 Darrell O. Ricke
Darrell O. Ricke
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Immunol., 24 February 2021
Sec. Vaccines and Molecular Therapeutics
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.640093
This article is part of the Research TopicImmunological Aspects of Vaccine SafetyView all 11 articles
COVID-19 (SARS-CoV-2) disease severity and stages varies from asymptomatic, mild flu-like symptoms, moderate, severe, critical, and chronic disease. COVID-19 disease progression include lymphopenia, elevated proinflammatory cytokines and chemokines, accumulation of macrophages and neutrophils in lungs, immune dysregulation, cytokine storms, acute respiratory distress syndrome (ARDS), etc. Development of vaccines to severe acute respiratory syndrome (SARS), Middle East Respiratory Syndrome coronavirus (MERS-CoV), and other coronavirus has been difficult to create due to vaccine induced enhanced disease responses in animal models. Multiple betacoronaviruses including SARS-CoV-2 and SARS-CoV-1 expand cellular tropism by infecting some phagocytic cells (immature macrophages and dendritic cells) via antibody bound Fc receptor uptake of virus. Antibody-dependent enhancement (ADE) may be involved in the clinical observation of increased severity of symptoms associated with early high levels of SARS-CoV-2 antibodies in patients. Infants with multisystem inflammatory syndrome in children (MIS-C) associated with COVID-19 may also have ADE caused by maternally acquired SARS-CoV-2 antibodies bound to mast cells. ADE risks associated with SARS-CoV-2 has implications for COVID-19 and MIS-C treatments, B-cell vaccines, SARS-CoV-2 antibody therapy, and convalescent plasma therapy for patients. SARS-CoV-2 antibodies bound to mast cells may be involved in MIS-C and multisystem inflammatory syndrome in adults (MIS-A) following initial COVID-19 infection. SARS-CoV-2 antibodies bound to Fc receptors on macrophages and mast cells may represent two different mechanisms for ADE in patients. These two different ADE risks have possible implications for SARS-CoV-2 B-cell vaccines for subsets of populations based on age, cross-reactive antibodies, variabilities in antibody levels over time, and pregnancy. These models place increased emphasis on the importance of developing safe SARS-CoV-2 T cell vaccines that are not dependent upon antibodies.
The SARS-CoV-2 virus is a unclassified betacoronavirus with sequenced genomes ranging from 29.8 to 29.9 k RNA bases. The SARS-CoV-2 genome encodes replicase proteins, structural proteins, and accessory proteins (1). The ORF1a and ORF1ab polyproteins are proteolytically cleaved into 16 non-structural proteins designated nsp1-16 (1). Like SARS, COVID-19 manifests as a virulent zoonotic virus in humans with currently 101,211,750 global cases and 2,183,169 deaths as of Jan. 28, 2021 (2). The details of SARS-CoV-2 infections and disease progression are still being worked out. One proposed step in COVID-19 disease progression involves the nucleocapsid protein binding to the prostaglandin-endoperoxide synthase 2 (PTGS2)/cyclooxygenase-2 (COX-2) promoter and upregulating expression resulting in elevated levels of prostaglandin E2 (PGE2) and other inflammatory molecules (3–5). Elevated PGE2 may be driving hyper-activation of mast cells associated with excess release of histamine and additional inflammatory molecules (5). COVID-19 is predicted to be a mast cell disease (6).
Zoonotic MERS-CoV, SARS-CoV-1, and SARS-CoV-2 are evolutionarily related with similarities in disease progression in humans. The mild variant first phase of viral progression generally presents with mild flu-like symptoms. For some individuals, infection progresses to a second moderate-severe variant phase. Progression to this phase coincidently coincides with timing of anticipated humoral immunity antibody response from memory B-cells for cross reactive antibodies. Coronavirus infection of phagocytic cells has been previously observed. MERS-CoV can infect monocyte-derived macrophages (MDMs), monocyte-derived dendric cells (MoDCs), and T cells (7, 8). In a mouse animal model, phagocytic cells contribute to the antibody-mediate elimination of SARS-CoV-1 (9). This process is expected for patients with mild symptoms who do not progress to moderate or severe disease. For patients with moderate and severe symptoms, pathophysiology is consistent with infection of phagocytic immune cells (immature MDMs and MoDCs). Chemokines attract additional dendritic cells and immature macrophages that are susceptible to infection leading to a possible infection amplifying cascade of phagocytic immune cells. For some patients with severe symptoms, excessive accumulation of macrophages contributes toward a storm of cytokines (10–12) and chemokines. These viruses also perturb the adaptive immune responses within infected individuals. Individuals with SARS have pronounced peripheral T cell lymphocytopenia with reduced CD4+ and CD8+ T cells (13, 14). MERS-CoV and SARS-CoV are associated with T cell apoptosis (15, 16). Infection of macrophages (17) and some T cells along with viral dysregulation of cellular pathways result in compromised innate and humoral immunity in patients in phase II (18). The possibility of migration throughput the body of infected immune cells and later high virus titer in blood can account for additional disease pathophysiology clinical observations observed for these viruses. Other disease differences may simply be the different population of cells with target host receptors angiotensin I converting enzyme 2 (ACE2) for SARS-CoV-1 and SARS-CoV-2 and dipeptidyl peptidase IV (DPP4) for MERS-CoV. The increased affinity of the SARS-CoV-2 Spike protein receptor-binding-domain (RBD) compared to SARS may account for the significant airborne transmission of SARS-CoV-2 (19). Also, neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity (20).
Characterizing variability of viral proteins can inform designing medical countermeasures (MCMs). For viral progeny, deleterious mutations are selected against (21). Neutral mutations (22) provide a framework for antigenic drift to facilitate escape from immune responses; these residues will continue to mutate over time. The critical-spacer model proposes that proteins have either amino acid residue side-chains critical for function or have variable side-chains while possibly function for positioning/folding of critical residues (23). The divergence model of protein evolution proposes that number of critical residues for a protein is consistent for evolutionarily closely related proteins (24). These concepts are applied to SARS-CoV-2 Spike (S) protein leveraging closely related coronavirus protein sequences to provide insights into viral vulnerabilities that can be leveraged in designing MCMs. The exposed domain of the Spike protein exhibits exposed surface areas with high variability. Increased risk for antibody-dependent enhancement (ADE) from antibodies targeting SARS-CoV-2, SARS-CoV-1, and MERS-CoV exposed residues is indicated by observed ADE in animal models and the antibody facilitated infection of phagocytic immune cells by coronaviruses (9, 25). In addition, SARS-CoV-2 antibodies bound to mast cells may also be involved in ADE for some MIS-C and MIS-A patients (26).
