 Anna Torun1
Anna Torun1 Piotr Trzonkowski
Piotr Trzonkowski Beata Pyrzynska
Beata Pyrzynska- 1Chair and Department of Biochemistry, Medical University of Warsaw, Warsaw, Poland
- 2Klarman Cell Observatory, Broad Institute of MIT and Harvard, Cambridge, MA, United States
- 3Department of Medical Immunology, Medical University of Gdansk, Gdansk, Poland
- 4Department of Gastroenterology, Hepatology, Feeding Disorders and Pediatrics, The Children’s Memorial Health Institute, Warsaw, Poland
The incidence and prevalence rate of chronic inflammatory disorders is on the rise in the pediatric population. Recent research indicates the crucial role of interactions between the altered intestinal microbiome and the immune system in the pathogenesis of several chronic inflammatory disorders in children, such as inflammatory bowel disease (IBD) and autoimmune diseases, such as type 1 diabetes mellitus (T1DM) and celiac disease (CeD). Here, we review recent knowledge concerning the pathogenic mechanisms underlying these disorders, and summarize the facts suggesting that the initiation and progression of IBD, T1DM, and CeD can be partially attributed to disturbances in the patterns of composition and abundance of the gut microbiota. The standard available therapies for chronic inflammatory disorders in children largely aim to treat symptoms. Although constant efforts are being made to maximize the quality of life for children in the long-term, sustained improvements are still difficult to achieve. Additional challenges are the changing physiology associated with growth and development of children, a population that is particularly susceptible to medication-related adverse effects. In this review, we explore new promising therapeutic approaches aimed at modulation of either gut microbiota or the activity of the immune system to induce a long-lasting remission of chronic inflammatory disorders. Recent preclinical studies and clinical trials have evaluated new approaches, for instance the adoptive transfer of immune cells, with genetically engineered regulatory T cells expressing antigen-specific chimeric antigen receptors. These approaches have revolutionized cancer treatments and have the potential for the protection of high-risk children from developing autoimmune diseases and effective management of inflammatory disorders. The review also focuses on the findings of studies that indicate that the responses to a variety of immunotherapies can be enhanced by strategic manipulation of gut microbiota, thus emphasizing on the importance of proper interaction between the gut microbiota and immune system for sustained health benefits and improvement of the quality of life of pediatric patients.
Introduction
The role of the immune system is to efficiently target diverse pathogens, such as viruses and bacteria, to keep cancer cells in check and avoid reactions against its own tissues and organs (1, 2). Inflammation is the defense mechanism of the body by which the immune system recognizes and removes harmful and foreign stimuli and initiates the healing process (3). There are two types of inflammation: acute and chronic (4). Acute inflammation starts rapidly, becomes severe in a short period of time and lasts for a few days (5), whereas chronic inflammation is slow and lasts for prolonged periods of time–from several months to years. Chronic inflammation can result from a failure in eliminating pathogenic organisms during acute inflammation, prolonged exposure to irritants or foreign materials, defects in the immune system, or autoimmune disorders (6).
During inflammation in response to foreign antigens, the immune cells of the tissue, such as macrophages and dendritic cells, release cytokines [e.g. interleukin-1 (IL-1) and tumor necrosis factor-α (TNF-α)] that stimulate the infiltration of circulating leukocytes (7, 8). In addition to the recruitment of leukocytes, the tissue immune cells also play a role in antigen removal by phagocytosis and serve as antigen-presenting cells (APCs) to lymphocytes (9). Neutrophils are the first leukocytes that enter the local injury site. They destroy the antigen by phagocytosis and release granules rich in enzymes, reactive oxygen species, and cytokines, such as IL-1, IL-6, and TNF-α (10, 11). Lymphocytes, including different types of T and B cells, are the next line of defense. They play a crucial role in inflammation by secreting cytokines, producing antibodies and immune complexes (12, 13). The production of inflammatory cytokines, growth factors, and enzymes during inflammation may lead to tissue damage and secondary repair processes (4).
Inflammation in autoimmune disorders is distinct; the immune system recognizes the normal components of the body as foreign antigens and attacks healthy tissues (2, 14). Auto-reactive T cells attack and damage specific tissues and organs. Several models for explaining molecular mechanisms triggering autoimmunity have been developed, including molecular mimicry, breach in central tolerance, non-specific bystander activation and persistent antigenic stimuli (15). A recent review provided a comprehensive summary of the concept of molecular mimicry and its potential involvement in different autoimmune diseases (16). The hypothesis of molecular mimicry of foreign antigens by the structures of the body assumes that two different molecules (such as foreign and self-peptide) have antigenic structures similar enough to be recognized by the same antibodies or T cells (antigenic cross-reaction). Regulatory T cells (Treg) are the major cell subset maintaining tolerance to self-antigens as they can potently suppress the over-activation of different immune cells, including effector T cells (Teff), B cells, natural killer (NK) cells, macrophages, and dendritic cells, and hence, they can maintain the balance between autoimmunity and self-tolerance (2, 17–19). Figure 1 summarizes factors that participate in the pathogenesis of chronic inflammatory disorders.
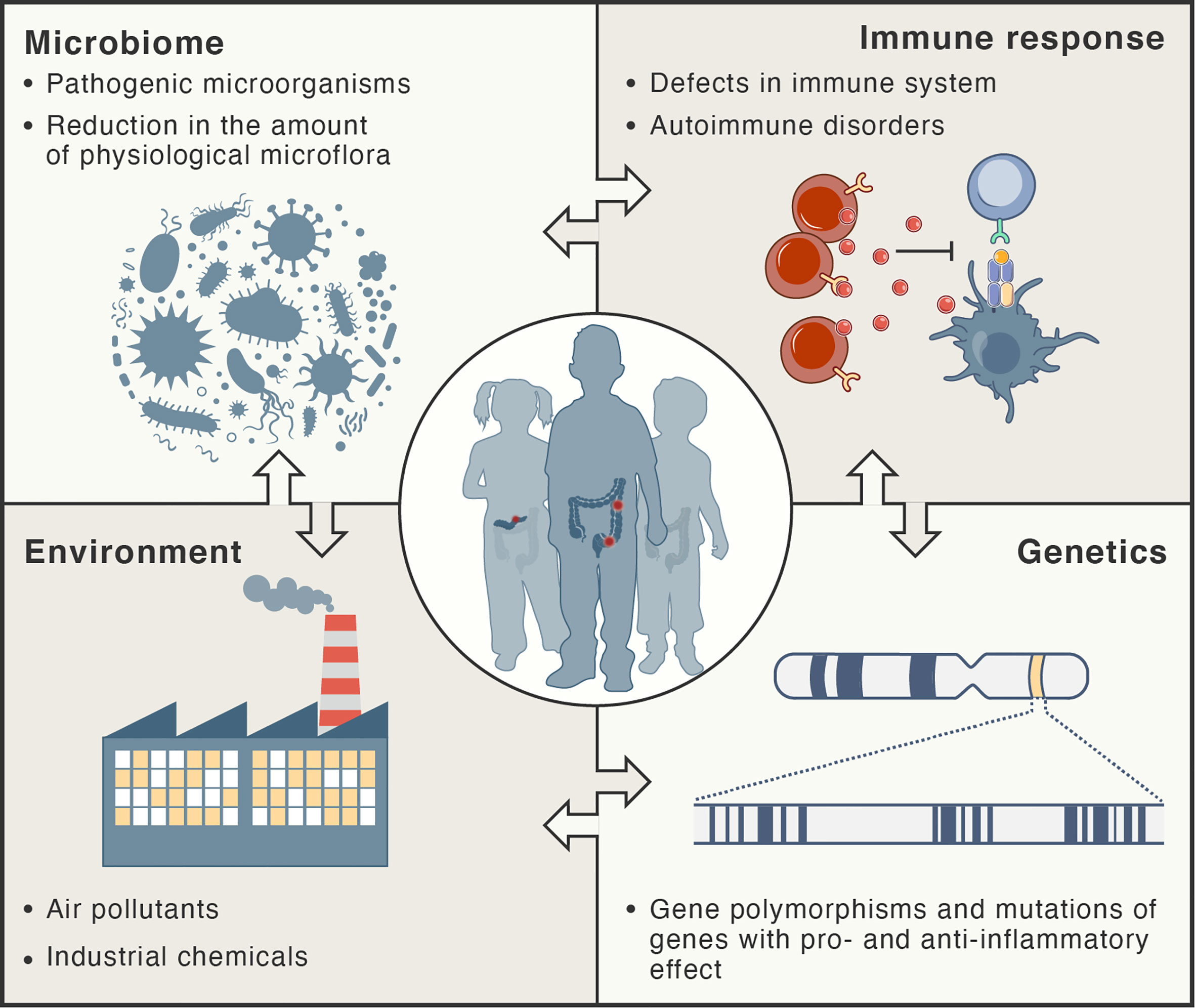
Figure 1 Risk factors for development of chronic inflammatory disorders. These types of diseases are thought to develop as result of complex interactions between the immune system, microbiome, and environment in genetically-susceptible hosts.
In this review, we discuss common chronic inflammatory disorders in children, such as type 1 diabetes mellitus (T1DM), celiac disease (CeD), and inflammatory bowel disease (IBD), with a focus on the role of microbiota in their pathogenesis. We describe current clinical approaches with manipulation of gut microbiota for pediatric therapy. Since many chronic inflammatory disorders arise from an imbalance between Teff cells and Treg cells, we point out the challenges in the development of effective therapies, disabling over-activated Teff cells, as well as the adoptive cell therapies, employing suppressive Treg cells.
Microbiota
The human intestine harbors approximately 1013 to 1014 commensal microorganisms, such as bacteria, viruses, and fungi, collectively termed as the microbiota (20). The intestinal microbiota maintains the integrity of the intestinal wall, protects against the overgrowth of pathogenic microorganisms by competing for the same nutrients and synthesizing protective substances, assists in food digestion, and produces vitamins and immunomodulating compounds such as short-chain fatty acids (SCFAs) (21–27). Although, some comprehensive reviews have summarized recent discoveries on the role of microorganisms in human health (28–30), in the following parts, we highlight a few important facts related to changes in microbiota composition and diversity during a life-time.
Microbial colonization of the human gut from maternal (for example, breast milk or birth canal) and non-maternal sources (for example, the diet and environment) and maturation of the gut barrier occur during early life (31–33). The contact with specific microbes during the first 6 months of life is considered as most crucial for the gut maturation (34). Finally, by three years of age, the microbiome profile exhibits the maturity similar to that found in adults (35, 36). Approximately 2000 bacterial species have been isolated from the intestine and most of them belong to four phyla: Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria. About 60% of the human gut bacteria belong to the phylum Firmicutes, most of which have gram-positive cell wall structure. They are present mainly in the mucus layer of the intestine. Bacteroidetes, the second most abundant phylum, constitutes about 30% of all bacteria in the gut and are gram-negative bacteria localized primarily in the gut lumen (37–39). The Firmicutes : Bacteroidetes ratio has been considered to be an indicator of the degree of maturation of the human gut microbiota and is lower in infants and the elderly compared to that in adults (40). Clostridium, Streptococcus, Ruminococcus, Lactobacillus, and Bifidobacterium (gram-positive bacteria) as well as Bacteroides and Escherichia (gram-negative bacteria) are the most prevalent bacterial genera in the human intestine (41). A recent large cohort study compared the stool microbiome of children and adults, revealing that children exhibit lower microbiota diversity with higher Bacteroides abundance and different metabolic pathways (42).
The presence of a large number of symbiotic microorganisms near the epithelial surface is an enormous challenge for the mucosal immune system because it must avoid harmful inflammatory responses to the symbionts, while preserving the ability to mount an immune response against pathogens (43). The interaction between gut microbiota and host cells is regulated by the immune system through pattern recognition receptors, including Toll-like and NOD-like receptors (44). Immunoglobulins (Ig) A and G are the predominant antibody isotypes contributing to intestinal barrier maintenance, microbiome selection, and decreased activation of innate immunity (45, 46). Microbiota-specific IgA and IgG are transmitted to newborns via maternal milk, to protect the neonatal intestine from bacterial translocation across the intestinal epithelium (46). Later in life, IgA and IgG are produced at mucosal sites by gut-associated lymphoid tissues (GALTs) and secreted into the intestinal lumen, where they limit the translocation of microorganisms into the body (47–49).
Recent findings associating colonization of the infant intestine by commensal microorganisms with the proper development and maturation of the immune system in mucosal tissues have been comprehensively reviewed (50–53). The early life seems to be a critical period in which immune system education takes place and the immune cells learn to tolerate commensal microbiota. Therefore, perturbed crosstalk between the microbiota and the immune system at this time can lead to serious life-lasting health defects. For example, an elevated risk of developing chronic inflammatory disorders, including T1DM (54, 55), CeD (56) and IBD (57), is associated with microbial dysbiosis in infants. Numerous studies using germ-free (GF) animals have provided insights into the mechanisms, by which the microbiota influences immune system development and maturation [reviewed in Ref (50, 52)]. Very limited exposure of the immune system to appropriate microbiota in early life leads to morphological abnormalities in GALTs, including Peyer’s patches, isolated lymphoid follicles and mesenteric lymph nodes (50). Functional impairment of the mucosal immune system in GF animals is related to decreased quantities of certain T cell subsets, including Teff and Treg cells, and decreased production of anti-microbial IgAs and IgGs. Such defects can be partially restored through gut colonization by a diverse microbial population (58–63). In contrast, the increased accumulation of invariant natural killer T (iNKT) cells in colons of GF animals has been detected and associated with susceptibility to colitis (64, 65). Interestingly, in mice the inhibition of iNKT cell expansion and susceptibility to colitis can be reversed only in the first 2 weeks of life, with the presence of Bacteroides fragilis in the colon (64, 65).
Perturbations in the Human Gut Microbiota
There are several factors that can influence intestinal homeostasis, including cesarean delivery, feeding infant formula instead of breastfeeding, antibiotics, diet, geography, and hygiene (66) (Figure 2). A higher degree of gut microbial diversity, with an increased abundance of Bacteroides, was observed in infants delivered vaginally than in those by C-sections (67, 68). These differences persisted up to the second year of life and are believed to result from the contact of the infant with vaginal microbiota (for example, Lactobacillus) of the mother during birth (68–71). Conversely, C-sections are associated with increased levels of intestinal pathogens such as Klebsiella, Citrobacter, and Escherichia coli (72, 73).
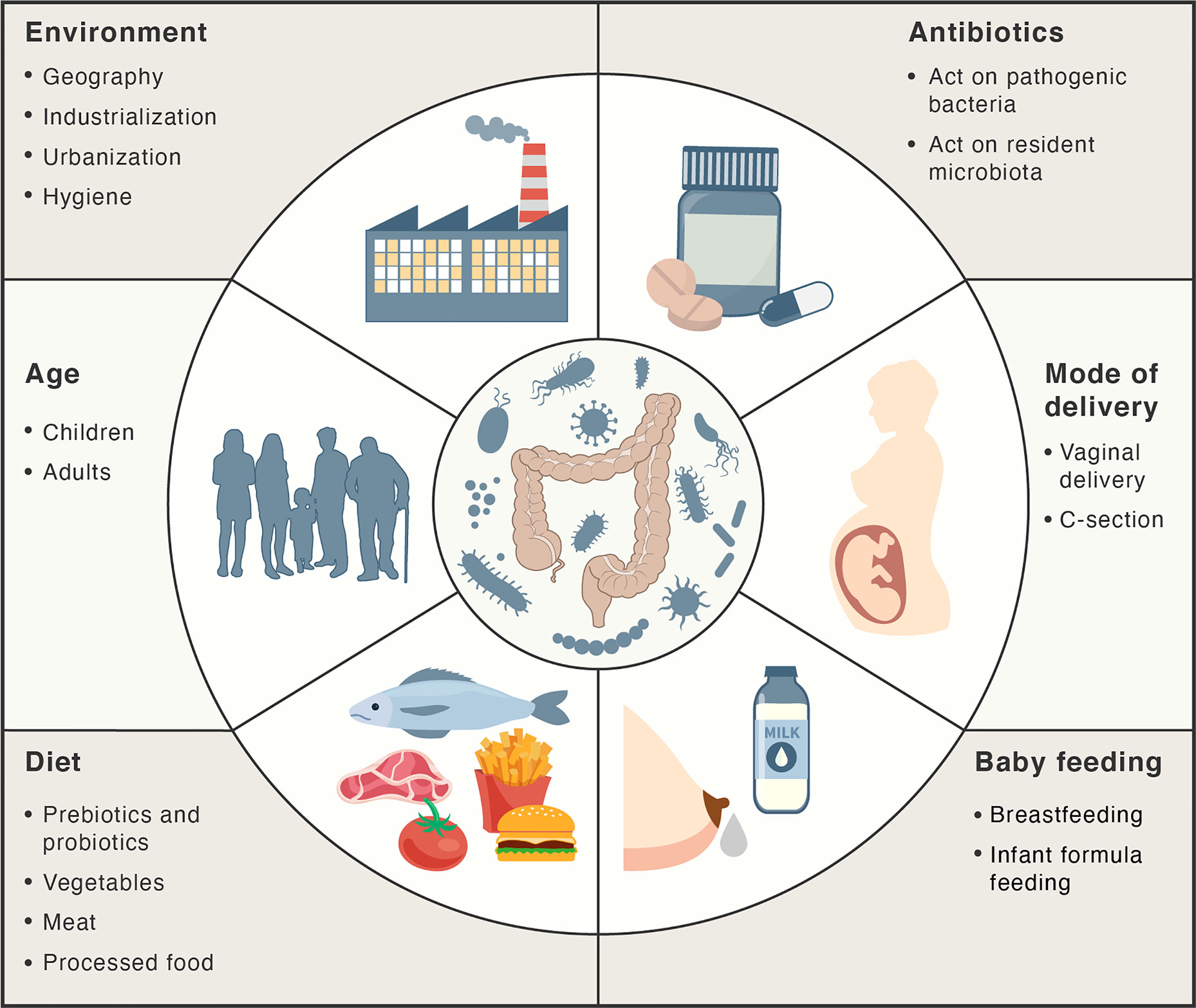
Figure 2 Main factors influencing the gut microbiota. Factors, such as mode of delivery, type of baby feeding, diet, age, environment and antibiotics may act positively or negatively on the intestinal microbiota composition and abundance.
Breastfeeding and antibiotics use are the most significant factors associated with shaping the development of microbiome structure during the first year of life. Breast milk plays a major role in limiting intestinal permeability and establishing a healthy gut barrier, as it contains a variety of nutrients, vitamins, macromolecules, and immunoglobulins (46, 74, 75). Moreover, it contains probiotic bacteria, such as Lactobacillus rhamnosus, L. gasseri, Lactococcus lactis, Leuconostoc mesenteroides, and Bifidobacteria (76, 77). The microbiota in breast milk induces immune tolerance, prevents infection, and participates in the maintenance of the epithelial barrier (76, 78). The oligosaccharides in breast milk act as prebiotic substrates for bacterial fermentation and contribute to the establishment of the infant gut microbiota (77). SCFAs that are released upon the breakdown of these oligosaccharides maintain intestinal integrity and minimize the growth of pathogenic microorganisms (79). These oligosaccharides have also been shown to inhibit the adhesion of pathogenic bacteria such as E. coli, Vibrio cholerae, and Salmonella fyris to epithelial cells (80). Oligosaccharide concentrations are higher in colostrum on the fourth day post childbirth (preterm mother’s milk) than that in mature milk at thirty days post childbirth, highlighting the importance of breastfeeding during the first days of life (81).
It has been reported that in formula-fed infants the microbiota exhibits a lower abundance of Firmicutes, Actinobacteria, and Bifidobacterium compared to breastfed infants (67, 68). Proper gut barrier maturation, manifested by the decrease in intestinal permeability, is particularly important in preterm infants, who are susceptible to necrotizing enterocolitis illness, resulting from a “leaky gut” and dysbiosis (82, 83). Importantly, gut barrier maturation in preterm infants can be induced by exclusive breastfeeding till the tenth day after birth (31, 82). The abundance of the members of Clostridiales in fecal microbiota seems to be associated with early breastfeeding (31). Moreover, breastfeeding decreases the incidence of gastrointestinal tract infections in infants (84).
Antibiotic treatments may also influence the microbiota by providing favorable conditions for the survival and overgrowth of pathogenic microorganisms in the intestine, such as S. typhimurium and Clostridium difficile (85, 86). It has been shown that oral antibiotics alter the gut microbiota for periods of time ranging from a few weeks to years (87–89). Increased antibiotic use, cleaner living conditions, and urbanization have also changed the exposure to different microorganisms. According to the hygiene hypothesis, decreased exposure to microbial antigens during early life could have a negative effect on the development of the adaptive immune response and may eventually lead to the development of autoimmune disorders. Evidence to support this hypothesis is the observation that both the temporal and geographic incidence of autoimmune and inflammatory diseases is seen to be parallel with the industrialization and urbanization of societies (90–92).
Modulation of Microbiota Composition
Recent advances in main microbiota-modulating methods have been described in details by Quigley and Gajula (93). The most common interventions include lifestyle modifications, such as diet changes, caloric restriction, and exercise, as well as clinical interventions, such as administration of probiotics, prebiotics, or antibiotics, and fecal microbiota transplantation (93). It is worth underlining that dietary changes, if sufficiently drastic, can rapidly change the gut microbiota, even in the span of 24 hours. Changes in dietary practices usually have long-term effects on the composition and functional capacity of the microbiota (94). It has been reported that diets high in fermentable plant sources lead to an increased abundance of Firmicutes, which metabolize dietary plant polysaccharides. In contrast, high-meat/low-plant diets lead to increased abundance of bile-tolerant microorganisms, such as Alistipes, Bilophila, and Bacteroides species, and a decreased abundance of Firmicutes (95).
Prebiotics, other important microbiota-modulatory factors, have been broadly defined by the International Scientific Association for Probiotics and Prebiotics as “substrates that are selectively utilized by host microorganisms conferring a health benefit” (96). The most commonly used are carbohydrate-based prebiotics, metabolizable by the gut microbiota, leading to selective stimulation of growth or activity of beneficial gut microorganisms. The fermentation products of prebiotics, mainly SCFAs, help to maintain the gut barrier integrity (97, 98).
Probiotics are live microorganisms that provide health benefits upon consumption by improving or restoring the gut flora. They exert their effects by reducing colonization by pathogenic microorganisms, enhancing mucus production, improving epithelium integrity, and balancing the interactions between gut bacteria and the immune system (99–101). In infants who receive probiotics, the changes induced by antibiotic treatment and cesarean delivery are reversed, and the normal composition and function of gut microbiota is restored (102–105).
The last method of modulating gut microbiota that we discuss here is fecal microbiota transplantation (FMT), which involves the transfer of fecal bacteria from a presumptively healthy donor into the gastrointestinal tract of affected patient, in order to treat microbial dysbiosis (106). This method reduces intestinal inflammation, as demonstrated by decreased levels of proinflammatory cytokines such as TNF-α, IL-1β, and interferon-γ (IFN-γ), and helps in restoring the intestinal homeostasis. It has been successfully used for treating recurrent and antibiotic-refractory C. difficile infections (107–110), even in pediatric patients (111).
Type 1 Diabetes Mellitus
T1DM is an autoimmune disease resulting from an inappropriate immune response that causes the destruction of insulin-secreting β-cells of the pancreatic islets, mediated mainly by autoreactive effector T cells [reviewed in (112)]. Destruction of pancreatic β-cells leads to the loss of endogenous insulin production, and results in impaired glucose metabolism. T1DM is usually diagnosed in children or young adults, but it can appear at any age (113, 114). With the increasing incidence of this disorder, the peak age at diagnosis has shifted to a younger age (115).
The major genetic risk factors involved in the development of T1DM are located within the class II human leukocyte antigen (HLA) region (116). The most important loci encode HLA-DR and HLA-DQ molecules that bind and present antigenic peptides to T cells. The high degree of polymorphism in the HLA region indicates that it is possible to mount an antibody response against a large number of constantly mutating microbial pathogens. Consequently, this wide diversity of recognized epitopes may cause the generation of antigenic cross-reactions due to molecular mimicry. Approximately 70% of T1DM cases carry HLA risk alleles (117, 118). The loci identified outside the HLA region are associated with polymorphisms of the insulin gene INS and lymphocyte protein tyrosine phosphatase PTPN22 gene, resulting in an increased risk of development of T1DM. A disease-associated genotype of the INS gene is associated with a poor expression of insulin in the thymus, leading to autoreactive INS-specific T cells not being destroyed during the thymic education of T cells (116, 119).
Autoantibodies that recognize insulin, glutamic acid decarboxylase 65, islet antigen 2, and zinc transporter 8 are most often present in the serum of patients with T1DM, and are the best characterized autoantibodies associated with T1DM (120). These autoantibodies are usually present before any dysglycemia or clinical symptoms appear (121). If only one of these major autoantibodies is present, the risk of T1DM is small; however, the presence of two or more autoantibodies indicates a high probability of developing the disease (122). Interestingly, children of mothers with T1DM have a relatively low genetic risk of developing T1DM themselves, as they are at half of the same genetic risk compared to children with a father who has T1DM. This observation shows that islet-specific autoantibodies found in the sera of mothers with T1DM transferred to the fetus do not damage the fetal pancreatic β-cells (123).
The diagnosis of diabetes is based on the measurement of blood glucose concentration and the presence of symptoms (124). Although in the early stages the disease is clinically silent, hyperglycemia and increased production of ketones from fatty acids eventually lead to polydipsia, polyuria, weight loss, and diabetic ketoacidosis (125). The chronic long-term complications associated with the disease include retinopathy, nephropathy, neuropathy, and cardiovascular disease (126, 127). T1DM during puberty appears to accelerate the development of complications (128).
Role of Gut Microbiota in T1DM
Recently, increasing incidence of T1DM in children within genetically stable populations has been observed (129), with less than 10% of genetically susceptible people developing clinical T1DM (130). This indicates that non-genetic factors also play an important role in the development of T1DM. Among the environmental factors that affect the development of T1DM microbiota composition, microbial infection and nutrition appear to be key factors. Recent reviews have addressed the involvement of dysbiosis in the pathogenesis of T1DM (131–135). Here we very briefly highlight some of the key findings.
Increased permeability of the intestine and changes in the composition of microbiota seem to be associated with T1DM pathogenesis (54, 136–143). Comparison of multiple islet autoantibodies-positive children with healthy controls demonstrated increased abundance of the Bacteroides species compared with Firmicutes species (137, 143, 144). The association of Bacteroides stercoris, B. fragilis, B. intestinalis, B. bifidum as well as Synergistetes taxa with diabetes was recently confirmed by machine-learning analyses of bacterial taxa and their metabolic pathways in children at T1DM onset (145). Autoantibody-positive children exhibit a low abundance of microbiota that produce butyrate (137, 141, 146). It has been reported that butyrate-induced mucin production is important for the integrity of the gut mucosa, and low levels of species that degrade mucin might imply an increased gut permeability (147). Moreover, a reduction in SCFA-producing bacteria was observed in T1DM at the onset of autoimmunity, which is associated with impaired gut barrier functions (148, 149). Decreased diversity of the intestinal microbiome has been reported in pediatric patients with β-cell autoimmunity and in those who progressed to clinical T1DM compared with non-seroconverted controls (137, 141). A marked drop in the diversity of the gut microbiota was also noted in infants genetically predisposed to T1DM during the time window between seroconversion and T1DM diagnosis (54). At the same time, a higher abundance of several pathobionts, including Ruminococcus gnavus and Streptococcus infantarius, and a lower abundance of bacteria known to counteract inflammation, such as Lachnospiraceae and Veillonellaceae, were detected in T1DM-affected patients. Moreover, there is a correlation between the changes in the gut microbiota and the pattern of T1DM progression, because the seroconverted group of patients revealed intermediate abundance of all these microorganisms compared to the non-converted and T1DM groups (54).
