 Satoshi Gando
Satoshi Gando Takeshi Wada
Takeshi Wada- 1Acute and Critical Center, Department of Acute and Critical Care Medicine, Sapporo Higashi Tokushukai Hospital, Sapporo, Japan
- 2Division of Acute and Critical Care Medicine, Department of Anesthesiology and Critical Care Medicine, Hokkaido University Faculty of Medicine, Sapporo, Japan
Thromboplasminflammation in coronavirus disease 2019 (COVID-19) coagulopathy consists of angiotensin II (Ang II)-induced coagulopathy, activated factor XII (FXIIa)- and kallikrein, kinin system-enhanced fibrinolysis, and disseminated intravascular coagulation (DIC). All three conditions induce systemic inflammation via each pathomechanism-developed production of inflammatory cytokines. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) downregulates angiotensin-converting enzyme 2, leading to an increase in Ang II levels. Ang II-induced coagulopathy comprising platelet activation, thrombin generation, plasminogen activator inhibitor-1 expression and endothelial injury causes thrombosis via the angiotensin II type 1 receptor. SARS-CoV-2 RNA and neutrophil extracellular trap (NET) DNA activate FXII, resulting in plasmin generation through FXIIa- and kallikrein-mediated plasminogen conversion to plasmin and bradykinin-induced tissue-type plasminogen activator release from the endothelium via the kinin B2 receptor. NETs induce immunothrombosis at the site of infection (lungs), through histone- and DNA-mediated thrombin generation, insufficient anticoagulation control, and inhibition of fibrinolysis. However, if the infection is sufficiently severe, immunothrombosis disseminates into the systemic circulation, and DIC, which is associated with the endothelial injury, occurs. Inflammation, and serine protease networks of coagulation and fibrinolysis, militate each other through complement pathways, which exacerbates three pathologies of COVID-19 coagulopathy. COVID-19 coagulopathy causes microvascular thrombosis and bleeding, resulting in multiple organ dysfunction and death in critically ill patients. Treatment targets for improving the prognosis of COVID-19 coagulopathy include thrombin, plasmin, and inflammation, and SARS-CoV-2 infection. Several drugs are candidates for controlling these conditions; however, further advances are required to establish robust treatments based on a clear understanding of molecular mechanisms of COVID-19 coagulopathy.
Introduction
Based on the knowledge of severe acute respiratory syndrome (SARS), analyses of full-length genome sequences of novel coronavirus revealed that they share 79.6% and 96% sequence identity with SARS coronavirus (SARS-CoV) and to bat coronavirus, respectively (1). These studies confirmed that novel coronaviruses use the same angiotensin-converting enzyme 2 (ACE2) as SARS-CoV as entry receptor to the infected cells (1, 2),. Accordingly, the novel coronavirus is named SARS-CoV-2 and the disease caused by SARS-CoV-2 is called coronavirus disease 2019 (COVID-19).
A rampage through the body (3). SARS-CoV-2 affects multiple organs in the body, from the brain to the blood, and is not limited to the lungs (3). Above all, the blood is one of the organs that has attracted much attention worldwide. Reports from China showed higher deterioration in platelet counts, coagulation and fibrinolysis markers, especially in D-dimer levels in non-survivors than in survivors of COVID-19 (4, 5). Tang et al. (6) showed a high incidence (71,4%) of disseminated intravascular coagulation (DIC) and abnormally high levels of fibrin/fibrinogen degradation products (FDP) and D-dimer in non-survivors of COVID-19. Following these studies, the International Society on Thrombosis and Haemostasis published interim guidance on COVID-19 coagulopathy (7). Since this announcement, an exceptionally high incidence of thromboembolic complications associated with abnormal coagulation parameters in COVID-19 has been spread (8). Importantly, bleeding complications were also high, and the odds ratio of D-dimer for prediction of bleeding was 3.56 in critically ill patients with SARS-CoV-2 infection (9). Contrary to the findings of Tang et al. (6), DIC was rare (2.2%) but was associated with significant bleeding in this study (9). The results coincide with the notion that DIC has long been acknowledged as a “thrombohemorrhagic disorder” (10). However, laboratory findings of early phase of COVID-19 show a moderate decrease in platelet counts, mild prolongation of prothrombin time, and high fibrinogen levels, which are different from DIC, and leads us to consider other pathomechanisms of COVID-19 coagulopathy (11).
The three key words of COVID-19 coagulopathy are inflammation (12), thrombosis, and bleeding. We tried to unify these key words explaining three viewpoints; ACE2 participated in angiotensin II (Ang II)-induced coagulopathy, hyperfibrinolysis, and DIC. Thromboplasminflammation in COVID-19 coagulopathy for diagnostic and therapeutic implications will be discussed based on these three viewpoints in this review.
Methods
PubMed was searched using terms (COVID-19 or SARS-CoV-2) and (ACE2 or angiotensin II, inflammation, coagulopathy, coagulation, fibrinolysis, thrombin, plasmin, treatment, guidance or guidelines) for studies published from January 1, 2020, to May 20, 2021. We manually checked the references of the selected articles to identify additional relevant articles. Articles relevant to the three keywords and viewpoints were selected and discussed.
Underlying Findings
Coronavirus Family and Coagulopathy
In addition to SARS-CoV and SARS-CoV-2, Middle East respiratory syndrome coronavirus (MERS-CoV), influenza viruses (including seasonal A and B, avian H5N1, and swine H1N1, etc.), and human coronavirus NL63 belong to the human coronavirus family. MERS-CoV uses human dipeptidyl peptidase-4 as the entry receptor, whereas the other viruses invade cells through ACE2 (13). Main cell types infected by these coronaviruses are airway epithelial cells and lung type II pneumocytes which cause pneumonia and, in severe cases, acute respiratory distress syndrome (ARDS). Importantly, all coronaviruses give rise to variety degree of thrombocytopenia, coagulation activation, and inhibition of fibrinolysis or hyperfibrinolysis, thus increasing the risk of local and systemic thrombosis, microvascular thrombosis, and bleeding (14, 15). Although similarities and differences in mechanisms of coagulofibrinolytic changes in coronaviruses have been discussed, ACE2 and the renin-angiotensin-aldosterone system (RAAS) is considered one of main mechanisms of coagulopathies in SARS-CoV, SARS-CoV-2, influenza virus, and human coronavirus NL63 infections (13–15).
SARS-CoV-2 Infection and the Renin-Angiotensin-Aldosterone System
The SARS-CoV-2 is a single-stranded RNA virus with an envelope that uses ACE2 as a cell entry receptor through envelope-anchored spike glycoprotein binding (1, 16). Spike glycoprotein drives the fusion of viral and infected cell membranes using S2 subunit through cleavage at the S1/S2 and S2’ sites of glycoprotein by furin and transmembrane protease serine type-2 (TMPRSS2), respectively (16, 17). The RAAS, which includes ACE2, has long been well known (Figure 1). Renin converts angiotensinogen to angiotensin I (Ang I), which is converted to Ang II by ACE. ACE2 degrades Ang I and Ang II to generate Ang(1-9) and Ang(1-7), which have anti-inflammatory and antithrombotic actions through the angiotensin II type 2 receptor (AT2R) and Mas receptor (18, 19). Conversely, Ang II has proinflammatory and prothrombotic properties binding to AT1R and secretes aldosterone from the adrenal cortex. Accordingly, RAAS is balanced by its two opposing properties of inflammation and coagulation by ACE2.
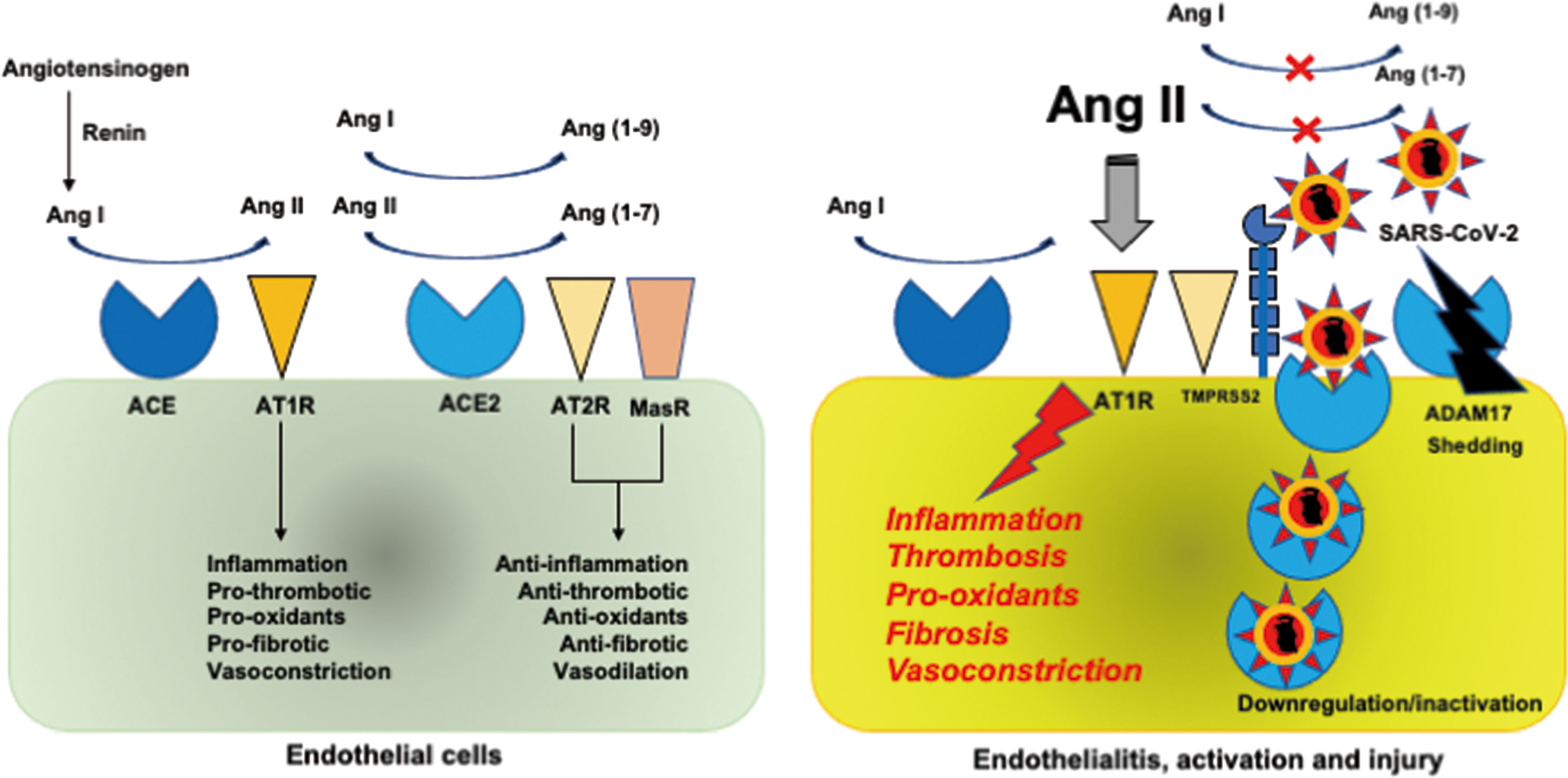
Figure 1 Renin-angiotensin-aldosterone system. (Left). Renin converts angiotensinogen to Ang I, which is converted to Ang II by ACE. ACE2 degrades Ang I and Ang II to generate Ang(1-9) and Ang(1-7), which have anti-inflammatory and antithrombotic actions through AT2R and MasR. Conversely, Ang II has proinflammatory and prothrombotic properties binding to AT1R and secretes aldosterone from the adrenal cortex. RAAS is balanced by its two opposed properties of inflammation and coagulation by ACE2. (Right). Binding of SARS-CoV-2 to ACE2 via the spike glycoprotein to TMPRSS2 results in the downregulation and inactivation of ACE2, leading to an increase in Ang II levels, which gives rise to inflammation and thrombosis through AT1R. Proteolysis and shedding of ACE2 resulting from ADAM17 upregulation also contributes to inactivation of ACE2. SARS-CoV-2 infection of endothelial cells causes endothelialitis associated with endothelial activation and injury. ACE, angiotensin-converting enzyme; ADAM17, a disintegrin and metalloprotease domain-containing protein 17; Ang, angiotensin; AT1R, angiotensin II type 1 receptor; MasR, Mas receptor; RAAS, renin-angiotensin-aldosterone system; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; TMPRSS2, transmembrane protease serine type 2.