SARS-CoV-2 spike protein sequence from GenBank entry MN908947.3 was searched against the non-redundant (nr) and PDB database using the NCBI BLASTP web interface. Hit protein sequences were downloaded. Protein multiple sequence alignments were created with the Dawn program (27). The Spike structure 6CRZ (28) was downloaded from RCSB PDB database (29). Dawn variation results were visualized with the Chimera program (30).
Dawn variation (V <n>) results for SARS-CoV-2 amino acid residues were classified as 650 V1 residues—dark green, 263 V2 residues—light green, 123 V3 residues—yellow, 107 V4 residues—light blue, and 152 V5+ residues—dark blue (Figure 1). The dark green residues represent candidate critical residues and the dark blue residues represent candidate spacer residues (Figure 1). Amino acid residues with conservative substitutions are also consider critical residues, and are colored light green in Figure 1; positions with > 95% of a single residue were included in this category to accommodate potential sequencing errors and possibly adaptative mutations. The V1+V2 residues represent 71% of the 1,295 Spike residues. The Spike protein exhibits regions of extensive variability of exposed surface residues (Figure 1).
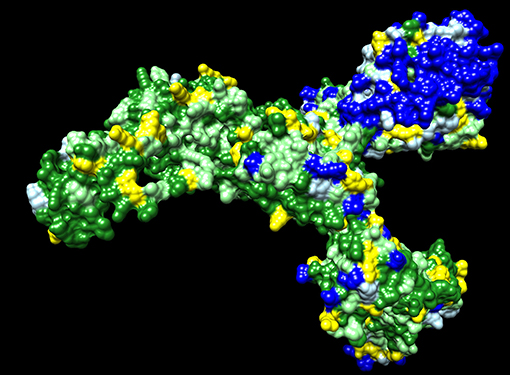
Figure 1. SARS-CoV-2 Spike protein variation results. Amino acid residue color code: dark green (critical residues—V1), light green (critical residues with conservative substitutions or variant in <10 sequences—V2, yellow (three variants—V3), light blue (four variants—V4; likely spacer residues), and blue (5+ variants—V5+; spacer residues).
The observed amino acid variations in SARS-CoV-2 proteins are consistent with expected natural variations in the context of random mutations and selection in the context of host immune responses. The Spike protein S1 extended domain shows the highest number of exposed surface highly variable residues (Figure 1). These spacer residues may function as exposed antigens for antibody responses with the possibility of suppressing immune responses to less immunogenic surface antigens. Many of these Spike protein antigens may lead to non-neutralizing antibodies. Mutations at these residues may provide antigenic drift to escape immune responses. As the COVID-19 pandemic continues, Spike mutation variants are accumulating resulting in the design of vaccine booster shots prior to initial population vaccinations (31). The Spike protein represents an evolving vaccine target with parallels to the annual influenza vaccine hemagglutinin and neuraminidase targets while the COVID-19 pandemic persists enabling rapid virus evolution in humans.
Coronaviruses have multiple approaches for infecting cells by direct receptor binding and by indirect antibody Fc uptake. The SARS-CoV-2 Spike protein contains receptor-binding domains (RBD) targeting human angiotensin I converting enzyme 2 (ACE2) (32, 33); this is the initial route for infecting host cells. To take advantage of antibody responses, coronaviruses also leverage antibody Fc uptake to infect some phagocytic immune cells (34). Coronaviruses use the Spike protein subunit 2 fusion peptide (FP), heptad repeat 1 (HR1), and heptad repeat 2 (HR2) to infect immune cells upon proteolytic cleavage of Spike within endosomes. HR1 and HR2 form a canonical 6-helix bundle involved in membrane fusion (35). Jaume et al. (34) found that antibody-mediated infection was dependent on Fc receptor II and not the endosomal/lysosomal pathway utilized by ACE2 targeting. Viral infection of complement receptor (CR) cells is an additional possible route of infecting cells expanding cellular tropism (36). This expanded cellular tropism mechanism provides coronaviruses like SARS-CoV-1, MERS-CoV, and SARS-CoV-2 with more than one cellular trophism for infecting host cells. This leads to the prediction that antibody mediated uptake of virus is the potential mechanism that induces ADE to cross-reactivity antibodies, maternally transferred antibodies (matAbs), and vaccines (37–40).
Macrophages play an important role in disease progression and possibly immune dysregulation for SARS and COVID-19. Lymphopenia is a common feature in patients with SARS (13, 41) and COVID-19 (42, 43). Direct infection of subpopulations of immune cells is possible if they express virus target receptors. Two receptors have been identified for SARS-CoV-1 including ACE2 (44) and C-type lectin domain family 4 member M (CLEC4M, CD209L, CD299, DC-SIGN2, DC-SIGNR, HP10347, and LSIGN) (45) with CLEC4M expressed in human lymph nodes (46). In a mouse model, depletion of CD4+ T cells resulted in an enhanced immune-mediated interstitial pneumonitis when challenged with SARS-CoV-1 (47). But, depletion of CD4+ and CD8+ T cells and antibodies enabled the innate defense mechanisms to control the SARS-CoV-1 virus without immune dysregulation (47). Similar results were also observed in mice with SARS-CoV-1 challenge, but treatment with liposomes containing clodronate, which deplete alveolar macrophages (AM), prevented immune deficient virus-specific T cell response (48). These studies point to an interplay between antibodies and macrophages in ADE responses in animal models. In a macaque model, anti-spike IgG causes acute lung injury by skewing macrophage response toward proinflammatory monocyte/macrophage recruitment and accumulation during acute SARS-CoV-1 infection (49). Blockade of in vitro human activated macrophages FcγR reduced proinflammatory cytokine production (49). CD169+ macrophages have ACE2 and are susceptible to SARS-CoV-2 infection (50). Both M1- and M2-type macrophages are susceptible to SARS-CoV-2 infection (51). These observations are likely linked by antibody-dependent enhancement of coronavirus infection of macrophages (34, 52). The pathophysiology of moderate and severe SARS and COVID-19 diseases fits a proposed model of antibody-dependent infection of macrophages as the key gate step in disease progression from mild to moderate and severe symptoms contributing to dysregulated immune responses (53) including apoptosis for some T cells/T cell lymphopenia, proinflammatory cascade with macrophage accumulation, and cytokine and chemokine accumulations in lungs with a cytokine storm in some patients. Infected phagocytic immune cells may enable the virus to spread to additional organs prior to viral sepsis (Figure 2).
