 Michele Fresneda Alarcon
Michele Fresneda Alarcon Zoe McLaren2
Zoe McLaren2 Helen Louise Wright
Helen Louise Wright
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 March 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.649693
This article is part of the Research Topic Neutrophil Functions in Host Immunity, Inflammation and Tissue Repair View all 24 articles
Dysregulated neutrophil activation contributes to the pathogenesis of autoimmune diseases including rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). Neutrophil-derived reactive oxygen species (ROS) and granule proteases are implicated in damage to and destruction of host tissues in both conditions (cartilage in RA, vascular tissue in SLE) and also in the pathogenic post-translational modification of DNA and proteins. Neutrophil-derived cytokines and chemokines regulate both the innate and adaptive immune responses in RA and SLE, and neutrophil extracellular traps (NETs) expose nuclear neoepitopes (citrullinated proteins in RA, double-stranded DNA and nuclear proteins in SLE) to the immune system, initiating the production of auto-antibodies (ACPA in RA, anti-dsDNA and anti-acetylated/methylated histones in SLE). Neutrophil apoptosis is dysregulated in both conditions: in RA, delayed apoptosis within synovial joints contributes to chronic inflammation, immune cell recruitment and prolonged release of proteolytic enzymes, whereas in SLE enhanced apoptosis leads to increased apoptotic burden associated with development of anti-nuclear auto-antibodies. An unbalanced energy metabolism in SLE and RA neutrophils contributes to the pathology of both diseases; increased hypoxia and glycolysis in RA drives neutrophil activation and NET production, whereas decreased redox capacity increases ROS-mediated damage in SLE. Neutrophil low-density granulocytes (LDGs), present in high numbers in the blood of both RA and SLE patients, have opposing phenotypes contributing to clinical manifestations of each disease. In this review we will describe the complex and contrasting phenotype of neutrophils and LDGs in RA and SLE and discuss their discrete roles in the pathogenesis of each condition. We will also review our current understanding of transcriptomic and metabolomic regulation of neutrophil phenotype in RA and SLE and discuss opportunities for therapeutic targeting of neutrophil activation in inflammatory auto-immune disease.
Systemic Lupus Erythematosus (SLE) is the archetypal autoimmune connective tissue disease, characterized by the production of multiple auto-antibodies [anti-nuclear antibodies (ANA), anti-double stranded DNA (dsDNA), anti-Sm/RNP, anti-Ro/La] and the consumption of complement (1, 2). It has the capacity to involve almost any organ system of the body resulting in protean and sometimes catastrophic consequences for patients. SLE disproportionately affects young women and is a condition with a broad spectrum of severity, ranging from mild joint involvement to life-threatening organ failure (3). Typical manifestations include systemic complaints, such as overwhelming and intrusive fatigue, brain fog, fever, swollen lymph nodes, mouth ulcers, chilblains, and weight loss (4). Rashes are common in SLE and may be transient or disfiguring; rashes are often triggered by sunlight and may be associated with hair loss and scarring. The characteristic malar flush of redness over the cheeks gives the name lupus (from wolf) and is likened to the appearance of a butterfly. Inflammation of joints, tendons, and muscle may cause arthritis, nodules, or contractures and give rise to disability and pain. Kidney disease including inflammation and immune complex deposition may occur which, if untreated, can lead to kidney failure and the need for dialysis and transplant (5, 6). Inflammation can develop in the pleural lining of the lungs, around the heart and may even affect the heart muscle and valves. SLE may also affect any part of the nervous system from brain and spinal cord to the peripheral nerves resulting in neurological problems, such as strokes, neuropathy, headache, visual loss, migraine, confusion, and acute psychosis (7). The disease may cause a fall in blood counts involving specific cell lines or indeed pancytopenia and may be associated with serious abnormalities of both clotting and bleeding. Repeated inflammatory insults, abnormal blood clotting and the consequence of treatment with high dose steroids and immunosuppression can also lead to chronic illness through damage accrual and increase the likelihood of infection, osteoporosis, premature ovarian failure, cardiovascular events such as atherosclerosis, stroke or heart attack, and malignancy. SLE is incurable and with modern treatment is still associated with an increased risk of mortality, and shortened life expectancy (3).
Rheumatoid arthritis (RA) is a chronic, autoimmune, systemic inflammatory condition associated typically with antibodies to rheumatoid factor (RF) and cyclic citrullinated peptides (anti-CCP or ACPA) (8). It is the commonest form of inflammatory arthritis and is characterized by inflammation of the tendon sheaths (tenosynovitis) and joint lining (synovitis) leading to growth of an inflammatory pannus which quickly erodes the joint cartilage and bone, causing recognizable deformities that were once commonplace in rheumatology clinics (9). Untreated or resistant to therapy, RA results in a symmetrical, deforming polyarthropathy. This leads to physical disability, progressive loss of function and as well as stiffness and pain. Extra-articular complications of the disease include interstitial lung disease, vasculitis, nodules, eye disease and an increased risk of cardiovascular disease, malignancy and osteoporosis (10, 11). Modern management is focused on a prompt diagnosis and early use of immunosuppressive treatments, including traditional and biologic disease-modifying anti-rheumatic drugs (DMARDs), with the aim of targeting disease remission. This has led to a reduction in the need for orthopedic surgery for patients with this form of arthritis, a move to out-patient based care as opposed to long hospitalizations, and a reduction in the systemic complications that can occur. However, RA still remains an incurable disease with treatments that rely on the long-term suppression of the immune system resulting in side effects and complications, including an increased risk of infection (9, 12).
Both RA and SLE are caused by a dysregulation of the innate and adaptive immune systems, including clonal expansion of auto-reactive lymphocytes, production of auto-antibodies and elevated production of multiple cytokines and other inflammatory mediators. Research into the underlying cause of both diseases focusses heavily on dysregulated T- and B-cell responses (9, 12, 13). However, it is inappropriately activated neutrophils that have the greatest potential to cause damage to local tissues, both due to their presence in high numbers at sites of inflammation and through release of their cytotoxic contents directly onto host tissues. Neutrophils are specialist cells of the innate immune system that normally play a major role in host defense against microorganisms through phagocytosis and generation of reactive oxygen species (ROS). Production of ROS within the phagosome occurs via the action of NADPH oxidase (NOX2) and myeloperoxidase (MPO) which, together with release of proteases from granules and vesicles into the phagosome, provide a defensive arsenal against a broad spectrum of microscopic pathogens. During infection, ROS and proteases may be released extracellularly causing local tissue damage at the site of infection (14). This damage is normally resolved by resident macrophages, which remove apoptotic neutrophils and damaged tissue as part of the normal process of inflammation resolution (15). Neutrophils are the most abundant leukocyte in humans, being produced by the bone marrow in huge numbers daily (estimated to be in the region of 5–10 × 1010 per day) (16, 17). Whilst the majority of neutrophils circulate in the blood (both free-flowing and marginated to the endothelial vessel walls), several populations of tissue neutrophils exist within healthy homeostasis, including within the lung, spleen and liver (18). These tissue neutrophils play a major role in surveillance and host defense, B-cell Ig-class switching, and phagocytosis of circulating bacteria, respectively. The liver is also a major site for efferocytosis and removal of neutrophils that have been involved in bacterial killing (18). Neutrophil release from the bone marrow, and subsequent homing back to bone marrow for efferocytosis at the end of their normal life span, is regulated by circadian expression of CXCR2 (receptor for CXCL2), the central clock gene BMAL1 and CXCR4 (receptor for CXCL12) (19), with granule content and the ability to produce NETs being the highest in the morning, decreasing throughout the day to reach the lowest levels by mid-afternoon in human neutrophils (20).
Neutrophils contribute to inflammation and tissue damage in inflammatory disease, when they become inappropriately activated by cytokines, chemokines and auto-antibodies (14). Auto-immune neutrophils function in a multitude of ways to direct the inflammatory response, including release of proteases which damage host tissue and activate soluble proteins (21), secretion of cytokines and chemokines which direct both the innate and adaptive immune responses (22), shedding of receptors, such as the interleukin-6 receptor to initiate trans-signaling (23, 24), release of neutrophil extracellular traps (NETs) providing a source of auto-antigens (25), and production of ROS (8, 26). This review will discuss the dysregulation of neutrophil activation in RA and SLE, two auto-immune diseases characterized by aberrant neutrophil activation. We will highlight how uncontrolled neutrophil activation contributes to the development of auto-immunity and disease progression, and summarize how inappropriate neutrophil activation might be targeted with therapeutics.
Neutrophils are terminally differentiated cells which, in the absence of inflammation, circulate in the blood for around 24–48 h before returning to the bone marrow and undergoing apoptosis (27). Constitutive neutrophil apoptosis is regulated by Bcl-2 family proteins: anti-apoptotic MCL1 and A1/BFL1, and pro-apoptotic BAX, BAK, and BID. Loss of MCL1 and BFL1 causes BAX:BAK pore formation in the mitochondrial membrane, releasing cytochrome c which along with APAF1 forms the apoptosome, leading to cleavage of caspases initiating apoptosis (14, 27–29). Neutrophil apoptosis may be delayed during inflammation by cytokines (e.g., GM-CSF, TNFα), bacterial lipopolysaccharide (LPS) and leukotrienes (e.g., leukotriene B4), which extend the life-span of neutrophils through stabilization or up-regulation of MCL1 and/or BFL1 (30–35). Aging neutrophils can be identified in circulation by decreased expression of L-selectin (CD62L) and CXCR2, and increased expression of CXCR4, the receptor for CXCL12; it is the increased signaling via CXCL12:CXCR4 which enables aged neutrophils to home back to the bone marrow to undergo apoptosis and clearance (19). Neutrophil apoptosis may be induced through activation of death-receptor signaling pathways, such as FASL, TRAIL, and TNFα which directly activate caspase-8 and caspase-3, and induce degradation of BID and BAX leading to mitochondrial release of cytochrome c (28, 36). Intriguingly, TNFα may be pro- or anti-apoptotic in vitro depending upon the concentration in media, with low concentrations delaying apoptosis and higher concentrations promoting apoptosis (37). Whilst not completely understood, this effect is believed to be mediated via the two TNF receptors (TNFR1 and TNFR2), with TNFR1 signaling anti-apoptotic activation of NF-κB, and TNFR2 activating death receptor signaling (38, 39).
In SLE, neutrophil apoptosis is enhanced (40–42) leading to increased apoptotic burden associated with development of anti-nuclear auto-antibodies (25, 43). A dysregulation between pro-apoptotic caspases and inhibitors of apoptosis (IAP1, IAP2, XIAP) may explain the enhanced apoptosis seen in SLE neutrophils in vitro (Figure 1) (41, 42, 44). Levels of pro-apoptotic TRAIL and FASL are also significantly higher in SLE serum, which can induce apoptosis in healthy neutrophils (41). Levels of GM-CSF are lower in SLE serum, and supplementation of SLE serum with physiological levels of GM-CSF can rescue the pro-apoptotic effect of SLE serum on healthy neutrophils (42). Incubation of apoptotic neutrophils with SLE PBMCs induces expression of interferon-alpha (IFNα) via a toll-like receptor (TLR)-dependent mechanism (45). SLE neutrophils also express nuclear antigens (dsDNA) at the plasma membrane, and this effect can be induced in healthy neutrophils incubated with SLE serum (46). As well as an increase in neutrophil apoptosis, defects in clearance of apoptotic cells by macrophages can also contribute to the accumulation of apoptotic debris in SLE (47).
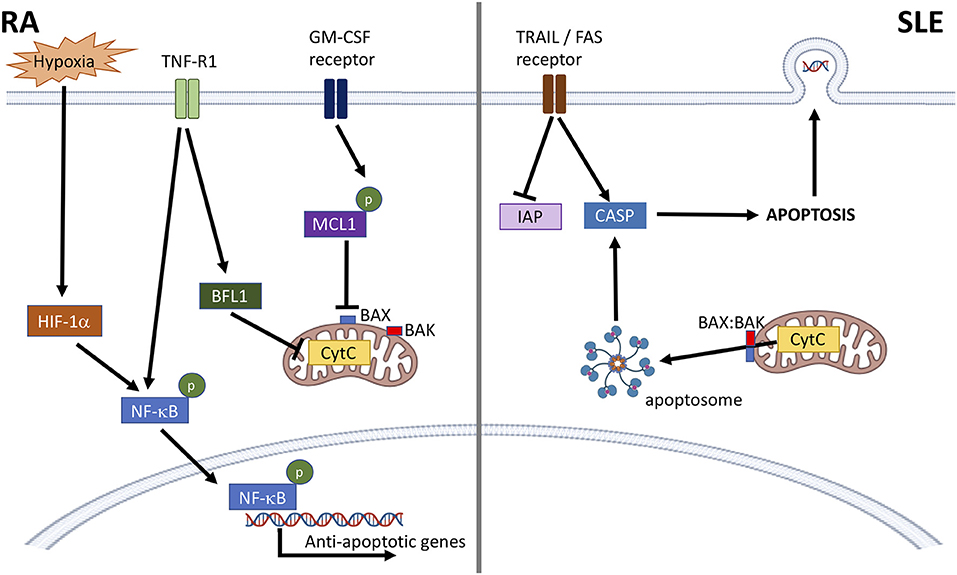
Figure 1. Dysregulation of neutrophil apoptosis in RA and SLE. In RA, anti-apoptotic factors, such as cytokines and hypoxia activate NF-κB and prevent mitochondrial cytochrome c (CytC) driven apoptosis through activation of BFL1, and stabilization of MCL1. This prevents BAX:BAK pore formation, CytC leakage and formation of the apoptosome. In SLE, activation of death receptors (e.g., TRAIL, FAS receptors) lowers levels of cellular inhibitors of apoptosis (IAP) and activates caspase-8 (CASP). BAX:BAK pore formation in the mitochondrial membrane releases CytC leading to apoptosome formation and activation of CASP-9 and CASP-3 leading to apoptosis. Nuclear antigens, including DNA, are also expressed at the plasma membrane.
Apoptosis is delayed in both blood and synovial fluid neutrophils from patients with RA (48, 49). The likely cause of this is inflammatory cytokines, such as GM-CSF, TNFα, IL1β, and interferons, elevated in both blood and synovial fluid (50), which have been demonstrated to delay neutrophil apoptosis in in vitro experiments (32–34, 37, 51–55). RA blood neutrophils express higher levels of MCL1, higher levels of phosphorylated NF-κB and lower levels of active caspase-9 compared to healthy controls (48). Delayed apoptosis in synovial fluid has also been attributed to other factors including lactoferrin and adenosine (56, 57). The hypoxic environment in RA synovial joints also plays a key role in delaying neutrophil apoptosis, via increased expression of MCL1 (Figure 1) (58). Hypoxia can also delay apoptosis via stabilization of hypoxia-inducible factor 1-alpha (HIF1-α) and activation of NF-κB (59), and regulates neutrophil retention at sites of inflammation, prolonging inflammation (60).
Microparticles (MPs) are small (0.2–2 μM) extracellular vesicles containing cargos of protein, lipids and nucleotides (e.g., RNA, miRNA) which are released from activated cells via budding and shedding of the cell membrane (61). MPs are distinct from other extracellular vesicles, i.e., exosomes and apoptotic bodies, by virtue of their size, cargo, membrane content, and mode of formation (61). MPs are taken up by neighboring cells through endocytosis, and their protein and RNA cargo can alter the recipient cell phenotype once released into the cytoplasm. They represent a novel form of paracellular communication and differ in composition depending upon the functional state of the originating cell. In SLE, MPs are annexin V+ and contain cell markers indicating both endothelial cell and neutrophil origins (62). SLE MPs activate plasmacytoid dendritic cells to produce a range of cytokines, including IFNα (62), and stimulate ROS production in autologous and healthy neutrophils (63). Acetylated histones within chromatin in MPs suggests the MPs are derived from apoptotic cells (62) and may be central to the stimulation of NOX2-independent NETs (64). The population of annexin V+/acetylated histone+ MPs appear to be specific to SLE and are not present in sera of healthy controls or individuals with RA (62). MPs released from RA neutrophils may have anti-inflammatory properties, activated through the pro-resolving protein Annexin-A1 (65, 66). Annexin-A1 is released from neutrophil granules following extravasation and is found in RA synovial fluid. This protein exerts its pro-resolving effect by promoting apoptosis and decreasing neutrophil:endothelial cell adhesion and extravasation (66, 67).
Neutrophils produce ROS via activation of the NADPH oxidase (NOX2) (68). NOX2 is a multi-component enzyme, which is assembled at the phagosomal and plasma membrane following cytokine priming (69, 70). Priming induces phosphorylation and mobilization of granular and cytosolic NOX2 components [p22phox and gp91phox which together comprise cytochrome b558, p40phox, p47phox, p67phox, and Rac (Rac-1 or Rac-2)] to the phagosomal and plasma membranes in readiness for phagosomal killing (70–73). Assembly of NOX2 at the plasma membrane leads to the release of ROS into the extracellular environment and is a major cause of damage to host tissue in RA and SLE. ROS production via NOX2 in primed neutrophils is triggered by activation of cell receptors [e.g., FcγR, complement receptors, f-Met-Leu-Phe (fMLP) receptor]. Activated NOX2 catalyses the reduction of oxygen to superoxide (O2-), an unstable oxygen radical which rapidly forms hydrogen peroxide (H2O2), the hydroxyl free radical (HO•) and/or peroxynitrite () depending on cellular conditions (68, 74). Hydrogen peroxide is the major substrate of myeloperoxidase (MPO), a neutrophil azurophilic granule enzyme implicated in the production of highly-reactive, secondary oxidants, such as hypochlorous, hypobromous, and hypothiocyanous acids which are potent anti-microbial agents that damage proteins, lipids and DNA (68, 75).