ACE2 is expressed in various cells of the whole body, including the lung epithelial cells, and endothelial cells (20). Binding of SARS-CoV-2 to ACE2 results in the downregulation and inactivation of ACE2, leading to an increase in Ang II levels (18). Liu et al. (21) confirmed the elevation of Ang II levels in parallel with the SARS-CoV-2 viral load in patients with COVID-19. Increased Ang II causes inflammation, thrombosis, and other negative effects on the body. Downregulation and inactivation of ACE2 occur mainly because of endocytosis through binding of the SARS-CoV-2 spike glycoprotein to ACE2 (1, 2, 16, 17). Proteolysis and shedding of ACE2 resulting from the upregulation of a disintegrin and metalloprotease domain-containing protein 17 (ADAM17) also contributes to ACE2 inactivation (22). The TMPRSS2-induced cleavage of ACE2 observed in SARS-CoV may play a role in shedding of ACE2 during SARS-CoV-2 infection (23). It has been speculated that there is an interplay between SARS-CoV-2 and interferons. Studies on SARS-CoV and human coronavirus NL-63 suggest that SARS-CoV-2 may induce interferon production, which downregulates ACE2 (24, 25). However, one study showed that interferon induces ACE2 expression (26). It was subsequently demonstrated that a truncated ACE2 isoform, not the full-length active ACE2, was induced by interferon (27). Therefore, the role of interferon in ACE2 downregulation in SARS-CoV-2 infection remains to be elucidated. Viral replication-dependent ACE2 downregulation during the culture of human coronavirus NL63 at optimal temperature suggests that viral load may have a role in the lack of availability of ACE2 in COVID-19 (28).
Pyroptosis and Necroptosis
Pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) are the ligands of innate immune system pattern recognition receptors such as toll-like receptors (TLRs), nucleotide-binding oligomerization domain-containing (NOD)-like receptors (NLRs), and retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs). PAMPs, such as bacterial lipopolysaccharide or viral RNA, induce the lytic programmed cell death processes called pyroptosis and necroptosis resulting in cell death and releases of DAMPs such as nucleic acids (DNA/RNA), histones, mitochondrial DNA, and high mobility group box 1 (HMGB1), all of which are involved in changes in inflammation, coagulation, and fibrinolysis (29, 30). Therefore, there is bidirectional interplay between PAMPs and DAMPs via pyroptosis and necroptosis.
Pyroptosis is caused by procaspase-1 and/or procaspase-11 (human capase-4 and -5) combining with NOD-like receptor family-involved pyrin domain-containing 3 (NLRP3) to generate the NLRP3 inflammasome, which then cleaves gasdermin D (GSDMD), releasing its N- and C-terminal products (GSDMD-N and GSDMD-C) through either direct activation of caspase-1 or indirectly through the recruitment of adaptor protein, apoptosis-associated speck-like protein containing a caspase activating and recruitment domain (ASC). GASDMD-N disrupt cell membrane by pore formation (29, 30).
The necroptosis pathway is induced by toll/interleukin-1 (IL-1) receptor homology domain-containing adaptor inducing interferon-β (TRIFF)-dependent activation of TLR3, and TLR4, and interferon receptor signaling. Autophosphorylation of receptor interacting serine/threonine kinase 1 (RIPK1) recruits RIPK3 resulting in mixed lineage kinase domain-like (MLKL) activation, leading to translocation of MLKL to plasma membranes followed by membrane disruption by permeabilization (29–31). Apoptotic caspases-3 and -8 mediate interplays among apoptosis, pyroptosis, and necroptosis (31). Although necroptosis occurs after impairment of apoptosis as backup mechanism, pyroptosis is considered as primary responses to cellular insult (30, 31).
Cell death programs are primarily physiological innate immune inflammatory responses against insults including infection through the protection from pathogen spreading by inducing death of infected cells, and through production of DAMPs, which evoke further inflammation and coagulation, in addition to PAMPs. However, when the insults are extensively severe, over-activated pyroptosis and necroptosis give rise to pathological phenomena (29, 32, 33). SARS-CoV-2 binding to ACE2, and Ang II-induced activation of AT1R, and activation of complement pathways by SARS-CoV-2 may induce assembly of the NLRP3 inflammasome (33–35), which contributes to the onset of cytokine storm, pneumonia, ARDS, thrombosis, DIC, and other types of organ dysfunction (35–37).
Cytokines
Viral RNA, function as PAMPs, and infected cell pyroptosis- and necroptosis-generated DAMPs bind to pattern recognition receptors such as TLR3, TLR7/TLR8, RLRs, and NLRP3, which induce the transcription of proinflammatory cytokines and type-1 interferon-α and -β (38, 39). High plasma levels of tumor necrosis factor-α (TNF-α), IL-1β, IL-6, and IL-8 have been confirmed in COVID-19, especially in non-survivors and in critically ill patients (4, 12, 40, 41). SARS-CoV-2-activated inflammatory cells, such as macrophages, monocytes, and neutrophils are thought to be the producing cells of these inflammatory cytokines (12, 42).
Platelets and Neutrophils Activation
DAMPs as well as inflammatory cytokines induce platelet activation and subsequent aggregation directly or through interactions with TLR2 and TLR4 expressed on the platelets, leading to platelet-neutrophil interactions (43–45). In addition, numerous viruses directly activate platelets via a variety of platelet surface receptors such as TLR7, FCγRII, and glycoprotein IIb/IIIa (46). Influenza virus, which is a single-stranded RNA virus, same as SARS-CoV-2, activates platelet via TLR7 (47). The activated platelets upregulate P-selectin from α-granules and express CD40 ligand (CD40L), leading to consequent their binding to P-selectin glycoprotein ligand-1 and CD40 on monocytes, neutrophils, and endothelial cells (48, 49). These processes release neutrophil extracellular traps (NETs) from neutrophils and express selectins and other adhesion molecules on endothelial cells.
Higher levels of soluble P-selectin and soluble CD40L in COVID-19 patients admitted to ICU than in non-ICU patients indicate platelet activation in severe COVID-19 (50). Lower platelet counts in non-survivors and severe cases of COVID-19 than in survivors and non-severe cases support platelet activation and its consumption (51, 52). Viruses can induce NET formation, which has roles in the pathogenesis of viral infection-induced systemic pathologies such as vasculitis and DIC (53). High-dimensional flow cytometric marker analysis revealed that neutrophil and platelet activations are associated with platelet-neutrophil aggregates in the blood of COVID-19 pneumonia patients (54). Myeloperoxidase-DNA and citrullinated histone H3, surrogate markers of NET formation, showed high levels in COVID-19, especially in patients on ventilators or admitted to the ICU (55, 56).
Taken together, SARS-CoV-2 induces activation and interactions of platelets and neutrophils, which give rise to NET release, associated with high levels of histones in the circulation.
Endothelial Cell Activation and Injury
TNF-α and IL-1 upregulate P-selectin and induce the expression of E-selectin on the endothelium and L-selectin on the neutrophils, leading to neutrophil and endothelial interactions, which accelerates the expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) in the endothelial cells (43, 57, 58). These processes promote activated neutrophils to adhere to endothelial cells secreting elastase, metalloproteases, myeloperoxidase, and reactive oxygen species, then induce endothelial injury (57, 59, 60). NETs containing histone directly injure endothelial cells. Sera from patients with high levels of histones showed reduced viability of cultured endothelial cells (61). Histones bind to endothelial cells and then induce Ca2+ influx and overload with consequent pore formation, leading to endothelial injury (62).
High levels of von Willebrand Factor antigen and its activity, and factor VIII (FVIII) activity in ICU-admitted COVID-19 patients indicate endothelial activation (50). Low survival provability in COVID-19 patients with elevated levels of soluble thrombomodulin, a marker of endothelial injury, suggest endothelial injury in severe COVID-19 cases (50). Scanning electron micrographs of pulmonary autopsy specimens revealed endothelial destruction and SARS-CoV-2 invasion in the endothelial cells, which suggest viral endothelialitis-induced endothelial injury (63).
Briefly, SARS-CoV-2, SARS-CoV-2-induced inflammatory cytokine expression, activated neutrophil-released granule-containing elastase, reactive oxygen species, and NETs with histones cause endothelial cell activation and injury.
Systemic Inflammation, Macro, and Microvascular Thrombosis
Large cohort studies on COVID-19 confirmed high temperature, and increased heart rate, respiratory rate, and white blood cell counts in severe cases and non-survivors, suggesting systemic inflammation with high levels of proinflammatory cytokines (4, 64). These have been recognized as systemic inflammatory response syndrome (SIRS) or cytokine storm, which are associated with systemic cytokine circulation, prothrombotic state, and endothelial dysfunction with increased permeability (10, 42, 65).
In addition to the high incidence of rapid arterial and deep venous thrombosis formation, microvascular fibrin thrombosis is a prominent characteristic of COVID-19 (7–9, 66). The first target of SARS-CoV-2 infection is the lung; therefore, many autopsy studies revealed fibrin microvascular thrombosis in arterioles, venules, and capillaries in the lungs (63, 67, 68). Pulmonary vascular occlusion from capillaries to medium-sized vessels with aggregated NETs were also confirmed (56). This stage of lung injury is called type L ARDS and is characterized by hypoxemia with low elastance (69). Importantly, in accordance with disease progression, thrombosis with neutrophilic plugs were present in the brain, heart, liver, kidneys, and spleen, in addition to the lungs (67), which suggests that initial immunothrombosis is disseminated throughout the body (70). SARS-CoV-2-developed pneumonia progresses to ARDS from exudative, proliferative to fibrotic stages showing hyaline membranes, septum thickening, alveolar and microvascular fibrin thrombosis, diffuse alveolar damage, and organizing fibrosis (68). This stage is called type H ARDS and is associated with the same hypoxemia level as that of type L, but is complicated by high elastance owing to diffuse alveolar damage (69). Type H is a classic typical ARDS, which has been known to usually complicate DIC (71).
Taken together, it is hypothesized that type L ARDS at initial infection stage represents pulmonary intravascular coagulopathy comprising immunothrombosis, which progresses to type H ARDS in severe cases at late stage and is associated with systemic microvascular thrombosis in addition to lung thrombosis (69, 72).
Bleeding
An important finding of an autopsy study was the coexistence of microvascular thrombosis and extensive hemorrhage in the lungs (68). Bleeding events rates in COVID-19 are unexpectedly high at 4.8%, and all patients diagnosed with DIC are associated with high-grade bleeding events with low fibrinogen levels (9). The results were confirmed in another study involving ICU patients, which showed 8.0% bleeding events and 43.3% thrombotic complications (73). A monocenter retrospective study of ICU-admitted patients showed that 21% were associated with hemorrhagic events (74).
Vaccine-Induced Immune Thrombotic Thrombocytopenia
Vaccine-induced immune thrombotic thrombocytopenia (VITT) in response to the recombinant adenoviral vector encoding the SARS-CoV-2 spike glycoprotein (ChAdOx1 nCov-19) should be considered an important finding related to COVID-19 coagulopathy (75). Cases of thrombosis and thrombocytopenia after vaccination with a replication-incompetent human adenovirus 26 vector vaccine (Ad26.COV2.S) have also been reported (76). Clinical features and laboratory data on instances of VITT resulting in fatal thromboembolic events, DIC, and hemorrhage associated with thrombocytopenia, low fibrinogen, and high D-dimer levels resemble autoimmune heparin-induced thrombocytopenia (75, 77). Platelet factor 4 (PF4)-heparin antibody positivity suggests that interplay between the vaccine and platelets and/or PF4 is a pathological mechanism of VITT (75). Further studies are needed to clarify pathomechanisms and establish diagnostic and therapeutic strategies for specific to VITT management and different from strategies used to address autoimmune heparin-induced thrombocytopenia (75).
Three Viewpoints
Ang II-Induced Coagulopathy
SARS-CoV-2 RNAemia was observed in 15% of COVID-19 patients (40). Another study confirmed extremely high frequency of viral RNAemia (78%) in critically ill COVID-19 patients (78). In addition, SARS-CoV-2 was detected in a variety of specimens, including blood (79). Autopsy findings showed systemic SARS-CoV-2 infected cells in multiple organs, including the lungs, heart, kidneys, gastrointestinal tracts, and endothelial cells (63, 67). Elevated Ang II levels being proportional to SARS-CoV-2 viral load are a result of the downregulation of ACE2 existing in a variety of infected organs. Ang II in the infected organs and in the circulation is likely to cause systemic effects on inflammation, platelet function, coagulation, and fibrinolysis through AT1R in COVID-19 (18–21, 80).
Platelets
Human platelets express AT1R, and potentiation by Ang II of adrenaline-induced platelet activation is mediated through this receptor (81, 82). Furthermore, another study demonstrated that platelets express ACE2 and TMPRSS2, and that binding of the SARS-CoV-2 spike glycoprotein to ACE2 on platelets could induce αIIb/β3 activation and P-selectin expression, leading to enhancement of thrombosis (83). Ang II infusion into healthy volunteers confirmed platelet activation by elevating plasma β-thromboglobulin levels and platelet surface expression of P-selectin (84). The high levels of Ang II, soluble P-selectin, and soluble CD40L in the circulation suggest the Ang II-induced platelet activation in COVID-19 (21, 50). Confirmation of soluble CD40L in COVID-19 patients with hyperactivated platelet indicate that activation of platelet contribute to activation of CD40-bearing neutrophils (85). Platelet activation, therefore, lead to interactions with neutrophils, resulting in NET formation (70).
