 Nafeesa Mehmood
Nafeesa Mehmood Liying Low
Liying Low Graham R. Wallace
Graham R. Wallace
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 May 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.648341
This article is part of the Research Topic Uveitis: Immunity, Genes and Microbes View all 23 articles
Behçet’s disease (BD) is a multisystem autoinflammatory condition characterized by mucosal ulceration, breakdown of immune privilege sites and vasculitis. A genetic basis for BD has been described in genome-wide and validation studies. Similarly, dysbiosis of oral and gut microbiomes have been associated with BD. This review will describe links between genetic polymorphisms in genes encoding molecules involved in gut biology and changes seen in microbiome studies. A potential decrease in bacterial species producing short chain fatty acids linked to mutations in genes involved in their production suggests a potential therapy for BD.
Behçet’s Disease (BD) is a multisystem autoinflammatory disorder characterized by inflammation at mucosal surfaces and break down of barriers at privileged sites including the eye, brain and joint (1). Many of the mucosal surfaces involved in BD host a community of microorganisms, and collectively their genetic content is known as the microbiome. The largest and most diverse of these microbial communities reside in the gut and have been shown to be important in the development and regulation of the immune system (2). Changes in the gut microbiome, also known as dysbiosis, have been associated with many human diseases including chronic kidney disease, diabetes, multiple sclerosis (3–5). The gut microbiome has also been shown to modulate the efficacy of immunotherapies in cancer patients (6, 7).
Since the first description of the disease triad in the 1930s, infectious microbial agents have always been speculated to be involved in the pathogenesis of BD, however, so far, no single pathogenic etiology has been found. Recent studies have investigated the association between the composition of the microbiomes and BD, which may provide a useful insight to the role of the microbiome in the disease process.
The gut microbiome profile in BD was first reported by Consolandi and associates, whereby they compared the fecal microbiota of 22 patients with 16 age-matched cohabitating individuals. Recruiting healthy controls who lived in the same house as the BD patients is of particular importance as it is known that several factors contribute to variations in gut microbiota composition, including environmental factors, geographical location, diet, age and host genetics (6). There was significant reduction in the alpha diversity and depletion of butyrate-producing bacteria, such as Roseburia and Subdoligranulum genera, in the gut microbiome of BD patients compared to healthy controls (Table 1). The short chain fatty acid (SCFA) profile, by chromatography-mass spectrometry (GC-MS), revealed a significant reduction of butyrate levels in fecal samples of BD patients. Butyrate is important in regulating the host immune system and is responsible for inhibiting intestinal pro-inflammatory cytokines and promoting T cell differentiation into a T regulatory cell (Treg) lineage. Thus, it can be hypothesized that the dysbiotic microbiota seen in BD patients could have an important role in the pathogenesis of BD, whereby a reduction in butyrate causes downstream effects of promoting immune-pathological T-effector responses, leading to gut inflammation (18). Interestingly, a similar pattern is seen in patients with inflammatory bowel disease (IBD) and oral administration of butyrate in experimental IBD mice, has shown to enhance Treg differentiation and reduce colitis (8, 19).
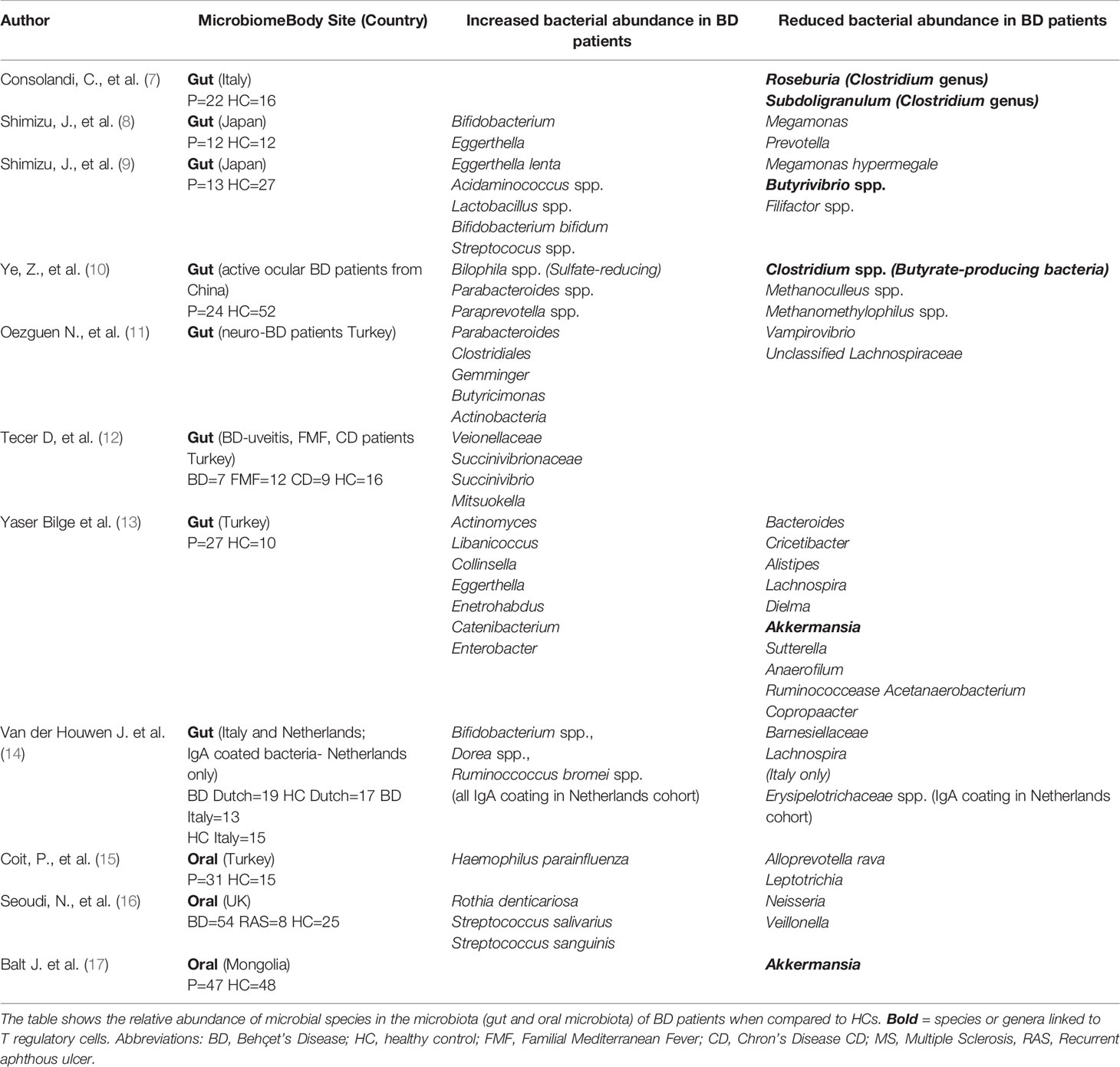
Table 1 Summary of microbiome dysbiosis in Behçet’s Disease patients compared to healthy controls or disease controls.