SLE neutrophils exhibit aberrant ROS production, with O2-, H2O2, and HO• being produced more rapidly and in higher levels than healthy individuals (76, 77). DNA, protein and lipid markers of intracellular oxidative stress are increased in SLE neutrophils (76), including 8-hydroxyguanosine, an oxidized self-DNA which may function as a damage-associated molecular pattern (DAMP) (78) and which is present in NETs (79). Neutrophils from SLE patients with circulating immune complexes or cytotoxic antibodies produce the highest O2- response to FcγR/complement receptor stimulation (80), and unstimulated ex vivo neutrophils from lupus nephritis patients have the highest levels of ROS production (77). SLE serum induces O2- generation in healthy neutrophils, with O2- production correlating positively with the presence of immune complexes and negatively with the concentration of complement (81). Many patients with SLE experience both flares and periods of inactive or quiet disease. Neutrophils from SLE patients in active flare often produce lower levels of ROS in vitro than those with inactive disease, possibly due to exhaustion of the neutrophils in vivo (76), or lower expression of Fcγ and complement receptors (82). As yet unidentified factors within SLE serum induce production of cytokines by human renal glomerular endothelial cells which promote neutrophil chemotaxis and adhesion (83). In patients with lupus nephritis, immune complexes become deposited within tissues in the kidney (6). This may arise through the binding of anti-nucleosomes and anti-C1q auto-antibodies to nucleosomes and C1q captured on the surface of glomerular endothelial cells by heparan sulfate (84). Neutrophils expressing FcγR2A adhere to immune complex deposits on the surface of glomerular capillaries, activating ROS production and release directly onto host tissue (85). This release of ROS is directly responsible for damage to glomeruli, including oxidation of DNA, lipids and proteins, and induction of apoptosis (86). Oxidized high-density lipoprotein (HDL), commonly found in SLE patients, is pro-inflammatory, driving production of IL-6 and TNF by macrophages, and lacking its normal, cardioprotective properties (87). A polymorphism in the gene for neutrophil cytosolic factor 1 (NCF1, rs201802880) in a sub-set of SLE patients is associated with lower ROS production by neutrophils (88). This polymorphism also increases expression of type 1 interferon-regulated genes (89), the importance of which will be discussed later in this review. Interestingly, the NCF1 (m1J) mutated mouse model of SLE has demonstrated a link between ROS deficiency and interferon-driven autoimmunity downstream of a deficient NOX2 complex (90).
In RA, both blood and synovial fluid neutrophils have an increased capacity to produce ROS (91, 92). ROS production within RA blood leads to modifications of immunoglobulin G (IgG) which are associated with increased immunogenicity and production of rheumatoid factor immune complexes (93). Within the RA joint IgG complexes, both soluble and embedded within synovial tissue, activate further ROS production by neutrophils via activation of FcγR2a and FcγR3b (Figure 2) (94, 95), and trigger degranulation of proteolytic enzymes including elastase and cathepsin G (8, 26, 56, 96–98). When this occurs at the articular surface a microenvironment of concentrated ROS, proteases and cytotoxic factors is formed, damaging the underlying structures (8). As well as damaging collagen fibers within cartilage, neutrophil granule proteases cause proteolytic cleavage and activation of proteins (matrix metalloproteinases, pro-cytokines/chemokines) and cleavage of soluble receptors to initiate trans-signaling (such as the IL-6 receptor) (8, 21, 23, 99–102). Additionally, ROS production within the joint disrupts oxidative homeostasis and drives adaptive immune responses to the synovial environment (68). ROS-induced MAPK and NF-κB activation in synovial fibroblasts activates production of pro-inflammatory prostaglandins by cyclooxygenase (COX)-2 (103). ROS also exert profound effects on the local T cell population, regulating differentiation, apoptosis and cytokine production (104, 105).
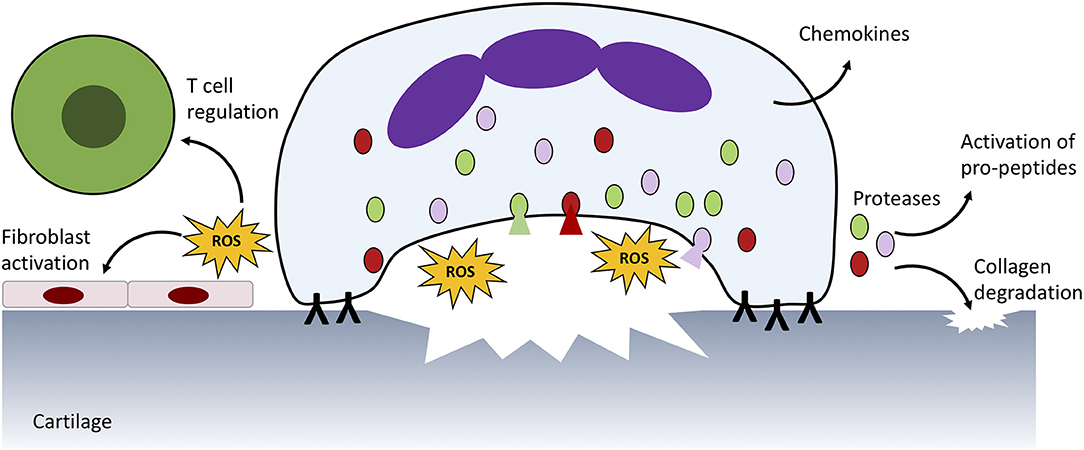
Figure 2. Neutrophil ROS production and protease release damages host tissue. In RA, immune complexes on the surface of the joint activate ROS production and protease release from granules (shaded red, green, and pink) which damages underlying cartilage and activates neighbouring immune cells and fibroblasts. Proteases also activate pro-peptides, such as cytokines produced by neutrophils and other infiltrating immune cells. A similar process is responsible for damage to blood vessels in SLE.
Neutrophil extracellular traps (NETs) are mesh like DNA structures decorated with histones, MPO and other antimicrobial proteins expelled from neutrophils in response to infectious or inflammatory stimuli (106). They are an alternative defense mechanism by which neutrophils trap and possibly kill microbes (106, 107). Whilst the early events that signal NET production rather than phagocytosis are unclear, at least two methods of chromatin decondensation leading to NET formation (NETosis) have been described: NOX2-dependent and NOX2-independent (108). NOX2-dependent NETosis, also termed suicidal NETosis, occurs via activation of NOX2 and production of intra-phagosomal ROS. This causes increased intracellular membrane permeability, movement of elastase to the nucleus and degradation of histones leading to chromatin decondensation and NET release (109). ROS production by NOX2 promotes the morphological changes that occur during NETosis (110) and inactivates caspases to block apoptosis pathways (109). NOX2-independent NETosis does not require the production of ROS by NOX2. In this case, mitochondrial ROS combine with increased intracellular calcium levels to activate peptidyl arginase deiminase (PAD) enzymes (e.g., PAD4) leading to hypercitrullination of histones, chromatin decondensation, and NET release (111, 112). Several inflammatory agents have been reported to induce NET release, including fMLP, IL-8, LPS, nitric oxide, and TNFα (113). Many proteins decorating NET are post-translationally modified, in particular histones which have been found to be methylated, acetylated (114) and citrullinated (111, 115–117) leading to speculation that NETs may be a source of auto-antigens in auto-immune disease (8). Recent work suggests that NETs (MPO:DNA and/or elastase:DNA complexes) detected in up to 79% of RA and up to 100% of SLE sera are generated in a NOX2-independent manner (118).
There is an ever increasing body of evidence to support the hypothesis that the externalization of double-stranded DNA and post-translationally modified proteins on NETs is implicated in the pathogenesis of SLE, through activation of interferon-producing plasmacytoid dendritic cells (pDCs) (Figure 3) (119) and damage to endothelial tissues and organs (25, 120). SLE sera cross-react in vitro with NET components (114, 121) and spontaneous NET production (NETosis) by SLE neutrophils ex vivo is also observed (120, 122). NET structures staining positive for DNA, elastase, MPO, and citrullinated histone H3 are found in cutaneous SLE lesions (123) and the kidney (120). Most strikingly the molecular targets of many of the almost 100 auto-antibodies associated with SLE, including those directed at nuclear DNA and nuclear proteins, can be detected in NETs (124). The majority of SLE patients will be positive for antibodies against ANA and/or dsDNA at some point in their disease course (2). Antibodies against histones (including acetylated and/or methylated histones) are also common (125–127). Recent proteomics analysis of NETs from SLE patients identified a number of histone proteins with acetylated, methylated and/or citrullinated residues (128), particularly in NOX2-independent NETs. A number of these, for example acetylated histone H2B (K21, K20), methylated histone H3 (K27) and acetylated histone H4 (K5, K8, K12, K16), correspond to known SLE auto-antibodies (114, 121, 129) and serum NET debris (130). Aside from histones, a number of rarer SLE auto-antibodies correspond to proteins identified on SLE NETs (128) including HMG-17 (131), catalase (132), lamin B1 and B2 (133–135), apolipoprotein A1 (136), cathelicidin (LL37) (137–139), annexin AI and α-enolase (140, 141). When directly compared with RA NETs, the levels of MPO, leukocyte elastase inhibitor and thymidine phosphorylase were higher in SLE NOX2-dependent NETs, whereas histones H1.0, H2B (type 1-J), H2B (type 2-F), and H4 were higher in SLE NOX2-independent NETs (128).
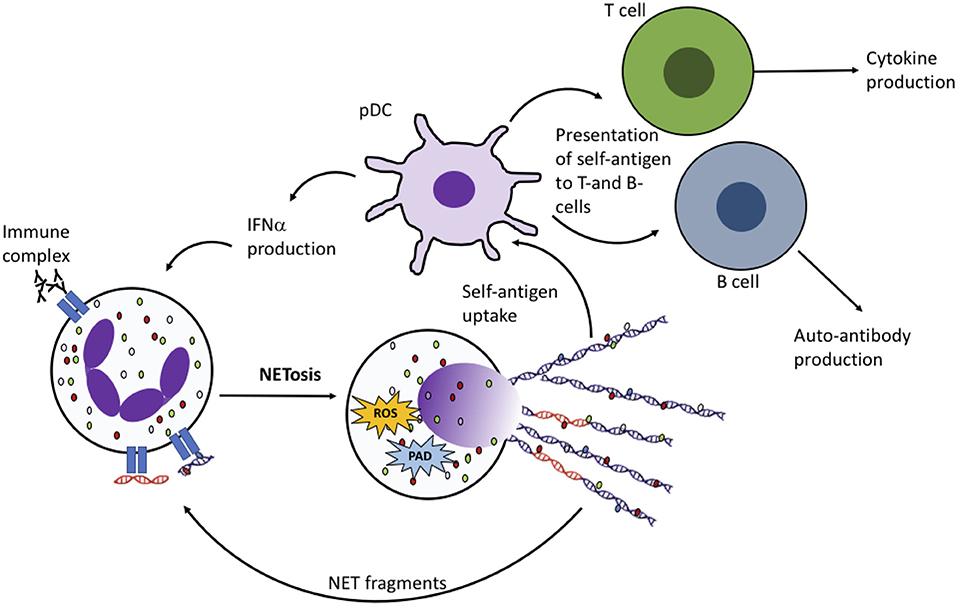
Figure 3. NET production in RA and SLE drives the auto-immune response. Fragments of DNA and proteins (including histones, MPO, elastase, HMGB1, LL37) are taken up by plasmacytoid dendritic cells (pDCs) and presented to auto-reactive B- and T-cells, leading to production of cytokines including interferon alpha (IFNα) and autoantibodies. NET fragments, including oxidized DNA (shown in orange) may also activate NET production by neighboring neutrophils.
Many NET proteins also correlate with SLE disease activity and play a direct role in tissue damage. MMP9 contained within SLE NETs activates endothelial MMP2, inducing endothelial dysfunction and apoptosis (142). Neutrophil gelatinase-associated lipocalin (NGAL) is both a urinary biomarker of lupus nephritis and a predictor of disease flare, although it is not known whether degranulating neutrophils or NETs are the source of NGAL in SLE urine (143–145). Auto-antibodies to NET proteins that become deposited within the glomeruli of patients with lupus nephritis (141) attract complement and leukocytes expressing Fcγ and complement receptors (including other neutrophils), activating the infiltrating cells causing further tissue damage, e.g., via ROS and protease degranulation (86, 146). In addition, excess NET production within the vasculature and glomeruli promotes vascular leakage and endothelial-to-mesenchymal transition (EndMT), a process that is associated with pathogenic fibrosis of tissues (147). NET production via PAD2 and PAD4 has also been shown to contribute to the development of atherosclerosis and vascular stiffness in murine lupus models (148, 149). Auto-antibodies against apolipoprotein A1 may neutralize the cardioprotective effect of the HDL complex, causing cardiovascular disease in SLE patients with anti-apolipoprotein A1 antibodies (136, 150). Whilst cathelicidin/LL37 may not play a direct role in tissue damage in SLE, its indirect roles include activation of type-I interferon production, activation of the inflammasome and further activation of NET production (137). The abundance of NET debris in SLE sera may be due to impairments in clearance mechanisms associated with tissue homeostasis. Both inhibitors of DNase-I, and anti-NET antibodies in SLE serum, have been proposed as mechanisms impairing the dismantling of NET structures. Impairment of DNase-I is also associated with kidney involvement in SLE (151).
Immune complexes in the sera of SLE patients may contain nucleic acids (Sm RNP RNA or DNA), which activate neutrophils to produce ROS and IL-8 via FcγR2a (152). This phenomenon is inhibited by chloroquine, a DMARD commonly used to treat SLE. SLE immune complexes often contain LL37:DNA complexes derived from NETs. LL-37:DNA complexes activate B cells via TLR9 leading to expansion of self-reactive memory B cells and IgG production (153). B cell activation by LL37:DNA can also be inhibited by chloroquine, suggesting an important role for endosomal TLR activation by NETs in the pathology of SLE. In a recent clinical study of 16 SLE patients, excessive NET production by was abrogated by combination therapy with rituximab (a B cell targeted therapy) and belimumab (anti-BLyS/BAFF) (154). Low disease activity, including a significant reduction in auto-antibody titers, was achieved in over 50% of patients in the study. Further analysis of sera from the patients in this study revealed that the combination of rituximab and belimumab significantly decreased anti-dsDNA, anti-histones, anti-nucleosomes and anti-C1q titers (155). In addition, the ability of patient sera to induce NET formation in vitro was decreased by around 75% following combination therapy with rituximab and belimumab (155).
In RA it is the exposure of citrullinated proteins on NETs that is a key driver of auto-immunity, leading to the development of anti-citrullinated peptide auto-antibodies (ACPA) (8, 156). NET debris can be detected in both serum and synovial fluid from RA patients (98, 118) and NET structures staining positive for CD15, elastase, MPO and citrullinated (cit) histone H3 can be seen in synovial biopsy tissues (156, 157). Many proteins in RA patient neutrophils are citrullinated via activation of PAD2 and PAD4 (158), including known auto-antibody targets: cit-actin, cit-histone H1.3, cit-histone H3, cit-vimentin (156, 158). PAD enzymes are present in synovial biopsies, localized with MPO in necrotic areas of synovial tissue that also contain large areas of citrullinated proteins (159), and PAD2 and PAD4 are present in NETs generated from ex vivo RA neutrophils (128). PADs are also found in high concentrations in synovial fluid alongside citrullinated proteins, such as α-enolase (160, 161). Proteomics analysis of RA NETs identified citrullinated forms of known auto-antibody targets, such as cit-α-enolase, cit-histone H2A, cit-histone H4, cit-vimentin (128), as well as acetylated and methylated histones in line with analysis of NET debris in RA serum (118). Proteomic analysis of synovial fluid from RA and spondyloarthritis (SpA) patients identified many neutrophil proteins present at significantly elevated concentrations in RA synovial fluid, including MPO, cathepsin G, annexin-A1, and NGAL. Although no difference was observed in the amount of cell-free DNA between RA and SpA synovial fluid, the levels of 21 NET proteins were elevated in RA SF, including histones H2A, H2B and H4, MMP9, elastase, and α-enolase (98). Whilst the levels of ACPA in RA serum do not appear to correlate with NET material, antibodies to NET material (ANETA) are significantly higher in seropositive RA patients (162).
Several mechanisms have been proposed explaining how NETs and NET proteins contribute to joint damage and disease activity in RA. Elastase within in NETs has been demonstrated to disrupt the cartilage matrix, triggering PAD2 release from fibroblast-like synoviocytes (FLS). Activated PAD2 causes citrullination of cartilage fragments which are then internalized by FLS and presented via MHC Class II to antigen specific T-cells leading to the production of ACPA in HLA-DRB1*04:01 transgenic mice (163, 164). Both MMP8 and MMP9, found in RA NETs, contribute to degradation of the cartilage matrix and are associated with increased mortality (100, 165, 166). NETs also contain enzymes which degrade aggrecan, another key structural component of cartilage (164). Citrullinated vimentin and aggrecan are preferentially recognized by T cells expressing the HLA-DRB1*04:01/04 allele (known as the “shared epitope”) (163, 167) inducing auto-antibody production (164). Auto-antibodies secreted by RA synovial B cells cross react with cit-fibrinogen, cit-histones H2A/H2B, and cit-vimentin, as well as NETs generated from RA blood and joint neutrophils (168). Presentation of citrullinated antigens to autoreactive T cells provides a molecular explanation for the strong association between the HLA-DRB1*04:01/04 allele and the development of RA (164, 167).
For many years, mature neutrophils were wrongly believed to be transcriptionally silent, and any changes in protein levels during activation were believed to be solely due to mobilization of internal stores during priming and/or membrane shedding rather than synthesis of new protein. However, there is now an increasing body of work demonstrating that neutrophil gene expression is dynamic, being rapidly regulated over short time points by exposure to inflammatory agents, such as TNFα, GM-CSF, IL-1β, LPS, and opsonized micro-particles (169–171), and over several hours by chromatin remodeling, for example in response to the TLR8 agonist resiquimod (R848) (172). Neutrophils have the capacity to express and secrete a wide range of inflammatory mediators, including interleukins (including IL-1α and−1β, IL-1RA, IL-6, IL-12, and IL-23), chemokines (CCL and CXCL family members), TNF superfamily members (including TNFα, BLyS/BAFF, APRIL, TRAIL, and RANKL), metabolites of arachidonic acid (leukotriene B4, prostaglandin E2, and thromboxane A2), and angiogenic factors, such as VEGF and HGF (8, 22, 173–177).
