
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 07 April 2021
Sec. T Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.644483
 Mukul Prasad1,2,3
Mukul Prasad1,2,3 Lukasz Wojciech1,3
Lukasz Wojciech1,3 Joanna Brzostek1,3,4†
Joanna Brzostek1,3,4† Jianfang Hu4†
Jianfang Hu4† Yen Leong Chua1,2,3
Yen Leong Chua1,2,3 Desmond Wai Hon Tung1,3
Desmond Wai Hon Tung1,3 Jiawei Yap1,3
Jiawei Yap1,3 Vasily Rybakin3,4†
Vasily Rybakin3,4† Nicholas R. J. Gascoigne1,2,3,4*
Nicholas R. J. Gascoigne1,2,3,4*Deletion of the gene for Themis affects T cell selection in the thymus, which would be expected to affect the TCR repertoire. We found an increased proportion of cells expressing Vα3.2 (TRAV9N-3) in the peripheral CD8+ T cell population in mice with germline Themis deficiency. Analysis of the TCRα repertoire indicated it was generally reduced in diversity in the absence of Themis, whereas the diversity of sequences using the TRAV9N-3 V-region element was increased. In wild type mice, Vα3.2+ cells showed higher CD5, CD6 and CD44 expression than non-Vα3-expressing cells, and this was more marked in cells from Themis-deficient mice. This suggested a virtual memory phenotype, as well as a stronger response to self-pMHC. The Vα3.2+ cells responded more strongly to IL-15, as well as showing bystander effector capability in a Listeria infection. Thus, the unusually large population of Vα3.2+ CD8+ T cells found in the periphery of Themis-deficient mice reflects not only altered thymic selection, but also allowed identification of a subset of bystander-competent cells that are also present in wild-type mice.
The inability to predict the foreign antigens that an organism will encounter in its lifetime creates a need for the immune system to generate and maintain a diverse T cell receptor repertoire (1–3). T cells develop in the thymus, where they rearrange T cell receptor (TCR) α and β genes and undergo positive and negative selection to ensure that only cells expressing TCRs which give an optimum response to the self-peptide MHC (pMHC) are allowed to leave the thymus. The TCR is formed by rearrangement of V(D)J elements, such that the binding site for the peptide MHC complex is formed from 3 complementarity determining regions (CDR1, 2, and 3) (4). Different TCR Vα gene segments have preferences for binding to, and therefore being preferentially selected by, either MHC-I or MHC-II. This selection bias is dictated by their CDR1 and CDR2 sequences (5–7) and can affect the CD4+:CD8+ ratio (8). Structural analysis of TCR-pMHC complexes indicates conserved sites in TCR CDR1 and 2 of Vα and Vβ that correspond to sites on the MHC α-helices (9), which can explain these biases of selection.
After thymic selection, T cells migrate to peripheral lymphoid organs. T cell survival in the periphery requires tonic TCR signaling from self-pMHC (10, 11). This tonic signaling is also important in shaping T cell potential to mount immune responses to foreign antigens (12) and for homeostatic proliferation (1). T cells undergo lymphopenia-induced proliferation (LIP) upon transfer into lymphopenic hosts, and this process requires interactions with self-pMHC as well as signals from cytokines such as IL-7 and IL-15 (13). The probability of a particular T cell to undergo LIP correlates with its affinity for self-pMHC (10). CD5 is a negative regulator of TCR signaling, and its expression reports the strength of interaction of TCR with self-pMHC (14, 15). CD5 expression also regulates responsiveness to IL-7 in naïve T cells (16). IL-7 and IL-15 are required for homeostatic maintenance of T cells in the periphery (17, 18) and IL-7 has a role in maintenance of the TCR repertoire diversity (19).
Memory cells, marked by CD44hi expression, are formed once an infection is cleared, and provide critical protection against re-encounter with the same pathogen. Apart from antigen induced memory cells, there are also “virtual” or “innate” memory phenotype (MP) cells (20, 21), which can also be identified by CD44hi expression (22) and which require IL-15 for maintenance (23, 24). These cells were thought to develop mostly due to LIP in a foreign antigen-independent manner (25, 26). However, recent analysis of TCR repertoire of CD8+ MP cells demonstrated that TCRs expressed by MP cells are distinct from these expressed by naïve CD8+ T cells, with MP clones showing higher reactivity to self pMHC (27, 28), suggesting a unique development program of at least part of the CD8+ MP population, controlled by self-pMHC. Not all cells that undergo proliferation in response to an infection or tumor are antigen specific. Non-antigen specific bystander T cells become activated by the cytokines produced during an infection (29), such as IL-15 induced by type I interferons (30). Bystander cells display effector functions (31, 32), and play a role in protection against chronic infections in humans (33–35).
The T cell lineage-restricted protein Themis regulates the threshold between positive and negative selection of T cells in the thymus (36–40). Themis interacts with and regulates the phosphatases Shp1 (Ptpn6) and Shp2 (Ptpn11), although the precise mechanism is controversial (39, 41–43). Themis deficiency affects the metabolic response of T cells to TCR stimulation (44). Post-selection deletion of Themis reduces the homeostatic response of peripheral CD8+ T cells to self-pMHC (45). Themis deficiency is predicted to alter TCR repertoire but altered TCR repertoire in Themis KO mice and its functional consequences have not yet been described. Here were report comparison of TCR repertoires of Themis KO and WT mice, and the discovery of an unusual virtual memory and bystander-competent CD8+ subpopulation bearing Vα3.2 TCR.
Themis–/–Foxp3-GFP, derived from B6.129S-Themistm1Gasc (36) crossed to B6.Cg-Foxp3tm2Tch/J (46), Themis+/+Foxp3-GFP (B6.Cg-Foxp3tm2Tch/J), Themisf/f.CD4-Cre– and Themisf/f.CD4-Cre+, Themisf/f.dLck-Cre– and Themisf/f.dLck-Cre+ (45), Rag1–/– and CD45.1 mice on C57BL/6 background were bred in our restricted flora (RF) facilities at Comparative Medicine, NUS. Mice were treated under Institutional Animal Care and Use Committee-approved guidelines in accordance with approved protocols.
Mice were euthanized and dissected. Peripheral (p)LN (pooled cervical, axillary, brachial, and inguinal LN) were excised and mashed upon a 70 μm cell strainer into 5 ml cRPMI. For cell surface staining, cells were spun at 1200 rpm at 4°C for 5 minutes and the resulting cell pellets were resuspended in 100 μl PBS with 0.5% BSA (FWB: FACS wash buffer), containing the fluorophore-conjugated antibodies’ dilutions (1:300) for the cell surface antigens and incubated on ice for 30 minutes in the dark. Cells were then centrifuged at 1200 rpm at 4°C for 5 minutes and resuspended in 300 μl of FWB for flow cytometry analysis. For biotinylated antibodies, staining with the antibody was followed by staining with 100 μl FWB containing BUV 395 labeled-streptavidin (BD Biosciences, New Jersey, USA) at 1:500 dilution for 30 minutes on ice. Cells were then centrifuged at 1200 rpm at 4°C for 5 minutes and resuspended in 300 μl of FWB for flow cytometry analysis.
For intracellular staining, the cells from the previous step were resuspended in 0.2 ml IC fixation buffer (eBiosciences, California, USA) while being vortexed, followed by an incubation at room temperature for 20 minutes. The cells were then washed twice with 2 ml 1X permeabilization buffer (eBiosciences, California, USA), resuspended in 100 μl FWB containing the fluorophore-conjugated antibodies’ dilutions (1:250) for the intracellular antigens and incubated at room temperature for 30 minutes. The cells were then washed once with 2 ml 1X permeabilization buffer and then with 2 ml FWB. The cells were then resuspended in 300 μl FWB for analysis on a flow cytometer. 25 μl Count Bright beads (Life Technologies, California, USA) were added to each sample for cell count analysis. Cells were analyzed on BD LSR Fortessa X-20 flow cytometer (BD Biosciences, New Jersey, USA). Flow cytometry data were analyzed using FlowJo software (Treestar, California, USA). All antibodies used for flow cytometry purposes are described in Table 1.
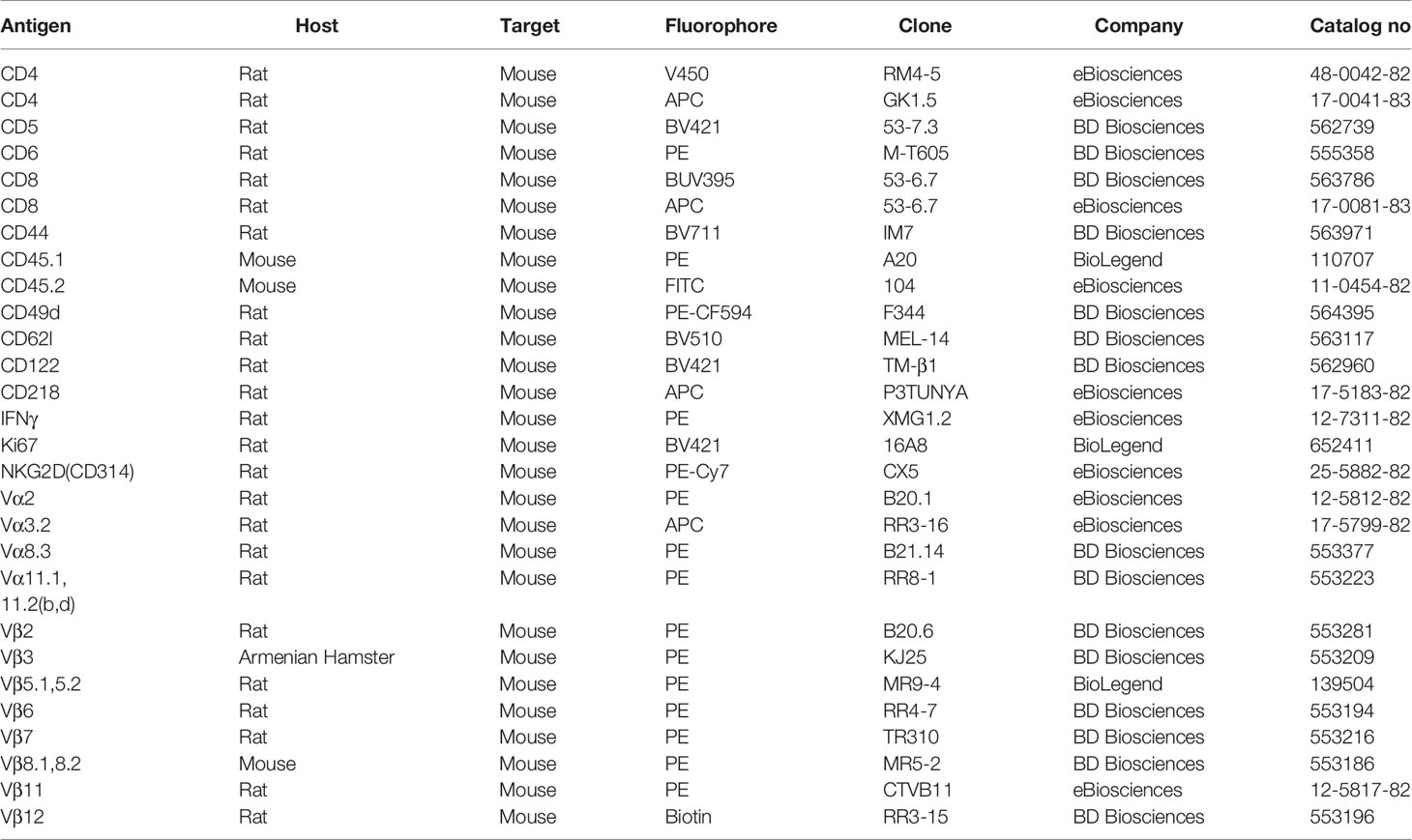
Table 1 Antibodies used for flow cytometry.
Cell pellets were washed twice with FWB for 5 minutes at 350g. The resulting cell pellet was vortexed after discarding the supernatant. 3 ml ice-cold ethanol was added to the cell pellet drop by drop while vortexing. Cells were votexed for another 30 seconds and then incubated at -20°C for an hour. The cells were then washed thrice with FWB. The resulting cells were then stained with Ki67 (BioLegend, California, USA) antibody at 1:1000 dilution and other cell surface antigens for 30 minutes at room temperature. The cells were then washed with 2 ml FWB and analyzed on a flow cytometer.
Mice were euthanized and dissected. Lymph nodes were excised and pooled together from several mice of the same genotype to prepare the samples for sorting. These were then mashed upon a 70 μm cell strainer into 5 ml cRPMI. The resulting cell suspensions were then spun down at 1200 rpm for 5 minutes at 4°C. The resulting cell pellet was surface stained by resuspending in 0.5 ml cRPMI per mouse containing fluorescently-conjugated antibodies at 1:500 dilution, followed by incubation at 4°C for 30 minutes on a shaker. They were then washed with cRPMI and then resuspended in 0.5 ml cRPMI per mouse for sorting. The cells were sorted on either Sy2000 (Sony Corporation, Tokyo, Japan) or Facsfusion (BD Biosciences, New Jersey, USA).
