 Nilesh M. Agalave
Nilesh M. Agalave Prapti H. Mody
Prapti H. Mody Thomas A. Szabo-Pardi
Thomas A. Szabo-Pardi Han S. Jeong
Han S. Jeong Michael D. Burton
Michael D. Burton- Neuroimmunology and Behavior Laboratory, Department of Neuroscience, School of Behavioral and Brain Sciences, Center for Advanced Pain Studies, University of Texas at Dallas, Richardson, TX, United States
Chemotherapy-induced peripheral neuropathy (CIPN) is a major dose-limiting side effect that occurs in up to 63% of patients and has no known effective treatment. A majority of studies do not effectively assess sex differences in the onset and persistence of CIPN. Here we investigated the onset of CIPN, a point of therapeutic intervention where we may limit, or even prevent the development of CIPN. We hypothesized that cap-dependent translation mechanisms are important in early CIPN development and the bi-directional crosstalk between immune cells and nociceptors plays a complementary role to CIPN establishment and sex differences observed. In this study, we used wild type and eIF4E-mutant mice of both sexes to investigate the role of cap-dependent translation and the contribution of immune cells and nociceptors in the periphery and glia in the spinal cord during paclitaxel-induced peripheral neuropathy. We found that systemically administered paclitaxel induces pain-like behaviors in both sexes, increases helper T-lymphocytes, downregulates cytotoxic T-lymphocytes, and increases mitochondrial dysfunction in dorsal root ganglia neurons; all of which is eIF4E-dependent in both sexes. We identified a robust paclitaxel-induced, eIF4E-dependent increase in spinal astrocyte immunoreactivity in males, but not females. Taken together, our data reveals that cap-dependent translation may be a key pathway that presents relevant therapeutic targets during the early phase of CIPN. By targeting the eIF4E complex, we may reduce or reverse the negative effects associated with chemotherapeutic treatments.
Introduction
Chemotherapy-induced peripheral neuropathy (CIPN) is one of the most prevalent and dose-limiting complications in chemotherapy patients. It is characterized by pain and numbness or a “pins and needles” feeling in the extremities (1–4). Approximately 70% of patients report the development of peripheral neuropathy during administration of chemotherapy, which leads to early treatment cessation and substandard regimens (5, 6). Additionally, over 30% of patients experience persistent neuropathy for months or years after treatment cessation, making CIPN a long-term morbidity in patients that survive cancer and other diseases (7).
There is limited data on sex differences in neuroimmune effects during CIPN. Both clinical and preclinical investigations have reported conflicting observations ranging from marked sexual dimorphisms to no sex differences during CIPN (8–11). There are few studies that investigate sex differences and the mechanisms involved in the development of CIPN. These findings necessitate a comprehensive and fully powered assessment of sex differences in the development of CIPN. Considering the recent interest in sex differences in chronic pain development and immune cell activation, it is purported that neuroimmune mechanisms vary between sexes. A benchmark paper in the pain field suggests sexual dimorphisms in the role of T-cells in pain initiation in females (12). Moreover, recent studies have shown that cytotoxic T-cells (CD8+) contribute to the resolution of CIPN (13, 14). A more recent study showed that “primed” CD8+ T-cells, previously exposed to a chemotherapy agent, could prevent CIPN development when transferred to a new host (15). Based on these previous studies, we decided to assess the role of discrete T-cell subtypes: CD4+(Th1, Th2), T regulatory (Tregs), Effector (Teff), and activated cytotoxic T-lymphocytes (CD8+) in the draining lymph nodes of male and female mice after induction of CIPN.
Immune cells recruited to the dorsal root ganglion (DRG) in turn can influence the activity of nociceptors through the release of inflammatory mediators (16, 17). This signaling could happen in tandem, in real-time, and this bi-directional crosstalk could be directly responsible for changes in nociceptors and immune cells, leading to changes in pain behaviors. These reciprocal interactions may contribute to maladaptive nociceptor plasticity if the homeostatic functioning of the cell-mediated immune response is dysregulated and has been shown to contribute to the development of CIPN (18–21)
Eukaryotic translation initiation factor 4E (eIF4E) is a cytosolic regulator that directs ribosomes to the cap structure of a subset of mRNAs to induce translation of proteins involved in inflammation and pain (22, 23). Cellular signaling cascades responsive to external stimuli such as the mitogen-associated protein kinase (MAPK) pathway and the mammalian target of rapamycin (mTOR) pathway directly feed into central cap-dependent translation in all cell types (24, 25). Regulation of eIF4E activation affects downstream signaling for production of a subset of pro-inflammatory cytokines involved in T-cell subsets activation, cellular metabolic pathways of nociceptors, and other components that regulate pain, inflammation, and behavior (26–33). While we have begun to assess the role of eIF4E phosphorylation in sensory neuron populations (34), a complete breakdown of sex and sensory neuron metabolic phenotyping has yet to occur along with phenotyping of immune cell subtypes during the development of CIPN. It is known that sensory neuron metabolism is exceptionally responsive to nerve injury and inflammation (35–37). CIPN is accompanied with changes in mitochondrial bioenergetics and an energy deficit in DRG neurons (38, 39). These changes can also alter ion signaling and other downstream metabolic processes to contribute to long-term development of nociceptor sensitivity and pain (40). The metabolic effect(s) paclitaxel has in late CIPN, has been assessed in sensory neurons from male rats only (38).
Therefore, we sought to elucidate the role of cap-dependent protein translation in CIPN-induced alterations that may be mediated by neuroimmune communication between activated immune cells and sensory neurons. We used eIF4E mice that harbor a point mutation at the serine 209 site on the eIF4E complex that codes alanine instead, designated as eIF4ES209A (41). This point mutation prevents phosphorylation of eIF4E, thus inhibiting its activation and consequently inhibiting cap-dependent protein translation (42, 43). Paclitaxel is one of many chemotherapeutics used in rodent models to mimic clinical CIPN (44). Given that CIPN has an elusive etiology and the diverse array of cell types that could participate in CIPN development, we investigated whether there was a sex-dependent role of eIF4E phosphorylation in DRG sensory neuron metabolism, local immune cell populations, peripheral T-cell subpopulations, and spinal microglia and astrocytes. We hypothesized that eIF4E phosphorylation regulates activation of specific macrophage and T-cell subpopulations, alters nociceptor metabolism, and glial activation during the early phase of paclitaxel-induced peripheral neuropathy.
Materials and Methods
Animals
Adult mice of both sexes (9-12 weeks old, 20-25g weight) were used for all experiments. eIF4ES209A mice were generated on a C57BL/6 background in the Sonenberg laboratory at McGill University as previously described (41). These were a gift and further bred to maintain genotypes at the University of Texas at (UT) Dallas vivarium to generate our experimental cohorts. In-house animals were weaned between 21 and 28 days of age and tail-clipped to verify genotypes. Both wild-type (WT) and eIF4ES209A gift mice were bred with breeder animals purchased from Jackson laboratory (WT). We used only homozygous eIF4ES209A mutant animals and WT littermates for experiments. All animals were housed at the UT Dallas vivarium with standard temperature (20-25°C) and a 12-hour light/dark cycle (lights on from 6 AM to 6 PM). Mice were group housed in polypropylene ventilated cages with 4-5 animals per cage and provided ad libitum access to food and water. All behavioral experiments and data analysis were carried out from 9am to 12pm during the light cycle. All experiments were performed in accordance with protocols and standard operating procedures (SOPs) approved by the University of Texas at Dallas Institutional Animal Care and Use Committee (IACUC) and the Institution Biosafety and Chemical Safety Committee. The experimenters were blinded to animal genotypes and treatment groups for all assays. A timeline of experiments and dosing regimen is shown in Figure 1.
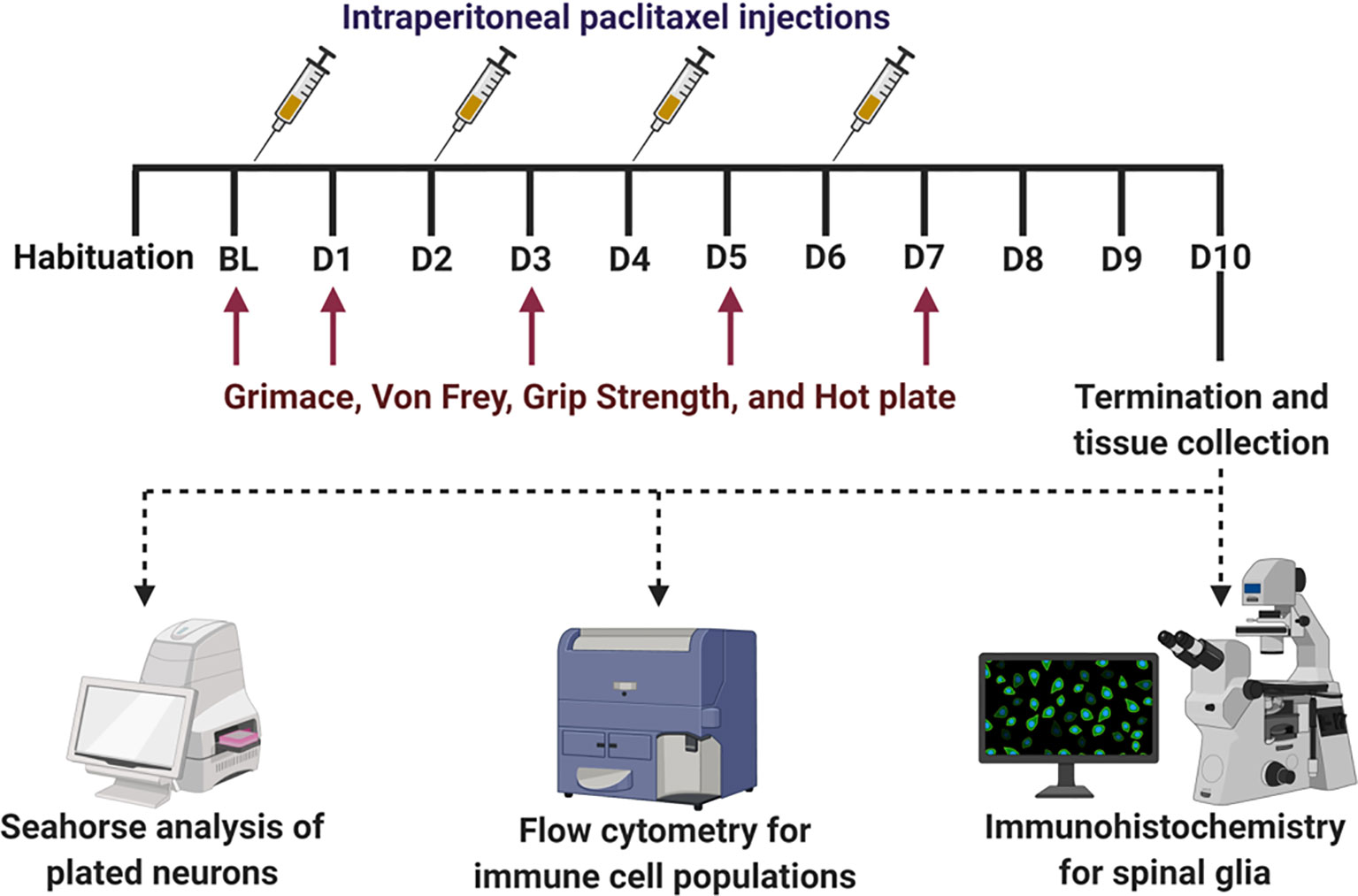
Figure 1 Experimental timeline with dosing regimen and terminal analysis. After habituation and baseline (BL) measures for behavior, all mice were subjected to intraperitoneal injection of paclitaxel (4 mg/kg per mouse) after BL i.e. on day 0, D2, D4, and D6 to induce peripheral neuropathy. Behavioral assays (mechanical hypersensitivity, grimace, grip strength, and thermal hypersensitivity) were measured on BL, D1, D3, D5, D7, and D9. On D10, animals were terminated and spinal cord, popliteal and inguinal lymph nodes, and DRG (L3 to T13) were collected. The lumbar portion of the spinal cord was used for immunohistochemical analyses for glia reactivity. The lymph nodes and DRG were used for flow cytometry analyses of immune cell populations. DRG neurons were plated and used for Seahorse analyses of bioenergetics.
Induction of Chemotherapy-Induced Peripheral Neuropathy (CIPN) With Administration of Paclitaxel
Paclitaxel (European pharmacopoeia (EP) Reference standard purchased from Sigma, Y0000698) was dissolved in a vehicle (1:1 ratio of Kolliphor oil (Sigma, C5135) and 100% ethanol), to prepare stock paclitaxel solution (5mg/mL stored at 4°C). Working stock was freshly prepared on injection days by diluting stock solution with sterile 1× PBS. Paclitaxel injections were administered intraperitoneally every other day for four (4mg/kg/day) injections to induce peripheral neuropathy (16mg/kg total dose per animal, Figure 1) as previously described (44, 45), these studies show little to no differences of 2mg/kg versus 4mg/kg in the development of painful CIPN.
Behavioral Experiments
Baseline readings for all behavior assays were taken twice, a day apart (days -1 and -3) before administration of the 1st paclitaxel injection on day 0. Behavioral assessment for spontaneous pain using the grimace scale, mechanical hypersensitivity, grip strength, and thermal hyperalgesia were performed on days 1, 3, 5, 7, and 9, in that order. This was important to maintain non-confounding observations of evoked pain behavior using touch or heat stimuli after noting spontaneous pain. All behavior datasets are additionally represented as effect size. Effect size here is determined by calculating the cumulative difference between the value for each time point and the baseline value. All effect values were added to get effect size represented as an absolute number for each of the animal groups (46).