Coagulation and Fibrinolysis
Ang II induces tissue factor mRNA and antigen expression on monocytes through AT1R (86). Ang II-mediated tissue factor expression in endothelial cells and vascular smooth muscle cells has also been confirmed (87, 88). Ang II accelerates arterial and venous thrombosis development via AT1R, which is associated with increased plasma levels of plasminogen activator inhibitor-1 (PAI-1) (89, 90). Furthermore, Ang II infusion to healthy volunteers confirmed the elevation of soluble P-selectin, thrombin and antithrombin complex and prothrombin fragment F1+2 in the plasma (84). These findings suggest that Ang II-induced tissue factor expression leads to systemic thrombin generation, resulting in thrombus formation in the vessels. The promotion of antithrombotic activity by ACE2 activation indirectly supports the idea that downregulation of ACE2 by SARS-CoV-2 participates the Ang II-induced tissue factor-dependent thrombin generation and thrombus formation (91). In vitro studies have confirmed that Ang II induced increases in PAI-1 mRNA expression and antigen levels in cultured media of endothelial cells with little changes in tissue-type plasminogen activator (t-PA) levels (87, 92), and these effects were mediated by AT1R (93). Ang II infusion into healthy volunteers result in rapid increases in PAI-1 antigen levels in the circulation (94).
Collectively, it is understood that activation of the tissue factor-dependent coagulation pathway and inhibition of fibrinolysis by Ang II participate in thrombus formation in COVID-19.
Endothelial Cells
In addition to PAI-1 production, Ang II has another action on endothelial cells expressing both AT1R and AT2R. Ang II upregulated the expression of ICAM-1 and VCAM-1 in cultured endothelial cells via AT1R (95, 96). Ang II also stimulated the secretion and rapid release of soluble ICAM-1 from endothelial cells both in cultured media and in circulation (96). Endothelialitis with endothelial injury due to SARS-CoV-2 infection may reduce thrombomodulin expression, which causes fibrin deposition as is observed in herpes infection (63, 97). These results suggest that Ang II-induced endothelial cell activation and injury are involved in the mechanisms of thrombus formation in SARS-CoV2 infection.
Taken together, Ang II-induced coagulopathy comprising platelet activation, thrombin generation, PAI-1 expression and endothelial injury participates in macro and microvascular thrombosis via AT1R in COVID-19. These processes are presented in Figure 2.
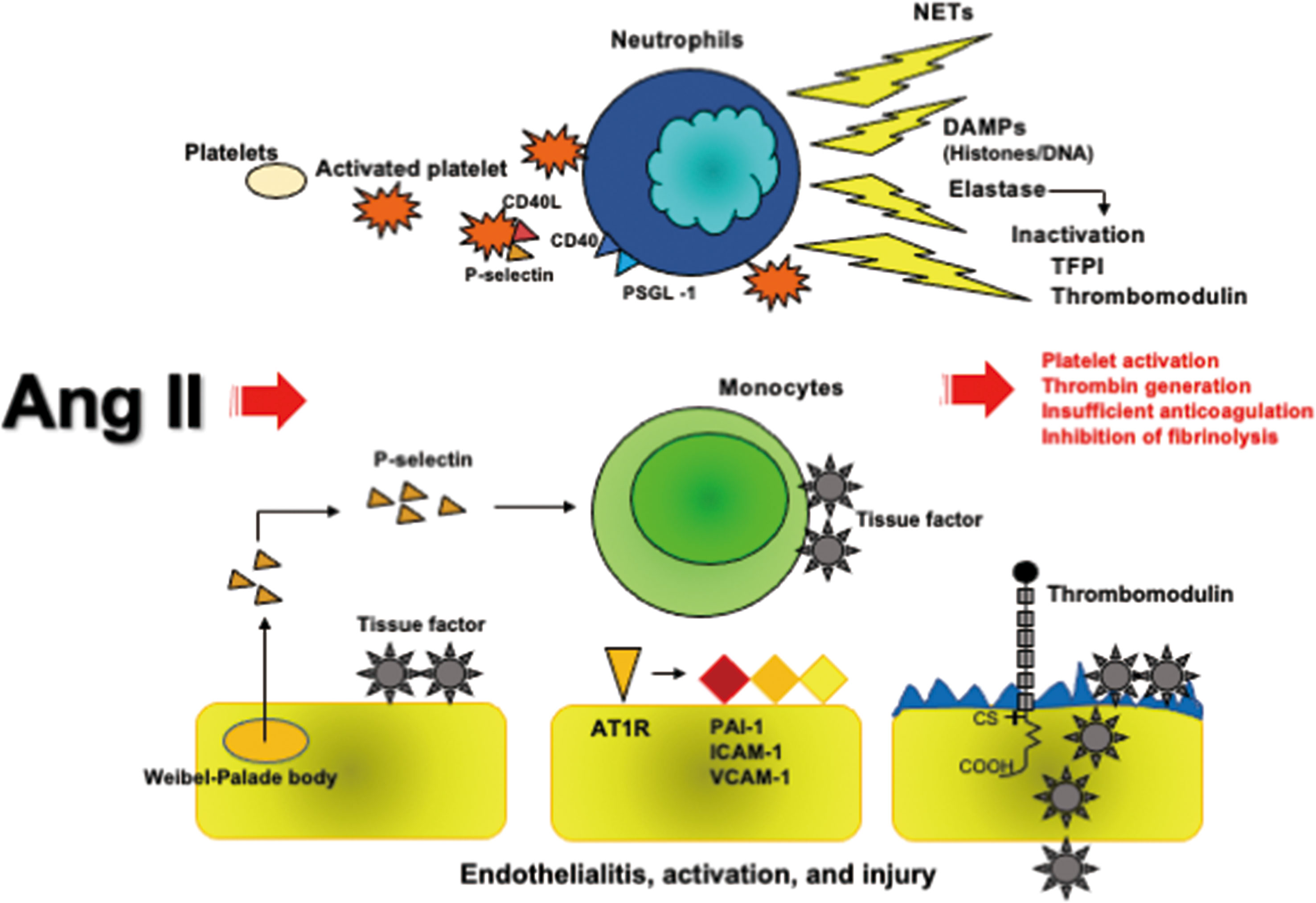
Figure 2 Ang II-induced coagulopathy. Ang II activates platelets expressing P-selectin and CD40L, consequently binding with CD40 and PSGL-1 expressed on neutrophils followed by NETs formation, which contain neutrophil DNA and histones decorated with elastase. Histones have a potent prothrombotic property and elastase cleaves to inactivate TFPI and thrombomodulin. Ang II and P-selectins express tissue factor on monocytes and endothelial cells. Ang II further induces the expressions of PAI-1, ICAM-1, and VCAM-1 on endothelial cells via AT1R. SARS-CoV-2-developed endothelialitis may reduce thrombomodulin expression like herpes simplex virus infection. All these processes caused by Ang II induce thrombus formation. Ang II, angiotensin II; AT1R, angiotensin II type 1 receptor; DAMPs, damage-associated molecular patterns; ICAM-1, intercellular adhesion molecule-1; NETs, neutrophil extracellular traps; PAI-1, plasminogen activator inhibitor; PSGL-1, P-selectin glycoprotein ligand-1; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; TFPI, tissue factor pathway inhibitor; VCAM-1, vascular cell adhesion molecule-1.
Systemic Hyperfibrinolysis
Activation of FXIIa-Dependent Contact and Kallikrein-Kinin Systems
The bidirectional interplay between the FXII-dependent contact system and kallikrein and kinin system (KKS) is deeply involved in inflammation, coagulation, and fibrinolysis. Virus-mediated direct activation of FXII has been presumed in COVID-19, as is observed in bacterial infection (98). Hepatitis C virus RNA directly binds to FXII and FXI and augments activation of these proteases, which supports the notion that SARS-CoV-2 RNA directly activates FXII, followed by KKS activation (99). DNA purified from polymorphonuclear leukocytes assembles and activates FXII and high molecular weight kininogen (HMWK), which induces the activation of prekallikrein followed by the release of bradykinin (100). Furthermore, binding of FXII and HMWK to NET fibers comprising neutrophil DNA followed by their activation was observed in the same experiment (100). Activated neutrophil-induced NET formation has been observed in COVID-19, as mentioned above. These results, therefore, indicate that both SARS-CoV-2 RNA and NETs are likely to activate FXII-dependent contact pathways and KKS (98).
Inflammation and Fibrinolysis
FXIIa triggers FXI-mediated coagulation activation and also reciprocally activates prekallikrein to produce kallikrein, followed by the production of bradykinin by cleaving HMWK. ACE hydrolyzes bradykinin to inactive products whilst metabolizing it to des-Arg9-bradykinin (DABK), which is broken down to inactive peptides by ACE2. DABK activates the kinin B1 receptor (KB1R), whereas bradykinin binds to KB2R, both of which induce inflammation through the production of proinflammatory cytokines, TNF-α, IL-1, IL-6, and IL-8, and reactive oxygen species (19, 101). SARS-CoV-2 downregulates ACE2 with consequent increases in Ang II and causes decreases in ACE activity through a negative feedback mechanism (18, 102). Reduction of ACE activity increases in bradykinin accumulation and ACE2 downregulation impairs DBAK inactivation, both of which give rise to activation of KB1R and KB2R, followed by enhancement of inflammation (19, 101) (Figure 3).
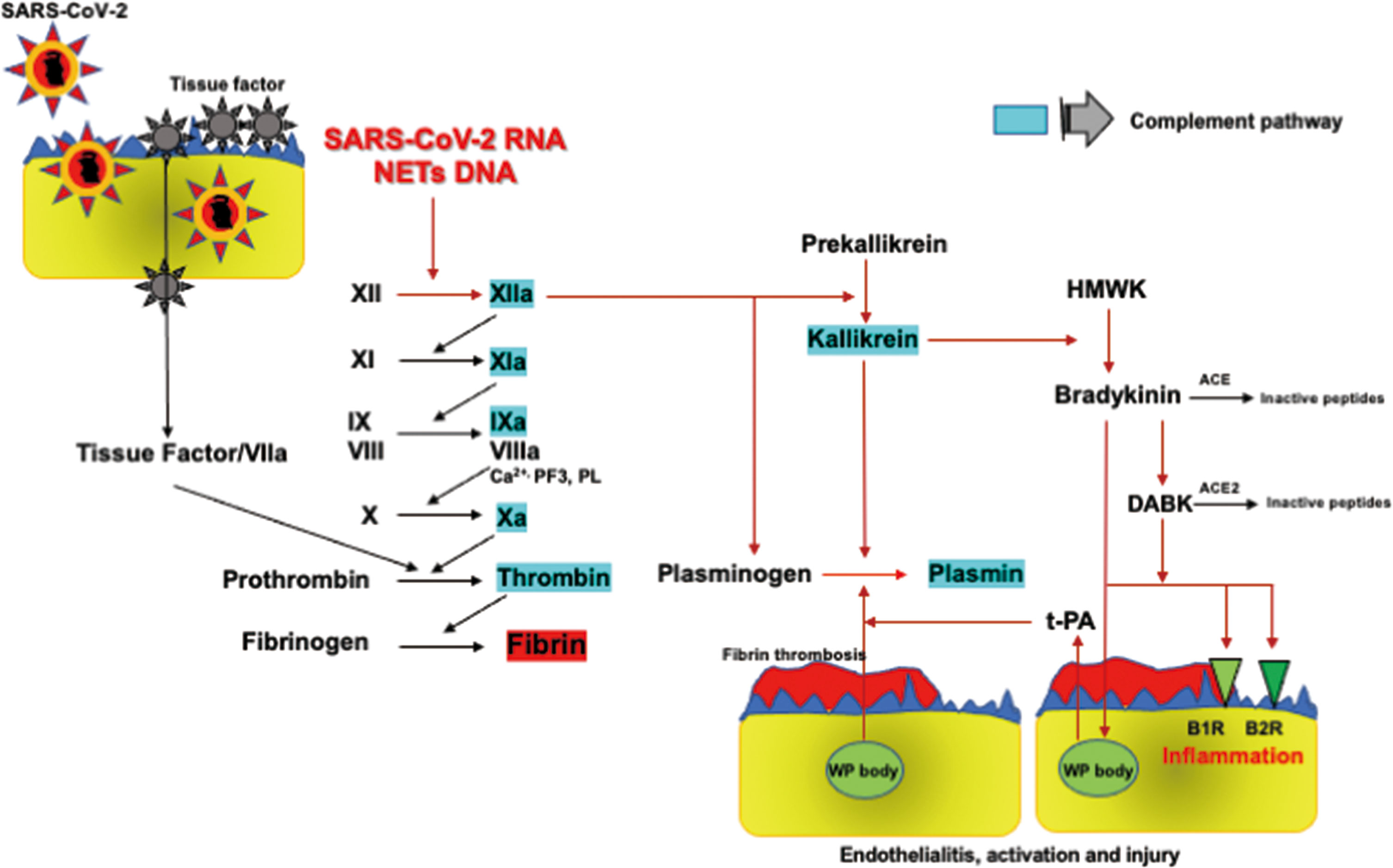
Figure 3 FXII-dependent contact activation system and KKS. SARS-CoV-2 RNA and NETs-containing DNA activate the FXII-dependent contact system, initiating both the contact coagulation pathway and KKS. FXIIa and kallikrein directly activate plasminogen to generate plasmin. Kallikrein cleaves HMWK to bradykinin, which is metabolized into DABK. Bradykinin and DABK induce inflammation producing inflammatory cytokines through KB1R and KB2R. Bradykinin also induces fibrinolysis through t-PA release from Weibel-Palade bodies in endothelial cells via KB2R, leading to plasmin formation. Therefore, bradykinin as well as FXIIa and kallikrein play central roles in inflammation and plasmin-induced fibrinolysis in COVID-19. ACE, angiotensin-converting enzyme; KB1R, kinin B1 receptor; KB2R, kinin B2 receptor; DABK, des-Arg9-bradykinin; HMWK, high molecular weight kininogen; KKS, kallikrein and kinin system; NETs, neutrophil extracellular traps; SARS-CoV2, severe acute respiratory syndrome coronavirus 2; t-PA, tissue-type plasminogen activator; WP body, Weibel-Palade body. Factors highlighted in blue activate complement pathways as shown in Figure 4.