Neutrophil gene expression is highly regulated during granulopoiesis (178–180), with transcripts for granule proteins (e.g., MPO, elastase, lactoferrin) becoming depleted once the mature protein has been packaged within granules in the maturing neutrophil (181). The content of neutrophil granules is determined by the order in which the granules develop: azurophilic granules first, followed by specific granules, then gelatinase granules, and finally secretory vesicles (182). Expression of neutrophil receptors (integrins, FcγR, cytokine receptors) is also highly controlled during granulopoiesis, with neutrophil markers CD16 (FcγR3b) and CD10 (neprilysin) for example only being expressed as the mature neutrophil is ready to exit the bone marrow (181, 183). Many neutrophil genes, including those coding for granule proteins, migratory proteins, chemokines, and chemokine receptors are also regulated by circadian rhythms (19). Dynamic changes in neutrophil gene expression also take place as cells migrate from the blood into inflamed tissues during inflammation, with transcripts for adhesion molecules decreasing and chemokine expression increasing at sites of inflammation (49, 184). Polymorphisms within expression quantitative trait loci (eQTLs) have been found in over 160 genes which play a fundamental role in every stage of neutrophil biology, from granulopoiesis to activation during infection and ultimately apoptosis. Several of these eQTLs are associated with known Mendelian disorders and inflammatory diseases (185).
Transcriptomic analysis of RA and SLE neutrophils has revealed a strong IFNα-induced gene expression signature in both conditions (186, 187). In RA, a high IFNα-regulated gene expression signature is a predictor of response to TNF inhibitor therapy (188). IFNα has recently been shown to regulate neutrophil activation in the presence of inflammatory cytokines (173) and in particular is involved in a switch in expression of chemokine genes. Whilst expression of genes for CCL and CXCL family chemokines is relatively low in blood neutrophils from RA patients with a high interferon gene expression signature, the expression of these chemokines increases significantly in RA synovial fluid neutrophils following migration to inflamed joints (49, 189–191). RA blood neutrophils also express mRNA for MHC Class II, with both RNA and MHC Class II protein being detected in RA synovial fluid neutrophils (192). Together this activated, synovial fluid neutrophil phenotype is responsible for driving activation of innate and adaptive immune cells in RA tissues. A polymorphism in the leukocyte phosphatase PTPN22 (R620W, rs2476601) enhances migration of RA neutrophils, and causes increased production of ROS following TNF-priming in vitro (193). Neutrophils from RA patients also express significantly higher levels of membrane proteinase-3 (mPR3) compared to healthy controls, an observation that is found in other neutrophil-driven autoimmune diseases, such as vasculitis, but not in T-cell driven type-I diabetes (194). In some RA patients, mRNA levels of granule genes (e.g., elastase, MPO) remain elevated even in mature blood neutrophils despite these transcripts normally being down-regulated during granulopoiesis (188). Expression of neutrophil granule genes is associated with non-response to TNF inhibitor therapy in RA but does not correlate with intracellular levels of granule proteins (188). Expression of membrane TNFα correlates with DAS28 and is decreased following successful treatment with TNF inhibitor therapy (48).
A global down-regulation of miRNA expression has been shown in RA blood and synovial fluid neutrophils compared to healthy controls (191). Decreased miRNA levels correlate with clinical parameters of disease including ACPA titer and DAS28 scores and is more pronounced in synovial fluid neutrophils. Targets of these down-regulated miRNAs include genes associated with cell migration, cell survival and inflammation, suggesting that a defect in miRNA expression may drive neutrophil-mediated inflammation within RA joints (191). An eQTL in the gene encoding PAD4 (PADI4, rs2240335-A) is associated with increased expression of PAD4 in neutrophils and is in almost complete linkage disequilibrium with the rs230188 SNP in PADI4 (LD r2 = 0.93) that confers an elevated risk of developing RA (185, 195).
In SLE, the IFNα-induced gene expression signature is believed to result from activation of pDCs by nuclear debris and NET fragments. The IFNα-induced gene expression signature can be detected in neutrophils, PBMCs and whole blood transcriptome analysis (187, 196–198), and in some studies has been shown to correlate with organ involvement and/or disease activity (198). However, a subset of SLE patients do not display this signature and indeed SLE patients are often described as being interferon “high” or “low” (196, 199). SLE neutrophil DNA is hypomethylated, especially near interferon-response genes (200). Some success in stratifying interferon-high patients to anti-IFNα therapy (anifrolumab) is evident (201), although the largest clinical trials of anifrolumab (TULIP-1 and TULIP-2) found no significant differences in the response to anti-interferon therapy between the interferon-high and interferon-low patient groups (202, 203). Expression of neutrophil genes within SLE whole blood has been attributed to the increase in the population of low density granulocytes (LDGs), discussed below (198), although proteomics analysis of SLE neutrophils and LDGs suggests that protein levels of MPO and other granule proteins is higher in SLE neutrophils (204). The expression of neutrophil granule protein genes in SLE whole blood is strongly associated with lupus nephritis (197, 205), vascular inflammation and cardiovascular involvement (206). SLE neutrophils also express high levels of BAFF/BLyS (198), the molecular target of belimumab which is the first biologic therapy developed to specifically target SLE (207).
Cellular metabolism is key regulator of neutrophil energy production, activation and function under conditions of both homeostasis and inflammation. The complex interplay of metabolic pathways and the utilization of shared metabolic intermediates has been the subject of great focus in immunology research as well as in diseases of metabolic dysfunction and aging (208–210). Complex changes in metabolic regulation within leukocytes occur during activation, when migration from blood to tissue or differentiation into tissue resident immune cells causes dramatic changes in nutrient and oxygen availability, placing sudden and high metabolic demands upon immune cells (211, 212). Fine-tuning of metabolism during an inflammatory response is a key for the generation of small molecule metabolites, such as ATP, NADPH, nucleotides, and amino acids, which are required rapidly and in high abundance during cellular activation (211, 212). Dysregulation of metabolic control has been described in inflammatory diseases, such as RA (213), where changes in T-cell glycolytic activity drives differentiation, hyperproliferation, and hypermigration of T-cell subsets (213–215).
Glucose metabolism has its greatest role in neutrophil energy production (Figure 4); neutrophils are known to rely on glycolysis to fuel their energy requirements (216), where the multi-step enzymatic conversion of glucose into pyruvate in the cytosol provides relatively low levels of ATP and NADH. Pyruvate would normally be oxidized by mitochondria through the tricarboxylic acid (TCA) cycle in aerobic conditions, however in neutrophils it is converted instead into lactate, enabling the generation of NAD+ for re-use in the glycolytic pathway. The first intermediate of glycolysis, glucose 6-phosphate (G6P), fuels the pentose phosphate pathway (PPP). NAPDH is produced in the oxidative phase of the PPP, maintaining NOX2 activity and ROS production, and is necessary for chromatin decondensation, NOX2-dependent NET formation and NET release (217). The non-oxidative step of the PPP generates nucleic acids and glycolytic precursors. Tight control over glucose metabolism and PPP activity is achieved by the glucose-6-phosphate transporter (G6PT)/G6Pase-β complex which maintains cellular energy homeostasis and functionality in neutrophils by limiting G6P availability in the cytoplasm. Dysfunction in glucose homeostasis due to defective G6PT activity in neutrophils impairs ROS production, calcium mobilization and chemotaxis (218, 219).
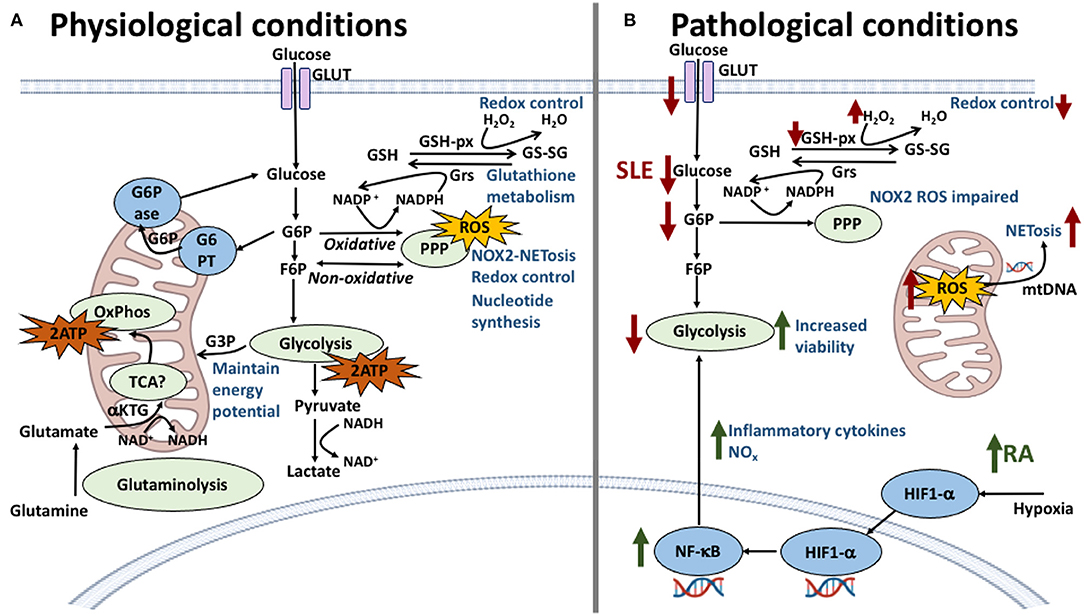
Figure 4. Neutrophil metabolic pathways and the effect of their dysregulation in SLE and RA. (A) In physiological conditions, glycolysis is the main energy producing pathway utilized by neutrophils; intermediate metabolites G6P and F6P are starter molecules for the pentose phosphate pathway (PPP), important in redox control, NOX2-dependent NETosis and ROS production. The oxidative stage of PPP produces NADPH which is used in the glutathione metabolism to reduce glutathione providing further redox capacity. Other energy producing pathways have been described, previously thought to be non-functional, such as the TCA cycle which connects glutaminolysis pathway and further regulates NOX2 complex by generating the reducing equivalents of NADPH. (B) In SLE and RA dysregulation of these pathways is responsible for the conditions observed at the cellular level. In SLE (red arrows), a lower expression of glucose transporters is met with lower levels of intracellular glucose, diminishing the energy output by glycolysis and compromising cellular viability. Furthermore, a decrease of G6P intermediates correlates with lower levels of NOX2-dependent ROS and decreased redox capacity. NOX2-independent ROS production in mitochondria is amplified in SLE neutrophils with increasing release of NETs containing mitochondrial DNA. In RA (green arrows), the inflammatory environment and hypoxic conditions increase the expression of HIF1-α which upregulates key glycolytic enzymes increasing energy production and viability. HIF1-α also is an upstream regulator of NF-κB which increases pro-inflammatory cytokine production therefore maintaining the inflammatory environment.
A number of other metabolic pathways have been described in neutrophils, including the Krebs/TCA cycle, oxidative phosphorylation (OXPHOS), and a fatty acid oxidation (FAO) pathway, all taking place within the mitochondria. Early experiments suggested that neutrophil mitochondria were not fully functionally active, having a dispensable role in neutrophil respiration and energy production. Mitochondrial density in human neutrophils is low and inhibitors of OXPHOS do not alter the rates of oxygen consumption or H2O2 production (220–222). However, the notion of metabolically inactive mitochondria was recently challenged by a study in mice which suggested that a metabolic shift from glycolysis to OXPHOS for energy sustainability was possible in neutrophils, both during changes that occur within the cancer tumor microenvironment, and in a number of neutrophil subsets (223). Mitochondria also have a wider and more important role in the regulation of aerobic glycolysis by maintaining the energy potential through complex III of the respiratory chain. Glycerol-3-phosphate (G3P), a by-product of glycolysis, can enter mitochondria where it is re-oxidized on the outer surface of the inner membrane, effectively maintaining membrane potential (224, 225). A further role for neutrophil mitochondrial metabolism in neutrophil function has been demonstrated, with mitochondrial activity being central to processes including ROS production, chemotaxis, and apoptosis (226–230). There is also increasing evidence of the utilization of the TCA cycle by neutrophils for energy metabolism under glucose-limited conditions (231) and during granulopoiesis (232). In addition, LDGs convert glutamate or proline to alpha-ketoglutarate (α-KG) to feed the TCA cycle and enable them to perform metabolically demanding neutrophil functions under conditions of glucose deprivation (233).
Parallel to glucose and mitochondrial metabolism, which are restricted to the cytosol and the mitochondria, glutaminolysis is a metabolic pathway that encompasses both environments and is tightly correlated with the phagocytic and bacterial killing ability of cultured neutrophils. Interestingly, neutrophils have been found to utilize more glutamine than other leukocytes including lymphocytes and macrophages (234). The precursor glutamine is not completely oxidized to generate ATP, and instead plays a role in the regulation and activation of the NOX2 complex, joining glucose metabolism in providing the means of rapidly generating the reducing equivalents of NADPH for the microbicidal NADPH oxidase (NOX2) system (235, 236). Glutaminolysis converts glutamine into TCA cycle metabolites, linking the two pathways together. Glutamine goes through a series of biochemical reactions where it is initially converted to glutamate, and then into α-ketoglutarate, which oxidizes NAD+ into NADH. At this point α-ketoglutarate enters the TCA cycle and is converted to malate which is then transported out of the mitochondria to be converted into pyruvate, which just like in glucose metabolism is converted into lactate to generate NAD+ (234, 237). Under conditions when glucose supply is limited, neutrophils can switch to the utilization of glutamine to meet their energetic needs (238). Furthermore, glutamine addition to cell culture medium of isolated neutrophils increases their phagocytic activity and rate of superoxide production (239). Glutamine has also been found to indirectly affect the functionality of neutrophils by modulating the production of IL-8, a neutrophil chemoattractant, in various cell types during activation (240, 241).
Within RA synovial fluid, a dynamic environment exists that controls neutrophil fate by a variety of anti- and pro-apoptotic factors. Low oxygen levels in the RA joint trigger a delay in apoptosis (58). HIF1-α is upregulated in hypoxic conditions by the family of oxygen sensing proteins known as prolyl hydroxylase domain enzymes (PHD1-3). HIF1-α exerts control over glycolysis by regulating the expression of key glycolytic enzymes, G3PDH and triosephosphate isomerse-1, providing a mechanism for the continued generation of ATP. This mechanism is an essential requirement for neutrophil functional responses to inflammation (242–244) and, by additionally directing the expression of the leukocyte β2 integrin CD18, it is critical for innate and adaptive immune responses (245). Studies have revealed that NF-κB is an important downstream effector of the HIF1-α-dependent response to hypoxia and that knockdown of the HIF1-α gene decreases glycolytic metabolism and induces cell death (59). RA synovial fluid contains significantly lower levels of glucose than OA synovial fluid, reflecting the high levels of cellular metabolism by infiltrating leukocytes and synovial tissues (246). Other metabolites within RA synovial fluid, including citrate, itaconate, and succinate, have been shown to regulate ROS production, cytokine expression and inflammasome activation in leukocytes (247–249), although the direct effect of these individual metabolites on synovial neutrophil function remains to be determined.
The abundance of apoptotic neutrophils in the blood along with their defective clearance and the propensity to form NETs contribute to the pathogenesis of SLE. Consistent studies reporting increased apoptosis, inflammatory phenotypes and mitochondrial defects in SLE neutrophils suggest that their cellular metabolism might be highly skewed (218, 250). SLE neutrophils have been found to have a decreased capacity for glucose uptake via defective expression of glucose transporters (GLUT-3 and GLUT-6) on the cell membrane compared to healthy neutrophils (250, 251). Limited glucose availability may pose a threat to cellular viability due to decreased glycolytic flux. This is true for other immune cells, such as lymphocytes, which enter BCL-2-regulated apoptosis when glycolytic flux decreases to levels that no longer sustain viability (252). Glucose availability is essential for the two primary pathways in activated neutrophils (glycolysis and PPP), and impaired glycolytic flux could explain the impaired NOX2-dependent ROS production seen in some SLE neutrophils, where glucose would be prioritized for energy production over ROS/NET production. Indeed, impairment of the G6PD/glucose flux in experimental models is directly associated with less NOX2 activity and with lower ROS production (253). Decreased levels of NOX2 ROS production may be compensated for by hyper-functional mitochondrial ROS production, which causes oxidation of unprotected mitochondrial DNA which is then extruded through NETosis (122). Increased mitochondrial ROS production and higher levels of O2-, H2O2, and HO• in SLE neutrophils (76, 77) are met by a decreased redox capacity compared to healthy controls (68, 251, 254). This is due to lower concentrations of glutathione which is critical for adequate redox capacity (255, 256). Neutrophils have a large capacity to keep glutathione in the reduced form via the activity of glutathione reductase (GRs) (257). A deficiency in this enzyme produces a more transient oxidative burst in response to bacteria (258). On the other hand, age-related impairment of glutathione peroxidase activity accounts for increased intracellular accumulation of hydrogen peroxide (259). Decreased levels of intracellular glutathione peroxidase (GSH-px), due to impaired GRs activity, leads to neutrophil dysfunction during conditions that are associated with chronic inflammation (260, 261). NOX2-independent ROS production in neutrophils is amplified by reagents affecting glutathione homeostasis (262). The wider impact of circulating SLE neutrophils in the modulation of other leukocyte activation is emphasized by the increased release of NETs containing mitochondrial DNA when compared to healthy donors; this was linked to chronic activation of pDCs and amplification of IFNα production (119, 263).
