 Patrick Schuhmachers
Patrick Schuhmachers Christian Münz
Christian Münz
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 23 March 2021
Sec. Viral Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.640918
This article is part of the Research Topic Humanized Mouse Models to Study Immune Responses to Human Infectious Organisms View all 11 articles
Epstein Barr virus (EBV) is one of the most successful pathogens in humans with more than 95% of the human adult population persistently infected. EBV infects only humans and threatens these with its potent growth transforming ability that readily allows for immortalization of human B cells in culture. Accordingly, it is also found in around 1-2% of human tumors, primarily lymphomas and epithelial cell carcinomas. Fortunately, however, our immune system has learned to control this most transforming human tumor virus in most EBV carriers, and it requires modification of EBV associated lymphomagenesis and its immune control by either co-infections, such as malaria, Kaposi sarcoma associated herpesvirus (KSHV) and human immunodeficiency virus (HIV), or genetic predispositions for EBV positive tumors to emerge. Some of these can be modelled in humanized mice that, therefore, provide a valuable platform to test curative immunotherapies and prophylactic vaccines against these EBV associated pathologies.
The Epstein Barr virus (EBV) or human herpesvirus 4 (HHV4) is a ubiquitous human γ-herpesvirus that persistently infects more than 95% of the human population (1). In Sub-Saharan Africa, this percentage is already reached at 2 years of age, while in Europe and the US one third of the population acquires EBV at a later age (2). This delayed primary EBV infection bears the risk to develop into infectious mononucleosis (IM), an immunopathology due to massive anti-viral CD8+ T cell expansion and the accompanying cytokine release (3). Especially CD8+ T cells that recognize lytic EBV antigens, expressed during the viral infection program that produces viral particles, are increased to high frequencies during IM (4). Even so IM resolves in most cases, alterations in the resulting EBV specific immune response might be the reason for elevated risks for EBV associated Hodgkin’s lymphoma and multiple sclerosis (MS) (5, 6).
In addition to immunopathologies due to altered or increased immune responses to EBV infection, this virus is primarily known for its oncogenic potential (7). It was originally discovered in endemic Burkitt’s lymphoma (BL) of Sub-Saharan African children (8, 9). Furthermore, EBV can be found in a subset of Hodgkin’s lymphoma (HL), diffuse large B cell lymphoma (DLBCL) and immunoblastic lymphomas during immune suppression after transplantation, such as post-transplantation lymphoproliferative disease (PTLD), or during HIV co-infection (10). These are mostly B cell lymphomas, but also EBV associated natural killer (NK)/T cell lymphomas can occur, often after prolonged uncontrolled EBV infection (11). In addition, EBV is associated with epithelial cell derived nasopharyngeal carcinoma (NPC) and 10% of gastric carcinomas. These EBV associated malignancies mainly express latent EBV antigens that are not involved in infectious virus production but contain at least one of the main two EBV oncogenes, nuclear antigen 2 (EBNA2) and latent membrane protein 1 (LMP1) (12, 13). Latent EBV infection follows a pre-latent phase upon B cell entry during which a set of both lytic and latent antigens are expressed in order to support B cell activation, proliferation and survival [as reviewed in (14)]. During latent infection, which is the default gene expression program upon B cell infection by EBV, up to 6 EBNAs, two LMPs as well as non-translated Epstein–Barr virus-encoded small RNAs (EBERs) and miRNAs are expressed. This gene expression program is thought to drive EBV infected B cells after viral transmission via saliva in sub-mucosal secondary lymphoid tissues like tonsils into activation and differentiation to memory B cells, in which EBV then persists for life (15). From this memory B cell compartment, in which EBV only expresses non-translated RNAs, lytic reactivation and infectious virus production occurs after plasma cell differentiation, presumably after encountering the cognate antigen of the B cell receptor of the infected cell (16). At submucosal secondary lymphoid tissues this might lead to viral shedding into saliva for transmission. The distinct B cell differentiation stages and their respective EBV gene expression patterns can also be found in the EBV associated malignancies, and the respective B cell lymphomas (BL, HL and DLBCL) increase in frequency during iatrogenic or HIV induced immune suppression (10, 17). This suggests that immune responses prevent transition from premalignant latent EBV infections to overt tumors. Indeed, primary immunodeficiencies that affect individual genes map to cytotoxic lymphocytes and their ability to kill EBV infected B cells as the most important component of EBV specific immune control (18–20). In order to interrogate the function of the in patients identified genes, dissect the contribution of viral genes with EBV mutants and characterize the influence of co-infections in vivo, preclinical mouse models with reconstituted human immune cells (humanized mice) have been developed and their contribution to a better understanding of EBV infection, oncogenesis and immune control will be summarized in this review.
To date humanized mice serve as a reliable model to study pathogens that exclusively target humans. In the past decades, several humanized mouse models were established that responded to infection with EBV and allowed for assessing the importance of host immune factors as well as viral proteins during an infection. In this review, we will primarily focus on NOD/Shi-scid/IL-2Rγnull (NOG) and NOD/LtSz-scid IL2Rγnull (NSG) mice with and without HLA-A2 transgenes as well as NSG or NOD/LtSz-scid mice implanted with human fetal liver and thymus tissue (BLT) that were all either neonatally or as adult mice reconstituted with human immune system components by transfer of human CD34+ hematopoietic progenitor cells (HPCs) or of cord blood often after CD34+ HPC depletion. Most studies with EBV infection have been performed in these particular humanized mouse models and these were consistently permissive for multiple EBV strains (21–25), mirroring acute infection as well as EBV associated lymphomagenesis of humans. A more complete overview of humanized mouse models was recently published (26). As is the case for humans, human B cells constitute the main reservoir for EBV in humanized mice, enabling viral replication and lymphoproliferation. EBV infection of humanized mice was therefore marked by viral loads in blood and secondary lymphoid organs (22, 24, 27, 28).
Analogously to acute symptomatic primary infection in humans (29–31), the number of NK cells in peripheral blood and spleen of humanized NSG mice increases starting at three weeks of EBV infection and peaks at week four. The NK cell response constitutes an important measure to prevent uncontrolled lytic EBV infection and to bridge the time until adaptive T cell responses are primed. Indicative of this is that depletion of NK populations in humanized NSG mice resulted in higher viral loads and tumor incidence (32, 33).
Initial control by NK cells is succeeded by priming and expansion of cytotoxic CD8+ T lymphocytes (CTLs) in peripheral blood. Those CTLs are mainly specific for lytic antigens (22, 25, 34, 35) and exhibit a cytolytic effector profile determined by high expression of activation molecules like HLA-DR or 2B4 and cytotoxic effector molecules such as Granzyme B (22, 25, 35). Consequently, expression of HLA-DR positively correlates with increasing viral loads in infected humanized NSG mice (36). In contrast to CTL expansion, CD4+ helper T cells do not expand to a similar degree which is why an inversion of the CD8 to CD4 T cell ratio is one of the hallmarks of EBV infection in humanized mice as is in humans suffering from IM (25, 34). Despite lower expansion rates and total numbers, CD4+ T cell help seems to be required to tackle the infection in humanized mice since CD4+ T cell depletion prior to infection results in higher viral loads (22). This corresponds to human data depicting a cytolytic effector function of EBV specific CD4+ T cells during infection (37, 38).