For the synthesis of the NGS libraries covering entire TCR repertoires, the total RNA was isolated from similar number of sorted SP CD8 thymocytes and peripheral CD8 T cells. The Vα3.2 TCR repertoires were retrieved from sorted Vα3.2+ CD8 SP thymocytes and Vα3.2+ CD8 peripheral T cells. The reverse transcriptase reaction was performed according to the previously published protocol (47) with template switching primers TAAGAGACAGCAACTACTACTGCrGrGrG (where r indicates ribonucleotide). Two rounds of amplification of the TCR’s cDNAs were performed using Q5® High-Fidelity DNA Polymerase (NEB, Massachusetts, USA) according to the manufacture instruction, with primers: tcgtcggcagcgtcagatgtgtataagagacagcaactactACTGC, GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGggtacacagcaggttctgg (first round), and CAAGCAGAAGACGGCATACGAGAT[i7]GTCTCGTGGGCTCGG AATGATACGGCGACCACCGAGATCTACAC[i5]TCGTCGGCAGCGTC where i7 and i5 represent Illumina Nextera V2 indexes (Illumina, California, USA). Libraries were purified using AMPure XP (Beckman Coulter, California, USA), and molar concentration of amplicons was quantified using Qubit DNA Assay (Thermo Fisher, Massachusetts, USA) and KAPA Library Quantification Kit (Kapa Biosystems, Massachusetts, USA). Sequencing of the TCR’s amplicons was performed on MiSeq platform using MiSeq Reagent Kits v2 (Illumina, California, USA).
Extraction of the sequences corresponding to the TCRs was performed using MiXCR platform (48). Further processing of data was done using VDJTools software (49) and Weblogo3 (50).
Sorted CD8+CD44lo cells were labeled with Cell Trace Violet (Life Technologies, California, USA). Cells were spun down and resuspended in PBS at a concentration of 2x106/ml. Cell Trace Violet was then added to the cell suspension at the concentration of 5 μM. Cells were vortexed immediately and incubated at 37°C for 10 minutes while vortexing every 5 minutes. After the incubation, media was added to quench the reaction at 5 times the original staining volume and further incubated for 5 minutes at 37°C. The cells were then spun down at 500 g for 5 minutes at 4°C. The cells were then used for the subsequent experiments.
After CTV labeling, live cells were counted and adjusted to the concentration of 1*106/ml. 100 μl of the cell suspension was seeded into a 96 well plate. For IL-15 stimulation, 100 μL of media with recombinant mouse IL-15 (Peprotech, New Jersey, USA) at the concentration of 2 μg/ml was added to the cells. The cells were incubated at 37°C for 5 days and then analyzed for proliferation by the CTV dilution on a flow cytometer. For IL-7/12/18 (Peprotech, New Jersey, USA) stimulation, 100 μl media with either IL-7 or a combination of either IL-7 and IL-12 or IL-7 and IL-18 (all at 100ng/ml) was added to the cells. The cells were incubated at 37°C for 7 days and then analyzed for proliferation by the CTV dilution on a flow cytometer.
After CTV labeling, live cells were counted and adjusted to the concentration of 2.5*106/ml. CD45.1 recipient mice were sublethally irradiated at 6 Gy. 0.2 ml cell suspension was injected retro-orbitally (intravenous i.v.) into either Rag1–/– or the sublethally irradiated CD45.1 mice. 5 recipient mice were injected per donor genotype and un-injected mice were used as controls. After one week, lymph nodes and spleens were excised from the CD45.1 mice or Rag1–/– mice after euthanizing them. The single cell suspensions from lymph nodes and spleens were stained for the congenic markers CD45.1, CD45.2 to distinguish donor and recipient cells. The proliferation of the donor cells was analyzed based upon CTV dilution as determined using a flow cytometer.
Themis–/– and Themis+/+ mice were infected with 104 colony-forming units of LM-OVA (51) via retro-orbital intravenous injection. The mice were euthanized at either day 4 or day 7 to analyze the bystander cells. Tetramer staining was done to gate out the antigen-specific cells. To assess their bystander activation, splenocytes were stimulated with IL-12 + IL-18 (Peprotech, New Jersey, USA) (100 ng/ml) for 6 hours in the presence of Brefeldin A (BD Biosciences, New Jersey, USA) at 1:500 dilution and stained for IFNγ via intracellular staining protocol mentioned above.
3.18 μl of PE labeled Streptavidin (1 mg/ml) (Life Technologies, California, USA) was added every 10 minutes for a total of 10 times to 10 μl of 2 mg/ml biotinylated H-2 Kb-OVA monomers in the dark. The tetramer was then used at 1:50 dilution for cell surface staining.
Prism (GraphPad Software, California, USA) and Excel (Microsoft Corporation, Washington, USA) were used for all statistical analysis and graphical representations. Normality of data was tested using Shapiro-Wilk test. All data sets were found to pass the normality test. Data are presented as means ± s.d., and we determined significance by two-sided Student’s t test. We considered a p-value of equal to or less than 0.05 as statistically significant.
Because Themis regulates thymocyte selection thresholds (39, 42), we predicted that Themis germline deletion would lead to changes in TCR repertoire. T cells from peripheral lymphoid organs (lymph nodes) were analyzed for any TCR bias resulting from Themis deficiency. The total lymphocyte pool was stained with specific TCR anti-Vα and -Vβ antibodies to analyze the TCR repertoire in Themis–/– and Themis+/+ mice. The biggest difference was in the proportion of Vα3.2+ CD8+ T cells (Figure 1A). Within the TCR Vα3 (TRAV9) family, Vα3.2 is found approximately two-fold more frequently in CD8+ cells than in CD4+ cells, whereas the other members are more frequently found in CD4+ cells (5, 7). The antibody RR3-16 was previously shown to recognize Vα3.2 rather than other Vα3 elements, recognizing Vα3 CDR1s with a phenylalanine at position 28 (5). This is only found in TRAV9N-3, not other TRAV9 family members [IMGT nomenclature and numbering (52)]. Thus the anti-Vα3.2 antibody RR3-16 identifies only T cells bearing a TRAV9N-3 TCR α-chain. Themis–/– mice had roughly three times higher proportion of Vα3.2+ CD8+ T cells compared to Themis+/+ mice (Figure 1A). Although there is an increase in their percentage in Themis–/– mice, the total Vα3.2+ CD8+ T cell counts were the same in Themis–/– and Themis+/+ mice (Figure 1B), despite the lymphopenia observed in Themis–/– mice (36–38). We also observed an increase in the proportion of Vβ5+ CD8+ T cells in Themis–/– mice as compared to Themis+/+ mice (Figure 1A). Based on this result, we predicted an increase in the proportion of CD8+ T cells expressing both Vα3.2 and Vβ5.1/5.2. Indeed, the percentage of these double positive cells was approximately three times higher in Themis–/– mice compared to Themis+/+ mice (Figure 1C). We did not observe differences in frequency of other TCR chains investigated, except a decrease in the proportion of Vα2+ CD4+ cells in Themis–/– mice relative to Themis+/+ mice (Supplementary Figure 1).
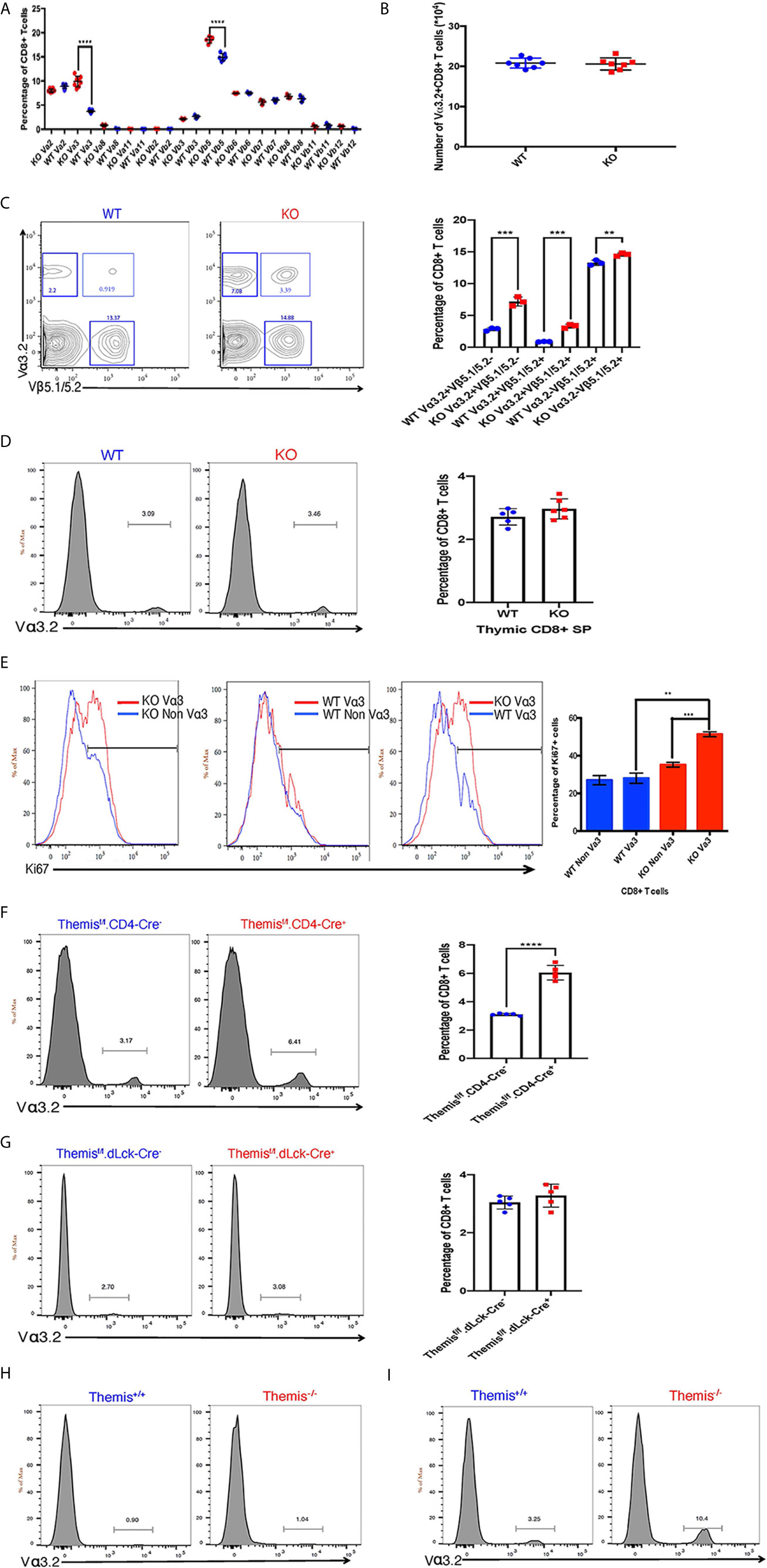
Figure 1 Changes in TCR repertoire of Themis–/– mice. (A) Proportion of various TCR α and β chains on CD8+ T cells in the periphery of Themis–/– (red) and Themis+/+ (blue) mice. (B) Absolute numbers of Vα3.2+ CD8+ T cells in the periphery of Themis–/– and Themis+/+ mice. (C) Proportion of Vα3.2+ Vβ5.1/5.2+ double-expressor CD8+ T cells in the periphery of Themis–/– and Themis+/+ mice. (D) Proportion of Vα3.2+ CD8+ SP T cells in the thymus of Themis–/– and Themis+/+ mice. (E) Proportion of Ki67+ cells amongst Vα3.2+ and non-Vα3.2+ CD8+ T cells in the periphery of Themis–/– and Themis+/+ mice. (F) Proportion of Vα3.2+ CD8+ T cells in the periphery of Themisf/f.CD4-Cre- and Themisf/f.CD4-Cre+ mice. (G) Proportion of Vα3.2+ CD8+ T cells in the periphery of Themisf/f.dLck-Cre- and Themisf/f.dLck-Cre+ mice. (H) Proportion of Vα3.2+ TCR on CD4+ T cells in the periphery of Themis–/– and Themis+/+ mice. (I) Proportion of Vα3.2+ TCR on CD8+ T cells in the periphery of Themis–/– and Themis+/+ mice. Data representative from three independent experiments with 4-5 biological replicates per genotype per experiment. **p < 0.01, ***p < 0.001, ****p < 0.0001 as determined by two-sided Student’s t-test. All error bars represent SDs.
