure a better quality of life and the reduction of CVID associated complications.
Common variable immunodeficiency (CVID) is the most common clinically significant primary immunodeficiency (PID) in adulthood (1). The diagnosis of CVID is established after the 4th year of age (but symptoms may be present earlier) and based on the following criteria (2): marked decrease of immunoglobulin (Ig)G, IgA and/or IgM as compared with age-related standard, impaired or absent antibody production, exclusion of secondary causes of hypogammaglobulinemia, and no evidence of profound T-cell deficiency. The European Society for Immunodeficiencies (ESID) diagnostic criteria (ESID Registry-Working definitions for clinical diagnosis of PID) are available from (http://esid.org/Working-Parties/Registry/Diagnosis-criteria).
Over 90% of CVID patients suffer from severe, recurrent, and sometimes chronic bacterial infections mainly of the respiratory and gastrointestinal tracts (3); other common clinical presentations are granulomatous diseases and unexplained polyclonal lymphoproliferation (3).
In recent years, the quality of life and prognosis of patients with CVID have improved thanks to advances in the management and prophylaxis of infections with antibacterial agents and immunoglobulin replacement therapy (IgRT). Simultaneously, there has been an increased awareness of autoimmunity as an emerging cause of morbidity and mortality (4).
Autoimmunity occurs in up to 30% of CVID patients (5–7), and it is frequently the presenting manifestation at the onset of immunodeficiency (1). In addition, in a study by Quinti et al. (8) autoimmunity was found in 17.4% of 224 patients with CVID, and in 2.3% of these patients, it was the only clinical manifestation at the diagnosis of CVID. Cytopenias are the most common autoimmune conditions described in CVID (6, 9, 10). Immune thrombocytopenia (ITP) is the most frequent autoimmune cytopenia, with a 7% to 14% prevalence, while the proportion of CVID patients with autoimmune hemolytic anemia (AIHA) and autoimmune neutropenia is 4-7% and 1%, respectively (6). Rheumatologic diseases observed in CVID patients are rheumatoid arthritis (2.6-3.6%), Sjögren’s syndrome (<1-4.2%), and systemic lupus erythematosus (<1%) (3). Other autoimmune conditions related to CVID are vitiligo (<1-3.9%), autoimmune thyroiditis (<1-3.9%), diabetes mellitus (<1-3.9%), multiple sclerosis (<1-3.9%), alopecia (1.1-1.6%), and pernicious anemia (<1-1.2%) (3).
Our study aims to characterize the clinical phenotype and immunological findings of CVID patients with associated autoimmune complications. CVID may indeed present a broad clinical spectrum, therefore preventing diagnostic delay may sometimes be very challenging (11), especially due to the lack of physicians’ awareness about some atypical manifestations of this disease.
A total of 95 adult patients (42 males and 53 females) diagnosed with CVID at the Division of Allergy and Clinical Immunology of the University of Naples Federico II, Naples, Italy, were enrolled in this retrospective cohort study. Clinical and laboratory data were retrospectively collected until December 2020. We included patients with a CVID diagnosis based on the ESID diagnostic criteria (available at http://esid.org/Working-Parties/Registry/Diagnosis-criteria), available data on sex, date of birth, age of onset, CVID diagnosis age, serum levels of immunoglobulins (IgG, IgA, and IgM) at diagnosis, and signature of the written informed consent. Patients under 18 years of age were excluded. Secondary causes of hypogammaglobulinemia (e.g., drugs, malignancies) were ruled out. We also excluded four CVID patients treated with rituximab for granulomatous and lymphocytic interstitial lung disease (GLILD). Patients diagnosed with CVID developing malignancies in clinical remission at the time of enrolment were included.
Less than 20% of cases of CVID patients have a known underlying genetic cause (12). In our cohort six patients underwent genetic tests, in four patients no genetic defects were found and two patients exhibit TACI mutations.
Clinical phenotypes were established according to Chapel 2012 (13, 14): i) no other disease-related complications, ii) cytopenias (thrombocytopenia/autoimmune hemolytic anemia/neutropenia), iii) polyclonal lymphoproliferation (granuloma/lymphoid interstitial pneumonitis/persistent unexplained lymphadenopathy), and iv) unexplained persistent enteropathy.
Splenomegaly and malignancies were excluded as phenotyping criteria according to Chapel 2012 (13, 14) and described separately. Indeed, splenic enlargement may be due to various causes rather than a relationship with the underlying disease (13), similarly, immunodeficiency could develop as a secondary clinical event of malignancies (i.e., lymphoid malignancies) (14).
In addition, we described other significant clinical features in our cohort, such as psoriasis, vitiligo, thyroiditis, coeliac disease, erythema nodosum, Raynaud’s phenomenon, alopecia, recurring oral ulcers, autoimmune gastritis, and primary biliary cholangitis.
Laboratory data included IgG, IgM, and IgA serum levels at the diagnosis and flow-cytometric analysis (BD FACS Canto II, Erembodegem, Belgium) of peripheral lymphocytes [CD3+, CD3+CD4+, CD3+CD8+, CD19+, CD4+CD25highCD127low T regulatory (Treg), CD19hiCD21loCD38lo B cells count, follicular helper T cells].
All patients had available data on sex, date of birth, age of onset, and CVID diagnosis age. Furthermore, serum levels of immunoglobulins (IgG, IgA, and IgM) at diagnosis, before IgRT treatment, were available for all 95 CVID patients (Figure 1).
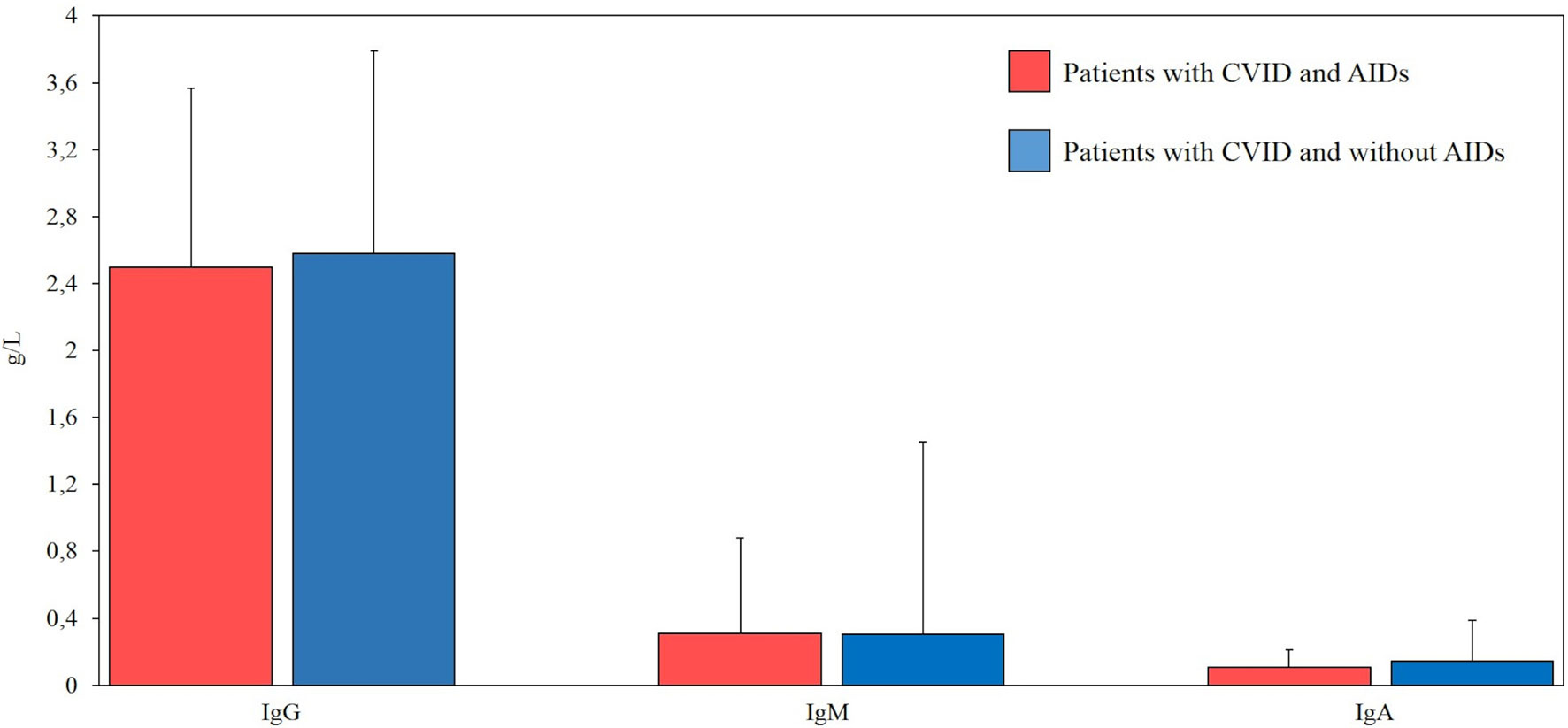
Figure 1 Comparison between serum levels of immunoglobulins (g/L) at diagnosis in our cohort of patients with common variable immunodeficiency (CVID) and autoimmune diseases (AIDs) (red bar, n=39) and CVID without AIDs (blue bar, n=56). Normal ranges (g/l) for IgG (7.37-16.07), IgM (0.40-2.30), IgA (0.70-4).