The peripheral blood population of neutrophils is not a homogeneous pool (264); several populations of neutrophils have been identified which circulate alongside mature neutrophils, including low-density granulocytes (LDGs, CD15high/CD14low/CD10+/−/CD16+), granulocytic myeloid-derived suppressor cells (G-MDSCs, CD11chigh/CD62Llow/CD11bhigh/CD16high/CD33low), and reverse migrated neutrophils (RM, CD54+/CD18high/CXCR1low) (265– 267). Tumor associated neutrophils (TANs), which may have a pro- or anti-tumor phenotype, are present in many cancers (266, 268, 269); their function is outside the scope of this review.
G-MDSCs are an immune regulatory neutrophil subtype that inhibit the activation and expansion of autologous T cells, via the production of ROS at the immune synapse (270). Expression of MAC-1 (integrin αMβ2) is essential to the suppressor function of G-MDSCs (270). G-MDSCs with the ability to inhibit T cell proliferation are present in RA blood and synovial fluid (271, 272) and have been studied to greater effect in murine models of auto-immune arthritis, where they have been shown to inhibit both T cell proliferation and differentiation of Th1 and Th17 cells, and promote Treg numbers (273–275). G-MDSCs are found at a higher proportion in the blood of SLE patients than in healthy controls (272). SLE G-MDSCs produce high levels of ROS and also have the ability to impair T cell expansion (276). G-MDSCs from lupus-prone mice produce more ROS and NETs than healthy mice, however the population of G-MDSCs is unable to expand under inflammatory conditions, suggesting the loss of G-MDSCs due to NETosis may contribute to the impaired resolution of inflammation in SLE (277).
Reverse migration of neutrophils from sites of inflammation back into the circulation has been observed in zebrafish, mice and humans (267). Zebrafish RM neutrophils remain functional and able to respond to a second inflammatory challenge (278). RM neutrophils express high levels of ICAM-1 (CD54) (279), and in mice have been shown to contribute to systemic inflammation (280). RM neutrophils represent around 1–2% of circulating blood neutrophils in RA patients and only around 0.25% of blood neutrophils in healthy individuals (279). RM RA neutrophils have lower levels of constitutive apoptosis and produce higher amounts of ROS than circulating blood neutrophils (279).
Low-density granulocytes (LDGs) were first reported in the blood of SLE patients in 1986 (281) but their function and pathological significance has only recently been explored. These cells, remaining in the peripheral blood mononuclear cell (PBMC) layer after density-gradient centrifugation, express cell-surface markers specific to mature neutrophils (CD15high/CD14low/CD10+/−/CD16+) (282, 283), whilst expressing mRNA transcripts characteristic of immature neutrophils (e.g., MPO, elastase) (120, 283, 284). High numbers of LDGs in SLE blood correlate with skin involvement, vasculitis, dsDNA titers and SLEDAI scores (120, 284, 285), and SLE LDGs have an increased tendency to form NETs in vitro (120). Un-stimulated SLE LDGs secrete increased amounts of IL-8 and IL-6 and have impaired phagocytic capacity (282). SLE LDGs also stimulate production of TNFα, TNFβ, and IFNγ by T cells (286). Recent work has revealed that the SLE LDG population is heterogeneous (mature CD10+ or immature CD10−), with significant differences in transcriptomic and epigenomic regulation of function and phenotype that correlates with clinical manifestations of the disease (187, 284). Mature CD10+ SLE LDGs express high amounts of mRNA and protein for interferon-regulated genes, whereas immature CD10− SLE LDGs express high amounts of mRNA for cell cycle genes (187, 204). CD10+ SLE LDGs undergo phagocytosis, chemotaxis and NETosis at higher levels than CD10− LDGs, which release MPO at higher amounts than CD10+ LDGs and normal density SLE neutrophils (187). It has been suggested that SLE LDGs undergo NETosis in response to the production of mtROS, with SLE LDG NETs containing mitochondrial DNA including oxidized DNA (8-oxo-2'-deoxyguanosine) which is strongly interferogenic (122). This phenomenon is also observed in chronic granulomatous disease LDGs, which lack functional NOX2 but can produce mtROS and NETs (122). SLE LDGs have stiffer biomechanical properties and are slower to migrate through microvascular mimetics in vitro (204). This may explain why LDGs are not found in affected tissues in SLE (284).
RA LDGs have a distinct transcriptome profile compared to RA neutrophils, expressing high levels of transcripts for granule proteins and cell cycle checkpoint genes, and lower levels of expression of apoptotic genes, cytokines, chemokines, and signaling receptors (283). The presence of LDGs in RA blood is unaffected by therapy, and LDG counts correlate with measures of disease activity (DAS28) (283). RA LDGs undergo lower levels of apoptosis in vitro after overnight culture; however whilst LDG apoptosis can be further delayed by GM-CSF, LDG apoptosis is unaffected by TNF-α. In addition, ROS production by TNFα-primed RA LDGs is lower than paired blood neutrophils, likely due to their lower expression of TNF-receptors (283). NET production by RA LDGs is not significantly different from paired neutrophils (283).
Whilst the main focus of investigation into the phenotype of LDGs has focused on SLE and to a lesser extent RA, their presence in the blood is not exclusive to rheumatic disease. LDGs have been identified in many other disease settings, including asthma, vasculitis, multiple sclerosis and chronic kidney disease (287–290). Indeed, they are even present in low numbers in healthy controls (291). Isolation of LDGs from blood is highly dependent upon the density of the isolation medium used (e.g., Ficoll, Percoll, Polymorphprep) (292) and this raises the question as to whether studies using different isolation protocols for preparation of neutrophils and LDGs from whole blood can be directly compared. There are mixed reports on the functionality of LDGs from healthy controls, and whether they have different immunological properties (e.g., T cell suppression) to normal density neutrophils and LDGs from inflammatory disease (291, 292). Another key question is whether LDGs represent a novel subset of neutrophils, or whether their phenotype reflects one of spectra of phenotypes that blood neutrophils may exhibit through functional plasticity. Evidence from RA and SLE (CD10− LDGs), where LDGs express cell cycle genes and transcripts for neutrophil granule proteins, suggests that these cells may have arisen from emergency granulopoiesis due to chronic inflammation (187, 283). Administration of LPS in vivo to healthy volunteers appears to support this conclusion, with an increase in immature CD16dim band cells being observed in blood 3h after LPS challenge (292). However, it is also possible to induce a low density phenotype from normal density neutrophils in vitro by activation with agents, such as fMLP, platelet activating factor, TNFα and LPS (291, 292), suggesting that LDGs may represent a subset of primed or activated neutrophils (292). Further work needs to be carried out to determine the true origin, phenotype and nature of LDGs and other neutrophil subsets, such as G-MDSCs (264).
In this review we have discussed the multitude of ways that inflammatory neutrophils drive inflammation in RA and SLE. This raises the potential to target dysregulated neutrophil activation with therapeutics in both diseases (293). In RA, neutrophil activation can be targeted by biologic DMARDs (bDMARDs), such as anti-TNF therapy, which has been demonstrated to decrease neutrophil membrane TNF expression and NF-κB activation (48). Newer orally available, small molecule therapies, such as JAK inhibitors have shown good efficacy in RA, as they target intracellular signaling via a number of cytokine receptors, including IFNα, IFNγ, GM-CSF, and IL-6 (294, 295). JAK inhibitors baricitinib (JAK1/2) and tofacitinib (JAK3/1) inhibit cytokine priming in neutrophils and can inhibit RA neutrophil migration and ROS production (296). In SLE, the bDMARD belimumab inhibits the cytokine BLyS/BAFF, a major source of which is activated neutrophils and LDGs (175, 198). Belimumab is one of only two drugs specifically licensed to treat SLE in the UK, the other being hydroxychloroquine.
Hydroxychloroquine, an anti-malarial already widely used to treat both RA and SLE, is a potent modulator of neutrophil function. It has been shown to inhibit neutrophilic inflammation into inflamed kidneys (297), inhibit NET production via inhibition of TLR9 (298), and block ROS and IL-8 production in response to RNA-containing immune complexes (152). Methotrexate, commonly used in both SLE and RA, inhibits cytokine-delayed neutrophil apoptosis, ROS production and leukotriene B4 synthesis (299–301). Glucocorticoids are frequently used in both RA and SLE to control disease flares. Prednisolone for example, rapidly disarms pro-inflammatory neutrophils and inhibit both ROS release and production of pro-inflammatory mediators (14, 302, 303).
Several newer therapies under development or in clinical trial also target neutrophil activation. Major activators of neutrophil production and priming, the colony stimulating factors (CSFs), are exciting targets for treatment of neutrophil-driven inflammatory diseases. Anti-GM-CSF (mavrilimumab) therapy has had success in RA clinical trials (294), and anti-G-CSF therapy is effective in treating murine arthritis, both inhibiting neutrophil migration into joints, and suppressing cytokine production (304). Neutrophil migration into inflammatory murine joints is also significantly decreased by inhibitors of CXCR1/CXCR2, the receptor for CXCL8 (IL-8) (305). This decrease in neutrophil infiltration is mirrored by lower disease activity and TNFα production within the joint (306). Bosutinib, an Abl/Src kinase inhibitor currently used to treat patients with chronic myeloid leukemia, inhibits neutrophil FcγR2A-induced ROS production, recruitment to glomerular capillaries and kidney injury in an immune complex-driven model of kidney disease (85) suggesting this may be a promising therapy to target neutrophilic damage in lupus nephritis.
Excess NET production represents an exciting prospect for therapeutic development, with the potential to break the chain leading to auto-antigen recognition, activation of pDCs, interferon production, auto-antibody production and damage to local tissues, such as cartilage and microvessels within the kidney. As mentioned earlier, a clinical trial of rituximab and belimumab inhibited NET production in SLE, and this was associated with lower auto-antibody titers (including lower anti-dsDNA and anti-histones) and a decrease in disease activity (154, 155). NET production may also be targeted by inhibitors of PAD4, MPO and neutrophil elastase. MPO and elastase inhibitors reduce neutrophil-driven inflammation in animal models of inflammatory disease and human respiratory disease (307, 308). PAD4 is an enzyme that catalyses the conversion of arginine to citrulline (116, 309). It plays an important role in chromatin decondensation during NETosis and is physically associated with the cytosolic subunits of the oxidative burst machinery in a way that regulates assembly of the active NOX2 complex (116, 309). Over the last few years, PAD4 has emerged as a potential therapeutic for the treatment of RA and SLE. Initially irreversible inhibitor compounds, such as F- and Cl-amide were found to inhibit PAD4 (310), and showed efficacy in RA and SLE models through the inhibition of NET production. Cl-amidine prevented development of atherosclerotic plaques, interferon production and immune complex deposition in the kidney in lupus-prone mice (148, 149, 311). However, their poorly understood involvement in this process was the driving force for discovering reversible inhibitors, such as GSK484 and GSK199. GSK484 is the more potent of the two and selectively targets PAD4. It inhibits citrullination in primary neutrophils and NET formation in both mouse and human neutrophils (312, 313). Currently novel agents targeting PAD4 are being developed, which have shown to decrease the levels of circulating NET DNA in serum (313, 314). However, complete inhibition of NET production may block an important neutrophil function that provides protection against infection (315). Indeed, PAD4 knock out mice are highly susceptible to developing systemic inflammation from bacterial keratitis, where NETs normally function to protect the host from infection at the expense of the cornea (316). PAD enzymes also have a key physiological role in regulating gene expression and cellular differentiation, therefore a more targeted approach to PAD/NET inhibition may be required for development of therapeutics (315).
In this review we have highlighted the way in which dysregulated neutrophil activation can contribute to the development and progression of RA and SLE. In particular, dysregulated apoptosis and NETosis lead to exposure of intracellular post-translationally modified proteins and DNA activating the adaptive immune response (interferon release, auto-antibody production) and inducing damage to tissues either directly or by activating neighboring cells. Neutrophil degranulation and ROS production damage local tissues and contribute to systemic inflammation. Aberrant neutrophil activation in RA and SLE is caused, in part, by a dysregulation of gene expression and metabolism, via different mechanisms specific to each disease. Targeting unwanted neutrophil activation in RA and SLE may be a promising avenue for investigation and may have fewer side effects than the broad-spectrum immunosuppressants often used to treat these life-limiting auto-immune conditions.
MF, ZM, and HW wrote, edited, and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
MF was funded by a Versus Arthritis and Masonic Charitable Foundation Ph.D. scholarship (Grant No. 22193). HW was funded by a Versus Arthritis Career Development Fellowship (Grant No. 21430).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Bengtsson AA, Ronnblom L. Systemic lupus erythematosus: still a challenge for physicians. J Intern Med. (2017) 281:52–64. doi: 10.1111/joim.12529
2. Isenberg DA, Manson JJ, Ehrenstein MR, Rahman A. Fifty years of anti-ds DNA antibodies: are we approaching journey's end? Rheumatology (Oxford). (2007) 46:1052–6. doi: 10.1093/rheumatology/kem112
3. Ward MM. Premature morbidity from cardiovascular and cerebrovascular diseases in women with systemic lupus erythematosus. Arthritis Rheum. (1999) 42:338–46. doi: 10.1002/1529-0131(199902)42:2<338::AID-ANR17>3.0.CO;2-U
4. Lisnevskaia L, Murphy G, Isenberg D. Systemic lupus erythematosus. Lancet. (2014) 384:1878–88. doi: 10.1016/S0140-6736(14)60128-8
5. Davidson A. What is damaging the kidney in lupus nephritis? Nat Rev Rheumatol. (2016) 12:143–53. doi: 10.1038/nrrheum.2015.159
6. Nishi H, Mayadas TN. Neutrophils in lupus nephritis. Curr Opin Rheumatol. (2019) 31:193–200. doi: 10.1097/BOR.0000000000000577
7. Hanly JG. Diagnosis and management of neuropsychiatric SLE. Nat Rev Rheumatol. (2014) 10:338–47. doi: 10.1038/nrrheum.2014.15
8. Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol. (2014) 10:593–601. doi: 10.1038/nrrheum.2014.80
9. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
10. Bucknall RC. Arthritis and inflammatory eye disease. Rheumatology (Oxford). (2005) 44:1207–9. doi: 10.1093/rheumatology/kei027
11. Roman MJ, Salmon JE. Cardiovascular manifestations of rheumatologic diseases. Circulation. (2007) 116:2346–55. doi: 10.1161/CIRCULATIONAHA.106.678334
12. McInnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet. (2017) 389:2328–37. doi: 10.1016/S0140-6736(17)31472-1
13. Zharkova O, Celhar T, Cravens PD, Satterthwaite AB, Fairhurst AM, Davis LS. Pathways leading to an immunological disease: systemic lupus erythematosus. Rheumatology (Oxford). (2017) 56:i55–66. doi: 10.1093/rheumatology/kew427
14. Wright HL, Moots RJ, Bucknall RC, Edwards SW. Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford). (2010) 49:1618–31. doi: 10.1093/rheumatology/keq045
15. Koh TJ, DiPietro LA. Inflammation and wound healing: the role of the macrophage. Expert Rev Mol Med. (2011) 13:e23. doi: 10.1017/S1462399411001943
16. Rosales C. Neutrophil: a cell with many roles in inflammation or several cell types? Front Physiol. (2018) 9:113. doi: 10.3389/fphys.2018.00113
17. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. (2010) 31:318–24. doi: 10.1016/j.it.2010.05.006
18. Hidalgo A, Chilvers ER, Summers C, Koenderman L. The neutrophil life cycle. Trends Immunol. (2019) 40:584–97. doi: 10.1016/j.it.2019.04.013
19. Adrover JM, Del Fresno C, Crainiciuc G, Cuartero MI, Casanova-Acebes M, Weiss LA, et al. A neutrophil timer coordinates immune defense and vascular protection. Immunity. (2019) 50:390–402 e310. doi: 10.1016/j.immuni.2019.01.002
20. Adrover JM, Aroca-Crevillen A, Crainiciuc G, Ostos F, Rojas-Vega Y, Rubio-Ponce A, et al. Programmed ‘disarming' of the neutrophil proteome reduces the magnitude of inflammation. Nat Immunol. (2020) 21:135–44. doi: 10.1038/s41590-019-0571-2
21. Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. (2006) 6:541–50. doi: 10.1038/nri1841
22. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. (2011) 11:519–31. doi: 10.1038/nri3024
23. Desgeorges A, Gabay C, Silacci P, Novick D, Roux-Lombard P, Grau G, et al. Concentrations and origins of soluble interleukin 6 receptor-alpha in serum and synovial fluid. J Rheumatol. (1997) 24:1510–6.
24. Hurst SM, Wilkinson TS, McLoughlin RM, Jones S, Horiuchi S, Yamamoto N, et al. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity. (2001) 14:705–14. doi: 10.1016/s1074-7613(01)00151-0
25. Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol. (2011) 7:691–9. doi: 10.1038/nrrheum.2011.132
26. Thieblemont N, Wright HL, Edwards SW, Witko-Sarsat V. Human neutrophils in auto-immunity. Semin Immunol. (2016) 28:159–73. doi: 10.1016/j.smim.2016.03.004
27. McCracken JM, Allen LA. Regulation of human neutrophil apoptosis and lifespan in health and disease. J Cell Death. (2014) 7:15–23. doi: 10.4137/JCD.S11038
28. El Kebir D, Filep JG. Modulation of neutrophil apoptosis and the resolution of inflammation through beta2 integrins. Front Immunol. (2013) 4:60. doi: 10.3389/fimmu.2013.00060
29. Dzhagalov I, St John A, He YW. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood. (2007) 109:1620–6. doi: 10.1182/blood-2006-03-013771
30. Klein JB, Buridi A, Coxon PY, Rane MJ, Manning T, Kettritz R, et al. Role of extracellular signal-regulated kinase and phosphatidylinositol-3 kinase in chemoattractant and LPS delay of constitutive neutrophil apoptosis. Cell Signal. (2001) 13:335–43. doi: 10.1016/s0898-6568(01)00151-6
31. Klein JB, Rane MJ, Scherzer JA, Coxon PY, Kettritz R, Mathiesen JM, et al. Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways. J Immunol. (2000) 164:4286–91. doi: 10.4049/jimmunol.164.8.4286
32. Derouet M, Thomas L, Cross A, Moots RJ, Edwards SW. Granulocyte macrophage colony-stimulating factor signaling and proteasome inhibition delay neutrophil apoptosis by increasing the stability of Mcl-1. J Biol Chem. (2004) 279:26915–21. doi: 10.1074/jbc.M313875200
33. Moulding DA, Quayle JA, Hart CA, Edwards SW. Mcl-1 expression in human neutrophils: regulation by cytokines and correlation with cell survival. Blood. (1998) 92:2495–502.