In a pilot 16S gut microbiome study of 12 Japanese patients with BD and 12 healthy controls (HC), Shimizu and colleagues reported a significant increase in the relative abundance of Bifidobacterium and Eggerthella, and decrease in the relative abundance of Megamonas and Prevotella in BD patients compared with controls, however there was correlation between the bacterial taxa and clinical manifestation of BD (9). (Table 1) Fecal soluble immunoglobulin A (sIgA) concentrations were also significantly increased in BD patients but did not correlate with any specific bacterial species (9). In a follow up study analysis of the taxonomic and predicted metagenome functional composition in the fecal sample of 13 Japanese patients with BD and 27 HC, the relative abundance of Lactobacillus, Bifidobacterium and Streptococcus spp. was significantly increased in patients with BD compared to HC, whilst Megamonas hypermegale and Butyrivibrio species were less abundant, which correlated with the previous study (Table 1). The latter species were suggested to produce SCFA, particularly butyrate and propionate, in the human intestine. Such a reduction in butyrate-producing-SCFA may be responsible for the skewed T cell differentiation, ie. an increase in Th17 with a concomitant reduction in Treg cells. Functional annotation analysis found links to the pentose phosphate pathway and inosine monophosphate biosynthesis, suggesting that changes in the microbiome could affect metabolic processes in patients with BD. Nevertheless, intestinal microbiome interactions with the immune response are complex and SCFA depletion may only be one of many contributing factors to BD pathogenesis (10).
Ye et al. compared the DNA extracted from fecal samples of 24 patients with active BD, who had only received topical steroids, and 52 gender, body mass index (BMI) and aged-matched healthy controls (HC) (11). Metagenomic analysis revealed significant differences in gut microbial composition between the two cohorts with sulfate-reducing bacteria (SRB) and opportunistic pathogens significantly enriched, and methanogens, Methanoculleus spp. and methanomethylophilus ssp. and butyrate-producing bacteria, Clostridium spp. were reduced in BD patients compared to controls. SRB are pro-inflammatory bacteria that can inhibit butyrate. The shortage of butyrate and methane, combined with an abundance of hydrogen sulphide, creates an atypical environment that may contribute to intestinal epithelial barrier damage. This may allow for subsequent invasion by Th1 and Th17 cells, reduction in Tregs, thus triggering downstream inflammatory processes within the gut in patients with BD. To address this Ye et al. used the animal model of experimental autoimmune uveitis in B10RIII mice. Mice were treated with antibiotics (ampicillin, neomycin, metronidazole and vancomycin) for 3 weeks, prior to daily transplantation of pooled fecal suspension either from patients with active BD or from healthy controls. After one week of daily fecal transplants, mice were immunized with an interphotoreceptor retinoid binding protein peptide (161-180) to induce EAU and the severity of uveitis was reported on day 14 following immunization. The results showed that severe uveitis (corneal oedema, posterior synechiae and infiltrates of inflammatory cells in retina, choroid and vitreous humor) was observed in the active BD-recipient group, whilst the HC-recipient group displayed a slight intraocular inflammatory reaction with very few inflammatory cells in the vitreous cavity, indicating that the components within the fecal samples from BD patients exacerbated the development of intraocular inflammation in these mice (11). This supports the hypothesis that gut microbiome composition may contribute to BD pathogenesis and severity.
In a novel study the gut microbiome composition of Turkish patients with neuro-BD was compared to patients with multiple sclerosis, an autoimmune condition, and to healthy controls. 16S rRNA gene sequencing revealed that neuro-BD patients had an increased relative abundance of Parabacteroides, Geminger and Prevotella, whilst Vampirovibrio and Lachnospiraceae were decreased (Table 1), although the differences did not reach statistical significance, probably due to the small cohort sizes. However, it is of interest that two neurological conditions were linked to different changes in the intestinal microbiota. Whether this is due to the different disease processes or treatment regimens for participants, as those with neuro-BD had commenced azathioprine (with or without colchicine treatment), whilst MS patients had commenced immunomodulatory drug treatment (drug treatment not specified), will have to be investigated further (12).
The gut microbiome was compared in patients with BD uveitis to patients with Familial Mediterranean Fever (FMF) and Crohn’s Disease (CD), both which have autoinflammatory processes, as well as healthy controls. In total seven patients with BD, 12 patients with FMF, 9 patients with CD and 16 healthy controls (HC) were analyzed by 16S rRNA gene sequencing. The results showed that Veionellaceae, Succinivibrionaceae families and Succinivibrio and Mitsuokella genera were enriched in patients with BD compared to the other groups. The relevance to uveitis is not yet elucidated (15).