In contrast to T cell responses, antibody mediated responses to EBV are not yet as well characterized in humanized mice. In humanized NOG and NSG mice, IgM responses to BFRF3 and EBNA1, respectively, were observed. Detection of EBV specific IgG antibodies, however, has proven to be more difficult. This drawback might arise from deficiencies in germinal center formation and therefore difficulties in antibody isotype class switching as well as inefficient B cell development in several humanized mouse systems (22, 39–43). There are, however, promising developments in the generation of humanized mice capable of mounting IgG responses to pathogens (44, 45).
Despite active immune control of EBV infection, lymphoma formation can be observed in humanized mice. The degree of lymphomagenesis is thereby dependent on the amount of viral particles with which the animal is challenged. Humanized NSG mice, for example, present in 20 – 30% of cases with disseminated lymphomas in spleen, liver, lymph nodes or kidney when challenged intraperitoneally with high dose EBV (105 infectious particles) for four to five weeks (46, 47). Other mouse models, as for example humanized BALB/c Rag2nullIL2rγnull SirpaNOD (BRGS) mice, present with higher lymphoma incidences of up to 75% while surviving four weeks of infection (48). Besides that, lymphomagenesis may be dependent on genomic or host immunologic alterations which will be discussed in the following sections.
Since the discovery that humanized mice are susceptible to EBV infection and allow for the identification of host immune factors in response to the pathogen, several groups started assessing the importance of viral genes during infection. For example, infection of a humanized BRGS mouse with type 1 or 2 EBV strains was examined. The two EBV strains differ mainly in their genetic sequence of the latent genes EBNA2, EBNA3A and EBNA3C (49–51) and, consequently, in their ability to transform infected cells in vitro. More importantly, in contrast to EBV type 1, EBV type 2 was additionally observed to be human T cell tropic which could explain findings of EBV related human T cell lymphomas (48). Coleman and colleagues were able to reproduce T cell tropism of EBV type 2 in vivo. However, the infection of humanized mice with either strain resulted in comparable degrees of viral replication and lymphomagenesis contrasting the in vitro findings. Lymphomas caused by both strains exhibited similar features, resembling diffuse large B cell lymphomas (DLBCL) and expressing all latent EBV gene products (48, 52). The strain specific differences that underlie in vivo infection seem, for now, to be limited to lower LMP1 expression levels and higher lytic activity in EBV type 2 infected animals (52). Still, the reason why the two EBV strains developed different strategies, remains elusive. In addition to EBV type 2 that is primarily found in Sub-Saharan Africa (53), also Asian EBV strains present with higher lytic EBV replication, and this also extends to infections of humanized mice (21, 54, 55).
Apart from defining differences between the virus strains, various gene loci of EBV type 1 were extensively studied in the past decades using humanized mice (Figure 1). White and colleagues investigated the contribution of EBNA3B to infection. In their study, they infected humanized NSG mice with the B95-8 (EBV type 1) strain of EBV which lacked the EBNA3B gene locus. Interestingly, the absence of EBNA3B led to higher tumor incidences in those mice which White and colleagues assigned to higher replicative activity of infected cells and a lower level of T cell infiltration into tumors due to decreased expression of chemo-attractants as for example CXCL9 and CXCL10 (56). A lower degree of T cell infiltration is thereby in line with EBNA3B being often targeted by T cells (57).
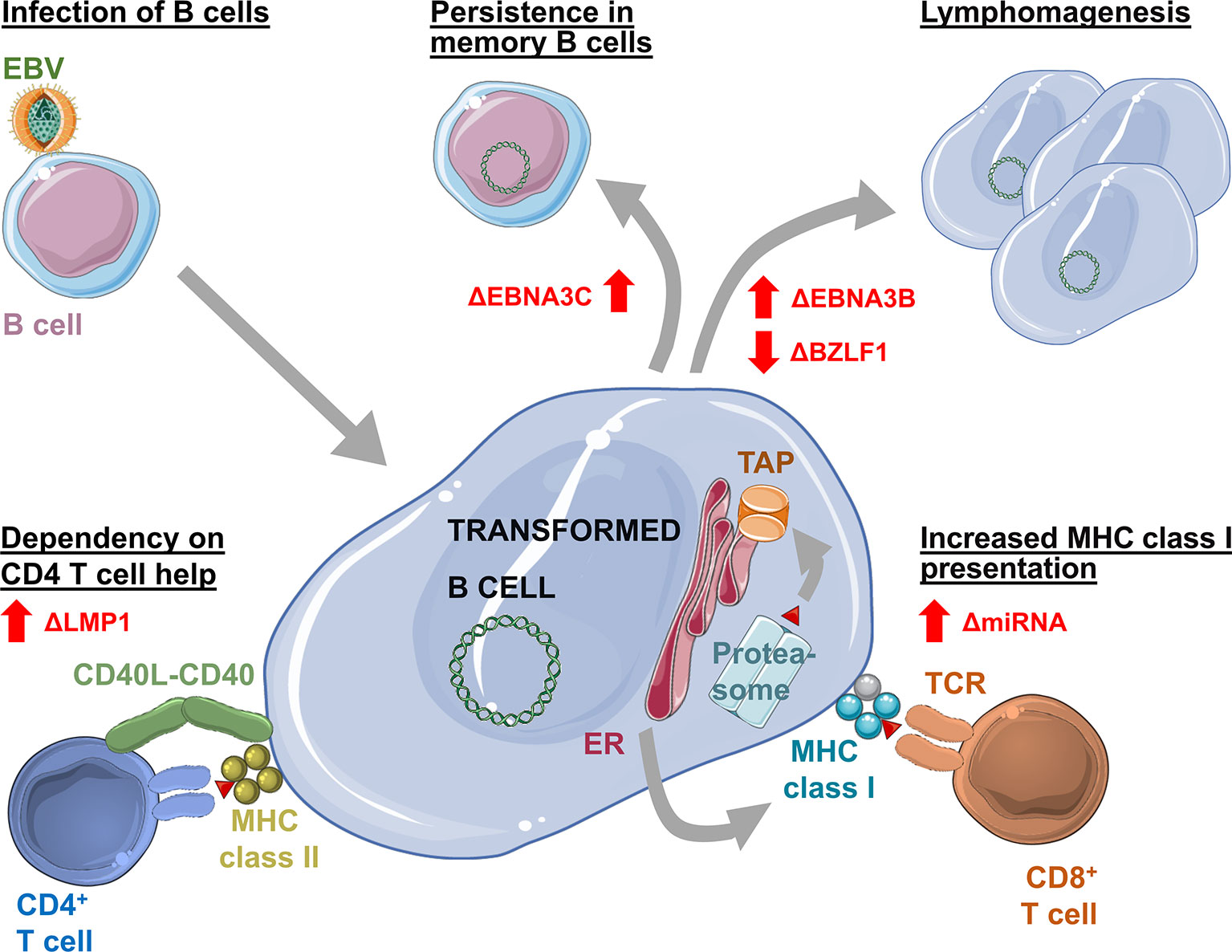
Figure 1 Changes in Epstein Barr virus (EBV) infection, pathogenesis and immune control as revealed by infection of humanized mice with mutant viruses. Elimination of EBNA3C (ΔEBNA3C) allows establishment of persistent EBV infection without transformation. Loss of EBNA3B (ΔEBNA3B) increases and loss of BZLF1 (ΔBZLF1) decreases EBV associated lymphomagenesis. Loss of viral miRNAs (ΔmiRNA) leads to increased expression of the antigen processing machinery for MHC class I presentation, allowing for improved immune control by CD8+ T cells. Deficiency in LMP1 (ΔLMP1) causes dependency on CD4+ T cell help for EBV induced B cell transformation.