34. Cross A, Moots RJ, Edwards SW. The dual effects of TNFalpha on neutrophil apoptosis are mediated via differential effects on expression of Mcl-1 and Bfl-1. Blood. (2008) 111:878–84. doi: 10.1182/blood-2007-05-087833
35. Petrin D, Turcotte S, Gilbert AK, Rola-Pleszczynski M, Stankova J. The anti-apoptotic effect of leukotriene B4 in neutrophils: a role for phosphatidylinositol 3-kinase, extracellular signal-regulated kinase and Mcl-1. Cell Signal. (2006) 18:479–87. doi: 10.1016/j.cellsig.2005.05.021
36. Geering B, Simon HU. Peculiarities of cell death mechanisms in neutrophils. Cell Death Differ. (2011) 18:1457–69. doi: 10.1038/cdd.2011.75
37. van den Berg JM, Weyer S, Weening JJ, Roos D, Kuijpers TW. Divergent effects of tumor necrosis factor alpha on apoptosis of human neutrophils. J Leukoc Biol. (2001) 69:467–73. doi: 10.1189/jlb.69.3.467
38. Murray J, Barbara JA, Dunkley SA, Lopez AF, Van Ostade X, Condliffe AM, et al. Regulation of neutrophil apoptosis by tumor necrosis factor-alpha: requirement for TNFR55 and TNFR75 for induction of apoptosis in vitro. Blood. (1997) 90:2772–83.
39. Nolan B, Kim R, Duffy A, Sheth K, De M, Miller C, et al. Inhibited neutrophil apoptosis: proteasome dependent NF-kappaB translocation is required for TRAF-1 synthesis. Shock. (2000) 14:290–4. doi: 10.1097/00024382-200014030-00008
40. Dransfield I, Buckle AM, Savill JS, McDowall A, Haslett C, Hogg N. Neutrophil apoptosis is associated with a reduction in CD16 (Fc gamma RIII) expression. J Immunol. (1994) 153:1254–63.
41. Midgley A, McLaren Z, Moots RJ, Edwards SW, Beresford MW. The role of neutrophil apoptosis in juvenile-onset systemic lupus erythematosus. Arthritis Rheum. (2009) 60:2390–401. doi: 10.1002/art.24634
42. Chiewchengchol D, Midgley A, Sodsai P, Deekajorndech T, Hirankarn N, Beresford MW, et al. The protective effect of GM-CSF on serum-induced neutrophil apoptosis in juvenile systemic lupus erythematosus patients. Clin Rheumatol. (2015) 34:85–91. doi: 10.1007/s10067-014-2800-2
43. Courtney PA, Crockard AD, Williamson K, Irvine AE, Kennedy RJ, Bell AL. Increased apoptotic peripheral blood neutrophils in systemic lupus erythematosus: relations with disease activity, antibodies to double stranded DNA, and neutropenia. Ann Rheum Dis. (1999) 58:309–14.
44. Midgley A, Mayer K, Edwards SW, Beresford MW. Differential expression of factors involved in the intrinsic and extrinsic apoptotic pathways in juvenile systemic lupus erythematosus. Lupus. (2011) 20:71–9. doi: 10.1177/0961203310382128
45. Midgley A, Thorbinson C, Beresford MW. Expression of Toll-like receptors and their detection of nuclear self-antigen leading to immune activation in JSLE. Rheumatology (Oxford). (2012) 51:824–32. doi: 10.1093/rheumatology/ker400
46. Midgley A, Beresford MW. Cellular localization of nuclear antigen during neutrophil apoptosis: mechanism for autoantigen exposure? Lupus. (2011) 20:641–6. doi: 10.1177/0961203310392421
47. Mahajan A, Herrmann M, Munoz LE. Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front Immunol. (2016) 7:35. doi: 10.3389/fimmu.2016.00035
48. Wright HL, Chikura B, Bucknall RC, Moots RJ, Edwards SW. Changes in expression of membrane TNF, NF-{kappa}B activation and neutrophil apoptosis during active and resolved inflammation. Ann Rheum Dis. (2011) 70:537–43. doi: 10.1136/ard.2010.138065
49. Wright HL, Lyon M, Chapman EA, Moots RJ, Edwards SW. Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species and neutrophil extracellular traps. Front Immunol. (2021) 11:584116. doi: 10.3389/fimmu.2020.584116
50. Wright HL, Bucknall RC, Moots RJ, Edwards SW. Analysis of SF and plasma cytokines provides insights into the mechanisms of inflammatory arthritis and may predict response to therapy. Rheumatology (Oxford). (2012) 51:451–9. doi: 10.1093/rheumatology/ker338
51. Watson RW, Rotstein OD, Parodo J, Bitar R, Marshall JC. The IL-1 beta-converting enzyme (caspase-1) inhibits apoptosis of inflammatory neutrophils through activation of IL-1 beta. J Immunol. (1998) 161:957–62.
52. Maianski NA, Roos D, Kuijpers TW. Bid truncation, bid/bax targeting to the mitochondria, and caspase activation associated with neutrophil apoptosis are inhibited by granulocyte colony-stimulating factor. J Immunol. (2004) 172:7024–30. doi: 10.4049/jimmunol.172.11.7024
53. Salamone G, Trevani A, Martinez D, Vermeulen M, Gamberale R, Fernandez-Calotti P, et al. Analysis of the mechanisms involved in the stimulation of neutrophil apoptosis by tumour necrosis factor-alpha. Immunology. (2004) 113:355–62. doi: 10.1111/j.1365-2567.2004.01973.x
54. Sakamoto E, Hato F, Kato T, Sakamoto C, Akahori M, Hino M, et al. Type I and type II interferons delay human neutrophil apoptosis via activation of STAT3 and up-regulation of cellular inhibitor of apoptosis 2. J Leukoc Biol. (2005) 78:301–9. doi: 10.1189/jlb.1104690
55. Parsonage G, Filer A, Bik M, Hardie D, Lax S, Howlett K, et al. Prolonged, granulocyte-macrophage colony-stimulating factor-dependent, neutrophil survival following rheumatoid synovial fibroblast activation by IL-17 and TNFalpha. Arthritis Res Ther. (2008) 10:R47–59. doi: 10.1186/ar2406
56. Wong SH, Francis N, Chahal H, Raza K, Salmon M, Scheel-Toellner D, et al. Lactoferrin is a survival factor for neutrophils in rheumatoid synovial fluid. Rheumatology (Oxford). (2009) 48:39–44. doi: 10.1093/rheumatology/ken412
57. Ottonello L, Cutolo M, Frumento G, Arduino N, Bertolotto M, Mancini M, et al. Synovial fluid from patients with rheumatoid arthritis inhibits neutrophil apoptosis: role of adenosine and proinflammatory cytokines. Rheumatology (Oxford). (2002) 41:1249–60. doi: 10.1093/rheumatology/41.11.1249
58. Cross A, Barnes T, Bucknall RC, Edwards SW, Moots RJ. Neutrophil apoptosis in rheumatoid arthritis is regulated by local oxygen tensions within joints. J Leukoc Biol. (2006) 80:521–8. doi: 10.1189/jlb.0306178
59. Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. (2005) 201:105–15. doi: 10.1084/jem.20040624
60. Elks PM, van Eeden FJ, Dixon G, Wang X, Reyes-Aldasoro CC, Ingham PW, et al. Activation of hypoxia-inducible factor-1alpha (Hif-1alpha) delays inflammation resolution by reducing neutrophil apoptosis and reverse migration in a zebrafish inflammation model. Blood. (2011) 118:712–22. doi: 10.1182/blood-2010-12-324186
61. Lawson C, Vicencio JM, Yellon DM, Davidson SM. Microvesicles and exosomes: new players in metabolic and cardiovascular disease. J Endocrinol. (2016) 228:R57–71. doi: 10.1530/JOE-15-0201
62. Dieker J, Tel J, Pieterse E, Thielen A, Rother N, Bakker M, et al. Circulating apoptotic microparticles in systemic lupus erythematosus patients drive the activation of dendritic cell subsets and prime neutrophils for NETosis. Arthritis Rheumatol. (2016) 68:462–72. doi: 10.1002/art.39417
63. Winberg LK, Jacobsen S, Nielsen CH. Microparticles from patients with systemic lupus erythematosus induce production of reactive oxygen species and degranulation of polymorphonuclear leukocytes. Arthritis Res Ther. (2017) 19:230. doi: 10.1186/s13075-017-1437-3
64. Rother N, Pieterse E, Lubbers J, Hilbrands L, van der Vlag J. Acetylated histones in apoptotic microparticles drive the formation of neutrophil extracellular traps in active lupus nephritis. Front Immunol. (2017) 8:1136. doi: 10.3389/fimmu.2017.01136
65. Headland SE, Jones HR, Norling LV, Kim A, Souza PR, Corsiero E, et al. Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci Transl Med. (2015) 7:315ra190. doi: 10.1126/scitranslmed.aac5608
66. Headland SE, Norling LV. The resolution of inflammation: principles and challenges. Semin Immunol. (2015) 27:149–60. doi: 10.1016/j.smim.2015.03.014
67. Perretti M, Solito E. Annexin 1 and neutrophil apoptosis. Biochem Soc Trans. (2004) 32:507–10. doi: 10.1042/BST0320507
68. Glennon-Alty L, Hackett AP, Chapman EA, Wright HL. Neutrophils and redox stress in the pathogenesis of autoimmune disease. Free Radic Biol Med. (2018) 125:25–35. doi: 10.1016/j.freeradbiomed.2018.03.049
69. Dang PM, Dewas C, Gaudry M, Fay M, Pedruzzi E, Gougerot-Pocidalo MA, et al. Priming of human neutrophil respiratory burst by granulocyte/macrophage colony-stimulating factor (GM-CSF) involves partial phosphorylation of p47(phox). J Biol Chem. (1999) 274:20704–8.
70. El-Benna J, Dang PM, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol. (2008) 30:279–89. doi: 10.1007/s00281-008-0118-3
71. Ward RA, Nakamura M, McLeish KR. Priming of the neutrophil respiratory burst involves p38 mitogen-activated protein kinase-dependent exocytosis of flavocytochrome b558-containing granules. J Biol Chem. (2000) 275:36713–9. doi: 10.1074/jbc.M003017200
72. Borregaard N, Heiple JM, Simons ER, Clark RA. Subcellular localization of the b-cytochrome component of the human neutrophil microbicidal oxidase: translocation during activation. J Cell Biol. (1983) 97:52–61.
73. Kjeldsen L, Sengelov H, Lollike K, Nielsen MH, Borregaard N. Isolation and characterization of gelatinase granules from human neutrophils. Blood. (1994) 83:1640–9.
74. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. (2014) 20:1126–67. doi: 10.1089/ars.2012.5149
75. Nauseef WM. Myeloperoxidase in human neutrophil host defence. Cell Microbiol. (2014) 16:1146–55. doi: 10.1111/cmi.12312
76. Elloumi N, Ben Mansour R, Marzouk S, Mseddi M, Fakhfakh R, Gargouri B, et al. Differential reactive oxygen species production of neutrophils and their oxidative damage in patients with active and inactive systemic lupus erythematosus. Immunol Lett. (2017) 184:1–6. doi: 10.1016/j.imlet.2017.01.018
77. Perazzio SF, Salomao R, Silva NP, Andrade LE. Increased neutrophil oxidative burst metabolism in systemic lupus erythematosus. Lupus. (2012) 21:1543–51. doi: 10.1177/0961203312461060
78. McConnell JR, Crockard AD, Cairns AP, Bell AL. Neutrophils from systemic lupus erythematosus patients demonstrate increased nuclear DNA damage. Clin Exp Rheumatol. (2002) 20:653–60.
79. Gehrke N, Mertens C, Zillinger T, Wenzel J, Bald T, Zahn S, et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity. (2013) 39:482–95. doi: 10.1016/j.immuni.2013.08.004
80. Alves CM, Marzocchi-Machado CM, Louzada-Junior P, Azzolini AE, Polizello AC, de Carvalho IF, et al. Superoxide anion production by neutrophils is associated with prevalent clinical manifestations in systemic lupus erythematosus. Clin Rheumatol. (2008) 27:701–8. doi: 10.1007/s10067-007-0768-x
81. Shingu M, Oribe M, Todoroki T, Tatsukawa K, Tomo-oka K, Yasuda M, et al. Serum factors from patients with systemic lupus erythematosus enhancing superoxide generation by normal neutrophils. J Invest Dermatol. (1983) 81:212–5.
82. Marzocchi-Machado CM, Alves CM, Azzolini AE, Polizello AC, Carvalho IF, Lucisano-Valim YM. Fcgamma and complement receptors: expression, role and co-operation in mediating the oxidative burst and degranulation of neutrophils of Brazilian systemic lupus erythematosus patients. Lupus. (2002) 11:240–8. doi: 10.1191/0961203302lu172oa
83. Russell DA, Markiewicz M, Oates JC. Lupus serum induces inflammatory interaction with neutrophils in human glomerular endothelial cells. Lupus Sci Med. (2020) 7:418. doi: 10.1136/lupus-2020-000418
84. O'Flynn J, Flierman R, van der Pol P, Rops A, Satchell SC, Mathieson PW, et al. Nucleosomes and C1q bound to glomerular endothelial cells serve as targets for autoantibodies and determine complement activation. Mol Immunol. (2011) 49:75–83. doi: 10.1016/j.molimm.2011.07.020
85. Nishi H, Furuhashi K, Cullere X, Saggu G, Miller MJ, Chen Y, et al. Neutrophil FcgammaRIIA promotes IgG-mediated glomerular neutrophil capture via Abl/Src kinases. J Clin Invest. (2017) 127:3810–26. doi: 10.1172/JCI94039
86. Flores-Mendoza G, Sanson SP, Rodriguez-Castro S, Crispin JC, Rosetti F. Mechanisms of tissue injury in lupus nephritis. Trends Mol Med. (2018) 24:364–78. doi: 10.1016/j.molmed.2018.02.003
87. Smith CK, Seto NL, Vivekanandan-Giri A, Yuan W, Playford MP, Manna Z, et al. Lupus high-density lipoprotein induces proinflammatory responses in macrophages by binding lectin-like oxidised low-density lipoprotein receptor 1 and failing to promote activating transcription factor 3 activity. Ann Rheum Dis. (2017) 76:602–11. doi: 10.1136/annrheumdis-2016-209683
88. Urbonaviciute V, Luo H, Sjowall C, Bengtsson A, Holmdahl R. Low production of reactive oxygen species drives systemic lupus erythematosus. Trends Mol Med. (2019) 25:826–35. doi: 10.1016/j.molmed.2019.06.001
89. Olsson LM, Johansson AC, Gullstrand B, Jonsen A, Saevarsdottir S, Ronnblom L, et al. A single nucleotide polymorphism in the NCF1 gene leading to reduced oxidative burst is associated with systemic lupus erythematosus. Ann Rheum Dis. (2017) 76:1607–13. doi: 10.1136/annrheumdis-2017-211287
90. Kelkka T, Kienhofer D, Hoffmann M, Linja M, Wing K, Sareila O, et al. Reactive oxygen species deficiency induces autoimmunity with type 1 interferon signature. Antioxid Redox Signal. (2014) 21:2231–45. doi: 10.1089/ars.2013.5828
91. Kundu S, Ghosh P, Datta S, Ghosh A, Chattopadhyay S, Chatterjee M. Oxidative stress as a potential biomarker for determining disease activity in patients with rheumatoid arthritis. Free Radic Res. (2012) 46:1482–9. doi: 10.3109/10715762.2012.727991
92. El Benna J, Hayem G, Dang PM, Fay M, Chollet-Martin S, Elbim C, et al. NADPH oxidase priming and p47phox phosphorylation in neutrophils from synovial fluid of patients with rheumatoid arthritis and spondylarthropathy. Inflammation. (2002) 26:273–8. doi: 10.1023/a:1021460517468
93. Rasheed Z. Hydroxyl radical damaged immunoglobulin G in patients with rheumatoid arthritis: biochemical and immunological studies. Clin Biochem. (2008) 41:663–9. doi: 10.1016/j.clinbiochem.2008.02.013
94. Fossati G, Bucknall RC, Edwards SW. Insoluble and soluble immune complexes activate neutrophils by distinct activation mechanisms: changes in functional responses induced by priming with cytokines. Ann Rheum Dis. (2002) 61:13–9. doi: 10.1136/ard.61.1.13
95. Robinson J, Watson F, Bucknall RC, Edwards SW. Activation of neutrophil reactive-oxidant production by synovial fluid from patients with inflammatory joint disease. Soluble and insoluble immunoglobulin aggregates activate different pathways in primed and unprimed cells. Biochem J. (1992) 286:345–51.
96. Edwards SW, Hughes V, Barlow J, Bucknall R. Immunological detection of myeloperoxidase in synovial fluid from patients with rheumatoid arthritis. Biochem J. (1988) 250:81–5.
97. Nzeusseu Toukap A, Delporte C, Noyon C, Franck T, Rousseau A, Serteyn D, et al. Myeloperoxidase and its products in synovial fluid of patients with treated or untreated rheumatoid arthritis. Free Radic Res. (2014) 48:461–5. doi: 10.3109/10715762.2014.886327
98. Birkelund S, Bennike TB, Kastaniegaard K, Lausen M, Poulsen TBG, Kragstrup TW, et al. Proteomic analysis of synovial fluid from rheumatic arthritis and spondyloarthritis patients. Clin Proteomics. (2020) 17:29. doi: 10.1186/s12014-020-09292-9
99. Baici A, Salgam P, Cohen G, Fehr K, Boni A. Action of collagenase and elastase from human polymorphonuclear leukocytes on human articular cartilage. Rheumatol Int. (1982) 2:11–6.