Assessment of Spontaneous Pain by Grimace Scale
Grimace is assessment of spontaneous pain using the observation of facial expression (47–49). Five parameters for facial grimace behavior were assessed: orbital tightening, ear position, cheek bulge, nose bulge, and whisker position. Each component was scored as follows: not present was given a score of “0”, moderately present was given a score of “1”, and obviously present was given a score of “2”. The score of the five components was averaged to give a mean grimace score (MGS).
Assessment of Mechanical Hypersensitivity by von Frey Filaments
Mechanical hypersensitivity was assessed using the previously described von Frey assay (50). Von Frey acrylic chamber (11cm long, 10 cm wide, and 4.5 cm height) with mesh floor, elevated 4.25 feet off the ground, were used for this assay. Animals were acclimated to the testing room and habituated to the von Frey apparatus and test environment for at least 1 hour prior to baseline and testing measurements. After 2 baseline recordings (one day rest in between the recordings), animals were randomly assigned to the different treatment groups. Mechanical hypersensitivity was determined by assessment of paw withdrawal threshold in response to the application of calibrated von Frey filaments (Stoelting, Illinois, USA) using the up-down method. A series of filaments with logarithmically incremental stiffness of 2.83, 3.22, 3.61, 3.84, 4.08, and 4.17 (converted to the 0.07, 0.16, 0.4, 0.6, 1, and 1.4 grams, respectively) were applied to the plantar surface of the hind paw and held for 2-3 seconds. A positive response was counted if there was a brisk withdrawal of the paw, paw shaking, and paw licking, or holding the paw in the air more than 2 seconds. The withdrawal threshold of both hind paws was measured and averaged.
Assessment of Muscle Dexterity by Grip Strength
Grip strength is a measure of functional pain in laboratory rodents and is measured with a grip strength test meter (IITC, California, USA). All animals were habituated to the apparatus for 5 minutes per animal, and were trained to grip the mesh wire surface prior to baseline or experiment. To measure, animals were gently placed on the rectangular wire meshed surface connected to the transducer in the grip strength meter and gently pulled away until they let go of the mesh. The maximal grip force was automatically measured by the transducer and recorded. Three trials were performed and averaged for both baseline measures and all experiments.
Assessment of Thermal Hyperalgesia by Hot Plate
Thermal sensitivity was assessed using the hot plate apparatus (IITC, California, USA). Animals were individually placed onto a surface maintained at 52°C, with their locomotion restricted by a Plexiglas chamber. Hind paw licking, shaking, and jumping was recorded as a positive response. Total time spent on the hot plate surface until a positive response was detected was measured and recorded as latency to response. The heating time cutoff was restricted to maximum 30 seconds to prevent tissue injury.
Immunohistochemistry
Animals were anesthetized with intraperitoneal injection of Ketamine/Xylazine (100 mg/kg) and intracardially perfused with 1x PBS solution followed by 4% paraformaldehyde (PFA). Lumbar spinal cord tissue was collected, post fixed in 4% PFA for 4 hours at 4°C and cryoprotected in 30% sucrose for 48 hours at 4°C. Tissues were embedded in optimal cutting temperature (OCT) (Fisher Scientific; 23-730-571) followed by cryo-sectioning into 20 μm sections that were mounted on positively charged glass slides (VWR, 48311-703). Mounted sections were treated with 1M HCL for 30 minutes followed by neutralization with 0.1 M sodium borate (pH 8.5) for 10 minutes. The slides were washed with 1x PBS and pre-incubated with 5% normal goat serum in 0.2% Triton X-100 in 1x PBS solution to block nonspecific binding. Subsequently, sections were incubated with the primary antibodies overnight at 4°C (see Table 1). The next day, sections were incubated in respective secondary antibodies and treated with DAPI solution (1:5000 dilution, Sigma-Aldrich D9542). Stained sections were covered with Gelvatol mounting medium and cover slips (51) and images were taken on a Zeiss Axio-observer 7 microscope (Jena, Germany) for quantitative analysis. Representative images were taken on an Olympus FV3000RS confocal laser scanning microscope (Shinjuku, Tokyo, Japan). Images acquired for quantitative analysis were taken using identical exposure times and representative images at identical laser power. Image analysis was done using FIJI (ImageJ) and Cell Sens version 3.1 (Olympus, Japan) software.
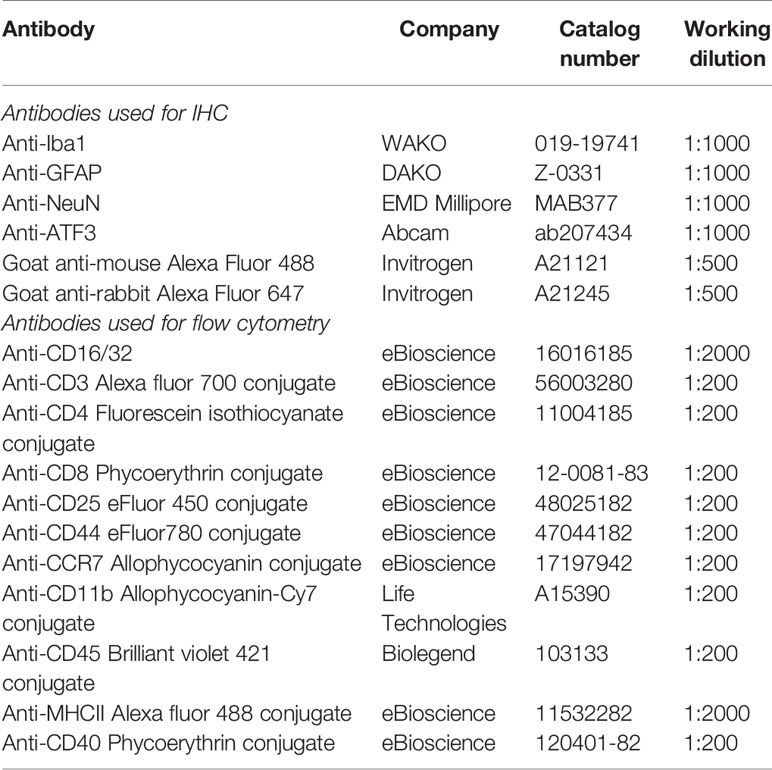
Table 1 Antibodies used for IHC and flow cytometry.
Flow Cytometry
Flow cytometric analysis of isolated immune cells from lymph node and dissociated DRG was performed based on previous protocols with few modifications (52). In brief, lymph nodes (popliteal and inguinal) and DRG (lumbar and thoracic, T13 to L5) were collected in ice cold sterile DPBS (Hyclone, Logan, UT). Lymph nodes were passed through a 70 micron nylon mesh with flow buffer (0.5% bovine serum albumin with 0.02% glucose) while DRG were first digested with enzymes (details described in section 2.6) and then passed through 70 micron nylon mesh. Resultant cell suspensions were centrifuged at 400 × g for 6 minutes at 4 °C. Cell pellets were washed with cold 1x PBS and resuspended in pre-chilled flow buffer. Fc receptors were blocked by anti-CD16/CD32 purified antibody. Cells from lymph nodes were incubated with anti-CD3-Alexa Flour 700, anti-CD4-Fluorescein isothiocyanate, anti-CD8 Phycoerythrin, anti-CD25-eFluor 450, anti-CD44-eFluor780, and anti-CCR7-Allophycocyanin (see Table 1 for antibody details). Cells from DRG were incubated with anti-CD11b-Allophycocyanin-Cy7, anti-CD45-Brillient violet 421, anti-MHCII-Alexa Fluor 488, and anti-CD40 Phycoerythrin antibodies. Appropriate compensation controls and isotypes were used for determination and gating. Cells were washed twice in ice cold flow buffer and resuspended in ice cold flow buffer. Thelper T-cells were initially identified with gating CD3 and CD4 cell surface markers, whereas cytotoxic T-cells were identified by gating for CD3 and CD8. These populations were further gated to identify activation and polarization (Th1 vs. Th2) with CD44 and CD25. CCR7 was also used to identify T-effector cells. Tissue activated macrophages were identified from CD45 separated cells with cell surface markers CD11b, MHC-II, and CD40. Stained samples were analyzed using a Becton-Dickinson Fortessa analyzer (Red Oaks, CA) and data were analyzed using FlowJo software (De Novo Software, Los Angeles, CA). For a complete list of antibodies used, please refer Table 1.
Primary Dorsal Root Ganglia (DRG) Culture and Cellular Energetics
On day 10, animals were deeply anesthetized with isoflurane and decapitated. DRG were dissected bilaterally starting from T13 to L5 and transferred to pre-chilled 1x PBS containing 1% penicillin/streptomycin (ThermoFisher Scientific, 15070063). Samples were centrifuged at 400 x g for 4 minutes. Supernatants were removed and DRG were treated with Collagenase A (Sigma, 10103586001) and incubated in water bath at 37°C for 20 minutes, followed with treatment of Collagenase D (Sigma, 1188866001) for another 20 minutes. Cells were centrifuged at 400 x g and pellet was resuspended in Enzyme T (soybean trypsin inhibitor made up in 1 part bovine serum albumin and 1 part DMEM/F12 media) to stop the enzymatic reaction. Digested tissues were triturated approximately 30 times using a 1 ml pipette tip and passed through the 70 micron nylon mesh, with a subsequent wash with DMEM/F12 media (supplemented with 10% Fetal bovine serum and 1% penicillin/streptomycin). Resultant suspension was centrifuged at 400 x g for 5 minutes and resuspended in DMEM/F12 medium. The number of cells was counted using a hemocytometer with trypan blue dye exclusion. For bioenergetic profile, 70,000 cells were seeded directly in respective XFp Cell Culture Miniplates (Agilent, 103025-100) coated with poly-D-Lysine (Millipore Sigma, P0899). Next day, XF Assay media (Seahorse Bioscience) supplemented with 2 mM GlutaMAX, 5 mM glucose (Sigma), 1 mM sodium pyruvate (Fisher Scientific, Loughborough, United Kingdom), and pH adjusted to pH 7.4, was warmed to 37°C. Cells were incubated with this Seahorse media for 1 hour at 37°C, 0% CO2 incubator. The media was replaced with the Seahorse XF calibrant solution for another hour before running the Mito Stress according to manufacturer’s recommended protocol (53).
Statistical Analysis
Data were analyzed using GraphPad Prism software (version 8.4) and expressed as mean ± standard error of the mean (SEM). For behavior, flow cytometry, and Seahorse datasets, two-way ANOVAs were performed followed by Tukey’s post-hoc for multiple comparisons. For Seahorse divided into phases of mitochondrial respiration, one-way ANOVA was performed with Sidak’s multiple comparison test. For IHC datasets, two-way ANOVA followed by Bonferroni’s post-hoc was performed for glia reactivity and Sidak’s post-hoc was used for ATF3. A p value of ≤ 0.05 was considered significant.
Results
Paclitaxel Induces Pain Like Behavior in Male and Female Mice, Which Is Mediated via eIF4E
Previously, it has been shown that an intraperitoneal injection regimen of paclitaxel leads to development of long-lasting mechanical hypersensitivity in WT male and female mice during later stages of CIPN (45, 54). We investigated whether cap-dependent translation was involved in earlier stages of CIPN development after intraperitoneal paclitaxel administration. We found a significant paclitaxel-dependent increase in mechanical hypersensitivity in WT mice of both sexes, compared to vehicle treatment (Figures 2A–D and Table 2). In the absence of eIF4E phosphorylation, this increase was observed only in females (Figures 2C, D and Table 2), not male mice (Figures 2A, B and Table 2). Compared to WT paclitaxel-treated mice, the eIF4ES209A paclitaxel-treated mice had significantly lower mechanical hypersensitivity (Figures 2B, D and Table 2) in both sexes. These findings indicate that eIF4E phosphorylation plays a protective role in the development of mechanical hypersensitivity after paclitaxel administration, but this protection is to a lesser extent in females. We assessed spontaneous pain by grimace and grip strength. Paclitaxel treatment induced more grimacing in male mice in an eIF4E-dependent manner (Figures 2E, F and Table 2). For females, spontaneous pain was higher with paclitaxel administration, but eIF4ES209A females had lower grimacing compared to WT (Figures 2G, H). This indicates that eIF4E affects pain perception associated with paclitaxel-induced peripheral neuropathy. There were no changes in the grip strength for either sex, genotype, or treatment (Figures 2I–L). Male WT mice showed the maximum latency of response to hot plate with paclitaxel treatment, with no difference in eIF4ES209A male mice (Figures 2M and Table 2), indicating that eIF4E is required for mediating response to thermal stimuli after paclitaxel. On the other hand, females of both genotypes did not show significant changes in latency with paclitaxel treatment compared to vehicle (Figures 2O, P and Table 2). Interestingly, eIF4ES209A females treated with paclitaxel had a significantly reduced latency of response to heat stimuli compared to their WT counterparts (Figure 2P and Table 2). Taken together, our data suggests that eIF4E is important for mechanisms involved in CIPN development early in both sexes.