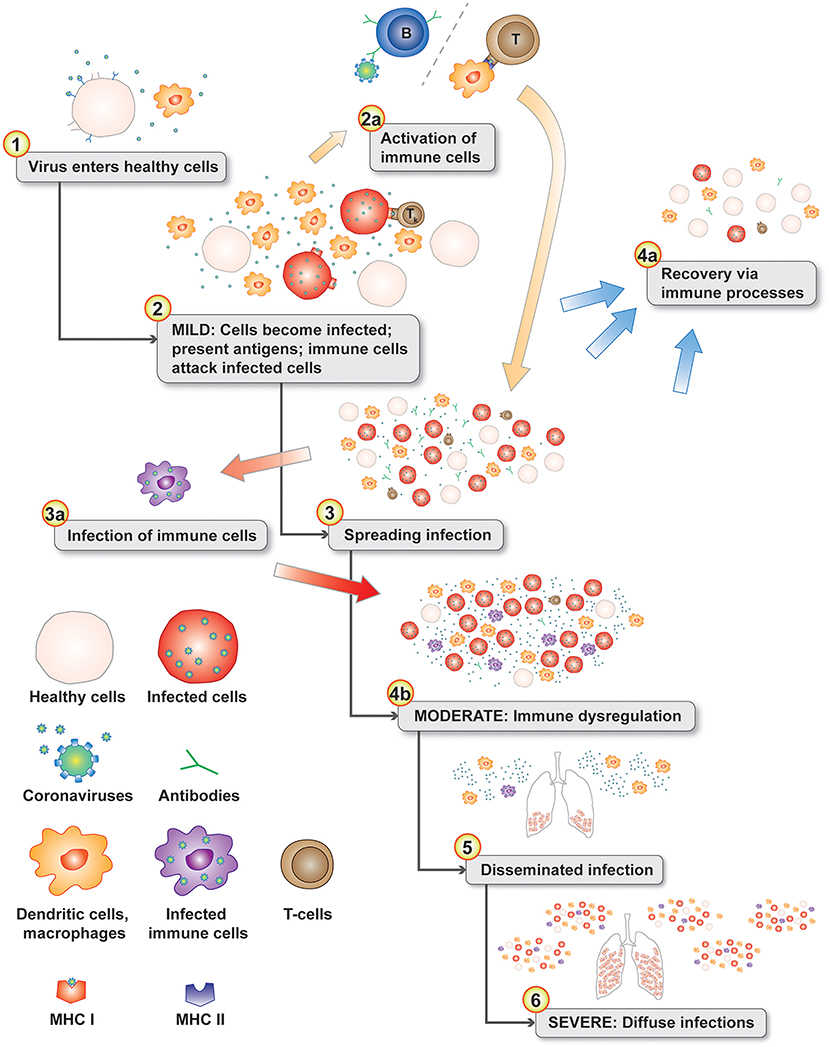
Figure 2. Disease progression model with normal immune responses during the initial mild symptoms phase (see 1–3). Antigen presenting cells migrate to the lymph nodes to activate T cells (2a). The progression gate to moderate and server disease is the infection of phagocytic immune cells (3a) leading to immune dysregulation (4b). In the lungs, chemokines attract additional dendritic cells and immature macrophages that are subsequently infected in an positive feedback-loop infection cascade (4b). Infected phagocytic immune cells disseminate throughout the body infecting additional organs (5 & 6). Levels of chemokine and cytokines in the lungs from infected cells can create a cytokine storm (6).
Antibody-dependent enhancement (ADE) may develop via more than one molecular mechanism. One model suggestions that antibody/Fc-receptor complex functionally mimics viral receptor enabling expanded host cell trophism of some phagocytic cells (54). Wan et al. (54) illustrate an antibody dosage effect for enhancing disease or inhibiting the virus dependent upon the antibody dosage. It is well-established that antibodies to one strain of a virus may be subneutralizing or non-neutralizing for viral infections of different strains (55–57). Infection of cells expressing Fc-gamma was shown for SARS-CoV-1 (58). A possible case of ADE was observed in a patient with a second SARS-CoV-2 infection (59). Early vaccine results show significant antibody responses by day 14 (60) which represents memory B-cell responses (i.e., original antigenic sin) with cross-reactivity antibodies from likely other coronavirus strain(s). Early high antibody responses are correlated with increased disease severity for both SARS (61) and COVID-19 (62–67). Wu et al. demonstrated that antibodies from COVID-19 patients enabled SARS-CoV-2 infections of Raji cells (lymphoma cells derived from B lymphocytes), K562 cells (derived from monocytes), and primary B cells (68). SARS-CoV-2 infection of some phagocytic cells (i.e., macrophages) may be a key gate step in disease progression for some patients.
Mast cells can degranulated by both IgE and IgG antibodies bound to Fc receptors (69). Cardiac injury is a common condition among hospitalized COVID-19 patients and is associated with higher risk of mortality (70). However, pathological manifestations of heart tissues found only scarce interstitial mononuclear inflammatory infiltrates without substantial myocardial damage (42). Myocardial injury significantly correlates with fatal outcome for COVID-19 (71). Multisystem inflammatory syndrome in children (MIS-C) and adults (MIS-A) associated with COVID-19 has appeared in areas following SARS-CoV-2 outbreaks. A model of MIS-C has been proposed where activation and degranulation of mast cells with Fc receptor-bound SARS-CoV-2 antibodies leads to increased histamine levels (26). This model is consistent with MIS-C in infants with maternally transferred antibodies (matAbs) (37–40) to SARS-CoV-2. SARS-CoV-2 nucleocapsid binding to PTGS2 prompter resulting in upregulated prostaglandin E2 (PGE2) in COVID-19 patients (4). Elevated PGE2 may be driving hyper-activated mast cells as an alternative mechanism driving increased histamine levels in older children and adults. These increased histamine levels are predicted to impede blood flow through cardiac capillaries due to constricted pericytes with increased risk for cardiac pathology due to cell death by anoxia and coronary artery aneurysms due to increased blood pressure (26). An instance of a 12 years old child with a previous asymptomatic COVID-19 infection developing MIS-C on likely second infection has been reported (72).