100. Van den Steen PE, Proost P, Grillet B, Brand DD, Kang AH, Van Damme J, et al. Cleavage of denatured natural collagen type II by neutrophil gelatinase B reveals enzyme specificity, post-translational modifications in the substrate, and the formation of remnant epitopes in rheumatoid arthritis. FASEB J. (2002) 16:379–89. doi: 10.1096/fj.01-0688com
101. Burkhardt H, Schwingel M, Menninger H, Macartney HW, Tschesche H. Oxygen radicals as effectors of cartilage destruction. Direct degradative effect on matrix components and indirect action via activation of latent collagenase from polymorphonuclear leukocytes. Arthritis Rheum. (1986) 29:379–87.
102. Shabani F, McNeil J, Tippett L. The oxidative inactivation of tissue inhibitor of metalloproteinase-1 (TIMP-1) by hypochlorous acid (HOCI) is suppressed by anti-rheumatic drugs. Free Radic Res. (1998) 28:115–23.
103. Onodera Y, Teramura T, Takehara T, Shigi K, Fukuda K. Reactive oxygen species induce Cox-2 expression via TAK1 activation in synovial fibroblast cells. FEBS Open Bio. (2015) 5:492–501. doi: 10.1016/j.fob.2015.06.001
104. Cemerski S, Cantagrel A, Van Meerwijk JP, Romagnoli P. Reactive oxygen species differentially affect T cell receptor-signaling pathways. J Biol Chem. (2002) 277:19585–93. doi: 10.1074/jbc.M111451200
105. Abimannan T, Peroumal D, Parida JR, Barik PK, Padhan P, Devadas S. Oxidative stress modulates the cytokine response of differentiated Th17 and Th1 cells. Free Radic Biol Med. (2016) 99:352–63. doi: 10.1016/j.freeradbiomed.2016.08.026
106. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
107. Brinkmann V, Goosmann C, Kuhn LI, Zychlinsky A. Automatic quantification of in vitro NET formation. Front Immunol. (2012) 3:413. doi: 10.3389/fimmu.2012.00413
108. Rosazza T, Warner J, Sollberger G. NET formation–mechanisms and how they relate to other cell death pathways. FEBS J. (2020). doi: 10.1111/febs.15589. [Epub ahead of print].
109. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. (2010) 191:677–91. doi: 10.1083/jcb.201006052
110. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. (2011) 21:290–304. doi: 10.1038/cr.2010.150
111. Neeli I, Khan SN, Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol. (2008) 180:1895–902. doi: 10.4049/jimmunol.180.3.1895
112. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci USA. (2015) 112:2817–22. doi: 10.1073/pnas.1414055112
113. Rohrbach AS, Slade DJ, Thompson PR, Mowen KA. Activation of PAD4 in NET formation. Front Immunol. (2012) 3:360. doi: 10.3389/fimmu.2012.00360
114. Liu CL, Tangsombatvisit S, Rosenberg JM, Mandelbaum G, Gillespie EC, Gozani OP, et al. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res Ther. (2012) 14:R25. doi: 10.1186/ar3707
115. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. (2009) 184:205–13. doi: 10.1083/jcb.200806072
116. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. (2010) 207:1853–62. doi: 10.1084/jem.20100239
117. Hemmers S, Teijaro JR, Arandjelovic S, Mowen KA. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS ONE. (2011) 6:e22043. doi: 10.1371/journal.pone.0022043
118. Pieterse E, Rother N, Yanginlar C, Gerretsen J, Boeltz S, Munoz LE, et al. Cleaved N-terminal histone tails distinguish between NADPH oxidase (NOX)-dependent and NOX-independent pathways of neutrophil extracellular trap formation. Ann Rheum Dis. (2018) 77:1790–8. doi: 10.1136/annrheumdis-2018-213223
119. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra19. doi: 10.1126/scitranslmed.3001180
120. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. (2011) 187:538–52. doi: 10.4049/jimmunol.1100450
121. Dieker J, Berden JH, Bakker M, Briand JP, Muller S, Voll R, et al. Autoantibodies against modified histone peptides in SLE patients are associated with disease activity and lupus nephritis. PLoS ONE. (2016) 11:e0165373. doi: 10.1371/journal.pone.0165373
122. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
123. Safi R, Al-Hage J, Abbas O, Kibbi AG, Nassar D. Investigating the presence of neutrophil extracellular traps in cutaneous lesions of different subtypes of lupus erythematosus. Exp Dermatol. (2019) 28:1348–52. doi: 10.1111/exd.14040
124. Sherer Y, Gorstein A, Fritzler MJ, Shoenfeld Y. Autoantibody explosion in systemic lupus erythematosus: more than 100 different antibodies found in SLE patients. Semin Arthritis Rheum. (2004) 34:501–37. doi: 10.1016/j.semarthrit.2004.07.002
125. Gripenberg M, Helve T, Kurki P. Profiles of antibodies to histones, DNA and IgG in patients with systemic rheumatic diseases determined by ELISA. J Rheumatol. (1985) 12:934–9.
126. Muller S, Bonnier D, Thiry M, Van Regenmortel MH. Reactivity of autoantibodies in systemic lupus erythematosus with synthetic core histone peptides. Int Arch Allergy Appl Immunol. (1989) 89:288–96.
127. Schett G, Steiner G, Smolen JS. Nuclear antigen histone H1 is primarily involved in lupus erythematosus cell formation. Arthritis Rheum. (1998) 41:1446–55. doi: 10.1002/1529-0131(199808)41:8<1446::AID-ART15>3.0.CO;2-6
128. Chapman EA, Lyon M, Simpson D, Mason D, Beynon RJ, Moots RJ, et al. Caught in a trap? Proteomic analysis of neutrophil extracellular traps in rheumatoid arthritis and systemic lupus erythematosus. Front Immunol. (2019) 10:423. doi: 10.3389/fimmu.2019.00423
129. van Bavel CC, Dieker JW, Kroeze Y, Tamboer WP, Voll R, Muller S, et al. Apoptosis-induced histone H3 methylation is targeted by autoantibodies in systemic lupus erythematosus. Ann Rheum Dis. (2011) 70:201–7. doi: 10.1136/ard.2010.129320
130. Pieterse E, Hofstra J, Berden J, Herrmann M, Dieker J, van der Vlag J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin Exp Immunol. (2015) 179:68–74. doi: 10.1111/cei.12359
131. Tzioufas AG, Boumba VA, Seferiadis K, Tsolas O, Moutsopoulos HM. Autoantibodies to HMG-17 nucleosomal protein in autoimmune rheumatic diseases. Correlation with systemic lupus erythematosus clinical activity and with antibodies to double-stranded DNA. Arthritis Rheum. (1993) 36:955–61.
132. Mansour RB, Lassoued S, Gargouri B, El Gaid A, Attia H, Fakhfakh F. Increased levels of autoantibodies against catalase and superoxide dismutase associated with oxidative stress in patients with rheumatoid arthritis and systemic lupus erythematosus. Scand J Rheumatol. (2008) 37:103–8. doi: 10.1080/03009740701772465
133. Brito J, Biamonti G, Caporali R, Montecucco C. Autoantibodies to human nuclear lamin B2 protein. Epitope specificity in different autoimmune diseases. J Immunol. (1994) 153:2268–77.
134. Reeves WH, Chaudhary N, Salerno A, Blobel G. Lamin B autoantibodies in sera of certain patients with systemic lupus erythematosus. J Exp Med. (1987) 165:750–62.
135. Senecal JL, Rauch J, Grodzicky T, Raynauld JP, Uthman I, Nava A, et al. Strong association of autoantibodies to human nuclear lamin B1 with lupus anticoagulant antibodies in systemic lupus erythematosus. Arthritis Rheum. (1999) 42:1347–53. doi: 10.1002/1529-0131(199907)42:7<1347::AID-ANR7>3.0.CO;2-#
136. Dinu AR, Merrill JT, Shen C, Antonov IV, Myones BL, Lahita RG. Frequency of antibodies to the cholesterol transport protein apolipoprotein A1 in patients with SLE. Lupus. (1998) 7:355–60. doi: 10.1191/096120398678920262
137. Kahlenberg JM, Kaplan MJ. Little peptide, big effects: the role of LL-37 in inflammation and autoimmune disease. J Immunol. (2013) 191:4895–901. doi: 10.4049/jimmunol.1302005
138. Sun CL, Zhang FZ, Li P, Bi LQ. LL-37 expression in the skin in systemic lupus erythematosus. Lupus. (2011) 20:904–11. doi: 10.1177/0961203311398515
139. Kienhofer D, Hahn J, Schubert I, Reinwald C, Ipseiz N, Lang SC, et al. No evidence of pathogenic involvement of cathelicidins in patient cohorts and mouse models of lupus and arthritis. PLoS ONE. (2014) 9:e115474. doi: 10.1371/journal.pone.0115474
140. Kretz CC, Norpo M, Abeler-Dorner L, Linke B, Haust M, Edler L, et al. Anti-annexin 1 antibodies: a new diagnostic marker in the serum of patients with discoid lupus erythematosus. Exp Dermatol. (2010) 19:919–21. doi: 10.1111/j.1600-0625.2010.01145.x
141. Bruschi M, Sinico RA, Moroni G, Pratesi F, Migliorini P, Galetti M, et al. Glomerular autoimmune multicomponents of human lupus nephritis in vivo: alpha-enolase and annexin AI. J Am Soc Nephrol. (2014) 25:2483–98. doi: 10.1681/ASN.2013090987
142. Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis. (2015) 74:1417–24. doi: 10.1136/annrheumdis-2013-204837
143. Aragon CC, Tafur RA, Suarez-Avellaneda A, Martinez MT, Salas AL, Tobon GJ. Urinary biomarkers in lupus nephritis. J Transl Autoimmun. (2020) 3:100042. doi: 10.1016/j.jtauto.2020.100042
144. El Shahawy MS, Hemida MH, Abdel-Hafez HA, El-Baz TZ, Lotfy AM, Emran TM. Urinary neutrophil gelatinase-associated lipocalin as a marker for disease activity in lupus nephritis. Scand J Clin Lab Invest. (2018) 78:264–8. doi: 10.1080/00365513.2018.1449242
145. Rubinstein T, Pitashny M, Levine B, Schwartz N, Schwartzman J, Weinstein E, et al. Urinary neutrophil gelatinase-associated lipocalin as a novel biomarker for disease activity in lupus nephritis. Rheumatology (Oxford). (2010) 49:960–71. doi: 10.1093/rheumatology/kep468
146. Mayadas TN, Rosetti F, Ernandez T, Sethi S. Neutrophils: game changers in glomerulonephritis? Trends Mol Med. (2010) 16:368–78. doi: 10.1016/j.molmed.2010.06.002
147. Pieterse E, Rother N, Garsen M, Hofstra JM, Satchell SC, Hoffmann M, et al. Neutrophil extracellular traps drive endothelial-to-mesenchymal transition. Arterioscler Thromb Vasc Biol. (2017) 37:1371–9. doi: 10.1161/ATVBAHA.117.309002
148. Knight JS, Zhao W, Luo W, Subramanian V, O'Dell AA, Yalavarthi S, et al. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest. (2013) 123:2981–93. doi: 10.1172/JCI67390
149. Liu Y, Carmona-Rivera C, Moore E, Seto NL, Knight JS, Pryor M, et al. Myeloid-specific deletion of peptidylarginine deiminase 4 mitigates atherosclerosis. Front Immunol. (2018) 9:1680. doi: 10.3389/fimmu.2018.01680
150. O'Neill SG, Giles I, Lambrianides A, Manson J, D'Cruz D, Schrieber L, et al. Antibodies to apolipoprotein A-I, high-density lipoprotein, and C-reactive protein are associated with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. (2010) 62:845–54. doi: 10.1002/art.27286
151. Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. (2010) 107:9813–8. doi: 10.1073/pnas.0909927107
152. Bonegio RG, Lin JD, Beaudette-Zlatanova B, York MR, Menn-Josephy H, Yasuda K. Lupus-associated immune complexes activate human neutrophils in an FcgammaRIIA-dependent but TLR-independent response. J Immunol. (2019) 202:675–83. doi: 10.4049/jimmunol.1800300
153. Gestermann N, Di Domizio J, Lande R, Demaria O, Frasca L, Feldmeyer L, et al. Netting neutrophils activate autoreactive B cells in lupus. J Immunol. (2018) 200:3364–71. doi: 10.4049/jimmunol.1700778
154. Kraaij T, Kamerling SWA, de Rooij ENM, van Daele PLA, Bredewold OW, Bakker JA, et al. The NET-effect of combining rituximab with belimumab in severe systemic lupus erythematosus. J Autoimmun. (2018) 91:45–54. doi: 10.1016/j.jaut.2018.03.003
155. van Dam LS, Osmani Z, Kamerling SWA, Kraaij T, Bakker JA, Scherer HU, et al. A reverse translational study on the effect of rituximab, rituximab plus belimumab, or bortezomib on the humoral autoimmune response in SLE. Rheumatology (Oxford). (2020) 59:2734–45. doi: 10.1093/rheumatology/kez623
156. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. (2013) 5:178ra140. doi: 10.1126/scitranslmed.3005580
157. Spengler J, Lugonja B, Ytterberg AJ, Zubarev RA, Creese AJ, Pearson MJ, et al. Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol. (2015) 67:3135–45. doi: 10.1002/art.39313
158. Romero V, Fert-Bober J, Nigrovic PA, Darrah E, Haque UJ, Lee DM, et al. Immune-mediated pore-forming pathways induce cellular hypercitrullination and generate citrullinated autoantigens in rheumatoid arthritis. Sci Transl Med. (2013) 5:209ra150. doi: 10.1126/scitranslmed.3006869
159. Turunen S, Huhtakangas J, Nousiainen T, Valkealahti M, Melkko J, Risteli J, et al. Rheumatoid arthritis antigens homocitrulline and citrulline are generated by local myeloperoxidase and peptidyl arginine deiminases 2, 3 and 4 in rheumatoid nodule and synovial tissue. Arthritis Res Ther. (2016) 18:239. doi: 10.1186/s13075-016-1140-9
160. Kinloch A, Lundberg K, Wait R, Wegner N, Lim NH, Zendman AJ, et al. Synovial fluid is a site of citrullination of autoantigens in inflammatory arthritis. Arthritis Rheum. (2008) 58:2287–95. doi: 10.1002/art.23618
161. Kinloch A, Tatzer V, Wait R, Peston D, Lundberg K, Donatien P, et al. Identification of citrullinated alpha-enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis Res Ther. (2005) 7:R1421–9. doi: 10.1186/ar1845
162. de Bont CM, Stokman MEM, Faas P, Thurlings RM, Boelens WC, Wright HL, et al. Autoantibodies to neutrophil extracellular traps represent a potential serological biomarker in rheumatoid arthritis. J Autoimmun. (2020) 113:102484. doi: 10.1016/j.jaut.2020.102484
163. Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol. (2017) 2:eaag3358. doi: 10.1126/sciimmunol.aag3358
164. Carmona-Rivera C, Carlucci PM, Goel RR, James EA, Brooks SR, Rims CR, et al. Neutrophil extracellular traps mediate articular cartilage damage and enhance cartilage component immunogenicity in rheumatoid arthritis. JCI Insight. (2020) 5:e139388. doi: 10.1172/jci.insight.139388
165. Mattey DL, Nixon NB, Dawes PT. Association of circulating levels of MMP-8 with mortality from respiratory disease in patients with rheumatoid arthritis. Arthritis Res Ther. (2012) 14:R204. doi: 10.1186/ar4042
166. Itoh T, Matsuda H, Tanioka M, Kuwabara K, Itohara S, Suzuki R. The role of matrix metalloproteinase-2 and matrix metalloproteinase-9 in antibody-induced arthritis. J Immunol. (2002) 169:2643–7. doi: 10.4049/jimmunol.169.5.2643
167. Scally SW, Petersen J, Law SC, Dudek NL, Nel HJ, Loh KL, et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med. (2013) 210:2569–82. doi: 10.1084/jem.20131241
168. Corsiero E, Bombardieri M, Carlotti E, Pratesi F, Robinson W, Migliorini P, et al. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann Rheum Dis. (2016) 75:1866–75. doi: 10.1136/annrheumdis-2015-208356
169. Wright HL, Thomas HB, Moots RJ, Edwards SW. RNA-seq reveals activation of both common and cytokine-specific pathways following neutrophil priming. PLoS ONE. (2013) 8:e58598. doi: 10.1371/journal.pone.0058598
170. de Kleijn S, Kox M, Sama IE, Pillay J, van Diepen A, Huijnen MA, et al. Transcriptome kinetics of circulating neutrophils during human experimental endotoxemia. PLoS ONE. (2012) 7:e38255. doi: 10.1371/journal.pone.0038255
171. Kobayashi SD, Voyich JM, Buhl CL, Stahl RM, DeLeo FR. Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: cell fate is regulated at the level of gene expression. Proc Natl Acad Sci USA. (2002) 99:6901–6. doi: 10.1073/pnas.092148299
172. Zimmermann M, Aguilera FB, Castellucci M, Rossato M, Costa S, Lunardi C, et al. Chromatin remodelling and autocrine TNFalpha are required for optimal interleukin-6 expression in activated human neutrophils. Nat Commun. (2015) 6:6061. doi: 10.1038/ncomms7061
173. Glennon-Alty L, Moots RJ, Edwards SW, Wright HL. Type-I interferon regulates cytokine-delayed neutrophil apoptosis, reactive oxygen species production and chemokine expression. Clin Exp Immunol. (2020) 203:151–9. doi: 10.1111/cei.13525
174. Chen M, Lam BK, Kanaoka Y, Nigrovic PA, Audoly LP, Austen KF, et al. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J Exp Med. (2006) 203:837–42. doi: 10.1084/jem.20052371
175. Scapini P, Carletto A, Nardelli B, Calzetti F, Roschke V, Merigo F, et al. Proinflammatory mediators elicit secretion of the intracellular B-lymphocyte stimulator pool (BLyS) that is stored in activated neutrophils: implications for inflammatory diseases. Blood. (2005) 105:830–7. doi: 10.1182/blood-2004-02-0564
176. Gilbert C, Poubelle PE, Borgeat P, Pouliot M, Naccache PH. Crystal-induced neutrophil activation: VIII. Immediate production of prostaglandin E2 mediated by constitutive cyclooxygenase 2 in human neutrophils stimulated by urate crystals. Arthritis Rheum. (2003) 48:1137–48. doi: 10.1002/art.10851
177. Yang CW, Unanue ER. Neutrophils control the magnitude and spread of the immune response in a thromboxane A2-mediated process. J Exp Med. (2013) 210:375–87. doi: 10.1084/jem.20122183
178. Hoogendijk AJ, Pourfarzad F, Aarts CEM, Tool ATJ, Hiemstra IH, Grassi L, et al. Dynamic transcriptome-proteome correlation networks reveal human myeloid differentiation and neutrophil-specific programming. Cell Rep. (2019) 29:2505–19 e2504. doi: 10.1016/j.celrep.2019.10.082
179. Rorvig S, Ostergaard O, Heegaard NH, Borregaard N. Proteome profiling of human neutrophil granule subsets, secretory vesicles, and cell membrane: correlation with transcriptome profiling of neutrophil precursors. J Leukoc Biol. (2013) 94:711–21. doi: 10.1189/jlb.1212619
180. Pedersen CC, Borup R, Fischer-Nielsen A, Mora-Jensen H, Fossum A, Cowland JB, et al. Changes in gene expression during G-CSF-induced emergency granulopoiesis in humans. J Immunol. (2016) 197:1989–99. doi: 10.4049/jimmunol.1502690
181. Theilgaard-Monch K, Jacobsen LC, Borup R, Rasmussen T, Bjerregaard MD, Nielsen FC, et al. The transcriptional program of terminal granulocytic differentiation. Blood. (2005) 105:1785–96. doi: 10.1182/blood-2004-08-3346