In addition to T cell infiltration, the importance of MHC class I restricted antigen presentation for EBV specific immune control by CD8+ T cells in humanized mice was demonstrated with a B95-8 virus that lacks the viral miRNAs (36). These compromise antigen processing for MHC class I presentation (58). In their absence EBV is more efficiently immune controlled in humanized mice in a CD8+ T cell dependent manner. In contrast to miRNA deficiency, EBER knock-out EBV infects humanized mice similarly to wild-type virus (59), except for the increased inflammation promoting potential of EBER2 of some Asian EBV strains (55).
Other studies on B95-8 viruses lacking the EBNA3A or EBNA3C gene locus highlighted that these genes are dispensable for establishment of persistent infection in vivo. Despite findings suggesting that EBNA3A or 3C knockout viruses are hardly able to transform B cells in vitro, persistence was established in secondary lymphoid organs and blood over a period of three months and T cell responses were mounted against the virus. Lack of EBNA3A or 3C, however, seems to result in decreased aggressiveness of EBV infection as viral loads were lower and the tumorigenic potential was presumably lost (60). Studies in cord blood reconstituted humanized NSG mice in which T cells might exert less immune control due to efficient inhibitory receptor engagement and in which higher frequencies of EBV associated lymphomas are observed (61), came to similar conclusions regarding a decreased aggressiveness in the absence of EBNA3A and 3C. However, EBNA3A mutants could not only establish persistent infection in this model, but tumorigenic potential was delayed and not lost. Still, both studies observed decreased LMP1 expression levels in infected cells in spleen and tumor tissue (60, 62). In addition, the use of a complete EBNA3A knockout virus (60) compared to an EBV mutant with only decreased EBNA3A expression (62) might explain differences in the viruses’ tumorigenic potential. In the same cord blood reconstituted humanized NSG mouse model, EBNA3C knockout EBV was able to cause lymphomas with lower frequency. Similar to EBNA3A hypomorphic EBV, lymphoma formation seemed to be delayed (52). Therefore, in humanized mice with diminished immune control, decreased EBNA3A or absent EBNA3C expression might still allow for the delayed development of EBV associated diffuse large B cell lymphomas.
Furthermore, LMP1 and 2 might also be dispensable for EBV infection in humanized mice (63, 64), but their absence delays lymphomagenesis. In the case of LMP1 deficiency the observed lymphoma formation required CD4+ T cell help (63).
Surprisingly also lytic EBV reactivation, at least early lytic gene expression, seems to promote lymphomagenesis in humanized mice. B95-8 EBV infection caused less tumors in the absence of the immediate early transactivation factor BZLF1 that induces lytic reactivation (23, 46). Vice versa, BZLF1 promotors that enhance lytic EBV infection are associated with increased lymphoma formation (65, 66). Infection of humanized mice with mutant EBV viruses can therefore reveal the function of genetic variability or viral gene products, enabling further insights into the life cycle of EBV in vivo.
In addition to testing mutant EBV viruses and different viral isolates in vivo, humanized mice also allow interrogation of human genetic variation, gene products and leucocyte compartments during EBV infection, oncogenesis and immune control. With regards to genetic variation IM and elevated antibody responses against EBV nuclear antigen 1 (EBNA1) have been found to synergize with the MHC class II molecule HLA-DRB1*1501 to increase risk for the development of MS (5). Indeed, HLA-DRB1*1501 restricted EBNA1 specific CD4+ T cell responses are also elevated in MS patients (67–69). However, despite elevated T cell responses to EBV infection in humanized mice that have been reconstituted from HLA-DRB1*1501 donors, these animals experience higher viral loads (70). Furthermore, the respective HLA-DRB1*1501 restricted CD4+ T cells that recognize EBV transformed B cells (lymphoblastoid cell lines or LCL) cross-react with myelin basic protein (MBP), an autoantigen in MS. These findings suggest that EBV is inefficiently immune controlled in the context of HLA-DRB1*1501 and that the resulting increased numbers of EBV infected B cells might stimulate in turn myelin antigen specific autoreactive T cell responses to cause MS.
Indeed CD4+ T cell responses seem essential to maintain efficient immune control of EBV in humanized mice. Both, iatrogenic immune suppression with tacrolimus (FK506) that mainly affects CD4+ T cell activation and expansion after EBV infection of humanized mice, and CD4+ T cell depletion by HIV co-infection leads to elevated viral loads and increased EBV associated B cell lymphoma formation (71, 72). During HIV co-infection CD8+ T cell depletion does not further increase EBV viral loads or lymphoma formation (72). This suggests that HIV induced CD4+ T cell depletion compromises T cell help to maintain protective CD8+ T cell function because CD8+ T cell depletion during only EBV infection of humanized mice significantly affects immune control (22, 36, 72, 73). Moreover, antibody mediated depletion of both CD4+ and CD8+ T cells increases viral loads and associated tumors in EBV infected humanized mice (22, 43). In addition, antibody blocking of 2B4, a co-stimulatory molecule on cytotoxic lymphocytes that uses SLAM-associated protein (SAP), and SAP is mutated in X-linked lymphoproliferative disease 1 (XLP1) thereby predisposing for EBV associated pathology, increases EBV viral loads and lymphomagenesis in humanized mice (73). 2B4 blocking on top of CD8+ T cell depletion does not lower EBV specific immune control further, suggesting that 2B4 is mainly required on CD8+ T cells to suppress EBV infection. SAP deficiency is, however, also associated with loss of invariant NKT cells (74) which have been shown to restrict EBV transformed B cells in humanized mice (75, 76). This could also contribute to compromised immune control of EBV in XLP1. Moreover, antibody blocking of PD-1, an inhibitory receptor on both effector and regulatory T cells, increases EBV loads and lymphomagenesis in infected humanized NSG mice engrafted with human CD34+ HPCs (26, 35). This loss of immune control correlates with immune suppressive cytokine and regulatory T cell amounts in anti-PD-1 treated and EBV infected humanized mice. Strikingly, blocking PD-1 and/or CTLA-4 in cord blood reconstituted humanized NSG mice resulted in decreased tumor formation (61). As cord blood reconstituted humanized NSG mice seem to develop weaker T cell mediated immune control after EBV infection and allow for higher frequencies of EBV associated lymphomas, especially after infection with the lytic M81 EBV strain, PD-1 and CTLA-4 blockade might be required to strengthen this immune control (61) which seems to be in part based on Vγ9Vδ2 T cells (77). Thus, composition of the T cell compartment and balance between regulatory and anti-viral T cells might determine the outcome of PD-1 and CTLA-4 blockade. However, genetic loss of CTLA-4 seems to be associated with EBV associate pathologies in some of the affected individuals (78). Similarly, PD-1 blockade seems to cause loss of EBV specific immune control and brain homing of EBV infected B cells in a subgroup of treated patients (79).