To find out whether these changes in proportions of Vα3.2+ CD8+ T cells in Themis–/– mice originate in the thymus, we analyzed the cell surface expression of Vα3.2 in CD8 SP thymocytes. The increase in the proportion of Vα3.2+ CD8+ T cells was only observed in the periphery of Themis–/– mice, and not in the thymus (Figure 1D). We hypothesized that the increase of Vα3.2+ CD8+ T cells reflected their increased homeostatic expansion in the periphery. We analyzed expression of the proliferation marker Ki67 to estimate homeostatic proliferation of CD8+ T cells in the periphery. We found that a higher proportion of Vα3.2+ CD8+ T cells from Themis–/– mice express Ki67, compared to Vα3.2– CD8+ T cells from Themis–/– mice or Vα3.2+ or Vα3.2– CD8+ T cells from Themis+/+ mice (Figure 1E). This suggests increased proliferation of Themis–/– Vα3.2+ CD8+ T cells in response to self-pMHC in the periphery.
Although the increase in proportion of Vα3.2+ CD8+ T cells was apparent only in the periphery of Themis–/– mice, we could not rule out that this phenotype is due to altered thymic selection in the absence of Themis. To investigate this, we used pre-selection (CD4-Cre) and post-selection (dLck-Cre) Themis conditional knockout mice (45). We found that only the pre-selection CD4-Cre based Themis deletion model showed increase in the proportion of Vα3.2+ CD8+ T cells in the periphery, relative to CD4-Cre– mice (Figure 1F), whereas the post-selection dLck-Cre deletion model had no changes relative to dLck-Cre– mice (Figure 1G). This shows that the phenomenon of increased proportion of Vα3.2+ CD8+ T cells has thymic origins and requires deletion of Themis before thymic selection.
As expected from previous studies (5–7), this TCR is more likely to be MHC-I restricted, as the prevalence of Vα3.2+ TCR is much higher in CD8+ T cells than CD4+ T cells in both Themis-sufficient and -deficient mice (Figures 1H, I).
To more precisely define the development of the TCR Vα3.2+ compartment in the absence of Themis, we analyzed Vα3.2 (i.e. TRAV9N-3) repertoires from SP CD8αβ+ thymocytes and CD8αβ+ lymph node T cells that developed in the Themis–/– and Themis+/+ mice. After the reconstruction of TCR sequences from raw NextGen sequence datasets, we were able to identify 437 and 599 different clonotypes originating from Themis+/+ and Themis–/– SP thymocytes, respectively (Figure 2A). The peripheral pools of TCRs in our database comprised 1751 unique clonotypes from the Themis+/+ and 2094 from the Themis–/– mice. Comparison of the SP thymocytes revealed 70 public TCRs that account for 7.2% shared repertoire between these two genotypes (Figure 2A, left). In peripheral T cells, the number of public clonotypes rose to 473, giving 12.3% of the common component (Figure 2A, right). As this estimate does not consider the abundance of the individual receptor in a given repertoire, results can be biased by a large number of singletons in the datasets. However, the calculation of the similarities between Themis+/+ and Themis–/– repertoires confirmed these observed trends (Figure 2B). Indeed, thymocyte-derived datasets were placed at a significant mathematical distance from each other (Themis+/+ and Themis–/– thymocytes). In the periphery (Themis+/+ and Themis–/– LN), these differences dwindled. A plausible explanation of this observed phenomenon is that analysis of the thymic repertoire is restricted to the single wave of the SP thymocytes passing through at that time. In contrast, the datasets from the lymphocytes depict a more prolonged process, which includes clonal or homeostatic proliferation together with accumulation of clones over time.
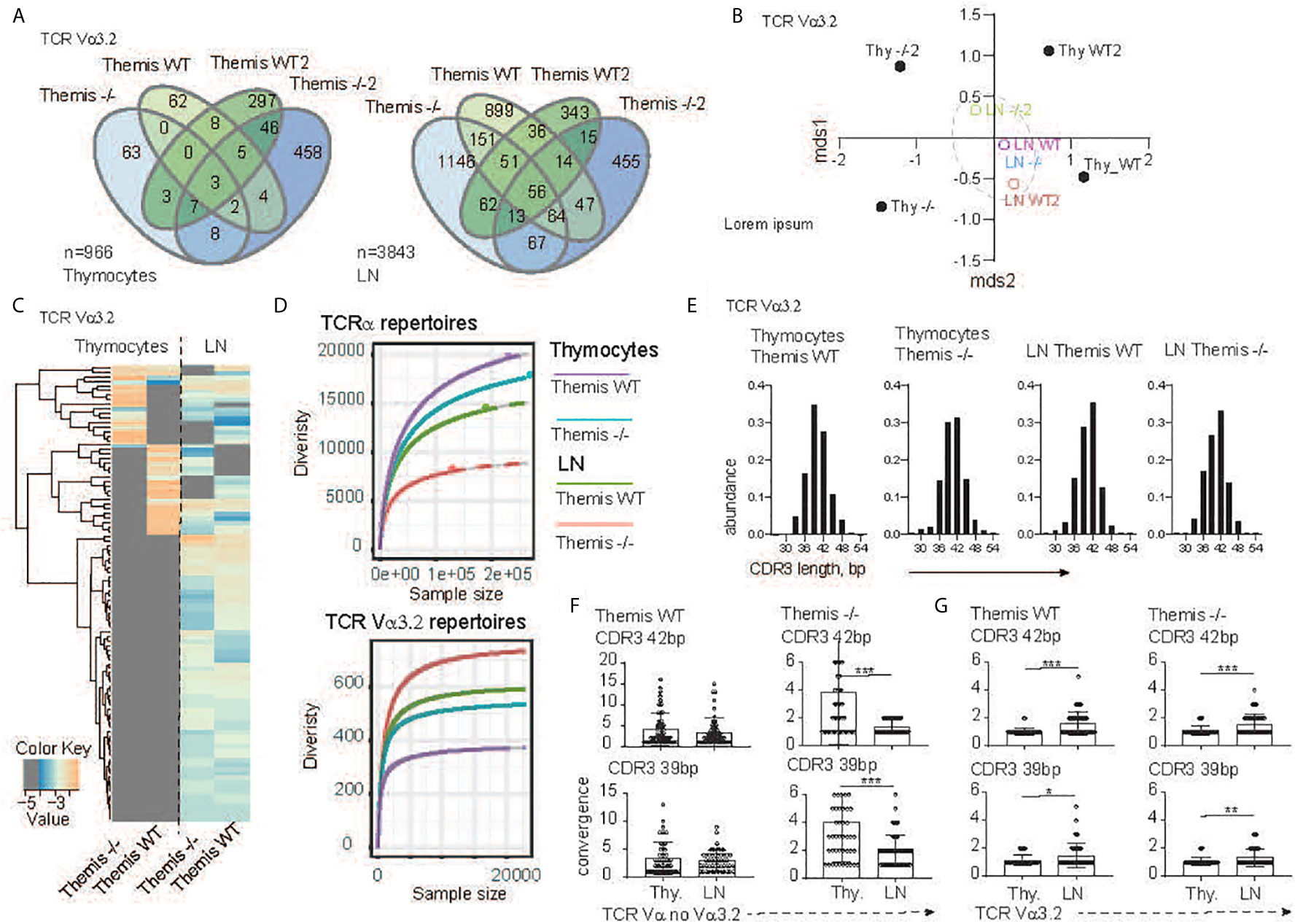
Figure 2 TCR sequencing analysis of the CD8 compartment from Themis+/+ and Themis–/–mice. (A) The Venn diagram depicts distribution of the individual TCR Vα3.2+ clonotypes within SP CD8+ thymocytes (left) and CD8+ lymphocytes (right) in Themis+/+ and Themis–/– mice. n indicates total number of detected clonotypes. (B) Dendrogram and non-metric multidimensional scaling (mds1 and mds2) ordination plot of Themis+/+ and Themis–/– TCR Vα3.2+ repertoire similarity. (C) Heatmap represents abundance of the individual TCR Vα3.2+ clonotypes in the SP thymocytes and lymphocytes in Themis+/+ and Themis–/– mice. (D) The repertoire diversity within thymocytes and peripheral T cell subsets. Upper graph. Diversity was calculated in the context of the entire TCRα repertoires. Lower panel analysis was restricted to the TCR Vα3.2+ (TRAV9N-3) compartment. Rarefaction curves were plotted based on a multinomial model (53) and extrapolated to the largest sample. (E) In silico spectratyping of the CDR3 region of the TCR Vα3.2+ compartments. CD8+ T cell populations and genotype are indicated on the top of each graph. TCR convergence estimated in the 50 most dominant clones with (F) non Vα3.2 and (G) Vα3.2+ TCRs representing 39 or 42 bp CDR3 lengths, respectively. TCR compartment, population and genotypes are indicated on the graphs. In all figures, data for each genotype were pooled from two individual experiments. Data were considered statistically significant when *p < 0.05, **p < 0.01, ***p < 0.001 as determined by for two-sided Student’s t-test with Welch’s correction.
Analysis of the individual clones’ distribution indeed revealed more similarities between the T cells’ repertoires in the lymph node environment (Figure 2C). Importantly, many of the unique TCRs found in the Themis+/+ and Themis–/– SP thymocytes acquire a public character in the peripheral lymphatic organs (Figure 2C). All these data collectively indicate that Vα3.2+ CD8+ T cell accumulation is a primary mechanism orchestrating peripheral repertoire development, and exclude Themis-dependent clonal deletion during the thymocytes’ progression. Themis deficiency affects the CD8+ compartment in both quantitative and qualitative ways (Figure 2D). Decrease of the total number of SP thymocytes and peripheral T cells in the Themis–/– model is accompanied by a substantial drop in TCR repertoire diversity within the CD8+ subsets when compared to the Themis+/+ counterpart (Figure 2D, upper panel). Paradoxically, these relationships were inverted when we analyzed only the TCR Vα3.2+ compartments. In other words, the Vα3.2 repertoire was more diverse in Themis-deficient LN CD8+ cells than Themis-sufficient LN CD8+ cells. These were both more diverse than the SP thymocytes Vα3.2 repertoire, but even there, the Themis-deficient cells had more diversity than Themis-sufficient cells (Figure 2D, lower panel). Peripheral expansion of individual T cells results in an overall decrease of TCR repertoire diversity in the LN (Figure 2D, upper panel).
As individual clonotypes proliferate, the expansion process reduces the proportion of different DNA sequences for a given amino acid sequence. We term this “TCR convergence”. Thus, to test whether the Vα3.2 clonal enrichment observed in the periphery was due to accumulation of different clones rather than to clonal expansion, we analyzed TCR convergence from thymus and lymph node-derived repertoires. Because all the repertoires were dominated by clones with 39 and 42 base pair-long CDR3s (Figure 2E), we restricted this estimation to the 50 most dominant clones representing a given CDR3 length. In the non-Vα3.2+ TCR repertoire from Themis+/+, convergence of individual TCRs was not significantly altered between thymus and LN (Figure 2F, left panel). In the same fractions of the CD8+ repertoire from Themis–/– mice, LN-derived TCR clonotypes had a reduced number of different DNA sequences in comparison to clonotypes associated with thymocytes (Figure 2F, right panel). This is likely due to LIP in the Themis-deficient mice. When we estimated TCR convergence in the Vα3.2+ T cell fraction, the result was strikingly different. Regardless of mouse genotype, the number of DNA sequences coding individual CDR3s increased in the peripheral repertoire (Figure 2G). These data strongly support the notion that the accumulation of Vα3.2+ T cells accounts for the repertoire enrichment in the periphery. Hence, we hypothesize that this phenomenon might be associated with better survival of the CD8+ Vα3.2+ clones, perhaps because of unique features of this TCR.
After finding that Themis deficiency induces an increase in the proportion of Vα3.2+ CD8+ T cells, we phenotyped these cells to try to understand their unique developmental and functional characteristics. Since the data from Ki67 staining indicated stronger proliferative responses to self-pMHC, we analyzed these cells for CD5 expression, a negative regulator of signaling that reports the strength of the interaction of TCR with self-pMHC (12, 14, 15). Another negative regulator of TCR signaling is CD6, a protein related to CD5. Although CD6 has not been shown to be directly involved in maintenance of homeostasis, CD6–/– mice show a very similar phenotype to CD5–/– mice, suggesting a similar function (54, 55). We therefore tested if CD6 can report signal from self-pMHC. Our data show that CD6 expression is correlated with that of CD5 within each of the thymocyte and lymphocyte populations analyzed, indicating that expression of CD6 and CD5 are regulated by a similar mechanism (Supplementary Figure 2A). Moreover, we found that reduction in signal from self pMHC through transfer of MHC class I-restricted OT-I CD8+ T cells into β2m deficient recipients resulted in decreased CD6 expression (Supplementary Figure 2B) in addition to the previously reported decrease in CD5 expression (56). These data strongly indicate that CD6 surface expression directly reports signal strength from self pMHC. CD44 is a memory and activation phenotypic marker (57), but has also been shown to correlate with CD5 expression (58–60). Thus, lymphocytes from Themis+/+ and Themis–/– mice were stained with CD5, CD6 and CD44-specific antibodies and analyzed by flow cytometry. Since Themis–/– mice have a higher proportion of CD44hi cells due to lymphopenia (36–38), we gated on CD44lo cells for our analysis of CD5, CD6 and CD44 expression (Supplementary Figure 2C). We observed that Vα3.2+ cells express more CD5 and CD6 than non Vα3.2-expressing cells in both Themis+/+ and Themis–/– mice, but the ratio of CD5 and CD6 expression between Vα3.2+ cells and non-Vα3.2 cells was even higher in Themis–/– mice (Figures 3A, B). We observed even more contrasting differences in expression of CD5 and CD6 between Vα3.2+ and non Vα3.2-expressing CD8 SP thymocytes (Figures 3C, D). These results suggest that Vα3.2+ cells receive stronger signal from self pMHC during development in the thymus, corroborating the findings from the conditional Themis deletion models, where the increased proportion of Vα3.2-expressing cells occurred if knockout happened before selection (CD4-Cre), but not if it occurred after selection (dLck-Cre) (Figures 1F, H). These cells also express more CD44 in the periphery (Figure 3E), indicating a virtual memory phenotype.