There were some missing data in the lymphocyte subsets counts. CD3+ were available for 26 (66%) patients with autoimmune complications, and for 25 (44%) CVID patients without autoimmune complications. CD8+ T cells and CD19+ cells were available for 26 (66%) patients with autoimmune complications, and for 23 (41%) CVID patients without autoimmune complications. CD4+CD25highCD127low Treg were available for 20 (51%) patients with autoimmune complications, and for 22 (39%) CVID patients without autoimmune complications. CD19hiCD21loCD38lo B cells were available for 19 (48%) patients with autoimmune complications and for 18 (32%) CVID patients without autoimmune complications. Follicular T helper cells were available for 15 (28%) patients with autoimmune complications and for 13 (23%) CVID patients without autoimmune complications. Cell counts are displayed as the absolute number of cell/microliter (cell/µL) (Figure 2).
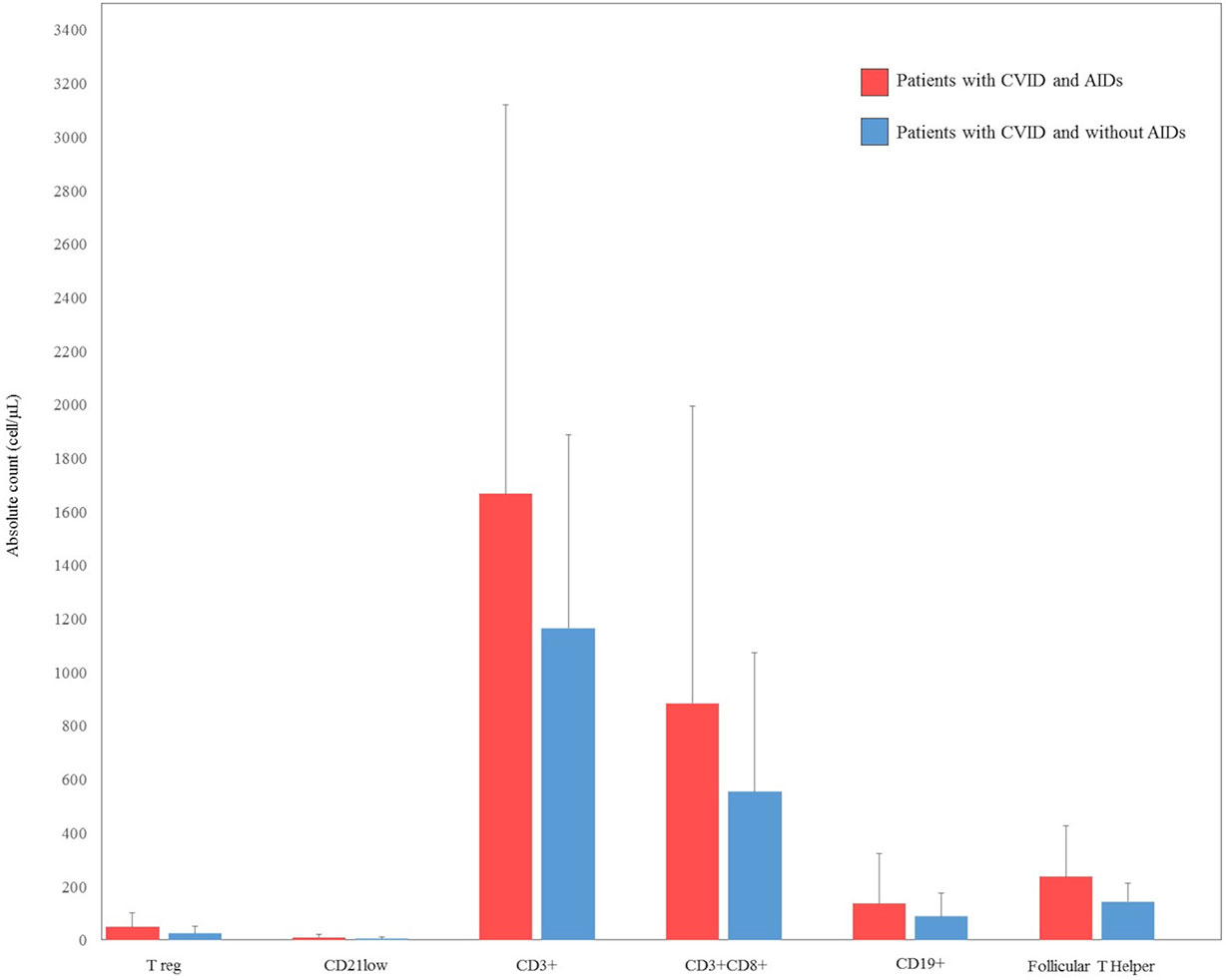
Figure 2 Comparison between lymphocyte count subsets at diagnosis in our cohort of patients with common variable immunodeficiency (CVID) and autoimmune diseases (AIDs) (red bar) and CVID without AIDs (blue bar). CD4+CD25highCD127low T regulatory (Treg), CD19hiCD21loCD38lo B cells (CD21low).
Due to the hypogammaglobulinemic condition and defect in specific antibodies response in CVID patients, autoantibodies were considered not significant for establishing the diagnosis of autoimmune diseases (15).
Data were analyzed using the software GraphPad Prism 8. Values were presented as frequency (number and percentage) and mean ± standard error of the mean (SEM). Comparisons of continuous variables between groups were performed with unpaired t-test, when applicable. A significance level of p ≤ 0.05 was assumed for all statistical evaluations.
95 CVID patients, 42 males (44.2%) and 53 females (55.8%), were enrolled in this study. The cohort was White-Caucasian. The average age at diagnosis was 52 years (24–83). The patients were followed for an average time of 11.56 years (1–35). 10 patients died during follow-up.
Thirty-nine patients (41%) showed autoimmune complications. Among them were 21 females (53.8%) and 18 males (46.2%). The frequency of the most common autoimmune manifestations in our cohort is summarized in Table 1 and Figure 3. Other autoimmune complications included coeliac disease, erythema nodosum, Raynaud’s phenomenon, alopecia, recurring oral ulcers, autoimmune gastritis, and primary biliary cholangitis (Table 1 and Figure 3).
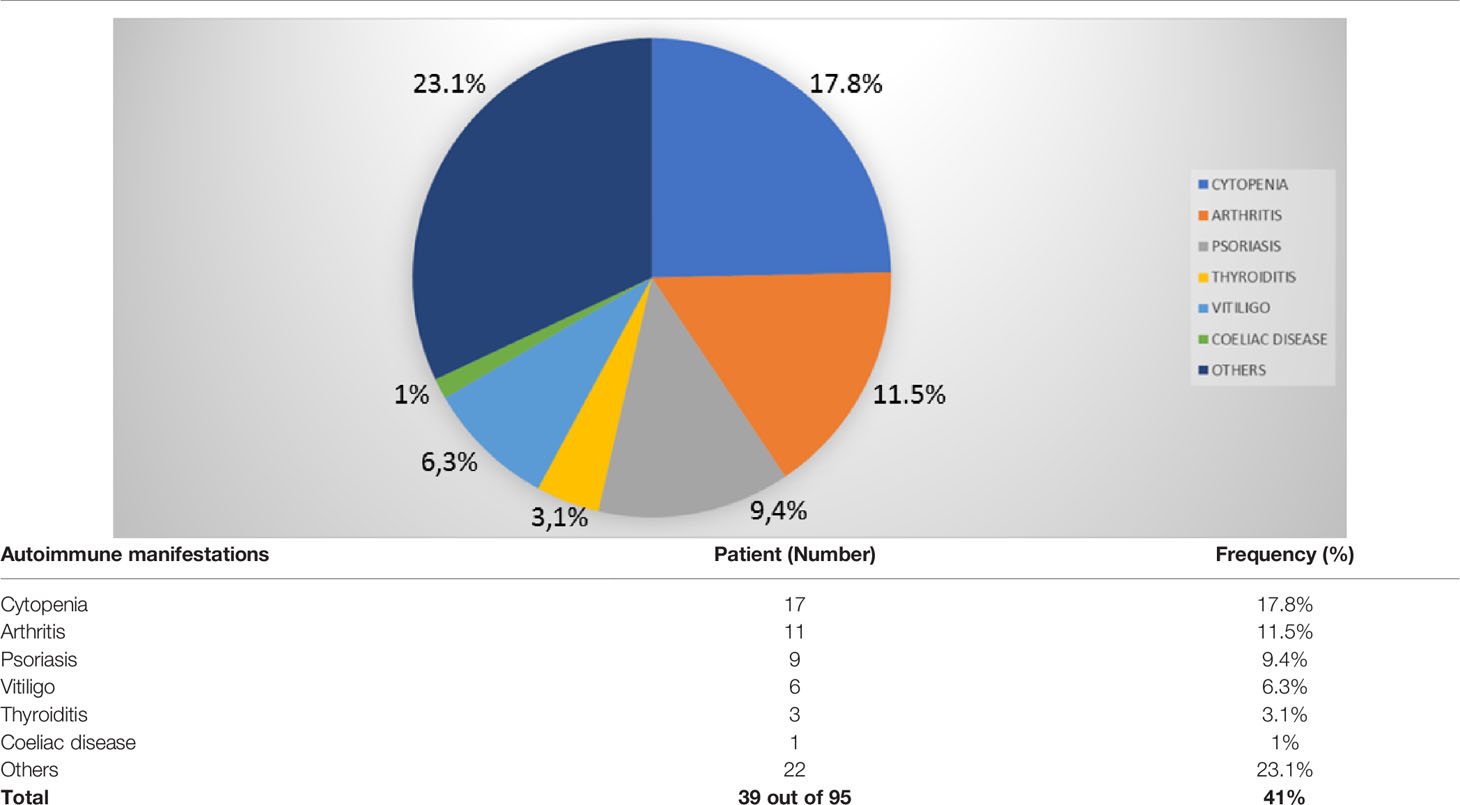
Table 1 Distribution of autoimmune disease in 95 CVID patients.