In addition to T cells, innate lymphocytes contribute to EBV specific immune control. Protection against EBV infection of humanized mice has been shown for NK, NKT and Vγ9Vδ2 T cells (32, 33, 75, 77, 80). Early differentiated NKG2A+KIR- NK cells restrict primarily lytic EBV replication and degranulate their cytotoxic machinery toward lytically EBV replicating BL cells (29, 32). In contrast, depletion of plasmacytoid dendritic cells, the main hematopoietic source of type I interferon (IFN) during viral infections, does not significantly influence EBV infection of humanized mice (81). Accordingly, type I IFN signaling deficiencies do not predispose for EBV associated pathologies (19). These studies demonstrate that humanized mice can be used to interrogate the role of genetic variations, of leucocyte compartments and of their receptors in EBV specific immune control.
In addition to HIV infection, some EBV associated malignancies are also observed during additional co-infections. Endemic BL can be primarily observed in geographic areas of holoendemic exposure to the malaria parasite Plasmodium falciparum (82, 83). In Sub-Saharan Africa, where Denis Burkitt described this tumor for the first time (84), Kaposi sarcoma associated herpesvirus (KSHV) or human herpesvirus 8 (HHV8) a another pathogen with which EBV interacts during co-infections is also highly prevalent (85). Both EBV and KSHV are found in the tumor cells of 90% of primary effusion lymphomas (PELs) (10). Moreover, KSHV infection seems to benefit from EBV co-infection for persistence (47, 86–88). Humanized mice that are infected with both KSHV and EBV develop B cell lymphomas with higher incidence (47). The developing lymphomas harbor both EBV and KSHV (Figure 2). They present with hallmarks of plasma cell differentiation that is characteristic for PELs (89). Interestingly, this plasma cell differentiation that is also in healthy EBV carriers associated with lytic EBV replication (16), causes elevated induction of at least early lytic EBV reactivation in PEL-like tumors of double-infected humanized mice (47). Co-infection with an EBV mutant that can no longer activate lytic infection (BZLF1 deficient EBV), reduces lymphomagenesis during EBV and KSHV co-infection of humanized mice, suggesting that the transactivated lytic EBV gene expression might contribute to conditioning of the tumor microenvironment for efficient growth (7).
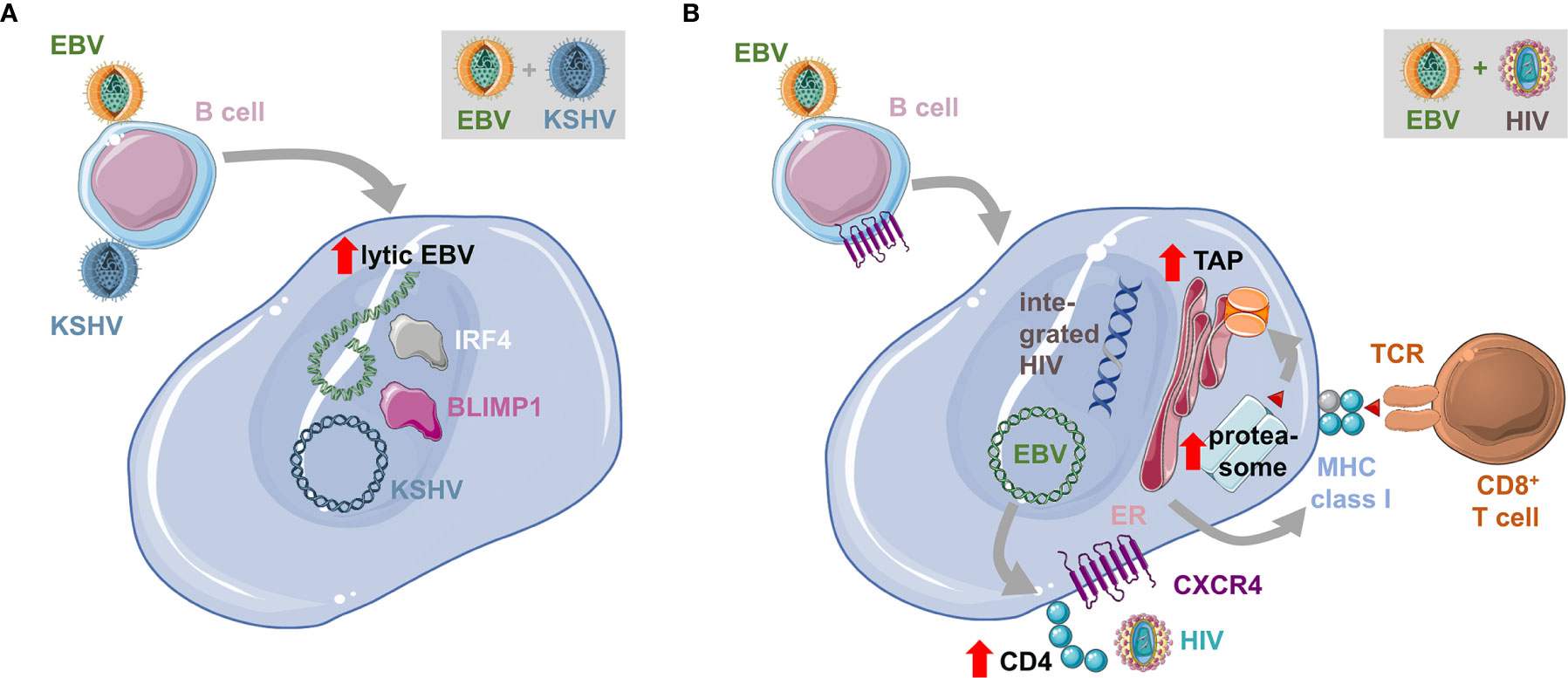
Figure 2 Influence of co-infections on EBV transformed B cells. (A) Co-infection with the Kaposi sarcoma associated herpesvirus (KSHV) transactivates lytic EBV replication and is associated with plasma cell differentiation, characterized for example by BLIMP1 and IRF4 expression. This leads to enhanced lymphomagenesis and the resulting tumors have characteristics of primary effusion lymphomas (PELs). (B) EBV infection of B cells up-regulates CD4 and with the sustained expression of CXCR4 renders EBV transformed B cells susceptible to infection with the human immunodeficiency virus (HIV). This leads to the integration of the reverse transcribed HIV genome into the host cell genome. Double-infected B cells up-regulate the antigen processing machinery for MHC class I presentation and are therefore efficiently immune controlled by CD8+ T cells.
HIV co-infection also does more than just suppress EBV specific immune control. It was noted that anti-retroviral therapy (ART) reduced immunoblastic EBV associated lymphomas in HIV infected individuals, while BL and HL frequencies did not decline (17). Similarly, in humanized mice HIV co-infection influences EBV infected B cells directly (72). It was found that HIV is capable to infect EBV transformed B cells due to CD4 up-regulation during EBV infection and maintained CXCR4 expression on human B cells (Figure 2). HIV also integrates and replicates in EBV infected B cells but alters their gene expression pattern for enhanced antigen processing toward MHC class I restricted antigen presentation. Accordingly, HIV and EBV infected B cells are efficiently recognized by both EBV and HIV specific CD8+ T cells and mainly accumulate in double-infected humanized mice after CD8+ T cell depletion. Future studies will need to show if double-infected B cells alter EBV associated lymphoma formation over a longer observation period, independent of HIV induced immune suppression.
