 Grace E. Ryan
Grace E. Ryan John E. Harris
John E. Harris Jillian M. Richmond
Jillian M. Richmond
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 03 May 2021
Sec. T Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.652191
This article is part of the Research TopicT Cells in Skin Inflammatory DisordersView all 9 articles
Tissue resident memory T cells (TRM) are a critical component of the immune system, providing the body with an immediate and highly specific response against pathogens re-infecting peripheral tissues. More recently, however, it has been demonstrated that TRM cells also form during autoimmunity. TRM mediated autoimmune diseases are particularly destructive, because unlike foreign antigens, the self-antigens are never cleared, continuously activating self-reactive TRM T cells. In this article, we will focus on how TRMs mediate disease in autoimmune skin conditions, specifically vitiligo, psoriasis, cutaneous lupus erythematosus, alopecia areata and frontal fibrosing alopecia.
Tissue resident memory T cells (TRM) are long-lived lymphocytes that reside in tissues and develop after a T cell-mediated immune response is initiated. Subpopulations of memory T cells can recirculate through the blood and lymphoid organs T recirculating memory cells (TRCM), though evidence suggests that true TRM have a reduced capacity for recirculation. Both CD4+ and CD8+ TRM have been described in skin and other tissues including the mucosa, lung, brain, GI tract and pancreas (1–5). Because of the very different locations that TRM are found, they are an extremely diverse population, showing specializations for their resident tissue. Mouse models reveal that TRMs function as sentinel-alarm cells that secrete cytokines and chemokines upon encountering their cognate antigen (6–8), with some infections such as vaccinia virus requiring TRM for optimal viral clearance (9). There is mounting evidence that TRM work together with other effector and memory T cell populations to provide tissue surveillance and clear infections (8, 10).
Compared to TRM in other tissues, skin TRM persist for years and express skin-specific homing antigens, such as cutaneous lymphocyte antigen (CLA) and CCR8. In addition, certain skin retention markers, including CD103 and CD69, are upregulated (11). Mouse models have demonstrated that TRM require IL-15 and TGFβ for their differentiation in the skin (12), and human TRM express their receptors (3, 13). The tissue-residency program that ensues involves downregulation of chemokine receptors involved in recirculation, including CCR7 and S1P1, as well as upregulation of key integrins, cytokine/growth factor receptors, and signaling molecules including CD69, CD103, CD49a, CD122 and PD-1, a pattern that is conserved in both mice and humans (5, 14–16). Skin TRM survival is dependent upon IL-15 and fatty acid metabolism (3, 17) (Figure 1).
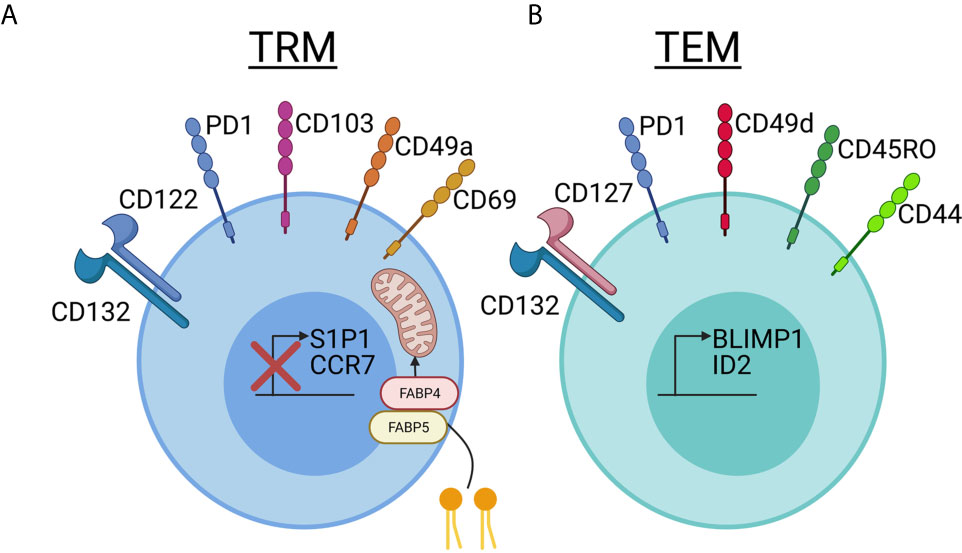
Figure 1 Comparing and contrasting tissue resident memory and effector memory T cell phenotypes in skin. (A) Tissue resident memory (TRM, left, blue) cells downregulate S1P1 and CCR7, which are used for recirculation in the blood and lymph, when they take up residency in skin. TRMs use CD69, which was previously thought to be a marker of recently activated T cells, to block the expression of S1P1 on the cell surface and prevent exit from the tissue. CD103 is an integrin family member that is involved in skin and mucosal homing. It is not required for skin retention, but is expressed at a much higher rate in TRM cells in the epidermis compared to T cells in the dermis. CD49a has recently been used to distinguish the CD8+CD103+CD49a+ TRM T cells involved in mediating disease in vitiligo from the CD8+CD103+CD49a- TRM cells involved in psoriasis. TRM cells survive long-term in the nutrient-poor conditions of the skin by metabolizing free fatty acids. Increased expression of FABP4 and FABP5, fatty-acid binding proteins, allows TRM cells to take up larger amounts of exogenous free fatty acids for energy. IL-15 is required for TRM differentiation in skin, and TRM bear the CD122 and CD132 receptor chains. Keratinocytes can present IL-15 to TRM in trans on the CD215 receptor chain. TRM cells increase expression of PD1, which has a known inhibitory role in immune activation, and is important in maintaining immune homeostasis. Reduced expression of PD-1 in TRMs is associated with inappropriate chronic inflammation. CD69, which was historically thought to be a T cell activation marker, is stably expressed on Trm. (B) T effector memory (TEM, right, green) cells can also express varying levels of PD-1, making this marker useful more for determining exhaustion and/or chronic inflammation rather than tissue residence. TEM seem to rely more on CD127/IL-7 as a survival factor, which is in contrast to the IL-15 dependency of TRM. More often, they express CD49d than CD49a, though CD49a may also be used as a marker of degranulation. CD49d can interact intracellularly with CD44 to promote signaling, though CD44 is often present on both TRM and TEM and is used as a marker of “antigen experienced” T cells. CD45RO has historically been used to identify TEM in blood, and this short isoform of CD45 plays a role in T cell activation and signaling though a variety of pathways including JAK/STAT and Src. The transcription factors BLMP1 and ID2 promote expression of TEM-associated markers. Created with BioRender.com.
In addition to anti-viral responses, TRM also develop in the context of xenobiotic and/or toxin exposure, allergies and autoimmune diseases. For example, fixed drug eruption is thought to be mediated by TRM (18). Type IV hypersensitivity to poison ivy, other contact dermatitis irritants or allergens induces TRM development (19). Healthy skin donors exhibit autoreactive T cell clones, which are thought to provide immune surveillance against tumors (3, 20). However, in autoimmune skin conditions, environmental and genetic factors precipitate inappropriate activation of autoreactive T cells and promote onset of autoimmunity. Further, a reduced ratio of Tregs to TRM and T effectors and/or reduced functionality of Tregs in skin tissue tips the balance towards development of autoimmunity (21–25), which may become recalcitrant or relapse following treatment cessation due to the persistence of autoreactive TRM. In this minireview, we will focus on TRM in autoimmune skin diseases, including discussions of how they may be targeted for durable treatment options.