Some studies showed that delivery by C-section can increase the risk of T1DM in infants (150–153), suggesting that microbiota transfer from the mother’s birth canal to the newborn may be protective against T1DM incidence in infants. Moreover, a higher T1DM risk was observed in children born through planned C-sections than in those born via unscheduled C-sections (154). Therefore, the high number of C-sections in developed countries may be partially responsible for the observed increased rate of diabetes (155). However, it should be emphasized that other studies found no increased cesarean delivery-associated T1DM risk (156, 157).
After delivery, breast milk is usually the first food that enters the gut of the newborn. It has been shown that breastfeeding lowers the risk of developing T1DM compared with formula feeding (158–160). Human milk oligosaccharides protect against autoimmune T1DM development in high-risk individuals (161). As mentioned above, breast milk has a significant effect on the composition of the gut microbiota of infants, and favors the dominance of Bifidobacterium because of its specific ability to degrade human milk oligosaccharides (162–164). The decrease in the proportion of Bifidobacterium, especially B. longum subsp. infantis, is temporally associated with the increased incidence of T1DM in childhood (165).
Several studies have reported that antibiotic use increases the risk of T1DM development, in contrast to antiviral or antifungal drugs (166–168). It was shown that exposure to a single antibiotic is not associated with higher diabetes risk, but taking two to five antibiotic courses is associated with an increase in diabetes risk (166). Another study found that the use of broad-spectrum antibiotics during the first 2 years of life is associated with an increased risk of T1DM during the later years of life (167). Moreover, the antibiotic-associated risk for T1DM is influenced by the mode of delivery, being higher in children delivered by a C-section than those delivered vaginally (167). However, it is important to note that other studies did not find a correlation between antibiotic use and T1DM (169, 170).
In agreement with the hygiene hypothesis, newborn infants from less-developed areas who are exposed to a wide range of microbial antigens and therefore receive strong immune signals are protected from diabetes (171).
Treatment of T1DM
Management of T1DM requires daily administration of exogenous insulin and frequent monitoring of blood glucose levels (172, 173). Despite ongoing technological advances in recombinant insulin, and technologies to deliver insulin and monitor blood glucose levels, the majority of affected patients cannot achieve the recommended glycemic targets (174–177). Particularly in children and young adults, blood glucose control, measured using glycated hemoglobin levels, is typically poor, reaching >8% in the majority when the desired levels are below 7.5% (174). As a result, patients remain at risk of acute and chronic long-term complications associated with the disease.
As multiple islet-specific autoantibodies are found in circulation from a few weeks up to 20 years before the clinical onset of this disease, there is a potential opportunity to prevent or postpone pancreatic β-cell loss. Therefore, other approaches are sought to maintain endogenous insulin secretion or impede the already-developed islet autoimmunity. These approaches include therapy with Treg cells (178–180), treatment with Fc receptor non-binding anti-CD3 monoclonal antibodies (teplizumab and otelixizumab) (181), and antigen-specific peptide immunotherapy that sequesters auto-antibodies (182–184).
Microbiota in T1DM Therapy
Modulation of the intestinal microbiome of affected individuals seems to be an obvious therapeutic approach for alleviating T1DM. The most promising T1DM treatments, leading to the improvement of the composition and diversity of gut microbiota include strategies such as increasing infant exposure to beneficial bacteria during early life, the transfer of gut microbiota (FMT approach) from presumptively healthy donors to T1DM-prone individuals, and administration of probiotics and prebiotics to replenish or repair the missing bacterial taxa [summarized in a recent review (185)]. It has been shown that probiotics administered in early infancy positively correlated with decreased islet-specific autoantibodies (186). Probiotic treatment improves overall islet function and gut islet immunomodulation and helps to control diabetes (187). Moreover, administration of the natural prebiotic, inulin-type fructans, improve gastrointestinal tract functions with a relative increase in Bifidobacterium species compared to the controls (188). In addition, experiments in mouse models of T1DM provide evidence that microbial metabolites, namely the SCFAs (products of fermentation by anaerobic intestinal microbiota), protect genetically-susceptible mice from developing diabetes (189). Nevertheless, the strategies for reshaping gut microbiota require further research exploration, since the “healthy” microbiota composition has not been precisely defined yet. Therefore, only limited conclusions concerning the directions toward modulation of the microbiota can be drawn for now (185).
Adoptive Therapy for T1DM
As mentioned above, Treg cells are the guardians of immune homeostasis and can inhibit autoreactive immune responses. In healthy individuals, Treg cells migrate to target tissues and suppress inflammation either by direct contact with antigen-presenting cells (such as dendritic cells), metabolic disruption and cytolysis of effector T cells (Teff), or secretion of soluble inhibitory cytokines that dampen the function of autoreactive cytotoxic cells (190–192). Importantly, the loss or defect in Treg cell function is implicated in the development of autoimmune diseases like T1DM (179), where the immunosuppressive function of Treg cells is impaired due to the reduction in the cell number, survival, or activity (190). The pathogenesis of diabetes is well mimicked in non-obese diabetic mouse models, where defects in function of Treg cells are also reported (193, 194). Hence, in order to establish immune tolerance, cellular therapies employing thymic Treg cells expanded in vitro, under conditions of good manufacturing practice (GMP), are emerging as potentially attractive therapeutic strategies. These cells have been extensively tested for the treatment of T1DM and have been shown to either prevent the development or reverse diabetes in preclinical studies (195, 196), and the first clinical trials with autologous polyclonal Treg cells expanded in vitro (178–180, 197, 198). Although no serious adverse effects were reported during the therapy (178, 179), an important concern is the in vivo stability of the Treg cell phenotype upon adoptive transfer. This feature of Treg cells is strictly dependent on the stable expression of the master transcription factor FOXP3 (199, 200).
Recently, a new experimental strategy has been designed to generate genetically engineered T cells from the patient that can function, persist and proliferate like normal Treg cells in vivo after infusion, enter the pancreas, and protect the function of islet cells (201). Gene editing techniques based on homology-directed repair were employed in this study. This strategy allows persistent and high expression of FOXP3 from the endogenous locus in patient CD4+ T cells, which is sufficient to convert them into functional Treg cells with sustained expression of canonical phenotypic markers and cytokine profiles (201) (Figure 3).
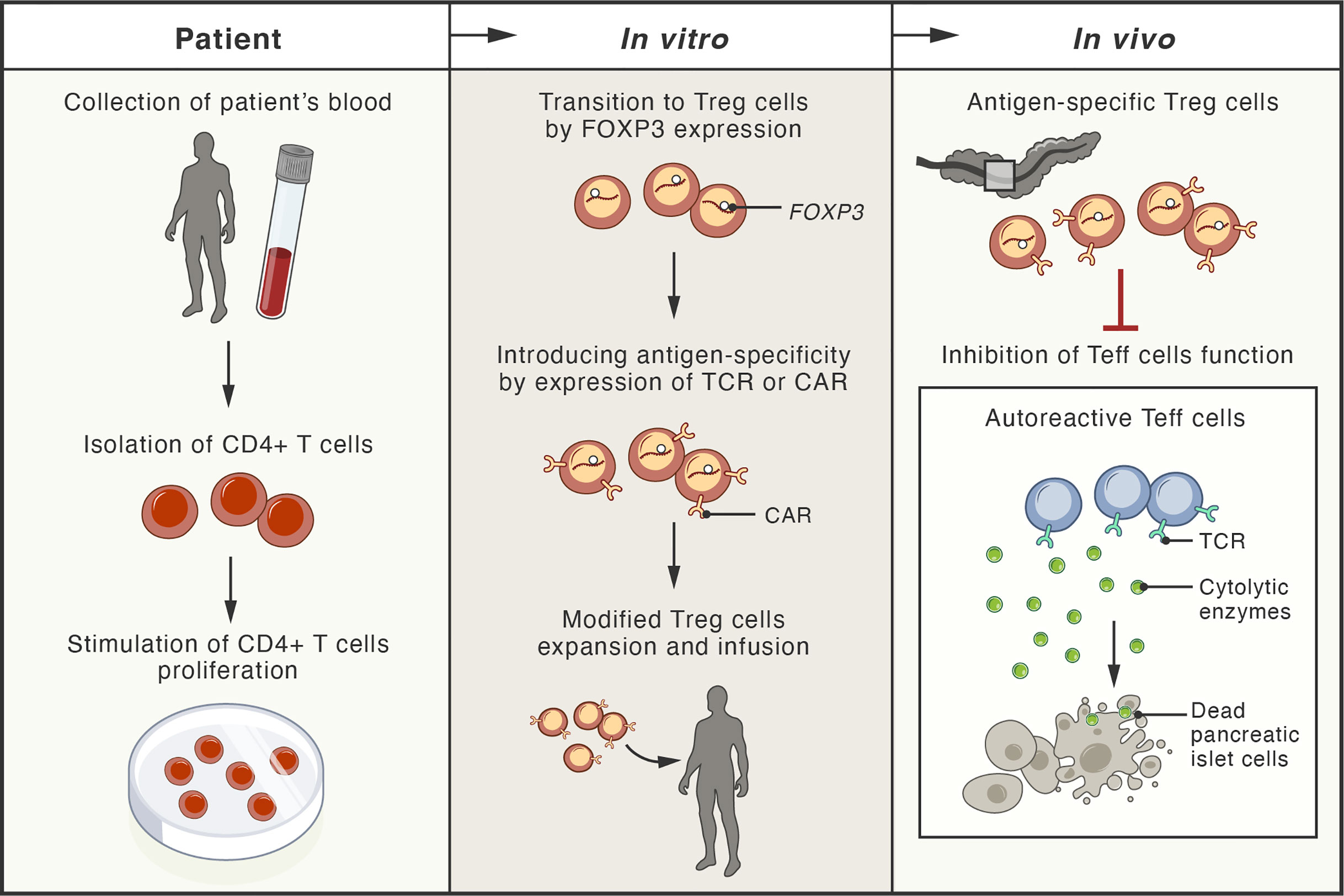
Figure 3 Schema of possible therapeutic approaches with adoptive cell therapies involving genetically modified regulatory T cells. The blood of patient with type 1 diabetes mellitus (T1DM) is used for isolation of CD4+ T cells. These cells, expanded in vitro, undergo transition to regulatory T cells (Treg) by genetic modification, leading to stable expression of the FOXP3 protein (master transcription factor of Treg cells). These cells are further modified by the expression of antigen-specific T-cell receptor (TCR) or chimeric antigen receptor (CAR). Upon infusion, the genetically modified Treg cells localize to the pancreas of the patient, where they inhibit the cytotoxic function of effector T cells (Teff), responsible for destruction of pancreatic islet cells.
The immunosuppressive functions of genetically engineered Treg cells have been demonstrated in a previously characterized murine model of inflammatory disease, where the injection of human Teff cells into immunodeficient NOD/SCID/gamma mice led to extensive Teff cells infiltration into numerous mice tissues followed by an immune response leading to severe xenogeneic graft-versus-host-like disease (202). In such a mouse model, the pre-injection of genetically engineered human Treg cells three days before injection of autologous Teff cells completely suppressed the activation of Teff cells and the development of graft-versus-host disease (201).
The specificity of immunosuppression executed by engineered Treg cells towards autologous pathogenic Teff cells can be enhanced by the expression of either the antigen-specific T-cell receptor (TCR) or the chimeric antigen receptor (CAR) on the surface of the Treg cells (203, 204). Studies using mouse models of autoimmune diabetes have demonstrated that therapy with antigen-specific Treg cells can improve the targeting of the adoptively transferred cells to specific tissues, and allow the specific recognition of common antigens in autoimmune diseases (195, 205, 206). Therefore, the antigen-specificity of Treg cells can improve their suppressive potency as well as the safety and efficacy of Treg cell-based therapies (201, 207).
However, some important considerations concerning the effect of transgenic expression of antigen-specific TCRs or CARs on Treg cell function should be kept in mind. It has been recently shown that the progression of T1DM is associated with reduced diversity of T-cell clones due to the expansion of clones with antigens-specific (mainly proinsulin-specific) TCRs in both autoaggressive Teff and suppressive Treg cell populations. This suggests that some common antigens stimulate the expansion of both T-cell subsets (208). Since the final outcome of the adoptive Treg cell therapy may depend on the balance between Teff and Treg subsets, the therapeutic Treg cells should theoretically be equipped with TCRs or CARs specific to antigens with higher affinity toward Treg cell populations only.
Controversies and Current ResearchGaps in Adoptive Therapies for T1DM and Potential Future Developments in This Field
The treatment of T1DM remains within the definition of ‘unmet medical need’ as there is no approved therapy stopping the progression of this disease and the only standard of care therapy is substitution with different forms of insulin injections. Undoubtedly, T1DM is the result of an immune-mediated destruction of pancreatic islets, but the primary trigger of this reaction is elusive. Until recently, the autoimmune background has been considered as the most probable mechanism (209), but recent reports also bring up the role of gut-associated microbiome and inflammation (210). The autoimmune background is suggested mainly by the linkage of T1DM with particular HLA haplotypes and organ-specific autoantibodies whose sensitivity and specificity allows them to be used as a laboratory marker of the diagnosis. On the other side, there are a number of autoantigens postulated triggers of autoimmune activation in T1DM, but none of them have been definitively confirmed as a single cause of the disease (211). Moreover, the trials with the use of these autoantigens as agents inducing tolerance in humans failed (212). The inflammatory background also remains unclear. Although the distinct pattern of gut bacteria in T1DM has been described, there is no direct evidence that the disease can be induced by these bacteria or cured by microbiome transplantation (149).
Having no clear target for precisely directed interventions, current attempts of the treatment focus on wider approaches. The most promising studies at the stage of clinical trials in humans cover the use of depletion agents, such as teplizumab (anti-CD3 antibody) (213), or adoptive transfer or induction of tolerogenic cells, such as polyclonal T regulatory cells (197). Briefly, depletion therapies are directed towards elimination (physical or functional) of autoreactive T effector cells, including islet-reactive T cells. The increase in the level of tolerogenic cells aims to switch off unwanted autoantigen presentation and autoimmune response by effector cells, even though the autoantigens are not clearly defined. These approaches come along with the pathogenesis of the disease as it is not only the autoimmunity itself, but rather an imbalance between effector and regulatory subsets, which is regarded as the factor facilitating the onset of T1DM (208). A common feature in this new wave of trials is the attempt to recruit patients who are in a very early stage of the disease, possibly at the asymptomatic phase. Some trials recruit healthy subjects susceptible to the development of the disease, such as members of the families of T1DM patients, offering the therapy as a kind of prophylaxis. These trials give the advantage of a less exacerbated immune process, probable lower numbers of autoantigens involved in the process before substantial epitope spread, and a higher proportion of the pancreas preserved. Novel diagnostic tools already allow us to identify such subjects (214). Scientifically, having insight into such early-phase trials, we might also be able to finally establish the autoantigens that are pivotal for the induction of the disease. It is highly possible that autoantigens change during the course of the disease due to the epitope spread, and particular stages of the disease may require interventions towards different autoantigens. Unfortunately, all these attempts leave behind patients with overt T1DM. It can be assumed that antigen-specific reactions are massive in these cases, and therefore they are much more difficult to harness (215). At the same time, the destruction of the islets is advanced to a high level at this stage. While exogenous insulin remains the only routinely available solution for these patients, they are good candidates for regenerative therapies, but these are slow and tedious to be adopted into clinics (216). Nevertheless, immune interventions might also be necessary here, as the autoimmune reactions are long-term memorized and can reappear many years after the onset of the disease as seen post islet allotransplantation in long-term T1DM patients (217).
Celiac Disease
CeD is a chronic autoimmune disease caused by a dysregulated immune response to gluten, characterized by remodeling of the small intestinal mucosa and villus atrophy. It is triggered by the ingestion of gluten in genetically predisposed individuals (218, 219). Gluten is a protein component of grains, consumed nowadays in significant quantities. While archeological research has provided evidence of grain consumption as early as 100,000 years ago (220), the more recent (10,000-20,000 years ago) domestication of grains, such as wheat, may have resulted in increased exposure to the gluten protein. Peptides that are the final products of partial gluten digestion can trigger increased gut permeability, gluten trafficking to the lamina propria, innate and adaptive immune response, and tissue damage (221–226). Tissue transglutaminase (tTG) has been identified as the autoantigen in CeD (227).
The genetic susceptibility to CeD is majorly contributed by HLA-DQ2 and/or -DQ8 (219), since the HLA-DQ molecules are responsible for the binding and presentation of gluten-deriving peptides to effector memory T cells that appear to drive the development of CeD (228). Apart from that, at least 39 non-HLA genes that predispose certain individuals to the disease have been identified, most of which are involved in inflammatory and immune responses (229). It is estimated that up to 40% of the general population carries the susceptibility genes, but the prevalence of CeD is only about 1% of the general population (230–232) and is higher among women (233, 234). The average age at diagnosis is the scholar age, which is between 6 and 9 years (235), but the disease can occur at any time from early childhood to old age.
Classical CeD is defined as CeD presenting with signs and symptoms of malabsorption: diarrhea, steatorrhea, weight loss, and growth failure. Other intestinal manifestations frequently described are bloating, aphthous stomatitis, alternating bowel habits, constipation, and gastroesophageal reflux disease. Classical malabsorptive symptoms of CeD are more commonly detected in the pediatric population. Up to two-thirds of cases exhibit classical presentations, and atypical symptoms, such as abdominal pain and poor growth, can be the chief complaint in the rest of the cases (236). Extraintestinal manifestations include osteopenia/osteoporosis, anemia, elevation of liver enzymes, and recurrent miscarriages in adults (237). Short stature is the most common extraintestinal manifestation in children, sometimes being the only clinical sign of the disease, and iron deficiency-induced anemia dominates in adults (238–240). Some extraintestinal manifestations are clearly correlated with the severity of intestinal damage (239, 241, 242). Anemia is associated with malabsorption of iron, vitamin B12, and folate (243, 244). Growth retardation is caused by nutrient malabsorption (245), and osteopenia may occur due to malabsorption of calcium and vitamin D and the consequent high bone turnover (246). Another specific manifestation of CeD is dermatitis herpetiformis, which is an itchy blistering skin disease typically observed on the elbows, knees, and buttocks, and occasionally in the scalp and upper back (247).
For the diagnosis of CeD, duodenal biopsy plus positive serological tests (anti-tTG antibodies, anti-endomysium antibodies (EmA), and deamidated gliadin peptide (DGP) antibodies) are the gold standard (248, 249). Pediatric patients with high titers (over ten times the cutoff) of anti-tTG antibodies, detectable EmA antibodies, HLA-DQ2/HLA-DQ8 positivity, and symptoms of CeD may skip the duodenal biopsy (230).
Role of Gut Microbiota in CeD
The incidence of CeD has increased in the past decades, suggesting the role of environmental factors in addition to gluten (250, 251). Patients with CeD present increased intestinal permeability; this may favor gluten sensitization as the characteristic adaptive immune response in patients with CeD takes place in the lamina propria (252). Disruption of the functional epithelial barrier of the intestine by opportunistic pathogens or infections may favor this condition [addressed in depth by a recent review (253)]. In patients with CeD, a shift toward a proinflammatory community and an increase of Proteobacteria and opportunistic pathogens, such as Neisseria or E. coli, as well as higher bacterial virulence genes have been observed (254–259). Bacteroides fragilis strains expressing metalloproteases, which have often been reported in patients with CeD, may lead to increased intestinal permeability, production of immunogenic peptides, and provoke an inflammatory response (260). Increased prevalence of pathogenic bacteria is also observed in the intestines of infants at risk of developing CeD (261).
CeD has been generally associated with alterations in the microbiome composition in both pediatric and adult populations (254, 261–264). Numerous studies have shown that the abundance of certain microbial taxa is significantly different in patients with CeD compared to that in healthy controls. Studies on intestinal biopsies and fecal samples have shown an increased abundance of Bacteroidetes (265) and Proteobacteria phyla (266), and a higher frequency of Clostridium (267), Bacteroides (265, 268), and Prevotella spp (269) in patients with CeD. The abundance of Lactobacillus and Bifidobacterium spp., on the contrary, is lower than that in controls (265, 270). It may be noted that Bifidobacterium can degrade proinflammatory gluten peptides and reduce their immunogenic potential (271, 272). Some lactobacilli have been shown to digest amylase-trypsin inhibitors, which are wheat proteins other than gluten that induce an innate immune response. Moreover, the administration of certain Lactobacillus species decreases both the permeability and inflammation stimulated by amylase-trypsin inhibitors (273).
Specific changes in the microbiota of patients with CeD have also been described and associated with poor clinical responsiveness to GFD (274) and the clinical manifestations of the disease (275). For example, compared to patients with other clinical features of CeD, the microbiota of patients with dermatitis herpetiformis is more similar to that of the controls (275).
A recent analysis of the gut microbiota and diet-related metabolites in a large cohort of children affected with CeD revealed specific microbiota signatures for both untreated individuals with new-onset CeD and individuals treated with GFD, with respect to the healthy controls (276). The authors confirmed the overabundance of the microbiota taxa mentioned above, such as Bacteroides. Additionally, they detected a lower abundance of Alistipes in children with new-onset CeD. The reduction in Megamonas, Ruminococcus, and Holdemanella was recognized as a consequence of the treatment with GFD. Importantly, the authors identified a lower abundance in 11 specific bacterial taxa considered as biomarkers of CeD, with Clostridium sensu stricto 1 as the most influential one (276).
Changes in the microbiome have also been correlated with alterations in microbiota-derived metabolites (270, 277, 278). It has been shown that patients with CeD have altered fecal SCFAs, the end products of fermentation of dietary fiber by the intestinal microbiota, and increased proteases (254, 279). Duodenal biopsies of patients with CeD show a high proteolytic activity that correlates with the proliferation of protease-producing pathogens such as Pseudomonas aeruginosa (254). P. aeruginosa cleaves gliadin peptides, whose immunogenicity can be reduced by lactobacilli found in non-CeD controls (254, 273). In addition to intestinal dysbiosis, the salivary microflora, which plays a role in hydrolyzing proline and glutamine-rich peptides, has also been shown to be altered in patients with CeD (280).
It has been shown that environmental factors, such as breastfeeding (281), influences the composition of the intestinal microbiota and may play a role in the development of CeD (282, 283). Oligosaccharides present in human milk enhance the gut barrier integrity by making the epithelium less vulnerable to bacteria-induced innate immunity (284). According to some studies, antibiotic exposure during the first year of life has been associated with an increased risk of developing CeD (285, 286), however, other reports have not found this type of causal association (170, 287). Some studies (234, 288), but not all (289), reported an association between changes in infant feeding practices and an increased incidence or earlier onset of CeD. In particular, a decreased duration of breastfeeding and the discontinuation of breastfeeding upon the introduction of gluten-containing complementary foods to the infant diet has been attributed to the “epidemic” of CeD in a Swedish population of children (234). The protective effect of concomitant breastfeeding and the introduction of gluten might result from the development of oral tolerance to gluten, enhanced by either breast milk-derived immunomodulatory factors or breast milk-stimulated infant gut colonization by beneficial microorganisms (290, 291). On the other hand, large amounts of gluten introduced to the diet of < 2-year-old children increases the risk of CeD (292).
Interestingly, several studies indicate that the HLA-DQ genotype may regulate gut colonization (261, 293, 294). The genotype of infants at risk of developing CeD influences the intestinal microbiota composition (294). Compared to control infants, neonates carrying the CeD-predisposing HLA haplotype show increased Firmicutes and Proteobacteria, and reduced Bacteroidetes and Actinobacteria. These patterns persist up to 2 years of age and are correlated with alterations in microbial metabolite production (295). For more details, concerning the association of changes in gut microbiota with risk of CeD development in children and adults, please see recent reviews (296–299).
Treatment of CeD
The only effective treatment available for CeD to resolve the symptoms associated with the disease and normalize the intestinal villi architecture is a strict gluten-free diet (GFD) (300–302). Generally, the clinical response to this treatment occurs much faster in children than in adults (238, 239). Problems arise when the GFD is not properly followed, which occurs mostly in adolescents (303). Seropositive patients with CeD may gain significant symptom improvement with administration of a mixture of two gluten-targeting recombinant proteases that decrease the immunogenicity of gluten in the small intestine by degrading it in the stomach (304, 305). Another novel oral agent, larazotide acetate, functions by regulating intestinal tight junctions, thereby preventing gluten from reaching the small intestinal submucosa and triggering an immune response (306).
Since the gluten antigen-specific memory T cells are the key players in the pathogenesis of CeD and they are able to persist in patient’s blood and gut for decades (307), new approaches to target such T cells can be considered as promising alternative therapeutic strategies for CeD. Antigen-specific immunotherapy, aimed at tolerance induction, is particularly relevant, since CeD is one of few autoimmune diseases where the antigens and driving pathogenic T cell responses are known (308). In order to induce immune tolerance, gradually escalating doses of disease-relevant antigens can be delivered by systemic or local administration. Different protocols for allergen desensitization have been developed for tolerogenic immunotherapies, with an aim to trigger either reprogramming of antigen-specific T cells into Treg cells or their clonal deletion by inducing cell death. The gluten antigens, delivered with gradual dose escalation, are presented to T cells in a tolerogenic manner. These approaches are either based on the systemic delivery of:
● Vaccines containing gluten-deriving antigenic peptides [such as Nexvax2; clinical trial NCT03644069 (309)],
● Synthetic nanoparticles coated with complexes consisting of antigenic peptides bound to MHC molecules (310),
● Nanoparticles loaded with the antigens, together with immunomodulatory agents [clinical trial NCT03486990 (311, 312)],
● Engineered erythrocytes, loaded with the antigens (313, 314)
or the delivery of antigenic peptides to mucosal tissue, mediated by genetically modified Lactobacillus lactis bacteria (315).
Additionally, methods to selectively eliminate disease-driving memory T cells have been tested, including restimulation-induced cell death (such as continuous activation of T cells with antigen and agonists of activating pathways (316), cytokine withdrawal-induced death (317), and selective induction of cell death by interfering with metabolic pathways of activated T cells (318). Figure 4 summarizes selected approaches for the treatment of CeD.