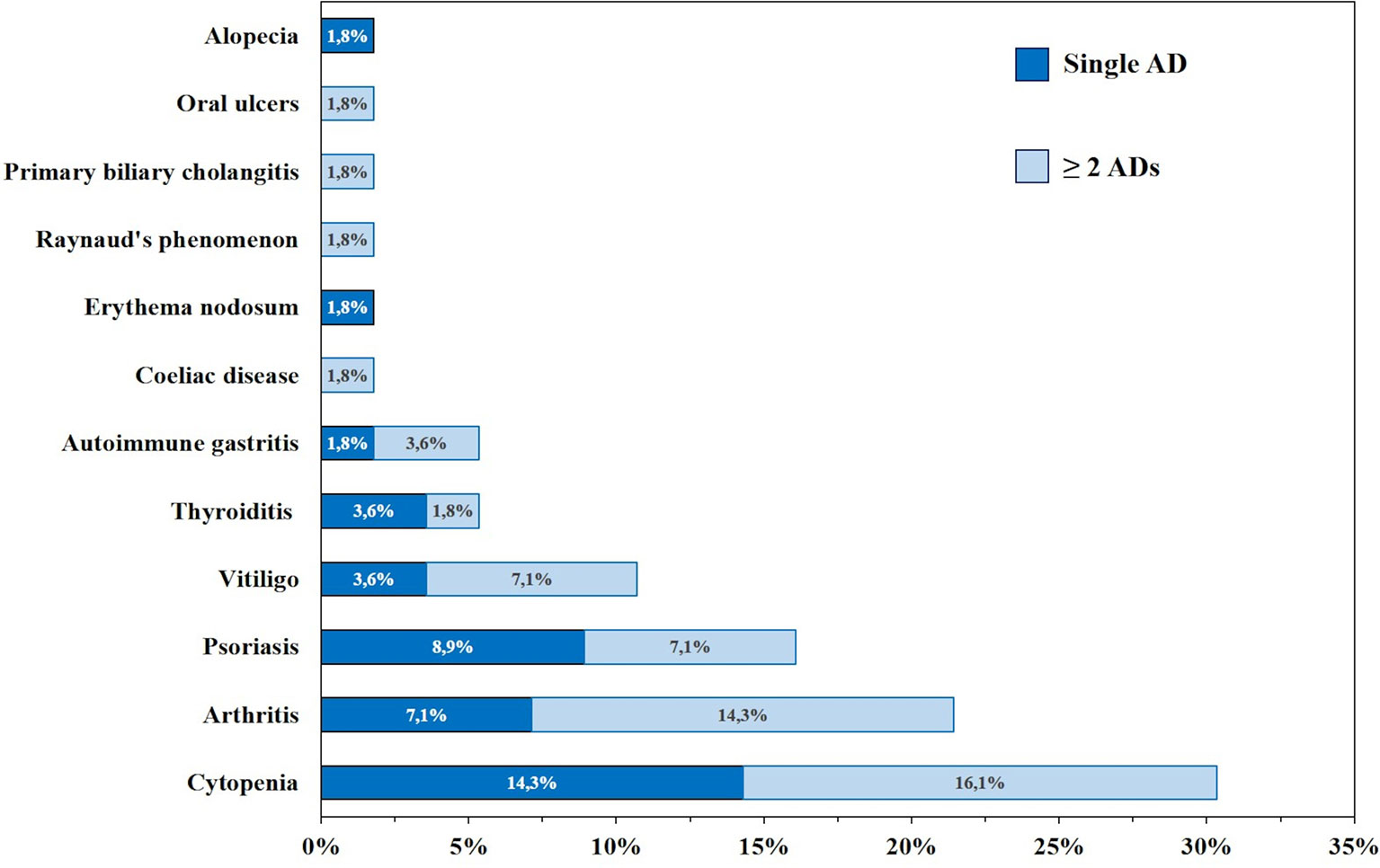
Figure 3 Frequency (%) of autoimmune manifestations presenting alone (dark blue) or in association (light blue) in patients with common variable immunodeficiency (CVID) (n=95).
Almost all patients (7 out of 9; 7%) affected by psoriasis presented a mild form. Two patients showed a diffuse cutaneous involvement, with nail psoriasis. One patient showed a severe form of erythrodermic psoriasis involving the entire body surface with palmoplantar psoriasis and onycholysis (Figure 4).
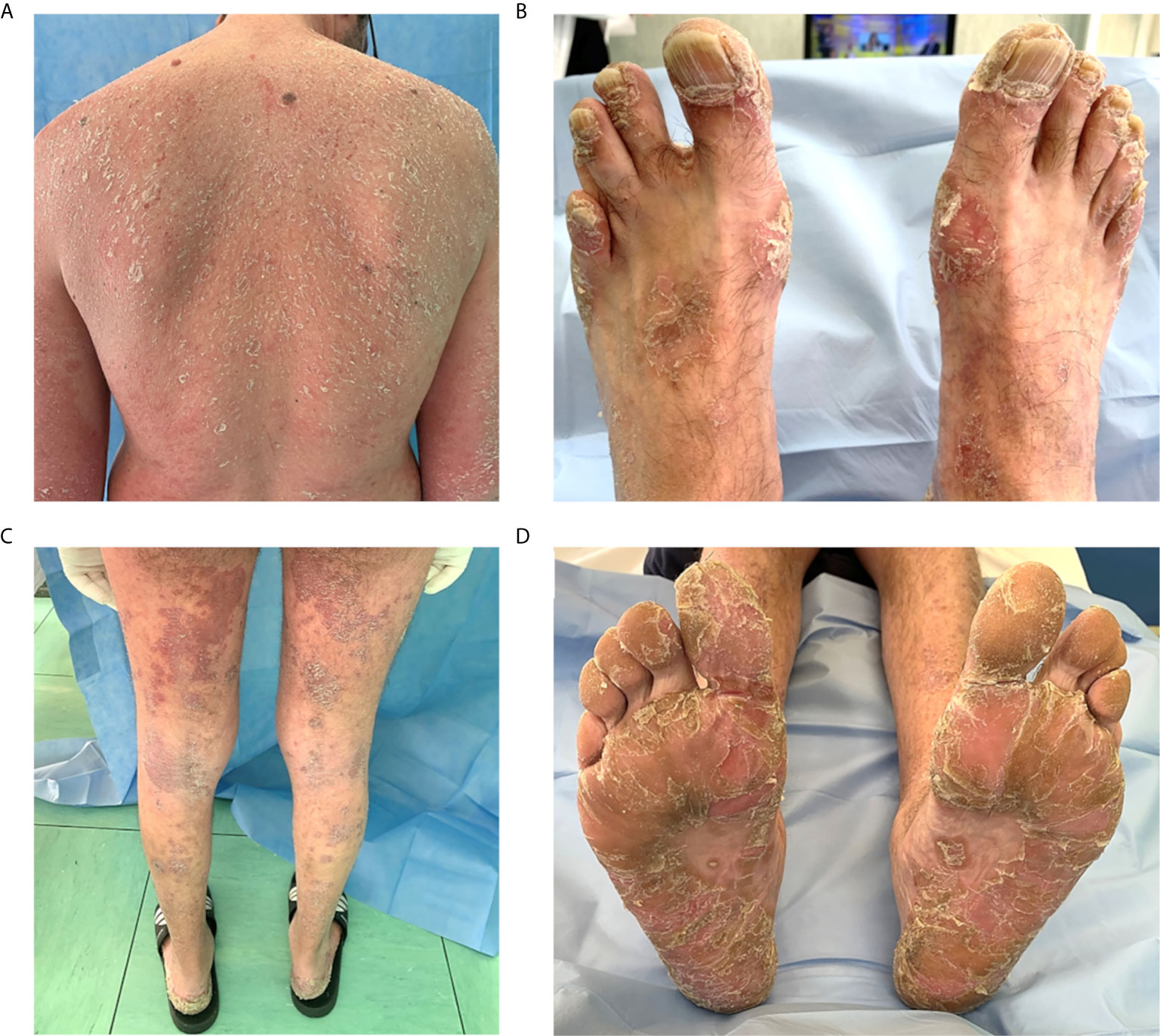
Figure 4 Severe psoriasis in a patient with common variable immunodeficiency. (A–D) Erythrodermic psoriasis diffuse to the entire body with a red and peeling rash. (B) Nail psoriasis is evident at toenails, causing pitting, discoloration and onycholysis. (C) Symmetrical erythematous plaques with sharp edges and covered with pearlescent scales, located on the posterior surface of the legs. (D) Palmoplantar psoriasis involving symmetrically palms of the hands and soles of the feet; squamae are the predominant lesions.
Among 11 patients presenting with arthritis (11.5%), nine patients showed a rheumatoid-like phenotype, and two patients were diagnosed with psoriatic arthritis, based on clinical, laboratory, and radiological findings (16, 17). We included in this study only patients with aseptic inflammatory arthritis (16, 17).