182. Borregaard N, Cowland JB. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood. (1997) 89:3503–21.
183. Elghetany MT. Surface antigen changes during normal neutrophilic development: a critical review. Blood Cells Mol Dis. (2002) 28:260–74. doi: 10.1006/bcmd.2002.0513
184. Lakschevitz FS, Visser MB, Sun C, Glogauer M. Neutrophil transcriptional profile changes during transit from bone marrow to sites of inflammation. Cell Mol Immunol. (2015) 12:53–65. doi: 10.1038/cmi.2014.37
185. Naranbhai V, Fairfax BP, Makino S, Humburg P, Wong D, Ng E, et al. Genomic modulators of gene expression in human neutrophils. Nat Commun. (2015) 6:7545. doi: 10.1038/ncomms8545
186. Wright HL, Thomas HB, Moots RJ, Edwards SW. Interferon gene expression signature in rheumatoid arthritis neutrophils correlates with a good response to TNFi therapy. Rheumatology (Oxford). (2015) 54:188–93. doi: 10.1093/rheumatology/keu299
187. Mistry P, Nakabo S, O'Neil L, Goel RR, Jiang K, Carmona-Rivera C, et al. Transcriptomic, epigenetic, and functional analyses implicate neutrophil diversity in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci USA. (2019) 116:25222–8. doi: 10.1073/pnas.1908576116
188. Wright HL, Cox T, Moots RJ, Edwards SW. Neutrophil biomarkers predict response to therapy with tumor necrosis factor inhibitors in rheumatoid arthritis. J Leukoc Biol. (2017) 101:785–95. doi: 10.1189/jlb.5A0616-258R
189. Schlenk J, Lorenz HM, Haas JP, Herrmann M, Hohenberger G, Kalden JR, et al. Extravasation into synovial tissue induces CCL20 mRNA expression in polymorphonuclear neutrophils of patients with rheumatoid arthritis. J Rheumatol. (2005) 32:2291–8.
190. Hatano Y, Kasama T, Iwabuchi H, Hanaoka R, Takeuchi HT, Jing L, et al. Macrophage inflammatory protein 1 alpha expression by synovial fluid neutrophils in rheumatoid arthritis. Ann Rheum Dis. (1999) 58:297–302. doi: 10.1136/ard.58.5.297
191. Arias de la Rosa I, Perez-Sanchez C, Ruiz-Limon P, Patino-Trives A, Torres-Granados C, Jimenez-Gomez Y, et al. Impaired microRNA processing in neutrophils from rheumatoid arthritis patients confers their pathogenic profile. Modulation by biological therapies. Haematologica. (2020) 105:2250–61. doi: 10.3324/haematol.2018.205047
192. Cross A, Bucknall RC, Cassatella MA, Edwards SW, Moots RJ. Synovial fluid neutrophils transcribe and express class II major histocompatibility complex molecules in rheumatoid arthritis. Arthritis Rheum. (2003) 48:2796–806. doi: 10.1002/art.11253
193. Bayley R, Kite KA, McGettrick HM, Smith JP, Kitas GD, Buckley CD, et al. The autoimmune-associated genetic variant PTPN22 R620W enhances neutrophil activation and function in patients with rheumatoid arthritis and healthy individuals. Ann Rheum Dis. (2015) 74:1588–95. doi: 10.1136/annrheumdis-2013-204796
194. Witko-Sarsat V, Lesavre P, Lopez S, Bessou G, Hieblot C, Prum B, et al. A large subset of neutrophils expressing membrane proteinase 3 is a risk factor for vasculitis and rheumatoid arthritis. J Am Soc Nephrol. (1999) 10:1224–33.
195. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. (2014) 506:376–81. doi: 10.1038/nature12873
196. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. (2003) 100:2610–5. doi: 10.1073/pnas.0337679100
197. Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell. (2016) 165:551–65. doi: 10.1016/j.cell.2016.03.008
198. Petri M, Fu W, Ranger A, Allaire N, Cullen P, Magder LS, et al. Association between changes in gene signatures expression and disease activity among patients with systemic lupus erythematosus. BMC Med Genomics. (2019) 12:4. doi: 10.1186/s12920-018-0468-1
199. Brohawn PZ, Streicher K, Higgs BW, Morehouse C, Liu H, Illei G, et al. Type I interferon gene signature test-low and -high patients with systemic lupus erythematosus have distinct gene expression signatures. Lupus. (2019) 28:1524–33. doi: 10.1177/0961203319885447
200. Coit P, Yalavarthi S, Ognenovski M, Zhao W, Hasni S, Wren JD, et al. Epigenome profiling reveals significant DNA demethylation of interferon signature genes in lupus neutrophils. J Autoimmun. (2015) 58:59–66. doi: 10.1016/j.jaut.2015.01.004
201. Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an anti-interferon-alpha receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. (2017) 69:376–86. doi: 10.1002/art.39962
202. Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. (2020) 382:211–21. doi: 10.1056/NEJMoa1912196
203. Furie R, Morand EF, Bruce IN, Manzi S, Kalunian K, Vital EM, et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): a randomised, controlled, phase 3 trial. Lancet Rheumatol. (2019) 1:E208–19. doi: 10.1016/S2665-9913(19)30098-0
204. Bashant KR, Aponte AM, Randazzo D, Rezvan Sangsari P, Wood AJ, Bibby JA, et al. Proteomic, biomechanical and functional analyses define neutrophil heterogeneity in systemic lupus erythematosus. Ann Rheum Dis. (2020) 80:209–18. doi: 10.1136/annrheumdis-2020-218338
205. Wither JE, Prokopec SD, Noamani B, Chang NH, Bonilla D, Touma Z, et al. Identification of a neutrophil-related gene expression signature that is enriched in adult systemic lupus erythematosus patients with active nephritis: clinical/pathologic associations and etiologic mechanisms. PLoS ONE. (2018) 13:e0196117. doi: 10.1371/journal.pone.0196117
206. Carlucci PM, Purmalek MM, Dey AK, Temesgen-Oyelakin Y, Sakhardande S, Joshi AA, et al. Neutrophil subsets and their gene signature associate with vascular inflammation and coronary atherosclerosis in lupus. JCI Insight. (2018) 3:e99276. doi: 10.1172/jci.insight.99276
207. Dubey AK, Handu SS, Dubey S, Sharma P, Sharma KK, Ahmed QM. Belimumab: First targeted biological treatment for systemic lupus erythematosus. J Pharmacol Pharmacother. (2011) 2:317–9. doi: 10.4103/0976-500X.85930
208. Mathis D, Shoelson SE. Immunometabolism: an emerging frontier. Nat Rev Immunol. (2011) 11:81–3. doi: 10.1038/nri2922
209. Ganeshan K, Chawla A. Metabolic regulation of immune responses. Annu Rev Immunol. (2014) 32:609–34. doi: 10.1146/annurev-immunol-032713-120236
210. Medzhitov R. Origin and physiological roles of inflammation. Nature. (2008) 454:428–35. doi: 10.1038/nature07201
211. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
212. Gaber T, Strehl C, Buttgereit F. Metabolic regulation of inflammation. Nat Rev Rheumatol. (2017) 13:267–79. doi: 10.1038/nrrheum.2017.37
213. Weyand CM, Goronzy JJ. Immunometabolism in early and late stages of rheumatoid arthritis. Nat Rev Rheumatol. (2017) 13:291–301. doi: 10.1038/nrrheum.2017.49
214. Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med. (2013) 210:2119–34. doi: 10.1084/jem.20130252
215. Yang Z, Shen Y, Oishi H, Matteson EL, Tian L, Goronzy JJ, et al. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med. (2016) 8:331ra338. doi: 10.1126/scitranslmed.aad7151
216. Sbarra AJ, Karnovsky ML. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J Biol Chem. (1959) 234:1355–62.
217. Azevedo EP, Rochael NC, Guimarães-Costa AB, De Souza-Vieira TS, Ganilho J, Saraiva EM, et al. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril- and phorbol 12-myristate 13-Acetate-induced neutrophil extracellular trap (NET) formation. J Biol Chem. (2015) 290:22174–83. doi: 10.1074/jbc.M115.640094
218. Chen LY, Shieh JJ, Lin B, Pan CJ, Gao JL, Murphy PM, et al. Impaired glucose homeostasis, neutrophil trafficking and function in mice lacking the glucose-6-phosphate transporter. Hum Mol Genet. (2003) 12:2547–58. doi: 10.1093/hmg/ddg263
219. Cooper MR, DeChatelet LR, McCall CE, LaVia MF, Spurr CL, Baehner RL. Complete deficiency of leukocyte glucose-6-phosphate dehydrogenase with defective bactericidal activity. J Clin Invest. (1972) 51:769–78. doi: 10.1172/JCI106871
220. Edwards SW. Biochemistry and Physiology of the Neutrophil. Cambridge: Cambridge University Press (1994).
221. Wilson PD, Rustin GJS, Smith GP, Peters TJ. Electron microscopic cytochemical localization of nucleoside phosphatases in normal and chronic granulocytic leukaemic human neutrophils. Histochem J. (1981) 13:73–84. doi: 10.1007/BF01005841
222. Hirsch JG, Fedorko ME. Ultrastructure of human leukocytes after simultaneous fixation with glutaraldehyde and osmium tetroxide and “postfixation” in uranyl acetate. J Cell Biol. (1968) 38:615–27. doi: 10.1083/jcb.38.3.615
223. Rice CM, Davies LC, Subleski JJ, Maio N, Gonzalez-Cotto M, Andrews C, et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat Commun. (2018) 9:5099. doi: 10.1038/s41467-018-07505-2
224. van Raam BJ, Sluiter W, de Wit E, Roos D, Verhoeven AJ, Kuijpers TW. Mitochondrial membrane potential in human neutrophils is maintained by complex III activity in the absence of supercomplex organisation. PLoS ONE. (2008) 3:e2013. doi: 10.1371/journal.pone.0002013
225. Zmijewski JW, Lorne E, Banerjee S, Abraham E. Participation of mitochondrial respiratory complex III in neutrophil activation and lung injury. Am J Physiol Lung Cell Mol Physiol. (2009) 296:L624–34. doi: 10.1152/ajplung.90522.2008
226. Fossati G, Moulding DA, Spiller DG, Moots RJ, White MRH, Edwards SW. The mitochondrial network of human neutrophils: role in chemotaxis, phagocytosis, respiratory burst activation, and commitment to apoptosis. J Immunol. (2003) 170:1964–72. doi: 10.4049/jimmunol.170.4.1964
227. Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, Kuijpers TW. Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ. (2004) 11:143–53. doi: 10.1038/sj.cdd.4401320
228. Vorobjeva N, Prikhodko A, Galkin I, Pletjushkina O, Zinovkin R, Sud'ina G, et al. Mitochondrial reactive oxygen species are involved in chemoattractant-induced oxidative burst and degranulation of human neutrophils in vitro. Eur J Cell Biol. (2017) 96:254–65. doi: 10.1016/j.ejcb.2017.03.003
229. Bao Y, Ledderose C, Seier T, Graf AF, Brix B, Chong E, et al. Mitochondria regulate neutrophil activation by generating ATP for autocrine purinergic signaling. J Biol Chem. (2014) 289:26794–803. doi: 10.1074/jbc.M114.572495
230. Bao Y, Ledderose C, Graf AF, Brix B, Birsak T, Lee A, et al. mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J Cell Biol. (2015) 210:1153–64. doi: 10.1083/jcb.201503066
231. Kumar S, Dikshit M. Metabolic insight of neutrophils in health and disease. Front Immunol. (2019) 10:2099. doi: 10.3389/fimmu.2019.02099
232. Six E, Lagresle-Peyrou C, Susini S, De Chappedelaine C, Sigrist N, Sadek H, et al. AK2 deficiency compromises the mitochondrial energy metabolism required for differentiation of human neutrophil and lymphoid lineages. Cell Death Dis. (2015) 6:e1856. doi: 10.1038/cddis.2015.211
233. Hsu BE, Tabariès S, Johnson RM, Andrzejewski S, Senecal J, Lehuédé C, et al. Immature low-density neutrophils exhibit metabolic flexibility that facilitates breast cancer liver metastasis. Cell Rep. (2019) 27:3902–15.e3906. doi: 10.1016/j.celrep.2019.05.091
234. Pithon-Curi TC, De Melo MP, Curi R. Glucose and glutamine utilization by rat lymphocytes, monocytes and neutrophils in culture: a comparative study. Cell Biochem Func. (2004) 22:321–6. doi: 10.1002/cbf.1109
235. Pithon-Curi TC, Trezena AG, Tavares-Lima W, Curi R. Evidence that glutamine is involved in neutrophil function. Cell Biochem Func. (2002) 20:81–6. doi: 10.1002/cbf.954
236. Sasaki E, Umeda T, Takahashi I, Arata K, Yamamoto Y, Tanabe M, et al. Effect of glutamine supplementation on neutrophil function in male judoists. Luminescence. (2013) 28:442–9. doi: 10.1002/bio.2474
237. Yang L, Venneti S, Nagrath D. Glutaminolysis: a hallmark of cancer metabolism. Annu Rev Biomed Eng. (2017) 19:163–94. doi: 10.1146/annurev-bioeng-071516-044546
238. Injarabian L, Devin A, Ransac S, Marteyn BS. Neutrophil metabolic shift during their lifecycle: impact on their survival and activation. Int J Mol Sci. (2020) 21:287. doi: 10.3390/ijms21010287
239. Furukawa S, Saito H, Inoue T, Matsuda T, Fukatsu K, Han I, et al. Supplemental glutamine augments phagocytosis and reactive oxygen intermediate production by neutrophils and monocytes from postoperative patients in vitro. Nutrition. (2000) 16:323–9. doi: 10.1016/s0899-9007(00)00228-8
240. Huang Y, Li N, Liboni K, Neu J. Glutamine decreases lipopolysaccharide-induced IL-8 production in Caco-2 cells through a non-NF-κB p50 mechanism. Cytokine. (2003) 22:77–83. doi: 10.1016/S1043-4666(03)00115-7
241. De Beaux AC, O'Riordain MG, Ross JA, Jodozi L, Carter DC, Fearon KCH. Glutamine-supplemented total parenteral nutrition reduces blood mononuclear cell interleukin-8 release in severe acute pancreatitis. Nutrition. (1998) 14:261–5. doi: 10.1016/S0899-9007(97)00477-2
242. Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, et al. The transcription factor HIF-1 plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. (2007) 21:1037–49. doi: 10.1101/gad.1529107
243. Watts ER, Walmsley SR. Inflammation and hypoxia: HIF and PHD isoform selectivity. Trends Mol Med. (2019) 25:33–46. doi: 10.1016/j.molmed.2018.10.006
244. Harris AJ, Mirchandani AS, Lynch RW, Murphy F, Delaney L, Small D, et al. IL4RA signaling abrogates hypoxic neutrophil survival and limits acute lung injury responses in vivo. Am J Respir Crit Care Med. (2019) 200:235–46. doi: 10.1164/rccm.201808-1599OC
245. Kong T, Eltzschig HK, Karhausen J, Colgan SP, Shelley CS. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of β2 integrin gene expression. Proc Natl Acad Sci USA. (2004) 101:10440–5. doi: 10.1073/pnas.0401339101
246. Anderson JR, Chokesuwattanaskul S, Phelan MM, Welting TJM, Lian LY, Peffers MJ, et al. (1)H NMR metabolomics identifies underlying inflammatory pathology in osteoarthritis and rheumatoid arthritis synovial joints. J Proteome Res. (2018) 17:3780–90. doi: 10.1021/acs.jproteome.8b00455
247. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. (2018) 556:113–7. doi: 10.1038/nature25986
248. Ryan DG, O'Neill LAJ. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. (2017) 591:2992–3006. doi: 10.1002/1873-3468.12744
249. Williams NC, O'Neill LAJ. A role for the krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation. Front Immunol. (2018) 9:141. doi: 10.3389/fimmu.2018.00141
250. Maratou E, Dimitriadis G, Kollias A, Boutati E, Lambadiari V, Mitrou P, et al. Glucose transporter expression on the plasma membrane of resting and activated white blood cells. Eur J Clin Invest. (2007) 37:282–90. doi: 10.1111/j.1365-2362.2007.01786.x
251. Li KJ, Wu CH, Hsieh SC, Lu MC, Tsai CY, Yu CL. Deranged bioenergetics and defective redox capacity in T lymphocytes and neutrophils are related to cellular dysfunction and increased oxidative stress in patients with active systemic lupus erythematosus. Clin Dev Immunol. (2012) 2012:548516. doi: 10.1155/2012/548516
252. MacIver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leuk Biol. (2008) 84:949–57. doi: 10.1189/jlb.0108024
253. Perner A, Nielsen SE, Rask-Madsen J. High glucose impairs superoxide production from isolated blood neutrophils. Intensive Care Med. (2003) 29:642–5. doi: 10.1007/s00134-002-1628-4
254. Huang WN, Tso TK, Huang HY. Enhanced oxidative status but not corresponding elevated antioxidative status by anticardiolipin antibody and disease activity in patients with systemic lupus erythematosus. Rheumatol Int. (2007) 27:453–8. doi: 10.1007/s00296-006-0233-1
255. Bilzer M, Lauteburg BH. Glutathione metabolism in activated human neutrophils: stimulation of glutathione synthesis and consumption of glutathione by reactive oxygen species. Eur J Clin Invest. (1991) 21:316–22. doi: 10.1111/j.1365-2362.1991.tb01376.x
256. Pravda J. Systemic lupus erythematosus: pathogenesis at the functional limit of redox homeostasis. Oxidat Med Cell Longev. (2019) 2019:1651724. doi: 10.1155/2019/1651724
257. Voetman AA, Loos JA, Roos D. Changes in the levels of glutathione in phagocytosing human neutrophils. Blood. (1980) 55:741–7.