Oral ulceration is an important manifestation of BD and maintains a specific microbiome. To investigate the involvement of the oral microbiome in BD pathogenesis, Coit et al. collected stimulated saliva samples from 31 BD patients and 15 HCs, after 5 minutes of paraffin chewing, between 9-11am to address circadian rhythm of saliva secretion. Microbial DNA was isolated and sequencing revealed a significant difference in bacterial communities between the two cohorts. Haemophilus parainfluenzae and Alloprevotella were increased in BD patients, whilst Leptotrichia and Clostridiales were decreased compared to control samples. H. parainfluenzae, a gram-negative bacterium, is commonly linked to endocarditis, meningitis, septicaemia and gingival inflammation. While use of immunosuppressants (cyclosporin, azathioprine and prednisolone) had an impact on abundances of bacteria, there was no differences in bacterial communities between drug treatments (16).
In a second study, the oral microbiome profiles of BD patients (active vs. inactive disease) were compared to patients with recurrent aphthous ulcers (RAS), used as disease control, and healthy individuals. In total, unstimulated saliva samples were collected from 54 patients with BD, 8 patients with RAS and 25 HC together with mucosal samples from some patients. The results showed differences between the cohorts, with Neisseria more commonly isolated from HC compared to BD patients, whereas Staphylococcus salivarius and Streptococcus sanguinis were more abundant in BD patients with active oral disease when compared to those with inactive disease, patients with RAS and HC. Candida albicans was more frequently isolated from salivary samples taken from orally inactive BD patients when compared to HCs and RAS, however this could be attributed to steroid treatment. Samples taken from ulcerated and non-ulcerated mucosa of active BD patients was compared to those from HCs and RAS and revealed Rothia dentocariosa as the abundant organism in non-ulcerated mucosa of active BD patients, with a trend toward Rothia mucilaginosa from the ulcer sites of active BD patients (17).
In a recent study the oral microbiome in Mongolian patients with BD was compared to healthy controls. Saliva samples were obtained from 47 patients and 48 healthy individuals and the V3-V4 region of 16S RNA was amplified. Specifically, Prevotella, Veillonella, Streptococcus, and Haemophilus were the most abundant species in all samples However, three genera including, Akkermansia were significantly lower in patients compared to controls (20).
These studies support a possible role for gut or oral dysbiosis in BD. Common features such as the reduction in butyrate producing species can be found in most studies, although different bacterial species are reported in different studies. Despite the evidence of microbial dysbiosis in BD patients, it is difficult to ascertain whether such differences are causative or reactive in BD. This is due to many confounding factors that could affect the microbiome including the type and duration of drug treatment, sampling and sequencing methodology, or host genetics. This could also be linked to the different manifestations of BD, which could only be addressed through large longitudinal studies.
To distinguish disease-associated determinants from country and methodology, the gut microbiomes of patients with BD from the Netherlands and Italy were analyzed by the same method and compared to each other and healthy controls. In both cohorts, alpha diversity were similar to controls. Beta-diversity analysis did not show clustering based on disease, however, there was segregation by country of origin. The Italian cohort showed a significant decrease in Barnesiellaceae and Lachnospira genera, which in the Netherlands cohort was only a trend. IgA coated of intestinal bacteria analyzed by RNA sequencing can identify distinct disease-associated species in fecal samples. Increased IgA coating of Bifidobacterium spp., Dorea spp., and Ruminoccoccus bromei spp. and reduced presence of IgA-coated Erysipelotrichaceae spp. in the Dutch BD cohort compared to healthy controls, although this was not analyzed in the Italian cohort (14). These data support the requirement for more, larger studies to determine the geographical and environmental changes compared to disease-induced changes in the BD gut microbiome.
Behçet’s Disease often involves variable disease manifestations at different body sites, such as the oral, skin, eyes, gastrointestinal, or nervous system, over time. To identify the clinical clusters of BD, Soejima and associates investigated the clinical phenotype of 657 patients with BD from the Yokohama City Hospital database and 6754 patients with BD from the Japanese Ministry of Health, Labour and Welfare database. They identified that gastrointestinal BD was associated with vascular but not eye involvement, while a second cluster had high incidence of eye involvement but no vascular or neurological involvement (13). Chronological analyses showed expansion of the gastrointestinal cluster in BD patients over 27 years in a relatively stable genetic cohort in Japan, indicating that environmental factors might be involved in the disease manifestation of BD.
Changes in the gut microbiome could drive the clinical phenotype in BD. To answer this, Yasar Bilge and associates compared the gut microbiome of 27 patients with BD (ocular, mucocutaneous, and vascular involvement), and 10 age- and sex-matched healthy controls. Linear discriminant analysis of this small patient cohort revealed a difference in the bacterial genera according to the manifestations of BD: Lachnospiraceae NK4A136 enriched in BD patients with eye involvement, Dialister, Intestinomonas, and Marvinbryantia enriched in BD patients with predominant mucocutaneous disease, Gemella in vascular BD. However, larger studies and functional relevance of such associations should be investigated (21).
This data, if validated in other patient cohorts, supports the stratification of individuals in relation to their gut microbiome profile (i.e. to determine if specific bacterial species are associated with specific manifestations or whether general alterations to the microbiome influences all manifestations). In either process, factors that may potentially influence the gut microbiome, such as diet, can be addressed by the potential of re-establishing a more balanced, symbiotic relationship might prove to be important in reducing the severity of active BD phases. To test this concept a randomized controlled trial the Modulation of Gut Microbiota through Nutritional Invention in Behçet’s Syndrome Patients (MAMBA) study, will recruit 90 patients with BD, to investigate the impact of three dietary interventions, a lacto-ovo-vegetarian diet, a Mediterranean diet, or a Mediterranean diet supplemented with butyrate on the severity of clinical manifestations of BD (22).
There is increasing evidence that the host genetic background shapes the gut microbiome profile (23, 24).
There are more than 20 genetic susceptibility loci that meet the threshold for genome-wide level of significance in patients with Behçet’s disease and several other loci that have been associated with BD (25). To address the potential role of genetics in BD gut dysbiosis we have selected a potential pathway associated with production of SCFA in the gut and mutations in the genes involved. Activation of Toll-like receptors on systemic dendritic cells induces the release of IL-23, which activates innate lymphoid cells to release IL-22 which leads to a rapid α (1,2)-fucosylation of small intestine epithelial cells via the release of fucosyltransferase 2 (FUT2). Fucosylated proteins enter into the lumen of the gut where they release fucose molecules which are metabolized by bacteria present. As stated, products from these bacteria include short chain fatty acids, including butyrate which supports T regulatory cells (26). This complex series of interactions involve several of the gene mutations in which have been associated with BD.