We also compared prevalence of autoimmune manifestations in our cohort and the general prevalence on Italian population; results are reported in Table 2. Clinical phenotypes were established according to Chapel 2012 classification (14).
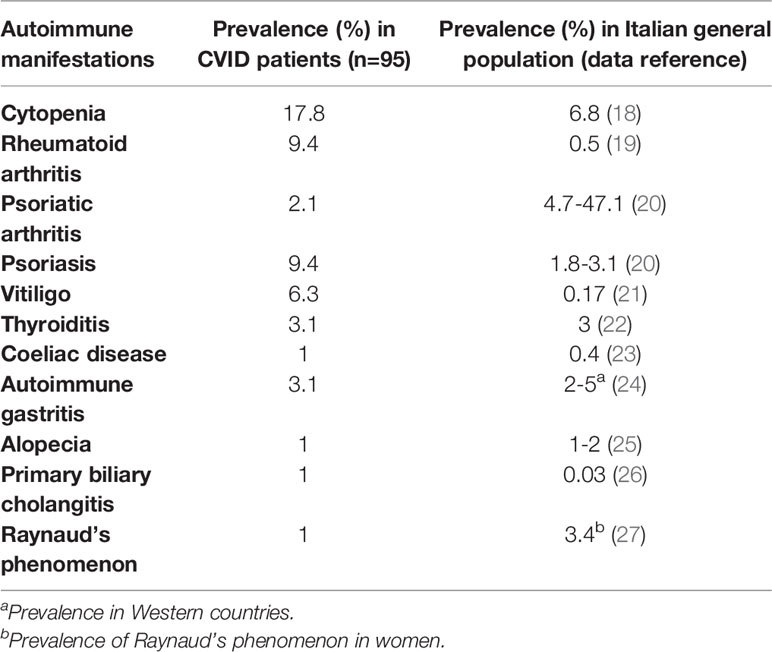
Table 2 Prevalence of autoimmune manifestations in our CVID cohort (n=95) vs Italian general population.
Phenotypes are not mutually exclusive as shown in Figure 5. Among patients with autoimmunity, 13 patients (13.6%) presented with the no other disease-related complications phenotype; 17 (17.8%) presented with the cytopenia phenotype; 22 (22.9%) presented with the polyclonal lymphoproliferation phenotype; 7 (7.3%) presented with the enteropathy phenotype.
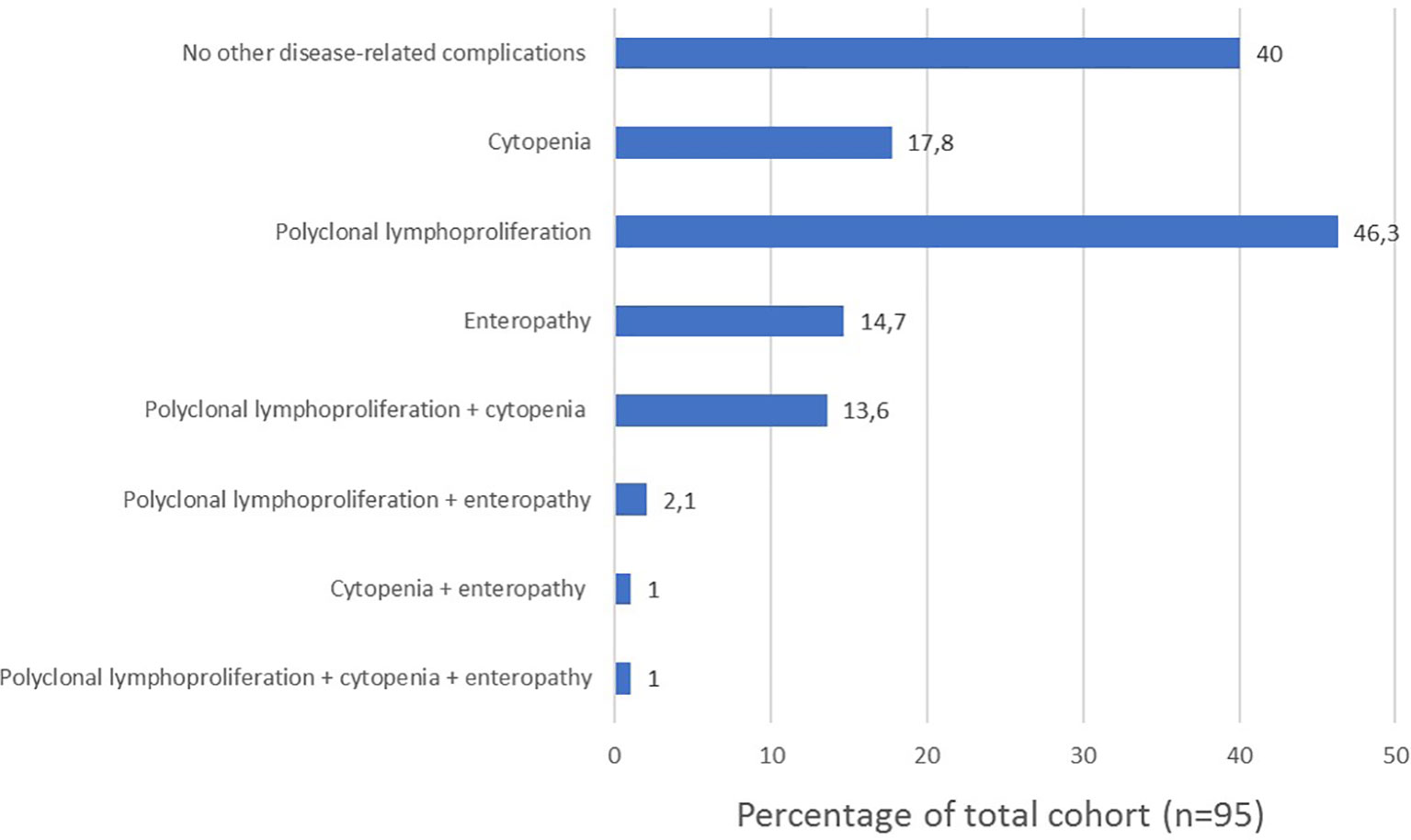
Figure 5 Frequency of clinical phenotypes and their overlap in our cohort (n=95).
Among patients with arthritis seven had no other disease-related complications (63.6%), three patients presented cytopenia, three patients polyclonal lymphoproliferation, and one patient had enteropathy. Clinical manifestations associated with autoimmune cytopenias were polyclonal lymphoproliferation (n=14, 82.3%), enteropathy (n=5, 29.4%), arthritis (n=3, 17.6%), thyroiditis (n=1, 5.8%), coeliac disease (n=1, 5.8%), autoimmune gastritis (n=1, 5.8%), vitiligo (n=1, 5.8%), and primary biliary cholangitis (n=1, 5.8%). 14 out of 95 (14.7%) patients presented malignancy during the follow-up. The most common malignancy in our cohort was non-Hodgkin’s lymphoma (n=4, 4.2%), followed by skin cancer (n=3, 3.15%; two patients with basal cell carcinoma and one patient with squamous cell carcinoma). Other malignancies found in our cohort were Hodgkin’s lymphoma, breast, gastric, thyroid, gallbladder, and nasopharyngeal cancers.
The overall prevalence of splenomegaly in our cohort was 47.3% (n=45) and it was found in 21 out of 39 patients (53.8%) with CVID and autoimmunity; most of the patients with splenomegaly had autoimmune cytopenias (15 out of 21; 71.4%). On the other hand, almost all patients with autoimmune cytopenia had splenomegaly (15 out of 17; 88%), in line with the observation that splenomegaly is more common in patients with cytopenias, although this association is not clearly understood (28). Granulomatous and lymphocytic interstitial lung disease (GLILD) was observed in 12 patients (12.6%).
All patients were treated with intravenous (76 patients) or subcutaneous (19 patients) IgRT (9, 29, 30). To manage rheumatologic complications four patients were treated with medium-low doses of glucocorticoids (≤10 mg/day), two patients with hydroxychloroquine (200-400 mg/day), two patients with sulfasalazine (2 g/day) and one patient, who failed therapy with hydroxychloroquine and cyclosporine (140 mg/day), was switched to methotrexate (7.5 mg/week) which led to symptom improvement.
Four patients with autoimmune thrombocytopenia started therapy with glucocorticoids up to at 1 mg/kg prednisone-equivalent. In addition, one patient who presented steroid-dependency underwent splenectomy and high-dose intravenous immunoglobulin (IVIG) (1–2 g/kg in 5 days) was administrated, with good clinical outcome. Clinical features of patients with CVID and autoimmunity are displayed in Table 3.
Table 3 Clinical features in our cohort of patients with CVID and autoimmune manifestations.
Serum immunoglobulin concentrations and CD3+, CD8+, CD4+CD25highCD127low, CD19, CD19hiCD21loCD38lo and follicular T helper cell counts were compared between CVID patients with and without autoimmune disorders. No statistically significant differences were observed in the immunoglobulin levels between the two groups of patients. There was no statistical difference in CD3+, CD8+, CD4+CD25highCD127low T reg, CD19, CD19hiCD21loCD38lo, and follicular T helper cells in CVID patients with or without autoimmune disorders. Laboratory findings of patients with CVID and autoimmune manifestations are summarized in Figures 1 and 2.