258. Yan J, Meng X, Wancket LM, Lintner K, Nelin LD, Chen B, et al. Glutathione reductase facilitates host defense by sustaining phagocytic oxidative burst and promoting the development of neutrophil extracellular traps. J Immunol. (2012) 188:2316–27. doi: 10.4049/jimmunol.1102683
259. Ito Y, Kajkenova O, Feuers RJ, Udupa KB, Desai VG, Epstein J, et al. Impaired glutathione peroxidase activity accounts for the age-related accumulation of hydrogen peroxide in activated human neutrophils. J Gerontol A Biol Sci Med Sci. (1998) 53:M169–75. doi: 10.1093/gerona/53a.3.m169
260. NaveenKumar SK, Hemshekhar M, Jagadish S, Manikanta K, Vishalakshi GJ, Kemparaju K, et al. Melatonin restores neutrophil functions and prevents apoptosis amid dysfunctional glutathione redox system. J Pineal Res. (2020) 69:e12676. doi: 10.1111/jpi.12676
261. Stojkov D, Amini P, Oberson K, Sokollik C, Duppenthaler A, Simon HU, et al. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J Cell Biol. (2017) 216:4073–90. doi: 10.1083/jcb.201611168
262. Reshetnikov V, Hahn J, Maueröder C, Czegley C, Munoz LE, Herrmann M, et al. Chemical tools for targeted amplification of reactive oxygen species in neutrophils. Front Immunol. (2018) 9:1827. doi: 10.3389/fimmu.2018.01827
263. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
264. Scapini P, Marini O, Tecchio C, Cassatella MA. Human neutrophils in the saga of cellular heterogeneity: insights and open questions. Immunol Rev. (2016) 273:48–60. doi: 10.1111/imr.12448
265. Grieshaber-Bouyer R, Nigrovic PA. Neutrophil heterogeneity as therapeutic opportunity in immune-mediated disease. Front Immunol. (2019) 10:346. doi: 10.3389/fimmu.2019.00346
266. Ng LG, Ostuni R, Hidalgo A. Heterogeneity of neutrophils. Nat Rev Immunol. (2019) 19:255–65. doi: 10.1038/s41577-019-0141-8
267. Nourshargh S, Renshaw SA, Imhof BA. Reverse migration of neutrophils: where, when, how, and why? Trends Immunol. (2016) 37:273–86. doi: 10.1016/j.it.2016.03.006
268. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
269. Lecot P, Sarabi M, Pereira Abrantes M, Mussard J, Koenderman L, Caux C, et al. Neutrophil heterogeneity in cancer: from biology to therapies. Front Immunol. (2019) 10:2155. doi: 10.3389/fimmu.2019.02155
270. Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest. (2012) 122:327–36. doi: 10.1172/JCI57990
271. Kurko J, Vida A, Glant TT, Scanzello CR, Katz RS, Nair A, et al. Identification of myeloid-derived suppressor cells in the synovial fluid of patients with rheumatoid arthritis: a pilot study. BMC Musculoskelet Disord. (2014) 15:281. doi: 10.1186/1471-2474-15-281
272. Zhu J, Chen S, Wu L, Wang R, Zheng S, Zhao D, et al. The expansion of myeloid-derived suppressor cells is associated with joint inflammation in rheumatic patients with arthritis. BioMed Res Int. (2018) 2018:5474828. doi: 10.1155/2018/5474828
273. Wang W, Jiao Z, Duan T, Liu M, Zhu B, Zhang Y, et al. Functional characterization of myeloid-derived suppressor cell subpopulations during the development of experimental arthritis. Eur J Immunol. (2015) 45:464–73. doi: 10.1002/eji.201444799
274. Park MJ, Lee SH, Kim EK, Lee EJ, Baek JA, Park SH, et al. Interleukin-10 produced by myeloid-derived suppressor cells is critical for the induction of Tregs and attenuation of rheumatoid inflammation in mice. Sci Rep. (2018) 8:3753. doi: 10.1038/s41598-018-21856-2
275. Li M, Zhu D, Wang T, Xia X, Tian J, Wang S. Roles of myeloid-derived suppressor cell subpopulations in autoimmune arthritis. Front Immunol. (2018) 9:2849. doi: 10.3389/fimmu.2018.02849
276. Ji J, Xu J, Zhao S, Liu F, Qi J, Song Y, et al. Myeloid-derived suppressor cells contribute to systemic lupus erythaematosus by regulating differentiation of Th17 cells and Tregs. Clin Sci (Lond). (2016) 130:1453–67. doi: 10.1042/CS20160311
277. Vlachou K, Mintzas K, Glymenaki M, Ioannou M, Papadaki G, Bertsias GK, et al. Elimination of granulocytic myeloid-derived suppressor cells in lupus-prone mice linked to reactive oxygen species-dependent extracellular trap formation. Arthritis Rheumatol. (2016) 68:449–61. doi: 10.1002/art.39441
278. Ellett F, Elks PM, Robertson AL, Ogryzko NV, Renshaw SA. Defining the phenotype of neutrophils following reverse migration in zebrafish. J Leukoc Biol. (2015) 98:975–81. doi: 10.1189/jlb.3MA0315-105R
279. Buckley CD, Ross EA, McGettrick HM, Osborne CE, Haworth O, Schmutz C, et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J Leukoc Biol. (2006) 79:303–11. doi: 10.1189/jlb.0905496
280. Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol. (2011) 12:761–9. doi: 10.1038/ni.2062
281. Hacbarth E, Kajdacsy-Balla A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheum. (1986) 29:1334–42.
282. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol. (2010) 184:3284–97. doi: 10.4049/jimmunol.0902199
283. Wright HL, Makki FA, Moots RJ, Edwards SW. Low-density granulocytes: functionally distinct, immature neutrophils in rheumatoid arthritis with altered properties and defective TNF signalling. J Leukoc Biol. (2017) 101:599–611. doi: 10.1189/jlb.5A0116-022R
284. Kegerreis BJ, Catalina MD, Geraci NS, Bachali P, Lipsky PE, Grammer AC. Genomic identification of low-density granulocytes and analysis of their role in the pathogenesis of systemic lupus erythematosus. J Immunol. (2019) 202:3309–17. doi: 10.4049/jimmunol.1801512
285. Midgley A, Beresford MW. Increased expression of low density granulocytes in juvenile-onset systemic lupus erythematosus patients correlates with disease activity. Lupus. (2016) 25:407–11. doi: 10.1177/0961203315608959
286. Rahman S, Sagar D, Hanna RN, Lightfoot YL, Mistry P, Smith CK, et al. Low-density granulocytes activate T cells and demonstrate a non-suppressive role in systemic lupus erythematosus. Ann Rheum Dis. (2019) 78:957–66. doi: 10.1136/annrheumdis-2018-214620
287. Fu J, Tobin MC, Thomas LL. Neutrophil-like low-density granulocytes are elevated in patients with moderate to severe persistent asthma. Ann Allergy Asthma Immunol. (2014) 113:635–40 e632. doi: 10.1016/j.anai.2014.08.024
288. Ui Mhaonaigh A, Coughlan AM, Dwivedi A, Hartnett J, Cabral J, Moran B, et al. Low density granulocytes in ANCA vasculitis are heterogenous and hypo-responsive to anti-myeloperoxidase antibodies. Front Immunol. (2019) 10:2603. doi: 10.3389/fimmu.2019.02603
289. Ostendorf L, Mothes R, van Koppen S, Lindquist RL, Bellmann-Strobl J, Asseyer S, et al. Low-density granulocytes are a novel immunopathological feature in both multiple sclerosis and neuromyelitis optica spectrum disorder. Front Immunol. (2019) 10:2725. doi: 10.3389/fimmu.2019.02725
290. Rodriguez-Carrio J, Carrillo-Lopez N, Ulloa C, Seijo M, Rodriguez-Garcia M, Rodriguez-Suarez C, et al. A subset of low density granulocytes is associated with vascular calcification in chronic kidney disease patients. Sci Rep. (2019) 9:13230. doi: 10.1038/s41598-019-49429-x
291. Hardisty GR, LLanwarne F, Minns D, Gillan JL, Davidson DJ, Gwyer Findley E, et al. Ultra-pure isolation of low density neutrophils casts doubt on their exceptionality in health and disease. bioRxiv. (2020). doi: 10.1101/2020.06.17.156588
292. Hassani M, Hellebrekers P, Chen N, van Aalst C, Bongers S, Hietbrink F, et al. On the origin of low-density neutrophils. J Leukoc Biol. (2020) 107:809–18. doi: 10.1002/JLB.5HR0120-459R
293. Nemeth T, Sperandio M, Mocsai A. Neutrophils as emerging therapeutic targets. Nat Rev Drug Discov. (2020) 19:253–75. doi: 10.1038/s41573-019-0054-z
294. Burmester GR, McInnes IB, Kremer J, Miranda P, Korkosz M, Vencovsky J, et al. A randomised phase IIb study of mavrilimumab, a novel GM-CSF receptor alpha monoclonal antibody, in the treatment of rheumatoid arthritis. Ann Rheum Dis. (2017) 76:1020–30. doi: 10.1136/annrheumdis-2016-210624
295. Taylor PC, Keystone EC, van der Heijde D, Weinblatt ME, Del Carmen Morales L, Reyes Gonzaga J, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med. (2017) 376:652–62. doi: 10.1056/NEJMoa1608345
296. Mitchell TS, Moots RJ, Wright HL. Janus kinase inhibitors prevent migration of rheumatoid arthritis neutrophils towards interleukin-8, but do not inhibit priming of the respiratory burst or reactive oxygen species production. Clin Exp Immunol. (2017) 189:250–8. doi: 10.1111/cei.12970
297. Tang TT, Lv LL, Pan MM, Wen Y, Wang B, Li ZL, et al. Hydroxychloroquine attenuates renal ischemia/reperfusion injury by inhibiting cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis. (2018) 9:351. doi: 10.1038/s41419-018-0378-3
298. Zhang S, Zhang Q, Wang F, Guo X, Liu T, Zhao Y, et al. Hydroxychloroquine inhibiting neutrophil extracellular trap formation alleviates hepatic ischemia/reperfusion injury by blocking TLR9 in mice. Clin immunol. (2020) 216:108461. doi: 10.1016/j.clim.2020.108461
299. Laurindo IM, Mello SB, Cossermelli W. Influence of low doses of methotrexate on superoxide anion production by polymorphonuclear leukocytes from patients with rheumatoid arthritis. J Rheumatol. (1995) 22:633–8.
300. Sperling RI, Benincaso AI, Anderson RJ, Coblyn JS, Austen KF, Weinblatt ME. Acute and chronic suppression of leukotriene B4 synthesis ex vivo in neutrophils from patients with rheumatoid arthritis beginning treatment with methotrexate. Arthritis Rheum. (1992) 35:376–84.
301. Weinmann P, Moura RA, Caetano-Lopes JR, Pereira PA, Canhao H, Queiroz MV, et al. Delayed neutrophil apoptosis in very early rheumatoid arthritis patients is abrogated by methotrexate therapy. Clin Exp Rheumatol. (2007) 25:885–7.
302. Wittkowski H, Foell D, af Klint E, De Rycke L, De Keyser F, Frosch M, et al. Effects of intra-articular corticosteroids and anti-TNF therapy on neutrophil activation in rheumatoid arthritis. Ann Rheum Dis. (2007) 66:1020–5. doi: 10.1136/ard.2006.061507
303. Liu L, Wang YX, Zhou J, Long F, Sun HW, Liu Y, et al. Rapid non-genomic inhibitory effects of glucocorticoids on human neutrophil degranulation. Inflamm Res. (2005) 54:37–41. doi: 10.1007/s00011-004-1320-y
304. Campbell IK, Leong D, Edwards KM, Rayzman V, Ng M, Goldberg GL, et al. Therapeutic targeting of the G-CSF receptor reduces neutrophil trafficking and joint inflammation in antibody-mediated inflammatory arthritis. J Immunol. (2016) 197:4392–402. doi: 10.4049/jimmunol.1600121
305. Cunha TM, Barsante MM, Guerrero AT, Verri WA Jr, Ferreira SH, Coelho FM, et al. Treatment with DF 2162, a non-competitive allosteric inhibitor of CXCR1/2, diminishes neutrophil influx and inflammatory hypernociception in mice. Br J Pharmacol. (2008) 154:460–70. doi: 10.1038/bjp.2008.94
306. Coelho FM, Pinho V, Amaral FA, Sachs D, Costa VV, Rodrigues DH, et al. The chemokine receptors CXCR1/CXCR2 modulate antigen-induced arthritis by regulating adhesion of neutrophils to the synovial microvasculature. Arthritis Rheum. (2008) 58:2329–37. doi: 10.1002/art.23622
307. Zheng W, Warner R, Ruggeri R, Su C, Cortes C, Skoura A, et al. PF-1355, a mechanism-based myeloperoxidase inhibitor, prevents immune complex vasculitis and anti-glomerular basement membrane glomerulonephritis. J Pharmacol Exp Ther. (2015) 353:288–98. doi: 10.1124/jpet.114.221788
308. Stockley R, De Soyza A, Gunawardena K, Perrett J, Forsman-Semb K, Entwistle N, et al. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir Med. (2013) 107:524–33. doi: 10.1016/j.rmed.2012.12.009
309. Zhou Y, An LL, Chaerkady R, Mittereder N, Clarke L, Cohen TS, et al. Evidence for a direct link between PAD4-mediated citrullination and the oxidative burst in human neutrophils. Sci Rep. (2018) 8:15228. doi: 10.1038/s41598-018-33385-z
310. Teo CY, Shave S, Chor AL, Salleh AB, Rahman MB, Walkinshaw MD, et al. Discovery of a new class of inhibitors for the protein arginine deiminase type 4 (PAD4) by structure-based virtual screening. BMC Bioinformatics. (2012) 13:S4. doi: 10.1186/1471-2105-13-S17-S4
311. Knight JS, Luo W, O'Dell AA, Yalavarthi S, Zhao W, Subramanian V, et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res. (2014) 114:947–56. doi: 10.1161/CIRCRESAHA.114.303312
312. Koushik S, Joshi N, Nagaraju S, Mahmood S, Mudeenahally K, Padmavathy R, et al. PAD4: pathophysiology, current therapeutics and future perspective in rheumatoid arthritis. Expert Opin Ther Targets. (2017) 21:433–47. doi: 10.1080/14728222.2017.1294160
313. Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol. (2015) 11:189–91. doi: 10.1038/nchembio.1735
314. Li M, Lin C, Deng H, Strnad J, Bernabei L, Vogl DT, et al. A novel peptidylarginine deiminase 4 (PAD4) inhibitor BMS-P5 blocks formation of neutrophil extracellular traps and delays progression of multiple myeloma. Mol Cancer Ther. (2020) 19:1530–8. doi: 10.1158/1535-7163.MCT-19-1020
315. Curran AM, Naik P, Giles JT, Darrah E. PAD enzymes in rheumatoid arthritis: pathogenic effectors and autoimmune targets. Nat Rev Rheumatol. (2020) 16:301–15. doi: 10.1038/s41584-020-0409-1
Keywords: neutrophils, rheumatoid arthritis, systemic lupus erythematosus, NETs, low density granulocytes, immunometabolism
Citation: Fresneda Alarcon M, McLaren Z and Wright HL (2021) Neutrophils in the Pathogenesis of Rheumatoid Arthritis and Systemic Lupus Erythematosus: Same Foe Different M.O. Front. Immunol. 12:649693. doi: 10.3389/fimmu.2021.649693
Received: 05 January 2021; Accepted: 12 February 2021;
Published: 04 March 2021.
Edited by:
Sonja Vermeren, University of Edinburgh, United KingdomReviewed by:
Marc Pouliot, Laval University, CanadaCopyright © 2021 Fresneda Alarcon, McLaren and Wright. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helen Louise Wright, aGx3cmlnaHRAbGl2ZXJwb29sLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.