Toll-like receptors (TLR) are a family of transmembrane sensors for bacteria, fungi and viruses that are a vital component of innate immunity. The strongest associations between TLR and BD is in with TLR4 which recognizes bacterial lipopolysaccharide. Nine single nucleotide polymorphisms previously detected in TLR4 by direct sequencing were analyzed for an association with BD in Korean patients. The most frequent haplotype, TAGCGGTAA, was significantly increased in HLA-B*51-positive patients compared with healthy control participants and potential linked to joint disease (27). This was supported by targeted exonic resequencing of specific genes identified in the original GWAS, from 2,461 BD cases compared with 2,458 controls where a SNP in TLR4 was significantly associated with BD (28). In buccal lesions from patients with BD, TLR expression was increased compared with healthy controls, however, a similar increase was seen in lesion tissue from patients with lichen planus or gingivitis, suggesting that this was a generalized inflammatory response as opposed to a BD-specific response. In these patients no significant associations were seen with TLR4, but a gain of function mutation in the TLR adaptor molecule, TIR-adaptor protein (TIRAP) was significantly associated with BD in UK, but not Middle Eastern, patients (29). This mutation is associated with an more potent cytokine response to TLR stimulation.
In a study of 400 patients with ocular BD compared to 600 healthy Han Chinese controls frequencies of genotypes of TLR2 rs2289318 and rs3804099 were significantly higher in ocular BD patients compared with controls. A validation cohort of 438 patients with BD uveitis and 1000 healthy controls confirmed the associations. TLR2 mRNA expression in PBMC was increased in healthy carriers of the certain genotypes, although this did not affect the release of TNF-α, IL-6, IL-10, and IL-1β could be detected (30).
In Tunisians patients with Behcet’s Disease expression of SNP in TLR9 (1486 T/C) was different between BD patients and healthy controls but did not reach statistical significance (31). In Chinese patients with BD TLR7 had an increased copy number and increased expression in PBMC compared to healthy controls (32).
These data support a role for bacterial sensing in BD.
Two major GWAS studies in Turkish and Japanese patients with BD identified SNP at the IL23R-IL12RB2 locus (rs924080 and rs12119179) respectively. Meta-analysis of both data sets further identified rs1495965 as associated with BD (33, 34). These associations have been supported in several other studies in different patient cohorts (35–39). These mutations have been linked to enhanced expression of IL-23R and potential upregulation of the IL-17 pathway.
Lymphocytes isolated from the intestinal mucosa of 8 patients with intestinal symptoms, that had started within 6 months of collection, were cloned and analyzed for surface phenotype and cytokine production. Results showed more CD4+ clones compared to CD8+ with the majority of both showing Th1 profile secreting IFN-γ and TNF-α, with a substantial population produced both IFN-γ and IL-17 (Th1/Th17 profile). Most of the T-cell clones showed perforin-mediated cytotoxicity and Fas–Fas Ligand-mediated pro-apoptotic activity. Therefore, in the early stages of intestinal BD both Th1 cytokines, IL-17 and cytotoxicity are involved in inflammation of the mucosal tissue. This data would support the use of anti-TNF treatment and potentially anti-IL-17 in controlling intestinal BD (40).
In support of treatment altering the Th17 pathway, twenty-seven patients who received interferon alfa-2a for active BD uveitis and Treg and Th17 cells were assessed. Treg, Th17 cell frequencies and Th17 RORγt expression were significantly elevated, although IL-10 concentration in Treg culture supernatants was significantly lower with cells from BD patients compared to controls. IFN alfa-2a treatment led to clinical remission in 70% of patients which correlated with a significant reversal in Treg and Th17 cell frequencies, Treg IL-10 and Th17 cytokine production (41).
IL-22 is a member of the IL-10 cytokine family that is involved in inflammatory processes. Th22-type T cell clones from ocular samples taken from Behçet’s disease patients with active uveitis, produced IL-22 and TNF-a but not IFN-γ or IL-17. IL-22 can affect epithelial and stromal cells and has been involved the pathogenesis of inflammatory skin disorders, such as psoriasis, atopic eczema, and allergic contact dermatitis (42). Healthy IL-22-deficient mice had altered microbiome, with decreased abundance of Lactobacillus, and increased levels of others. Induction of experimental colitis caused more sever disease in deficient mice compared to controls. When the microbiome from IL-22 deficient mice was transmitted to wild-type animals along this led to an increased susceptibility to colitis, suggesting an important role for IL-22 the balance between immunity and colonic microbiota (43). Therefore, although no polymorphisms in IL22 have been reported in BD, a potential role in pathogenesis is likely.
Genome-wide array analysis of Iranian patients with BD and controls identified an association with five coding polymorphisms of the gene encoding fucosyltransferase -2 (FUT2), including the rs601338 nonsense variant which, in Caucasians, determines the secretion of the H antigen (precursor of the ABO blood group antigens) in body fluids and on the intestinal mucosa. These results were validated by comparison with GWAS analysis of Turkish patients with BD. A non-secretor phenotype is suggested to link with intestinal glycosylation (44). The association with FUT2 polymorphisms and BD was supported in an larger study of three populations. The rs601338 A allele, in Turkish and Iranian patients and the rs1047781 T allele in Japanese patients for which homozygosity leads to an ABO non-secretor phenotype were confirmed in this study. These non-secretor genotypes are also associated with Crohn’s disease risk (45).