The close relationships among the contact activation system, KKS, and fibrinolytic activation have long been acknowledged. FXIIa directly changes plasminogen to plasmin, and plasmin then reciprocally activates FXIIa under the condition of reduction of the α2-plasmin inhibitor (103–105). FXIIa further potentiates fibrinolytic properties through the inactivation of PAI-1 (106). FXIIa-produced kallikrein from prekallikrein also activates plasminogen into plasmin (107). Bradykinin plays a central role in the cross-talk between inflammation and fibrinolysis through the expression of inflammatory cytokines and t-PA release from endothelial cells via KB2R (108, 109). Influenza RNA virus induces hyperfibrinolysis, showing increased plasmin generation associated with high FDP and D-dimer levels, and low fibrinogen levels. In addition, a high FDP/D-dimer ratio of approximately 3.3 suggests fibrin(ogen)olysis due to plasmin-induced hyperfibrinolysis. Increased plasmin promotes inflammation and viral replication, which are thought to be due to fibrinolysis of immunothrombosis (110). These findings indicate that plasmin plays a pivotal role in the pathogenesis and inflammatory responses to influenza virus infection. A high FDP/D-dimer ratio of about 7.5 with low fibrinogen levels in non-survivors suggests plasmin-induced fibrin(ogen)olysis in SARS-CoV-2 infection (6). A meta-analysis of the human lung transcriptional landscape showed that ACE2 and TMPRSS2 were co-expressed with those of AT1R, kininogen, kallikrein, and fibrinogen in alveolar cells and that the t-PA gene was expressed in lung endothelial cells (111). These results support the idea of close relationships among SARS-CoV-2, ACE2, KKS, RAAS, coagulation, and fibrinolysis.
In brief, the FXII contact activation system and KKS are involved in inflammation and plasmin generation-mediated hyperfibrinolysis through insufficient ACE2, which may be an important pathomechanism in COVID-19 coagulopathy.
DIC
The clinical characteristics of COVID-19 coagulopathy are distinct from DIC because of moderate decrease in platelet counts, mild prolongation of prothrombin time, and extremely high fibrinogen levels despite systemic thrombin and plasmin generation (50, 112, 113). Two meta-analyses, including 1,210 and 4,889 COVID-19 patients, showed that prevalence of DIC were 4.3% and 6.2%, respectively (114, 115). Pre-stage of DIC diagnosed with sepsis-induced coagulopathy score was 16.2% (114). Notably, DIC patients showed a 26.2-times higher incidence of death (log risk ratio, 3.267; confidence interval: 2.19-4.34) (115). These studies indicate the importance of high severity and late stage of the disease in developing DIC, and showed that certain patients with COVID-19 coagulopathy may proceed to fatal DIC with bleeding (6, 9, 115).
Cytokines
Inflammatory cytokines activate platelets and induce the expression of P-selectin, which results in tissue factor expression on monocytes and interactions with neutrophils, leading to neutrophil activation with NET release (10). Three important cytokines involved in these processes are TNF-α, IL-1, and IL-6, which were increased in COVID-19. TNF-α releases t-PA from endothelial cells followed by persistent elevation of PAI-1 playing roles in both the induction and inhibition of fibrinolysis, while IL-6 has the most relevant roles in the activation of coagulation (10, 116). IL-1 induces dysregulation of the anticoagulation pathways in addition to generating thrombin (10, 117).
Platelets
Platelet activation-induced P-selectin expression is a strong trigger for monocyte tissue factor expression that is a main mechanism of developing DIC (10). In severe COVID-19 patients, increased platelet activation with P-selectin expression and αIIb/β3 signaling gave rise to platelet aggregation with monocytes followed by increased tissue factor expression on monocytes (118). Moreover, D-dimer levels showed significant correlations with platelet activation for P-selectin and monocyte tissue factor expression (118), which indicate that platelet activation-induced tissue factor expression on monocytes is associated with COVID-19 coagulopathy, leading to DIC in critical cases (10, 119).
NLRP3 Inflammasome
Either SARS-CoV-2 itself or its RNA activate NLRP3 inflammasome via ACE2-Ang II-AT1R axis. This results in production of IL-1β and IL-18, which are involved in development of DIC (33–35). Platelet NLRP3 inflammasome regulates αIIb/β3 integrin outside-in signaling via IL-1β production, contributing to platelet activation, aggregation and thrombus formation (36). Activation of monocyte/macrophage inflammasome, and subsequent GASDMD-dependent pyroptosis-released tissue factor in the forms of microparticles, resulted in systemic blood coagulation and massive thrombosis, which were associated with high lethality in mice (37). Tissue factor-dependent coagulation activation as a main mechanism of DIC has been well acknowledged (10), therefore, inflammasome activation may be involved in the development of DIC in patients with COVID-19 (37).
Histones and NETs
Recent advances in the pathophysiology of DIC demonstrate the importance of pyroptosis- and necroptosis-derived histones and NETs comprising neutrophil histones, DNA, and elastase, which also act as a source of histones (120, 121). Histones and NETs trigger inflammatory cytokines release, and also initiate coagulation by tissue factor expression on monocytes and endothelial cells and by FXII activation, which is amplified by reduced anticoagulant factors and impaired fibrinolysis. Platelets are also activated by histones and NETs, leading to procoagulant phenotype via P-selectin expression (120, 122). Thrombin generation is extremely enhanced by prothrombinase comprising FVa and FXa, while Abrams et al. (123) found that histones act as substitutes for prothrombinase to promote FXa cleavage of prothrombin to form active thrombin and initiate DIC. In vivo infusion of histones causes DIC in mice associated with thrombocytopenia, consumption coagulopathy and organ dysfunction (123, 124). DIC patients, especially non-survivors, showed significantly higher histone levels than non-DIC patients, and NET formation independently predicted DIC and death in critically ill patients (123–125).
In brief, high histone levels and multiple evidence of NETs formation in various tissues and organs in COVID-19 indicate that DIC is likely to develop in critical SARS-CoV-2 infections.
Complement Pathways
The close interplays between complement pathways, coagulation and fibrinolysis systems play pivotal roles in the development of inflammation and DIC. Both C3a and C5a induce activation of coagulation, insufficient anticoagulation systems, and inhibition of fibrinolysis, which are the three major pathomechanisms of DIC (10, 126, 127) (Figure 4). C5a activates neutrophils, monocytes/macrophages, and endothelial cells through the C5a receptor and C5-like receptor 2, leading to inflammation, DIC, and the development of organ dysfunction (126, 127). Autopsy findings showing deposition of the membrane attack complex (C5b-9), C4d, and mannose-binding lectin (MBL)-associated serine protease 2 (MASP2) co-localized with SARS-CoV-2 spike glycoproteins in the microvasculature indicating the activation of complement pathways (128). Above studies suggest that activation of the complement pathway-generated C3a and C5a contributes to the development of DIC associated with systemic inflammation and organ dysfunction in COVID-19.
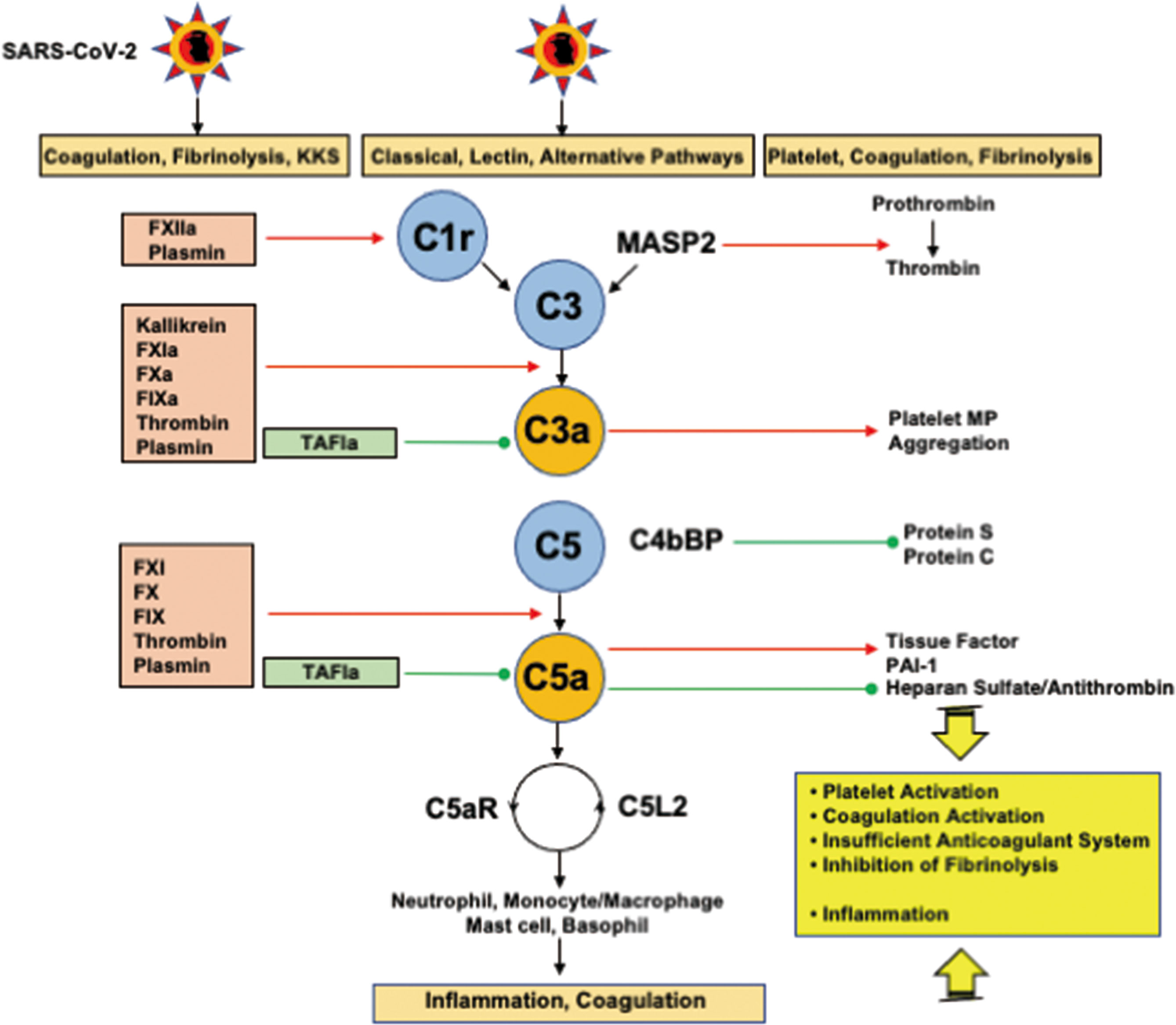
Figure 4 Interplays among coagulation, fibrinolysis, complement pathways, and inflammation. FXIIa and plasmin activate C1r to initiate the classical pathway. In addition to kallikrein, FXIa, FXa, FIXa, thrombin, and plasmin cleave C3 and C5 to generate C3a and C5a. Importantly, kallikrein, thrombin, and plasmin are able to directly activate C3, and thrombin can generate C5a without the participation of C3. TAFIa is the only molecule that blocks C3a and C5a. MASP2 and MAC (not shown in the figure) cleave prothrombin into thrombin. C3a generates platelet procoagulant microparticles and induce platelet aggregation. C5a expresses tissue factor and PAI-1 on neutrophils and mast cells, respectively through C5aR, and inhibits the antithrombin-mediated anticoagulation pathway through the shedding of glycocalyx heparan sulfate. Complex formation between C4bBP and protein S impairs the functional property of protein S, a cofactor of protein C, resulting in a decrease in the conversion of protein C to activated protein C. C5a finally induces inflammation via inflammatory cells through C5aR and C5L2. These mutual actions of the serine protease network contribute to inflammation and thrombus formation at the site of infection, if the infection is sufficiently severe, disseminating to the systemic circulation, and DIC associated with SIRS ensues. C5aR, C5a receptor; C5L2, C5-like receptor2; C4bBP, C4b binding protein; DIC, disseminated intravascular coagulation; MAC, membrane attack complex; MASP, mannose-binding lectin-associated serine protease 2; PAI-1, plasminogen activator inhibitor-1; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; SIRS, systemic inflammatory response syndrome; TAFIa, activated thrombin-activatable fibrinolysis inhibitor. Red arrows indicate activation and green lines with circle-head indicate inhibition.
Phenotypes
DIC consists of thrombotic and fibrinolytic phenotypes (10, 122). DIC is a thrombotic phenotype with microvascular fibrin thrombosis associated with consumption coagulopathy (10, 129). DIC with a fibrinolytic phenotype is defined as the coexistence of DIC and systemic pathologic fibrin(gen)olysis. In other words, condition in which the same insult simultaneously causes DIC and systemic pathologic fibrin(gen)olysis is DIC with a fibrinolytic phenotype, which is usually associated with microvascular thrombosis and oozing-type bleeding (122). In COVID-19, DIC with hyperfibrinolysis due to pathologic activation of FXII and KKS belongs to the fibrinolytic phenotype.