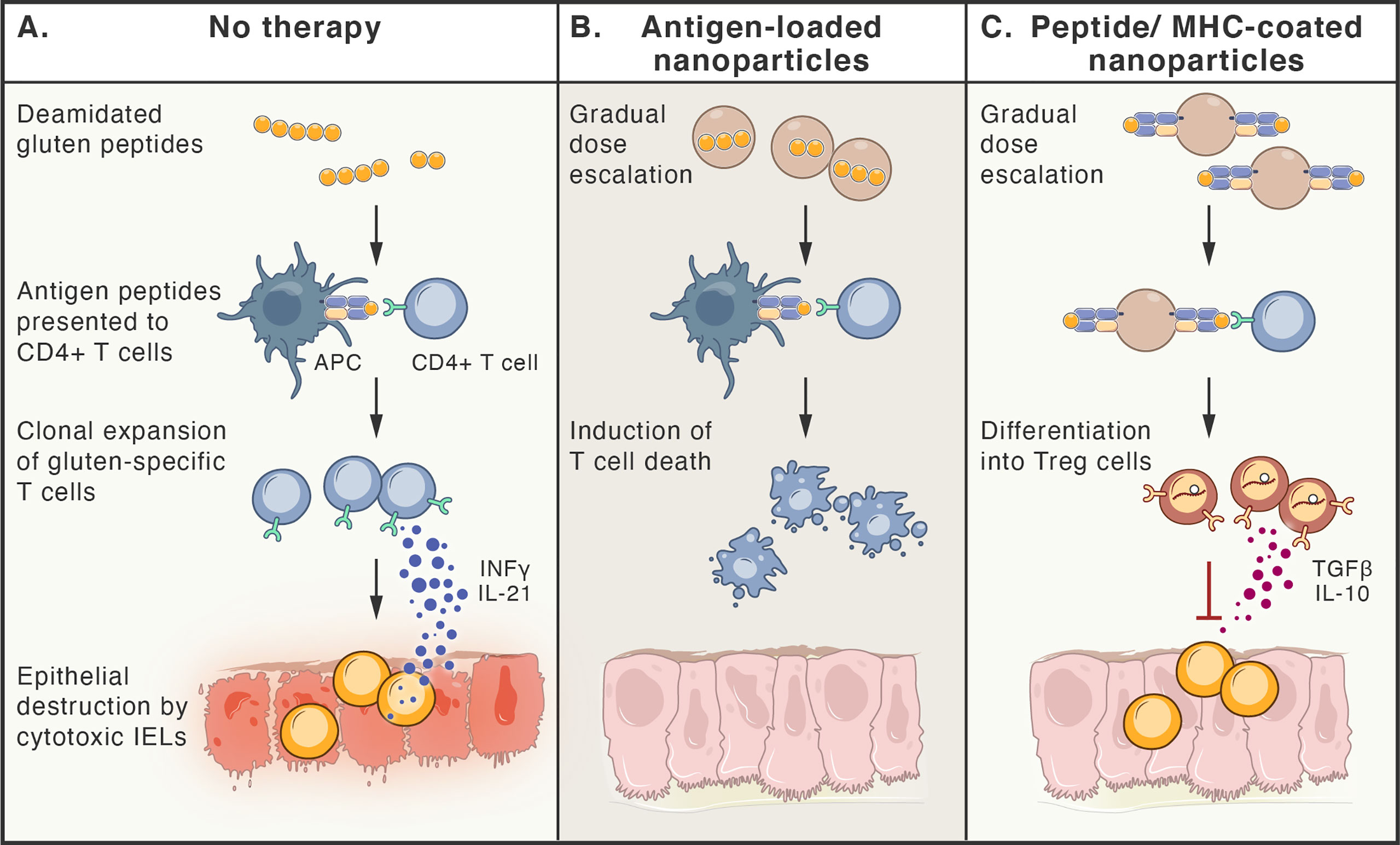
Figure 4 Antigen-specific therapies aiming at tolerance induction in celiac disease. (A). Peptides deriving from poorly digested gluten, enter lamina propria, where they are taken up by antigen presenting cells (APCs). Only HLA-DQ2 or -DQ8 molecules are able to bind antigenic gluten-deriving peptides and present them to gluten-specific CD4+T cells. These cells become activated, proliferate, release cytokines (e.g. IFN-γ and IL-21) and migrate to the lamina propria, where they induce cytotoxic function of intraepithelial lymphocytes (IELs), that kill epithelial cells. (B, C). Therapy-induced tolerance is based on the gradual dose escalation of delivered gluten peptides (e.g. released from nanoparticles or bound to MHC molecules on nanoparticles). Presentation of antigens to gluten-specific CD4+T cells in a tolerogenic manner, leads to either death of these T cell clones (B) or their differentiation to Treg cells (C), that are able to inhibit immune reaction by multiple ways, including release of TGF-β and IL-10.
Microbiota in CeD Therapy
Compliance with the gluten-free diet (GFD) is difficult (319), and thus, alternative therapies are more feasible (320). The use of probiotics has been suggested as a supplemental treatment for patients with refractory CeD (321). Specific Lactobacillus strains have been shown to reduce the immunotoxicity of gluten (322), and probiotic cocktails including L. rhamnosus have been shown to improve intestinal barrier function (323). B. breve and B. longum have been shown to have anti-inflammatory properties in children with CeD (324). In the future, findings on gluten-degrading activities by specific microorganisms may open new possibilities for a probiotics-based complementary therapy of CeD. Many clinical trials involving the use of probiotics to treat and prevent CeD are promising (325), but more studies are needed to better understand the exact mechanisms linking dysbiosis and probiotics with the onset and development of this disease. The studies comparing the fecal samples and duodenal biopsies, obtained from patients with CeD versus healthy individuals, have shown a severe alteration of gut microbiota (270, 326, 327). When patients with CeD were treated with GFD, the increased concentration of bacteria was reduced to that in the healthy population, which suggests the influence of diet on intestinal microbiota. However, in most studies only partial modulation of the microbiota was observed on the GFD (328, 329). In vitro studies to assess the use of probiotics in the treatment of CeD, have demonstrated that selected Lactobacilli strains, when added to sourdough fermentation, lyse the proline/glutamine-rich gluten peptides, reduce the gluten concentration to <10 ppm (gluten-free), and decrease their immunotoxicity (330). Four strains of Lactobacilli (L. ruminis, L. Johndoni, L. amylovorus, L. salivaris), capable of degrading and reducing the immunotoxicity of gliadin peptides, were identified from the proximal gastrointestinal tract of pigs (331). In a study including 20 patients with CeD, receiving hydrolyzed wheat gluten bread (containing Lactobacillus alimentaris, L. brevis, L. sanfranciscenis, L. Hilgardi) for six days, no significant increase in IFN-γ responses, compared to healthy controls, was found (332). In another study, that challenged CeD patients in remission for 2 months with Lactobacilli predigested gluten, no worsening of symptoms, histological structure of the small intestine or serological marker was found (333). This outcome suggests that Lactobacilli-derived peptidase was capable of completely degrading gluten and reducing its immunotoxicity in CeD (334). These studies indicate that the addition of probiotics, rich in Lactobacilli spp., may alleviate the consequences of accidental or contaminant gluten exposure (333). Dysbiosis in CeD is associated with abnormal tight junctions and increased intestinal permeability. Lindfors et al. studied the effects of probiotics on human colon cells and demonstrated that B. lactis decreased intestinal permeability, and the effect was dependent on the probiotic’s dose (272). Moreover, Bifidobacteria downregulated in vitro proinflammatory cytokines production induced by gliadins (271) or by fecal samples from patients with CeD. This suggests that Bifodobacterial strains can reverse the effects of CeD-associated microbiota (335). Lactobacillus rhamnosus GG strain decreased gliadin peptide-induced changes in intercellular junction proteins and gliadin-induced enteropathy in rats. Similar beneficial effects of probiotic B.longum CECT 7347 were observed in the small bowel of weaning animals fed gliadin. These observations may suggest that early administration of probiotics can have a protective effect on the intestinal mucosa (336). All the above-mentioned studies indicate a beneficial effect of probiotics on digestion of gliadin peptides, intestinal barrier, and immune system, as well as on intestinal mucosa (337–339).
Inflammatory Bowel Disease
IBD is a group of chronic, relapsing, and remitting inflammatory conditions that primarily affect the gastrointestinal tract (340–342). Traditionally, it has been divided into Crohn’s disease (CD), ulcerative colitis (UC), and IBD unclassified (IBDU). It begins most commonly during adolescence and young adulthood, as up to 25% of IBD patients are below 18 years of age (343–346).
The cause of IBD remains poorly understood; however, research shows the involvement of genetics, microbiome, environment, and immune system (347). The disease has a complex multifactorial etiology with genetic defects in pathways associated with the immune system, epithelial barrier, and infections (57, 347, 348). Interestingly, among numerous genetic loci associated with IBD risk, there is also the PTPN22 gene (349), one of the previously mentioned risk factors for T1DM. In contrast to diabetes, the autoimmunity-associated polymorphic variant of the PTPN22 protein seems to be protective against IBD risk, an effect associated with alterations in the gut microbiota composition in humans (350) and mouse models of colitis (351). There is an overlap in the genetic susceptibility to CD and UC, with patients frequently having subtypes of both diseases in their family history (347, 352, 353).
CD and UC have typical features that can aid diagnosis. CD can occur from the mouth to the anus, and it is patchy, transmural, granulomatous, and may have stricturing or penetrating (fistulating) features (354, 355). In addition, 20% of children with CD have perianal involvement, such as skin tags, fissures, fistulas, and abscesses (356). Conversely, UC is a disease of the colonic and rectal mucosa that usually does not lead to fibrotic strictures or perianal disease (357). IBDU, which does not fit into the other groups, is more common in children (358). Extraintestinal manifestations of IBD include dermatologic conditions, such as erythema nodosum and pyoderma gangrenosum, arthritis, growth failure, osteoporosis, and anemia. Approximately 20% of children present with extraintestinal manifestations of IBD, such as growth failure, anemia, and perianal disease, as the only initial features (359).
For IBD diagnosis, intestinal endoscopy with biopsy remains the standard (355). Examination of the stool is done for inconspicuous blood and pathogens. Fecal calprotectin, a neutrophil-derived protein with elevated concentrations during intestinal inflammation, is a useful biomarker (360, 361).
Breastfeeding is known to protect against the development of IBD, with greater benefits accruing from a longer duration of breastfeeding. In addition, high fiber and fruit intake is associated with a decreased risk for CD, and a high intake of vegetables is associated with a decreased risk for UC. Diets high in fats and meat are associated with an increased risk of IBD (95, 362, 363).
Role of Gut Microbiota in IBD
Multiple lines of evidence support the important role of the microbiota in the initiation and progression of IBD (364), highlighting reduced microbial diversity in the guts of both pediatric and adult patients (365–367).
Previous methods for pathobiont discovery in IBD patients focused on identifying intestinal bacteria bound to IgA (368, 369). Currently, nucleic acids sequencing analysis of IgG-bound microbes is considered a more clinically relevant method (370), since in IBD patients, the level of IgGs in the gut lumen is elevated (371), and IgGs seem to be more specific toward pathogens than IgAs (372). Analysis of the microbiome in pediatric IBD patients revealed increased IgG binding by invasive strains, such as Burkholderia cepacia, Flavonifractor plautii and Rumminococcus sp., while IgG binding of non-invasive Pseudomonas protogens was reduced (370). Other studies reported alterations in certain genera of the phylum Firmicutes and increased abundance of Enterobacteriaceae species in adults affected by IBD (373, 374). Some studies have also shown changes in Bacteroides spp (375). These changes are generally more pronounced in patients with CD than in patients with UC (376). Numerous other disease-specific changes in the microbiome have been recently reported in IBD patients (377, 378). Several pathogens, including S. enterica, Shigella flexneri, Yersinia enterocolitica, and V. cholerae may participate in IBD pathogenesis because they produce mucin-degrading enzymes (379–382), and they may break down the intestinal mucosal barrier that reduces the contact between microorganisms and the epithelial cell surface. In this manner, pathogens may increase the susceptibility of IBD development (383, 384). In addition, decreased numbers of SCFA-producing microorganisms, such as Clostridium spp. and Faecalibacterium prausnitzii, have been observed in patients with IBD (373, 385–387). The decreased abundance of Faecalibacterium prausnitzii in the ileum is associated with an increased risk of postoperative recurrence of ileal CD and endoscopic recurrence at six months (385). Roseburia spp. and F. prausnitzii, members of the Firmicutes phylum, are among the most beneficial microbes (385, 388). Besides producing SCFAs by fiber fermentation, these microbes also secrete several anti-inflammatory metabolites (389). SCFAs, particularly butyrate, promote the development of Treg cells and mucus production to downregulate inflammatory signaling pathways (390, 391).
Another feature of IBD is the reduction of the tryptophan metabolite, indoleacrylic acid, produced by several Peptostreptococcus spp., which also promotes the function of the mucosal barrier and reduces inflammatory responses (392, 393). Some great reviews addressing the complex interplay between important dietary factors and microbiota profiles in health and IBD have been recently published (394–396).
Although the differences in microbiota composition may indeed reflect IBD-specific changes, one must keep in mind that the microbiota composition exhibits significant heterogeneity between presumptively healthy individuals, even within the same person, when it is assessed at different time points (366, 397). This conclusion underlines the need for large cohort studies, that may differentiate between disease-dependent and –independent, as well as disease activity-dependent changes in microbiota composition. A recent large cohort longitudinal study ranking the main factors affecting microbiota variance in IBD patients listed geographic location, CD diagnosis, history of surgical resection, consumption of alcohol, presence of UC, medications taken, and dietary habits as the most significant (366). A recent machine-learning analysis uncovered an additional level of complexity in the relationship between the gut microbiome and disease activity, reporting microbiome influence on gene regulation (transcriptomic profile) of the host in large groups of patients with colorectal cancer, IBD and related Irritable Bowel Syndrome. Moreover, the regulation of different host signaling pathways by the gut microbiota seems to be disease-specific [https://doi.org/10.1101/2021.03.29.437589 and (398)].
Treatment of IBD
Current therapeutic strategies focus on treating IBD relapses and prolonging remission (340–342). For the induction of remission, exclusive enteral nutrition, corticosteroids, or anti-TNF-α antibodies (infliximab and adalimumab) are used (399, 400). Exclusive enteral nutrition, which is a nutritionally complete elemental and polymeric formula diet that contains no solid food, is the first-line option for induction of remission in pediatric CD (341), because it is as effective as corticosteroids in inducing remission, but without the side effects associated with corticosteroid therapy (401–403). Once remission has been established, it is maintained with the following agents: 5-aminosalicylate, thiopurines or anti-TNF-α antibodies. New-generation monoclonal therapies include vedolizumab and ustekinumab. Surgical intervention is often required in both UC and CD, as seen in 10% and 25% of children prior to the age of 18 years, respectively (404, 405). Unfortunately, there is no permanent cure for the disease. Medications currently available are extremely effective, but have significant potential toxicity, including increased infection risk, steroid toxicity, and increased risk of malignancy.
Since the loss of immune homeostasis due to defects in the number and suppressive function of Treg cells has been documented in IBD (406), alternative therapies employing adoptive Treg cell transfer have been explored in numerous studies using mouse models of colitis (407–410). In particular, Treg cells secreting anti-inflammatory cytokine IL-10 have been shown to partially prevent the development of the disease (411, 412), while the disruption of IL-10 expression in Treg cells, residing in the intestinal mucosa, led to the development of spontaneous colitis in mice (413). Therefore, numerous approaches for the expansion of human Treg cells in vitro, under GMP conditions, have been explored, including application of rapamycin and an agonist of retinoic acid receptor (414–416). These agents are also being used in ongoing phase I/IIa clinical trials (TRIBUTE; NCT03185000), initiated for the evaluation of therapeutic adoptive transfer of Treg cells in patients with CD. Another successful strategy included the injection of expanded Treg cells (treated with retinoic acid receptor agonist) together with recombinant IL-2. In this study, an immunodeficient mouse model of CD with subcutaneously implanted human intestine tissue was used (417). Patients-derived Treg cells were expanded in vitro and adoptively transferred to the mice, where they efficiently homed to the human small bowel. In vitro-expanded Treg cells were also effective in suppressing the function of Teff cells isolated from inflamed Crohn’s mucosa (417). The proper tissue-specific migration and gut homing of Treg cells are critical factors influencing the success of Treg cell-based therapies. Integrin α4/β7 and CCR9 protein have been identified as gut-homing receptors that can be simply induced during Treg cells expansion by addition of retinoic acid (RA) to the cell culture (418). In another interesting approach, researchers engineered dendritic cells (DCs) to produce high concentrations of both RA and 1,25-dihydroxyvitamin D (1,25(OH)2D). Since 1,25(OH)2D induces expression of FOXP3 and IL-10, while RA stimulates expression of gut-homing receptors in T cells, the approach led to the induction of Treg cells in vivo, in peripheral lymphoid tissues, and subsequent homing of Treg cells in the intestine followed by stable suppression of intestinal inflammation in a mouse model of colitis (419) (Figure 5).
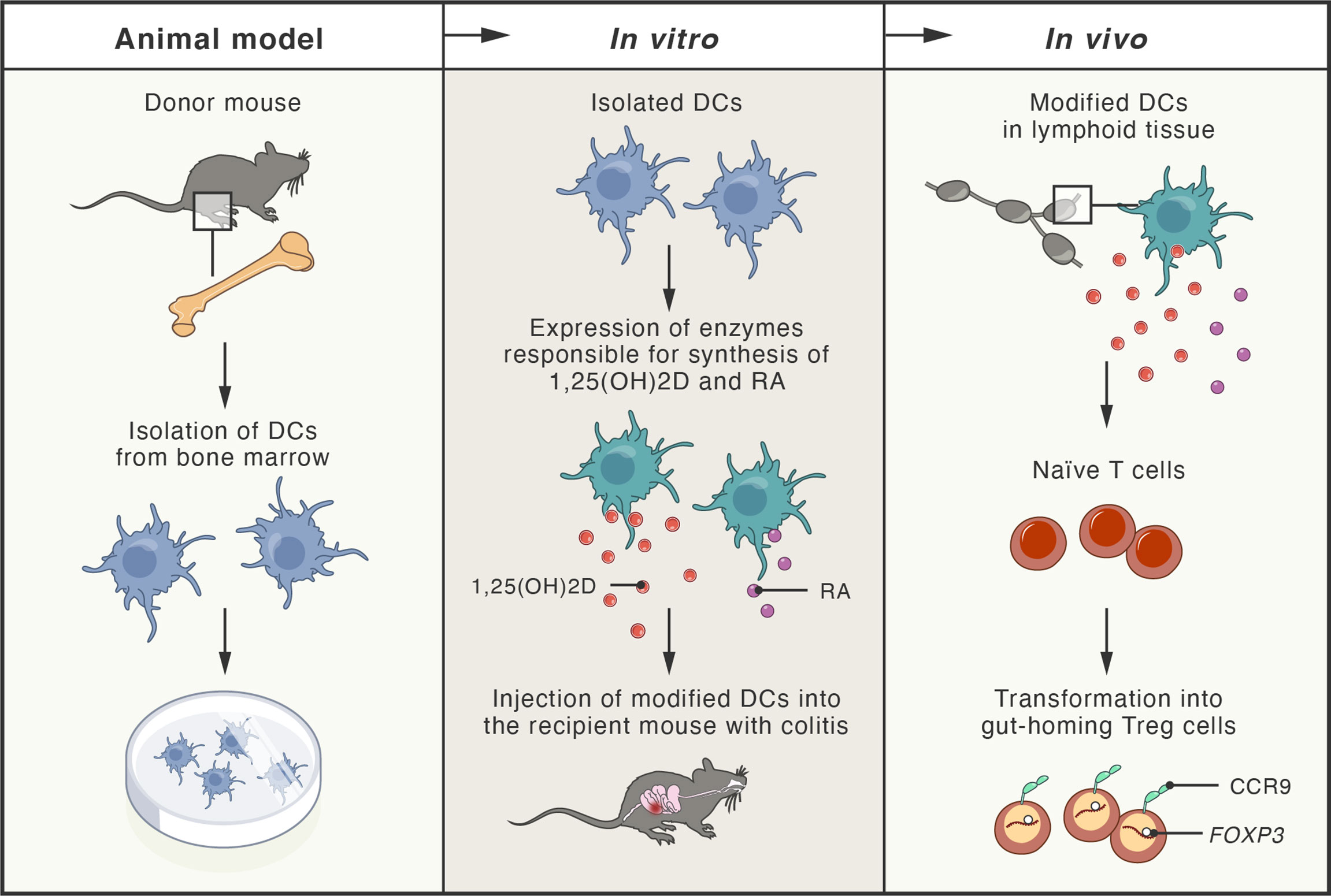
Figure 5 Schema of new therapeutic approach tested in mouse models of colitis. Dendritic cells (DCs) obtained from the bone marrow of donor mice were genetically modified to express enzymes responsible for production of 1,25-dihydroxyvitamin D (1,25(OH)2D; active vitamin D metabolite) and retinoic acid (RA; active vitamin A metabolite). Modified DCs were transferred to the recipient mice with experimentally-induced colitis. In peripheral lymphoid tissues, 1,25(OH)2D and RA (produced by the modified DCs), induced naïve T cells transformation into gut-homing regulatory T cells (Treg), by induction of FOXP3 and CCR9 (gut-homing receptor). Treg cells efficiently homed to the intestine, where they inhibited inflammation and colitis.
An important consideration in therapeutic strategies employing Treg cells is the antigen specificity of their TCRs. The polyclonal population of Treg cells, residing in the intestine, relies on the broad reactivity of its TCRs, which recognize a broad spectrum of microbiota-derived antigens, increasing the likelihood of TCR activation and proper maintenance of immune tolerance and intestinal homeostasis. Not surprisingly, the limited TCR repertoire in mice results in the development of spontaneous colitis (420). On the other hand, for the therapy of experimental colitis in mice, the antigen-specific Treg cells are more potent than polyclonal Treg cells (421, 422). An interesting approach with such antigen-specific Treg cells has been reported in phase I/IIa clinical trials for adults (CATS1 study) using Treg cells that specifically recognize a common food antigen of ovalbumin (423). The ovalbumin-enriched diet, in the form of a meringue cake, was used by the patients with refractory CD to facilitate the local activation of therapeutic Treg cells. This approach led to partial remission with mild adverse effects.
Microbiota in IBD Therapy
Administration of probiotics, including Lactobacillus or Bifidobacterium spp., has been successful in some bowel disorders (424). Oral treatment with the probiotic E. coli Nissle 1917 has been shown to maintain disease remission in patients with UC (425). Probiotic E. coli Nissle 1917 can secrete microcins with antimicrobial activity, and suppress competing Enterobacteriaceae that may exacerbate gut inflammation (426). A mixture containing eight probiotic organisms has been proven to be beneficial as an addition to standard therapy for patients with UC (427). However, at present, probiotics are not recommended in the guidelines on the management of UC because of insufficient high quality evidence (428). It has also been shown that fecal stream diversion decreases inflammation in ileal CD (429). Despite some encouraging data, FMT (described in more details in the next paragraph) remains an investigational treatment that is used only in clinical trials (430). Moreover, antibiotics can induce remission and prevent relapse in patients with IBD (431, 432). However, despite the benefits of antibiotics for the treatment of CD and UC, their use in modifying the microbiota is limited by their inability to selectively eliminate pathogenic bacteria without affecting beneficial microorganisms, particularly with prolonged or repeated courses (433, 434). Currently, antibiotics are recommended to treat diseases complicated by infection (abscesses, bacterial overgrowth, Clostridium difficile) or perianal fistulising disease (428). Exclusive enteral nutrition, which is the dietary intervention used in CD, quickly alters the microbiota composition and effectively reduces intestinal inflammation in pediatric patients (435). The mechanism by which this diet induces remission in CD remains unclear, but it may promote the growth of beneficial microorganisms or the depletion of pathobionts. Unfortunately, there is still a lack of knowledge about the identity of IBD-causing pathogens that trigger inflammation in genetically susceptible individuals.
The outcome of new therapeutic strategies, such as adoptive therapy with Treg cells, is likely to be modulated by the intestinal microbiota, which represents a substantial antigen load in the gastrointestinal tract. In healthy individuals, Treg cells residing in the intestinal lamina propria provide tolerance towards the gut microbiota and their suppressive capacity is enhanced during intestinal inflammation (436, 437).
Controversies and Current Research Gaps in Modulation of Microbiota Composition and Potential Future Developments in This Field
Recent advances in microbial genomic sequencing and other biology techniques allow for novel insights into the potential contribution of the gut microbiota to health and diseases (438). Consequently, changes in the composition and functionality of the gut microbiota have been found in an increasing number of diseases (439–442). However, it remains unclear whether dysbiosis is a cause, a consequence, or incidental to the disease. FMT is currently gaining increasing clinical and research importance. At present, FMT is recommended to treat recurrent Clostridioides difficile infection (CDI), but there is a growing number of ongoing trials exploring its other potential therapeutic indications (443). One of them is IBD. An altered microbiome has been described as one of the factors contributing to the pathogenesis of IBD, but it is still unclear whether this is a cause or effect of the gut inflammation in Crohn’s disease (CD) and ulcerative colitis (UC) (433, 444). The safety and efficacy of FMT in patients with both IBD and CDI have been assessed prospectively in the NCT03106844 study. Currently, the research continues to advance toward exploring this treatment for IBD. Four randomized controlled trials, which assessed the use of FMT in UC, have been published, and in three of them a significantly increased rate of both clinical and endoscopic remission in UC patients, receiving FMT compared to those receiving placebo, were reported (445–448). In a recent Cochrane systematic review, an overall remission rate at week 8, across these four studies was 37% (n=52/140) in patients receiving FMT, compared to 18% (n=24/137) in those receiving placebo (relative risk 2.03; 95% confidence interval: 1.07–3.86) (449). The highest rates of steroid-free response and remission were reported in the single study that used anaerobic conditions for FMT preparation, suggesting that this can be a relevant factor. One study demonstrated better outcomes with one donor of the stool than with other donors. This observation may indicate that donor selection may be much more important in UC than in CDI. However, microbial characteristics for an optimal donor in IBD have not been well defined, and the hypotheses mentioned above require further studies. Since the role of the gut microbiota in the pathogenesis of UC is under intense investigation, a future study will focus on exploring whether successful donors have a gut microbiota particularly enriched in specific microbial strains that are absent in the gut in UC. Overall, the existing evidence suggests that FMT may be potentially useful for treating mild to moderate UC. When it comes to CD, only studies on relatively small adult and pediatric cohorts have been published. A recent meta-analysis of eleven studies, including four case reports and seven cohort studies, reported an overall 50.5% (n=42/83) rate of clinical remission in CD patients receiving FMT (450). However, the heterogeneity of disease activity and FMT administration protocols of the included studies limited the drawing of conclusions. A double-blind randomized controlled trial, evaluating the efficacy of FMT in adults with CD is ongoing (NCT03078803).
The FMT can also be considered a potential therapeutic approach in T1DM. The composition of the gut microbiota in T1DM-affected children or individuals at high risk of developing T1DM is different from that in healthy individuals (138, 146, 451–453). Additionally, in many cases, changes in microbiota composition have been detected before the first symptoms of T1DM (146, 452), suggesting a functional association between changes in microbiota and the disease onset. In a recent trial (NTR3697), the positive health effects of FMT in ten young patients (18-30 years old) with new-onset T1DM were detected (454), providing a basis for future clinical trials. On the other hand, standardization of transferred microorganisms used in the FMT approach may be necessary, and further profiling of microbiota composition in “effective” donors is urgently needed. The data published so far indicate that adverse events of FMT occur in about 20% of patients, however, most of them are mild (455). Only one case of serious adverse events, myasthenia gravis, has been reported (456). According to current ECCO/ESPGHAN guidelines for the medical management of pediatric CD, both probiotics and FMT are neither recommended for induction nor remission maintenance (457). It seems that in contrast to CDI, frequent transplantation is necessary for FMT to be effective for any chronic disorder. It has been reported that improved microbial diversity can persist for several weeks, but does not persist after 1 year (448, 458). The pathophysiology of chronic illness is very complex and will likely require more long-term FMT or controlling inflammation in the recipient intestine to facilitate the engraftment [broadly reviewed in Ref (459)].