In summary, the development of humanized mice revolutionized the study of pathogens exclusive to humans such as EBV. The possibility to use small rodents instead of endangered New World monkeys (90, 91) greatly facilitated the research on host immune factors and viral genes during infection. Various humanized mouse models are consistently permissive for infection with and allow replication of EBV. Even more promising, these models mimic human lymphoproliferative diseases and the reaction of human immune system components to infection. In addition, host and virus genetic alterations and their implications in disease outcome may be more extensively studied in humanized mice which may not be comparably easy in humans. Further studies on host susceptibility factors like HLA-DRB1*1501 are needed to assess additional groups at risk of developing EBV associated diseases and to develop personalized treatments in the future. Similarly, the interplay between EBV and other co-infecting pathogens is difficult to assess in humans but may be elucidated in humanized mice. Thus, with humanized mice it became feasible to study not only contributions of single immune cell types or single molecules to infection outcome but also to investigate the importance of virus and host genetics. The findings resulting from infection models in humanized mice thereby enable applied research on EBV vaccines, or the development of new treatments against EBV induced lymphomas.
Despite these advantages there are still limitations to each of the humanized mouse models employed. The biggest limitation with respect to EBV infection is that humanized mice do not support infection of oropharyngeal epithelia. In humans the infection starts in the oropharynx with a complex interplay between submucosal B cells and polarized epithelia. Therefore, EBV’s complete life cycle cannot be mimicked thus far. Furthermore, most models failed to mount proper antibody responses to EBV which is why their contribution to infection could not yet be well studied using a mouse model. Reports on new humanized mouse models with improved seroconversion to IgG, however, seem promising in solving this problem (44, 92). A further limitation of the presented EBV infection models is that in most cases lymphomas with expression of all latent EBV genes develop. As a result, the in humans more prominent HL or BL with restricted latent gene expression cannot currently be modelled in humanized mice and therefore such models still have to be developed. Along these lines infection with EBNA2 deficient EBV has recently been reported to cause lymphomas with some HL characteristics and might be further explored to gain insights into this EBV associated malignancy (93). With further developments of humanized mice in the upcoming years we may be able to reveal even minor host and viral genetic as well as host immune factors that contribute to control of EBV infection. Thus, we may be able to use the gained knowledge to design vaccines or therapies against this most transforming human tumor virus.
PS and CM wrote the manuscript. All authors contributed to the article and approved the submitted version.
Our research is supported by Cancer Research Switzerland (KFS-4962-02-2020), CRPP-PrecisionMS and HMZ ImmunoTargET of the University of Zurich, the Cancer Research Center Zurich, the Baugarten Foundation, the Sobek Foundation, the Swiss Vaccine Research Institute, Roche, Novartis, Novartis Foundation (20B099), and the Swiss National Science Foundation (310030B_182827, 310030L_197952/1 and CRSII5_180323).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ehlers B, Spiess K, Leendertz F, Peeters M, Boesch C, Gatherer D, et al. Lymphocryptovirus phylogeny and the origins of Epstein-Barr virus. J Gen Virol (2010) 91:630–42. doi: 10.1099/vir.0.017251-0
2. Dunmire SK, Verghese PS, Balfour HH Jr. Primary Epstein-Barr virus infection. J Clin Virol (2018) 102:84–92. doi: 10.1016/j.jcv.2018.03.001
3. Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med (2010) 362:1993–2000. doi: 10.1056/NEJMcp1001116
4. Callan MF, Tan L, Annels N, Ogg GS, Wilson JD, O’Callaghan CA, et al. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus In vivo. J Exp Med (1998) 187:1395–402. doi: 10.1084/jem.187.9.1395
5. Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol (2017) 13:25–36. doi: 10.1038/nrneurol.2016.187
6. Hjalgrim H, Askling J, Rostgaard K, Hamilton-Dutoit S, Frisch M, Zhang JS, et al. Characteristics of Hodgkin’s lymphoma after infectious mononucleosis. N Engl J Med (2003) 349:1324–32. doi: 10.1056/NEJMoa023141
7. Münz C. Latency and lytic replication in the oncogenesis of the Epstein Barr virus. Nat Rev Micobiol (2019) 17:691–700. doi: 10.1038/s41579-019-0249-7
8. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet (1964) 1:702–3. doi: 10.1016/S0140-6736(64)91524-7
9. Epstein MA, Henle G, Achong BG, Barr YM. Morphological and biological studies on a virus in cultured lymphoblasts from Burkitt’s lymphoma. J Exp Med (1964) 121:761–70. doi: 10.1084/jem.121.5.761
10. Shannon-Lowe C, Rickinson A. The Global Landscape of EBV-Associated Tumors. Front Oncol (2019) 9:713. doi: 10.3389/fonc.2019.00713
11. Fournier B, Boutboul D, Bruneau J, Miot C, Boulanger C, Malphettes M, et al. Rapid identification and characterization of infected cells in blood during chronic active Epstein-Barr virus infection. J Exp Med (2020) 217:e20192262. doi: 10.1084/jem.20192262
12. Kempkes B, Ling PD. EBNA2 and Its Coactivator EBNA-LP. Curr Top Microbiol Immunol (2015) 391:35–59. doi: 10.1007/978-3-319-22834-1_2
13. Kieser A, Sterz KR. The Latent Membrane Protein 1 (LMP1). Curr Top Microbiol Immunol (2015) 391:119–49. doi: 10.1007/978-3-319-22834-1_4
14. Kalla M, Hammerschmidt W. Human B cells on their route to latent infection-early but transient expression of lytic genes of Epstein-Barr virus. Eur J Cell Biol (2012) 91:65–9. doi: 10.1016/j.ejcb.2011.01.014
15. Babcock JG, Hochberg D, Thorley-Lawson AD. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity (2000) 13:497–506. doi: 10.1016/S1074-7613(00)00049-2
16. Laichalk LL, Thorley-Lawson DA. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J Virol (2005) 79:1296–307. doi: 10.1128/JVI.79.2.1296-1307.2005
