 Wen-Juan Zhang
Wen-Juan Zhang Shu-Juan Chen1
Shu-Juan Chen1 Su-Zhen Wu
Su-Zhen Wu Hui Wang
Hui Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 11 June 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.643149
Fibrosis is the final common pathway of inflammatory diseases in various organs. The inflammasomes play an important role in the progression of fibrosis as innate immune receptors. There are four main members of the inflammasomes, such as NOD-like receptor protein 1 (NLRP1), NOD-like receptor protein 3 (NLRP3), NOD-like receptor C4 (NLRC4), and absent in melanoma 2 (AIM2), among which NLRP3 inflammasome is the most studied. NLRP3 inflammasome is typically composed of NLRP3, ASC and pro-caspase-1. The activation of inflammasome involves both “classical” and “non-classical” pathways and the former pathway is better understood. The “classical” activation pathway of inflammasome is that the backbone protein is activated by endogenous/exogenous stimulation, leading to inflammasome assembly. After the formation of “classic” inflammasome, pro-caspase-1 could self-activate. Caspase-1 cleaves cytokine precursors into mature cytokines, which are secreted extracellularly. At present, the “non-classical” activation pathway of inflammasome has not formed a unified model for activation process. This article reviews the role of NLRP1, NLRP3, NLRC4, AIM2 inflammasome, Caspase-1, IL-1β, IL-18 and IL-33 in the fibrogenesis.
Fibrosis is a common stage in the progression of various organ inflammatory diseases. Its “typical” feature is the deposition of collagen and the formation of extracellular matrix (ECM) (1). The common pathological process of fibrogenesis is that after endogenous/exogenous factors damage organs, macrophages (Mø) in organs activate and release a large number of cytokines, such as transforming growth factor-β (TGF-β) and interleukin-1β (IL-1β) (2). These cytokines directly convert the intrinsic cells in the organs into fibroblasts through receptors on the surface of the cell membrane, leading to the activation of intrinsic cells, producing a large amount of collagen and ECM, and forming the fibrosis (3).
The inflammasomes are intracellular complexes composed of multiple proteins as important components of the innate immune system (4). The inflammasomes are widely expressed in the cytoplasm of the cells, including immune and non-immune cells (5, 6). The immune cells mainly include monocytes (M)/Mø, B cells, T cells, and dendritic cells (DCs) (7–10). The non-immune cells mainly include hepatic stellate cells (HSCs), fibroblasts/myofibroblast (MF), endothelial cells (ECs), and parenchymal cells (PCs) (11–15). The backbone proteins of inflammasomes can recognize the dual signals through pattern recognition receptors (PRRs) on the surface of the cell membrane. The first signal is extracellular pathogen-associated molecular patterns (PAMPs) and the second signal is intracellular damage-associated molecular patterns (DAMPs) (4, 16). The skeleton protein recruits apoptosis-associated speck-like protein containing a CARD (ASC) and pro-cysteinyl aspartate specific proteinase-1 (pro-caspase-1) to form NOD-like receptors (NLRs) and AIM2-like receptors (ALRs) as the main family members of inflammasome complexes (16, 17). The inflammasome complexes induce cells to produce cytokines and cause cell death (4, 16). Cumulative evidences show that the inflammasomes are involved in the fibrogenesis of various organs (18–21). Therefore, it is necessary to elucidate the process of inflammasomes, in particular the canonical pathways for identification of new therapeutic targets for the treatment of fibrosis.
According to the activation of cysteinyl aspartate specific proteinase (Caspase) during the formation of inflammasomes, inflammasomes are classified into “classical” and “non-classical” inflammasomes. The “classical” inflammasome mainly activate Caspase-1, while the “non-classical” inflammasome mostly activate other Caspases other than Caspase-1 (22). The “classical” inflammasome involved NOD-like receptors were divided into four classes (NODs, NLRPs, NLRC4 and NLRC5) based on the nucleotide-binding oligomerization domain (NOD, also known as NACHT) (22). (1) NODs, including NOD1-5 and MHC class II transactivator (CIITA). (2) NOD-like receptor proteins (NLRPs), also known as leucine-rich repeat domain proteins (NACHT, LRR and PYD domains-containing proteins, NALPs), including NALP1-14. (3) NOD-like receptor C4 (NLRC4), including IL-1β-converting enzyme-protease-activating factor (IPAF) and neuronal apoptosis inhibitor protein (NAIP). (4) NOD-like receptor C5 (NLRC5), also namely NOD27. In addition, AIM2 belongs to non-NLRs (22). In addition, “non-classical” inflammasome are not clearly classified.
NOD-like receptors (NLRs) are mainly composed of a carboxyl (C) terminal, a central, and an amino acid (N) terminal domain (23). The C-terminus includes a leucin rich repeat (LRR), the center domain includes NACHT, and the N-terminus includes a pyrin domain (PYD) containing a caspase recruitment domain (CARD)/baculoviral inhibitor of apoptosis repeat (BIR)/acidic transactivator (24). The C-terminal LRR recognizes the ligand; the central NACHT hydrolyzes adenosine triphosphate (ATP) by activating a deoxy-ribonucleoside triphosphate (dNTP) enzyme; the N-terminal CARD interacts with the adaptor protein through CARD-CARD to activate downstream signals (22) (Figure 1).
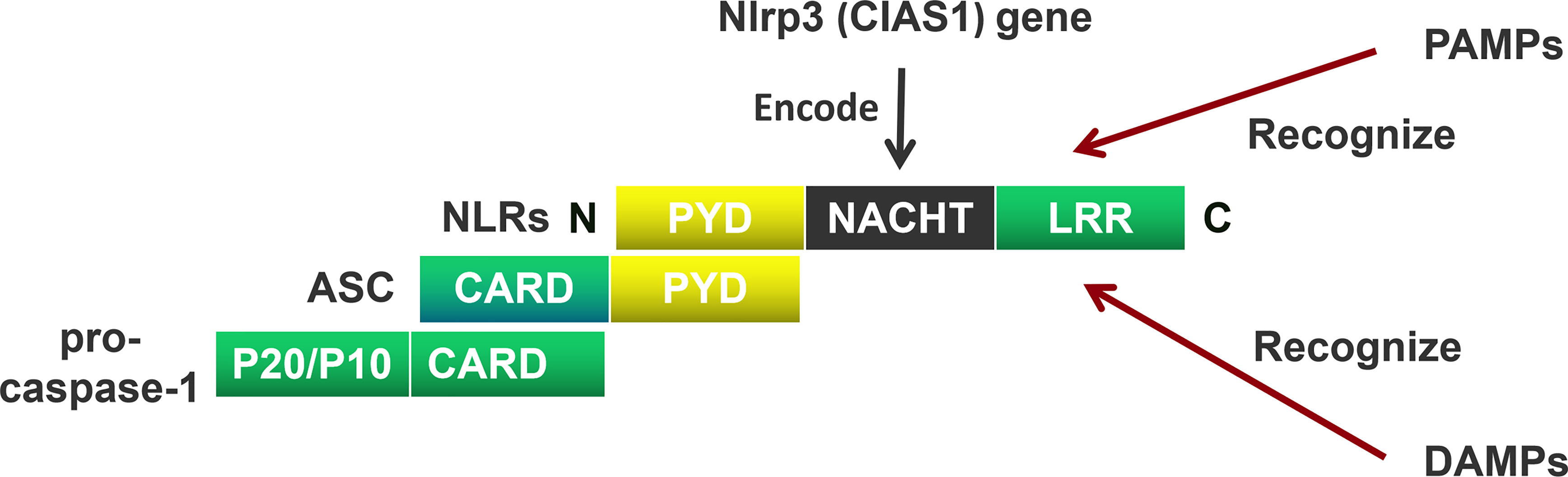
Figure 1 The Composition of the NOD-like receptors (NLRs). NOD-like receptors (NLRs) are mainly composed of a carboxyl (C) terminal, a central terminal, and an amino acid (N) terminal. The C-terminus includes a leucin rich repeat (LRR), the center terminus includes NACHT, and the N-terminus includes a pyrin domain (PYD). The C-terminal LRR recognizes the pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), the central-terminal NACHT encodes by Nlrp3 (CIAS1) gen. The N-terminal CARD interacts with the adaptor protein through PYD-PYD. Apoptosis-associated speck-like protein containing a CARD (ASC) recruits pro-cysteinyl aspartate specific proteinase-1 (pro-caspase-1) through CARD domain to activate downstream signals.
The activation of “classical” inflammasome is commonly reported, which usually requires “dual signals” (22). The “first signal” is that the activation signals of toll-like receptors (TLRs), such as Chlamydia pneumoniae/Schistosoma mansoni (S. mansoni), which induce the expression of inflammasomes (25, 26). The “second signal” is composed to the ligands of inflammasomes, such as PAMPs/DAMPs, which induce the activation of inflammasomes. The activation of “classical” inflammasome is mainly that NLRP3 serves as the central skeleton of the inflammasome, and ASC acts as a linker protein connecting NLRP3 with the pro-caspase I, forming inflammasome complexes (4, 16). After activation of the inflammasomes, they depend on Caspase-1 to produce mature IL-1β, IL-18 and IL-33 (22). IL-1β and IL-18 exert biological functions by binding to IL-1/18 receptors (IL-1/18Rs) (27, 28). IL-33 mainly induces Th2 cells to release IL-13 and IL-5 (29). To date, there are few reports on the activation of “non-classical” inflammasome.
The effects of inflammasomes are also generally divided into “classical” and “non-classical” types. The “classical” effect is that the inflammasomes dependent-Caspase-1 induces cells to secrete pro-inflammatory cytokines, removing pathogens and endogenous death signals (30). The “non-classical” effect is that inflammasome components are independent of inflammasome complexes and directly regulate biological processes, such as cell proliferation, gene transcription and translation, and tumor formation (31). The “non-classical” effect of NOD-like receptor protein 3 (NLRP3) is that TGF-β participates in the fibrogenesis through epithelial-mesenchymal transition (EMT) (30).
NOD-like receptor protein 1 (NLRP1/NALP1) is called the first inflammasome and exerts its biological activity as an inflammasome complex (32). Toxins and muramyl dipeptide (MDP) as PAMPs lead to the outflow of intracellular potassium ions (K+), activating NLRP1 inflammasome and inducing IL-1β secretion by Mø (33).
NLRP1 has been reported to be involved in myocardial fibrogenesis in pressure overload rats (34). NLRP1 mediates myocardial fibrogenesis in mice via mitogen-activated protein kinase (MAPK), nuclear factor-κB (NF-κB), and TGF-β/Smad (34). NLRP1 also mediates rat fibrogenesis through TGF-β1/Smad (18). TGF-β1 induces rat cardiac fibroblasts (CFs) to express NLRP1, and through the nuclear translocation of Smad2 and Smad3, promotes the conversion of CFs to MF, leading to ECM deposition and fibrosis (18).
NLRP3, also known as NALP3, is usually used as the backbone protein of NLRP3 inflammasome, and forms a complex with ASC and pro-caspase-1, leading to the activation of NLRP3 inflammasome (16).