Vitiligo is characterized by distinct patchy white spots present on patients’ skin, which are caused by CD8+ T cells inappropriately targeting melanocytes for destruction. Many studies have implicated both genetic and environmental factors are important in the pathogenesis of vitiligo (26). Familial clustering of vitiligo suggested that there was a genetic component before genetic studies were available. Alkhateeb et al. reported in 2003 that approximately 20% of people with vitiligo reported at least one affected first-degree relative (27). Since then, genome-wide association studies (GWAS) have identified multiple genes associated with a higher risk for developing vitiligo, including genes involved in both innate and adaptive immunity (28, 29). It is now recognized that melanocytes in vitiligo patients have a reduced ability to manage cellular stress, making them more vulnerable to environmental stressors such as UV radiation and certain chemicals (30, 31). Cytokines and danger associated molecules released in response to states of high stress in the skin attract and activate immune cells (32–34). In addition, the IFNγ-CXCR3 pathway has been determined to be critical to the migration of autoreactive CD8+ T cells in vitiligo in mice and humans, and correlates with disease progression and severity (35–39). While treatments are available for vitiligo patients, the white spots rapidly reappear in the same location if therapy is stopped, demonstrating that memory persists in the skin even after treatment. For example, JAK/STAT signaling is recognized as a key mediator in many of the effector functions of TRM cells, including the production of cytokines and inflammation inducers. JAK inhibitors interrupt the IFNγ signaling pathway and cause skin repigmentation; however, depigmentation recurs when treatment is stopped (40). Azzolino et al. recently reported that established TRM numbers in skin in a mouse model of vitiligo are not affected by Jak inhibitors, thus providing an explanation for why JAKi do not provide durable treatment responses (41).
T cells in vitiligo patients react to melanocyte self-antigens including gp100/Pmel-17, melan-A/MART-1, tyrosinase and tyrosinase-related proteins 1 & 2 (42–47). Recent studies have characterized melanocyte-specific CD8+ TRMs from patients with active and stable vitiligo using blister biopsies and punch biopsies (3, 4, 48). Approximately 80% of the melanocyte-specific CD8+ T cells express TRM markers, including CD69 single positive cells (which may be actively differentiating, or recirculating cells) and CD69/CD103 double positive cells (3). CD49a, a marker that distinguishes a subset of CD8+ CD103+ T cells with the cytotoxic potential to produce IFNγ, is expressed in half of epidermal and less than a quarter of dermal CD8+ CD103+ T cells in patients with vitiligo. Comparatively, in healthy skin, it is expressed by one third of epidermal and less than 5% of dermal CD8+ CD103+ T cells (4). In all patients with vitiligo, these cells were enriched in the skin; moreover, in patients with active disease, there was a significant increase in the number of T cells as compared to those with stable disease (36), particularly CD69+ (3). Boniface et al. found co-expression of CXCR3 on CD69+CD103+ melanocyte-specific T cells in vitiligo skin at a higher frequency than psoriasis or healthy skin (48). CD8+ TRM are also present in depigmenting lesions in post-melanoma-immunotherapy-associated vitiligo (49).
We examined factors required for maintenance of these autoreactive TRM in the skin during vitiligo, with the hope of identifying new durable treatments targeting autoreactive TRM. Based on Mackay et al, which demonstrated TRM require IL-15 for their differentiation, we tested whether blocking this cytokine pathway could deplete TRM. Treating mice with anti-CD122 antibodies, which block IL-15 signaling and thus TRM proliferation and survival, provided long-lasting repigmentation (8) (Figure 2A). Other labs have mapped additional roles IL-15 plays in vitiligo. Chen et al. demonstrated that oxidative stress can result in increased IL-15 transpresentation on CD215 by keratinocytes (34). Jacquemin et al. demonstrated that IL-15 can induce NKG2D expression on effector memory CD8+ T cells (57), which, based on our FTY720 blockade data demonstrating that TRM are not sufficient for maintaining depigmentation in mice (8), supports a role for effector memory T cells as the melanocyte killers. IL-15 blocking antibodies are now being explored as a potential durable therapy for vitiligo patients (NCT04338581; Table S1).
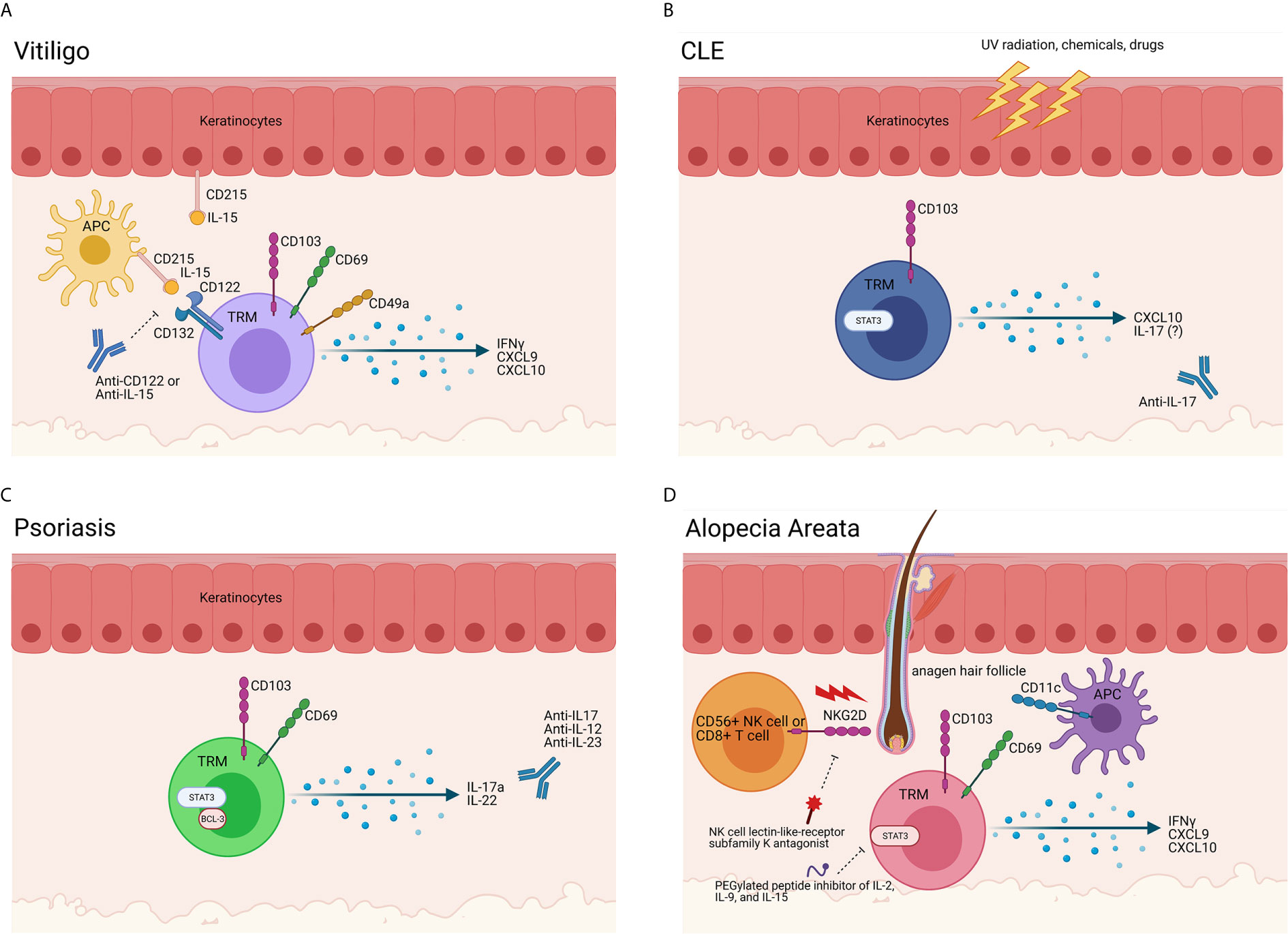
Figure 2 Evidence of TRM in autoimmune skin disease. (A) TRM in vitiligo. Melanocyte-specific TRM are present in vitiligo lesional skin. These cells express CD49a, CD69, CD103, and CD122 and produce IFNγ and chemokines upon encountering cognate antigen, such as gp100/Pmel-17, melan-A/MART-1, tyrosinase and/or tyrosinase-related proteins 1 & 2 presented on HLA-A2. Antigen-presenting cells (APCs) and keratinocytes also play a role in vitiligo immunopathogenesis, as they release cytokines in response to a myriad of stimuli and stressors. APCs produce IL-15 and trans-present it on CD215 to T cells, which bear the CD122 and CD132 chains of the IL-15 receptor. In vitiligo, our data support that TRM act as sentinel/alarm cells to induce recall responses against melanocytes. Blocking CD122 deprives the TRM of the IL-15 survival signal, thereby inhibiting their function and/or reducing their numbers to provide a durable treatment. A clinical trial is currently enrolling to test the potential of a commercially available anti-IL-15 antibody for vitiligo (NCT04338581). (B) TRM cells in CLE. Triggers