The process by which FUT2 polymorphisms influence the gut microbiome is not yet clear. Secretor mothers produce milk containing α1-2 fucosylated human milk oligosaccharides and several strains of bacteria in the infant gut have the capacity to utilize these oligosaccharides (46). In vaginally born infants, the mother’s secretor status had no effect on the infants’ microbiome. By comparison, in children delivered by caesarean section several differences were seen in infants from secretor mothers compared to non-secretor mothers. In particular, Bifidobacteria were depleted and Enterococci increased among the caesarean-born infants of non-secretor mothers. Akkermansia was increased in infants of secretor mothers. In a study of 14 non-secretor (FUT2 rs601338 genotype AA) and 57 secretor (genotypes GG and AG) adult individuals of western European descent, the gut microbiota was analyzed. Results showed a several differences between the two groups with nonsecretors presenting lower species richness than secretors and different enterotypes in the two groups (46). This data supports the concept that FUT2 polymorphism can influence the composition of the gut microbiota and variation between individuals (47). By comparison, the association of rs601338 and intestinal microbiota composition was analyzed in 1,190 healthy individuals, and the results showed no association of the FUT2 genotype, secretor status and microbial alpha diversity, microbial composition or inferred microbial function after correction for multiple testing (48).
Non-secretors, who are homozygous for the loss-of-function alleles of FUT2 gene, have increased susceptibility to Crohn’s disease (CD). From 39 healthy individuals, 75 endoscopic lavage samples were profiled for microbiome, proteome and metabolome composition based on FUT2 genotype. Metagenomic analysis revealed differences in energy metabolism in the microbiome of non-secretor and heterozygote individuals, in particular enrichment of carbohydrate, vitamin and lipid metabolism, and depletion of amino-acid biosynthesis and metabolism. Metabolomic analysis of human specimens showed changes of many metabolites related to the different genotype groups. These results support compositional and functional differences in the microbiota of non-secretors that may be relevant for CD susceptibility (49).
Taken together polymorphisms in all genes encoding proteins in the pathway TLR-IL-23-IL-22- FUT2 have all been reported in patients with BD. Systemic activation of this pathway could lead to a reduction in beneficial SCFA either by increased targeting of SCFA-producing species or via reduced fucosylation and SCFA generation. This is one potential pathway initiated by systemic activation of tissue dendritic cells. The best direct evidence of cause and effect remains the induction of uveitis in B10RIII mice following transplantation of fecal material from patients with active BD (11). However, it is not known what induced the changes in the microbiome of these patients nor what processes drove ocular inflammation even in this genetically susceptible mouse model.
A possible link between gut and oral microbiome dysbiosis is supported by several studies and the involvement of the FUT2 pathway and gene polymorphisms in molecules involved may be relevant. However, there are several aspects that must be addressed. First, that different bacterial species and genera have been identified in the different studies. While there may be a commonality in decreased abundance of species linked to short chain fatty acid regulation that could link the studies more work needs to be done in larger cohorts with the same collection and preparation procedures. Second, certain demographic analysis does not support a link between manifestations of BD form example ocular involvement not associated with intestinal BD (13). Thirdly, many patients with BD do not have intestinal-BD, therefore there is no direct association between gut inflammation, the microbiome and other manifestations of BD. Fourth, several other sites affected by BD including oral and genital mucosa and