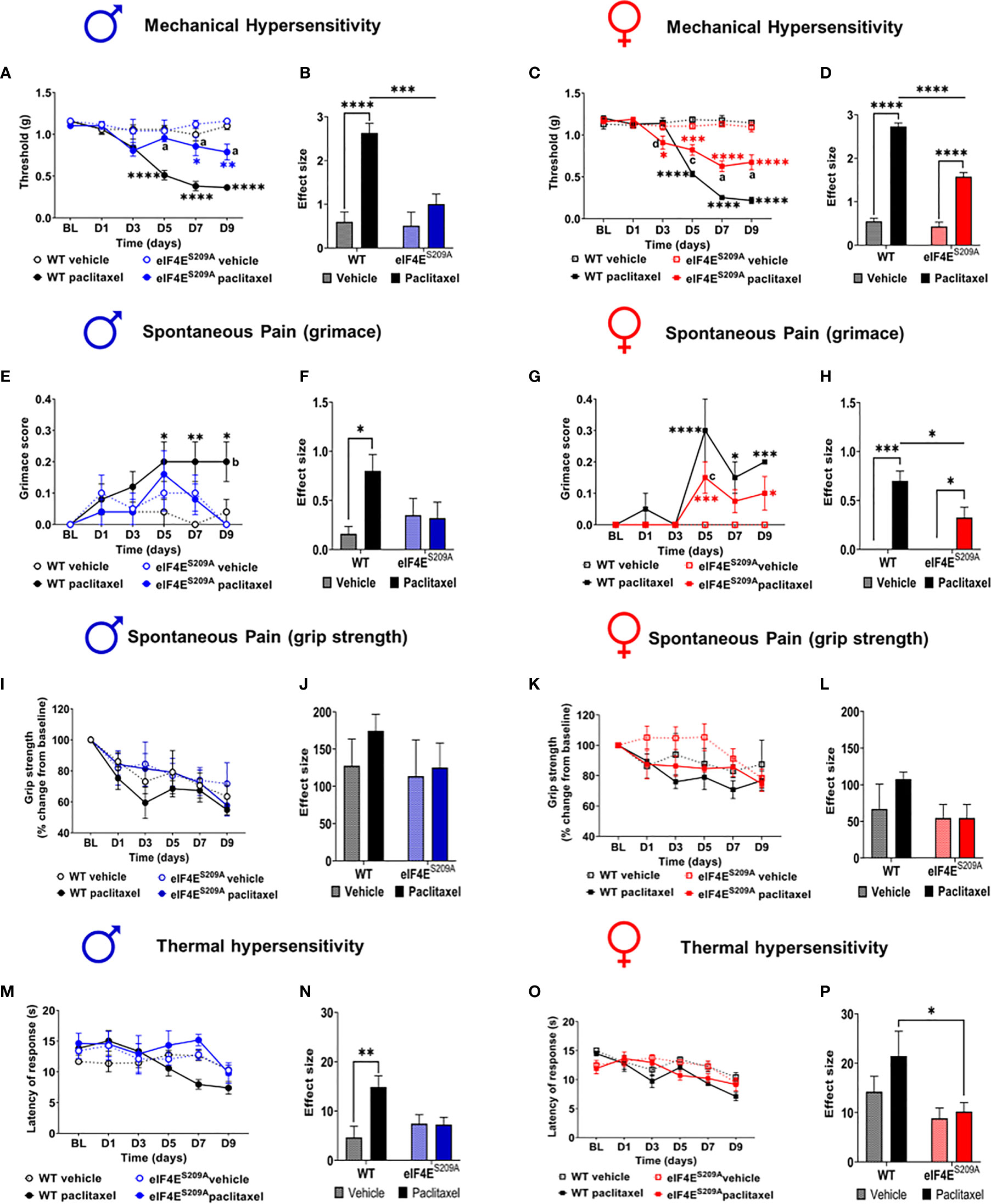
Figure 2 Paclitaxel induces pain like behavior in male and female mice, which is eIF4E dependent. (A) Mechanical hypersensitivity in male WT and eIF4ES209A mice. Blue asterisks *p 0.0475, ** p 0.0014 for eIF4E vehicle VS paclitaxel, Black asterisks ****p<0.0001 for WT vehicle VS paclitaxel, a=p<0.0001 for WT paclitaxel VS eIF4E paclitaxel. (B) Effect size here is determined by calculating the cumulative difference between the value for each time point and the baseline value. Data for male mice shown as effect size, ****p<0.0001, ***p 0.0004. (C) Mechanical hypersensitivity in female WT and eIF4ES209A mice. Red asterisks *p 0.0261 , ***p 0.003 for eIF4E vehicle VS paclitaxel, Black asterisks ****p<0.0001 for WT vehicle VS paclitaxel, a=p<0.0001, c=p 0.0029, d=p 0.0286 for WT paclitaxel VS eIF4E paclitaxel. (D) Data for female mice shown as effect size, ****p<0.0001. (E) Spontaneous pain assessment by grimace score in male mice. *p 0.0495, **p 0.008 for WT vehicle VS paclitaxel. (F) Grimace scores shown as effect size for male mice, *p 0.0302. (G) Grimace scores for female mice. *p 0.0153 for D7 WT vehicle VS paclitaxel, 0.0328 for D9 for eIF4E vehicle VS paclitaxel, ***p 0.0004 for D5 for eIF4E vehicle VS paclitaxel, 0.0009 for D9 for WT vehicle VS paclitaxel, ****p<0.0001 for WT vehicle VS paclitaxel. (H) Grimace scores shown as effect size for female mice, *p 0.0249 compared for treatment, *p 0.0293 compared for genotype, ***p 0.0005. (I) Grip strength for male mice. (J) Grip strength for males shown as effect size. (K) Grip strength for female mice. (L) Grip strength in females shown as effect size. (M) Thermal hypersensitivity in male mice. (N) Thermal hypersensitivity in males shown as effect size. **p 0.0072. (O) Thermal hypersensitivity in female mice. (P) Thermal hypersensitivity in females shown as effect size, *p 0.0453. All data are presented as mean ± standard error of the mean, males: n = 6 mice per group; females: n = 12 mice per group. Two-way ANOVA was performed with Tukey’s post-hoc for multiple comparisons. For all line graphs, asterisks indicate significant differences between vehicle VS paclitaxel-treated for each genotype at indicated time points. “a” (p < 0.0001), “b” (p < 0.001), “c” (p < 0.01), and “d” (p < 0.05) depict significant difference between paclitaxel-treated WT and eIF4ES209A mice at indicated time points.
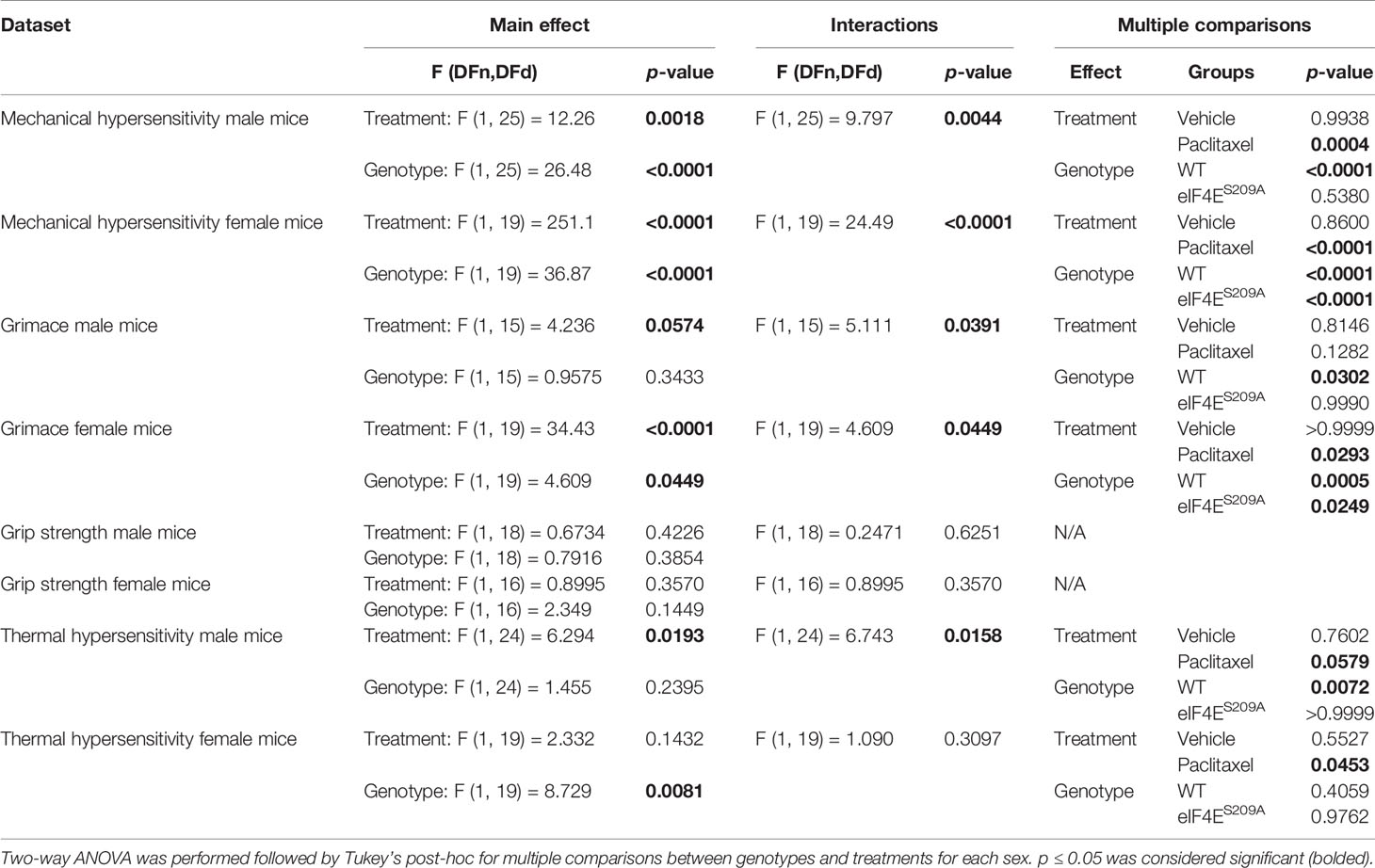
Table 2 Statistical values for behavior data analysis.
Paclitaxel Dysregulates CD4+ and CD8+ T-Cell Subpopulations in an eIF4E-Dependent Manner
An obvious early contribution of adaptive immune cells in the development of paclitaxel-induced peripheral neuropathy has not been shown till date. Given that the paclitaxel was administered peripherally, T-cells are activated in the lymph nodes, and neuropathy develops in the limbs, we chose to study subpopulations of T-cells from the draining (popliteal and inguinal) lymph nodes that are CD4+ (Thelper) CD8+, CD25+/- and CD44+/- (activated T-cells), CCR7+ (effector T-cells) (Figures 3A–F). We found a significant increase in CD4+ T-cells in WT female mice injected with paclitaxel, but not males (Figures 3A, B, E and Table 3). The CD4+ subset was further analyzed for CCR7, the marker for lymphocyte homing to lymph nodes as well as T-cell priming (55, 56). The CD4+CD25-CD44+ subset was significantly reduced in eIF4ES209A females treated with paclitaxel compared to WT (Figure 3G, Table 3). The CD4+CCR7+ subpopulation was significantly higher in eIF4ES209A mice of both sexes independent of paclitaxel treatment (Figures 3C, F and Table 3). This suggests that inhibiting cap-dependent translation upregulates T-cell priming and activation of T cells. We found significant downregulation of CD8+ T-cells in female eIF4ES209A mice compared to WT, irrespective of treatment (Figures 3G–L, Table 3). This population was unchanged in male mice (Figure 3H). CD8+CCR7+ T-cells, i.e. activated memory T-cells were higher in eIF4ES209A mice (Figures 3I, L and Table 3) regardless of sex or treatment. CD8+CD44+CD25- T-cells were increased in paclitaxel-treated eIF4ES209A male mice compared to WT males treated with paclitaxel (Figure 3J and Table 3). There were no differences in female mice for this subpopulation (Figure 3M and Table 3). Overall, our data shows that paclitaxel induces activation and/or expansion of T-helper, memory T, and effector T-cells, which is modulated in an eIF4E-dependent manner.