Virus vaccines can use live-attenuated virus strains, inactivated (killed) virus, protein subunit, messenger ribonucleic acid (mRNA), or deoxyribonucleic acid (DNA) vaccine. Antibodies induced by vaccines can be neutralizing or non-neutralizing. Non-neutralizing antibodies can contribute to anti-viral activities with mechanisms including antibody-medicated complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP) [reviewed (73)]. The yearly influenza vaccine induces both neutralizing and non-neutralizing antibodies that provide projection against the strains in the vaccine and closely related strains. Vaccine-associated enhanced disease (VAED) can result when there are multiple circularizing serotypes of virus [e.g., Dengue fever (55–57)] or when the virus uses antibodies for expanded host cell trophism of phagocytic immune cells.
Many of the viruses associated with ADE have cell membrane fusion mechanisms (38). For influenza A H1N1, vaccine-induced cross-reactive anti-HA2 antibodies in a swine model promote virus fusion causing vaccine-associated enhanced respiratory disease (VAERD) (74). ADE was observed for the respiratory syncytial virus (RSV) in the Bonnet monkey model (37). Van Erp et al. (37) recommends avoidance of induction of respiratory syncytial virus (RSV) non-neutralizing antibodies or subneutralizing antibodies to avoid ADE. ADE has been observed in multiple SARS-CoV-1 animal models. In a mouse model, attempts to create vaccines for SARS-CoV-1 lead to pulmonary immunopathology upon challenge with SARS-CoV-1 (75, 76); these vaccines included inactivated whole viruses, inactivated viruses with adjuvant, and a recombinant DNA spike (S) protein vaccine in a virus-like particle (VLP) vaccine. Severe pneumonia was observed in mice vaccinated with nucleocapsid protein after challenge with SARS-CoV-1 (77). Enhanced hepatitis was observed in a ferret model with a vaccine with recombinant modified vaccinia virus Ankara (rMVA) expressing the SARS-CoV-1 Spike protein (78). ADE was observed for rhesus macaques with SARS-CoV-1 vaccine (79). SARS-CoV-1 ADE is mediated by spike protein antibodies (80). Antibodies to the SARS-CoV-1 spike protein can mediate viral entry via Fc receptor-expressing cells in a dose-dependent manner (54). Jaume et al. (34) point out the potential pitfalls associated with immunizations against SARS-CoV-1 Spike protein due to Fc mediate infection of immune cells. This leads to the prediction that new attempts to create either SARS-CoV-1 vaccines, MERS-CoV vaccines (81), or SARS-CoV-2 vaccines have potentially higher risks for inducing ADE in humans facilitated by antibody infection of phagocytic immune cells. This potential ADE risk is independent of the vaccine technology (82) or targeting strategy selected due to predicted phagocytic immune cell infections upon antibody uptake. For MERS patients, the seroconversion rate increased with disease severity (83). Severe clinical worsening for SARS patients occurs concurrently with timing of IgG seroconversion (84). Clinical evidence of early high IgG responses in SARS patients is correlated with disease progression (85) and severity (62–67). Antibody treatments for critically ill COVID-19 patients have been halted due to a potential safety signal and unfavorable risk-benefit profile (86). Current SARS-CoV-2 vaccines appear to be providing protection with high antibody titers; the possibility of ADE risks associated with waning titers of antibodies over time remains unknown.
Convalescent plasma therapy takes the antibodies from a recovering patient and provides them to patients with active infections. COVID-19 results for convalescent plasma therapy appear to have mixed results with no statistically significant improvement in randomized clinical trials (87, 88): in a trial, no significant difference in 28-days mortality (15.7 vs 24.0% odds ratio: 0.59, p = 0.30) was observed in a randomized trial (87); and, in the PLACID trial, progression to severe disease or all-cause mortality at 28 days occurred in 44 (19%) convalescent plasma arm vs. 41 (18%) control arm (risk ratio 1.04) (88). Neither trial mentions antibody-dependent enhancement in context of progression to severe disease or all-cause mortality. For SARS, a higher discharge rate was observed amount patients who were given convalescent plasma before day 14 of illness (58.3%) vs. after 14 days (15.6%), p < 0.001; the mortality rate for the second group was 21.9% which was higher than the all SARS-related mortality rate in Hong Kong of 17% (89); while this looks promising for most patients, the increased mortality above the regional average observed for patients after 14 days of illness should be noted.
Analyzing the Cryo-EM structures of MERS-CoV and SARS-CoV-1 spike (S) glycoproteins, Yuan et al. (90) suggest that the fusion peptide (FP) and the heptad repeat 1 region (HR1) are potential targets for eliciting broadly neutralizing antibodies based on exposure on the surface of the stem region, with no N-linked glycosylation sites in this region, and sequence conservation. Antibodies that interrupt virus-cell fusion will likely block the infection of immune cells using Fc-mediated uptake of virus (34). This has been demonstrated for SARS-CoV-1 for antibodies to the HR2 region (91–93). Likewise, SARS-CoV-2 antibodies that block cell fusion are likely to not share the same ADE risk of other SARS-CoV-2 antibodies.
B cell vaccines that target the Spike protein cell fusion mechanisms have the highest chance of raising neutralizing antibodies with minimal or no ADE risk due to antibody binding sterically blocking cell fusion. Antibodies targeting other portions of the Spike protein or other SARS-CoV-2 exposed proteins may enable infection of phagocytic immune cells even if they are neutralizing.
T cell vaccines that target SARS-CoV-2 replicase proteins have the highest change of avoiding viral escape by antigenic variation and accumulation of mutations in variable residues. Lisziewicz and Lori (94) described an approach for developing a T cell COVID-19 vaccine. EpiVax EPV-CoV19 (95) is an example COVID-19 T cell vaccine.