Currently, there are three hypotheses regarding the activation of NLRP3 inflammasome. (1) K+ outflow hypothesis: ATP recognizes P2X7 purinergic receptor (P2X7R) on the cell membrane, opens the ion channel, and leads to K+ outflow, recruiting ubiquitinated connexin to punch holes in the cell membrane. The PAMPs enters the cells and promotes the binding of the catalytic domain of NIMA-related kinase 7 (NEK7) to NLRP3 and activates NLRP3 inflammasome (16, 35). (2) Hypothesis of reactive oxygen species (ROS): Streptozotocin (STZ), bleomycin (BLM) and statins first damage mitochondria (36, 37), and then activate phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt), c-Jun N-terminal kinase (JNK), and p38/MAPK/extracellular signal-regulated protein kinase (ERK) pathway, respectively, reactivates NADPH oxidase 4 (NOX4), leading to ROS activation (38, 39). ROS induces dissociation of thioredoxin and thioredoxin interacting protein (TXNIP). TXNIP directly activates NLRP3 inflammasome (22). In addition, ROS also induces the conversion of mitochondrial DNA (mtDNA) into oxidized form (ox-mtDNA), which, as the ligand of NLRP3, directly binds and activates NLRP3, activating NLRP3 inflammasome (40). (3) Lysosomal damage hypothesis: the crystals/macromolecules (22, 41), such as beta amyloid, monosodium urate (MSU), airborne particles and cholesterol, activate NADPH oxidase through chemical response, which damages the lysosome, releasing cathepsin B, and activating NLRP3 inflammasome (22, 42). These three hypotheses may explain the activation of NLRP3 inflammasome by some stimulants, but not explain all the activation of NLRP3 inflammasome.
Aside from the above three hypotheses, there are five ways to activate NLRP3 inflammasome. (1) Sodium ion (Na+) inflow: Epithelial sodium channels (ENaC) on the surface of the cell membrane are opened to allow Na+ inflow, leading to K+ outflow, and activating the NLRP3 inflammasome (43). (2) Chloride (Cl-) outflow: chloride intracellular channels (CLIC) act as the downstream of the K+ outflow-mitochondrial ROS axis. ROS induces the transfer of CLIC to the cell membrane, leading to Cl- outflow. The outflow of Cl- enables NEK7 to bind to NLRP3 and promotes the assembly and activation of NLRP3 inflammasome (44). (3) Calcium ion (Ca2+) accumulation: Phospholipase C hydrolyzes phosphatidylinositol-4,5-diphosphate to form diacyl glycerol (DAG) and inositol trisphosphate (InsP3). InsP3 binds to the InsP3 receptor on the endoplasmic reticulum membrane, causing the endoplasmic reticulum to release Ca2+, resulting in an increase in intracellular Ca2+. Ca2+ is recognized by calcium-sensing receptor (CASR) and activates NLRP3 inflammasome (45). (4) Inhibition of autophagy: STZ cooperates with thioacetamide (TAA) through adenosine monophosphate-activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR) pathway inhibition autophagy effect, leading to activation of NLRP3 inflammasome (46). (5) Cyclic adenosine monophosphate (cAMP) reduction: CASR leads to a decrease in intracellular cAMP, weakens the binding capacity of cAMP and NLRP3, and activates NLRP3 inflammasome (45) (Figure 2).
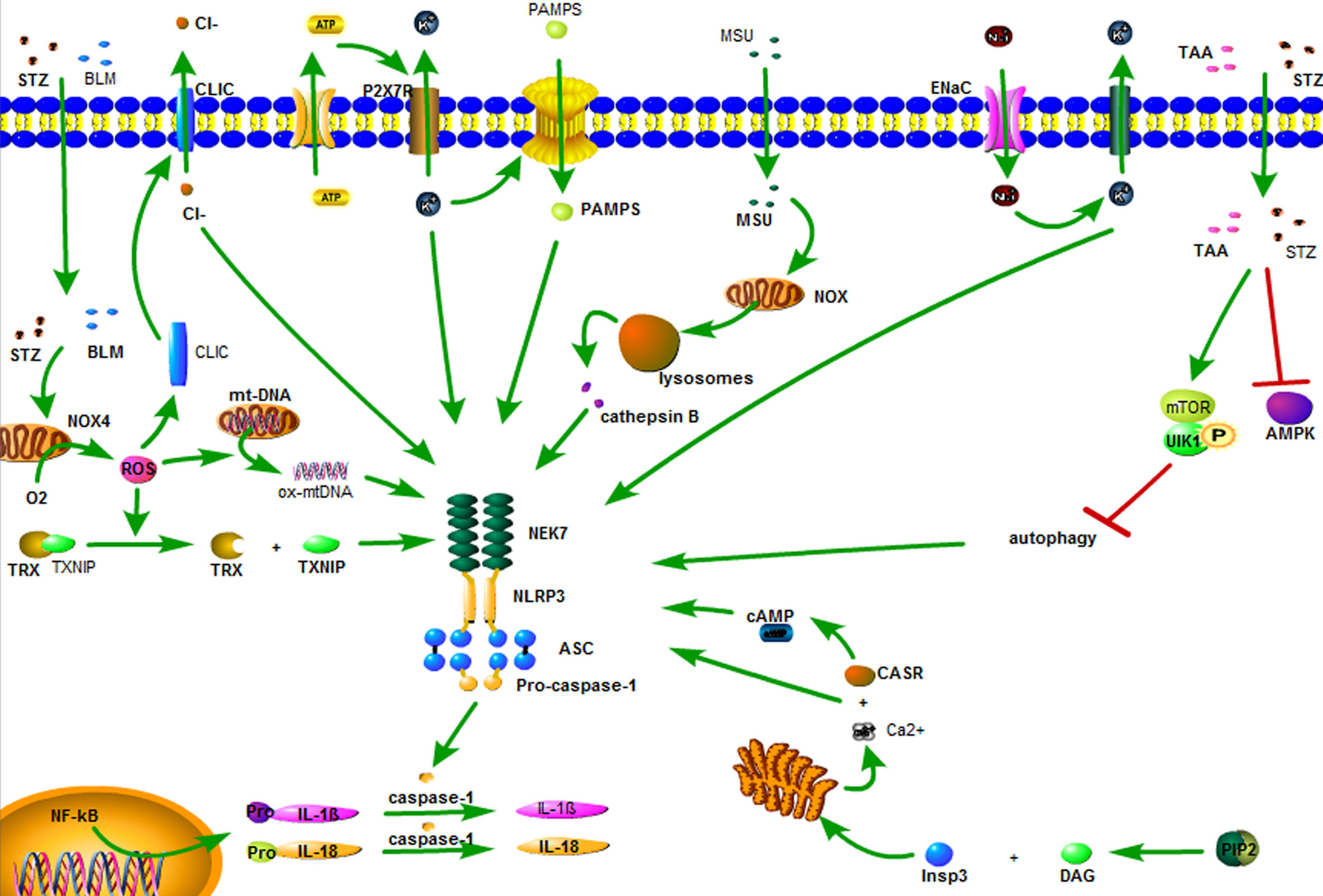
Figure 2 The activation of the NLRP3 inflammasome. ATP recognizes P2X7 purinergic receptor (P2X7R) on the cell membrane, opens the ion channel, and leads to K+ outflow, recruiting ubiquitinated connexin to punch holes in the cell membrane. The PAMPs enters the cells and promotes the binding of the catalytic domain of NIMA-related kinase 7 (NEK7) to NLRP3 and activates NLRP3 inflammasome. Streptozotocin (STZ), bleomycin (BLM) and statins first damage mitochondria, and then activate NADPH oxidase 4 (NOX4), leading to ROS activation. ROS induces dissociation of thioredoxin and thioredoxin interacting protein (TXNIP). TXNIP directly activates NLRP3 inflammasome. In addition, ROS also induces the conversion of mitochondrial DNA (mtDNA) into oxidized form (ox-mtDNA), which, as the ligand of NLRP3, directly binds and activates NLRP3, activating NLRP3 inflammasome. The crystals/macromolecules, such as monosodium urate (MSU) activates NADPH oxidase through chemical reaponse, which damages the lysosome, releasing cathepsin B, activating NLRP3 inflammasome. Epithelial sodium channels (ENaC) on the surface of the cell membrane are opened to allow Na+ inflow, leading to K+ outflow, and activating the NLRP3 inflammasome. Chloride intracellular channels (CLIC) act as the downstream of the K+ outflow-mitochondrial ROS axis. ROS induces the transfer of CLIC to the cell membrane, leading to Cl- outflow. The outflow of Cl- enables NEK7 to bind to NLRP3 and promotes the assembly and activation of NLRP3 inflammasome. Phospholipase C hydrolyzes phosphatidylinositol-4,5-diphosphate to form diacyl glycerol (DAG) and inositol trisphosphate (InsP3). InsP3 binds to the InsP3 receptor on the endoplasmic reticulum membrane, causing the endoplasmic reticulum to release Ca2+, resulting in an increase in intracellular Ca2+. Ca2+ is recognized by calcium-sensing receptor (CASR) and activates NLRP3 inflammasome. STZ cooperates with thioacetamide (TAA) through adenosine monophosphate-activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR) pathway inhibition autophagy effect, leading to activation of NLRP3 inflammasome. CASR leads to a decrease in intracellular cAMP, weakens the binding capacity of cAMP and NLRP3, and activates NLRP3 inflammasome.
Liver fibrosis is a common stage of chronic liver injury caused by multiple factors (47). The factors involved in liver fibrogenesis include chemical factors, metabolic factors and infectious factors (48). The chemical factors include ethanol and tetracycline. The metabolic factors include high-fat diet (HFD) and non-alcoholic fatty. The infectious factors include hepatitis B virus (HBV), schistosomes such S. mansoni and S. japonicum (48). Among them, the infectious factor as PAMPs, after acting on the livers, first destroys the liver cells, the NLRP3 inflammasomes in the liver cells is activated, leading to hepatocyte necrosis (49). The necrotic liver cells release DAMPs, which can activate Kupffer cells (KCs) (11). The KCs recognize PAMPs through TLRs on the one hand, and induce the expressions of NLRP3 inflammasome-related pathway components such as NLRP3, pro-caspase-1, and pro-IL-1β through TLRs-NF-κB pathways (16, 22). On the other hand, the KCs recognize DAMPs, which can directly damage mitochondria and cause them to release ROS (50). As the upstream signal of NLRP3, ROS activates NLRP3 through the ROS-TXNIP pathway (51). ROS also promotes the transfer of high mobility group box 1 (HMGB1) from the nucleus to the cytoplasm (52). As DAMPs, HMGB1 can also activate NLRP3 through the TLR4-NF-κB pathways (53). After NLRP3 is activated, NLRP3 forms NLRP3 inflammasome together with ASC and pro-caspase-1 (54). Nuclear factor erythroid 2-related factor 2 (Nrf2) is an important transcription factor that regulates cellular anti-oxidative stress (55). Under physiological conditions, the cytoplasmic protein chaperone molecule Kelch-like ECH-associated protein 1 (Keap1) in KCs binds to Nrf2 and makes it appear to be inhibited (56). When mitochondria release ROS, Nrf2 dissociates from Keap1 and moves into the nucleus, and combines with the antioxidant response element (ARE) to activate the antioxidant enzyme heme oxygenase-1 (HO-1) expression to inhibit the activation of ROS/NLRP3 inflammasome pathways (57). The antioxidant response cannot resist the oxidation response, which leads to the KCs activation (58). Activated KCs activate HSCs by releasing TGF-β and IL-1β (59). HSCs also have the activation of NLRP3 inflammasome and the self-activation of pro-caspase-1 to form mature Caspase-1 (60). Caspase-1 in HSCs can also catalyze the maturation of pro-IL-1β to form IL-1β and release it outside the cell to form a positive feedback effect (60). Activated KCs recruit monocytes in peripheral blood by releasing CC motif chemokine ligand 2 (CCL2), CCL5, and monocyte chemotactic protein-1 (MCP-1), enlarging inflammatory responses (61, 62). The enlarged inflammatory responses continue to activate HSCs, causing HSCs to express α-smooth muscle actin (α-SMA) and Collagen I, leading to ECM deposition and eventually progressing into liver fibrosis (11) (Figure 3).