such as UV radiation, infection, hormones, drugs and chemicals create an apoptotic and inflammatory environment (50). In lupus-prone individuals, antigen-presenting cells (APCs) migrate to the site of injury and ingest the cell debris and possible autoantigens. When the APCs present the autoantigens to self-reactive T cells and B cells, the autoimmune response is initiated, and disease occurs (51). Specifically, CD103+ TRM cells in the skin are positively correlated to disease severity and recurrence in patients. While TRM and T cell antigen specificities in lupus nephritis were recently described (52, 53), specificities of skin autoreactive T cells are unknown. A trial blocking anti-IL-17 antibody for DLE is recruiting (NCT03866317). (C) TRM cells in Psoriasis. CD69+ CD103+ TRM are present in psoriasis lesions. The production of cytokines such as IL-17a, IL-22, and Bcl-3 by these TRM cells lead to aberrant keratinocyte proliferation and differentiation. A recent study described melanocyte-specific T cells in psoriasis patients, though other specificities are unknown (54). Many trials for IL-17 family cytokine blockade, including IL-12/23, are ongoing. (D) TRM cells in Alopecia Areata (AA) and Cicatricial Alopecia (CA). NKG2D+ CD8+ T cells begin the disease process, breaching the immune privileged hair follicle through the activation of T cells and dendritic cells. Though the exact role of TRM cells in the pathogenesis of AA and FFA is unknown, it is clear that the production of IFNγ, CXCL9, and CXCL10 by T cells in the hair follicle is crucial in the development in disease. While clonal expansions of CD8 T cells have been detected with TCR sequencing (55), the antigen specificities of TRM in alopecias are still largely unknown (56). Trials for NK cell receptor and IL-2/9/15 inhibition are planned (NCT03532958 and NCT04740970). Created with BioRender.com.
Cutaneous lupus erythematosus (CLE) is a spectrum of autoimmune skin diseases encompassing several clinical subtypes, all of which are characterized by interface dermatitis, or inflammation at the dermal-epidermal junction (58). The most well-known manifestation of CLE is the characteristic erythematous malar rash, which is associated with systemic lupus erythematosus (SLE). Other CLE subtypes can present without any systemic involvement, with different entities conferring different levels of risk of developing SLE. Genetic factors and environmental stressors both contribute to the etiology of CLE (Figure 2B).
The T cell specificities in CLE skin are largely unknown, but SLE studies identified T cells reactive to nucleosomes/histones (59), which can induce anti-dsDNA antibody production (60). A recent study characterized antigen specificities of T cells in the kidney in lupus nephritis patients, and found that they recognize SmD1, RNP70, histone, Ro, La, and nuclear antigen (52, 61). A population of CD8+ CD103+ tissue-resident memory T cells was identified and correlated positively with disease severity in patients (53). Interestingly, tofacitinib, a JAK3>1>>2 inhibitor, was effective in preventing disease progression in mouse models of lupus nephritis and suppressed the development of CD8+ TRM cells. Thus, the microenvironment that the TRM cells create in lupus nephritis appears to be crucial for disease progression.
Many studies have shown aberrant T cell signaling contributes to the pathogenesis of CLE (62), including expression of cytotoxic markers characteristic of T cell function (63, 64). However, it is only recently that TRM cells have been linked to disease progression: In the skin, increased TRM were found in CLE patients refractory to antimalarials (65). JAK/STAT inhibitors have also demonstrated efficacy in treating CLE: In a case study of a patient with chilblain lupus, a subtype of CLE, the JAK 1/2-kinase inhibitor ruxolitinib provided a rapid response. When ruxolitinib was discontinued, the skin lesions re-appeared, but upon re-starting the drug they resolved completely (66). Thus, similar to vitiligo studies, TRM in CLE are not affected permanently by Jak inhibitors, and disease recurs once treatment is stopped. Examination of skin biopsies taken from the patient revealed that ruxolitinib inhibited the production of CLE-typical chemokines, specifically CXCL10, supporting the idea that TRM cytokine production and downstream signaling drives recruitment of recirculating cytotoxic cells to the skin, as was demonstrated in vitiligo mouse models described above. Another study identified IL-17+ cells by immunohistochemistry (67); a trial assessing IL-17 blockade for discoid lupus erythematosus is recruiting (Table S1).
Psoriasis is a chronic and relapsing inflammatory skin disease mediated by T cells. As in other autoimmune skin diseases, the relapsing nature of psoriasis suggested the presence of immune memory in the skin. Using a humanized mouse model of psoriasis, transplantation of unaffected skin from a patient with psoriasis was grafted onto mice deficient in type I and type II interferons and the recombination activating gene 2, (IFNG-/- IFNAR-/- RAG2-/-) and the skin graft spontaneously developed psoriasis. Thus, the immune cells already resident in the grafted skin were sufficient for inducing disease (68). Moreover, E-selectin inhibitors are not efficacious in preventing psoriasis, further supporting that nonmigratory immune cells can mediate disease (69). However, the question of whether TRM are sufficient, or whether they also require TRCM, has not yet definitively been addressed, because the resident immune cells in skin graft experiments may have given rise to TRCM, and there may be redundant pathways in addition to E-selectin that mediate skin recruitment in psoriasis.