Discussion
Chronic inflammatory disorders are often managed with symptomatic therapy, and eventually lead to multiple downstream sequelae (460–462). Despite many similarities between this group of diseases in children and adults, pediatric onset often presents with atypical features (460, 463, 464). Pediatric patients are particularly vulnerable to adverse effects of drugs because the number of clinical trials in this group of patients is limited, and drug absorption and metabolism are more variable and less predictable (465, 466). An additional challenge in pediatric care is achieving normal growth, puberty, and access to education. The impact of drugs on child growth and development is still not fully understood. Further studies are needed to better understand the etiology and pathogenesis of these inflammatory disorders, and to identify preventive strategies, alternative or complementary therapies, and prevent complications. The interaction between genetic and environmental factors is crucial in the development of chronic inflammatory disorders (467). Continued research on the specific role played by the gut microbiota and the complex interactions between microorganisms and the host will help to address these issues better (468). Data on the importance of the interactions between microbiota and the human immune system, the crosstalk between various species of microbes, and details on the communications between various organs and their microbiota is still lacking (469, 470).
Some novel technologies employing genetic engineering of immune cells, as well as adoptive transfer of modified patient immune cells to treat the disease, are being extensively tested in laboratories and clinical trials (471–473). The therapies involving engineered Treg cells that are expected to specifically suppress the proliferation and production of inflammatory cytokines by cytotoxic Teff cells in the pancreas and stop the destruction of pancreatic islet cells, or at least slow the progression of T1DM (201), are particularly promising. However, several considerations should be kept in mind when using these therapies, including the antigen specificity of the transferred engineered immune cells, and their interactions with the gut microbiota that may affect the therapy outcome in multiple ways (474, 475). Advances in scientific research must always be seen in the context of clinical care, outcomes, and prognosis for children with diseases, such as T1DM, CeD, and IBD.
The gut microbiota should be certainly considered as one of the potential factors that can modulate the responses to immunotherapies. The outcome of new therapeutic strategies, such as adoptive therapy with Treg cells, is likely to be modulated by the intestinal microbiota, which represents a substantial antigen load in the gastrointestinal tract. In healthy individuals, Treg cells residing in the intestinal lamina propria provide tolerance towards the gut microbiota and their suppressive capacity is enhanced during intestinal inflammation (436, 437). However, since adoptive cell transfer is a relatively novel therapy for autoimmune diseases, currently, little is known about the influence of gut microbiota on the outcome of adoptive therapy with Treg cells. On the other hand, progress has recently been made in understanding the role of gut microbiota in modulating CAR-T cell outcome in cancer therapy and demonstrated that gut microbiota can influence the balance between activity of effector CAR-T cells and suppressive activity of Treg cells [reviewed in Ref (476)]. By analogy, since the predominant function of adoptively-transferred Treg cells in therapy of autoimmune diseases is to control activity of autoreactive effector T cells, one can expect that the gut microbiota may also influence this process.
Concluding Remarks
Undoubtedly, the microbiota significantly influences health status. Although the origin of diseases described in this review differ, an improper balance in microbiota content seems to be common to the progression of all of them. The inflammation behind untolerated or an altered composition of the microbiome is currently attributed to the syndromes closely associated with the alimentary tract but also with many other diseases. The discovery of the interaction between saprophytic bacteria and our tissues has increased our understanding of the pathogenesis of many diseases. It was once overwhelming to think that the gut bacteria might contribute so heavily to our daily well-being. Fortunately, this knowledge allows us to modulate the microbiome, which has become a novel way to treat many diseases. The awareness that the microbiome influences us continuously is also acknowledged with many novel therapies, and notably in those that are disease-modifying. The microbiome as a factor affecting the efficacy of biologics or cell therapies is now considered a good and cheap way to improve such responses. Bacteria have been our commensals for a long time, and the reciprocal benefits of this symbiotic relationship should not be a surprise. Further exploration of the role of the microbiome in our life will definitely give us many novel tools for disease treatment.
Author Contributions
All authors participated in manuscript preparation, including text (AT, BP, JK, PT, AH) and figures (AH, AT, BP) preparation. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Science Centre (NCN, Poland) with grant no: 2016/23/B/NZ5/02622.
Conflict of Interest
PT is co-inventor of patents related to presented content and stakeholder of POLTREG venture (company commercializing therapy based on Treg cells). Medical University of Gdansk received payments and royalties for the license to presented content.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing.
Abbreviations
APC, antigen-presenting cell; CAR, chimeric antigen receptor; CD, Crohn’s disease; CDI, Clostridioides difficile infection; CeD, celiac disease; DCs, dendritic cells; DGP, deamidated gliadin peptide; EmA, anti-endomysium antibodies; FMT, fecal microbiota transplantation; GF, germ-free; GFD, gluten-free diet; GMP, good manufacturing practice; GALT, gut-associated lymphoid tissue; HLA, human leukocyte antigen; IBD, Inflammatory bowel disease; IBDU, IBD unclassified; Ig, immunoglobulin; IFN-γ, interferon-γ; IL, interleukin; NK, natural killer; RA, retinoic acid; SCFA, short-chain fatty acid; T1DM, type 1 diabetes mellitus; TCR, T-cell receptor; Teff, effector T cells; TNF-α, tumor necrosis factor-α; Treg, regulatory T cells; tTG, tissue transglutaminase; UC, ulcerative colitis.
References
1. Paludan SR, Pradeu T, Masters SL, Mogensen TH. Constitutive Immune Mechanisms: Mediators of Host Defence and Immune Regulation. Nat Rev Immunol (2021) 21(3):137–50. doi: 10.1038/s41577-020-0391-5
2. Cheng M, Anderson MS. Thymic Tolerance as a Key Brake on Autoimmunity. Nat Immunol (2018) 19(7):659–64. doi: 10.1038/s41590-018-0128-9
3. Netea MG, Balkwill F, Chonchol M, Cominelli F, Donath MY, Giamarellos-Bourboulis EJ, et al. A Guiding Map for Inflammation. Nat Immunol (2017) 18(8):826–31. doi: 10.1038/ni.3790
4. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory Responses and Inflammation-Associated Diseases in Organs. Oncotarget (2018) 9(6):7204–18. doi: 10.18632/oncotarget.23208
6. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic Inflammation in the Etiology of Disease Across the Life Span. Nat Med (2019) 25(12):1822–32. doi: 10.1038/s41591-019-0675-0
7. Na YR, Stakenborg M, Seok SH, Matteoli G. Macrophages in Intestinal Inflammation and Resolution: A Potential Therapeutic Target in IBD. Nat Rev Gastroenterol Hepatol (2019) 16(9):531–43. doi: 10.1038/s41575-019-0172-4
8. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat Rev Immunol (2020) 20(1):7–24. doi: 10.1038/s41577-019-0210-z
9. Gaudino SJ, Kumar P. Cross-Talk Between Antigen Presenting Cells and T Cells Impacts Intestinal Homeostasis, Bacterial Infections, and Tumorigenesis. Front Immunol (2019) 10:360. doi: 10.3389/fimmu.2019.00360
10. Soehnlein O, Steffens S, Hidalgo A, Weber C. Neutrophils as Protagonists and Targets in Chronic Inflammation. Nat Rev Immunol (2017) 17(4):248–61. doi: 10.1038/nri.2017.10
11. Ng LG, Ostuni R, Hidalgo A. Heterogeneity of Neutrophils. Nat Rev Immunol (2019) 19(4):255–65. doi: 10.1038/s41577-019-0141-8
12. Fang P, Li X, Dai J, Cole L, Camacho JA, Zhang Y, et al. Immune Cell Subset Differentiation and Tissue Inflammation. J Hematol Oncol (2018) 11(1):97. doi: 10.1186/s13045-018-0637-x
13. Hisamatsu T, Erben U, Kuhl AA. The Role of T-Cell Subsets in Chronic Inflammation in Celiac Disease and Inflammatory Bowel Disease Patients: More Common Mechanisms or More Differences? Inflammation Intest Dis (2016) 1(2):52–62. doi: 10.1159/000445133
14. Duan L, Rao X, Sigdel KR. Regulation of Inflammation in Autoimmune Disease. J Immunol Res (2019) 2019:7403796. doi: 10.1155/2019/7403796
15. Fujinami RS, von Herrath MG, Christen U, Whitton JL. Molecular Mimicry, Bystander Activation, or Viral Persistence: Infections and Autoimmune Disease. Clin Microbiol Rev (2006) 19(1):80–94. doi: 10.1128/CMR.19.1.80-94.2006
16. Rojas M, Restrepo-Jimenez P, Monsalve DM, Pacheco Y, Acosta-Ampudia Y, Ramirez-Santana C, et al. Molecular Mimicry and Autoimmunity. J Autoimmun (2018) 95:100–23. doi: 10.1016/j.jaut.2018.10.012
17. Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T. Regulatory T Cells: How do They Suppress Immune Responses? Int Immunol (2009) 21(10):1105–11. doi: 10.1093/intimm/dxp095
18. Bluestone JA, Bour-Jordan H, Cheng M, Anderson M. T Cells in the Control of Organ-Specific Autoimmunity. J Clin Invest (2015) 125(6):2250–60. doi: 10.1172/JCI78089
19. Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front Immunol (2019) 10:43. doi: 10.3389/fimmu.2019.00043
20. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature (2010) 464(7285):59–65. doi: 10.1038/nature08821
21. Sommer F, Backhed F. The Gut Microbiota–Masters of Host Development and Physiology. Nat Rev Microbiol (2013) 11(4):227–38. doi: 10.1038/nrmicro2974
22. Bibbo S, Ianiro G, Giorgio V, Scaldaferri F, Masucci L, Gasbarrini A, et al. The Role of Diet on Gut Microbiota Composition. Eur Rev Med Pharmacol Sci (2016) 20(22):4742–9.
23. Ducarmon QR, Zwittink RD, Hornung BVH, van Schaik W, Young VB, Kuijper EJ. Gut Microbiota and Colonization Resistance Against Bacterial Enteric Infection. Microbiol Mol Biol Rev (2019) 83(3):e00007–19. doi: 10.1128/MMBR.00007-19
24. Odenwald MA, Turner JR. The Intestinal Epithelial Barrier: A Therapeutic Target? Nat Rev Gastroenterol Hepatol (2017) 14(1):9–21. doi: 10.1038/nrgastro.2016.169
25. Woo V, Alenghat T. Host-Microbiota Interactions: Epigenomic Regulation. Curr Opin Immunol (2017) 44:52–60. doi: 10.1016/j.coi.2016.12.001
26. Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, et al. Bifidobacteria Can Protect From Enteropathogenic Infection Through Production of Acetate. Nature (2011) 469(7331):543–7. doi: 10.1038/nature09646
27. Sun M, Wu W, Liu Z, Cong Y. Microbiota Metabolite Short Chain Fatty Acids, GPCR, and Inflammatory Bowel Diseases. J Gastroenterol (2017) 52(1):1–8. doi: 10.1007/s00535-016-1242-9
28. Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R. Current Understanding of the Human Microbiome. Nat Med (2018) 24(4):392–400. doi: 10.1038/nm.4517
29. Gibiino G, Ianiro G, Cammarota G, Gasbarrini A. The Gut Microbiota: Its Anatomy and Physiology Over a Lifetime. Minerva Gastroenterol Dietol (2017) 63(4):329–36. doi: 10.23736/S1121-421X.17.02405-9
30. Rinninella E, Raoul P, Cintoni M, Franceschi F, Miggiano GAD, Gasbarrini A, et al. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem Across Age, Environment, Diet, and Diseases. Microorganisms (2019) 7(1):14. doi: 10.3390/microorganisms7010014
31. Ma B, McComb E, Gajer P, Yang H, Humphrys M, Okogbule-Wonodi AC, et al. Microbial Biomarkers of Intestinal Barrier Maturation in Preterm Infants. Front Microbiol (2018) 9:2755. doi: 10.3389/fmicb.2018.02755
32. Suzuki K, Ha SA, Tsuji M, Fagarasan S. Intestinal IgA Synthesis: A Primitive Form of Adaptive Immunity That Regulates Microbial Communities in the Gut. Semin Immunol (2007) 19(2):127–35. doi: 10.1016/j.smim.2006.10.001
33. Williams AM, Probert CS, Stepankova R, Tlaskalova-Hogenova H, Phillips A, Bland PW. Effects of Microflora on the Neonatal Development of Gut Mucosal T Cells and Myeloid Cells in the Mouse. Immunology (2006) 119(4):470–8. doi: 10.1111/j.1365-2567.2006.02458.x
34. Hill CJ, Lynch DB, Murphy K, Ulaszewska M, Jeffery IB, O’Shea CA, et al. Evolution of Gut Microbiota Composition From Birth to 24 Weeks in the INFANTMET Cohort. Microbiome (2017) 5(1):4. doi: 10.1186/s40168-017-0240-3
35. Rodriguez JM, Murphy K, Stanton C, Ross RP, Kober OI, Juge N, et al. The Composition of the Gut Microbiota Throughout Life, With an Emphasis on Early Life. Microb Ecol Health Dis (2015) 26:26050. doi: 10.3402/mehd.v26.26050
36. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human Gut Microbiome Viewed Across Age and Geography. Nature (2012) 486(7402):222–7. doi: 10.1038/nature11053
37. Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human Genetics Shape the Gut Microbiome. Cell (2014) 159(4):789–99. doi: 10.1016/j.cell.2014.09.053
38. Van den Abbeele P, Belzer C, Goossens M, Kleerebezem M, De Vos WM, Thas O, et al. Butyrate-Producing Clostridium Cluster XIVa Species Specifically Colonize Mucins in an In Vitro Gut Model. ISME J (2013) 7(5):949–61. doi: 10.1038/ismej.2012.158
39. Jandhyala SM, Talukdar R, Subramanyam C, Vuyyuru H, Sasikala M, Nageshwar Reddy D. Role of the Normal Gut Microbiota. World J Gastroenterol (2015) 21(29):8787–803. doi: 10.3748/wjg.v21.i29.8787
40. Mariat D, Firmesse O, Levenez F, Guimaraes V, Sokol H, Dore J, et al. The Firmicutes/Bacteroidetes Ratio of the Human Microbiota Changes With Age. BMC Microbiol (2009) 9:123. doi: 10.1186/1471-2180-9-123
41. Conlon MA, Bird AR. The Impact of Diet and Lifestyle on Gut Microbiota and Human Health. Nutrients (2014) 7(1):17–44. doi: 10.3390/nu7010017
42. Radjabzadeh D, Boer CG, Beth SA, van der Wal P, Kiefte-De Jong JC, Jansen MAE, et al. Diversity, Compositional and Functional Differences Between Gut Microbiota of Children and Adults. Sci Rep (2020) 10(1):1040. doi: 10.1038/s41598-020-57734-z
43. Hills RD Jr., Pontefract BA, Mishcon HR, Black CA, Sutton SC, Theberge CR. Gut Microbiome: Profound Implications for Diet and Disease. Nutrients (2019) 11(7):1613. doi: 10.3390/nu11071613
44. Sirisinha S. Insight Into the Mechanisms Regulating Immune Homeostasis in Health and Disease. Asian Pac J Allergy Immunol (2011) 29(1):1–14.
45. Peterson DA, McNulty NP, Guruge JL, Gordon JI. IgA Response to Symbiotic Bacteria as a Mediator of Gut Homeostasis. Cell Host Microbe (2007) 2(5):328–39. doi: 10.1016/j.chom.2007.09.013
46. Koch MA, Reiner GL, Lugo KA, Kreuk LS, Stanbery AG, Ansaldo E, et al. Maternal IgG and IgA Antibodies Dampen Mucosal T Helper Cell Responses in Early Life. Cell (2016) 165(4):827–41. doi: 10.1016/j.cell.2016.04.055
47. Sait LC, Galic M, Price JD, Simpfendorfer KR, Diavatopoulos DA, Uren TK, et al. Secretory Antibodies Reduce Systemic Antibody Responses Against the Gastrointestinal Commensal Flora. Int Immunol (2007) 19(3):257–65. doi: 10.1093/intimm/dxl142
48. Pabst O. New Concepts in the Generation and Functions of Iga. Nat Rev Immunol (2012) 12(12):821–32. doi: 10.1038/nri3322
49. Castro-Dopico T, Clatworthy MR. Igg and Fcgamma Receptors in Intestinal Immunity and Inflammation. Front Immunol (2019) 10:805. doi: 10.3389/fimmu.2019.00805
50. Gensollen T, Iyer SS, Kasper DL, Blumberg RS. How Colonization by Microbiota in Early Life Shapes the Immune System. Science (2016) 352(6285):539–44. doi: 10.1126/science.aad9378
51. Laforest-Lapointe I, Arrieta MC. Patterns of Early-Life Gut Microbial Colonization During Human Immune Development: An Ecological Perspective. Front Immunol (2017) 8:788. doi: 10.3389/fimmu.2017.00788
52. Al Nabhani Z, Eberl G. Imprinting of the Immune System by the Microbiota Early in Life. Mucosal Immunol (2020) 13(2):183–9. doi: 10.1038/s41385-020-0257-y
53. Blaser MJ. The Theory of Disappearing Microbiota and the Epidemics of Chronic Diseases. Nat Rev Immunol (2017) 17(8):461–3. doi: 10.1038/nri.2017.77
54. Kostic AD, Gevers D, Siljander H, Vatanen T, Hyotylainen T, Hamalainen AM, et al. The Dynamics of the Human Infant Gut Microbiome in Development and in Progression Toward Type 1 Diabetes. Cell Host Microbe (2015) 17(2):260–73. doi: 10.1016/j.chom.2015.01.001
55. Vatanen T, Kostic AD, d’Hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in Microbiome Lps Immunogenicity Contributes to Autoimmunity in Humans. Cell (2016) 165(4):842–53. doi: 10.1016/j.cell.2016.04.007
56. Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ. Dysbiosis of the Gut Microbiota in Disease. Microb Ecol Health Dis (2015) 26:26191. doi: 10.3402/mehd.v26.26191
57. Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, et al. The Treatment-Naive Microbiome in New-Onset Crohn’s Disease. Cell Host Microbe (2014) 15(3):382–92. doi: 10.1016/j.chom.2014.02.005
58. Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, et al. Gut Immune Maturation Depends on Colonization With a Host-Specific Microbiota. Cell (2012) 149(7):1578–93. doi: 10.1016/j.cell.2012.04.037
59. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of Colonic Regulatory T Cells by Indigenous Clostridium Species. Science (2011) 331(6015):337–41. doi: 10.1126/science.1198469
60. Atarashi K, Umesaki Y, Honda K. Microbiotal Influence on T Cell Subset Development. Semin Immunol (2011) 23(2):146–53. doi: 10.1016/j.smim.2011.01.010
61. Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, et al. Mucosal Immunology. Individual Intestinal Symbionts Induce a Distinct Population of RORgamma(+) Regulatory T Cells. Science (2015) 349(6251):993–7. doi: 10.1126/science.aaa9420
62. Hapfelmeier S, Lawson MA, Slack E, Kirundi JK, Stoel M, Heikenwalder M, et al. Reversible Microbial Colonization of Germ-Free Mice Reveals the Dynamics of IgA Immune Responses. Science (2010) 328(5986):1705–9. doi: 10.1126/science.1188454
63. Al Nabhani Z, Dulauroy S, Marques R, Cousu C, Al Bounny S, Dejardin F, et al. A Weaning Reaction to Microbiota is Required for Resistance to Immunopathologies in the Adult. Immunity (2019) 50(5):1276–88.e5. doi: 10.1016/j.immuni.2019.02.014
64. Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, et al. Microbial Exposure During Early Life has Persistent Effects on Natural Killer T Cell Function. Science (2012) 336(6080):489–93. doi: 10.1126/science.1219328
65. An D, Oh SF, Olszak T, Neves JF, Avci FY, Erturk-Hasdemir D, et al. Sphingolipids From a Symbiotic Microbe Regulate Homeostasis of Host Intestinal Natural Killer T Cells. Cell (2014) 156(1-2):123–33. doi: 10.1016/j.cell.2013.11.042
66. Walker WA. Initial Intestinal Colonization in the Human Infant and Immune Homeostasis. Ann Nutr Metab (2013) 63(Suppl 2):8–15. doi: 10.1159/000354907
67. Stewart CJ, Ajami NJ, O’Brien JL, Hutchinson DS, Smith DP, Wong MC, et al. Temporal Development of the Gut Microbiome in Early Childhood From the TEDDY Study. Nature (2018) 562(7728):583–8. doi: 10.1038/s41586-018-0617-x
68. Azad MB, Konya T, Maughan H, Guttman DS, Field CJ, Chari RS, et al. Gut Microbiota of Healthy Canadian Infants: Profiles by Mode of Delivery and Infant Diet at 4 Months. CMAJ (2013) 185(5):385–94. doi: 10.1503/cmaj.121189
69. Jakobsson HE, Abrahamsson TR, Jenmalm MC, Harris K, Quince C, Jernberg C, et al. Decreased Gut Microbiota Diversity, Delayed Bacteroidetes Colonisation and Reduced Th1 Responses in Infants Delivered by Caesarean Section. Gut (2014) 63(4):559–66. doi: 10.1136/gutjnl-2012-303249
70. Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery Mode Shapes the Acquisition and Structure of the Initial Microbiota Across Multiple Body Habitats in Newborns. Proc Natl Acad Sci USA (2010) 107(26):11971–5. doi: 10.1073/pnas.1002601107
71. Pandey PK, Verma P, Kumar H, Bavdekar A, Patole MS, Shouche YS. Comparative Analysis of Fecal Microflora of Healthy Full-Term Indian Infants Born With Different Methods of Delivery (Vaginal vs Cesarean): Acinetobacter Sp. Prevalence in Vaginally Born Infants. J Biosci (2012) 37(6):989–98. doi: 10.1007/s12038-012-9268-5
72. Dogra S, Sakwinska O, Soh SE, Ngom-Bru C, Bruck WM, Berger B, et al. Dynamics of Infant Gut Microbiota are Influenced by Delivery Mode and Gestational Duration and Are Associated With Subsequent Adiposity. mBio (2015) 6(1):e02419–14. doi: 10.1128/mBio.02419-14
73. Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, et al. Factors Influencing the Composition of the Intestinal Microbiota in Early Infancy. Pediatrics (2006) 118(2):511–21. doi: 10.1542/peds.2005-2824
74. Mosca F, Gianni ML. Human Milk: Composition and Health Benefits. Pediatr Med Chir (2017) 39(2):155. doi: 10.4081/pmc.2017.155
75. Patki S, Patki U, Patil R, Indumathi S, Kaingade P, Bulbule A, et al. Comparison of the Levels of the Growth Factors in Umbilical Cord Serum and Human Milk and Its Clinical Significance. Cytokine (2012) 59(2):305–8. doi: 10.1016/j.cyto.2012.04.010
76. Parigi SM, Eldh M, Larssen P, Gabrielsson S, Villablanca EJ. Breast Milk and Solid Food Shaping Intestinal Immunity. Front Immunol (2015) 6:415. doi: 10.3389/fimmu.2015.00415
77. Moossavi S, Miliku K, Sepehri S, Khafipour E, Azad MB. The Prebiotic and Probiotic Properties of Human Milk: Implications for Infant Immune Development and Pediatric Asthma. Front Pediatr (2018) 6:197. doi: 10.3389/fped.2018.00197
78. Rogier EW, Frantz AL, Bruno ME, Wedlund L, Cohen DA, Stromberg AJ, et al. Lessons From Mother: Long-Term Impact of Antibodies in Breast Milk on the Gut Microbiota and Intestinal Immune System of Breastfed Offspring. Gut Microbes (2014) 5(5):663–8. doi: 10.4161/19490976.2014.969984
79. Aakko J, Kumar H, Rautava S, Wise A, Autran C, Bode L, et al. Human Milk Oligosaccharide Categories Define the Microbiota Composition in Human Colostrum. Benef Microbes (2017) 8(4):563–7. doi: 10.3920/BM2016.0185
80. Coppa GV, Zampini L, Galeazzi T, Facinelli B, Ferrante L, Capretti R, et al. Human Milk Oligosaccharides Inhibit the Adhesion to Caco-2 Cells of Diarrheal Pathogens: Escherichia Coli, Vibrio Cholerae, and Salmonella Fyris. Pediatr Res (2006) 59(3):377–82. doi: 10.1203/01.pdr.0000200805.45593.17
81. Gabrielli O, Zampini L, Galeazzi T, Padella L, Santoro L, Peila C, et al. Preterm Milk Oligosaccharides During the First Month of Lactation. Pediatrics (2011) 128(6):e1520–31. doi: 10.1542/peds.2011-1206
82. Jin YT, Duan Y, Deng XK, Lin J. Prevention of Necrotizing Enterocolitis in Premature Infants - An Updated Review. World J Clin Pediatr (2019) 8(2):23–32. doi: 10.5409/wjcp.v8.i2.23
83. Fox TP, Godavitarne C. What Really Causes Necrotising Enterocolitis? ISRN Gastroenterol (2012) 2012:628317. doi: 10.5402/2012/628317
84. Duijts L, Jaddoe VW, Hofman A, Moll HA. Prolonged and Exclusive Breastfeeding Reduces the Risk of Infectious Diseases in Infancy. Pediatrics (2010) 126(1):e18–25. doi: 10.1542/peds.2008-3256
85. Blaser MJ. Antibiotic Use and its Consequences for the Normal Microbiome. Science (2016) 352(6285):544–5. doi: 10.1126/science.aad9358
86. Keeney KM, Yurist-Doutsch S, Arrieta MC, Finlay BB. Effects of Antibiotics on Human Microbiota and Subsequent Disease. Annu Rev Microbiol (2014) 68:217–35. doi: 10.1146/annurev-micro-091313-103456
87. Dethlefsen L, Huse S, Sogin ML, Relman DA. The Pervasive Effects of an Antibiotic on the Human Gut Microbiota, as Revealed by Deep 16S rRNA Sequencing. PloS Biol (2008) 6(11):e280. doi: 10.1371/journal.pbio.0060280
88. Jernberg C, Lofmark S, Edlund C, Jansson JK. Long-Term Ecological Impacts of Antibiotic Administration on the Human Intestinal Microbiota. ISME J (2007) 1(1):56–66. doi: 10.1038/ismej.2007.3
89. Jakobsson HE, Jernberg C, Andersson AF, Sjolund-Karlsson M, Jansson JK, Engstrand L. Short-Term Antibiotic Treatment has Differing Long-Term Impacts on the Human Throat and Gut Microbiome. PloS One (2010) 5(3):e9836. doi: 10.1371/journal.pone.0009836
90. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide Incidence and Prevalence of Inflammatory Bowel Disease in the 21st Century: A Systematic Review of Population-Based Studies. Lancet (2018) 390(10114):2769–78. doi: 10.1016/S0140-6736(17)32448-0
91. Bach JF, Chatenoud L. The Hygiene Hypothesis: An Explanation for the Increased Frequency of Insulin-Dependent Diabetes. Cold Spring Harb Perspect Med (2012) 2(2):a007799. doi: 10.1101/cshperspect.a007799