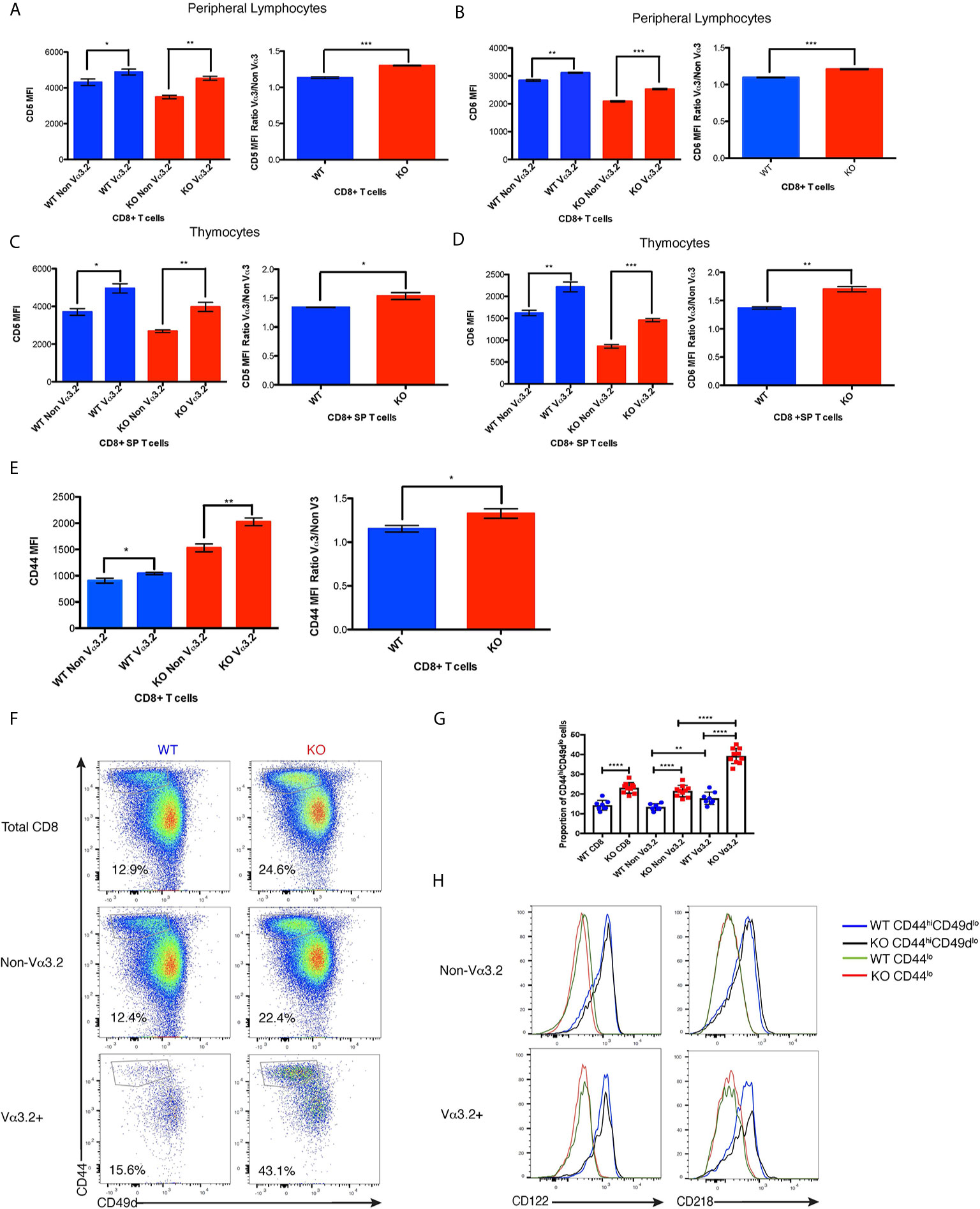
Figure 3 Phenotypic profile of Vα3.2+ CD8+ T cells in Themis–/– and Themis+/+ mice. (A) CD5 expression on Vα3.2+ and non-Vα3.2+ CD8+ T cells in the periphery of Themis–/– and Themis+/+ mice. (B) CD6 expression on Vα3.2+ and non-Vα3.2+ CD8+ T cells in the periphery of Themis–/– and Themis+/+ mice. (C) CD5 expression on Vα3.2+ and non-Vα3.2+ CD8+ SP thymocytes from Themis–/– and Themis+/+ mice. (D) CD6 expression on Vα3.2+ and non-Vα3.2+ CD8+ SP thymocytes from Themis–/– and Themis+/+ mice. (E) CD44 expression on Vα3.2+ and non-Vα3.2+ CD8+ T cells in the periphery of Themis–/– and Themis+/+ mice. (F) Representative FACS plots for CD44 vs CD49d staining in CD8+ T cells in the periphery of Themis–/– and Themis+/+ mice. (G) Bar plots showing summary of proportion of CD44hiCD49dlo cells in CD8+ T cells in the periphery of Themis–/– and Themis+/+ mice. (H) Histogram of CD122 and CD218 staining in CD44hiCD49dlo vs CD44lo populations of Vα3.2+ and non-Vα3.2+ CD8+ T cells in the periphery of Themis–/– and Themis+/+ mice. Data are representative of three independent experiments with 4-5 biological replicates per genotype per experiment. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 as determined by two-sided Student’s t-test. All error bars represent SDs.
As virtual memory phenotype cells have been classically defined as CD44hiCD49dlo (24), we wanted to check whether this population would be over-represented in the Vα3.2+ cells. We observed that Vα3.2+ CD8+ T cells had a higher proportion of CD44hiCD49dlo cells than non Vα3.2-expressing cells in both Themis+/+ and Themis–/– mice (Figures 3F, G). This difference was much more enhanced in Themis–/– mice, where almost 40% of the Vα3.2+ cells were CD44hiCD49dlo as compared to only 20% of the non Vα3.2-expressing cells (Figures 3F, G). Since virtual memory cells have been shown to be dependent on cytokine signaling for survival (24), we looked at the expression of CD122 (common gamma chain receptor for IL-2 and IL-15) and CD218 (IL-18 receptor). We observed that only the CD44hiCD49dlo population expressed these markers (Figure 3H). Since a higher proportion of Vα3.2+ CD8+ T cells were CD44hiCD49dlo, and the CD44hiCD49dlo cells express CD122 and CD218, this confirms the preferentially virtual memory phenotype of the Vα3.2+ CD8+ T cells.
The unique behavior of the TCR Vα3.2 (TRAV9N-3) repertoire and the phenotype of the Vα3.2-expressing cells raised the possibility of TCR-driven mechanisms orchestrating the function and peripheral molding of this CD8+ T cell compartment. The physical features of Vα segments, in particular CDR1 and CDR2, determine interactions with the α-helices of the MHC-I (5, 7, 9). At the same time, the increased amount of surface CD5 and CD6 in TCR Vα3.2+ CD8+ cells suggests enhanced interaction of these cells with self-pMHC-I. Hence, CDR3 might be involved in increased self-ligand recognition by the Vα3.2-carrying T cells (61, 62).
To shed more light on this issue, we looked more closely into the peptide sequences representing CDR3 regions. Besides TRAV9N-3 (Vα3.2), we selected TCRs that use TRAV14 (Vα2), TRAV6 (Vα4), and TRAV12 (Vα8) as a representative fraction of the lymph node- and thymus-derived CD8+ TCRα repertoire (Figure 4A, Supplementary Figure 3A). We restricted datasets to those TCRs with 14 amino acid-long CDR3 as these were most abundant across the investigated repertoires (Figure 2E). Interestingly, CDR3 sequences in Vα3.2 (TRAV9N-3) TCRs showed a distinctive physical makeup (Figure 4B). Estimated overall hydropathicity revealed that the CDR3s of the Vα3.2 TCRs were much more hydrophobic than those found in the other investigated repertoires. These trends were observed in both thymus and LN-derived sequences, but the differences were much stronger in the peripheral repertoires (Figure 4B upper panel). Similarly, CDR3s associated with Vα3.2 were more positively charged than their counterparts from non-Vα3.2 TCRs (Figure 4B lower panel).
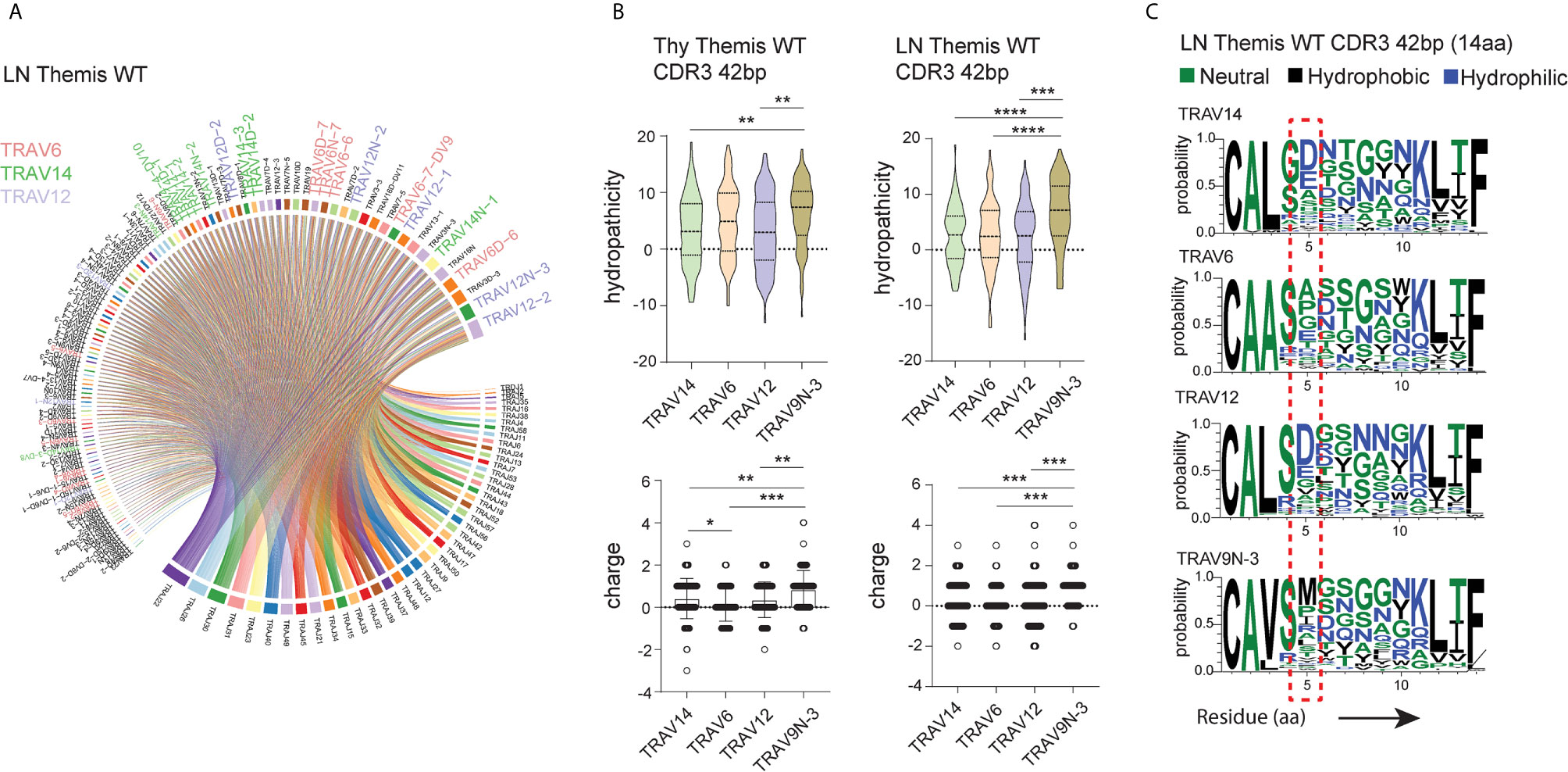
Figure 4 Unique physical features of CDR3 of Vα3.2+ CD8+ T cells. (A) Vα and Jα segment usage in the peripheral CD8+ TCRα repertoire. TRAV12, TRAV6, and TRAV14 segments are highlighted: lavender, plum, and green, respectively. (B) Hydrophobicity (upper panel) and charge (lower panel) of the CDR3 regions from Vα3.2 (TRAV9N-3) and a representative fraction of non-Vα3.2 receptors. Each Vα-associated group is represented by the 100 most dominant clonotypes from 14 amino acid-long CDR3 compartment. (C) Distribution of the amino acids within CDR3 regions. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 as determined by two-sided Student’s t-test with Welch’s correction.