Given past data on multiple SARS-CoV-1 and MERS-CoV vaccine efforts have failed due to ADE in animal models (75, 81), it is reasonable to hypothesize a similar ADE risk for SARS-CoV-2 antibodies and vaccines. ADE risks may be associated with antibody level (which can wane over time after vaccination) and also if the antibodies are derived from prior exposures to other coronaviruses. In addition, ADE with mast cells likely plays a role in MIS-C for infants and possibly older MIS-C and MIS-A patients. While expanded trophism of SARS-CoV-2 represents a possible ADE risk in the subset of COVID-19 patients with disease progression beyond the mild disease stage.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
DR conceived of the presented ideas, analyzed the data, and wrote the manuscript.
This material is based upon work supported by the Under Secretary of Defense for Research and Engineering under Air Force Contract No. FA8702-15-D-0001. Any opinions, findings, conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the Under Secretary of Defense for Research and Engineering.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledges the Department of Defense(DoD), Defense Threat Reduction Agency(DTRA), and The Joint Science and Technology Office(JSTO) of the Chemical and Biological Defense Program (CBDP) for their support under the Discovery of Medical countermeasures Against Novel Entities (DOMANE) initiative. We acknowledges Nora Smith for literature search assistance and Irene Stapleford for graphic art assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.640093/full#supplementary-material
1. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. (2020) 92:418–23. doi: 10.1002/jmv.26234
2. Coronavirus COVID-19 Global Cases by Johns Hopkins CSSE. Available online at: https://gisanddata.maps.arcgis.com/apps/opsdashboard/index.html#/bda7594740fd40299423467b48e9ecf6 (accessed January 28, 2021).
3. Yan X, Hao Q, Mu Y, Timani KA, Ye L, Zhu Y, et al. Nucleocapsid protein of SARS-CoV activates the expression of cyclooxygenase-2 by binding directly to regulatory elements for nuclear factor-kappa B and CCAAT/enhancer binding protein. Int J Biochem Cell Biol. (2006) 38:1417–28. doi: 10.1016/j.biocel.2006.02.003
4. Hong W, Chen Y, You K, Tan S, Wu F, Tao J, et al. Celebrex adjuvant therapy on COVID-19: an experimental study. Front Pharmacol. (2020) 11:1795. doi: 10.1101/2020.05.05.20077610
5. Tomera K, Malone R, Kittah J. Hospitalized COVID-19 patients treated with celecoxib and high dose famotidine adjuvant therapy show significant clinical responses. SSRN [Preprint]. (2020) doi: 10.2139/ssrn.3646583
6. Malone RW, Tisdall P, Fremont-Smith P, Liu Y, Huang X-P, White KM, et al. COVID-19: famotidine, histamine, mast cells, and mechanisms. Res Square [Preprint]. (2020) doi: 10.21203/rs.3.rs-30934/v1
7. Chu H, Zhou J, Wong BH-Y, Li C, Chan JF-W, Cheng Z-S, et al. Middle east respiratory syndrome coronavirus efficiently infects human primary t lymphocytes and activates the extrinsic and intrinsic apoptosis pathways. J Infect Dis. (2015) 213:904–14. doi: 10.1093/infdis/jiv380
8. Zhou J, Chu H, Chan JF-W, Yuen K-Y. Middle East respiratory syndrome coronavirus infection: virus-host cell interactions and implications on pathogenesis. Virol J. (2015) 12:218. doi: 10.1186/s12985-015-0446-6
9. Yasui F, Kohara M, Kitabatake M, Nishiwaki T, Fujii H, Tateno C, et al. Phagocytic cells contribute to the antibody-mediated elimination of pulmonary-infected SARS coronavirus. Virology. (2014) 454-455:157–68. doi: 10.1016/j.virol.2014.02.005
10. Huang K-J, Su I-J, Theron M, Wu Y-C, Lai S-K, Liu C-C, et al. An interferon-γ-related cytokine storm in SARS patients. J Med Virol. (2005) 75:185–94. doi: 10.1002/jmv.20255
11. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. (2012) 76:16–32. doi: 10.1128/MMBR.05015-11
12. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. (2017) 39:529–39. doi: 10.1007/s00281-017-0629-x
13. Wong RSM, Wu A, To KF, Lee N, Lam CWK, Wong CK, et al. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. BMJ. (2003) 326:1358–62. doi: 10.1136/bmj.326.7403.1358
14. Li T, Qiu Z, Zhang L, Han Y, He W, Liu Z, et al. Significant changes of peripheral t lymphocyte subsets in patients with severe acute respiratory syndrome. J Infect Dis. (2004) 189:648–51. doi: 10.1086/381535
15. Yang Y, Xiong Z, Zhang S, Yan Y, Nguyen J, Ng B, et al. Bcl-xL inhibits T-cell apoptosis induced by expression of SARS coronavirus E protein in the absence of growth factors. Biochem J. (2005) 392(Pt 1):135–43. doi: 10.1042/BJ20050698
16. Li G, Fan Y, Lai Y, Han T, Li Z, Zhou P, et al. Coronavirus infections and immune responses. J Med Virol. (2020) 92:424–32. doi: 10.1002/jmv.25685
17. Yip MS, Leung NHL, Cheung CY, Li PH, Lee HHY, Daëron M, et al. Antibody-dependent infection of human macrophages by severe acute respiratory syndrome coronavirus. Virol J. (2014) 11:82. doi: 10.1186/1743-422X-11-82
18. Gu J, Korteweg C. Pathology and pathogenesis of severe acute respiratory syndrome. Am J Pathol. (2007) 170:1136–47. doi: 10.2353/ajpath.2007.061088
19. Tai W, He L, Zhang X, Pu J, Voronin D, Jiang S, et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cel Mol Immunol. (2020) 17:613–20. doi: 10.1038/s41423-020-0400-4
20. Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. (2020) 370:856. doi: 10.1126/science.abd2985
22. Kimura M. Evolutionary rate at the molecular level. Nature. (1968) 217:624–6. doi: 10.1038/217624a0
23. Bottema CDK, Ketterling RP, Li S, Yoon H-S, III JAP, Sommer SS. Missense mutations and evolutionary conservation of amino acids: evidence that many of the amino acids in factor IX function as “spacer” elements. Am J Hum Genet. (1991) 49:820–38.