92. Itoh A, Ridgway WM. Targeting Innate Immunity to Downmodulate Adaptive Immunity and Reverse Type 1 Diabetes. Immunotargets Ther (2017) 6:31–8. doi: 10.2147/ITT.S117264
93. Quigley EMM, Gajula P. Recent Advances in Modulating the Microbiome. F1000Res (2020) 9:46. doi: 10.12688/f1000research.20204.1
94. Zhang N, Ju Z, Zuo T. Time for Food: The Impact of Diet on Gut Microbiota and Human Health. Nutrition (2018) 51-52:80–5. doi: 10.1016/j.nut.2017.12.005
95. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet Rapidly and Reproducibly Alters the Human Gut Microbiome. Nature (2014) 505(7484):559–63. doi: 10.1038/nature12820
96. Gibson GR, Hutkins R, Sanders ME, Prescott SL, Reimer RA, Salminen SJ, et al. Expert Consensus Document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) Consensus Statement on the Definition and Scope of Prebiotics. Nat Rev Gastroenterol Hepatol (2017) 14(8):491–502. doi: 10.1038/nrgastro.2017.75
97. Roberfroid MB. Prebiotics: Preferential Substrates for Specific Germs? Am J Clin Nutr (2001) 73(2 Suppl):406S–9S. doi: 10.1093/ajcn/73.2.406s
98. Goncalves P, Araujo JR, Di Santo JP. A Cross-Talk Between Microbiota-Derived Short-Chain Fatty Acids and the Host Mucosal Immune System Regulates Intestinal Homeostasis and Inflammatory Bowel Disease. Inflammation Bowel Dis (2018) 24(3):558–72. doi: 10.1093/ibd/izx029
99. Gareau MG, Sherman PM, Walker WA. Probiotics and the Gut Microbiota in Intestinal Health and Disease. Nat Rev Gastroenterol Hepatol (2010) 7(9):503–14. doi: 10.1038/nrgastro.2010.117
100. Ohland CL, Macnaughton WK. Probiotic Bacteria and Intestinal Epithelial Barrier Function. Am J Physiol Gastrointest Liver Physiol (2010) 298(6):G807–19. doi: 10.1152/ajpgi.00243.2009
101. Sanders ME. Impact of Probiotics on Colonizing Microbiota of the Gut. J Clin Gastroenterol (2011) 45(Suppl):S115–9. doi: 10.1097/MCG.0b013e318227414a
102. Korpela K, Salonen A, Vepsalainen O, Suomalainen M, Kolmeder C, Varjosalo M, et al. Probiotic Supplementation Restores Normal Microbiota Composition and Function in Antibiotic-Treated and in Caesarean-Born Infants. Microbiome (2018) 6(1):182. doi: 10.1186/s40168-018-0567-4
103. Kuitunen M, Kukkonen K, Juntunen-Backman K, Korpela R, Poussa T, Tuure T, et al. Probiotics Prevent IgE-Associated Allergy Until Age 5 Years in Cesarean-Delivered Children But Not in the Total Cohort. J Allergy Clin Immunol (2009) 123(2):335–41. doi: 10.1016/j.jaci.2008.11.019
104. Forster CS, Hsieh MH, Cabana MD. Perspectives From the Society for Pediatric Research: Probiotic Use in Urinary Tract Infections, Atopic Dermatitis, and Antibiotic-Associated Diarrhea: An Overview. Pediatr Res (2020). doi: 10.1038/s41390-020-01298-1
105. Kallio S, Kukkonen AK, Savilahti E, Kuitunen M. Perinatal Probiotic Intervention Prevented Allergic Disease in a Caesarean-Delivered Subgroup at 13-Year Follow-Up. Clin Exp Allergy (2019) 49(4):506–15. doi: 10.1111/cea.13321
106. Quraishi MN, Shaheen W, Oo YH, Iqbal TH. Immunological Mechanisms Underpinning Faecal Microbiota Transplantation for the Treatment of Inflammatory Bowel Disease. Clin Exp Immunol (2020) 199(1):24–38. doi: 10.1111/cei.13397
107. Quraishi MN, Widlak M, Bhala N, Moore D, Price M, Sharma N, et al. Systematic Review With Meta-Analysis: The Efficacy of Faecal Microbiota Transplantation for the Treatment of Recurrent and Refractory Clostridium Difficile Infection. Aliment Pharmacol Ther (2017) 46(5):479–93. doi: 10.1111/apt.14201
108. Hvas CL, Dahl Jorgensen SM, Jorgensen SP, Storgaard M, Lemming L, Hansen MM, et al. Fecal Microbiota Transplantation Is Superior to Fidaxomicin for Treatment of Recurrent Clostridium Difficile Infection. Gastroenterology (2019) 156(5):1324–32.e3. doi: 10.1053/j.gastro.2018.12.019
109. Jiang ZD, Ajami NJ, Petrosino JF, Jun G, Hanis CL, Shah M, et al. Randomised Clinical Trial: Faecal Microbiota Transplantation for Recurrent Clostridum Difficile Infection - Fresh, or Frozen, or Lyophilised Microbiota From a Small Pool of Healthy Donors Delivered by Colonoscopy. Aliment Pharmacol Ther (2017) 45(7):899–908. doi: 10.1111/apt.13969
110. Ooijevaar RE, van Nood E, Goorhuis A, Terveer EM, van Prehn J, Verspaget HW, et al. Ten-Year Follow-Up of Patients Treated With Fecal Microbiota Transplantation for Recurrent Clostridioides Difficile Infection From a Randomized Controlled Trial and Review of the Literature. Microorganisms (2021) 9(3):548. doi: 10.3390/microorganisms9030548
111. Kronman MP, Nielson HJ, Adler AL, Giefer MJ, Wahbeh G, Singh N, et al. Fecal Microbiota Transplantation Via Nasogastric Tube for Recurrent Clostridium Difficile Infection in Pediatric Patients. J Pediatr Gastroenterol Nutr (2015) 60(1):23–6. doi: 10.1097/MPG.0000000000000545
112. Pugliese A. Autoreactive T Cells in Type 1 Diabetes. J Clin Invest (2017) 127(8):2881–91. doi: 10.1172/JCI94549
113. Buzzetti R, Zampetti S, Maddaloni E. Adult-Onset Autoimmune Diabetes: Current Knowledge and Implications for Management. Nat Rev Endocrinol (2017) 13(11):674–86. doi: 10.1038/nrendo.2017.99
114. Parikka V, Nanto-Salonen K, Saarinen M, Simell T, Ilonen J, Hyoty H, et al. Early Seroconversion and Rapidly Increasing Autoantibody Concentrations Predict Prepubertal Manifestation of Type 1 Diabetes in Children at Genetic Risk. Diabetologia (2012) 55(7):1926–36. doi: 10.1007/s00125-012-2523-3
115. Harjutsalo V, Sjoberg L, Tuomilehto J. Time Trends in the Incidence of Type 1 Diabetes in Finnish Children: A Cohort Study. Lancet (2008) 371(9626):1777–82. doi: 10.1016/S0140-6736(08)60765-5
116. Robertson CC, Rich SS. Genetics of Type 1 Diabetes. Curr Opin Genet Dev (2018) 50:7–16. doi: 10.1016/j.gde.2018.01.006
117. Roep BO, Tree TI. Immune Modulation in Humans: Implications for Type 1 Diabetes Mellitus. Nat Rev Endocrinol (2014) 10(4):229–42. doi: 10.1038/nrendo.2014.2
118. Hummel M, Bonifacio E, Schmid S, Walter M, Knopff A, Ziegler AG. Brief Communication: Early Appearance of Islet Autoantibodies Predicts Childhood Type 1 Diabetes in Offspring of Diabetic Parents. Ann Intern Med (2004) 140(11):882–6. doi: 10.7326/0003-4819-140-11-200406010-00009
119. Pociot F, Akolkar B, Concannon P, Erlich HA, Julier C, Morahan G, et al. Genetics of Type 1 Diabetes: What’s Next? Diabetes (2010) 59(7):1561–71. doi: 10.2337/db10-0076
120. Lampasona V, Liberati D. Islet Autoantibodies. Curr Diabetes Rep (2016) 16(6):53. doi: 10.1007/s11892-016-0738-2
121. Ling Q, Lu J, Li J, Xu Q, Zhu D, Bi Y. Risk of Beta-Cell Autoimmunity Presence for Progression to Type 1 Diabetes: A Systematic Review and Meta-Analysis. J Autoimmun (2018) 86:9–18. doi: 10.1016/j.jaut.2017.09.012
122. Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, et al. Seroconversion to Multiple Islet Autoantibodies and Risk of Progression to Diabetes in Children. JAMA (2013) 309(23):2473–9. doi: 10.1001/jama.2013.6285
123. Harjutsalo V, Reunanen A, Tuomilehto J. Differential Transmission of Type 1 Diabetes From Diabetic Fathers and Mothers to Their Offspring. Diabetes (2006) 55(5):1517–24. doi: 10.2337/db05-1296
124. American Diabetes A. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2018. Diabetes Care (2018) 41(Suppl 1):S13–27. doi: 10.2337/dc18-S002
125. Dabelea D, Rewers A, Stafford JM, Standiford DA, Lawrence JM, Saydah S, et al. Trends in the Prevalence of Ketoacidosis at Diabetes Diagnosis: The SEARCH for Diabetes in Youth Study. Pediatrics (2014) 133(4):e938–45. doi: 10.1542/peds.2013-2795
126. Bergenstal RM. Glycemic Variability and Diabetes Complications: Does it Matter? Simply Put, There Are Better Glycemic Markers! Diabetes Care (2015) 38(8):1615–21. doi: 10.2337/dc15-0099
127. Virk SA, Donaghue KC, Cho YH, Benitez-Aguirre P, Hing S, Pryke A, et al. Association Between Hba1c Variability and Risk of Microvascular Complications in Adolescents With Type 1 Diabetes. J Clin Endocrinol Metab (2016) 101(9):3257–63. doi: 10.1210/jc.2015-3604
128. Cho YH, Craig ME, Donaghue KC. Puberty as an Accelerator for Diabetes Complications. Pediatr Diabetes (2014) 15(1):18–26. doi: 10.1111/pedi.12112
129. Patterson CC, Harjutsalo V, Rosenbauer J, Neu A, Cinek O, Skrivarhaug T, et al. Trends and Cyclical Variation in the Incidence of Childhood Type 1 Diabetes in 26 European Centres in the 25 Year Period 1989-2013: A Multicentre Prospective Registration Study. Diabetologia (2019) 62(3):408–17. doi: 10.1007/s00125-018-4763-3
130. Achenbach P, Bonifacio E, Koczwara K, Ziegler AG. Natural History of Type 1 Diabetes. Diabetes (2005) 54(Suppl 2):S25–31. doi: 10.2337/diabetes.54.suppl_2.S25
131. Bibbo S, Dore MP, Pes GM, Delitala G, Delitala AP. Is There a Role for Gut Microbiota in Type 1 Diabetes Pathogenesis? Ann Med (2017) 49(1):11–22. doi: 10.1080/07853890.2016.1222449
132. Sohail MU, Althani A, Anwar H, Rizzi R, Marei HE. Role of the Gastrointestinal Tract Microbiome in the Pathophysiology of Diabetes Mellitus. J Diabetes Res (2017) 2017:9631435. doi: 10.1155/2017/9631435
133. Abdellatif AM, Sarvetnick NE. Current Understanding of the Role of Gut Dysbiosis in Type 1 Diabetes. J Diabetes (2019) 11(8):632–44. doi: 10.1111/1753-0407.12915
134. Zhou H, Sun L, Zhang S, Zhao X, Gang X, Wang G. Evaluating the Causal Role of Gut Microbiota in Type 1 Diabetes and Its Possible Pathogenic Mechanisms. Front Endocrinol (Lausanne) (2020) 11:125. doi: 10.3389/fendo.2020.00125
135. Dedrick S, Sundaresh B, Huang Q, Brady C, Yoo T, Cronin C, et al. The Role of Gut Microbiota and Environmental Factors in Type 1 Diabetes Pathogenesis. Front Endocrinol (Lausanne) (2020) 11:78. doi: 10.3389/fendo.2020.00078
136. Pinto E, Anselmo M, Calha M, Bottrill A, Duarte I, Andrew PW, et al. The Intestinal Proteome of Diabetic and Control Children Is Enriched With Different Microbial and Host Proteins. Microbiology (2017) 163(2):161–74. doi: 10.1099/mic.0.000412
137. de Goffau MC, Luopajarvi K, Knip M, Ilonen J, Ruohtula T, Harkonen T, et al. Fecal Microbiota Composition Differs Between Children With Beta-Cell Autoimmunity and Those Without. Diabetes (2013) 62(4):1238–44. doi: 10.2337/db12-0526
138. de Goffau MC, Fuentes S, van den Bogert B, Honkanen H, de Vos WM, Welling GW, et al. Aberrant Gut Microbiota Composition at the Onset of Type 1 Diabetes in Young Children. Diabetologia (2014) 57(8):1569–77. doi: 10.1007/s00125-014-3274-0
139. Endesfelder D, Zu Castell W, Ardissone A, Davis-Richardson AG, Achenbach P, Hagen M, et al. Compromised Gut Microbiota Networks in Children With Anti-Islet Cell Autoimmunity. Diabetes (2014) 63(6):2006–14. doi: 10.2337/db13-1676
140. Alkanani AK, Hara N, Gottlieb PA, Ir D, Robertson CE, Wagner BD, et al. Alterations in Intestinal Microbiota Correlate With Susceptibility to Type 1 Diabetes. Diabetes (2015) 64(10):3510–20. doi: 10.2337/db14-1847
141. Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. Gut Microbiome Metagenomics Analysis Suggests a Functional Model for the Development of Autoimmunity for Type 1 Diabetes. PloS One (2011) 6(10):e25792. doi: 10.1371/journal.pone.0025792
142. Soyucen E, Gulcan A, Aktuglu-Zeybek AC, Onal H, Kiykim E, Aydin A. Differences in the Gut Microbiota of Healthy Children and Those With Type 1 Diabetes. Pediatr Int (2014) 56(3):336–43. doi: 10.1111/ped.12243
143. Davis-Richardson AG, Ardissone AN, Dias R, Simell V, Leonard MT, Kemppainen KM, et al. Bacteroides Dorei Dominates Gut Microbiome Prior to Autoimmunity in Finnish Children at High Risk for Type 1 Diabetes. Front Microbiol (2014) 5:678. doi: 10.3389/fmicb.2014.00678
144. Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, et al. Toward Defining the Autoimmune Microbiome for Type 1 Diabetes. ISME J (2011) 5(1):82–91. doi: 10.1038/ismej.2010.92
145. Biassoni R, Di Marco E, Squillario M, Barla A, Piccolo G, Ugolotti E, et al. Gut Microbiota in T1DM-Onset Pediatric Patients: Machine-Learning Algorithms to Classify Microorganisms as Disease Linked. J Clin Endocrinol Metab (2020) 105(9):e3114–26. doi: 10.1210/clinem/dgaa407
146. Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The Human Gut Microbiome in Early-Onset Type 1 Diabetes From the TEDDY Study. Nature (2018) 562(7728):589–94. doi: 10.1038/s41586-018-0620-2
147. Davis-Richardson AG, Triplett EW. A Model for the Role of Gut Bacteria in the Development of Autoimmunity for Type 1 Diabetes. Diabetologia (2015) 58(7):1386–93. doi: 10.1007/s00125-015-3614-8
148. Pozuelo M, Panda S, Santiago A, Mendez S, Accarino A, Santos J, et al. Reduction of Butyrate- and Methane-Producing Microorganisms in Patients With Irritable Bowel Syndrome. Sci Rep (2015) 5:12693. doi: 10.1038/srep12693
149. de Groot PF, Belzer C, Aydin O, Levin E, Levels JH, Aalvink S, et al. Distinct Fecal and Oral Microbiota Composition in Human Type 1 Diabetes, An Observational Study. PloS One (2017) 12(12):e0188475. doi: 10.1371/journal.pone.0188475
150. Bonifacio E, Warncke K, Winkler C, Wallner M, Ziegler AG. Cesarean Section and Interferon-Induced Helicase Gene Polymorphisms Combine to Increase Childhood Type 1 Diabetes Risk. Diabetes (2011) 60(12):3300–6. doi: 10.2337/db11-0729
151. Lee HY, Lu CL, Chen HF, Su HF, Li CY. Perinatal and Childhood Risk Factors for Early-Onset Type 1 Diabetes: A Population-Based Case-Control Study in Taiwan. Eur J Public Health (2015) 25(6):1024–9. doi: 10.1093/eurpub/ckv059
152. Clausen TD, Bergholt T, Eriksson F, Rasmussen S, Keiding N, Lokkegaard EC. Prelabor Cesarean Section and Risk of Childhood Type 1 Diabetes: A Nationwide Register-Based Cohort Study. Epidemiology (2016) 27(4):547–55. doi: 10.1097/EDE.0000000000000488
153. Andersen V, Moller S, Jensen PB, Moller FT, Green A. Caesarean Delivery and Risk of Chronic Inflammatory Diseases (Inflammatory Bowel Disease, Rheumatoid Arthritis, Coeliac Disease, and Diabetes Mellitus): A Population Based Registry Study of 2,699,479 Births in Denmark During 1973-2016. Clin Epidemiol (2020) 12:287–93. doi: 10.2147/CLEP.S229056
154. Black M, Bhattacharya S, Philip S, Norman JE, McLernon DJ. Planned Cesarean Delivery at Term and Adverse Outcomes in Childhood Health. JAMA (2015) 314(21):2271–9. doi: 10.1001/jama.2015.16176
155. Betran AP, Ye J, Moller AB, Zhang J, Gulmezoglu AM, Torloni MR. The Increasing Trend in Caesarean Section Rates: Global, Regional and National Estimates: 1990-2014. PloS One (2016) 11(2):e0148343. doi: 10.1371/journal.pone.0148343
156. Stene LC, Magnus P, Lie RT, Sovik O, Joner G, Norwegian Childhood Diabetes Study G. No Association Between Preeclampsia or Cesarean Section and Incidence of Type 1 Diabetes Among Children: A Large, Population-Based Cohort Study. Pediatr Res (2003) 54(4):487–90. doi: 10.1203/01.PDR.0000081301.25600.5D
157. Ievins R, Roberts SE, Goldacre MJ. Perinatal Factors Associated With Subsequent Diabetes Mellitus in the Child: Record Linkage Study. Diabetes Med (2007) 24(6):664–70. doi: 10.1111/j.1464-5491.2007.02147.x
158. Rosenbauer J, Herzig P, Giani G. Early Infant Feeding and Risk of Type 1 Diabetes Mellitus-a Nationwide Population-Based Case-Control Study in Pre-School Children. Diabetes Metab Res Rev (2008) 24(3):211–22. doi: 10.1002/dmrr.791
159. Lund-Blix NA, Dydensborg Sander S, Stordal K, Nybo Andersen AM, Ronningen KS, Joner G, et al. Infant Feeding and Risk of Type 1 Diabetes in Two Large Scandinavian Birth Cohorts. Diabetes Care (2017) 40(7):920–7. doi: 10.2337/dc17-0016
160. Cardwell CR, Stene LC, Ludvigsson J, Rosenbauer J, Cinek O, Svensson J, et al. Breast-Feeding and Childhood-Onset Type 1 Diabetes: A Pooled Analysis of Individual Participant Data From 43 Observational Studies. Diabetes Care (2012) 35(11):2215–25. doi: 10.2337/dc12-0438
161. Xiao L, Van’t Land B, Engen PA, Naqib A, Green SJ, Nato A, et al. Human Milk Oligosaccharides Protect Against the Development of Autoimmune Diabetes in NOD-Mice. Sci Rep (2018) 8(1):3829. doi: 10.1038/s41598-018-22052-y
162. Turroni F, Milani C, Duranti S, Ferrario C, Lugli GA, Mancabelli L, et al. Bifidobacteria and the Infant Gut: An Example of Co-Evolution and Natural Selection. Cell Mol Life Sci (2018) 75(1):103–18. doi: 10.1007/s00018-017-2672-0
163. Lugli GA, Duranti S, Milani C, Mancabelli L, Turroni F, Alessandri G, et al. Investigating Bifidobacteria and Human Milk Oligosaccharide Composition of Lactating Mothers. FEMS Microbiol Ecol (2020) 96(5). doi: 10.1093/femsec/fiaa049
164. Bunesova V, Lacroix C, Schwab C. Fucosyllactose and L-fucose Utilization of Infant Bifidobacterium Longum and Bifidobacterium Kashiwanohense. BMC Microbiol (2016) 16(1):248. doi: 10.1186/s12866-016-0867-4
165. Insel R, Knip M. Prospects for Primary Prevention of Type 1 Diabetes by Restoring a Disappearing Microbe. Pediatr Diabetes (2018) 19(8):1400–6. doi: 10.1111/pedi.12756
166. Boursi B, Mamtani R, Haynes K, Yang YX. The Effect of Past Antibiotic Exposure on Diabetes Risk. Eur J Endocrinol (2015) 172(6):639–48. doi: 10.1530/EJE-14-1163
167. Clausen TD, Bergholt T, Bouaziz O, Arpi M, Eriksson F, Rasmussen S, et al. Broad-Spectrum Antibiotic Treatment and Subsequent Childhood Type 1 Diabetes: A Nationwide Danish Cohort Study. PloS One (2016) 11(8):e0161654. doi: 10.1371/journal.pone.0161654
168. Raisanen L, Viljakainen H, Sarkkola C, Kolho KL. Perinatal Risk Factors for Pediatric Onset Type 1 Diabetes, Autoimmune Thyroiditis, Juvenile Idiopathic Arthritis, and Inflammatory Bowel Diseases. Eur J Pediatr (2021). doi: 10.1007/s00431-021-03987-3
169. Tapia G, Stordal K, Marild K, Kahrs CR, Skrivarhaug T, Njolstad PR, et al. Antibiotics, Acetaminophen and Infections During Prenatal and Early Life in Relation to Type 1 Diabetes. Int J Epidemiol (2018) 47(5):1538–48. doi: 10.1093/ije/dyy092
170. Kemppainen KM, Vehik K, Lynch KF, Larsson HE, Canepa RJ, Simell V, et al. Association Between Early-Life Antibiotic Use and the Risk of Islet or Celiac Disease Autoimmunity. JAMA Pediatr (2017) 171(12):1217–25. doi: 10.1001/jamapediatrics.2017.2905
171. Kallionpaa H, Laajala E, Oling V, Harkonen T, Tillmann V, Dorshakova NV, et al. Standard of Hygiene and Immune Adaptation in Newborn Infants. Clin Immunol (2014) 155(1):136–47. doi: 10.1016/j.clim.2014.09.009
172. American Diabetes A. 13. Children and Adolescents: Standards of Medical Care in Diabetes-2021. Diabetes Care (2021) 44(Suppl 1):S180–S99. doi: 10.2337/dc21-S013
173. American Diabetes A. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2021. Diabetes Care (2021) 44(Suppl 1):S111–S24. doi: 10.2337/dc21-S009
174. Miller KM, Foster NC, Beck RW, Bergenstal RM, DuBose SN, DiMeglio LA, et al. Current State of Type 1 Diabetes Treatment in the U.S.: Updated Data From the T1D Exchange Clinic Registry. Diabetes Care (2015) 38(6):971–8. doi: 10.2337/dc15-0078
175. American Diabetes A. 6. Glycemic Targets: Standards of Medical Care in Diabetes-2021. Diabetes Care (2021) 44(Suppl 1):S73–84. doi: 10.2337/dc21-S006
176. DuBose SN, Hermann JM, Tamborlane WV, Beck RW, Dost A, DiMeglio LA, et al. Obesity in Youth With Type 1 Diabetes in Germany, Austria, and the United States. J Pediatr (2015) 167(3):627–32.e1-4. doi: 10.1016/j.jpeds.2015.05.046
177. McKnight JA, Wild SH, Lamb MJ, Cooper MN, Jones TW, Davis EA, et al. Glycaemic Control of Type 1 Diabetes in Clinical Practice Early in the 21st Century: An International Comparison. Diabetes Med (2015) 32(8):1036–50. doi: 10.1111/dme.12676
178. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 Diabetes Immunotherapy Using Polyclonal Regulatory T Cells. Sci Transl Med (2015) 7(315):315ra189. doi: 10.1126/scitranslmed.aad4134
179. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, et al. Administration of CD4+CD25highCD127- Regulatory T Cells Preserves Beta-Cell Function in Type 1 Diabetes in Children. Diabetes Care (2012) 35(9):1817–20. doi: 10.2337/dc12-0038
180. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juscinska J, et al. Therapy of Type 1 Diabetes With CD4(+)CD25(High)CD127-Regulatory T Cells Prolongs Survival of Pancreatic Islets - Results of One Year Follow-Up. Clin Immunol (2014) 153(1):23–30. doi: 10.1016/j.clim.2014.03.016
181. Daifotis AG, Koenig S, Chatenoud L, Herold KC. Anti-CD3 Clinical Trials in Type 1 Diabetes Mellitus. Clin Immunol (2013) 149(3):268–78. doi: 10.1016/j.clim.2013.05.001
182. Alleva DG, Maki RA, Putnam AL, Robinson JM, Kipnes MS, Dandona P, et al. Immunomodulation in Type 1 Diabetes by NBI-6024, An Altered Peptide Ligand of the Insulin B Epitope. Scand J Immunol (2006) 63(1):59–69. doi: 10.1111/j.1365-3083.2005.01705.x
183. Thrower SL, James L, Hall W, Green KM, Arif S, Allen JS, et al. Proinsulin Peptide Immunotherapy in Type 1 Diabetes: Report of a First-in-Man Phase I Safety Study. Clin Exp Immunol (2009) 155(2):156–65. doi: 10.1111/j.1365-2249.2008.03814.x
184. Alhadj Ali M, Liu YF, Arif S, Tatovic D, Shariff H, Gibson VB, et al. Metabolic and Immune Effects of Immunotherapy With Proinsulin Peptide in Human New-Onset Type 1 Diabetes. Sci Transl Med (2017) 9(402):eaaf7779. doi: 10.1126/scitranslmed.aaf7779
185. Gradisteanu Pircalabioru G, Corcionivoschi N, Gundogdu O, Chifiriuc MC, Marutescu LG, Ispas B, et al. Dysbiosis in the Development of Type I Diabetes and Associated Complications: From Mechanisms to Targeted Gut Microbes Manipulation Therapies. Int J Mol Sci (2021) 22(5):2763. doi: 10.3390/ijms22052763
186. Uusitalo U, Liu X, Yang J, Aronsson CA, Hummel S, Butterworth M, et al. Association of Early Exposure of Probiotics and Islet Autoimmunity in the TEDDY Study. JAMA Pediatr (2016) 170(1):20–8. doi: 10.1001/jamapediatrics.2015.2757
187. Groele L, Szajewska H, Szypowska A. Effects of Lactobacillus Rhamnosus GG and Bifidobacterium Lactis Bb12 on Beta-Cell Function in Children With Newly Diagnosed Type 1 Diabetes: Protocol of a Randomised Controlled Trial. BMJ Open (2017) 7(10):e017178. doi: 10.1136/bmjopen-2017-017178
188. Healey G, Murphy R, Butts C, Brough L, Whelan K, Coad J. Habitual Dietary Fibre Intake Influences Gut Microbiota Response to an Inulin-Type Fructan Prebiotic: A Randomised, Double-Blind, Placebo-Controlled, Cross-Over, Human Intervention Study. Br J Nutr (2018) 119(2):176–89. doi: 10.1017/S0007114517003440
189. Marino E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, et al. Gut Microbial Metabolites Limit the Frequency of Autoimmune T Cells and Protect Against Type 1 Diabetes. Nat Immunol (2017) 18(5):552–62. doi: 10.1038/ni.3713
190. Bacchetta R, Gambineri E, Roncarolo MG. Role of Regulatory T Cells and FOXP3 in Human Diseases. J Allergy Clin Immunol (2007) 120(2):227–35; quiz 36-7. doi: 10.1016/j.jaci.2007.06.023
191. Samy ET, Parker LA, Sharp CP, Tung KS. Continuous Control of Autoimmune Disease by Antigen-Dependent Polyclonal CD4+CD25+ Regulatory T Cells in the Regional Lymph Node. J Exp Med (2005) 202(6):771–81. doi: 10.1084/jem.20041033
192. Vignali DA, Collison LW, Workman CJ. How Regulatory T Cells Work. Nat Rev Immunol (2008) 8(7):523–32. doi: 10.1038/nri2343
193. Holohan DR, Van Gool F, Bluestone JA. Thymically-Derived Foxp3+ Regulatory T Cells are the Primary Regulators of Type 1 Diabetes in the non-Obese Diabetic Mouse Model. PloS One (2019) 14(10):e0217728. doi: 10.1371/journal.pone.0217728
194. Fousteri G, Jasinski J, Dave A, Nakayama M, Pagni P, Lambolez F, et al. Following the Fate of One Insulin-Reactive CD4 T Cell: Conversion Into Teffs and Tregs in the Periphery Controls Diabetes in NOD Mice. Diabetes (2012) 61(5):1169–79. doi: 10.2337/db11-0671
195. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In Vitro-Expanded Antigen-Specific Regulatory T Cells Suppress Autoimmune Diabetes. J Exp Med (2004) 199(11):1455–65. doi: 10.1084/jem.20040139
196. Presa M, Chen YG, Grier AE, Leiter EH, Brehm MA, Greiner DL, et al. The Presence and Preferential Activation of Regulatory T Cells Diminish Adoptive Transfer of Autoimmune Diabetes by Polyclonal Nonobese Diabetic (Nod) T Cell Effectors Into NSG Versus NOD-scid Mice. J Immunol (2015) 195(7):3011–9. doi: 10.4049/jimmunol.1402446
197. Marek-Trzonkowska N, Mysliwiec M, Iwaszkiewicz-Grzes D, Gliwinski M, Derkowska I, Zalinska M, et al. Factors Affecting Long-Term Efficacy of T Regulatory Cell-Based Therapy in Type 1 Diabetes. J Transl Med (2016) 14(1):332. doi: 10.1186/s12967-016-1090-7
198. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of Ex Vivo Expanded T Regulatory Cells in Adults Transplanted With Umbilical Cord Blood: Safety Profile and Detection Kinetics. Blood (2011) 117(3):1061–70. doi: 10.1182/blood-2010-07-293795