Cheuk et al. demonstrated that the marker CD49a identifies two distinct populations of CD8+ CD103+ TRM cells. While vitiligo disease is driven by CD49a+ TRM cells producing IFNγ, CD49a- TRM cells in psoriasis mediate disease through the production of interleukin-17 (IL-17) (4, 70), (Figure 2C). The identification of CD49a- IL-17+ T cells as critical mediators of disease fits with the previous success of anti-IL-17 drugs that mediate successful amelioration of disease (UptoDate). The FDA has approved several monoclonal antibodies to IL-17 for the treatment of psoriasis: tildrakizumab, guselkumab, risankizumab, secukinumab, ixekizumab, and brodalamub (Table S1). Scientists have also developed drugs to target IL-23, a cytokine produced by myeloid dendritic cells to activate IL-17-producing CD49a- TRM cells as well as non-TRM IL-17-producing cells (71). Nevertheless, IL-17 blockade may require long-term administration, as it is blocking a molecule made by the TRM rather than targeting TRM themselves. In a clinical study of ten patients with psoriasis receiving secukinumab for 24 weeks, Fujiyama et al. demonstrated that while there was a significant decrease in CD8+ CD103+ cells in lesional skin, there was only a slight decrease in CD8+ CD103+ CD49- cells, suggesting that TRM cells are preserved (72).
While CD8+ CD103+ TRM cells make up the majority of the T cell infiltrate in psoriatic lesions, CD4+ TRM cells are also present in low numbers in the epidermis and dermis (73). Like CD8+ TRM, CD4+ TRM are identified by the marker CD69+. However, unlike CD8+ TRM, CD4+ TRM cells show variability in the expression of CD103, and the majority of CD4+ TRM do not express it. CD4+ TRM were further distinguished from CD8+ TRM cells by Cheuk et al, who demonstrated that epidermal CD4+ TRM cells in resolved psoriatic lesions produced IL-22 while CD8+ T cells mainly produced IL-17A. IL-22 is a pro-inflammatory cytokine that promotes the expression of multiple chemokines in the skin (74). In addition, through the STAT3 pathway, IL-22 promotes the proliferation and decreases the differentiation of keratinocytes, a hallmark of psoriatic skin (75). Using immunohistochemistry, Tohyama et al. reported that the production of IL-17A by CD8+ TRM cells and IL-22 by CD4+ TRM cells correlated with Bcl-3 production, IL-22-induced gene expression, and expression of other genes associated with psoriasis in lesional skin as compared to healthy control skin. By targeting Bcl-3, both CD4+ and CD8+ TRM disease pathways in psoriasis could be interrupted (76). It is now understood that expression of Bcl-3 promotes CD4+ T cell survival and is necessary for the proper development of Th1, Th2, and Th17 cells (77). While Bcl-3 inhibitors have been extensively studied as potential therapeutics in cancer, their role as a therapeutic in psoriasis or other skin diseases has not yet been explored.
Although blocking the psoriatic microenvironment produced by TRM cells has proven effective in treating disease, scientists are also looking at how to directly target TRM cells. Pan et al. examined the metabolic pathways of mouse CD8+ TRM cells in order to elucidate how TRM cells survive in the nutrient-poor conditions of the skin. In both mouse models and skin biopsies from patients with psoriatic lesions, it was found that TRM T cells required exogenous free fatty acids to survive in the skin. This characteristic is unique to TRM T cells and could be a potential target for treatment for skin diseases mediated by TRM T cells (17).
The hair follicle has long been recognized as an immune privileged site, as it protects the hair from potential autoreactive immune attacks through a system of complex regulation and suppression of the immune system. When this system breaks down, hair loss can result (78–80). Alopecia areata (AA) and cicatricial alopecia (CA) are common, inflammatory hair loss conditions. While CA is scarring and has a distinct clinical presentation of hair loss along the frontotemporal hairline and eyebrows, AA can present variably, ranging from a single patch of hair loss to total body hair loss and is non-scarring (56, 81). A combination of genetics and environmental triggers have been implicated in the pathogenesis of both alopecias.
CA is a diverse group of scarring hair loss encompassing several clinical variants including frontal fibrosing alopecia (FFA), lichen planopilaris (LPP) and central centrifugal cicatricial alopecia (CCCA). FFA and LPP most commonly occur in postmenopausal Caucasian women, however there have been a few cases in younger women (82). Autoimmune thyroid disease is the most common comorbid autoimmune disorder with FFA and LPP. In contrast, CCCA almost exclusively occurs in African American women (83), and Type 2 diabetes is the most common comorbidity.
AA affects people of all different ages, sexes, and ethnic backgrounds (84). To date, the immunopathogenesis of AA is the most well-characterized of the alopecias: AA is driven by CD8+ T cells and natural killer (NK) cells as evidenced by both mouse models and studies of ex vivo human tissues (80). NKG2D+ cells are central to pathogenesis (85), as NKG2D activates both CD8+ T cells and NK cells to promote destruction of the hair follicle. A clinical trial targeting NK cell lectin-like-receptor subfamily K is currently enrolling (Table S1).
While anagen hair follicle epithelium is the target of CD8+ T cells in AA, FFA is mediated by destruction of stem cells in the hair follicle (86). Targets in CCCA have not been well-characterized, and future studies of alopecia in patients with skin of color are warranted to understand nuances in immunopathogenesis and potential novel treatment options.
Del Duca et al. recently determined that lesional skin from AA and FFA patients has significantly more CD8+ cytotoxic T cells, CD11c+ dendritic cells, and CD103+ CD69+ TRM cells when compared to nonlesional skin (87). In particular, FFA was shown to have a significant upregulation of the IFNγ/CXCL9/CXCL10 pathway and the JAK-STAT pathway (88), (Figure 2D). In a recent case series, Yang et al. reported significant clinical improvement as measured using LPPAI scoring in a patient treated with the JAK inhibitor (JAKi) tofacitinib. This patient had previously failed monthly intralesional triamcinolone, topical steroids, doxycycline, and hydroxychloroquine (89). Similarly, AA patients have successfully been treated with JAKi (90–94). However, it is important to note that, like vitiligo, alopecia recurs upon cessation of JAKi (40). The known recurrence of AA and FFA, the confirmed upregulation of CD103+CD69+ TRM cells, and the reported clinical significance of JAKi support the crucial role of TRM cells in disease pathogenesis (50). A new clinical trial for a selective and simultaneous inhibitor of cytokines IL-2, IL-9, and IL-15 is enrolling for alopecia, which may have an impact on TRM (Table S1).
The complex roles of TRM cells in autoimmune skin diseases are just beginning to be understood. However, it is clear that TRM play significant roles in disease reactivation and flares, and must be targeted if we hope to achieve long-lasting and durable treatment options for patients. The tremendous strides in the last ten years in understanding the pathophysiology and treatment of autoimmune skin disease is incredibly exciting, emphasizing not only how much we still have to learn about these diseases, but also the enormous impact it will have on patient survival and quality of life.
GR: original draft and figure drafts. JR and GR: literature search. GR and JR: digital artwork generation. JR and JH: editing. All authors contributed to the article and approved the submitted version.