17. Totonchy J, Cesarman E. Does persistent HIV replication explain continued lymphoma incidence in the era of effective antiretroviral therapy? Curr Opin Virol (2016) 20:71–7. doi: 10.1016/j.coviro.2016.09.001
18. Damania B, Münz C. Immunodeficiencies that predispose to pathologies by human oncogenic gamma-herpesviruses. FEMS Microbiol Rev (2019) 43:181–92. doi: 10.1093/femsre/fuy044
19. Latour S, Fischer A. Signaling pathways involved in the T-cell-mediated immunity against Epstein-Barr virus: Lessons from genetic diseases. Immunol Rev (2019) 291:174–89. doi: 10.1111/imr.12791
20. Tangye SG, Latour S. Primary immunodeficiencies reveal the molecular requirements for effective host defense against EBV infection. Blood (2020) 135:644–55. doi: 10.1182/blood.2019000928
21. Tsai MH, Lin X, Shumilov A, Bernhardt K, Feederle R, Poirey R, et al. The biological properties of different Epstein-Barr virus strains explain their association with various types of cancers. Oncotarget (2017) 8:10238–54. doi: 10.18632/oncotarget.14380
22. Strowig T, Gurer C, Ploss A, Liu YF, Arrey F, Sashihara J, et al. Priming of protective T cell responses against virus-induced tumors in mice with human immune system components. J Exp Med (2009) 206:1423–34. doi: 10.1084/jem.20081720
23. Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J Virol (2011) 85:165–77. doi: 10.1128/JVI.01512-10
24. Yajima M, Imadome K, Nakagawa A, Watanabe S, Terashima K, Nakamura H, et al. A new humanized mouse model of Epstein-Barr virus infection that reproduces persistent infection, lymphoproliferative disorder, and cell-mediated and humoral immune responses. J Infect Dis (2008) 198:673–82. doi: 10.1086/590502
25. Shultz LD, Saito Y, Najima Y, Tanaka S, Ochi T, Tomizawa M, et al. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r gamma(null) humanized mice. Proc Natl Acad Sci U S A (2010) 107:13022–7. doi: 10.1073/pnas.1000475107
26. Stripecke R, Münz C, Schuringa JJ, Bissig KD, Soper B, Meeham T, et al. Innovations, challenges, and minimal information for standardization of humanized mice. EMBO Mol Med (2020) 12:e8662. doi: 10.15252/emmm.201708662
27. Islas-Ohlmayer M, Padgett-Thomas A, Domiati-Saad R, Melkus MW, Cravens PD, Martin Mdel P, et al. Experimental infection of NOD/SCID mice reconstituted with human CD34+ cells with Epstein-Barr virus. J Virol (2004) 78:13891–900. doi: 10.1128/JVI.78.24.13891-13900.2004
28. Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA, et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med (2006) 12:1316–22. doi: 10.1038/nm1431
29. Azzi T, Lunemann A, Murer A, Ueda S, Beziat V, Malmberg KJ, et al. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood (2014) 124:2533–43. doi: 10.1182/blood-2014-01-553024
30. Williams H, McAulay K, Macsween KF, Gallacher NJ, Higgins CD, Harrison N, et al. The immune response to primary EBV infection: a role for natural killer cells. Br J Haematol (2005) 129:266–74. doi: 10.1111/j.1365-2141.2005.05452.x
31. Hendricks DW, Balfour HH Jr., Dunmire SK, Schmeling DO, Hogquist KA, Lanier LL. Cutting edge: NKG2ChiCD57+ NK cells respond specifically to acute infection with cytomegalovirus and not Epstein-Barr virus. J Immunol (2014) 192:4492–6. doi: 10.4049/jimmunol.1303211
32. Chijioke O, Muller A, Feederle R, Barros MH, Krieg C, Emmel V, et al. Human natural killer cells prevent infectious mononucleosis features by targeting lytic Epstein-Barr virus infection. Cell Rep (2013) 5:1489–98. doi: 10.1016/j.celrep.2013.11.041
33. Landtwing V, Raykova A, Pezzino G, Beziat V, Marcenaro E, Graf C, et al. Cognate HLA absence in trans diminishes human NK cell education. J Clin Invest (2016) 126:3772–82. doi: 10.1172/JCI86923
34. Balfour HH Jr., Odumade OA, Schmeling DO, Mullan BD, Ed JA, Knight JA, et al. Behavioral, virologic, and immunologic factors associated with acquisition and severity of primary epstein-barr virus infection in university students. J Infect Dis (2013) 207:80–8. doi: 10.1093/infdis/jis646
35. Chatterjee B, Deng Y, Holler A, Nunez N, Azzi T, Vanoaica LD, et al. CD8+ T cells retain protective functions despite sustained inhibitory receptor expression during Epstein-Barr virus infection in vivo. PLoS Pathog (2019) 15:e1007748. doi: 10.1371/journal.ppat.1007748
36. Murer A, Ruhl J, Zbinden A, Capaul R, Hammerschmidt W, Chijioke O, et al. MicroRNAs of Epstein-Barr Virus Attenuate T-Cell-Mediated Immune Control In Vivo. MBio (2019) 10:e01941–18. doi: 10.1128/mBio.01482-19
37. Landais E, Saulquin X, Scotet E, Trautmann L, Peyrat MA, Yates JL, et al. Direct killing of Epstein-Barr virus (EBV)-infected B cells by CD4 T cells directed against the EBV lytic protein BHRF1. Blood (2004) 103:1408–16. doi: 10.1182/blood-2003-03-0930
38. Meckiff BJ, Ladell K, McLaren JE, Ryan GB, Leese AM, James EA, et al. Primary EBV Infection Induces an Acute Wave of Activated Antigen-Specific Cytotoxic CD4+ T Cells. J Immunol (2019) 203:1276–87. doi: 10.4049/jimmunol.1900377
39. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol (2014) 32:364–72. doi: 10.1038/nbt.2858
40. Lang JL, Kelly M, Freed BM, McCarter MD, Kedl RM, Torres RM, et al. Studies of Lymphocyte Reconstitution in a Humanized Mouse Model Reveal a Requirement of T Cells for Human B Cell Maturation. J Immunol (2013) 190:2090–101. doi: 10.4049/jimmunol.1202810
41. Watanabe Y, Takahashi T, Okajima A, Shiokawa M, Ishii N, Katano I, et al. The analysis of the functions of human B and T cells in humanized NOD/shi-scid/gammac(null) (NOG) mice (hu-HSC NOG mice). Int Immunol (2009) 21:843–58. doi: 10.1093/intimm/dxp050
42. Gurer C, Strowig T, Brilot F, Pack M, Trumpfheller C, Arrey F, et al. Targeting the nuclear antigen 1 of Epstein-Barr virus to the human endocytic receptor DEC-205 stimulates protective T-cell responses. Blood (2008) 112:1231–9. doi: 10.1182/blood-2008-03-148072
43. Yajima M, Imadome K, Nakagawa A, Watanabe S, Terashima K, Nakamura H, et al. T cell-mediated control of Epstein-Barr virus infection in humanized mice. J Infect Dis (2009) 200:1611–5. doi: 10.1086/644644