The coexistence of immunodeficiency with autoimmunity may appear oxymoronic, in particular it is unclear how autoantibodies can be produced in a state of antibody deficiency (1). On the one hand, the production of antibodies of patients with CVID is severely impaired or completely absent, and, on the other hand, patients with CVID may show features consistent with an overactive immune system (31, 32). To understand this relationship, it must be considered that the immune system is in constant balance with stimulatory and inhibitory factors that affect common immune cells (31). In addition, it has been shown that the development of autoimmunity in these patients is the consequence of a lower efficacy of the self-tolerance mechanisms, linked in turn to alterations of the immune-regulatory mechanisms (32). Based on this suggested pathogenetical mechanism, several research groups have tried to identify some common immunophenotipical features in patients with CVID associated with autoimmune manifestations. The composition of the B cell compartment had been previously used to classify CVID patients to predict clinical phenotype and complications (7, 33–36). An expansion in CD21low B cells has been found in a subgroup of CVID patients with autoimmunity by several research groups (7, 35, 37), as found in other autoimmune diseases (38, 39). Rakhmanov et al. (40) demonstrated that in CVID, CD21low B cells are a polyclonal, pre-activated, partially autoreactive, functionally attenuated B cell population with preferential enrichment in peripheral tissues, like the bronchoalveolar space of CVID or the synovium of rheumatoid arthritis patients. In addition, CD21low B cells produce significantly higher amounts of IgM as compared to näive B cells upon stimulation with CD40L, IL-2 and IL-10, and higher IgM levels have been associated with the development of autoimmunity in CVID patients (5, 7, 28, 41). Several studies have demonstrated that T cells play a significant role in CVID pathogenesis, overtaking the old-fashioned concept that CVID is exclusively due to B cell defects (4), and their abnormalities have been analyzed in patients with associated autoimmune diseases. CVID patients with autoimmunity generally present with lower total T cells than those without autoimmunity (4, 42). Among T cell subtypes, autoimmunity has particularly been correlated with a reduction in Treg cells and an increase in T follicular helper CD4+ cells (43, 44). Indeed, patients with CVID and autoimmune cytopenia show irregularly shaped hyperplastic germinal centers with an increased number of circulating T follicular helper cells, which likely play a role in developing autoreactive B cells (31, 45). In our cohort, we compared Ig levels or lymphocyte subsets between CVID patients with or without autoimmune disorders or arthritis, but we did not find any statistical difference between these groups. However, there was a large amount of missing data for lymphocyte subsets counts, and this could represent an important limitation of our analysis and should be considered in the interpretation of our data. Despite that, it should be emphasized that other previously reported analysis of the immunophenotypic parameters in patients with CVID-associated autoimmune diseases leaded to controversial results. For instance, Gutierrez et al. (46) did not demonstrate significant differences concerning IgA and IgM levels, CD19+ B-cell counts and CD4/CD8 ratio between groups of patients with CVID associated with rheumatologic manifestations and patients with CVID without autoimmune complications.
The prevalence of autoimmune diseases in our study population was 41% (8). Fischer et al. (4) demonstrated that autoimmune and inflammatory diseases are much more frequent in patients with PIDs than in the general population. Data from the ESID registry analyzing a cohort of 2,700 CVID patients reported that especially autoimmune cytopenia was 700 times more prevalent in CVID patients than the general population (47). In addition, it is frequently the presenting manifestation at the onset of immunodeficiency (1) preceding CVID diagnosis by several years in up to 60% of patients (48). ITP is the most common autoimmune cytopenia in CVID with a 7% to 14% prevalence, while AIHA and autoimmune neutropenia respectively occur in 7% and 1% of CVID patients (6). Cytopenia has been reported as the only autoimmune condition associated with a decreased survival, therefore, phenotyping CVID patients, it is considered separately from autoimmunity in general (14, 42).
Our whole cohort was classified according to Chapel 2012 clinical phenotypes (14). The phenotype overlap we described align with those observed by Chapel et al. (13, 14). The authors analyzed data from 334 patients with CVID from 7 European centers to distinguish clinical phenotypes and observed that 83% of the patients had only one clinical phenotype.
However, other research groups have investigated CVID clinical features association (Table 4) and reported that phenotype overlap might be more common. The most contrasting results were obtained by Selenius et al. (49) analyzing a Finnish cohort of 132 patients (106 diagnosed with “probable” and 26 with “possible” CVID) and describing the concomitance of multiple phenotypes in 73% of patients with probable CVID (49). The most common phenotype overlap we observed was the association between polyclonal lymphoproliferation and cytopenia phenotype. Feuille et al. (32) analyzed data from the United States Immunodeficiency Network (USIDNET) Registry to characterize phenotypes associated with autoimmune cytopenias in 990 patients with CVID. The authors found that CVID patients with autoimmune cytopenia were more likely to have other CVID-associated non-infectious complications (OR= 2.9; 95%-CI: 1.9–4.6, P<0.001), including lymphoproliferation, lymphomas, granulomatous disease, hepatic disease, interstitial lung diseases, enteropathy, and organ-specific autoimmunity. These results align with those observed in our study (Tables 1 and 3).
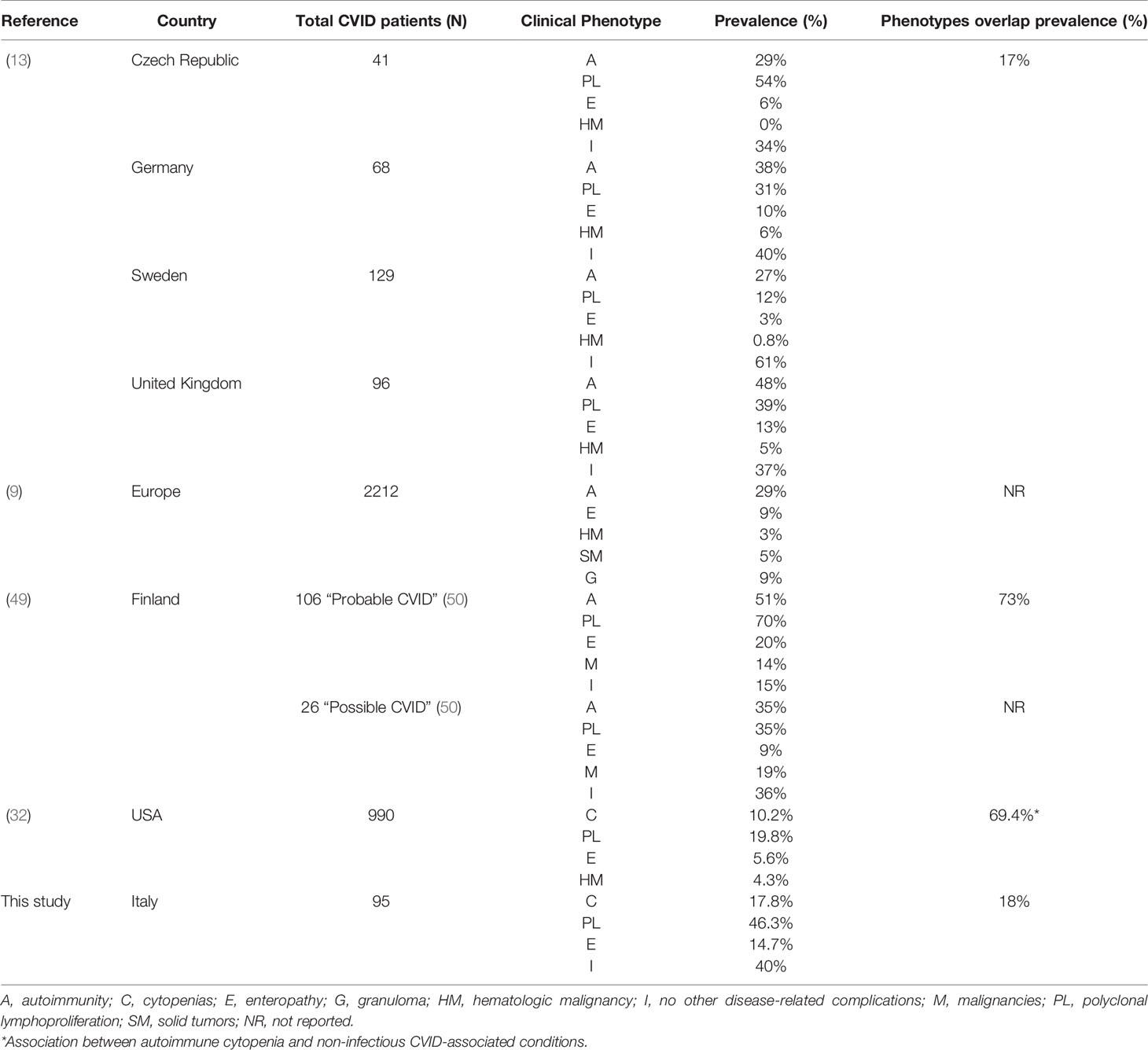
Table 4 Prevalence of CVID clinical phenotypes and their overlap in geographically different cohorts.