GR was supported by the UMass Research & Curriculum Exploration program. JR is supported by a Career Development Award from the Dermatology Foundation, a Target Identification in Lupus Award from the Lupus Research Alliance and a Concept Award from the US Department of Defense Lupus Research Program #LR190030. JH is supported by NIH R01 AR069114, R61 AR070302, the Hartford Foundation, and a Calder Research Scholar Award from the American Skin Association. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The views expressed in this article are those of the authors and may not reflect the official policy or position of the Department of the Army, Department of Defense, or the U.S. Government.
JH is the founder of Villaris Therapeutics and NIRA Biosciences. JH and JR are inventors on patent application #62489191, “Diagnosis and Treatment of Vitiligo” which covers targeting IL-15 and TRM for the treatment of vitiligo; and on patent application #15/851,651, “Anti-human CXCR3 antibodies for the Treatment of Vitiligo” which covers targeting CXCR3 for the treatment of vitiligo. JR is a reviewer for Frontiers Immunology journal.
Figures were created with BioRender.com
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.652191/full#supplementary-material
1. Collins N, Jiang X, Zaid A, Macleod BL, Li J, Park CO, et al. Skin CD4+ Memory T Cells Exhibit Combined Cluster-Mediated Retention and Equilibration With the Circulation. Nat Commun (2016) 7:11514. doi: 10.1038/ncomms11514
2. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human Tissue-Resident Memory T Cells are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep (2017) 20:2921–34. doi: 10.1016/j.celrep.2017.08.078
3. Richmond JM, Strassner JP, Zapata L, Garg M, Riding RL, Refat MA, et al. Antibody Blockade of IL-15 Signaling Has the Potential to Durably Reverse Vitiligo. Sci Transl Med (2018) 10(450):eaam7710. doi: 10.1126/scitranslmed.aam7710
4. Cheuk S, Schlums H, Gallais Sérézal I, Martini E, Chiang SC, Marquardt N, et al. Cd49a Expression Defines Tissue-Resident Cd8+ T Cells Poised for Cytotoxic Function in Human Skin. Immunity (2017) 46:287–300. doi: 10.1016/j.immuni.2017.01.009
5. Weisberg SP, Carpenter DJ, Chait M, Dogra P, Gartrell-Corrado RD, Chen AX, et al. Tissue-Resident Memory T Cells Mediate Immune Homeostasis in the Human Pancreas Through the PD-1/PD-L1 Pathway. Cell Rep (2019) 29:3916–32.e5. doi: 10.1016/j.celrep.2019.11.056
6. Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and Alarm Function of Resident Memory Cd8+ T Cells. Nat Immunol (2013) 14:509–13. doi: 10.1038/ni.2568
7. Shin H, Iwasaki A. A Vaccine Strategy That Protects Against Genital Herpes by Establishing Local Memory T Cells. Nature (2012) 491:463–7. doi: 10.1038/nature11522
8. Richmond JM, Strassner JP, Rashighi M, Agarwal P, Garg M, Essien KI, et al. Resident Memory and Recirculating Memory T Cells Cooperate to Maintain Disease in a Mouse Model of Vitiligo. J Invest Dermatol (2018) 139(4):P769–78. doi: 10.1016/j.jid.2018.10.032
9. Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin Infection Generates Non-Migratory Memory CD8+ T(Rm) Cells Providing Global Skin Immunity. Nature (2012) 483:227–31. doi: 10.1038/nature10851
10. Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, et al. Human Skin is Protected by Four Functionally and Phenotypically Discrete Populations of Resident and Recirculating Memory T Cells. Sci Transl Med (2015) 7:279ra39. doi: 10.1126/scitranslmed.3010302
11. Tokura Y, Phadungsaksawasdi P, Kurihara K, Fujiyama T, Honda T. Pathophysiology of Skin Resident Memory T Cells. Front Immunol (2020) 11:618897. doi: 10.3389/fimmu.2020.618897
12. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon M-L, et al. The Developmental Pathway for CD103+CD8+ Tissue-Resident Memory T Cells of Skin. Nat Immunol (2013) 14:1294. doi: 10.1038/ni.2744
13. Mohammed J, Beura LK, Bobr A, Astry B, Chicoine B, Kashem SW, et al. Stromal Cells Control the Epithelial Residence of DCs and Memory T Cells by Regulated Activation of TGF-β. Nat Immunol (2016) 17:414–21. doi: 10.1038/ni.3396
14. Schenkel JM, Masopust D. Tissue-Resident Memory T Cells. Immunity (2014) 41:886–97. doi: 10.1016/j.immuni.2014.12.007
15. Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, et al. Antigen-Independent Differentiation and Maintenance of Effector-Like Resident Memory T Cells in Tissues. J Immunol (2012) 188:4866–75. doi: 10.4049/jimmunol.1200402
16. Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJC, et al. Distribution and Compartmentalization of Human Circulating and Tissue-Resident Memory T Cell Subsets. Immunity (2013) 38:187–97. doi: 10.1016/j.immuni.2012.09.020
17. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, et al. Survival of Tissue-Resident Memory T Cells Requires Exogenous Lipid Uptake and Metabolism. Nature (2017) 543:252–6. doi: 10.1038/nature21379
18. Shiohara T, Mizukawa Y, Teraki Y. Pathophysiology of Fixed Drug Eruption: The Role of Skin-Resident T Cells. Curr Opin Allergy Clin Immunol (2002) 2:317–23. doi: 10.1097/00130832-200208000-00005
19. Gamradt P, Laoubi L, Nosbaum A, Mutez V, Lenief V, Grande S, et al. Inhibitory Checkpoint Receptors Control Cd8+ Resident Memory T Cells to Prevent Skin Allergy. J Allergy Clin Immunol (2019) 143:2147–57.e9. doi: 10.1016/j.jaci.2018.11.048
20. De Jong A, Peña-Cruz V, Cheng T-Y, Clark RA, Van Rhijn I, Moody DB. CD1a-Autoreactive T Cells are a Normal Component of the Human αβ T Cell Repertoire. Nat Immunol (2010) 11:1102–9. doi: 10.1038/ni.1956
21. Ujiie H. Regulatory T Cells in Autoimmune Skin Diseases. Exp Dermatol (2019) 28:642–6. doi: 10.1111/exd.13535