24. Ricke DO. Divergence model of protein evolution. bioRxiv [Preprint]. (2016). doi: 10.1101/045930
25. Maier HJ, Britton P. Involvement of autophagy in coronavirus replication. Viruses. (2012) 4:3440–51. doi: 10.3390/v4123440
26. Ricke DO, Gherlone N, Fremont-Smith P, Tisdall P, Fremont-Smith M. Kawasaki disease, multisystem inflammatory syndrome in children: antibody-induced mast cell activation hypothesis. J Pediatrics Pediatr Med. (2020) 4:1–7. doi: 10.29245/2578-2940/2020/2.1157
27. Ricke DO, Shcherbina A. Dawn: rapid large-scale protein multiple sequence alignment and conservation analysis. In: IEEE High Performance Extreme Computing Conference (HPEC). Waltham, MA (2015). doi: 10.1109/HPEC.2015.7322463
28. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. (2020) 2020:eabb2507. doi: 10.1101/2020.02.11.944462
29. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. (2000) 28:235–42. doi: 10.1093/nar/28.1.235
30. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. (2004) 25:1605–12. doi: 10.1002/jcc.20084
31. Liu A. First Moderna, Now Pfizer-BioNTech Working on Booster Shot Amid Rise of COVID-19 Variants. (2021). Available online at: https://www.fiercepharma.com/pharma/first-moderna-now-pfizer-biontech-also-working-booster-shot-amid-rise-covid-19-variants (accessed January 28, 2021).
32. Xu X, Chen P, Wang J, Feng J, Zhou H, Li X, et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. (2020) 63:457–60. doi: 10.1007/s11427-020-1637-5
33. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for lineage B β-coronaviruses, including 2019-nCoV. Nat Microbiol. (2020) 5:562–9. doi: 10.1038/s41564-020-0688-y
34. Jaume M, Yip MS, Cheung CY, Leung HL, Li PH, Kien F, et al. Anti-severe acute respiratory syndrome coronavirus spike antibodies trigger infection of human immune cells via a pH- and cysteine protease-independent FcγR pathway. J Virol. (2011) 85:10582–97. doi: 10.1128/JVI.00671-11
35. Gao J, Lu G, Qi J, Li Y, Wu Y, Deng Y, et al. Structure of the fusion core and inhibition of fusion by a heptad repeat peptide derived from the S protein of Middle East respiratory syndrome coronavirus. J Virol. (2013) 87:13134–40. doi: 10.1128/JVI.02433-13
36. Wang FS, Chu FL, Jin L, Li YG, Zhang Z, Xu D, et al. Acquired but reversible loss of erythrocyte complement receptor 1 (CR1, CD35) and its longitudinal alteration in patients with severe acute respiratory syndrome. Clin Exp Immunol. (2005) 139:112–9. doi: 10.1111/j.1365-2249.2005.02681.x
37. van Erp EA, van Kasteren PB, Guichelaar T, Ahout IML, de Haan CAM, Luytjes W, et al. In vitro enhancement of respiratory syncytial virus infection by maternal antibodies does not explain disease severity in infants. J Virol. (2017) 91:e00851–17. doi: 10.1128/JVI.00851-17
38. Smatti MK, Al Thani AA, Yassine HM. Viral-induced enhanced disease illness. Front Microbiol. (2018) 9:2991. doi: 10.3389/fmicb.2018.02991
39. Jares Baglivo S, Polack FP. The long road to protect infants against severe RSV lower respiratory tract illness. F1000Res. (2019) 8:F1000 Faculty Rev−610. doi: 10.12688/f1000research.18749.1
40. Winarski KL, Tang J, Klenow L, Lee J, Coyle EM, Manischewitz J, et al. Antibody-dependent enhancement of influenza disease promoted by increase in hemagglutinin stem flexibility and virus fusion kinetics. Proc Natl Acad Sci USA. (2019) 116:15194. doi: 10.1073/pnas.1821317116
42. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
43. Guan W-J, Ni Z-Y, Hu Y, Liang W-H, Ou C-Q, He J-X, et al. Clinical characteristics of 2019 novel coronavirus infection in China. N Engl J Med. (2020) 382:1708–20. doi: 10.1101/2020.02.06.20020974
44. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. (2003) 426:450–4. doi: 10.1038/nature02145
45. Jeffers SA, Tusell SM, Gillim-Ross L, Hemmila EM, Achenbach JE, Babcock GJ, et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc Natl Acad Sci USA. (2004) 101:15748. doi: 10.1073/pnas.0403812101
46. Liu H, Yu W, Liou L-Y, Rice AP. Isolation and characterization of the human DC-SIGN and DC-SIGNR promoters. Gene. (2003) 313:149–59. doi: 10.1016/S0378-1119(03)00674-7
47. Chen J, Lau YF, Lamirande EW, Paddock CD, Bartlett JH, Zaki SR, et al. cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J Virol. (2010) 84:1289. doi: 10.1128/JVI.01281-09
48. Zhao J, Zhao J, Van Rooijen N, Perlman S. Evasion by stealth: inefficient immune activation underlies poor T cell response and severe disease in SARS-CoV-infected mice. PLoS Pathog. (2009) 5:e1000636. doi: 10.1371/journal.ppat.1000636
49. Liu L, Wei Q, Lin Q, Fang J, Wang H, Kwok H, et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight. (2019) 4:e123158. doi: 10.1172/jci.insight.123158