Dermatological involvement including alopecia totalis, lichen planus, and vitiligo has occasionally been reported in CVID (31). Psoriasis typically affects approximately 2% to 4% of the general population (51) and it has been considered at length an uncommon manifestation in CVID, seen in less than 1% of patients (52). However, data regarding frequency and features of psoriasis in CVID patients were previously limited to few case reports. Gualdi et al. (53) reported for the first time that psoriasis prevalence was much higher in the CVID patients (9/47 patients; 19.14%) than the general population. The cause of this higher prevalence is not well-known but contributing factors may include T-cell imbalance or other immune-related dysregulation mechanisms (53). A recent one-year observational prospective study carried out by our research group (54) performed a complete dermatological examination on 58 patients with CVID describing psoriasis in 13 out of 58 patients (22%). Interestingly almost all patients (12 of 13; 92%) presented a mild form of psoriasis, possibly due to the immunomodulatory effects of IgRT on dendritic cells, regulatory T cells, B cells, and NK cells functions, which are involved in the pathogenesis of the dermatosis (54, 55). In addition, marked clinical improvement in psoriasis has been observed after the initiation of IgRT, which has a significant role in the management of psoriasis in CVID patients.
Rheumatologic disorders are also frequently described in CVID patients (46) and chronic inflammatory arthritis occurs in approximately 3% of CVID patients (6, 10, 41, 46). The most frequent joint manifestation in CVID is aseptic, possibly immune-mediated arthritis (1-10%), characterized by a symmetrical involvement of limited or several joints, especially knees, ankles, and hands (56). However, septic arthritis should always be excluded, as patients with CVID can often present arthritis due to extracellular bacteria, encapsulated bacteria (Streptococcus pneumoniae, Haemophilus influenzae, and Staphylococcus aureus), or even atypical bacteria (Mycoplasma hominis, Mycoplasma pneumoniae, Mycoplasma salivarium, and Ureaplasma urealyticum), as well as enteroviruses (16, 17). Differential diagnosis should also include amyloidosis; indeed, in CVID patients with recurrent infections, amyloidosis is a potential cause of joint inflammation (31).
Accordingly, the most common rheumatologic condition in our CVID patients was inflammatory arthritis, but we also reported erythema nodosum and Raynaud’s phenomenon in 2 out of 39 patients. Conventional radiography and Magnetic Resonance Imaging are useful diagnostic tools to assess the diagnosis and to identify erosive and non-erosive arthritis, as described in one patient included in our study (Figure 6). On the contrary, antinuclear antibodies and rheumatoid factor are typically absent in CVID (31). Synovial biopsies show synovial hyperplasia and capillary proliferation; the classic synovial infiltrates of B lymphocytes and polymorphonuclear leukocytes are usually absent, while the T infiltrates are mainly composed of CD8+ T cells (57).
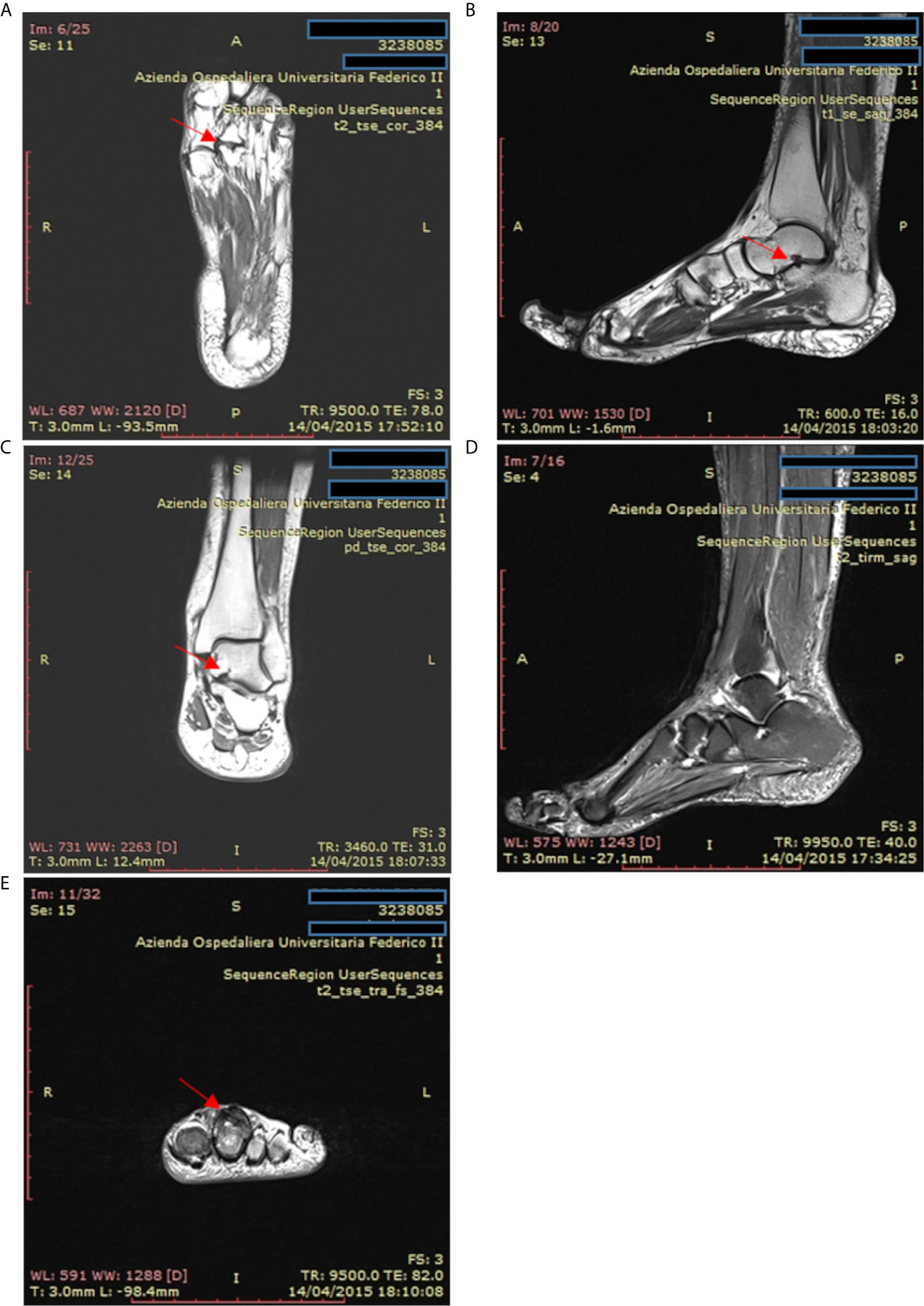
Figure 6 Magnetic resonance imaging of the ankle and foot in a patient with psoriatic arthritis and common variable immunodeficiency. (A) T2-weighted coronal magnetic resonance images of the left foot in a patient with psoriatic arthritis. Erosions are present at the second metatarsal-phalangeal joint (arrow). (B, C) T1-weighted sagittal axis and coronal magnetic resonance images showing cystic erosion (7 mm) of the floor of the sinus tarsi (arrow). (D) T2-weighted sagittal magnetic resonance images of foot and ankle showing joint effusion in the tibiotarsic compartment and enthesitis at the Achilles tendon insertion. (E) T2-weighted axial magnetic resonance images of the fingers from a patient with psoriatic arthritis exhibiting flexor and extensor tenosynovitis at the second finger (arrow).
The treatment of rheumatological disease in CVID is almost the same as that of primary autoimmune disease. Glucocorticoids represent the first line of treatment for all the complications of CVID associated with immune dysregulation. Other treatment strategies, administrated in combination with IgRT (9, 29, 30), include Disease Modifying Antirheumatic Drugs (DMARDs) (e.g., hydroxychloroquine, methotrexate, azathioprine, and mycophenolate mofetil) or new biotechnological therapies [e.g., monoclonal antibodies targeting Tumor Necrosis Factor-alpha (TNF-alpha)] (58). In addition, over the last 5-10 years, rituximab (RTX) has proven to be an effective and relatively safe second-line therapy for both autoimmune and non-malignant lymphoproliferative manifestations (59). Abatacept (a fusion protein of the extracellular domain of CTLA-4 and human IgG1, which prevents antigen-presenting cells from delivering the co-stimulatory signal) has also been used to treat CVID associated autoimmune cytopenia with promising results (60, 61).
In conclusion, autoimmune manifestations, especially cytopenia, and inflammatory diseases are much more frequent in patients with PIDs than in the general population. For this reason, patients with autoimmune anemia, thrombocytopenia, or both should always be screened for PIDs, including CVID. Early recognition and tailored treatments of autoimmune conditions are pivotal to ensure a better quality of life and the reduction of complications associated with CVID. Indeed, thanks to the recent molecular and genetic findings in this immunodeficiency, more targeted approaches are available through precision medicine therapy. For this reason, physicians should raise their awareness about the autoimmune manifestation of PIDs to avoid diagnostic delays and ensure adequate pharmacological therapies.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
This study was performed in accordance with the principles of the Helsinki Declaration. Written informed consent was obtained from every subject involved in this study. An informed written consent was obtained for the use of human images (Figure 4).
IM, FG, AdP, GS, and FR participated in planning the study, drafting the article, critical revision of the article for important intellectual content, and final approval of the article. AP, MW, and CR participated in drafting the article, analysis, and interpretation of data. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AIHA, Autoimmune hemolytic anemia; BAFF-R, B cell-activating factor receptor; CD, Cluster of differentiation; CID, Combined immunodeficiency; CVID, Common variable immunodeficiency; CTLA-4, Cytotoxic T-lymphocyte antigen 4; DMARDs, Disease-modifying anti-rheumatic drugs; ESID, European Society for Immunodeficiency; GLILD, Granulomatous and Lymphocytic Interstitial Lung Disease; ICOS, Inducible T Cell Costimulator; ICOS-L, Inducible T Cell Costimulator Ligand; IgRT, Immunoglobulin replacement therapy; Ig, immunoglobulin; ITP, Immune thrombocytopenia; IVIG, Intravenous immunoglobulin; LRBA, Lipopolysaccharide-responsive beige-like anchor protein; PID, Primary immunodeficiency; SEM, Standard error of the mean; pDCs, Plasmacytoid dendritic cell; RTX, Rituximab; TACI, Transmembrane activator and the calcium-modulator and cyclophilin ligand interactor; TLR, Toll-like receptor, TNF-alpha, Tumor Necrosis Factor-alpha; Treg, T regulatory cells.