44. Li Y, Masse-Ranson G, Garcia Z, Bruel T, Kok A, Strick-Marchand H, et al. A human immune system mouse model with robust lymph node development. Nat Methods (2018) 15:623–30. doi: 10.1038/s41592-018-0071-6
45. Jangalwe S, Shultz LD, Mathew A, Brehm MA. Improved B cell development in humanized NOD-scid IL2Rgamma(null) mice transgenically expressing human stem cell factor, granulocyte-macrophage colony-stimulating factor and interleukin-3. Immun Inflamm Dis (2016) 4:427–40. doi: 10.1002/iid3.124
46. Antsiferova O, Müller A, Rämer P, Chijioke O, Chatterjee B, Raykova A, et al. Adoptive transfer of EBV specific CD8+ T cell clones can transiently control EBV infection in humanized mice. PLoS Pathog (2014) 10:e1004333. doi: 10.1371/journal.ppat.1004333
47. McHugh D, Caduff N, Barros MHM, Rämer P, Raykova A, Murer A, et al. Persistent KSHV infection increases EBV-associated tumor formation in vivo via enhanced EBV lytic gene expression. Cell Host Microbe (2017) 22:61–73. doi: 10.1016/j.chom.2017.06.009
48. Coleman CB, Lang J, Sweet LA, Smith NA, Freed BM, Pan Z, et al. Epstein-Barr Virus Type 2 Infects T Cells and Induces B Cell Lymphomagenesis in Humanized Mice. J Virol (2018) 92:e00813-818. doi: 10.1128/JVI.00813-18
49. Adldinger HK, Delius H, Freese UK, Clarke J, Bornkamm GW. A putative transforming gene of Jijoye virus differs from that of Epstein-Barr virus prototypes. Virology (1985) 141:221–34. doi: 10.1016/0042-6822(85)90253-3
50. Dambaugh T, Hennessy K, Chamnankit L, Kieff E. U2 region of Epstein-Barr virus DNA may encode Epstein-Barr nuclear antigen 2. Proc Natl Acad Sci U S A (1984) 81:7632–6. doi: 10.1073/pnas.81.23.7632
51. Sample J, Young L, Martin B, Chatman T, Kieff E, Rickinson A, et al. Epstein-Barr virus types 1 and 2 differ in their EBNA-3A, EBNA-3B, and EBNA-3C genes. J Virol (1990) 64:4084–92. doi: 10.1128/JVI.64.9.4084-4092.1990
52. Romero-Masters JC, Ohashi M, Djavadian R, Eichelberg MR, Hayes M, Bristol JA, et al. An EBNA3C-deleted Epstein-Barr virus (EBV) mutant causes B-cell lymphomas with delayed onset in a cord blood-humanized mouse model. PLoS Pathog (2018) 14:e1007221. doi: 10.1371/journal.ppat.1007221
53. Correia S, Bridges R, Wegner F, Venturini C, Palser A, Middeldorp JM, et al. Sequence Variation of Epstein-Barr Virus: Viral Types, Geography, Codon Usage, and Diseases. J Virol (2018) 92:e01132-18. doi: 10.1128/JVI.01132-18
54. Tsai MH, Raykova A, Klinke O, Bernhardt K, Gartner K, Leung CS, et al. Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep (2013) 5:458–70. doi: 10.1016/j.celrep.2013.09.012
55. Li Z, Tsai MH, Shumilov A, Baccianti F, Tsao SW, Poirey R, et al. Epstein-Barr virus ncRNA from a nasopharyngeal carcinoma induces an inflammatory response that promotes virus production. Nat Microbiol (2019) 4:2475–86. doi: 10.1038/s41564-019-0546-y
56. White RE, Ramer PC, Naresh KN, Meixlsperger S, Pinaud L, Rooney C, et al. EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J Clin Invest (2012) 122:1487–502. doi: 10.1172/JCI58092
57. Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD. The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol (2015) 33:787–821. doi: 10.1146/annurev-immunol-032414-112326
58. Albanese M, Tagawa T, Bouvet M, Maliqi L, Lutter D, Hoser J, et al. Epstein-Barr virus microRNAs reduce immune surveillance by virus-specific CD8+ T cells. Proc Natl Acad Sci U S A (2016) 113:E6467–75. doi: 10.1073/pnas.1605884113
59. Gregorovic G, Boulden EA, Bosshard R, Karstegl CE, Skalsky R, Cullen BR, et al. Epstein-Barr viruses deficient in EBER RNAs give higher LMP2 RNA expression in lymphoblastoid cell lines and efficiently establish persistent infection in humanized mice. J Virol (2015) 89:11711–4. doi: 10.1128/JVI.01873-15
60. Murer A, McHugh D, Caduff N, Kalchschmidt JS, Barros MH, Zbinden A, et al. EBV persistence without its EBNA3A and 3C oncogenes in vivo. PLoS Pathog (2018) 14:e1007039. doi: 10.1371/journal.ppat.1007039
61. Ma SD, Xu X, Jones R, Delecluse HJ, Zumwalde NA, Sharma A, et al. PD-1/CTLA-4 Blockade Inhibits Epstein-Barr Virus-Induced Lymphoma Growth in a Cord Blood Humanized-Mouse Model. PLoS Pathog (2016) 12:e1005642. doi: 10.1371/journal.ppat.1005642
62. Romero-Masters JC, Ohashi M, Djavadian R, Eichelberg MR, Hayes M, Zumwalde NA, et al. An EBNA3A-Mutated Epstein-Barr Virus Retains the Capacity for Lymphomagenesis in a Cord Blood-Humanized Mouse Model. J Virol (2020) 94:e02168–19. doi: 10.1128/JVI.02168-19
63. Ma SD, Xu X, Plowshay J, Ranheim EA, Burlingham WJ, Jensen JL, et al. LMP1-deficient Epstein-Barr virus mutant requires T cells for lymphomagenesis. J Clin Invest (2015) 125:304–15. doi: 10.1172/JCI76357
64. Ma SD, Tsai MH, Romero-Masters JC, Ranheim EA, Huebner SM, Bristol J, et al. LMP1 and LMP2A collaborate to promote Epstein-Barr virus (EBV)-induced B cell lymphomas in a cord blood-humanized mouse model but are not essential. J Virol (2017) 91:e01928-16. doi: 10.1128/JVI.01928-16
65. Ma SD, Yu X, Mertz JE, Gumperz JE, Reinheim E, Zhou Y, et al. An Epstein-Barr Virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. J Virol (2012) 86:7976–87. doi: 10.1128/JVI.00770-12
66. Bristol JA, Djavadian R, Albright ER, Coleman CB, Ohashi M, Hayes M, et al. A cancer-associated Epstein-Barr virus BZLF1 promoter variant enhances lytic infection. PLoS Pathog (2018) 14:e1007179. doi: 10.1371/journal.ppat.1007179
67. Lünemann JD, Edwards N, Muraro PA, Hayashi S, Cohen JI, Münz C, et al. Increased frequency and broadened specificity of latent EBV nuclear antigen-1-specific T cells in multiple sclerosis. Brain (2006) 129:1493–506. doi: 10.1093/brain/awl067
68. Lünemann JD, Jelcic I, Roberts S, Lutterotti A, Tackenberg B, Martin R, et al. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-g and IL-2. J Exp Med (2008) 205:1763–73. doi: 10.1084/jem.20072397
69. Wang J, Jelcic I, Muhlenbruch L, Haunerdinger V, Toussaint NC, Zhao Y, et al. HLA-DR15 Molecules Jointly Shape an Autoreactive T Cell Repertoire in Multiple Sclerosis. Cell (2020) 183:1264–81.e20. doi: 10.1016/j.cell.2020.09.054
70. Zdimerova H, Murer A, Engelmann C, Raykova A, Deng Y, Gujer C, et al. Attenuated immune control of Epstein-Barr virus in humanized mice is associated with the multiple sclerosis risk factor HLA-DR15. Eur J Immunol (2021) 51:64-75. doi: 10.1002/eji.202048655