skin harbor their own microbiome, which will need to be analyzed for association with manifestations of BD. Fifth, there is a significant non-genetic component in Behçet’s Disease. As stated, the MAMBA study (22) will address dietary influences on the gut microbiome. Recently the numbers of new patients being diagnosed with BD in Japan and Korea have been steadily decreasing (50, 51). Whether such changes involve factors such as a move to a more westernized diet would be interesting to address. Effects of different treatment regimes on the gut microbiome should be investigated. Finally, there are other gene loci that have been associated with BD that could impact on the composition of or the response to the gut microbiome, including HLA-B*51 and IL-10 and cells such as natural killer cells which can produce IL-10 when stimulated. Moreover, IL-10 production by Treg has been linked to Akkermansia which is altered in several studies.
In conclusion, while it is clear that dysbiosis of the gut microbiome is associated with Behçet’s disease and a role for known genetic mutations can be linked to these changes (Figure 1), further research is required to understand the combinations involved in the manifestations of this complex disease.
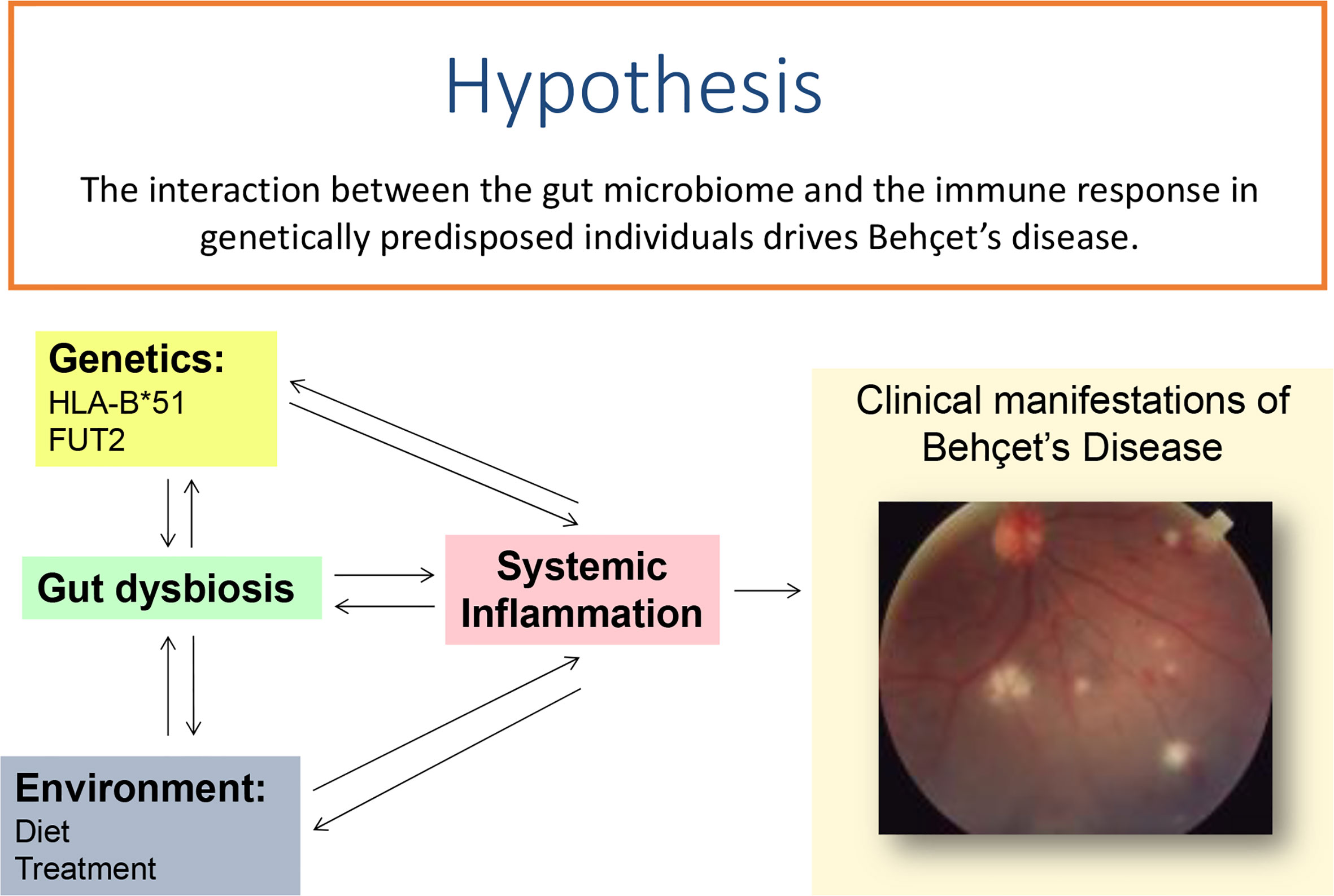
Figure 1 Systemic inflammation leading to clinical features in Behçet’s disease may either be initiated by of causing changes in the gut microbiome in patients who are genetically predisposed to the condition.
All authors contributed equally to this manuscript. All authors contributed to the article and approved the submitted version.
Liying Low is funded by a Fight for Sight Clinical Research Fellowship (Ref 1840/1841).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Leccese P, Alpsoy E. Behçet’s Disease: An Overview of Etiopathogenesis. Front Immunol (2019) 10:1067. doi: 10.3389/fimmu.2019.01067
2. Belkaid Y, Harrison OJ. Homeostatic Immunity and the Microbiota. Immunity (2017) 46:562–76. doi: 10.1016/j.immuni.2017.04.008
3. Massey W, Brown JM. The Gut Microbial Endocrine Organ in Type 2 Diabetes. Endocrinol (United States) (2021) 162:bqaa235. doi: 10.1210/endocr/bqaa235
4. Kishikawa T, Ogawa K, Motooka D, Hosokawa A, Kinoshita M, Suzuki K, et al. A Metagenome-Wide Association Study of Gut Microbiome in Patients With Multiple Sclerosis Revealed Novel Disease Pathology. Front Cell Infect Microbiol (2020) 10:585973. doi: 10.3389/fcimb.2020.585973
5. Horai R, Caspi RR. Microbiome and Autoimmune Uveitis. Front Immunol (2019) 10:232. doi: 10.3389/fimmu.2019.00232
6. Gupta VK, Paul S, Dutta C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front Microbiol (2017) 8:1162. doi: 10.3389/fmicb.2017.01162
7. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut Microbiome Modulates Response to Anti-PD-1 Immunotherapy in Melanoma Patients. Science (2018) 359:97–103. doi: 10.1126/science.aan4236
8. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg Induction by a Rationally Selected Mixture of Clostridia Strains From the Human Microbiota. Nature (2013) 500:232–6. doi: 10.1038/nature12331
9. Shimizu J, Kubota T, Takada E, Takai K, Fujiwara N, Arimitsu N, et al. Bifidobacteria Abundance-Featured Gut Microbiota Compositional Change in Patients With Behcet’s Disease. PloS One (2016) 11:e0153746. doi: 10.1371/journal.pone.0153746
10. Shimizu J, Kubota T, Takada E, Takai K, Fujiwara N, Arimitsu N, et al. Relative Abundance of Megamonas Hypermegale and Butyrivibrio Species Decreased in the Intestine and Its Possible Association With the T Cell Aberration by Metabolite Alteration in Patients With Behcet’s Disease. Clin Rheumatol (2019) 38:1437–45. doi: 10.1007/s10067-018-04419-8
11. Ye Z, Zhang N, Wu C, Zhang X, Wang Q, Huang X, et al. A Metagenomic Study of the Gut Microbiome in Behcet’s Disease. Microbiome (2018) 6:135. doi: 10.1186/s40168-018-0520-6
12. Oezguen N, Yalcinkaya N, Kücükali CI, Dahdouli M, Hollister EB, Luna RA, et al. Microbiota Stratification Identifies Disease-Specific Alterations in Neuro-Behçet’s Disease and Multiple Sclerosis. Clin Exp Rheumatol (2019) 37 Suppl 121:58–66.