To determine the source of these unique characteristics, we analyzed the distribution of amino acids within the CDR3s. In the Vα3.2+ TCRs, the fifth position was more frequently occupied by hydrophobic amino acids (Figure 4C). Methionine in this position was restricted to Vα3.2 TCRs, accounting for around 30% of the entire pool of clonotypes within the TRAV9N-3 repertoire. Isoleucine and leucine in the fifth position were not specific to Vα3.2 CDR3s, but they were much more abundant in the Vα3.2 CDR3 pool, together representing another 30% of the TRAV9N-3 repertoire. In contrast, negatively charged residues glutamic acid and, much more abundantly, aspartic acid, were frequent in the fifth position in the non-Vα3.2 TCRs (Supplementary Figure 3B). Finally, to test whether these observations were restricted to the investigated fraction of the repertoires, we analyzed the overall hydropathicity and charge of CDR3s (datasets were not restricted to the particular CDR3 length) from Vα3.2 and the entire non-Vα3.2+ TCR pool. Again, CDR3 from Vα3.2 showed a more hydrophobic and more positively charged repertoire (Supplementary Figure 3C).
Increased CD5, CD6, and CD44 surface expression suggested self-reactivity and better survival of Vα3.2+ CD8+ T cells in the periphery. Previous studies have reported that CD5hi cells are hyperresponsive to in vitro stimulation with IL-7, and that proliferative responses to IL-7 by CD5hi cells were increased upon addition of pro-inflammatory cytokines such as IL-12 and IL-18 (16, 63, 64). Therefore, we hypothesized that Vα3.2+ CD8+ T cells would be more responsive to cytokine signaling than non-Vα3.2+ CD8+ T cells, specially for Themis-deficient CD8+ T cells. To test this hypothesis, we sorted naïve CD8+ T cells from Themis–/– and Themis+/+ mice and stimulated them in vitro in an antigen-independent manner with IL-7+IL-12, or IL-7+IL-18. Vα3.2+ CD8+ T cells proliferated better than non-Vα3.2+ CD8+ T cells, in response to IL-7+IL-18 stimulation. This difference between the two populations was stronger for Themis-deficient CD8+ T cells (Figures 5A–C). In response to IL-7+IL-12 stimulation, Themis–/– CD8+ T cells showed reduced proliferation in comparison to Themis+/+ CD8+ T cells. However, we observed significantly higher proliferation of Themis–/– but not Themis+/+ Vα3.2+ cells compared to non-Vα3.2+ cells (Figures 5D–F). These results indicate that Themis–/– Vα3.2+ CD8+ T cells respond more strongly to cytokines as compared to Themis–/– Vα3.2- CD8+ T cells, resulting in enhanced homeostatic proliferation.
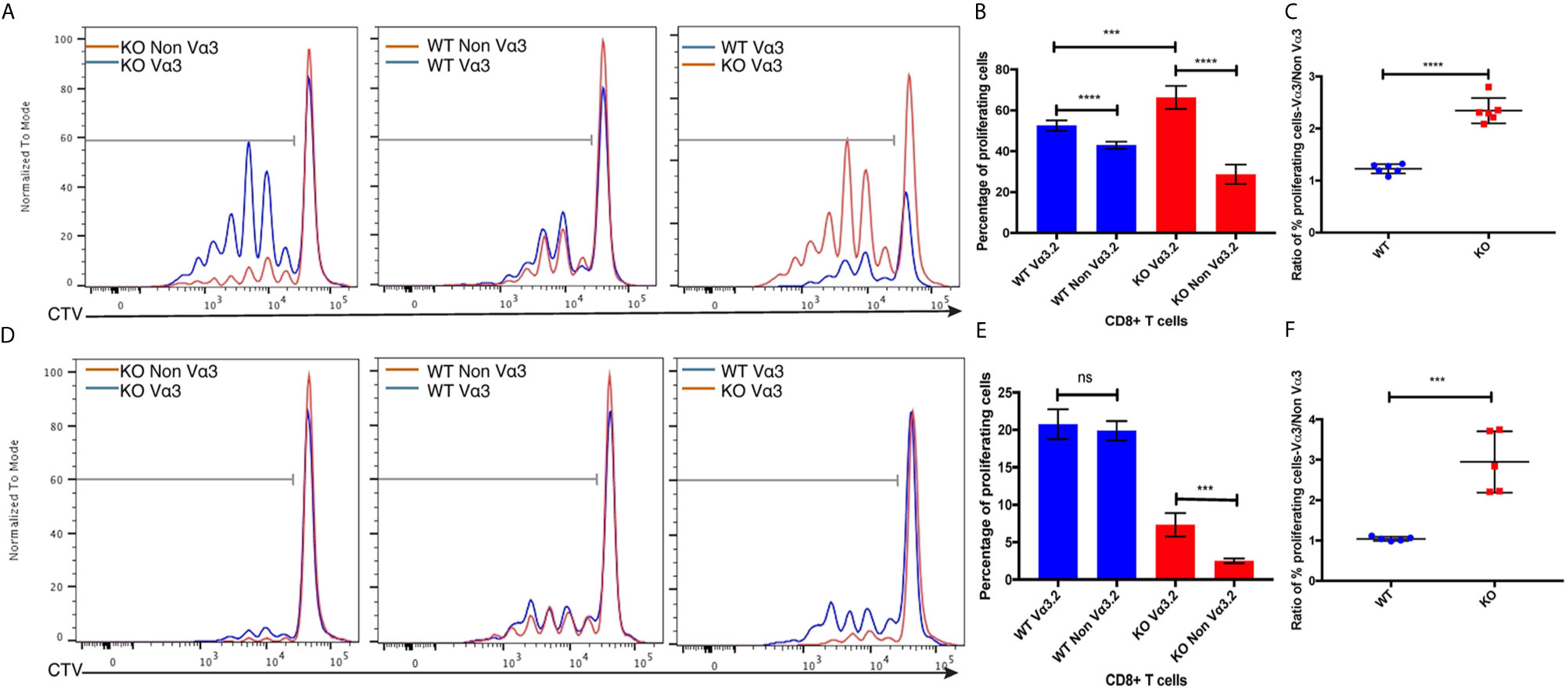
Figure 5 Responses of Vα3.2+ CD8+ T cells to pro-inflammatory cytokine stimulation in vitro. (A) Proliferation of Vα3.2+ and non-Vα3.2+ naïve CD8 T cells from Themis–/– and Themis+/+ mice in response to IL-7 + IL-18 stimulation. (B) Histogram summary of the proliferative responses to IL-17 + IL-18 stimulation. (C) Ratio of % proliferating Vα3.2+ and non-Vα3.2+ CD8+ T cells upon IL-7 + IL-18 stimulation. (D) Proliferation of Vα3.2+ and non-Vα3.2+ naïve CD8+ T cells from Themis–/– and Themis+/+ mice in response to IL-7 + IL-12 stimulation. (E) Histogram summary of the proliferative responses to IL-7 + IL-12 stimulation. (F) Ratio of % proliferating Vα3.2+ and non-Vα3.2+ CD8+ T cells upon IL-7 + IL-12 stimulation. Data representative from three independent experiments with 4-5 biological replicates per genotype per experiment. nsnot significant, ***p < 0.001, ****p < 0.0001 as determined by two-sided Student’s t-test. All error bars represent SDs.
To investigate the peripheral increase of Vα3.2+ CD8+ T cells and their LIP potential, we injected sorted naïve CD8+ (CD44lo) T cells from Themis–/– and Themis+/+ mice (CD45.2) into sublethally irradiated CD45.1 recipients. After a week, the recipients were euthanized and lymph nodes and spleens were analyzed. Overall, Themis+/+ CD8+ T cells showed enhanced LIP compared to Themis–/– CD8+ T cells (Figure 6), but in Themis–/– mice, the Vα3.2+ cells were slightly more proliferative than the non-Vα3.2+ cells. These results suggest better survival of Vα3.2+ cells in a lymphopenic environment, and explain the increase in the proportion of Vα3.2+ cells observed in the periphery of Themis–/– mice. We did not see any differences in LIP between Vα3.2+ and non-Vα3.2+ CD8+ T cells injected into Rag1–/– hosts (Supplementary Figure 4), possibly because proliferation in Rag1–/– mice is in response to the gut microbiome rather than the lymphopenia itself (65).
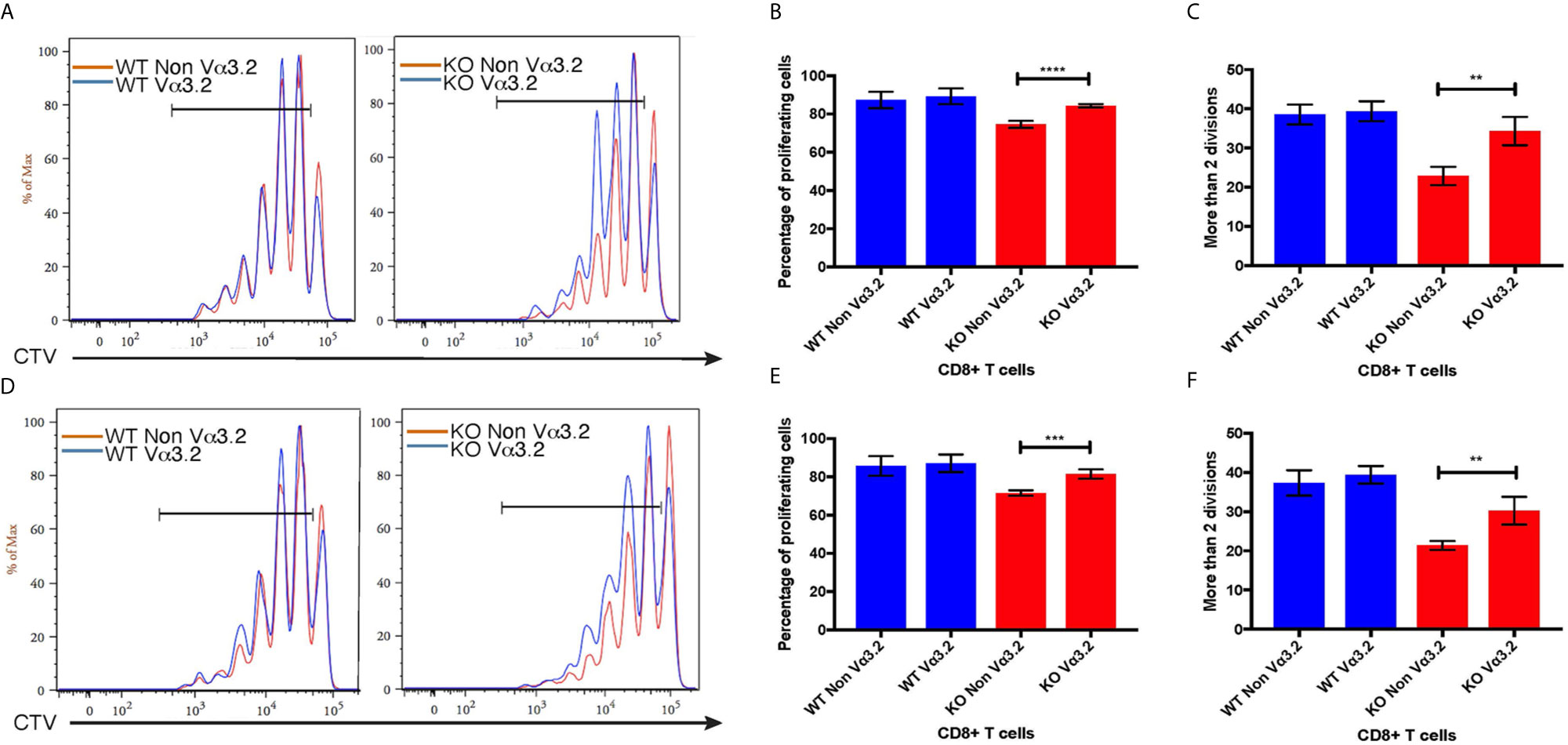
Figure 6 Proliferative responses of Vα3.2+ CD8+ T cells in lymphopenic hosts. Proliferation of Vα3.2+ and non-Vα3.2+ naïve CD8+ T cells from Themis–/– and Themis+/+ mice in (A) lymph nodes and (D) spleen of sublethally irradiated CD45.1hosts. Histogram summary of the proliferation responses in (B) lymph nodes and (E) spleen of sublethally irradiated CD45.1 hosts. Proportion of Vα3.2+ and non-Vα3.2+ CD8+ T cells from Themis–/– and Themis+/+ mice that had more than two divisions in (C) lymph nodes and (F) spleen of sublethally irradiated CD45.1 hosts. Data are representative from three independent experiments with 4-5 biological replicates per genotype per experiment. nsnot significant, **p < 0.01, ***p < 0.001, ****p < 0.0001 as determined by two-sided Student’s t-test. All error bars represent SDs.