71. Caduff N, McHugh D, Murer A, Ramer P, Raykova A, Landtwing V, et al. Immunosuppressive FK506 treatment leads to more frequent EBV-associated lymphoproliferative disease in humanized mice. PLoS Pathog (2020) 16:e1008477. doi: 10.1371/journal.ppat.1008477
72. McHugh D, Myburgh R, Caduff N, Spohn M, Kok YL, Keller CW, et al. EBV renders B cells susceptible to HIV-1 in humanized mice. Life Sci Alliance (2020) 3:e202000640. doi: 10.26508/lsa.202000640
73. Chijioke O, Marcenaro E, Moretta A, Capaul R, Munz C. The SAP-dependent 2B4 receptor mediates CD8+ T cell dependent immune control of Epstein Barr virus infection in mice with reconstituted human immune system components. J Infect Dis (2015) 212:803–7. doi: 10.1093/infdis/jiv114
74. Pasquier B, Yin L, Fondaneche MC, Relouzat F, Bloch-Queyrat C, Lambert N, et al. Defective NKT cell development in mice and humans lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product. J Exp Med (2005) 201:695–701. doi: 10.1084/jem.20042432
75. Yuling H, Ruijing X, Li L, Xiang J, Rui Z, Yujuan W, et al. EBV-induced human CD8+ NKT cells suppress tumorigenesis by EBV-associated malignancies. Cancer Res (2009) 69:7935–44. doi: 10.1158/0008-5472.CAN-09-0828
76. Chung BK, Tsai K, Allan LL, Zheng DJ, Nie JC, Biggs CM, et al. Innate immune control of EBV-infected B cells by invariant natural killer T cells. Blood (2013) 122:2600–8. doi: 10.1182/blood-2013-01-480665
77. Zumwalde NA, Sharma A, Xu X, Ma S, Schneider CL, Romero-Masters JC, et al. Adoptively transferred Vgamma9Vdelta2 T cells show potent antitumor effects in a preclinical B cell lymphomagenesis model. JCI Insight (2017) 2:e93179. doi: 10.1172/jci.insight.93179
78. Schwab C, Gabrysch A, Olbrich P, Patino V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 CTLA-4-insufficient individuals. J Allergy Clin Immunol (2018) 142:1932–46. doi: 10.1016/j.jaci.2018.02.055
79. Johnson DB, McDonnell WJ, Gonzalez-Ericsson PI, Al-Rohil RN, Mobley BC, Salem JE, et al. A case report of clonal EBV-like memory CD4+ T cell activation in fatal checkpoint inhibitor-induced encephalitis. Nat Med (2019) 25:1243–50. doi: 10.1038/s41591-019-0523-2
80. Xiang Z, Liu Y, Zheng J, Liu M, Lv A, Gao Y, et al. Targeted activation of human Vgamma9Vdelta2-T cells controls epstein-barr virus-induced B cell lymphoproliferative disease. Cancer Cell (2014) 26:565–76. doi: 10.1016/j.ccr.2014.07.026
81. Gujer C, Murer A, Muller A, Vanoaica D, Sutter K, Jacque E, et al. Plasmacytoid dendritic cells respond to Epstein-Barr virus infection with a distinct type I interferon subtype profile. Blood Adv (2019) 3:1129–44. doi: 10.1182/bloodadvances.2018025536
82. Thorley-Lawson DA, Allday MJ. The curious case of the tumour virus: 50 years of Burkitt’s lymphoma. Nat Rev Microbiol (2008) 6:913–24. doi: 10.1038/nrmicro2015
83. Rochford R, Cannon MJ, Moormann AM. Endemic Burkitt’s lymphoma: a polymicrobial disease? Nat Rev Microbiol (2005) 3:182–7. doi: 10.1038/nrmicro1089
84. Burkitt D. A sarcoma involving the jaws in African children. Br J Surg (1958) 46:218–23. doi: 10.1002/bjs.18004619704
85. Cesarman E, Damania B, Krown SE, Martin J, Bower M, Whitby D. Kaposi sarcoma. Nat Rev Dis Primers (2019) 5:9. doi: 10.1038/s41572-019-0060-9
86. Faure A, Hayes M, Sugden B. How Kaposi’s sarcoma-associated herpesvirus stably transforms peripheral B cells towards lymphomagenesis. Proc Natl Acad Sci U S A (2019) 116:16519–28. doi: 10.1073/pnas.1905025116
87. Labo N, Marshall V, Miley W, Davis E, McCann B, Stolka KB, et al. Mutual detection of Kaposi’s sarcoma-associated herpesvirus and Epstein-Barr virus in blood and saliva of Cameroonians with and without Kaposi’s sarcoma. Int J Cancer (2019) 145:2468–77. doi: 10.1002/ijc.32546
88. Sallah N, Miley W, Labo N, Carstensen T, Fatumo S, Gurdasani D, et al. Distinct genetic architectures and environmental factors associate with host response to the gamma2-herpesvirus infections. Nat Commun (2020) 11:3849. doi: 10.1038/s41467-020-17696-2
89. Klein U, Gloghini A, Gaidano G, Chadburn A, Cesarman E, Dalla-Favera R, et al. Gene expression profile analysis of AIDS-related primary effusion lymphoma (PEL) suggests a plasmablastic derivation and identifies PEL-specific transcripts. Blood (2003) 101:4115–21. doi: 10.1182/blood-2002-10-3090
90. Epstein MA, Morgan AJ, Finerty S, Randle BJ, Kirkwood JK. Protection of cottontop tamarins against Epstein-Barr virus-induced malignant lymphoma by a prototype subunit vaccine. Nature (1985) 318:287–9. doi: 10.1038/318287a0
91. Cleary ML, Epstein MA, Finerty S, Dorfman RF, Bornkamm GW, Kirkwood JK, et al. Individual tumors of multifocal EB virus-induced malignant lymphomas in tamarins arise from different B-cell clones. Science (1985) 228:722–4. doi: 10.1126/science.2986287
92. Yu H, Borsotti C, Schickel JN, Zhu S, Strowig T, Eynon EE, et al. A novel humanized mouse model with significant improvement of class-switched, antigen-specific antibody production. Blood (2017) 129:959–69. doi: 10.1182/blood-2016-04-709584
93. Li C, Romero-Masters JC, Huebner S, Ohashi M, Hayes M, Bristol JA, et al. EBNA2-deleted Epstein-Barr virus (EBV) isolate, P3HR1, causes Hodgkin-like lymphomas and diffuse large B cell lymphomas with type II and Wp-restricted latency types in humanized mice. PLoS Pathog (2020) 16:e1008590. doi: 10.1371/journal.ppat.1008590
Keywords: cytotoxic lymphocytes, human immunodeficiency virus (HIV), Kaposi sarcoma associated herpesvirus (KSHV), HLA-DRB1*1501, mutant Epstein Barr viruses (EBVs)
Citation: Schuhmachers P and Münz C (2021) Modification of EBV Associated Lymphomagenesis and Its Immune Control by Co-Infections and Genetics in Humanized Mice. Front. Immunol. 12:640918. doi: 10.3389/fimmu.2021.640918
Received: 12 December 2020; Accepted: 08 March 2021;
Published: 23 March 2021.
Edited by:
Yan Li, Nanjing University, ChinaReviewed by:
Benjamin E. Gewurz, Brigham and Women’s Hospital and Harvard Medical School, United StatesCopyright © 2021 Schuhmachers and Münz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Münz, Y2hyaXN0aWFuLm11ZW56QHV6aC5jaA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.