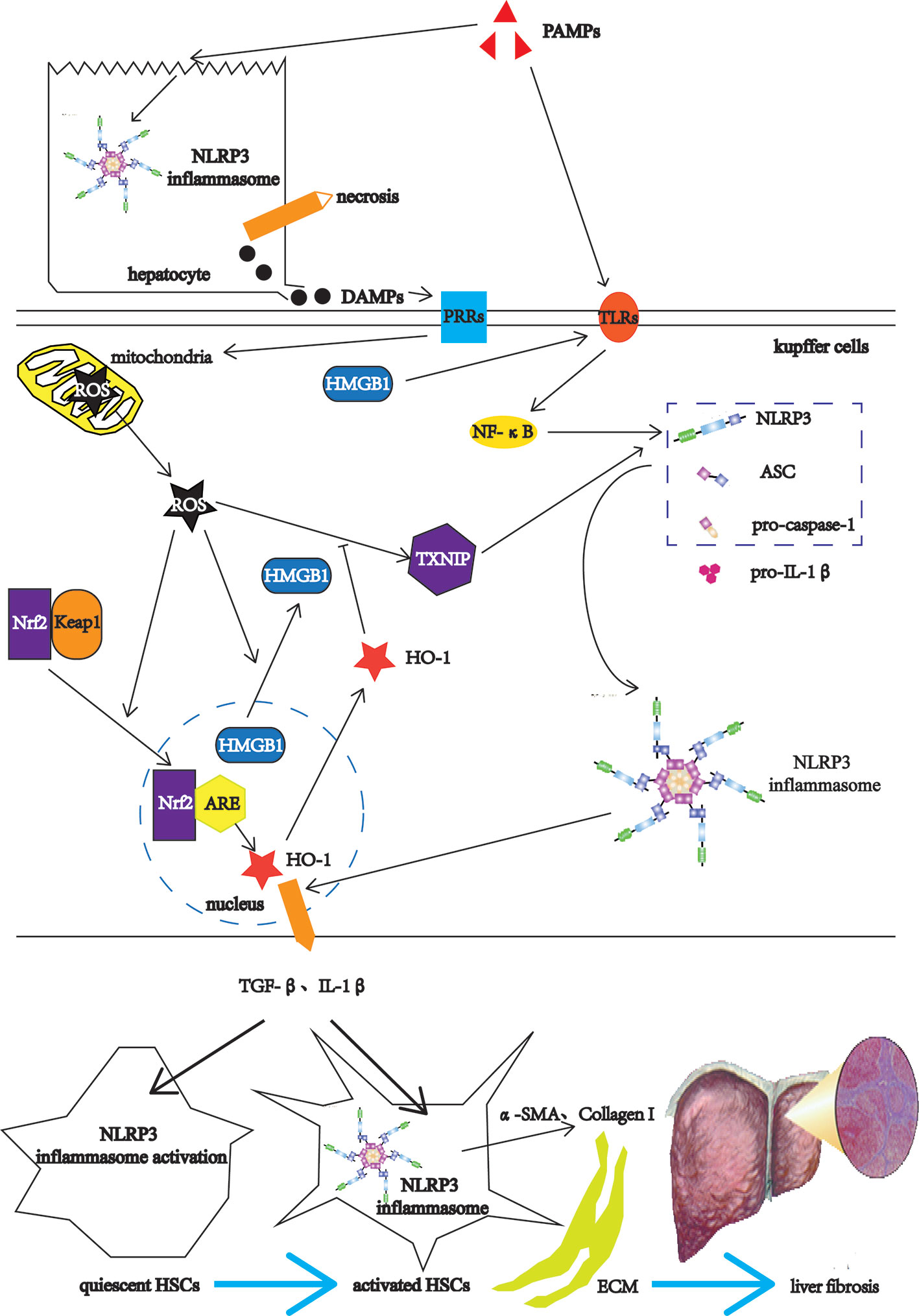
Figure 3 The NLRP3 inflammasome in the liver fibrosis. After the PAMPs act on the livers, they destroy the hepatocytes, and activate the NLRP3 inflammasomes in hepatocytes, leading to hepatocyte necrosis. The necrotic liver cells release DAMPs, which can activate Kupffer cells (KCs). The KCs recognize PAMPs through TLRs on the one hand, and induce the expressions of NLRP3 inflammasome-related pathway components such as NLRP3, pro-caspase-1, and pro-IL-1β through TLRs-NF-κB pathways. On the other hand, the KCs recognize DAMPs, which can directly damage mitochondria and cause them to release ROS. As the upstream signal of NLRP3, ROS activates NLRP3 through the ROS-TXNIP pathway. ROS also promotes the transfer of high mobility group box 1 (HMGB1) from the nucleus to the cytoplasm. The HMGB1 can also activate NLRP3 through the TLR4-NF-κB pathways. After NLRP3 is activated, NLRP3 forms NLRP3 inflammasome together with ASC and pro-caspase-1. Nuclear factor erythroid 2-related factor 2 (Nrf2) is an important transcription factor that regulates cellular anti-oxidative stress. Under physiological conditions, the cytoplasmic protein chaperone molecule Kelch-like ECH-associated protein 1 (Keap1) in KCs binds to Nrf2 and makes it appear to be inhibited. When mitochondria release ROS, Nrf2 dissociates from Keap1 and moves into the nucleus, and combines with the antioxidant response element (ARE) to activate the antioxidant enzyme heme oxygenase-1 (HO-1) expression to inhibit the activation of ROS/NLRP3 inflammasome pathways. The antioxidant response cannot resist the oxidation response, which leads to the KCs activation. Activated KCs activate HSCs by releasing TGF-β and IL-1β, which activate the NLRP3 inflammasome in HSCs. The activated HSCs express α-smooth muscle actin (α-SMA) and Collagen I, leading to ECM deposition and eventually progressing into liver fibrosis.
S. japonicum, MCD and angiotensin II (Ang II) activate NLRP3 inflammasome through lysosomal damage, inducing oxidative responses (63, 64). The NLRP3 inflammasome mediated Smad3, causes HSCs to express α-SMA, leading to liver fibrosis (19, 65). The formation of liver fibrosis is closely related to the liver-gut axis (66). The PAMPs (such as LPS) in the leakage of chronic liver disease can activate NF-κB through TLRs on the surface of KCs, promote the activation of NLRP3 inflammasomes, and induce the generation of pro-inflammatory signals (such as: IL-1β, IL-18, IL-6, etc.) (67). These pro-inflammatory signals activate HSCs through cytokine receptors (CKRs)/myeloid differentiation factor 88 (MyD88), leading to liver fibrosis-related molecules matrix metalloproteinases (MMP) and tissue inhibitor of metalloproteinases 1 (TIMP) imbalance, promote ECM deposition and form liver fibrogenesis (27, 68) (Figure 4). The PAMPs produced by the imbalance of the intestinal flora and the increase in intestinal permeability can be transferred to the liver from the intestine through the bloodstream, which is similar to the effect of PAMPs from chronic liver diseases (69). MCC950, an inhibitor of NLRP3 inflammasome activation, could block the activation of NLRP3 inflammasome, reduce the production of TGF-β and Collagen I, and delay progression of liver fibrogenesis (64, 70). However, systemic knock-in of NLRP3 gene in mice accelerates the progression of liver fibrogenesis by promoting the activation of NLRP3 inflammasome, inducing hepatocyte pyrolysis, forming severe liver tissue inflammation (49). The hepatocytes can also directly participate in liver fibrogenesis (71). Professor Li et al. reported that Ang II generates hydrogen peroxide (H2O2) through NOX4 by acting on the angiotensin II type-1 receptor (AT1R) on the surface of hepatocytes (71). The H2O2 activates the NLRP3 inflammasome to produce IL-1β. IL-1β induces the phosphorylation of Smad2/3 to promote the transformation of hepatocytes to EMT, expressing Collagen I, and forming liver fibrosis (71).
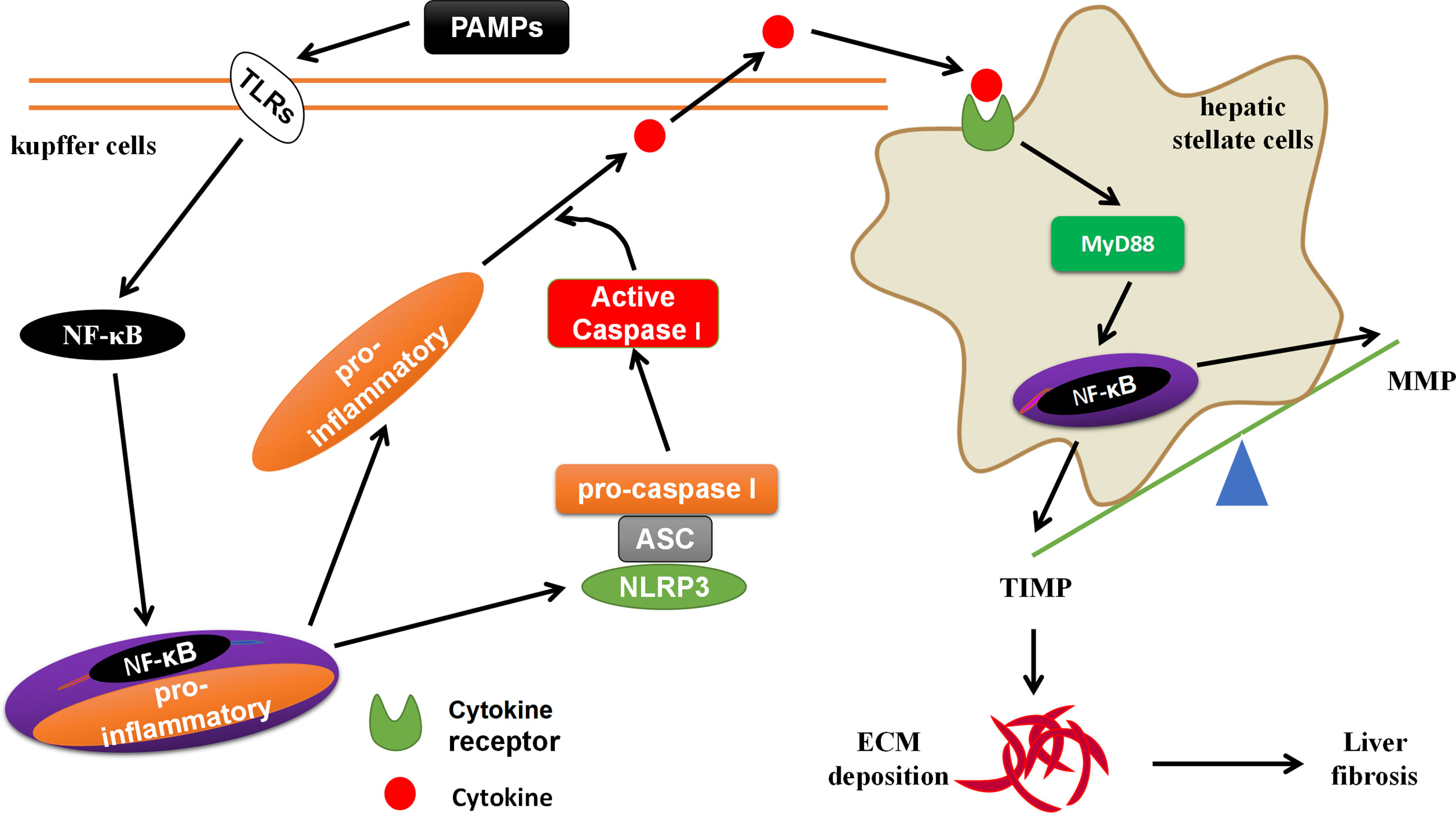
Figure 4 The crosstalk between KCs and HSCs. The PAMPs in the leakage of chronic liver disease can activate NF-κB through TLRs on the surface of KCs, promote the activation of NLRP3 inflammasomes, and induce the generation of pro-inflammatory signals. These pro-inflammatory signals activate HSCs through cytokine receptors (CKRs)/myeloid differentiation factor 88 (MyD88)/NF-κB, leading to liver fibrosis-related molecules matrix metalloproteinases (MMP) and tissue inhibitor of metalloproteinases 1 (TIMP) imbalance, promote ECM deposition and form liver fibrogenesis.