The virtual memory phenotype (CD44hi) Vα3.2+ cells also suggested better responses to IL-15 stimulation, as IL-15 is required for maintenance of the memory pool (66). Thus, we tested the response of naïve CD8+ T cells from Themis–/– and Themis+/+ mice to IL-15. In response to IL-15 stimulation in vitro, Vα3.2+ CD8+ T cells proliferated more than non-Vα3.2+ CD8+ T cells (Figures 7A, B), leading to an increase in the proportion of Vα3.2+ cells. This increase in proportion was enhanced and much more obvious in Themis–/– cells compared to Themis+/+ cells (Figure 7C).
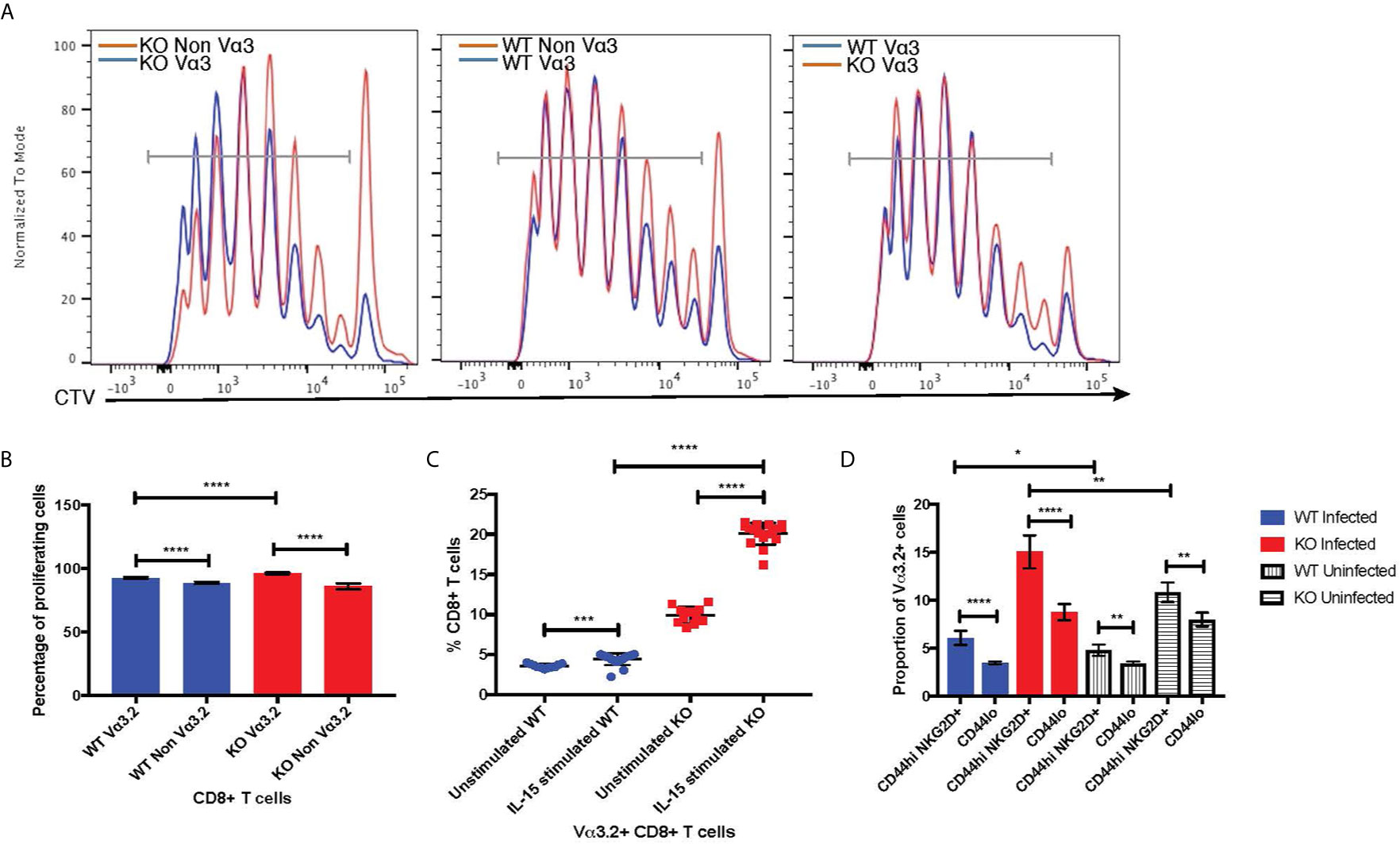
Figure 7 Bystander potential of Vα3.2+ CD8+ T cells. (A) Proliferation of Vα3.2+ and Vα3.2- naïve CD8+ T cells from Themis–/– and Themis+/+ mice in response to IL-15 stimulation. (B) Histogram summary of the proliferative responses to IL-15 stimulation. (C) Proportion of Vα3.2+ and non-Vα3.2+ CD8 T cells upon IL-15 stimulation. (D) Proportions of Vα3.2+ T cells in the bystander (CD8+ Tetramerneg CD44hi NKG2D+) and control (CD8+ Tetramerneg CD44lo) population on day 4 of LM-OVA infection in uninfected and infected Themis–/– and Themis+/+ mice. Data are representative from three independent experiments with 3-4 biological replicates per genotype per experiment. nsnot significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 as determined by two-sided Student’s t-test. All error bars represent SDs.
Increased response to IL-15 stimulation indicates the potential of Vα3.2+ CD8+ T cells to be bystander cells. These help antigen specific cells during an infection, by becoming activated in a non-antigen specific manner by cytokines produced in the local milieu and performing effector functions to help clear the infection (67). To investigate the bystander potential in an infection model, we infected Themis–/– and Themis+/+ mice with a genetically engineered strain of Listeria monocytogenes which expresses ovalbumin (LM-OVA) (51) and sacrificed them at day 4 to preclude antigen specific responses (68). We confirmed the absence of CD8+ T cells specific for OVA-derived SIINFEKL peptide at day 4 using tetramer staining; whereas SIINFEKL-specific CD8+ T cells were abundant on day 7 post infection (Supplementary Figure 5A). Bystander cells were defined as CD8+ Tetramerneg CD44hi NKG2D+ (Supplementary Figures 5B, C) (68). We analyzed the proportion of Vα3.2+ cells in this population, and compared with the proportion of Vα3.2+ cells in the CD8+ Tetramerneg CD44lo population as a control. We observed a higher proportion of Vα3.2+ cells in the bystander population compared to the control (Figure 7D). This effect was observed in Themis-sufficient and –deficient mice, but the magnitude was bigger in Themis–/– mice, possibly due to their higher proportion of Vα3.2+ cells and a higher proportion of CD44hi cells. We identified some CD8+ Tetramerneg CD44hi NKG2D+ bystander phenotype cells in uninfected mice, but this population was clearly increased upon infection. To test the bystander potential of the CD8+ Tetramerneg CD44hi NKG2D+ cells from the infected and uninfected mice, we measured cytokine production in a non antigen-dependent manner, after IL-12+IL-18 stimulation for 6 hours (68, 69). Only bystander cells from mice infected with LM-OVA were able to produce IFN-γ, showing that the bystander effect was real and induced by infection (Supplementary Figure 5D). However, we did not observe any statistically significant differences between Vα3.2+ cells and non-Vα3.2+ cells in cytokine production.
The processes of negative and positive selection that shape the TCR repertoire occur in the thymus. Themis has been shown to be involved in these processes (36–39). Thus, we hypothesized that the TCR repertoire of Themis–/– mice might be different than that of Themis+/+ mice. Our results show that there are indeed changes in TCR repertoire in Themis–/– mice. We observed that the proportion of Vα3.2+ CD8+ T cells and Vα3.2+ Vβ5.1/5.2+ cells was triple or more in the periphery of Themis–/– mice than Themis+/+ mice. We have previously found in rearranging TCR minigene mice that co-expression of Vα3.2 and Vβ5.2 is a favored combination (70). This increase in the number of CD8+ T cells carrying Vα3.2 TCR occurred in the periphery, as the proportion of these cells in the thymus were similar between Themis–/– and Themis+/+ mice. This peripheral increase is corroborated by their expression of Ki67, a marker of S, G2 and M phases of the cell cycle, and therefore a proliferation marker. This indicates that these CD8+ T cells are undergoing proliferation in the lymphopenic niche of Themis–/– mice, possibly in response to the same self-pMHC ligands on which they are positively selected in the thymus. As expected, we also observed that both Vα3.2+ and Vβ5.1/5.2+ TCRs were preferentially expressed in CD8+ T cells rather than CD4+ T cells. This is due to preferential selection on MHC-I rather than MHC-II after binding of CDR1 and CDR2 regions of these TCRs to MHC α-helices (5, 6). The quantitative changes in the proportion of the CD8+ Vα3.2+ and non-Vα3.2+ compartments in Themis–/– and Themis+/+ mice were accompanied by a substantial increase in TCR repertoire diversity of the Vα3.2+ cells in the Themis–/– model. The Vα3.2 clonotype enrichment in the thymus and peripheral lymphatic organs of Themis–/– mice might indicate resistance of these cells to clonal deletion in the thymus and better survival in the periphery. Interestingly, observed trends in the thymic and peripheral CD8+ Vα3.2+ repertoire mirrored phenomena attributed to the central commitment and peripheral reshaping of the CD4+ regulatory (Treg) subset. Like the CD8+ Vα3.2+ compartment, Tregs retain a highly diverse TCR repertoire in the periphery (71, 72). Importantly, Treg selection relies on recognizing self‐antigen in the thymus (73), and self‐antigen recognition constitutes a crucial factor in further remolding of this T cell subset in the periphery (74, 75).
We analyzed the expression of CD5 on Vα3.2+ CD8+ T cells, as CD5 reports the strength of interaction with self-peptide MHC (14) and response from cytokine signals (60). Although T cells from Themis–/– mice had lower CD5 compared to T cells from Themis+/+ mice (76), we found that Vα3.2+ CD8+ T cells showed higher expression of CD5 than non-Vα3.2+ cells in both Themis–/– and Themis+/+ mice, and that the ratio of CD5 MFI between Vα3.2+ and Vα3.2– CD8+ T cells was higher in Themis–/– mice than Themis+/+ mice, both in the thymus and the periphery. Similar results were observed for CD6 surface expression, which we verified as a marker of signal strength from self pMHC. Thus phenotypic differences between Vα3.2+ and Vα3.2– CD8+ T cells begin in the thymus. We also observed higher expression of the memory marker CD44 in peripheral Vα3.2+ CD8+ T cells compared to non-Vα3.2+ CD8+ T cells. CD44 expression has been shown to increase upon cell division in lymphopenic environments (20, 21), again indicating increased LIP by Vα3.2+ T cells. Data from the expression of these molecules suggest that Vα3.2+ T cells interact more productively with self-pMHC compared to non-Vα3.2-expressing T cells, which could explain why they are better at proliferating in the periphery, as indicated by their Ki67 expression. Taken together with our CD5 and CD6 data, it suggests that Vα3.2+ T cells are highly self-reactive. This notion was further supported by the analysis of CDR3 peptide sequences of Vα3.2+ and non-Vα3.2+ TCRs in the context of their physical features. Distribution of the hydrophobic residues within CDR3 constitutes unique physical makeup of Vα3.2+ TCRs indicating enhanced self-reactivity within this CD8+ T cell fraction (62).
T cells with high CD5 and CD44 expression have better responses to cytokine signaling (57, 60). We observed that, in response to IL-7+12/18 stimulation in vitro, we observed higher proliferative responses from Vα3.2+ CD8+ T cells compared to non-Vα3.2-expressing CD8+ T cells from Themis–/– mice. This again indicates better survival and homeostatic potential of these cells. When we injected sorted T cells into sublethally irradiated host mice, we observed that Vα3.2+ CD8+ T cells from Themis–/– mice showed enhanced LIP compared to their non-Vα3.2-expressing counterparts, but we did not see the same effect when we injected these cells into Rag1–/– hosts. This is not surprising as it has been reported that irradiated hosts are able to accumulate more donor cells than Rag1–/– hosts, because proliferation in Rag1–/– hosts is driven primarily by bacteria from the microbiome, since Rag1–/– hosts are severely immunodeficient (65). The enhanced LIP of Vα3.2+ CD8+ T cells from Themis–/– mice cells shows that they are better at survival in the periphery and possibly indicates their proliferation and maintenance by integration of strong interactions with self-peptide MHC and cytokine signaling.