ARDS and the Three Viewpoints
SARS-CoV-2 invades lung tissue through abundantly expressed ACE2 on type II pneumocytes and endothelial cells, leading to Ang II-induced inflammation and coagulopathy. SARS-CoV-2 also gives rise to innate immune inflammation associated with leukocytes and endothelial cell activation, increasing coagulation and fibrinolysis associated with complement activation. The net results of these events include local production of inflammatory cytokines (130) and local neutrophil activation-induced immunothrombosis (56, 131) associated with pulmonary hemorrhage (68, 132). These events may explain the pathogenesis of type L ARDS (69). If SARS-CoV-2 viral loads are sufficiently high, this type of ARDS may progress to type H ARDS associated with SIRS, due to cytokine storm (42, 69, 133) and systemic microvascular thrombosis (67). A few of these cases develop DIC with a fibrinolytic phenotype characterized by increased bleeding (6, 9, 114, 115, 134). The clinical features of SARS-CoV-2 ARDS thus include increased inflammation, coagulation (involving thrombin), and fibrinolysis (involving plasmin), which can be called thromboplasminflammation. These processes are illustrated in Figure 5.
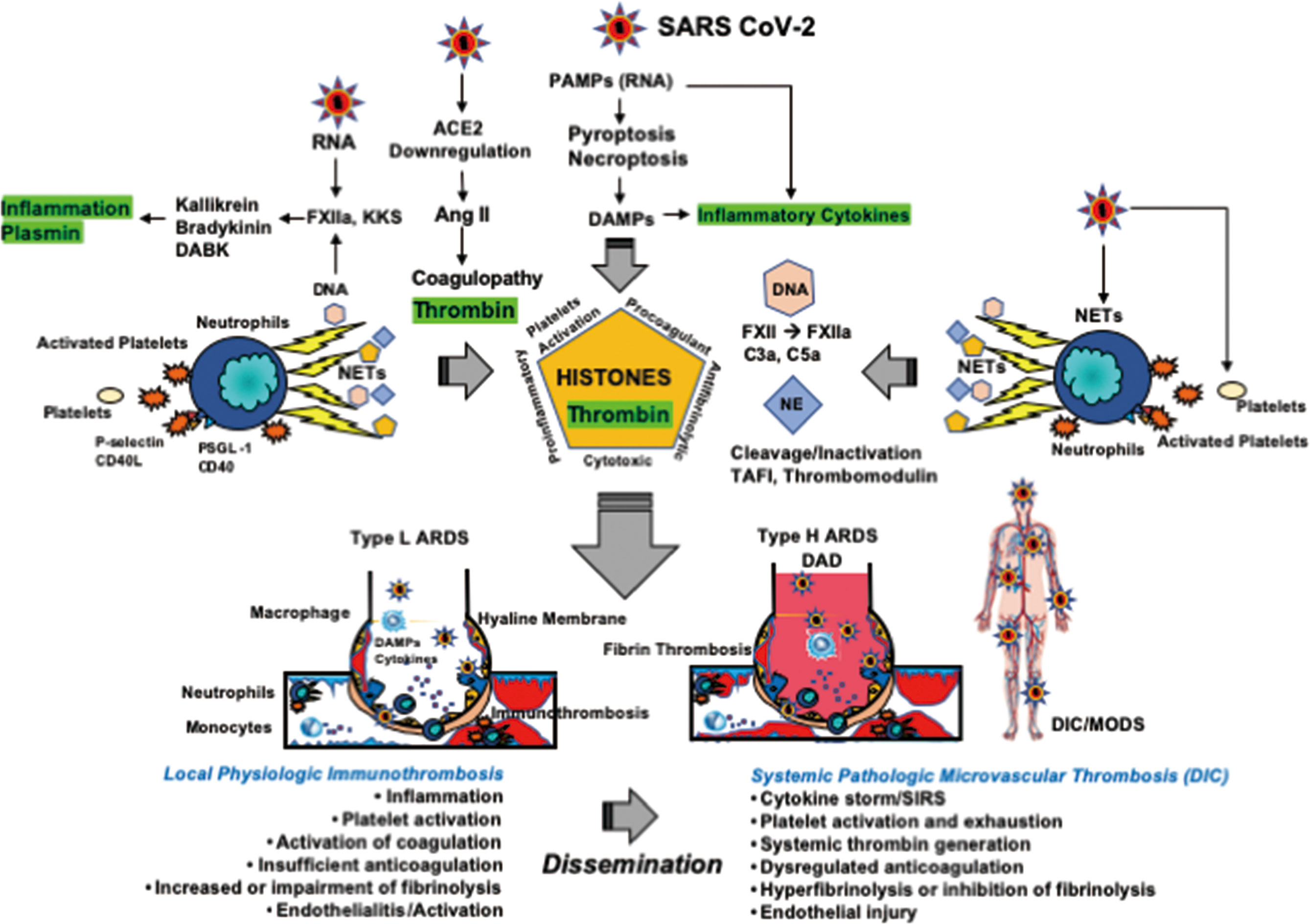
Figure 5 Thromboplasminflammation in COVID-19 coagulopathy and ARDS. NETs consisting of neutrophil DNA, histones, and elastase play a central role in immunothrombosis at the site of infection. DNA activates FXII to form FXIIa followed by activations of the KKS and complement pathways. Histones induce platelet activation and enhance coagulation through tissue factor expression and reduction in endogenous anticoagulants. Histones also have antifibrinolytic properties. Histones elicit inflammation by releasing TNF-α, IL-1, and IL-6 from storage pools and through NF-κB and inflammasome activations. Histones also show direct cytotoxic actions by changing intracellular Ca2+ load. Neutrophil elastase contributes to insufficient anticoagulation via the cleavage of TFPI and endothelial thrombomodulin. Therefore, the results of NETs releases are inflammation, activation of coagulation, insufficient anticoagulation, and impairment of fibrinolysis, which forms immunothrombosis at the site of infection. SARS-CoV-2 primarily targets the lungs and are entrapped by immunothrombosis, leading to type L ARDS; namely, pneumonia associated with VA/Q mismatch-induced hypoxemia with low elastance, also be called pulmonary intravascular coagulation. If the SARS-CoV-2 viral load is critical, immunothrombosis disseminates to the systemic circulation and DIC associated with SIRS ensues. At this stage, the lungs develop to type H ARDS showing diffuse alveolar damage associated with alveolar and intravascular fibrin microthrombosis, which give rise to right-to-left shunt-induced severe hypoxemia with high elastance. Mutual interactions between DIC and ARDS have been well acknowledged, which contribute to MODS. Of note, DIC is usually thrombotic due to thrombin generation and PAI-1 expression. Therefore, stimulation and inhibition of fibrinolysis are both dependent on the balance between t-PA/plasmin and PAI-1. ACE, angiotensin-converting enzyme; Ang, angiotensin; ARDS, acute respiratory distress syndrome; DABK, des-Arg9-bradykinin; DAD, diffuse alveolar damage; DAMPs, damage-associated molecular patterns; DIC, disseminated intravascular coagulation; KKS, kallikrein and kinin system; NE, neutrophil elastase; NETs, neutrophil extracellular traps; NF-κB, nuclear factor κB; MODS, multiple organ dysfunction syndrome; PAI-1, plasminogen activator inhibitor-1; PAMPs, pathogen-associated molecular patterns; PSGL-1, P-selectin glycoprotein ligand-1; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; SIRS, systemic inflammatory response syndrome; TAFI, thrombin-activatable fibrinolysis inhibitor; t-PA, tissue-type plasminogen activator.
Management
Monitoring
Systemic inflammation due to proinflammatory cytokines is monitored by SIRS criteria, which discriminate 87.9% of severe sepsis patients with poor prognosis, and the number of SIRS criteria met showed proportional increases in adjusted odds of death (65). The recommended monitoring includes platelet counts, prothrombin time, fibrinogen, D-dimer, and FDP levels (7). Low platelet counts and high fibrinogen and D-dimer levels were associated with the severity of COVID-19, while prothrombin time and activated partial thromboplastin time showed no correlation with severity risk, which may be explained by increased FVIII and fibrinogen levels (8). Non-survivors in the late phase of COVID-19 showed a prominent reduction in fibrinogen associated with marked prolongation of prothrombin time, high levels of FDP, and D-dimer (6). Extremely high FDP/D-dimer ratios associated with DIC in non-survivors suggest systemic pathologic fibrin(ogen)olysis, which indicates the need for monitoring of the FDP, FDP/D-dimer ratio, and DIC score in critically ill COVID-19 coagulopathy (6). Specific and sensitive criteria should be used properly for DIC diagnosis, keeping in mind the bias in fibrinogen and prothrombin time (129, 135).
Treatment
The first systematic review concluded that new evidence on thromboembolism in COVID-19 does not warrant a change in the present strategies on thromboprophylaxis with either unfractionated heparin or low molecular weight heparin among hospitalized patients (136, 137). For therapeutic anticoagulation, unfractionated heparin, low molecular weight heparin, or direct oral anticoagulants are recommended with a variety of regimens (136). Systematic review and meta-analyses have led to proposal of anticoagulation for prevention and therapeutic management of thrombosis in COVID-19 (138). During our analyses of published data, eight sets of guidance or guidelines have been published by a variety of societies including the World Health Organization. In addition, one multicenter international prospective registration, and four prospective randomized controlled studies have been published (139, 140). The data registry curating the largest population datasets suggests increased bleeding risk among the general population, and better results using anticoagulation therapy in those with respiratory failure requiring invasive ventilation (140). However, robust guidelines for anticoagulation in clinical practice are still needed.
From the viewpoint of three pathophysiologies of COVID-19 coagulopathy, a theoretical rationale exists in antithrombin and recombinant human thrombomodulin for COVID-19 coagulopathy. Both drugs can control inflammation. In addition, antithrombin inhibits FXIIa, FXIa, FXa, FIXa, FVIIa, thrombin, kallikrein, and C1s in the complement system (135). The target proteases of recombinant human thrombomodulin are thrombin via inactivation of FVa and FVIIIa. Furthermore, recombinant human thrombomodulin inhibits plasmin through activated thrombin-activatable fibrinolysis inhibitor, which further inactivates bradykinin, C3a, and C5a (10, 124, 127, 141). The binding of recombinant human thrombomodulin to histones protected platelet aggregation, fibrin thrombosis, and organ dysfunction in histone-induced DIC (124). The protease inhibitor nafamostat mesylate potently inhibits TMPRSS2, blocking spike glycoprotein-mediated fusion of the virus (142). Nafamostat mesylate is also able to inhibit other proteases such as thrombin and plasmin. Controlling plasmin is important because plasmin enhances the infectivity of SARS-CoV-2 by cleaving to the spike glycoprotein (143). All three drugs, locally approved for the treatment of DIC, may be useful for COVID-19 coagulopathy. The other promising drugs are those targeting histones and NETs, such as an anti-histone antibody, DNAse, non-anticoagulant heparin, and peptidylarginine deaminase 4 inhibitor (120). Lastly, ACE2 is a potential therapeutic target for COVID-19 coagulopathy (144).
Thromboplasminflammation
COVID-19 coagulopathy is a disease of thromboplasminflammation consisting of Ang II-induced coagulopathy, hyperfibrinolysis by FXIIa and KKS, and DIC, all of which are associated with inflammation. Therefore, the main pathomechanisms of COVID-19 comprise overproduction of both thrombin and plasmin associated with inflammation. This could be termed thromboplasminflammation, which we define as coexistence of dysregulated thrombin generation with inflammation and systemic pathological plasmin generation in an insult. Although mechanisms are different from COVID-19 coagulopathy, coexistence of dysregulated thrombin generation and systemic pathological plasmin generation associated with fibrin(ogen)olysis has been acknowledged from the past (145).
Immunothrombosis, local thrombin generation associated with inflammation, is physiological thrombus formation at the site of infection (lungs in the case of SARS-CoV-2 infection) to kill microorganisms and inhibit their spread, in addition to impairing release of PAMPs and DAMPs, with limited host damage (70). If the infection is sufficiently severe, local immunothrombosis disseminate into systemic circulation, leading to the pathological condition called thromboinflammation, in which bidirectional interplay between thrombin and inflammation play pivotal roles (70, 122, 141). COVID-19 coagulopathy meets the definition of thromboinflammation; in addition, pathological plasmin plays important roles in COVID-19 coagulopathy. In other words, thromboinflammation and pathological plasmin generation coexist in thromboplasminflammation in COVID-19 coagulopathy.
Open Questions and Future Perspective
A major problem of many studies on COVID-19 coagulopathy is the lack of appropriate time sequence from the onset to the death and insufficient evaluation of disease severity using objective scores such as acute physiology and chronic health evaluation or sequential organ failure assessment. This review showed that Ang II-induced coagulopathy, hyperfibrinolysis, and DIC are three major pathomechanisms of COVID-19 coagulopathy; however, at what stage (early or late) and disease severity (mild or severe), the disease causes these three conditions – and if it is simultaneous or separate - could not be elucidated. In the late stage of severe COVID-19 coagulopathy, many confounders such as sepsis may affect the coagulopathy.