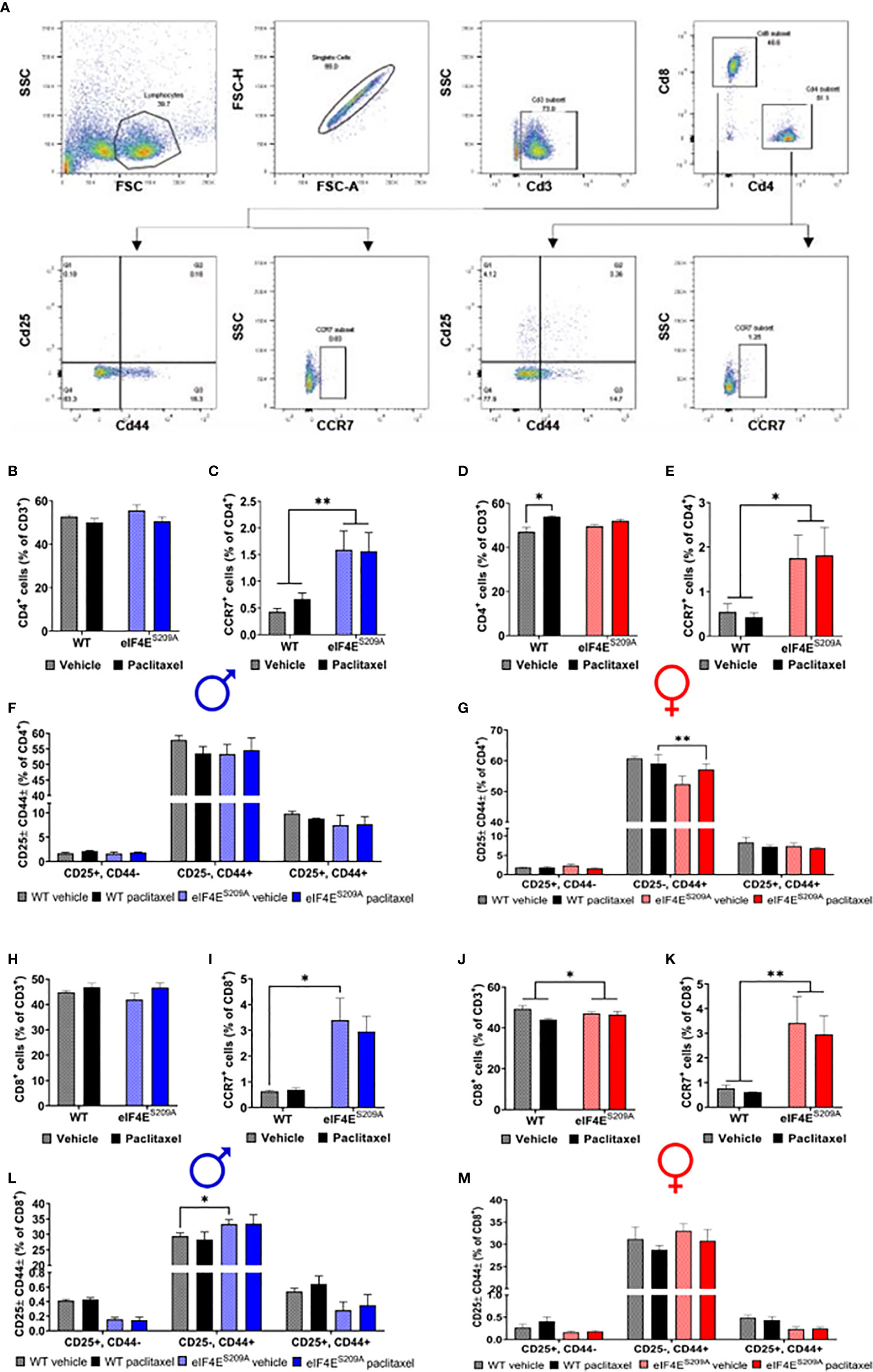
Figure 3 Paclitaxel dysregulates CD4+ and CD8+ T-cell subpopulations in an eIF4E-dependent manner. (A) Gating strategy used for flow cytometry of lymphocytes from popliteal and inguinal lymph nodes. T-cells were separated from the whole lymphoid cells population using CD3. CD3+ cells were then gated for CD4+ T-cell and CD8+ T-cells. For each of these subsets, cells were further gated for CCR7 or CD25 and CD44. (B) Quantification of CD4+ T-cells from the CD3 population from male mice. (C) CCR7+ T-cells from isolated CD4+ population in males, **p 0.0063. (D) CD4+ cells gated for CD25 and/or CD44 in males. (E) Quantification of CD4+ T-cells from the CD3+ population from female mice, *p 0.0101. (F) CCR7+ T-cells from isolated CD4+ population in female mice, *p 0.0250. (G) CD4+ cells gated for CD25 and/or CD44 in female mice, **p 0.0011. (H) Quantification of CD8+ T-cells from the CD3+ population from male mice. (I) CCR7+ T-cells from isolated CD8+ population in males, *p 0.0433. (J) CD8+ cells gated for CD25 and/or CD44 in males, *p 0.0396. (K) Quantification of CD8+ T-cells from the CD3 population from female mice, *p 0.041. (L) CCR7+ T-cells from isolated CD8+ population in female mice, **p 0.0097. (M) CD8+ cells gated for CD25 and/or CD44 in female mice. All data are presented as mean ± standard error of the mean, n = 3 mice each for male/female WT vehicle-treated and WT paclitaxel groups, n = 4 mice each for male/female eIF4ES209A vehicle-treated and paclitaxel. Two-way ANOVA was performed with Tukey’s post-hoc for multiple comparisons.
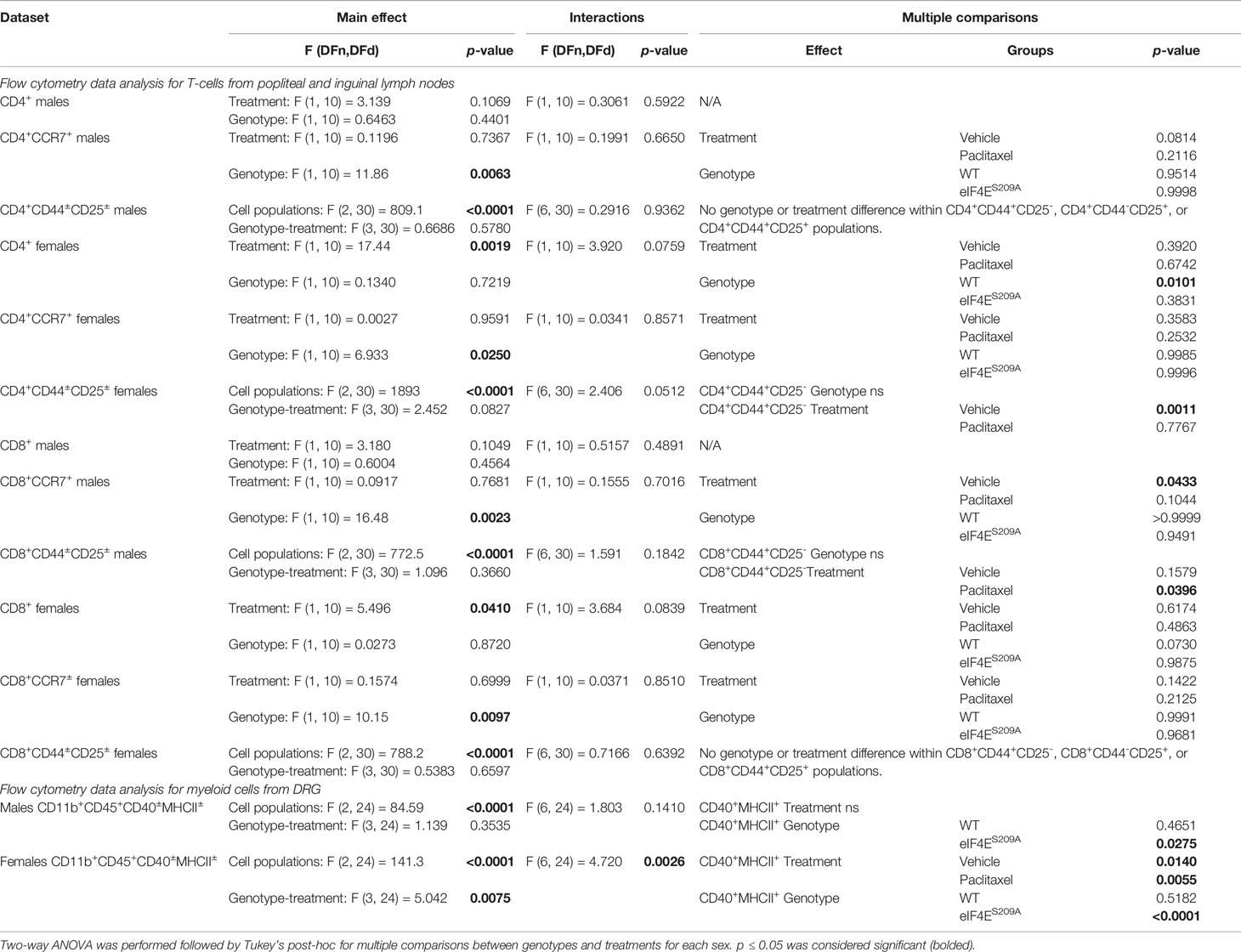
Table 3 Statistical values for flow cytometry data analysis.
Paclitaxel Induces Increased Infiltration of Activated Myeloid Cells in DRG in Absence of eIF4E Phosphorylation
We further investigated if peripheral neuropathy leads to the recruitment of macrophages into the DRG. No studies to date have shown the recruitment of the macrophages and the activation state of macrophages in DRG after CIPN. We looked for the infiltration and activation of macrophages using the CD11b+-CD45+ and MHCII+-CD40+ cells respectively in male and female mice. We identified macrophages within DRG by gating for CD11b+ and CD45+ cells. This population was further gated for MHCII+ and CD40+ to assess activated macrophage subsets (Figure 4A). We found that CD11b+-CD45+-CD40+-MHC-II- and CD11b+-CD45+-CD40--MHC-II+ populations were unchanged in both sexes with paclitaxel or lack of eIF4E (Figures 4B, C and Table 3), indicative of no effect on dendritic cells or tissue-resident macrophages. We found a significant increase in CD11b+-CD45+-CD40+-MHC-II+ cells in male and female eIF4ES209A mice treated with paclitaxel compared to vehicle. Additionally, in female mice, this subpopulation was significantly higher in paclitaxel-treated eIF4ES209A animals compared to paclitaxel-treated WT (Figure 4C and Table 3). This indicates that antigen-presenting cells (APCs) are upregulated with paclitaxel treatment in absence of eIF4E. Interestingly, female eIF4ES209A vehicle-treated mice had a significantly lower proportion of activated APCs compared to WT (Table 3). This shows that cap-dependent translation may be facilitating the recruitment of activated myeloid cells as a result of paclitaxel treatment, which in turn could be activating or expanding the T-cell populations.
Figure 4 Paclitaxel induces activation of antigen-presenting myeloid cells in eIF4ES209A mice DRG. (A) Gating strategy for isolating myeloid APCs from mouse DRG. Total cells were gated for CD11b and CD45 to separate out immune cells. The CD11b+ CD45+ subset was then gated for MHC-II+ and/or CD40+ cells. (B) Quantification of CD40+ and/or MHC-II+ cells from male mice, *p 0.0275. (C) Quantification of CD40+ and/or MHC-II+ cells from female mice, *p 0.014, **p 0.0055, ****p<0.0001. All data are presented as mean ± standard error of the mean, n = 3 mice each for male/female WT vehicle-treated and WT paclitaxel groups, n = 4 mice each for male/female eIF4ES209A vehicle-treated and paclitaxel. Two-way ANOVA was performed with Tukey’s post-hoc for multiple comparisons.
Paclitaxel Alters Mitochondrial Function of DRG Neurons in an eIF4E-Dependent Manner
We investigated mitochondrial function in DRG neurons to examine if paclitaxel changed the bioenergetic profiles (Figures 5A–Q and Table 4) in an eIF4E-dependent manner. The overall oxygen consumption rate (OCR) was lower in paclitaxel-treated eIF4ES209A males and females compared to paclitaxel-treated WT (Figures 5B, C, J, K and Table 4) whereas the overall extracellular acidification rate (ECAR) was higher with paclitaxel treatment in both sexes and lower in paclitaxel-treated eIF4ES209A males compared to paclitaxel-treated WT males (Figures 5H, I, P, Q and Table 4). This suggests that lack of eIF4E causes a shift in mitochondrial respiration with paclitaxel treatment. We divided the different phases of the OCR bioenergetic profile into basal respiration (before oligomycin treatment), ATP production (after oligomycin treatment and before FCCP – 2-[2-[4-(trifluoromethoxy)phenyl]hydrazinylidene]-propanedinitrile treatment), maximal respiration (after FCCP and before antimycin A and rotenone), and non-mitochondrial respiration or reserve capacity (Figure 5A). For males, we found a significant increase in the basal respiration and non-mitochondrial respiration after paclitaxel treatment in WT, but not eIF4ES209A mice (Figures 5D, G and Table 4), but no change in ATP turnover or maximal respiration (Figures 5E, F and Table 4). For female mice, we found that basal respiration and non-mitochondrial respiration was increased after paclitaxel treatment for both genotypes (Figures 5L, O and Table 4). ATP turnover and maximal respiration was also higher in WT females treated with paclitaxel compared to vehicle and in eIF4ES209A females compared to paclitaxel-treated WT (Figures 5M, N and Table 4). Taken together, our data shows that sensory neurons in the DRG undergo an excited mitochondrial shift with paclitaxel treatment in an eIF4E-dependent manner in both sexes. Paclitaxel increased all aspects of mitochondrial respiration in females but did not affect ATP turnover and maximal respiration in males, which indicates divergent effects of paclitaxel in eIF4E absence in both sexes.