22. Ali N, Rosenblum MD. Regulatory T Cells in Skin. Immunology (2017) 152:372–81. doi: 10.1111/imm.12791
23. Richmond JM, Strassner JP, Essien KI. Harris JE. T-Cell Positioning by Chemokines in Autoimmune Skin Diseases. Immunol Rev (2019) 289:186–204. doi: 10.1111/imr.12762
24. Asothai R, Anand V, Das D, Antil PS, Khandpur S, Sharma VK, et al. Distinctive Treg Associated CCR4-CCL22 Expression Profile With Altered Frequency of Th17/Treg Cell in the Immunopathogenesis of Pemphigus Vulgaris. Immunobiology (2015) 220:1129–35. doi: 10.1016/j.imbio.2015.06.008
25. Das D, Akhtar S, Kurra S, Gupta S, Sharma A. Emerging Role of Immune Cell Network in Autoimmune Skin Disorders: An Update on Pemphigus, Vitiligo and Psoriasis. Cytokine Growth Factor Rev (2019) 45:35–44. doi: 10.1016/j.cytogfr.2019.01.001
26. Rashighi M, Harris JE. Vitiligo Pathogenesis and Emerging Treatments. Dermatol Clin (2017) 35:257–65. doi: 10.1016/j.det.2016.11.014
27. Alkhateeb A, Fain PR, Thody A, Bennett DC, Spritz RA. Epidemiology of Vitiligo and Associated Autoimmune Diseases in Caucasian Probands and Their Families. Pigment Cell Res (2003) 16:208–14. doi: 10.1034/j.1600-0749.2003.00032.x
28. Spritz RA, Gowan K, Bennett DC, Fain PR. Novel Vitiligo Susceptibility Loci on Chromosomes 7 (AIS2) and 8 (AIS3), Confirmation of SLEV1 on Chromosome 17, and Their Roles in an Autoimmune Diathesis. Am J Hum Genet (2004) 74:188–91. doi: 10.1086/381134
29. Zhang X-J, Chen J-J, Liu J-B. The Genetic Concept of Vitiligo. J Dermatol Sci (2005) 39:137–46. doi: 10.1016/j.jdermsci.2005.06.004
30. Jimbow K, Chen H, Park J-S, Thomas PD. Increased Sensitivity of Melanocytes to Oxidative Stress and Abnormal Expression of Tyrosinase-Related Protein in Vitiligo. Br J Dermatol (2001) 144:55–65. doi: 10.1046/j.1365-2133.2001.03952.x
31. He Y, Li S, Zhang W, Dai W, Cui T, Wang G, et al. Dysregulated Autophagy Increased Melanocyte Sensitivity to H2O2-Induced Oxidative Stress in Vitiligo. Sci Rep (2017) 7:42394. doi: 10.1038/srep42394
32. Toosi S, Orlow SJ, Manga P. Vitiligo-Inducing Phenols Activate the Unfolded Protein Response in Melanocytes Resulting in Upregulation of IL6 and IL8. J Invest Dermatol (2012) 132:2601–9. doi: 10.1038/jid.2012.181
33. Kroll TM, Bommiasamy H, Boissy RE, Hernandez C, Nickoloff BJ, Mestril R, et al. 4-Tertiary Butyl Phenol Exposure Sensitizes Human Melanocytes to Dendritic Cell-Mediated Killing: Relevance to Vitiligo. J Invest Dermatol (2005) 124:798–806. doi: 10.1111/j.0022-202X.2005.23653.x
34. Chen X, Guo W, Chang Y, Chen J, Kang P, Yi X, et al. Oxidative Stress-Induced Il-15 Trans-Presentation in Keratinocytes Contributes to CD8+ T Cells Activation Via JAK-STAT Pathway in Vitiligo. Free Radic Biol Med (2019) 139:80–91. doi: 10.1016/j.freeradbiomed.2019.05.011
35. Harris JE, Harris TH, Weninger W, Wherry EJ, Hunter CA, Turka LA. A Mouse Model of Vitiligo With Focused Epidermal Depigmentation Requires Ifn-γ for Autoreactive Cd8+ T-Cell Accumulation in the Skin. J Invest Dermatol (2012) 132:1869–76. doi: 10.1038/jid.2011.463
36. Strassner JP, Rashighi M, Ahmed Refat M, Richmond JM, Harris JE. Suction Blistering the Lesional Skin of Vitiligo Patients Reveals Useful Biomarkers of Disease Activity. J Am Acad Dermatol (2017) 76:847–55.e5. doi: 10.1016/j.jaad.2016.12.021
37. Rashighi M, Agarwal P, Richmond JM, Harris TH, Dresser K, Su M-W, et al. CXCL10 is Critical for the Progression and Maintenance of Depigmentation in a Mouse Model of Vitiligo. Sci Transl Med (2014) 6:223ra23. doi: 10.1126/scitranslmed.3007811
38. Richmond JM, Masterjohn E, Chu R, Tedstone J, Youd ME, Harris JE. Cxcr3 Depleting Antibodies Prevent and Reverse Vitiligo in Mice. J Invest Dermatol (2017) 137:982–5. doi: 10.1016/j.jid.2016.10.048
39. Richmond JM, Bangari DS, Essien KI, Currimbhoy SD, Groom JR, Pandya AG, et al. Keratinocyte-Derived Chemokines Orchestrate T-Cell Positioning in the Epidermis During Vitiligo and May Serve as Biomarkers of Disease. J Invest Dermatol (2017) 137:350–8. doi: 10.1016/j.jid.2016.09.016
40. Harris JE, Rashighi M, Nguyen N, Jabbari A, Ulerio G, Clynes R, et al. Rapid Skin Repigmentation on Oral Ruxolitinib in a Patient With Coexistent Vitiligo and Alopecia Areata (Aa). J Am Acad Dermatol (2016) 74:370–1. doi: 10.1016/j.jaad.2015.09.073
41. Azzolino V, Zapata L, Garg M, Gjoni M, Riding RL, Strassner JP, et al. Jak Inhibitors Reverse Vitiligo in Mice But Do Not Deplete Skin Resident Memory T Cells. J Invest Dermatol (2020) 141(1):182–4.e1. doi: 10.1016/j.jid.2020.04.027
42. Palermo B, Campanelli R, Garbelli S, Mantovani S, Lantelme E, Brazzelli V, et al. Specific Cytotoxic T Lymphocyte Responses Against Melan-a/MART1, Tyrosinase and gp100 in Vitiligo by the Use of Major Histocompatibility Complex/Peptide Tetramers: The Role of Cellular Immunity in the Etiopathogenesis of Vitiligo. J Invest Dermatol (2001) 117:326–32. doi: 10.1046/j.1523-1747.2001.01408.x
43. Sugita S, Takase H, Taguchi C, Imai Y, Kamoi K, Kawaguchi T, et al. Ocular Infiltrating Cd4+ T Cells From Patients With Vogt-Koyanagi-Harada Disease Recognize Human Melanocyte Antigens. Invest Ophthalmol Vis Sci (2006) 47:2547–54. doi: 10.1167/iovs.05-1547
44. van den Boorn JG, Konijnenberg D, Dellemijn TAM, Wietze Van Der Veen JP, Bos JD, Melief CJM, Vyth-Dreese FA, Luiten RM, et al. Autoimmune Destruction of Skin Melanocytes by Perilesional T Cells From Vitiligo Patients. J Invest Dermatol (2009) 129:2220–32. doi: 10.1038/jid.2009.32
45. Le Poole IC, van den Wijngaard RM, Westerhof W, Das PK. Presence of T Cells and Macrophages in Inflammatory Vitiligo Skin Parallels Melanocyte Disappearance. Am J Pathol (1996) 148:1219–28.