13. Soejima Y, Kirino Y, Takeno M, Kurosawa M, Takeuchi M, Yoshimi R, et al. Changes in the Proportion of Clinical Clusters Contribute to the Phenotypic Evolution of Behçet’s Disease in Japan. Arthritis Res Ther (2021) 23:49. doi: 10.1186/s13075-020-02406-6
14. van der Houwen TB, van Laar JAM, Kappen JH, van Hagen PM, de Zoete MR, van Muijlwijk GH, et al. Behçet’s Disease Under Microbiotic Surveillance? a Combined Analysis of Two Cohorts of Behçet’s Disease Patients. Front Immunol (2020) 11:1192. doi: 10.3389/fimmu.2020.01192
15. Tecer D, Gogus F, Kalkanci A, Erdogan M, Hasanreisoglu M, Ergin Ç, et al. Succinivibrionaceae is Dominant Family in Fecal Microbiota of Behçet’s Syndrome Patients With Uveitis. PloS One (2020) 15(10):e0241691. doi: 10.1371/journal.pone.0241691
16. Coit P, Mumcu G, Ture-Ozdemir F, Unal AU, Alpar U, Bostanci N, et al. Sequencing of 16S Rrna Reveals a Distinct Salivary Microbiome Signature in Behçet’s Disease. Clin Immunol (2016) 169:28–35. doi: 10.1016/j.clim.2016.06.002
17. Seoudi N, Bergmeier LA, Drobniewski F, Paster B, Fortune F. The Oral Mucosal and Salivary Microbial Community of Behçet’s Syndrome and Recurrent Aphthous Stomatitis. J Oral Microbiol (2015) 7:27150. doi: 10.3402/jom.v7.27150
18. Consolandi C, Turroni S, Emmi G, Severgnini M, Fiori J, Peano C, et al. Behçet’s Syndrome Patients Exhibit Specific Microbiome Signature. Autoimmun Rev (2015) 14:269–76. doi: 10.1016/j.autrev.2014.11.009
19. Moco S, Candela M, Chuang E, Draper C, Cominetti O, Montoliu I, et al. Systems Biology Approaches for Inflammatory Bowel Disease: Emphasis on Gut Microbial Metabolism. Inflammation Bowel Dis (2014) 20:2104–14. doi: 10.1097/MIB.0000000000000116
20. Balt J, Uehara O, Abiko Y, Jamyanjav B, Jav S, Nagasawa T, et al. Alteration of Oral Flora in Mongolian Patients With Behçet’s Disease: A Multicentre Study. Clin Exp Rheumatol (2020) 38(Suppl 127(5):80–5.
21. Yasar Bilge NS, Pérez Brocal V, Kasifoglu T, Bilge U, Kasifoglu N, Moya A, et al. Intestinal Microbiota Composition of Patients With Behçet’s Disease: Differences Between Eye, Mucocutaneous and Vascular Involvement. the Rheuma-BIOTA Study. Clin Exp Rheumatol (2020) 38 Suppl 127:60–8. doi: 10.1136/annrheumdis-2020-eular.3626
22. Pagliai G, Dinu M, Fiorillo C, Becatti M, Turroni S, Emmi G, et al. Modulation of Gut Microbiota Through Nutritional Interventions in Behçet’s Syndrome Patients (the MAMBA Study): Study Protocol for a Randomized Controlled Trial. Trialss (2020) 21:511. doi: 10.1186/s13063-020-04444-6
23. Olivares M, Neef A, Castillejo G, De Palma G, Varea V, Capilla A, et al. The HLA-DQ2 Genotype Selects for Early Intestinal Microbiota Composition in Infants At High Risk of Developing Coeliac Disease. Gut (2015) 64:406–17. doi: 10.1136/gutjnl-2014-306931
24. Zhernakova A, Kurilshikov A, Bonder MJ, Tigchelaar EF, Schirmer M, Vatanen T, et al. Population-Based Metagenomics Analysis Reveals Markers for Gut Microbiome Composition and Diversity. Science (2016) 352:565–56. doi: 10.1126/science.aad3369
25. Ortiz-Fernández L, Sawalha AH. Genetics of Behçet’s Disease: Functional Genetic Analysis and Estimating Disease Heritability. Front Med (2021) 8:625710. doi: 10.3389/fmed.2021.625710
26. Pickard JM, Maurice CF, Kinnebrew MA, Abt MC, Schenten D, Golovkina TV, et al. Rapid Fucosylation of Intestinal Epithelium Sustains Host-Commensal Symbiosis in Sickness. Nature (2014) 514:638641. doi: 10.1038/nature13823
27. Horie Y, Meguro A, Ota M, Kitaichi N, Katsuyama Y, Takemoto Y, et al. Association of TLR4 Polymorphisms With Behçet’s Disease in a Korean Population. Rheumatology (2009) 48:638–42. doi: 10.1093/rheumatology/kep077
28. Kirino Y, Zhou Q, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahie E, et al. Targeted Resequencing Implicates the Familial Mediterranean Fever Gene MEFV and the Toll-Like Receptor 4 Gene TLR4 in Behçet Disease. Proc Natl Acad Sci USA (2013) 110:8134–9. doi: 10.1073/pnas.1306352110
29. Durrani O, Banahan K, Sheedy FJ, McBride L, Ben-Chetrit E, Greiner K, et al. Tirap Ser180Leu Polymorphism is Associated With Behçet’s Disease. Rheumatology (2011) 50:1760–5. doi: 10.1093/rheumatology/ker200
30. Fang J, Hu R, Hou S, Ye Z, Xiang Q, Qi J, et al. Association of TLR2 Gene Polymorphisms With Ocular Behcet’s Disease in a Chinese Han Population. Invest Ophthalmol Vis Sci (2013) 54:8384–92. doi: 10.1167/iovs.13-12878
31. Dhifallah IB, Lachheb J, Houman H, Hamzaoui K. Toll-Like-Receptor Gene Polymorphisms in a Tunisian Population With Behçet’s Disease. Clin Exp Rheumatol (2009) 27(Suppl 53):S58–62.