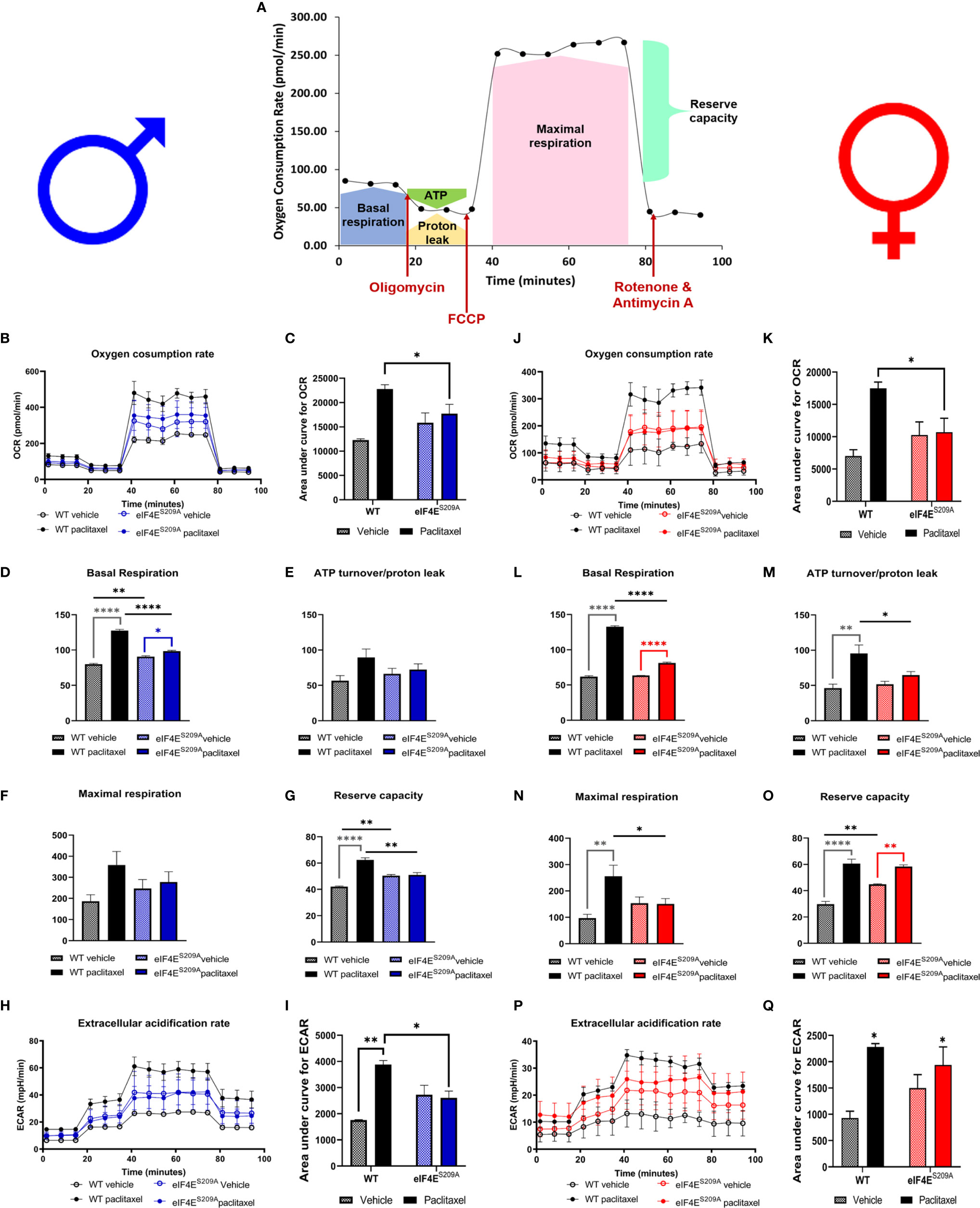
Figure 5 Paclitaxel alters mitochondrial function of DRG neurons in an eIF4E-dependent manner. Dissociated dorsal root ganglion (DRG) neurons from WT and eIF4ES209A male and female mice were analyzed by the Seahorse Mito Stress test. (A) Graphic showing different phases of mitochondrial respiration. Adapted from the Agilent Seahorse XFp Cell Mito Test Kit User Guide published by Agilent Technologies. (B) Oxygen consumption rate (OCR) through phases of the Mito Stress test for male mice. (C) OCR data in male mice shown as area under the curve (AUC), *p 0.0136. (D-G) OCR data for male mice broken down into phases of mitochondrial respiration. Basal respiration: *p 0.0134, **p 0.0018, ****p<0.0001. Reserve capacity: **p 0.0063 for WT vs eIF4ES209A vehicle, **p 0.001 for WT vs eIF4ES209A paclitaxel. (H) Extracellular acidification rate (ECAR) for male mice. (I) ECAR data in male mice shown as AUC, *p 0.0303 for WT vs eIF4ES209A vehicle, **p 0.0021 for WT vehicle vs WT paclitaxel groups. (J) OCR through phases of the Mito Stress test for female mice. (K) OCR data in female mice shown as AUC, *p 0.0455. (L–O) OCR data for female mice broken down into phases of mitochondrial respiration. Basal respiration: ****p<0.0001, ATP turnover: *p 0.0481, **p 0.0021, maximal respiration: *p 0.0417, **p 0.0011, reserve capacity: **p 0.0037 for WT vs eIF4ES209A vehicle, **p 0.074 for eIF4ES209A vehicle vs eIF4ES209A paclitaxel, ****p<0.0001. (P) ECAR for female mice. (Q) ECAR data in female mice shown as AUC, *p 0.0116 for both genotypes vehicle vs paclitaxel-treated groups. All data are presented as mean ± standard error of the mean, n = 3-4 mice each for male/female WT vehicle-treated and WT paclitaxel groups, n = 4-7 mice each for male/female eIF4ES209A vehicle-treated and paclitaxel. For analyzing AUC data, two-way ANOVA was performed with Tukey’s post-hoc for multiple comparisons. For analyzing each phase of mitochondrial respiration, an ordinary one-way ANOVA was used with Sidak’s post-hoc for multiple comparisons.
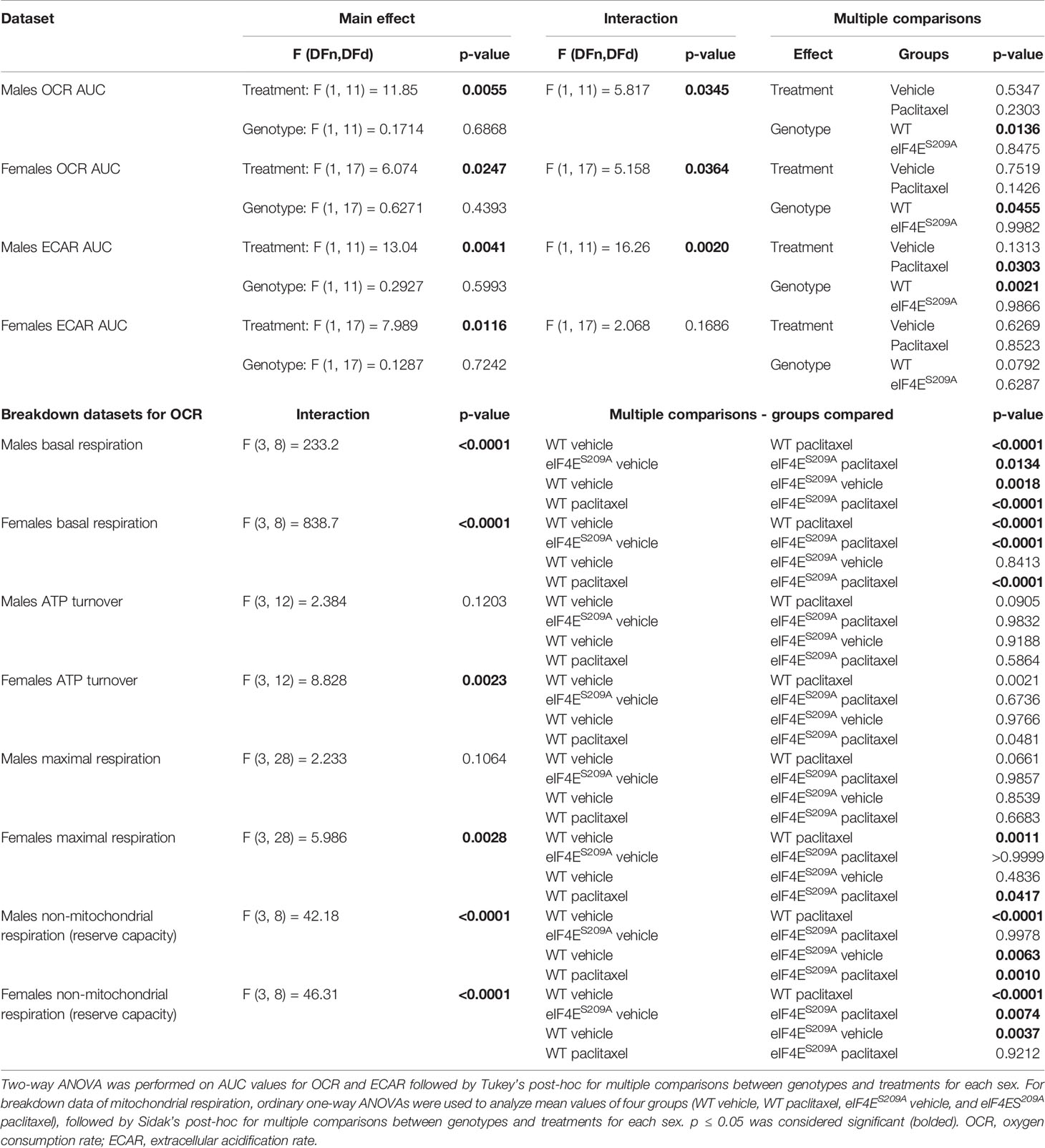
Table 4 Statistical values for DRG neuron mitochondrial respiration.
Sensory Neuron Damage by Paclitaxel Is Similar for Both Sexes and Genotypes
Previous studies have shown no association between sickness behavior or change in the body weight of the animal with 16mg/Kg dose of paclitaxel. Moreover, other researchers have used even higher doses of paclitaxel to induce peripheral neuropathy (57, 58). We assessed number of DRG neurons positive for activating transcription factor 3 (ATF3). The number of ATF3+ neurons were significantly higher in the DRG of paclitaxel-treated animals indicating damage; however, there were no sex-based or genotype differences observed (Figure 6 and Table 5). The increase is similar to previously published literature, albeit at a later time point (59, 60), thus we conclude that this higher dose does not comparatively alter DRG neurons at day 10 after our regimen.
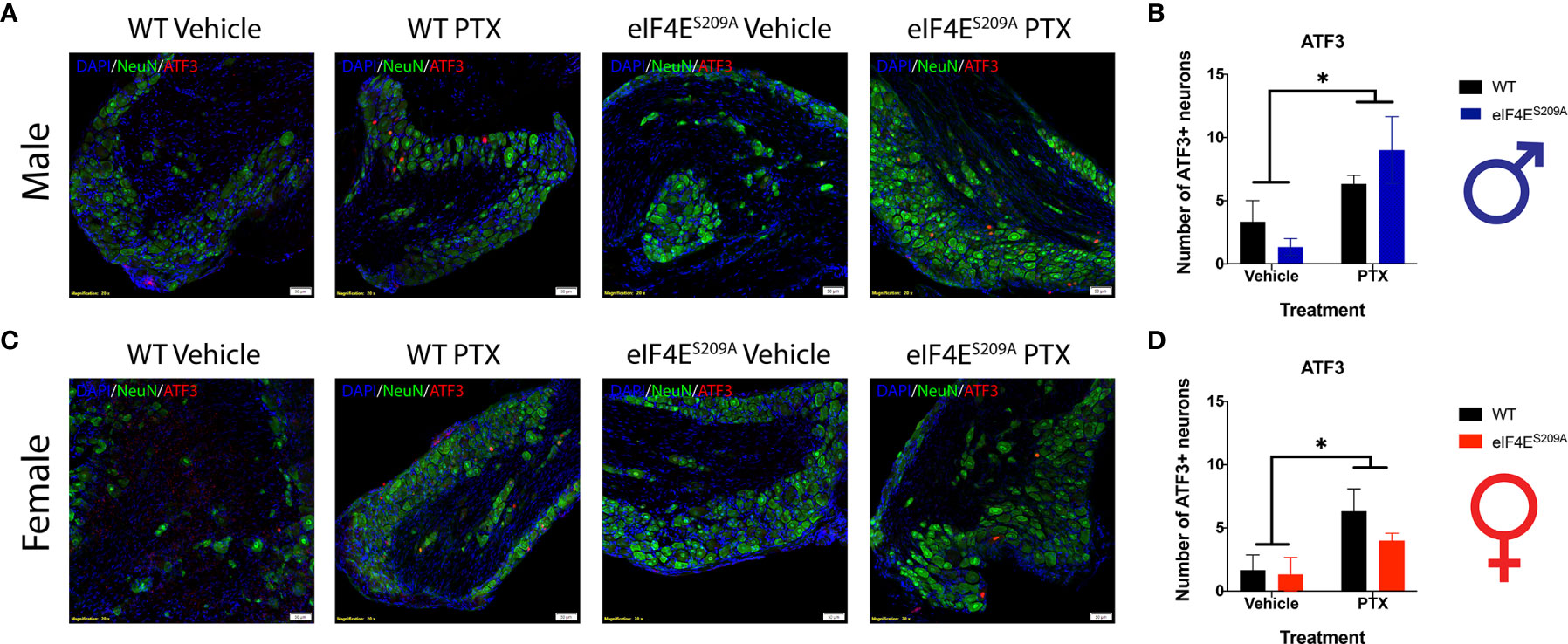
Figure 6 ATF3+ neurons are observed in DRG of all animals subjected to Paclitaxel. (A, B) Representative composite images showing ATF3+ cells (red) and NeuN+ cells (green) for male mice and their quantitation, respectively. *p 0.0114 for males’ vehicle vs paclitaxel groups. (C, D) Representative composite images showing ATF3+ cells (red) and NeuN+ cells (green) for female mice and their quantitation, respectively. *p 0.0218 for females for vehicle vs paclitaxel groups. All sections were counterstained with DAPI (blue). Data represented as mean and SEM. Two-way ANOVA was performed with Sidak’s post-hoc for multiple comparisons; n=3 for all groups. Magnification 20X, scale bar 50µm. PTX – paclitaxel.