Hepatic stellate cells (HSCs), as a key cell type of the liver, are involved in the development of liver fibrogenesis by HSCs activation (11). The soluble egg antigen (SEA) of Schistosoma japonicum activates the NLRP3 inflammasome in HSCs by activating spleen tyrosine kinase (Syk), C-type lectin receptor Dectin-1 and JNK pathway (72). Ang II up-regulates mir-21 expression by targeting Smad7 and Spry1. On the one hand, mir-21 inhibits Smad7 by targeting and releases the inhibitory effect of Smad7 on Smad2/3, leading to the activation of Smad2/3/NOX4/ROS (38). On the other hand, mir-21 inhibits Spry1 by targeting and releases releases the inhibitory effect of Spry1 on ERK, which activates the ERK/NF-κB signaling pathway, leading to the activation of NLRP3 inflammasomes (38). After the mouse primary HSCs or hepatic stellate cell lines (LX-2/HSC-T6) are treated with exogenous stimulants, such as MSU/bacterial RNA, NLRP3 inflammasome can be activated and induce HSCs to secrete IL-1β (70, 73). IL-1β interacts with IL-1Rs on the membrane of HSCs, which activates NF-κB and causes TGF-β expression. TGF-β induces the expression of α-SMA and Collagen I through the TGF-βR on the cell membrane of HSCs (70, 73). In addition to IL-1β and TGF-β, tumor necrosis factor (TNF) and IL-17 also play a role in the comparable way (74).
Most views suggest that NLRP3 inflammasome participates in liver fibrogenesis in an “indirect” manner with the activation of other signals (3, 38, 74). However, there have been reported that NLRP3 inflammasome is independent of cytokines, and is directly expressed and activated in HSCs, and is involved in liver fibrogenesis in a “direct” manner with HSCs activation (75). Basing on above reports, the “indirect” and “direct” manners are coexist, and the “indirect” manner plays a major role in liver fibrogenesis.
Fructose activates NLRP3 inflammasome by inducing ROS production (75). NLRP3 inflammasome reactivates Smad2/3, leading to cardiac fibrosis (76). CFs are the key cells of cardiac fibrosis and are involved in the development of cardiac fibrogenesis (77). After TGF-β was administered to mouse primary CFs, CFs differentiated significantly (75). After the primary CFs of NLRP3-/- mice were isolated and were added TGF-β, the differentiation of CFs is weakened (76). The results suggest that NLRP3 plays an important role in the differentiation of CFs. In addition to NLRP3 participating in the differentiation of CFs as a single molecule, it also participates in cardiac fibrogenesis via the activation of NLRP3 inflammasome and the product IL-1β (78). IL-1β plays a role in promoting cardiac fibrogenesis through the conversion of CFs to MF (79).
Lipopolysaccharide (LPS) and BLM activate NLRP3 inflammasome via ROS (80, 81). NLRP3 inflammasome lead to pulmonary fibrosis through the IL-1β/IL-1Rs/MyD88/NF-κB signaling pathway (81). Lung fibroblasts are key cells for pulmonary fibrosis (82). After isolation of mouse primary lung fibroblasts, BLM was added to the mouse primary lung fibroblasts, and it was found that NLRP3 inflammasome regulate IL-1β via miR-155, leading to lung fibrosis (83, 84). It can be seen that IL-1β plays an important role in the formation of pulmonary fibrogenesis. In addition to IL-1β, TGF-β and platelet derived growth factor-AA (PDGF-AA) have the comparable function (84). In recent years, lung ECs have been the focus of research on pulmonary fibrogenesis. The studies have found that NLRP3 inflammasome transforms ECs into EMT, forming pulmonary fibrosis (85).
Adenine diet and unilateral ureteral obstruction (UUO) can both induce NLRP3 inflammasome activation via ROS (86, 87). Recently, there have also been reports of NLRP3 inflammasome-dependent NF-κB activation after major nephrectomy (88). NLRP3 inflammasome activates the T cells and induces renal fibrogenesis through the IL-23/IL-17 axis (86). Recently, the report showed that MCC950 was given too late to sufficiently block renal inflammation, and not to delaying the progression of renal fibrogenesis (87).
Endothelial cells (ECs) have also received attention in renal fibrogenesis. After TGF-β was administered to tubular epithelial cells (TECs) in mice, NLRP3 expression was increased and NLRP3 transformed TECs into EMT, and then into MF through the phosphorylation of Smad2/3, resulting in increased expression of α-SMA and matrix metalloprotein 9 (MMP9). After TGF-β was given to the primary TECs of NLPR3-/-mice, the NLRP3 expression was decreased, the phosphorylation of Smad2/3 was decreased, and the expression of α-SMA and MMP9 was decreased (14). The above reports display that NLRP3 promotes the conversion of TECs to renal fibrosis through the TGF-β/Smad pathway.
NLRP3 inflammasome is also involved in pancreatic fibrogenesis caused by bombesin, peritoneal fibrogenesis caused by methylglyoxal (MGO), cystic fibrogenesis caused by Aspergillus fumigatus (A. fumigatus)/Pseudomonas aeruginosa (P. aeruginosa), and bladder fibrogenesis caused by bladder opening obstruction (27, 89–91). Using the same NLPR3-/- mice, it was found that peritoneal fibrosis was reduced after MGO was administration (90). The reports demonstrate that the NLRP3 is a key molecule in fibrogenesis.
The NLRC4 inflammasome is usually activated by the flagellin of gram-positive and gram-negative bacteria and endolin of type III secretion system (T3SS) derived from gram-negative bacteria (92, 93). For example, NAIP5 in mice is activated by bacterial flagellin, while NAIP in humans is activated by the needle-like subunits of T3SS (92, 93). However, the activation mechanisms are unclear. In A. fumigatus or P. aeruginosa infected mice, NLRC4 expression depended on cystic fibrosis transmembrane conductance regulator (CFTR) reached a peak at 7 days (27). However, it has also been reported that NLRC4 produces IL-1R antagonist (IL-1Ra) via NF-κB, to bind IL-1β, delaying the progression of fibrogenesis (27). Furthermore, NLRC4 has also been reported to promote liver cell regeneration and reverse liver fibrosis (20). But the molecular mechanism in which NLRC4 plays a negative role in fibrogenesis remains to be studied.
AIM2 inflammasome is composed of oligosaccharides and PYD domains (17). AIM2 inflammasome recognizes the double-stranded DNA (dsDNA) in the cell through the oligosaccharide domain, and then bind to ASC through the PYD domain, leading to pro-caspase-1 self-activation (17). After Peripheral blood mononuclear cells (PBMCs) were treated with Poly (dA: dT), the expression of AIM2 inflammasome was increased (21). AIM2 inflammasome dependent on Caspase-4 induces IL-1α secretion by PBMCs. IL-1α binds to IL-1αR, inducing TGF-β secretion by PBMCs (21). TGF-β is a key factor in the fibrogenesis (94). It can be concluded that AIM2 inflammasome participates in the development of fibrogenesis.
NOD-like receptor protein 6 (NLRP6) is mainly expressed in the small intestine, large intestine, and liver (95). NLRP6, as a special functional protein in the NLRs family, has a “negatively regulation” to liver fibrogenesis (96, 97). In vivo experiments found that in allogeneic hematopoietic stem cell transplantation (Allo-HSCT) mice, NLRP6 inhibits liver fibrogenesis through the activation of p38/MAPK, NF-κB and NLRP3 inflammasome, respectively (96). In vitro experiments found that NLRP6 inhibited the proliferation and activation of LX-2 cells, and by enhancing the expression of magnesium ion-dependent protein phosphatase 1A (PPM1A), it inhibited the phosphorylation of Smad2/3 and reduced the expression of Collagen I and Collagen III (97).
NOD-like receptor C5 (NLRC5) belongs to the largest member of the NLRs family and is expressed in the cytoplasm and nucleus of most cells (98). NLRC5 is also involved in the development of fibrogenesis. NLRC5 expression is present in liver tissues of patients with cirrhosis and also found in CCl4 treated mice (99, 100). In addition, TGF-β regulates Smad2/3 and NF-κB via NLRC5, induces LX-2 activation and expresses α-SMA and Collagen I (99, 100).
Caspase-1 is mainly used as the activation product of “classical” inflammasome and is involved in the fibrogenesis (101). Caspase-1 catalyzes maturation of pro-IL-1β and secretion of IL-1β (22). IL-1β has a pro-fibrotic effect, and is usually involved in the fibrogenesis in combination with IL-1βRs on the surface of resident cells (102). In BLM-induced pulmonary fibrogenesis mice, the inhibitor of Caspase-1, YVAD-fmk, delays the progression of pulmonary fibrogenesis (81). Once the production of caspase-1 was blocked by YVAD-fmk, the interaction between NLRP3 and ASC, ASC and pro-caspase-1 was weakened (81). It suggested that YVAD-fmk inhibits the production of caspase-1, hinders the formation of NLRP3 inflammasome, and delays the progression of pulmonary fibrogenesis. Similar reports have been displayed in S. japonicum infection and HFD-induced liver fibrogenesis (63, 103).
IL-1β is mainly secreted by activated M, Mø, and DCs (104). At present, there are three main types of IL-1β secretion mechanisms. (1) ATP causes K+ outflow and Ca2+ inflow, then activated phospholipase C and phospholipase A2, resuting in cells to secrete IL-1β (22). (2) IL-1β secretion after inflammasome formation and activation (27). (3) The Caspase-4 and Caspase-1 are activated sequentially, and induce PBMCs to secrete IL-1β (105). IL-1β binds to IL-1Rs on the surface of cell membranes, and promotes pro-IL-1β transcription and translation to produce IL-1β (22). IL-1β could also promote hepatocyte apoptosis, activate M and neutrophils, leading to fibrosis (106).
As a key signal for leading to fibrogenesis, IL-1β plays a role in promoting fibrogenesis by binding to IL-1βRs (102). IL-1β and IL-1βRs are in a dynamic equilibrium. The agonists of IL-1βRs can promote fibrogenesis through IL-1β (107), but the antagonists of which can prevent the fibrogenesis of IL-1β promotion by reducing IL-1βRs (27). IL-1β promotes fibrogenesis through TGF-β, ERK1/2, c-Jun, and PI3K/Akt, respectively (108). IL-1β also promotes renal stromal cells (SCs) through the IL-1 receptors-IL-1R-related kinase 4 (IRAK4) -protocogene (MYC) transcription factor axis, to expresses platelet-derived growth factor receptor (PDGFR) (102). PDGFR, in combination with PDGF, promotes the appreciation and migration of SCs, leading to the deposition of ECM and the formation of renal fibrosis (109).