In response to IL-15 stimulation, the proportion of Vα3.2+ CD8+ T cells from Themis–/– mice doubled in number compared to non-Vα3.2-expressing CD8+ T cells from Themis–/– mice. We saw only a slight increase in the frequency of Vα3.2+ T cells upon IL-15 stimulation in Themis+/+ mice. This higher response to IL-15 stimulation in the Themis–/– mice indicates the potential of Vα3.2+ T cells to be bystanders, which help the antigen specific cells during an infection. Bystander cells become activated in a non-antigen specific manner by the cytokines produced locally, and perform effector functions to help clear the infection. Bystander cells display increased lytic capabilities and are found to be recruited to the sites of infection such as the lungs during an influenza infection (67). In recent reports, bystander cells have been shown to be involved in restraining HIV reservoir (34) and implicated in the immune response to Covid-19 (35). We tested this bystander potential in an LM-OVA model, where mice were sacrificed 4 days after infection, such that no OVA-specific cells could be detected, and bystander cells were gated based on CD44 and NKG2D expression as in previous reports (68). We observed higher proportions of Vα3.2+ CD8+ T cells in the bystander population. This phenomenon was amplified in Themis–/– mice, and these cells were able to produce IFN-γ upon IL-12+IL-18 stimulation in an antigen independent manner, demonstrating their bystander potential.
Overall, this work shows that the TCR repertoire is generally reduced in diversity in the absence of Themis. However, there was an unpredicted effect on T cells with a certain TCR Vα region: cells expressing Vα3.2 (TRAV9N-3), were even more frequent than usual in the CD8+ population in Themis-deficient mice. Their TCRα CDR3 repertoire was increased in Themis-deficient CD8+ peripheral T cells. Moreover, they had an unusual phenotype that indicated a stronger stimulation by self pMHC, higher responsiveness to cytokines, and an effector memory and bystander phenotype. The bystander phenotype was borne out functionally in cells responding to Listeria infection. Bystander T cells are commonly found during viral infections (34, 35) and tumor micro-environments (77), so a better understanding of such cytokine-responsive T cell populations with unique TCRs could help to harness them for cellular therapy against infections.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/, GSE162963.
The animal study was reviewed and approved by National University of Singapore Institutional Animal Care and Use Committee (NUS IACUC).
MP, LW, and JB performed the literature review, planned the experiments, and did data interpretation. MP performed most of the experiments. LW performed the TCR sequencing and CDR3 analysis. JB assisted in the Listeria infection experiments and performed the CD6 validation experiments. YC and DT assisted in the mouse breeding and irradiation of mice. JY generated the biotinylated H-2 Kb-OVA monomers. VR and JH were involved in optimization of the experimental protocols. NG supervised the study and did data interpretation. All the authors contributed to writing and editing the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by the Singapore Ministry of Health’s National Medical Research Council under its CBRG/0097/2015 and by the Singapore Ministry of Education’s grant 2014-T2-1-136, to NG. Work performed at Scripps Research was supported by NIH grants AI073870 and DK094173 to NG.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. P. Hutchinson and Mr. G. Teo (NUS Immunology Program Flow Cytometry Laboratory) for helping with cell sorting.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.644483/full#supplementary-material
Supplementary Figure 1 | Proportion of various TCR α and β chains on CD4+ T cells in Themis–/– (red) and Themis+/+ (blue) mice. Data are representative from three independent experiments with 4-5 biological replicates per genotype per experiment. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 as determined by two-sided Student’s t-test. All error bars represent SDs.
Supplementary Figure 2 | (A) Correlation between CD5 and CD6 cell surface expression on peripheral naïve CD4+ (CD25–, CD44low) and CD8+ (CD44low) T cells. CD5 and CD6 MFI on 10% cells with highest (CD5high) and lowest (CD5low) cell surface expression. Data from 8 mice, pooled from 2 independent experiments. (B) Sorted naïve (CD44low) OT-I CD8+ T cells were transferred into β2m WT or KO recipients, followed by CD5 and CD6 surface staining of lymphocytes 24h later. Data from 1 experiments, using 3 (β2m KO) and 6 (WT) recipient mice. (C) Gating strategy for CD44lo cells from Themis–/– and Themis+/+ mice. Data are representative from three independent experiments with 4-5 biological replicates per genotype per experiment. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 as determined by two-sided Student’s t-test. All error bars represent SDs.
Supplementary Figure 3 | (A) Vα and Jα segment usage in the thymic SP CD8 TCRα repertoire. (B) Distribution of the amino acids within CDR3 regions. (C) Hydrophobicity (upper panel) and charge (lower panel) of the CDR3 regions from Vα3.2+ (TRAV9N-3) and non-Vα3.2 receptors. 100 most dominant clones from entire repertoires were analyzed.
Supplementary Figure 4 | Proliferation of Vα3.2+ and non-Vα3.2+ naïve CD8+ T cells from Themis–/– and Themis+/+ mice in (A) lymph nodes and (B) spleen of Rag1–/– hosts. Histogram summary of the proliferation responses in (C) lymph nodes and (D) spleen of Rag1–/– hosts. Proportion of Vα3.2+ and non-Vα3.2+ CD8+ T cells from Themis–/– and Themis+/+ mice that had more than two divisions in (E) lymph nodes and (F) spleen of Rag1–/– hosts. Data representative from two independent experiments with 4-5 biological replicates per genotype per experiment. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 as determined by two-sided Student’s t-test. All error bars represent SDs.
Supplementary Figure 5 | (A) Representative histograms of CD8+ Tetramer+ cells in uninfected and infected Themis–/– and Themis+/+ mice on day 4 and day 7 of LM-OVA infection. (B) Gating strategy for bystander cells. (C) Representative FACS plots of bystander cells in uninfected and infected Themis–/– and Themis+/+ mice on day 4 of LM-OVA infection. (D) Proportions of Vα3.2+ and non-Vα3.2+ CD8+ T cells from uninfected and LM-OVA- infected Themis–/– and Themis+/+ mice which were IFN-γ+ upon IL-12+18 stimulation. Data are representative from three independent experiments with 3-4 biological replicates per genotype per experiment.
1. Goldrath AW, Bevan MJ. Selecting and maintaining a diverse T-cell repertoire. Nature (1999) 402:6–13. doi: 10.1038/35005508
2. Starr TK, Jameson SC, Hogquist KA. Positive and Negative selection of T cells. Annu Rev Immunol (2003) 21:139–76. doi: 10.1146/annurev.immunol.21.120601.141107
3. Gascoigne NR, Palmer E. Signaling in thymic selection. Curr Opin Immunol (2011) 23:207–12. doi: 10.1016/j.coi.2010.12.017
4. Davis MM. Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature (1988) 334:395–402. doi: 10.1038/334395a0
5. Sim BC, Zerva L, Greene MI, Gascoigne NRJ. Control of MHC Restriction by TCR Valpha CDR1 and CDR2. Science (1996) 273:963–6. doi: 10.1126/science.273.5277.963
6. Sim BC, Lo D, Gascoigne NR. Preferential expression of TCR V alpha regions in CD4/CD8 subsets: class discrimination or co-receptor recognition? Immunol Today (1998) 19:276–82. doi: 10.1016/s0167-5699(98)01257-2
7. Sim BC, Wung JL, Gascoigne NR. Polymorphism within a TCRAV family influences the repertoire through class I/II restriction. J Immunol (1998) 160:1204–11.
8. Sim BC, Aftahi N, Reilly C, Bogen B, Schwartz RH, Gascoigne NR, et al. Thymic skewing of the CD4/CD8 ratio maps with the T-cell receptor alpha-chain locus. Curr Biol (1998) 8:701–4. doi: 10.1016/s0960-9822(98)70276-3
9. Garcia KC, Adams JJ, Feng D, Ely LK. The molecular basis of TCR germline bias for MHC is surprisingly simple. Nat Immunol (2009) 10:143–7. doi: 10.1038/ni.f.219
10. Kieper WC, Burghardt JT. Surh CD. A Role for TCR Affinity in Regulating Naive T Cell Homeostasis. J Immunol (2004) 172:40–4. doi: 10.4049/jimmunol.172.1.40
11. Sprent CDSJ. Homeostasis of Naive and Memory T Cells. Immunity (2008) 29:848–62. doi: 10.1016/j.immuni.2008.11.002
12. Fulton RB, Hamilton SE, Xing Y, Best JA, Goldrath AW, Hogquist KA, et al. The TCR’s sensitivity to self peptide–MHC dictates the ability of naive CD8+ T cells to respond to foreign antigens. Nat Immunol (2014) 16:107–17. doi: 10.1038/ni.3043
13. Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, et al. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci USA (2001) 98:8732–7. doi: 10.1073/pnas.161126098
14. Tarakhovsky A, Kanner S, Hombach J, Ledbetter J, Muller W, Killeen N, et al. A role for CD5 in TCR-mediated signal transduction and thymocyte selection. Science (1995) 269:535–7. doi: 10.1126/science.7542801
15. Wong P, Barton GM, Forbush KA, Rudensky AY. Dynamic tuning of T cell reactivity by self-peptide-major histocompatibility complex ligands. J Exp Med (2001) 193:1179–87. doi: 10.1084/jem.193.10.1179
16. Cho J-H, Kim H-O, Surh CD. Sprent J. T Cell Receptor-Dependent Regulation of Lipid Rafts Controls Naive CD8+ T Cell Homeostasis. Immunity (2010) 32:214–26. doi: 10.1016/j.immuni.2009.11.014
17. Surh CD, Sprent J. Homeostatic T Cell Proliferation. J Exp Med (2000) 192:F9–F14. doi: 10.1084/jem.192.4.F9
18. Sprent J, Surh CD. Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol (2011) 12:478–84. doi: 10.1038/ni.2018
19. Palmer MJ, Mahajan VS, Chen J, Irvine DJ, Lauffenburger DA. Signaling thresholds govern heterogeneity in IL-7-receptor-mediated responses of naïve CD8+ T cells. Immunol Cell Biol (2011) 89:581–94. doi: 10.1038/icb.2011.5
20. Kieper WC, Jameson SC. Homeostatic expansion and phenotypic conversion of naïve T cells in response to self peptide/MHC ligands. Proc Natl Acad Sci USA (1999) 96:13306–11. doi: 10.1073/pnas.96.23.13306
21. Cho BK, Rao VP, Ge Q, Eisen HN, Chen J. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J Exp Med (2000) 192:549–56. doi: 10.1084/jem.192.4.549
22. Goldrath AW, Bevan MJ. Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity (1999) 11:183–90. doi: 10.1016/s1074-7613(00)80093-x
23. Lee J-Y, Hamilton SE, Akue AD, Hogquist KA, Jameson SC. Virtual memory CD8 T cells display unique functional properties. Proc Natl Acad Sci USA (2013) 110:13498–503. doi: 10.1073/pnas.1307572110
24. Sosinowski T, White JT, Cross EW, Haluszczak C, Marrack P, Gapin L, et al. CD8α+ dendritic cell trans presentation of IL-15 to naive CD8+ T cells produces antigen-inexperienced T cells in the periphery with memory phenotype and function. J Immunol (2013) 190:1936–47. doi: 10.4049/jimmunol.1203149
25. Akue AD, Lee J-Y, Jameson SC. Derivation and maintenance of virtual memory CD8 T cells. J Immunol (2012) 188:2516–23. doi: 10.4049/jimmunol.1102213
26. Jameson SC, Lee YJ, Hogquist KA. Innate memory T cells. Adv Immunol (2015) 126:173–213. doi: 10.1016/bs.ai.2014.12.001
27. Drobek A, Moudra A, Mueller D, Huranova M, Horkova V, Pribikova M, et al. Strong homeostatic TCR signals induce formation of self-tolerant virtual memory CD8 T cells. EMBO J (2018) 37:3236. doi: 10.15252/embj.201798518