50. Chen y, Feng Z, Diao B, Wang R, Wang G, Wang C, et al. The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) directly decimates human spleens and lymph nodes. medRxiv [Preprint]. (2020) doi: 10.1101/2020.03/27.20045427
51. Boumaza A, Gay L, Mezouar S, Diallo AB, Michel M, Desnues B, et al. Monocytes and macrophages, targets of SARS-CoV-2: the clue for Covid-19 immunoparalysis. bioRxiv [Preprint]. (2020). doi: 10.1101/2020.09.17.300996
52. Dandekar AA, Perlman S. Immunopathogenesis of coronavirus infections: implications for SARS. Nat Rev Immunol. (2005) 5:917–27. doi: 10.1038/nri1732
53. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type i interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. (2016) 19:181–93. doi: 10.1016/j.chom.2016.01.007
54. Wan Y, Shang J, Sun S, Tai W, Chen J, Geng Q, et al. Molecular Mechanism for Antibody-Dependent Enhancement of Coronavirus Entry. J Virol. (2020) 94:e02015–19. doi: 10.1128/JVI.02015-19
55. Guzman MG, Alvarez M, Rodriguez-Roche R, Bernardo L, Montes T, Vazquez S, et al. Neutralizing antibodies after infection with dengue 1 virus. Emerg Infect Dis. (2007) 13:282–6. doi: 10.3201/eid1302.060539
56. Dejnirattisai W, Jumnainsong A, Onsirisakul N, Fitton P, Vasanawathana S, Limpitikul W, et al. Cross-reacting antibodies enhance dengue virus infection in humans. Science. (2010) 328:745. doi: 10.1126/science.1185181
57. Katzelnick LC, Gresh L, Halloran ME, Mercado JC, Kuan G, Gordon A, et al. Antibody-dependent enhancement of severe dengue disease in humans. Science. (2017) 358:929. doi: 10.1126/science.aan6836
58. Yeh C-S, Yang J-Y, Liu W-T, Huang JC, Chen Y-MA, Wang S-F. SARS coronavirus has antibody-dependent enhancement (ADE) effect through the autologous antibodies against envelope spikes on Fcγ receptor expressing cells. J Virus Erad. (2016) 2:48. doi: 10.1016/S2055-6640(20)31216-4
59. Tillett RL, Sevinsky JR, Hartley PD, Kerwin H, Crawford N, Gorzalski A, et al. Genomic evidence for reinfection with SARS-CoV-2: a case study. Lancet Infect Dis. (2020) S1473-3099:30764-7. doi: 10.2139/ssrn.3680955
60. Jackson LA, Anderson EJ, Rouphael NG, Roberts PC, Makhene M, Coler RN, et al. An mRNA vaccine against SARS-CoV-2 — preliminary report. N Engl J Med. (2020) 383:1920–31. doi: 10.1056/NEJMoa2022483
61. Lee N, Chan PKS, Ip M, Wong E, Ho J, Ho C, et al. Anti-SARS-CoV IgG response in relation to disease severity of severe acute respiratory syndrome. J Clin Virol. (2006) 35:179–84. doi: 10.1016/j.jcv.2005.07.005
62. Liu X, Wang J, Xu X, Liao G, Chen Y, Hu C-H. Patterns of IgG and IgM antibody response in COVID-19 patients. Emerg Microbes Infect. (2020) 9:1269–74. doi: 10.1080/22221751.2020.1773324
63. Chen W, Zhang J, Qin X, Wang W, Xu M, Wang L-F, et al. SARS-CoV-2 neutralizing antibody levels are correlated with severity of COVID-19 pneumonia. Biomed Pharmacother. (2020) 130:110629. doi: 10.1016/j.biopha.2020.110629
64. Young BE, Ong SWX, Ng LFP, Anderson DE, Chia WN, Chia PY, et al. Viral dynamics and immune correlates of COVID-19 disease severity. Clin Infect Dis. (2020) 2020:ciaa1280. doi: 10.1093/cid/ciaa1280
65. Pujadas E, Chaudhry F, McBride R, Richter F, Zhao S, Wajnberg A, et al. SARS-CoV-2 viral load predicts COVID-19 mortality. Lancet Respir Med. (2020) 8:e70. doi: 10.1016/S2213-2600(20)30354-4
66. Luo YR, Chakraborty I, Yun C, Wu AHB, Lynch KL. Kinetics of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) antibody avidity maturation and association with disease severity. Clin Infect Dis. (2020). doi: 10.1093/cid/ciaa1389. [Epub ahead of print].
67. Fajnzylber J, Regan J, Coxen K, Corry H, Wong C, Rosenthal A, et al. SARS-CoV-2 viral load is associated with increased disease severity and mortality. Nat Commun. (2020) 11:5493. doi: 10.21203/rs.3.rs-43878/v1
68. Wu F, Yan R, Liu M, Liu Z, Wang Y, Luan D, et al. Antibody-dependent enhancement (ADE) of SARS-CoV-2 infection in recovered COVID-19 patients: studies based on cellular and structural biology analysis. medRxiv [Preprint]. (2020). doi: 10.1101/2020.10.08.20209114
69. Tkaczyk C, Okayama Y, Woolhiser MR, Hagaman DD, Gilfillan AM, Metcalfe DD. Activation of human mast cells through the high affinity IgG receptor. Mol Immunol. (2002) 38:1289–93. doi: 10.1016/S0161-5890(02)00077-9
70. Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. (2020) doi: 10.1001/jamacardio.2020.0950
71. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. (2020) 5:811–18. doi: 10.1001/jamacardio.2020.1017
72. Jordan J. MIS-C cases in children connected to COVID-19 surface in Northeast Ohio 2021 Available online at: https://fox8.com/news/coronavirus/inflammatory-syndrome-cases-in-children-connected-to-covid-19-surface-in-northeast-ohio/ (accessed February 2, 2021).