IL-18, also known as interferon-γ inducing factor (IGIF), is usually expressed in a variety of cells as pro-IL-18 (110). In addition to being cleaved by “classical” Caspase-1, IL-18 is also cleaved by “non-classical” protease 3/Caspase-3 (111, 112). Pro-IL-18 is cleaved into mature IL-18 and secreted extracellularly by the cells (4). Most studies report that IL-18 has a profibrotic effect. There are currently three main ways to promote fibrogenesis. (1) IL-18 induces Th1 cells produce IFN-γ and IL-13, causing fibrosis (113). (2) Ischemia-reperfusion injury induces Mø to M2-type cells through IL-18, forming fibrosis (114). (3) IL-18 recruits T cells and Mø via chemokines, and transforms Mø into MF, resulting in fibrosis (115). After administration of IL-18 inhibitors, T cells and Mø decreased and the transformation of Mø into MF slowed (114).
A few publications report that IL-18 has an anti-fibrotic effect. The expression of IL-18 in serum and liver tissue was induced by DNA vaccine of IL-18, reducing schistosome-associated liver fibrosis (SSLF) (116). IL-18 was transfected into S. japonicum-infected hepatocytes, and hepatocytes expressed IL-18. IL-18 reverses the conversion of Th1 to Th2, improving SSLF (117). The anti-fibrotic effect of IL-18 mainly occurs in SSLF, and it may be related to the pathogenic way of S. japonicum. Whether IL-18 exerts the effect of promoting fibrogenesis or suppressing fibrogenesis remains to be proved experimentally.
IL-33, also known as the 11th member of the IL-1 family (IL-1F11), is usually expressed in the nucleus of ECs, fibroblasts and immune cells in the form of pro-IL-33 (118). When cell death or tissue damage occurs, pro-IL-33 is cleaved by Caspase-1/Caspase-3/Caspase-7 to mature IL-33 (118). IL-33 is secreted outside the cell as an “alarmin” and participates in tissue homeostasis by Th2 cells (29).
IL-33 promotes fibrogenesis in two main ways. (1) Pro-fibrosis effect of IL-33/ST2 signal axis: In liver fibrogenesis caused by S. japonicum infected with mice, the expressions of IL-33 and ST2 in the liver are increased (119). ST2 is a ligand of IL-33 (114). IL-33 is dependent on ST2 for MCD diet-induced liver fibrogenesis (120). In addition, IL-33/ST2 is also involved in BLM and E. coli-induced fibrogenesis (121). In BLM-induced fibrogenesis, IL-33/ST2 through B7 homology 3 (B7H3), polarizes bone marrow (BM) cells to M2 cells, secreting IL-13 and TGF-β (122, 123). (2) IL-33 cooperated with other molecules to promote fibrogenesis: IL-33, IL-25 and thymic stromal lymphopoietin (TSLP) involved in secondary pulmonary fibrogenesis caused by S. mansoni (124).
In vitro experiments showed that P. aeruginosa induced the IL-33 expression in the cystic fibrosis airway epithelial cell line (CFTRdelF508) (125). IL-33 promotes human primary eosinophils to express IL-13; IL-13 induces the MF in the intestine to express Collagen (126). The renal tubular cell line (HK-2) was pretreated with IL-33shRNA, then treated with hypoxia and reoxygenation, and α-SMA and Collagen I expressions were reduction (127).
There are many reports about the role of NLRP3 inflammasome in the fibrogenesis, but there is insufficient evidence on how NLRP3 inflammasome regulate fibrogenesis. NLRP1 and AIM2 inflammasomes are rarely studied in the fibrogenesis and need to understand the phenomenon from the molecular mechanism. More and more studies suggest it is very important that NLRC4 inflammasome in the fibrogenesis, but the molecular mechanism remains to be experimentally elucidated. The role of other inflammasomes in the fibrogenesis has also been reported, such as the role of NLRP6/NLRC5 in the fibrogenesis. IL-1β, IL-18, and IL-33, as activation products of inflammasomes, usually participate in fibrogenesis with other signaling pathways. However, the effect of IL-18 on the fibrogenesis is still controversial, and more experiments are needed to determine whether IL-18 promotes fibrogenesis or inhibits fibrogenesis. In summary, the mechanism of inflammasomes is not completely clear, and the relationship with fibrogenesis deserve more in-depth investigations. The solution of these investigations helps to clarify the role of inflammasomes in fibrogenesis and find new targets for the treatment of fibrosis. Through these targets, drugs focusing on inflammasome-associated molecules are developed to treat fibrotic diseases. Therefore, more in-depth researches centering on inflammasome and fibrosis are necessary.
W-JZ, HW and S-ZW conceived, performed and designed the topics. W-JZ gathered and read papers, as well as wrote the first draft of the manuscript. S-JC, HW and S-ZW corrected and validated the manuscript. S-JC drew the figures. All authors contributed to the article and approved the submitted version.
This work was financially supported by the grants from The Open Project of Key Laboratory of Prevention and treatment of cardiovascular and cerebrovascular diseases, Ministry of Education (No. XN202016).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Yin-Ming Liang and Ji-Sheng Hu for revision.
NLRP1, NOD-like receptor protein 1; NLRP3, NOD-like receptor protein 3; NLRC4, NOD-like receptor C4; AIM2, absent in melanoma 2; ECM, extracellular matrix; mø, macrophages; TGF-β, transforming growth factor-β; IL-1β, interleukin-1β; M, monocytes; DCs, dendritic cells; HSCs, hepatic stellate cells; MF, myofibroblast; ECs, endothelial cells; PCs, parenchymal cells; PRRs, pattern recognition receptor; PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; ASC, apoptosis-associated speck-like protein containing a CARD; pro-caspase-1, pro-cysteinyl aspartate specific proteinase-1; NLRs, NOD-like receptors; ALRs, AIM2-like receptors; Caspase, cysteinyl aspartate specific proteinase; NOD, nucleotide-binding oligomerization domain; NLRPs, NOD-like receptor proteins; NALPs, NACHT, LRR and PYD domains-containing proteins; IPAF, IL-1β-converting enzyme-protease-activating factor; NAIP, neuronal apoptosis inhibitor protein; NLRC5, NOD-like receptor C5; LRR, leucin rich repeat; CARD, caspase recruitment domain; PYD, pyrin domain; BIR, baculoviral inhibitor of apoptosis repeat; dNTP, deoxy-ribonucleoside triphosphate; ATP, adenosine triphosphate; C. pneumoniae, Chlamydia pneumoniae; S. mansoni, Schistosoma mansoni; IL-1/18Rs, interleukin-1/18 receptors; EMT, epithelial-mesenchymal transition; MDP, muramyl dipeptide; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor-κB; CFs, cardiac fibroblasts; P2X7R, P2X7 purinergic receptor; NEK7, NIMA-related kinase 7; ROS, reactive oxygen species; STZ, streptozotocin; BLM, bleomycin; PI3K, phosphatidylinositol 3-kinase; Akt, protein kinase B; JNK, c-Jun N-terminal kinase; ERK, extracellular signal-regulated protein kinase; NOX4, NADPH oxidase 4; TXNIP, thioredoxin interacting protein; mtDNA, mitochondrial DNA; ox-mtDNA, oxidized form; MSU, monosodium urate; ENaC, epithelial sodium channels; CLIC, chloride intracellular channels; DAG, diacyl glycerol; InsP3, inositol trisphosphate; CASR, calcium-sensing receptor; TAA, thioacetamide; AMPK, adenosine monophosphate-activated protein kinase; mTOR, mammalian target of rapamycin; cAMP, cyclic AMP; CCl4, carbon tetrachloride; DMN, dimethylnitrosamine; DEN, diethylnitrosamine; HFD, high-fat diet; MCD, methionine/choline-deficient diet; HBV, hepatitis B virus; S. japonicum, Schistosoma japonicum; KCs, kupffer cells; CCL2, CC motif chemokine ligand 2; α-SMA, α-smooth muscle actin; E. coli, Escherichia coli; TNF, tumor necrosis factor; LPS, lipopolysaccharide; PDGF-AA, platelet derived growth factor-AA; UUO, unilateral ureteral obstruction; TECs, tubular epithelial cells; MMP9, matrix metalloprotein 9; MGO, methylglyoxal; A. fumigatus, Aspergillus fumigatus; P. aeruginosa, Pseudomonas aeruginosa; T3SS, type III secretion system; CFTR, cystic fibrosis transmembrane conductance regulator; IL-1Ra, IL-1R antagonist; dsDNA, double-stranded DNA; PBMCs, peripheral blood mononuclear cells; DENV, dengue virus; IRAK4, IL-1 receptor-associated kinase 4; SCs, stromal cells; PDGFR, platelet-derived growth factor receptor; IGIF, interferon-γ inducing factor; SSLF, schistosome-associated liver fibrosis; IL-1F11, interleukin-1 family 11; B7H3, B7 homology 3; BM, bone marrow; TSLP, thymic stromal lymphopoietin; NLRP6, NOD-like receptor protein 6; Allo-HSCT, allogeneic hematopoietic stem cell transplantation.

1. Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity (2016) 44(3):450–62. doi: 10.1016/j.immuni.2016.02.015
2. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver Fibrosis and Repair: Immune Regulation of Wound Healing in a Solid Organ. Nat Rev Immunol (2014) 14(3):181–94. doi: 10.1038/nri3623
3. Mack M. Inflammation and Fibrosis. Matrix Biol (2018) 68-69:106–21. doi: 10.1016/j.matbio.2017.11.010
4. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci (2019) 20(13):E3328. doi: 10.3390/ijms20133328
5. Lang T, Lee JPW, Elgass K, Pinar AA, Tate MD, Aitken EH, et al. Macrophage Migration Inhibitory Factor Is Required for NLRP3 Inflammasome Activation. Nat Commun (2018) 9(1):2223. doi: 10.1038/s41467-018-04581-2
6. Sun Q, Loughran P, Shapiro R, Shrivastava IH, Antoine DJ, Li T, et al. Redox-Dependent Regulation of Hepatocyte Absent in Melanoma 2 Inflammasome Activation in Sterile Liver Injury in Mice. Hepatology (2017) 65(1):253–68. doi: 10.1002/hep.28893
7. Jakubzick CV, Randolph GJ, Henson PM. Monocyte Differentiation and Antigen-Presenting Functions. Nat Rev Immunol (2017) 17(6):349–62. doi: 10.1038/nri.2017.28
8. Rawlings DJ, Metzler G, Wray-Dutra M, Jackson SW. Altered B Cell Signalling in Autoimmunity. Nat Rev Immunol (2017) 17(7):421–36. doi: 10.1038/nri.2017.24
9. Moulton VR, Tsokos GC. T Cell Signaling Abnormalities Contribute to Aberrant Immune Cell Function and Autoimmunity. J Clin Invest (2015) 125(6):2220–7. doi: 10.1172/JCI78087
10. Durai V, Murphy KM. Functions of Murine Dendritic Cells. Immunity (2016) 45(4):719–36. doi: 10.1016/j.immuni.2016.10.010
11. Higashi T, Friedman SL, Hoshida Y. Hepatic Stellate Cells as Key Target in Liver Fibrosis. Adv Drug Deliv Rev (2017) 121:27–42. doi: 10.1016/j.addr.2017.05.007
12. Le Bras A. Dynamics of Fibroblast Activation in the Infarcted Heart. Nat Rev Cardiol (2018) 15(7):379. doi: 10.1038/s41569-018-0025-9
13. Falke LL, Gholizadeh S, Goldschmeding R, Kok RJ, Nguyen TQ. Diverse Origins of the Myofibroblast-Implications for Kidney Fibrosis. Nat Rev Nephrol (2015) 11(4):233–44. doi: 10.1038/nrneph.2014.246
14. Wang W, Wang X, Chun J, Vilaysane A, Clark S, French G, et al. Inflammasome-Independent NLRP3 Augments TGF-β Signaling in Kidney Epithelium. J Immunol (2013) 190(3):1239–49. doi: 10.4049/jimmunol.1201959
15. Wang Q, Chou X, Guan F, Fang Z, Lu S, Lei J, et al. Enhanced Wnt Signalling in Hepatocytes Is Associated With Schistosoma Japonicum Infection and Contributes to Liver Fibrosis. Sci Rep (2017) 7(1):230. doi: 10.1038/s41598-017-00377-4
16. He Y, Hara H, Núñez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
17. Wang B, Yin Q. AIM2 Inflammasome Activation and Regulation: A Structural Perspective. J Struct Biol (2017) 200(3):279–82. doi: 10.1016/j.jsb.2017.08.001
18. Zong J, Zhang H, Li FF, Liang K, Liu JL, Xu LH, et al. NLRP1 Promotes TGF-β1-Induced Myofibroblast Differentiation in Neonatal Rat Cardiac Fibroblasts. J Mol Histol (2018) 49(5):509–18. doi: 10.1007/s10735-018-9789-9
19. Cai SM, Yang RQ, Li Y, Ning ZW, Zhang LL, Zhou GS, et al. Angiotensin-(1-7) Improves Liver Fibrosis by Regulating the NLRP3 Inflammasome Via Redox Balance Modulation. Antioxid Redox Signal (2016) 24(14):795–812. doi: 10.1089/ars.2015.6498
20. DeSantis DA, Ko CW, Wang L, Lee P, Croniger CM. Constitutive Activation of the NLRC4 Inflammasome Prevents Hepatic Fibrosis and Promotes Hepatic Regeneration After Partial Hepatectomy. Mediators Inflamm (2015) 2015:909827. doi: 10.1155/2015/909827
21. Terlizzi M, Molino A, Colarusso C, Donovan C, Imitazione P, Somma P, et al. Activation of the AIM2 Inflammasome in Peripheral Blood Mononuclear Cells From Idiopathic Pulmonary Fibrosis Patients Leads to the Release of Pro-Fibrotic Mediators. Front Immunol (2018) 9:670. doi: 10.3389/fimmu.2018.00670
22. Szabo G, Csak T. Inflammasomes in Liver Diseases. J Hepatol (2012) 57(3):642–54. doi: 10.1016/j.jhep.2012.03.035
23. Lu A, Wu H. Structural Mechanisms of Inflammasome Assembly. FEBS J (2015) 282(3):435–44. doi: 10.1111/febs.13133
24. Hu Z, Chai J. Structural Mechanisms in NLR Inflammasome Assembly and Signaling. Curr Top Microbiol Immunol (2016) 397:23–42. doi: 10.1007/978-3-319-41171-2_2
25. He X, Mekasha S, Mavrogiorgos N, Fitzgerald KA, Lien E, Ingalls RR. Inflammation and Fibrosis During Chlamydia Pneumoniae Infection Is Regulated by IL-1 and the NLRP3/ASC Inflammasome. J Immunol (2010) 184(10):5743–54. doi: 10.4049/jimmunol.0903937
26. Ritter M, Gross O, Kays S, Ruland J, Nimmerjahn F, Saijo S, et al. Schistosoma Mansoni Triggers Dectin-2, Which Activates the Nlrp3 Inflammasome and Alters Adaptive Immune Responses. Proc Natl Acad Sci USA (2010) 107(47):20459–64. doi: 10.1073/pnas.1010337107
27. Iannitti RG, Napolioni V, Oikonomou V, De Luca A, Galosi C, Pariano M, et al. IL-1 Receptor Antagonist Ameliorates Inflammasome-Dependent Inflammation in Murine and Human Cystic Fibrosis. Nat Commun (2016) 7:10791. doi: 10.1038/ncomms10791
28. Wawrocki S, Druszczynska M, Kowalewicz-Kulbat M, Rudnicka W. Interleukin 18 (IL-18) as a Target for Immune Intervention. Acta Biochim Pol (2016) 63(1):59–63. doi: 10.18388/abp.2015_1153
29. Kotsiou OS, Gourgoulianis KI, Zarogiannis SG. IL-33/ST2 Axis in Organ Fibrosis. Front Immunol (2018) 9:2432. doi: 10.3389/fimmu.2018.02432
30. Lorenz G, Darisipudi MN, Anders HJ. Canonical and Non-Canonical Effects of the NLRP3 Inflammasome in Kidney Inflammation and Fibrosis. Nephrol Dial Transplant (2014) 29(1):41–8. doi: 10.1093/ndt/gft332
31. Rathinam VA, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell (2016) 165(4):792–800. doi: 10.1016/j.cell.2016.03.046
32. Chavarria-Smith J, Vance RE. The NLRP1 Inflammasomes. Immunol Rev (2015) 265(1):22–34. doi: 10.1111/imr.12283
33. Yu CH, Moecking J, Geyer M, Masters SL. Mechanisms of NLRP1-Mediated Autoinflammatory Disease in Humans and Mice. J Mol Biol (2018) 430(2):142–52. doi: 10.1016/j.jmb.2017.07.012
34. Zong J, Li FF, Liang K, Dai R, Zhang H, Yan L, et al. Nuclear Localization Leucine-Rich-Repeat Protein 1 Deficiency Protects Against Cardiac Hypertrophy by Pressure Overload. Cell Physiol Biochem (2018) 48(1):75–86. doi: 10.1159/000491664
35. He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 Is an Essential Mediator of NLRP3 Activation Downstream of Potassium Efflux. Nature (2016) 530(7590):354–7. doi: 10.1038/nature16959
36. Wu M, Han W, Song S, Du Y, Liu C, Chen N, et al. NLRP3 Deficiency Ameliorates Renal Inflammation and Fibrosis in Diabetic Mice. Mol Cell Endocrinol (2018) 478:115–25. doi: 10.1016/j.mce.2018.08.002
37. Stout-Delgado HW, Cho SJ, Chu SG, Mitzel DN, Villalba J, El-Chemaly S, et al. Age-Dependent Susceptibility to Pulmonary Fibrosis Is Associated With NLRP3 Inflammasome Activation. Am J Respir Cell Mol Biol (2016) 55(2):252–63. doi: 10.1165/rcmb.2015-0222OC
38. Ning ZW, Luo XY, Wang GZ, Li Y, Pan MX, Yang RQ, et al. MicroRNA-21 Mediates Angiotensin II-Induced Liver Fibrosis by Activating NLRP3 Inflammasome/IL-1β Axis Via Targeting Smad7 and Spry1. Antioxid Redox Signal (2017) 27(1):1–20. doi: 10.1089/ars.2016.6669
39. Gong Z, Zhou J, Zhao S, Tian C, Wang P, Xu C, et al. Chenodeoxycholic Acid Activates NLRP3 Inflammasome and Contributes to Cholestatic Liver Fibrosis. Oncotarget (2016) 7(51):83951–63. doi: 10.18632/oncotarget.13796
40. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome During Apoptosis. Immunity (2012) 36(3):401–14. doi: 10.1016/j.immuni.2012.01.009
41. Zheng R, Tao L, Jian H, Chang Y, Cheng Y, Feng Y, et al. NLRP3 Inflammasome Activation and Lung Fibrosis Caused by Airborne Fine Particulate Matter. Ecotoxicol Environ Saf (2018) 163:612–9. doi: 10.1016/j.ecoenv.2018.07.076
42. Sun B, Wang X, Ji Z, Wang M, Liao YP, Chang CH, et al. NADPH Oxidase-Dependent NLRP3 Inflammasome Activation and its Important Role in Lung Fibrosis by Multiwalled Carbon Nanotubes. Small (2015) 11(17):2087–97. doi: 10.1002/smll.201402859
43. Scambler T, Jarosz-Griffiths HH, Lara-Reyna S, Pathak S, Wong C, Holbrook J, et al. ENaC-Mediated Sodium Influx Exacerbates NLRP3-Dependent Inflammation in Cystic Fibrosis. Elife (2019) 8:e49248. doi: 10.7554/eLife.49248
44. Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, et al. CLICs-Dependent Chloride Efflux Is an Essential and Proximal Upstream Event for NLRP3 Inflammasome Activation. Nat Commun (2017) 8(1):202. doi: 10.1038/s41467-017-00227-x
45. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, et al. The Calcium-Sensing Receptor Regulates the NLRP3 Inflammasome Through Ca2+ and Camp. Nature (2012) 492(7427):123–7. doi: 10.1038/nature11588
46. Wang Q, Wei S, Zhou S, Qiu J, Shi C, Liu R, et al. Hyperglycemia Aggravates Acute Liver Injury by Promoting Liver-Resident Macrophage NLRP3 Inflammasome Activation Via the Inhibition of AMPK/mTOR-Mediated Autophagy Induction. Immunol Cell Biol (2020) 98(1):54–66. doi: 10.1111/imcb.12297
47. Campana L, Iredale JP. Regression of Liver Fibrosis. Semin Liver Dis (2017) 37(1):1–10. doi: 10.1055/s-0036-1597816
48. Yanguas SC, Cogliati B, Willebrords J, Maes M, Colle I, van den Bossche B, et al. Experimental Models of Liver Fibrosis. Arch Toxicol (2016) 90(5):1025–48. doi: 10.1007/s00204-015-1543-4
49. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 Inflammasome Activation Results in Hepatocyte Pyroptosis, Liver Inflammation, and Fibrosis in Mice. Hepatology (2014) 59(3):898–910. doi: 10.1002/hep.26592
50. Martínez-Torres AC, Reyes-Ruiz A, Calvillo-Rodriguez KM, Alvarez-Valadez KM, Uscanga-Palomeque AC, Tamez-Guerra RS, et al. Immunepotent CRP Induces DAMPs Release and ROS-Dependent Autophagosome Formation in HeLa and MCF-7 Cells. BMC Cancer (2020) 20(1):647. doi: 10.1186/s12885-020-07124-5
51. Liu Y, Lou G, Li A, Zhang T, Qi J, Ye D, et al. AMSC-Derived Exosomes Alleviate Lipopolysaccharide/D-Galactosamine-Induced Acute Liver Failure by miR-17-Mediated Reduction of TXNIP/NLRP3 Inflammasome Activation in Macrophages. EBioMedicine (2018) 36:140–50. doi: 10.1016/j.ebiom.2018.08.054
52. Geng Y, Ma Q, Liu YN, Peng N, Yuan FF, Li XG, et al. Heatstroke Induces Liver Injury Via IL-1β and HMGB1-Induced Pyroptosis. J Hepatol (2015) 63(3):622–33. doi: 10.1016/j.jhep.2015.04.010
53. Liu X, Lu B, Fu J, Zhu X, Song E, Song Y. Amorphous Silica Nanoparticles Induce Inflammation Via Activation of NLRP3 Inflammasome and HMGB1/TLR4/Myd88/NF-κB Signaling Pathway in HUVEC Cells. J Hazard Mater (2021) 404(Pt B):124050. doi: 10.1016/j.jhazmat.2020.124050
54. Swanson KV, Deng M, Ting JP. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
55. Silva-Palacios A, Ostolga-Chavarría M, Zazueta C, Königsberg M. Nrf2: Molecular and Epigenetic Regulation During Aging. Ageing Res Rev (2018) 47:31–40. doi: 10.1016/j.arr.2018.06.003
56. Kopacz A, Kloska D, Forman HJ, Jozkowicz A, Grochot-Przeczek A. Beyond Repression of Nrf2: An Update on Keap1. Free Radic Biol Med (2020), 157:63–74. doi: 10.1016/j.freeradbiomed.2020.03.023
57. Hennig P, Garstkiewicz M, Grossi S, Di Filippo M, French LE, Beer HD. The Crosstalk Between Nrf2 and Inflammasomes. Int J Mol Sci (2018) 19(2):562. doi: 10.3390/ijms19020562
58. Liu WH, Shi LS, Chung MC, Chang TC, Lee SY. Antcamphin M Inhibits TLR4-Mediated Inflammatory Responses by Upregulating the Nrf2/HO-1 Pathway and Suppressing the NLRP3 Inflammasome Pathway in Macrophages. Am J Chin Med (2019) 47(7):1611–26. doi: 10.1142/S0192415X19500824
59. Ma PF, Gao CC, Yi J, Zhao JL, Liang SQ, Zhao Y, et al. Cytotherapy With M1-Polarized Macrophages Ameliorates Liver Fibrosis by Modulating Immune Microenvironment in Mice. J Hepatol (2017) 67(4):770–9. doi: 10.1016/j.jhep.2017.05.022
60. Inzaugarat ME, Johnson CD, Holtmann TM, McGeough MD, Trautwein C, Papouchado BG, et al. NLRP3 Inflammasome Activation in Hepatic Stellate Cells Induces Murine Liver Fibrosis. Hepatology (2019) 69(2):845–59. doi: 10.1002/hep.30252
61. Gao J, Wei B, Liu M, Hirsova P, Sehrawat TS, Cao S, et al. Endothelial p300 Promotes Portal Hypertension and Hepatic Fibrosis Through C-C Motif Chemokine Ligand 2-Mediated Angiocrine Signaling. Hepatology (2020) 10.1002/hep.31617. doi: 10.1002/hep.31617
62. Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. An Essential Role for Monocyte Chemoattractant Protein-1 in Alcoholic Liver Injury: Regulation of Proinflammatory Cytokines and Hepatic Steatosis in Mice. Hepatology (2011) 54(6):2185–97. doi: 10.1002/hep.24599
63. Meng N, Xia M, Lu YQ, Wang M, Boini KM, Li PL, et al. Activation of NLRP3 Inflammasomes in Mouse Hepatic Stellate Cells During Schistosoma J Infection. Oncotarget (2016) 7(26):39316–31. doi: 10.18632/oncotarget.10044
64. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 Inflammasome Blockade Reduces Liver Inflammation and Fibrosis in Experimental NASH in Mice. J Hepatol (2017) 66(5):1037–46. doi: 10.1016/j.jhep.2017.01.022
65. Han X, Song J, Lian LH, Yao YL, Shao DY, Fan Y, et al. Ginsenoside 25-OCH3-PPD Promotes Activity of LXRs to Ameliorate P2X7R-Mediated NLRP3 Inflammasome in the Development of Hepatic Fibrosis. J Agric Food Chem (2018) 66(27):7023–35. doi: 10.1021/acs.jafc.8b01982
66. Albillos A, de Gottardi A, Rescigno M. The Gut-Liver Axis in Liver Disease: Pathophysiological Basis for Therapy. J Hepatol (2020) 72(3):558–77. doi: 10.1016/j.jhep.2019.10.003
67. Zou J, Wang SP, Wang YT, Wan JB. Regulation of the NLRP3 Inflammasome With Natural Products Against Chemical-Induced Liver Injury. Pharmacol Res (2021) 164:105388. doi: 10.1016/j.phrs.2020.105388
68. Roderfeld M. Matrix Metalloproteinase Functions in Hepatic Injury and Fibrosis. Matrix Biol (2018) 68-69:452–62. doi: 10.1016/j.matbio.2017.11.011
69. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu Rev Pathol (2020) 15:493–518. doi: 10.1146/annurev-pathmechdis-012419-032847
70. Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, et al. Inflammasome-Mediated Regulation of Hepatic Stellate Cells. Am J Physiol Gastrointest Liver Physiol (2009) 296(6):G1248–57. doi: 10.1152/ajpgi.90223.2008
71. Zhang LL, Huang S, Ma XX, Zhang WY, Wang D, Jin SY, et al. Angiotensin(1-7) Attenuated Angiotensin II-Induced Hepatocyte EMT by Inhibiting NOX-Derived H2O2-Activated NLRP3 Inflammasome/IL-1β/Smad Circuit. Free Radic Biol Med (2016) 97:531–43. doi: 10.1016/j.freeradbiomed.2016.07.014
72. Lu YQ, Zhong S, Meng N, Fan YP, Tang WX. NLRP3 Inflammasome Activation Results in Liver Inflammation and Fibrosis in Mice Infected With Schistosoma Japonicum in a Syk-Dependent Manner. Sci Rep (2017) 7(1):8120. doi: 10.1038/s41598-017-08689-1
73. Wang H, Liu S, Wang Y, Chang B, Wang B. NLRP3 Inflammasome Activation by Escherichia Coli RNA Induces TGF-β 1 Secretion in Hepatic Stellate Cells. Bosn J Basic Med Sci (2016) 16(2):126–31. doi: 10.17305/bjbms.2016.699
74. Wree A, McGeough MD, Inzaugarat ME, Eguchi A, Schuster S, Johnson CD, et al. NLRP3 Inflammasome Driven Liver Injury and Fibrosis: Roles of IL-17 and TNF in Mice. Hepatology (2018) 67(2):736–49. doi: 10.1002/hep.29523
75. Kang LL, Zhang DM, Ma CH, Zhang JH, Jia KK, Liu JH, et al. Cinnamaldehyde and Allopurinol Reduce Fructose-Induced Cardiac Inflammation and Fibrosis by Attenuating CD36-Mediated TLR4/6-IRAK4/1 Signaling to Suppress NLRP3 Inflammasome Activation. Sci Rep (2016) 6:27460. doi: 10.1038/srep27460
76. Bracey NA, Gershkovich B, Chun J, Vilaysane A, Meijndert HC, Wright JR Jr, et al. Mitochondrial NLRP3 Protein Induces Reactive Oxygen Species to Promote Smad Protein Signaling and Fibrosis Independent From the Inflammasome. J Biol Chem (2014) 289(28):19571–84. doi: 10.1074/jbc.M114.550624
77. Zhang H, Tian L, Shen M, Tu C, Wu H, Gu M, et al. Generation of Quiescent Cardiac Fibroblasts From Human Induced Pluripotent Stem Cells for In Vitro Modeling of Cardiac Fibrosis. Circ Res (2019) 125(5):552–66. doi: 10.1161/CIRCRESAHA.119.315491
78. Li X, Geng J, Zhao J, Ni Q, Zhao C, Zheng Y, et al. Trimethylamine N-Oxide Exacerbates Cardiac Fibrosis Via Activating the NLRP3 Inflammasome. Front Physiol (2019) 10:866. doi: 10.3389/fphys.2019.00866
79. Gan W, Ren J, Li T, Lv S, Li C, Liu Z, et al. The SGK1 Inhibitor EMD638683, Prevents Angiotensin II-Induced Cardiac Inflammation and Fibrosis by Blocking NLRP3 Inflammasome Activation. Biochim Biophys Acta Mol Basis Dis (2018) 1864(1):1–10. doi: 10.1016/j.bbadis.2017.10.001
80. Li Y, Li H, Liu S, Pan P, Su X, Tan H, et al. Pirfenidone Ameliorates Lipopolysaccharide-Induced Pulmonary Inflammation and Fibrosis by Blocking NLRP3 Inflammasome Activation. Mol Immunol (2018) 99:134–44. doi: 10.1016/j.molimm.2018.05.003
81. Song C, He L, Zhang J, Ma H, Yuan X, Hu G, et al. Fluorofenidone Attenuates Pulmonary Inflammation and Fibrosis Via Inhibiting the Activation of NALP3 Inflammasome and IL-1β/IL-1R1/Myd88/NF-κB Pathway. J Cell Mol Med (2016) 20(11):2064–77. doi: 10.1111/jcmm.12898
82. Ligresti G, Caporarello N, Meridew JA, Jones DL, Tan Q, Choi KM, et al. CBX5/G9a/H3K9me-Mediated Gene Repression Is Essential to Fibroblast Activation During Lung Fibrosis. JCI Insight (2019) 5(12):1–20. doi: 10.1172/jci.insight.127111
83. Artlett CM, Sassi-Gaha S, Hope JL, Feghali-Bostwick CA, Katsikis PD. Mir-155 Is Overexpressed in Systemic Sclerosis Fibroblasts and Is Required for NLRP3 Inflammasome-Mediated Collagen Synthesis During Fibrosis. Arthritis Res Ther (2017) 19(1):144. doi: 10.1186/s13075-017-1331-z
84. Wang X, Sun B, Liu S, Xia T. Structure Activity Relationships of Engineered Nanomaterials in Inducing NLRP3 Inflammasome Activation and Chronic Lung Fibrosis. NanoImpact (2017) 6:99–108. doi: 10.1016/j.impact.2016.08.002
85. Lv Z, Wang Y, Liu YJ, Mao YF, Dong WW, Ding ZN, et al. NLRP3 Inflammasome Activation Contributes to Mechanical Stretch-Induced Endothelial-Mesenchymal Transition and Pulmonary Fibrosis. Crit Care Med (2018) 46(1):e49–58. doi: 10.1097/CCM.0000000000002799
86. Chi HH, Hua KF, Lin YC, Chu CL, Hsieh CY, Hsu YJ, et al. IL-36 Signaling Facilitates Activation of the NLRP3 Inflammasome and IL-23/IL-17 Axis in Renal Inflammation and Fibrosis. J Am Soc Nephrol (2017) 28(7):2022–37. doi: 10.1681/ASN.2016080840
87. Ludwig-Portugall I, Bartok E, Dhana E, Evers BD, Primiano MJ, Hall JP, et al. An NLRP3-Specific Inflammasome Inhibitor Attenuates Crystal-Induced Kidney Fibrosis in Mice. Kidney Int (2016) 90(3):525–39. doi: 10.1016/j.kint.2016.03.035