28. Miller CH, Klawon DEJ, Zeng S, Lee V, Socci ND, Savage PA. Eomes identifies thymic precursors of self-specific memory-phenotype CD8+ T cells. Nat Immunol (2020) 21:567–77. doi: 10.1038/s41590-020-0653-1
29. Koschella M, Voehringer D, Pircher H. CD40 Ligation In Vivo Induces Bystander Proliferation of Memory Phenotype CD8 T Cells. J Immunol (2004) 172:4804–11. doi: 10.4049/jimmunol.172.8.4804
30. Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science (1996) 272:1947–50. doi: 10.1126/science.272.5270.1947
31. Monjazeb AM, Tietze JK, Grossenbacher SK, Hsiao H-H, Zamora AE, Mirsoian A, et al. Bystander Activation and Anti-Tumor Effects of CD8+ T Cells Following Interleukin-2 Based Immunotherapy Is Independent of CD4+ T Cell Help. PloS One (2014) 9:e102709–13. doi: 10.1371/journal.pone.0102709
32. Rivino L, Kumaran EA, Thein T-L, Too CT, Gan VCH, Hanson BJ, et al. Virus-specific T lymphocytes home to the skin during natural dengue infection. Sci Transl Med (2015) 7:278ra35–278ra35. doi: 10.1126/scitranslmed.aaa0526
33. Chen AM, Khanna N, Stohlman SA, Bergmann CC. Virus-specific and bystander CD8 T cells recruited during virus-induced encephalomyelitis. J Virol (2005) 79:4700–8. doi: 10.1128/JVI.79.8.4700-4708.2005
34. Jin J-H, Huang H-H, Zhou M-J, Li J, Hu W, Huang L, et al. Virtual memory CD8+ T cells restrain the viral reservoir in HIV-1-infected patients with antiretroviral therapy through derepressing KIR-mediated inhibition. Cell Mol Immunol (2020) 376:49. doi: 10.1038/s41423-020-0408-9
35. Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Wu JE, et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science (2020) 369:eabc8511. doi: 10.1126/science.abc8511
36. Fu G, Vallée S, Rybakin V, McGuire MV, Ampudia J, Brockmeyer C, et al. Themis controls thymocyte selection through regulation of T cell antigen receptor–mediated signaling. Nat Immunol (2009) 10:848–56. doi: 10.1038/ni.1766
37. Johnson AL, Aravind L, Shulzhenko N, Morgun A, Choi S-Y, Crockford TL, et al. Themis is a member of a new metazoan gene family and is required for the completion of thymocyte positive selection. Nat Immunol (2009) 10:831–9. doi: 10.1038/ni.1769
38. Lesourne R, Uehara S, Lee J, Song K-D, Li L, Pinkhasov J, et al. Themis, a T cell-specific protein important for late thymocyte development. Nat Immunol (2009) 10:840–7. doi: 10.1038/ni.1768
39. Fu G, Casas J, Rigaud S, Rybakin V, Lambolez F, Brzostek J, et al. Themis sets the signal threshold for positive and negative selection in T-cell development. Nature (2013) 504:441–5. doi: 10.1038/nature12718
40. Gascoigne NRJ, Rybakin V, Acuto O, Brzostek J. TCR Signal Strength and T Cell Development. Annu Rev Cell Dev Biol (2016) 32:327–48. doi: 10.1146/annurev-cellbio-111315-125324
41. Paster W, Bruger AM, Katsch K, Grégoire C, Roncagalli R, Fu G, et al. A THEMIS: SHP1 complex promotes T-cell survival. EMBO J (2015) 34:393–409. doi: 10.15252/embj.201387725
42. Choi S, Warzecha C, Zvezdova E, Lee J, Argenty J, Lesourne R, et al. THEMIS enhances TCR signaling and enables positive selection by selective inhibition of the phosphatase SHP-1. Nat Immunol (2017) 18:433–41. doi: 10.1038/ni.3692
43. Mehta M, Brzostek J, Chen EW, Tung DWH, Chen S, Sankaran S, et al. Themis-associated phosphatase activity controls signaling in T cell development. Proc Natl Acad Sci USA (2018) 115:E11331–40. doi: 10.1073/pnas.1720209115
44. Prasad M, Brzostek J, Gautam N, Balyan R, Rybakin V, Gascoigne NRJ. Themis regulates metabolic signaling and effector functions in CD4+ T cells by controlling NFAT nuclear translocation. Cell Mol Immunol (2020) 17:364. doi: 10.1038/s41423-020-00578-4
45. Brzostek J, Gautam N, Zhao X, Chen EW, Mehta M, Tung DWH, et al. T cell receptor and cytokine signal integration in CD8+ T cells is mediated by the protein Themis. Nat Immunol (2020) 21:186–98. doi: 10.1038/s41590-019-0570-3
46. Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, et al. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol (2007) 8:359–68. doi: 10.1038/ni1445
47. Matz M, Shagin D, Bogdanova E, Britanova O, Lukyanov S, Diatchenko L, et al. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res (1999) 27:1558–60. doi: 10.1093/nar/27.6.1558
48. Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods (2015) 12:380–1. doi: 10.1038/nmeth.3364
49. Shugay M, Bagaev DV, Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV, et al. VDJtools: Unifying Post-analysis of T Cell Receptor Repertoires. PloS Comput Biol (2015) 11:e1004503. doi: 10.1371/journal.pcbi.1004503
50. Crooks GE, Hon G, Chandonia J-M, Brenner SE. WebLogo: a sequence logo generator. Genome Res (2004) 14:1188–90. doi: 10.1101/gr.849004
51. Pope C, Kim SK, Marzo A, Masopust D, Williams K, Jiang J, et al. Organ-specific regulation of the CD8 T cell response to Listeria monocytogenes infection. J Immunol (2001) 166:3402–9. doi: 10.4049/jimmunol.166.5.3402
52. Lefranc M-P, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, et al. IMGT®, the international ImMunoGeneTics information system® 25 years on. Nucleic Acids Res (2015) 43:D413–22. doi: 10.1093/nar/gku1056
53. Colwell RK, Chao A, Gotelli NJ, Lin SY, Mao CX, Chazdon RL, et al. Models and estimators linking individual-based and sample-based rarefaction, extrapolation and comparison of assemblages. J Plant Ecol (2012) 5:3–21. doi: 10.1093/jpe/rtr044
54. Orta-Mascaró M, Consuegra-Fernández M, Carreras E, Roncagalli R, Carreras-Sureda A, Alvarez P, et al. CD6 modulates thymocyte selection and peripheral T cell homeostasis. J Exp Med (2016) 213:1387–97. doi: 10.1084/jem.20151785
55. Gimferrer I, Farnós M, Calvo M, Mittelbrunn M, Enrich C, Sánchez-Madrid F, et al. The Accessory Molecules CD5 and CD6 Associate on the Membrane of Lymphoid T Cells. J Biol Chem (2003) 278:8564–71. doi: 10.1074/jbc.M209591200
56. Azzam HS, Grinberg A, Lui K, Shen H, Shores EW, Love PE. CD5 Expression Is Developmentally Regulated By T Cell Receptor (TCR) Signals and TCR Avidity. J Exp Med (1998) 188:2301–11. doi: 10.1084/jem.188.12.2301
57. Ponta H, Sherman L, Herrlich PA. CD44: From adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol (2003) 4:33–45. doi: 10.1038/nrm1004
58. Mitnacht R, Tacke M, Hünig T. Expression of cell interaction molecules by immature rat thymocytes during passage through the CD4+8+ compartment: developmental regulation and induction by T cell receptor engagement of CD2, CD5, CD28, CD11a, CD44 and CD53. Eur J Immunol (1995) 25:328–32. doi: 10.1002/eji.1830250204
59. Kassiotis G, Zamoyska R, Stockinger B. Involvement of avidity for major histocompatibility complex in homeostasis of naive and memory T cells. J Exp Med (2003) 197:1007–16. doi: 10.1084/jem.20021812
60. Herndler-Brandstetter D, Brunner S, Weiskopf D, van Rijn R, Landgraf K, Dejaco C, et al. Post-thymic regulation of CD5 levels in human memory T cells is inversely associated with the strength of responsiveness to interleukin-15. Hum Immunol (2011) 72:627–31. doi: 10.1016/j.humimm.2011.03.028
61. Ritmahan W, Kesmir C, Vroomans RMA. Revealing factors determining immunodominant responses against dominant epitopes. Immunogenetics (2020) 72:109–18. doi: 10.1007/s00251-019-01134-9
62. Stadinski BD, Shekhar K, Gómez-Touriño I, Jung J, Sasaki K, Sewell AK, et al. Hydrophobic CDR3 residues promote the development of self-reactive T cells. Nat Immunol (2016) 17:946–55. doi: 10.1038/ni.3491
63. Walsh MC, Pearce EL, Cejas PJ, Lee J, Wang L-S, Choi Y. IL-18 synergizes with IL-7 to drive slow proliferation of naive CD8 T cells by costimulating self-peptide-mediated TCR signals. J Immunol (2014) 193:3992–4001. doi: 10.4049/jimmunol.1400396
64. Goplen NP, Saxena V, Knudson KM, Schrum AG, Gil D, Daniels MA, et al. IL-12 Signals through the TCR To Support CD8 Innate Immune Responses. J Immunol (2016) 197:2434–43. doi: 10.4049/jimmunol.1600037
65. Kieper WC, Troy A, Burghardt JT, Ramsey C, Lee JY, Jiang H-Q, et al. Recent immune status determines the source of antigens that drive homeostatic T cell expansion. J Immunol (2005) 174:3158–63. doi: 10.4049/jimmunol.174.6.3158
66. Zhang X, Sun S, Hwang I, Tough DF, Sprent J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity (1998) 8:591–9. doi: 10.1016/s1074-7613(00)80564-6
67. Sckisel GD, Tietze JK, Zamora AE, Hsiao H-H, Priest SO, Wilkins DEC, et al. Influenza infection results in local expansion of memory CD8 +T cells with antigen non-specific phenotype and function. Clin Exp Immunol (2013) 175:79–91. doi: 10.1111/cei.12186
68. Chu T, Tyznik AJ, Roepke S, Berkley AM, Woodward-Davis A, Pattacini L, et al. Bystander-Activated Memory CD8+ T Cells Control Early Pathogen Load in an Innate-like, NKG2D-Dependent Manner. Cell Rep (2013) 3:701–8. doi: 10.1016/j.celrep.2013.02.020
69. Freeman BE, Hammarlund E, Raué H-P, Slifka MK. Regulation of innate CD8+ T-cell activation mediated by cytokines. Proc Natl Acad Sci USA (2012) 109:9971–6. doi: 10.1073/pnas.1203543109
70. Rybakin V, Westernberg L, Fu G, Kim H-O, Ampudia J, Sauer K, et al. Allelic Exclusion of TCR α-Chains upon Severe Restriction of Vα Repertoire. PloS One (2014) 9:e114320–15. doi: 10.1371/journal.pone.0114320
71. Leung MWL, Shen S, Lafaille JJ. TCR-dependent differentiation of thymic Foxp3+ cells is limited to small clonal sizes. J Exp Med (2009) 206:2121–30. doi: 10.1084/jem.20091033
72. Föhse L, Suffner J, Suhre K, Wahl B, Lindner C, Lee C-W, et al. High TCR diversity ensures optimal function and homeostasis of Foxp3+ regulatory T cells. Eur J Immunol (2011) 41:3101–13. doi: 10.1002/eji.201141986
73. Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol (2001) 2:301–6. doi: 10.1038/86302
74. Romagnoli P, Hudrisier D, van Meerwijk JPM. Preferential recognition of self antigens despite normal thymic deletion of CD4(+)CD25(+) regulatory T cells. J Immunol (2002) 168:1644–8. doi: 10.4049/jimmunol.168.4.1644
75. Wong J, Obst R, Correia-Neves M, Losyev G, Mathis D, Benoist C. Adaptation of TCR repertoires to self-peptides in regulatory and nonregulatory CD4+ T cells. J Immunol (2007) 178:7032–41. doi: 10.4049/jimmunol.178.11.7032
76. Zvezdova E, Mikolajczak J, Garreau A, Marcellin M, Rigal L, Lee J, et al. Themis1 enhances T cell receptor signaling during thymocyte development by promoting Vav1 activity and Grb2 stability. Sci Signal (2016) 9:ra51–1. doi: 10.1126/scisignal.aad1576
Keywords: bystander activation, CD8 T cell, self-reactive, themis, T cell receptor
Citation: Prasad M, Wojciech L, Brzostek J, Hu J, Chua YL, Tung DWH, Yap J, Rybakin V and Gascoigne NRJ (2021) Expansion of an Unusual Virtual Memory CD8+ Subpopulation Bearing Vα3.2 TCR in Themis-Deficient Mice. Front. Immunol. 12:644483. doi: 10.3389/fimmu.2021.644483
Received: 21 December 2020; Accepted: 19 March 2021;
Published: 07 April 2021.
Edited by:
Yun-Cai Liu, Tsinghua University, ChinaReviewed by:
Taishin Akiyama, RIKEN Yokohama, JapanCopyright © 2021 Prasad, Wojciech, Brzostek, Hu, Chua, Tung, Yap, Rybakin and Gascoigne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas R. J. Gascoigne, bWljbnJqZ0BudXMuZWR1LnNn
†Present address: Joanna Brzostek, Biologie III, University Freiburg, Freiburg, Germany Jianfang Hu, Process Development Department, Poseida San Diego, CA, United States Vasily Rybakin, Repertoire Immune Medicines, Cambridge, MA, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.