199. Allan SE, Alstad AN, Merindol N, Crellin NK, Amendola M, Bacchetta R, et al. Generation of Potent and Stable Human CD4+ T Regulatory Cells by Activation-Independent Expression of FOXP3. Mol Ther (2008) 16(1):194–202. doi: 10.1038/sj.mt.6300341
200. Hori S, Nomura T, Sakaguchi S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
201. Honaker Y, Hubbard N, Xiang Y, Fisher L, Hagin D, Sommer K, et al. Gene Editing to Induce FOXP3 Expression in Human CD4(+) T Cells Leads to a Stable Regulatory Phenotype and Function. Sci Transl Med (2020) 12(546):eaay6422. doi: 10.1126/scitranslmed.aay6422
202. King MA, Covassin L, Brehm MA, Racki W, Pearson T, Leif J, et al. Human Peripheral Blood Leucocyte Non-Obese Diabetic-Severe Combined Immunodeficiency Interleukin-2 Receptor Gamma Chain Gene Mouse Model of Xenogeneic Graft-Versus-Host-Like Disease and the Role of Host Major Histocompatibility Complex. Clin Exp Immunol (2009) 157(1):104–18. doi: 10.1111/j.1365-2249.2009.03933.x
203. Brusko TM, Koya RC, Zhu S, Lee MR, Putnam AL, McClymont SA, et al. Human Antigen-Specific Regulatory T Cells Generated by T Cell Receptor Gene Transfer. PloS One (2010) 5(7):e11726. doi: 10.1371/journal.pone.0011726
204. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-Specific Regulatory T Cells Generated With a Chimeric Antigen Receptor. J Clin Invest (2016) 126(4):1413–24. doi: 10.1172/JCI82771
205. Tarbell KV, Petit L, Zuo X, Toy P, Luo X, Mqadmi A, et al. Dendritic Cell-Expanded, Islet-Specific CD4+ Cd25+ CD62L+ Regulatory T Cells Restore Normoglycemia in Diabetic NOD Mice. J Exp Med (2007) 204(1):191–201. doi: 10.1084/jem.20061631
206. Imam S, Prathibha R, Dar P, Almotah K, Al-Khudhair A, Hasan SA, et al. eIF5A Inhibition Influences T Cell Dynamics in the Pancreatic Microenvironment of the Humanized Mouse Model of Type 1 Diabetes. Sci Rep (2019) 9(1):1533. doi: 10.1038/s41598-018-38341-5
207. Friedmann-Morvinski D, Bendavid A, Waks T, Schindler D, Eshhar Z. Redirected Primary T Cells Harboring a Chimeric Receptor Require Costimulation for Their Antigen-Specific Activation. Blood (2005) 105(8):3087–93. doi: 10.1182/blood-2004-09-3737
208. Gliwinski M, Iwaszkiewicz-Grzes D, Woloszyn-Durkiewicz A, Tarnowska M, Zalinska M, Hennig M, et al. Proinsulin-Specific T Regulatory Cells may Control Immune Responses in Type 1 Diabetes: Implications for Adoptive Therapy. BMJ Open Diabetes Res Care (2020) 8(1):e000873. doi: 10.1136/bmjdrc-2019-000873
209. Eisenbarth G. Type I Diabetes Mellitus. A Chronic Autoimmune Disease. N Engl J Med (1986) 314(21):1360–8. doi: 10.1056/NEJM198605223142106
210. Korsgren S, Molin Y, Salmela K, Lundgren T, Melhus A, Korsgren O. On the Etiology of Type 1 Diabetes: A New Animal Model Signifying a Decisive Role for Bacteria Eliciting an Adverse Innate Immunity Response. Am J Pathol (2012) 181(5):1735–48. doi: 10.1016/j.ajpath.2012.07.022
211. Kent SC BJ. Narrowing in on the Anti- β Cell-Specific T Cells: Looking ‘Where the Action Is’. Curr Opin Endocrinol Diabetes Obes (2017) 24(2):98–102. doi: 10.1097/MED.0000000000000323
212. Skyler JS, Brown D, Chase HP, Collier E, Cowie C, Eisenbarth GS, et al. Effects of Insulin in Relatives of Patients With Type 1 Diabetes Mellitus. N Engl J Med (2002) 346(22):1685–91. doi: 10.1056/NEJMoa012350
213. Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N Engl J Med (2019) 381(7):603–13. doi: 10.1056/NEJMoa1902226
214. Ferrat LA, Vehik K, Sharp SA, Lernmark Å, Rewers MJ, She JX, et al. A Combined Risk Score Enhances Prediction of Type 1 Diabetes Among Susceptible Children. Nat Med (2020) 26(8):1247–55. doi: 10.1038/s41591-020-0930-4
215. Iwaszkiewicz-Grzes D, Gliwinski M, Eugster A, Piotrowska M, Dahl A, Marek-Trzonkowska N, et al. Antigen-Reactive Regulatory T Cells Can be Expanded In Vitro With Monocytes and Anti-CD28 and Anti-CD154 Antibodies. Cytotherapy (2020) 22(11):629–41. doi: 10.1016/j.jcyt.2020.07.001
216. Van Hulle F, De Groot K, Stangé G, Suenens K, De Mesmaeker I, De Paep DL, et al. Formation of Amyloid in Encapsulated Human Pancreatic and Human Stem Cell-Generated Beta Cell Implants. Am J Transplant (2020). doi: 10.1111/ajt.16398
217. Huurman VA, Hilbrands R, Pinkse GG, Gillard P, Duinkerken G, van de Linde P, et al. Cellular Islet Autoimmunity Associates With Clinical Outcome of Islet Cell Transplantation. PloS One (2008) 3(6):e2435. doi: 10.1371/journal.pone.0002435
218. Lundin KE, Qiao SW, Snir O, Sollid LM. Coeliac Disease - From Genetic and Immunological Studies to Clinical Applications. Scand J Gastroenterol (2015) 50(6):708–17. doi: 10.3109/00365521.2015.1030766
219. van Heel DA, Franke L, Hunt KA, Gwilliam R, Zhernakova A, Inouye M, et al. A Genome-Wide Association Study for Celiac Disease Identifies Risk Variants in the Region Harboring IL2 and IL21. Nat Genet (2007) 39(7):827–9. doi: 10.1038/ng2058
220. Mercader J. Mozambican Grass Seed Consumption During the Middle Stone Age. Science (2009) 326(5960):1680–3. doi: 10.1126/science.1173966
221. Silano M, Vincentini O, De Vincenzi M. Toxic, Immunostimulatory and Antagonist Gluten Peptides in Celiac Disease. Curr Med Chem (2009) 16(12):1489–98. doi: 10.2174/092986709787909613
222. Shan L, Molberg O, Parrot I, Hausch F, Filiz F, Gray GM, et al. Structural Basis for Gluten Intolerance in Celiac Sprue. Science (2002) 297(5590):2275–9. doi: 10.1126/science.1074129
223. Jelinkova L, Tuckova L, Cinova J, Flegelova Z, Tlaskalova-Hogenova H. Gliadin Stimulates Human Monocytes to Production of IL-8 and TNF-Alpha Through a Mechanism Involving NF-Kappab. FEBS Lett (2004) 571(1-3):81–5. doi: 10.1016/j.febslet.2004.06.057
224. Lammers KM, Khandelwal S, Chaudhry F, Kryszak D, Puppa EL, Casolaro V, et al. Identification of a Novel Immunomodulatory Gliadin Peptide That Causes Interleukin-8 Release in a Chemokine Receptor CXCR3-Dependent Manner Only in Patients With Coeliac Disease. Immunology (2011) 132(3):432–40. doi: 10.1111/j.1365-2567.2010.03378.x
225. Picarelli A, Di Tola M, Sabbatella L, Anania MC, Di Cello T, Greco R, et al. 31-43 Amino Acid Sequence of the Alpha-Gliadin Induces Anti-Endomysial Antibody Production During In Vitro Challenge. Scand J Gastroenterol (1999) 34(11):1099–102. doi: 10.1080/003655299750024896
226. Pagliari D, Urgesi R, Frosali S, Riccioni ME, Newton EE, Landolfi R, et al. The Interaction Among Microbiota, Immunity, and Genetic and Dietary Factors Is the Condicio Sine Qua non Celiac Disease Can Develop. J Immunol Res (2015) 2015:123653. doi: 10.1155/2015/123653
227. Hall EJ, Batt RM. Dietary Modulation of Gluten Sensitivity in a Naturally Occurring Enteropathy of Irish Setter Dogs. Gut (1992) 33(2):198–205. doi: 10.1136/gut.33.2.198
228. Jabri B, Sollid LM. T Cells in Celiac Disease. J Immunol (2017) 198(8):3005–14. doi: 10.4049/jimmunol.1601693
229. Trynka G, Hunt KA, Bockett NA, Romanos J, Mistry V, Szperl A, et al. Dense Genotyping Identifies and Localizes Multiple Common and Rare Variant Association Signals in Celiac Disease. Nat Genet (2011) 43(12):1193–201. doi: 10.1038/ng.998
230. Husby S, Koletzko S, Korponay-Szabo IR, Mearin ML, Phillips A, Shamir R, et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition Guidelines for the Diagnosis of Coeliac Disease. J Pediatr Gastroenterol Nutr (2012) 54(1):136–60. doi: 10.1097/MPG.0b013e31821a23d0
231. Rubio-Tapia A, Ludvigsson JF, Brantner TL, Murray JA, Everhart JE. The Prevalence of Celiac Disease in the United States. Am J Gastroenterol (2012) 107(10):1538–44. doi: 10.1038/ajg.2012.219
232. Singh P, Arora A, Strand TA, Leffler DA, Catassi C, Green PH, et al. Global Prevalence of Celiac Disease: Systematic Review and Meta-Analysis. Clin Gastroenterol Hepatol (2018) 16(6):823–36.e2. doi: 10.1016/j.cgh.2017.06.037
233. Liu E, Lee HS, Aronsson CA, Hagopian WA, Koletzko S, Rewers MJ, et al. Risk of Pediatric Celiac Disease According to HLA Haplotype and Country. N Engl J Med (2014) 371(1):42–9. doi: 10.1056/NEJMoa1313977
234. Ivarsson A, Myleus A, Norstrom F, van der Pals M, Rosen A, Hogberg L, et al. Prevalence of Childhood Celiac Disease and Changes in Infant Feeding. Pediatrics (2013) 131(3):e687–94. doi: 10.1542/peds.2012-1015
235. Kivela L, Kaukinen K, Lahdeaho ML, Huhtala H, Ashorn M, Ruuska T, et al. Presentation of Celiac Disease in Finnish Children Is No Longer Changing: A 50-Year Perspective. J Pediatr (2015) 167(5):1109–15.e1. doi: 10.1016/j.jpeds.2015.07.057
236. Vivas S, Ruiz de Morales JM, Fernandez M, Hernando M, Herrero B, Casqueiro J, et al. Age-Related Clinical, Serological, and Histopathological Features of Celiac Disease. Am J Gastroenterol (2008) 103(9):2360–5. doi: 10.1111/j.1572-0241.2008.01977.x
237. Volta U, Caio G, Stanghellini V, De Giorgio R. The Changing Clinical Profile of Celiac Disease: A 15-Year Experience (1998-2012) in an Italian Referral Center. BMC Gastroenterol (2014) 14:194. doi: 10.1186/s12876-014-0194-x
238. Jericho H, Sansotta N, Guandalini S. Extraintestinal Manifestations of Celiac Disease: Effectiveness of the Gluten-Free Diet. J Pediatr Gastroenterol Nutr (2017) 65(1):75–9. doi: 10.1097/MPG.0000000000001420
239. Nurminen S, Kivela L, Huhtala H, Kaukinen K, Kurppa K. Extraintestinal Manifestations Were Common in Children With Coeliac Disease and Were More Prevalent in Patients With More Severe Clinical and Histological Presentation. Acta Paediatr (2019) 108(4):681–7. doi: 10.1111/apa.14324
240. Iughetti L, Bulgarelli S, Forese S, Lorini R, Balli F, Bernasconi S. Endocrine Aspects of Coeliac Disease. J Pediatr Endocrinol Metab (2003) 16(6):805–18. doi: 10.1515/JPEM.2003.16.6.805
241. Rubio-Tapia A, Hill ID, Kelly CP, Calderwood AH, Murray JA. American College of G. ACG Clinical Guidelines: Diagnosis and Management of Celiac Disease. Am J Gastroenterol (2013) 108(5):656–76. doi: 10.1038/ajg.2013.79
242. Deora V, Aylward N, Sokoro A, El-Matary W. Serum Vitamins and Minerals at Diagnosis and Follow-up in Children With Celiac Disease. J Pediatr Gastroenterol Nutr (2017) 65(2):185–9. doi: 10.1097/MPG.0000000000001475
243. Halfdanarson TR, Litzow MR, Murray JA. Hematologic Manifestations of Celiac Disease. Blood (2007) 109(2):412–21. doi: 10.1182/blood-2006-07-031104
244. Baydoun A, Maakaron JE, Halawi H, Abou Rahal J, Taher AT. Hematological Manifestations of Celiac Disease. Scand J Gastroenterol (2012) 47(12):1401–11. doi: 10.3109/00365521.2012.706828
245. Nurminen S, Kivela L, Taavela J, Huhtala H, Maki M, Kaukinen K, et al. Factors Associated With Growth Disturbance at Celiac Disease Diagnosis in Children: A Retrospective Cohort Study. BMC Gastroenterol (2015) 15:125. doi: 10.1186/s12876-015-0357-4
246. Kamycheva E, Goto T, Camargo CA Jr. Celiac Disease is Associated With Reduced Bone Mineral Density and Increased FRAX Scores in the US National Health and Nutrition Examination Survey. Osteoporos Int (2017) 28(3):781–90. doi: 10.1007/s00198-016-3791-4
247. Rubio-Tapia A, Kyle RA, Kaplan EL, Johnson DR, Page W, Erdtmann F, et al. Increased Prevalence and Mortality in Undiagnosed Celiac Disease. Gastroenterology (2009) 137(1):88–93. doi: 10.1053/j.gastro.2009.03.059
248. Volta U, Granito A, Fiorini E, Parisi C, Piscaglia M, Pappas G, et al. Usefulness of Antibodies to Deamidated Gliadin Peptides in Celiac Disease Diagnosis and Follow-Up. Dig Dis Sci (2008) 53(6):1582–8. doi: 10.1007/s10620-007-0058-0
249. Volta U, Fabbri A, Parisi C, Piscaglia M, Caio G, Tovoli F, et al. Old and New Serological Tests for Celiac Disease Screening. Expert Rev Gastroenterol Hepatol (2010) 4(1):31–5. doi: 10.1586/egh.09.66
250. Liu E, Dong F, Baron AE, Taki I, Norris JM, Frohnert BI, et al. High Incidence of Celiac Disease in a Long-term Study of Adolescents With Susceptibility Genotypes. Gastroenterology (2017) 152(6):1329–36.e1. doi: 10.1053/j.gastro.2017.02.002
251. Abadie V, Sollid LM, Barreiro LB, Jabri B. Integration of Genetic and Immunological Insights Into a Model of Celiac Disease Pathogenesis. Annu Rev Immunol (2011) 29:493–525. doi: 10.1146/annurev-immunol-040210-092915
252. Menard S, Lebreton C, Schumann M, Matysiak-Budnik T, Dugave C, Bouhnik Y, et al. Paracellular Versus Transcellular Intestinal Permeability to Gliadin Peptides in Active Celiac Disease. Am J Pathol (2012) 180(2):608–15. doi: 10.1016/j.ajpath.2011.10.019
253. Sanchez D, Hoffmanova I, Szczepankova A, Habova V, Tlaskalova-Hogenova H. Contribution of Infectious Agents to the Development of Celiac Disease. Microorganisms (2021) 9(3):547. doi: 10.3390/microorganisms9030547
254. Caminero A, McCarville JL, Galipeau HJ, Deraison C, Bernier SP, Constante M, et al. Duodenal Bacterial Proteolytic Activity Determines Sensitivity to Dietary Antigen Through Protease-Activated Receptor-2. Nat Commun (2019) 10(1):1198. doi: 10.1038/s41467-019-09037-9
255. D’Argenio V, Casaburi G, Precone V, Pagliuca C, Colicchio R, Sarnataro D, et al. Metagenomics Reveals Dysbiosis and a Potentially Pathogenic N. Flavescens Strain in Duodenum of Adult Celiac Patients. Am J Gastroenterol (2016) 111(6):879–90. doi: 10.1038/ajg.2016.95
256. De Palma G, Nadal I, Medina M, Donat E, Ribes-Koninckx C, Calabuig M, et al. Intestinal Dysbiosis and Reduced Immunoglobulin-Coated Bacteria Associated With Coeliac Disease in Children. BMC Microbiol (2010) 10:63. doi: 10.1186/1471-2180-10-63
257. Sanchez E, Nadal I, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Reduced Diversity and Increased Virulence-Gene Carriage in Intestinal Enterobacteria of Coeliac Children. BMC Gastroenterol (2008) 8:50. doi: 10.1186/1471-230X-8-50
258. Sanchez E, Ribes-Koninckx C, Calabuig M, Sanz Y. Intestinal Staphylococcus Spp. and Virulent Features Associated With Coeliac Disease. J Clin Pathol (2012) 65(9):830–4. doi: 10.1136/jclinpath-2012-200759
259. Sjoberg V, Sandstrom O, Hedberg M, Hammarstrom S, Hernell O, Hammarstrom ML. Intestinal T-cell Responses in Celiac Disease - Impact of Celiac Disease Associated Bacteria. PloS One (2013) 8(1):e53414. doi: 10.1371/journal.pone.0053414
260. Sanchez E, Laparra JM, Sanz Y. Discerning the Role of Bacteroides Fragilis in Celiac Disease Pathogenesis. Appl Environ Microbiol (2012) 78(18):6507–15. doi: 10.1128/AEM.00563-12
261. Olivares M, Benitez-Paez A, de Palma G, Capilla A, Nova E, Castillejo G, et al. Increased Prevalence of Pathogenic Bacteria in the Gut Microbiota of Infants at Risk of Developing Celiac Disease: The PROFICEL Study. Gut Microbes (2018) 9(6):551–8. doi: 10.1080/19490976.2018.1451276
262. Girbovan A, Sur G, Samasca G, Lupan I. Dysbiosis a Risk Factor for Celiac Disease. Med Microbiol Immunol (2017) 206(2):83–91. doi: 10.1007/s00430-017-0496-z
263. Chander AM, Yadav H, Jain S, Bhadada SK, Dhawan DK. Cross-Talk Between Gluten, Intestinal Microbiota and Intestinal Mucosa in Celiac Disease: Recent Advances and Basis of Autoimmunity. Front Microbiol (2018) 9:2597. doi: 10.3389/fmicb.2018.02597
264. Sanchez E, De Palma G, Capilla A, Nova E, Pozo T, Castillejo G, et al. Influence of Environmental and Genetic Factors Linked to Celiac Disease Risk on Infant Gut Colonization by Bacteroides Species. Appl Environ Microbiol (2011) 77(15):5316–23. doi: 10.1128/AEM.00365-11
265. Collado MC, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Specific Duodenal and Faecal Bacterial Groups Associated With Paediatric Coeliac Disease. J Clin Pathol (2009) 62(3):264–9. doi: 10.1136/jcp.2008.061366
266. Verdu EF, Galipeau HJ, Jabri B. Novel Players in Coeliac Disease Pathogenesis: Role of the Gut Microbiota. Nat Rev Gastroenterol Hepatol (2015) 12(9):497–506. doi: 10.1038/nrgastro.2015.90
267. Ou G, Hedberg M, Horstedt P, Baranov V, Forsberg G, Drobni M, et al. Proximal Small Intestinal Microbiota and Identification of Rod-Shaped Bacteria Associated With Childhood Celiac Disease. Am J Gastroenterol (2009) 104(12):3058–67. doi: 10.1038/ajg.2009.524
268. Schippa S, Iebba V, Barbato M, Di Nardo G, Totino V, Checchi MP, et al. A Distinctive ‘Microbial Signature’ in Celiac Pediatric Patients. BMC Microbiol (2010) 10:175. doi: 10.1186/1471-2180-10-175
269. De Palma G, Cinova J, Stepankova R, Tuckova L, Sanz Y. Pivotal Advance: Bifidobacteria and Gram-negative Bacteria Differentially Influence Immune Responses in the Proinflammatory Milieu of Celiac Disease. J Leukoc Biol (2010) 87(5):765–78. doi: 10.1189/jlb.0709471
270. Di Cagno R, De Angelis M, De Pasquale I, Ndagijimana M, Vernocchi P, Ricciuti P, et al. Duodenal and Faecal Microbiota of Celiac Children: Molecular, Phenotype and Metabolome Characterization. BMC Microbiol (2011) 11:219. doi: 10.1186/1471-2180-11-219
271. Laparra JM, Sanz Y. Bifidobacteria Inhibit the Inflammatory Response Induced by Gliadins in Intestinal Epithelial Cells Via Modifications of Toxic Peptide Generation During Digestion. J Cell Biochem (2010) 109(4):801–7. doi: 10.1002/jcb.22459
272. Lindfors K, Blomqvist T, Juuti-Uusitalo K, Stenman S, Venalainen J, Maki M, et al. Live Probiotic Bifidobacterium Lactis Bacteria Inhibit the Toxic Effects Induced by Wheat Gliadin in Epithelial Cell Culture. Clin Exp Immunol (2008) 152(3):552–8. doi: 10.1111/j.1365-2249.2008.03635.x
273. Caminero A, McCarville JL, Zevallos VF, Pigrau M, Yu XB, Jury J, et al. Lactobacilli Degrade Wheat Amylase Trypsin Inhibitors to Reduce Intestinal Dysfunction Induced by Immunogenic Wheat Proteins. Gastroenterology (2019) 156(8):2266–80. doi: 10.1053/j.gastro.2019.02.028
274. Wacklin P, Laurikka P, Lindfors K, Collin P, Salmi T, Lahdeaho ML, et al. Altered Duodenal Microbiota Composition in Celiac Disease Patients Suffering From Persistent Symptoms on a Long-Term Gluten-Free Diet. Am J Gastroenterol (2014) 109(12):1933–41. doi: 10.1038/ajg.2014.355
275. Wacklin P, Kaukinen K, Tuovinen E, Collin P, Lindfors K, Partanen J, et al. The Duodenal Microbiota Composition of Adult Celiac Disease Patients is Associated With the Clinical Manifestation of the Disease. Inflammation Bowel Dis (2013) 19(5):934–41. doi: 10.1097/MIB.0b013e31828029a9
276. Zafeiropoulou K, Nichols B, Mackinder M, Biskou O, Rizou E, Karanikolou A, et al. Alterations in Intestinal Microbiota of Children With Celiac Disease at the Time of Diagnosis and on a Gluten-free Diet. Gastroenterology (2020) 159(6):2039–51.e20. doi: 10.1053/j.gastro.2020.08.007
277. De Angelis M, Vannini L, Di Cagno R, Cavallo N, Minervini F, Francavilla R, et al. Salivary and Fecal Microbiota and Metabolome of Celiac Children Under Gluten-Free Diet. Int J Food Microbiol (2016) 239:125–32. doi: 10.1016/j.ijfoodmicro.2016.07.025
278. Serena G, Yan S, Camhi S, Patel S, Lima RS, Sapone A, et al. Proinflammatory Cytokine Interferon-Gamma and Microbiome-Derived Metabolites Dictate Epigenetic Switch Between Forkhead Box Protein 3 Isoforms in Coeliac Disease. Clin Exp Immunol (2017) 187(3):490–506. doi: 10.1111/cei.12911
279. Tjellstrom B, Stenhammar L, Hogberg L, Falth-Magnusson K, Magnusson KE, Midtvedt T, et al. Gut Microflora Associated Characteristics in Children With Celiac Disease. Am J Gastroenterol (2005) 100(12):2784–8. doi: 10.1111/j.1572-0241.2005.00313.x
280. Francavilla R, Ercolini D, Piccolo M, Vannini L, Siragusa S, De Filippis F, et al. Salivary Microbiota and Metabolome Associated With Celiac Disease. Appl Environ Microbiol (2014) 80(11):3416–25. doi: 10.1128/AEM.00362-14
281. Akobeng AK, Heller RF. Assessing the Population Impact of Low Rates of Breast Feeding on Asthma, Coeliac Disease and Obesity: The Use of a New Statistical Method. Arch Dis Child (2007) 92(6):483–5. doi: 10.1136/adc.2006.097667
282. Lionetti E, Castellaneta S, Francavilla R, Pulvirenti A, Tonutti E, Amarri S, et al. Introduction of Gluten, HLA Status, and the Risk of Celiac Disease in Children. N Engl J Med (2014) 371(14):1295–303. doi: 10.1056/NEJMoa1400697
283. Vriezinga SL, Auricchio R, Bravi E, Castillejo G, Chmielewska A, Crespo Escobar P, et al. Randomized Feeding Intervention in Infants at High Risk for Celiac Disease. N Engl J Med (2014) 371(14):1304–15. doi: 10.1056/NEJMoa1404172
284. Wang C, Zhang M, Guo H, Yan J, Liu F, Chen J, et al. Human Milk Oligosaccharides Protect Against Necrotizing Enterocolitis by Inhibiting Intestinal Damage Via Increasing the Proliferation of Crypt Cells. Mol Nutr Food Res (2019) 63(18):e1900262. doi: 10.1002/mnfr.201900262
285. Dydensborg Sander S, Nybo Andersen AM, Murray JA, Karlstad O, Husby S, Stordal K. Association Between Antibiotics in the First Year of Life and Celiac Disease. Gastroenterology (2019) 156(8):2217–29. doi: 10.1053/j.gastro.2019.02.039
286. Canova C, Zabeo V, Pitter G, Romor P, Baldovin T, Zanotti R, et al. Association of Maternal Education, Early Infections, and Antibiotic Use With Celiac Disease: A Population-Based Birth Cohort Study in Northeastern Italy. Am J Epidemiol (2014) 180(1):76–85. doi: 10.1093/aje/kwu101
287. Marild K, Ye W, Lebwohl B, Green PH, Blaser MJ, Card T, et al. Antibiotic Exposure and the Development of Coeliac Disease: A Nationwide Case-Control Study. BMC Gastroenterol (2013) 13:109. doi: 10.1186/1471-230X-13-109
288. Radlovic NP, Mladenovic MM, Lekovic ZM, Stojsic ZM, Radlovic VN. Influence of Early Feeding Practices on Celiac Disease in Infants. Croat Med J (2010) 51(5):417–22. doi: 10.3325/cmj.2010.51.417
289. Welander A, Tjernberg AR, Montgomery SM, Ludvigsson J, Ludvigsson JF. Infectious Disease and Risk of Later Celiac Disease in Childhood. Pediatrics (2010) 125(3):e530–6. doi: 10.1542/peds.2009-1200
290. Verhasselt V. Oral Tolerance in Neonates: From Basics to Potential Prevention of Allergic Disease. Mucosal Immunol (2010) 3(4):326–33. doi: 10.1038/mi.2010.25
291. Verhasselt V. Neonatal Tolerance Under Breastfeeding Influence: The Presence of Allergen and Transforming Growth Factor-Beta in Breast Milk Protects the Progeny From Allergic Asthma. J Pediatr (2010) 156(2 Suppl):S16–20. doi: 10.1016/j.jpeds.2009.11.015
292. Ivarsson A, Hernell O, Stenlund H, Persson LA. Breast-Feeding Protects Against Celiac Disease. Am J Clin Nutr (2002) 75(5):914–21. doi: 10.1093/ajcn/75.5.914
293. Palma GD, Capilla A, Nova E, Castillejo G, Varea V, Pozo T, et al. Influence of Milk-Feeding Type and Genetic Risk of Developing Coeliac Disease on Intestinal Microbiota of Infants: The PROFICEL Study. PloS One (2012) 7(2):e30791. doi: 10.1371/journal.pone.0030791
294. Olivares M, Neef A, Castillejo G, Palma GD, Varea V, Capilla A, et al. The HLA-DQ2 Genotype Selects for Early Intestinal Microbiota Composition in Infants at High Risk of Developing Coeliac Disease. Gut (2015) 64(3):406–17. doi: 10.1136/gutjnl-2014-306931
295. Sellitto M, Bai G, Serena G, Fricke WF, Sturgeon C, Gajer P, et al. Proof of Concept of Microbiome-Metabolome Analysis and Delayed Gluten Exposure on Celiac Disease Autoimmunity in Genetically At-Risk Infants. PloS One (2012) 7(3):e33387. doi: 10.1371/journal.pone.0033387
296. Olshan KL, Leonard MM, Serena G, Zomorrodi AR, Fasano A. Gut Microbiota in Celiac Disease: Microbes, Metabolites, Pathways and Therapeutics. Expert Rev Clin Immunol (2020) 16(11):1075–92. doi: 10.1080/1744666X.2021.1840354
297. Han T, Li J. Gut Microbiota as a New Player in Children With Celiac Disease. J Gastroenterol Hepatol (2021) 36(1):39–40. doi: 10.1111/jgh.15322
298. Valitutti F, Cucchiara S, Fasano A. Celiac Disease and the Microbiome. Nutrients (2019) 11(10). doi: 10.3390/nu11102403
299. Sacchetti L, Nardelli C. Gut Microbiome Investigation in Celiac Disease: From Methods to its Pathogenetic Role. Clin Chem Lab Med (2020) 58(3):340–9. doi: 10.1515/cclm-2019-0657
300. Lebwohl B, Ludvigsson JF, Green PH. Celiac Disease and Non-Celiac Gluten Sensitivity. BMJ (2015) 351:h4347. doi: 10.1136/bmj.h4347
301. Ludvigsson JF, Ciacci C, Green PH, Kaukinen K, Korponay-Szabo IR, Kurppa K, et al. Outcome Measures in Coeliac Disease Trials: The Tampere Recommendations. Gut (2018) 67(8):1410–24. doi: 10.1136/gutjnl-2017-314853
302. Moreno ML, Cebolla A, Munoz-Suano A, Carrillo-Carrion C, Comino I, Pizarro A, et al. Detection of Gluten Immunogenic Peptides in the Urine of Patients With Coeliac Disease Reveals Transgressions in the Gluten-Free Diet and Incomplete Mucosal Healing. Gut (2017) 66(2):250–7. doi: 10.1136/gutjnl-2015-310148
303. Meyer S, Rosenblum S. Activities, Participation and Quality of Life Concepts in Children and Adolescents With Celiac Disease: A Scoping Review. Nutrients (2017) 9(9). doi: 10.3390/nu9090929
304. Murray JA, Kelly CP, Green PHR, Marcantonio A, Wu TT, Maki M, et al. No Difference Between Latiglutenase and Placebo in Reducing Villous Atrophy or Improving Symptoms in Patients With Symptomatic Celiac Disease. Gastroenterology (2017) 152(4):787–98.e2.