88. Wen Y, Pan MM, Lv LL, Tang TT, Zhou LT, Wang B, et al. Artemisinin Attenuates Tubulointerstitial Inflammation and Fibrosis Via the NF-κB/NLRP3 Pathway in Rats With 5/6 Subtotal Nephrectomy. J Cell Biochem (2019) 120(3):4291–300. doi: 10.1002/jcb.27714
89. Zhang GX, Wang MX, Nie W, Liu DW, Zhang Y, Liu HB. P2X7R Blockade Prevents NLRP3 Inflammasome Activation and Pancreatic Fibrosis in a Mouse Model of Chronic Pancreatitis. Pancreas (2017) 46(10):1327–35. doi: 10.1097/MPA.0000000000000928
90. Hishida E, Ito H, Komada T, Karasawa T, Kimura H, Watanabe S, et al. Crucial Role of NLRP3 Inflammasome in the Development of Peritoneal Dialysis-Related Peritoneal Fibrosis. Sci Rep (2019) 9(1):10363. doi: 10.1038/s41598-019-46504-1
91. Hughes FM Jr, Sexton SJ, Jin H, Govada V, Purves JT. Bladder Fibrosis During Outlet Obstruction Is Triggered Through the NLRP3 Inflammasome and the Production of IL-1β. Am J Physiol Renal Physiol (2017) 313(3):F603–10. doi: 10.1152/ajprenal.00128.2017
92. Duncan JA, Canna SW. The NLRC4 Inflammasome. Immunol Rev (2018) 281(1):115–23. doi: 10.1111/imr.12607
93. Vance RE. The NAIP/NLRC4 Inflammasomes. Curr Opin Immunol (2015) 32:84–9. doi: 10.1016/j.coi.2015.01.010
94. Nigdelioglu R, Hamanaka RB, Meliton AY, O’Leary E, Witt LJ, Cho T, et al. Transforming Growth Factor (TGF)-β Promotes De Novo Serine Synthesis for Collagen Production. J Biol Chem (2016) 291(53):27239–51. doi: 10.1074/jbc.M116.756247
95. Levy M, Shapiro H, Thaiss CA, Elinav E. NLRP6: A Multifaceted Innate Immune Sensor. Trends Immunol (2017) 38(4):248–60. doi: 10.1016/j.it.2017.01.001
96. Li M, Chen Y, Shi J, Ju W, Qi K, Fu C, et al. NLRP6 Deficiency Aggravates Liver Injury After Allogeneic Hematopoietic Stem Cell Transplantation. Int Immunopharmacol (2019) 74:105740. doi: 10.1016/j.intimp.2019.105740
97. Zhu Y, Ni T, Deng W, Lin J, Zheng L, Zhang C, et al. Effects of NLRP6 on the Proliferation and Activation of Human Hepatic Stellate Cells. Exp Cell Res (2018) 370(2):383–8. doi: 10.1016/j.yexcr.2018.06.040
98. Kobayashi KS, van den Elsen PJ. NLRC5: A Key Regulator of MHC Class I-Dependent Immune Responses. Nat Rev Immunol (2012) 12(12):813–20. doi: 10.1038/nri3339
99. Xu T, Ni MM, Xing L, Li XF, Meng XM, Huang C, et al. NLRC5 Regulates TGF-β1-Induced Proliferation and Activation of Hepatic Stellate Cells During Hepatic Fibrosis. Int J Biochem Cell Biol (2016) 70:92–104. doi: 10.1016/j.biocel.2015.11.010
100. Liu X, Wu Y, Yang Y, Li W, Huang C, Meng X, et al. Role of NLRC5 in Progression and Reversal of Hepatic Fibrosis. Toxicol Appl Pharmacol (2016) 294:43–53. doi: 10.1016/j.taap.2016.01.012
101. Miao NJ, Xie HY, Xu D, Yin JY, Wang YZ, Wang B, et al. Caspase-11 Promotes Renal Fibrosis by Stimulating IL-1β Maturation Via Activating Caspase-1. Acta Pharmacol Sin (2019) 40(6):790–800. doi: 10.1038/s41401-018-0177-5
102. Otto G. IL-1β Switches on Kidney Fibrosis. Nat Rev Nephrol (2018) 14(8):475. doi: 10.1038/s41581-018-0026-2
103. Morrison MC, Mulder P, Salic K. Intervention With a Caspase-1 Inhibitor Reduces Obesity-Associated Hyperinsulinemia, Non-Alcoholic Steatohepatitis and Hepatic Fibrosis in LDLR-/-. Leiden Mice. Int J Obes (Lond) (2016) 40(9):1416–23. doi: 10.1038/ijo.2016.74
104. El Kasmi KC, Vue PM, Anderson AL, Devereaux MW, Ghosh S, Balasubramaniyan N, et al. Macrophage-Derived IL-1β/Nf-κB Signaling Mediates Parenteral Nutrition-Associated Cholestasis. Nat Commun (2018) 9(1):1393. doi: 10.1038/s41467-018-03764-1
105. Cheung KT, Sze DM, Chan KH, Leung PH. Involvement of Caspase-4 in IL-1β Production and Pyroptosis in Human Macrophages During Dengue Virus Infection. Immunobiology (2018) 223(4-5):356–64. doi: 10.1016/j.imbio.2017.10.044
106. Amir M, Czaja MJ. Inflammasome-Mediated Inflammation and Fibrosis: It Is More Than Just the IL-1β. Hepatology (2018) 67(2):479–81. doi: 10.1002/hep.29491
107. Barlo NP, van Moorsel CH, Korthagen NM, Heron M, Rijkers GT, Ruven HJ, et al. Genetic Variability in the IL1RN Gene and the Balance Between IL-1 Receptor Agonist and IL-1β in Idiopathic Pulmonary Fibrosis. Clin Exp Immunol (2011) 166(3):346–51. doi: 10.1111/j.1365-2249.2011.04468.x
108. Zhang M, Guo Y, Fu H, Hu S, Pan J, Wang Y, et al. Chop Deficiency Prevents UUO-Induced Renal Fibrosis by Attenuating Fibrotic Signals Originated From HMGB1/TLR4/NF-κB/IL-1β Signaling. Cell Death Dis (2015) 6:e1847. doi: 10.1038/cddis.2015.206
109. Klinkhammer BM, Floege J, Boor P. PDGF in Organ Fibrosis. Mol Aspects Med (2018) 62:44–62. doi: 10.1016/j.mam.2017.11.008
110. Artlett CM. The Role of the NLRP3 Inflammasome in Fibrosis. Open Rheumatol J (2012) 6:80–6. doi: 10.2174/1874312901206010080
111. Sugawara S, Uehara A, Nochi T, Yamaguchi T, Ueda H, Sugiyama A, et al. Neutrophil Proteinase 3-Mediated Induction of Bioactive IL-18 Secretion by Human Oral Epithelial Cells. J Immunol (2001) 167(11):6568–75. doi: 10.4049/jimmunol.167.11.6568
112. Pirhonen J. Regulation of IL-18 Expression in Virus Infection. Scand J Immunol (2001) 53(6):533–9. doi: 10.1046/j.1365-3083.2001.00939.x
113. Hayashi N, Yoshimoto T, Izuhara K, Matsui K, Tanaka T, Nakanishi K. T Helper 1 Cells Stimulated With Ovalbumin and IL-18 Induce Airway Hyperresponsiveness and Lung Fibrosis by IFN-γ and IL-13 Production. Proc Natl Acad Sci USA (2007) 104(37):14765–70. doi: 10.1073/pnas.0706378104
114. Liang H, Xu F, Zhang T, Huang J, Guan Q, Wang H, et al. Inhibition of IL-18 Reduces Renal Fibrosis After Ischemia-Reperfusion. BioMed Pharmacother (2018) 106:879–89. doi: 10.1016/j.biopha.2018.07.031
115. Xiao H, Li H, Wang JJ, Zhang JS, Shen J, An XB, et al. IL-18 Cleavage Triggers Cardiac Inflammation and Fibrosis Upon β-Adrenergic Insult. Eur Heart J (2018) 39(1):60–9. doi: 10.1093/eurheartj/ehx261
116. Tian Z, Wang XY, Zhou YF, Feng QM, Zhang SJ, Yin TQ, et al. Schistosoma Japonicum scFv-IL18 Fusion DNA Ameliorates Hepatic Fibrosis in Schistosomiasis-Infected Mice Via Improving Local Concentration of IL-18 in Liver. Exp Parasitol (2013) 134(4):447–54. doi: 10.1016/j.exppara2013.05.002
117. Zhang LH, Pan JP, Yao HP, Sun WJ, Xia DJ, Wang QQ, et al. Intrasplenic Transplantation of IL-18 Gene-Modified Hepatocytes: An Effective Approach to Reverse Hepatic Fibrosis in Schistosomiasis Through Induction of Dominant Th1 Response. Gene Ther (2001) 8(17):1333–42. doi: 10.1038/sj.gt.3301524
118. Liu X, Xiao Y, Pan Y, Li H, Zheng SG, Su W. The Role of the IL-33/ST2 Axis in Autoimmune Disorders: Friend or Foe? Cytokine Growth Factor Rev (2019) 50:60–74. doi: 10.1016/j.cytogfr.2019.04.004
119. Li ZY, Xiao L, Lin G, Tang J, Chen Y, Chen L, et al. Contribution of Tissue Transglutaminase to the Severity of Hepatic Fibrosis Resulting From Schistosoma Japonicum Infection Through the Regulation of IL-33/ST2 Expression. Parasit Vectors (2019) 12(1):302. doi: 10.1186/s13071-019-3542-4
120. Gao Y, Liu Y, Yang M, Guo X, Zhang M, Li H, et al. IL-33 Treatment Attenuated Diet-Induced Hepatic Steatosis But Aggravated Hepatic Fibrosis. Oncotarget (2016) 7(23):33649–61. doi: 10.18632/oncotarget.9259
121. Fanny M, Nascimento M, Baron L, Schricke C, Maillet I, Akbal M, et al. The IL-33 Receptor ST2 Regulates Pulmonary Inflammation and Fibrosis to Bleomycin. Front Immunol (2018) 9:1476. doi: 10.3389/fimmu.2018.01476
122. Nakashima T, Liu T, Hu B, Wu Z, Ullenbruch M, Omori K, et al. Role of B7H3/IL-33 Signaling in Pulmonary Fibrosis-Induced Profibrogenic Alterations in Bone Marrow. Am J Respir Crit Care Med (2019) 200(8):1032–44. doi: 10.1164/rccm.201808-1560OC
123. Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 Promotes ST2-Dependent Lung Fibrosis by the Induction of Alternatively Activated Macrophages and Innate Lymphoid Cells in Mice. J Allergy Clin Immunol (2014) 134(6):1422–32. doi: 10.1016/j.jaci.2014.05.011
124. Vannella KM, Ramalingam TR, Borthwick LA, Barron L, Hart KM, Thompson RW, et al. Combinatorial Targeting of TSLP, IL-25, and IL-33 in Type 2 Cytokine-Driven Inflammation and Fibrosis. Sci Transl Med (2016) 8(337):337ra365. doi: 10.1126/scitranslmed.aaf1938
125. Farias R, Rousseau S. The TAK1→IKKBETA→TPL2→MKK1/MKK2 Signaling Cascade Regulates IL-33 Expression in Cystic Fibrosis Airway Epithelial Cells Following Infection by Pseudomonas Aeruginosa. Front Cell Dev Biol (2016) 3:87. doi: 10.3389/fcell.2015.00087
126. Masterson JC, Capocelli KE, Hosford L, Biette K, McNamee EN, de Zoeten EF, et al. Eosinophils and IL-33 Perpetuate Chronic Inflammation and Fibrosis in a Pediatric Population With Stricturing Crohn’s Ileitis. Inflamm Bowel Dis (2015) 21(10):2429–40. doi: 10.1097/MIB.0000000000000512
Keywords: inflammasome, fibrosis, NLRP3, caspase-1, IL-1β
Citation: Zhang W-J, Chen S-J, Zhou S-C, Wu S-Z and Wang H (2021) Inflammasomes and Fibrosis. Front. Immunol. 12:643149. doi: 10.3389/fimmu.2021.643149
Received: 17 December 2020; Accepted: 24 May 2021;
Published: 11 June 2021.
Edited by:
Juan Carlos Cutrin, University of Turin, ItalyReviewed by:
Ankit Saxena, National Institutes of Health (NIH), United StatesCopyright © 2021 Zhang, Chen, Zhou, Wu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Wang, d2FuZ2h1aUB4eG11LmVkdS5jbg==; Su-Zhen Wu, d3VzdXpoZW4yMDA1QDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.