1. Agarwal S, Cunningham-Rundles C. Autoimmunity in Common Variable Immunodeficiency. Ann Allergy Asthma Immunol (2019) 123:454–60. doi: 10.1016/j.anai.2019.07.014
2. Geha RS, Notarangelo LD, Casanova JL, Chapel H, Conley ME, Fischer A, et al. Primary Immunodeficiency Diseases: An Update From the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol (2007) 120:776–94. doi: 10.1016/j.jaci.2007.08.053
3. Salzer U, Warnatz K, Peter HH. Common Variable Immunodeficiency: An Update. Arthritis Res Ther (2012) 14:223. doi: 10.1186/ar4032
4. Fischer A, Provot J, Jais JP, Alcais A, Mahlaoui N. Members of The Cfpidsg. Autoimmune and Inflammatory Manifestations Occur Frequently in Patients With Primary Immunodeficiencies. J Allergy Clin Immunol (2017) 140:1388–93.e8. doi: 10.1016/j.jaci.2016.12.978
5. Cunningham-Rundles C, Bodian C. Common Variable Immunodeficiency: Clinical and Immunological Features of 248 Patients. Clin Immunol (1999) 92:34–48. doi: 10.1006/clim.1999.4725
6. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and Mortality in Common Variable Immune Deficiency Over 4 Decades. Blood (2012) 119:1650–7. doi: 10.1182/blood-2011-09-377945
7. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass Trial: Defining Subgroups in Common Variable Immunodeficiency. Blood (2008) 111:77–85. doi: 10.1182/blood-2007-06-091744
8. Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al. Long-Term Follow-Up and Outcome of a Large Cohort of Patients With Common Variable Immunodeficiency. J Clin Immunol (2007) 27:308–16. doi: 10.1007/s10875-007-9075-1
9. Gathmann B, Mahlaoui N, Gerard L, Oksenhendler E, Warnatz K, Schulze I, et al. Clinical Picture and Treatment of 2212 Patients With Common Variable Immunodeficiency. J Allergy Clin Immunol (2014) 134:116–26. doi: 10.1016/j.jaci.2013.12.1077
10. Maglione PJ. Autoimmune and Lymphoproliferative Complications of Common Variable Immunodeficiency. Curr Allergy Asthma Rep (2016) 16:19. doi: 10.1007/s11882-016-0597-6
11. Graziano V, Pecoraro A, Mormile I, Quaremba G, Genovese A, Buccelli C, et al. Delay in Diagnosis Affects the Clinical Outcome in a Cohort of Cvid Patients With Marked Reduction of Iga Serum Levels. Clin Immunol (2017) 180:1–4. doi: 10.1016/j.clim.2017.03.011
12. De Valles-Ibanez G, Esteve-Sole A, Piquer M, Gonzalez-Navarro EA, Hernandez-Rodriguez J, Laayouni H, et al. Evaluating the Genetics of Common Variable Immunodeficiency: Monogenetic Model and Beyond. Front Immunol (2018) 9:636. doi: 10.3389/fimmu.2018.00636
13. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common Variable Immunodeficiency Disorders: Division Into Distinct Clinical Phenotypes. Blood (2008) 112:277–86. doi: 10.1182/blood-2007-11-124545
14. Chapel H, Lucas M, Patel S, Lee M, Cunningham-Rundles C, Resnick E, et al. Confirmation and Improvement of Criteria for Clinical Phenotyping in Common Variable Immunodeficiency Disorders in Replicate Cohorts. J Allergy Clin Immunol (2012) 130:1197–8.e9. doi: 10.1016/j.jaci.2012.05.046
15. Azizi G, Ziaee V, Tavakol M, Alinia T, Yazdai R, Mohammadi H, et al. Approach to the Management of Autoimmunity in Primary Immunodeficiency. Scand J Immunol (2017) 85:13–29. doi: 10.1111/sji.12506
16. Ardeniz O, Gulbahar O, Mete N, Cicek C, Basoglu OK, Sin A, et al. Chlamydia Pneumoniae Arthritis in a Patient With Common Variable Immunodeficiency. Ann Allergy Asthma Immunol (2005) 94:504–8. doi: 10.1016/S1081-1206(10)61122-2
17. Steuer A, Franz A, Furr PM, Taylor-Robinson D, Webster AD, Hughes GR. Common Variable Immunodeficiency Presenting as a Mycoplasma Hominis Septic Arthritis. J Infect (1996) 33:235–7. doi: 10.1016/s0163-4453(96)92441-x
18. Galdarossa M, Vianello F, Tezza F, Allemand E, Treleani M, Scarparo P, et al. Epidemiology of Primary and Secondary Thrombocytopenia: First Analysis of an Administrative Database in a Major Italian Institution. Blood Coagul Fibrinolysis (2012) 23:271–7. doi: 10.1097/MBC.0b013e328351882d
19. Rossini M, Rossi E, Bernardi D, Viapiana O, Gatti D, Idolazzi L, et al. Prevalence and Incidence of Rheumatoid Arthritis in Italy. Rheumatol Int (2014) 34:659–64. doi: 10.1007/s00296-014-2974-6
20. Prignano F, Rogai V, Cavallucci E, Bitossi A, Hammen V, Cantini F. Epidemiology of Psoriasis and Psoriatic Arthritis in Italy-A Systematic Review. Curr Rheumatol Rep (2018) 20:43. doi: 10.1007/s11926-018-0753-1
21. Ingordo V, Gentile C, Iannazzone SS, Cusano F, Naldi L. Vitiligo and Autoimmunity: An Epidemiological Study in a Representative Sample of Young Italian Males. J Eur Acad Dermatol Venereol (2011) 25:105–9. doi: 10.1111/j.1468-3083.2010.03696.x
22. Filippi U, Brizzolara R, Venuti D, Cesarone A, Maritati VA, Podesta M, et al. Prevalence of Post-Partum Thyroiditis in Liguria (Italy): An Observational Study. J Endocrinol Invest (2008) 31:1063–8. doi: 10.1007/BF03345653
23. Volta U, Bellentani S, Bianchi FB, Brandi G, De Franceschi L, Miglioli L, et al. High Prevalence of Celiac Disease in Italian General Population. Dig Dis Sci (2001) 46:1500–5. doi: 10.1023/a:1010648122797
24. Coati I, Fassan M, Farinati F, Graham DY, Genta RM, Rugge M. Autoimmune Gastritis: Pathologist’s Viewpoint. World J Gastroenterol (2015) 21:12179–89. doi: 10.3748/wjg.v21.i42.12179
25. Megiorni F, Pizzuti A, Mora B, Rizzuti A, Garelli V, Maxia C, et al. Genetic Association of HLA-DQB1 and HLA-DRB1 Polymorphisms With Alopecia Areata in the Italian Population. Br J Dermatol (2011) 165:823–7. doi: 10.1111/j.1365-2133.2011.10466.x
26. Marzioni M, Bassanelli C, Ripellino C, Urbinati D, Alvaro D. Epidemiology of Primary Biliary Cholangitis in Italy: Evidence From a Real-World Database. Dig Liver Dis (2019) 51:724–9. doi: 10.1016/j.dld.2018.11.008
27. De Angelis R, Salaffi F, Grassi W. Raynaud’s Phenomenon: Prevalence in an Italian Population Sample. Clin Rheumatol (2006) 25:506–10. doi: 10.1007/s10067-005-0077-1
28. Boileau J, Mouillot G, Gerard L, Carmagnat M, Rabian C, Oksenhendler E, et al. Autoimmunity in Common Variable Immunodeficiency: Correlation With Lymphocyte Phenotype in the French DEFI Study. J Autoimmun (2011) 36:25–32. doi: 10.1016/j.jaut.2010.10.002
29. Chapel H, Cunningham-Rundles C. Update in Understanding Common Variable Immunodeficiency Disorders (Cvids) and the Management of Patients With These Conditions. Br J Haematol (2009) 145:709–27. doi: 10.1111/j.1365-2141.2009.07669.x
30. Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection Outcomes in Patients With Common Variable Immunodeficiency Disorders: Relationship to Immunoglobulin Therapy Over 22 Years. J Allergy Clin Immunol (2010) 125:1354–60.e4. doi: 10.1016/j.jaci.2010.02.040
31. Xiao X, Miao Q, Chang C, Gershwin ME, Ma X. Common Variable Immunodeficiency and Autoimmunity–An Inconvenient Truth. Autoimmun Rev (2014) 13:858–64. doi: 10.1016/j.autrev.2014.04.006
32. Feuille EJ, Anooshiravani N, Sullivan KE, Fuleihan RL, Cunningham-Rundles C. Autoimmune Cytopenias and Associated Conditions in CVID: A Report From the USIDNET Registry. J Clin Immunol (2018) 38:28–34. doi: 10.1007/s10875-017-0456-9
33. Driessen GJ, Van Zelm MC, Van Hagen PM, Hartwig NG, Trip M, Warris A, et al. B-Cell Replication History and Somatic Hypermutation Status Identify Distinct Pathophysiologic Backgrounds in Common Variable Immunodeficiency. Blood (2011) 118:6814–23. doi: 10.1182/blood-2011-06-361881