305. Syage JA, Murray JA, Green PHR, Khosla C. Latiglutenase Improves Symptoms in Seropositive Celiac Disease Patients While on a Gluten-Free Diet. Dig Dis Sci (2017) 62(9):2428–32. doi: 10.1007/s10620-017-4687-7
306. Leffler DA, Kelly CP, Green PH, Fedorak RN, DiMarino A, Perrow W, et al. Larazotide Acetate for Persistent Symptoms of Celiac Disease Despite a Gluten-Free Diet: A Randomized Controlled Trial. Gastroenterology (2015) 148(7):1311–9 e6. doi: 10.1053/j.gastro.2015.02.008
307. Risnes LF, Christophersen A, Dahal-Koirala S, Neumann RS, Sandve GK, Sarna VK, et al. Disease-Driving CD4+ T Cell Clonotypes Persist for Decades in Celiac Disease. J Clin Invest (2018) 128(6):2642–50. doi: 10.1172/JCI98819
308. Sollid LM, Qiao SW, Anderson RP, Gianfrani C, Koning F. Nomenclature and Listing of Celiac Disease Relevant Gluten T-Cell Epitopes Restricted by HLA-DQ Molecules. Immunogenetics (2012) 64(6):455–60. doi: 10.1007/s00251-012-0599-z
309. Goel G, King T, Daveson AJ, Andrews JM, Krishnarajah J, Krause R, et al. Epitope-Specific Immunotherapy Targeting CD4-Positive T Cells in Coeliac Disease: Two Randomised, Double-Blind, Placebo-Controlled Phase 1 Studies. Lancet Gastroenterol Hepatol (2017) 2(7):479–93. doi: 10.1016/S2468-1253(17)30110-3
310. Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Synthetic Nanoparticles for Vaccines and Immunotherapy. Chem Rev (2015) 115(19):11109–46. doi: 10.1021/acs.chemrev.5b00109
311. Kishimoto TK, Maldonado RA. Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance. Front Immunol (2018) 9:230. doi: 10.3389/fimmu.2018.00230
312. Pearson RM, Podojil JR, Shea LD, King NJC, Miller SD, Getts DR. Overcoming Challenges in Treating Autoimmuntity: Development of Tolerogenic Immune-Modifying Nanoparticles. Nanomedicine (2019) 18:282–91. doi: 10.1016/j.nano.2018.10.001
313. Pishesha N, Bilate AM, Wibowo MC, Huang NJ, Li Z, Deshycka R, et al. Engineered Erythrocytes Covalently Linked to Antigenic Peptides Can Protect Against Autoimmune Disease. Proc Natl Acad Sci USA (2017) 114(12):3157–62. doi: 10.1073/pnas.1701746114
314. Grimm AJ, Kontos S, Diaceri G, Quaglia-Thermes X, Hubbell JA. Memory of Tolerance and Induction of Regulatory T Cells by Erythrocyte-Targeted Antigens. Sci Rep (2015) 5:15907. doi: 10.1038/srep15907
315. Cook DP, Gysemans C, Mathieu C. Lactococcus Lactis As a Versatile Vehicle for Tolerogenic Immunotherapy. Front Immunol (2017) 8:1961. doi: 10.3389/fimmu.2017.01961
316. Majri SS, Fritz JM, Villarino AV, Zheng L, Kanellopoulou C, Chaigne-Delalande B, et al. Stat5b: A Differential Regulator of the Life and Death of CD4(+) Effector Memory T Cells. J Immunol (2018) 200(1):110–8. doi: 10.4049/jimmunol.1701133
317. Zheng L, Li J, Lenardo M. Restimulation-Induced Cell Death: New Medical and Research Perspectives. Immunol Rev (2017) 277(1):44–60. doi: 10.1111/imr.12535
318. Geltink RIK, Kyle RL, Pearce EL. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu Rev Immunol (2018) 36:461–88. doi: 10.1146/annurev-immunol-042617-053019
319. Valitutti F, Trovato CM, Montuori M, Cucchiara S. Pediatric Celiac Disease: Follow-Up in the Spotlight. Adv Nutr (2017) 8(2):356–61. doi: 10.3945/an.116.013292
320. Norsa L, Tomba C, Agostoni C, Branchi F, Bardella MT, Roncoroni L, et al. Gluten-Free Diet or Alternative Therapy: A Survey on What Parents of Celiac Children Want. Int J Food Sci Nutr (2015) 66(5):590–4. doi: 10.3109/09637486.2015.1064872
321. Pisapia L, Camarca A, Picascia S, Bassi V, Barba P, Del Pozzo G, et al. Hla-DQ2.5 Genes Associated With Celiac Disease Risk are Preferentially Expressed With Respect to non-Predisposing HLA Genes: Implication for Anti-Gluten T Cell Response. J Autoimmun (2016) 70:63–72. doi: 10.1016/j.jaut.2016.03.016
322. Traykova D, Schneider B, Chojkier M, Buck M. Blood Microbiome Quantity and the Hyperdynamic Circulation in Decompensated Cirrhotic Patients. PloS One (2017) 12(2):e0169310. doi: 10.1371/journal.pone.0169310
323. De Angelis M, Rizzello CG, Fasano A, Clemente MG, De Simone C, Silano M, et al. VSL3 Probiotic Preparation has the Capacity to Hydrolyze Gliadin Polypeptides Responsible for Celiac Sprue. Biochim Biophys Acta (2006) 1762(1):80–93. doi: 10.1016/j.bbadis.2005.09.008
324. Primec M, Micetic-Turk D, Langerholc T. Analysis of Short-Chain Fatty Acids in Human Feces: A Scoping Review. Anal Biochem (2017) 526:9–21. doi: 10.1016/j.ab.2017.03.007
325. Serena G, Kelly CP, Fasano A. Nondietary Therapies for Celiac Disease. Gastroenterol Clin North Am (2019) 48(1):145–63. doi: 10.1016/j.gtc.2018.09.011
326. Nistal E, Caminero A, Herran AR, Arias L, Vivas S, de Morales JM, et al. Differences of Small Intestinal Bacteria Populations in Adults and Children With/Without Celiac Disease: Effect of Age, Gluten Diet, and Disease. Inflammation Bowel Dis (2012) 18(4):649–56. doi: 10.1002/ibd.21830
327. Bodkhe R, Shetty SA, Dhotre DP, Verma AK, Bhatia K, Mishra A, et al. Comparison of Small Gut and Whole Gut Microbiota of First-Degree Relatives With Adult Celiac Disease Patients and Controls. Front Microbiol (2019) 10:164. doi: 10.3389/fmicb.2019.00164
328. Cenit MC, Olivares M, Codoner-Franch P, Sanz Y. Intestinal Microbiota and Celiac Disease: Cause, Consequence or Co-Evolution? Nutrients (2015) 7(8):6900–23. doi: 10.3390/nu7085314
329. Pecora F, Persico F, Gismondi P, Fornaroli F, Iuliano S, de’Angelis GL, et al. Gut Microbiota in Celiac Disease: Is There Any Role for Probiotics? Front Immunol (2020) 11:957. doi: 10.3389/fimmu.2020.00957
330. Rizzello CG, De Angelis M, Di Cagno R, Camarca A, Silano M, Losito I, et al. Highly Efficient Gluten Degradation by Lactobacilli and Fungal Proteases During Food Processing: New Perspectives for Celiac Disease. Appl Environ Microbiol (2007) 73(14):4499–507. doi: 10.1128/AEM.00260-07
331. Duar RM, Clark KJ, Patil PB, Hernandez C, Bruning S, Burkey TE, et al. Identification and Characterization of Intestinal Lactobacilli Strains Capable of Degrading Immunotoxic Peptides Present in Gluten. J Appl Microbiol (2015) 118(2):515–27. doi: 10.1111/jam.12687
332. Mandile R, Picascia S, Parrella C, Camarca A, Gobbetti M, Greco L, et al. Lack of Immunogenicity of Hydrolysed Wheat Flour in Patients With Coeliac Disease After a Short-Term Oral Challenge. Aliment Pharmacol Ther (2017) 46(4):440–6. doi: 10.1111/apt.14175
333. Greco L, Gobbetti M, Auricchio R, Di Mase R, Landolfo F, Paparo F, et al. Safety for Patients With Celiac Disease of Baked Goods Made of Wheat Flour Hydrolyzed During Food Processing. Clin Gastroenterol Hepatol (2011) 9(1):24–9. doi: 10.1016/j.cgh.2010.09.025
334. Francavilla R, De Angelis M, Rizzello CG, Cavallo N, Dal Bello F, Gobbetti M. Selected Probiotic Lactobacilli Have the Capacity to Hydrolyze Gluten Peptides During Simulated Gastrointestinal Digestion. Appl Environ Microbiol (2017) 83(14):e00376–17. doi: 10.1128/AEM.00376-17
335. Medina M, De Palma G, Ribes-Koninckx C, Calabuig M, Sanz Y. Bifidobacterium Strains Suppress In Vitro the Pro-Inflammatory Milieu Triggered by the Large Intestinal Microbiota of Coeliac Patients. J Inflammation (Lond) (2008) 5:19. doi: 10.1186/1476-9255-5-19
336. Olivares M, Laparra M, Sanz Y. Oral Administration of Bifidobacterium Longum CECT 7347 Modulates Jejunal Proteome in an In Vivo Gliadin-Induced Enteropathy Animal Model. J Proteomics (2012) 77:310–20. doi: 10.1016/j.jprot.2012.09.005
337. D’Arienzo R, Maurano F, Luongo D, Mazzarella G, Stefanile R, Troncone R, et al. Adjuvant Effect of Lactobacillus Casei in a Mouse Model of Gluten Sensitivity. Immunol Lett (2008) 119(1-2):78–83. doi: 10.1016/j.imlet.2008.04.006
338. D’Arienzo R, Stefanile R, Maurano F, Mazzarella G, Ricca E, Troncone R, et al. Immunomodulatory Effects of Lactobacillus Casei Administration in a Mouse Model of Gliadin-Sensitive Enteropathy. Scand J Immunol (2011) 74(4):335–41. doi: 10.1111/j.1365-3083.2011.02582.x
339. Laparra JM, Olivares M, Gallina O, Sanz Y. Bifidobacterium Longum CECT 7347 Modulates Immune Responses in a Gliadin-Induced Enteropathy Animal Model. PloS One (2012) 7(2):e30744. doi: 10.1371/journal.pone.0030744
340. Dignass A, Van Assche G, Lindsay JO, Lemann M, Soderholm J, Colombel JF, et al. The Second European Evidence-Based Consensus on the Diagnosis and Management of Crohn’s Disease: Current Management. J Crohns Colitis (2010) 4(1):28–62. doi: 10.1016/j.crohns.2009.12.002
341. Ruemmele FM, Veres G, Kolho KL, Griffiths A, Levine A, Escher JC, et al. Consensus Guidelines of ECCO/ESPGHAN on the Medical Management of Pediatric Crohn’s Disease. J Crohns Colitis (2014) 8(10):1179–207. doi: 10.1016/j.crohns.2014.04.005
342. Turner D, Levine A, Escher JC, Griffiths AM, Russell RK, Dignass A, et al. Management of Pediatric Ulcerative Colitis: Joint ECCO and ESPGHAN Evidence-Based Consensus Guidelines. J Pediatr Gastroenterol Nutr (2012) 55(3):340–61. doi: 10.1097/MPG.0b013e3182662233
343. Oliveira SB, Monteiro IM. Diagnosis and Management of Inflammatory Bowel Disease in Children. BMJ (2017) 357:j2083. doi: 10.1136/bmj.j2083
344. Ponder A, Long MD. A Clinical Review of Recent Findings in the Epidemiology of Inflammatory Bowel Disease. Clin Epidemiol (2013) 5:237–47. doi: 10.2147/CLEP.S33961
345. Benchimol EI, Bernstein CN, Bitton A, Carroll MW, Singh H, Otley AR, et al. Trends in Epidemiology of Pediatric Inflammatory Bowel Disease in Canada: Distributed Network Analysis of Multiple Population-Based Provincial Health Administrative Databases. Am J Gastroenterol (2017) 112(7):1120–34. doi: 10.1038/ajg.2017.97
346. Kaplan GG. The Global Burden of IBD: From 2015 to 2025. Nat Rev Gastroenterol Hepatol (2015) 12(12):720–7. doi: 10.1038/nrgastro.2015.150
347. Khor B, Gardet A, Xavier RJ. Genetics and Pathogenesis of Inflammatory Bowel Disease. Nature (2011) 474(7351):307–17. doi: 10.1038/nature10209
348. Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association Analyses Identify 38 Susceptibility Loci for Inflammatory Bowel Disease and Highlight Shared Genetic Risk Across Populations. Nat Genet (2015) 47(9):979–86. doi: 10.1038/ng.3359
349. Diaz-Gallo LM, Espino-Paisan L, Fransen K, Gomez-Garcia M, van Sommeren S, Cardena C, et al. Differential Association of Two PTPN22 Coding Variants With Crohn’s Disease and Ulcerative Colitis. Inflammation Bowel Dis (2011) 17(11):2287–94. doi: 10.1002/ibd.21630
350. Yilmaz B, Spalinger MR, Biedermann L, Franc Y, Fournier N, Rossel JB, et al. The Presence of Genetic Risk Variants Within PTPN2 and PTPN22 Is Associated With Intestinal Microbiota Alterations in Swiss IBD Cohort Patients. PloS One (2018) 13(7):e0199664. doi: 10.1371/journal.pone.0199664
351. Spalinger MR, Schmidt TS, Schwarzfischer M, Hering L, Atrott K, Lang S, et al. Protein Tyrosine Phosphatase non-Receptor Type 22 Modulates Colitis in a Microbiota-Dependent Manner. J Clin Invest (2019) 129(6):2527–41. doi: 10.1172/JCI123263
352. Childers RE, Eluri S, Vazquez C, Weise RM, Bayless TM, Hutfless S. Family History of Inflammatory Bowel Disease Among Patients With Ulcerative Colitis: A Systematic Review and Meta-Analysis. J Crohns Colitis (2014) 8(11):1480–97. doi: 10.1016/j.crohns.2014.05.008
353. Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, Ahmad T, et al. Inherited Determinants of Crohn’s Disease and Ulcerative Colitis Phenotypes: A Genetic Association Study. Lancet (2016) 387(10014):156–67. doi: 10.1016/S0140-6736(15)00465-1
354. O’Donoghue DP, Dawson AM. Crohn’s Disease in Childhood. Arch Dis Child (1977) 52(8):627–32. doi: 10.1136/adc.52.8.627
355. North American Society for Pediatric Gastroenterology H, Nutrition, Colitis Foundation of A, Bousvaros A, Antonioli DA, Colletti RB, Dubinsky MC, Glickman JN, Gold BD, et al. Differentiating Ulcerative Colitis From Crohn Disease in Children and Young Adults: Report of a Working Group of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the Crohn’s and Colitis Foundation of America. J Pediatr Gastroenterol Nutr (2007) 44(5):653–74. doi: 10.1097/MPG.0b013e31805563f3
356. Kugathasan S, Judd RH, Hoffmann RG, Heikenen J, Telega G, Khan F, et al. Epidemiologic and Clinical Characteristics of Children With Newly Diagnosed Inflammatory Bowel Disease in Wisconsin: A Statewide Population-Based Study. J Pediatr (2003) 143(4):525–31. doi: 10.1067/S0022-3476(03)00444-X
357. Danese S, Fiocchi C. Ulcerative Colitis. N Engl J Med (2011) 365(18):1713–25. doi: 10.1056/NEJMra1102942
358. Levine A, Koletzko S, Turner D, Escher JC, Cucchiara S, de Ridder L, et al. ESPGHAN Revised Porto Criteria for the Diagnosis of Inflammatory Bowel Disease in Children and Adolescents. J Pediatr Gastroenterol Nutr (2014) 58(6):795–806. doi: 10.1097/MPG.0000000000000239
359. Griffiths AM. Specificities of Inflammatory Bowel Disease in Childhood. Best Pract Res Clin Gastroenterol (2004) 18(3):509–23. doi: 10.1016/j.bpg.2004.01.002
360. Henderson P, Anderson NH, Wilson DC. The Diagnostic Accuracy of Fecal Calprotectin During the Investigation of Suspected Pediatric Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. Am J Gastroenterol (2014) 109(5):637–45. doi: 10.1038/ajg.2013.131
361. Lin JF, Chen JM, Zuo JH, Yu A, Xiao ZJ, Deng FH, et al. Meta-Analysis: Fecal Calprotectin for Assessment of Inflammatory Bowel Disease Activity. Inflammation Bowel Dis (2014) 20(8):1407–15. doi: 10.1097/MIB.0000000000000057
362. Bernstein CN, Rawsthorne P, Cheang M. Blanchard Jf. A Population-Based Case Control Study of Potential Risk Factors for IBD. Am J Gastroenterol (2006) 101(5):993–1002. doi: 10.1111/j.1572-0241.2006.00381.x
363. Hou JK, Abraham B, El-Serag H. Dietary Intake and Risk of Developing Inflammatory Bowel Disease: A Systematic Review of the Literature. Am J Gastroenterol (2011) 106(4):563–73. doi: 10.1038/ajg.2011.44
364. Xavier RJ, Podolsky DK. Unravelling the Pathogenesis of Inflammatory Bowel Disease. Nature (2007) 448(7152):427–34. doi: 10.1038/nature06005
365. Alipour M, Zaidi D, Valcheva R, Jovel J, Martinez I, Sergi C, et al. Mucosal Barrier Depletion and Loss of Bacterial Diversity Are Primary Abnormalities in Paediatric Ulcerative Colitis. J Crohns Colitis (2016) 10(4):462–71. doi: 10.1093/ecco-jcc/jjv223
366. Clooney AG, Eckenberger J, Laserna-Mendieta E, Sexton KA, Bernstein MT, Vagianos K, et al. Ranking Microbiome Variance in Inflammatory Bowel Disease: A Large Longitudinal Intercontinental Study. Gut (2021) 70(3):499–510. doi: 10.1136/gutjnl-2020-321106
367. Michail S, Durbin M, Turner D, Griffiths AM, Mack DR, Hyams J, et al. Alterations in the Gut Microbiome of Children With Severe Ulcerative Colitis. Inflammation Bowel Dis (2012) 18(10):1799–808. doi: 10.1002/ibd.22860
368. Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, et al. Immunoglobulin A Coating Identifies Colitogenic Bacteria in Inflammatory Bowel Disease. Cell (2014) 158(5):1000–10. doi: 10.1016/j.cell.2014.08.006
369. Leake I. Ibd: Microbial Drivers of IBD Identified by Levels of IgA Coating. Nat Rev Gastroenterol Hepatol (2014) 11(11):642. doi: 10.1038/nrgastro.2014.166
370. Armstrong H, Alipour M, Valcheva R, Bording-Jorgensen M, Jovel J, Zaidi D, et al. Host Immunoglobulin G Selectively Identifies Pathobionts in Pediatric Inflammatory Bowel Diseases. Microbiome (2019) 7(1):1. doi: 10.1186/s40168-018-0604-3
371. Harmsen HJ, Pouwels SD, Funke A, Bos NA, Dijkstra G. Crohn’s Disease Patients Have More IgG-Binding Fecal Bacteria Than Controls. Clin Vaccine Immunol (2012) 19(4):515–21. doi: 10.1128/CVI.05517-11
372. Benckert J, Schmolka N, Kreschel C, Zoller MJ, Sturm A, Wiedenmann B, et al. The Majority of Intestinal IgA+ and IgG+ Plasmablasts in the Human Gut Are Antigen-Specific. J Clin Invest (2011) 121(5):1946–55. doi: 10.1172/JCI44447
373. Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-Phylogenetic Characterization of Microbial Community Imbalances in Human Inflammatory Bowel Diseases. Proc Natl Acad Sci USA (2007) 104(34):13780–5. doi: 10.1073/pnas.0706625104
374. Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H. Spatial Organization and Composition of the Mucosal Flora in Patients With Inflammatory Bowel Disease. J Clin Microbiol (2005) 43(7):3380–9. doi: 10.1128/JCM.43.7.3380-3389.2005
375. Gophna U, Sommerfeld K, Gophna S, Doolittle WF, Veldhuyzen van Zanten SJ. Differences Between Tissue-Associated Intestinal Microfloras of Patients With Crohn’s Disease and Ulcerative Colitis. J Clin Microbiol (2006) 44(11):4136–41. doi: 10.1128/JCM.01004-06
376. Yilmaz B, Juillerat P, Oyas O, Ramon C, Bravo FD, Franc Y, et al. Microbial Network Disturbances in Relapsing Refractory Crohn’s Disease. Nat Med (2019) 25(2):323–36. doi: 10.1038/s41591-018-0308-z
377. Pascal V, Pozuelo M, Borruel N, Casellas F, Campos D, Santiago A, et al. A Microbial Signature for Crohn’s Disease. Gut (2017) 66(5):813–22. doi: 10.1136/gutjnl-2016-313235
378. Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, et al. Gut Microbiome Structure and Metabolic Activity in Inflammatory Bowel Disease. Nat Microbiol (2019) 4(2):293–305. doi: 10.1038/s41564-018-0306-4
379. Arabyan N, Park D, Foutouhi S, Weis AM, Huang BC, Williams CC, et al. Salmonella Degrades the Host Glycocalyx Leading to Altered Infection and Glycan Remodeling. Sci Rep (2016) 6:29525. doi: 10.1038/srep29525
380. Henderson IR, Czeczulin J, Eslava C, Noriega F, Nataro JP. Characterization of Pic, a Secreted Protease of Shigella Flexneri and Enteroaggregative Escherichia Coli. Infect Immun (1999) 67(11):5587–96. doi: 10.1128/IAI.67.11.5587-5596.1999
381. Mantle M, Rombough C. Growth in and Breakdown of Purified Rabbit Small Intestinal Mucin by Yersinia Enterocolitica. Infect Immun (1993) 61(10):4131–8. doi: 10.1128/IAI.61.10.4131-4138.1993
382. Szabady RL, Yanta JH, Halladin DK, Schofield MJ, Welch RA. Taga is a Secreted Protease of Vibrio Cholerae That Specifically Cleaves Mucin Glycoproteins. Microbiol (Reading) (2011) 157(Pt 2):516–25. doi: 10.1099/mic.0.044529-0
383. Macpherson AJ, Slack E, Geuking MB, McCoy KD. The Mucosal Firewalls Against Commensal Intestinal Microbes. Semin Immunopathol (2009) 31(2):145–9. doi: 10.1007/s00281-009-0174-3
384. Belkaid Y, Hand TW. Role of the Microbiota in Immunity and Inflammation. Cell (2014) 157(1):121–41. doi: 10.1016/j.cell.2014.03.011
385. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, et al. Faecalibacterium Prausnitzii Is an Anti-Inflammatory Commensal Bacterium Identified by Gut Microbiota Analysis of Crohn Disease Patients. Proc Natl Acad Sci USA (2008) 105(43):16731–6. doi: 10.1073/pnas.0804812105
386. Aujnarain A, Mack DR, Benchimol EI. The Role of the Environment in the Development of Pediatric Inflammatory Bowel Disease. Curr Gastroenterol Rep (2013) 15(6):326. doi: 10.1007/s11894-013-0326-4
387. Khan MT, Duncan SH, Stams AJ, van Dijl JM, Flint HJ, Harmsen HJ. The Gut Anaerobe Faecalibacterium Prausnitzii Uses an Extracellular Electron Shuttle to Grow at Oxic-Anoxic Interphases. ISME J (2012) 6(8):1578–85. doi: 10.1038/ismej.2012.5
388. Neyrinck AM, Possemiers S, Druart C, Van de Wiele T, De Backer F, Cani PD, et al. Prebiotic Effects of Wheat Arabinoxylan Related to the Increase in Bifidobacteria, Roseburia and Bacteroides/Prevotella in Diet-Induced Obese Mice. PloS One (2011) 6(6):e20944. doi: 10.1371/journal.pone.0020944
389. Velasquez-Manoff M. Gut Microbiome: The Peacekeepers. Nature (2015) 518(7540):S3–11. doi: 10.1038/518S3a
390. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal Microbe-Derived Butyrate Induces the Differentiation of Colonic Regulatory T Cells. Nature (2013) 504(7480):446–50. doi: 10.1038/nature12721
391. Gaudier E, Jarry A, Blottiere HM, de Coppet P, Buisine MP, Aubert JP, et al. Butyrate Specifically Modulates MUC Gene Expression in Intestinal Epithelial Goblet Cells Deprived of Glucose. Am J Physiol Gastrointest Liver Physiol (2004) 287(6):G1168–74. doi: 10.1152/ajpgi.00219.2004