46. Maezawa N, Yano A. Two Distinct Cytotoxic T Lymphocyte Subpopulations in Patients With Vogt-Koyanagi-Harada Disease That Recognize Human Melanoma Cells. Microbiol Immunol (1984) 28:219–31. doi: 10.1111/j.1348-0421.1984.tb00673.x
47. Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. Tyrosinase Family Proteins are Antigens Specific to Vogt-Koyanagi-Harada Disease. J Immunol (2000) 165:7323–9. doi: 10.4049/jimmunol.165.12.7323
48. Boniface K, Jacquemin C, Darrigade A-S, Dessarthe B, Martins C, Boukhedouni N, et al. Vitiligo Skin is Imprinted With Resident Memory Cd8 T Cells Expressing Cxcr3. J Invest Dermatol (2018) 138:355–64. doi: 10.1016/j.jid.2017.08.038
49. Nakashima C, Ishida Y, Nakagawa K, Irie H, Hirata M, Kataoka T, et al. Identification of CD 49a+ CD 8+ Resident Memory T Cells in Vitiligo-Like Lesions Associated With Nivolumab Treatment for Melanoma. J Eur Acad Dermatol Venereol (2020) 2020(34):e50–115. doi: 10.1111/jdv.15970
50. Cevasco NC, Bergfeld WF, Remzi BK, de Knott HR. A Case-Series of 29 Patients With Lichen Planopilaris: The Cleveland Clinic Foundation Experience on Evaluation, Diagnosis, and Treatment. J Am Acad Dermatol (2007) 57:47–53. doi: 10.1016/j.jaad.2007.01.011
51. Kirchhof MG, Dutz JP. The Immunopathology of Cutaneous Lupus Erythematosus. Rheum Dis Clin North Am (2014) 40:455–74. doi: 10.1016/j.rdc.2014.04.006
52. Abdirama D, Tesch S, Grießbach A-S, von Spee-Mayer C, Humrich JY, Stervbo U, et al. Nuclear Antigen-Reactive Cd4+ T Cells Expand in Active Systemic Lupus Erythematosus, Produce Effector Cytokines, and Invade the Kidneys. Kidney Int (2020) 99(1):P238–46. doi: 10.1016/j.kint.2020.05.051
53. Zhou M, Guo C, Li X, Huang Y, Li M, Zhang T, et al. Jak/Stat Signaling Controls the Fate of CD8CD103 Tissue-Resident Memory T Cell in Lupus Nephritis. J Autoimmun (2020) 109:102424. doi: 10.1016/j.jaut.2020.102424
54. Arakawa A, Siewert K, Stöhr J, Besgen P, Kim S-M, Rühl G, et al. Melanocyte Antigen Triggers Autoimmunity in Human Psoriasis. J Exp Med (2015) 212:2203–12. doi: 10.1084/jem.20151093
55. de Jong A, Jabbari A, Dai Z, Xing L, Lee D, Li MM, et al. High-Throughput T Cell Receptor Sequencing Identifies Clonally Expanded Cd8+ T Cell Populations in Alopecia Areata. JCI Insight (2018) 139(19):e121949. doi: 10.1172/jci.insight.121949
56. Olayinka J, Richmond JM. Immunopathogenesis of Alopecia Areata. Curr Res Immunol (2021) 2:7–11. doi: 10.1016/j.crimmu.2021.02.001
57. Jacquemin C, Martins C, Lucchese F, Thiolat D, Taieb A, Seneschal J, et al. Nkg2d Defines a Subset of Skin Effector Memory Cd8 T Cells With Proinflammatory Functions in Vitiligo. J Invest Dermatol (2020) 140:1143–53.e5. doi: 10.1016/j.jid.2019.11.013
58. Garelli CJ, Refat MA, Nanaware PP, Ramirez-Ortiz ZG, Rashighi M, Richmond JM. Current Insights in Cutaneous Lupus Erythematosus Immunopathogenesis. Front Immunol (2020) 11:1353. doi: 10.3389/fimmu.2020.01353
59. Lu L, Kaliyaperumal A, Boumpas DT, Datta SK. Major Peptide Autoepitopes for Nucleosome-Specific T Cells of Human Lupus. J Clin Invest (1999) 104:345–55. doi: 10.1172/JCI6801
60. Voll RE, Roth EA, Girkontaite I, Fehr H, Herrmann M, Lorenz H-M, et al. Histone-Specific Th0 and Th1 Clones Derived From Systemic Lupus Erythematosus Patients Induce Double-Stranded DNA Antibody Production. Arthritis Rheumatism: Off J Am Coll Rheumatol (1997) 40:2162–71. doi: 10.1002/art.1780401210
61. Oke V, Wahren-Herlenius M. Cutaneous Lupus Erythematosus: Clinical Aspects and Molecular Pathogenesis. J Intern Med (2013) 273:544–54. doi: 10.1111/joim.12057
62. Katsuyama T, Tsokos GC, Moulton VR. Aberrant T Cell Signaling and Subsets in Systemic Lupus Erythematosus. Front Immunol (2018) 9:1088. doi: 10.3389/fimmu.2018.01088
63. Wenzel J, Zahn S, Bieber T, Tüting T. Type I Interferon-Associated Cytotoxic Inflammation in Cutaneous Lupus Erythematosus. Arch Dermatol Res (2009) 301:83–6. doi: 10.1007/s00403-008-0892-8
64. Grassi M, Capello F, Bertolino L, Seia Z. Identification of Granzyme B-Expressing CD-8-Positive T Cells in Lymphocytic Inflammatory Infiltrate in Cutaneous Lupus Erythematosus and in Dermatomyositis. Clin (2009) 34(8):910–4. doi: 10.1111/j.1365-2230.2009.03297.x
65. Zeidi M, Desai K, Chen KL, Kim H, Lim RK, Werth VP. Increased Tissue-Resident Memory T (T-Rm) Cells and STAT3 Expression in Cutaneous Lupus Erythematosus Patients Refractory to Antimalarials. J Invest Dermatol 139(5):S174. doi: 10.1016/j.jid.2019.03.1080
66. Klaeschen AS, Wolf D, Brossart P, Bieber T, Wenzel J. Jak Inhibitor Ruxolitinib Inhibits the Expression of Cytokines Characteristic of Cutaneous Lupus Erythematosus. Exp Dermatol (2017) 26:728–30. doi: 10.1111/exd.13253
67. Tanasescu C, Balanescu E, Balanescu P, Olteanu R, Badea C, Grancea C, et al. Il-17 in Cutaneous Lupus Erythematosus. Eur J Intern Med (2010) 21:202–7. doi: 10.1016/j.ejim.2010.03.004
68. Mak RKH, Hundhausen C, Nestle FO. Progress in Understanding the Immunopathogenesis of Psoriasis. Actas Dermosifiliogr (2009) 100(Suppl 2):2–13. doi: 10.1016/s0001-7310(09)73372-1
69. Bhushan M, Bleiker TO, Ballsdon AE, Allen MH, Sopwith M, Robinson MK, et al. Anti-E-selectin is Ineffective in the Treatment of Psoriasis: A Randomized Trial. Br J Dermatol (2002) 146:824–31. doi: 10.1046/j.1365-2133.2002.04743.x
70. Kurihara K, Fujiyama T, Phadungsaksawasdi P, Ito T, Tokura Y. Significance of IL-17A-producing Cd8cd103 Skin Resident Memory T Cells in Psoriasis Lesion and Their Possible Relationship to Clinical Course. J Dermatol Sci (2019) 95:21–7. doi: 10.1016/j.jdermsci.2019.06.002