34. Yong PF, Tarzi M, Chua I, Grimbacher B, Chee R. Common Variable Immunodeficiency: An Update on Etiology and Management. Immunol Allergy Clin North Am (2008) 28:367–86. doi: 10.1016/j.iac.2008.01.001
35. Warnatz K, Denz A, Drager R, Braun M, Groth C, Wolff-Vorbeck G, et al. Severe Deficiency of Switched Memory B Cells (CD27(+)IgM(-)IgD(-)) in Subgroups of Patients With Common Variable Immunodeficiency: A New Approach to Classify a Heterogeneous Disease. Blood (2002) 99:1544–51. doi: 10.1182/blood.v99.5.1544
36. Piqueras B, Lavenu-Bombled C, Galicier L, Bergeron-Van Der Cruyssen F, Mouthon L, Chevret S, et al. Common Variable Immunodeficiency Patient Classification Based on Impaired B Cell Memory Differentiation Correlates With Clinical Aspects. J Clin Immunol (2003) 23:385–400. doi: 10.1023/a:1025373601374
37. Arumugakani G, Wood PM, Carter CR. Frequency of Treg Cells is Reduced in CVID Patients With Autoimmunity and Splenomegaly and Is Associated With Expanded CD21lo B Lymphocytes. J Clin Immunol (2010) 30:292–300. doi: 10.1007/s10875-009-9351-3
38. Agematsu K, Nagumo H, Shinozaki K, Hokibara S, Yasui K, Terada K, et al. Absence of IgD-CD27(+) Memory B Cell Population in X-Linked Hyper-IgM Syndrome. J Clin Invest (1998) 102:853–60. doi: 10.1172/JCI3409
39. Park JY, Shcherbina A, Rosen FS, Prodeus AP, Remold-O’donnell E. Phenotypic Perturbation of B Cells in the Wiskott-Aldrich Syndrome. Clin Exp Immunol (2005) 139:297–305. doi: 10.1111/j.1365-2249.2005.02693.x
40. Rakhmanov M, Keller B, Gutenberger S, Foerster C, Hoenig M, Driessen G, et al. Circulating CD21low B Cells in Common Variable Immunodeficiency Resemble Tissue Homing, Innate-Like B Cells. Proc Natl Acad Sci USA (2009) 106:13451–6. doi: 10.1073/pnas.0901984106
41. Abolhassani H, Amirkashani D, Parvaneh N, Mohammadinejad P, Gharib B, Shahinpour S, et al. Autoimmune Phenotype in Patients With Common Variable Immunodeficiency. J Investig Allergol Clin Immunol (2013) 23:323–9.
42. Gereige JD, Maglione PJ. Current Understanding and Recent Developments in Common Variable Immunodeficiency Associated Autoimmunity. Front Immunol (2019) 10:2753. doi: 10.3389/fimmu.2019.02753
43. Genre J, Errante PR, Kokron CM, Toledo-Barros M, Camara NO, Rizzo LV. Reduced Frequency of CD4(+)CD25(HIGH)FOXP3(+) Cells and Diminished FOXP3 Expression in Patients With Common Variable Immunodeficiency: A Link to Autoimmunity? Clin Immunol (2009) 132:215–21. doi: 10.1016/j.clim.2009.03.519
44. Arandi N, Mirshafiey A, Abolhassani H, Jeddi-Tehrani M, Edalat R, Sadeghi B, et al. Frequency and Expression of Inhibitory Markers of CD4(+) Cd25(+) FOXP3(+) Regulatory T Cells in Patients With Common Variable Immunodeficiency. Scand J Immunol (2013) 77:405–12. doi: 10.1111/sji.12040
45. Romberg N, Chamberlain N, Saadoun D, Gentile M, Kinnunen T, Ng YS, et al. CVID-Associated TACI Mutations Affect Autoreactive B Cell Selection and Activation. J Clin Invest (2013) 123:4283–93. doi: 10.1172/JCI69854
46. Gutierrez MJ, Sullivan KE, Fuleihan R, Consortium U, Bingham CO 3rd. Phenotypic Characterization of Patients With Rheumatologic Manifestations of Common Variable Immunodeficiency. Semin Arthritis Rheumatol (2018) 48:318–26. doi: 10.1016/j.semarthrit.2018.02.013
47. Odnoletkova I, Kindle G, Quinti I, Grimbacher B, Knerr V, Gathmann B, et al. The Burden of Common Variable Immunodeficiency Disorders: A Retrospective Analysis of the European Society for Immunodeficiency (ESID) Registry Data. Orphanet J Rare Dis (2018) 13:201. doi: 10.1186/s13023-018-0941-0
48. Patuzzo G, Barbieri A, Tinazzi E, Veneri D, Argentino G, Moretta F, et al. Autoimmunity and Infection in Common Variable Immunodeficiency (CVID). Autoimmun Rev (2016) 15:877–82. doi: 10.1016/j.autrev.2016.07.011
49. Selenius JS, Martelius T, Pikkarainen S, Siitonen S, Mattila E, Pietikainen R, et al. Unexpectedly High Prevalence of Common Variable Immunodeficiency in Finland. Front Immunol (2017) 8:1190. doi: 10.3389/fimmu.2017.01190
50. Conley ME, Notarangelo LD, Etzioni A. Diagnostic Criteria for Primary Immunodeficiencies. Representing Pagid (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol (1999) 93:190–7. doi: 10.1006/clim.1999.4799
51. Fotiadou C, Lazaridou E, Ioannides D. Management of Psoriasis in Adolescence. Adolesc Health Med Ther (2014) 5:25–34. doi: 10.2147/AHMT.S36672
52. Jindal AK, Rawat A, Sharma A, Dogra S, Suri D, Singh S. Psoriasis: An Unusual Autoimmune Manifestation in a Boy With Common Variable Immunodeficiency. Indian Dermatol Online J (2017) 8:292–4. doi: 10.4103/2229-5178.209610
53. Gualdi G, Lougaris V, Baronio M, Vitali M, Tampella G, Moratto D, et al. Burden of Skin Disease in Selective Iga Deficiency and Common Variable Immunodeficiency. J Investig Allergol Clin Immunol (2015) 25:369–71.
54. Megna M, Pecoraro A, Balato N, Villani A, Crescenzi L, Balato A, et al. Psoriasis in a Cohort of Patients With Common Variable Immunodeficiency. Br J Dermatol (2019) 180:935–6. doi: 10.1111/bjd.17408
55. Paquin-Proulx D, Sandberg JK. Persistent Immune Activation in CVID and the Role of IVIg in Its Suppression. Front Immunol (2014) 5:637. doi: 10.3389/fimmu.2014.00637
56. Swierkot J, Lewandowicz-Uszynska A, Chlebicki A, Szmyrka-Kaczmarek M, Polanska B, Jankowski A, et al. Rheumatoid Arthritis in a Patient With Common Variable Immunodeficiency: Difficulty in Diagnosis and Therapy. Clin Rheumatol (2006) 25:92–4. doi: 10.1007/s10067-005-1141-6
57. Fernandez-Castro M, Mellor-Pita S, Citores MJ, Munoz P, Tutor-Ureta P, Silva L, et al. Common Variable Immunodeficiency in Systemic Lupus Erythematosus. Semin Arthritis Rheumatol (2007) 36:238–45. doi: 10.1016/j.semarthrit.2006.09.005
58. Luijten RK, Fritsch-Stork RD, Bijlsma JW, Derksen RH. The Use of Glucocorticoids in Systemic Lupus Erythematosus. After 60 Years Still More an Art Than Science. Autoimmun Rev (2013) 12:617–28. doi: 10.1016/j.autrev.2012.12.001
59. Pecoraro A, Crescenzi L, Galdiero MR, Marone G, Rivellese F, Rossi FW, et al. Immunosuppressive Therapy With Rituximab in Common Variable Immunodeficiency. Clin Mol Allergy (2019) 17:9. doi: 10.1186/s12948-019-0113-3
60. Uzel G, Karanovic D, Su H, Rump A, Agharahimi A, Holland S, et al. Management of Cytopenias in CTLA4 Haploinsufficiency Using Abatacept and Sirolimus. Blood (2018) 132:2409–. doi: 10.1182/blood-2018-99-120185
Keywords: common variable immunodeficiency, autoimmunity, arthritis, cytopenia, psoriasis
Citation: Mormile I, Punziano A, Riolo CA, Granata F, Williams M, de Paulis A, Spadaro G and Rossi FW (2021) Common Variable Immunodeficiency and Autoimmune Diseases: A Retrospective Study of 95 Adult Patients in a Single Tertiary Care Center. Front. Immunol. 12:652487. doi: 10.3389/fimmu.2021.652487
Received: 12 January 2021; Accepted: 15 June 2021;
Published: 05 July 2021.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Mikko Risto Juhana Seppänen, Helsinki University Central Hospital, FinlandCopyright © 2021 Mormile, Punziano, Riolo, Granata, Williams, de Paulis, Spadaro and Rossi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Spadaro, c3BhZGFyb0B1bmluYS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.