Trauma-, sepsis-, and ischemia-induced coagulopathy is an innate immune and inflammatory responses to these insults. If the insults are sufficiently severe, dysregulated inflammatory and coagulofibrinolytic response, namely DIC, ensues. The main differences in COVID-19 coagulopathy are considered to be a SARS-CoV-2-induced deterioration in the RAAS and KKS systems and direct SARS-CoV-2 infection to endothelial cells, both of which give rise to thrombosis associated with endothelialitis-induced activation and injury of the endothelium. However, clear molecular differences in the pathophysiologies of coagulopathy between viral and bacterial infections, between COVID-19 coagulopathy and other established coagulopathies of sepsis, trauma, and ischemia-reperfusion, and differences in coagulopathy within coronavirus family remain to be clarified. Although endothelialitis, endothelial activation and injury have been confirmed in COVID-19 coagulopathy, very few studies showed dynamics of anticoagulation or antifibrinolysis molecules such as antithrombin, protein C, protein S, tissue factor pathway inhibitor, and α2-plasmin inhibitor, which need to be investigated. Lastly, appropriateness of fibrinolytic and antifibrinolytic therapies for COVID-19 is still unknown. Based on the elucidation of clear molecular mechanisms of COVID-19 coagulopathy, further advances are required to establish robust treatment strategies for COVID-19 coagulopathy.
Conclusions
The three pathomechanisms of COVID-19 coagulopathy are Ang II-induced coagulopathy, hyperfibrinolysis due to FXII and KKS activation, and DIC, which elicit thromboplasminflammation, leading to systemic inflammation, activation of coagulation and fibrinolysis associated with organ dysfunction, bleeding, and poor outcome (Figure 5). Controlling thrombin, plasmin, and inflammation are key to improving COVID-19 coagulopathy.
Author Contributions
SG and TW equally contributed to this review. SG and TW planned, cowrote, improved, revised, and approved this review. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by JSPS KAKENHI (Grant-in-Aid (C) 2020, 20K09280).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Glossary

References
1. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A Pneumonia Outbreak Associated With a New Coronavirus of Probable Bat Origin. Nature (2020) 579:270–3. doi :10.1038/s41586-020-2012-7
2. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-Converting Enzyme 2 Is a Functional Receptor for the SARS Coronavirus. Nature (2003) 426:450–4. doi: 10.1038/nature02145
3. Wadman M, Couzin-Frankel J, Kaiser J, Matacic C. A Rampage Through the Body. Science (2020) 368:356–60. doi: 10.1126/science.368.6489.356
4. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients With COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
5. Liao D, Zhou F, Luo L, Xu M, Wang H, Xia J, et al. Haematological Characteristics and Risk Factors in the Classification and Prognosis Evaluation of COVID-19: A Retrospective Cohort Study. Lancet Haematol (2020) 7:e671–8. doi: 10.1016/S2352-3026(20)30217-9
6. Tang N, Li D, Wang X, Sun Z. Abnormal Coagulation Parameters Are Associated With Poor Prognosis in Patients With Novel Coronavirus Pneumonia. J Thromb Haemost (2020) 18:844–7. doi: 10.1111/jth.14768
7. Thachil J, Tang N, Gando S, Falanga A, Cattaneo M, Levi M, et al. ISTH Interim Guidance on Recognition and Management of Coagulopathy in COVID-19. J Thromb Haemost (2020) 18:1023–6. doi: 10.1111/jth.14810
8. Lin J, Yan H, Chen H, He C, Lin C, He H, et al. Covid-19 and Coagulation Dysfunction in Adults: A Systematic Review and Meta-Analysis. J Med Virol (2021) 93:934–44. doi: 10.1002/jmv.26346
9. Al-Samkari H, Karp Leaf RS, Dzik WH, Carlson JC, Fogerty AE, Waheed A, et al. COVID and Coagulation: Bleeding and Thrombotic Manifestations of SARS-CoV2 Infection. Blood (2020) 136:489–500. doi: 10.1182/blood.2020006520
10. Gando S, Levi M, Toh CH. Disseminated Intravascular Coagulation. Nat Rev Dis Primers (2016) 2:16037. doi: 10.1038/nrdp.2016.37
11. Levi M, Hunt BJ. Thrombosis and Coagulopathy in COVID-19: An Illustrated Review. Res Pract Thromb Haemost (2020) 4:744–51. doi: 10.1002/rth2.12400
12. Moore JB, June CH. Cytokine Release Syndrome in Severe COVID-19. Science (2020) 368:473–4. doi: 10.1126/science.abb8925
13. Gralinski LE, Baric RS. Molecular Pathology of Emerging Coronavirus Infections. J Pathol (2015) 235:185–95. doi: 10.1002/path.4454
14. Giannis D, Ziogas IA, Gianni P. Coagulation Disorders in Coronavirus Infected Patients: COVID-19, SARS-Cov-1, MERS-CoV and Lessons From the Past. J Clin Virol (2020) 127:104362. doi: 10.1016/j.jcv.2020.104362
15. Goeijenbier M, van Wissen M, van de Weg C, Jong E, Gerdes VE, Meijers JC, et al. Review: Viral Infections and Mechanisms of Thrombosis and Bleeding. J Med Virol (2012) 84:1680–96. doi: 10.1002/jmv.23354
16. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-Cov-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181:271–280.e278. doi :10.1016/j.cell.2020.02.052
17. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell (2020) 181:281–92.e286. doi :10.1016/j.cell.2020.02.058
18. Verdecchia P, Cavallini C, Spanevello A, Angeli F. The Pivotal Link Between ACE2 Deficiency and SARS-CoV-2 Infection. Eur J Intern Med (2020) 76:14–20. doi: 10.1016/j.ejim.2020.04.037
19. Chung MK, Karnik S, Saef J, Bergmann C, Barnard J, Lederman MM, et al. Sars-CoV-2 and ACE2: The Biology and Clinical Data Settling the ARB and ACEI Controversy. EBioMedicine (2020) 58:102907. doi: 10.1016/j.ebiom.2020.102907
20. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J Pathol (2004) 203:631–7. doi: 10.1002/path.1570
21. Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and Biochemical Indexes From 2019-nCoV Infected Patients Linked to Viral Loads and Lung Injury. Sci China Life Sci (2020) 63:364–74. doi: 10.1007/s11427-020-1643-8
22. Patel VB, Clarke N, Wang Z, Fan D, Parajuli N, Basu R, et al. Angiotensin II Induced Proteolytic Cleavage of Myocardial ACE2 Is Mediated by TACE/ADAM-17: A Positive Feedback Mechanism in the RAS. J Mol Cell Cardiol (2014) 66:167–76. doi: 10.1016/j.yjmcc.2013.11.017
23. Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pöhlmann S. TMPRSS2 and ADAM17 Cleave ACE2 Differentially and Only Proteolysis by TMPRSS2 Augments Entry Driven by the Severe Acute Respiratory Syndrome Coronavirus Spike Protein. J Virol (2014) 88:1293–307. doi: 10.1128/JVI.02202-13
24. de Lang A, Osterhaus AD, Haagmans BL. Interferon-Gamma and Interleukin-4 Downregulate Expression of the SARS Coronavirus Receptor ACE2 in Vero E6 Cells. Virology (2006) 353:474–81. doi: 10.1016/j.virol.2006.06.011
25. Glowacka I, Bertram S, Herzog P, Pfefferle S, Steffen I, Muench MO, et al. Differential Downregulation of ACE2 by the Spike Proteins of Severe Acute Respiratory Syndrome Coronavirus and Human Coronavirus NL63. J Virol (2010) 84:1198–205. doi: 10.1128/JVI.01248-09
26. Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 Receptor ACE2 is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets Across Tissues. Cell (2020) 181:1016–35.e1019. doi :10.1016/j.cell.2020.04.035
27. Onabajo OO, Banday AR, Stanifer ML, Yan W, Obajemu A, Santer DM, et al. Interferons and Viruses Induce a Novel Truncated ACE2 Isoform and Not the Full-Length SARS-CoV-2 Receptor. Nat Genet (2020) 52:1283–93. doi: 10.1038/s41588-020-00731-9
28. Dijkman R, Jebbink MF, Deijs M, Milewska A, Pyrc K, Buelow E, et al. Replication-Dependent Downregulation of Cellular Angiotensin-Converting Enzyme 2 Protein Expression by Human Coronavirus NL63. J Gen Virol (2012) 93:1924–9. doi: 10.1099/vir.0.043919-0
29. Amarante-Mendes GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R, Bortoluci KR. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front Immunol (2018) 9:2379. doi: 10.3389/fimmu.2018.02379
30. Frank D, Vince JE. Pyroptosis Versus Necroptosis: Similarities, Differences, and Crosstalk. Cell Death Differ (2019) 26:99–114. doi: 10.1038/s41418-018-0212-6
31. Schwarzer R, Laurien L, Pasparakis M. New Insights Into the Regulation of Apoptosis, Necroptosis, and Pyroptosis by Receptor Interacting Protein Kinase 1 and Caspase-8. Curr Opin Cell Biol (2020) 63:186–93. doi: 10.1016/j.ceb.2020.02.004
32. Khoury MK, Gupta K, Franco SR, Liu B. Necroptosis in the Pathophysiology of Disease. Am J Pathol (2020) 190:272–85. doi: 10.1016/j.ajpath.2019.10.012
33. de Rivero Vaccari JC, Dietrich WD, Keane RW, de Rivero Vaccari JP. The Inflammasome in Times of COVID-19. Front Immunol (2020) 11:583373. doi: 10.3389/fimmu.2020.583373
34. Zhao M, Bai M, Ding G, Zhang Y, Huang S, Jia Z, et al. Angiotensin II Stimulates the NLRP3 Inflammasome to Induce Podocyte Injury and Mitochondrial Dysfunction. Kidney Dis (Basel) (2018) 4:83–94. doi: 10.1159/000488242
35. Ratajczak MZ, Kucia M. Sars-CoV-2 Infection and Overactivation of Nlrp3 Inflammasome as a Trigger of Cytokine “Storm” and Risk Factor for Damage of Hematopoietic Stem Cells. Leukemia (2020) 34:1726–9. doi: 10.1038/s41375-020-0887-9
36. Qiao J, Wu X, Luo Q, Wei G, Xu M, Wu Y, et al. NLRP3 Regulates Platelet Integrin αiibβ3 Outside-in Signaling, Hemostasis and Arterial Thrombosis. Haematologica (2018) 103:1568–76. doi: 10.3324/haematol.2018.191700
37. Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome Activation Triggers Blood Clotting and Host Death Through Pyroptosis. Immunity (2019) 50:1401–11.e1404. doi :10.1016/j.immuni.2019.04.003
38. Jorgensen I, Miao EA. Pyroptotic Cell Death Defends Against Intracellular Pathogens. Immunol Rev (2015) 265:130–42. doi: 10.1111/imr.12287
39. Jensen S, Thomsen AR. Sensing of RNA Viruses: A Review of Innate Immune Receptors Involved in Recognizing RNA Virus Invasion. J Virol (2012) 86:2900–10. doi: 10.1128/JVI.05738-11
40. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
41. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of Immune Response in Patients With COVID-19 in Wuhan, China. Clin Infect Dis (2020) 71:762–8. doi: 10.1093/cid/ciaa248
42. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the Eye of the Cytokine Storm. Microbiol Mol Biol Rev (2012) 76:16–32. doi: 10.1128/MMBR.05015-11
43. Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, et al. Endothelial Cells in Physiology and in the Pathophysiology of Vascular Disorders. Blood (1998) 91:3527–61.