71. Hawkes JE, Yan BY, Chan TC, Krueger JG. Discovery of the IL-23/IL-17 Signaling Pathway and the Treatment of Psoriasis. J Immunol (2018) 201:1605–13. doi: 10.4049/jimmunol.1800013
72. Fujiyama T, Umayahara T, Kurihara K, Shimauchi T, Ito T, Aoshima M, et al. Skin Infiltration of Pathogenic Migratory and Resident T Cells is Decreased by Secukinumab Treatment in Psoriasis. J Invest Dermatol (2020) 140:2073–6.e6. doi: 10.1016/j.jid.2020.02.024
73. Owczarczyk Saczonek A, Krajewska-Włodarczyk M, Kasprowicz-Furmańczyk M, Placek W. Immunological Memory of Psoriatic Lesions. Int J Mol Sci (2020) 21(2):625. doi: 10.3390/ijms21020625
74. Aujla SJ, Kolls JK. Il-22: A Critical Mediator in Mucosal Host Defense. J Mol Med (2009) 87:451–4. doi: 10.1007/s00109-009-0448-1
75. Eyerich K, Dimartino V, Cavani A. Il-17 and IL-22 in Immunity: Driving Protection and Pathology. Eur J Immunol (2017) 47:607–14. doi: 10.1002/eji.201646723
76. Tohyama M, Shirakata Y, Hanakawa Y, Dai X, Shiraishi K, Murakami M, et al. Bcl-3 Induced by IL-22 Via STAT3 Activation Acts as a Potentiator of Psoriasis-Related Gene Expression in Epidermal Keratinocytes. Eur J Immunol (2018) 48:168–79. doi: 10.1002/eji.201747017
77. Mufazalov IA, Kuschmann J, Andruszewski D, Masri J, Gabriel LA, Adams P, et al. Balanced Bcl-3 Expression in Murine Cd4 T Cells is Required for Generation of Encephalitogenic Th17 Cells. Eur J Immunol (2017) 47:1335–41. doi: 10.1002/eji.201746933
78. Harries MJ, Meyer K, Chaudhry I, E Kloepper J, Poblet E, Griffiths CE, et al. Lichen Planopilaris is Characterized by Immune Privilege Collapse of the Hair Follicle’s Epithelial Stem Cell Niche. J Pathol (2013) 231:236–47. doi: 10.1002/path.4233
79. Christoph T, Müller-Röver S, Audring H, Tobin DJ, Hermes B, Cotsarelis G, et al. The Human Hair Follicle Immune System: Cellular Composition and Immune Privilege. Br J Dermatol (2000) 142:862–73. doi: 10.1046/j.1365-2133.2000.03464.x
80. Paus R, Bertolini M. The Role of Hair Follicle Immune Privilege Collapse in Alopecia Areata: Status and Perspectives. J Investig Dermatol Symp Proc (2013) 16:S25–7. doi: 10.1038/jidsymp.2013.7
81. Iorizzo M, Tosti A. Frontal Fibrosing Alopecia: An Update on Pathogenesis, Diagnosis, and Treatment. Am J Clin Dermatol (2019) 20:379–90. doi: 10.1007/s40257-019-00424-y
82. Manatis-Lornell A, Okhovat J-P, Marks DH, Hagigeorges D, Senna MM. Comorbidities in Patients With Lichen Planopilaris: A Retrospective Case-Control Study. J Am Acad Dermatol (2020) 83(1):205–8. doi: 10.1016/j.jaad.2019.07.018
83. Kyei A, Bergfeld WF, Piliang M, Summers P. Medical and Environmental Risk Factors for the Development of Central Centrifugal Cicatricial Alopecia: A Population Study. Arch Dermatol (2011) 147:909–14. doi: 10.1001/archdermatol.2011.66
84. Strazzulla LC, Wang EHC, Avila L, Lo Sicco K, Brinster N, Christiano AM, et al. Alopecia Areata: Disease Characteristics, Clinical Evaluation, and New Perspectives on Pathogenesis. J Am Acad Dermatol (2018) 78:1–12. doi: 10.1016/j.jaad.2017.04.1141
85. Petukhova L, Duvic M, Hordinsky M, Norris D, Price V, Shimomura Y, et al. Genome-Wide Association Study in Alopecia Areata Implicates Both Innate and Adaptive Immunity. Nature (2010) 466:113–7. doi: 10.1038/nature09114
86. Tziotzios C, Stefanato CM, Fenton DA, Simpson MA, McGrath JA. Frontal Fibrosing Alopecia: Reflections and Hypotheses on Aetiology and Pathogenesis. Exp Dermatol (2016) 25:847–52. doi: 10.1111/exd.13071
87. Del Duca E, Ruano Ruiz J, Pavel AB, Dutt Sanyal R, Song T, Gay-Mimbrera J, et al. Frontal Fibrosing Alopecia Shows Robust Th1 and JAK3 Skewing. Br J Dermatol (2020) 183(6):1083–93. doi: 10.1111/bjd.19040
88. Gay-Mimbrera J, Ruano J, Pavel AB, Sanyal RD, Zhang N, Estrada YD, et al. Molecular Profiling of Frontal Fibrosing Alopecia (Ffa) Reveals TH1 and JAK-STAT Up-Regulation With No Suppression of Hair Keratins. J Invest Dermatol 138(5):S225. doi: 10.1016/j.jid.2018.03.1339
89. Yang CC, Khanna T, Sallee B, Christiano AM, Bordone LA. Tofacitinib for the Treatment of Lichen Planopilaris: A Case Series. Dermatol Ther (2018) 31:e12656. doi: 10.1111/dth.12656
90. Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, et al. Alopecia Areata is Driven by Cytotoxic T Lymphocytes and is Reversed by JAK Inhibition. Nat Med (2014) 20:1043–9. doi: 10.1038/nm.3645
91. Morris GM, Nahmias ZP, Kim BS. Simultaneous Improvement of Alopecia Universalis and Atopic Dermatitis in a Patient Treated With a JAK Inhibitor. JAAD Case Rep (2018) 4:515–7. doi: 10.1016/j.jdcr.2017.12.016
92. Jabbari A, Dai Z, Xing L, Cerise JE, Ramot Y, Berkun Y, et al. Reversal of Alopecia Areata Following Treatment With the JAK1/2 Inhibitor Baricitinib. EBioMedicine (2015) 2:351–5. doi: 10.1016/j.ebiom.2015.02.015
93. Kennedy Crispin M, Ko JM, Craiglow BG, Li S, Shankar G, Urban JR, et al. Safety and Efficacy of the JAK Inhibitor Tofacitinib Citrate in Patients With Alopecia Areata. JCI Insight (2016) 1:e89776. doi: 10.1172/jci.insight.89776
Keywords: resident memory T cell (TRM), cutaneous lupus erythematosus (CLE), vitiligo, psoriasis, alopecia, dermatology, autoimmunity
Citation: Ryan GE, Harris JE and Richmond JM (2021) Resident Memory T Cells in Autoimmune Skin Diseases. Front. Immunol. 12:652191. doi: 10.3389/fimmu.2021.652191
Received: 11 January 2021; Accepted: 06 April 2021;
Published: 03 May 2021.
Edited by:
Kenji Kabashima, Kyoto University, JapanReviewed by:
Koji Yasutomo, Tokushima University, JapanCopyright © 2021 Ryan, Harris and Richmond. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Grace E. Ryan, Z3JhY2UucnlhbkB1bWFzc21lZC5lZHU=; Jillian M. Richmond, amlsbGlhbi5yaWNobW9uZEB1bWFzc21lZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.