73. Sedova ES, Scherbinin DN, Lysenko AA, Alekseeva SV, Artemova EA, Shmarov MM. Non-neutralizing antibodies directed at conservative influenza antigens. Acta Naturae. (2019) 11:22–32. doi: 10.32607/20758251-2019-11-4-22-32
74. Khurana S, Loving CL, Manischewitz J, King LR, Gauger PC, Henningson J, et al. Vaccine-Induced Anti-HA2 antibodies promote virus fusion and enhance influenza virus respiratory disease. Sci Transl Med. (2013) 5:200ra114. doi: 10.1126/scitranslmed.3006366
75. Tseng C-T, Sbrana E, Iwata-Yoshikawa N, Newman PC, Garron T, Atmar RL, et al. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS ONE. (2012) 7:e35421. doi: 10.1371/journal.pone.0035421
76. Bolles M, Deming D, Long K, Agnihothram S, Whitmore A, Ferris M, et al. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J Virology. (2011) 85:12201–15. doi: 10.1128/JVI.06048-11
77. Yasui F, Kai C, Kitabatake M, Inoue S, Yoneda M, Yokochi S, et al. Prior immunization with severe acute respiratory syndrome (SARS)-associated coronavirus (SARS-CoV) nucleocapsid protein causes severe pneumonia in mice infected with SARS-CoV. J Immunol. (2008) 181:6337. doi: 10.4049/jimmunol.181.9.6337
78. Weingartl H, Czub M, Czub S, Neufeld J, Marszal P, Gren J, et al. Immunization with modified vaccinia virus ankara-based recombinant vaccine against severe acute respiratory syndrome is associated with enhanced hepatitis in ferrets. J Virol. (2004) 78:12672. doi: 10.1128/JVI.78.22.12672-12676.2004
79. Wang Q, Zhang L, Kuwahara K, Li L, Liu Z, Li T, et al. Immunodominant SARS coronavirus epitopes in humans elicited both enhancing and neutralizing effects on infection in non-human primates. ACS Infect Dis. (2016) 2:361–76. doi: 10.1021/acsinfecdis.6b00006
80. Wang S-F, Tseng S-P, Yen C-H, Yang J-Y, Tsao C-H, Shen C-W, et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem Biophys Res Commun. (2014) 451:208–14. doi: 10.1016/j.bbrc.2014.07.090
81. Agrawal AS, Tao X, Algaissi A, Garron T, Narayanan K, Peng B-H, et al. Immunization with inactivated Middle East Respiratory Syndrome coronavirus vaccine leads to lung immunopathology on challenge with live virus. Hum Vaccin Immunother. (2016) 12:2351–6. doi: 10.1080/21645515.2016.1177688
82. Rauch S, Jasny E, Schmidt KE, Petsch B. New vaccine technologies to combat outbreak situations. Front Immunol. (2018) 9:1963. doi: 10.3389/fimmu.2018.01963
83. Ko J-H, Müller MA, Seok H, Park GE, Lee JY, Cho SY, et al. Serologic responses of 42 MERS-coronavirus-infected patients according to the disease severity. Diagn Microbiol Infect Dis. (2017) 89:106–11. doi: 10.1016/j.diagmicrobio.2017.07.006
84. Peiris JSM, Chu CM, Cheng VCC, Chan KS, Hung IFN, Poon LLM, et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. (2003) 361:1767–72. doi: 10.1016/S0140-6736(03)13412-5
85. Hsueh P-R, Hsiao C-H, Yeh S-H, Wang W-K, Chen P-J, Wang J-T, et al. Microbiologic characteristics, serologic responses, and clinical manifestations in severe acute respiratory syndrome, Taiwan. Emerg Infect Dis. (2003) 9:1163–7. doi: 10.3201/eid0909.030367
86. May B. Regeneron Halts Enrollment of Critically Ill Patients in COVID-19 Antibody Trial BioSpace. (2020). Avilable online at: https://www.biospace.com/article/regeneron-halts-enrollment-in-covid-19-trial-following-safety-signal-in-critically-ill-patients/ (accessed January 28, 2021).
87. Li L, Zhang W, Hu Y, Tong X, Zheng S, Yang J, et al. Effect of convalescent plasma therapy on time to clinical improvement in patients with severe and life-threatening covid-19: a randomized clinical trial. JAMA. (2020) 324:460–70. doi: 10.1001/jama.2020.12607
88. Agarwal A, Mukherjee A, Kumar G, Chatterjee P, Bhatnagar T, Malhotra P. Convalescent plasma in the management of moderate covid-19 in adults in India: open label phase II multicentre randomised controlled trial (PLACID Trial). BMJ. (2020) 371:m3939. doi: 10.1136/bmj.m3939
89. Cheng Y, Wong R, Soo YOY, Wong WS, Lee CK, Ng MHL, et al. Use of convalescent plasma therapy in SARS patients in Hong Kong. Eur J Clin Microbiol Infect Dis. (2005) 24:44–6. doi: 10.1007/s10096-004-1271-9
90. Yuan Y, Cao D, Zhang Y, Ma J, Qi J, Wang Q, et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat Commun. (2017) 8:15092. doi: 10.1038/ncomms15092
91. Lip K-M, Shen S, Yang X, Keng C-T, Zhang A, Oh H-LJ, et al. Monoclonal antibodies targeting the HR2 domain and the region immediately upstream of the HR2 of the S protein neutralize in vitro infection of severe acute respiratory syndrome coronavirus. J Virol. (2006) 80:941. doi: 10.1128/JVI.80.2.941-950.2006
92. Tripet B, Kao DJ, Jeffers SA, Holmes KV, Hodges RS. Template-based coiled-coil antigens elicit neutralizing antibodies to the SARS-coronavirus. J Struct Biol. (2006) 155:176–94. doi: 10.1016/j.jsb.2006.03.019
93. Keng ECT, Zhang A, Shen S, Lip K-M, Fielding B, Tan T, et al. Amino acids 1055 to 1192 in the s2 region of severe acute respiratory syndrome coronavirus s protein induce neutralizing antibodies: implications for the development of vaccines and antiviral agents. J Virol. (2005) 79:3289–96. doi: 10.1128/JVI.79.6.3289-3296.2005
94. Lisziewicz J, Lori F. Precision COVID-19 vaccine with companion diagnostics. Prec Nanomed. (2020) 3:487–94. doi: 10.33218/001c.12561
95. EPV-CoV19 EpiVax's T Cell Epitope-Driven vaccine for COVID-19 (2020). Available online at: https://epivax.com/pipeline/epv-cov19 (accessed January 28, 2021).
Keywords: antibody dependent enhancement, ADE, COVID-19, SARS-CoV-2, multisystem inflammatory syndrome, MIS-C, antibody dependent enhancement
Citation: Ricke DO (2021) Two Different Antibody-Dependent Enhancement (ADE) Risks for SARS-CoV-2 Antibodies. Front. Immunol. 12:640093. doi: 10.3389/fimmu.2021.640093
Received: 10 December 2020; Accepted: 03 February 2021;
Published: 24 February 2021.
Edited by:
Michael Vajdy, EpitoGenesis, United StatesReviewed by:
Gregor Ebert, Walter and Eliza Hall Institute of Medical Research, AustraliaCopyright © 2021 Ricke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Darrell O. Ricke, ZGFycmVsbC5yaWNrZUBsbC5taXQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.