32. Fang J, Chen L, Tang J, Hou S, Liao D, Ye Z, et al. Association Between Copy Number Variations of TLR7 and Ocular Behçet’s Disease in a Chinese Han Population. Investig Ophthalmol Vis Sci (2015) 56:1517–23. doi: 10.1167/iovs.14-15030
33. Remmers EF, Cosan F, Kirino Y, Ombrello MJ, Abaci N, Satorius C, et al. Genome-Wide Association Study Identifies Variants in the MHC Class I, IL10, and IL23R-IL12RB2 Regions Associated With Behçet’s Disease. Nat Genet (2010) 42:698–702. doi: 10.1038/ng.625
34. Mizuki N, Meguro A, Ota M, Ohno S, Shiota T, Kawagoe T, et al. Genome-Wide Association Studies Identify Il23r-IL12RB2 and IL10 as Behçet’s Disease Susceptibility Loci. Nat Genet (2010) 42:703–6. doi: 10.1038/ng.624
35. Xavier JM, Shahram F, Davatchi F, Rosa A, Crespo J, Abdollahi BS, et al. Association Study of IL10 and IL23r-IL12RB2 in Iranian Patients With Behçet’s Disease. Arthritis Rheum (2012) 64:2761–72. doi: 10.1002/art.34437
36. Çavuş F, Ulusoy C, Örçen A, Gül A, Tüzün E, Vural B. Increased IL-23 Receptor, Tnf-Alpha and IL-6 Expression in Individuals With the IL23R-IL12RB2 Locus Polymorphism. Immunol Lett (2014) 160:96–8. doi: 10.1016/j.imlet.2014.01.001
37. Kang EH, Kim S, Park MY, Choi JY, Choi IA, Kim MJ, et al. Behçet’s Disease Risk Association Fine-Mapped on the IL23R-IL12RB2 Intergenic Region in Koreans. Arthritis Res Ther (2017) 19(1):227–36. doi: 10.1186/s13075-017-1435-5
38. Kramer M, Hasanreisoglu M, Weiss S, Kumova D, Schaap-Fogler M, Guntekin-Ergun S, et al. Single-Nucleotide Polymorphisms in IL23R-IL12RB2 (Rs1495965) are Highly Prevalent in Patients With Behcet’s Uveitis and Vary Between Populations. Ocul Immunol Inflamm (2019) 27:766–73. doi: 10.1080/09273948.2018.1467463
39. Yu H, Zheng M, Zhang L, Li H, Zhu Y, Cheng L, et al. Identification of Susceptibility Snps in IL10 and IL23R-IL12RB2 for Behçet’s Disease in Han Chinese. J Allergy Clin Immunol (2017) 139(2):621–7. doi: 10.1016/j.jaci.2016.05.024
40. Emmi G, Silvestri E, Della BC, Grassi A, Benagiano M, Cianchi F, et al. Cytotoxic Th1 and Th17 Cells Infiltrate the Intestinal Mucosa of Behcet Patients and Exhibit High Levels of TNF-Alpha in Early Phases of the Disease. Med (United States) (2016) 95:e5516. doi: 10.1097/MD.0000000000005516
41. Albayrak O, Oray M, Can F, Kirimli GU, Gul A, Tugal-Tutkun I, et al. Effect of Interferon Alfa-2a Treatment on Adaptive and Innate Immune Systems in Patients With Behçet Disease Uveitis. Investig Ophthalmol Vis Sci (2019) 60:52–63. doi: 10.1167/iovs.18-25548
42. Sugita S, Kawazoe Y, Imai A, Kawaguchi T, Horie S, Keino H, et al. Role of IL-22– and TNF-Alpha–Producing Th22 Cells in Uveitis Patients With Behçet’s Disease. J Immunol (2013) 190:5799–808. doi: 10.4049/jimmunol.1202677
43. Zenewicz LA, Yin X, Wang G, Elinav E, Hao L, Zhao L, et al. Il-22 Deficiency Alters Colonic Microbiota to Be Transmissible and Colitogenic. J Immunol (2013) 190:5306–12. doi: 10.4049/jimmunol.1300016
44. Xavier JM, Shahram F, Sousa I, Davatchi F, Matos M, Abdollahi BS, et al. Fut2: Filling the Gap Between Genes and Environment in Behçet’s Disease? Ann Rheum Dis (2015) 74:618–24. doi: 10.1136/annrheumdis-2013-204475
45. Takeuchi M, Mizuki N, Meguro A, Ombrello MJ, Kirino Y, Satorius C, et al. Dense Genotyping of Immune-Related Loci Implicates Host Responses to Microbial Exposure in Behçet’s Disease Susceptibility. Nat Genet (2017) 49:438–43. doi: 10.1038/ng.3786
46. Korpela K, Salonen A, Hickman B, Kunz C, Sprenger N, Kukkonen K, et al. Fucosylated Oligosaccharides in Mother’s Milk Alleviate the Effects of Caesarean Birth on Infant Gut Microbiota. Sci Rep (2018) 8:13757. doi: 10.1038/s41598-018-32037-6
47. Wacklin P, Tuimala J, Nikkilä J, Tims S, Mäkivuokko H, Alakulppi N, et al. Faecal Microbiota Composition in Adults is Associated With the FUT2 Gene Determining the Secretor Status. PloS One (2014) 9:e94863. doi: 10.1371/journal.pone.0094863
48. Turpin W, Bedrani L, Espin-Garcia O, Xu W, Silverberg MS, Smith MI, et al. Fut2 Genotype and Secretory Status are Not Associated With Fecal Microbial Composition and Inferred Function in Healthy Subjects. Gut Microbes (2018) 9:357–68. doi: 10.1080/19490976.2018.1445956
49. Tong M, McHardy I, Ruegger P, Goudarzi M, Kashyap PC, Haritunians T, et al. Reprograming of Gut Microbiome Energy Metabolism by the FUT2 Crohn’s Disease Risk Polymorphism. ISME J (2014) 8:2193–206. doi: 10.1038/ismej.2014.64
50. Goto H, Mochizuki M, Yamaki K, Kotake S, Usui M, Ohno S. Epidemiological Survey of Intraocular Inflammation in Japan. Jpn J Ophthalmol (2007) 51:41–4. doi: 10.1007/s10384-006-0383-4
Keywords: Behçet’s disease, microbiome, genetic polymorphism, gut, butyrate
Citation: Mehmood N, Low L and Wallace GR (2021) Behçet’s Disease—Do Microbiomes and Genetics Collaborate in Pathogenesis? Front. Immunol. 12:648341. doi: 10.3389/fimmu.2021.648341
Received: 31 December 2020; Accepted: 12 April 2021;
Published: 21 May 2021.
Edited by:
Peizeng Yang, First Affiliated Hospital of Chongqing Medical University, ChinaReviewed by:
Alexander Kurilshikov, University Medical Center Groningen, NetherlandsCopyright © 2021 Mehmood, Low and Wallace. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Graham R. Wallace, Zy5yLndhbGxhY2VAYmhhbS5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.