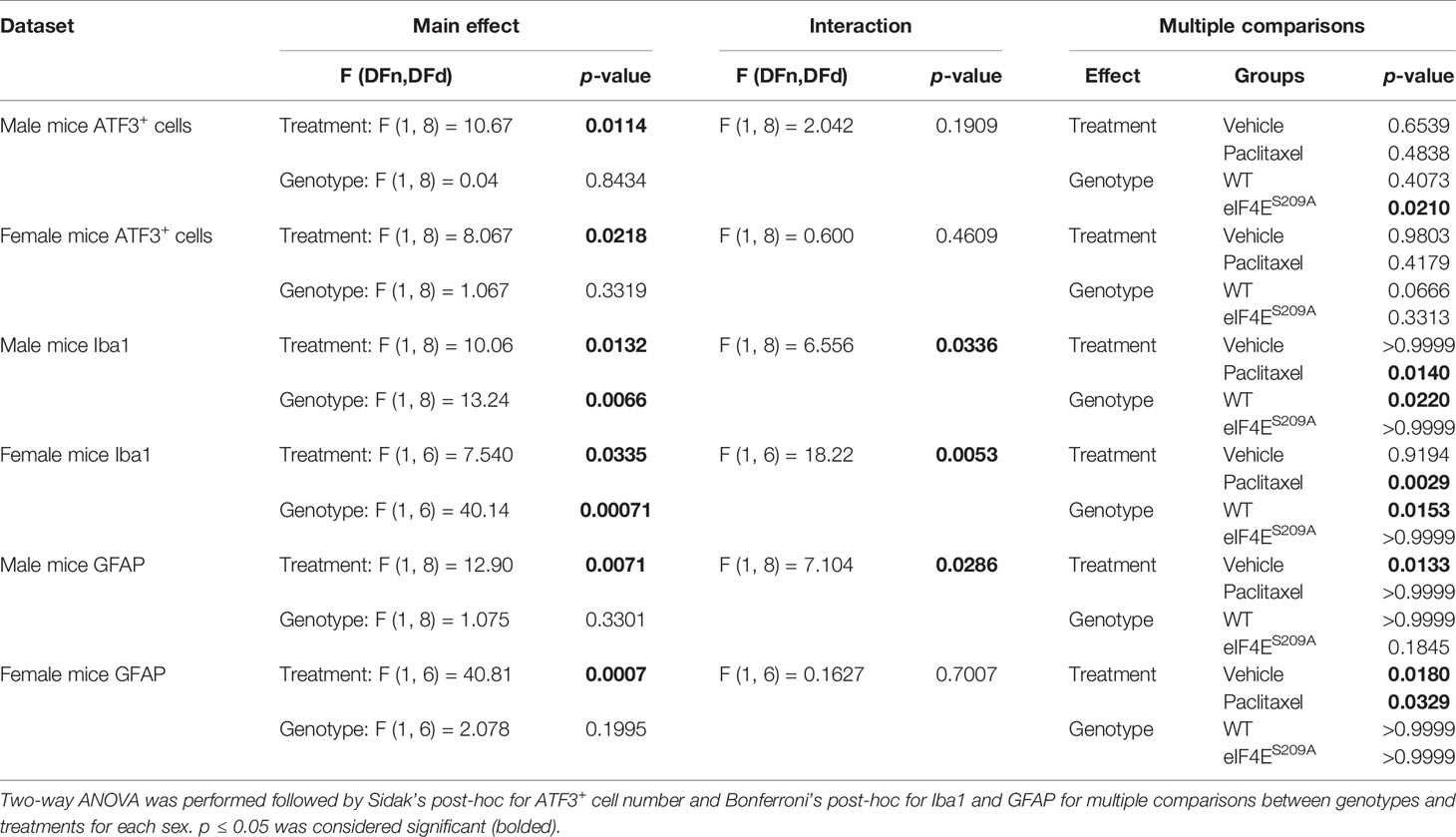
Table 5 Statistical values for IHC data analysis.
Spinal Glia Reactivity Is Altered in a Sex-Dependent Manner With Paclitaxel Treatment
Previous data has shown activation of microglia and astrocytes at different phases of CIPN (61–63). Here, we investigated whether the eIF4E complex plays a role in the activation of microglia and astrocytes in the spinal cord. We found a significant increase in immunoreactivity of microglia and astrocytes in both male and female WT mice (Figure 7). Iba1 fluorescence was significantly higher in paclitaxel-treated WT mice of both sexes compared to vehicle (Figures 7A–C and Table 5), indicating that paclitaxel induced microglial activation. This increase was lacking for eIF4ES209A animals for whom the Iba1 immunoreactivity stayed at WT vehicle levels. In eIF4ES209A females, there was a decrease in Iba1 fluorescence with paclitaxel treatment compared to vehicle (Figure 7C). For astrocytes, females lacking cap-dependent translation had increased GFAP fluorescence after paclitaxel compared to vehicle (Figures 7D, F, G, and H, and Table 5), which was different from the males where the GFAP fluorescence was at the same level as seen for the WT vehicle group (Figures 7D, E). This data shows that paclitaxel may mediate activation of astrocytes via an eIF4E-independent pathway in females but modulates microglia activation via eIF4E in both sexes and astrocyte activation in males.
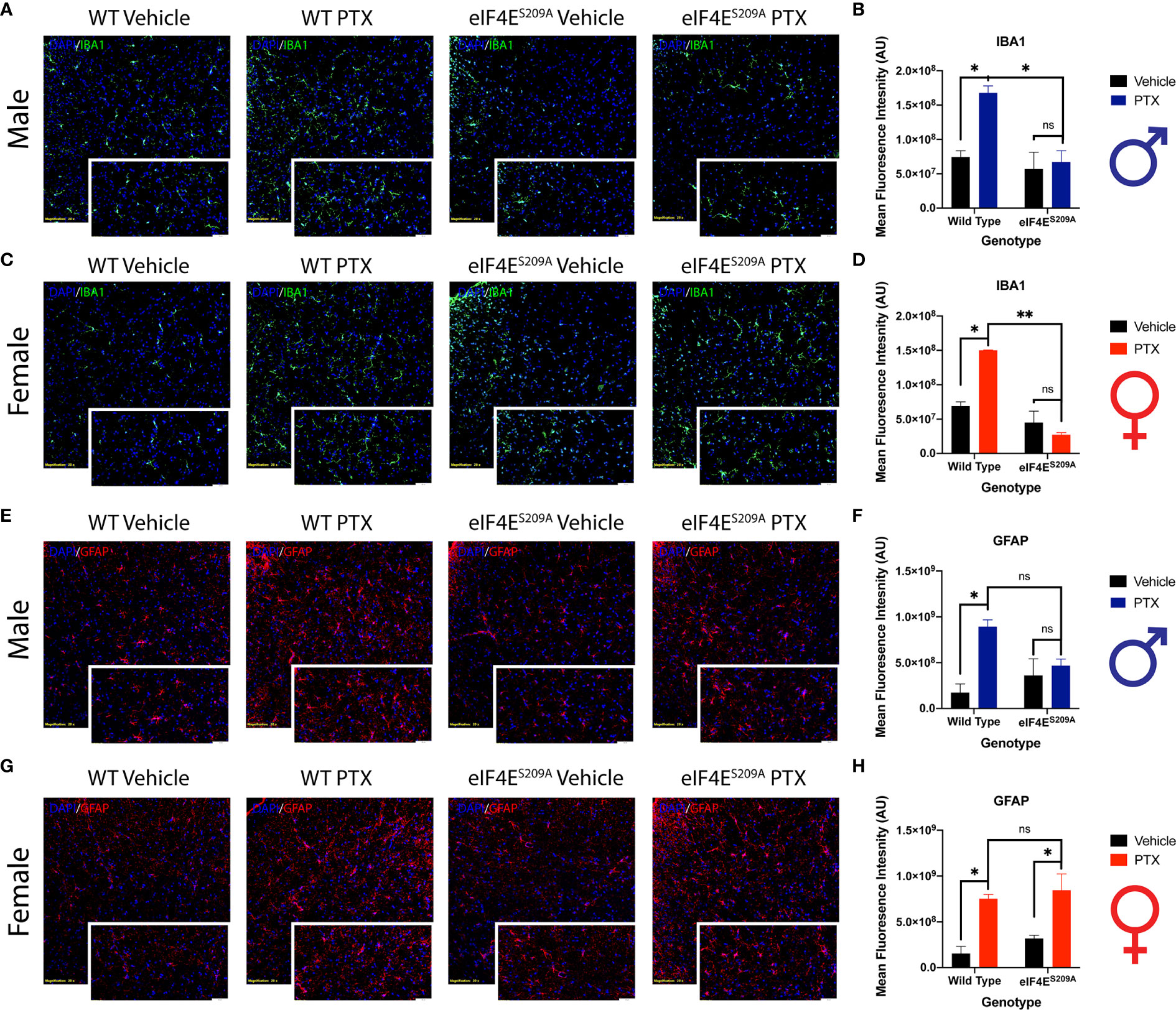
Figure 7 Spinal glia reactivity is altered in a sex-dependent manner with paclitaxel treatment. (A) Immunohistochemistry for male microglia on lumbar spinal cord. Blue represents DAPI for cell nuclei, green represents Iba1 for microglia. Magnification 20X, scale bar 50µm. (B) Quantification of Iba1 fluorescence intensity from male mice, *p 0.022 for WT vehicle vs WT paclitaxel groups, *p 0.014 for WT paclitaxel vs eIF4ES209A paclitaxel groups. (C) Immunohistochemistry for female microglia on lumbar spinal cord. Blue represents DAPI for cell nuclei, green represents Iba1 for microglia. Magnification 20X, scale bar 50µm. (D) Quantification of Iba1 fluorescence intensity from female mice, *p 0.0153, **p 0.0029. (E) Immunohistochemistry for male astrocytes on lumbar spinal cord. Blue represents DAPI for cell nuclei, red represents GFAP for astrocytes. Magnification 20X, scale bar 50µm. (F) Quantification of GFAP fluorescence intensity from male mice, *p 0.0133. (G) Immunohistochemistry for female astrocytes on lumbar spinal cord. Blue represents DAPI for cell nuclei, red represents GFAP for astrocytes. Magnification 20X, scale bar 50µm. (H) Quantification of GFAP fluorescence intensity from female mice, *p 0.018 for WT vehicle vs WT paclitaxel groups, *p 0.0329 for eIF4ES209A vehicle vs eIF4ES209A paclitaxel groups. Data are presented as mean ± standard error of the mean, males: n = 3 mice per group; females n = 3 mice per group. Two-way ANOVA was performed with Bonferroni’s post-hoc for multiple comparisons. ns, not significant.
Discussion
The current study begins to uncover the importance of eIF4E phosphorylation in the development of CIPN and associated neuroimmune consequences in male and female mice. In our study, we found an eIF4E-dependent increase of mechanical and thermal hypersensitivity in both sexes, upregulated T-helper and reduced cytotoxic T-lymphocytes in draining lymph nodes in female but not male mice, whereas activated macrophages in DRG were significantly increased in eIF4E but not WT mice of both sexes. In addition to this, DRG neuronal mitochondrial function was significantly higher in WT mice but not in eIF4E mice of both sexes. Microglia were significantly activated in an eIF4E-dependent manner with paclitaxel treatment in WT mice of both sexes; however, astrocytes were activated in an eIF4E-dependent manner with paclitaxel treatment only in male mice. Female eIF4E mice showed significant astrocyte activation.
Our study was designed and powered with the intent to appropriately assess sex differences in neuroimmune interactions during CIPN development—a question left unanswered in previous literature. We found an eIF4E-dependent increase in spontaneous pain as well as mechanical and thermal hypersensitivity after paclitaxel. This suggests that the pain-like behavior changes due to paclitaxel are mediated via the central cap-dependent translation pathway that is common across diverse cell types, with more pronounced effects in female mice.
The cell bodies of primary afferent neurons reside in the dorsal root ganglia. The persistent signals from nerve terminals activate the machinery and signaling cascades in the nucleus and cytoplasm of the neuronal soma (64–66). Hyper-excited nociceptor neurons recruit immune cells by secreting chemokines, neurotransmitters, and/or other neuropeptides (16, 67). It has been shown that paclitaxel treatment recruits macrophages into the DRG (20). Our findings corroborate this finding at an earlier time point and add to this knowledge by characterizing the macrophage population as CD45+CD11b+CD40+MHC-II+ i.e. activated APCs. It has been shown that such infiltration may be beneficial to recruit Th2 anti-inflammatory cells at later time points (62, 68). We have presented evidence that paclitaxel injection leads to a significant increase in the activated APC population in eIF4E males and females, but not in WT mice. Thus, a lack of eIF4E phosphorylation or cap-dependent translation exacerbates the immune cell response and DRG infiltration during CIPN development.