392. Wlodarska M, Luo C, Kolde R, d’Hennezel E, Annand JW, Heim CE, et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe (2017) 22(1):25–37.e6. doi: 10.1016/j.chom.2017.06.007
393. Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, et al. CARD9 Impacts Colitis by Altering Gut Microbiota Metabolism of Tryptophan Into Aryl Hydrocarbon Receptor Ligands. Nat Med (2016) 22(6):598–605. doi: 10.1038/nm.4102
394. Armstrong H, Mander I, Zhang Z, Armstrong D, Wine E. Not All Fibers Are Born Equal; Variable Response to Dietary Fiber Subtypes in IBD. Front Pediatr (2020) 8:620189. doi: 10.3389/fped.2020.620189
395. Castro F, de Souza HSP. Dietary Composition and Effects in Inflammatory Bowel Disease. Nutrients (2019) 11(6):1398. doi: 10.3390/nu11061398
396. Levine A, Rhodes JM, Lindsay JO, Abreu MT, Kamm MA, Gibson PR, et al. Dietary Guidance From the International Organization for the Study of Inflammatory Bowel Diseases. Clin Gastroenterol Hepatol (2020) 18(6):1381–92. doi: 10.1016/j.cgh.2020.01.046
397. Halfvarson J, Brislawn CJ, Lamendella R, Vazquez-Baeza Y, Walters WA, Bramer LM, et al. Dynamics of the Human Gut Microbiome in Inflammatory Bowel Disease. Nat Microbiol (2017) 2:17004. doi: 10.1038/nmicrobiol.2017.4
398. Mars RAT, Yang Y, Ward T, Houtti M, Priya S, Lekatz HR, et al. Longitudinal Multi-Omics Reveals Subset-Specific Mechanisms Underlying Irritable Bowel Syndrome. Cell (2020) 182(6):1460–73.e17. doi: 10.1016/j.cell.2020.08.007
399. Kaur A, Goggolidou P. Ulcerative Colitis: Understanding Its Cellular Pathology Could Provide Insights Into Novel Therapies. J Inflammation (Lond) (2020) 17:15. doi: 10.1186/s12950-020-00246-4
400. Na SY, Moon W. Perspectives on Current and Novel Treatments for Inflammatory Bowel Disease. Gut Liver (2019) 13(6):604–16. doi: 10.5009/gnl19019
401. Lee D, Baldassano RN, Otley AR, Albenberg L, Griffiths AM, Compher C, et al. Comparative Effectiveness of Nutritional and Biological Therapy in North American Children With Active Crohn’s Disease. Inflammation Bowel Dis (2015) 21(8):1786–93. doi: 10.1097/MIB.0000000000000426
402. Borrelli O, Cordischi L, Cirulli M, Paganelli M, Labalestra V, Uccini S, et al. Polymeric Diet Alone Versus Corticosteroids in the Treatment of Active Pediatric Crohn’s Disease: A Randomized Controlled Open-Label Trial. Clin Gastroenterol Hepatol (2006) 4(6):744–53. doi: 10.1016/j.cgh.2006.03.010
403. Grover Z, Muir R, Lewindon P. Exclusive Enteral Nutrition Induces Early Clinical, Mucosal and Transmural Remission in Paediatric Crohn’s Disease. J Gastroenterol (2014) 49(4):638–45. doi: 10.1007/s00535-013-0815-0
404. Ashton JJ, Versteegh HP, Batra A, Afzal NA, King A, Griffiths DM, et al. Colectomy in Pediatric Ulcerative Colitis: A Single Center Experience of Indications, Outcomes, and Complications. J Pediatr Surg (2016) 51(2):277–81. doi: 10.1016/j.jpedsurg.2015.10.077
405. Blackburn SC, Wiskin AE, Barnes C, Dick K, Afzal NA, Griffiths DM, et al. Surgery for Children With Crohn’s Disease: Indications, Complications and Outcome. Arch Dis Child (2014) 99(5):420–6. doi: 10.1136/archdischild-2013-305214
406. Maul J, Loddenkemper C, Mundt P, Berg E, Giese T, Stallmach A, et al. Peripheral and Intestinal Regulatory CD4+ CD25(High) T Cells in Inflammatory Bowel Disease. Gastroenterology (2005) 128(7):1868–78. doi: 10.1053/j.gastro.2005.03.043
407. Mottet C, Uhlig HH, Powrie F. Cutting Edge: Cure of Colitis by CD4+CD25+ Regulatory T Cells. J Immunol (2003) 170(8):3939–43. doi: 10.4049/jimmunol.170.8.3939
408. Izcue A, Hue S, Buonocore S, Arancibia-Carcamo CV, Ahern PP, Iwakura Y, et al. Interleukin-23 Restrains Regulatory T Cell Activity to Drive T Cell-Dependent Colitis. Immunity (2008) 28(4):559–70. doi: 10.1016/j.immuni.2008.02.019
409. Coombes JL, Robinson NJ, Maloy KJ, Uhlig HH, Powrie F. Regulatory T Cells and Intestinal Homeostasis. Immunol Rev (2005) 204:184–94. doi: 10.1111/j.0105-2896.2005.00250.x
410. Ogino H, Nakamura K, Iwasa T, Ihara E, Akiho H, Motomura Y, et al. Regulatory T Cells Expanded by Rapamycin In Vitro Suppress Colitis in an Experimental Mouse Model. J Gastroenterol (2012) 47(4):366–76. doi: 10.1007/s00535-011-0502-y
411. Foussat A, Cottrez F, Brun V, Fournier N, Breittmayer JP, Groux H. A Comparative Study Between T Regulatory Type 1 and CD4+CD25+ T Cells in the Control of Inflammation. J Immunol (2003) 171(10):5018–26. doi: 10.4049/jimmunol.171.10.5018
412. Powrie F, Leach MW, Mauze S, Menon S, Caddle LB, Coffman RL. Inhibition of Th1 Responses Prevents Inflammatory Bowel Disease in Scid Mice Reconstituted With CD45RBhi Cd4+ T Cells. Immunity (1994) 1(7):553–62. doi: 10.1016/1074-7613(94)90045-0
413. Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. Regulatory T Cell-Derived interleukin-10 Limits Inflammation at Environmental Interfaces. Immunity (2008) 28(4):546–58. doi: 10.1016/j.immuni.2008.02.017
414. Golovina TN, Mikheeva T, Brusko TM, Blazar BR, Bluestone JA, Riley JL. Retinoic Acid and Rapamycin Differentially Affect and Synergistically Promote the Ex Vivo Expansion of Natural Human T Regulatory Cells. PloS One (2011) 6(1):e15868. doi: 10.1371/journal.pone.0015868
415. Scotta C, Esposito M, Fazekasova H, Fanelli G, Edozie FC, Ali N, et al. Differential Effects of Rapamycin and Retinoic Acid on Expansion, Stability and Suppressive Qualities of Human CD4(+)CD25(+)FOXP3(+) T Regulatory Cell Subpopulations. Haematologica (2013) 98(8):1291–9. doi: 10.3324/haematol.2012.074088
416. Goldberg R, Scotta C, Cooper D, Nissim-Eliraz E, Nir E, Tasker S, et al. Correction of Defective T-Regulatory Cells From Patients With Crohn’s Disease by Ex Vivo Ligation of Retinoic Acid Receptor-Alpha. Gastroenterology (2019) 156(6):1775–87. doi: 10.1053/j.gastro.2019.01.025
417. Canavan JB, Scotta C, Vossenkamper A, Goldberg R, Elder MJ, Shoval I, et al. Developing In Vitro Expanded CD45RA+ Regulatory T Cells as an Adoptive Cell Therapy for Crohn’s Disease. Gut (2016) 65(4):584–94. doi: 10.1136/gutjnl-2014-306919
418. Hoeppli RE, MacDonald KN, Leclair P, Fung VCW, Mojibian M, Gillies J, et al. Tailoring the Homing Capacity of Human Tregs for Directed Migration to Sites of Th1-Inflammation or Intestinal Regions. Am J Transplant (2019) 19(1):62–76. doi: 10.1111/ajt.14936
419. Xu Y, Cheng Y, Baylink DJ, Wasnik S, Goel G, Huang M, et al. In Vivo Generation of Gut-Homing Regulatory T Cells for the Suppression of Colitis. J Immunol (2019) 202(12):3447–57. doi: 10.4049/jimmunol.1800018
420. Nishio J, Baba M, Atarashi K, Tanoue T, Negishi H, Yanai H, et al. Requirement of Full TCR Repertoire for Regulatory T Cells to Maintain Intestinal Homeostasis. Proc Natl Acad Sci USA (2015) 112(41):12770–5. doi: 10.1073/pnas.1516617112
421. Elinav E, Waks T, Eshhar Z. Redirection of Regulatory T Cells With Predetermined Specificity for the Treatment of Experimental Colitis in Mice. Gastroenterology (2008) 134(7):2014–24. doi: 10.1053/j.gastro.2008.02.060
422. Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z. Suppression of Murine Colitis and its Associated Cancer by Carcinoembryonic Antigen-Specific Regulatory T Cells. Mol Ther (2014) 22(5):1018–28. doi: 10.1038/mt.2014.41
423. Desreumaux P, Foussat A, Allez M, Beaugerie L, Hebuterne X, Bouhnik Y, et al. Safety and Efficacy of Antigen-Specific Regulatory T-Cell Therapy for Patients With Refractory Crohn’s Disease. Gastroenterology (2012) 143(5):1207–17.e2. doi: 10.1053/j.gastro.2012.07.116
424. Ford AC, Quigley EM, Lacy BE, Lembo AJ, Saito YA, Schiller LR, et al. Efficacy of Prebiotics, Probiotics, and Synbiotics in Irritable Bowel Syndrome and Chronic Idiopathic Constipation: Systematic Review and Meta-Analysis. Am J Gastroenterol (2014) 109(10):1547–61. doi: 10.1038/ajg.2014.202
425. Kruis W, Fric P, Pokrotnieks J, Lukas M, Fixa B, Kascak M, et al. Maintaining Remission of Ulcerative Colitis With the Probiotic Escherichia Coli Nissle 1917 Is as Effective as With Standard Mesalazine. Gut (2004) 53(11):1617–23. doi: 10.1136/gut.2003.037747
426. Sassone-Corsi M, Nuccio SP, Liu H, Hernandez D, Vu CT, Takahashi AA, et al. Microcins Mediate Competition Among Enterobacteriaceae in the Inflamed Gut. Nature (2016) 540(7632):280–3. doi: 10.1038/nature20557
427. Ghouri YA, Richards DM, Rahimi EF, Krill JT, Jelinek KA, DuPont AW. Systematic Review of Randomized Controlled Trials of Probiotics, Prebiotics, and Synbiotics in Inflammatory Bowel Disease. Clin Exp Gastroenterol (2014) 7:473–87. doi: 10.2147/CEG.S27530
428. Lamb CA, Kennedy NA, Raine T, Hendy PA, Smith PJ, Limdi JK, et al. British Society of Gastroenterology Consensus Guidelines on the Management of Inflammatory Bowel Disease in Adults. Gut (2019) 68(Suppl 3):s1–s106. doi: 10.1136/gutjnl-2019-318484
429. Rutgeerts P, Goboes K, Peeters M, Hiele M, Penninckx F, Aerts R, et al. Effect of Faecal Stream Diversion on Recurrence of Crohn’s Disease in the Neoterminal Ileum. Lancet (1991) 338(8770):771–4. doi: 10.1016/0140-6736(91)90663-A
430. Konig J, Siebenhaar A, Hogenauer C, Arkkila P, Nieuwdorp M, Noren T, et al. Consensus Report: Faecal Microbiota Transfer - Clinical Applications and Procedures. Aliment Pharmacol Ther (2017) 45(2):222–39. doi: 10.1111/apt.13868
431. Thia KT, Mahadevan U, Feagan BG, Wong C, Cockeram A, Bitton A, et al. Ciprofloxacin or Metronidazole for the Treatment of Perianal Fistulas in Patients With Crohn’s Disease: A Randomized, Double-Blind, Placebo-Controlled Pilot Study. Inflammation Bowel Dis (2009) 15(1):17–24. doi: 10.1002/ibd.20608
432. Prantera C, Lochs H, Grimaldi M, Danese S, Scribano ML, Gionchetti P, et al. Rifaximin-Extended Intestinal Release Induces Remission in Patients With Moderately Active Crohn’s Disease. Gastroenterology (2012) 142(3):473–81 e4. doi: 10.1053/j.gastro.2011.11.032
433. Khan KJ, Ullman TA, Ford AC, Abreu MT, Abadir A, Marshall JK, et al. Antibiotic Therapy in Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. Am J Gastroenterol (2011) 106(4):661–73. doi: 10.1038/ajg.2011.72
434. Wang SL, Wang ZR, Yang CQ. Meta-Analysis of Broad-Spectrum Antibiotic Therapy in Patients With Active Inflammatory Bowel Disease. Exp Ther Med (2012) 4(6):1051–6. doi: 10.3892/etm.2012.718
435. Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe (2017) 22(2):247. doi: 10.1016/j.chom.2017.07.011
436. Yang BH, Hagemann S, Mamareli P, Lauer U, Hoffmann U, Beckstette M, et al. Foxp3(+) T Cells Expressing RORgammat Represent a Stable Regulatory T-cell Effector Lineage With Enhanced Suppressive Capacity During Intestinal Inflammation. Mucosal Immunol (2016) 9(2):444–57. doi: 10.1038/mi.2015.74
437. Whibley N, Tucci A, Powrie F. Regulatory T Cell Adaptation in the Intestine and Skin. Nat Immunol (2019) 20(4):386–96. doi: 10.1038/s41590-019-0351-z
438. Barko PC, McMichael MA, Swanson KS, Williams DA. The Gastrointestinal Microbiome: A Review. J Vet Intern Med (2018) 32(1):9–25. doi: 10.1111/jvim.14875
439. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A Metagenome-Wide Association Study of Gut Microbiota in Type 2 Diabetes. Nature (2012) 490(7418):55–60. doi: 10.1038/nature11450
440. Pascale A, Marchesi N, Marelli C, Coppola A, Luzi L, Govoni S, et al. Microbiota and Metabolic Diseases. Endocrine (2018) 61(3):357–71. doi: 10.1007/s12020-018-1605-5
441. Jackson MA, Verdi S, Maxan ME, Shin CM, Zierer J, Bowyer RCE, et al. Gut Microbiota Associations With Common Diseases and Prescription Medications in a Population-Based Cohort. Nat Commun (2018) 9(1):2655. doi: 10.1038/s41467-018-05184-7
442. Tong Y, Marion T, Schett G, Luo Y, Liu Y. Microbiota and Metabolites in Rheumatic Diseases. Autoimmun Rev (2020) 19(8):102530. doi: 10.1016/j.autrev.2020.102530
443. Allegretti JR, Mullish BH, Kelly C, Fischer M. The Evolution of the Use of Faecal Microbiota Transplantation and Emerging Therapeutic Indications. Lancet (2019) 394(10196):420–31. doi: 10.1016/S0140-6736(19)31266-8
444. Schirmer M, Franzosa EA, Lloyd-Price J, McIver LJ, Schwager R, Poon TW, et al. Dynamics of Metatranscription in the Inflammatory Bowel Disease Gut Microbiome. Nat Microbiol (2018) 3(3):337–46. doi: 10.1038/s41564-017-0089-z
445. Rossen NG, Fuentes S, van der Spek MJ, Tijssen JG, Hartman JH, Duflou A, et al. Findings From a Randomized Controlled Trial of Fecal Transplantation for Patients With Ulcerative Colitis. Gastroenterology (2015) 149(1):110–8 e4. doi: 10.1053/j.gastro.2015.03.045
446. Moayyedi P, Surette MG, Kim PT, Libertucci J, Wolfe M, Onischi C, et al. Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology (2015) 149(1):102–9 e6. doi: 10.1053/j.gastro.2015.04.001
447. Paramsothy S, Kamm MA, Kaakoush NO, Walsh AJ, van den Bogaerde J, Samuel D, et al. Multidonor Intensive Faecal Microbiota Transplantation for Active Ulcerative Colitis: A Randomised Placebo-Controlled Trial. Lancet (2017) 389(10075):1218–28. doi: 10.1016/S0140-6736(17)30182-4
448. Costello SP, Hughes PA, Waters O, Bryant RV, Vincent AD, Blatchford P, et al. Effect of Fecal Microbiota Transplantation on 8-Week Remission in Patients With Ulcerative Colitis: A Randomized Clinical Trial. JAMA (2019) 321(2):156–64. doi: 10.1001/jama.2018.20046
449. Imdad A, Nicholson MR, Tanner-Smith EE, Zackular JP, Gomez-Duarte OG, Beaulieu DB, et al. Fecal Transplantation for Treatment of Inflammatory Bowel Disease. Cochrane Database Syst Rev (2018) 11:CD012774. doi: 10.1002/14651858.CD012774.pub2
450. Paramsothy S, Paramsothy R, Rubin DT, Kamm MA, Kaakoush NO, Mitchell HM, et al. Faecal Microbiota Transplantation for Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. J Crohns Colitis (2017) 11(10):1180–99. doi: 10.1093/ecco-jcc/jjx063
451. Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, et al. Gut Microbiota in Children With Type 1 Diabetes Differs From That in Healthy Children: A Case-Control Study. BMC Med (2013) 11:46. doi: 10.1186/1741-7015-11-46
452. Hagopian WA, Erlich H, Lernmark A, Rewers M, Ziegler AG, Simell O, et al. The Environmental Determinants of Diabetes in the Young (TEDDY): Genetic Criteria and International Diabetes Risk Screening of 421 000 Infants. Pediatr Diabetes (2011) 12(8):733–43. doi: 10.1111/j.1399-5448.2011.00774.x
453. Leiva-Gea I, Sanchez-Alcoholado L, Martin-Tejedor B, Castellano-Castillo D, Moreno-Indias I, Urda-Cardona A, et al. Gut Microbiota Differs in Composition and Functionality Between Children With Type 1 Diabetes and MODY2 and Healthy Control Subjects: A Case-Control Study. Diabetes Care (2018) 41(11):2385–95. doi: 10.2337/dc18-0253
454. de Groot P, Nikolic T, Pellegrini S, Sordi V, Imangaliyev S, Rampanelli E, et al. Faecal Microbiota Transplantation Halts Progression of Human New-Onset Type 1 Diabetes in a Randomised Controlled Trial. Gut (2021) 70(1):92–105. doi: 10.1136/gutjnl-2020-322630
455. Gutin L, Piceno Y, Fadrosh D, Lynch K, Zydek M, Kassam Z, et al. Fecal Microbiota Transplant for Crohn Disease: A Study Evaluating Safety, Efficacy, and Microbiome Profile. United Eur Gastroenterol J (2019) 7(6):807–14. doi: 10.1177/2050640619845986
456. Ding X, Li Q, Li P, Zhang T, Cui B, Ji G, et al. Long-Term Safety and Efficacy of Fecal Microbiota Transplant in Active Ulcerative Colitis. Drug Saf (2019) 42(7):869–80. doi: 10.1007/s40264-019-00809-2
457. van Rheenen PF, Aloi M, Assa A, Bronsky J, Escher JC, Fagerberg UL, et al. The Medical Management of Paediatric Crohn’s Disease: An ECCO-ESPGHAN Guideline Update. J Crohns Colitis (2020). doi: 10.1093/ecco-jcc/jjaa161
458. Jacob V, Crawford C, Cohen-Mekelburg S, Viladomiu M, Putzel GG, Schneider Y, et al. Single Delivery of High-Diversity Fecal Microbiota Preparation by Colonoscopy Is Safe and Effective in Increasing Microbial Diversity in Active Ulcerative Colitis. Inflammation Bowel Dis (2017) 23(6):903–11. doi: 10.1097/MIB.0000000000001132
459. Danne C, Rolhion N, Sokol H. Recipient Factors in Faecal Microbiota Transplantation: One Stool Does Not Fit All. Nat Rev Gastroenterol Hepatol (2021). doi: 10.1038/s41575-021-00441-5
460. Chiang JL, Maahs DM, Garvey KC, Hood KK, Laffel LM, Weinzimer SA, et al. Type 1 Diabetes in Children and Adolescents: A Position Statement by the American Diabetes Association. Diabetes Care (2018) 41(9):2026–44. doi: 10.2337/dci18-0023
461. de Souza HSP, Fiocchi C, Iliopoulos D. The IBD Interactome: An Integrated View of Aetiology, Pathogenesis and Therapy. Nat Rev Gastroenterol Hepatol (2017) 14(12):739–49. doi: 10.1038/nrgastro.2017.110
462. Guandalini S. The Approach to Celiac Disease in Children. Int J Pediatr Adolesc Med (2017) 4(3):124–7. doi: 10.1016/j.ijpam.2017.08.002
463. Jericho H, Guandalini S. Extra-Intestinal Manifestation of Celiac Disease in Children. Nutrients (2018) 10(6):755. doi: 10.3390/nu10060755
464. Rosen MJ, Dhawan A, Saeed SA. Inflammatory Bowel Disease in Children and Adolescents. JAMA Pediatr (2015) 169(11):1053–60. doi: 10.1001/jamapediatrics.2015.1982
465. Elzagallaai AA, Greff M, Rieder MJ. Adverse Drug Reactions in Children: The Double-Edged Sword of Therapeutics. Clin Pharmacol Ther (2017) 101(6):725–35. doi: 10.1002/cpt.677
466. Alomar MJ. Factors Affecting the Development of Adverse Drug Reactions (Review Article). Saudi Pharm J (2014) 22(2):83–94. doi: 10.1016/j.jsps.2013.02.003
467. Zuo T, Kamm MA, Colombel JF, Ng SC. Urbanization and the Gut Microbiota in Health and Inflammatory Bowel Disease. Nat Rev Gastroenterol Hepatol (2018) 15(7):440–52. doi: 10.1038/s41575-018-0003-z
468. Ruff WE, Greiling TM, Kriegel MA. Host-Microbiota Interactions in Immune-Mediated Diseases. Nat Rev Microbiol (2020) 18(9):521–38. doi: 10.1038/s41579-020-0367-2
469. Lazar V, Ditu LM, Pircalabioru GG, Gheorghe I, Curutiu C, Holban AM, et al. Aspects of Gut Microbiota and Immune System Interactions in Infectious Diseases, Immunopathology, and Cancer. Front Immunol (2018) 9:1830. doi: 10.3389/fimmu.2018.01830
470. Yoo JY, Groer M, Dutra SVO, Sarkar A, McSkimming DI. Gut Microbiota and Immune System Interactions. Microorganisms (2020) 8(10):1587. doi: 10.3390/microorganisms8101587
471. Li D, Li X, Zhou WL, Huang Y, Liang X, Jiang L, et al. Genetically Engineered T Cells for Cancer Immunotherapy. Signal Transduct Target Ther (2019) 4:35. doi: 10.1038/s41392-019-0070-9
472. Pavlovic K, Tristan-Manzano M, Maldonado-Perez N, Cortijo-Gutierrez M, Sanchez-Hernandez S, Justicia-Lirio P, et al. Using Gene Editing Approaches to Fine-Tune the Immune System. Front Immunol (2020) 11:570672. doi: 10.3389/fimmu.2020.570672
473. Waldman AD, Fritz JM, Lenardo MJ. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat Rev Immunol (2020) 20(11):651–68. doi: 10.1038/s41577-020-0306-5
474. Mukhatayev Z, Ostapchuk YO, Fang D, Le Poole IC. Engineered Antigen-Specific Regulatory T Cells for Autoimmune Skin Conditions. Autoimmun Rev (2021) 20(3):102761. doi: 10.1016/j.autrev.2021.102761
475. Rana J, Biswas M. Regulatory T Cell Therapy: Current and Future Design Perspectives. Cell Immunol (2020) 356:104193. doi: 10.1016/j.cellimm.2020.104193
Keywords: chronic inflammatory disorders, pediatric diseases, microbiota, immune homeostasis, adoptive cell therapy, regulatory T cells
Citation: Torun A, Hupalowska A, Trzonkowski P, Kierkus J and Pyrzynska B (2021) Intestinal Microbiota in Common Chronic Inflammatory Disorders Affecting Children. Front. Immunol. 12:642166. doi: 10.3389/fimmu.2021.642166
Received: 15 December 2020; Accepted: 24 May 2021;
Published: 07 June 2021.
Edited by:
Chang H. Kim, University of Michigan, United StatesCopyright © 2021 Torun, Hupalowska, Trzonkowski, Kierkus and Pyrzynska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beata Pyrzynska, YmVhdGEucHlyenluc2thQHd1bS5lZHUucGw=