44. Fuchs TA, Bhandari AA, Wagner DD. Histones Induce Rapid and Profound Thrombocytopenia in Mice. Blood (2011) 118:3708–14. doi: 10.1182/blood-2011-01-332676
45. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular Histones Promote Thrombin Generation Through Platelet-Dependent Mechanisms: Involvement of Platelet TLR2 and TLR4. Blood (2011) 118:1952–61. doi: 10.1182/blood-2011-03-343061
46. Assinger A. Platelets and Infection - An Emerging Role of Platelets in Viral Infection. Front Immunol (2014) 5:649. doi: 10.3389/fimmu.2014.00649
47. Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, et al. The Role of Platelets in Mediating a Response to Human Influenza Infection. Nat Commun (2019) 10:1780. doi: 10.1038/s41467-019-09607-x
48. Semple JW, Italiano JE Jr., Freedman J. Platelets and the Immune Continuum. Nat Rev Immunol (2011) 11:264–74. doi: 10.1038/nri2956
49. Jenne CN, Kubes P. Platelets in Inflammation and Infection. Platelets (2015) 26:286–92. doi: 10.3109/09537104.2015.1010441
50. Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19-Associated Coagulopathy: Evidence From a Single-Centre, Cross-Sectional Study. Lancet Haematol (2020) 7:e575–82. doi: 10.1016/S2352-3026(20)30216-7
51. Yang X, Yang Q, Wang Y, Wu Y, Xu J, Yu Y, et al. Thrombocytopenia and Its Association With Mortality in Patients With COVID-19. J Thromb Haemost (2020) 18:1469–72. doi: 10.1111/jth.14848
52. Lippi G, Plebani M, Henry BM. Thrombocytopenia Is Associated With Severe Coronavirus Disease 2019 (COVID-19) Infections: A Meta-Analysis. Clin Chim Acta (2020) 506:145–8. doi: 10.1016/j.cca.2020.03.022
53. Schönrich G, Raftery MJ. Neutrophil Extracellular Traps Go Viral. Front Immunol (2016) 7:366. doi: 10.3389/fimmu.2016.00366
54. Nicolai L, Leunig A, Brambs S, Kaiser R, Weinberger T, Weigand M, et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation (2020) 142:1176–89. doi: 10.1161/CIRCULATIONAHA.120.048488
55. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil Extracellular Traps in COVID-19. JCI Insight (2020) 5(11):e138999. doi: 10.1172/jci.insight.138999
56. Leppkes M, Knopf J, Naschberger E, Lindemann A, Singh J, Herrmann I, et al. Vascular Occlusion by Neutrophil Extracellular Traps in COVID-19. EBioMedicine (2020) 58:102925. doi: 10.1016/j.ebiom.2020.102925
57. McGill SN, Ahmed NA, Christou NV. Endothelial Cells: Role in Infection and Inflammation. World J Surg (1998) 22:171–8. doi: 10.1007/s002689900366
58. Bevilacqua MP. Endothelial-Leukocyte Adhesion Molecules. Annu Rev Immunol (1993) 11:767–804. doi: 10.1146/annurev.iy.11.040193.004003
59. Weiss SJ. Tissue Destruction by Neutrophils. N Engl J Med (1989) 320:365–76. doi: 10.1056/NEJM198902093200606
60. Aird WC. Endothelial Cell Dynamics and Complexity Theory. Crit Care Med (2002) 30:S180–185. doi: 10.1097/00003246-200205001-00002
61. Cheng Z, Abrams ST, Alhamdi Y, Toh J, Yu W, Wang G, et al. Circulating Histones Are Major Mediators of Multiple Organ Dysfunction Syndrome in Acute Critical Illnesses. Crit Care Med (2019) 47:e677–84. doi: 10.1097/CCM.0000000000003839
62. Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating Histones Are Mediators of Trauma-Associated Lung Injury. Am J Respir Crit Care Med (2013) 187:160–9. doi: 10.1164/rccm.201206-1037OC
63. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
64. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med (2020) 382:1708–20. doi: 10.1056/NEJMoa2002032
65. Kaukonen KM, Bailey M, Pilcher D, Cooper DJ, Bellomo R. Systemic Inflammatory Response Syndrome Criteria in Defining Severe Sepsis. N Engl J Med (2015) 372:1629–38. doi: 10.1056/NEJMoa1415236
66. Al-Ani F, Chehade S, Lazo-Langner A. Thrombosis Risk Associated With COVID-19 Infection. A Scoping Review Thromb Res (2020) 192:152–60. doi: 10.1016/j.thromres.2020.05.039
67. Schurink B, Roos E, Radonic T, Barbe E, Bouman CSC, de Boer HH, et al. Viral Presence and Immunopathology in Patients With Lethal COVID-19: A Prospective Autopsy Cohort Study. Lancet Microbe (2020) 1:e290–299. doi: 10.1016/S2666-5247(20)30144-0
68. Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, Vander Heide RS. Pulmonary and Cardiac Pathology in African American Patients With COVID-19: An Autopsy Series From New Orleans. Lancet Respir Med (2020) 8:681–6. doi: 10.1016/S2213-2600(20)30243-5
69. Gattinoni L, Chiumello D, Caironi P, Busana M, Romitti F, Brazzi L, et al. Covid-19 Pneumonia: Different Respiratory Treatments for Different Phenotypes? Intensive Care Med (2020) 46:1099–102. doi: 10.1007/s00134-020-06033-2
70. Engelmann B, Massberg S. Thrombosis as an Intravascular Effector of Innate Immunity. Nat Rev Immunol (2013) 13:34–45. doi: 10.1038/nri3345
71. Welty-Wolf KE, Carraway MS, Ortel TL, Piantadosi CA. Coagulation and Inflammation in Acute Lung Injury. Thromb Haemost (2002) 88:17–25. doi: 10.1055/s-0037-1613147
72. McGonagle D, O’Donnell JS, Sharif K, Emery P, Bridgewood C. Immune Mechanisms of Pulmonary Intravascular Coagulopathy in COVID-19 Pneumonia. Lancet Rheumatol (2020) 2:e437–45. doi: 10.1016/S2665-9913(20)30121-1
73. Shah A, Donovan K, McHugh A, Pandey M, Aaron L, Bradbury CA, et al. Thrombotic and Haemorrhagic Complications in Critically Ill Patients With COVID-19: A Multicentre Observational Study. Crit Care (2020) 24:561. doi: 10.1186/s13054-020-03260-3
74. Fraissé M, Logre E, Pajot O, Mentec H, Plantefève G, Contou D. Thrombotic and Hemorrhagic Events in Critically Ill COVID-19 Patients: A French Monocenter Retrospective Study. Crit Care (2020) 24:275. doi: 10.1186/s13054-020-03025-y
75. Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, Eichinger S. Thrombotic Thrombocytopenia After ChAdOx1 nCov-19 Vaccination. N Engl J Med (2021) 384(22):2092–101. doi: 10.1056/NEJMoa2104840
76. See I, Su JR, Lale A, Woo EJ, Guh AY, Shimabukuro TT, et al. US Case Reports of Cerebral Venous Sinus Thrombosis With Thrombocytopenia After Ad26.Cov2.s Vaccination, March 2 to April 21, 2021. JAMA (2021) 30:e217517. doi: 10.1001/jama.2021.7517
77. Greinacher A, Selleng K, Warkentin TE. Autoimmune Heparin-Induced Thrombocytopenia. J Thromb Haemost (2017) 15:2099–114. doi: 10.1111/jth.13813
78. Bermejo-Martin JF, González-Rivera M, Almansa R, Micheloud D, Tedim AP, Domínguez-Gil M, et al. Viral RNA Load in Plasma Is Associated With Critical Illness and a Dysregulated Host Response in COVID-19. Crit Care (2020) 24:691. doi: 10.1186/s13054-020-03398-0
79. Peng L, Liu J, Xu W, Luo Q, Chen D, Lei Z, et al. Sars-CoV-2 Can be Detected in Urine, Blood, Anal Swabs, and Oropharyngeal Swabs Specimens. J Med Virol (2020) 92:1676–380. doi: 10.1002/jmv.25936
80. Miesbach W. Pathological Role of Angiotensin II in Severe Covid-19. TH Open (2020) 4:e138–44. doi: 10.1055/s-0040-1713678
81. Crabos M, Bertschin S, Bühler FR, Rogg H, Evéquoz D, Eberhard M, et al. Identification of AT1 Receptors on Human Platelets and Decreased Angiotensin II Binding in Hypertension. J Hypertens (1993) Suppl 11:S230–231. doi: 10.1097/00004872-199312050-00094
82. Ding YA, MacIntyre DE, Kenyon CJ, Semple PF. Potentiation of Adrenaline-Induced Platelet Aggregation by Angiotensin II. Thromb Haemost (1985) 54:717–20. doi: 10.1055/s-0038-1660105
83. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. Sars-CoV-2 Binds Platelet ACE2 to Enhance Thrombosis in COVID-19. J Hematol Oncol (2020) 13:120. doi: 10.1186/s13045-020-00954-7
84. Larsson PT, Schwieler JH, Wallén NH. Platelet Activation During Angiotensin II Infusion in Healthy Volunteers. Blood Coagul Fibrinolysis (2000) 11:61–9. doi: 10.1097/00001721-200011010-00007
85. Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, et al. Platelets Can Associate With SARS-Cov-2 RNA and Are Hyperactivated in COVID-19. Circ Res (2020) 127:1404–18. doi: 10.1161/CIRCRESAHA.120.317703
86. He M, He X, Xie Q, Chen F, He S. Angiotensin II Induces the Expression of Tissue Factor and Its Mechanism in Human Monocytes. Thromb Res (2006) 117:579–90. doi: 10.1016/j.thromres.2005.04.033
87. Nishimura H, Tsuji H, Masuda H, Kasahara T, Yoshizumi M, Sugano T, et al. The Effects of Angiotensin Metabolites on the Regulation of Coagulation and Fibrinolysis in Cultured Rat Aortic Endothelial Cells. Thromb Haemost (1999) 82:1516–21. doi: 10.1055/s-0037-1614864
88. Taubman MB, Marmur JD, Rosenfield CL, Guha A, Nichtberger S, Nemerson Y. Agonist-Mediated Tissue Factor Expression in Cultured Vascular Smooth Muscle Cells. Role of Ca2+ Mobilization and Protein Kinase C Activation. J Clin Invest (1993) 91:547–52. doi: 10.1172/JCI116234
89. Kamińska M, Mogielnicki A, Stankiewicz A, Kramkowski K, Domaniewski T, Buczko W, et al. Angiotensin II Via AT1 Receptor Accelerates Arterial Thrombosis in Renovascular Hypertensive Rats. J Physiol Pharmacol (2005) 56:571–85.
90. Mogielnicki A, Chabielska E, Pawlak R, Szemraj J, Buczko W. Angiotensin II Enhances Thrombosis Development in Renovascular Hypertensive Rats. Thromb Haemost (2005) 93:1069–76. doi: 10.1160/TH04-10-0701
91. Fraga-Silva RA, Sorg BS, Wankhede M, Dedeugd C, Jun JY, Baker MB, et al. ACE2 Activation Promotes Antithrombotic Activity. Mol Med (2010) 16:210–5. doi: 10.2119/molmed.2009.00160
92. Vaughan DE, Lazos SA, Tong K. Angiotensin II Regulates the Expression of Plasminogen Activator Inhibitor-1 in Cultured Endothelial Cells. A Potential Link Between the Renin-Angiotensin System and Thrombosis. J Clin Invest (1995) 95:995–1001. doi: 10.1172/JCI117809
93. Nakamura S, Nakamura I, Ma L, Vaughan DE, Fogo AB. Plasminogen Activator Inhibitor-1 Expression Is Regulated by the Angiotensin Type 1 Receptor In Vivo. Kidney Int (2000) 58:251–9. doi: 10.1046/j.1523-1755.2000.00160.x
94. Ridker PM, Gaboury CL, Conlin PR, Seely EW, Williams GH, Vaughan DE. Stimulation of Plasminogen Activator Inhibitor In Vivo by Infusion of Angiotensin II. Evidence of a Potential Interaction Between the Renin-Angiotensin System and Fibrinolytic Function. Circulation (1993) 87:1969–73. doi: 10.1161/01.CIR.87.6.1969
95. Tummala PE, Chen XL, Sundell CL, Laursen JB, Hammes CP, Alexander RW, et al. Angiotensin II Induces Vascular Cell Adhesion Molecule-1 Expression in Rat Vasculature: A Potential Link Between the Renin-Angiotensin System and Atherosclerosis. Circulation (1999) 100:1223–9. doi: 10.1161/01.CIR.100.11.1223
96. Pastore L, Tessitore A, Martinotti S, Toniato E, Alesse E, Bravi MC, et al. Angiotensin II Stimulates Intercellular Adhesion Molecule-1 (ICAM-1) Expression by Human Vascular Endothelial Cells and Increases Soluble ICAM-1 Release In Vivo. Circulation (1999) 100:1646–52. doi: 10.1161/01.CIR.100.15.1646
97. Key NS, Vercellotti GM, Winkelmann JC, Moldow CF, Goodman JL, Esmon NL, et al. Infection of Vascular Endothelial Cells With Herpes Simplex Virus Enhances Tissue Factor Activity and Reduces Thrombomodulin Expression. Proc Natl Acad Sci USA (1990) 87:7095–9. doi: 10.1073/pnas.87.18.7095
98. Oehmcke-Hecht S, Köhler J. Interaction of the Human Contact System With Pathogens-An Update. Front Immunol (2018) 9:312. doi: 10.3389/fimmu.2018.00312
99. Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, et al. Extracellular RNA Constitutes a Natural Procoagulant Cofactor in Blood Coagulation. Proc Natl Acad Sci USA (2007) 104:6388–93. doi: 10.1073/pnas.0608647104