A prior study performed only in male mice showed an increase in CD4+ T-cells at paclitaxel post-injection day 7 (14). We found a similar increase of CD4+ T-cells in female WT mice with paclitaxel treatment. Additionally, a novel finding was that CD8+ T-cells were decreased in female but not in male WT mice during early phase of CIPN development. This indicates sex differences in T-cell activation in the lymph nodes, which would affect the subsets of T-cells that would infiltrate other tissues such as DRG (69). A previously published study showed increased numbers of CD3+, helper T-cells, and cytotoxic T-lymphocytes after paclitaxel on day 7 in mice DRG (70). Our dataset adds niche T-cell population characterization to this previously published literature. T-cell subset activation in the lymph nodes is highly dependent upon the antigen-presenting cells naïve T-cells encounter (71). Once activated, these subsets follow their divergent fates of redistribution to tissues in need of immune response. Thus, characterizing different subsets of T-cells in the lymph nodes allows us to determine what populations we can expect in interfaces where neuroimmune communication can occur, such as the meninges or DRG. T-cells are also important in the transition from acute to chronic pain (15). By showing alterations in specialized T-cell subsets, our study raises important questions about the implications for establishment of CIPN and its resolution. Animals lacking eIF4E had higher proportion of memory T-cells in both sexes. There were no changes in the effector T-cell population for the lymph nodes. These alterations in the activation and proportion of T-cell subsets and APCs suggest that although affective pain behavior is similar in male and female mice after development of CIPN, the mechanism driving the pain phenotypes in both sexes involves varied contribution from innate and adaptive immune cells.
Basal respiration and non-mitochondrial respiration were higher in male DRG neurons treated with paclitaxel compared to vehicle, which is contrary to a previous report for rats (38). We have further shown that the lack of eIF4E does not change this trend, even though oxygen consumption is lower, suggesting that paclitaxel may induce mitochondrial function independent of cap-dependent translation regulation. In females, while the effects on basal respiration follow the same trend as the males, paclitaxel seems to mediate an increase in non-mitochondrial respiration independent of eIF4E. Female DRG neurons also had significantly higher ATP turnover and maximal respiration with paclitaxel treatment in WT. Considering that eIF4E female mice show higher mechanical hypersensitivity with paclitaxel and that their DRG neuron ATP turnover and maximal respiration is higher, this may be indicative of the severity of the pain phenotype. Whereas for males, the increased basal respiration may reflect the pain phenotype in general. To our knowledge, this is the first study to show sex differences in mitochondrial bioenergetics in the premise of cap-dependent translation for paclitaxel-induced peripheral neuropathy. These differences seen early during CIPN development, may be important for identifying strategies to prevent its establishment in both sexes. Thus, it is necessary to understand the particular mitochondrial pathways downstream of eIF4E to identify potential targets to prevent CIPN. Alternatively, therapies that address restoration of normal mitochondrial function and morphology are also promising for either sex.
Previously published data has shown that paclitaxel leads to spinal activation of astrocytes but not microglia at different phases of paclitaxel-induced peripheral neuropathy in mouse and rat models (72, 73). In contrast, we found that both microglia and astrocytes are significantly activated after paclitaxel injection in male mice, which is eIF4E dependent. However, the astrocyte reactivity increase in female mice was independent of eIF4E, suggesting that paclitaxel may have alternate mechanisms of astrocyte activation in females. Activated glia are known to secrete a number of pro-inflammatory mediators that act directly on CNS neurons, which can sensitize them if subjected to constant stimulation over extended periods (64). This has implications for the establishment of pain phenotypes post paclitaxel treatment. It has also been shown that intraperitoneal paclitaxel can activate astrocytes and mediate mechanical allodynia in the 1st hour, possibly by miniscule amounts crossing the blood-brain barrier (62). Thus, the glia activation seen 10 days post our paclitaxel regimen may be important for onset as well as establishment of CIPN through cap-dependent translation in males but via other pathways in females.
Regulation of cap-dependent translation happens via multiple different pathways. This is partly related to the expression of cell-specific surface receptors such as TrkA/B, gp130, mGLuR1/5, and insulin receptors and this influences which pathway is activated in response to which stimuli (74–76). In behavioral experiments, we assessed the role of eIf4E for regulation of brain behaviors i.e the interaction of all cells in the body after insult, but in the molecular experiments, we target specific populations of cells to understand how they individually change as a result of eIF4E manipulation and paclitaxel treatment. Our study provides novel insight into how cap-dependent translation dysregulation of immune or neuronal cells may be important for sex-divergent mechanisms of early CIPN. But comprehensive future studies would still be required to further deduce the cell-specific responses after paclitaxel treatment in the absence of cap-dependent translation.
Overall, our data suggest that although pain-like behavior during the early development of the CIPN are similar, the mechanisms and cell types involved in engendering these behaviors are different in males and females. The differences in immune cell populations and activation of glia indicate that separate upstream pathways may regulate cap-dependent translation in both sexes and thus lead to changed downstream outcomes. The central player i.e. the eIF4E complex can thus be a valuable target for preventing establishment of CIPN and limiting associated pain.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by University of Texas at Dallas Institute for Animal Care and Use Committee (IACUC).
Author Contributions
Methodology, NA. Data analysis and curation, NA, PM, TS-P, and HJ. Writing manuscript and drawing figures, NA, PM, TS-P, and HJ. MB conceptualized the study and participated and supervised in all aspects of the study and manuscript preparation. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the NIH/NINDS, grant number K22NS096030 (MB), the University of Texas System STARS program research support grant (MB), the American Pain Society Future Leaders Grant (MB), and the Rita Allen Foundation Grant (MB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Luz R. Barron, Brandon T. Lane, and Carlos Salinas for their technical contributions. We would also like to thank all current and former lab members for the assistance and input on this manuscript. Graphics created with Biorender.com.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.642420/full#supplementary-material
References
1. Cata J, Weng H R, Lee NB, Reuben J, Dougherty MP. Clinical and experimental finding in humans and animals with chemotherapy-induced peripheral neuropathy. Minerva Anestesiol (2006) 72(3):151–69.
2. Quasthoff S, Hartung HP. Chemotherapy-induced peripheral neuropathy. J Neurol (2002) 249(1):9–17. doi: 10.1007/pl00007853
3. Windebank AJ, Grisold W. Chemotherapy-induced neuropathy. J Peripher Nerv Syst (2008) 13(1):27–46. doi: 10.1111/j.1529-8027.2008.00156.x
4. Kannarkat G, Lasher EE, Schiff D. Neurologic complications of chemotherapy agents. Curr Opin Neurol (2007) 20(6):719–25. doi: 10.1097/WCO.0b013e3282f1a06e
5. Pachman DR, Barton DL, Swetz KM, Loprinzi CL. Troublesome symptoms in cancer survivors: fatigue, insomnia, neuropathy, and pain. J Clin Oncol (2012) 30(30):3687–96. doi: 10.1200/JCO.2012.41.7238
6. Seretny M, Currie GL, Sena ES, Ramnarine S, Grant R, MacLeod MR, et al. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain (2014) 155(12):2461–70. doi: 10.1016/j.pain.2014.09.020
7. Ibrahim EY, Ehrlich BE. Prevention of chemotherapy-induced peripheral neuropathy: A review of recent findings. Crit Rev Oncol Hematol (2020) 145:102831. doi: 10.1016/j.critrevonc.2019.102831
8. Hwang BY, Kim ES, Kim CH, Kwon JY, Kim HK. Gender differences in paclitaxel-induced neuropathic pain behavior and analgesic response in rats. Korean J Anesthesiol (2012) 62(1):66–72. doi: 10.4097/kjae.2012.62.1.66
9. Naji-Esfahani H, Vaseghi G, Safaeian L, Pilehvarian AA, Abed A, Rafieian-Kopaei M. Gender differences in a mouse model of chemotherapy-induced neuropathic pain. Lab Anim (2016) 50(1):15–20. doi: 10.1177/0023677215575863
10. Gewandter JS, Kleckner AS, Marshall JH, Brown JS, Curtis LH, Bautista J, et al. Chemotherapy-induced peripheral neuropathy (CIPN) and its treatment: an NIH Collaboratory study of claims data. Supportive Care Cancer (2020) 28(6):2553–62. doi: 10.1007/s00520-019-05063-x
11. Ferrari LF, Araldi D, Green PG, Levine JD. Marked sexual dimorphism in neuroendocrine mechanisms for the exacerbation of paclitaxel-induced painful peripheral neuropathy by stress. Pain (2020) 161(4):865–74. doi: 10.1097/j.pain.0000000000001798
12. Sorge RE, Mapplebeck JCS, Rosen S, Beggs S, Taves S, Alexander JK, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci (2015) 18(8):1081–3. doi: 10.1038/nn.4053
13. Krukowski K, Eijkelkamp N, Laumet G, Hack CE, Li Y, Dougherty PM, et al. CD8+ T Cells and Endogenous IL-10 Are Required for Resolution of Chemotherapy-Induced Neuropathic Pain. J Neurosci (2016) 36(43):11074–83. doi: 10.1523/JNEUROSCI.3708-15.2016
14. Makker PG, Duffy SS, Lees JG, Perera CJ, Tonkin RS, Butovsky O, et al. Characterisation of Immune and Neuroinflammatory Changes Associated with Chemotherapy-Induced Peripheral Neuropathy. PloS One (2017) 12(1):e0170814. doi: 10.1371/journal.pone.0170814
15. Laumet G, Edralin JD, Dantzer R, Heijnen CJ, Kavelaars A. Cisplatin educates CD8+ T cells to prevent and resolve chemotherapy-induced peripheral neuropathy in mice. Pain (2019) 160(6):1459–68. doi: 10.1097/j.pain.0000000000001512
16. Pinho-Ribeiro FA, Verri WA Jr, Chiu IM. Nociceptor Sensory Neuron-Immune Interactions in Pain and Inflammation. Trends Immunol (2017) 38(1):5–19. doi: 10.1016/j.it.2016.10.001
17. McMahon SB, La Russa F, Bennett DL. Crosstalk between the nociceptive and immune systems in host defence and disease. Nat Rev Neurosci (2015) 16(7):389–402. doi: 10.1038/nrn3946
18. Moalem G, Xu K, Yu L. T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience (2004) 129(3):767–77. doi: 10.1016/j.neuroscience.2004.08.035
19. Siau C, Xiao W, Bennett GJ. Paclitaxel- and vincristine-evoked painful peripheral neuropathies: loss of epidermal innervation and activation of Langerhans cells Vol. 201. Experimental Neurology (2006) p. 507–14. doi: 10.1016/j.expneurol.2006.05.007
20. Zhang H, Li Y, de Carvalho-Barbosa M, Kavelaars A, Heijnen CJ, Albrecht PJ, et al. Dorsal Root Ganglion Infiltration by Macrophages Contributes to Paclitaxel Chemotherapy-Induced Peripheral Neuropathy. J Pain (2016) 17(7):775–86. doi: 10.1016/j.jpain.2016.02.011
21. Luo X, Huh Y, Bang S, He Q, Zhang L, Matsuda M, et al. Macrophage Toll-like Receptor 9 Contributes to Chemotherapy-Induced Neuropathic Pain in Male Mice. J Neurosci (2019) 39(35):6848–64. doi: 10.1523/jneurosci.3257-18.2019
22. Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell (2009) 136(4):731–45. doi: 10.1016/j.cell.2009.01.042
23. Khoutorsky A, Price TJ. Translational Control Mechanisms in Persistent Pain. Trends Neurosci (2018) 41(2):100–14. doi: 10.1016/j.tins.2017.11.006
24. Shveygert M, Kaiser C, Bradrick SS, Gromeier M. Regulation of Eukaryotic Initiation Factor 4E (eIF4E) Phosphorylation by Mitogen-Activated Protein Kinase Occurs through Modulation of Mnk1-eIF4G Interaction▿. Mol Cell Biol (2010) 30(21):5160–7. doi: 10.1128/mcb.00448-10
25. Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature (2012) 485(7396):109–13. doi: 10.1038/nature11083
26. Nikolcheva T, Pyronnet S, Chou SY, Sonenberg N, Song A, Clayberger C, et al. A translational rheostat for RFLAT-1 regulates RANTES expression in T lymphocytes. J Clin Invest (2002) 110(1):119–26. doi: 10.1172/jci15336
27. Herdy B, Jaramillo M, Svitkin YV, Rosenfeld AB, Kobayashi M, Walsh D, et al. Translational control of the activation of transcription factor NF-κB and production of type I interferon by phosphorylation of the translation factor eIF4E. Nat Immunol (2012) 13(6):543–50. doi: 10.1038/ni.2291
28. Pashenkov MV, Balyasova LS, Dagil YA, Pinegin BV. The Role of the p38-MNK-eIF4E Signaling Axis in TNF Production Downstream of the NOD1 Receptor. J Immunol (2017) 198(4):1638–48. doi: 10.4049/jimmunol.1600467
29. Moy JK, Khoutorsky A, Asiedu MN, Black BJ, Kuhn JL, Barragán-Iglesias P, et al. The MNK–eIF4E Signaling Axis Contributes to Injury-Induced Nociceptive Plasticity and the Development of Chronic Pain. J Neurosci (2017) 37(31):7481–99. doi: 10.1523/jneurosci.0220-17.2017
30. Amorim IS, Kedia S, Kouloulia S, Simbriger K, Gantois I, Jafarnejad SM, et al. Loss of eIF4E Phosphorylation Engenders Depression-like Behaviors via Selective mRNA Translation. J Neurosci (2018) 38(8):2118–33. doi: 10.1523/JNEUROSCI.2673-17.2018
31. Amorim IS, Lach G, Gkogkas CG. The Role of the Eukaryotic Translation Initiation Factor 4E (eIF4E) in Neuropsychiatric Disorders. Front Genet (2018) 9:1–9. doi: 10.3389/fgene.2018.00561
32. Moy JK, Kuhn JL, Szabo-Pardi TA, Pradhan G, Price TJ. eIF4E phosphorylation regulates ongoing pain, independently of inflammation, and hyperalgesic priming in the mouse CFA model. Neurobiol Pain (2018) 4:45–50. doi: 10.1016/j.ynpai.2018.03.001
33. Mody PH, Dos Santos NL, Barron LR, Price TJ, Burton MD. eIF4E phosphorylation modulates pain and neuroinflammation in the aged. Geroscience (2020) 42(6):1663–74. doi: 10.1007/s11357-020-00220-1
34. Megat S, Ray PR, Moy JK, Lou TF, Barragan-Iglesias P, Li Y, et al. Nociceptor Translational Profiling Reveals the Ragulator-Rag GTPase Complex as a Critical Generator of Neuropathic Pain. J Neurosci (2019) 39(3):393–411. doi: 10.1523/JNEUROSCI.2661-18.2018
35. Munoz FM, Gao R, Tian Y, Henstenburg BA, Barrett JE, Hu H. Neuronal P2X7 receptor-induced reactive oxygen species production contributes to nociceptive behavior in mice. Sci Rep (2017) 7(1):3539. doi: 10.1038/s41598-017-03813-7
36. de Campos Lima T, Santos D, Lemes JBP, Chiovato LM, Lotufo C. Hyperglycemia induces mechanical hyperalgesia and depolarization of the resting membrane potential of primary nociceptive neurons: Role of ATP-sensitive potassium channels. J Neurol Sci (2019) 401:55–61. doi: 10.1016/j.jns.2019.03.025
37. Luu W, Bjork J, Salo E, Entenmann N, Jurgenson T, Fisher C, et al. Modulation of SUR1 KATP Channel Subunit Activity in the Peripheral Nervous System Reduces Mechanical Hyperalgesia after Nerve Injury in Mice. Int J Mol Sci (2019) 20(9):1–17. doi: 10.3390/ijms20092251
38. Duggett NA, Griffiths LA, Flatters SJL. Paclitaxel-induced painful neuropathy is associated with changes in mitochondrial bioenergetics, glycolysis, and an energy deficit in dorsal root ganglia neurons. Pain (2017) 158(8):1499–508. doi: 10.1097/j.pain.0000000000000939
39. Singhmar P, Huo X, Li Y, Dougherty PM, Mei F, Cheng X, et al. Orally active Epac inhibitor reverses mechanical allodynia and loss of intraepidermal nerve fibers in a mouse model of chemotherapy-induced peripheral neuropathy. Pain (2018) 159(5):884–93. doi: 10.1097/j.pain.0000000000001160
40. Yilmaz E, Watkins SC, Gold MS. Paclitaxel-induced increase in mitochondrial volume mediates dysregulation of intracellular Ca2+ in putative nociceptive glabrous skin neurons from the rat. Cell Calcium (2017) 62:16–28. doi: 10.1016/j.ceca.2017.01.005
41. Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, et al. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc Natl Acad Sci U.S.A. (2010) 107(32):14134–9. doi: 10.1073/pnas.1005320107
42. Moy JK, Khoutorsky A, Asiedu MN, Dussor G, Price TJ. eIF4E Phosphorylation Influences Bdnf mRNA Translation in Mouse Dorsal Root Ganglion Neurons. Front Cell Neurosci (2018) 12:29:29. doi: 10.3389/fncel.2018.00029
43. Hershey JW, Sonenberg N, Mathews MB. Principles of translational control: an overview. Cold Spring Harb Perspect Biol (2012) 4(12):1–10. doi: 10.1101/cshperspect.a011528
44. Toma W, Kyte SL, Bagdas D, Alkhlaif Y, Alsharari SD, Lichtman AH, et al. Effects of paclitaxel on the development of neuropathy and affective behaviors in the mouse. Neuropharmacology (2017) 117:305–15. doi: 10.1016/j.neuropharm.2017.02.020
45. Smith SB, Crager SE, Mogil JS. Paclitaxel-induced neuropathic hypersensitivity in mice: responses in 10 inbred mouse strains. Life Sci (2004) 74(21):2593–604. doi: 10.1016/j.lfs.2004.01.002
46. Hassler SN, Kume M, Mwirigi JM, Ahmad A, Shiers S, Wangzhou A, et al. The cellular basis of protease-activated receptor 2-evoked mechanical and affective pain. JCI Insight (2020) 5(11):1–17. doi: 10.1172/jci.insight.137393
47. Langford DJ, Bailey AL, Chanda ML, Clarke SE, Drummond TE, Echols S, et al. Coding of Facial Expressions of Pain in the Laboratory Mouse. Nat Methods (2010) 7(6):447–9. doi: 10.1038/nmeth.1455
48. Matsumiya LC, Sorge RE, Sotocinal SG, Tabaka JM, Wieskopf JS, Zaloum A, et al. Using the Mouse Grimace Scale to reevaluate the efficacy of postoperative analgesics in laboratory mice. J Am Assoc Lab Anim Sci (2012) 51(1):42–9.
49. Avona A, Burgos-Vega C, Burton MD, Akopian AN, Price TJ, Dussor G. Dural Calcitonin Gene-Related Peptide Produces Female-Specific Responses in Rodent Migraine Models. J Neurosci (2019) 39(22):4323–31. doi: 10.1523/jneurosci.0364-19.2019
50. Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods (1994) 53(1):55–63. doi: 10.1016/0165-0270(94)90144-9
51. Harlow E, Lane D. Mounting samples in gelvatol or mowiol. CSH Protoc (2006) 2006(1). doi: 10.1101/pdb.prot4461
52. Johnson SD, Young MR. Indomethacin Treatment of Mice with Premalignant Oral Lesions Sustains Cytokine Production and Slows Progression to Cancer. Front Immunol (2016) 7:379. doi: 10.3389/fimmu.2016.00379
53. Rogers GW, Burroughs SE, Dranka BP. Direct Cellular Metabolism Measurements. Genet Eng Biotechnol News (2019) 39(3):48–50. doi: 10.1089/gen.39.03.12
54. Inyang KE, McDougal TA, Ramirez ED, Williams M, Laumet G, Kavelaars A, et al. Alleviation of paclitaxel-induced mechanical hypersensitivity and hyperalgesic priming with AMPK activators in male and female mice. Neurobiol Pain (2019) 6:100037. doi: 10.1016/j.ynpai.2019.100037
55. Korbecki J, Grochans S, Gutowska I, Barczak K, Baranowska-Bosiacka I. CC Chemokines in a Tumor: A Review of Pro-Cancer and Anti-Cancer Properties of Receptors CCR5, CCR6, CCR7, CCR8, CCR9, and CCR10 Ligands. Int J Mol Sci (2020) 21(20):1–34. doi: 10.3390/ijms21207619
56. Trebst C, Brunhorn K, Lindner M, Windhagen A, Stangel M. Expression of chemokine receptors on peripheral blood mononuclear cells of patients with immune-mediated neuropathies treated with intravenous immunoglobulins. Eur J Neurol (2006) 13(12):1359–63. doi: 10.1111/j.1468-1331.2006.01521.x
57. Galmarini CM, Bouchet BP, Falette N, Vila L, Lamblot C, Audoynaud C, et al. Weekly administration of paclitaxel induces long-term aneugenicity in nude mice. Cancer Biol Ther (2007) 6(3):377–82. doi: 10.4161/cbt.6.3.3713
58. Huehnchen P, Boehmerle W, Springer A, Freyer D, Endres M. A novel preventive therapy for paclitaxel-induced cognitive deficits: preclinical evidence from C57BL/6 mice. Trans Psychiatry (2017) 7(8):1–11. doi: 10.1038/tp.2017.149
59. Peters CM, Jimenez-Andrade JM, Jonas BM, Sevcik MA, Koewler NJ, Ghilardi JR, et al. Intravenous paclitaxel administration in the rat induces a peripheral sensory neuropathy characterized by macrophage infiltration and injury to sensory neurons and their supporting cells. Exp Neurol (2007) 203(1):42–54. doi: 10.1016/j.expneurol.2006.07.022
60. Li Y, Yin C, Liu B, Nie H, Wang J, Zeng D, et al. Transcriptome profiling of long noncoding RNAs and mRNAs in spinal cord of a rat model of paclitaxel-induced peripheral neuropathy identifies potential mechanisms mediating neuroinflammation and pain. J Neuroinflamm (2021) 18(1):1–22. doi: 10.1186/s12974-021-02098-y
61. Robinson CR, Zhang H, Dougherty PM. Astrocytes, but not microglia, are activated in oxaliplatin and bortezomib-induced peripheral neuropathy in the rat. Neuroscience (2014) 274:308–17. doi: 10.1016/j.neuroscience.2014.05.051
62. Liu X, Tonello R, Ling Y, Gao YJ, Berta T. Paclitaxel-activated astrocytes produce mechanical allodynia in mice by releasing tumor necrosis factor-alpha and stromal-derived cell factor 1. J Neuroinflamm (2019) 16(1):209. doi: 10.1186/s12974-019-1619-9
63. Ochi-ishi R, Nagata K, Inoue T, Tozaki-Saitoh H, Tsuda M, Inoue K. Involvement of the chemokine CCL3 and the purinoceptor P2X7 in the spinal cord in paclitaxel-induced mechanical allodynia. Mol Pain (2014) 10:53. doi: 10.1186/1744-8069-10-53
64. Matsuda M, Huh Y, Ji RR. Roles of inflammation, neurogenic inflammation, and neuroinflammation in pain. J Anesth (2019) 33(1):131–9. doi: 10.1007/s00540-018-2579-4
65. Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med (2010) 16(11):1267–76. doi: 10.1038/nm.2234
66. Weyer AD, Zappia KJ, Garrison SR, O'Hara CL, Dodge AK, Stucky CL. Nociceptor Sensitization Depends on Age and Pain Chronicity. Eneuro (2016) 3(1):26. doi: 10.1523/eneuro.0115-15.2015
67. Kandasamy R, Price TJ. The Pharmacology of Nociceptor Priming. Handb Exp Pharmacol (2015) 227:15–37. doi: 10.1007/978-3-662-46450-2_2
68. Tonello R, Xie W, Lee SH, Wang M, Liu X, Strong JA, et al. Local Sympathectomy Promotes Anti-inflammatory Responses and Relief of Paclitaxel-induced Mechanical and Cold Allodynia in Mice. Anesthesiology (2020) 132(6):1540–53. doi: 10.1097/ALN.0000000000003241
69. Huang S, Ziegler CGK, Austin J, Mannoun N, Vukovic M, Ordovas-Montanes J, et al. Lymph nodes are innervated by a unique population of sensory neurons with immunomodulatory potential. Cell (2021) 184(2):441–59. doi: 10.1016/j.cell.2020.11.028
70. Liu X-J, Zhang Y, Liu T, Xu Z-Z, Park C-K, Berta T, et al. Nociceptive neurons regulate innate and adaptive immunity and neuropathic pain through MyD88 adapter. Cell Res (2014) 24(11):1374–7. doi: 10.1038/cr.2014.106
71. Bousso P. T-cell activation by dendritic cells in the lymph node: lessons from the movies. Nat Rev Immunol (2008) 8(9):675–84. doi: 10.1038/nri2379
72. Li Y, Zhang H, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM. Toll-like receptor 4 signaling contributes to Paclitaxel-induced peripheral neuropathy. J Pain (2014) 15(7):712–25. doi: 10.1016/j.jpain.2014.04.001
73. Zhang H, Yoon SY, Zhang H, Dougherty PM. Evidence that spinal astrocytes but not microglia contribute to the pathogenesis of Paclitaxel-induced painful neuropathy. J Pain (2012) 13(3):293–303. doi: 10.1016/j.jpain.2011.12.002
74. Uttam S, Wong C, Price TJ, Khoutorsky A. eIF4E-Dependent Translational Control: A Central Mechanism for Regulation of Pain Plasticity. Front Genet (2018) 9:1–10. doi: 10.3389/fgene.2018.00470
75. Mamane Y, Petroulakis E, Rong L, Yoshida K, Ler LW, Sonenberg N. eIF4E–from translation to transformation. Oncogene (2004) 23(18):3172–9. doi: 10.1038/sj.onc.1207549
Keywords: Dorsal Root Ganglia, astrocyte, microglia, T-cell, mitochondrial respiration, eIF4E, sex differences, neuroimmune
Citation: Agalave NM, Mody PH, Szabo-Pardi TA, Jeong HS and Burton MD (2021) Neuroimmune Consequences of eIF4E Phosphorylation on Chemotherapy-Induced Peripheral Neuropathy. Front. Immunol. 12:642420. doi: 10.3389/fimmu.2021.642420
Received: 16 December 2020; Accepted: 18 March 2021;
Published: 12 April 2021.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Michaela Semeraro, Assistance Publique Hopitaux De Paris, FranceTemugin Berta, University of Cincinnati, United States
Copyright © 2021 Agalave, Mody, Szabo-Pardi, Jeong and Burton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael D. Burton, TWljaGFlbC5CdXJ0b25AVVREYWxsYXMuZWR1
†These authors have contributed equally to this work