100. Oehmcke S, Mörgelin M, Herwald H. Activation of the Human Contact System on Neutrophil Extracellular Traps. J Innate Immun (2009) 1:225–30. doi: 10.1159/000203700
101. Meini S, Zanichelli A, Sbrojavacca R, Iuri F, Roberts AT, Suffritti C, et al. Understanding the Pathophysiology of COVID-19: Could the Contact System Be the Key? Front Immunol (2020) 11:2014. doi: 10.3389/fimmu.2020.02014
102. Clotet-Freixas S, Soler MJ, Palau V, Anguiano L, Gimeno J, Konvalinka A, et al. Sex Dimorphism in ANGII-Mediated Crosstalk Between ACE2 and ACE in Diabetic Nephropathy. Lab Invest (2018) 98:1237–49. doi: 10.1038/s41374-018-0084-x
103. Goldsmith GH Jr., Saito H, Ratnoff OS. The Activation of Plasminogen by Hageman Factor (Factor XII) and Hageman Factor Fragments. J Clin Invest (1978) 62:54–60. doi: 10.1172/JCI109113
104. Braat EA, Dooijewaard G, Rijken DC. Fibrinolytic Properties of Activated FXII. Eur J Biochem (1999) 263:904–11. doi: 10.1046/j.1432-1327.1999.00593.x
105. Kaplan AP, Austen KF. A Prealbumin Activator of Prekallikrein. II. Derivation of Activators of Prekallikrein From Active Hageman Factor by Digestion With Plasmin. J Exp Med (1971) 133:696–712. doi: 10.1084/jem.133.4.696
106. Tanaka A, Suzuki Y, Sugihara K, Kanayama N, Urano T. Inactivation of Plasminogen Activator Inhibitor Type 1 by Activated Factor XII Plays a Role in the Enhancement of Fibrinolysis by Contact Factors In-Vitro. Life Sci (2009) 85:220–5. doi: 10.1016/j.lfs.2009.05.014
107. Colarusso C, Terlizzi M, Pinto A, Sorrentino R. A Lesson From a Saboteur: High-MW Kininogen Impact in Coronavirus-Induced Disease 2019. Br J Pharmacol (2020) 177(21):4866–72. doi: 10.1111/bph.15154
108. Brown NJ, Nadeau JH, Vaughan DE. Selective Stimulation of Tissue-Type Plasminogen Activator (t-PA) In Vivo by Infusion of Bradykinin. Thromb Haemost (1997) 77:522–5. doi: 10.1055/s-0038-1656000
109. Brown NJ, Gainer JV, Murphey LJ, Vaughan DE. Bradykinin Stimulates Tissue Plasminogen Activator Release From Human Forearm Vasculature Through B(2) Receptor-Dependent, NO Synthase-Independent, and Cyclooxygenase-Independent Pathway. Circulation (2000) 102:2190–6. doi: 10.1161/01.CIR.102.18.2190
110. Berri F, Rimmelzwaan GF, Hanss M, Albina E, Foucault-Grunenwald ML, Lê VB, et al. Plasminogen Controls Inflammation and Pathogenesis of Influenza Virus Infections Via Fibrinolysis. PloS Pathog (2013) 9:e1003229. doi: 10.1371/journal.ppat.1003229
111. Sidarta-Oliveira D, Jara CP, Ferruzzi AJ, Skaf MS, Velander WH, Araujo EP, et al. Sars-CoV-2 Receptor is Co-Expressed With Elements of the Kinin-Kallikrein, Renin-Angiotensin and Coagulation Systems in Alveolar Cells. Sci Rep (2020) 10:19522. doi: 10.1038/s41598-020-76488-2
112. Umemura Y, Yamakawa K, Kiguchi T, Nishida T, Kawada M, Fujimi S. Hematological Phenotype of COVID-19-Induced Coagulopathy: Far From Typical Sepsis-Induced Coagulopathy. J Clin Med (2020) 9:2875. doi: 10.3390/jcm9092875
113. Ranucci M, Sitzia C, Baryshnikova E, Di Dedda U, Cardani R, Martelli F, et al. Covid-19-Associated Coagulopathy: Biomarkers of Thrombin Generation and Fibrinolysis Leading the Outcome. J Clin Med (2020) 9(11):3487. doi: 10.3390/jcm9113487
114. Uaprasert N, Moonla C, Sosothikul D, Rojnuckarin P, Chiasakul T. Systemic Coagulopathy in Hospitalized Patients With Coronavirus Disease 2019: A Systematic Review and Meta-Analysis. Clin Appl Thromb Hemost (2021) 27:1076029620987629. doi: 10.1177/1076029620987629
115. Jin S, Jin Y, Xu B, Hong J, Yang X. Prevalence and Impact of Coagulation Dysfunction in COVID-19 in China: A Meta-Analysis. Thromb Haemost (2020) 120:1524–35. doi: 10.1055/s-0040-1714369
116. van der Poll T, de Jonge E, Levi M. Regulatory Role of Cytokines in Disseminated Intravascular Coagulation. Semin Thromb Hemost (2001) 27:639–51. doi: 10.1055/s-2001-18868
117. Boermeester MA, van Leeuwen PA, Coyle SM, Wolbink GJ, Hack CE, Lowry SF. Interleukin-1 Blockade Attenuates Mediator Release and Dysregulation of the Hemostatic Mechanism During Human Sepsis. Arch Surg (1995) 130:739–48. doi: 10.1001/archsurg.1995.01430070061012
118. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CRR, et al. Platelet Activation and Platelet-Monocyte Aggregate Formation Trigger Tissue Factor Expression in Patients With Severe COVID-19. Blood (2020) 136:1330–41. doi: 10.1182/blood.2020007252
119. Eslamifar Z, Behzadifard M, Soleimani M, Behzadifard S. Coagulation Abnormalities in SARS-CoV-2 Infection: Overexpression Tissue Factor. Thromb J (2020) 18:38. doi: 10.1186/s12959-020-00250-x
120. Alhamdi Y, Toh CH. Recent Advances in Pathophysiology of Disseminated Intravascular Coagulation: The Role of Circulating Histones and Neutrophil Extracellular Traps. F1000Res (2017) 6:2143. doi: 10.12688/f1000research.12498.1
121. Ito T, Nakahara M, Masuda Y, Ono S, Yamada S, Ishikura H, et al. Circulating Histone H3 Levels are Increased in Septic Mice in a Neutrophil-Dependent Manner: Preclinical Evaluation of a Novel Sandwich ELISA for Histone H3. J Intensive Care (2018) 6:79. doi: 10.1186/s40560-018-0348-y
122. Gando S, Otomo Y. Local Hemostasis, Immunothrombosis, and Systemic Disseminated Intravascular Coagulation in Trauma and Traumatic Shock. Crit Care (2015) 19:72. doi: 10.1186/s13054-015-0735-x
123. Abrams ST, Su D, Sahraoui Y, Lin Z, Cheng Z, Nesbitt K, et al. Assembly of Alternative Prothrombinase by Extracellular Histones Initiate and Disseminate Intravascular Coagulation. Blood (2021) 137:103–14. doi: 10.1182/blood.2019002973
124. Nakahara M, Ito T, Kawahara K, Yamamoto M, Nagasato T, Shrestha B, et al. Recombinant Thrombomodulin Protects Mice Against Histone-Induced Lethal Thromboembolism. PloS One (2013) 8:e75961. doi: 10.1371/journal.pone.0075961
125. Abrams ST, Morton B, Alhamdi Y, Alsabani M, Lane S, Welters ID, et al. A Novel Assay for Neutrophil Extracellular Trap Formation Independently Predicts Disseminated Intravascular Coagulation and Mortality in Critically Ill Patients. Am J Respir Crit Care Med (2019) 200:869–80. doi: 10.1164/rccm.201811-2111OC
126. Rittirsch D, Flierl MA, Ward PA. Harmful Molecular Mechanisms in Sepsis. Nat Rev Immunol (2008) 8:776–87. doi: 10.1038/nri2402
127. Gando S. Role of Fibrinolysis in Sepsis. Semin Thromb Hemost (2013) 39:392–9. doi: 10.1055/s-0033-1334140
128. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Transl Res (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
129. Taylor FB Jr., Toh CH, Hoots WK, Wada H, Levi M. Towards Definition, Clinical and Laboratory Criteria, and a Scoring System for Disseminated Intravascular Coagulation. Thromb Haemost (2001) 86:1327–30. doi: 10.1055/s-0037-1616068
130. Pandolfi L, Fossali T, Frangipane V, Bozzini S, Morosini M, D’Amato M, et al. Broncho-Alveolar Inflammation in COVID-19 Patients: A Correlation With Clinical Outcome. BMC Pulm Med (2020) 20:301. doi: 10.1186/s12890-020-01343-z
131. Dentone C, Vena A, Loconte M, Grillo F, Brunetti I, Barisione E, et al. Bronchoalveolar Lavage Fluid Characteristics and Outcomes of Invasively Mechanically Ventilated Patients With COVID-19 Pneumonia in Genoa, Italy. BMC Infect Dis (2021) 21:353. doi: 10.1186/s12879-021-06015-9
132. Satturwar S, Fowkes M, Farver C, Wilson AM, Eccher A, Girolami I, et al. Postmortem Findings Associated With SARS-Cov-2: Systematic Review and Meta-Analysis. Am J Surg Pathol (2021) 45:587–603. doi: 10.1097/PAS.0000000000001650
133. Udomsinprasert W, Jittikoon J, Sangroongruangsri S, Chaikledkaew U. Circulating Levels of Interleukin-6 and Interleukin-10, But Not Tumor Necrosis Factor-Alpha, as Potential Biomarkers of Severity and Mortality for COVID-19: Systematic Review With Meta-Analysis. J Clin Immunol (2021) 41:11–22. doi: 10.1007/s10875-020-00899-z
134. Lang CN, Dettinger JS, Berchtold-Herz M, Utzolino S, Bemtgen X, Zotzmann V, et al. Intracerebral Hemorrhage in COVID-19 Patients With Pulmonary Failure: A Propensity Score-Matched Registry Study. Neurocrit Care (2021) 1–9. doi: 10.1007/s12028-021-01202-7
135. Gando S, Saitoh D, Ogura H, Fujishima S, Mayumi T, Araki T, et al. A Multicenter, Prospective Validation Study of the Japanese Association for Acute Medicine Disseminated Intravascular Coagulation Scoring System in Patients With Severe Sepsis. Crit Care (2013) 17:R111. doi: 10.1186/cc12783
136. Flaczyk A, Rosovsky RP, Reed CT, Bankhead-Kendall BK, Bittner EA, Chang MG. Comparison of Published Guidelines for Management of Coagulopathy and Thrombosis in Critically Ill Patients With COVID 19: Implications for Clinical Practice and Future Investigations. Crit Care (2020) 24:559. doi: 10.1186/s13054-020-03273-y
137. Maldonado E, Tao D, Mackey K. Antithrombotic Therapies in COVID-19 Disease: A Systematic Review. J Gen Intern Med (2020) 35:2698–706. doi: 10.1007/s11606-020-05906-y
138. McBane RD 2nd, Torres Roldan VD, Niven AS, Pruthi RK, Franco PM, Linderbaum JA, et al. Anticoagulation in COVID-19: A Systematic Review, Meta-Analysis, and Rapid Guidance From Mayo Clinic. Mayo Clin Proc (2020) 95:2467–86. doi: 10.1016/j.mayocp.2020.08.030
139. Leentjens J, van Haaps TF, Wessels PF, Schutgens REG, Middeldorp S. Covid-19-Associated Coagulopathy and Antithrombotic Agents-Lessons After 1 Year. Lancet Haematol (2021) S2352–3026(21):00105–8. doi: 10.1016/S2352-3026(21)00105-8
140. Santoro F, Núñez-Gil IJ, Viana-Llamas MC, Maroun Eid C, Romero R, Fernández Rozas I, et al. Anticoagulation Therapy in Patients With Coronavirus Disease 2019: Results From a Multicenter International Prospective Registry (Health Outcome Predictive Evaluation for Corona Virus Disease 2019 [Hope-Covid19]). Crit Care Med (2021) 49:e624–33. doi: 10.1097/CCM.0000000000005010
141. Jackson SP, Darbousset R, Schoenwaelder SM. Thromboinflammation: Challenges of Therapeutically Targeting Coagulation and Other Host Defense Mechanisms. Blood (2019) 133:906–18. doi: 10.1182/blood-2018-11-882993
142. Yamamoto M, Kiso M, Sakai-Tagawa Y, Iwatsuki-Horimoto K, Imai M, Takeda M, et al. The Anticoagulant Nafamostat Potently Inhibits SARS-Cov-2 S Protein-Mediated Fusion in a Cell Fusion Assay System and Viral Infection In Vitro in a Cell-Type-Dependent Manner. Viruses (2020) 12:629. doi: 10.3390/v12060629
143. Ji HL, Zhao R, Matalon S, Matthay MA. Elevated Plasmin(ogen) as a Common Risk Factor for COVID-19 Susceptibility. Physiol Rev (2020) 100:1065–75. doi: 10.1152/physrev.00013.2020
144. Zoufaly A, Poglitsch M, Aberle JH, Hoepler W, Seitz T, Traugott M, et al. Human Recombinant Soluble ACE2 in Severe COVID-19. Lancet Respir Med (2020) 8:1154–8. doi: 10.1016/S2213-2600(20)30418-5
Keywords: COVID-19, inflammation, plasmin, SARS-CoV-2, thrombin
Citation: Gando S and Wada T (2021) Thromboplasminflammation in COVID-19 Coagulopathy: Three Viewpoints for Diagnostic and Therapeutic Strategies. Front. Immunol. 12:649122. doi: 10.3389/fimmu.2021.649122
Received: 03 January 2021; Accepted: 28 May 2021;
Published: 11 June 2021.
Edited by:
Hugo Caire Castro-Faria-Neto, Oswaldo Cruz Foundation (Fiocruz), BrazilReviewed by:
Kenichi Okuda, University of North Carolina at Chapel Hill, United StatesDipyaman Ganguly, Indian Institute of Chemical Biology (CSIR), India
Copyright © 2021 Gando and Wada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Satoshi Gando, Z2FuZG9pY291ZEBpY2xvdWQuY29t; Takeshi Wada, dHdhZGExQG1lZC5ob2t1ZGFpLmFjLmpw